

Summary
Fibrolamellar hepatocellular carcinoma (FLHCC) is driven by J-PKAcα, a kinase fusion chimera of the J-domain of DnaJB1 with PKAcα, the catalytic subunit of Protein Kinase A (PKA). Here we report the crystal structures of the chimeric fusion RIα2:J-PKAcα2 holoenzyme formed by J-PKAcα and the PKA regulatory (R) subunit RIα, and the wild type (wt) RIα2:PKAcα2 holoenzyme. The chimeric and wt RIα holoenzymes have quaternary structures different from the previously solved wt RIβ and RIIβ holoenzymes. The wt RIα holoenzyme showed the same configuration as the chimeric RIα2:J-PKAcα2 holoenzyme and a distinct second conformation. The J-domains are positioned away from the symmetrical interface between the two RIα:J-PKAcα heterodimers in the chimeric fusion holoenzyme and are highly dynamic. The structural and dynamic features of these holoenzymes enhance our understanding of the fusion chimera protein J-PKAcα that drives FLHCC as well as the isoform specificity of PKA.
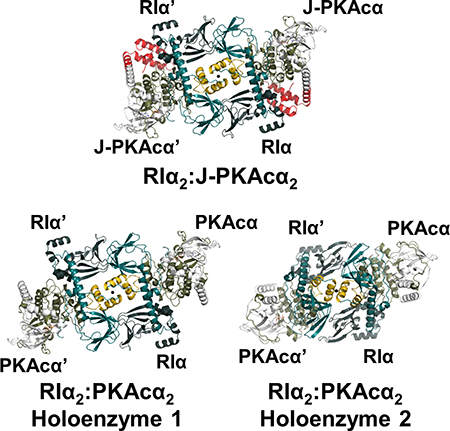
Graphical Abstract
eTOC Blurb
• Fibrolamellar hepatocellular carcinoma is driven by J-PKAcα, a kinase fusion chimera of the J-domain of DnaJB1 with PKAcα, the catalytic subunit of PKA. Here we report the crystal structures of the chimeric RIα2:J-PKAcα2 holoenzyme formed by J-PKAcα and the PKA regulatory (R) subunit RIα, and the wild-type RIα2:PKAcα2 holoenzyme.
Introduction
FLHCC is a rare liver cancer that predominantly affects adolescent and young adults with no history of liver disease (Craig et al., 1980; Eggert et al., 2013; Honeyman et al., 2014; Kakar et al., 2005; Lalazar and Simon, 2018; Torbenson, 2012). It does not respond well to chemotherapy and the overall five year survival rate of FLHCC patients is only 30–45% (El-Serag and Davila, 2004; Kakar et al., 2005; Katzenstein et al., 2003; Lim et al., 2014; Mavros et al., 2012; Weeda et al., 2013). The chimeric gene DNAJB1-PRKACA, ubiquitously and exclusively found in almost all FLHCC patients, is the result of a ~400 kb deletion in one copy of chromosome 19 (Darcy et al., 2015; Engelholm et al., 2017; Honeyman et al., 2014; Kastenhuber et al., 2017; Oikawa et al., 2015; Riggle et al., 2016a, 2016b; Simon et al., 2015). This produces an enzymatically active chimeric protein J-PKAcα. The tumor is driven not by the deletion but by the formation of the J-PKAcα fusion protein, and the tumorigenicity of J-PKAcα is dependent on its kinase activity (Kastenhuber et al., 2017). The fusion chimera protein has the first 69 residues of the N-terminus of DnaJB1, namely the J-domain, and the C-terminal 336 residues of PKAcα (Cheung et al., 2015; Honeyman et al., 2014) (Figure 1A). In its inactive state in cells, PKA exists as a holoenzyme composed of two catalytic subunits and one regulatory (R) subunit homodimer (Taylor et al., 2012). Cyclic adenosine monophosphate (cAMP) binding to the R subunits unleashes the PKAcα activity. Each R subunit is composed of an N-terminal dimerization/docking (D/D) domain followed by a flexible linker and two tandem highly conserved cyclic nucleotide-binding domains (CNB-A and CNB-B) (Figure 1A). There are four functionally non-redundant R isoforms, RIα, RIβ, RIIα, and RIIβ with similar domain organization (Taylor et al., 2012). The engineered R:PKAcα heterodimers where one PKAcα subunit is bound to a truncated monomeric form of the R subunit all appear to be very similar (Boettcher et al., 2011; Ilouz et al., 2012; Zhang et al., 2012). However, when the two R:PKAcα heterodimers, linked to the D/D domain by the flexible linkers are assembled into holoenzymes, each forms a unique symmetry-related interface between the two heterodimers and thus creates isoform-specific quaternary structures, as shown by the solved structures of the RIβ and RIIβ holoenzymes and the RIα holoenzyme model (Boettcher et al., 2011; Ilouz et al., 2012; Zhang et al., 2012) (Figures S1A and S1B). Among the four R isoforms, RIα can be considered as a master regulator for PKA signaling in mammalian cells. Deletion of RIα, for example, is embryonically lethal in mice and leads to unregulated PKA activity (Amieux et al., 1997). RIα also compensates when other R subunits are depleted or when PKAcα is overexpressed (Amieux and McKnight, 2002). It is the only upregulated R isoform in FLHCC cancer cells (Riggle et al., 2016b; Simon et al., 2015). Haploinsufficiency of RIα leads to a wide range of disease states, including Carney Complex (CNC) disease (Linglart et al., 2012; Park et al., 2012; Veugelers et al., 2004) as the other R subunit isoforms cannot compensate (Greene et al., 2008). Interestingly, recent studies (Graham et al., 2017; Terracciano et al., 2004) identified three individual patients with FLHCC and a personal history of CNC disease although the majority of CNC patients have no history of FHLCC.
Figure 1. Overall structure of the chimeric RIα2:J-PKAcα2 holoenzyme.

(A) Domain organization and color coding of J-PKAcα and RIα subunits.
(B) Structure of the holoenzyme. One heterodimer is labeled as RIα:J-PKAcα and its two-fold symmetry mate is labeled as RIα’:J- PKAcα’. The two-fold axis position is shown as a solid black circle.
(C) B factor analysis of the holoenzyme.
(D) The J-domain is in close proximity to the CNB-B domain of RIα.
Structural studies of the J-PKAcα chimera (Cheung et al., 2015) showed that it has all the structural hallmarks of wt PKAcα with the conserved bilobal kinase core shared by all kinase superfamily members. The only structural alteration is the fused J-domain, which replaces the myristylation motif (residues 1–14). In the crystal structure the J-domain is tucked underneath the C-lobe of the conserved kinase core (Cheung et al., 2015). However, in molecular dynamics (MD) simulations and NMR assays the fused J-domain explores a large diffusional space (Tomasini et al., 2018). Though J-PKAcα is overexpressed relative to PKAcα in FLHCC cells (Honeyman et al., 2014; Simon et al., 2015), overexpression of PKAcα alone is insufficient to recapitulate the oncogenic effect of J-PKAcα (Kastenhuber et al., 2017). Compensatory expression of RIα mRNA and protein were detected in FLHCC tumors while both the mRNA and protein levels of RIIβ are down-regulated (Riggle et al., 2016b; Simon et al., 2015). J-PKAcα can interact with truncated RIα and RIIβ to form R:J-PKAcα heterodimers in vitro (Cheung et al., 2015), suggesting that both wt PKAcα and the chimeric J-PKAcα can form holoenzymes. To understand how PKA signaling might be disrupted by the FLHCC chimera it is essential to appreciate the architecture of the chimeric and wt holoenzymes as well as knowledge of their dynamics. In this study, we show that the J-PKAcα chimera is inhibited by full-length RIα and capable of forming the canonical holoenzyme with activation still under the control of cAMP. We report the crystal structures of the oncogenic RIα chimeric holoenzyme and the wt holoenzyme at 3.66 Å and 4.75 Å resolution, respectively (Figures S1C). To explore whether the addition of the J-domain affects the conformational landscape of each holoenzyme, we furthermore report on MD simulations of the chimeric and wt RIα holoenzymes. We found states where the J-domain of J-PKAcα is able to interact with the C-terminal CNB-B domain of the RIα subunits; however, in the majority of MD states, the J-domain was dynamic and rotated away from the R:PKAcα interface. Altogether, these structural and dynamic descriptions of the driver of FLHCC enhance our understanding the molecular mechanism of this disease as well as our understanding of the dynamic allosteric mechanisms that couple cAMP binding to PKA activation.
