

Abstract
Two-component systems are essential for bacteria to sense, respond, and adapt to changing environments, such as elevation of Cu(I)/Ag(I) ions in the periplasm. In Escherichia coli, the CusS-CusR two-component system upregulates the cusCFBA genes under increased periplasmic Cu(I)/Ag(I) concentrations to help maintain metal ion homeostasis. The CusS histidine kinase is a homodimeric integral membrane protein that binds to periplasmic Cu(I)/Ag(I) and transduces a signal to its cytoplasmic kinase domain. However, the mechanism of how metal binding in the periplasm activates autophosphorylation in the cytoplasm is unknown. Here, we report that only one of the two metal ion binding sites in CusS enhances dimerization of the sensor domain. Utilizing nanodisc technology to study full-length CusS, we show that metal-induced dimerization in the sensor domain triggers kinase activity in the cytoplasmic domain. We also investigated autophosphorylation in the cytoplasmic domain of CusS and phosphotransfer between CusS and CusR. In vitro analyses show that CusS autophosphorylates its conserved H271 residue at the N1 position of the histidine imidazole. The phosphoryl group is removed by the response regulator CusR in a reaction that requires a conserved aspartate at position 51. Functional analyses in vivo of CusS and CusR variants with mutations in the autophosphorylation or phosphoacceptor residues suggest that the phosphotransfer event is essential for metal resistance in E. coli. Biochemical analysis shows that the CusS dimer autophosphorylates using a cis mechanism. Our results support a signal transduction model in which rotation and bending movements in the cytoplasmic domain maintain the mode of autophosphorylation.
INTRODUCTION
Bacteria live in a variety of environments and therefore have developed mechanisms to respond to changing conditions. Two-component systems (TCSs) are the predominant signaling systems used in prokaryotes to sense, respond, and adapt to a variety of stimuli in the environment (1,2). A prototypical TCS is comprised of an integral membrane histidine kinase (HK) and its cognate cytoplasmic response regulator (RR) (3). Typically, HKs have a variable extracellular sensor domain that is connected to the conserved cytoplasmic kinase domain through transmembrane helices. Upon sensing of environmental stimuli by the extracellular sensor domain, a signal is transmitted to the cytoplasmic domain initiating autophosphorylation on a conserved and specific His residue. In most prokaryotic systems, the RRs are the terminal component of the signaling pathway that function as phosphorylation-activated switches to adapt to stimuli changes. The RR catalyzes the phosphoryl transfer from the phosphohistidine of the HK to its conserved Asp in the regulatory domain. The signaling pathway consists of three phosphotransfer reactions. First, a HK homodimer uses ATP to autophosphorylate a conserved histidine residue. Second, the phosphoryl group on the phosphorylated HK (HK~P) is transferred to a conserved aspartate residue on a cognate RR. Third, the phosphorylated RR (RR~P) is dephosphorylated by an intrinsic or HK-induced RR~P autophosphatase activity. When phosphorylated, the RR~P interacts with genes or protein targets triggering cellular response to the stimuli (1,4). The mechanism of signal propagation from the sensor domain to the kinase domain resulting in changes in activity has been an active area of study.
While copper is an essential element for most organisms, only a small amount of intracellular copper is needed and excess amounts can be toxic even at low levels due to its redox properties (5–8). Silver is not required for any biological process, but it shares similar chemical and ligand binding properties with Cu(I), and it is toxic at even lower concentrations (9). Therefore, organisms have developed mechanisms to maintain copper and silver ion homeostasis in the intracellular environment through acquisition, sequestration, and efflux of metal ions. E. coli survives copper and silver stress in part by pumping out excess metals through the CusCFBA efflux pump; the CusS-CusR TCS regulates the expression of cusCFBA genes involved in maintaining metal ion homeostasis in cells (7,10).
CusS is a histidine kinase with a periplasmic sensor domain, two transmembrane (TM) α-helices, and a cytoplasmic domain. The periplasmic sensor domain of CusS senses increased levels of Cu(I)/Ag(I) and transmits a signal to the cytoplasmic kinase domain upon binding Cu(I)/Ag(I). The cytoplasmic domain of CusS consists of three domains: a “histidine kinase, adenylyl cyclases, methyl-accepting proteins, phosphatases” (HAMP) domain, a dimerization and histidine phosphotransfer (DHp) domain, and a catalytic and ATP binding domain (CA) (1). The HAMP domain consists of two amphipathic helices with coiled-coil properties that form a homodimeric, four-helical, parallel coiled-coil structure (11,12). The DHp domain includes two α-helices that mediate homodimerization by forming a four-helix bundle (1,13). The CA domain typically forms an α/β sandwich that binds ATP and catalyzes autophosphorylation of a conserved His residue in the DHp domain (14). The H271 residue of CusS corresponds to the highly conserved histidine residue found in all histidine kinases, and is the likely site of phosphorylation in this kinase, but its functional role has not yet been demonstrated in any in vitro or in vivo studies. CusR is the downstream response regulator of CusS, which consists of a receiver domain and an effector domain. Upon phosphorylation of the conserved aspartate, D51, in the receiver domain, CusR is activated and functions as a transcriptional activator to upregulate the transcription of the cusCFBA genes (7).
Protein phosphorylation is one of the most common post-translational modifications among all living things, including animals, plants, fungi, bacteria, and archaea (15–17). There are nine known phosphoamino acids and they can be divided into four groups: O-phosphates (phosphoserine, phosphotyrosine, phosphothreonine), N-phosphates (phosphoarginine, phosphohistidine, phospholysine), acyl phosphates (phosphoaspartate, phosphoglutamate), and S-phosphates (phosphocysteine) (18). In bacteria, kinases that are part of TCSs frequently modify His and Asp residues to generate phosphohistidine (pHis) and phosphoaspartate (pAsp). pHis is unique among the phosphoamino acids because it can be phosphorylated on either the N1 or N3 position on the imidazole ring generating 1-pHis or 3-pHis, respectively (17). When compared to the most studied O-phosphates, pHis has a higher ΔG of hydrolysis (−12 to −13 kcal/mol for pHis versus −6.5 to −9.5 kcal/mol for O-phosphates) (17). In HKs, the high energy nature of pHis would mean that the phosphorylation lifetime may be much more transient than that of an O-phosphate. Dephosphorylation of the HK can occur through direct hydrolysis or through transfer of the phosphoryl group to the RR. Similarly, phosphorylated aspartate residues in RRs are generally more prone to hydrolysis than O-phosphates and are often a relatively short lived species (19). Because of the ease with which HKs and RRs can be phosphorylated and dephosphorylated, these types of modification are a flexible mechanism for cells to respond to external stimuli and environmental conditions.
It was previously shown that the cusS gene has an important role in silver resistance and regulation of copper homeostasis, and that the sensor domain of CusS [CusS(39–187)] interacts directly with Ag(I) and undergoes a conformational change and dimerizes upon metal binding (20,21). The crystal structure of the silver-bound periplasmic sensor domain of E. coli CusS [Ag(I)-CusS((39–187)] reveals a homodimer with four Ag(I) per dimer. Two Ag(I) are bound at the dimer interface and the other two Ag(I) are bound internally within each separate domain (22). Functional studies demonstrated that the interface binding sites are more important than the internal binding sites for metal ion resistance in E. coli (22). Although the crystal structure has given insight on the molecular basis of metal binding in CusS(39–187), the relevance of the metal binding residues and metal binding sites in metal-induced dimerization and signal transduction to the kinase core in E. coli remains unclear.
HKs are known to exist and function as homodimers (23), and thus it was expected that the sensor domains would form dimers in solutions when expressed as truncated proteins (24). However, among extracellular sensor domains, only CitA and DcuS are confirmed to dimerize and do so weakly in solution (Kd of 9.7 ± 3.7 mM) (25,26). The sensor domain of E. coli TorS dimerizes in solution with Kd of 49.6 µM (27). The sensor domains of the HKs DcuS, DctB, CitA, and NarX exist as monomers during purification (26,28,29). The effects of ligands on dimerization of the sensor domains have been studied in a few cases, and ligand-induced dimerization in the sensor domains of HKs has been shown for CusS as well as CitA and NarX (20,28,29). In prototypical HKs, the N-and C-terminal helices of the sensor domain are expected to be contiguous with the transmembrane helices. Therefore, ligand-induced dimerization could result in conformational changes in the periplasm that are transduced to the cytoplasmic helices and result in changes in kinase activity. However, in the case of CusS with two separate ligand binding sites in the dimer, how each site contributes to the dimerization and activation of the kinase is unknown. The hypothesis is that metal-induced dimerization of the periplasmic sensor domain of CusS is the trigger for signal transduction to the kinase core and thus activates cusCFBA genes for metal resistance.
The complete signal transduction mechanism of HKs has been challenging to determine due to the difficulties in studying membrane proteins and their insolubility in aqueous systems. Here, we describe the solubilization and purification of CusS and the self-assembly of CusS in nanodiscs, which we used to study the auto-kinase properties in more detail. Unlike liposomes, nanodiscs provide a soluble and native-like phospholipid bilayer environment that provides stability and accessibility of the target membrane protein (30). They also allow better control of the oligomeric state of the target membrane protein (31,32). A nanodisc is composed of two amphipathic membrane scaffold protein (MSP) molecules that encircle a plug of lipid bilayer like a belt, shielding the hydrophobic lipid acyl chain and making the nanodisc particles monodisperse and homogeneous (30,31). The self-assembly of the target membrane protein and nanodisc is achieved by gradual removal of detergent from the mixture of detergent-solubilized protein and nanodisc. The end result is the target membrane protein, such as CusS, embedded in the nanodisc.
Here, we have further investigated the relationship between metal binding and kinase activation of CusS. We examined the metal binding properties of each CusS metal binding site. Using in vitro crosslinking experiments we show that the interface binding site is responsible for metal-induced dimerization of CusS(39–187) while the internal binding site is not. We have also successfully self-assembled full-length CusS into nanodiscs and shown that it is stable and active. To investigate the role of each metal binding site in kinase activation, the nanodiscs containing CusS variants were also tested for their autophosphorylation activities. We show that only the interface metal binding site is essential for triggering autophosphorylation. Furthermore, we have used monoclonal anti-pHis antibodies to investigate the catalytic mechanism of the cytoplasmic kinase core of CusS (CusScp). We found that CusS is phosphorylated at the N1 position on its conserved H271 and that the conserved D51 on CusR is required for dephosphorylation of CusS, likely through phosphotransfer to this residue. Mutagenesis combined with in vivo complementation assays were performed to further investigate the functional significance of the phosphoacceptor residues in both CusS (H271) and CusR (D51). Biochemical studies have shown that CusScp undergoes autophosphorylation via a cis mechanism, i.e. phosphorylation and catalytic activity occur within the same subunit. This finding contradicts a previous published hypothesis that all bacterial HKs were universally expected to autophosphorylate in trans. These findings have provided us an opportunity to further understand and to propose a model for the signal transduction mechanism of CusS.
