

ABSTRACT
Cyclins associate with cyclin-dependent serine/threonine kinase 1 (CDK1) to generate the M phase-promoting factor (MPF) activity essential for progression through mitosis and meiosis. Although cyclin B1 (CCNB1) is required for embryo development, previous studies concluded that CCNB2 is dispensable for cell cycle progression. Given previous findings of high Ccnb2 mRNA translation rates in prophase-arrested oocytes, we re-evaluated the role of this cyclin during meiosis. Ccnb2−/− oocytes underwent delayed germinal vesicle breakdown and showed defects during the metaphase-to-anaphase transition. This defective maturation was associated with compromised Ccnb1 and Moloney sarcoma oncogene (Mos) mRNA translation, delayed spindle assembly and increased errors in chromosome segregation. Given these defects, a significant percentage of oocytes failed to complete meiosis I because the spindle assembly checkpoint remained active and anaphase-promoting complex/cyclosome function was inhibited. In vivo, CCNB2 depletion caused ovulation of immature oocytes, premature ovarian failure, and compromised female fecundity. These findings demonstrate that CCNB2 is required to assemble sufficient pre-MPF for timely meiosis re-entry and progression. Although endogenous cyclins cannot compensate, overexpression of CCNB1/2 rescues the meiotic phenotypes, indicating similar molecular properties but divergent modes of regulation of these cyclins.
KEY WORDS: Cyclins, Fertility, MPF, Meiosis, Mouse oocytes
Summary: The altered timing of maturation caused by CyclinB2 loss results in aneuploidy, ovulation of immature oocytes and decreased fecundity, indicating that CyclinB2 is essential for oocyte re-entry and progression through meiosis.
INTRODUCTION
Successful completion of the two meiotic cell divisions is essential for gamete development and fertility. Re-entry of fully grown oocytes into meiosis requires assembly of the M phase-promoting factor (MPF) and activation of its kinase activity (Masui and Markert, 1971). This complex subsequently phosphorylates a large number of protein substrates, triggering dissolution of the nuclear membrane, chromosome condensation and spindle assembly (Morgan, 2007). Once proper chromosome-to-microtubule attachment is attained, rapid inactivation of MPF is necessary for the transition to anaphase. This inactivation depends on the switch-like activation of the anaphase-promoting complex/cyclosome (APC/C), followed by ubiquitylation and degradation of cyclins and, therefore, inactivation of MPF (Thornton and Toczyski, 2006; Yang and Ferrell, 2013). Concomitant degradation of securin leads to activation of separase, which cleaves cohesins, allowing separation of the bivalents in anaphase (Lane et al., 2012). Given the asymmetrical position of the spindle in oocytes, telophase results in the extrusion of a small polar body.
MPF comprises two classes of proteins that orchestrate progression through both M-phases of mitosis and meiosis: a family of cyclin-dependent serine/threonine kinases (CDKs) and their binding partners, cyclins (Morgan, 2007). Although three M-phase Ccnb mRNAs are present in mammals (Ccnb1, Ccnb2 and Ccnb3), most of the molecular properties of the CDK1/CCNB complex are based on observations of the CDK1/CCNB1 heterodimer. However, other cyclins, such as CCNB2, also interact with CDK1, activating its phosphotransferase activity (Jackman et al., 1995). In somatic cells, CCNB1 and B2 are thought to be localized in different subcellular compartments (Jackman et al., 1995). Whereas CCNB1 is either soluble or interacts with microtubules, CCNB2 is associated with the cellular membrane. During mitosis, CCNB1 translocates into the nucleus, whereas CCNB2 remains sequestered in the cytoplasm (Jackman et al., 1995).
Although all three cyclin mRNAs are detected in mouse oocytes, the CCNB1/CDK1 complex is generally regarded as the major driver of meiosis progression. Little is known about the role of CCNB3 during oocyte maturation, with the exception of one report that suggests its requirement in meiosis I (Zhang et al., 2015). Early attempts to define CCNB2 function either by genetic inactivation of the gene or by knockdown with antisense RNAs did not produce clear phenotypes, leading to the conclusion that CCNB2 is dispensable for both mitosis and meiosis, possibly because of compensation by CCNB1 (Brandeis et al., 1998; Ledan et al., 2001). However, recent findings repropose a role for CCNB2 during mouse meiotic divisions. In experiments investigating the function of the spindle component NDC80/HEC1 during meiotic prophase, knockdown of CCNB2 with morpholino oligonucleotides (MO) markedly decreased mouse oocyte meiotic re-entry (Gui and Homer, 2013). Similar findings were reported in a recent study investigating CCNB1 function in oocytes (Li et al., 2018). Recent data also demonstrated that the rates of translation of the two major CCNBs, B1 and B2, are regulated in distinct fashions during meiotic prophase (Han et al., 2017). Ccnb1 mRNA is expressed with three distinct 3′-untranslated regions (UTRs) of different lengths; translation of the two longer mRNA variants is repressed during meiotic prophase and a third, short variant, is constitutively translated (Yang et al., 2017). CCNB1 protein is detectable in meiotic prophase, albeit at levels that are low compared with M-phase. During prometaphase, the translation of the two longer variants is activated and drives the large accumulation of the CCNB1 protein (Yang et al., 2017). Although two Ccnb2 mRNA variants are also detected, the rate of translation of Ccnb2 mRNA is high during prophase I and the protein is readily detectable, a feature that is reminiscent of that reported in frog oocytes (Piqué et al., 2008).
The above findings suggest that CCNB2 is considerably more abundant than CCNB1 in prophase I germinal vesicle (GV)-arrested oocytes. Prompted by these observations, we further investigated the function of CCNB2 during mouse oocyte meiotic progression. Using previously generated Ccnb2−/− mice (Brandeis et al., 1998), we show that oocytes deficient in CCNB2 have compromised nuclear maturation, resulting in the subfertility of these mice. Thus, we conclude that CCNB2 has a significant role during mouse oocyte meiosis, which cannot be compensated for by endogenous CCNB1.
RESULTS
Distinct Ccnb1 and Ccnb2 mRNA translation rates define the accumulation pattern of the two cyclins at the prophase-to-metaphase transition
We previously reported that the patterns of Ccnb1 and Ccnb2 mRNA loading onto ribosomes in full-grown mouse oocytes differ considerably (Han et al., 2017). Here, we confirm and extend this initial observation with a detailed time-course experiment monitoring the loading of ribosomes onto these two mRNAs in GV-arrested oocytes and during progression through metaphase I (MI) (Fig. 1A,B). The overall mRNA levels for the two cyclins were comparable and stable (Fig. 1A,B). However, although little Ccnb1 mRNA loading onto ribosomes was detected in GV-arrested oocytes, Ccnb2 mRNA loading was considerably higher. These indirect measures of translation were corroborated by mining data sets assessing the poly(A)-tail length of mRNAs in GV-arrested oocytes (Morgan et al., 2017). Ccnb2 mRNA had a significantly longer poly(A)-tail compared with Ccnb1 (Fig. 1C), and increased poly(A)-tail length is associated with increased rates of translation (Tay et al., 2000; Reyes and Ross, 2016).
Fig. 1.

Translation of Ccnb1 and Ccnb2 mRNAs is differentially regulated during meiotic maturation in mouse oocytes. (A,B) RNA-Seq was performed using mRNA extracts from total cell lysate (total mRNA) or after immunoprecipitation of HA-tagged ribosomes (ribosome-bound mRNA) from oocytes arrested in prophase with cilostamide (time 0) or collected 2, 4, 6 and 8 h after meiotic resumption. Counts per million (CPM) of mapped reads are reported for Ccnb1 (A) and Ccnb2 (B); average CPMs of two independent biological replicates with range are reported. (C) Poly(A) tail lengths of the Ccnb1 and Ccnb2 mRNAs in GV oocytes. The data were mined from PMID: 28792939 and are reported as binned values of up to 80 (A) nucleotides. (D) Rates of translation of Ccnb1 and Ccnb2 mRNA variants in prophase I. Oocytes were injected with a 1:1 mix of oligo-adenylated YFP-(Ccnb2-short, Ccnb2-long, Ccnb1-short or Ccnb1-long) 3′UTR and polyadenylated mCherry. Rate of translation in GV-arrested oocytes were calculated with a 3 h window at a sampling rate of 15 min. Student's t-tests were performed for statistical significance; ***P<0.0001. (E) Translational efficiency of Ccnb1 and Ccnb2 mRNA was calculated by dividing the CPMs of ribosome-bound mRNA by the CPMs of total mRNA. Four biological replicates were used for these calculations. (F) To evaluate the absolute concentration of cyclins in GV-arrested oocytes, western blots were performed using cell lysates and cyclin levels were quantified by interpolating from a standard curve of known concentrations of CCNB1 and CCNB2 recombinant proteins. Calculated concentrations are reported as the mean and s.d. of three independent biological replicates. ns, not significant.