Results
Overall structure of the FLHCC driver RIα2:J-PKAcα2 chimeric fusion holoenzyme
The complex of the full-length RIα and J-PKAcα chimera was formed in vitro by mixing the individually purified subunits followed by gel filtration (Figures S2A and S2B). The full-length holoenzyme structure was determined at 3.66 Å resolution (Figures 1B and S2C and Table 1). Each asymmetric unit (ASU) contains one holoenzyme molecule consisting of an RIα homodimer and two chimeric J-PKAcα subunits, thus the chimeric holoenzyme has the same stoichiometry as the previously published wt holoenzymes (Taylor et al., 2012). The presence of the J-domain does not prevent formation of the holoenzymes, and is positioned away from the symmetrical interface between the two RIα:J-PKAcα heterodimers in the holoenzyme. The J-domain can be easily accommodated spatially in the holoenzyme complex; there appears to be no steric constraints. The interface between the two heterodimers in the chimeric holoenzyme is strictly two-fold symmetry-related and created solely by the two RIα subunits, which pack against each other in an antiparallel orientation that includes a four-helical bundle involving the N3A motifs of the RIα subunits (Figure 1B). The PKAcα part of the chimera is almost identical to the PKI-bound wt PKAcα structure (Zheng et al., 1993), with a Cα root mean square deviation (RMSD) of 0.42 Å. The only structural alteration is a more linear and extended A-helix fused with the J-domain (Figure S2D). Additionally, J-PKAcα in the chimeric holoenzyme is superimposable to the previously reported (Cheung et al., 2015) structure of the PKI-bound chimera with a Cα RMSD of 0.39 Å (Figure S2E). The fused J-domain is similarly tucked underneath the C-lobe, and the contact area for the J-domain in the chimera is ~380 Å2. The J-domain in the chimeric holoenzyme has significantly higher temperature factors (B factor) than the rest of the holoenzyme, even at this medium resolution, suggesting that it retains a high degree of flexibility in the holoenzyme, similar to its PKI-bound state in solution based on NMR experiments (Tomasini et al., 2018) (Figure 1C and Table S1). The heterodimer in the chimeric holoenzyme is also structurally similar to the previously solved R:PKAcα heterodimers (Figure S2F) (Taylor et al., 2012), showing the J-domain fusion to the PKAcα does not alter the PKAcα interactions with the RIα subunit. The J-domains in the holoenzyme locate close to the CNB-B domain of the adjacent RIα subunit, with the shortest Cα atoms distance at ~8 Å (Figure 1D). Residues 1–91 of RIα are missing in the electron density although by SDS-PAGE and silver staining, we validated that full-length RIα and J-PKAcα are present in the protein crystal (Figures S2A and S2B). This absence of electron density for the D/D domain and part of the following N-linker is likely related to the flexible nature of this region (Li et al., 2000).
Table 1.
Data Collection and Refinement Statistics
| RIα2:J-PKAcα2 | RIα2:PKAcα2 | |
|---|---|---|
| Data collection | ||
| Space group | P6522 | P212121 |
| No. of molecules in one asymmetric unit | 1 | 2 |
| Cell dimensions | ||
| a, b, c (Å) | 166.50, 166.50, 332.70 | 140.50, 186.16, 186.67 |
| α, β, γ (°) | 90, 90, 120 | 90, 90, 90 |
| Resolution (Å) | 50–3.66 (3.79–3.66) | 50–4.75 (4.92–4.75) |
| Rsym (%) | 13.5 (49.8) | 10.9 (41.2) |
| I / σI | 29.5 (8.7) | 16.3 (2.7) |
| Completeness (%) | 100.0 (100.0) | 97.2 (80.5) |
| Redundancy | 21.3 (22.2) | 6.7 (4.6) |
| Refinement | ||
| Resolution (Å) | 50–3.66 | 50–4.75 |
| No. reflections | 30810 | 24239 |
| Rwork / Rfreea (%) | 20.0/25.0 | 21.2/25.5 |
| No. atoms | ||
| Protein | 11292 | 20336 |
| Ligand/ion | 66 | None |
| Water | None | None |
| B-factors | ||
| Protein | 110.69 | 269.92 |
| Ligand/ion | 91.06 | None |
| R.m.s deviations | ||
| Bond lengths (Å) | 0.003 | 0.014 |
| Bond angles (°) | 0.623 | 1.570 |
Values in parentheses are for the highest resolution shell.
Rfree was calculated by using a 5% of randomly selected reflections.
Highly dynamic J-domains in the chimeric fusion RIα2:J-PKAcα2 holoenzyme
Small Angle X-ray Scattering (SAXS) results (Figure S3) are in general consistent with the observed shape of the chimeric holoenzyme in solution. The calculated solution scattering data from the crystal structure fit to the SAXS solution experimental data reasonably well with a χ2 of 1.44. The chimeric holoenzyme in solution displays larger Rg and Dmax values (Figure S3D and Table S2) than those calculated for the crystal structure. The dynamic D/D domain with the N-linker regions or the dynamic nature of the J-domain may account for the observed larger dimension of the chimeric RIα holoenzyme in SAXS experiments compared to the crystal structure (Figure S3D).
Previous MD simulations of isolated J-PKAcα (Tomasini et al., 2018) identified two representative conformational states (Figure 2A). In the highest occupied state, the J-domain was positioned beneath the C-lobe of the kinase core in a J-in state, which is similar to what we observe in our chimeric holoenzyme structure. A second state showed the J-domain rotated away from the core to form an extended J-out conformation, and this flexibility of the J-domain was confirmed by NMR studies (Tomasini et al., 2018). To probe the possible motions of the J-domain in the chimeric holoenzyme, we performed three 1 μs MD simulations of the chimeric holoenzyme starting from either the crystal structure, or from a conformation with the J-domain modeled onto the holoenzyme crystal structure in the J-in or J-out state. The simulations from the chimeric crystal structure showed the majority of conformations in an extended J-out state, and far from the RIα subunit (Figure 2B). This is in contrast to the simulation performed with free J-PKAcα (Tomasini et al., 2018), where the J-in state was the highest occupied state. In the simulation started from the J-in state model, the J-domain from one chimera rotated to an extended conformation while that of the other remained in a J-in state (Figure S4A) to form stable interactions with its adjacent R subunit (Figure S4B). The minimum distances between Cα atoms in the J-domain to any Cα atom in the adjacent RIα subunit over all simulations ranged from 5.1 Å to 31.2 Å, emphasizing the flexibility of the J-domain (Figure 2C). In the simulation starting from the J-out state model, the J-domains of both chimeric subunits remained in the J-out state throughout the 1 μs simulation and did not show any interaction with the R subunits (Figure S4C). The calculated data from the three final MD simulation conformations of the chimeric holoenzyme (Figure S5 and Table S2), with one copy or both of the J-domain sampling the “out” state, are generally in agreement with the experimentally obtained SAXS solution data despite the lack of electron density for the D/D domain. The Rg and Dmax values of these three MD simulation conformations of the chimeric holoenzyme are also closer to the SAXS solution data than that of the crystal structure, suggesting the extended J-out state is a likely conformation of the J-domain in the chimeric holoenzyme in solution.
Figure 2. Dynamic conformations of J-domains in the chimeric holoenzyme during MD simulations.

(A) Three different simulations were initiated from: the chimeric crystal structure (crystal), the J-in and J-out states. θ1 and θ2 are angles defined to probe the dynamics of the J-domain.
(B) Top: Orientation of the J-domain for both copies of the chimera in the RIα2:J-PKAcα2 holoenzyme, as given by θ1 and θ2 over a 1 μs simulation of the chimeric holoenzyme starting from the crystal structure. The red ‘x’ indicates the position of the J-domain at the beginning of the simulation. Darker colors indicate later in time. Larger values of θ2 indicate a conformation in which the J-domain is tucked underneath the PKAcα core while smaller values indicate an extended conformation. Bottom: Initial and final conformations of the RIα2:J-PKAcα2 simulation started from the crystal structure.
(C) Minimum Cα distances between the J-domain and the adjacent RIα subunit for the three simulations. Solid and dotted lines indicate each copy of the J-domain in the holoenzyme respectively.
