

Abstract
The cellular membrane constitutes one of the most fundamental compartments of a living cell, where key processes such as selective transport of material and exchange of information between the cell and its environment are mediated by proteins that are closely associated with the membrane. The heterogeneity of lipid composition of biological membranes and the effect of lipid molecules on the structure, dynamics, and function of membrane proteins are now widely recognized. Characterization of these functionally important lipid-protein interactions with experimental techniques is however still prohibitively challenging. Molecular dynamics (MD) simulations offer a powerful complementary approach with sufficient temporal and spatial resolutions to gain atomic-level structural information and energetics on lipid-protein interactions. In this review, we aim to provide a broad survey of MD simulations focusing on exploring lipid-protein interactions and characterizing lipid-modulated protein structure and dynamics that have been successful in providing novel insight into the mechanism of membrane protein function.
Graphical Abstract
1. Introduction: Biological Roles of the Cellular Membrane
Biological membranes provide effective diffusion barriers that serve not only to separate the interior of a cell from its surroundings, but also to define distinct compartments within the cell.1–3 At the same time, owing the many embedded proteins, they allow for selective and controlled traffic of material and processing of information reaching the cellular membranes. Apart from transport of nutrients into the cell and export of waste material to the outside, these barriers also allow for establishment of electrochemical gradients between different compartments with important biological consequences, e.g., ionic gradients that control the function of excitabile cells, differential electrochemical gradients in mitochondria, which is essential to energy interconversion, and formation of highly acidic environments in lysosomes which are required for disposal of targeted material.3
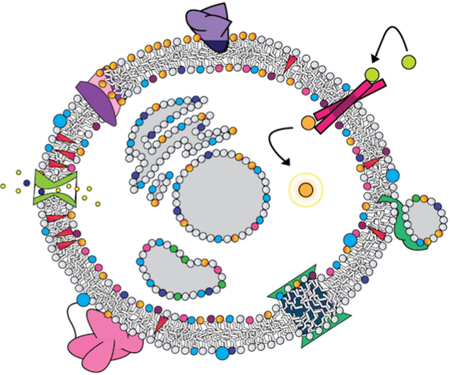
Biological membranes are complex;1 while predominantly composed of phospholipids, cellular membranes can contain a dizzyingly complex array of components with a high variability depending on location.4,5 Beyond phospholipids, common components of biological membranes include sphingolipids, sterols, carbohydrates attached through glycosylation,4 and perhaps most importantly, membrane proteins.3,4 The compartmentalization of the interior of the cell, and of its organelles, cannot be maintained through mere formation of a lipid barrier. Active regulation by membrane proteins is required to maintain distinct conditions on either side of the barrier, as well as to make the dynamic changes required as the cell and organism face changing environmental situations (Figure 1).6–10
Figure 1.
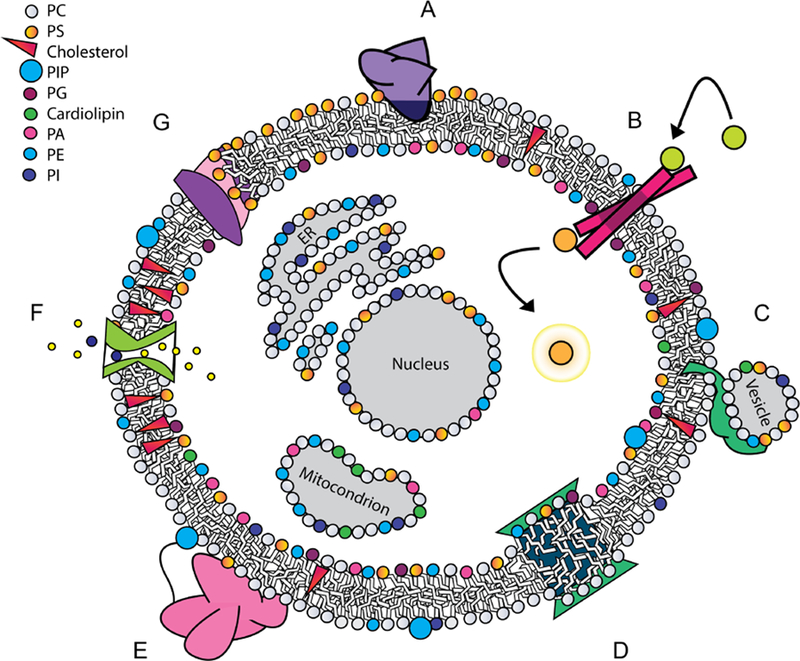
Proteins engage with lipids in diverse modes, many of which have functional significance. (A) Peripheral binding with a hydrophobic anchor, which can be lipid-specific, in this case to PS; (B) integral receptor involved in transmembrane signaling; (C) protein that induces vesicle fusion; (D) integral protein that induces local curvature through hydrophobic mismatch; (E) peripheral protein tethered to a membrane lipid, while its globular domain interacts with the interfacial region without embedding in the membrane core; (F) channel embedded in the membrane controlling ion transport across the membrane, while interacting with cholesterol; (G) transport of lipids across a membrane by a phospholipid scramblase.
Estimates of the genome suggest that 25% of proteins overall are membrane proteins.11 Membrane proteins may pass across the full length of the bilayer, or interact in a more peripheral manner.3,12 They can directly influence the structure13 and even the composition of the cellular membrane, allowing for asymmetric membrane compositions to be maintained.6,7,14 Critically, membrane proteins need to also allow for controlled signaling to take place across the physical barrier of the cellular membrane3 (Figure 1).
The structure and function of membrane proteins can be regulated by lipid bilayers and specific interactions with its lipid constituents.15–17 Specific binding events are known to modulate structure and function of membrane proteins,15 regulating key biochemical pathways such as blood coagulation.16 Some membrane proteins are even regulated by stereoisomer-specific binding to particular phospholipids.18
Many processes regulated through protein-lipid interactions have direct implications for broader human health and disease.16,17,19 In addition, the cellular membrane is a site of engagement for proteins involved in a wide variety of disease conditions. For instance, amyloid fibril formation is thought to be spurred by anionic membranes.20 Membrane binding proteins are also directly involved in a number of microbial attack mechanisms. A variety of toxins bind to cellular membranes, where they can either interfere with channels and receptors,21 or directly cause pore formation.22 Engagement of proteins with lipids is a key first step to viral infection, and lipid composition has been shown to be coupled to other steps in the viral replication life cycle.23,24
As the myriad ways in which lipids modulate protein function have become known, there has been great interest in gaining atomic-level information on the underlying molecular interactions. A variety of experimental techniques have been mustered to gather information on interactions between proteins and lipids (Figure 2). Functional techniques involve measuring the protein binding or turnover properties with different membrane model systems, such as liposomes,34 nanodiscs,35 supported lipid bilayers36 or lipid monolayers.37
Figure 2.