During meiotic progression, few or no changes in Ccnb2 message loading onto the ribosomes were detected up to MI, whereas a major increase in Ccnb1 mRNA ribosome association occurred during MI (Fig. 1A). This differential pattern of translation was in good accordance with data from previous experiments using luciferase reporters, including with the 3′UTRs of the two mRNAs (Han et al., 2017). We also have shown that alternate polyadenylation signal usage (APA) has a major role in defining the 3′UTR length and translation rate of Ccnb1 mRNA (Yang et al., 2017). Given that the Ccnb2 mRNA 3′UTR also contains an internal polyadenylation signal, we compared the translation rate of the two 3′UTR variants. Ccnb1 3′UTR short and long constructs were used as a control. The rates of translation of the two Ccnb2 3′UTR reporters were comparable in GV-arrested oocytes (Fig. 1D). However, the rate of translation driven by the long Ccnb1 3′UTR was considerably lower than that of either Ccnb2 3′UTR (Fig. 1D). Only the rate of translation of the short Ccnb1 3′UTR approximated those of either Ccnb2 3′UTR. These rates were in good agreement with the calculated translation efficiency of the mRNA, defined as the amount of mRNA bound to the ribosome divided by the total amount of mRNA (Fig. 1E). Given that previous experiments indicated comparable degradation rates of the two proteins during prophase I (Han et al., 2017), we hypothesized that CCNB2 accumulates in GV-arrested oocytes at higher levels than does CCNB1. We quantified the amount of CCNB1 and CCNB2 protein in GV-arrested oocytes by interpolating from a standard curve constructed with known concentrations of recombinant protein (Fig. S1A,B) and found levels of CCNB2 to be five times higher than those of CCNB1 (CCNB1: 7.4 pg/oocyte and CCNB2: 38.2 pg/oocyte) (Fig. 1F). During oocyte maturation, CCNB1 accumulation increased, whereas CCNB2 levels remained relatively unchanged, suggesting a shift in the stoichiometry of the CCNB/CDK1 complex during meiotic progression.
Ccnb2−/− female mice display decreased fecundity
Together with a previous report (Gui and Homer, 2013), the above findings are at odds with the widely held notion that CCNB2 is dispensable for oocyte maturation. Therefore, we re-evaluated the fertility phenotype of the previously generated Ccnb2−/− mice (Brandeis et al., 1998).
Whereas both Ccnb2+/− and Ccnb2−/− mice were fertile and produced pups, Ccnb2−/− mice had decreased fecundity (Fig. 2A). Fecundity declined rapidly with age, suggesting premature ovarian failure (Fig. 2A and Fig. S1C,D). This phenotype was not the result of the embryonic lethality of the Ccnb2−/− pups because there was no statistically significant deviation from the expected Mendelian ratio when heterozygous males and females were mated (Fig. S1E), suggesting that CCNB2 is dispensable for embryo development. Furthermore, Ccnb2−/− females generated fewer pups even when crossed with Ccnb2+/+ males, indicating that the subfertility was associated with the female. Although there was no statistical difference between the number of pups generated by crossing Ccnb2−/− females with Ccnb2+/+ or Ccnb2−/− males, there was a tendency for homozygous mating to produce fewer pups. This suggest a role for CCNB2 in male spermatogenesis, which has previously been proposed (Tang et al., 2017). The female fecundity phenotype might be caused by delayed puberty and/or ovarian or oocyte dysfunction. Delayed puberty was ruled out because the pregnancy rates of mated Ccnb2−/− females did not improve over a 9-month period. Furthermore, the age at first pregnancy of Ccnb2+/+ and Ccnb2−/− females was comparable (Fig. S1F). Consistent with the original report (Brandeis et al., 1998), whereas Ccnb2+/− pups were indistinguishable from their Ccnb2+/+ littermates, the Ccnb2−/− pups were significantly smaller, weighing an average of 1.4±0.35 g less than their Ccnb2+/+ siblings at 21 days of age (Fig. 2B).
Fig. 2.

Ccnb2−/− mice show compromised fecundity. (A) Cumulative number of pups per female derived from different mating schemes. Mating schemes and number of pairs were as follows: +/+♂×+/+♀, n=20; +/−♂×+/−♀, n=35; −/−♂×−/−♀, n=6; +/+♂×−/−♀, n=6. Student's t-tests were performed between +/−♂×+/−♀ and −/−♂×−/−♀ (red asterisks) or +/−♂×+/−♀ and +/+♂×−/−♀ (blue asterisks); each point is the mean±s.e.m. of the number of pups obtained; **P<0.01, ***P<0.001. Breeding was initiated when the mice reached 8 weeks of age. (B) Pup body weights from +/−♂×+/−♀ matings were recorded 21 days after birth. The weight of each mouse was normalized for the average weight of the litter and plotted according to their genotype as medians with interquartile range. Student's t-tests were performed for statistical significance; ***P<0.0002. (C) Representative histological sections of ovaries from Ccnb2+/+ and Ccnb2−/− mice. Follicle stages were scored and representative pictures of pre-antral, early antral (when multiple cavities have not coalesced in a single antrum), late antral (when a single, large antrum is present) follicles and corpora lutea are shown. (D) Follicle counts were performed on pairs of ovaries from three biological replicates for each group and are plotted as mean±s.d. ns, not significant.
The adult ovarian morphology of Ccnb2−/− mice was unremarkable. Follicles at various developmental stages (e.g. pre-antral, early antral, late antral and corpora lutea) were counted and no differences in follicle number at any stage were found between the two genotypes (Fig. 2C,D). Approximately 48 h after equine chorionic gonadotropin (eCG) injection, ovaries were dissected out, and weighed before oocyte retrieval. There was no difference in ovarian weight or in the number of oocytes retrieved (Fig. S1G,H), suggesting similar responsiveness to gonadotropin stimulation. Also, there was no difference in oocyte diameter between the Ccnb2+/+ and Ccnb2−/− groups (Fig. S1I). Two chromatin conformations have been described for oocytes retrieved from antral follicles: immature nonsurrounded-nucleolus (NSN) and mature nucleolus-surrounded (SN). Although both types are competent to resume meiosis, SN oocytes are developmentally competent (Zuccotti et al., 1998). Quantification of the two conformations showed that >95% of oocytes retrieved from both Ccnb2+/+ and Ccnb2−/− ovaries were SN (Fig. S1J). Thus, all the morphological parameters investigated indicated that Ccnb2−/− oocytes develop normally up to the GV-arrest stage. To further assess ovarian function and monitor follicle maturation and/or ovulation potential, prepubertal females were injected with eCG followed by hCG, a regimen that induces ovulation. Slightly fewer oocytes were retrieved from Ccnb2−/− females (Fig. S1K).
Timing of G2/M transition is aberrant in Ccnb2−/− oocytes
Western blot analysis of Ccnb2+/− and Ccnb2−/− oocyte extracts revealed a gene dosage-dependent decrease in CCNB2 protein (Fig. 3A). Recently, it was reported that oocyte-specific knockout of CCNB1 results in the overexpression of CCNB2 (Li et al., 2018); however, CCNB1 expression was not affected in Ccnb2−/− oocytes (Fig. S2A,B). Also, loss of CCNB2 did not affect CDK1 protein levels, indicating no effect on either the synthesis or stability of the kinase moiety (Fig. S2B). To understand the contribution of CCNB2 to the overall CDK1 activity in GV-arrested oocytes, we measured CDK1 kinase activity using two independent strategies. Extracts from Ccnb2+/+ and Ccnb2−/− oocytes were incubated with a CDK1 substrate glutathione-S-transferase-protein phosphatase 1 (GST-PP1) and phosphorylation was measured by phospho-specific antibodies (pT320-PP1) (Daldello et al., 2015; Lewis et al., 2013). There was a significant decrease in CDK1 activity in Ccnb2−/− oocyte extracts compared with Ccnb2+/+ oocyte extracts (Fig. 3B,C and Fig. S2C). MPF activity was also measured in intact oocytes using a previously described CDK1-FRET reporter assay (Gavet and Pines, 2010a,b). In GV-arrested Ccnb2−/− oocytes, the FRET signal was significantly decreased compared with Ccnb2+/+ oocytes (Fig. S2D).
Fig. 3.
