

Pharmacological inhibition of β-catenin/BCL9 interaction modulates Treg cells and improves immunotherapy.
Abstract
The Wnt/β-catenin (β-cat) pathway plays a critical role in cancer. Using hydrocarbon-stapled peptide technologies, we aim to develop potent, selective inhibitors targeting this pathway by disrupting the interaction of β-cat with its coactivators B-cell lymphoma 9 (BCL9) and B-cell lymphoma 9-like (B9L). We identified a set of peptides, including hsBCL9CT-24, that robustly inhibits the activity of β-cat and suppresses cancer cell growth. In animal models, these peptides exhibit potent anti-tumor effects, favorable pharmacokinetic profiles, and minimal toxicities. Markedly, these peptides promote intratumoral infiltration of cytotoxic T cells by reducing regulatory T cells (Treg) and increasing dendritic cells (DCs), therefore sensitizing cancer cells to PD-1 inhibitors. Given the strong correlation between Treg infiltration and APC mutation in colorectal cancers, it indicates our peptides can reactivate anti-cancer immune response suppressed by the oncogenic Wnt pathway. In summary, we report a promising strategy for cancer therapy by pharmacological inhibition of the Wnt/β-cat signaling.
INTRODUCTION
Numerous studies have implicated the canonical Wnt pathway, a tightly regulated signal transduction system required for embryonic development and tissue homeostasis, in tumorigenesis and metastasis (1, 2). The canonical Wnt pathway is activated by Wnt ligands binding to the Frizzled family of cell surface receptors, which stabilizes β-catenin (β-cat) by preventing phosphorylation-dependent protein ubiquitination and degradation by the adenomatous polyposis coli (APC). In many cancers, loss-of-function mutations in the APC components and gain-of-function mutations of β-cat can enable β-cat to escape degradation and accumulate in the nucleus, where it engages lymphoid enhancer factor/T cell factor (LEF/TCF) to induce expression of genes that promote cell survival, proliferation, and migration (1, 2). To achieve an enhanced transcription activity, β-cat binds to its coactivator, B cell lymphoma 9 (BCL9), and its homolog, B cell lymphoma 9–like (B9L) (3).
Aberrant activation of the Wnt pathway is associated with initiation and progression of a wide range of human epithelial malignancies (1, 2, 4). Colorectal cancer (CRC), for instance, is tightly connected to Wnt signaling, as over 80% of human CRCs have genomic alterations in the Wnt pathway components—primarily APC and β-cat mutations. Approximately half of all CRCs display heightened expression of BCL9 (5). Furthermore, the Wnt pathway is elevated in colon cancer stem cells (CSCs), which are known to exhibit enhanced resistance to apoptosis and a heightened ability to migrate and invade, therefore playing a major role in colon cancer metastasis (1). In addition, the Wnt pathway is involved in breast and lung cancers. Accumulation of stabilized β-cat is a common feature in breast cancer and is correlated with poor prognosis (6). Loss of Wnt1 results in significant depletion of breast CSCs, thus inhibiting tumor growth and metastatic progression (7). Likewise, the Wnt pathway is frequently up-regulated in lung cancers; over 75% of lung cancers exhibit overexpression of DVL-3, a positive regulator of Wnt signaling. Activation of Wnt signaling can synergize with other essential pathways to drive lung cancer progression. It has been shown that induction of Wnt pathway genes increases tumorigenesis in a KRAS-driven lung cancer model, while inhibition of the Wnt pathway leads to tumor regression and reduced metastasis. As expected, the expression of Wnt pathway components predicts poor prognosis of lung cancer patients (8).
Recently, the role of Wnt signaling in immune evasion of cancer cells has emerged in many carcinomas, including melanoma and CRC. In melanoma, activation of tumor-intrinsic β-cat can lead to T cell exclusion by inhibiting intratumoral dendritic cells (DCs) (9, 10). In a CRC mouse model, loss of APC has been linked to decreases in intratumoral CD8+ T cells and promotion of cancer progression (9). Therefore, it is speculated that Wnt pathway inhibitors can overcome resistance to immunotherapies and exhibit synergistic effects when combined with immuno-oncological regimens.
While appealing and promising, it remains a major challenge to develop nontoxic, tumor-specific Wnt pathway inhibitors with strong potency for clinical application. A number of Wnt pathway inhibitors have been found to exhibit substantial adverse effects in animal studies and clinical trials (2, 11). At present, few Wnt inhibitors have actively undergone clinical trials, and these drug candidates are likely to be problematic due to their mechanisms of action (11). The first class of Wnt inhibitors blocks upstream events of the pathway and includes LGK-974 (discontinued), OMP-54F28 (phase 1), and OMP-18R5 (phase 1) (2, 11). LGK-974 inhibits the secretion and production of Wnt ligands, while the OMP series inhibits the Wnt ligand receptor Frizzled. Drug candidates in this category cannot confer inhibitory effects to cancer cells that harbor APC, Axin, or β-cat mutations, because β-cat activation is nearly independent of Wnt/Frizzled (11). Moreover, targeting Wnt/Frizzled has been shown to cause substantial toxicity in clinical trials (11). The second class of Wnt inhibitors diminishes β-cat transcriptional activity by disrupting the interaction between β-cat and its coactivators. This strategy aims to target β-cat directly rather than its upstream factors in the Wnt pathway, thus appearing to be a more promising approach. However, β-cat interacts with the majority of its protein partners through the same binding surface—for example, TCF, APC, and E-cadherin span the entire central arm of β-cat—except for BCL9/B9L, which targets β-cat’s N-terminal arm with essentially no overlap (2). Thus, inhibitors of β-cat’s common binding surface have exhibited substantial adverse effects in animal studies and clinical trials, likely due to interference with off-target interactions. For example, disruption of the interaction between LEF/TCF and β-cat by small molecules is known to elicit serious side effects, including severe bone marrow hypoplasia, anemia, and generalized wasting of treated mice—likely due to the presence of quinone reactive groups in compounds and interrupted homeostatic Wnt signaling in normal hematopoietic and intestinal stem cells (12). PRI-724, a small-molecule inhibitor disrupting the interaction between CBP and β-cat, is under phase 2 trials with daily infusion, but adverse side effects have been reported (11). Thus, there is an unmet medical need to develop a potent, specific β-cat inhibitor targeting the Wnt pathway at the transcriptional level with optimal pharmacokinetics (PK) and tolerable safety profiles.
In a previous study, we developed a stabilized α helix of BCL9 B (SAH-BCL9B) that dissociates the β-cat/BCL9 complex and selectively suppresses β-cat activity (5). However, the SAH-BCL9B’s in vitro potency required further optimization to reach an IC50 (half maximal inhibitory concentration) in the nanomolar range. Moreover, the pharmacological features of this initial peptide, including PK and toxicokinetics (TK), remained unclear. To advance this BCL9-targeted peptide toward clinical use, we conducted extensive chemical modifications and optimizations on the original peptide, discovering a set of potent and selective peptides that targets oncogenic Wnt activity through inhibition of β-cat activity. The newly developed hsBCL9CT series exhibits markedly more robust antitumor efficacy in comparison to the original peptide, SAH-BCL9B, and has encouraging pharmacological features: favorable PK and toxicological properties, improved in vivo half-life, and excellent cell permeability. The hsBCL9CT series can overcome resistance to immune checkpoint therapies in multiple cancer models, showing synergistic antitumor effects with anti–PD-1 (programmed cell death protein 1) antibodies (Abs). Mechanistically, we found that hsBCL9CT-24 can modulate the tumor microenvironment (TME) by inhibiting regulatory T (Treg) cell infiltration via the transforming growth factor–β (TGF-β)–CCL20/CCL22 axis while promoting DC infiltration via the CCL4 axis. These changes, in turn, increase intratumoral infiltration of cytotoxic CD8+ T cells, effector CD8+ cells, and effector CD4+ cells. Given these advantages, the hsBCL9CT peptides could provide promising new therapeutic avenues to treat human cancers.
RESULTS
Identification of hsBCL9CT lead peptides via customized screenings and chemical optimizations
SAH-BCL9B, a cell-permeable all-hydrocarbon α-helical peptide of the BCL9 homology domain 2 (BCL9-HD2), was previously shown to exhibit promising inhibitory effects on the Wnt pathway (5). To enhance in vitro potency and affinity of this peptide, we applied staple incorporation technologies and conducted lead optimization studies in three steps: (i) domain mapping of the full-length BCL9-HD2, (ii) introduction of point mutations in the active region to optimize overall peptide charge, and (iii) optimization of terminal modification and staple sites. On the basis of these design criteria, hydrocarbon stapled BCL9-HD2 (hsBCL9) peptides were designed (13), and 20 overlapping stapled peptides of 7- to 8-mer (hsBCL9CT-01 to hsBCL9CT-20) spanning the 351– to 374–amino acid region of BCL9-HD2 were synthesized to select the most active peptide. Among them, hsBCL9CT-20 exhibited the best profile in in vitro assays and was selected for further modification by addition of known peptide tags and terminal modifications to improve peptide permeability and solubility (14). Structure-activity relationship analyses were conducted to characterize the newly generated peptide series, and hsBCL9CT-24 was found to demonstrate the most potent in vitro activity, exhibiting stronger binding affinity to β-cat in comparison to BCL9-HD2A (control peptide from BCL9-HD2; table S1).
β-Cat binding affinity and the competitive activity of candidate peptides bearing sequence alterations were then determined by two biochemical assays conducted under optimized conditions: homogeneous time resolved fluorescence (HTRF) binding assays and amplified luminescence proximity homogeneous assays (ALPHA) (see Methods for details). The sequences of all control peptides and hsBCL9-related derivatives are included in table S1.
HTRF was first used to determine fluorescence resonance energy transfer between donor and acceptor molecules in close proximity, thus assaying protein-protein interactions and binding affinity in a robust and sensitive manner (15). Both biotinylated BCL9 peptide, conjugated with streptavidin-XL655, and histidine-tagged β-cat, conjugated with Eu-labeled monoclonal Ab, were used. β-Cat protein and biotinylated BCL9-HD2 peptide were cross-titrated to determine conditions leading to optimal, concentration-dependent fluorescent readout (fig. S1, A and B). With an average Z factor (Z′) of 0.8, which fell within the range indicating excellent assay performance and robustness, final concentrations of β-cat and BCL9-HD2 were established to maintain high assay signal and sensitivity. The customized HTRF assay yielded a Kd (dissociation constant) value of 38.3 nM for β-cat/BCL9-HD2A binding (fig. S1C), while hsBCL9CT-24 yielded a Kd value of 4.21 nM (fig. S1D). For comparison, SAH-BCL9B exhibited a Kd value of 192.3 nM (fig. S1E), suggesting that hsBCL9CT-24 has a greatly improved binding affinity for β-cat over SAH-BCL9B.
Subsequently, robust ALPHA competition assays were developed to determine the potency of inhibitors in disrupting the β-cat/BCL9 interaction; assay settings were further modified from previously reported analytical conditions (16). Streptavidin-coated donor beads were conjugated via polyethylene glycol (PEG) linker to biotinylated BCL9-HD2 peptide, while the opposing binding partner, β-cat, was recognized directly by an Ab conjugated to the acceptor bead. The interaction between donor and acceptor beads in standard ALPHA assay converts ambient oxygen to singlet molecular oxygen, with emission resulting if the beads interact within a 200-nm range. Our strategy allows greater surface area of β-cat/BCL9 binding and presumably greater signal intensity (16). Briefly, we applied cross-titrations of β-cat protein, biotinylated BCL9 peptide, and anti–β-cat Ab to optimize signal-to-background ratios of the assay. Constructs BCL9-HD2A and BCL9-HD2B were created to develop optimal assay conditions with a 180-fold preference for signal over background, a marked improvement over previously reported ALPHA assay methods (16). A sigmoidal dose-response curve was then fitted to establish ideal β-cat concentrations (fig. S1F). Kd determination in ALPHA binding assay of BCL9-HD2A and BCL9-HD2B peptides resulted in Kd values of 14.29 and 4.33 nM, respectively (fig. S1G). BCL9-HD2B cross-titrated with 25 and 100 nM β-cat protein also yielded Kd values of approximately 2.25 and 1.89 nM, respectively, and was selected as the peptide control in ALPHA competition assays (fig. S1H). With this assay, the binding affinities of hsBCL9CT-1 to hsBCL9CT-20 were measured, and hsBCL9CT-20 displayed the strongest effect (fig. S1I). The binding affinity of hsBCL9CT-24 was then assessed via ALPHA binding assay, showing a low Kd value of 4.73 nM (Fig. 1A). In comparison, SAH-BCL9B demonstrated a Kd value of 141.6 nM (fig. S1J) and IC50 of 135 nM in recombinant glutathione S-transferase (GST)–tagged β-cat and His-BCL9 assay (5). Other peptides (hsBCL9CT-31 and hsBCL9CT-32; table S1) generated from the hsBCL9CT-24 scaffold also demonstrated marked potency in disrupting the β-cat/BCL9 interaction (fig. S1, K and L).