Experimental techniques that yield information on protein-lipid interactions. (A) Electron crystallography showing lipid-mediated crystal packing of aquaporin-0. Reprinted with permission from ref 25. Copyright 2011 Elsevier. (B) X-ray crystallography revealing phospholipid arrangement around a Ca2+-ATPase. Adapted with permission from ref 26. Copyright 2017 Springer Nature. (C) Cryo-EM revealing PIP bound in TRPv1 calcium channel. Adapted with permission from ref 27. Copyright 2016 Springer Nature. (D) NMR spectroscopy determining the tilt angle of transmembrane helices in lipid bilayer. Reprinted with permission from ref 28. Copyright 2017 Yamamoto et al. Licensed under a Creative Commons Attribution 4.0 International License. (E) Continuous-wave EPR discovering different conformations of the C2 domain of protein kinase C with respect to the membrane in the absence or the presence of PIP2 lipid. Adapted with permission from ref 29. Copyright 2008 American Chemical Society. (F) Time-resolved IR spectroscopy monitoring simultaneously the conformation of lipid and protein. Adapted with permission from ref 30. Published by National Academy of Sciences. (G) Single-molecule imaging distinguishing between transient and stable lipid-protein binding events. Reprinted with permission from ref 31. Copyright 2016 American Chemical Society. (H) Biochemical assays describing how the binding of a GLA domain to a PS-containing membrane affects blood coagulation kinetics. Adapted with permission from ref 32. Copyright 2011 American Society for Biochemistry and Molecular Biology. (I) Negative-staining electron microscopy capturing the interaction between M1 protein and lipid bilayer at the influenza virus A budding neck. Adapted with permission from ref 33. Copyright 2016 John Wiley and Sons.
Depending on the protein studied, relevant physicochemical or biochemical properties can be measured with techniques such as optical spectroscopy,38,39 calorimetry,40,41 electrochemistry,42,43 mass spectrometry,44,45 magnetic resonance,46,47 etc. On the other hand, structural techniques measure the size and shape of protein-lipid assembly with varying degrees of spatial resolution. For example, by connecting a transmembrane protein to a fluorescent label, super-resolution fluorescence microscopy can directly visualize the shape of an organelle membrane envelope as well as the protein distribution in it,48–51 approximately corresponding to a resolution of 10–100 nm. In the 1–10 nm regime, spectroscopy methods such as Förster/fluorescence resonance energy transfer (FRET)52 and double electron-electron resonance (DEER)53 can detect large conformational transitions of membrane transporters during their catalytic cycles.54 In comparison, small angle X-ray/neutron scattering55,56 and atomic force microscopy (AFM)57,58 can monitor the global changes in cellular membranes. Zooming further in, X-ray crystallography,59 nuclear magnetic resonance (NMR)60 and electron cryo-microscopy (cryo-EM)27,61 can yield atomic details of protein-lipid interactions.
These techniques have greatly enriched our knowledge of protein-lipid interactions, but obtaining data at both high spatial and temporal resolutions has proven prohibitively challenging. While X-ray crystallography and cryo-EM provide detailed structural information (Figure 3), dynamic information is lacking. In addition, it is unusual to be able to co-crystallize the protein with more than a handful of lipids, thus giving little information on the effect of their interactions within the larger bilayer context. NMR, on the other hand, provides dynamic information, but only for relatively small protein systems. Fluorescence techniques can glean information at the 10–100 nm resolution without atomic details. Functional assays are excellent in examining whether specific lipid-protein interactions have a major impact on the ultimate function, but often cannot provide information on the underlying molecular mechanisms.
Figure 3.
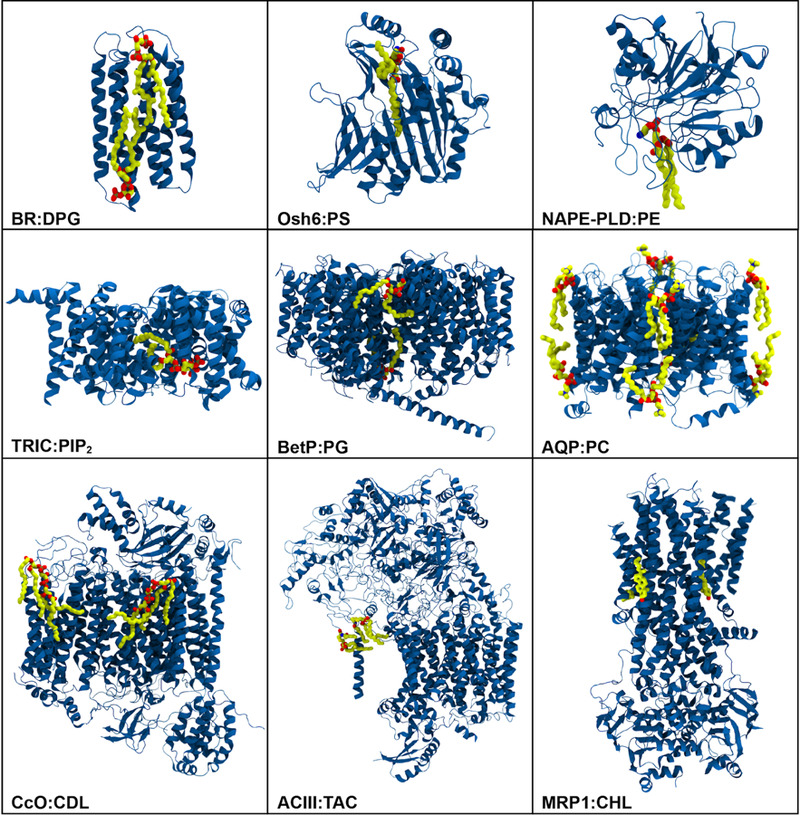
Representative structures of membrane proteins (blue) resolved experimentally with various types of lipids (carbon: yellow, oxygen: red, nitrogen: blue): bacteriorhodopsin (BR) with diphytanylglycerol (DPG) (PDB:2BRD), oxysterol-binding homology protein 6 (Osh6) with phosphatidylserine (PS) (PDB:4B2Z), human phospholipase D (NAPE-PLD) with phosphatidylethanolamine (PE) (PDB:4QN9), trimeric intracellular cation (TRIC) channel with phosphatidylinositol bisphosphate (PIP2) (PDB:5EGI), betaine transporter (BetP) with phosphatidylglycerol (PG) (PDB:4C7R), aquaporin-0 (AQP) with phosphatidylcholine (PC) (PDB:2B6O), cytochrome c oxidase (CcO) with cardiolipin (CDL) (PDB:5XDX), alternative complex III (ACIII) with triacylated cysteine (TAC) (PDB:6BTM), ATP-binding cassette (ABC) transporter MRP1 with cholesterol (CHL) (PDB:6BHU).
Molecular dynamics (MD) simulation and its related methods hold great promise in characterizing the structural and dynamical aspects of lipid-protein interactions critical to membrane protein function.62,63 Atomistic MD simulations allow for temporal resolutions of as high as 1 fs and spatial resolutions of sub-angstrom level. It is nowadays possible to simulate membrane protein interactions with lipid bilayers of complex compositions and in increasingly realistic environments. While challenges remain, particularly in the ability to obtain sufficient sampling of processes in the cellular environment, simulations have already allowed us to characterize critical lipid-protein interactions in membrane environments.62
In this article, we aim to review simulation studies that have given insight into lipid-protein interactions, particularly those with functional implications. The focus of this review is on MD studies where a protein was simulated in the presence of explicit lipids and where lipid-protein interactions and their structural, dynamical, or functional ramifications were analyzed and reported. First, we will provide an overview of major simulation techniques used in computational studies of biological membranes, namely, atomistic (all-atom (AA) or united atom (UA)), coarse-grained (CG), and multiscale descriptions, and modeling techniques used to embed/insert membrane-associated proteins into a lipid bilayer. Then, results obtained through simulations will be detailed in the subsequent two sections, divided into interactions between lipids and integral membrane proteins and peripheral membrane proteins. We will then discuss specific lipids that play special roles in modulating protein structure and function. Finally, effects of proteins on membrane structure as captured through simulations will be reviewed.