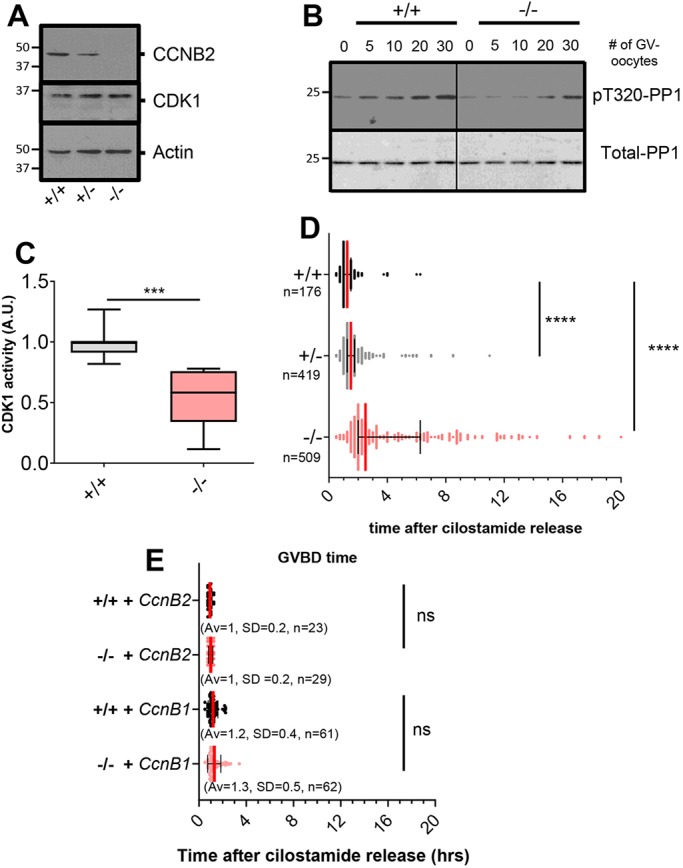
Aberrant timing of meiotic resumption in oocytes depleted of CCNB2 results from defective pre-MPF. (A) Western blot performed on lysates of 150 oocytes from Ccnb2+/+, +/− and −/− mice. (B) Kinase assays were performed using increasing numbers of oocytes from Ccnb2+/+ (+/+) or Ccnb2−/− (−/−) mice and a GST-PP1 fragment as a substrate. T320 PP1 phosphorylation was detected by a phospho-specific antibody (pT320-PP1). The level of total substrate loaded was evaluated by Ponceau S staining (Total-PP1). (C) Quantification of six independent kinase assays. pT320-PP1/Total-PP1 ratios from Ccnb2−/− oocytes were expressed as fold changes over their matched Ccnb2+/+ controls. Signals are expressed as median with interquartile range. A Student's t-test was performed to determine statistical significance; ***P=0.0007. (D) Time of GVBD was determined through brightfield images acquired every 15 min up to 24 h. Times are plotted as medians with interquartile ranges, averages (Av), s.d. and number of oocytes (n) observed from seven independent experiments. A nonparametric Mann–Whitney test was performed to evaluate statistical significance; ****P<0.0001. (E) Oocytes were injected with mRNA encoding either Ccnb1-mCherry or Ccnb2-mCherry and, after 3 h incubation, were released in cilostamide-free medium. GVBD time and statistical significance were determined as in (D); times are reported as medians with interquartile ranges, Av, s.d. and number of oocytes (n) scored from two independent experiments. ns, not significant.
Given that the above data were consistent with decreased pre-MPF activity, we investigated whether spontaneous maturation was affected in Ccnb2−/− oocytes. Whereas Ccnb2+/+ and Ccnb2+/− oocytes resumed meiosis in a synchronous manner [GV breakdown (GVBD) time=1.3±0.7 h and 1.8±1.2 h, respectively, mean±standard deviation (s.d.)], meiotic re-entry of Ccnb2−/− oocytes was significantly delayed (GVBD time=4.4±3.7 h) and highly asynchronous (Bartlett's test, P<0.0001). The time of GVBD of each Ccnb2−/− oocyte was inversely correlated with the endogenous CDK1 activity at GV (Fig. S2E), whereas there was no correlation between GVBD time and oocyte diameter (Fig. S2F). Treatment of Ccnb2+/+ oocytes with the CDK1-specific inhibitor, dinaciclib, revealed a dose-dependent delay and increased variability in GVBD time (Fig. S2G), partially phenocopying the loss of CCNB2. If decreased MPF activity were the sole mechanism for delayed meiotic resumption, overexpression of cyclins should rescue the null phenotype. Indeed, injection of either Ccnb2-mCherry or Ccnb1-mCherry (Fig. 3E and Fig. S2H,I) in Ccnb2−/− oocytes restored the GVBD time to that of Ccnb2+/+ oocytes, and oocytes expressing higher levels of either cyclin underwent GVBD earlier (Fig. S2J). Of note, both cyclin constructs translocated into the nucleus with identical kinetics (Fig. S2K). These findings strongly support the hypothesis that CCNB2 protein accumulation in prophase I is required to generate sufficient CDK activity for timely re-entry into meiosis, and that oocytes with decreased pre-MPF activity resume meiosis in a delayed and asynchronous fashion.
CDK1-dependent, but not PKA-dependent, events are disrupted in Ccnb2−/− oocytes
To define the molecular defects associated with CCNB2 depletion during the G2/M transition, we examined the timing of CDC25B translocation. We previously showed that, in mouse oocytes, CDC25B import into the nucleus is one of the first detectable events following the decrease in cAMP (Oh et al., 2010), the signal that maintains oocyte meiotic arrest (Conti et al., 2002; Mehlmann, 2005). Therefore, we injected oocytes with CDC25B-(phosphatase dead)-Yellow Fluorescent Protein (YFP) to follow the kinetics of CDC25B translocation in intact oocytes (Fig. 4A). As previously described, Ccnb2+/+ oocytes matured synchronously, whereas Ccnb2−/− oocyte maturation was delayed and asynchronous (Fig. S3A). In both groups, CDC25B translocation continued until the oocytes underwent GVBD, at which point CDC25B diffused throughout the cytoplasm (Fig. 4A). There was no significant difference in the rate of CDC25B translocation between Ccnb2+/+ and Ccnb2−/− oocytes (Fig. 4C). Given that GVBD time was delayed in Ccnb2−/− oocytes, CDC25B accumulation in the nucleus continued for longer periods of time, resulting in increased reporter signal in the nucleus (Fig. S3B). This difference in the CDC25B nuclear/cytoplasmic ratio was not because of differences in the amount of reporter expressed (Fig. S3C). These measurements document that the rate of CDC25B import was not affected by the decrease in MPF activity and that PKA downregulation occurred normally in Ccnb2−/− oocytes. Remarkably, CDC25B translocation alone was not sufficient to trigger GVBD in Ccnb2−/− oocytes.
Fig. 4.

The rate of WEE1B but not CDC25 translocation is decreased on CccnB2−/− oocytes. Oocytes were injected with inactive Cdc25B-YFP (A) or Wee1B-YFP (B) and, after overnight incubation, were released in cilostamide-free medium. Brightfield and YFP images were acquired every 5 min for 20 h. (A,B) Images of a Ccnb2+/+ oocyte and of two examples of Ccnb2−/− oocytes. The red box marks the time of GVBD. Scale bars: 20 µm. (C) Rates of CDC25-YFP or (D) WEE1B-YFP translocation were calculated from individual oocytes as the slope of the linear regression of the nuclear/cytoplasmic ratios. Rates are expressed as medians with interquartile range and the number of oocytes from two independent experiments is reported for each condition. Student's t-tests were performed to assess statistical significance; ****P<0.0001. ns, not significant.
CDC25B translocation is followed by CCNB1 import into, and WEE1B (WEE1 homolog 2; WEE2 – Mouse Genome Informatics) export out of, the nucleus preceding GVBD (Oh et al., 2010). Effects on CCNB1 import could not be measured in Ccnb2−/− oocytes because of the rescuing effect of the reporter (Fig. 3E). However, YFP-tagged WEE1B export from the nucleus occurred over a wide range of time, consistent with the variable timing of GVBD (Fig. 4B and Fig. S3D). Ccnb2−/− oocytes showed significantly decreased WEE1B translocation rates compared with Ccnb2+/+ oocytes (Fig. 4D); WEE1B translocation rate was inversely correlated to GVBD time (Fig. S3E). We previously demonstrated that WEE1B export is dependent on CDK1 activity (Oh et al., 2010). Therefore, the decreased WEE1B export rate observed in Ccnb2−/− oocytes indicated slower CDK1 activation. We tracked CDK1 activation in live oocytes using the Förster resonance energy transfer (FRET) approach. The rate of CDK1 activation, measured by the Hill slope of FRET activation before GVBD, was significantly decreased in Ccnb2+/− and Ccnb2−/− oocytes (Fig. S3F,G). These results indicate that, in the absence of CCNB2, CDK1 activation is no longer switch like, but becomes gradual, resulting in inefficient WEE1B export and delayed GVBD.
Translation of key cell cycle components is defective in Ccnb2−/− oocytes
Consistent with findings in Xenopus oocytes, the translational program in mouse oocyte is dependent, at least in part, on CDK1 activity (Ballantyne et al., 1997; Han et al., 2017). Given that CDK1 activation was probably defective in Ccnb2−/− oocytes, we tested whether CDK1-dependent translation was affected. Oocytes were co-injected with mRNA encoding mCherry (loading control) and a YFP reporter fused to either Ccnb1-long 3′UTR or Moloney sarcoma oncogene (Mos) 3′UTR. The accumulation of YFP and mCherry in individual oocytes was recorded throughout meiotic maturation and signals were expressed as ratios of YFP/mCherry (Fig. S4A,B). The rates of translation of the two reporters were calculated before (0-2 h) and after (4-8 h) GVBD (Fig. 5A,B). As previously shown, the translation of both YFP-Ccnb1-long 3′UTR and YFP-Mos 3′UTR increased during meiosis in Ccnb2+/+ oocytes. In the absence of CCNB2, the translational activation varied widely in the Ccnb2−/− group, with some oocytes showing a protein synthesis pattern similar to that of Ccnb2+/+ oocytes, whereas others showed no or reduced translation activation for both reporters (Fig. 5A,B). Furthermore, Ccnb2−/− oocytes unable to extrude the first polar body had reduced rates of translation compared with Ccnb2−/− oocytes able to complete meiosis I (Fig. S4C,D).