Fig. 1. In vitro profiling of hsBCL9CT-24.
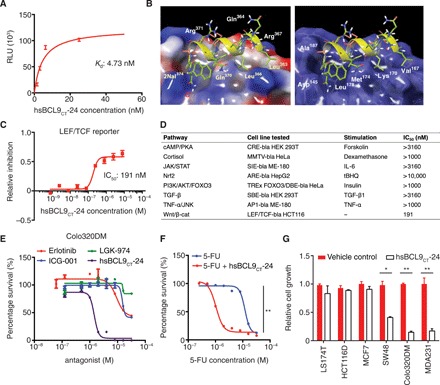
(A) Customized ALPHA assay detecting protein binding interactions between biotinylated hsBCL9CT-24 and His-tagged β-cat. RLU, relative luciferase units. (B) Docking hsBCL9CT-24 into the β-cat hydrophobic pocket (Protein Data Bank: 3SL9) performed by GlideXP Maestro Schrodinger. In the left panel, green labels represent hydrophobic binding sites, while white labels denote hydrophilic amino acids. In the right panel, white labels illustrate amino acids in the β-cat hydrophobic pocket. (C) β-cat reporter assay conducted in LEF/TCF-bla HCT116 cells (Ser45 deletion in one allele of the CTNNB1 gene) treated with hsBCL9CT-24 (IC50 = 191 nM). (D) Table summarizing reporter assay results, denoting hsBCL9CT-24’s specificity in Wnt/β-cat inhibition (IC50 = 191 nM) and lack of off-target effects in other signaling cascades (IC50 > 1000 nM for all). (E) Dose-response curves showing inhibitory effects of the indicated molecules on growth of the Colo320DM cell line: hsBCL9CT-24 (IC50 = 1.45 μM) compared to ICG-001 (IC50 = 15.03 μM), LGK-974 (IC50 = 18.46 μM), and erlotinib (IC50 = 10 μM). (F) Dose-response curves showing Colo320DM cells treated with 5-fluorouracil (5-FU) or 5-FU combined with 2 μM hsBCL9CT-24. Addition of 2 μM hsBCL9CT-24 lowered the IC50 of 5-FU from 12.1 μM to 1 μM. **P < 0.01, two-way analysis of variance (ANOVA). (G) BrdU cell proliferation assay of colon cancer lines (LS174T, HCT116D, SW48, and Colo320DM) and breast cancer cell lines (MDA231 and MCF7) treated with 8 μM hsBCL9CT-24 over 24 hours. *P < 0.05, **P < 0.01, unpaired Student’s t test. Results were denoted as means ± SEM for assays performed in triplicate and repeated twice.
A simulation of the stapled peptide’s selective β-cat docking mechanism was created, and this protein-protein interaction was further investigated by identifying key binding sites in relevant hydrophobic regions (Fig. 1B). To determine whether potency of the hsBCL9CT series can be abolished by these critical amino acid mutations, we rationally designed several hsBCL9CT derivatives (hsBCL9M1 to hsBCL9M7; table S1) to incorporate point mutations and serve as negative control analogs (fig. S1, M to T). Amino acids L366, I369, and L373 present in BCL9-HD2 were shown to drive hydrophobic interactions binding to helices 2 and 3 of the armadillo repeat 1 of β-cat (table S1) (5). A series of mutants with single amino acid alterations for L366 (M5), I369 (M6), and L373 (M7) were found to have similar circular dichroism spectra when compared to hsBCL9CT-24 (fig. S1U), suggesting that the change of these amino acids did not cause substantial alteration of the helical peptide structure. However, these mutants have compromised potency in blocking the interaction between β-cat and BCL9 when compared to wild-type hsBCL9CT-24 (fig. S1Y), demonstrating that these amino acids in hsBCL9CT-24 are essential for full function. In addition to M5 to M7, mutants with multiple amino acid substitutions (M1 to M4) were tested and found to display much lower activity when compared to hsBCL9CT-24 (fig. S1, V to Y). In summary, we identified a series of hsBCL9CT peptides that exhibits robust inhibition against the interaction between β-cat and BCL9.
hsBCL9CT peptides show strong inhibition of Wnt/β-cat pathway activity
We next examined whether these peptides displayed potent inhibitory effects on the Wnt/β-cat pathway. First, the hsBCL9CT peptides were found to be highly permeable and readily taken up by treated cells (fig. S2A). β-Cat transcriptional activity was then measured by LEF/TCF reporter assay in HCT116 cells (Ser45 deletion in one allele of the CTNNB1 gene), and cell viability was determined by cell viability assay in Colo320DM cells. The HCT116 cell line was selected for its aberrant Wnt signaling activation (caused by β-cat mutation), while the Colo320DM cell line was selected for its proliferative dependency on β-cat and BCL9 (4, 5, 17). As expected, hsBCL9CT-24 demonstrated more potent inhibition of β-cat transcriptional activity (Fig. 1C and fig. S2, B and C) when compared to SAH-BCL9B (fig. S2D). Notably, the newly developed peptide exhibited much stronger inhibitory effects on β-cat than the other two Wnt inhibitors currently in clinical trials, ICG-001 (PRI-724 analog) and LGK-974 (fig. S2, E and F) (11). Alteration of essential amino acids abolished hsBCL9CT-24’s function (fig. S2, G to J, and table S1). Furthermore, in coimmunoprecipitation assay, the newly developed peptide displayed an elevated ability to block the interaction between β-cat and BCL9/B9L (fig. S2, K to N). To examine whether hsBCL9CT-24 displayed off-target effects, we conducted multiple reporter assays to investigate whether the peptide could abrogate non-Wnt signaling cascades, including cyclic adenosine monophosphate (cAMP)/protein kinase A (PKA), cortisol, Janus kinase (JAK)/signal transducer and activator of transcription (STAT), Nrf2, phosphatidylinositol 3-kinase (PI3K)/AKT/FOXO3, TGF-β, and tumor necrosis factor–α (TNF-α)/c-Jun N-terminal kinase (JNK) signaling pathways (Fig. 1D and fig. S3, A to G). Notably, hsBCL9CT-24 showed negligible effects on these pathways, with IC50 of >1000 nM.
Consistent with results from LEF/TCF reporter assays, hsBCL9CT-24 proved to be more cytotoxic than the original SAH-BCL9B in Colo320DM cells (fig. S3H), while mutations of key amino acids abolished the effects of hsBCL9CT-24 and hsBCL9CT-35 (fig. S3, I to L). Notably, hsBCL9CT-24 was 12-fold more effective than ICG-001 and LGK-974 in suppressing the cell growth of Colo320DM, a BCL9- and β-cat–dependent cell line (Fig. 1E) (4, 5). In addition to Wnt pathway inhibitors, hsBCL9CT-24 demonstrated greater potency than other targeted therapeutic agents such as erlotinib (Fig. 1E). When tested in combination with a common CRC chemotherapy drug, hsBCL9CT-24 was able to sensitize colon cancer cells to 5-fluorouracil (5-FU) treatment (Fig. 1F). To further confirm specificity of the hsBCL9CT peptide series, we applied the peptides to both Wnt/β-cat–dependent and Wnt/β-cat–independent cancer cells. The growth of five Wnt/β-cat–dependent cell lines (SW48, Colo320DM, MDA231, 4T1, and CT26) was inhibited by hsBCL9CT-24, while four Wnt/β-cat–independent cell lines (MCF7, LS174T, HCT116D, and RKO) were not affected by hsBCL9CT-24, further indicating that the anticancer effects of hsBCL9CT-24 are mediated by blockage of the Wnt/β-cat pathway (Fig. 1G and fig. S3L) (5, 17).
Together, the hsBCL9CT series’ targeted inhibition of the oncogenic Wnt/β-cat pathway exhibited exceptional inhibitory effects on the growth of cancer cells. In addition, the anticancer effects of these optimized peptides demonstrated much greater efficacy in BCL9/β-cat–dependent cells compared to other Wnt inhibitors currently under clinical trial investigation.
hsBCL9CT lead peptides demonstrate favorable PK and TK profiles
In addition to in vitro efficacy, substantial improvement of pharmacological features was a major goal of our peptide optimization. First, the PK and TK properties of hsBCL9CT-24 and hsBCL9CT-35, two lead peptides in the hsBCL9CT series, were evaluated. Both peptides were serially administered in female BALB/c nude mice by intravenous (i.v.), subcutaneous (s.c.), intraperitoneal (i.p.), or intramuscular (i.m.) injections, with mice demonstrating clear dose-dependent trends in PK parameters such as area under the curve (AUC; table S2A). Previous reports have described the ability of double cross-linked peptides to improve half-lives, and this phenomenon was confirmed by hsBCL9CT-35, a double cross-linked peptide that displayed a longer average half-life than hsBCL9CT-24, a single cross-linked peptide (18). The elimination kinetics of hsBCL9CT-24 demonstrated durable plasma half-lives of 1.4, 3.6, 13.0, and 10.6 hours via i.v., i.p., s.c., and i.m. injections, respectively, while hsBCL9CT-35’s half-lives were 7.03 and 9.81 hours via i.v. and i.p. injections, respectively. hsBCL9CT-24 had a bioavailability (F) of 80% (i.p.) and 56% (s.c.), while hsBCL9CT-35 showed a bioavailability of 100% (i.p.). Both hsBCL9CT-24 and hsBCL9CT-35 demonstrated favorable bioavailability and half-life properties, indicating the feasibility of daily dosing (fig. S4, A and E, and table S2A). hsBCL9CT-24 administered at 5, 10, and 15 mg/kg via i.v. injection yielded a Cmax in plasma ranging from 41 to 102 μM, up to 500-fold greater than the compound’s in vitro IC50 in LEF/TCF Wnt reporter assay, and demonstrated AUC values in plasma spanning from 76 to 160 hours × nmol/ml (table S2B). In addition, hsBCL9CT-24 had low rates of clearance, ranging from 0.0397 to 0.0576 liter/hour per kg, and low steady-state volume of distribution (Vss), ranging from 0.0692 to 0.0946 liter/kg. Both the low clearance and extended half-life of hsBCL9CT peptides in mice suggest that efficacious exposure levels are achievable at therapeutic doses in humans.
Subsequently, a dose-escalating study was performed to examine the toxicity profile of hsBCL9CT-24 in the BALB/c model. Mice were administered vehicle control or hsBCL9CT-24 at 10, 15, or 20 mg/kg via i.p. injections, once daily (QD) over 14 days. Increasing dose-dependent AUClast and AUCINF values were observed, and other parameters such as Tmax, Cmax, and terminal t1/2 were assessed across all cohorts (table S2, C and D). Food consumption loss and body weight reduction were both determined in all hsBCL9CT-24 treatment groups throughout this period, with less severity observed in the 10 mg/kg cohort (fig. S4B). Beginning on day 6, all mice treated with hsBCL9CT-24 showed a steady recovery in food consumption and body weight, suggesting an acclimation to drug treatment, and animals recovered to near-baseline levels with continued treatment by the end of the study (fig. S4, B and F). All animals survived the duration of study, and minimal toxicity was observed in the brain, lung, kidney, liver, heart, and spleen tissues of the hsBCL9CT-24–treated animals, as gauged by histology analysis (fig. S4C).
Serum chemistry and hematology studies were performed after a 2-week treatment period, and there was no difference found between animals treated with vehicle control and hsBCL9CT-24 (fig. S4I). Following i.p. administration, systemic exposure to hsBCL9CT-24 at all three dosing concentrations was performed. Aside from the 10 mg/kg cohort, in which AUC on day 14 was much lower than AUC on day 1, the 15 and 20 mg/kg cohorts displayed similar AUC values between days 1 and 14, indicating that drug-induced immunogenicity and metabolism induction may be unlikely to occur after long-term treatment (fig. S4, G and H).