We will not include simulations of pure lipid bilayers, or studies in which a protein was simulated in a bilayer context, but no examination was made of interactions between the protein and lipids. As the focus is on lipid-protein interactions, we will generally not discuss simulations merely using implicit membrane models. Simulations of peptides are generally excluded from this review, except in cases were a truncated peptide was used to gain insight into a larger protein system, e.g., in studies of Ras linkers. Studies discussed were mostly performed from the late 1990s to the present. Some earlier simulations, which are too short to give information on protein-lipid phenomena, but are of interest for history of development of protein-lipid simulations, are covered in our historical discussion in the Computational Methods section.
2. Computational Methods to Characterize Lipid-protein Interactions
Biomolecular phenomena take place on a range of temporal and spatial resolutions. As with experimental methods, the choice of computational method is determined by the type of phenomena to be studied. Most computational studies of protein-lipid interactions use classical MD simulations, which employ Newton’s equations of motion to describe the dynamics of particles (e.g., atoms) in a biomolecular system. Several packages have been developed to apply MD to biomolecular systems.64–66 A variety of molecular representations, including atomistic (all-atom and united-atom), coarse-grained (CG) and multiscale hybrid models, have been employed to investigate lipid-protein interactions (Figure 4). All of these methods use the basic approximation of representing molecules as a set of interaction sites (e.g., atoms), with the number of atoms in each interaction site defining the resolution of the method.
Figure 4.
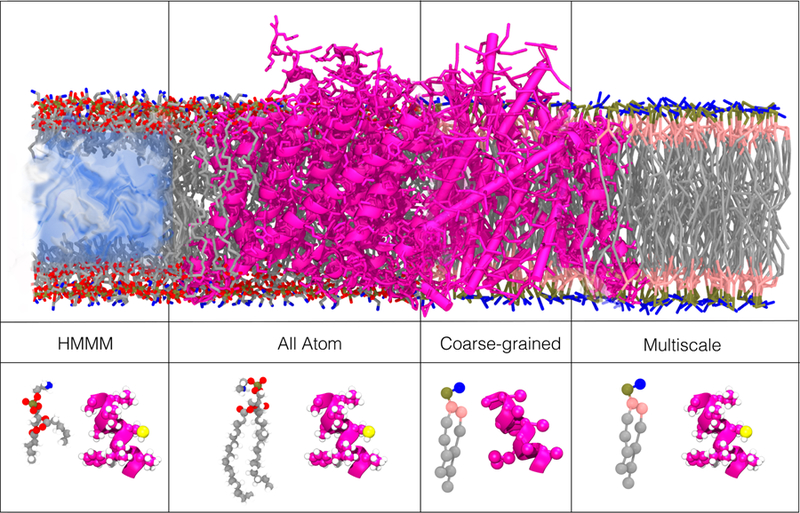
Examples of common resolutions/representations used in the investigation of lipid-protein interactions. The upper panels illustrate different representations for a membrane-embedded ClC channel (PDB:1OTS). The lower panels illustrate each of the representations using a single phosphatidylserine (PS) lipid and a short alpha helix (ClC channel, PDB:1OTS). All-atom (AA) simulations use one interaction-site per atom. In coarse-grained (CG) simulations, several atoms are grouped into one interaction site. Multiscale simulations use a combination of resolutions either in the same simulation or in sequence. The HMMM (highly mobile membrane mimetic) model uses truncated lipids and a membrane core of inorganic solvent to increase lateral diffusion of membrane lipids.
All-atom (AA) models employ one interaction site per atom, thereby providing high resolution information on molecular interactions (Figure 4). They are limited, however, to the microsecond (μs) timescale and to small system sizes on the order of tens of nanometers (Figure 5). In a related representation known as the united atom (UA) model, non-polar hydrogen atoms and the heavy atoms to which they are bonded are represented by one interaction site. Currently, the UA model has comparable limitations to the AA model in length and timescale.
Figure 5.
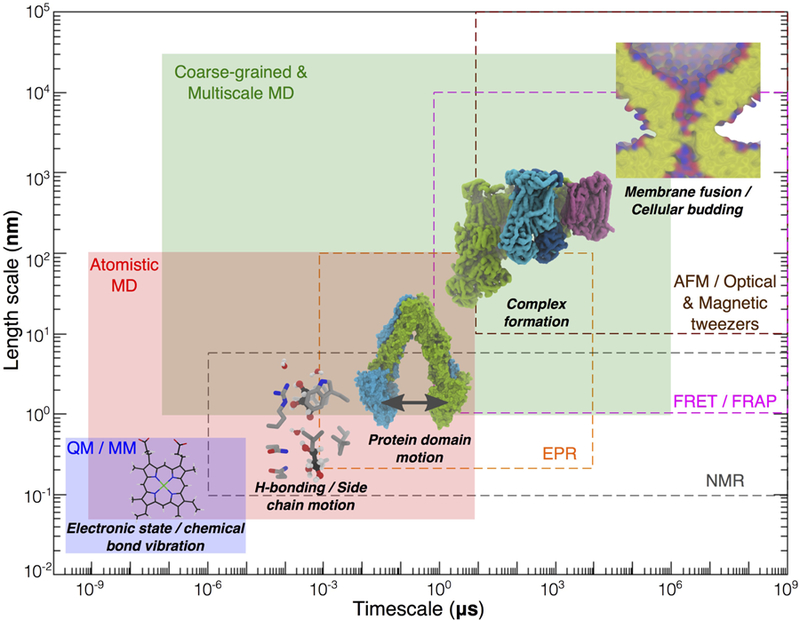
Scope of methods in describing the dynamics of chemical and biological processes. Effective length and time scales of all-atom (AA) MD, quantum/molecular mechanics (QM/MM) and reduced representation/multiscale simulations (e.g., CG) are in the shaded boxes, while those of experimental techniques (nuclear magnetic resonance (NMR), electron paramagnetic resonance (EPR), Förster resonance energy transfer (FRET), fluorescence recovery after photobleaching (FRAP), atomic force microscopy (AFM), and magnetic and optical tweezers) are in the dashed boxes. The structure of heme highlights electronic state and chemical bond vibration. Different molecular systems are chosen to exemplify the scope of simulation and computational studies. Permeation of water and glycerol molecules through the E. coli glycerol facilitator protein (structure taken from PDB:1FX8) involves intermolecular contacts (e.g., H-bonds) and side chain motions of amino acids lining the pore. ABC transporters undergo large-scale protein domain motions during the transport cycle. The structure of a mitochondrial aerobic respiratory super-complex67 (PDB:5J4Z) illustrates the slow process of formation of multiprotein complexes. Fusion of membranes of two cellular compartments illustrates structural changes of a cell or an organelle, representing one of the slowest processes targeted by computational studies of membrane systems.
The highly mobile membrane mimetic (HMMM) model (Figure 4) is an example of an approximate AA model for lipids which allows for enhanced sampling of lipid-protein interactions through increased lipid diffusion while maintaining an AA representation for the headgroup. CG models reduce the complexity of simulated systems (Figure 4) by using one interaction site to represent multiple atoms, allowing simulations to access slow biomolecular events on the scale of up to milliseconds (Figure 5). Multiscale simulations, in which multiple resolutions are combined in order to take advantage of faster sampling in coarser representations while also preserving a detailed description in at least part of the system (Figure 4), allow for the study of events for which multiple time and length scales may be relevant.
In the following section, we will describe development of these methods from their early (see Section 2.1) to their modern successes. We will first discuss AA and UA simulations, then move to CG, multiscale methods and HMMM. Finally, we will discuss methods which have been developed to facilitate proper embedding of proteins into the membrane, an important initial step in simulating membrane proteins and studying lipid-protein interactions.
2.1. Early MD Simulations of Biological Membranes
The earliest simulations of membranes go back to the 1980s. Since that time, a synergistic combination of growth in computational power and methodological advances has made previously unattainable phenomena, such as membrane fusion, within reach of computational studies (Figure 5).