Experimental procedures
Strain and plasmids constructions—
The CusS(39–187) wild-type and mutant plasmids used in this study are shown in Supplemental Table S1. The previously described pTXB3CusSs plasmid (20) was used as the starting plasmid to generate the plasmids for expression of the sensor domains used in this study. In the previous study, the intein expression vector was chosen to increase the solubility of CusS(39–187). In our study, the Ser residue adjacent to the intein cleavage site was mutated to Ala (S192A) to increase cleavage efficiency (New England Biolabs manual E6901) using the QuikChange II XL Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA). Thus, after cleavage of the intein tag, there is a C-terminal EGSA sequence artifact. Strep-tag II was added to the N-terminus of CusS(39–187) by mutagenesis following the protocol of Liu & Naismith (33). This plasmid encoding the wild-type CusS sensor domain is named pStrep-cusS(39–187). Mutations of the relevant metal binding residues were introduced into pStrep cusS(39–187) using the QuikChange II XL Site-Directed Mutagenesis Kit. Mutations H42A/F43I/H176A were made to eliminate the interface binding sites creating pStrep-cusS(39–187)-AIA. Mutations M133I/M135I/H145A were made to eliminate the internal binding sites creating pStrep-cusS(39–187)-IIA. The final purified proteins are CusS(39–187) containing an N-terminal Strep tag, C-terminal EGSA residues after intein cleavage, and the desired mutations. All plasmid genes for protein products were sequenced to verify accuracy. Plasmids pcusS, pcusS-AIA, and pcusS-IIA, which produce the full-length CusS and variants, were previously made and described (22). Plasmid pcusS-H271A containing a mutation disrupting the catalytic H271 residue was made using the QuikChange II XL Site-Directed Mutagenesis Kit. The final full-length CusS protein contains a C-terminal 6xHis tag.
TMHMM transmembrane topology prediction software predicted the cytoplasmic domain of CusS (CusScp) to consist of amino acids 208 through 480. The cusScp gene encoding amino acids 208–480 was amplified using oligonucleotides containing sites for NheI and XhoI restriction endonucleases. The amplified product was digested using NheI and XhoI enzymes and ligated into pET21b(+). The correct final product pcusScp-short was verified by DNA sequencing. The final version of CusScp includes N-terminal MAS residues as a starting Met and a cloning artifact, and C-terminal LE residues as a cloning artifact before the 6xHis tag. The protein produced from this construct is named CusScp-short. The plasmid to produce CusScp with C-terminal Strep-Myc-Strep-FLAG-HA-Strep tags, pcusScp-long, was synthesized by Bio Basic (Amherst, NY) in the pET28b(+) plasmid between the NcoI and NotI sites. The protein produced from this construct is named CusScp-long. The plasmids pcusScp-short and pcusScp-long were used as templates for mutagenesis using the QuikChange II XL Site-Directed Mutagenesis Kit. Constructs were made with mutations of the catalytic histidine residue H271A (CusScp-H271A), the ATP binding site N386A/N414A double mutant (CusScp-AA), and both catalytic histidine and ATP binding site H271A/N386A/N414A triple mutant (CusScp-AAA).
The cusR gene encoding amino acids 2–227 was amplified using oligonucleotides containing sites for the NdeI and XhoI restriction endonucleases. The amplified product was digested using NdeI and XhoI enzymes and ligated into pET22b(+). The correct final product pcusR was verified by DNA sequencing. The CusR expressed from this plasmid includes an N-terminal starting methionine and C-terminal LE residues as a cloning artifact before the 6xHis tag. The pcusR construct was used as a template to generate the mutant pcusR-D51A, where the catalytic aspartate residue was mutated to alanine.
The bacterial strains used for in vivo complementation assays are listed in Supplemental Table S2. The strains containing knockouts of cusR or cusS are from the Keio collection (34). Using the lambda-Red-mediated gene recombination technique as described previously (22,35), double knockout strains of cusS/cueO (named ΔcusS) (22) or cusR/cueO (named ΔcusR) (this study) were created. For growth experiments, the WT strain was transformed with either pET21b(+) or pET22b(+) empty vector, the ΔcusS strain was transformed with pET21b(+), pcusS, or pcusS-H271A, and the ΔcusR strain was transformed with pET22b(+), pcusR, or pcusR-D51A.
Purification of N-terminal Strep-tagged CusS(39–187) wild-type and mutants—
Expression and purification of Strep-tagged CusS(39–187) and mutants CusS(39–187)-AIA and CusS(39–187)-IIA were performed as previously described (20) with a few changes. Following the overnight on-column cleavage, the eluted fractions were loading onto a Strep-tactin resin affinity column (IBA, Germany), washed with Buffer W (100 mM Tris pH 8.0, 150 mM NaCl), and protein was eluted with Buffer E (Buffer W + 2.5 mM desthiobiotin) in 4 mL fractions. Purity of protein fractions was detected by SDS-PAGE. Fractions that showed a significant amount of other protein products were further purified using a HiPrep Sephacryl S-100 26/60 column previously equilibrated with Buffer W. Pure CusS(39–187) wild-type or mutants were then pooled and concentrated to 0.5–1 mg/mL. Protein was dialyzed against 50 mM HEPES pH 7.5 for in vitro crosslinking. Protein concentrations were determined using the Bradford assay.
To obtain Ag(I)-loaded CusS(39–187) wild-type and mutants for in vitro crosslinking, dialysis was performed at 4°C in 50 mM HEPES pH 7.5 containing a 5-fold molar excess of AgNO3 three times. After equilibration, excess and unbound metals were removed by dialysis against 50 mM HEPES pH 7.5 buffer at 4°C three times. Dialysis was performed using Slide-A-Lyzer dialysis cassettes. After dialysis, the protein concentrations were determined using Bradford assay.
In vitro chemical crosslinking with BS3—
The amine reactive crosslinking reagent BS3 (bis[sulfosuccinimidyl] suberate) was purchased from Thermo Scientific Pierce (Rockford, IL). A 25 mM stock of the crosslinker BS3 was prepared in water. The crosslinking reactions were carried out in 50 mM HEPES buffer pH 7.5 with a final reaction volume of 45 µL. A 50-fold molar excess of crosslinker BS3 (0.5 mM) was added to 10 µM apo or Ag(I)-bound CusS(39–187) wild-type and mutants. The reactions were incubated on ice for 2 hr and the reactions were quenched by adding 1 M ammonium bicarbonate to final concentration of 20 mM. Equal amounts of each protein were loaded onto SDS gels. The crosslinked products were separated by SDS-PAGE and visualized by silver staining and Western blot using anti-Strep tag antibody, and the bands were quantified using ImageJ.
Expression and purification of membrane scaffold protein MSP1D1—
pMSP1D1 plasmid was purchased from Addgene (plasmid # 20061) and used for bacterial expression of the membrane scaffold protein for nanodisc formation (31). This construct contains MSP1D1 protein in pET28a vector with an N-terminal 7xHis tag followed by TEV cleavage site. MSP1D1 has a deletion of the first 11 amino acids, which are not required for nanodisc formation. For expression of MSP1D1, E. coli BL21(DE3) cells were transformed with pMSP1D1 and grown in LB containing 34 µg/mL kanamycin at 37°C until OD600 of 0.8–1.0, then 1 mM IPTG was added and the culture was continued to grow for another 5 hr prior to harvest.
MSP1D1 was prepared as previously described (30,36) with some modifications. To purify MSP1D1, cells were resuspended in binding buffer (50 mM Tris pH 8.0, 500 mM NaCl, 20 mM imidazole) containing 1 mM phenylmethylsulfonyl fluoride (PMSF), 4 µg/mL of aprotinin, leupeptin, pepstatin (protease inhibitors), and 1% Triton X-100 at 4°C for 1 hr. The cells were then lysed using a Branson sonicator (ten 30-sec rounds with 1 min pause in between). Deoxyribonuclease I (DNaseI) was added to a final concentration of 10 µg/mL and the lysate was mixed for another 30 min prior to centrifugation to remove the cell debris. The supernatant was applied to Ni2+ Sepharose resin equilibrated in binding buffer, washed with washing buffer (binding buffer containing 1% Triton X-100), and proteins were eluted with 400 mM imidazole in final buffer (20 mM Tris pH 8.0, 200 mM NaCl). The recovery of proteins was checked by SDS-PAGE and fractions containing MSP1D1 were pooled and concentration was determined spectrophotometrically using the extinction coefficient ε = 21430 M−1 cm-1.
The N-terminal 7xHis tag on MSP1D1 was removed by treatment with TEV protease. The 7xHis tag cleaved MSP1D1 is referred as MSP1D1(−). TEV protease was added to MSP1D1 protein at 1 mg TEV/10 mg MSP1D1 in cleavage buffer (50 mM Tris pH 8.0, 0.5 mM EDTA, 1 mM DTT). The mixture was incubated at room temperature overnight (~20–24 hr). To remove the EDTA and DTT, the mixture was dialyzed against final buffer at 4°C. The 6xHis-TEV protease, cleaved 7xHis tag, and uncleaved MSP1D1 were removed by applying the mixture onto a Ni2+ column. The MSP1D1(−) appears in the flow through and washes. The MSP1D1(−) is ~2.7 kDa smaller than the intact MSP1D1 and was distinguished by SDS-PAGE as MSP1D1(−) migrates slightly faster. The recovery and purity of MSP1D1(−) was checked by SDS-PAGE and the fractions containing pure MSP1D1(−) were collected and concentrated. The protein concentration was measured using a Bradford assay.
Preparation of detergent solubilized E. coli phospholipids—
E. coli lipids were prepared by placing 2 mL of 25 mg/mL E. coli polar lipids (Avanti Polar Lipids Inc.) in a glass vial, evaporating the solvent and drying the lipid in a vacuum desiccator overnight. The dried lipid was resuspended in 1.6 mL of 50 mM HEPES pH 7.5, 200 mM NaCl buffer containing 48 mg/mL n-octyl-β-glucopyranoside (OGP) and solubilized at 4°C for 1 hr or until the solution turned clear. The OGP-solubilized lipid was extruded through 0.2 µm polycarbonate filter 13 times using the mini-extruder (Avanti Polar Lipids Inc.). The filtered lipid was then aliquoted, frozen in liquid N2, and stored at −80°C. The lipid concentration was determined using a phosphorus assay as previously described (30).
CusR and CusR-D51A were purified as described for the CusScp-short homodimer.
Expression and purification of histidine kinase CusS wild-type and mutants—
The overexpression of full-length CusS proteins was performed in an auto-inducing condition in the absence of IPTG as described by Studier (37) with some changes as detailed below. E. coli BL21(DE3) cells were transformed with plasmids containing genes encoding for CusS, CusS-H271A, CusS-AIA, and CusS-IIA (Supp. Table S1). Overnight cultures of each strain were grown in LB media containing 100 µg/mL ampicillin at 37°C. Fresh LB media containing 100 µg/mL ampicillin and 1X 5052 mixture (0.5% glycerol, 0.05% glucose, 0.2% lactose) was inoculated with the overnight culture and grown at 37°C. Once the OD600 reached ~0.8, the temperature was lowered to room temperature (~22°C) and growth was continued overnight (20–24 hr).
To solubilize the membrane protein CusS, cells were resuspended in 50 mM Tris pH 8 containing 1 mM PMSF, 4 µg/mL of protease inhibitors, and lysozyme. Lysis was achieved by sonication at output 7 (ten 30-sec rounds with 1 min pause in between). DNaseI was then added to a final concentration of 10 µg/mL to remove any DNA, and lysate was mixed for another 30 min. The lysate was fractionated by ultracentrifugation at 125,000 x g for 1 hr into membrane fraction (pellet) and cytosolic fraction (supernatant). The pellet containing the membrane fraction was resuspended in binding buffer containing 1 mM MgCl2, and solubilized with 2% Empigen BB at 4°C for 1.5 hr. The mixture was ultracentrifuged at 125,000 x g for 1 hr and the supernatant containing the solubilized membrane protein CusS was collected.