Fig. 5.

MI spindle formation and activation of Ccnb1 and Mos translation are disrupted in Ccnb2−/− mice. (A,B) Oocytes were injected with a 1:1 mix of polyadenylated mCherry and either YFP-Ccnb1-long 3′UTR (A) or YFP-Mos 3′UTR (B). After overnight incubation, oocytes were released in cilostamide-free medium, and brightfield, YFP and mCherry images were acquired every 15 min for 24 h. YFP signals were normalized by maximal mCherry signals (YFP/mCherry). The normalized rate of YFP accumulation was calculated for each oocyte before (0-2 h) and after (4-6 h) GVBD. Rates were plotted as the median (red) and interquartile range. Student's t-tests were used to evaluate statistical significance; **P=0.0058, ****P<0.0001. The number of oocytes from three independent experiments is reported for each condition. (C) Oocytes were released in cilostamide-free medium and fixed for 8 h after meiotic resumption. The spindle, chromatin and kinetochores were visualized with β-tubulin 488 antibody, DAPI and CREST, respectively. Images show oocytes arrested in prophase I, GVBD without a spindle, early spindle (tubulin organized into a sphere surrounded by chromosomes) and bipolar MI spindle. Scale bars: 10 µm. (D) Oocytes were scored for maturation stage and data are presented as the percentage of total oocytes. The number of oocytes from two independent experiments observed for each group is reported.
It is well established that spindle formation requires protein synthesis, in particular, the accumulation of CCNB1, which is necessary to increase MPF activity (Davydenko et al., 2013; Hampl and Eppig, 1995; Winston, 1997). Given that Ccnb2−/− oocytes had decreased pre-MPF activity and decreased rates of YFP-Ccnb1-long 3′UTR accumulation, we predicted that the timing of spindle formation would be delayed in Ccnb2−/− oocytes. Eight hours after meiotic resumption, >90% of Ccnb2+/+ oocytes had assembled a MI bipolar spindle, whereas only 10% of the Ccnb2−/− oocytes showed this configuration (Fig. 5C,D). Together, these findings support a role of CCNB2 in the CDK1-dependent translation of CCNB1 and MOS and timely assembly of the MI spindle.
Delayed MI/anaphase I transition in Ccnb2−/− oocytes is associated with defective APC activity and persistent activation of the SAC
To define whether additional defects in meiotic progression are associated with CCNB2 depletion, we examined the ability of Ccnb2−/− oocytes to complete meiosis I. First polar body extrusion (PBE I) was both significantly delayed and decreased in these oocytes (Fig. 6A). Furthermore, delayed GVBD time was associated with an inability to complete meiosis I (Fig. S5A). Only 30% of the Ccnb2−/− oocytes reached metaphase II (MII) with a well-formed spindle and chromosomes aligned along the metaphase plate, whereas the rest of the Ccnb2−/− oocytes were arrested in MI or at telophase I (Fig. 6B,C). This phenotype was not the result of the in vitro culture conditions because in vivo ovulated Ccnb2−/− oocytes also clearly showed compromised progression to MII (Fig. S5B).
Fig. 6.

A subpopulation of Ccnb2−/− oocytes fails to complete meiosis I because of altered APC activation. Oocytes were released in cilostamide-free medium and brightfield images were captured every 15 min. (A) The cumulative PBE times were plotted and Student's t-tests between Ccnb2+/+ and Ccnb2−/− oocytes performed (red asterisks); *P<0.05, **P<0.01. Bars represent the s.e.m.; the number of oocytes from seven independent experiments are reported. (B,C) Oocytes were released in cilostamide-free medium and fixed after 24 h. The spindle and the chromatin were visualized with β-tubulin 488 antibody and DAPI, respectively. (B) Images of oocytes arrested in prophase I, MI, telophase I and MII. Scale bars: 10 µm. (C) Oocytes were scored for maturation stage [reported in (B)] and data were reported as the percentage of total oocytes from three independent experiments. (D) Oocytes were injected with mRNA encoding the APC substrate Securin-YFP and, after 17 h incubation, released in cilostamide-free medium. The securin-YFP level was measured every 15 min. Oocytes from three independent experiments were observed. (E,F) Oocytes were injected with mRNA encoding either Ccnb1-mCherry (E) or Ccnb2-mCherry (F) and, after 3 h incubation, released in cilostamide-free medium. CCNB1-mCherry or CCNB2-mCherry levels were measured every 15 min. Oocytes from three and two independent experiments were observed, respectively.
A compromised MI/anaphase I transition might result from defects in the activation of the APC/CDC20 complex, which promotes the degradation of securin and cyclins. Oocytes were injected with Securin-YFP mRNA to monitor the kinetics of APC activation in live oocytes. Securin degradation was delayed and inefficient in Ccnb2−/− oocytes, occurring at a rate that was 50% slower than that of Ccnb2+/+ oocytes (Fig. 6D and Fig. S5C).
CDK1 activates APC directly and indirectly (Adhikari et al., 2014; Golan et al., 2002; Lahav-Baratz et al., 1995; Qiao et al., 2016; Yang and Ferrell, 2013), and CDK1 activity is crucial for satisfaction of the spindle assembly checkpoint (SAC) (Lara-Gonzalez et al., 2012). Using the FRET probe, we measured changes in CDK1 activity of single oocytes between 2 and 6 h after GVBD. Ccnb2−/− oocytes that would eventually extrude the first polar body had increased CDK1 activity similar to that of Ccnb2+/+ oocytes, whereas CDK1 activation was decreased in Ccnb2−/− oocytes unable to complete meiosis I (Fig. S5D). Moreover, APC activation was rescued by overexpression of either CCNB1 (Fig. 6E and Fig. S5E) or CCNB2 (Fig. 6F and Fig. S5E) in Ccnb2−/− oocytes.
We investigated whether the failure of some Ccnb2−/− oocytes to progress through anaphase I might be because of the presence of unattached chromosomes and an active SAC. Mitotic arrest deficient 2 (MAD2; MAD2L1 – Mouse Genome Informatics) colocalization with the kinetochore, visualized by CREST antibody, has been used as a proxy for SAC activation (Collins et al., 2015; Gui and Homer, 2012). Ccnb2+/+ oocytes displayed low MAD2 signals on the kinetochores at both MI (7 h) and MII (24 h), indicating that the SAC had been satisfied (Fig. 7A,B). Pharmacological depolymerization of microtubules with nocodazole strongly activated the SAC in Ccnb2+/+ oocytes, and MAD2 localized on the spindle at most of the kinetochores (Fig. 7A,B). Ccnb2−/− oocytes that reached MII displayed MAD2 levels on the kinetochores that were comparable to those of Ccnb2+/+ oocytes arrested in MII (Fig. 7A,B). Conversely, Ccnb2−/− oocytes unable to complete meiosis I after 24 h displayed significantly higher levels of MAD2 on the kinetochores compared with Ccnb2+/+ MI oocytes (Fig. 7A,B). This suggests that the MI-arrested Ccnb2−/− oocytes did not transition to anaphase I because the SAC was still active. The interval between GVBD and PBE I was considerably longer in Ccnb2−/− oocytes than in Ccnb2+/+ oocytes (Ccnb2+/+: 7.8±2.3 h; Ccnb2−/−: 10.6±2.8 h) (Fig. 7C). Given that oocytes might tolerate some unattached kinetochores and still progress through anaphase I (Lane et al., 2012), SAC was blocked with reversine, an inhibitor of monopolar spindle 1 (MPS1) that prevents MAD2 localization onto the kinetochores (Tipton et al., 2013). Pharmacological SAC inhibition shortened meiosis I in both Ccnb2+/+ (5.6±0.7 h) and Ccnb2−/− (5.1±1.1 h) oocytes, and almost all the oocytes completed meiosis I in a comparable amount of time (Fig. 7C). Thus, the persistent SAC activity in Ccnb2−/− oocytes was the cause of delayed PBE I timing and/or the arrest in MI. Given that the SAC functions to delay anaphase until all chromosomes are attached to prevent aneuploidy, we investigated whether oocytes able to satisfy the SAC and enter meiosis II had the correct number of chromosomes. We performed an in situ chromosome spread with monastrol and counted the kinetochores in single oocytes, as previously published (Chiang and Lampson, 2013). Whereas <10% of the Ccnb2+/+ oocytes were aneuploid (kinetochore number deviating from 20 pairs), over 20% of Ccnb2−/− oocytes were aneuploid (Fig. 7D). Although reversine increased the rate of aneuploidy in both groups, Ccnb2−/− oocytes displayed a greater degree of aneuploidy (Fig. 7D). Together, these findings indicate that Ccnb2−/− oocytes were less efficient in satisfying the SAC, leading to defective APC activation and, ultimately, a delayed, or failure to, exit from MI. Incomplete nuclear maturation and increased aneuploidy are the most probable causes of decreased litter size in these mice.