To provide a proper description of protein-lipid interactions, modeling explict lipids in a bilayer environment was critical. The earliest simulations of highly simplified bilayers, using UA models, were performed in the 1980s,68 with special methods employed to avoid the need for explicit solvent.69,70 The models were intended to replicate the behavior of a decanoate-decanol-water system, which had been previously characterized experimentally.69,70
Simulations were carried out by van der Ploeg et al for 80 ps on bilayers consisting of 2×16 and 2×64 decanoate molecules respectively, with periodicity in only two dimensions.69,70 Each of ten interaction sites in the decanoate chain was assigned one of three functional group types, with Lennard-Jones potential terms varied to reproduce correct behavior.69,70 The terminal interaction sites of the decanoate chains were modeled to resemble lipid headgroups in their behavior, with all other interaction-site types modeled as methylene groups or terminal methyl groups.69,70 In order to model realistic bilayer interactions in the absence of solvent, harmonic potentials were used to restrain the headgroup position of decanoate molecules and approximate their behavior in solution.69 These simulations allowed for characterization of lipid bilayer order parameters and tilt of lipid molecules in the membrane, and demonstrating the applicability of MD simulations to biological membranes (Figure 6A).69,70
Figure 6.
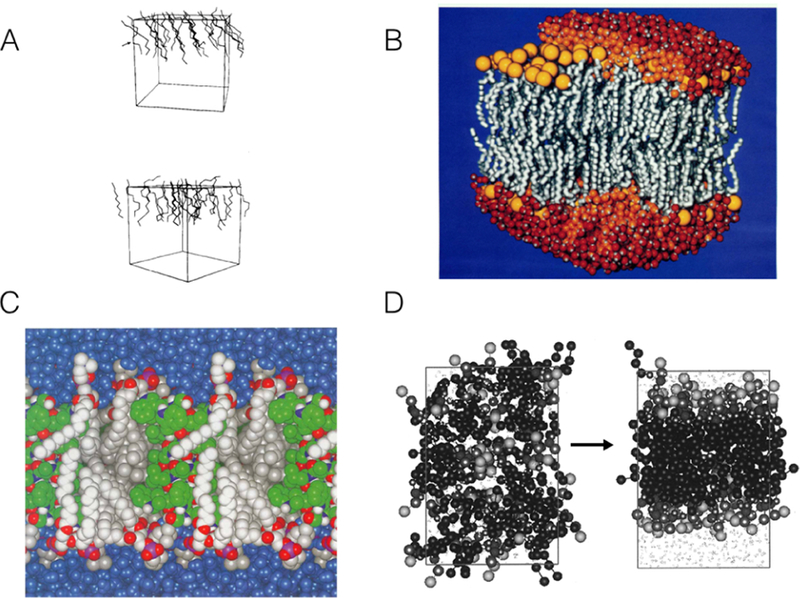
Early simulations of lipid bilayers. (A) Snapshot of a united-atom (UA), unsolvated model bilayer system (top and bottom leaflets), simulated for 80 ps. Manipulation of Lennard-Jones parameters and use of harmonic restraints on the “headgroup” interaction sites allowed the model lipids to reproduce behavior of a decanoate-decanol-water system. Only the upper leaflet is shown for clarity.69 Reprinted with permission from ref 69. Copyright 1982 AIP Publishing. (B) Fully atomistic bilayer simulation of 200 phospholipids, 120 ps in length, fully solvated and ionized.71 Lipid tails shown in grey, headgroups in yellow, and water molecules in orange and red. Reprinted with permission from ref 71. Copyright 1993 American Chemical Society. (C) Fully atomistic, solvated simulation of gramicidin A channel in a bilayer of 16 lipids, 500 ps in length. Protein in green, lipids in silver, water in blue.72 Adapted with permission from ref 72. Copyright 1994 National Academy of Sciences. (D) CG simulations showing spontaneous bilayer formation for the first CG model of a phospholipid bilayer.73 Adapted with permission from ref 73. Copyright 2001 American Chemical Society.
Later simulations began to introduce greater realism to the model systems. An over 3,000 atom, 100–ps simulation added water and ions to the UA decanoate-decanol model, as well as providing a fully atomistic description of the headgroups with partial atomic charges.74 An AA, DLPE system of 2×24 lipids and 553 water molecules was later simulated for 200 ps, improving bilayer modeling through the use of full phospholipids.75 A simulation with DMPC of equivalent size allowed for comparison of differences in headgroup interactions for DLPE and DMPC.76 An additional simulation studied lateral diffusion of DPPC in a solvated bilayer of 72 lipids.77 The first sufficiently large and detailed system simulated to be potentially useful for studying protein-lipid interactions was a set of two 200–lipid POPC bilayer simulations solvated with approximately 5,000 water molecules and simulated for 120 ps (Figure 6B).71
The earliest protein-lipid simulations used the methods of van der Ploeg et al69,70 to examine interactions of the simplified decanoate molecule with α-helical polypeptides.78 The peptides studied were polyglycine and glycophorin. In one simulation, the glycophorin structure was kept α-helical, while the second simulation was performed allowing conformational changes to occur.78 In the second simulation, deviations from an α-helical conformation were found.78 Order parameters of lipids were assessed as a function of distance to the midplane of the membrane. This study effectively explored membrane effects on the core region of the protein. However, the short timescale of the simulation as well as the drastically simplified membrane model limited the conclusions.
Later simulations were performed in the 1990s on the gramicidin A channel in a lipid bilayer (Figure 6C). This channel proved an ideal case to study because of the small size of the protein and the large body of available experimental information which could be compared to the simulation results.72 In the earliest simulation, the channel was inserted in a solvated 16–lipid DMPC bilayer and simulated for 500 ps. NMR order parameters were found to be in good agreement with experiment.72 The authors hypothesized that interactions between a tryptophan residue and the glycerol moiety of the lipid bilayer observed during their simulation might be important for stabilization of the protein-membrane interface. Later simulations of the channel were run for 1000 ps (1 ns), allowing for additional analysis of lipid-protein interactions79 and characterization of a Na+ binding site.80 In one of the earliest studies examining membrane effects of a peripheral protein, phospholipase A2 was simulated at the surface of a monolayer, allowing for desolvation of lipid molecules in the proximity of the protein to be assessed.81
At this point, characterization of lipid-protein interactions was still hampered by the short timescales accessible by atomistic simulations. Thus during this period non-atomic membrane models were used in combination with atomic-level protein representations to study phenomena that occur over longer timescales. This included the use of Lennard-Jones membrane models to study ion binding in gramicidin A82 and a synthetic ion channel in an octane/water system for 1 ns.83 Biphasic systems of this type have continued to be used to study slow phenomena, such as peripheral protein binding.84 While these systems lose ability to describe interactions in detail, they allow for efficient sampling of protein positioning in a membrane-like environment.
Interest in achieving longer timescales spurred development of CG models of phospholipid bilayers (Figure 6D).73,85 In the earliest CG lipid bilayer simulation, DMPC was modeled using six bead types. One interaction site represented each, respectively, of the choline group, phosphate group, glycerol backbone, ester groups, and two types of alkane interaction sites. Simulation of the system composed of 50 DMPC lipids and 428 CG water molecules was sufficiently fast to allow for spontaneous formation of the bilayer (Figure 6).73
In the remainder of this section, we follow the development of simulation methodologies for protein-lipid interactions beyond these early attempts to their modern incarnations. We will first discuss AA and CG simulations and their force field characterization, describing how these methods gradually improved to allow for more accurate descriptions of lipid bilayers at longer timescales and with greater diversity of bilayer compositions. Multiscale simulation methods will then be described. A discussion will follow, of the development and the use of a specialized membrane model allowing for enhanced lipid diffusion and consequently increased sampling of lipid-protein interactions, the highly mobile membrane mimetic (HMMM) model. Finally, we will detail a key step involved in setting up simulations of membrane proteins, namely, how to embed proteins within the lipid bilayer, which is a non-trivial process particularly in the absence of detailed experimental data.