Our previous studies have reported on SAH-BCL9B’s specific inhibition of oncogenic Wnt activity without changing homeostatic function, including Wnt-mediated intestinal tissue homeostasis (5). Here, s.c. injection with hsBCL9CT-24 at 30 mg/kg over 7 days revealed comparable histological features of bone marrow and intestinal tissue in C57BL/6 mice when compared to vehicle control (fig. S4D). Repeated treatment with hsBCL9CT-35 at 30 or 40 mg/kg resulted in good tolerability and no obvious toxicological alterations in the intestine and bone marrow, consistent with our previous findings for SAH-BCL9B (fig. S4K) (5). These results suggest that hsBCL9CT-24 and its derivatives induce negligible adverse effects in vivo.
Preliminary experiments were then conducted to test the stability and purity across three batches of hsBCL9CT-24 synthesized at different times. PK parameters were investigated in BALB/c mice administered hsBCL9CT-24 (5 mg/kg) from all three lots via i.v. or i.p. injections over 36 hours, which yielded consistent results (table S3A). The solubility of hsBCL9CT-24 was then analyzed in water, dimethyl sulfoxide (DMSO), and phosphate-buffered saline (PBS), with purity assessed by high-performance liquid chromatography (HPLC) in varying conditions over 1 month, showing consistency across all fields (table S3, B and C). Plasma stability assays demonstrated that hsBCL9CT-24 exhibited 90.28% remaining after 60 min, while procaine control exhibited only 0.02%; the half-life (T1/2) value of hsBCL9CT-24 in plasma was 307.51 min, approximately 60 times over procaine (table S3D). In summary, the lead peptides of the hsBCL9CT series, including hsBCL9CT-24 and hsBCL9CT-35, have promising PK and TK profiles for further study.
hsBCL9CT-24 shows robust antitumor efficacy across multiple in vivo models
The newly generated peptides’ remarkable improvement in PK enabled us to test in vivo efficacy via common drug delivery approaches, including i.p. and i.v. injections, which was not previously feasible with SAH-BCL9B. The antitumor effects of hsBCL9CT-24 were evaluated in three models: (i) the Colo320DM human CRC xenograft model, (ii) a CRC patient-derived xenograft (PDX) model, and (iii) the CT26 syngeneic model (19).
hsBCL9CT-24 was first tested in the Colo320DM xenograft model. Following s.c. inoculation of tumor cells, nude mice were treated with vehicle control or hsBCL9CT-24 (5, 10, or 15 mg/kg) by i.v. injection, QD for 14 days. Peptide administration resulted in dose-dependent reduction of tumor volume, with tumor growth inhibition (TGI) reaching 49.7 and 58.8% in the 10 and 15 mg/kg cohorts, respectively (Fig. 2A). Expression of CD44 and VEGFA, two downstream targets of Wnt/β-cat signaling with pathological significance, markedly decreased upon hsBCL9CT-24 treatment (Fig. 2B and fig. S4, K to M).
Fig. 2. hsBCL9CT-24 inhibits tumor growth and Wnt pathway activity in multiple CRC mouse models.
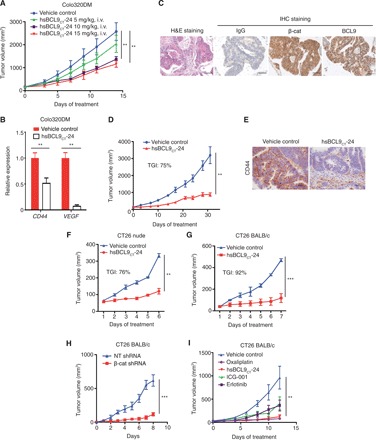
(A) Dose-escalation study of hsBCL9CT-24 treatment in the Colo320DM xenograft model. Four cohorts of female BALB/c nude mice (n = 4 per cohort) were administered vehicle control or hsBCL9CT-24 (5, 10, or 15 mg/kg) via i.v. injection, QD over 14 days. Tumor sizes are displayed as means ± SEM (**P < 0.01). (B) Quantitative reverse transcription polymerase chain reaction (qRT-PCR) measurement of CD44 and VEGF in the Colo320DM tumors following hsBCL9CT-24 treatment (**P < 0.01). (C) Representative images of immunohistochemistry (IHC) staining for β-cat and BCL9 in a CRC patient-derived tumor tissue. H&E, hematoxylin and eosin. (D) Tumor samples in (C) were inoculated in NOD/SCID mice and treated with vehicle control or hsBCL9CT-24 at 15 mg/kg via i.p. injection, QD for 31 days (n = 8 per cohort, **P < 0.01). (E) Representative images of IHC staining for CD44 expression in the tumors from (D). Scale bar, 100 μm. (F) CT26 cells were inoculated in BALB/c nude mice before treatment with vehicle control or hsBCL9CT-24 (n = 6 per cohort) at 25 mg/kg via i.p. injection, QD for 5 days (**P < 0.01). (G) CT26 cells were inoculated in BALB/c mice before treatment with vehicle control or hsBCL9CT-24 at 25 mg/kg via i.p. injection (n = 6 per cohort), QD for 6 days (***P < 0.001). (H) CT26 cells transduced with nontargeting (NT) shRNA or β-cat shRNA were inoculated in BALB/c mice (n = 8 per cohort, ***P < 0.001); day 0 started from 10 days after tumor inoculation. (I) CT26 cells were inoculated in BALB/c mice and treated with either vehicle control, hsBCL9CT-24, or ICG-001 at 25 mg/kg via i.p. injection, erlotinib (25 mg/kg) via oral QD, or oxaliplatin (5 mg/kg) via i.p. and QD over 12 days (n = 4 per cohort, **P < 0.01). Results were denoted as means ± SEM for experiments performed in triplicate, and each experiment was repeated twice. Statistical significance of differences between groups was determined by two-way ANOVA for all tumor growth assays.
Next, hsBCL9CT-24 was tested in PDX models of human CRC. Ten patient-derived CRC tissue samples were stained for β-cat, BCL9, c-Myc, and CD44 (fig. S5A). Most samples exhibited heightened expression of these proteins, confirming the close relevance of the Wnt/β-cat pathway to CRC development. One sample that exhibited strong staining of both BCL9 and β-cat was selected for further investigation (Fig. 2C). The CRC tumor was sliced into fragments and implanted s.c. into the right flanks of nonobese diabetic/severe combined immunodeficient (NOD/SCID) mice; tumor-bearing animals were treated with vehicle control or hsBCL9CT-24 at 15 mg/kg for 31 days. hsBCL9CT-24 significantly reduced tumor volume relative to control throughout the study period, with a TGI of 75% by day 31 (Fig. 2D). Increased apoptosis and decreased cell proliferation were observed in the hsBCL9CT-24–treated tumors compared to the control-treated tumors {48.16% tumor cells were Ki67+ in the control group versus 22.37% in the hsBCL9CT-24 group; 1.67% tumor cells were TUNEL+ (terminal deoxynucleotidyl transferase dUTP nick end labeling positive) in the control group versus 3.94% in the hsBCL9CT-24 group; fig. S5, B to D}. Consistently, CD44 expression was reduced in tumors treated with hsBCL9CT-24 (Fig. 2E).
Last, hsBCL9CT-24 was assessed in the CT26 mouse CRC model established in both immunocompetent (regular BALB/c) and immunodeficient (nude) mice. In nude mice, hsBCL9CT-24 exhibited a comparable potency against the growth of CT26 as compared to the Colo320DM and PDX models, with a TGI of 76% upon 7-day treatment (Fig. 2F). Unexpectedly, the same dose of hsBCL9CT-24 treatment almost fully blocked tumor formation of CT26 in BALB/c mice, which have standard immune systems (TGI of 92% at day 6; Fig. 2G). To confirm that this antitumor effect was Wnt pathway dependent, β-cat expression was suppressed via short hairpin RNA (shRNA), and CT26 tumors with reduced levels of β-cat exhibited marked reduction in tumor growth (Fig. 2H). Consistently, treatment with hsBCL9CT-24 significantly prolonged the survival of animals (fig. S5F). The parental peptide SAH-BCL9B, when delivered by i.p. injection, displayed mild antitumor effects (fig. S5E), while with the same dose, hsBCL9CT-24 proved similar to oxaliplatin and much more effective than ICG-001 and erlotinib (Fig. 2I). Together, hsBCL9CT-24 and its derivatives exhibit robust antitumor effects in vivo by targeting oncogenic β-cat/BCL9 signaling, with effects more pronounced in immunocompetent animals.
Activation of the Wnt pathway correlates with Treg cell infiltration in human cancers
The notable antitumor effects of hsBCL9CT-24 in immunocompetent mice implicate that the regulation of the immune system plays an important, yet unclear, role in mediating the function of hsBCL9CT-24. The oncogenic Wnt pathway has been shown to promote tumorigenicity by maintaining a procancer TME (9, 10, 20). Thus, we examined whether activation of the Wnt pathway correlated with infiltration of immune cells in the TME of CRCs. The amount of CD8+ T cells was significantly reduced in APC-mutant CRCs, indicating that the activation of the Wnt pathway suppressed anticancer immunity in human CRC (Fig. 3A). Unexpectedly, no co-reduction in infiltration of activated DCs, potent activators of CD8+ T cell mobilization and activation, was observed (Fig. 3B and fig. S6A). In contrast, infiltration of Treg cells, potent inhibitors of CD8+ T cells, markedly increased in APC-mutant CRC samples (Fig. 3C). These findings suggest a correlation among aberrant Wnt pathway activation, Treg cell infiltration, and CD8+ T cell inhibition in CRC. To further understand the link between Wnt signaling and Treg cells, we analyzed the correlation between expression of three T cell markers—CD4, CD25 (IL2-RA), and FOXP3 and Wnt pathway genes in a set of 287 human CRC cases. In human colon cancer, coexpression of the above T cell markers is the hallmark of hyper-suppressive Treg cells, which are correlated with poor prognosis of CRC patients (21). The majority of Wnt pathway genes (MolSigDB; see Methods for details) were positively correlated with the expression of CD4, FOXP3, and CD25 in human CRC samples (fig. S6B). Further analysis by hierarchical clustering identified a list of 37 Wnt pathway genes strongly associated with the expression of all three markers (Fig. 3D). To further examine these results, human CRC tumors were stratified into two categories, Treg-hi (CD4-hi, CD25-hi, and FOXP3-hi) and Treg-lo (CD4-lo, CD25-lo, and FOXP3-lo), and the same Wnt pathway genes were markedly up-regulated in the Treg-hi cohort (Fig. 3E and fig. S6C). Last, these results were validated with immunohistochemistry (IHC) staining for β-cat, FOXP3, and CD8 in freshly collected human CRC samples. Treg cell infiltration, as indicated by FOXP3 expression, was markedly up-regulated in tissue samples with β-cat overexpression, while the amount of CD8+ T cells was down-regulated (Fig. 3F). Notably, the positive correlation between Wnt signaling and Treg cell infiltration remained consistent across multiple types of human cancers, including rectal, lung squamous cell carcinoma and adenocarcinoma, and triple-negative breast cancer (fig. S6, D to G). Together, these results demonstrate that the activation of the Wnt pathway is strongly correlated with Treg cell infiltration in human cancers.
Fig. 3. Activation of the Wnt pathway is associated with Treg cell infiltration in human CRC.