Isoform-specific interface between the RIα:J-PKAcα heterodimers
The interface between the chimeric heterodimers is solely created by the antiparallel alignment of the CNB-A and CNB-B domains in the RIα dimer, with a contact area of ~970 Å2 (Figure 3A). Wedged against each other from the two-fold symmetry-related RIα subunits are the two CNB-A N3A motifs (Figure S2C) which include the αN and αA helices as well as the connecting 310-loop. A similar N3A-N3A’ interface was first reported in the cAMP-bound RIα homodimeric RIα2(cAMP)4 structure (Figure 3B) (Bruystens et al., 2014). Each αA-helix is perpendicular to the opposing αN’-helix, thus creating a rectangular shaped four-helical bundle interface. The RIα-RIα’ interface also contains two identical salt bridge contacts between E179 in the RIα CNB-A domain and R315’ and R340’ in the RIα’ CNB-B’ domain (Figure 3A). Similar to the cAMP-bound RIα dimer (Bruystens et al., 2014), the N3A-N3A’ helical bundle is mostly hydrophobic, involving residues M123, Y120 and F148 from each N3A motif. These hydrophobic interactions are generally stable throughout the course of the MD simulations. The helical bundle with its two-fold symmetry also includes two identical hydrogen bond networks.
Figure 3. Interactions of the two RIα:J-PKAcα heterodimers in the chimeric holoenzyme.

(A) Overall interface of the two heterodimers consists of a large N3A-N3A’ interface and two identical small interfaces with salt bridges. Sequence alignment of the N3A motifs from different R isoforms is shown at the right bottom. Interface residues at the N3A motif are labeled in red. CNC mutations are marked with asterisks.
(B) The N3A-N3A’ four-helical bundle acts as a structural anchor during cAMP activation. The αB/C-helix and the CNB-B domain of RIα undergo dramatic conformational changes upon cAMP binding, while the N3A-N3A’ helical bundle is almost unaltered except the move-out of the 310-loops shown by the red arrows. The superimposition of the N3A-N3A’ interfaces in the chimeric holoenzyme and the cAMP-bound RIα dimer (gray, PDB ID 4MX3) is shown in the dashed circle.
Residues Y120 and K121 in the αN-helix form hydrogen bonds with N142’, S145’ and D149’ in the αA’-helix. While not directly involved in interactions at the N3A-N3A’ interface, R144 in the αA-helix forms hydrogen bonds with the backbone oxygens of F136 and L139 from the 310-loop. These hydrogen bonds break during cAMP activation as a consequence of outward motion of the 310-loop. Mutations of residues R144 and S145 are associated with CNC disease, which creates a holoenzyme that is poorly regulated and more easily activated by cAMP (Park et al., 2012). Substitutions of R144, S145 and N3A interface residues Y120 and F148 caused increased sensitivity for cAMP activation of the corresponding RIα holoenzymes and reduced cooperativity for cAMP binding (Bruystens et al., 2014). The Hill Coefficient for R144S and S145G were reduced to 1.4–1.5 while the Hill Coefficient was 1.0–1.1 for the Y120A and F148A mutants. The N3A-N3A’ helical bundle was also seen in truncated RIα monomer structures as an interaction site for crystal packing (Badireddy et al., 2011; Wu et al., 2004a, 2004b). However, this interface is not observed in any structures associated with RIIα or RIIβ. Sequence alignment also shows that RII subunits lack most of the key residues involved in forming the N3A-N3A’ interface (Figure 3A), emphasizing again that the N3A-N3A’ four-helical bundle is isoform-specific.
The overall structure of the N3A-N3A’ helical bundle is also conserved in the cAMP-bound RIα homodimer (Figure 3B). Thus, the N3A-N3A’ bundle likely serves as a structural anchor and contributes to the activation of the holoenzyme by cAMP activation and the following dissociation of R and C subunits. By contrast, the extended αB/C-helix that connects the CNB-A and CNB-B domains in the holoenzyme adopts a bent configuration in the cAMP-bound RIα homodimer with the CNB-B domain rotated dramatically to a position underneath the relatively stable CNB-A domain (Figure 3B). Moreover, the R315’-E179-R340’ salt bridge interactions observed in the holoenzyme (Figure 3A) between the RIα dimer become broken in the cAMP-bound RIα homodimer.
Overall structure of wt RIα2:PKAcα2 demonstrates two distinct holoenzyme conformations
To determine if the structure of the chimeric holoenzyme is unique to the fusion chimera protein, the wt RIα holoenzyme was formed in vitro by mixing the individually purified subunits followed by gel filtration, and its structure was determined at 4.75 Å resolution. It required different crystallization conditions (Figure 4 and Table 1) and has a distinct space group (P212121) compared to the chimeric RIα2:J-PKAcα2 holoenzyme and the previously solved structures of the wt tetrameric RIβ2:PKAcα2 or RIIβ2:PKAcα2 holoenzymes (Ilouz et al., Zhang et al., 2012). Each ASU contains four RIα:PKAcα heterodimers. The presence of full-length proteins in the crystals was confirmed by SDS gel analysis and silver staining, as described earlier (Figures S6A and S6B). Analysis of the crystal packing showed that each ASU has two RIα2:PKAcα2 holoenzyme molecules with distinct quaternary structures. While holoenzyme 1 has a conformation almost identical to the chimeric holoenzyme (Figures 4A, S6C and S6D), holoenzyme 2 has a much smaller N3A-N3A’ interface with an area of ~370 Å2 created only by the αN-helices (Figures 4B and S6D) and thus has a conformation distinct from holoenzyme 1. Both of the holoenzyme molecules contain two RIα:PKAcα heterodimers with a rotational twofold symmetry through the N3A-N3A’ interface. The heterodimers of RIα:PKAcα are almost identical in the two different tetrameric holoenzyme conformations (Figure S6E) with a Cα RMSD of 0.26 Å and also resemble the previously published structure of a truncated RIα(91–379):PKAcα heterodimer (Figure S6F) with a Cα RMSD of 0.86 Å (Kim et al., 2007). Similar to that in the chimeric holoenzyme, in both of the wt conformations, CNB-A is juxta-positioned against CNB-B’ thus supporting the enhanced allostery that is associated with the RIα2:PKAcα2 holoenzyme compared to the RIα:PKAcα heterodimer (Taylor et al., 2012). RIα competition assay results showed that the chimera and wt PKAcα have similar ability for RIα association (Figure 4C). In addition, the chimeric and wt RIα holoenzymes have no significant differences in cAMP activation nor its cooperativity (Figure 4C).
Figure 4. Interactions of the two RIα:PKAcα heterodimers in the wt holoenzyme.

(A) Interface of the two heterodimers in the wt holoenzyme 1. Domain organization and color coding of the PKAcα and RIα subunits are shown on the top.
(B) Interface of the two heterodimers in the wt holoenzyme 2.
(C) Fluorescence polarization assays to measure RIα inhibition (left) by PKAcα (red) and J-PKAcα (blue) as well as holoenzyme activation by cAMP (right). All data points are mean ± s.d. (n = 3 independent experiments).
The PKAcα subunits in holoenzyme 2 become closer to the heterodimer interface and to the symmetry-related RIα subunit than in holoenzyme 1 (Figure S6G). During the MD simulation, PKAcα in holoenzyme 2 is capable of interacting further with RIα’ (Figure S6H). MD simulations of each of the two conformations of the wt RIα2:PKAcα2 holoenzyme indicates that over the 1 μs of the simulation they are stable and do not interconvert (Figure S7A). The interfacial area between the RIα dimer in the wt holoenzyme 1 crystal structure resembles that of the chimeric holoenzyme simulations. The contact area in holoenzyme 2 is slightly increased (Figure S7B), which is largely due to a slight rotation of the RIα-RIα’ interface during the MD simulation.
Isoform-specific quaternary structures of PKA holoenzymes
The chimeric and wt RIα holoenzymes have quaternary structures different from the previously solved wt RIβ and RIIβ holoenzymes, even though the structures of all PKA heterodimers are remarkably similar (Figure 5). The quaternary structure isoform diversity is essential for each holoenzyme to create a distinct signaling hub that can respond to local levels of second messengers such as cAMP, and allows formation of distinct macromolecular complexes with local substrates and accessory proteins at different cellular sites.
Figure 5. Structural comparison of PKA holoenzymes.