Fig. 7.

A population of Ccnb2−/− oocytes arrests in MI because of persistent SAC activity. (A,B) Oocytes were released in cilostamide-free medium and fixed at the indicated times. Where specified, oocytes were treated with nocodazole 15 min before fixation. MAD2 and CREST, and chromatin were visualized with specific antibodies and DAPI, respectively. (A) Representative pictures are presented for each condition. Scale bars: 10 µm (B) The amount of MAD2 localized at each kinetochore was quantified by measuring the MAD2/CREST ratio. Signals are plotted as the mean±s.e.m. The number of kinetochores analyzed from two independent experiment is reported. Student's t-tests were used to evaluate statistical significance; ns: not significant, **** indicates P<0.0001. (C) Oocytes were released in the absence or presence of 100 nM reversine. The time of GVBD and PBE were determined through brightfield images acquired every 15 min for 24 h. The length of meiosis I was calculated as the time between GVBD and PBE. Five independent experiments are included. (D,E) Oocytes were matured for 20 h with or without 100 nM reversine. In situ chromosome spreads were performed by treating oocytes for 2 h with 100 µM monastrol. Oocytes were then fixed and immunofluorescence stained for kinetochores using CREST (green) and for chromosomes using DAPI (red). Confocal pictures were obtained every 0.2 µm and maximal intensity projections were produced with ImageJ. Kinetochores were quantified throughout the Z-stacks Scale bars: 5 µm. (E) Two independent experiments were analyzed and Fisher exact tests were used to evaluate statistical significance; *P<0.05.
DISCUSSION
Our findings conclusively established that CCNB2 has an indispensable role during mouse oocyte meiosis, being involved in both the G2/M and the MI/anaphase I transitions. CCNB2 contributed to pre-MPF activity during prophase and was required to generate sufficient MPF to progress through maturation in a timely and efficient fashion (Fig. 3). Moreover, loss of CCNB2 was associated with compromised progression through MI, with ovulation of immature oocytes and/or oocytes with increased aneuploidy, and compromised developmental competence, resulting in the decreased fecundity of Ccnb2−/− females (Fig. 2).
Ccnb2 mRNA was translated at a higher rate than Ccnb1 mRNA in prophase (Fig. 1A,B), confirming our previous polysomal array data (Han et al., 2017). We previously reported similar rates of degradation for the two proteins in GV-arrested oocytes in the presence of cycloheximide (Han et al., 2017). Taken together, these data indicate that the CCNB2 protein accumulates at higher levels than the CCNB1 protein at this time. We confirmed this experimentally, showing that there was five times more CCNB2 than CCNB1 in prophase oocytes (Fig. 1F). These different rates of accumulation probably afford variable rates of CDK1 activation at the G2/M and MI/anaphase I transitions.
CCNB2 was crucial to generate sufficient levels of pre-MPF in GV oocytes. By using both intact cell and cell-free kinase assays, we showed that Ccnb2−/− oocytes had decreased CDK1 activity compared with Ccnb2+/+ oocytes (Fig. 3B and Fig. S2E). Given this decreased pre-MPF activity, conversion of pre-MPF to MPF was also affected. This was confirmed by measuring MPF activation via a FRET probe (Fig. S3F,G) and by measuring the kinetics of WEE1B export from the nucleus, an event shown to be CDK1 dependent (Fig. 4B) (Oh et al., 2010). CDK1 activation in Ccnb2−/− oocytes no longer displayed a switch-like property as in Ccnb2+/+, but rather increased slowly over a long period of time. As a consequence of this gradual increase, GVBD became an inefficient and error-prone process (Fig. 3D). The high variability in the time of GVBD in Ccnb2−/− oocytes, or when the CDK1 activity was blocked with small-molecule inhibitors in Ccnb2+/+ oocytes, suggested that the threshold of CDK1 activity required to undergo GVDB varies among individual oocytes. This pattern of CCNB2 accumulation allowed enough pre-MPF activity to ensure that every oocyte reached the MPF threshold required to promptly undergo GVBD. Therefore, this unique pattern of CCNB2 accumulation is crucial to establish the ‘poised state’ that characterizes the rapid re-entry into meiosis of mouse oocytes.
The oocyte phenotype associated with the inactivation of the Ccnb2 gene could be explained by a defect in oocyte growth and/or defective follicle development. However, the following data reject this possibility. First, no significant deviations from Ccnb2+/+ were observed in ovary weight and morphology, or follicle number and their development. Oocyte number and diameter were also normal, and no significant correlation was established between oocyte size and penetrance of the phenotype. More importantly, evaluation of the NSN/SN state of the chromatin showed full competence with few NSN oocytes. This chromatin conformation is a sensitive indicator of oocyte competence to mature to MII. Finally, a review of published data characterizing growing oocytes does support the view that the Ccnb2−/− oocytes are fully grown. CDK1 accumulates progressively during oocyte growth and lower levels of this kinase should be detected in oocytes with compromised follicle development. Instead, Ccnb2−/− oocytes showed CDK1 protein levels comparable to those of fully grown Ccnb2+/+ oocytes. Previous studies have not revealed major defects in the timing of meiotic re-entry in oocytes from early antral follicles (Firmani et al., 2018), which is instead the hallmark of the Ccnb2−/− phenotype. A word a caution is that the Ccnb2 gene is inactivated globally and possible defects in the somatic compartment cannot be formally excluded. The fact that mice show a phenotype of premature ovarian failure could be explained by the degeneration of the defective oocytes, but also might be caused by defects in the somatic compartment associated with aging.
Previous attempts to deplete oocytes of CCNB2 with MO resulted in an 80% decrease in meiotic re-entry 3 h after 3-isobutyl-1-methylxanthine (IBMX) release (Gui and Homer, 2013) and led the authors to conclude that CCNB2-depleted oocytes do not re-enter meiosis. Our data showed instead that meiotic re-entry was delayed, but not abolished. However, in the study by Gui and Homer, only the first 3 h of meiotic resumption were reported and, therefore, any further delay in maturation might have been unnoticed. Similarly, Li et al. observed a decrease in meiotic maturation in Ccnb2−/− oocytes. Again, only the first 3 h of maturation were reported.
We detected additional phenotypes by further assessing the role of CCNB2 throughout meiotic maturation. Depletion of CCNB2 disrupted the normal increase in CDK1 activity that occurs during metaphase (Fig. S5D). This defective CDK1 activation might not only be directly because of the absence of the CDK1/CCNB2 complex, but also indirectly caused by a decrease in the CDK1/CCNB1 complex, as suggested by the decreased rate of Ccnb1 mRNA translation in a subset of Ccnb2−/− oocytes (Fig. 5). Additionally, the decreased translation of Mos mRNA probably affects the positive feedback between the extracellular signal-related (ERK) pathway and MPF (Nebreda and Ferby, 2000) (Fig. 5).
Deficient CDK1 activity in MI had several consequences. There was a delay in spindle assembly (Fig. 5C), which recapitulates previous experiments using pharmacological inhibition of CDK1 (Davydenko et al., 2013). Similarly, SAC inactivation was incomplete in the Ccnb2−/− oocytes, leading to defective APC activation. Indeed, stable microtubule attachments to the kinetochores have been shown to depend on the increase in CDK1 activity (Davydenko et al., 2013). In agreement with these findings, disruption of CDK1 activity, via depletion of CCNB2, resulted in persistent MAD2 loading onto the kinetochores (Fig. 7B). Inhibition of the checkpoint with reversine restored the ability of the oocyte to progress to anaphase (Fig. 7C). Downstream of SAC satisfaction is the activation of APC/C. Our data indicated that, without CCNB2, there was a major delay in APC activation, as measured by securin degradation (Fig. 6D). In Ccnb2−/− oocytes, APC activation was not switch like as in Ccnb2+/+ oocytes, but was instead prolonged and gradual, in agreement with the idea that cooperative CDK1 activation is required for full APC activation (Yang and Ferrell, 2013). Thus, in Ccnb2−/− oocytes, the switch-like entry into and exit from meiosis I was lost, resulting in nonsynchronous, delayed, and sometimes failed MI/anaphase I transitions. The prolonged maturation time of the Ccnb2−/− oocytes could lead to a phenotype similar to that which is described for oocyte aging, which results in compromised developmental competence (Demond et al., 2016). In addition, the increased aneuploidy rate detected was associated with compromised ability to sustain embryo development. All these defects could explain the decreased fecundity of Ccnb2−/− females.