2.2. Atomic-level Simulations
Atomistic simulations use interaction sites to describe individual atoms. Two levels of atomistic simulations have been used to study protein-lipid interactions, all-atom (AA) models in which every atom is assigned an interaction site, and united-atom (UA) models in which all atoms except non-polar hydrogens are represented as individual interaction sites. Non-polar hydrogens and the heavy-atoms they are bound to are assigned a single interaction site in UA models.
A perennial area of interest for the development of MD simulations are the force fields used to set characteristics of the molecules being simulated, such as bonds, angles, dihedrals, and partial charges. Parameters are derived by first using quantum mechanical (QM) calculations, and then fitting parameters with molecular mechanics (MM) to be consistent with the QM and experimental results. The quality of the force field determines the quality of the simulation results. Development of AA force fields for proteins can be traced to the 1980s.86–88 Popular force fields developed during this period which reproduced important characteristics of proteins and are still used for modern simulations, include CHARMM,89–91 AMBER,92,93 GROMOS,94 and OPLS.95,96 Early versions of each were UA force fields.86,88 Due to increases in computational power and limitations inherent to the UA approach, AA force fields have since become the standard.86,88 Only the GROMOS force fi is still UA, although it forms the basis of additional AA force fields.86,88
Inclusion of lipids into AA force fields proved initially challenging, due to both the liquid-crystalline characteristics of lipid bilayers and the paucity of high-resolution structural information for lipids.87 In the early 2000s, when AA simulations had become standard for proteins, many lipid simulations were performed with UA force fields.87 The AA CHARMM and the UA Berger force field parameters for lipids were both commonly used during this period. A number of simulations using CHARMM22 for lipids97 reproduced experimental results, including a membrane channel within a bilayer.72 The initial MD simulations for lipids in the CHARMM22 force field were 100 ps in length, and a number of limitations became evident upon extending the timescale to 800 ps.98 The surface tension of the CHARMM22 bilayer was too high, causing the surface area to collapse and the bilayer to become gel-like unless constant area was imposed during the simulation or a surface tension designed to yield the experimental surface area applied. In addition, order parameters of lipid atoms near the water-bilayer interface were in error for CHARMM22 bilayer simulations.98 Notably, the UA Berger lipids did not suffer from these problems.99
CHARMM27100 improved on CHARMM22 lipids through refinement of the Lennard-Jones and torsional parameters of the alkane moiety, as well as the torsional and partial charge parameters of the phosphate moiety.98 It was used for a range of benchmark studies with simulation times reaching 100 ns.98 CHARMM27 was also found to describe lipid-protein interactions.98 At longer timescales, however, a systematic overestimation of the chain order parameters became evident. CHARM27r101 resolved this problem, but issues with C2 order parameters were still evident. It was still not advisable to carry out simulations using the NPT ensamble. CHARMM36102,103 yielded the correct headgroup surface area and correct chain order parameters using NPT conditions. (Figure 7)
Figure 7.
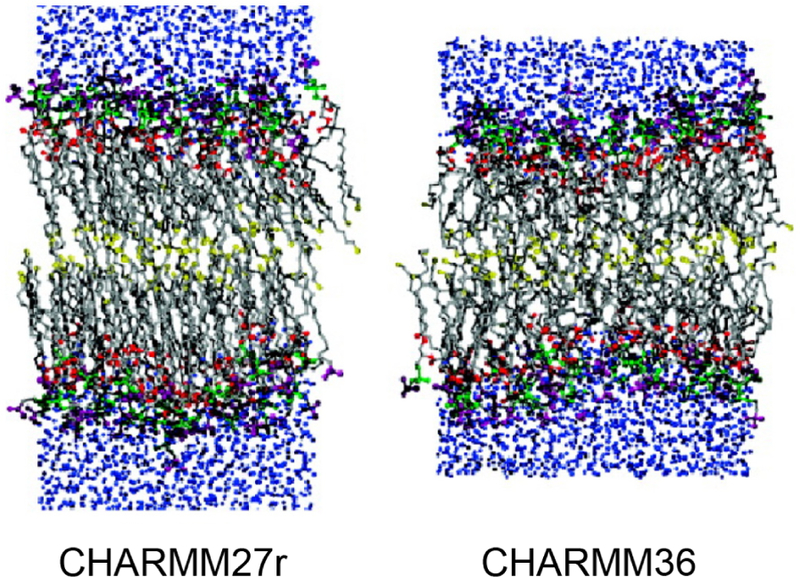
Illustration of a key improvement to simulations of lipid bilayers resulting from changes to the CHARMM36 force field as compared to CHARMM27r. In CHARMM27r, the bilayer phase transitioned inappropriately to gel phase, while CHARMM36 maintains liquid-crystalline phase.98 Reprinted with permission from ref 98. Copyright 2011 American Chemical Society.
As of 2006, the only lipid force fields commonly in use were the CHARMM AA and Berger UA force fields.104 Berger lipids,99 as well as the later Stockholm Lipids (Slipids)105,106 and additional force fi supporting lipids,107 have been used in conjunction with the AMBER, CHARMM, and OPLS force fi representing the protein portion. Berger lipids are UA lipid models, but have also been used with AA models.108,109 Use of UA was originally attractive because it resulted in a 60% reduction of pairwise interactions to be calculated. Studies have examined protein-lipid interactions for Berger lipids with other protein force fields,104,109 with overestimation of interactions between protein and lipid tails found for one pairing.104 A 2016 study which compared CHARMM36, Berger, and Slipids in studying a microbial peptide found that membranes with Berger lipids were prone to pore formation, an effect not found in the newer CHARMM and Slipids.110
AMBER force field lipid parameters have also been improved in recent years. The Lipid11 modular lipid force field for AMBER111 was developed using the General AMBER Force Field (GAFF112). It is designed to function more in the manner of a protein force field; rather than developing parameters for entire lipids, parameters were designed for head groups and tails which could then be combined.111 Use of an additional surface tension term was required for Lipid11 in order to prevent a phase transition during simulations. The Lipid14 updated modular force field no longer required this additional term.113,114 Lipid headgroup and tail charges were modified as well as Lennard-Jones and torsion parameters for alkane chains.113 The first systematic parameterization of lipid parameters in OPLS-AA was provided at the same time Lipid14 was released.115,116 In the initial parameterization effort, lipids showed a transition to crystalline phase at temperatures above the main phase transition temperature, but reparameterization of the hydrocarbon torsional potentials and Lennard-Jones parameters resolved this issue.116
One significant approximation made by both UA and AA force fields is that atoms operate as interaction sites with fixed charges. In reality, these atoms are electronically polarized. Polarizable force fields such as DRUDE, in comparison, include polarization effects. Models used to incorporate these effects include the inducible point dipole model and the fluctuating charge model.117 Early versions of polarizable force fields were implemented in the 1970s118,119 and began to be studied “intensively” in the 1990s.117 The multipole-based polarizable force field AMOEBA was found to provide improved descriptions of structural and thermodynamic properties of peptides and proteins.120,121 The DRUDE force field has been successfully implemented for both proteins122 and zwitterionic PC and PE lipids123,124 for simulations up to μs in length.125 While DRUDE-polarizable CHARMM has been used to study membrane interactions of ionizable arginine sidechains,126 polarizable force fields are yet to be extensively used to study lipid-protein interactions.