(A) Side-by-side view of heterodimers at the same orientation: structures of RIα:J-PKAcα, RIα:PKAcα, RIβ:PKAcα (PDB ID 4DIN) and RIIβ:PKAcα (PDB ID 3TNP) heterodimers in the respective PKA holoenzymes.
(B) Structure comparison of the chimeric RIα2:J-PKAcα2 and wt RIα, RIβ and RIIβ holoenzymes.
The RIα holoenzymes and the view of CNB-B movement in these holoenzymes reported in this study (Figures 1 and 4) are distinct from the earlier model of the RIα holoenzyme that was based on crystal packing of two truncated RIα(73–244):PKAcα heterodimers (Boettcher et al., 2011). The earlier RIα holoenzyme model showed the two R:PKAcα heterodimers have cross talks between the CNB-A domain of one R: PKAcα dimer with the PKAcα’ of the other dimer and also allowed the modeled-in CNB-B domain movement. Such mobility of the CNB-B domain is consistent with previously obtained SAXS data of the RIα(91–379):PKAcα heterodimer and led to a suggestion that the CNB-B domain of RIα is mobile and moves away from PKAcα with Gly235 serving as a hinge point (Cheng et al., 2009). Recent studies have shown that CNB-B domain flexibility is linked to cAMP activation in the RIα(91–379):PKAcα truncated heterodimer (Hirakis et al., 2017; Barros et al., 2017). However, this view of CNB-B movement in the holoenzyme is different from the packing observed here in the full-length chimeric and wt RIα holoenzymes (Figures 1 and 4) where the CNB-B domain interacts with the opposite CNB-A’ domain; this interaction would prevent the suggested hinge motion in the holoenzyme. Consistent with our full-length RIα holoenzyme structure, MD simulations monitoring the dynamics of the αB/C-helix indicate it to be stable in the full-length holoenzyme with a near linear average of ~162° in all simulations (Figure S7C).
Effects of the J-domain on PKAcα function
The discovery that J-PKAcα is an oncogenic driver of FLHCC and thus a therapeutic target represents a significant breakthrough for FLHCC research (Honeyman et al., 2014). The fusion of the J-domain to PKAcα (Honeyman et al., 2014; Kastenhuber et al., 2017) may lead to alterations in kinase activity, substrates, dynamics, location or regulation at the level of the kinase subunit, holoenzyme and/or even higher molecular complexity level. As shown in the RIα competition as well as the cAMP activation assays, no significant differences were observed in terms of RIα association with either the chimera or wt PKAcα (Figure 4C) and the addition of the J-domain does not impact the sensitivity of the chimeric holoenzyme to cAMP activation (Figure 4C). As suggested by a thermostability assay (Figure 6A), the dynamic J-domain does not introduce a significant destabilizing effect on the chimera, nor on the chimeric RIα holoenzyme (Table S3). Similarly, J-PKAcα displayed unaltered binding affinities for ATP and inhibitor peptide (Figure 6B). Additionally, in agreement with previous reports (Cheung et al., 2015), the chimeric protein was slightly more active than its wt counterpart with unchanged enzymatic efficiency as shown by kcat/Km values (Figure 6C), suggesting that the J-domain may affect PKAcα enzyme dynamics.
Figure 6. Stability, ATP binding and kinetic studies of J-PKAcα and PKAcα.

(A) Stability of J-PKAcα and PKAcα (apo, with ATP, and/or with PKI binding) measured by thermofluor assay.
(B) ATP and PKI binding affinities of J-PKAcα (blue) and wt PKAcα (red). ATP binding (left) and PKI binding (right) were measured by thermofluor and fluorescence anisotropy assay respectively.
(C) Steady-state kinetics of phosphotransfer reaction of J-PKAcα (blue) and wt PKAcα (red). All data points are mean ± s.d. (n = 3 (panels a and b) and 2 (panel c) independent experiments).
Discussion
The oncogenic J-PKAcα has been crystallized here in one of its most important physiological states where it is associated in a holoenzyme complex with the RIα subunit. This structure demonstrates that the N-terminal fusion does not interfere with the general organization of the R2:PKAcα2 holoenzyme, and this also has relevance for the various PKAcα isoforms some of which have large extensions at the N-terminus (Søberg et al., 2017). Comparing the conformational states of the wt and chimeric RIα holoenzymes that display some specific interfaces may guide the development of drugs that selectively target not only to the J-domain and catalytic core to directly block chimera activity, but also regions present only at the holoenzyme level to block holoenzyme activation. The presence of alternate conformations of the holoenzymes may constitute a way to target the chimera selectively, as the conserved activity site of the wt PKAcα and the chimeric fusion J-PKAcα have little structural differences and the enzyme function is barely affected by the J-domain fusion. The enhanced dynamics of the chimeric holoenzyme may also expose some sites that are otherwise too transient to target. It may also be possible to trap a dynamic state independent of whether the holoenzyme is dissociated or not. Using a strategy that simultaneously blocks the activity of the oncogenic driver kinase and/or its holoenzyme dissociation would significantly reduce the possibility that a random mutation in the driver enables the tumor cells to escape treatment. RIα is a critical master switch for regulating PKA activity in cells, and it is likely that unregulated PKA activity is important, at least in part, for driving FLHCC. The importance of RIα is further supported by the recent finding that in a few rare cases, CNC mutations in RIα can drive FLHCC. Most CNC mutations, including the haplo-insufficiency caused by nonsense mediated decay of the RIα messenger RNA, do not drive FLHCC, so the unregulated phenotype associated with CNC is not in itself sufficient to explain these rare CNC mutations that are associated with FLHCC. It is not yet known how expression of the J-PKAcα fusion chimera causes FLHCC transformation. It could be the consequence of altered phosphorylation in the transformed cells, modified localization of the fusion chimera, changes in kinase dynamics, or altered interactions with other proteins. In the crystal structure of the chimeric fusion RIα2:J-PKAcα2 holoenzyme, the presence of the J-domain does not prevent formation of the holoenzymes, and the fusion region between the catalytic subunit and the J-domain, is not at the symmetrical interface in the holoenzyme between the two RIα:J-PKAcα heterodimers (Figure 1). Thus, rather than affecting the PKAcα interactions with the regulatory subunits, it is possible that addition of the J-domain alters the conformational landscape of the chimeric fusion holoenzymes, impacting interactions with other molecules. The higher B-factors in the J-domain suggested a large degree of conformational flexibility (Figure 1C and Table S1). MD simulations and SAXS experiments indicate a wide range in the conformational diversity of the J-domain appendage both in isolated J-PKAcα and in the holoenzyme. This could potentially directly influence enzyme dynamics or substrate specificity through allosteric networks or indirectly through holoenzyme interaction with other proteins, which affects substrate targets. It will be important in the future to further explore the detailed effects of J-domain fusion on the kinetics of PKA-substrate assembly and disassembly as the J-domain appendage likely influences enzyme kinetics through allosteric networks or holoenzyme interaction with other proteins or substrates. At this point it is not clear how the presence of the J-domain influences the function of the isolated PKAcα and PKA holoenzymes in cells. Thermostability assay (Figure 6A and Table S3) results showed the dynamic J-domain does not introduce a significant destabilizing effect on the fusion chimera nor on the chimeric RIα holoenzyme. However, it is not clear the lifetime of the fusion chimera is altered by the presence of the J-domain in cells. Changes to the lifetime of the fusion chimera or chimeric holoenzyme would lead to different numbers of kinase and, potentially, contribute to altered localization and protein-protein interactions. The wt PKAcα is also myristylated at its N-terminus and we have shown previously that this can be important for targeting the RIIβ holoenzymes to membranes (Zhang et al., 2015). This acylation site is missing in the fusion chimera protein and may also contribute to dysfunctional PKA signaling. Interestingly, the striking similarity in the overall structures and biochemical properties of the wt and chimeric RIα holoenzymes suggests the specificity of chimeric holoenzyme and its role in FLHCC need to be further sought at another level. Additionally, PKA holoenzyme is often assembled by scaffold proteins, referred to as A Kinase Anchoring Proteins (AKAPs), at specific cellular sites with ion channels and other regulatory enzymes such as phosphatases and phosphodiesterases to form macromolecular signaling complexes. It will be extremely important to elucidate how the conformational state and abundance of the different holoenzymes in the tumor cells and the holoenzymes communicate with their neighbors and substrates. In particular, it is important to determine how these macromolecular assemblies are altered in FLHCC by comparing paired tumor and adjacent normal liver samples. Understanding in detail how J-PKAcα signaling pathways drive disease will shed light on understanding its transformation to FLHCC and is expected to improve diagnosis and therapeutic treatment for this cancer.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ping Zhang (ping.zhang@nih.gov).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
E. coli BL21 (DE3) pLysS and E. coli BL21 (DE3) cells, which were grown in regular LB media, were used for the expression of all recombinant proteins in this study.