CCNB1 and CCNB2 diverged early on during evolution and are present in both animals and plants. CCNB3 is expressed in animals, but not in plants, and its involvement in gametogenesis has been reported in Drosophila, Caenorhabditis elegans, and the mouse (Jacobs et al., 1998; Tarailo-Graovac and Chen, 2012; Zhang et al., 2015). Three cyclins are present in human and mouse and essentially all mammals express CCNB2. Two hypotheses have been proposed to explain the evolution of multiple cyclins involved in cell cycle progression (Murray, 2004). One view proposes that the binding of different cyclins to CDK1 conveys distinct biochemical properties to the complex, resulting in different substrate specificity. The opposing view is that cyclins in complex with CDKs have essentially the same biochemical properties, but differ in the pattern of activation in space and time during the cell cycle. Our data are in line with the second model. The phenotype described is consistent with an indispensable function for CCNB2 during mouse oocyte meiosis that cannot be compensated for by endogenous levels of CCNB1. However, overexpression of exogenous CCNB1 rescued the phenotype because of CCNB2 depletion during both the GV/MI (Fig. 3E) and the MI/anaphase I transitions (Fig. 6E). In a reciprocal approach using Ccnb1−/− mice, endogenous CCNB2 is overexpressed, and this alone drives oocyte progression through meiosis I, but the oocytes could not progress to MII; however, overexpression of exogenous CCNB2 rescues the MII entry (Li et al., 2018). Thus, these complementary studies indicate that the two cyclins have overlapping functions at the molecular level but are not functionally redundant because of the different time of expression. These findings also emphasize the importance of the post-transcriptional program in regulating meiotic divisions. However, and despite our efforts to express recombinant proteins to levels comparable to endogenous ones, we cannot exclude the possibility that subtle differences in the biochemical properties of the two cyclins are present, but masked by the ectopic expression. Of note, it has been reported that CCNB2 remains in the cytoplasm during mitosis, whereas CCNB1 is translocated in the nucleus (Jackman et al., 1995). In mouse oocytes, we found no differences between CCNB1 and CCNB2 because both tagged proteins were translocated to the nucleus at G2/M. It remains to be determined whether subtle differences in localization of the endogenous proteins can be detected by more sensitive approaches.
Given our findings, we propose that CCNB2-depleted oocytes could be used as a model for defective CDK1 activation throughout meiosis, a condition that might be a significant cause of meiotic maturation block and infertility in humans. Moreover, the increased aneuploidy in the CCNB2-depleted oocytes establishes a link between defective activation of CDK1 and errors in chromosome segregation. This possibility should be explored further in those instances of increased aneuploidy in humans, including those associated with aging.
MATERIALS AND METHODS
Mice, oocyte collection, and microinjection
All experimental procedures involving mice were approved by the Institutional Animal Care and Use Committee at UCSF (protocol: AN101432). Ccnb2−/− mice were a kind gift from Dr Tim Hunt (Institute of Cancer Research, London, UK) (Brandeis et al., 1998). The conditional RiboTag mice used in this study were previously described (Sousa Martins et al., 2016). C57BL/6 female mice (21-24-days old) were primed with 5 units of eCG and sacrificed 44-48 h later to collect GV-arrested oocytes. For the collection of MII-arrested oocytes, females were primed with 5 units of eCG; after 48 h, they were injected with hCG, and after 13 h, sacrificed for egg retrieval. Cumulus-enclosed oocytes from antral follicles were isolated, and mechanically denuded in HEPES modified Minimum Essential Medium Eagle (Sigma-Aldrich, M2645) supplemented with 1 µM cilostamide (Calbiochem, 231085). When specified, oocytes were microinjected with 5-10 pl of mRNA. Oocytes were then cultured at 37°C with 5% CO2 in Minimum Essential Media (MEM)-α medium (Gibco, 12561-056) supplemented with 0.2 mM sodium pyruvate, 75 µg/ml penicillin, 10 µg/ml streptomycin, 3 mg/ml bovine serum albumin (BSA), and 1 µM cilostamide for 3 h or 16 h, as indicated in the figure legends. For each independent experiment, oocytes from at least three different mice were used.
Plasmid construct and mRNA preparation
(C483S)-CDC25B and (K237A)-WEE1B coding sequences were cloned upstream of the YPet coding sequence. The CCNB1 and CCNB2 open reading frame sequences were obtained by sequencing oocyte cDNA and cloned upstream of the mCherry coding sequence. The Ccnb1, Ccnb2 and Mos 3′UTR sequences were obtained in the same manner and cloned downstream of the YPet coding sequence. All constructs were prepared in the pCDNA 3.1 vector containing a T7 promoter and fidelity was confirmed by DNA sequencing. mRNA of all the reporters was in vitro transcribed with the mMESSAGE mMACHINE T7 Transcription Kit (Ambion, AM1344); when specified, polyadenylation (150-200 nt) was achieved using the Poly(A) Tailing Kit (Ambion, AM1350). All messages were purified using the MEGAclear Kit (Ambion, AM1908). mRNA concentrations were measured by NanoDrop and message integrity was evaluated by electrophoresis.
Time-lapse microscopy, analysis of protein translocation and YFP-3′UTR translation
Time-lapse experiments were performed using a Nikon Eclipse T2000-E equipped with a mobile stage and an environmental chamber at 37°C and 5% CO2 and the following settings: filter set, dichroic mirror YFP/CFP/mCherry 69008BS; Ypet channel (Ex: S500/20×49057 Em: D535/30 m 47281); and mCherry channel (Ex: 580/25×49829 Em: 632/60 m). (C483S)-CDC25B-YFP, (K237A)-WEE1B-YFP, securin-YFP or CCNB1-mCherry were injected at 300 ng/µl. After injection, oocytes were incubated for 16 h to allow expression of the probes. Nuclear and total probe ratios were calculated after subtraction of the background. Rates of translocation were calculated as the slope of the line obtained by linear regression. YFP-3′UTR reporters were co-injected with polyadenylated mCherry at 12.5 µg/µl each. After injection, oocytes were incubated for 16 h to allow expression of the probes. YFP signals were normalized by the plateaued level of the mCherry signal to correct for injection levels. Rates were calculated with YFP/mCherry ratios as the slope of the curve obtained by linear regression of the time points indicated.
RiboTag-immunoprecipitation and RNA-Seq
Oocytes were collected in minimal volumes (5-10 µl) of 0.1% polyvinylpyrrolidone (PVP) in PBS, flash frozen in liquid nitrogen, and stored at −80°C. Samples were thawed, randomly pooled to yield a total of 200 oocytes per time point per replicate, and 300 µl supplemented homogenization buffer (sHB) was added to each pooled sample. The homogenates were then vortexed for 30 s, flash frozen in liquid nitrogen, and allowed to thaw at room temperature (RT); this was repeated twice. Finally, the homogenates were centrifuged for 10 min at maximum speed at 4°C and the supernatant (IP soup) was collected in new tubes. Meanwhile, the appropriate volume (50 µl per sample) of Dynabeads Protein G (Invitrogen) was washed three times in 500 µl HB on a rotor at 4°C for 5 min per wash. An additional two washes were performed with 500 µl sHB on a rotor at 4°C for 10 min per wash. The final wash solution was removed and the beads were eluted in the original volume of sHB and kept on ice. 20 µl cleaned beads were added to each IP soup to pre-clear on a rotor at 4°C for 1 h. The beads were removed via a magnetic rack and 15 µl of IP soup was collected from each sample in 200 µl of RLT buffer (Qiagen) to serve as the input samples. Input samples were frozen and kept at −80°C until RNA extraction. 3 µl (3 µg) anti-hemagglutinin (HA).11 epitope tag antibody (BioLegend, 901501) was added to each of the remaining IP soups and all samples were incubated on a rotor at 4°C for 4 h. 30 µl cleaned beads were then added to the samples and incubated overnight on a rotor at 4°C. The beads (now bound by HA-tagged ribosomes and the associated mRNAs) were washed five times in 1 ml of wash buffer with 1 M urea (uWB) on a rotor at 4°C for 10 min per wash. The final uWB wash was removed and 250 µl RLT buffer was added to each sample and vortexed for 30 s. RNA extraction was performed following the RNeasy Plus Micro Kit protocol (Qiagen, 74034). Samples were eluted in 10 µl of RNAse-free water. RNA samples were sent to the Gladstone Institutes Genomics Core for quality control using Bioanalyzer (Agilent) and cDNA library preparation with the Ovation RNA-Seq System V2 (NuGEN). Samples were sequenced using the HiSeq400 platform.
Western blot
Oocytes were collected in 0.1% PVP in Dulbecco's PBS (DPBS) and then boiled for 5 min at 95°C in 1× Laemmli Sample Buffer (Bio-Rad) supplemented with β-mercaptoethanol. Lysates were resolved in 10% Laemmli gels and transferred onto supported nitrocellulose membranes. Membranes were incubated in buffer containing the primary antibody overnight at 4°C; the antibodies and dilutions used were as follows: CCNB2, 1:1000 (R&D Systems, AF6004); CCNB1, 1:500 (Abcam, ab72); β-actin, 1:1000 (Abcam, ab8227); CDK1, 1:1000 (Cell Signaling, 9112); CPEB1, 1:1000 (Abcam, ab73287); T320-pp1, 1:30,000 (Abcam, ab62334); GST, 1:10,000 (Sigma, 16-209). Recombinant CCNB1 (Abcam, ab128445) or recombinant CCNB2 (My BioSource, MBS1451430) were serial diluted to produce a standard curve.