To purify CusS, the supernatant was applied onto a Ni2+ column, washed with HEPES binding buffer (50 mM HEPES pH 7.5, 200 mM NaCl, 20 mM imidazole) containing 0.05% n-dodecyl-β-D-maltoside (DDM), and eluted with 400 mM imidazole in HEPES final buffer (50 mM HEPES pH 7.5, 200 mM NaCl, 0.05% DDM). The recovery and purity of proteins were checked by SDS-PAGE and the fractions containing CusS were pooled and concentrated. The protein was then dialyzed against HEPES final buffer at 4°C. The protein concentrations were determined spectrophotometrically using the extinction coefficient ε = 24410 M−1 cm-1. Four CusS variants were purified: CusS (wild-type protein), CusS-H271A, CusS-AIA, and CusS-IIA.
Reconstitution and purification of nanodiscs containing wild-type and mutant CusS membrane proteins—
The nanodisc system is a three-component system which consists of a membrane scaffold protein, lipids, and the membrane protein of interest. All the components are mixed together in appropriate molar ratios and kept solubilized in detergent. Upon removal of detergent, the self-assembly of the membrane protein embedded in the nanodiscs is initiated. Here, the three components used were MSP1D1(−), OGP-solubilized E. coli lipids, and CusS proteins (CusS (wild-type), CusS-H271A, CusS-AIA, and CusS-IIA) in DDM. DDM was kept at 0.9 mM above its CMC value (0.12 mM in 0.2 M NaCl), to ensure the solubilization of phospholipids and membrane proteins. In accordance with previous established protocols, the MSP1D1(−), E. coli lipids, DDM, and CusS were combined in a molar ratio of 1:65:6:0.1 (30,38). The final concentrations of the components in the mixture were 150 µM MSP1D1(−), 9.75 mM E. coli lipids, 0.9 mM DDM, and 15 µM CusS. The mixture was incubated on ice for 1 hr. To promote the self-assembly of nanodiscs containing CusS, the detergent was slowly removed by adding 200 mg Biobeads SM2 (Bio-Rad) per 1 mL mixture and incubated with gentle shaking at 4°C for 4 hr. The mixture was then transferred into fresh 200 mg/mL Biobeads SM2 and continued incubation at 4°C overnight. The mixture was collected for further purification to separate nanodiscs containing CusS from empty nanodiscs. The mixture was applied onto a Ni2+ column, washed with HEPES binding buffer, and eluted with 400 mM imidazole in HEPES binding buffer. The recovery and purity of CusS in nanodiscs was checked by SDS-PAGE, and fractions containing CusS were pooled and concentrated to 0.3 mg/mL (~3 µM CusS). Finally, CusS was dialyzed against 50 mM HEPES pH 7.5 buffer at 4°C. CusS concentrations were determined by Bradford assays. Four nanodisc-embedded CusS variants were reconstituted and purified: CusS, CusS-H271A, CusS-AIA, and CusS-IIA.
Autophosphorylation assay of CusS variants in nanodiscs—
Due to solubility issues of AgNO3, the CusS in nanodiscs was dialyzed against 50 mM HEPES pH 7.5 and the kinase buffer was modified to be 50 mM HEPES pH 7.5, 50 mM KNO3, 20 mM Mg(NO3)2 (HEPES kinase buffer). The CusS wild-type and mutants were diluted to 0.2 mg/mL (~1.8 µM CusS) in HEPES kinase buffer. Three reactions were prepared for each CusS protein: apo-CusS with the addition of ATP (apo, + ATP); silver bound CusS with the addition of ATP (+ Ag, + ATP); and silver bound CusS without ATP [+ Ag, (−)]. For reactions with silver, AgNO3 was added to a final concentration of 400 µM and incubated at room temperature for 30 min, prior to the addition of ATP. The autophosphorylation reactions were initiated by adding ATP at a final concentration of 2 mM and incubated at room temperature for 1 hr. The reactions were terminated by adding 4X SDS sample buffer (pH 8.8) and analyzed by SDS-PAGE followed by two-color Western blot analysis.
CusScp-short and CusScp-long expression and purification—
For expression of the homodimer CusScp-short and CusScp-long, E. coli BL21(DE3) cells were transformed with pcusScp-short or pcusScp-long and grown in LB containing 100 µg/mL ampicillin or 30 µg/mL kanamycin respectively. For the isolation of heterodimers of CusScp-short and CusScp-long, E. coli BL21(DE3) cells were co-transformed with varied combinations of pcusScp-short and pcusScp-long plasmids, followed by selection on the basis of double resistance to 100 µg/mL ampicillin and 30 µg/mL kanamycin. Cultures were grown in LB media containing the appropriate antibiotics at 37°C until OD600 of 0.5–0.7, then 1 mM IPTG was added and the culture was continued to grow for another 5 hr.
To purify the homodimers CusScp-short or CusScp-long, cells were resuspended in binding buffer containing 1 mM PMSF and 4 µg/mL protease inhibitors. Lysis was achieved using a Branson sonicator and the cell debris was removed by centrifugation. The supernatant was applied to Ni2+ Sepharose 6 Fast Flow resin (GE Healthcare) equilibrated in binding buffer. The column was then washed with washing buffer (binding buffer containing 0.5 % Triton X-100), followed by elution with 6 mL of each 0.1, 0.2, and 0.5 mM imidazole in final buffer. The recovery and purity of the proteins were checked by SDS-PAGE. The purified protein fractions were then pooled and dialyzed against final buffer. The protein concentration was determined using a Bradford assay.
The isolation of the heterodimers was achieved using both Ni2+ Sepharose and Strep-tactin Sepharose resin. The supernatant was first loaded onto Ni2+ column, then washed and eluted as described above. Both the CusScp-short homodimer and CusScp-short-CusScp-long heterodimer were in the eluted fractions from the Ni2+ column. To isolate the CusScp-short-CusScp-long heterodimer, the eluted fractions from the first step were loaded onto Strep-tactin resin. The CusScp-short homodimer was in the flow-through, and the CusScp-short-CusScp-long heterodimer was eluted with final buffer containing 2.5 mM desthiobiotin. The recovery and purity of the proteins were checked by SDS-PAGE. The purified protein fractions were pooled and dialyzed against final buffer. The protein concentration was determined using a Bradford assay.
Autophosphorylation assay of homodimer and heterodimer CusScp—
Purified CusScp was diluted to 25 µM with kinase buffer (50 µM Tris pH 8.0, 50 mM KCl, and 10 mM MgCl2). The autophosphorylation reaction was initiated by adding ATP at a final concentration of 1 mM. The reaction was performed at 37°C for times ranging from 0 min to 2 hr (or unless stated otherwise) and terminated by adding an equal volume of 2X SDS sample buffer (pH 8.8). The samples were then analyzed by SDS-PAGE followed by Western blot as described below.
Phosphotransfer to CusR by CusScp~P—
CusScp was autophosphorylated as described above at 37°C for 30 min, then was mixed with CusR at molar ratio of 1:1 and incubated together at 37°C for times ranging from 0 to 5 min, unless stated otherwise. The reaction was terminated by adding 2X SDS sample buffer. The samples were analyzed by SDS-PAGE followed by Western blotting as described below.
Two-color dot blot and Western blot analysis to detect phosphohistidine—
Dot blot analysis was used to detect the phosphorylation position on CusScp-short with anti-N1-phosphohistidine (anti-1-pHis, clone SC1–1, EMD Millipore) and anti-N3-phosphohistidine (anti-3-pHis, clone SC39–6, EMD Millipore) antibodies (39). The purified CusScp was diluted to 10 µM with kinase buffer and the autophosphorylation reaction was initiated by adding ATP to final concentration of 0.4 mM. The reaction was incubated at 37°C for 30 min. Samples (2 µL) taken before and after addition of ATP were spotted on MeOH-activated polyvinylidene difluoride (PVDF) membrane and allowed to dry at room temperature for 1 hr. The membrane was blocked in blocking buffer (PBS, 0.1% Tween-20, 3% BSA) at room temperature for 1 hr or overnight at 4°C, followed by incubation in primary antibodies diluted in blocking buffer at room temperature for 2 hr (rabbit anti-1-pHis mixed with mouse anti-6xHis tag, or rabbit anti-3-pHis mixed with mouse anti-6xHis tag; diluted anti-1-pHis and anti-3-pHis to 1:1,000, diluted mouse anti-6xHis tag to 1:4,000). After 3 × 5 min washes in PBST (PBS + 0.1% Tween-20), the membrane was incubated in secondary antibodies at room temperature for 1 hr (goat anti-rabbit IR 800 mixed with goat anti-mouse IR 680; diluted both 1:15,000 in PBST). The membrane was washed and imaged using an Odyssey Infrared Imaging System (LI-COR Biosciences).
For Western blot analysis, after proteins were separated by SDS-PAGE, they were transferred to PVDF membrane at constant 150 mA for 2 hr using semi-dry transfer cell (Bio-Rad). Dual-detection Western analysis was performed as described above, with the exception that the primary antibodies used were rabbit anti-1-pHis and mouse anti-6xHis tag (diluted 1:1,000 and 1:4,000 respectively in blocking buffer).
Cell survival of BW25113 knock-out cells with or without plasmids on CuSO4-containing plates—
In vivo complementation assays were done as previously described (22). Briefly, cells were grown in LB media containing 100 µg/mL ampicillin at 37°C for 3.5 hr. Cell densities were normalized to OD600 of 0.8 and serial dilutions from 1 to 10−7 were made from this culture. All dilutions were spotted twice onto LB agar plates containing 100 µg/mL ampicillin, 1 mM IPTG, and a range of CuSO4 concentrations (0–3 mM). Plates were incubated at 37°C for 24 hr, then cell growth was scored on a scale of 0–8, with 8 representing cells that grew in the last dilution (10−7) and 0 representing no cell growth. The scores were converted to % cell survival, with 100% survival representing cell growth out to 10−6 and 10−7 dilutions, as previously described (22). All growth experiments were conducted a minimum of three times.
For cell survival assays without plasmids, cells were grown, plated, and scored the same way as described above except neither antibiotic or IPTG were added.
Determination of intracellular copper accumulation in CuSO4-containing media—
Samples for GFAAS were prepared as previously described (22). Briefly, cells were grown in LB media without any antibiotics. When cells reached an OD600 = 0.8–0.9, 1 mM CuSO4 was added and growth was continued at 37°C. Cell aliquots were collected before adding CuSO4 as 0 hr samples, and then aliquots were collected at 4 hrs after addition of CuSO4. Cells were normalized to a final OD600 of 0.8 and centrifuged to obtain cell pellets. The cell pellets were washed three times with sterile 1X PBS containing 1 mM EDTA and then dried at 75°C for 2 hr. To each pellet, 1 mL of 70% trace metals nitric acid was added and cells were mineralized at 80°C for 2 hr. Prior to sample loading in the GFAAS, samples were diluted to a final nitric acid concentration of 4% with nano-pure water. A standard calibration curve was generated using 0 to 40 ppb Cu, with final R2 ≥ 0.99. The concentration of copper for each strain was determined as an average of three replicates. Statistical significance was determined using t test analysis.
Results
Dimerization of the sensor domain of CusS is driven by metal ion binding to the interface—
Most HKs are known to exist and function as homodimers and that signal transduction occurs within dimeric assemblies. However, the dimerization of the isolated sensor domains is relatively weak, and many sensor domains are monomers in solution when expressed as truncated proteins (24). Sedimentation velocity analytical ultracentrifugation and in vitro crosslinking experiments have shown that apo-CusS(39–187) is primarily monomeric in solution, and that the addition of Ag(I) enhances the dimerization of CusS(39–187) (20). The crystal structure of the sensor domain of CusS shows that there are silver ions at two distinct binding sites in the sensor domain, one at the dimer interface and one in a loop region solely within one domain (22). It was also shown that the metal ion binding residues at the dimer interface are conserved within orthologs and that this site is more important for metal resistance in E. coli (22). We hypothesize that dimerization, which may lead to kinase activation, is largely driven by metal ion binding to the interface metal binding site. To investigate this hypothesis, in vitro crosslinking was performed on the apo and Ag(I)-bound CusS(39–187) wild-type and mutants that eliminate the metal ion binding sites.