METHOD DETAILS
Protein expression, purification and crystallization
Bovine wt full-length RIα was expressed in Escherichia coli (E. coli) BL21 (DE3) pLysS as described before (Barros et al., 2017). The protein expression was induced at 16 °C overnight by 0.5 mM isopropyl β-D-thiogalactoside (IPTG) when the cell density reached 0.6–0.8 OD600. Ammonium sulfate precipitation was performed on the cell lysate supernatant by adding 26 grams of crushed ammonium sulfate per 100 ml of supernatant at 4 °C. The ammonium sulfate precipitates were dissolved in lysis buffer containing 50 mM MES (pH 5.8), 100 mM NaCl, 2 mM EGTA, 2 mM EDTA and 5 mM DTT, supplemented with cOmplete™ protease inhibitor cocktail tablets from Sigma and 10 μM 3-isobutyl-1-methylxanthine phosphodiesterase inhibitor, and batch bound to the cAMP resin overnight. The resin was rinsed with lysis buffer plus 600 mM NaCl, and the protein was eluted from the resin with lysis buffer plus 25 mM cGMP and then concentrated and purified by gel filtration in the buffer containing 50 mM MES (pH 5.8), 200 mM NaCl, 2 mM EGTA, 2 mM EDTA and 5 mM DTT through Hiload 16/600 Superdex 200 pg size exclusion column. Both human full-length J-PKAcα and PKAcα were engineered with an N-terminal His6SUMO tag. The constructs were then transformed into E. coli BL21 (DE3) for protein expression. The starter cultures were grown in LB media with 50 μg/mL kanamycin overnight at 37 °C and then 1:100 diluted into the same media. The cultures were grown at 37 °C until the cell density reached 0.5–0.6 OD600, after which the temperature was lowered to 24 °C and protein expression was induced overnight by adding IPTG to a final concentration of 0.5 mM. Cells were harvested by centrifugation, resuspended in lysis buffer containing 20 mM Tris-HCl pH 8.0, 100 mM NaCl, 5 mM β-mercaptoethanol and lysed by microfluidizer. The lysates were centrifuged, and collected supernatants were incubated with Ni-nitrilotriacetic (Ni-NTA) agarose beads overnight at 4 °C. The beads were rinsed with lysis buffer and then 10X bed volume of wash buffer (lysis buffer plus 20 mM imidazole). The proteins were eluted with 3X bed volume of elution buffer 1 (lysis buffer plus 50 mM imidazole) and elution buffer 2 (lysis buffer plus 100 mM imidazole). The eluates were spin dialyzed into the lysis buffer, after which NP-40 was added to them to a final concentration of 0.1%, and subjected to U1P1 (an engineered SUMO protease) digestion for 1 h at 25 °C at a molar ratio of 200:1 (protein:enzyme) to remove the His6SUMO tag. The cleaved tag and the protease were then removed from the proteins using Ni-NTA beads. Then the full-length RIα2:J-PKAcα2 and RIα2:PKAcα2 holoenzymes were formed by mixing RIα with J-PKAcα or PKAcα in a 1:1.5 molar ratio and spin dialyzed into a holoenzyme buffer containing 50 mM MOPS pH 7.0, 50 mM NaCl, 1 mM TCEP, 1 mM MgCl2 and 0.1 mM ATP. The formed complexes were loaded onto Hiload 16/600 Superdex 200 pg size exclusion column preequilibrated with the same buffer. Proteins from the peak fractions corresponding to the holoenzymes were collected, concentrated to ~10 mg/mL and subjected to extensive crystallization screening or used for biochemical assays. Crystallization was conducted at 20 °C using the hanging drop vapor diffusion method by mixing the protein and precipitants at a ratio of 1:1. The RIα2:J-PKAcα2 crystals were grown in a buffer containing 100 mM NaCl, 16–18% pentaerythritol propoxylate and 10% dimethyl sulfoxide and to their final size in ~2 weeks. The RIα2:PKAcα2 crystals were grown in a buffer containing 100 mM HEPES sodium-MOPS (acid) pH 7.5, 90 mM NPS (30 mM sodium nitrate, 30 mM sodium phosphate dibasic, 30 mM ammonium sulfate), 40–42% Precipitant Mix 2 (40% ethylene glycol; 20% PEG 8000), 3% D-(+)-glucose monohydrate and to their final size in ~3 weeks.
Structure determination
Diffraction data were collected at the 22ID beamline of the Advanced Photon Source (APS), Argonne National Laboratory (ANL). Data were indexed, integrated and scaled using the HKL2000 program (Otwinowski et al., 1997). The best RIα2:J-PKAcα2 and RIα2:PKAcα2 holoenzyme crystals diffracted to 3.66 and 4.75 Å, respectively. The initial phase of RIα2:J-PKAcα2 was determined using program PHASER (McCoy et al., 2007) with the structures of PKAcα Δexon1 (from PDB ID 4WB8) (Cheung et al., 2015) and RIα (from PDB ID 2QCS) (Barros et al., 2017) as search models. Refinement of the molecular replacement model was performed with PHENIX (Adams et al., 2010) and COOT (Emsley et al., 2004) alternatively. Initially, three rounds of Cartesian, individual B-factors, atomic occupancies and Cartesian simulated annealing (start temperature 5,000 K) refinement were performed in PHENIX, with the restraints of torsion-angle non-crystallographic symmetry (NCS), reference models and secondary structures. The reference models were J-PKAcα (from PDB ID 4WB7) (Cheung et al., 2015) and RIα (from PDB ID 2QCS) (Barros et al., 2017). In addition, stereochemistry and atomic displacement parameters weights were optimized during the refinement. The final refinement protocol included three rounds of Cartesian, individual B-factors and atomic occupancies refinement. The final RIα2:J-PKAcα2 model has 92.5% of residues in the favored Ramachandran region and 7.5% in the allowed region. The initial phase of RIα2:PKAcα2 was determined using program PHASER with the refined structure of the J-domain omitted RIα:J-PKAcα as the search model. Refinement of the molecular replacement model was carried out with REFMAC5 (Murshudov et al., 2011), PHENIX and COOT. First, rigid body refinement was performed using REFMAC5. Then 10 rounds of Cartesian, group B-factors (single residues were divided into mainchain and sidechain), atomic occupancies and Cartesian simulated annealing (start temperature 5,000 K) refinement were performed in PHENIX, with the restraints of global NCS, reference models (from PDB ID 2QCS) and secondary structures. The final refinement protocol included three rounds of Cartesian, individual B-factors and atomic occupancies refinement with the global NCS restraint. The final RIα2:PKAcα2 model has 84.6% of residues in the favored Ramachandran region and 15.4% in the allowed region. Data collection and refinement statics are summarized in Table 1. Models were evaluated using the MolProbity web server (molprobity.biochem.duke.edu/).
Small angle X-ray scattering (SAXS) experiment
SAXS measurements were performed at the 12ID-B beamline of APS, ANL. Photon energy was 13.3 KeV, and sample-to-detector distance was 3.6 m. To minimize radiation damage, thirty image frames were recorded with an exposure time of 1–2 s for each buffer and sample solution using a flow cell. The 2D images were reduced to 1D scattering profiles, and then grouped by sample and averaged using the MatLab software package at the beamlines. Concentration series measurements for the same sample were carried out to remove the scattering contribution due to interparticle interactions and to extrapolate the data to infinite dilution. The concentrations were 0.5, 0.7 and 0.9 mg/ml for RIα2:J-PKAcα2 in the buffer containing 50 mM MOPS pH 7.0, 50 mM NaCl, 1 mM TCEP, 1 mM MgCl2 and 0.1 mM ATP. The buffer background subtraction and intensity extrapolation to infinite dilution were carried out using NCI in-house developed MatLab script NCI-SAXS. Theoretical scattering profiles were generated from crystal structure and models and compared with the experimental SAXS data at q < 0.5 Å−1 using the CRYSOL software (Svergun et al., 1995). The pair-distance distribution function P(r) and maximum dimension (Dmax) were generated using GNOM (Svergun et al., 1992).