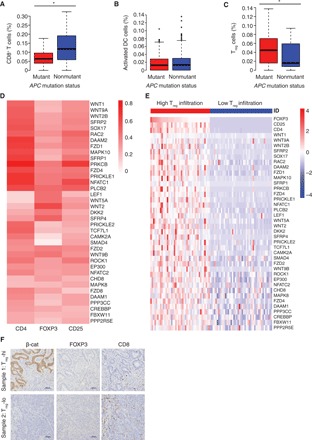
The correlation between infiltration of (A) CD8+ T cells, (B) activated DCs, and (C) Treg cells and APC gene mutation status in human CRC samples was analyzed by CIBERSORT (*P < 0.05). (D) Relative heat map of expression correlation between CD4, CD25, and FOXP3 with a subset of Wnt pathway genes in 287 colon cancer tumors. (E) Relative heat map of Wnt pathway gene expression in colon cancer tumors stratified by Treg cell infiltration level [Treg-hi (CD4-hi CD25-hi FOXP3-hi) versus Treg-lo (CD4-lo CD25-lo FOXP3-lo)]. High infiltration is denoted by the top quartile, while low infiltration is composed of the bottom quartile of the 287 tumors. Each column represents one sample, with samples arranged according to FOXP3 expression level. (F) IHC staining for β-cat, FOXP3, and CD8 in two representative human CRC samples.
hsBCL9CT-24 reduces intratumoral infiltration of Treg cells by inhibition of CCL20 and CCL22
Given the strong clinical relevance between Treg cell infiltration and Wnt pathway activation, we next examined whether blockage of Wnt signaling with hsBCL9CT-24 can functionally modulate Treg cells in the cancer immunologic microenvironment. In the CT26 syngeneic CRC model, treatment of hsBCL9CT-24 led to a marked decrease of intratumoral Treg cell count in the overall CD45+ T cell populations (Fig. 4, A and B). Similarly, hsBCL9CT-35 treatment also decreased Treg cell infiltration in the CT26 tumors (fig. S7A). Consistent with the colon cancer model, reduction of Treg cell infiltration was also observed in the LLC1 (lung cancer) and 4T1 (breast cancer) tumors when treated with hsBCL9CT-24 (Fig. 4, C and D). Notably, hsBCL9CT-24 effectively regulated Treg cells in both BALB/c and C57BL/6 backgrounds. To dissect how hsBCL9CT-24 affects Treg cells in the TME, we conducted coculture studies. The presence of CT26 cancer cells pretreated with hsBCL9CT-24 did not affect survival of Treg cells. However, coculture of hsBCL9CT-24–treated CT26 cells reduced the migration of Treg cells by twofold in an in vitro transwell assay (Fig. 4E). Consistent with these results, coculture of CT26 cancer cells transduced with shRNA targeting β-cat also effectively reduced the migration of Treg cells, further suggesting that activation of the Wnt signaling in cancer cells is required for intratumoral mobilization of Treg cells (Fig. 4F and fig. S7B). Mechanistically, blockage of the Wnt pathway by hsBCL9CT-24 inhibited the expression of TGFB1 in CT26 cells, consequently leading to reduction of CCL20 and CCL22, two TGF-β–dependent chemokines critical for Treg cell recruitment into the TME (Fig. 4, G and H) (22, 23). Collectively, these data indicate that hsBCL9CT-24–mediated blockage of the Wnt pathway suppresses intratumoral infiltration of Treg cells in cancer.
Fig. 4. hsBCL9CT-24 reduces Treg cell infiltration by inhibition of CCL20 and CCL22 in cancer.
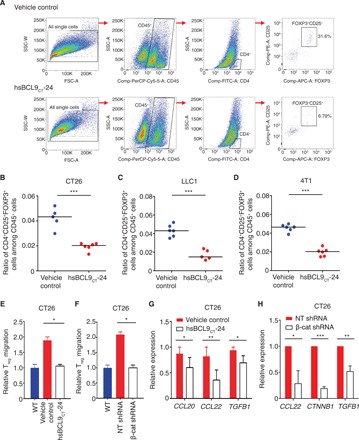
(A) BALB/c mice inoculated with CT26 tumors were treated with vehicle control or hsBCL9CT-24 (20 mg/kg) via i.p. injection, QD over 14 days. Representative flow panels of CD4+CD25+FOXP3+ T cells (Treg) are shown. (B) Ratio of CD4+CD25+FOXP3+ cells among the CD45+ cell populations in the tumors from (A) (***P < 0.001). Mice bearing (C) LLC1 tumors or (D) 4T1 tumors were treated with vehicle control or hsBCL9CT-24 (20 mg/kg) via i.p. injection, QD over 14 days. Percentages of CD25+FOXP3+ Treg cells among the CD45+ cell populations in the respective tumors are shown (***P < 0.001). (E) Migration of freshly isolated Treg cells cocultured with CT26 cells pretreated with or without hsBCL9CT-24 (5 μM) for 24 hours (*P < 0.05). (F) Migration of freshly isolated Treg cells cocultured with CT26 cells transduced with NT shRNA or β-cat shRNA (*P < 0.05). WT, wild type. (G) qRT-PCR measurement of CCL20 (*P < 0.05), CCL22 (**P < 0.01), and TGFB1 (*P < 0.05) in CT26 cells treated with or without hsBCL9CT-24 (5 μM) for 24 hours. (H) qRT-PCR measurement of CCL22 (*P < 0.05), CTNNB1 (***P < 0.001), and TGFB1 (**P < 0.01) expression in CT26 cells transduced with NT shRNA or β-cat shRNA. Results were denoted as means ± SEM for experiments performed in triplicate. Each experiment was repeated twice, and the statistical significance of differences between groups was determined by unpaired Student’s t test.
hsBCL9CT-24 increases cytotoxic Teff cells and overcomes resistance to PD-1 inhibitors
Treg cells have been shown to promote tumor growth by negating the function of cytotoxic CD8+ T cells. In contrast, CD8+ T cells can be primed and activated by intratumoral DCs to inhibit tumor growth. Wnt pathway modulation of DC infiltration was not observed in CRC samples—distinct from melanoma (Fig. 3B). However, inhibition of the Wnt pathway by hsBCL9CT-24 functionally increased intratumoral infiltration of CD103+CD11c+ DCs in CT26 tumors (Fig. 5A and fig. S7C). A similar phenomenon was observed in CT26 tumors with reduced levels of β-cat (Fig. 5B). This recruitment of DCs was likely the result of up-regulation of CCL4, a chemokine that promotes DC homing to tumors (Fig. 5, C and D). Furthermore, because of concurrent reduction of Treg cells and increase in DCs, the amount of infiltrating CD8+ T cells escalated upon hsBCL9CT-24 treatment (Fig. 5, E and F). These CD8+ T cells were also more active and cytotoxic, as denoted by granzyme B positivity (Fig. 5, G and H, and fig. S7D). Treatment using hsBCL9CT-24 led to an over 10-fold increase in effector CD8+ cells, as indicated by expression patterns of CD44+CD62L− (Fig. 5, I and J, and fig. S7E). Alongside elevation of CD8+ cell counts, several other anticancer immune cell populations, including effector CD4+ T cells and T helper 17 (TH17) cells, also surged upon hsBCL9CT-24 treatment (fig. S7, F to H), indicating a comprehensive induction of anticancer immunity in the TME given hsBCL9CT-24 treatment. These results are notable, given previous literary reports that CD4+ and CD8+ effector cell populations are the primary factors determining tumor growth (24).
Fig. 5. hsBCL9CT-24 reactivates anticancer immunity and overcomes resistance to PD-1 inhibitors.
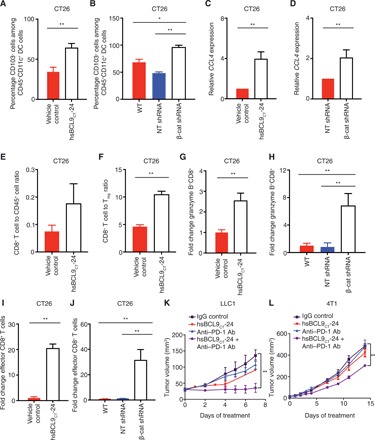
(A) BALB/c mice inoculated with CT26 tumors were treated with vehicle control or hsBCL9CT-24 (20 mg/kg) via i.p. injection, QD over 14 days (n = 4 per cohort). The percentage of CD103+ cells among CD45+CD11c+ DC cells in tumor was analyzed (**P < 0.01). (B) Percentage of CD103+ cells among CD45+CD11c+ DC cells in wild-type (transduced without shRNA), NT shRNA, or β-cat shRNA transduced CT26 tumors (*P < 0.05, **P < 0.01). (C) qRT-PCR measurement of CCL4 expression in CT26 cells treated with vehicle or hsBCL9CT-24 at 5 μM for 24 hours (**P < 0.01). (D) qRT-PCR measurement of CCL4 expression in CT26 cells transduced with NT shRNA or β-cat shRNA (**P < 0.01). (E) Fluorescence-activated cell sorting analysis of the ratio of CD8+ to CD45+ T cells in the tumors described in (A). (F) Ratio of CD8+ cytotoxic T cells over Treg cells in the tumors described in (A) (**P < 0.01). (G) Fold change of granzyme B+CD8+ T cells among the overall CD8+ T cell population before and after hsBCL9CT-24 treatment (**P < 0.01). (H) Fold change of granzyme B+CD8+ T cells among overall CD8+ T cell population in CT26 WT, NT shRNA, and β-cat shRNA transduced tumors (**P < 0.01). (I) Fold change of CD8+CD44+CD62L− cells (effector CD8+ cells) among the overall CD8+ T cell population before and after hsBCL9CT-24 treatment (**P < 0.01). (J) Fold change of effector CD8+ cells among the overall CD8+ T cell population in CT26 WT, NT shRNA, and β-cat shRNA transduced tumors (**P < 0.01). Statistical significance of differences between groups was determined by unpaired Student’s t test. (K) Combination treatment of hsBCL9CT-24 and anti–PD-1 Ab resulted in almost complete regression in the LLC1 model. C57BL/6 mice were inoculated with LLC1 cells via single flank implantation and treated with immunoglobulin G (IgG), hsBCL9CT-24 (25 mg/kg, QD), anti–PD-1 Ab [twice weekly (BIW)], and hsBCL9CT-24 + anti–PD-1 Ab as indicated after tumor volume reached 30 mm3 (n = 4 per cohort) (**P < 0.01). (L) Combination treatment of hsBCL9CT-24 and anti–PD-1 Ab resulted in significant tumor reduction in the 4T1 model. BALB/c mice were inoculated with 4T1 cells via mammary gland inoculation and treated with IgG, hsBCL9CT-24 (20 mg/kg) QD, anti–PD-1 Ab, BIW, and hsBCL9CT-24 + anti–PD-1 Ab as indicated after tumor volume reached 20 mm3 (n = 4 per cohort) (**P < 0.01). Statistical significance of differences in tumor growth assays was determined by two-way ANOVA. Results were denoted as means ± SEM for experiments performed in triplicate, and each experiment was repeated twice.
Notably, intratumoral effector T (Teff) cell inadequacy is a key reason why some tumors are intrinsically resistant to immune checkpoint therapies such as anti–PD-1 inhibitors (25, 26). Both 4T1 and LLC1 tumors are relatively recalcitrant to PD-1 blockade. We therefore examined whether treatment using hsBCL9CT-24 could overcome this resistance through immune-editing functions. While anti–PD-1 Ab alone was unable to inhibit tumor growth in both mouse models, combinatory treatment with hsBCL9CT-24 and anti–PD-1 Ab markedly reduced LLC1 and 4T1 tumor growth (Fig. 5, K and L, and fig. S7, I and J). Although anti–PD-1 Ab alone only led to ~30% TGI in the CT26 model, combinatory treatment with hsBCL9CT-24 and anti–PD-1 Ab resulted in over 95% TGI (fig. S7K). These results suggest that blockage of the Wnt pathway by hsBCL9CT-24 can overcome resistance to anti–PD-1 therapy by increasing active, cytotoxic Teff cell infiltration. The combined treatment of Wnt inhibitors and immunotherapies can provide synergistic antitumor effects in otherwise untreatable cancers.
DISCUSSION
The oncogenic Wnt pathway promotes tumorigenicity by two primary mechanisms: It directly expedites cancer cell growth and suppresses apoptosis, and indirectly promotes tumor formation by negating the function of anticancer immunity. Thus, Wnt pathway inhibitors can kill two birds with one stone and represent an extremely appealing development strategy in cancer therapy. Unfortunately, the development of viable Wnt pathway inhibitors has been a major challenge for decades. In this study, we identified a set of stapled peptides that can potently inhibit β-cat activity by specifically targeting its interaction with the coactivator BCL9. These peptides are effective in suppressing tumor growth across multiple animal models and exhibit favorable pharmacological features and low toxicity in major organs. Because of their potent inhibition of oncogenic Wnt pathways, these peptides can overcome cancer cell resistance to immune checkpoint inhibitors and synergize with the latter to provide robust antitumor effects.