In the in vitro crosslinking studies, the oligomerization of CusS(39–187) wild-type and variants was probed using the homobifunctional amine-reactive crosslinker BS3 and analyzed on SDS-PAGE followed by Western blot analysis using an anti-Strep tag antibody. The CusS(39–187)-AIA variant disrupts the interface metal ion binding site by converting the metal ion coordinating residues His42, Phe43, and His176 to Ala, Ile and Ala, respectively. Similarly, the CusS(39–187)-IIA variant removes the internal metal ion binding site through alteration of Met133, Met135, and His145 to Ile, Ile, and Ala, respectively. Western blot analysis (Fig. 1A) shows that apo CusS(39–187) and variants migrate at the same position in both the absence and presence of crosslinker, with a small amount of higher molecular weight species observed in the presence of crosslinker. Upon addition of Ag(I), significant increase in the higher molecular weight bands (**) in the presence of crosslinker are observed for CusS(39–187) and CusS(39–187)-IIA, but not for CusS(39–187)-AIA. The relative densities of the dimers (**) were calculated and normalized to the apo proteins (Fig. 1B). This analysis shows that Ag(I)-bound CusS(39–187) and CusS(39–187)-IIA have more intense higher molecular weight bands compared to the apo state, indicating more dimers formed. CusS(39–187)-AIA has a similar intensity for the higher molecular weight band when compared to the apo and Ag(I)-bound proteins. Similar banding patterns were observed in the silver-stained gel with similar density profiles (Supp. Fig. S1). The results show that the interface metal ion binding site contributes more significantly to ligand-induced dimerization of CusS(39–187) than the internal metal ion binding site.
Figure 1.

In vitro crosslinking reactions of apo and Ag(I)- CusS(39–387) wild-type and mutants. Apo- and Ag(I)- CusS(39–387) samples (10 μM) in the presence and absence of BS3 crosslinker were analyzed by Western analysis. (A) The Western blot analysis was performed using anti-Strep tag antibody. The expected monomer (~18 kDa) CusS(39–387) is indicated by (*) and the expected dimer (~36 kDa) is indicated by (**). Experiment was repeated twice; a representative experiment is shown. (B) The relative densities of the dimers (**) were calculated and normalized to the apo proteins. Student’s t test was used to calculate the P value. Error bars indicate standard deviations of duplicate experiments.
CusS is phosphorylated on N1 of H271—
Upon phosphorylation, HKs produce phosphohistidine (pHis), which is unique among phosphoamino acids because there are two biologically relevant isomers that can occur depending on which of the imidazole nitrogens, N1 or N3, is phosphorylated (17). Different kinases have different preferences for phosphorylation positions. To determine the position of phosphorylation, as well as the amino acid that is phosphorylated, autophosphorylation reactions were performed on constructs of CusS consisting of the isolated cytoplasmic region (CusScp) with a C-terminal 6xHis tag. Phosphorylation of CusScp on either the N1 or N3 position of histidine was probed by dot blot analyses using primary antibodies specific to 1-and 3-pHis (39).
An autophosphorylation reaction was performed in kinase buffer containing the divalent metal ion Mg2+ and ATP, then a two-color dot blot analysis was performed. The pHis substrate signal has a green color, while the 6xHis tag signal, which indicates the presence of the protein, is red. A yellow color indicates the presence of both the pHis signal and the His tag signal. As shown in Figure 2A, pHis is detected only on the anti-1-pHis blot and not the anti-3-pHis blot, indicating that phosphorylation occurs on the N1 position of the histidine in CusScp. Figure 2B shows a time course of CusScp phosphorylation analyzed by Western blot using anti-1-pHis antibody. Autophosphorylation occurs rapidly, within 5 minutes of the start of the reaction, and the maximum level of phosphorylation was reached within 30 min.
Figure 2.
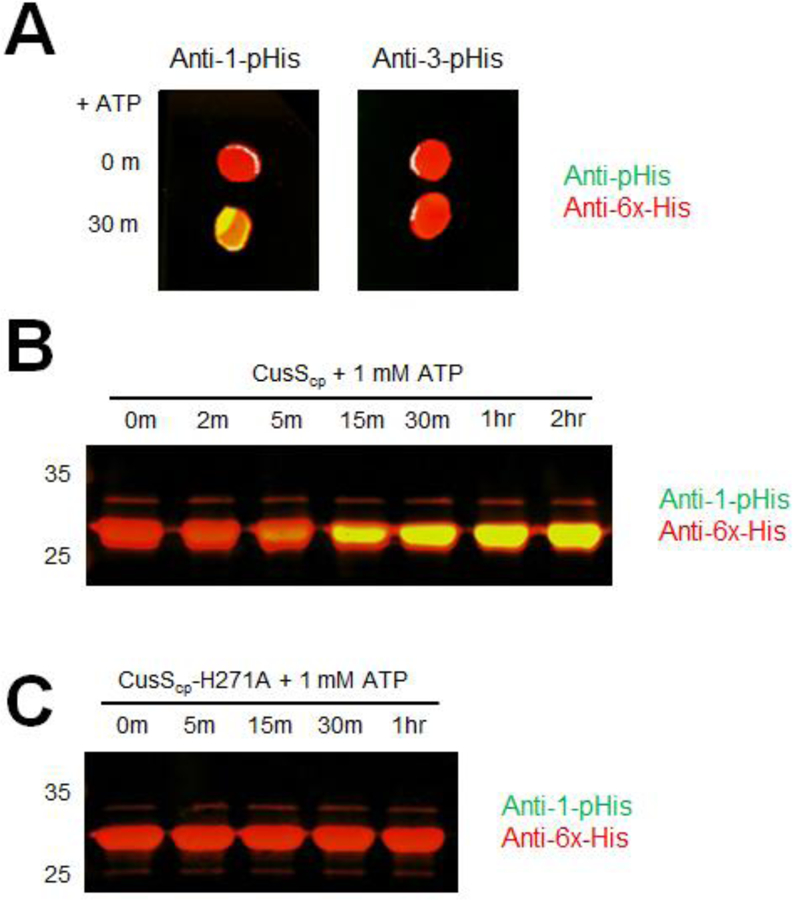
In vitro autophosphorylation of CusScp wild-type and CusScp-H271A. (A) Dot blot analysis of CusScp. CusScp was diluted to 10 μM in kinase buffer and autophosphorylation was initiated by adding ATP and incubated at 37°C. Samples were collected at 0 and 30 min, then 2 μL of each was spotted on PVDF membrane and probed with anti-1-pHis and anti-His tag antibodies simultaneously (left), or with anti-3-pHis and anti-His tag antibodies simultaneously (right). (B) CusScp (25 μM) was diluted in kinase buffer containing ATP to a final concentration of 1 mM, and reactions were incubated at 37°C. Samples were collected at the indicated times (2 min to 2 hr). (C) CusScp-H271A (25 μM) was diluted in kinase buffer containing ATP to a final concentration of 1 mM, and reactions were incubated at 37°C. Samples were collected at the indicated times (5 min to 1 hr). The reactions were terminated by adding 2X SDS sample buffer and were loaded onto SDS-PAGE. After electrophoresis, the proteins were transferred onto PVDF membranes and analyzed by Western blot probed with anti-1-pHis and anti-His-tag antibodies simultaneously. For all panels, each experiment was repeated at least three times; a representative experiment is shown.
Sequence alignments of the cytoplasmic regions of CusS homologs show that there is a conserved histidine at position 271 of E. coli CusS. Based on its conservation, it is likely that this histidine is the site of phosphorylation. To test whether this histidine is important for phosphorylation, a variant of CusScp was generated in which H271 was mutated to alanine (CusScp-H271A). A probe with the anti-1-pHis antibody following the kinase assay showed no detectable signal from pHis even up to a 1 hour timepoint, confirming the importance of this residue in autophosphorylation (Fig. 2C).
Loss of the interface binding site eliminates autophosphorylation—
CusS senses and binds to Ag(I) in the periplasmic domain, then transduces a signal to the cytoplasmic domain to initiate autophosphorylation. To examine autophosphorylation in vitro, we prepared full-length CusS solubilized in nanodiscs and performed kinase assays in the absence and presence of Ag(I). After adding AgNO3, autophosphorylation was initiated by the addition of ATP, then the samples were incubated at room temperature for 1 hr. The extent of autophosphorylation was analyzed by Western blot probed by the anti-1-pHis antibody. As shown in Figure 3, the most autophosphorylation was observed for CusS when both Ag(I) and ATP are present. This suggests that Ag(I) binding increases the kinase activity of CusS and that autophosphorylation is largely ATP-dependent. In the absence of Ag(I) though in the presence of ATP, less autophosphorylation was observed for CusS. To ensure that the pHis signal detected was from autophosphorylation on the conserved H271, this histidine was mutated to an alanine. Kinase activity was completely abolished in CusS-H271A under all conditions, including when both Ag(I) and ATP are present (Fig. 3).
Figure 3.
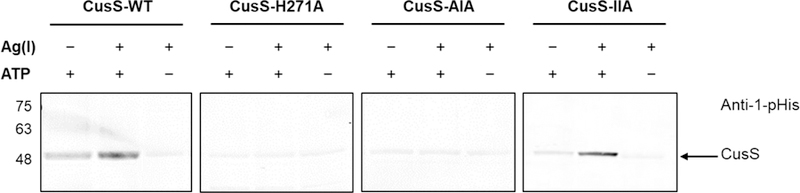
Autophosphorylation of the full-length histidine kinase CusS in nanodiscs. Each protein CusS-WT, CusS-H271A, CusS-AIA, and CusS-IIA was diluted in HEPES kinase buffer to a final concentration of 0.2 mg/mL. The absence (−) and presence (+) of Ag(I) or ATP in the reactions are indicated. Reactions were incubated at room temperature for 1 hr. Samples were separated by SDS-PAGE and analyzed by Western blot using anti-1-pHis antibody. Experiment was repeated twice; a representative experiment is shown.
To further investigate the role of the metal binding sites and metal-induced dimerization in CusS kinase activity, mutations of the interface binding site (CusS-AIA) and internal binding site (CusS-IIA) were generated in the full-length CusS. These protein variants were embedded into nanodiscs. Autophosphorylation assays were then performed on both CusS-AIA and CusS-IIA nanodisc samples and analyzed as described for the wild-type protein. When the interface binding site is mutated (CusS-AIA), no increase of pHis signal was observed in the presence of both Ag(I) and ATP (Fig. 3). However, when only the internal binding site is mutated (CusS-IIA), an increase in pHis signal was detected when Ag(I) and ATP were added. This result implies that the interface binding site is more crucial for CusS kinase activity.
CusS undergoes cis autophosphorylation—
Autophosphorylation in a dimeric complex can occur potentially either within a single monomer of the dimer or across the dimeric species, which are referred to as cis or trans phosphorylation, respectively. To test whether CusS autophosphorylation occurs in cis or trans, we performed autophosphorylation assays of different combinations of wild-type and mutant heterodimers. Heterodimers of wild-type and mutants were formed by using the co-expression method as described in Casino et al (4) for the histidine kinase HK853. When residues N380 and D411 in HK853 are mutated, phosphorylation is prevented by disrupting the interaction with ATP in the ATP binding site (4). Based on a sequence alignment of CusScp and HK853, the corresponding residues in E. coli CusS are N386 and N414. Three CusS variants were generated: one in which the phosphoacceptor His271 is replaced by Ala (CusScp-H271A), a second that contains a double mutation (N386A/N414A, referred to as CusScp-AA) that prevents autophosphorylation by disrupting ATP binding, and the third is a triple mutant that combines both of the previous alterations (H271A/N386A/N414A, referred to as CusScp-AAA). Kinase assays confirm that these constructs show no autophosphorylation (Fig. 4).