Kinase activity assay
The enzymatic activity of wt PKAcα or J-PKAcα was measured spectrophotometrically with a coupled enzyme assay (Cook et al., 1982). The ADP formation is coupled to the pyruvate kinase (PK) and lactate dehydrogenase (LDH) reactions. The reaction rate is determined by following the decrease in absorbance at 340 nm at 25 °C on a Photodiode Array Lambda 465 UV/Vis Spectrophotometer (PerkinElemer). The Michaelis-Menten parameters for ATP were determined by fixing Kemptide substrate (LRRASLG) at saturating concentrations while varying the concentrations of ATP. Reactions were pre-equilibrated at room temperature and initiated by adding ATP. The kinase reaction mixture contained 100 mM MOPS pH 7.1, 50 mM KCl, 6 mM phosphoenolpyruvate, 0.5 mM nicotinamide adenine dinucleotide (NADH), 100 μM of Kemptide, 15 units of LDH, 7 units of PK, and varying concentrations of ATP from 0 to 250 μM. MgCl2 was present in a constant 1 mM excess over ATP. The data was analyzed and fitted to the Michaelis-Menten equation using SigmaPlot software.
Inhibitor peptide PKI binding assay
Fluorescence anisotropy was used to measure PKI to PKAcα or J-PKAcα. 0.9 nM FAM-labeled PKI (5–24) peptide was mixed with 0–2000 nM PKAcα or J-PKAcα in buffer containing 20 mM MOPS pH 7.0, 150 mM NaCl, 10 mM MgCl2, 1 mM ATP, and 0.01% Triton X-100. Fluorescence anisotropy was measured by using GENios Pro micro-plate reader (Tecan) in black flat-bottom costar assay plates with 485 nm excitation and 535 nm emission. The data was analyzed and fitted to the anisotropy single association hyperbolic equation using Prism software.
RIα competition assay for catalytic subunit binding
Fluorescence polarization assay was used to measure the competition of RIα subunit with IP20 for wt PKAcα or J-PKAcα. 2 nM N-terminus FAM-labeled PKI peptide (5–24), and 10 nM PKAcα or J-PKAcα were mixed in the buffer containing 20 mM HEPES pH 7.0, 75 mM KCl, 0.005% Triton X-100, 10 mM MgCl2, 1 mM ATP, and 1 mM DTT. Two-fold serial dilutions of RIα from 30 nM to 0 nM were added to the PKI-bound catalytic subunits, followed by fluorescence polarization measurements using GENios Pro micro-plate reader (Tecan) in black flat-bottom costar assay plates with 485 nm excitation and 535 nm emission. The data was analyzed and fitted to the EC50 dose-response equation using Prism software.
Stability assay
ThermoFluor assay was used to measure the stabilities of apo PKAcα or J-PKAcα subunits and its ATP and/or peptide binding forms. The reaction was conducted with 5 μM of proteins in 45 μL of the buffer containing 20 mM MOPS pH 7.0, 150 mM NaCl. Ligands were used at the following concentrations 1 mM ATP, 10 mM MgCl2, and 25 μM PKI peptide (5–24). For each ligand, triplicate reactions were measured in a 96-well plate. After proteins and ligands were mixed and incubated for 5 min on ice, 5 μL of 200X SYPRO Orange dye was added to each reaction. The samples were heated from 20 to 85 °C with a 0.5 °C/min heating rate by using CFX96 Real-Time PCR Detection System (Bio-Rad) in temperature scanning mode. The fluorescence signals were measured using the ROX channel.
ATP binding assay
ATP dissociation constants were determined using the ThermoFluor assay. Similar condition as thermostability assay was used for ATP binding. The reactions were carried out in the buffer containing 20 mM MOPS pH 7.0, 150 mM NaCl with a range of ATP concentrations from 0 to 0.75 mM. After mixed with PKAcα or J-PKAcα, and incubated for 5 min on ice, 5 μL of 200X SYPRO Orange dye was added to each reaction. The samples were heated from 20 to 85 °C with a 0.5 °C/min heating rate by using CFX96 Real-Time PCR Detection System (Bio-Rad) in temperature scanning mode. The final concentration 4.5 μM of catalytic subunits was used to fit the data. The fluorescence signals were measured using the ROX channel. Each melting temperature was recorded and plotted versus ATP concentration.
PKA cAMP activation assay
Fluorescence polarization assay was used to measure the activation of wt and chimeric RIα holoenzymes. 2 nM N-terminus FAM-labeled PKI peptide (5–24), 7.2 nM RIα2, and 12 nM catalytic subunit (wt or chimera) were mixed in the buffer containing 20 mM HEPES pH 7.0, 75 mM KCl, 0.005% Triton X-100, 10 mM MgCl2, 1 mM ATP and 1 mM DTT. To activate PKA catalytic subunits, 2-fold serial dilutions of cAMP from 3000 nM to 0 nM were added. The fluorescence polarization was measure by using GENios Pro micro-plate reader (Tecan) in black flat-bottom costar assay plates with 485 nm excitation and 535 nm emission. The data was analyzed and fitted to the EC50 dose-response equation using Prism software.
Molecular dynamics simulations
MD simulations were performed to probe the dynamics of the RIα holoenzyme complexes. As previous simulations of the isolated J-PKAcα indicated a wide ensemble of conformations for the J-domain appendage (Tomasini et al., 2018), we performed three different simulations of RIα2:J-PKAcα2 with differing initial positions of the J-domain: the crystal structure, a J-in state model in which the J-domain was positioned close to the core of the catalytic subunit, and a J-out state model in which the J-domain was rotated away from the core of the catalytic subunit and the R:J-PKAcα interface. The J-domain conformations of the J-in and J-out states were those found in Tomasini et al. (Tomasini et al., 2018) as the top two representative conformations in a series of simulations performed on the isolated J-PKAcα. These two conformations of the J-domain were modeled onto the RIα2:J-PKAcα2 crystal structure. A similar methodology was used to model the first 14 amino acids and myristoylation motif which were missing from both conformations of RIα2:PKAcα2.
Structures were processed using the Protein Preparation Wizard in Maestro, solvated in a rectangular box with ~60,000 SPC waters and 150 mM sodium and chloride ions. Simulations were performed using the Desmond MD Package (Bowers et al., 2006) using the OPLS3 force field (Harder et al., 2016). Each system was subject to energy minimization using the steepest decent method succeeded by 100 ps of Brownian Dynamics simulation at constant volume and a temperature of 10 K with heavy atoms constrained. Subsequent equilibration included a 12 ps simulation at constant volume and at 10 K with heavy atoms restrained, followed by a 12 ps simulation at constant pressure with heavy atoms restrained, and finally a heating simulation in which the restraints were gradually relaxed and the system heated to 300 K over 24 ps. For production runs, the temperature was kept at 300 K using a Nose-Hoover Chain thermostat with a relaxation time of 1 ps (Martyna et al., 1992). The pressure was controlled at 1 bar using the Martyna-Tobias-Klein barostat with a relaxation time of 2 ps (Tuckerman et al., 2006). An integration time-step of 2 fs was used. Production simulations were performed for 1 μs saving system snapshots every 25 ps.
QUANTIFICATION AND STATISTICAL ANALYSIS
All the biochemical/biophysical assays in this study were performed in triplicates from two to three independent experiments (n = 2–3). Average values (mean) and standard deviations (SD) were calculated from those two to three independent experiments using Microsoft Excel. No methods were used to determine if the data met assumptions of the statistical approach.
Supplementary Material
Highlights.
Structures of chimeric RIα2:J-PKAcα2 and wildtype RIα2:PKAcα2 holoenzymes are described
The J-domains in RIα2:J-PKAcα2 are highly dynamic
RIα2:PKAcα2 has two distinct conformations
The RIα holoenzyme structures differ from the wildtype RIβ and RIIβ holoenzymes
Acknowledgments
We thank Drs. Alexandr Kornev, Di Xia, and Kylie Walters for critical reading of the manuscript and helpful discussions. We acknowledge use of the SAXS Core facility of Center for Cancer Research (CCR), National Cancer Institute (NCI) which is funded by FNLCR contract HHSN261200800001E and the intramural research program of the NIH, NCI, CCR. X-ray diffraction and SAXS data were collected at the 22ID and 12ID-B beamlines of the Advanced Photon Source, Argonne National Laboratory, respectively. We thank the Biophysics Resource in the Structural Biophysics Laboratory, NCI at Frederick for assistance. This work was supported by the National Institutes of Health grant GM34921 (S.S.T.), the Department of Defense grant CA160446 (S.M.S), and NIH grant 5R56CA207929 (S.M.S.), and the Intramural Research Program of the NIH, NCI, CCR (Zhang lab).