Blockage of the Wnt pathway by disrupting the binding of BCL9 to β-cat is advantageous over many current strategies in targeting the Wnt pathway, as it can circumvent several frequently encountered challenges. First, BCL9/B9L is predominantly found in tumor tissues; over 50% of CRC samples overexpress BCL9 when compared to normal intestine and colon tissues (4, 5). Consistently, genetic deletion of BCL9 and B9L in the murine gut results in no overt phenotypic consequences, indicating that blockade of BCL9 function may not be harmful to normal colon cells (27). Thus, BCL9/B9L-based inhibitors can lead to cancer-specific targeting in CRC, therefore providing an improved safety profile over other β-cat inhibitors, such as LEF/TCF- or CBP-based inhibitors that affect normal tissue due to their ubiquitous expression in cancer and normal tissues. Second, the unique mode of interaction between BCL9/B9L and β-cat provides specificity of inhibition. BCL9 binds to helices 1 and 2 of β-cat, which are distinct from those of LEF/TCF, CBP, or retinoic acid; β-cat interacts with the majority of its binding partners through its entire length, while BCL9/B9L targets β-cat’s N-terminal arm, a binding region that is partially shared with E-cadherin’s region V binding domain (28). Previous studies have shown that the simultaneous mutation of L156A and L159A in β-cat disrupts only BCL9 binding, but not cadherin binding, indicating that key β-cat/BCL9 hydrophobic residues likely do not interfere with the interaction between β-cat and E-cadherin (3). Therefore, targeting the HD2 domain of BCL9/B9L provides a valuable opportunity to block oncogenic β-cat activity specifically in cancer cells. We did not observe substantial adverse effects of hsBCL9CT-24 in major organs. While treatment of hsBCL9CT-24 caused mild loss of body weight and food intake early on, these adverse effects can be reversed in time without termination of treatment. This interesting acclimation implies that our peptide does not affect the stem cell pool of the intestine, therefore representing a major advance in comparison to many other Wnt pathway inhibitors.
Development of all-hydrocarbon stabilized peptides is an ideal approach to interrupting the interaction between BCL9’s HD2 domain and helices 2 and 3 of β-cat. In developing the hsBCL9CT series, we applied α-helix stabilization strategies via all-hydrocarbon cross-linking, which was first described in 2000 by Verdine and colleagues (29). The introduction of an all-hydrocarbon staple has been found to confer high levels of α-helical content associated with a 5- to 5000-fold increase in target affinity, strong protection from proteolytic degradation, robust cell penetration by endocytic vesicle trafficking, extension of in vivo half-life, and specific antagonism of protein-protein interactions in cultured cells (13). These inhibitors may also use energy-dependent pinocytosis to facilitate cell uptake (30). A number of these benefits are exhibited in an all-hydrocarbon stapled peptide currently in clinical trials (clinicaltrial.gov identifier: NCT02909972), targeting p53/MDM2/MDMX interaction for cancer therapy (31). Currently, the all-hydrocarbon stapled peptide of BCL9-HD2 is the only in vivo–validated stapled peptide approach that mimics the actual binding interface between BCL9 and β-cat without requiring a deep pocket surface (5). Thus, all-hydrocarbon stapling is one of the few options that can address the challenges in targeting the β-cat/BCL9 interaction. The emergence of this and other peptide (14) and small molecule–based approaches (32, 33) to targeting the β-cat/BCL9 interface highlights the clinical translation potential of this pharmacological strategy to achieve efficacious and tolerable Wnt inhibitors.
Development of viable Wnt inhibitors could spur more therapeutic options for cancers that are currently difficult to target, especially those with aberrant activation of the Wnt pathway. One of the best examples is CRC; over 90% of sporadic CRCs contain mutations in Wnt signaling, several components of which function as either tumor suppressors or oncogenes. Nearly half of all CRCs develop to late stage by diagnosis, and there are limited therapeutic options. Patients are routinely administered FOLFOX/FOLFIRI chemotherapy or biological regimens, including epidermal growth factor receptor (EGFR)/vascular endothelial growth factor (VEGF) inhibitors such as Avastin and Erbitux (34, 35). However, only 50% of biomarker-matched patients (wild-type KRAS) respond to either drug, demonstrating a huge unmet medical need for new therapeutics (36). Given the prevalence of Wnt pathway activation in CRC, the hsBCL9CT peptide series could be a promising new approach to targeting CRC. To further enhance hsBCL9CT’s efficacy in clinical applications, additional studies should be conducted to identify biomarkers (e.g., certain Wnt pathway components) that can select patients more likely to respond to BCL9-targeted therapy.
In addition to targeted therapy, immune checkpoint inhibitors have recently emerged and appear highly promising for cancer treatment. Unfortunately, while a small subset of patients displays unprecedented, durable response to immunotherapy, the majority remain unresponsive. It has been shown in human CRC that high frequencies of genomic disorder (e.g., MSI-high) could be predictive of patients’ response to immunotherapy (37). However, only a small subset of CRC patients are considered MSI-high (38). In this study, we found that tumor infiltration of Treg cells is significantly increased in CRC patients harboring APC mutations. Tumor infiltration of Treg cells is a hallmark of immune suppression in many cancers. Therefore, our findings suggest that one can effectively enhance anticancer immunity in the majority of CRC patients by blocking Wnt signaling, because over 80% of all CRC patients have APC mutations (38). Using hsBCL9CT-24, we found that blockage of the oncogenic Wnt pathway can reduce Treg cell infiltration and overcome resistance to anti–PD-1 Ab in multiple cancer models. These findings are consistent with recent reports that Wnt pathway activation is linked to primary resistance in immunotherapies (26). A recent report has also claimed that apoptotic Treg cells can significantly affect Teff cell exhaustion by secreting certain cell components (39). The use of hsBCL9CT-24 peptide primarily reduces Treg cell infiltration without affecting Treg cell survival, making it an ideal strategy to reactivate intratumoral Teff cells and transform a “cold” immune microenvironment into a “hot” one.
In addition to Treg cells, DCs represent another important component of the immune system that modulates cancer progression. While it has been shown in melanoma that tumor-intrinsic Wnt signaling can inhibit the activation and infiltration of DCs and therefore promote immune evasion of cancer cells (9, 10, 20), we did not observe a significant correlation between DC infiltration and APC mutations in CRCs. Despite these emerging tissue specificities, blockage of the Wnt pathway in cancer cells by hsBCL9CT-24 can simultaneously suppress Treg cell infiltration while increasing DC infiltration through different mechanisms, resulting in a marked elevation of active CD8+ T cells in colon cancer models. As expected, a decrease of CD8+ cytotoxic T cells is associated with activation of β-cat in human CRC (38) and directly contributes to immune evasion of cancer cells. Our peptides could be applied to reset this disordered immune TME in CRC.
Although the Wnt pathway is a well-documented signaling cascade that causes growth and proliferation in numerous cancers, no Wnt inhibitors in clinical stages or beyond have demonstrated robust specificity and minimal tissue toxicity due to the critical nature of canonical Wnt signaling. Here, we report a set of stapled peptides that can effectively target the Wnt/β-cat pathway to suppress tumor growth and reactivate anticancer immunity. Given their favorable pharmacological features, minimal toxicity, and robust anticancer efficacy, hsBCL9CT-24 and the associated peptide series may be used to treat a wide range of human cancers.
METHODS
Schrodinger Maestro modeling
Molecular model generation and visualization was achieved through Schrodinger Maestro 10.4, performed according to the standard protocol. BCL9, β-cat, and TCF4 (3SL9) protein structures were imported from PDB (Protein Data Bank), and the β-cat molecule was optimized for glide docking with Protein Preparation Wizard (Prep Wizard). hsBCL9CT-24 was constructed in Maestro and converted to a three-dimensional structure through LigPrep, retaining stereochemical properties. Subsequently, glide and receptor grid generation were applied at the β-cat N′ helices 2 and 3 regions, followed by glide and ligand docking of hsBCL9CT-24 at its binding site for visualization.
Peptide synthesis
Peptides were produced in AnaSpec, CA, according to previous protocols (5). Synthesis and purification of peptides were measured by analytical HPLC and mass spectrometry (MS).
Cell culture and T cell expansion
Cell lines 4T1, Colo320DM, SW48, HCT116D, CT26, LLC1, LS174T, MCF7, MDA231, and RKO were purchased from the American Type Culture Collection (ATCC) and cultured according to the supplier’s recommendations, supplemented with 10% fetal bovine serum (FBS) and antibiotics. Treg cells were expanded and cultured with the T Cell Activation/Expansion Kit for mice (130-093-627, Applied Miltenyi Biotec) according to the supplier’s instructions. Cells (2 × 106) were suspended with 20 μl of Anti-Biotin MACSiBead Particles in 2 ml of RPMI 1640 (SH30809.01, Applied HyClone), supplemented with 10% FBS (10099-141, Applied Gibco), interleukin-2 (IL-2) (1000 U/ml, 402-ML-020, Applied R&D Systems), and TGF-β (5 ng/ml, Applied R&D Systems, code no.: 7666-MB-005) per well in a 24-well plate. Cells were seeded 1 day before treatment with the respective hsBCL9CT peptides and incubated for 24 hours at 37°C and 5% CO2, with DMSO vehicle as a peptide control.
Lentivirus production and infection
Lentiviruses were prepared according to the manufacturer’s protocol (GIPZ Lentiviral shRNA, Dharmacon). CT26, Treg, and CD8+ cells were infected by β-cat shRNA lentiviruses according to the manufacturer’s protocol using the Dharmacon GIPZ Lentiviral shRNA Kit (GE Healthcare). The nontargeting (NT) lentiviral shRNA construct expresses an shRNA sequence with no substantial homology to any mammalian transcript, providing a negative control. Cells were infected with NT, β-cat shRNA1, and β-cat shRNA2 lentiviruses as described in our previous studies, and β-cat protein level was evaluated by immunoblotting (5).
Cell proliferation and cell viability assays
Cell proliferation of CD8+, CT26, Treg, 4T1, CT26, LS174T, MCF7, MDA231, and RKO cells was analyzed by bromodeoxyuridine (BrdU) enzyme-linked immunosorbent assay (ELISA) (Roche) according to the manufacturer’s instructions. Cells (5000 per well) were seeded in 50 μl of Opti-MEM medium (Invitrogen), while 0.5 μl of each compound at 200× was diluted in 50 μl of 2% FBS Opti-MEM medium. BrdU labeling solution (10 μl per well) was added if the cells were cultured in 100 μl per well (final concentration, 10 μM BrdU), and cells were reincubated for an additional 2 to 24 hours at 37°C. Sample absorbance was measured using an ELISA reader at 450 nm (reference wavelength of 690 nm). Each experiment was repeated three times.
Cell viability of Colo320DM and CT26 was determined (CellTiter-Glo, Promega) according to the manufacturer’s protocol. Cells (5000 per well) were seeded in 50 μl of Opti-MEM medium (Invitrogen), while 0.5 μl of each compound at 200× was diluted in 50 μl of 2% FBS Opti-MEM medium. The assay plate was incubated for 72 hours at 5% CO2 and 37°C. CellTiterGlo reagent (100 μl) was added, and 100 μl of this final mixture was analyzed for luminescence according to the manufacturer’s recommendations. Assay readout was determined by Enspire (PerkinElmer). Each experiment was repeated three times.
Testing compounds included hsBCL9CT-24 [trifluoroacetic acid (TFA) salts], LGK-974 (Selleck), ICG-001 (Selleck), erlotinib (LC Laboratories), 5-FU (ApexBio), and oxaliplatin (ApexBio). GraphPad Prism was used for data analysis.
LEF/TCF reporter assay
Reporter activity was measured with the SelectScreen Cell-based Pathway Profiling Service (Life Technologies) with GeneBLAzer. hsBCL9CT-24 (TFA, HCl, and C2H3O2 salts), hsBCL9CT-35, ICG-001 (Selleck), and LGK-974 (Selleck) dose-dependent inhibition curves were analyzed in Wnt/β-cat signaling pathways via Reporter CellSensor LEF-TCF-bla HCT116 (Invitrogen). All reported data represent triplicate averages.
