

Atypical heterochromatin forms on telomeres and correlates with ALT activities.
Abstract
Alternative lengthening of telomeres, or ALT, is a recombination-based process that maintains telomeres to render some cancer cells immortal. The prevailing view is that ALT is inhibited by heterochromatin because heterochromatin prevents recombination. To test this model, we used telomere-specific quantitative proteomics on cells with heterochromatin deficiencies. In contrast to expectations, we found that ALT does not result from a lack of heterochromatin; rather, ALT is a consequence of heterochromatin formation at telomeres, which is seeded by the histone methyltransferase SETDB1. Heterochromatin stimulates transcriptional elongation at telomeres together with the recruitment of recombination factors, while disrupting heterochromatin had the opposite effect. Consistently, loss of SETDB1, disrupts telomeric heterochromatin and abrogates ALT. Thus, inhibiting telomeric heterochromatin formation in ALT cells might offer a new therapeutic approach to cancer treatment.
INTRODUCTION
The replicative life span of a eukaryotic cell is correlated with telomere shortening. Telomere shortening below a critical length results in the activation of proliferative checkpoints and cell senescence or apoptosis. Cancer cells, as part of the process of transformation, acquire dedicated mechanisms for maintaining telomeres above this critical length and are thus effectively immortal. In most human cancers, telomerase is reactivated, permitting a lengthening of the shortest telomeres. However, in ~15% of cancers, telomeres are maintained by a telomerase-independent mechanism that relies on homologous recombination and amplification of telomeric DNA. This pathway is called alternative lengthening of telomeres (ALT). ALT telomeres colocalize with nuclear bodies formed by the promyelocytic leukemia (PML) protein in a structure termed the ALT-associated PML body (1). Recombination promoting factors, such as the SMC5/6 complex (2), and the prolonged binding of the replication protein A1 (RPA1) (3) are important for ALT maintenance. ALT can be induced by perturbing canonical chromatin assembly pathways, since the knockdown of anti-silencing factor 1, which is a major histone H3/H4 chaperone, is sufficient to trigger ALT in cancer cell lines (4). More than 90% of ALT-positive tumors have inactivating mutations in the alpha thalassemia/mental retardation syndrome X-linked (ATRX) gene (5). Besides mutations in the ATRX pathway, no other mutation has been linked to ALT in human cancer (6). Nevertheless, loss of ATRX alone is not sufficient to trigger ALT (7), which suggests that ALT has other, as yet unknown, determinants. As ATRX is a heterochromatin binding factor that belongs to the SWItch/Sucrose Non-Fermentable family of chromatin remodeling proteins (8), it was inferred that mutation of ATRX results in defective heterochromatin, and this defective heterochromatin is linked to ALT activation (9).
The principle marker of heterochromatin is an enrichment of trimethylation of histone H3 on lysine 9 (H3K9me3). Multiple proteins have H3K9me3-binding chromodomains. Principle among them is heterochromatin protein 1 (HP1), which exists in isoforms α, β, and γ. HP1 recruitment by H3K9me3 is critical for heterochromatin as it, in turn, recruits multiple proteins that contribute to heterochromatin formation and function (10). H3K9me3 can also directly interact with ATRX (11, 12), a protein that is required to suppress ALT.
H3K9me3 and heterochromatin are present at pericentromeres. Pericentric heterochromatin formation is controlled by the two redundant histone methyltransferases, the suppressor of variegation 3-9 homologs (SUV39H1 and SUV39H2, hereafter termed as SUV39H) (13). The loss of Suv39h leads to a loss of H3K9me3 at pericentromeres and to the appearance of typical ALT features at telomeres, such as increased telomere recombination and increased formation of ALT-associated PML bodies (14, 15). As SUV39H is a master regulator of heterochromatin formation, the prevailing mode was that SUV39H-mediated heterochromatin protects telomeres and therefore opposes ALT activation (16). In human cell lines, a long noncoding RNA transcribed from telomeres, known as telomeric repeat–containing RNA (TERRA), has been proposed to contribute to telomeric heterochromatin formation (17), and long telomeres bear heterochromatic marks that repress TERRA (18). Moreover, ALT telomeric chromatin is decondensed and displays decreased H3K9me3, suggesting a link between ALT and defective heterochromatin function (19). However, recent chromatin immunoprecipitation (ChIP)–sequencing meta-analyses challenge those findings, and rather suggest that human telomeres generally do not bear H3K9me3 and are euchromatic, while telomeres from an ALT cell line have H3K9me3 enrichment (20).
There are currently no molecular mechanisms linking defective heterochromatin formation and ALT activation. Here, we used a quantitative locus-specific proteomic technique, proteomics of isolated chromatin segments (PICh), to analyze telomere responses to defective heterochromatin. We found that telomeric heterochromatin is distinct from pericentromeric heterochromatin because it depends on the activity of the SET Domain Bifurcated 1 (SETDB1) histone methyltransferase and not on SUV39H for the deposition of H3K9me3. Moreover, we found that ALT does not rely on the loss of heterochromatin at telomeres, but it rather relies on heterochromatin formation at telomeres.
RESULTS
SETDB1 catalyses H3K9me3 addition on telomeres
To better understand the relation between heterochromatin dysfunction and the appearance of ALT features, we first sought to determine how telomere heterochromatin forms in mouse embryonic stem cells (mESCs). mESCs have low levels of telomeric heterochromatin (21), can extend their telomeres by recombination (22), and their ALT characteristics are increased upon Suv39h knockout (KO) in these cells (15). We used proteomics of PICh to capture telomeric chromatin from mESCs (23) and analyzed bound proteins by mass spectrometry (MS). We performed two independent experiments and identified more than 2000 proteins, among which 350 were reproducibly enriched. These included the shelterin and the CST complex (CTC1, STN1, and TEN1), which form abundant telomere-specific complexes (Fig. 1A and table S1). These also included low-abundance telomeric chromatin components, such as telomerase (TERT) and its cofactor TELOMERASE CAJAL BODY PROTEIN 1 (TCAB1) (Fig. 1A). Despite this high sensitivity, we found no evidence of SUV39H on telomeres. It is unlikely that the PICh approach fails to identify SUV39H at telomeres for technical reasons, as we readily identified SUV39H at pericentromeres using this technique (21). Instead, we identified three other H3K9 methyltransferases: G9A, GLP, and SETDB1 (Fig. 1A). As G9A and GLP can only mono- and dimethylate H3K9 (24), our result suggests that SETDB1 generates H3K9me3 on telomeres.
Fig. 1. SETDB1 installs H3K9me3 on mESC telomeres.
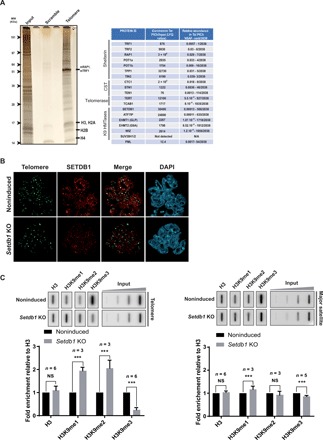
(A) Left: Silver staining of telomeric proteins from wild-type mESCs. Molecular weights (MW) are on the left of the gel. Right: Table of selected telomere proteins from PICh MS data highlighting shelterin enrichment, CST complex enrichment, telomerase enrichment, and enrichment of the H3 lysine 9 methyltransferases and their cofactors. Ranking is according to the normalized spectral abundance of each factor. (B) SETDB1 (red) immunostaining combined with telomere FISH staining (green) in noninduced wild-type mESCs or Setdb1 KO induced by 4 days of tamoxifen treatment. (C) Top: ChIP experiments using antibodies raised against H3 and mono-, di-, and trimethylated H3K9 to monitor H3K9 methylation at heterochromatin regions upon the loss of Setdb1. Twenty percent of the immunoprecipitated DNA was blotted and probed with a telomere-specific probe (left) or a major satellite-specific probe (right). Inputs of 0.01, 0.05, 0.25, and 1.25% were loaded. Bottom: Quantifications representing the fold enrichment of H3K9 methylation at telomere or pericentromere in Setdb1 knockout cells normalized to total H3 signal and the input relative to noninduced wild-type mESCs. ***P < 0.005, Student’s t test; NS, not significant.
We performed immunofluorescence on mESCs using telomere-specific probes and SETDB1 antibodies to monitor SETDB1 subnuclear localization. Endogenous SETDB1 colocalized with a subset of telomeres in most nuclei of wild-type mESCs (Fig. 1B). To confirm the presence of SETDB1 at telomeres, we used a conditional knockout mESC line, in which Setdb1 is abrogated upon tamoxifen treatment (25). SETDB1 becomes almost undetectable by immunoblotting 3 days after inducing Setdb1 disruption (fig. S1A). The SETDB1 signals at telomeres were strongly reduced upon Setdb1 KO induction (Fig. 1B), indicating that SETDB1 is a component of mESCs telomeric chromatin and that it is specifically enriched there.