Figure 4.

Sequence alignment of CusScp and autophosphorylation of CusScp variants. (A) Sequence alignment of the cytoplasmic domain of E. coli CusS (residues 208–480) with the cytoplasmic domain of Thermotoga maritima HK853 (residues 232–489). The conserved catalytic His residues are highlighted in bold in a red box. The residues highlighted in blue boxes are the residues involved in ATP binding. (B) CusScp wild-type, CusScp-H271A, CusScp-N386A/N414A (CusScp-AA), and CusScp-H271A/N386A/N414A (CusScp-AAA) (25 μM) were diluted in kinase buffer containing ATP at a final concentration of 1 mM, and reactions were incubated at 37°C, samples were collected at 0 and 30 min. The autophosphorylation reactions were terminated by adding 2X SDS sample buffer and were loaded onto SDS-PAGE, followed by Western blot analysis using anti-1-pHis and anti-His tag antibodies simultaneously.
To distinguish each subunit of the heterodimers, we utilized the subunit mass difference between the “short” form of CusScp with C-terminal 6xHis tag (CusScp-short) and the “long” form of CusScp with a C-terminal Strep-Myc-Strep-FLAG-HA-Strep tag (CusScp-long). We also prepared both forms as either wild-type or the desired mutants. To form the CusScp-short and CusScp-long homodimers, cells were transformed with plasmids expressing each construct individually and proteins were purified by Ni2+ Sepharose resin and Strep-tactin Sepharose resin, respectively. A cartoon depiction of homo-and heterodimers and the expected and experimental outcomes of autophosphorylation is shown in Fig 5A, using a representation similar to that originally created by Casino et al (4). To generate different combinations of heterodimers (Fig. 5A, column 1), CusScp-short and CusScp-long forms were co-expressed and purified by both Ni2+ and Strep-tactin Sepharose resins. The heterodimers were soluble and functional, and they migrated in native-PAGE at an intermediate position between the bands of the CusScp-short and CusScp-long homodimers (Fig. 5B). CusScp-long homodimer was soluble and functional as shown by the phosphorylation assay analyzed by both native-and SDS-PAGE (Fig. 5C and D).
Figure 5.

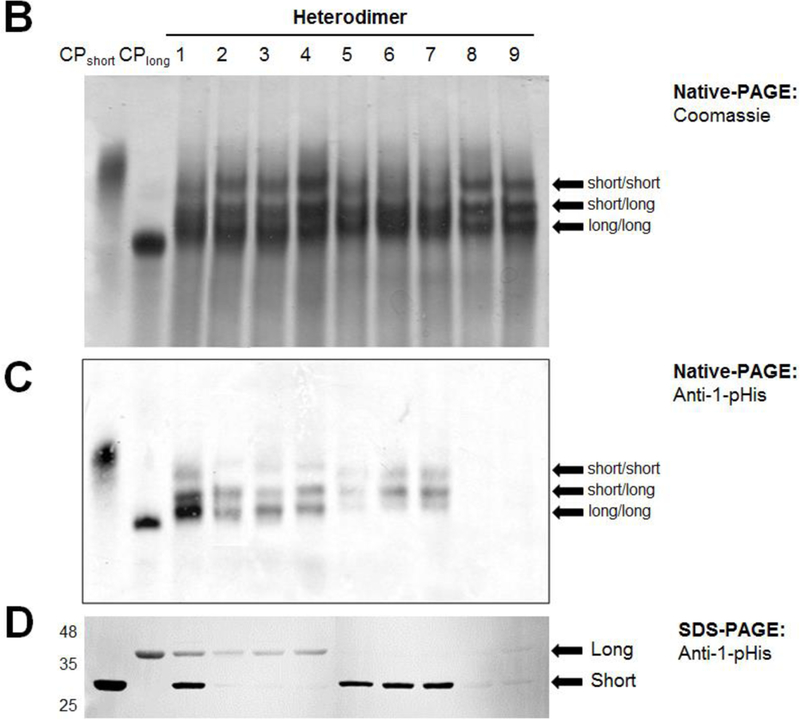
CusScp autophosphorylates using a cis mechanism. (A) Cartoons of the expected and experimental results of autophosphorylation studies with homodimers and heterodimers of CusScp. The first column identifies wild-type CusScp-short homodimers (CPshort), wild-type CusScp-long (CPlong), and total of nine different heterodimers numbered 1–9, which are either of wild-type or carrying mutations with disrupted phosphoacceptor His or ATP binding (indicated by a red circle crossed with a line). CusScp-short is represented by blue rectangles and CusScp-long is represented by gray rectangles. Phosphorylated CusScp is shown in red rectangles and labeled with a P. The second and third columns depict the expected phosphorylation pattern for cis and trans phosphorylation, respectively. The last column summarizes the phosphorylation patterns observed in our experiments. (B) Native-PAGE analysis of the homodimers and heterodimers revealed by Coomassie staining. (C) Autophosphorylation analysis by native-PAGE followed by Western blotting using an anti-1-pHis antibody. (D) Autophosphorylation analysis by SDS-PAGE followed by Western blotting using an anti-1-pHis antibody. Labels short/short, short/long, and long/long were used on the native-PAGE analyses to identify the homodimer or heterodimer. For SDS-PAGE, short and long were used to identify each subunit.
The phosphorylation assays for the homodimers and heterodimers were initiated by adding ATP, then were analyzed by Western blotting of both native-PAGE and SDS-PAGE gels. Figure 5A shows the expected results for either cis or trans phosphorylation of the different construct combinations, and the phosphorylated subunits are colored in red. For instance, for heterodimers 4 and 7, phosphorylation can only occur through cis phosphorylation; and for heterodimers 8 and 9, phosphorylation can only occur through trans phosphorylation. The SDS-PAGE results (Fig. 5D) were compared with the expected outcomes (Fig. 5A). Figure 5D shows a pHis signal from heterodimers 4 and 7, and no phosphorylation from heterodimers 8 and 9, which is consistent with cis phosphorylation. The other heterodimers show phosphorylation profiles consistent with cis phosphorylation as well. Cis phosphorylation was also confirmed by Western blots following native-PAGE, in which pHis signals were detected on the corresponding heterodimers shown by the intermediate bands (Fig. 5C).
CusR rapidly dephosphorylates CusS—
The cognate pair of the histidine kinase CusS is the response regulator CusR. To investigate the effects of CusR on CusS phosphorylation, we analyzed CusS phosphorylation using the anti-1-pHis antibody in the presence of CusR. First, CusScp was phosphorylated with ATP to generate phosphorylated CusScp (CusScp~P). As depicted in Figure 6A, when equal amounts of CusR are added to the CusScp~P sample, no yellow band is detected, indicating the loss of pHis signal. Loss of the phosphorylated CusS species occurred within 0.5 min. To ensure the loss of signal wasn’t due to the dilution of the CusScp~P sample, an equal volume of kinase buffer was added to the CusScp~P sample as a control. No change was seen in this sample (Fig. 6A, “buffer” lane).
Figure 6.
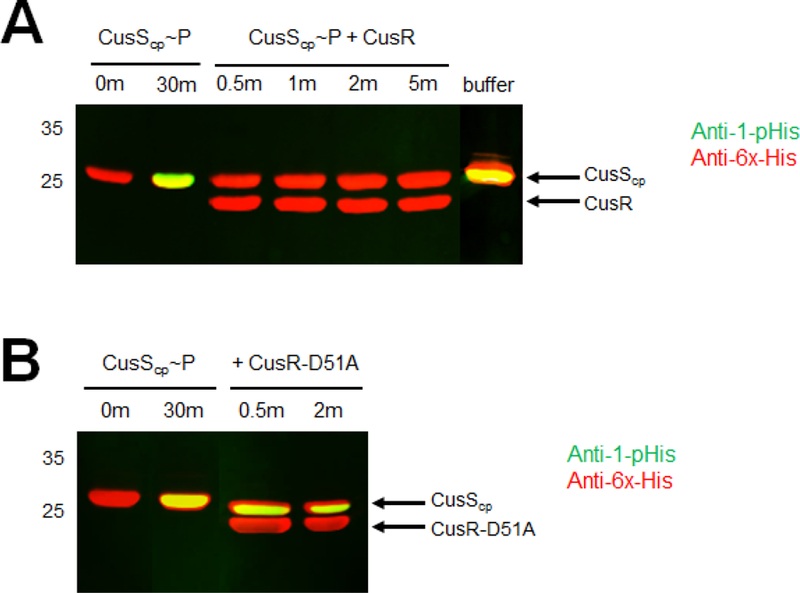
In vitro CusR dephosphorylation of CusScp~P. The phosphorylated CusScp (CusScp~P) was prepared by incubation with 1 mM ATP in kinase buffer at 37°C for 30 min. Equimolar amounts of CusScp~P and CusR wild-type or mutant were mixed and incubated. Samples were collected at the indicated times and stopped by adding 2X SDS sample buffer. Proteins were separated by SDS-PAGE, followed by Western blot analysis using anti-1-pHis and anti-His tag antibodies simultaneously. Each experiment was repeated at least three times; a representative experiment is shown.
Based on sequence alignments, the phosphorylated aspartate in CusR is expected to be D51. To test the impact of this site on CusS dephosphorylation, D51 of CusR was mutated to an alanine (CusR-D51A) and the assay was performed by mixing CusScp~P with an equimolar amount of purified CusR-D51A. The pHis signal on CusScp~P persisted when mixed with CusR-D51A (Fig. 6B), indicating that this CusR mutant lacks the ability to dephosphorylate CusS. This result likely reflects an inability to transfer the phosphoryl group from CusS to CusR when D51 is mutated.
CusS H271 and CusR D51 are essential for survival at elevated copper concentrations—
The conserved residues CusS-H271 and CusR-D51 are the putative phosphorylation sites involved in the His-Asp phosphotransfer pathway to activate the downstream response. The importance of the cusS gene in copper resistance has been previously shown, where the deletion of chromosomal cusS led to a decrease in copper tolerance and increase of minimum inhibitory concentration value to silver (21). To investigate the role of the phosphoacceptor sites in metal resistance by CusS and CusR in vivo, E. coli cells expressing full-length CusS, CusS-H271A, CusR, and CusR-D51A were evaluated for survival on CuSO4-containing media (Fig. 7A and 7B). The background strain (WT) has a chromosomal deletion of cueO (ΔcueO), a multicopper oxidase that converts Cu(I) to Cu(II) (40). Thus with this ΔcueO background, we are able to observe growth phenotype even under aerobic conditions (41). To characterize the CusS and CusR variants, the ΔcusS strain contains deletions of both the cueO and cusS genes and the ΔcusR strain contains deletions of both the cueO and cusR genes (Supp. Table S2). The WT strain was transformed with either pET21b(+) or pET22b(+) empty vector, the ΔcusS strain was transformed with plasmids for expression of cusS wild-type (pcusS) or cusS-H271A mutant (pcusS-H271A), and the ΔcusR strain was transformed with plasmids for expression of cusR wild-type (pcusR) or cusR-D51A mutant (pcusR-D51A). After growth, cells were scored based on survival at each dilution spot and the phenotypes are reported as % cell survival at each CuSO4 concentration.