CellSensor LEF/TCF-bla HCT116 cells contain a β-lactamase reporter gene under the control of the β-cat/LEF/TCF response element stably integrated into HCT116 cells (Life Technologies). LEF/TCF-bla HCT116 cells were shown to constitutively express β-lactamase, which can be further stimulated with mouse Wnt3a. There are seven copies of the LEF/TCF consensus binding sequences, and this cell line can be used to detect agonists and antagonists of the Wnt/β-cat signaling pathway. In the studies reported here, IC50 determination used a standard 10-point titration curve with threefold dilution, according to the best practices for accurate IC50 determination reported in the literature (40). LEF-TCF-bla HCT116 cells were thawed and prepared as described above for the Activator Screen. Thirty-two microliters of cell suspension was added to each well of a 384-well poly-d-lysine assay plate. Cells in assay media were then incubated for 16 to 24 hours in the plate at 37°C/5% CO2 in a humidified incubator. Four microliters of a 10× serial dilution of ICG-001 (control inhibitor starting concentration of 25,000 nM) or other compounds was added to appropriate wells of the plate, before 4 μl of assay media was added to each well to bring the final assay volume to 40 μl. The plate was incubated for 5 hours at 37°C/5% CO2 in a humidified incubator, and 8 μl of 1 μM substrate loading solution was added to each well before each plate was incubated for 2 hours at room temperature. Plates were read on a fluorescence plate reader, and each experiment was repeated three times.
Data analysis: Background-Subtracted Fluorescence (Fl = Fluorescence Intensity): Fl Sample − Fl Cell-Free Ctrl; Emission Ratio (using values corrected for background fluorescence): Coumarin Emission (460 nm)/Fluorescein Emission (530 nm); Response Ratio (Act. = Activation): Emission Ratio Compound/Emission RatioNo Act. Ctrl
Immunoblotting
Immunoblotting was performed as described in our previous studies, using the following primary Abs: BCL9 (ab37305, Abcam), BCL9L (AF4967, R&D Systems), β-cat (CAT5-H10, Zymed), fluorescein isothiocyanate (FITC; ab19224, Abcam), and β-actin (ab8226, Abcam) (5). Horseradish peroxidase (HRP)–conjugated secondary Abs were purchased from Santa Cruz Biotechnology and SouthernBiotech. Each experiment was repeated three times.
Coimmunoprecipitation
Coimmunoprecipitation was performed as previously described (5). Briefly, cells were lysed in 50 mM tris, 150 mM NaCl, and 1% CHAPS buffer containing protease and phosphatase inhibitors. Lysates were precleared with protein A/G PLUS-Agarose beads (Santa Cruz Biotechnology) for 3 hours, followed by overnight incubation at 4°C with the respective Abs. Agarose A/G beads were then added for 4 hours, pelleted, and washed as described (5). Each experiment was repeated three times.
IHC studies and scoring
IHC staining was performed on FFPE (formalin-fixed, paraffin-embedded) tissue samples under the standard protocol (ChemPartner). Samples were collected during passage of 10 CRC PDX models in Colo320DM BALB/c nude mice, administered hsBCL9CT-24 treatment over 21 days. Additional analyses were also performed on tumor samples from CT26 BALB/c syngeneic mice administered hsBCL9CT-24. Patient samples were collected from 10 adenocarcinoma patients (moderate, metastatic, and poor differentiation) according to the standard protocol for optimal selection of candidates tested in PDX mouse models.
Sections at 4 μm in thickness were prepared and dried in thermotanks before staining, followed by deparaffinization and rehydration via sequential washing with xylene, graded ethanol, and PBS. Samples were then subjected to high temperature–induced epitope retrieval in target retrieval solution [10 mM citrate buffer (pH 6.0)], 3% hydrogen peroxide blocking buffer (in PBS), and Ultra V Block. During protein blocking, slides were treated with 5% goat serum in 1× tris-buffered saline, 0.05% Tween 20 for 30 min at room temperature. Slides were incubated overnight at 4°C with 200 μl of the following primary Abs: β-cat (Abcam, ab32572, 1:1000), BCL9 (Abcam, ab37305, 1:200), c-Myc (Abcam, ab86356, 1:50), CD4 (Affymetrix eBioscience,149766, 1:100), CD8α (Affymetrix eBioscience, 140808, 1:200), CD44 (Abcam, ab157107, 1:500), FOXP3 (Spring Bioscience, M3974, 1:25), Ki67 (Abcam, ab15580, 1:200), vascular endothelial growth factor A (VEGFA) (5 μg/ml, LS-B10263, LS Biosciences), PD-1 (5.69 μg/ml, LS-B2519, LS Biosciences), or rabbit immunoglobulin G (IgG; Beyotime, A7016) as negative control. Slides were then incubated with HRP-conjugated secondary Abs for 30 min at room temperature: EnVision+/HRP Rabbit Ab (Dako 4003, lot 10069185), rabbit anti-FITC Ab conjugated with HRP (Hypoxyprobe HP2-100), or peroxidase-conjugated Affinipure rabbit anti-rat Ab (Jackson ImmunoResearch 312-035-003, lot 107245). Color development was performed with DAB Chromogen (Dako), and sections were then counterstained with Harris hematoxylin (Leica), differentiated with HCl-alcohol (70%) for 1 to 2 s, dehydrated, and mounted for detection (UltraVision Quanto Detection System).
Sample scoring was determined independently by two observers blinded to clinical data. Each specimen received a composite score assessing both the percentage of stained cells (0 to 100%) and staining intensity. The size of the positively stained compartment was estimated and classified on a five-point positive range score as follows: grade 0, 0 to 5%; grade 1, 6 to 25%; grade 2, 26 to 50%; grade 3, 51 to 75%; and grade 4, >75%. The positive extent score was determined as follows: 0, no staining; 1, light yellow; 2, brown; and 3, dark brown. The positive range score and positive extent score were subsequently added, yielding a composite quantitative score: <2, negative (−); 2 to 3, slight positive (+); 4 to 5, moderately positive (++); and 6 to 7, strongly positive (+++). The investigator was blinded to the group and labeling allocation during the experiment. Representative images were selected and depicted in figures at ×40 magnification as indicated (viewed via an inverted microscope: Olympus CKX31SF).
TUNEL assay
TUNEL assay was performed according to the standard protocol (R&D system). Briefly, slides were baked for 60 min at 60°C, deparaffinized, and rehydrated through xylene and graded alcohol incubation. Tissues were pretreated with protein digestion enzyme or proteinase K (20 μg/ml) for 15 min, washed, and quenched in 3% H2O2 in PBS. Equilibration buffer was applied for 10 s, followed by incubation with 55 μl of TdT enzyme in a humidified chamber at 37°C for 1 hour. Stop/wash buffer and anti-digoxigen conjugate were subsequently applied, and color was developed in peroxidase substrate before washing in a coplin jar. Counterstaining was performed with hematoxylin and 0.25% HCl-alcohol before dehydration and mounting for visualization. TUNEL slides were scanned by Aperio ScanScope and divided into four quadrants in the software; two of eight random rectangular regions of size 500 μm × 500 μm (0.25 mm2) were selected and counted for positively stained cells. The investigator was blinded to the group and labeling allocation during the experiment.
Transwell migration assay
In vitro cell migration assays were performed with Transwell chambers (8 μm pore diameter; Corning Costar), as described in our previous studies (5). CD8+ cells were individually analyzed, as were Treg cells, which were expanded as described above and cocultured with CT26 cells. The reported data represent the average of three independent experiments performed in triplicate. Each experiment was repeated three times.
Flow cytometry analysis
At experiment termination of syngeneic murine models, tumors were cut and digested in a digestion cocktail (collagenase and deoxyribonuclease). Harvested cells in aliquots of up to 1 × 106 cells per 100 μl were dispensed into fluorescence-activated cell sorting tubes and stained for flow cytometry according to the standard protocol (20, 22). CD4, CD8, FOXP3, and Treg cells analyses were performed as the following (9): Tumor cell suspensions were incubated with CD16/32 Ab (BioLegend) at 4°C to block FC receptors. Cells were then stained with CD4-FITC (eBioscience, clone GK1.5), CD8a-allophycocyanin (eBioscience, clone 53-6.7), CD3–peridinin chlorophyll protein (PerCP)–Cy5.5 (BioLegend), FOXP3-allophycocyanin (eBioscience), CD25-phycoerythrin (PE) (BioLegend), CD45-PeCP.Cy5.5 (eBioscience), CD44-PE (eBioscience), CD62L-eFluor 450 (eBioscience), interferon-γ (IFN-γ)–PE (PeproTech), or granzyme B–eFluor 450 (eBioscience). Data acquisition was performed with FACSAria I followed by analyses with FlowJo software (Tree Star). To identify Treg cells, the following gating strategies were used: (i) selection of live single-cell leukocytes [side scatter (SSC)–W/H low, anti-CD45+], (ii) selection of CD4+ T cells (anti-CD4+, anti-CD8−), and (iii) selection of CD25+FOXP3+ Treg cells (anti-CD25+, anti-FOXP3+). The investigator was blinded to the group and labeling allocation during the experiment. To identify TH17 cells, the following gating strategies were used: (i) selection of live single-cell leukocytes [side scatter (SSC)–W/H low, anti-CD45+] and (ii) selection of CD194+CD196+ T cells (anti-CD194+, anti-CD196+), CD194–Brilliant Violet 421 (BioLegend), and CD196-PE (eBioscience). To identify DCs, the following gating strategies were used: (i) selection of lymphocyte cells, (ii) selection of CD11c+ lymphocytes and CD11c+ eFluor 450 (eBioscience), and (iii) selection of CD103+ T cells (anti-CD103) and CD103-PE (eBioscience). Each data point was performed in triplicate, and each experiment was repeated twice.
Efficacy of hsBCL9CT peptides in mouse xenograft models
Treatment was conducted in accordance with the standard protocol and regulations (The Association for Assessment and Accreditation of Laboratory Animal Care International) approval, and mice were routinely monitored for any effects of tumor growth and treatments on normal behavior such as mobility, visual estimation of food and water consumption, body weight gain/loss, eye/hair matting, and other abnormal effects. Tumor size and volume (V = 0.5ab2, where a and b are the long and short diameters of the tumor, respectively) were recorded every other day in two dimensions via caliper measurements. The T/C percent value was calculated by taking the ratio of the mean volume in treated and control groups on any given day.
Colo320DM colon cancer cells (ATCC) were cultured according to the standard protocol (5) at Viva Biotech and harvested for s.c. inoculation (5 × 106 cells in PBS) in the right flank region of BALB/c female nude mice (SLAC, SCXK2013-0018, certification 20130018040083) at 6 to 8 weeks of age. Mice without developing tumors were excluded before grouping. A total of 16 mice were separated into four randomized cohorts (n = 4), with treatment beginning 14 days after inoculation (mean tumor size of 156.78 mm3). These xenograft murine models were treated by i.v. injection with vehicle control (2.5% DMSO in sterile water) or hsBCL9CT-24 peptide (5, 10, and 15 mg/kg) with daily doses over 14 days. Tumor weight was measured at the end of study.
PDX murine models were created in NOD/SCID male mice according to the standard protocol (SLAC, SCXK2013-0018, certification 2013001812168) in two randomized arms (n = 8; Viva Biotech). CRC patient tumors selected for the xenograft model demonstrated high expression levels of β-cat, BCL9, c-Myc, and CD44 in IHC staining and were sectioned into 3 mm × 3 mm × 3 mm fragments before s.c. implantation in the right flank region. Treatment was administered 18 days following transplantation (mean tumor size of 134.77 mm3) via i.v. injection with vehicle control (4% ethanol, 8% Tween 80 in PBS) or hsBCL9CT-24 peptide (15 mg/kg), with i.p. daily doses over 31 days. Tumor mass was collected and fixed in 10% formalin at the conclusion of study for further analyses. The investigator was blinded to the group allocation during the experiment. Statistical analysis was determined by one-way analysis of variance (ANOVA) (GraphPad Prism), and differences between groups were considered significant when the P value was less than 0.05.
MTD and antitumor efficacy in syngeneic mouse models
All experiments were approved by IACUC and performed with strict adherence to a series of Document and SOP relative to animal ethics and welfare. Mice were housed in specific pathogen–free conditions (ChemPartner, purchased from Vital River Lab Animal Technology Co. Ltd.). Two hours following the final treatment, mice were euthanized and necropsied for further study, including cardiac puncture for 500 μl of blood, tumor tissue collection, and major organ separation and fixation.