To determine whether SETDB1 can generate H3K9me3 on telomeres, we immunoprecipitated chromatin from wild-type and Setdb1-negative mESCs with H3K9me3, H3K9me2, and H3K9me1 ChIP-grade antibodies and monitored H3K9 methylation on telomeres and on pericentromeres as a control. Loss of Setdb1 reduced H3K9me3 and increased H3K9me1 and H3K9me2 on telomeres. Loss of Setdb1 did not greatly affect H3K9 methylation at pericentromeres, indicating that SETDB1 action is telomere specific (Fig. 1C).
We analyzed previously published SUV39H and SETDB1 ChIP-sequencing results from mESCs (26, 27). SUV39H was not enriched on telomeres of mESCs, but SETDB1 was (fig. S1B). Our data demonstrate that telomeric and pericentromeric H3K9me3 heterochromatin marks are derived from SETDB1 and SUV39H, respectively.
Moreover, neither SETDB1 nor SUV39H was detected in telomere purifications from several types of differentiated somatic cells and tissues, including in mouse embryonic fibroblasts (table S2). Instead, we detected G9A and/or GLP and found that H3K9me2 was more prevalent (fig. S1C), suggesting that telomeric H3K9me3 might be developmentally regulated.
Distinct heterochromatin regions react differently to Suv39h loss
SUV39H is thought to maintain telomeric heterochromatin in mESCs (15, 28, 29). We monitored H3K9 methylation at telomeres in wild-type mESCs and Suv39h-knockout derivatives. The H3K9me3 mark was lost at pericentromeres (13), and there was a robust increase in H3K9me3 at telomeres (~3.5-fold greater than wild-type levels) in Suv39h-negative mESCs (Fig. 2A). To rule out potential ChIP discrepancies arising from the use of antibodies, we used MS as an orthogonal means to analyze histone modifications at both pericentromeres and telomeres (21, 30). MS confirmed a ~2.5-fold increase in the H3K9me3 mark on telomeric nucleosomes in Suv39h-negative cells, while H3K9me3 was undetectable on pericentromeric nucleosomes. We observed a substantial gain in the Polycomb-dependent H3K27me3 mark at Suv39h-negative pericentromeres, consistent with a Polycomb redeployment to this region to maintain silencing (31). In contrast, we could not detect H3K27me3 at telomeres (Fig. 2B). We performed immunoblotting with H3K9me3 and H3K27me3 antibodies on telomere and pericentromere-purified nucleosome preparations from Suv39h-negative mESCs. We detected a substantial H3K27me3 signal on Suv39h-negative pericentromeric nucleosomes, which otherwise had no detectable H3K9me3. Suv39h-negative telomeric nucleosomes had higher levels of H3K9me3 than wild-type telomeres (fig. S2A).
Fig. 2. Telomeres and pericentromeres respond differently to Suv39h loss in mESCs.
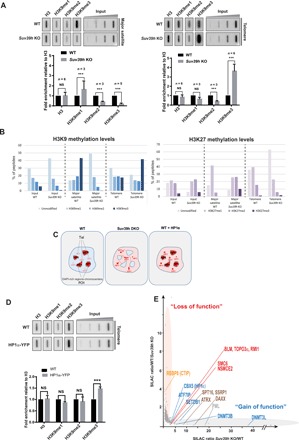
(A) Top: ChIP experiments using antibodies raised against H3 and mono-, di-, and trimethylated H3K9 to monitor H3K9 methylation upon Suv39h loss. Blotted DNA was probed with a telomere-specific probe (right) or a major satellite-specific probe (left). Bottom: Quantifications representing the fold enrichment of H3K9 methylation at major satellites (pericentromere) or telomeres normalized to the total H3 signal and the input relative to wild-type (WT) mESCs. (B) Quantification of relative methylation of histone tails obtained from PICh-purified telomeres and pericentromeres by quantitative MS. H3K9 methylation (left) and H3K27 methylation (right). (C) Model explaining how heterochromatin formation is stimulated by increased HP1α availability at telomeres. Interphase nuclei are represented. Tel, telomere; PCH, pericentromeric heterochromatin organized in chromocenters (DAPI-rich regions). As a result of H3K9me3 erasure at PCH in Suv39h-negative cells, HP1α is delocalized and becomes available to bind to SETDB1-induced heterochromatin, which includes telomeres. Increasing HP1α availability in normal cells by a slight HP1α overexpression results increased heterochromatin formation at telomeres. ***P < 0.005. (D) Left: ChIP experiments using antibodies raised against H3 and mono-, di-, and trimethylated H3K9 in wild-type mESCs overexpressing HP1α–yellow fluorescent protein (YFP). The immunoprecipitated DNA was blotted and probed with a telomere probe. Right: Graph of quantifications of representing the fold enrichment of H3K9 methylation at telomere normalized to total H3 signal and the input relative to wild-type cells. (E) qPICh plots highlighting major protein losses (orange oval) and gains (gray oval) upon SUV39H withdrawal in mESCs. NS, not significant.
How SUV39H loss results in the recruitment of excess H3K9me3 to telomeres is not clear, given that SUV39H is not present on telomeres in wild-type cells. We reasoned that SETDB1 recruitment or activity on telomeres might be hyperstimulated in Suv39h-negative cells and that this would increase telomeric H3K9me3. Since HP1α binding is known to induce local SETDB1-dependent heterochromatinization in mESCs (32, 33), increased telomeric HP1α could stimulate SETDB1 recruitment further and thus increase heterochromatin formation at telomeres upon loss of Suv39h (Fig. 2C). In normal cells, a large pool of HP1 proteins binds to pericentromeric heterochromatin in an H3K9me3-dependent fashion (34). However, as H3K9me3 is absent at pericentromeric regions in Suv39h-negative cells, the pericentromeric HP1 pool is displaced. This occurs without any change in the overall abundance of HP1 (35). We therefore hypothesized that some of the released HP1α becomes available to bind to telomeres in Suv39h-negative mESCs. To test whether increased HP1α availability can stimulate heterochromatin formation on telomeres, we used fluorescence cell sorting to establish a wild-type mESC population that slightly overexpresses an HP1α–yellow fluorescent protein fusion (fig. S2B). HP1α overexpression was sufficient to increase H3K9me3 on telomeres (Fig. 2D) but not at pericentromeres (fig. S2C). Conversely, efficient HP1α knockdown in Suv39h KO cells led to a reduction in telomeric H3K9me3 (fig. S2D). These observations, along with the previous observation that pericentromeres mostly rely on SUV39H, suggest that HP1 displacement stimulates H3K9me3 at telomeres in mESCs.
To confirm that telomeres have more SETDB1-dependent heterochromatin in Suv39h-negative cells, we used a quantitative PICh (qPICh) approach, which combines PICh with SILAC (stable isotope labeling with amino acids in cell culture) to obtain an unbiased measurement of variations in telomeric protein composition between two distinct samples (21). HP1α, SETDB1, and SETDB1-associated factors all increased at the telomeres of Suv39h cells (Fig. 2E). We confirmed using immunofluorescence that there were more HP1α bound to telomeres in Suv39h KO cells (fig. S2E; see Materials and Methods). This relocalization of HP1α to telomeres thus explains why more H3K9me3 forms at telomeres in the absence of Suv39h. These results also suggest that pericentromeric heterochromatin perturbations affect other heterochromatin regions indirectly, by altering the balance of available heterochromatin proteins, such as HP1α (Fig. 2C).
SETDB1 allows the recruitment of ATRX and histone chaperones to telomeric heterochromatin
Our results indicate that SETDB1 is responsible for heterochromatin formation at telomeres. We therefore analyzed the consequences of disrupting Setdb1 on telomere biology. Telomere DNA damage was not altered (fig. S3A). Thus, we analyzed the consequences on steady-state telomere composition. As Setdb1-negative mESCs stop proliferating 7 days after Setdb1 knockout (25), we performed qPICh 4 days after inducing Setdb1 knockout, when the cell cycle profiles, and thus proliferation, of wild-type and Setdb1-negative mESCs were comparable, as determined by fluorescence-activated cell sorting (FACS) (fig. S3B).
The lack of telomeric heterochromatin in Setdb1-knockout cells was associated with both losses and gains of proteins. There was an increased telomeric association of both G9A and GLP and their cofactor widely-interspaced zinc finger-containing protein (WIZ) in the Setdb1 knockout (Fig. 3A and fig. S3C). G9A and GLP are involved in H3K9 mono- and dimethylation, potentially explaining the gain in telomeric H3K9me1/2 upon SETDB1 withdrawal (Fig. 1C). HP1α and ATRX, which both have dedicated H3K9me3-binding domains, had strongly reduced telomeric binding (Fig. 3A) also consistent with a loss of H3K9me3 at telomeres in the Setdb1 knockout (Fig. 1C). We used immunoblots to confirm that ATRX no longer binds to telomeres upon Setdb1 knockout (Fig. 3B). Overall, many factors with methylated lysine–binding domains were lost from telomeres upon SETDB1 withdrawal (including DNA methyltransferases DNMT3A/B and L; Fig. 3C). Telomeric compositional changes in Setdb1-knockout cells were the reverse of those seen in the Suv39h knockout; proteins that were lost from telomeres upon Setdb1 knockout were gained on telomeres in Suv39h-negative cells and vice versa (compare Figs. 2E and 3A). As the bound factors correlate with the reciprocal changes in telomeric H3K9me3 levels between the two knockouts presenting strongly reduced or excessive heterochromatin formation, it seems likely that changes in the level of bound factors result from changes in telomeric H3K9me3 levels.