Figure 7.

Cell survival assays and copper accumulation in BW25113 cells. (A) BW25113ΔcueO cells transformed with pET21b (WT, black) and BW25113ΔcueOΔcusS cells transformed with pET21b (ΔcusS, gray), pET21b-cusS (ΔcusS/pcusS green), or pET21b-cusS-H271A (ΔcusS/pcusS-H271A, red), (B) BW25113ΔcueO cells transformed with pET22b (WT, black) and BW25113ΔcueOΔcusR cells transformed with pET22b (ΔcusR, gray), pET22b-cusR (ΔcusR/pcusR, green) or pET22b-cusR-D51A (ΔcusR/pcusR-D51A, red), and (C) ) BW25113ΔcueO (WT, black), BW25113ΔcueOΔcusS (green) and BW25113ΔcueOΔcusR (gray) cells were grown at 37°C on LB agar plates containing 100 μg/mL ampicillin, 1 mM IPTG, and various concentrations of CuSO4 (0–3 mM) for 24 hr. Growth was scored relative to 100% cell survival, which is defined as cell growth in the 10−6 and 10−7 dilutions. Error bars indicate standard deviations of triplicate experiments. (D) Copper accumulation in cells 0 hr (left) and 4 hr (right) after the addition of 1 mM CuSO4. Copper levels are calculated as nanograms of copper per 108 cells. Each bar represents the average of three replicates and the standard deviations are indicated by the error bars. Asterisks (*) indicate a statistically significant difference from WT with a P of < 0.05 (Student’s t test).
As shown in Figure 7A, the WT strain shows an impaired growth phenotype at 2.75 mM CuSO4, whereas the ΔcusS strain shows an impaired growth at 1.25 mM CuSO4 and has no growth after 2 mM CuSO4. When wild-type CusS is provided on a plasmid in the ΔcusS strain (ΔcusS/pcusS), the growth defect is partially rescued. Although full complementation is not observed, the % cell survival is significantly higher than the ΔcusS strain, with growth observed up to 2.5 mM CuSO4. This is consistent with previous findings (22). When the putative catalytic His residue was mutated to Ala (pcusS-H271A), the cell growth is disrupted at 1.25 mM CuSO4 and no growth is observed after 1.75 mM CuSO4, which is lower % cell survival than ΔcusS. To ensure that the full-length CusS wild-type and CusS-H271A mutant proteins were expressed at the similar levels from the pET21b(+) plasmids, Western blot analysis was performed by detecting the expressed proteins with an anti-6xHis tag antibody. Both CusS wild-type and CusS-H271A mutants are expressed from the plasmid at similar levels, and no His-tagged CusS was detected from the WT strain or ΔcusS with the pET21b(+) empty vector (Supp. Fig. S2A).
The effects of CusR variants on cells’ ability to survive copper challenges was also tested (Fig. 7B). The WT strain experiences susceptibility at 2.75 mM CuSO4, while the ΔcusR strain shows growth impairment at 1.25 mM CuSO4 and shows no growth after 2 mM CuSO4. When ΔcusR was complemented with wild-type CusR from a plasmid (ΔcusR/pcusR), an almost full complementation is observed. When the phosphoacceptor Asp was mutated to an Ala (ΔcusR/pcusR-D51A), the % cell survival is considerably decreased and is not significantly different than the ΔcusR strain. Western blot analysis shows that the expression levels of wild-type CusR and CusR-D51A are similar, and no expression of His-tagged CusR was detected from the WT and ΔcusR with pET22b(+) empty vector (Supp. Fig. S2B).
Loss of CusR more significantly impairs survival at high copper concentrations than loss of CusS—
The TCS CusS-CusR works together through histidyl-aspartyl-phosphotransfer and results in the upregulation of cusCFBA genes involved in Cu(I)/Ag(I) resistance in E. coli. As a phosphorelay pair, the expectation is that both the CusR and CusS proteins contribute to the cell ability to survive elevated levels of copper. In order to compare the copper susceptibility of strains harboring deletions of cusS or cusR, growth assays were performed with identical conditions for these strains on copper-containing media. The WT strain does not show any decreased tolerance to copper until 2.5 mM CuSO4 and shows no growth after 2.75 mM CuSO4 (Fig. 7C). The ΔcusS strain shows a phenotype at 1.25 mM CuSO4 with no cell growth after 2.5 mM CuSO4, while the ΔcusR strain shows decreased copper tolerance at 1.5 mM CuSO4 and shows no cell growth after 2.25 mM CuSO4. This result shows that loss of CusR reduces the ability of E. coli to survive at higher copper concentrations more significantly than the loss of CusS.
To study the effect of these double knock-out strains on copper accumulation in E. coli, intracellular copper concentrations of these cells were measured using a graphite furnace atomic absorption spectrometer (GFAAS). Cells were grown aerobically in copper-containing media, and time point samples at 0 and 4 hr were collected and analyzed. No significant copper was detected in the 0 hr samples, but after 4 hrs of copper exposure, ΔcusS cells had an increase in copper accumulation with a 2-fold increase compared to that of the WT strain (Fig. 7D). The ΔcusR strain had a larger increase in the copper accumulation with a 3.5-fold increase compared to the WT strain, and a 2-fold increase compared to the ΔcusS strain.
Discussion
The cusRS genes encode a two-component system with CusS as the membrane-bound histidine kinase and CusR as the cytoplasmic response regulator (10,42). The histidine kinase CusS senses and binds to Cu(I)/Ag(I) ions in the periplasm and transduces a signal to its kinase domain, initiating ATP-dependent autophosphorylation on a histidine residue. In vitro studies have shown that CusS senses and binds to Ag(I) directly through the periplasmic sensor domain (20). Upon Ag(I) binding, the periplasmic domain of CusS undergoes a conformational change and dimerization is enhanced (20). However, many questions regarding CusS signal transduction and autophosphorylation mechanisms remain largely unanswered. Here, studies were conducted to understand the metal binding properties of CusS and to connect how metal binding in the periplasm triggers kinase activity in the cytoplasmic domain.
Upon sensing an environmental signal, two-component systems function through autophosphorylation on the conserved His of the HK, followed by phosphotransfer to a conserved Asp on its cognate RR. However, due to the instability of pHis and the technical challenges for such analysis, the mechanism of phosphorylation of HKs remains understudied. Histidine can be phosphorylated on either N1 or N3 position on the imidazole ring, with the 3-pHis isomer being more favorable than 1-pHis because it is thermodynamically more stable (17). In bacterial TCS, several HKs are phosphorylated at the N3 position of the conserved His residues, including Salmonella typhimurium CheA and Thermotoga maritima HK853 (4,43). Our dot blot analyses using monoclonal antibodies shows that CusScp is phosphorylated at the N1 position on the histidine imidazole (Fig. 2A), which overturns the previous assumption that all bacterial HKs autophosphorylate at the N3 position and shows that there is variation among bacterial HKs (1,4,43). Moreover, we have confirmed that the highly conserved H271 on CusS is the phosphorylation site. Addition of CusR to a solution containing phosphorylated CusS results in a rapid loss of the pHis signal, presumably due to phosphotransfer to CusR. The conserved aspartate, D51, on CusR is necessary for the dephosphorylation event. This phosphotransfer event between CusS-H271 and CusR-D51 is crucial for the metal resistance response in E. coli (Fig. 7). It has been previously shown that the rate of HK dephosphorylation and RR phosphorylation are different among TCSs (44). Rapid dephosphorylation of CusS in the presence of CusR (within 30 seconds) implies that the TCS CusS-CusR is able to respond quickly to elevated levels of Cu(I) or Ag(I) to help promote a prompt response.
Based on the copper survival studies, it was observed that copper tolerance is lower in the absence of cusR gene (Fig. 7C) and the absence of cusR causes higher accumulation of copper in the cells (Fig. 7D). These results imply that the cusR gene is the more important component of the TCS pair in metal tolerance and metal homeostasis. Moreover, it has been shown that CusS only phosphorylates CusR specifically, while CusR can be phosphorylated by three other non-cognate HKs: BarA, UhpB, and YedV (44). Therefore, the higher copper tolerance in the ΔcusS strain than the ΔcusR strain might be due to the possibility of CusR being activated by other HKs when CusS is not present. More crosstalk studies remain to be done to determine the role of other HKs and their effect in metal resistance.
Early studies of the HKs, such as E. coli EnvZ, NtrB, CheA and AtoS, demonstrated that they autophosphorylate in trans, in which the CA domain of one subunit phosphorylates the conserved His on the DHp domain from the other subunit (13,45–47). Therefore, all HKs were assumed to universally autophosphorylate through a trans mechanism. However, it was later shown that bacterial HKs can also autophosphorylate using a cis mechanism, in which the CA domain of one subunit phosphorylates the conserved His on the DHp domain of the same subunit. In 2009 cis phosphorylation was reported in Thermotoga maritima HK853 and Staphylococcus aureus PhoR, and later E. coli ArcB was also found to utilize a cis mechanism in 2010 (4,48). Here, we demonstrate that CusScp undergoes autophosphorylation by a cis mechanism (Fig. 5), which further confirms that despite high homology among HKs, different mechanisms of autophosphorylation can occur. It was reported that the handedness of the DHp loop may be a functionally relevant determinant of the cis or trans autophosphorylation mechanism, with right-handedness undergoing trans phosphorylation and left-handedness in cis (13). Therefore, because we have observed cis phosphorylation, we predict that the DHp loop of CusS is left-handed and that the DHp α2 should be positioned to the left of α1 (Supp. Fig. S3B). Additionally, we speculate that our results support a model in which rotational movements in the DHp and CA domain maintain the kinase state.
The sensing of Cu(I)/Ag(I) through the sensor domain of CusS transduces signal to the cytoplasmic kinase core through the transmembrane helices and the HAMP linker domain. Although HKs exist and function as a homodimer, the truncated periplasmic sensor domain does not always exist in a dimeric form (25,26,28,29). The truncated CusS(39–187) is also largely monomeric in solution and the presence of Ag(I) enhances the dimerization of CusS(39–187). However, how metal binding and metal-induced dimerization in the sensor domain of CusS activates autophosphorylation activity in the cytoplasmic domain is not known. In this study, the roles of the different metal binding sites (interface and internal binding sites) in ligand-induced dimerization and autophosphorylation were investigated. The in vitro crosslinking experiments show that the interface binding site contributes to the dimerization of CusS(39–187) in the presence of Ag(I) while the internal binding site does not (Fig. 1).. To investigate how stimuli sensing leads to signal transduction, we applied nanodisc technology to study the full-length membrane protein CusS in a native-like lipid bilayer environment. Full-length CusS variants embedded in the nanodiscs were active and underwent autophosphorylation on the conserved H271 upon Ag(I) binding. Furthermore, when the interface binding site was mutated, autophosphorylation was not observed (Fig. 3). Overall, this body of work concludes that metal-induced dimerization through the interface binding site is important for signal transduction that triggers autophosphorylation in CusS.