Cultured CT26 colon cancer cells (ATCC) were harvested, resuspended in PBS at 1 × 106 cells/ml with viability of >90%, and s.c. implanted in the right flank of female, 6- to 8-week, pathogen-free BALB/c mice (1 × 106 cells/ml). Any mouse without a developing tumor was excluded before grouping. Two randomized cohorts (n = 8) with tumor size between 80 and 120 mm3 were administered vehicle control (i.p., 4% ethanol, 8% Tween 80 in PBS) or hsBCL9CT-24 (20 mg/kg, i.p., QD) 14 days after inoculation. Four randomized cohorts (n = 5) with tumor size between 30 and 50 mm3 were administered vehicle control or inhibitor; hsBCL9CT-24 and ICG-001 were dosed at 25 mg/kg i.p. QD, and erlotinib was dosed at 25 mg/kg orally QD for 12 days in the CT26 BALB/c mouse model. In the combination arm, mice were treated with hsBCL9CT-24 24 hours before administration of anti–PD-1 Ab (Bio X Cell, BE0146-25MG, InVivoMAb anti-mouse PD-1, clone RMP1-14). In the LLC1 lung cancer model, 5-week female C57BL/6 mice were inoculated with LLC1 cells via single flank implantation and treated as indicated after tumor volume reached 30 mm3 (n = 4 per cohort). BALB/c mice were inoculated with 4T1 cells via mammary gland inoculation and were treated as indicated after tumor volume reached 20 mm3. Mice were grouped into four randomized cohorts (n = 4) and given IgG control, anti–PD-1 Ab [10 mg/kg, i.p., twice weekly (BIW)], hsBCL9CT-24 (20 mg/kg, QD), or combination arm [hsBCL9CT-24 (20 mg/kg, QD) + anti–PD-1 Ab] via i.p. injection. In the combination arm, mice were treated with hsBCL9CT-24 24 hours before administration of anti–PD-1 Ab (10 mg/kg, BIW). Each study was repeated twice.
Tumor volume was measured every day or three times per week using digital calipers and calculated on the basis of the following formula: tumor volume = (length × width2)/2. Murine body weights were recorded after tumor measurement, and relative change in body weight (RCBW) of each mouse was calculated according to the following formula: RCBW% = (BWi − BW0)/BW0 × 100%, in which BWi represents the body weight of a mouse at day i, and BW0 represents the body weight at day 0. Dosing was immediately terminated once a mouse experienced ≥15% loss in body weight. Tumor growth inhibition rate (TGI%) per dosing group was calculated according to the following formula: TGI% = [1 − (TVi − TV0)/(TVvi − TVv0)] × 100%, in which TVi represents the mean tumor volume of a dosing group on a specific day, TV0 is the mean tumor volume of a dosing group on day 0, TVvi is the mean tumor volume of vehicle group on a specific day, and TVv0 is the mean tumor volume of vehicle group on day 0. The investigator was blinded to the group allocation during the experiment. Statistical analyses of murine body weight, RCBW, and tumor volume per group were completed via Microsoft Excel 2007. Tumor size of >1000 mm3 was set as the endpoint, and tumor/organ samples were collected from selected mice at the end of the study.
PK profiling
PK analysis of hsBCL9CT-24 and hsBCL9CT-35 was conducted in 35 male ICR outbred mice (Shanghai SLAC Laboratory Animal Co. Ltd.) in accordance with the standard protocol (Viva Biotech) and ethical regulations (IACUC). hsBCL9CT-24 and hsBCL9CT-35 were administered in the tail vein of murine models through i.v. bolus and i.p. injections. Positive control vehicles were prepared with 10% DMSO and 90% deionized H2O to generate a mixed solvent, with peptides tested at nominal concentrations of 0.5 mg/ml (incubated at room temperature before administration). Blood samples (300 μl) were collected periodically at the retro-orbital vein during 0.25, 1, 2, 4, 6, 7, 12, and 24 hours after dose, followed by plasma separation for pending bioanalysis. Additional details are described in Supplementary Materials and Methods.
Analytic concentrations were determined through LC-MS/MS (Agilent 1100 HPLC, AB Sciex API 4000). The internal standard osalmid (Sigma), acetonitrile (MeCN), and all solvents and chemicals met or exceeded analytical grade standards. MS was completed through electrospray ionization and positive MRM (multi reaction monitor) scans, while chromatographic conditions were set as follows: column [Thermo Betasil C18 (2.1 mm × 50 mm, 5 μm)], mobile phase A (0.1% HCOOH in H2O), mobile phase B (0.1% HCOOH in MeCN), elution rate (500 μl/min), temperature (40°C), and injection volume (15 μl). Standard stock solution was prepared with an analyte (1 mg/ml) in DMSO (70% MeCN in UP H2O), with calibration samples serially diluted to 10, 50, 100, 500, 1000, 2000, and 3000 ng/ml, while quality control final concentrations were spread over 40, 800, 1600, and 2400 ng/ml. The investigator was blinded to the group allocation during the experiment. Standard PK parameters were calculated by noncompartmental analysis modules in WinNonlin Professional v5.2 (Pharsight, U.S. Food and Drug Administration–certified) and included area under curve (AUClast and AUCINF), terminal half-life [terminal t1/2 = ln(2)/λz], maximum plasma concentration (Cmax), time to reach maximum plasma concentration (Tmax), clearance (CL), volume of distribution (Vz), apparent volume of distribution at steady state (Vss), and mean residence time from time of dosing to infinity (MRTINF). Bioavailability (F) was calculated as follows: F = [(AUCint(i.p.) × dosei.v.)/(AUCint(i.v.) × dosei.p.)] × 100%. Each data point was performed in triplicate, and each experiment was repeated twice.
Analysis of gene expression in human cancers
Tumor sample gene expression data were obtained from The Cancer Genome Atlas (TCGA), including 287 colon adenocarcinoma (COAD) samples, 95 rectum adenocarcinoma (READ) samples, 517 lung adenocarcinoma (LUAD) samples, 501 lung squamous cell carcinoma (LUSC) samples, and 97 triple-negative breast cancer (TNBC) samples. Tumor purity and estimation of CD4+ or CD8+ infiltration in COAD, READ, LUAD, LUSC, and BRCA were performed as previously described (41). Wnt pathway genes were downloaded from the Broad Institute for analyses at the following link: http://software.broadinstitute.org/gsea/msigdb/cards/KEGG_WNT_SIGNALING_PATHWAY.
In COAD, Spearman correlation analysis was conducted to determine the correlation of each Wnt pathway gene with the Treg cell markers CD4, CD25, and FOXP3 across the 287 samples. Subsequently, hierarchical clustering was conducted according to the correlation coefficient of each Wnt pathway gene and Treg cell marker. A subset of Wnt pathway genes was found to be positively correlated with the expression of all three Treg cell markers. The expression correlation between genes within this subset and Treg cell markers was then calculated in the READ, LUAD, LUSC, and TNBC tumor samples. To build a heat map of the expression of these Wnt pathway genes in COAD, the TCGA COAD tumor samples were first stratified according to Treg cell infiltration (determined by the expression level of Treg cell markers CD4, CD25, and FOXP3). Expression of the Wnt pathway genes was then plotted for tumors with high Treg cell infiltration (Treg-hi; top quartile of Treg cell marker expression), tumors with low Treg cell infiltration (Treg-lo; bottom quartile of Treg cell marker expression), and others (either top or bottom quartile of Treg cell marker expression).
APC mutation data were obtained from TCGA cbioportal (www.cbioportal.org), and CIBERSORT (https://cibersort.stanford.edu) was used to estimate tumor immune cell infiltration. COAD samples were separated into two APC mutated and nonmutated cohorts, and t test was used to evaluate CD8+ T cell and Treg cell infiltration difference between APC mutated and nonmutated samples.
Statistical analysis
Data were presented as means ± SEM unless otherwise indicated. Data were analyzed by Student’s t test when comparing two groups. Data of multiple-dose drug treatment and tumor growth were analyzed by two-way ANOVA. Survival times of mouse groups were compared using log-rank test and analyzed for 30 days spanning tumor sizes from 100 to 1000 mm3 in CT26 syngeneic mice as treated above.
Supplementary Material
Acknowledgments
We thank LabCentral (Cambridge, MA, USA) and Harvard Life Lab (Boston, MA, USA) for providing laboratory spaces to perform some preliminary in vitro studies and WntRx for providing materials to perform some preliminary biochemical experiments. Funding: This study was funded by Projects on Science and Technology Commission of Shanghai: 18ZR1403900 (to D.Z.), the National Natural Science Foundation of China (81872895 to D.Z.), the National Natural Science Foundation of China (31871369 to Y.-X.F.), the Zhejiang Provincial Natural Science Foundation of China (LD19H160002 to Y.-X.F.), and the National Overseas Talent Program (to D.Z. and Y.-X.F.). Author contributions: M.F. and L.X. performed cell culture, expansion, migration, proliferation, and quantitative reverse transcription polymerase chain reaction analysis. J.Q.J. executed in vitro studies and prepared the manuscript. T.X. and S.M. contributed bioinformatics analysis. X.W. performed cancer cell proliferation analysis. J.C. performed flow cytometry analysis and in vivo studies. M.L. designed immunology studies and wrote the manuscript. X.H. performed the FACS study in LLC1 model. C.C. and A.X.Z. analyzed the data and provided pathology support for IHC. S.R. executed computational chemistry studies. Y.-X.F. designed in vivo studies, analyzed the data, and wrote the manuscript. D.Z. designed the studies, executed experimentation, oversaw and analyzed the data, and wrote the manuscript. Competing interests: C.C., D.Z., and Y.-X.F. were consultants of WntRx Pharmaceuticals. The other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. The datasets generated during and/or analyzed during the current study are available in the figshare repository at https://doi.org/10.6084/m9.figshare.5624863.v1.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/5/eaau5240/DC1
Supplementary Materials and Methods
Fig. S1. Biochemical profile and LC-MS analysis of hsBCL9CT peptides.
Fig. S2. Cellular uptake, Wnt reporter, and coimmunoprecipitation analysis of hsBCL9CT peptides.
Fig. S3. Selectivity in multiple signaling pathways and antiproliferation assay of hsBCL9CT peptides.
Fig. S4. PK, toxicology, and histology analysis of hsBCL9CT-24 and hsBCL9CT-35.
Fig. S5. IHC staining in patient tumor tissues and PDX tumor models.
Fig. S6. Wnt pathway gene expression is correlated with CD4+CD25+FOXP3+ Treg cell infiltration in cancers.
Fig. S7. Effects of hsBCL9CT-24 treatment on anticancer immune cells.
Table S1. Sequences of hsBCL9CT peptides and related derivatives.
Table S2. PK and TK profiles of hsBCL9CT-24 and hsBCL9CT-35 in mice.
Table S3. Additional PK, solubility, and stability investigations with hsBCL9CT-24.