The steady increase in available computing power and development of more efficient algorithms, AA simulations are now routinely performed up to μs in length and accurately predict a variety of phenomena, including sidechain-lipid interactions. Slow processes, however, continue to pose challenging cases to AA simulations. For example, large conformational changes involved in the mechanisms of membrane transporters often take place over μs or longer. Processes such as protein complex formation are still too slow to produce replicate data sets. Due to the expense of AA simulations, non-equilibrium and biasing methods such as steered molecular dynamics (SMD)127,128 have been developed to allow for more extensive sampling of slow events.
2.3. Coarse-grained and Multiscale Simulations
Even though AA simulations have been successful in addressing a wide range of biological questions, their application is still limited to relatively small simulation systems and to fast biological processes.129 This led to the development of coarse-grained (CG) methods, which assume various levels of reduced representations of the molecular system to enhance the computing efficiency, thus allowing the investigation of much larger systems and longer timescales.130–138
During the past few decades, several CG models employing different force fields and sampling schemes have been developed, aiming at reducing the number of degrees of freedom of modeled systems and allowing for longer timesteps in MD simulations. For proteins, one extreme example of the simplified models is the simple lattice HP model which contains only two types of beads, representing the hydrophobic and polar amino acids, respectively.139,140 Structurally, more realistic models such as SICHO141 were designed to replace each amino acid side chain with the corresponding pseudo-atom bead. Although these early studies featured with crude representations of low resolution lack accuracy, they provided a strong foundation for the development of more accurate CG models. For example, using one or two pseudo-atoms to approximate the geometry of amino acid backbones and side chains, intermediate resolution models such as CABS142 and UNRES143 provide more realistic representation of protein structure and enable the characterization of more protein features. More recently, by introducing only a subtle level of simplification, the PRIMO model144,145 developed by Feig and coworkers allowed for a high-resolution representation of proteins closer to atomistic level, while gaining noticeable simulation speedup in comparison to AA simulations. More importantly, the PRIMO model enables productive studies of membrane protein dynamics in implicit membrane environments.146 Another high-resolution CG model that is widely used is the Rosetta model147 developed by Baker and coworkers, which combines initial CG modeling with atomistic refinement and is specifically designed for protein structure prediction.
The MARTINI model148,150–152 (Figure 8A) developed by Marrink and coworkers is the most popular CG model for membrane simulations. Its scope was extended from the original focus on lipid molecules to other biological molecules such as peptides and proteins, and has been continuously developed to be implemented in various simulation packages to investigate diverse aspects of protein dynamics and lipid-protein interactions.153–157 The MARTINI model uses a four-to-one mapping scheme, with each pseudo-atom bead of protein/lipid, water, or ion representing approximately four heavy atoms and the associated hydrogens, four water molecules, and an ion with its fi hydration shell, respectively. This straightforward mapping scheme of MARTINI enables effective conversion of simulation systems between the AA and CG resolutions. CG models remove the fastest vibrational degrees of freedom (hydrogen bond vibrations), thus smoothing out the energy landscape in comparison to their AA counterparts. The smoothed energy landscape effectively enhances the sampling of conformational space, making CG modeling a promising tool for quantitative characterization of complex molecular processes such as lipid-protein interactions (Figure 8B). While speeding up the sampling of the configuration space is the main advantage of CG models, the speed-up is not uniform for all degrees of freedom, hindering the calibration of effective timescale of CG simulations.158 Furthermore, the reduction of degrees of freedom also affects the thermodynamic properties of a modeled system, particularly shifting the balance between enthalpy and entropy. In other words, although the free energy differences may be accurately estimated by a CG model, the enthalpic and entropic contributions may not be accurate.157
Figure 8.
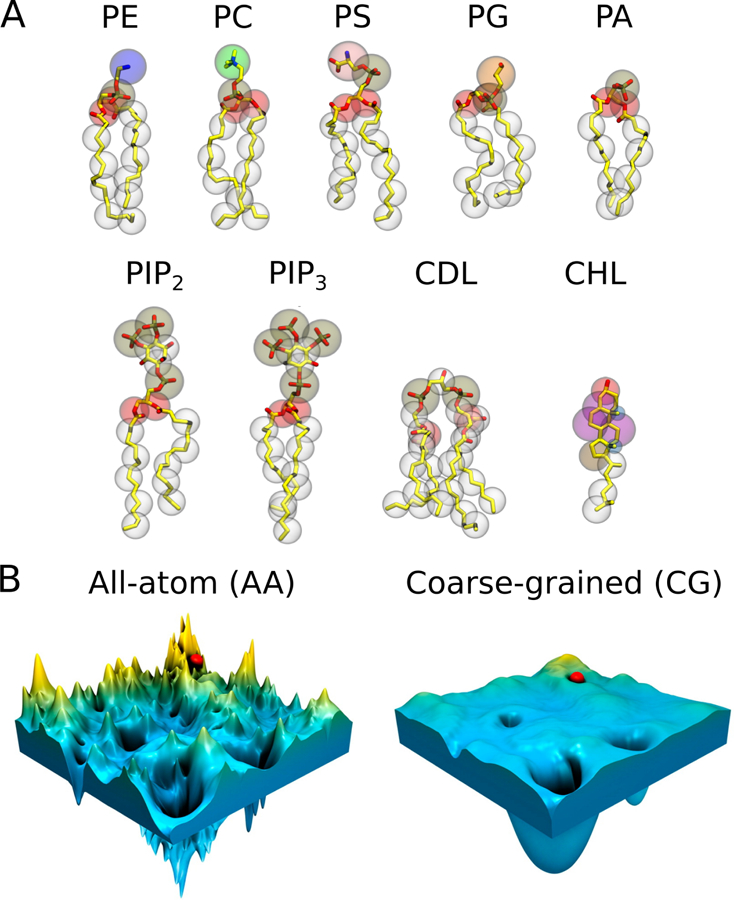
(A) CG representations of common lipids in MARTINI,148 overlaid on the corresponding AA topologies (hydrogen atoms are omitted for clarity). The CG beads are shown as transparent vdW spheres. Each bead represents about four heavy atoms with the associated hydrogen atoms. (B) Schematic illustration of the rugged and complex energy landscape of an AA model (left) compared to the smooth surface in a CG model (right). The smoothening of the CG energy landscape helps to avoid trapping in the local energy minima while searching for the global minimum. Adapted with permission from ref 149. Copyright 2016 American Chemical Society.
The MARTINI CG model has proven powerful in studying the interactions between membrane proteins and lipids, e.g., in proper placement of lipids around integral proteins (Section 3), association of peripheral proteins to the membrane surface (Section 4), and the detection of specific lipid binding sites on proteins (Section 5). MARTINI is also able to model oligomerization and aggregation processes of membrane proteins, which are sometimes mediated by specific lipid molecules in the membrane.
Structure prediction studies showed that the accuracy of CG models can be significantly improved by refining the final structures with AA simulations.159,160 This strategy thus has been applied in many studies, and was often called “multiscale modeling” in that CG simulations were first used to facilitate the modeling efficiency and AA simulations were then followed to gain more accurate details. The reconstruction of AA models from CG models involves backbone rebuilding and side chain adjustment, which have been realized by many programs employing diverse algorithms.149
A real multiscale model should allow the coexistence of multiple resolutions in the same simulation system simultaneously and reliable algorithms to enable the transfer of information between the resolutions.161–166 Multiscale modeling benefits from the efficient computing of the CG part of the system, while preserving a higher level of details for other parts when needed. This feature makes multiscale approaches powerful and appealing tools in characterizing diverse biological systems. One important application of multiscale modeling is the investigation of interactions between membrane proteins and the surrounding solvents and lipids. As an example of a multiscale approach, in the PACE model,167–169 proteins are represented by united atoms while solvent and lipids are described by CG particles.