Despite these significant findings, some important questions about the CusS signal transduction and autophosphorylation mechanism remain largely unanswered due to lack of structural data on the full-length CusS or the cytoplasmic domain of CusS. It is expected that Cu(I)/Ag(I) binding in the periplasmic sensor domain may lead to structural changes in the proximal transmembrane domain. There are four basic mechanisms proposed for signal transduction by transmembrane helices: association/dissociation (translation in the plane of the membrane), piston movement (translation perpendicular to the membrane), rotation along an axis parallel to the membrane (pivot motion), and rotation perpendicular to the membrane (12,28). Piston sliding movement has been proposed in chemotactic receptor Tar and in several HKs such as DcuS, CitA, NarX, and NarQ (26,28,29,49–51). From alignments of the structures of apo-and ligand-bound sensor domains, the positions of the N- and C-terminal helices were compared. A downward displacement of the C-terminal helix was observed in aspartate receptor Tar upon aspartate binding, and a downward displacement of the N-terminal helix was observed in HK NarX upon nitrate binding (29,52,53). In CitA, an upward displacement of the C-terminal helix of the sensor domain was observed upon citrate binding, thus pulling the TM2 in an upward piston movement (28). Recently, the structures of the sensor, TM, and HAMP domains of nitrate sensor NarQ have further provided insights into the signaling mechanism through the TM, in which the binding of nitrate induces rearrangements and piston-like shifts of the TM helices which causes helical rotation at the output helices on the HAMP domain (51). Future experiments aiming to obtain a 3D structure of full-length CusS(39–187) might be helpful in determining any displacements on the N- and/or C-terminal helices in the sensor domain of CusS, since these helices are expected to be contiguous with the transmembrane helices (22).
The full cytoplasmic domain of the prototypic HK CusS consists of a HAMP linker domain followed by DHp and CA domains. During the last few years, a number of structures of truncated domains have shed light into various aspects of signal transduction mechanism of the TCSs. However, no structure of the full cytoplasmic region of prototypic (HAMP-containing) HKs was reported until recently. A 3D structure of the full cytoplasmic domain of E. coli CpxA (CpxAHDC), a HK that regulates Cpx signaling system that regulates an envelope stress response, was solved in the active kinase state (54). In this paper, the authors reported that the HAMP domain modulates the segmental helical mobility of the central HK α-helices to promote a strong helical bending movement and dynamical asymmetry that characterizes the kinase-active state. They emphasized that the helical bending was caused by S238 and highly conserved P253 residues (Supp. Fig. S3A). The first kink at the S238 was probably required to avoid steric clash at a layer of polar residues (Q239-Q240) and the second kink is caused by the proline residue. There is asymmetry in the structure, in which one CA is in the active state and the other one is inactive. These asymmetric kinase-competent states have also been biochemically demonstrated for NRII and HK853 (55,56), and structurally predicted for DesK, VicK, and EnvZ chimeras (56–58). In all structures of HK-RR complexes, such as HK853-RR468, the HK component is symmetric and thus symmetric conformations of HKs maybe related with either phosphotransfer or phosphatase reaction (4,56). Overall, it is now believed that the symmetry-asymmetry transitions are important for function (56,59).
Based on the high homology and conservation of the kinase domains among HKs, it is likely that CusScp will have fold similar to other HKs. The amino acid sequence of CusScp was submitted to the Robetta server for 3D structure prediction (60). The top predicted structure, shown in Supplemental Figure S3B, was built using the crystal structure of the cytoplasmic domain of E. coli CpxAHDC (PDB entry 4BIU) (54). The HK CpxAHDC shares significant homology with HK CusScp with 31.1% identity and 47.9% similarity (Supp. Fig. S3A). Based on the predicted structure of CusScp, the HAMP domain forms a parallel four-helix bundle with two helices from each monomer. The DHp domain is formed by two long helices (α1 and α2), where α1 contains the conserved catalytic His residue and is continuous from α2 of HAMP domain. The CA domain has an α/β sandwich fold made up by five β strands and three α helices. The S238 and P253 residues of CpxAHDC correspond to R261 and P276 of CusScp (Supp Fig S3A). Therefore, it is feasible that R261 and P276 residues may cause helical bending in the top half of α1 on the DHp domain as observed in the CpxAHDC structure, and asymmetrically positioning the CA domain closer to the H271 within the same subunit. This could be critical since the distance between the β phosphate of the ADP molecule and H271 on CusS is ~21.3 Å in the predicted structure.
Structural evidence has suggested that communication between the sensor and kinase domains in HK signaling is mediated by subtle structural changes along the dimer interface and that there may be related aspects of symmetry and asymmetry. The ligand-bound state of a histidine kinase sensor generates kinase activity for many receptors and phosphatase activity for others. While we can use symmetry-asymmetry transitions to describe the functional states of the kinase domain, we can also use it to describe the sensor domain. Structures of HK sensor domains in apo and ligand-bound states have been solved, and from the analysis of these known structures, it is suggested that symmetric sensor domains lead to kinase activity. For example, apo TorS-TorT is asymmetric and the kinase domain is in the phosphatase state; upon binding to the ligand trimethylamine-N-oxide, TorS-TorT is symmetric and the kinase domain is in the kinase state (61). Based on these studies, it is plausible to hypothesize that symmetric extracellular arrangement triggers an asymmetric cytoplasmic arrangement, and the asymmetric extracellular arrangement triggers a symmetric cytoplasmic arrangement (56,61).
Based on our work on CusS and the work of others on other histidine kinases, we propose the following features for the mechanism of the E. coli histidine kinase CusS (Fig. 8): At low concentrations of Cu(I)/Ag(I) in the periplasm, the sensor domain of CusS is in an asymmetric apo state (Fig. 8, left). In the apo CusS state, the cytoplasmic domain is in a symmetric, kinase-inactive arrangement. When the Cu(I)/Ag(I) levels are elevated, the sensor domain of CusS binds to metal ions through its interface and internal binding sites forming a symmetric arrangement (Fig. 8, right). Ligand-induced dimerization of the sensor domain through the interface binding sites triggers piston-like movement in the transmembrane domain, which then transduces a signal down to the cytoplasmic domain. This signal triggers conformational changes in the HAMP domain. Consequently, it stresses the top half of DHp α1 helix (between the R261 and P276 residues shown in green), promoting segmental helical motions that result in a dynamical asymmetry between the CA domains. One of the CA domains is mobile and brings the ATP nucleotide closer to the phosphoacceptor H271 on the same subunit, initiating a cis autophosphorylation reaction. The other CA domain remains in an inactive conformation. To confirm this model, further experiments focusing on obtaining 3D structures of the cytoplasmic domain of CusS in the absence and presence of the nucleotide will be useful in understanding the different conformations of the cytoplasmic domain at its active and inactive states.
Figure 8.
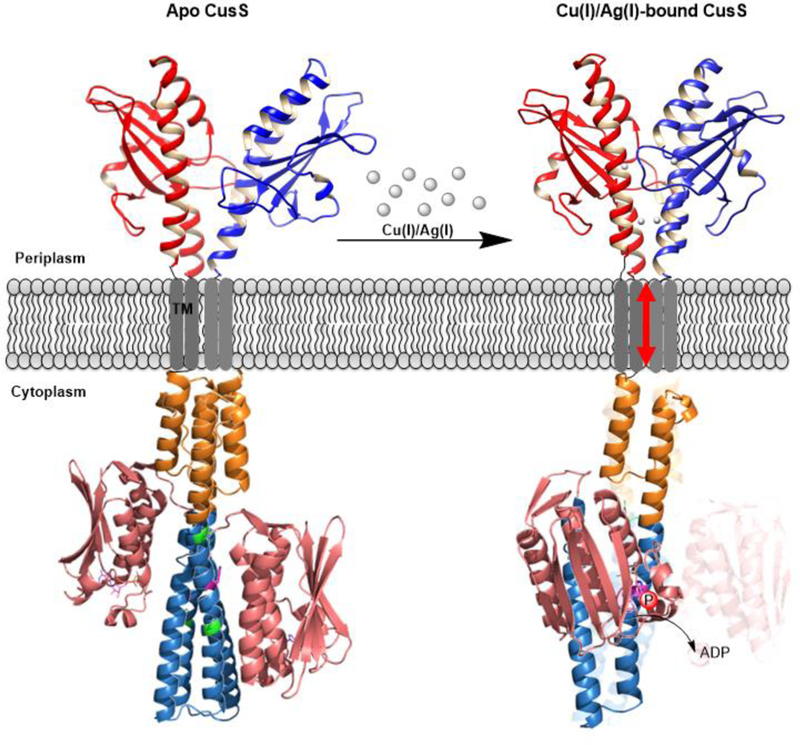
Proposed model for CusS signal transduction mechanism via symmetry-asymmetry transitions. Structure of the sensor domain of CusS is from the solved crystal structure of Ag(I)-CusS(39–17) (PDB entry 5KU5) (22). Structure of the cytoplasmic domain is the predicted 3D structure from Robetta (60). In apo CusS (left) the sensor domain is in an asymmetric arrangement, while the cytoplasmic is in a symmetric arrangement and is in the inactive state. Upon elevation of Cu(I)/Ag(I) levels in the periplasm, the sensor domain binds to the metal ions. In Cu(I)/Ag(I)-bound CusS (right), the sensor domain of CusS dimerizes upon metal binding through the interface binding site and adopts a symmetric arrangement. The dimerization triggers piston-like movement in the TM domain, which causes helical bending in the DHp domain (teal) between the residues shown in green and swings the CA domain (salmon pink) closer to the catalytic H271 on the same subunit. The transferred phosphate is shown by red circles with label “P”.
Supplementary Material
Acknowledgments
Funding
This work was supported by National Institutes of Health grant GM079192 to MMM. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations
- HK
histidine kinase
- HK~P
phosphorylated HK
- MSP
membrane scaffold protein
- pAsp
phosphoaspartate
- pHis
phosphohistidine
- RR
response regulator
- RR~P
phosphorylated RR
- TCS
two component system
- TM
transmembrane
- HAMP
histidine kinase, adenylyl cyclases, methyl-accepting proteins, phosphatases
- DHp
dimerization and histidine phosphotransfer
- CA
catalytic and ATP binding domain
Footnotes
Competing Interests
The Authors declare there are no competing interests associated with the manuscript.