Footnotes
Declaration of Interests
Authors declare no competing interests.
DATA AND SOFTWARE AVAILABILITY
Coordinates and structure factors have been deposited in the Protein Data Bank with accession numbers 6BYR (RIα2:J-PKAcα2) and 6BYS (RIα2:PKAcα2).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX : a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr Sect D Biol Crystallogr. 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amieux PS, and McKnight GS (2002). The essential role of RI alpha in the maintenance of regulated PKA activity. Ann. N. Y. Acad. Sci 968, 75–95. [DOI] [PubMed] [Google Scholar]
- Amieux PS, Cummings DE, Motamed K, Brandon EP, Wailes LA, Le K, Idzerda RL, and Stanley McKnight G (1997). Compensatory regulation of RIα protein levels in protein kinase A mutant mice. J. Biol. Chem 272, 3993–3998. [DOI] [PubMed] [Google Scholar]
- Badireddy S, Yunfeng G, Ritchie M, Akamine P, Wu J, Kim CW, Taylor SS, Qingsong L, Swaminathan K, and Anand GS (2011). Cyclic AMP analog blocks kinase activation by stabilizing inactive conformation: conformational selection highlights a new concept in allosteric inhibitor design. Mol. Cell. Proteomics 10, M110.004390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barros E, Malmstrom RD, Nourbakhsh K, Del Rio JC, Kornev AP, Taylor SS, and Amaro RE (2017). Electrostatic Interactions as Mediators in the Allosteric Activation of Protein Kinase A RIα. Biochemistry 56, 1536–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boettcher AJ, Wu J, Kim C, Yang J, Bruystens J, Cheung N, Pennypacker JK, Blumenthal DA, Kornev AP, and Taylor SS (2011). Realizing the Allosteric Potential of the Tetrameric Protein Kinase A RIα Holoenzyme. Structure 19, 265–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers KJ, Chow DE, Xu H, Dror RO, Eastwood MP, Gregersen BA, Klepeis JL, Kolossvary I, Moraes MA, Sacerdoti FD, et al. (2006). Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In ACM/IEEE SC 2006 Conference (SC’06), (IEEE; ), pp. 43–43. [Google Scholar]
- Brown SHJ, Wu J, Kim C, Alberto K, and Taylor SS (2009). Novel Isoform-specific Interfaces Revealed by PKA RIIβ Holoenzyme Structures. J Mol Biol 393, 1070–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruystens JGH, Wu J, Fortezzo A, Kornev AP, Blumenthal DK, and Taylor SS (2014). PKA RIα homodimer structure reveals an intermolecular interface with implications for cooperative cAMP binding and Carney complex disease. Structure 22, 59–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CY, Yang J, Taylor SS, and Blumenthal DK (2009). Sensing domain dynamics in protein kinase A-Iα complexes by solution x-ray scattering. J. Biol. Chem 284, 35916–35925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung J, Ginter C, Cassidy M, Franklin MC, Rudolph MJ, Robine N, Darnell RB, and Hendrickson WA (2015). Structural insights into mis-regulation of protein kinase A in human tumors. Proc. Natl. Acad. Sci. U. S. A 112, 1374–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook PF, Neville ME, Vrana KE, Hartl FT, and Roskoski R (1982). Adenosine cyclic 3’,5’-monophosphate dependent protein kinase: kinetic mechanism for the bovine skeletal muscle catalytic subunit. Biochemistry 21, 5794–5799. [DOI] [PubMed] [Google Scholar]
- Craig JR, Peters RL, Edmondson HA, and Omata M (1980). Fibrolamellar carcinoma of the liver: A tumor of adolescents and young adults with distinctive clinico-pathologic features. Cancer 46, 372–379. [DOI] [PubMed] [Google Scholar]
- Darcy DG, Chiaroni-Clarke R, Murphy JM, Honeyman JN, Bhanot U, LaQuaglia MP, and Simon SM (2015). The genomic landscape of fibrolamellar hepatocellular carcinoma: whole genome sequencing of ten patients. Oncotarget 6, 755–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggert T, McGlynn KA, Duffy A, Manns MP, Greten TF, and Altekruse SF (2013). Epidemiology of fibrolamellar hepatocellular carcinoma in the USA, 2000–10. Gut 62, 1667–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Serag HB, and Davila JA (2004). Is fibrolamellar carcinoma different from hepatocellular carcinoma? A US population-based study. Hepatology 39, 798–803. [DOI] [PubMed] [Google Scholar]
- Emsley P, and Cowtan K (2004). Coot : model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Engelholm LH, Riaz A, Serra D, Dagnæs-Hansen F, Johansen JV, Santoni-Rugiu E, Hansen SH, Niola F, and Frödin M (2017). CRISPR/Cas9 Engineering of Adult Mouse Liver Demonstrates That the Dnajb1 – Prkaca Gene Fusion Is Sufficient to Induce Tumors Resembling Fibrolamellar Hepatocellular Carcinoma. Gastroenterology 153, 1662–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham R, Lackner K, Terracciano L, González-Cantú Y, Maleszewski JJ, Greipp PT, Simon SM, and Torbenson MS (2017). Fibrolamellar Carcinoma in the Carney Complex: PRKAR1A Loss Instead of the Classic DNAJB1-PRKACA Fusion. Hepatology 68, 1441–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene EL, Horvath AD, Nesterova M, Giatzakis C, Bossis I, and Stratakis CA (2008). In vitro functional studies of naturally occurring pathogenic PRKAR1A mutations that are not subject to nonsense mRNA decay. Hum. Mutat 29, 633–639. [DOI] [PubMed] [Google Scholar]
- Harder E, Damm W, Maple J, Wu C, Reboul M, Xiang JY, Wang L, Lupyan D, Dahlgren MK, Knight JL, et al. (2016). OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput 12, 281–296. [DOI] [PubMed] [Google Scholar]
- Hirakis SP, Malmstrom RD, and Amaro RE (2017). Molecular Simulations Reveal an Unresolved Conformation of the Type IA Protein Kinase A Regulatory Subunit and Suggest Its Role in the cAMP Regulatory Mechanism. Biochemistry 56, 3885–3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honeyman JN, Simon EP, Robine N, Chiaroni-Clarke R, Darcy DG, Lim IIP, Gleason CE, Murphy JM, Rosenberg BR, Teegan L, et al. (2014). Detection of a Recurrent DNAJB1-PRKACA Chimeric Transcript in Fibrolamellar Hepatocellular Carcinoma. Science 343, 1010–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilouz R, Bubis J, Wu J, Yim YY, Deal MS, Kornev AP, Ma Y, Blumenthal DK, and Taylor SS (2012). Localization and quaternary structure of the PKA RIβ holoenzyme. Proc. Natl. Acad. Sci. U. S. A 109, 12443–12448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakar S, Burgart LJ, Batts KP, Garcia J, Jain D, and Ferrell LD (2005). Clinicopathologic features and survival in fibrolamellar carcinoma: Comparison with conventional hepatocellular carcinoma with and without cirrhosis. Mod. Pathol 18, 1417–1423. [DOI] [PubMed] [Google Scholar]
- Kastenhuber ER, Lalazar G, Houlihan SL, Tschaharganeh DF, Baslan T, Chen C-C, Requena D, Tian S, Bosbach B, Wilkinson JE, et al. (2017). DNAJB1-PRKACA fusion kinase interacts with β-catenin and the liver regenerative response to drive fibrolamellar hepatocellular carcinoma. Proc. Natl. Acad. Sci. U. S. A 13076–13084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzenstein HM, Krailo MD, Malogolowkin MH, Ortega JA, Qu W, Douglass EC, Feusner JH, Reynolds M, Quinn JJ, Newman K, et al. (2003). Fibrolamellar hepatocellular carcinoma in children and adolescents. Cancer 97, 2006–2012. [DOI] [PubMed] [Google Scholar]
- Kim C, Cheng CY, Saldanha SA, and Taylor SS (2007). PKA-I holoenzyme structure reveals a mechanism for cAMP-dependent activation. Cell 130, 1032–1043. [DOI] [PubMed] [Google Scholar]