Different flavors of multiscale models have been developed, with the majority defining fixed boundaries between different resolutions. The adaptive resolution method, however, allows a selected part of the system to change the granularity during the simulation.170–175 One representative method developed recently is AdResS,175 which contains a strict atomistic region, a strict CG region, and an interfacial region that allows for particle exchange between different resolutions. The method offers atomistic-level description at the active sites and a desirable CG resolution for the remaining simulation region. The integration and exchange of information between the different resolutions remain the key limiting factors for multiscale modeling, and better and faster algorithms that allow for more efficient and reliable, on-the-fly exchange of resolutions are still in great demand.
2.4. HMMM Simulations
Lipid diffusion at ambient temperatures is on the order of 10−8cm2s−1. As AA simulations of membrane proteins are typically run in the 100 ns–1 μs range, orders of magnitude greater sampling would be required to allow diffusive equilibration to occur. This makes many membrane-associated processes, such as spontaneous lipid mixing and membrane insertion of peripheral proteins, difficult to simulate. The highly mobile membrane mimetic (HMMM) model was developed to accelerate lipid diffusion while maintaining atomic-level details for the headgroup region (Figure 9B).176 HMMM uses lipids that are identical to AA lipids except that the acyl tail is truncated. The space between the truncated lipid tails is then filled with an organic solvent such as DCLE (dichloroethane) to reproduce some of the characteristics of the hydrophobic core of a lipid bilayer.176
Figure 9.

Spontaneous binding and insertion of the factor VII GLA domain to anionic membranes captured by HMMM. (A) The binding of GLA domains (purple trace) to PS headgroups is mediated by bound Ca2+ ions (purple spheres) and basic sidechains (green licorice). Reprinted Cover Image with permission from ref 176. Copyright 2012 Elsevier. (B) Diffusion of lipid phosphorus atoms in a 10-ns simulation of a full DOPS lipid bilayer (left) with a 1-ns simulation of an HMMM PS lipid bilayer (right). Despite an order-of-magnitude shorter simulation, the HMMM model captures much larger lateral diffusion and mixing of lipids. Reprinted with permission from ref 176. Copyright 2012 Elsevier.
It was demonstrated that a triphasic system containing truncated PS lipids, DCLE solvent, and water would spontaneously form a core layer of DCLE with lipids at the perimeter and water in the external region.176 The spontaneous bilayer formation occurred within 20 ns, with over 80% of initially water-submerged lipids reaching the DCLE interface within 10 ns.176 Five systems were tested with area per lipid ranging from 68 Å2 to 294 Å2, yielding lipid diffusion constants at least 10 times that of full-length lipids.176 Structural analysis showed that the HMMM model reproduced key membrane elements, such as degree of hydration and counter-ion penetration.176 To test efficacy of the method with a protein system, the coagulation Factor VII GLA domain, which had previously been studied using AA simulation with the goal of understanding its membrane binding,177 (Figure 9A) was chosen. In AA simulation, however, it was not possible to model spontaneous binding of the protein. Using HMMM, the GLA domain bound spontaneously to PS-HMMM membranes in ten independent, resulting in a converged model of the membrane-bound GLA-domain.176
Since the introduction of the HMMM model, the method has been applied to a wide variety of membrane-associated systems.176,178–197 Furthermore, the energetics of amino acid partitioning into the bilayer were assessed.181 PMFs calculated for sidechain insertion into the interfacial region showed that HMMM reproduced accurate results of AA and CG PMFs obtained for insertion into full lipid bilayers.181 The HMMM with a DCLE core, however, did not accurately describe core energetics for protein partitioning.181 HMMM has been integrated into the CHARMM-GUI input generator, allowing for convenient generation of HMMM bilayers with a variety of lipid compositions.190
HMMM has been used to study a wide variety of membrane proteins, including coagulation factor GLA domains,189 talin,182 and cytochrome P450.178 It has also been employed to study transmembrane domains183 and the insertion process of lipids into the membrane.180 Use of HMMM has allowed for extensive sampling of lipid-protein interactions following spontaneous binding of proteins with a high degree of lipid specificity,189 something that would be difficult to sample with full lipids in either AA or CG simulations.
While HMMM was highly proficient in modeling of peripheral proteins, energetic differences between DCLE solvent and a natural membrane core region can result in deformations of complex, multi-helix transmembrane proteins.193 Interactions between protein sidechains and the solvent are overly favorable, resulting in intercalation of solvent molecules between protein structural elements.193 Vermaas et al, attempting to overcome this difficulty, developed in silico solvents for use in HMMM simulations. These molecules, which do not exist in physical reality, are custom-parameterized to mimic the membrane core while retaining liquid properties.193 The solvents were demonstrated to allow for improved simulation of transmembrane proteins in HMMM, although some problems still remain.193 Currently, HMMM still has limited ability to simulate transmembrane proteins with multiple transmembrane helices. Furthermore, HMMM cannot be used to accurately represent energetics and processes at the membrane core, as the properties of core solvents still differ significantly from those of the acyl chains in the lipid bilayer core. In addition, certain types of lipids, such as sphingolipids and sterols, have not been tested in HMMM to ensure it reproduces characteristics of full membranes composed of these lipids.
2.5. Membrane Embedding Methods
Prior to simulating a protein-lipid system, proteins must be placed in or on the membrane. While the process of membrane binding and insertion can, in principle, be done by performing long MD simulations, especially for peripheral proteins, it is often too slow and has to be done in advance. Due to the heterogeneous nature of biological membranes, alternative approaches such as implicit solvent/membrane models are rather rudimentary, lacking crucial information such as water-protein and lipid-protein interactions.198 For explicit lipid membranes, complications such as membrane curvature and undulation, lipid entanglement and protein structural changes may simply be artifacts caused by poor placement of the protein, rendering the necessity to properly prepare the initial system. A variety of techniques have been developed to construct membrane-embedded protein complexes; some of them use MD as a method to refine the placement along the process.199–203
2.5.1. Methods to Predict Protein Position in a Lipid Bilayer
As different proteins have different shapes, amino acid compositions, and membrane insertion depths, visual inspection alone may not be enough to correctly identify hydrophobic belts or protein sections exposed to the hydrophobic part of the membrane. Spatial arrangement of proteins in membranes can be predicted using available algorithms whose results agree well with experimentally determined tilt angles within a particular membrane thickness.204–207 PPM (Position of Proteins in Membrane)205 is one of the popular resources for rapid evaluation of the positions of transmembrane and monotopic proteins in a lipid bilayer and is also available through a web-based interface.204,208 In this method, a lipid bilayer is represented by a hydrophobic slab and its interfacial regions. PPM performs grid-based scanning to minimize the global transfer energy of a protein, treated as a rigid body, from water to the hydrophobic core of the bilayer. A very similar algorithm and a predecessor of PPM is IMPALA (Integral Membrane Protein and Lipid Association), which performs energetic optimization of protein’s position in a bilayer composed of lipid acyl chains.207,209
Other commonly used algorithms are MEMEMBED210 and LAMBADA.211 MEMEMBED uses direct search and genetic algorithms to align α-helical and β-barrel transmembrane proteins to a model membrane.210 LAMBADA performs grid-based scanning to search for energetically minimal protein positioning determined by hydrophobic scores,211 and provides an input for InflateGRO, an automatic membrane embedding tool (described below).