REFERENCES AND NOTES
- 1.Clevers H., Nusse R., Wnt/β-catenin signaling and disease. Cell 149, 1192–1205 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Polakis P., Drugging Wnt signalling in cancer. EMBO J. 31, 2737–2746 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sampietro J., Dahlberg C. L., Cho U. S., Hinds T. R., Kimelman D., Xu W., Crystal structure of a β-catenin/BCL9/Tcf4 complex. Mol. Cell 24, 293–300 (2006). [DOI] [PubMed] [Google Scholar]
- 4.Mani M., Carrasco D. E., Zhang Y., Takada K., Gatt M. E., Dutta-Simmons J., Ikeda H., Diaz-Griffero F., Pena-Cruz V., Bertagnolli M., Myeroff L. L., Markowitz S. D., Anderson K. C., Carrasco D. R., BCL9 promotes tumor progression by conferring enhanced proliferative, metastatic, and angiogenic properties to cancer cells. Cancer Res. 69, 7577–7586 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takada K., Zhu D., Bird G. H., Sukhdeo K., Zhao J.-J., Mani M., Lemieux M., Carrasco D. E., Ryan J., Horst D., Fulciniti M., Munshi N. C., Xu W., Kung A. L., Shivdasani R. A., Walensky L. D., Carrasco D. R., Targeted disruption of the BCL9/β-catenin complex inhibits oncogenic Wnt signaling. Sci. Transl. Med. 4, 148ra117 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin S.-Y., Xia W., Wang J. C., Kwong K. Y., Spohn B., Wen Y., Pestell R. G., Hung M.-C., β-catenin, a novel prognostic marker for breast cancer: Its roles in cyclin D1 expression and cancer progression. Proc. Natl. Acad. Sci. U.S.A. 97, 4262–4266 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jang G.-B., Kim J.-Y., Cho S.-D., Park K.-S., Jung J.-Y., Lee H.-Y., Hong I.-S., Nam J.-S., Blockade of Wnt/β-catenin signaling suppresses breast cancer metastasis by inhibiting CSC-like phenotype. Sci. Rep. 5, 12465 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stewart D. J., Wnt signaling pathway in non–small cell lung cancer. J. Natl. Cancer Inst. 106, djt356 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Spranger S., Bao R., Gajewski T. F., Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 523, 231–235 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Spranger S., Dai D., Horton B., Gajewski T. F., Tumor-residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell 31, 711–723.e4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kahn M., Can we safely target the WNT pathway? Nat. Rev. Drug Discov. 13, 513–532 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sukhdeo K., Mani M., Zhang Y., Dutta J., Yasui H., Rooney M. D., Carrasco D. E., Zheng M., He H., Tai Y.-T., Mitsiades C., Anderson K. C., Carrasco D. R., Targeting the β-catenin/TCF transcriptional complex in the treatment of multiple myeloma. Proc. Natl. Acad. Sci. U.S.A. 104, 7516–7521 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Verdine G. L., Hilinski G. J., Stapled peptides for intracellular drug targets. Methods Enzymol. 503, 3–33 (2012). [DOI] [PubMed] [Google Scholar]
- 14.Kawamoto S. A., Coleska A., Ran X., Yi H., Yang C.-Y., Wang S., Design of triazole-stapled BCL9 α-helical peptides to target the β-catenin/B-cell CLL/lymphoma 9 (BCL9) protein–protein interaction. J. Med. Chem. 55, 1137–1146 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Degorce F., Card A., Soh S., Trinquet E., Knapik G. P., Xie B., HTRF: A technology tailored for drug discovery –a review of theoretical aspects and recent applications. Curr. Chem. Genomics 3, 22–32 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang M., Wisniewski J. A., Ji H., AlphaScreen selectivity assay for β-catenin/B-cell lymphoma 9 inhibitors. Anal. Biochem. 469, 43–53 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Lau T., Chan E., Callow M., Waaler J., Boggs J., Blake R. A., Magnuson S., Sambrone A., Schutten M., Firestein R., Machon O., Korinek V., Choo E., Diaz D., Merchant M., Polakis P., Holsworth D. D., Krauss S., Costa M., A novel tankyrase small-molecule inhibitor suppresses APC mutation–driven colorectal tumor growth. Cancer Res. 73, 3132–3144 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Bird G. H., Madani N., Perry A. F., Princiotto A. M., Supko J. G., He X., Gavathiotis E., Sodroski J. G., Walensky L. D., Hydrocarbon double-stapling remedies the proteolytic instability of a lengthy peptide therapeutic. Proc. Natl. Acad. Sci. U.S.A. 107, 14093–14098 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lechner M. G., Karimi S., Barry-Holson K., Angell T., Murphy K., Church C., Ohlfest J., Hu P., Epstein A., Immunogenicity of murine solid tumor models as a defining feature of in vivo behavior and response to immunotherapy. J. Immunother. 36, 477–489 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xiao Q., Wu J., Wang W.-J., Chen S., Zheng Y., Yu X., Meeth K., Sahraei M., Bothwell A. L. M., Chen L., Bosenberg M., Chen J., Sexl V., Sun L., Li L., Tang W., Wu D., DKK2 imparts tumor immunity evasion through β-catenin-independent suppression of cytotoxic immune-cell activation. Nat. Med. 24, 262–270 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang X., Kelaria S., Kerstetter J., Wang J., The functional and prognostic implications of regulatory T cells in colorectal carcinoma. J. Gastrointest. Oncol. 6, 307–313 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cook K. W., Letley D. P., Ingram R. J. M., Staples E., Skjoldmose H., Atherton J. C., Robinson K., CCL20/CCR6-mediated migration of regulatory T cells to the Helicobacter pylori-infected human gastric mucosa. Gut 63, 1550–1559 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang P., Li Q.-J., Feng Y., Zhang Y., Markowitz G. J., Ning S., Deng Y., Zhao J., Jiang S., Yuan Y., Wang H.-Y., Cheng S.-Q., Xie D., Wang X.-F., TGF-β-miR-34a-CCL22 signaling-induced Treg cell recruitment promotes venous metastases of HBV-positive hepatocellular carcinoma. Cancer Cell 22, 291–303 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kilinc M. O., Gu T., Harden J. L., Virtuoso L. P., Egilmez N. K., Central role of tumor-associated CD8+ T effector/memory cells in restoring systemic antitumor immunity. J. Immunol. 182, 4217–4225 (2009). [DOI] [PubMed] [Google Scholar]
- 25.Kim J. M., Chen D. S., Immune escape to PD-L1/PD-1 blockade: Seven steps to success (or failure). Ann. Oncol. 27, 1492–1504 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Sharma P., Hu-Lieskovan S., Wargo J. A., Ribas A., Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 168, 707–723 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deka J., Wiedemann N., Anderle P., Murphy-Seiler F., Bultinck J., Eyckerman S., Stehle J.-C., André S., Vilain N., Zilian O., Robine S., Delorenzi M., Basler K., Aguet M., Bcl9/Bcl9l are critical for Wnt-mediated regulation of stem cell traits in colon epithelium and adenocarcinomas. Cancer Res. 70, 6619–6628 (2010). [DOI] [PubMed] [Google Scholar]
- 28.Hoggard L. R., Zhang Y., Zhang M., Panic V., Wisniewski J. A., Ji H., Rational design of selective small-molecule inhibitors for β-catenin/B-cell lymphoma 9 protein–protein interactions. J. Am. Chem. Soc. 137, 12249–12260 (2015). [DOI] [PubMed] [Google Scholar]
- 29.Schafmeister C. E., Po J., Verdine G. L., An all-hydrocarbon cross-linking system for enhancing the helicity and metabolic stability of peptides. J. Am. Chem. Soc. 122, 5891–5892 (2000). [Google Scholar]
- 30.van den Berg A., Dowdy S. F., Protein transduction domain delivery of therapeutic macromolecules. Curr. Opin. Biotechnol. 22, 888–893 (2011). [DOI] [PubMed] [Google Scholar]
- 31.Chang Y. S., Graves B., Guerlavais V., Tovar C., Packman K., To K.-H., Olson K. A., Kesavan K., Gangurde P., Mukherjee A., Baker T., Darlak K., Elkin C., Filipovic Z., Qureshi F. Z., Cai H., Berry P., Feyfant E., Shi X. E., Horstick J., Allen Annis D., Manning A. M., Fotouhi N., Nash H., Vassilev L. T., Sawyer T. K., Stapled α–helical peptide drug development: A potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy. Proc. Natl. Acad. Sci. U.S.A. 110, E3445–E3454 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.dela Roche M., Rutherford T. J., Gupta D., Veprintsev D. B., Saxty B., Freund S. M., Bienz M., An intrinsically labile α-helix abutting the BCL9-binding site of β-catenin is required for its inhibition by carnosic acid. Nat. Commun. 3, 680 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wisniewski J. A., Yin J., Teuscher K. B., Zhang M., Ji H., Structure-based design of 1,4-dibenzoylpiperazines as β-catenin/B-cell lymphoma 9 protein–protein interaction inhibitors. ACS Med. Chem. Lett. 7, 508–513 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Giantonio B. J., Catalano P. J., Meropol N. J., O’Dwyer P. J., Mitchell E. P., Alberts S. R., Schwartz M. A., Benson A. B. III, Bevacizumab in combination with oxaliplatin, fluorouracil, and leucovorin (FOLFOX4) for previously treated metastatic colorectal cancer: Results from the Eastern Cooperative Oncology Group Study E3200. J. Clin. Oncol. 25, 1539–1544 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Sobrero A. F., Maurel J., Fehrenbacher L., Scheithauer W., Abubakr Y. A., Lutz M. P., Vega-Villegas M. E., Eng C., Steinhauer E. U., Prausova J., Lenz H.-J., Borg C., Middleton G., Kröning H., Luppi G., Kisker O., Zubel A., Langer C., Kopit J., Burris H. A. III, EPIC: Phase III trial of cetuximab plus irinotecan after fluoropyrimidine and oxaliplatin failure in patients with metastatic colorectal cancer. J. Clin. Oncol. 26, 2311–2319 (2008). [DOI] [PubMed] [Google Scholar]
- 36.Di Nicolantonio F., Arena S., Tabernero J., Grosso S., Molinari F., Macarulla T., Russo M., Cancelliere C., Zecchin D., Mazzucchelli L., Sasazuki T., Shirasawa S., Geuna M., Frattini M., Baselga J., Gallicchio M., Biffo S., Bardelli A., Deregulation of the PI3K and KRAS signaling pathways in human cancer cells determines their response to everolimus. J. Clin. Invest. 120, 2858–2866 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boland P. M., Ma W. W., Immunotherapy for colorectal cancer. Cancers 9, 50 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grasso C. S., Giannakis M., Wells D. K., Hamada T., Mu X. J., Quist M., Nowak J. A., Nishihara R., Qian Z. R., Inamura K., Morikawa T., Nosho K., Abril-Rodriguez G., Connolly C., Escuin-Ordinas H., Geybels M. S., Grady W. M., Hsu L., Hu-Lieskovan S., Huyghe J. R., Kim Y. J., Krystofinski P., Leiserson M. D. M., Montoya D. J., Nadel B. B., Pellegrini M., Pritchard C. C., Puig-Saus C., Quist E. H., Raphael B. J., Salipante S. J., Shin D. S., Shinbrot E., Shirts B., Shukla S., Stanford J. L., Sun W., Tsoi J., Upfill-Brown A., Wheeler D. A., Wu C. J., Yu M., Zaidi S. H., Zaretsky J. M., Gabriel S. B., Lander E. S., Garraway L. A., Hudson T. J., Fuchs C. S., Ribas A., Ogino S., Peters U., Genetic mechanisms of immune evasion in colorectal cancer. Cancer Discov. 8, 730–749 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maj T., Wang W., Crespo J., Zhang H., Wang W., Wei S., Zhao L., Vatan L., Shao I., Szeliga W., Lyssiotis C., Liu J. R., Kryczek I., Zou W., Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat. Immunol. 18, 1332–1341 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Turner R. J., Charlton S. J., Assessing the minimum number of data points required for accurate IC50 determination. Assay Drug Dev. Technol. 3, 525–531 (2005). [DOI] [PubMed] [Google Scholar]
- 41.Li B., Severson E., Pignon J., Zhao H., Li T., Novak J., Jiang P., Shen H., Aster J. C., Rodig S., Signoretti S., Liu J. S., Liu X. S., Comprehensive analyses of tumor immunity: Implications for cancer immunotherapy. Genome Biol. 17, 174 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/5/eaau5240/DC1
Supplementary Materials and Methods
Fig. S1. Biochemical profile and LC-MS analysis of hsBCL9CT peptides.
Fig. S2. Cellular uptake, Wnt reporter, and coimmunoprecipitation analysis of hsBCL9CT peptides.
Fig. S3. Selectivity in multiple signaling pathways and antiproliferation assay of hsBCL9CT peptides.
Fig. S4. PK, toxicology, and histology analysis of hsBCL9CT-24 and hsBCL9CT-35.
Fig. S5. IHC staining in patient tumor tissues and PDX tumor models.
Fig. S6. Wnt pathway gene expression is correlated with CD4+CD25+FOXP3+ Treg cell infiltration in cancers.
Fig. S7. Effects of hsBCL9CT-24 treatment on anticancer immune cells.
Table S1. Sequences of hsBCL9CT peptides and related derivatives.
Table S2. PK and TK profiles of hsBCL9CT-24 and hsBCL9CT-35 in mice.
Table S3. Additional PK, solubility, and stability investigations with hsBCL9CT-24.