Immunofluorescence
Oocytes were fixed in DPBS supplemented with 0.1% Triton X-100 and 2% formaldehyde (Sigma, 28908) for 30 min. After three 10-min washes with blocking buffer, the oocytes were incubated overnight in blocking buffer (1× PBS, 0.3% BSA, 0.01% Tween), then permeabilized for 15 min in DPBS supplemented with 0.3% BSA and 0.1% Triton X-100. After three 10-min washes with blocking buffer, oocytes were incubated for 1 h in primary antibody diluted in blocking buffer. The antibodies used were as follows: β-tubulin-488, 1:100 (Cell Signaling Technology, 3623); CREST, 1:200 (ImmunoVision, HCT-0100); and MAD2, 1:200 (Dr Rey-Huei Chen, Academia Sinica, Taipei). After three 10-min washes with blocking buffer, the membrane was incubated for 1 h with the appropriate secondary antibody, 1:500 (Alexa-568 goat anti-human or Alexa-488 goat anti-rabbit). Oocytes were washed for 10 min in blocking buffer for three washes and mounted with VECTASHIELD Mounting Medium with DAPI (Vector, H-1200). Pictures were acquired with a confocal Nikon C1SI equipped with an X60 oil immersion lens.
In vitro CDK1 kinase assay
Oocytes were collected in 30 µl of 2X kinase buffer (100 mM Hepes, 30 mM MgCl2, 2 mM EGTA, 10 mM CaCl2, 2 mM DTT, 2 µg/ml leupeptin, 2 µg/ml aprotinin, 2 µM okadaic acid). Oocytes were lysed by freezing and thawing twice in liquid nitrogen. Extracts were incubated at 30°C for 15 min in the presence of 0.1 mM ATP, 10 mM DTT, 2 µM okadaic acid, and 2 µg of recombinant peptide GST-PP1 as the substrate. PP1-GST was produced as previously described (Daldello et al., 2015). Reactions were stopped by adding Laemmli sample buffer and boiling at 95°C for 5 min. CDK1 activity was measured by quantifying the western blot signal of phosphorylated T320 of the PP1-GST substrate.
In situ chromosome spread
In situ chromosome spreads were performed following a published protocol (Chiang and Lampson, 2013). Oocytes were treated with monastrol for 2 h at 37°C. Immunofluorescence staining was perform as described earlier. Z-stacks were acquired every 0.2 μm throughout the chromatin volume using an inverted confocal microscope (Eclipse Ti-E, Nikon) equipped with a spinning disk (CSU-X1, Yokogawa Electric Corporation) and 100×/NA 1.45 objective. Exposure time was kept at 200 µs for both 405 nm (DAPI) and 561 nm (CREST). Images were processed using ImageJ.
FRET experiment
The CDK1 FRET sensor (2327) was a gift from Dr Jonathon Pines (The Institute of Cancer Research, London, UK) (Addgene plasmid # 26064). Oocytes were injected with 5-10 pl of FRET sensor mRNA at 300 ng/µl and, after 16 h incubation, the fluorescence level was quantified as described earlier. Signal intensities of YFP/YFP, CFP/CFP and CFP/YFP channels were subtracted from the background. The CFP/YFP channel was corrected for the YFP bleed-through and FRET was calculated as (CFP/YFP)/(CFP/CFP).
Data processing, quantification and statistical analysis
Quality checks of RNA-Seq reads were performed using FastQC and reads were trimmed with Trimmomatic. Alignment of the reads to the mouse genome was performed using Hisat2, .bam files were sorted and indexed using Samtools, and count files were generated by HTSeq. TMM normalization and statistical analyses of the RNA-Seq data were performed with edgeR.
MAD2/CREST signals were quantified with Fiji. Student's t-tests and nonparametric Mann–Whitney tests were performed using GraphPad Prism 8.
Note added in proof
At the time of submission of the revised article, a report describing the role of Ccnb3 during mouse oocyte maturation was published (Karasu et al., 2019). This report showed that CCNB3 is required for efficient exit from MI.
Supplementary Material
Acknowledgements
We thank Dr Tim Hunt and Dr Jonathon Pines for sharing the Ccnb2−/− mice, and Dr Rey-Huei Chen for the gift of the MAD2 antibody. The authors are indebted to Dr Sophie Dumont for the helpful discussion and advice on the SAC measurements.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: E.M.D., M.C.; Investigation: E.M.D., X.G.L., C.-R.Y.; Resources: J.K., M.C.; Writing - original draft: E.M.D., M.C.; Writing - review & editing: E.M.D., X.G.L., C.-R.Y., M.C.; Supervision: M.C.; Funding acquisition: E.M.D., M.C.
Funding
These studies were supported by the National Institutes of Health (R01 GM097165 and GM116926 to M.C.). E.M.D. is supported by a fellowship from the Lalor Foundation. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.172734.supplemental
References
- Adhikari D., Diril M. K., Busayavalasa K., Risal S., Nakagawa S., Lindkvist R., Shen Y., Coppola V., Tessarollo L., Kudo N. R. et al. (2014). Mastl is required for timely activation of APC/C in meiosis I and Cdk1 reactivation in meiosis II. J. Cell Biol. 206, 843-853. 10.1083/jcb.201406033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballantyne S., Daniel D. L. and Wickens M. (1997). A dependent pathway of cytoplasmic polyadenylation reactions linked to cell cycle control by c-mos and CDK1 activation. Mol. Biol. Cell 8, 1633-1648. 10.1091/mbc.8.8.1633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandeis M., Rosewell I., Carrington M., Crompton T., Jacobs M. A., Kirk J., Gannon J. and Hunt T. (1998). Cyclin B2-null mice develop normally and are fertile whereas cyclin B1-null mice die in utero. Proc. Natl. Acad. Sci. USA 95, 4344-4349. 10.1073/pnas.95.8.4344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang T. and Lampson M. A. (2013). Counting Chromosomes in Intact Eggs. In Mammalian Oocyte Regulation (ed. Homer H. A.), pp. 249-253. Totowa, NJ: Humana Press. [DOI] [PubMed] [Google Scholar]
- Collins J. K., Lane S. I. R., Merriman J. A. and Jones K. T. (2015). DNA damage induces a meiotic arrest in mouse oocytes mediated by the spindle assembly checkpoint. Nat. Commun. 6, 8553 10.1038/ncomms9553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti M., Andersen C. B., Richard F., Mehats C., Chun S. Y., Horner K., Jin C. and Tsafriri A. (2002). Role of cyclic nucleotide signaling in oocyte maturation. Mol. Cell. Endocrinol. 187, 153-159. 10.1016/S0303-7207(01)00686-4 [DOI] [PubMed] [Google Scholar]
- Daldello E. M., Le T., Poulhe R., Jessus C., Haccard O. and Dupré A. (2015). Control of Cdc6 accumulation by Cdk1 and MAPK is essential for completion of oocyte meiotic divisions in Xenopus. J. Cell Sci. 128, 2482-2496. 10.1242/jcs.166553 [DOI] [PubMed] [Google Scholar]
- Davydenko O., Schultz R. M. and Lampson M. A. (2013). Increased CDK1 activity determines the timing of kinetochore-microtubule attachments in meiosis I. J. Cell Biol. 202, 221-229. 10.1083/jcb.201303019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demond H., Trapphoff T., Dankert D., Heiligentag M., Grümmer R., Horsthemke B. and Eichenlaub-Ritter U. (2016). Preovulatory aging in vivo and in vitro affects maturation rates, abundance of selected proteins, histone methylation pattern and spindle integrity in murine oocytes. PLoS ONE 11, e0162722 10.1371/journal.pone.0162722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firmani L. D., Uliasz T. F. and Mehlmann L. M. (2018). The switch from cAMP-independent to cAMP-dependent arrest of meiotic prophase is associated with coordinated GPR3 and CDK1 expression in mouse oocytes. Dev. Biol. 434, 196-205. 10.1016/j.ydbio.2017.12.014 [DOI] [PubMed] [Google Scholar]
- Gavet O. and Pines J. (2010a). Progressive activation of CyclinB1-Cdk1 coordinates entry to mitosis. Dev. Cell 18, 533-543. 10.1016/j.devcel.2010.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavet O. and Pines J. (2010b). Activation of cyclin B1–Cdk1 synchronizes events in the nucleus and the cytoplasm at mitosis. J. Cell Biol. 189, 247-259. 10.1083/jcb.200909144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golan A., Yudkovsky Y. and Hershko A. (2002). The cyclin-ubiquitin ligase activity of cyclosome/APC is jointly activated by protein kinases Cdk1-cyclin B and Plk. J. Biol. Chem. 277, 15552-15557. 10.1074/jbc.M111476200 [DOI] [PubMed] [Google Scholar]