Fig. 3. Loss of Setdb1 disrupts telomeric binding of methylated lysine readers, histone chaperones, and recombination factors.
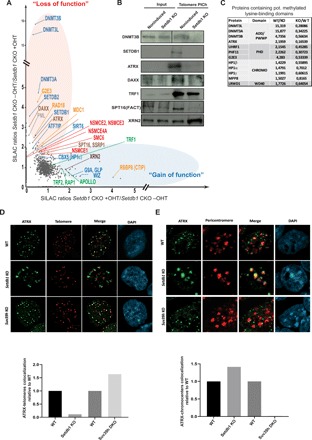
(A) qPICh plots showing changes in protein composition at telomeres upon SEDTB1 removal in mESCs. Major protein losses (in the red oval) and gains (in the blue oval) are highlighted. (B) Immunoblots using the indicated antibodies of purified telomeric extracts by PICh from noninduced wild-type cells or Setdb1 KO induced after 4 days of treatment with tamoxifen. An anti-SPT16 antibody was used to analyze FACT recruitment to telomeres. (C) qPICh SILAC in cell culture ratio values of proteins lost that contain putative methylated lysine-binding domains in Setdb1 KO mESC. (D) Top: ATRX (green) immunostaining combined with telomere fluorescence in situ hybridization staining (red) in wild-type or Setdb1- and Suv39h-negative mESCs. Bottom: Quantification of the loss or the gain of colocalization of ATRX-telomere signals relative to wild-type colocalization values. (E) Top: ATRX (green) immunostaining combined with pericentromere fluorescence in situ hybridization (FISH) (red). Bottom: Quantification of the loss or the gain of colocalization of ATRX-pericentromere signal relative to the wild-type colocalization values.
ATRX, which is involved in repressing ALT, principally colocalized with SETDB1-methylated telomeres and with SUV39H-methylated pericentromeres. As pericentromeres lose H3K9me3 in Suv39h-negative mESCs, ATRX was excluded from pericentromeres and colocalized with telomeres. Conversely, as telomeres lose H3K9me3 in Setdb1-knockout cells, ATRX was excluded from telomeres and mostly colocalized with pericentromeres (Fig. 3, D to E).
We confirmed the close correlation between H3K9me3 enrichment and ATRX binding by ChIP using an ATRX-specific antibody (fig. S3, D and E). Moreover, the histone chaperone DAXX (a partner of ATRX) and the chaperone facilitates transcription (FACT) complex are reduced at telomeres in Setdb1-negative mESCs (Fig. 3, A and B). DAXX and the FACT complex also have an increased association with telomeres in Suv39h-negative mESCs (Fig. 2E), which suggests that chromatin assembly/disassembly processes at telomeres are perturbed upon SETDB1 removal and stimulated upon SUV39H removal.
Telomeric heterochromatin is atypical and stimulates transcriptional elongation
ATRX binds telomeres only when they are heterochromatinized and is known to be a suppressor of ALT. We thus hypothesized that the heterochromatin status of telomeres might somehow correlate with ALT activation. ALT has recently been correlated with increased TERRA accumulation (36), so we set out to explore ALT by focusing on telomere transcription. The FACT histone chaperone is critical to promote transcriptional elongation (37). Reduced FACT in Setdb1-knockout cells (Fig. 3, A and B) could be indicative of defective telomere transcription. We also observed increased binding of the 5′-3′ exoribonuclease 2 (XRN2) transcription termination factor to telomeres without SETDB1 (Fig. 3, A and B), again consistent with perturbed elongation. De novo DNMT3A, DNMT3B, and DNMT3L (Figs. 2E and 3A) were the major proteins lost on Setdb1-knockout telomeres and the major ones gained on Suv39h-knockout telomeres. We confirmed that DNMT3B no longer binds to telomeres in Setdb1-knockout cells by immunoblots (Fig. 3, A and B). We looked at non-CpG methylation of telomeres, as asymmetric DNA methylation is widespread in mESCs (38), but did not find any (on the 5′-CCCTAA-3′ strand; fig. S4, A and B), suggesting that DNMT3B recruitment might play another role. H3K36me3 has recently been shown to recruit DNMT3B to promote transcriptional elongation and/or to prevent intragenic initiation (39, 40). Therefore, changes in the recruitment of all these factors upon Setdb1 or Suv39h knockout suggested that there might be a loss or a gain in H3K36me3 and a measurable impact on TERRA transcription. Although heterochromatin loss generally facilitates RNA production rather than disrupting it, we observe the opposite effect on telomeres upon altering H3K9me3 levels. In wild-type cells, TERRAs usually appear as a 0.1- to 10-kb smear of 5′-UUAGGG-3′ containing RNA species on Northern blots. In addition to the TERRA smear, small TERRA species consistently appeared in Setdb1-negative cells (Fig. 4A). Shorter TERRAs in Setdb1-negative cells are unlikely to result from increased degradation of longer RNA species, since we do not see a corresponding decrease in the levels of longer TERRAs. We also measured other hallmarks associated with transcription in Setdb1-negative cells by ChIP and immunoblot. H3K36me3 and phosphorylated serine 2 on RNA polymerase II are chromatin features associated with RNA polymerase II processivity. Both were significantly reduced at Setdb1-knockout telomeres (Fig 4, B and C), suggesting that elongation is defective and/or that cryptic initiation occurs in the absence of SETDB1/H3K9me3 at telomeres. Thus, we infer that telomeric heterochromatin stimulates long TERRA production rather than inhibiting it. In full agreement with this, telomeres in Suv39h-negative cells accumulate more telomeric RNAs (fig. S4C), and they have more heterochromatin and higher H3K36 trimethylation than wild-type cells (fig. S4D). Thus, telomeric heterochromatin is atypical because it is coenriched both with H3K9me3 and H3K36me3, and this combination might stimulate transcription at telomeres.
Fig. 4. Telomeric H3K9me3 levels correlate with telomere transcription.
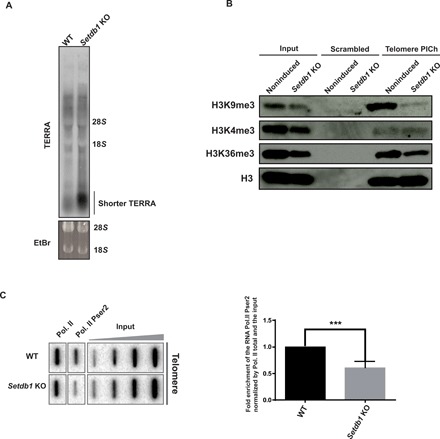
(A) TERRA Northern blot of wild-type or Setdb1-knockout cells. TERRAs were detected with (CCCTAA)6-radiolabeled probes. Ethidium bromide (EtBr) signals were used as loading controls. Signal intensities for TERRA species below 1000 nucleotides in wild-type versus Setdb1 KO lanes were compared. (B) Immunoblots of scramble or telomere PICh for the analysis of the enrichment of several histone modifications H3K9me3, H3K4me3, and H3K36me3 at telomeres upon SETDB1 removal. (C) ChIP experiments using antibodies raised against total RNA polymerase II (Pol. II) and against its phosphorylated serine 2 (Pser2) of its C-terminal domain in noninduced wild-type and Setdb1 knockout mESCs. Twenty percent of the immunoprecipitated DNA was blotted and probed with a telomere-specific probe. Inputs of 0.01, 0.05, 0.25, and 1.25% were loaded. Bottom: Quantifications representing the fold enrichment of the CTD phosphorylation at serine 2 of the RNA polymerase II in Setdb1 knockout cells normalized by the total RNA polymerase II signals at telomeres and the input relative to the wild type. ***P = 0.005, Student’s t test.