References
- 1.Stock AM, Robinson VL, and Goudreau PN (2000) Two-component signal transduction. Annu Rev Biochem 69, 183–215 [DOI] [PubMed] [Google Scholar]
- 2.West AH, and Stock AM (2001) Histidine kinases and response regulator proteins in two-component signaling systems. Trends Biochem Sci 26, 369–376 [DOI] [PubMed] [Google Scholar]
- 3.Stock JB, Stock AM, and Mottonen JM (1990) Signal transduction in bacteria. Nature 344, 395–400 [DOI] [PubMed] [Google Scholar]
- 4.Casino P, Rubio V, and Marina A (2009) Structural insight into partner specificity and phosphoryl transfer in two-component signal transduction. Cell 139, 325–336 [DOI] [PubMed] [Google Scholar]
- 5.Rensing C, and Grass G (2003) Escherichia coli mechanisms of copper homeostasis in a changing environment. FEMS Microbiol Rev 27, 197–213 [DOI] [PubMed] [Google Scholar]
- 6.Beswick PH, Hall GH, Hook AJ, Little K, McBrien DC, and Lott KA (1976) Copper toxicity: evidence for the conversion of cupric to cuprous copper in vivo under anaerobic conditions. Chem Biol Interact 14, 347–356 [DOI] [PubMed] [Google Scholar]
- 7.Outten FW, Huffman DL, Hale JA, and O’Halloran TV (2001) The independent cue and cus systems confer copper tolerance during aerobic and anaerobic growth in Escherichia coli. J Biol Chem 276, 30670–30677 [DOI] [PubMed] [Google Scholar]
- 8.Kimura T, and Nishioka H (1997) Intracellular generation of superoxide by copper sulphate in Escherichia coli. Mutat Res 389, 237–242 [DOI] [PubMed] [Google Scholar]
- 9.Franke S, Grass G, and Nies DH (2001) The product of the ybdE gene of the Escherichia coli chromosome is involved in detoxification of silver ions. Microbiology 147, 965–972 [DOI] [PubMed] [Google Scholar]
- 10.Munson GP, Lam DL, Outten FW, and O’Halloran TV (2000) Identification of a copper-responsive two-component system on the chromosome of Escherichia coli K-12. J Bacteriol 182, 5864–5871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Butler SL, and Falke JJ (1998) Cysteine and disulfide scanning reveals two amphiphilic helices in the linker region of the aspartate chemoreceptor. Biochemistry 37, 10746–10756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hulko M, Berndt F, Gruber M, Linder JU, Truffault V, Schultz A, Martin J, Schultz JE, Lupas AN, and Coles M (2006) The HAMP domain structure implies helix rotation in transmembrane signaling. Cell 126, 929–940 [DOI] [PubMed] [Google Scholar]
- 13.Ashenberg O, Keating AE, and Laub MT (2013) Helix bundle loops determine whether histidine kinases autophosphorylate in cis or in trans. J Mol Biol 425, 1198–1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tanaka T, Saha SK, Tomomori C, Ishima R, Liu D, Tong KI, Park H, Dutta R, Qin L, Swindells MB, Yamazaki T, Ono AM, Kainosho M, Inouye M, and Ikura M (1998) NMR structure of the histidine kinase domain of the E. coli osmosensor EnvZ. Nature 396, 88–92 [DOI] [PubMed] [Google Scholar]
- 15.Khorchid A, and Ikura M (2006) Bacterial histidine kinase as signal sensor and transducer. Int J Biochem Cell Biol 38, 307–312 [DOI] [PubMed] [Google Scholar]
- 16.Kobir A, Shi L, Boskovic A, Grangeasse C, Franjevic D, and Mijakovic I (2011) Protein phosphorylation in bacterial signal transduction. Biochim Biophys Acta 1810, 989–994 [DOI] [PubMed] [Google Scholar]
- 17.Kee JM, and Muir TW (2012) Chasing phosphohistidine, an elusive sibling in the phosphoamino acid family. ACS Chem Biol 7, 44–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duclos B, Marcandier S, and Cozzone AJ (1991) Chemical properties and separation of phosphoamino acids by thin-layer chromatography and/or electrophoresis. Methods Enzymol 201, 10–21 [DOI] [PubMed] [Google Scholar]
- 19.Page SC, Immormino RM, Miller TH, and Bourret RB (2016) Experimental Analysis of Functional Variation within Protein Families: Receiver Domain Autodephosphorylation Kinetics. J Bacteriol 198, 2483–2493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gudipaty SA, and McEvoy MM (2014) The histidine kinase CusS senses silver ions through direct binding by its sensor domain. Biochim Biophys Acta 1844, 1656–1661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gudipaty SA, Larsen AS, Rensing C, and McEvoy MM (2012) Regulation of Cu(I)/Ag(I) efflux genes in Escherichia coli by the sensor kinase CusS. FEMS Microbiol Lett 330, 30–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Affandi T, Issaian AV, and McEvoy MM (2016) The Structure of the Periplasmic Sensor Domain of the Histidine Kinase CusS Shows Unusual Metal Ion Coordination at the Dimeric Interface. Biochemistry 55, 5296–5306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Z, and Hendrickson WA (2010) Structural characterization of the predominant family of histidine kinase sensor domains. J Mol Biol 400, 335–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheung J, and Hendrickson WA (2010) Sensor domains of two-component regulatory systems. Curr Opin Microbiol 13, 116–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reinelt S, Hofmann E, Gerharz T, Bott M, and Madden DR (2003) The structure of the periplasmic ligand-binding domain of the sensor kinase CitA reveals the first extracellular PAS domain. J Biol Chem 278, 39189–39196 [DOI] [PubMed] [Google Scholar]
- 26.Cheung J, and Hendrickson WA (2008) Crystal structures of C4-dicarboxylate ligand complexes with sensor domains of histidine kinases DcuS and DctB. J Biol Chem 283, 30256–30265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moore JO, and Hendrickson WA (2009) Structural analysis of sensor domains from the TMAO-responsive histidine kinase receptor TorS. Structure 17, 1195–1204 [DOI] [PubMed] [Google Scholar]
- 28.Sevvana M, Vijayan V, Zweckstetter M, Reinelt S, Madden DR, Herbst-Irmer R, Sheldrick GM, Bott M, Griesinger C, and Becker S (2008) A ligand-induced switch in the periplasmic domain of sensor histidine kinase CitA. J Mol Biol 377, 512–523 [DOI] [PubMed] [Google Scholar]
- 29.Cheung J, and Hendrickson WA (2009) Structural analysis of ligand stimulation of the histidine kinase NarX. Structure 17, 190–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boldog T, Li M, and Hazelbauer GL (2007) Using Nanodiscs to create water-soluble transmembrane chemoreceptors inserted in lipid bilayers. Methods Enzymol 423, 317–335 [DOI] [PubMed] [Google Scholar]
- 31.Denisov IG, Grinkova YV, Lazarides AA, and Sligar SG (2004) Directed self-assembly of monodisperse phospholipid bilayer Nanodiscs with controlled size. J Am Chem Soc 126, 3477–3487 [DOI] [PubMed] [Google Scholar]
- 32.Bayburt TH, and Sligar SG (2010) Membrane protein assembly into Nanodiscs. FEBS Lett 584, 1721–1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu H, and Naismith JH (2008) An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol 8, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, and Mori H (2006) Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2, 2006 0008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Datsenko KA, and Wanner BL (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97, 6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ritchie TK, Grinkova YV, Bayburt TH, Denisov IG, Zolnerciks JK, Atkins WM, and Sligar SG (2009) Chapter 11 - Reconstitution of membrane proteins in phospholipid bilayer nanodiscs. Methods Enzymol 464, 211–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Studier FW (2005) Protein production by auto-induction in high density shaking cultures. Protein Expr Purif 41, 207–234 [DOI] [PubMed] [Google Scholar]
- 38.Marty MT, Das A, and Sligar SG (2012) Ultra-thin layer MALDI mass spectrometry of membrane proteins in nanodiscs. Anal Bioanal Chem 402, 721–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fuhs SR, Meisenhelder J, Aslanian A, Ma L, Zagorska A, Stankova M, Binnie A, Al-Obeidi F, Mauger J, Lemke G, Yates JR 3rd, and Hunter T (2015) Monoclonal 1- and 3-Phosphohistidine Antibodies: New Tools to Study Histidine Phosphorylation. Cell 162, 198–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grass G, and Rensing C (2001) CueO is a multi-copper oxidase that confers copper tolerance in Escherichia coli. Biochem Biophys Res Commun 286, 902–908 [DOI] [PubMed] [Google Scholar]
- 41.Franke S, Grass G, Rensing C, and Nies DH (2003) Molecular analysis of the copper-transporting efflux system CusCFBA of Escherichia coli. J Bacteriol 185, 3804–3812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blattner FR, Plunkett G 3rd, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, and Shao Y (1997) The complete genome sequence of Escherichia coli K-12. Science 277, 1453–1462 [DOI] [PubMed] [Google Scholar]
- 43.Surette MG, Levit M, Liu Y, Lukat G, Ninfa EG, Ninfa A, and Stock JB (1996) Dimerization is required for the activity of the protein histidine kinase CheA that mediates signal transduction in bacterial chemotaxis. J Biol Chem 271, 939–945 [DOI] [PubMed] [Google Scholar]
- 44.Yamamoto K, Hirao K, Oshima T, Aiba H, Utsumi R, and Ishihama A (2005) Functional characterization in vitro of all two-component signal transduction systems from Escherichia coli. J Biol Chem 280, 1448–1456 [DOI] [PubMed] [Google Scholar]
- 45.Ninfa EG, Atkinson MR, Kamberov ES, and Ninfa AJ (1993) Mechanism of autophosphorylation of Escherichia coli nitrogen regulator II (NRII or NtrB): trans-phosphorylation between subunits. J Bacteriol 175, 7024–7032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Swanson RV, Bourret RB, and Simon MI (1993) Intermolecular complementation of the kinase activity of CheA. Mol Microbiol 8, 435–441 [DOI] [PubMed] [Google Scholar]
- 47.Filippou PS, Kasemian LD, Panagiotidis CA, and Kyriakidis DA (2008) Functional characterization of the histidine kinase of the E. coli two-component signal transduction system AtoS-AtoC. Biochim Biophys Acta 1780, 1023–1031 [DOI] [PubMed] [Google Scholar]
- 48.Pena-Sandoval GR, and Georgellis D (2010) The ArcB sensor kinase of Escherichia coli autophosphorylates by an intramolecular reaction. J Bacteriol 192, 1735–1739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yeh JI, Biemann HP, Prive GG, Pandit J, Koshland DE Jr., and Kim SH (1996) High-resolution structures of the ligand binding domain of the wild-type bacterial aspartate receptor. J Mol Biol 262, 186–201 [DOI] [PubMed] [Google Scholar]
- 50.Ottemann KM, Xiao W, Shin YK, and Koshland DE Jr. (1999) A piston model for transmembrane signaling of the aspartate receptor. Science 285, 1751–1754 [DOI] [PubMed] [Google Scholar]
- 51.Gushchin I, Melnikov I, Polovinkin V, Ishchenko A, Yuzhakova A, Buslaev P, Bourenkov G, Grudinin S, Round E, Balandin T, Borshchevskiy V, Willbold D, Leonard G, Buldt G, Popov A, and Gordeliy V (2017) Mechanism of transmembrane signaling by sensor histidine kinases. Science 356 [DOI] [PubMed] [Google Scholar]
- 52.Chervitz SA, and Falke JJ (1995) Lock on/off disulfides identify the transmembrane signaling helix of the aspartate receptor. J Biol Chem 270, 24043–24053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chervitz SA, and Falke JJ (1996) Molecular mechanism of transmembrane signaling by the aspartate receptor: a model. Proc Natl Acad Sci U S A 93, 2545–2550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mechaly AE, Sassoon N, Betton JM, and Alzari PM (2014) Segmental helical motions and dynamical asymmetry modulate histidine kinase autophosphorylation. PLoS Biol 12, e1001776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jiang P, Peliska JA, and Ninfa AJ (2000) Asymmetry in the autophosphorylation of the two-component regulatory system transmitter protein nitrogen regulator II of Escherichia coli. Biochemistry 39, 5057–5065 [DOI] [PubMed] [Google Scholar]
- 56.Casino P, Miguel-Romero L, and Marina A (2014) Visualizing autophosphorylation in histidine kinases. Nat Commun 5, 3258. [DOI] [PubMed] [Google Scholar]
- 57.Albanesi D, Martin M, Trajtenberg F, Mansilla MC, Haouz A, Alzari PM, de Mendoza D, and Buschiazzo A (2009) Structural plasticity and catalysis regulation of a thermosensor histidine kinase. Proc Natl Acad Sci U S A 106, 16185–16190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang C, Sang J, Wang J, Su M, Downey JS, Wu Q, Wang S, Cai Y, Xu X, Wu J, Senadheera DB, Cvitkovitch DG, Chen L, Goodman SD, and Han A (2013) Mechanistic insights revealed by the crystal structure of a histidine kinase with signal transducer and sensor domains. PLoS Biol 11, e1001493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bhate MP, Molnar KS, Goulian M, and DeGrado WF (2015) Signal transduction in histidine kinases: insights from new structures. Structure 23, 981–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim DE, Chivian D, and Baker D (2004) Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res 32, W526–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Moore JO, and Hendrickson WA (2012) An asymmetry-to-symmetry switch in signal transmission by the histidine kinase receptor for TMAO. Structure 20, 729–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.