- Gui L. and Homer H. (2012). Spindle assembly checkpoint signalling is uncoupled from chromosomal position in mouse oocytes. Development 139, 1941-1946. 10.1242/dev.078352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui L. and Homer H. (2013). Hec1-dependent cyclin B2 stabilization regulates the G2-M transition and early prometaphase in mouse oocytes. Dev. Cell 25, 43-54. 10.1016/j.devcel.2013.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampl A. and Eppig J. J. (1995). Translational regulation of the gradual increase in histone H1 kinase activity in maturing mouse oocytes. Mol. Reprod. Dev. 40, 9-15. 10.1002/mrd.1080400103 [DOI] [PubMed] [Google Scholar]
- Han S. J., Martins J. P. S., Yang Y., Kang M. K., Daldello E. M. and Conti M. (2017). The translation of cyclin B1 and B2 is differentially regulated during mouse oocyte reentry into the meiotic cell cycle. Sci. Rep. 7, 14077 10.1038/s41598-017-13688-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman M., Firth M. and Pines J. (1995). Human cyclins B1 and B2 are localized to strikingly different structures: B1 to microtubules, B2 primarily to the Golgi apparatus. EMBO J. 14, 1646-1654. 10.1002/j.1460-2075.1995.tb07153.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs H. W., Knoblich J. A. and Lehner C. F. (1998). Drosophila Cyclin B3 is required for female fertility and is dispensable for mitosis like Cyclin B. Genes Dev. 12, 3741-3751. 10.1101/gad.12.23.3741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karasu M. E., Bouftas N., Keeney S. and Wassmann K. (2019). Cyclin B3 promotes anaphase I onset in oocyte meiosis. J. Cell Biol. 218, 1265-1281. 10.1083/jcb.201808091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahav-Baratz S., Sudakin V., Ruderman J. V. and Hershko A. (1995). Reversible phosphorylation controls the activity of cyclosome-associated cyclin-ubiquitin ligase. Proc. Natl. Acad. Sci. USA 92, 9303-9307. 10.1073/pnas.92.20.9303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane S. I. R., Yun Y. and Jones K. T. (2012). Timing of anaphase-promoting complex activation in mouse oocytes is predicted by microtubule-kinetochore attachment but not by bivalent alignment or tension. Development 139, 1947-1955. 10.1242/dev.077040 [DOI] [PubMed] [Google Scholar]
- Lara-Gonzalez P., Westhorpe F. G. and Taylor S. S. (2012). The spindle assembly checkpoint. Curr. Biol. 22, R966-R980. 10.1016/j.cub.2012.10.006 [DOI] [PubMed] [Google Scholar]
- Ledan E., Polanski Z., Terret M.-E. and Maro B. (2001). Meiotic maturation of the mouse oocyte requires an equilibrium between cyclin B synthesis and degradation. Dev. Biol. 232, 400-413. 10.1006/dbio.2001.0188 [DOI] [PubMed] [Google Scholar]
- Lewis C. W., Taylor R. G., Kubara P. M., Marshall K., Meijer L. and Golsteyn R. M. (2013). A western blot assay to measure cyclin dependent kinase activity in cells or in vitro without the use of radioisotopes. FEBS Lett. 587, 3089-3095. 10.1016/j.febslet.2013.08.003 [DOI] [PubMed] [Google Scholar]
- Li J., Tang J.-X., Cheng J.-M., Hu B., Wang Y.-Q., Aalia B., Li X.-Y., Jin C., Wang X.-X., Deng S.-L. et al. (2018). Cyclin B2 can compensate for Cyclin B1 in oocyte meiosis I. J. Cell Biol. 217, 3901-3911. 10.1083/jcb.201802077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masui Y. and Markert C. L. (1971). Cytoplasmic control of nuclear behavior during meiotic maturation of frog oocytes. J. Exp. Zool. 177, 129-145. 10.1002/jez.1401770202 [DOI] [PubMed] [Google Scholar]
- Mehlmann L. M. (2005). Stops and starts in mammalian oocytes: recent advances in understanding the regulation of meiotic arrest and oocyte maturation. Reproduction 130, 791-799. 10.1530/rep.1.00793 [DOI] [PubMed] [Google Scholar]
- Morgan D. O. (2007). The Cell Cycle: Principles of Control. London, MA: Published by New Science Press in association with Oxford University Press; Distributed inside North America by Sinauer Associates, Publishers. [Google Scholar]
- Morgan M., Much C., DiGiacomo M., Azzi C., Ivanova I., Vitsios D. M., Pistolic J., Collier P., Moreira P. N., Benes V. et al. (2017). mRNA 3′ uridylation and poly(A) tail length sculpt the mammalian maternal transcriptome. Nature 548, 347-351. 10.1038/nature23318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray A. W. (2004). Recycling the cell cycle. Cell 116, 221-234. 10.1016/S0092-8674(03)01080-8 [DOI] [PubMed] [Google Scholar]
- Nebreda A. R. and Ferby I. (2000). Regulation of the meiotic cell cycle in oocytes. Curr. Opin. Cell Biol. 12, 666-675. 10.1016/S0955-0674(00)00150-2 [DOI] [PubMed] [Google Scholar]
- Oh J. S., Han S. J. and Conti M. (2010). Wee1B, Myt1, and Cdc25 function in distinct compartments of the mouse oocyte to control meiotic resumption. J. Cell Biol. 188, 199-207. 10.1083/jcb.200907161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piqué M., López J. M., Foissac S., Guigó R. and Méndez R. (2008). A combinatorial code for CPE-mediated translational control. Cell 132, 434-448. 10.1016/j.cell.2007.12.038 [DOI] [PubMed] [Google Scholar]
- Qiao R., Weissmann F., Yamaguchi M., Brown N. G., VanderLinden R., Imre R., Jarvis M. A., Brunner M. R., Davidson I. F., Litos G. et al. (2016). Mechanism of APC/C CDC20 activation by mitotic phosphorylation. Proc. Natl. Acad. Sci. USA 113, E2570-E2578. 10.1073/pnas.1604929113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes J. M. and Ross P. J. (2016). Cytoplasmic polyadenylation in mammalian oocyte maturation: oocyte cytoplasmic polyadenylation. Wiley Interdiscip. Rev. RNA 7, 71-89. 10.1002/wrna.1316 [DOI] [PubMed] [Google Scholar]
- Sousa Martins J. P., Liu X., Oke A., Arora R., Franciosi F., Viville S., Laird D. J., Fung J. C. and Conti M. (2016). DAZL and CPEB1 regulate mRNA translation synergistically during oocyte maturation. J. Cell Sci. 129, 1271-1282. 10.1242/jcs.179218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J.-X., Li J., Cheng J.-M., Hu B., Sun T.-C., Li X.-Y., Batool A., Wang Z.-P., Wang X.-X., Deng S.-L. et al. (2017). Requirement for CCNB1 in mouse spermatogenesis. Cell Death Dis. 8, e3142 10.1038/cddis.2017.555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarailo-Graovac M. and Chen N. (2012). Proper Cyclin B3 dosage is important for precision of metaphase-to-anaphase onset timing in Caenorhabditis elegans. G3 (Bethesda) 2, 865-871. 10.1534/g3.112.002782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay J., Hodgman R. and Richter J. D. (2000). The control of Cyclin B1 mRNA translation during mouse oocyte maturation. Dev. Biol. 221, 1-9. 10.1006/dbio.2000.9669 [DOI] [PubMed] [Google Scholar]
- Thornton B. R. and Toczyski D. P. (2006). Precise destruction: an emerging picture of the APC. Genes Dev. 20, 3069-3078. 10.1101/gad.1478306 [DOI] [PubMed] [Google Scholar]
- Tipton A. R., Ji W., Sturt-Gillespie B., Bekier M. E., Wang K., Taylor W. R. and Liu S.-T. (2013). Monopolar spindle 1 (MPS1) kinase promotes production of closed MAD2 (C-MAD2) conformer and assembly of the mitotic checkpoint complex. J. Biol. Chem. 288, 35149-35158. 10.1074/jbc.M113.522375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winston N. J. (1997). Stability of cyclin B protein during meiotic maturation and the first mitotic cell division in mouse oocytes. Biol. Cell 89, 211-219. 10.1111/j.1768-322X.1997.tb01009.x [DOI] [PubMed] [Google Scholar]
- Yang Q. and Ferrell J. E. (2013). The Cdk1–APC/C cell cycle oscillator circuit functions as a time-delayed, ultrasensitive switch. Nat. Cell Biol. 15, 519-525. 10.1038/ncb2737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Yang C.-R., Han S. J., Daldello E. M., Cho A., Martins J. P. S., Xia G. and Conti M. (2017). Maternal mRNAs with distinct 3′ UTRs define the temporal pattern of Ccnb1 synthesis during mouse oocyte meiotic maturation. Genes Dev. 31, 1302-1307. 10.1101/gad.296871.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T., Qi S.-T., Huang L., Ma X.-S., Ouyang Y.-C., Hou Y., Shen W., Schatten H. and Sun Q.-Y. (2015). Cyclin B3 controls anaphase onset independent of spindle assembly checkpoint in meiotic oocytes. Cell Cycle Georget. Tex 14, 2648-2654. 10.1080/15384101.2015.1064567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccotti M., Giorgi Rossi P., Martinez A., Garagna S., Forabosco A. and Redi C. A. (1998). Meiotic and developmental competence of mouse antral oocytes. Biol. Reprod. 58, 700-704. 10.1095/biolreprod58.3.700 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.