Telomeric heterochromatin promotes the appearance of ALT features
We next investigated the impact of heterochromatin gain or loss on the appearance of ALT-associated features. A distinctive feature of ALT telomeres is their association with recombination-promoting factors. Setdb1-negative cells had reduced association of several recombination/ALT-promoting factors, including RPA1, breast cancer type 1 susceptibility protein (BRCA1), and the SMC5/6 complex (Fig. 5A). We confirmed reduced BRCA1 association to telomeres by ChIP upon Setdb1 knockout (fig. S5A). These factors and other recombination factors well known to contribute to ALT [topoisomerase IIIα, bloom helicase (BLM), and RecQ-mediated genome instability protein 1 (RMI1)] increased in Suv39h-negative cells, which have excess heterochromatin at telomeres (Fig. 2, A and E). This suggests that heterochromatin might be an essential feature of ALT, stimulating both transcription and recombination. To test the requirement for heterochromatin in recombination, we measured telomere sister exchange events in wild-type and Setdb1- and Suv39h-knockout cells. In Setdb1-negative cells, there was no major effect of losing telomeric H3K9me3 on exchange events. However, the increased heterochromatin at telomeres in the absence of Suv39h correlated with increased telomeric exchanges (Fig. 5B). We then looked for the formation of ALT-associated PML bodies, another ALT characteristic. In wild-type mESCs, telomeres partially colocalize with PML bodies, and the PML protein was enriched in our telomere preparations (Fig. 1A and table S1). PML association with telomeres was substantially diminished upon Setdb1 knockout (Fig. 3A), and we confirmed this by immunofluorescence (Fig. 5C). By contrast, telomere-PML colocalization substantially increased in the Suv39h knockout, whose telomeres are more heterochromatic (Fig. 5C). We tested whether those changes could be explained by changes in the number of detectable telomere signals in the different backgrounds but did not measure any difference (fig. S5B). Increased telomere-PML colocalization in Suv39h knockout cells was due to increased telomeric heterochromatin because efficiently reducing SETDB1 by short hairpin RNA (shRNA) in Suv39h-knockout cells (fig. S5C) resulted in a significant reduction in PML association to telomeres (fig. S5D). We also counted less telomere sister exchange in the double Suv39h knockout; SETDB1 knockdown, albeit this reduction, was not significant (fig. S5E). It could be that the remaining SETDB1 protein after knockdown is sufficient to maintain telomere sister exchanges in this context.
Fig. 5. Atypical heterochromatin correlates with the appearance of ALT characteristics.
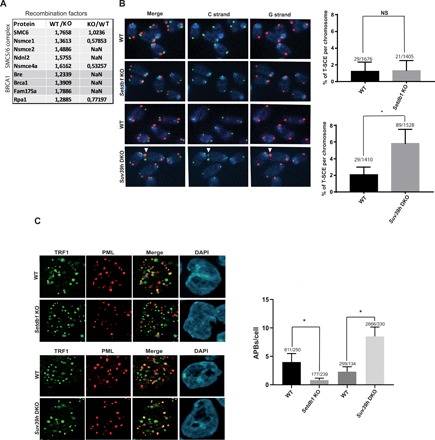
(A) qPICh SILAC in cell culture ratio values of recombination factors lost in Setdb1 KO mESCs. (B) Chromosome orientation fluorescence in situ hybridization of metaphase chromosomes from wild-type and Setdb1- and Suv39h-negative cells. In green, the C-rich strand and in red the G-rich strand. Quantification of the T-SCE defined by an exchange both of the lagging and the leading strand. (C) Coimmunostaining of PML (red) and TRF1 (green) and quantification of PML body-telomere colocalization (APBs) per cells in wild-type, and Setdb1- or Suv39h-negative cells. *P < 0.05; NS, not significant.
C-circles are relevant ALT markers in human cancer cells. We thus measured C-circle accumulation in mESCs (fig. S5F). Wild-type mESCs have significant levels of C-circles, and these levels are increased upon Suv39h knockout, consistent with an increased ALT activity. C-circles are also increased in Setdb1 knockout cells. Since C-circles have also been found without any other ALT marker upon replication stress (41), it could be that replication stress occurs at Setdb1 KO telomeres, without any other sign of ALT activation. Consistently, Setdb1 knockout telomeres are slightly shorter (fig. S5G), such as those seen when telomeres undergo replication stress without ALT activation (41). In line with our results showing that heterochromatin promotes ALT features, the telomeres are slightly longer in Suv39h-negative cells (fig. S5G), as already described (15). Thus, we conclude that increased telomeric heterochromatinization drives the recruitment of ALT-promoting factors and the appearance of distinctive ALT features on telomeres.
ATRX prevents recombination only when telomeres are heterochromatic
The concomitant increase in ATRX recruitment with heterochromatinization did not prevent increased recombination and transcription. This suggests that ATRX levels are limiting and not sufficient to fully protect telomeres from recombining. Since ATRX is usually inactivated in ALT tumors and cell lines, and since the removal of ATRX is not sufficient to drive ALT in human cancer cells, we reasoned that ATRX might only protect against ALT when telomeres are heterochromatic. If this were the case, then disrupting ATRX in the context of heterochromatic telomeres would be sufficient to promote ALT features. To test this, we knocked down ATRX in wild-type mESCs and in Suv39h-negative mESCs, both of which have heterochromatic telomeres. The loss of ATRX was sufficient to trigger significantly more telomere sister exchange in both backgrounds (fig. S6A). As the loss of telomeric heterochromatin disrupts ATRX recruitment without triggering telomere recombination, we hypothesized that ATRX can only suppress ALT when telomeric SETDB1 and H3K9me3 are present. To test this idea, we knocked down ATRX in the Setdb1 knockout (fig. S6B) and did not observe increased telomere-sister chromatid exchanges (T-SCE, fig. S6C). Since ATRX recruitment is H3K9me3 dependent, we conclude that ATRX is required to prevent recombination only when telomeres are heterochromatic.
SETDB1 disruption inhibits ALT in human cancer cells
The ALT characteristics in our mouse models strongly correlate with heightened heterochromatin formation on telomeres. Therefore, we tested whether heterochromatin would be important for ALT maintenance in ALT cell lines derived from human tumors. We examined whether SETDB1 is present on human ALT telomeres. We found substantial accumulation of SETDB1 specifically at ALT telomeres in WI38-VA13, U2-OS, and MFH148, which are three different human ALT cell lines (Fig. 6A). Telomere-SETDB1 colocalization was only partial, suggesting dynamic association. In contrast, there was no SETDB1 at telomeres in the ALT-negative control cell line MFH152 (42).
Fig. 6. SETDB1 controls ALT maintenance in human tumor cell lines.
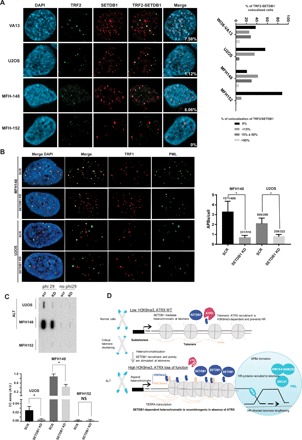
(A) Left: Coimmunostaining of SETDB1 (red) with TRF2 (green) in human ALT cell lines (VA13, U2OS, and MFH148) versus a human non-ALT cell line as a control (MFH152) cells. Right: Quantification of the percentage of SETDB1-TRF2 colocalization events. (B) Left: Coimmunostaining of TRF1 (red) with PML (green) to measure ALT-associated PML body formation upon SETDB1 knockdown in two ALT cell line U2OS and MFH148. Right: Quantification APB number per cell. *P < 0.05. (C) Top: C-circle assay in two ALT-positive cell lines and one negative cell line upon Scramble (SCR) or SETDB1 knockdown. (D) Proposed model derived from our main findings: SETDB1 mediates heterochromatin at telomeres, and the recruitment of ATRX is dependent of H3K9me3 deposition and prevents HR. SETDB1-dependent heterochromatin is recombinogenic and promotes ALT features in the absence of ATRX. A.U., arbitrary units; NS, not significant.
To test whether SETDB1 is responsible for heterochromatin formation on human ALT telomeres, we measured telomeric H3K9me3 by ChIP in two distinct ALT cell lines in which SETDB1 levels were reduced by specific shRNAs (fig. S7A). H3K9me3 levels were significantly reduced in these cells (fig. S7B). Next, to examine SETDB1 involvement in ALT maintenance, we also efficiently knocked down SETDB1 by small interfering RNA (siRNA) treatments (fig. S7C). This knockdown significantly suppressed ALT phenotypes, as telomeres were no longer colocalized with PML nuclear bodies, and there was less C-circles (Fig. 6, B and C). Like in Setdb1 KO mESCs, these cells had a similar cell cycle profile as the wild-type parental cells (fig. S7D), but they quickly stopped proliferating and died 6 days after efficient knockdown. In contrast, reducing SUV39H1 (fig. S7E) has no impact on telomere-PML colocalization and on C-circle production (fig.S7, F and G), suggesting that, like in mouse cells, SETDB1, not SUV39H, is responsible for ALT telomeric heterochromatin formation in human. Reducing SETDB1 dosage had no significant impact on overall ALT telomere length, as measured by telomere restriction fragment analyses (fig. S7H). This result was also observed by others upon removal of several critical ALT regulators (43–45).