- Lalazar G, and Simon SM (2018). Fibrolamellar Carcinoma: Recent Advances and Unresolved Questions on the Molecular Mechanisms. Semin. Liver Dis 38, 51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Gangal Milind, Jones JM, Deich Jason, Lovett Kimberly E., Taylor Susan S., A., and Johnson David A. (2000). Consequences of cAMP and Catalytic-Subunit Binding on the Flexibility of the A-Kinase Regulatory Subunit. Biochemistry 39, 15626–15632. [DOI] [PubMed] [Google Scholar]
- Lim I, Farber B, and LaQuaglia M (2014). Advances in Fibrolamellar Hepatocellular Carcinoma: A Review. Eur. J. Pediatr. Surg 24, 461–466. [DOI] [PubMed] [Google Scholar]
- Linglart A, Fryssira H, Hiort O, Holterhus P-M, Perez de Nanclares G, Argente J, Heinrichs C, Kuechler A, Mantovani G, Leheup B, et al. (2012). PRKAR1A and PDE4D Mutations Cause Acrodysostosis but Two Distinct Syndromes with or without GPCR-Signaling Hormone Resistance. J. Clin. Endocrinol. Metab 97, E2328–E2338. [DOI] [PubMed] [Google Scholar]
- Martyna GJ, Klein ML, and Tuckerman M (1992). Nosé-Hoover chains: The canonical ensemble via continuous dynamics. Cite as. J. Chem. Phys 97, 2635–2643. [Google Scholar]
- Mavros MN, Mayo SC, Hyder O, and Pawlik TM (2012). A Systematic Review: Treatment and Prognosis of Patients with Fibrolamellar Hepatocellular Carcinoma. J. Am. Coll. Surg 215, 820–830. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ, and IUCr (2007). Phaser crystallographic software. J. Appl. Crystallogr 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Skubák P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, and Vagin AA (2011). REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D. Biol. Crystallogr 67, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oikawa T, Wauthier E, Dinh TA, Selitsky SR, Reyna-Neyra A, Carpino G, Levine R, Cardinale V, Klimstra D, Gaudio E, et al. (2015). Model of fibrolamellar hepatocellular carcinomas reveals striking enrichment in cancer stem cells. Nat. Commun. 6, 8070. doi: 10.1038/ncomms9070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, and Minor W (1997). [20] Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Park KU, Kim H-S, Lee SK, Jung W-W, and Park Y-K (2012). Novel Mutation in PRKAR1A in Carney Complex. Korean J. Pathol 46, 595–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riggle KM, Riehle KJ, Kenerson HL, Turnham R, Homma MK, Kazami M, Samelson B, Bauer R, McKnight GS, Scott JD, et al. (2016a). Enhanced cAMP-stimulated protein kinase A activity in human fibrolamellar hepatocellular carcinoma. Pediatr. Res 80, 110–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riggle KM, Turnham R, Scott JD, Yeung RS, and Riehle KJ (2016b). Fibrolamellar Hepatocellular Carcinoma: Mechanistic Distinction From Adult Hepatocellular Carcinoma. Pediatr. Blood Cancer 63, 1163–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon EP, Freije CA, Farber BA, Lalazar G, Darcy DG, Honeyman JN, Chiaroni-Clarke R, Dill BD, Molina H, Bhanot UK, et al. (2015). Transcriptomic characterization of fibrolamellar hepatocellular carcinoma. Proc. Natl. Acad. Sci. U. S. A 112, E5916–E5925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Søberg K, Moen LV, Skålhegg BS, and Laerdahl JK (2017). Evolution of the cAMP-dependent protein kinase (PKA) catalytic subunit isoforms. PLoS One 12: e0181091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svergun DI (1992). Determination of the Regnlarization Parameter in Indirect-Transform Methods Using Perceptual Criteria. J. Appl 25, 495–503. [Google Scholar]
- Svergun D, Barberato C, Koch MHJ, and IUCr (1995). CRYSOL – a Program to Evaluate X-ray Solution Scattering of Biological Macromolecules from Atomic Coordinates. J. Appl. Crystallogr 28, 768–773. [Google Scholar]
- Taylor SS, Ilouz R, Zhang P, and Kornev AP (2012). Assembly of allosteric macromolecular switches: lessons from PKA. Nat. Rev. Mol. Cell Biol 13, 646–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terracciano LM, Tornillo L, Avoledo P, Von Schweinitz D, Kühne T, and Bruder E (2004). Fibrolamellar hepatocellular carcinoma occurring 5 years after hepatocellular adenoma in a 14-year-old girl: a case report with comparative genomic hybridization analysis. Arch. Pathol. Lab. Med 128, 222–226. [DOI] [PubMed] [Google Scholar]
- Tomasini MD, Wang Y, Karamafrooz A, Li G, Beuming T, Gao J, Taylor SS, Veglia G, and Simon SM (2018). Conformational Landscape of the PRKACA-DNAJB1 Chimeric Kinase, the Driver for Fibrolamellar Hepatocellular Carcinoma. Sci. Rep 8, 720. doi: 10.1038/s41598-017-18956-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torbenson M (2012). Fibrolamellar carcinoma: 2012 update. Scientifica (Cairo). 2012, 743790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veugelers M, Wilkes D, Burton K, McDermott DA, Song Y, Goldstein MM, La Perle K, Vaughan CJ, O’Hagan A, Bennett KR, et al. (2004). Comparative PRKAR1A genotype-phenotype analyses in humans with Carney complex and prkar1a haploinsufficient mice. Proc. Natl. Acad. Sci. U. S. A 101, 14222–14227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuckerman ME, Alejandre J, López-Rendón R, Jochim AL, and Martyna GJ (2006). A Liouville-operator derived measure-preserving integrator for molecular dynamics simulations in the isothermal–isobaric ensemble. J. Phys. A. Math. Gen 39, 5629–5651. [Google Scholar]
- Weeda VB, Murawski M, McCabe AJ, Maibach R, Brugières L, Roebuck D, Fabre M, Zimmermann A, Otte JB, Sullivan M, et al. (2013). Fibrolamellar variant of hepatocellular carcinoma does not have a better survival than conventional hepatocellular carcinoma--results and treatment recommendations from the Childhood Liver Tumour Strategy Group (SIOPEL) experience. Eur. J. Cancer 49, 2698–2704. [DOI] [PubMed] [Google Scholar]
- Wu J, Jones JM, Xuong NH, Ten Eyck LF, and Taylor SS (2004a). Crystal structures of RIα subunit of cyclic adenosine 5′-monophosphate (cAMP)-dependent protein kinase complexed with (R p)-adenosine 3′,5′-cyclic monophosphothioate and (S p)-adenosine 3′,5′-cyclic monophosphothioate, the phosphothioate analogues of cAMP. Biochemistry 43, 6620–6629. [DOI] [PubMed] [Google Scholar]
- Wu J, Brown S, Xuong N-H, and Taylor SS (2004b). RIalpha subunit of PKA: a cAMP-free structure reveals a hydrophobic capping mechanism for docking cAMP into site B. Structure 12, 1057–1065. [DOI] [PubMed] [Google Scholar]
- Wu J, Brown SHJ, von Daake S, and Taylor SS (2007). PKA type IIalpha holoenzyme reveals a combinatorial strategy for isoform diversity. Science 318, 274–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Smith-Nguyen EV, Keshwani MM, Deal MS, Kornev AP, and Taylor SS (2012). Structure and Allostery of the PKA RII Tetrameric Holoenzyme. Science 335, 712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Ye F, Bastidas AC, Kornev AP, Wu J, Ginsberg MH, and Taylor SS (2015). An Isoform-Specific Myristylation Switch Targets Type II PKA Holoenzymes to Membranes. Structure 23, 1563–1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J, Trafny EA, Knighton DR, Xuong NH, Taylor SS, Ten Eyck LF, and Sowadski JM (1993). 2.2 A refined crystal structure of the catalytic subunit of cAMP-dependent protein kinase complexed with MnATP and a peptide inhibitor. Acta Crystallogr. D. Biol. Crystallogr. 49, 362–365. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.