We conclude that ALT requires the combined effects of losing ATRX and of increased levels of the heterochromatin mark H3K9me3 at telomeres. Increased H3K9me3 is due to SETDB1 recruitment. H3K9me3 maintains telomeric transcriptional elongation and triggers the recruitment of recombination factors and the recruitment of ATRX, which keeps ALT in check. Overall, these results demonstrate that heterochromatin not only is present at ALT telomeres but also is important for the ALT phenotype.
DISCUSSION
Here, we demonstrate a crucial role of SETDB1 in enforcing heterochromatin formation at telomere and controlling ALT. We found that SETDB1-dependent heterochromatin is recombinogenic: Excessive heterochromatin formation by SETDB1 promotes the appearance of ALT characteristics, whereas the loss of SETDB1 suppresses ALT (Fig. 6D).
Telomeric heterochromatin formation was initially proposed to be catalyzed by SUV39H (15). Our complementary data show that this is unlikely. SUV39H forms heterochromatin at pericentromeres but not at telomeres. However, in cells lacking SUV39H, the loss of H3K9me3 induces the displacement of a large pool of HP1 from pericentromeres. HP1 is relocalized in part at telomeres where it further stimulates SETDB1 recruitment and increases H3K9me3 formation. We found that this excessive heterochromatin stimulates ALT-associated PML body formation, TERRA transcription, and the recruitment of recombination factors. On the other hand, the loss of SETDB1 disrupts telomeric heterochromatin and suppresses associated ALT features.
Given the importance of ATRX in suppressing ALT, the question of how it is recruited to telomeres is critical. We found that ATRX binding to telomeres primarily depends on H3K9me3. That ATRX recruitment to telomeres depends on H3K9me3 also explains why the removal of ATRX is not sufficient to induce ALT in most human cell lines, since in most of these cells, telomeres are presumably not heterochromatic (20) and, therefore, are poorly transcribed and not recombinogenic. It is also implicit from our results that telomeres must become heterochromatic during cell crisis and that SETDB1 must be (hyper)active in ALT tumors. As inhibiting heterochromatin suppresses the ALT pathway in tumor cells, small-molecule inhibitors for histone methyltransferases such as SETDB1 might represent a potential therapeutic strategy for disrupting ALT in human cancers.
Telomeric heterochromatin is atypical because it is enriched both in the marks H3K9me3 and H3K36me3. Concomitant enrichment of these two marks has already been observed on the last exon of a subset of zinc finger genes. Zinc finger sequences are trinucleotide repeat regions embedded in genes, which are rapidly evolving (46). Like ALT telomeres, these genes are bound by SETDB1, transcribed, and prone to recombine. It is tempting to speculate that ALT and zinc finger gene evolution underlie similar SETDB1-dependent mechanisms.
MATERIALS AND METHODS
Cell culture
Suv39h KO mESCs and control mESCs (mixed background 129/SV-C57B1/6J) were given by T. Jenuwein, and the Setdb1 conditional knockout cells were given by Y. Shinkai. ESCs were grown on 0.2% (w/v) gelatin-coated dishes in Dulbecco’s modified Eagle’s medium (DMEM) containing high glucose, 15% (v/v) fetal calf serum, 2 mM l-glutamine, 0.1 mM β-mercaptoethanol, 1× nonessential amino acids, and leukemia inhibitory factor (LIF). No antibiotics were added. Setdb1 was knocked down by treating Setdb1 conditional knockout cells for 5 days with 800 nM hydroxytamoxifen (OHT) in culture medium. Human cell lines were grown in DMEM containing high glucose, 10% (v/v) fetal calf serum, and 2 mM l-glutamine in an incubator at 37°C with 5% CO2.
Lentiviral infection
Depletions of the endogenous ATRX or HP1α or SETDB1 in mESCs or human cells were achieved using lentiviral vectors pLKO-shATRX910 [ATRX MISSION shRNA SHCLNG-NM_009530 TRCN0000081910 (CCGGCCTGTCACTTTCACCTCTCAACTCGAGTTGAGAGGTGAAAG TGACAGGTTTTTG)] or pLKO-shHP1α (HP1α MISSION shRNA SHCLNG-NM_007626 TRCN0000071050) or pLKO-shSETDB1 in mouse (SETDB1 MISSION shRNA SHCLNG-NM_018877 TRCN0000339743, TRCN0000092976, and TRCN0000092975) or human (SETDB1 MISSION shRNA SHCLNG-NM_012432 TRCN0000276105 and TRCN0000276169) expressing an shRNA against ATRX or HP1α or SETDB1. Lentiviral particles were generated in human embryonic kidney 293 T cells by cotransfection of the pLKO plasmid targeting ATRX or HP1α or SETDB1 or control green fluorescent protein with the plasmids pCMV.dR8.91 and pCMV.VSVg. Infection was performed in the presence of polybrene (5 μg/ml) for 24 hours followed by selection with puromycine (1 to 2 μg/ml) or blasticidine (15 μg/ml). For Setdb1 conditional knockout cells, the puromycin resistance gene on the plasmid pLKO-sh ATRX81910 was changed by the blasticidine resistance gene.
RNA interference
All siRNAs were from Dharmacon: D-001810-10-05 ON-TARGETplus Non-targeting Control Pool for scramble and Accell Human SETDB1 (9869) E-020070-00-0010 or SUV39H1 E-009604-00-0010 siRNA SMARTpool for SETDB1 or SUV39H1 targeting. Human cells at 60% confluency in six-well plates were transfected twice at day 1 and at day 4 with Lipofectamine RNAiMAX (Life Technologies) and an siRNA concentration of 90 pM per well. Cells were collected at day 7 for immunofluorescence, C-circle assays, and FACS analyses.
Immunofluorescence, FISH, and microscopy
Cells were grown on coverslips and processed, as previously described (23). For fluorescence in situ hybridization (FISH) staining, cells were incubated for 45 min in 2× SSC at 72°C and then dehydrated with ethanol and air-dried. DNA was denatured at 82°C for 2 min and incubated with 50 mM of the appropriate probe overnight at 37°C. Cells were washed twice with 2× SSC for 20 min at 42°C. To detect the biotinylated major satellite probes, slides were incubated with a streptavidin Alexa Fluor 488 conjugate for 1 hour at room temperature. Last, DNA was stained with 1/1000 4′,6-diamidino-2-phenylindole (DAPI), and slides were mounted with ProLong Gold (Life Technologies).
Slides were analyzed using a Zeiss Axiovert Apotome microscope. Statistical analysis was performed using the Prism software with a Student’s t test. Asterisks denote P values below 0.05, and “NS” denotes nonsignificant statistical test.
PICh
The PICh experiment procedure was adapted for telomeric regions using 0.25 μM telomere desthiobiotin-TEG-4XC18 spacers, 5′TtAgGgTtAgGgTtAgGgTtAgGgt-3′–specific locked nucleic acid (LNA) probes, and ≈8 × 108 mESCs following the previously described protocol (23). qPICh was performed, as previously described (21). For PICh/Western blot, 6 mg of chromatin was piched for each condition, hybridized with 12.5 μl of 100 μM telomeric probes, and captured with 500 μl of C1 MyOne beads. Eluted PICh material was resuspended in 60 μl of reverse cross-link solution, and 5 to 12 μl of PICh samples were processed for Western blotting.
Chromatin immunoprecipitations
ChIP assays were performed, as previously described (23). Cells were cross-linked with 1% formaldehyde for 10 min at room temperature and quenched by adding glycine to a final concentration of 0.125 M for 5 min at room temperature. After washing in cold phosphate-buffered saline (PBS) and scrapping, cells were resuspended in five volumes of cold hypotonic buffer [10 mM Hepes (pH 7.9), 10 mM KCl, and 1.5 mM MgCl2] for 10 min at 4°C and lysed with 15 strokes with a tight pestle. The human cell chromatin was extracted as follows: Cell pellets were incubated for 5 min at 4°C with 15 ml of cold buffer A [20 mM Hepes (pH 7.9), 10 mM EDTA, 0.5 mM EGTA, and 0.25% Triton X-100] and washed with 15 ml of cold buffer B [50 mM Hepes (pH 7.9), 150 mM NaCl, 1 mM EDTA, and 0.5 mM EGTA] for 5 min at 4°C. Chromatin samples were resuspended in three volumes of sonication buffer [50 mM Hepes (pH 7.9), 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.1% Na-deoxycholate, and 0.5% Na-laurylsarkosine]. A total of 3 ml of chromatin was sonicated using a Misonix S-4000 sonicator with a high-power probe and 40% output, processed 15 s ON and 45 s OFF until the resulting chromatin was fragmented to a median fragment size of ∼250 nucleotides as assayed by agarose gel electrophoresis. Three hundred to 500 μg of chromatin was used per ChIP and incubated overnight at 4°C with 5 to 10 μg of antibodies. Protein-antibody complexes were captured with 100 μl of protein A or protein G dynabeads for 30 min at room temperature or for 2 hours at 4°C. Beads were washed three times with low-salt buffer [10 mM Hepes (pH 7.9), 150 mM NaCl, 2 mM EDTA, and 1% Triton X-100], two times with LiCl Buffer [20 mM Hepes (pH 7.9), 150 to 250 mM LiCl, 2 mM EDTA, 1% Triton X-100, 0.1% Na-deoxycholate, and 0.5% Na-laurylsarkosine], and once with TE [10 mM tris (pH 7.4) and 1 mM EDTA] and lastly eluted in 250 μl of elution buffer [50 mM tris-HCl (pH 8), 50 mM NAHCO3, 1% SDS, and 1 mM EDTA] for 20 s at 65°C. After reversal of formaldehyde cross-links at 65°C overnight and ribonuclease A (RNAse A) treatment at 37°C following by proteinase K digestion at 55°C, DNA fragments were extracted with phenol:chlorophorme and precipitated in ethanol with sodium acetate. For slot blot, DNA was denatured at 99°C in 0.4 M NaOH and 10 mM EDTA 10 min. Twenty percent of the immunoprecipitated DNA was spotted on a positively charged nylon Hybond-XL membrane and probed with a telomere-specific probe or a major satellite-specific probe. Inputs of 0.01, 0.05, 0.25, and 1.25% were loaded.
DNA was ultraviolet (UV)–cross-linked to the membrane at 0.14 J/cm2 and labeled with the appropriate radioactive probes in hybridization buffer for at least 4 hours at 37°C. After two washes with 2× SSC 0.1% SDS for 30 min at 37°C, the membrane was exposed for various times (2 hours to overnight) to the Phosphor screen. The screen was read by Phosphor Imager, and the images were analyzed using ImageQuant software. The telomeric or pericentric enrichment for histone marks were normalized by the total enrichment of H3 and the input. Statistical analysis was performed using the Prism software with a Student’s t test. Asterisks denote P values below to 0.05, and “NS” denotes nonsignificant statistical test.
Northern blot assay
RNAs were extracted with TRIzol:chloroform and precipitated with isopropanol following standard procedures. Northern blots were performed with the Ambion NorthernMax Kit, and manufacturer’s instructions were followed. All the buffers and powders were provided by the kit. A total of 7 μg of RNA was denatured at 65°C and loaded onto a 1% agarose denaturation gel (formaldehyde) about 6 mm thick. The gel was run at ~5 V/cm. RNA was transferred to a positively charged nylon membrane using downward capillary transfer for 4 hours. RNA was UV–cross-linked to the membrane at 0.21 J/cm2 and air-dried and hybridized with the appropriate radiolabeled probes ON at 37°C. The membrane was exposed for various times (2 hours to overnight) to the Phosphor screen, and the screen was read by Phosphor Imager.
Terminal restriction fragment for telomere length analysis
Cells were extracted in DNA lysis buffer [100 mM NaCl, 10 mM tris (pH 8), 10 mM EDTA, and 0.5% SDS] treated with RNAse A for 1 hour at 37°C, with proteinase K overnight at 55°C. A phenol-chloroform step allowed the isolation of DNA. DNA was precipitated with ethanol and sodium acetate without centrifugation (“jellyfish” method). A total of 1.5 μg of human genomic DNA were digested with the restriction enzymes Hinf I and Rsa I at 37°C for 4 hours. For mESCs, 1% agarose plugs with 0, 5 × 106 cells were prepared and digested with proteinase K in lysis buffer [100 mM EDTA (pH 8), 0.2% sodium deoxycholate, 1% sarkosyl, and proteinase K (1 mg/ml)] for 24 hours at 37°C. Plugs were digested with Dpn II 12 hours at 37°C. Pulse field gel electrophoresis (Rotaphor 8 Biometra) was used to separate telomere fragments according to manufacturer’s recommendations. Human DNA samples were loaded in 0.9% agarose gel in 0.5× tris-borate EDTA (TBE) and run at 180 V (120° angle) for 23 hours at 13°C, and plugs with mESCs were loaded in 0.9% gel in 0.5× TBE and run with the following parameters: 24-hour interval, 100 to 10 s log, 120° to 110° linear angle, 200- to 150-V log voltage, and 13°C. The gel with separated DNA fragments was transferred on a positively charged nylon membrane by Southern blotting. Briefly, the gel was treated with 0.25 N HCl for depurination for 10 min. The gel was incubated in denaturation solution (0.5 M NaOH and 1.5 M NaCl) for 30 min and in neutralization solution [0.5 M tris (pH 7) and 1.5 M NaCl] for 30 min. DNA was transferred by capillary overnight in 20× SSC. DNA was UV–cross-linked to the membrane at 0.120 J, air-dried, and hybridized with a telomeric radiolabeled probes (CCCTAA)6. The membrane was exposed to the Phosphor screen and revealed by Phosphor Imager.
Fluorescence-activated cell sorting
FACS analysis was performed using a standard protocol. Cells were fixed in 70% cold ethanol overnight at −20°C. The next day, 2 × 106 fixed cells were washed in PBS and incubated in propidium iodide (10 μg/ml; Sigma-Aldrich, P4170) and RNAse A (0.2 mg/ml) at 37°C for 1 hour. Samples were diluted 1:5, thoroughly mixed on a vortex mixer to achieve single-cell suspensions, and measured by flow cytometric analysis (FACSCalibur, Becton Dickinson).
Immunoblots
The protein samples were separated on 7.5% tris-glycine polyacrylamide gels (Bio-Rad) (SPT16, XRN2, SETDB1, ATRX, and DNMT3B) or 12% polyacrylamide gels [telomere repeat binding factor 1 (TRF1), TRF2, histones, and their modifications]. Proteins were transferred to nitrocellulose membranes using the transfer-blot Turbo Transfer system (Bio-Rad). Primary and secondary horseradish peroxidase–conjugated antibodies were incubated by standard Western blotting procedures and detected by adding ECL chemiluminescent solution (PerkinElmer). Signals were captured on autoradiography films (Amersham).
Chromosome orientation FISH
Sixty percent of confluent ES cells were incubated with BrdU and BrdC (30 and 10 μM, respectively) simultaneously for 12 hours. Colcemid (0.2 μg/ml) was added to the cells in fresh medium for 2 to 4 hours. The medium was kept, and cells were detached with trypsin and pooled with the precedent discarded medium. Cells were centrifuged, and the supernatant discarded. Four milliliters of prewarmed hypotonic solution (75 mM KCl) was added slowly to the cell pellets and incubated for 20 min at 37°C. Samples were prefixed with 1 ml of fresh methanol/acetic acid (3:1), centrifuged, fixed with 5 ml of fresh methanol/acetic acid (3:1), and stored overnight at 4°C before spreading on clean and cold slides pretreated with methanol. After spreading, slides were air-dried overnight. For the chromosome orientation FISH, the slides were rehydrated for 5 min in PBS, treated with RNAse A (0.5 mg/ml) in PBS for 10 min at 37°C, washed quickly with PBS, stained with Hoechst 3358 (0.5 μg/ml) in PBS for 15 min at room temperature, and washed quickly with PBS. Slides were flooded with 60 μl of PBS and exposed to 365 nm of UV light (1 cm away from the UV source) at room temperature for 30 min. Slides were washed quickly in PBS and air-dried before digestion with Exonuclease III (3 U/μl) in its buffer for 10 min at room temperature. Treated slides were washed two times 5 min with PBS, dehydrated in successive ethanol washes (70, 80, 90, and 100%), and air-dried. Peptide nucleic acid (PNA) probes CY3 (CCCTAA)6 (100 mM diluted 1:100) were hybridized in PNA hybridization buffer [70% formamide, 20 mM tris (pH 7.4), and blocking reagent (Roche)] at room temperature for 2 hours. Slides were washed two times for 15 min in wash buffer PNA [70% formamide and 20 mM tris (pH 7.4)] and three times for 5 min in wash buffer 3 [50 mM tris (pH 7.4), 150 mM NaCl, and 0.05% Tween 20]. Slides were dehydrated with successive ethanol washes. LNA probes Alexa Fluor 488 (TTAGGG)6 (100 mM diluted 1:100) in LNA hybridization buffer (50% formamide, 2× SSC, and blocking reagent) for 2 hours at room temperature. Slides were washed two times for 15 min with wash buffer LNA (50% formamide and 2× SSC) and three times for 5 min with wash buffer 3. Slides were costained with DAPI and analyzed by a microscope.
C-circle assay
Genomic DNA was extracted with the kit Purelink Genomic DNA extraction (Invitrogen) according the manufacturer’s instructions and eluted in 10 mM tris (pH 7.8). A total of 30 ng of DNA was incubated in master mix containing bovine serum albumin (0.2 mg/ml); 0.1% Tween 20; 1 mM each of deoxyadenosine triphosphate, deoxycytidine triphosphate, deoxyguanosine triphosphate, and deoxythymidine triphosphate; 4 μM dithiothreitol; φ29 buffer 1×; and φ29 DNA polymerase in a volume final of 20 μl and incubated at 30°C for 8 hours and then at 65°C for 20 min. Each assay was performed with and without the φ29 DNA polymerase. A slot blot analysis in nondenatured condition was done to quantify each CC assay. The 20 μl of CC assay was diluted in 300 μl of 2× SSC and slot-blotted on positively charged membrane Hybond-XL (Amersham). After UV cross-linking (0.120 J), the membrane was hybridized for a minimum of 6 hours at 37°C in Perfect Hybridization Plus Buffer (Sigma-Aldrich) with labeled 32P-(CCCTAA)6 telomeric probe. The membrane was exposed for various times (2 hours to overnight) to the Phosphor screen and read by Phosphor Imager. Statistical analysis was performed using the Prism software with a Student’s t test. Asterisks denote P values below to 0.05, and “NS” denotes nonsignificant statistical test.
Radiolabeling probes
Telomeric, major satellite, and TERRA probes were labeled with using the T4 Polynucleotide Kinase kit. A total of 50 pmol of DNA was labeled with γ [32P ATP] by the PNK enzyme for 1 hour at 37°C.
Actin probes were labeled by using the Prime-a-Gene Promega kit. A total of 50 pmol of DNA was labeled with α [32P CTP] by the DNA polymerase I for 1 hour at room temperature. Probes were purified on microspin G25 columns (GE Healthcare no. 27-5325-01).
Supplementary Material
Acknowledgments
The BRCA1 antibody is a gift from S. Namekawa (Children’s Hospital, Cincinatti). The TRF1 antibody was a gift from T. de Lange (Rockefeller University). The HP1α-YFP construct is from Florence Cammas (IRCM, Montpellier). Suv39h DKO cells are from T. Jenuwein, and Setdb1 KO cells are from Y. Shinkai. Funding: The Dejardin laboratory was supported by grants from ARC equipe labelisee 2016, the ERC CoG METACHROM, from INCA, from the Fondation Schlumberger pour l’education et la recherche, and from Merck (MSD). S.K. and A.B. were supported by a Labex Epigenmed PhD fellowship, and M.G. was supported by the University of Montpellier and ARC. Work in the Imhof laboratory is supported by grants from the DFG (CRC1064-Z3). Work in the Luco laboratory was supported by grants from the ATIP/AVENIR and ANR. Work in the Wellinger laboratory is supported by a grant from the CIHR (FDN 154315) and the Canada research chair in telomere biology. E.A. was supported by the EpiGeneSys European Network. Author contributions: Conceptualization: S.K., M.G., and J.D.; methodology: S.K. and M.G.; investigation: S.K., M.G., A.B., N.S., S.S., S.I., T.K.B., R.J.W., E.B., R.F.L., E.A., A.I, S.U., and E.B.; writing: S.K., M.G., and J.D.; visualization: S.K., M.G., A.B., N.S., and J.D.; funding acquisition: J.D. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/5/eaav3673/DC1
Fig. S1. SETDB1 telomeric binding is developmentally regulated.
Fig. S2. HP1α stimulates SETDB1-dependent heterochromatin formation at telomeres.
Fig. S3. ATRX binds heterochromatic telomeres.
Fig. S4. Telomeric heterochromatin stimulates transcriptional elongation.
Fig. S5. SETDB1-dependent heterochromatin promotes the recruitment of recombination factors and the appearance of ALT features.
Fig. S6. ATRX prevents recombination when telomeres are heterochromatic.
Fig. S7. Loss of SETDB1 and not of SUV39H promotes ALT features.
Fig. S8. Full membranes from which ChIP figures were prepared.
Table S1. List of factors enriched in the telomere PICh performed with wild-type ESCs.
Table S2. List of factors enriched in the telomere PICh performed with wild-type mouse embryonic fibroblast cells.
REFERENCES AND NOTES
- 1.Yeager T. R., Neumann A. A., Englezou A., Huschtscha L. I., Noble J. R., Reddel R. R., Telomerase-negative immortalized human cells contain a novel type of promyelocytic leukemia (PML) body. Cancer Res. 59, 4175–4179 (1999). [PubMed] [Google Scholar]
- 2.Potts P. R., Yu H., The SMC5/6 complex maintains telomere length in ALT cancer cells through SUMOylation of telomere-binding proteins. Nat. Struct. Mol. Biol. 14, 581–590 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Flynn R. L., Cox K. E., Jeitany M., Wakimoto H., Bryll A. R., Ganem N. J., Bersani F., Pineda J. R., Suva M. L., Benes C. H., Haber D. A., Boussin F. D., Zou L., Alternative lengthening of telomeres renders cancer cells hypersensitive to ATR inhibitors. Science 347, 273–277 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O’Sullivan R. J., Arnoult N., Lackner D. H., Oganesian L., Haggblom C., Corpet A., Almouzni G., Karlseder J., Rapid induction of alternative lengthening of telomeres by depletion of the histone chaperone ASF1. Nat. Struct. Mol. Biol. 21, 167–174 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heaphy C. M., de Wilde R. F., Jiao Y., Klein A. P., Edil B. H., Shi C., Bettegowda C., Rodriguez F. J., Eberhart C. G., Hebbar S., Offerhaus G. J., McLendon R., Rasheed B. A., He Y., Yan H., Bigner D. D., Oba-Shinjo S. M., Marie S. K. N., Riggins G. J., Kinzler K. W., Vogelstein B., Hruban R. H., Maitra A., Papadopoulos N., Meeker A. K., Altered telomeres in tumors with ATRX and DAXX mutations. Science 333, 425 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lovejoy C. A., Li W., Reisenweber S., Thongthip S., Bruno J., de Lange T., de S., Petrini J. H. J., Sung P. A., Jasin M., Rosenbluh J., Zwang Y., Weir B. A., Hatton C., Ivanova E., Macconaill L., Hanna M., Hahn W. C., Lue N. F., Reddel R. R., Jiao Y., Kinzler K., Vogelstein B., Papadopoulos N., Meeker A. K.; ALT Starr Cancer Consortium , Loss of ATRX, genome instability, and an altered DNA damage response are hallmarks of the alternative lengthening of telomeres pathway. PLOS Genet. 8, e1002772 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Napier C. E., Huschtscha L. I., Harvey A., Bower K., Noble J. R., Hendrickson E. A., Reddel R. R., ATRX represses alternative lengthening of telomeres. Oncotarget 6, 16543–16558 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ishov A. M., Vladimirova O. V., Maul G. G., Heterochromatin and ND10 are cell-cycle regulated and phosphorylation-dependent alternate nuclear sites of the transcription repressor Daxx and SWI/SNF protein ATRX. J. Cell Sci. 117, 3807–3820 (2004). [DOI] [PubMed] [Google Scholar]
- 9.Voon H. P. J., Collas P., Wong L. H., Compromised telomeric heterochromatin promotes ALTernative lengthening of telomeres. Trends Cancer 2, 114–116 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Nozawa R. S., Nagao K., Masuda H. T., Iwasaki O., Hirota T., Nozaki N., Kimura H., Obuse C., Human POGZ modulates dissociation of HP1α from mitotic chromosome arms through Aurora B activation. Nat. Cell Biol. 12, 719–727 (2010). [DOI] [PubMed] [Google Scholar]
- 11.Dhayalan A., Tamas R., Bock I., Tattermusch A., Dimitrova E., Kudithipudi S., Ragozin S., Jeltsch A., The ATRX-ADD domain binds to H3 tail peptides and reads the combined methylation state of K4 and K9. Hum. Mol. Genet. 20, 2195–2203 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iwase S., Xiang B., Ghosh S., Ren T., Lewis P. W., Cochrane J. C., Allis C. D., Picketts D. J., Patel D. J., Li H., Shi Y., ATRX ADD domain links an atypical histone methylation recognition mechanism to human mental-retardation syndrome. Nat. Struct. Mol. Biol. 18, 769–776 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peters A. H., O’Carroll D., Scherthan H., Mechtler K., Sauer S., Schöfer C., Weipoltshammer K., Pagani M., Lachner M., Kohlmaier A., Opravil S., Doyle M., Sibilia M., Jenuwein T., Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell 107, 323–337 (2001). [DOI] [PubMed] [Google Scholar]
- 14.Benetti R., Gonzalo S., Jaco I., Schotta G., Klatt P., Jenuwein T., Blasco M. A., Suv4-20h deficiency results in telomere elongation and derepression of telomere recombination. J. Cell Biol. 178, 925–936 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.García-Cao M., O’Sullivan R., Peters A. H., Jenuwein T., Blasco M. A., Epigenetic regulation of telomere length in mammalian cells by the Suv39h1 and Suv39h2 histone methyltransferases. Nat. Genet. 36, 94–99 (2004). [DOI] [PubMed] [Google Scholar]
- 16.Blasco M. A., The epigenetic regulation of mammalian telomeres. Nat. Rev. Genet. 8, 299–309 (2007). [DOI] [PubMed] [Google Scholar]
- 17.Deng Z., Norseen J., Wiedmer A., Riethman H., Lieberman P. M., TERRA RNA binding to TRF2 facilitates heterochromatin formation and ORC recruitment at telomeres. Mol. Cell 35, 403–413 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arnoult N., Van Beneden A., Decottignies A., Telomere length regulates TERRA levels through increased trimethylation of telomeric H3K9 and HP1α. Nat. Struct. Mol. Biol. 19, 948–956 (2012). [DOI] [PubMed] [Google Scholar]
- 19.Episkopou H., Draskovic I., Van Beneden A., Tilman G., Mattiussi M., Gobin M., Arnoult N., Londoño-Vallejo A., Decottignies A., Alternative lengthening of Telomeres is characterized by reduced compaction of telomeric chromatin. Nucleic Acids Res. 42, 4391–4405 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cubiles M. D., Barroso S., Vaquero-Sedas M. I., Enguix A., Aguilera A., Vega-Palas M. A., Epigenetic features of human telomeres. Nucleic Acids Res. 46, 2347–2355 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saksouk N., Barth T. K., Ziegler-Birling C., Olova N., Nowak A., Rey E., Mateos-Langerak J., Urbach S., Reik W., Torres-Padilla M. E., Imhof A., Déjardin J., Simboeck E., Redundant mechanisms to form silent chromatin at pericentromeric regions rely on BEND3 and DNA methylation. Mol. Cell 56, 580–594 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Zalzman M., Falco G., Sharova L. V., Nishiyama A., Thomas M., Lee S. L., Stagg C. A., Hoang H. G., Yang H. T., Indig F. E., Wersto R. P., Ko M. S. H., Zscan4 regulates telomere elongation and genomic stability in ES cells. Nature 464, 858–863 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Déjardin J., Kingston R. E., Purification of proteins associated with specific genomic loci. Cell 136, 175–186 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shinkai Y., Tachibana M., H3K9 methyltransferase G9a and the related molecule GLP. Genes Dev. 25, 781–788 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matsui T., Leung D., Miyashita H., Maksakova I. A., Miyachi H., Kimura H., Tachibana M., Lorincz M. C., Shinkai Y., Proviral silencing in embryonic stem cells requires the histone methyltransferase ESET. Nature 464, 927–931 (2010). [DOI] [PubMed] [Google Scholar]
- 26.Bulut-Karslioglu A., De La Rosa-Velázquez I. A., Ramirez F., Barenboim M., Onishi-Seebacher M., Arand J., Galán C., Winter G. E., Engist B., Gerle B., O'Sullivan R. J., Martens J. H., Walter J., Manke T., Lachner M., Jenuwein T., Suv39h-dependent H3K9me3 marks intact retrotransposons and silences LINE elements in mouse embryonic stem cells. Mol. Cell 55, 277–290 (2014). [DOI] [PubMed] [Google Scholar]
- 27.Karimi M. M., Goyal P., Maksakova I. A., Bilenky M., Leung D., Tang J. X., Shinkai Y., Mager D. L., Jones S., Hirst M., Lorincz M. C., DNA methylation and SETDB1/H3K9me3 regulate predominantly distinct sets of genes, retroelements, and chimeric transcripts in mescs. Cell Stem Cell 8, 676–687 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.He Q., Kim H., Huang R., Lu W., Tang M., Shi F., Yang D., Zhang X., Huang J., Liu D., Songyang Z., The Daxx/Atrx complex protects tandem repetitive elements during DNA hypomethylation by promoting H3K9 trimethylation. Cell Stem Cell 17, 273–286 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Udugama M., M Chang F. T., Chan F. L., Tang M. C., Pickett H. A., R McGhie J., Mayne L., Collas P., Mann J. R., Wong L. H., Histone variant H3.3 provides the heterochromatic H3 lysine 9 tri-methylation mark at telomeres. Nucleic Acids Res. 43, 10227–10237 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jasencakova Z., Scharf A. N., Ask K., Corpet A., Imhof A., Almouzni G., Groth A., Replication stress interferes with histone recycling and predeposition marking of new histones. Mol. Cell 37, 736–743 (2010). [DOI] [PubMed] [Google Scholar]
- 31.Déjardin J., Switching between epigenetic states at pericentromeric heterochromatin. Trends Genet. 31, 661–672 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Ayyanathan K., Lechner M. S., Bell P., Maul G. G., Schultz D. C., Yamada Y., Tanaka K., Torigoe K., Rauscher F. J. III, Regulated recruitment of HP1 to a euchromatic gene induces mitotically heritable, epigenetic gene silencing: A mammalian cell culture model of gene variegation. Genes Dev. 17, 1855–1869 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hathaway N. A., Bell O., Hodges C., Miller E. L., Neel D. S., Crabtree G. R., Dynamics and memory of heterochromatin in living cells. Cell 149, 1447–1460 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lachner M., O’Carroll D., Rea S., Mechtler K., Jenuwein T., Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 410, 116–120 (2001). [DOI] [PubMed] [Google Scholar]
- 35.Lehnertz B., Ueda Y., Derijck A. A. H. A., Braunschweig U., Perez-Burgos L., Kubicek S., Chen T., Li E., Jenuwein T., Peters A. H., Suv39h-mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr. Biol. 13, 1192–1200 (2003). [DOI] [PubMed] [Google Scholar]
- 36.Arora R., Lee Y., Wischnewski H., Brun C. M., Schwarz T., Azzalin C. M., RNaseH1 regulates TERRA-telomeric DNA hybrids and telomere maintenance in ALT tumour cells. Nat. Commun. 5, 5220 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Orphanides G., Wu W. H., Lane W. S., Hampsey M., Reinberg D., The chromatin-specific transcription elongation factor FACT comprises human SPT16 and SSRP1 proteins. Nature 400, 284–288 (1999). [DOI] [PubMed] [Google Scholar]
- 38.Ramsahoye B. H., Biniszkiewicz D., Lyko F., Clark V., Bird A. P., Jaenisch R., Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc. Natl. Acad. Sci. U.S.A. 97, 5237–5242 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baubec T., Colombo D. F., Wirbelauer C., Schmidt J., Burger L., Krebs A. R., Akalin A., Schübeler D., Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature 520, 243–247 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Neri F., Rapelli S., Krepelova A., Incarnato D., Parlato C., Basile G., Maldotti M., Anselmi F., Oliviero S., Intragenic DNA methylation prevents spurious transcription initiation. Nature 543, 72–77 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Poole L. A., Zhao R., Glick G. G., Lovejoy C. A., Eischen C. M., Cortez D., SMARCAL1 maintains telomere integrity during DNA replication. Proc. Natl. Acad. Sci. U.S.A. 112, 14864–14869 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marzec P., Armenise C., Pérot G., Roumelioti F. M., Basyuk E., Gagos S., Chibon F., Déjardin J., Nuclear-receptor-mediated telomere insertion leads to genome instability in ALT cancers. Cell 160, 913–927 (2015). [DOI] [PubMed] [Google Scholar]
- 43.Tsai H. J., Huang W. H., Li T. K., Tsai Y. L., Wu K. J., Tseng S. F., Teng S. C., Involvement of topoisomerase III in telomere-telomere recombination. J. Biol. Chem. 281, 13717–13723 (2006). [DOI] [PubMed] [Google Scholar]
- 44.Zeng S., Xiang T., Pandita T. K., Gonzalez-Suarez I., Gonzalo S., Harris C. C., Yang Q., Telomere recombination requires the MUS81 endonuclease. Nat. Cell Biol. 11, 616–623 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dilley R. L., Verma P., Cho N. W., Winters H. D., Wondisford A. R., Greenberg R. A., Break-induced telomere synthesis underlies alternative telomere maintenance. Nature 539, 54–58 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Emerson R. O., Thomas J. H., Adaptive evolution in zinc finger transcription factors. PLOS Genet. 5, e1000325 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/5/eaav3673/DC1
Fig. S1. SETDB1 telomeric binding is developmentally regulated.
Fig. S2. HP1α stimulates SETDB1-dependent heterochromatin formation at telomeres.
Fig. S3. ATRX binds heterochromatic telomeres.
Fig. S4. Telomeric heterochromatin stimulates transcriptional elongation.
Fig. S5. SETDB1-dependent heterochromatin promotes the recruitment of recombination factors and the appearance of ALT features.
Fig. S6. ATRX prevents recombination when telomeres are heterochromatic.
Fig. S7. Loss of SETDB1 and not of SUV39H promotes ALT features.
Fig. S8. Full membranes from which ChIP figures were prepared.
Table S1. List of factors enriched in the telomere PICh performed with wild-type ESCs.
Table S2. List of factors enriched in the telomere PICh performed with wild-type mouse embryonic fibroblast cells.