2.5.2. Methods to Assemble Proteins in a Lipid Bilayer
Once the position and orientation of the protein in the membrane are approximated, it can be translated into a membrane patch. An immediate problem is the collision between lipid molecules and the protein, which may not be resolved by simple energy minimization protocols.199 Overlapping lipid molecules can be identified and deleted using commonly used molecular viewers, such as VMD,214 CHIMERA215 and PyMol (Figure 10A). Then, MD simulations can be performed to optimize lipid packing around the protein. Still, this simple procedure may result in large gaps in lipid packing as most proteins contain large degrees of asymmetry in their lateral surface area along the membrane normal.216
Figure 10.
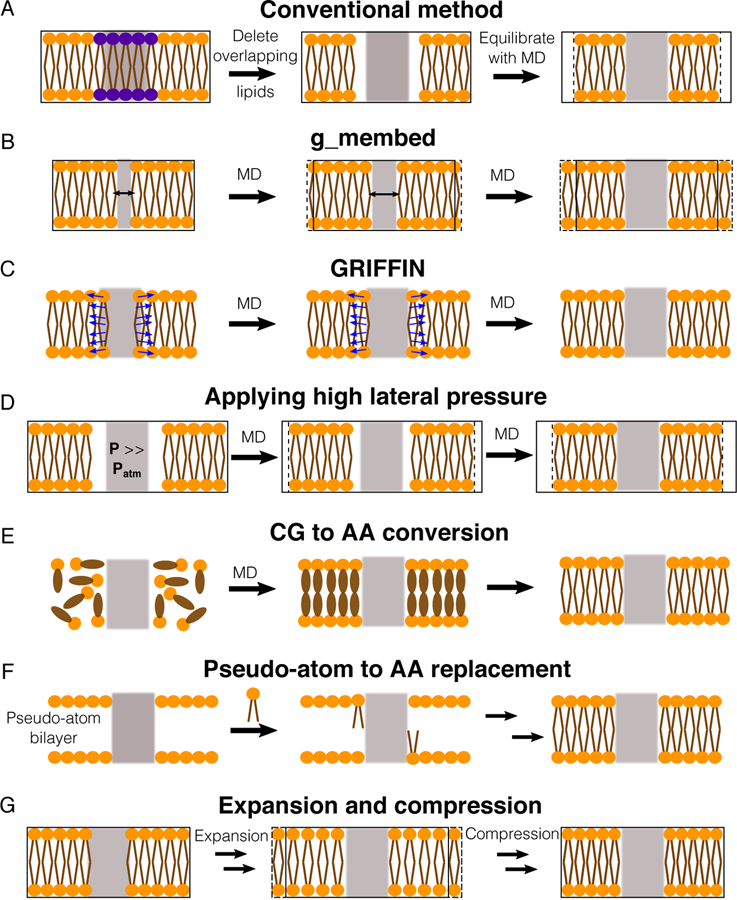
Methods for assembling proteins in membranes. Proteins, lipid head groups and lipid tails are represented by gray rectangles, orange circles and brown lines respectively. Black-solid boxes represent the original dimension of a simulated system, whereas dashed boxes represent changes during the optimization process. (A) The simplest way to optimize lipid packing is to delete lipid molecules colliding the protein and then perform an MD simulation until the system reaches optimal dimensions. (B) g_membed applies a repulsive force to gradually grow the protein to its targeted dimension.201 (C) GRIFFIN applies a repulsive field to carve out lipid molecules inside the protein grid.202 (D) A simulation, in which a high pressure is applied to laterally swallow the protein in the bilayer.203 (E) Flooding simulations can be used to probe lipid binding sites at a CG level first and then transform the assembled complex into an AA model.212 (F) One approach is to first place pseudo atoms or beads of targeted lipid types in a bilayer encompassing the protein according to their cross-sectional areas and then replace the beads with lipid conformations selected from previous MD simulations.72,79,213 (G) Another approach is to perform a series of expansion and compression simulations of the membrane and scaling of lipid size.199
Other MD-based protocols have been used for assembling a protein in a membrane. The tool g_membed201 applies a repulsive force to create a hole at a designated position of the membrane embedded protein, and then gradually grows the protein from its originally scaled down representation to its real dimension while pushing away overlapping lipid molecules (Figure 10B). Another method, named GRIFFIN (GRId-based Force Field INput),202 adds a repulsive field to the membrane section occupied by the protein to carve out an empty volume needed for optimal protein placement (Figure 10C). Besides the use of repulsive forces, the assembly process can be done through pressure-induced simulations. In an approach proposed by Javanainen,203 a simulation is carried out in vacuum, with positional restraints applied to the protein and normal restraints applied to maintain the geometry of the lipid molecules, under a high pressure (up to ~1,000 atm) to push the protein into the bilayer (Figure 10D). As many proteins interact with specific lipids, the assembly of a protein in a membrane constituted of multiple lipid types requires more attention as lipid binding affects the protein structure. A conventional approach to probe lipid binding sites is to flood the system with CG lipid molecules. MemProtMD212 is an automated pipeline, which performs a 1-μs flooding simulation of CG lipid molecules to assemble a CG protein-embedded membrane and then converts the generated complex to an AA model (Figure 10E). A major problem resulted from the conversion of low (CG) to high (AA) resolution models can be the potential entanglement between proteins and lipids (e.g., ring piercing between lipid acyl chains and aromatic amino acids), which can be solved by applying alchemical soft-core potentials to the affected molecules.217
Automatic and more systematic methods are being used in constructing a protein-embedded membrane complex. CHARMM-GUI213,218–220 is the most widely used automatic builder for assembling membrane proteins and complex membrane systems, including bacterial outer membranes.221 This builder aligns the protein in a membrane using coordinates retrieved from the OPM (Orientations of Proteins in Membranes) database204,208 or provided by users. Lipid molecules are then placed according to the cross-sectional areas of the protein and the lipid molecules. Following the protocols developed by Woolf and Roux72,79 (Figure 10F), the lipid assembly is done by first placing pseudo atoms (large vdW spheres) representing lipid molecules around the protein and then substituting those particles with the lipid molecules randomly selected from a library of lipid conformations collected from MD simulations.
Another automatic builder is InflateGRO,199 which is implemented in GROMACS and used in conjunction with LAMBADA211 (Figure 10G). Once the protein is aligned with a pre-built membrane, lipid molecules within a defined lipid phosphorus-protein Cα distance cutoff are deleted. To completely remove clashes between lipid molecules and the protein, InflateGRO performs a series of lateral expansions of the membrane to allow the translation of the colliding lipid molecules. It then performs a series of compressions and energy minimizations to bring the membrane back to its original dimension to accommodate lipid packing.
3. Functional Lipid-protein Interactions in Integral Membrane Proteins
Integral membrane proteins span the lipid bilayer with at least one transmembrane domain. They constitute an integral component of biological membranes and are involved in a wide range of important biochemical and physiological processes, such as energy transduction, neuronal communication and immune response, making them critical drug targets for a variety of diseases.222–225 Some major classes of integral membrane proteins include channels, transporters and receptors, whose structure and function are tightly associated with their lipid environments.
Channels and transporters facilitate the passage of chemical species, particularly polar and charged molecules, across the hydrophobic core of the membrane.226–231 Channels are modulated by various membrane-associated factors, including the membrane potential, ligand binding, and mechanical stress of a local membrane environment. Structural transitions are crucial for the activation (gating) of channels, upon which the open pore allows for rapid permeation of substrate molecules sometimes at rates close to the diffusion limit. For transporters, the turnover rate is much slower than channels, due to more pronounced conformational changes involved in their functional cycle. The transition between structural conformations typically involve large movements of transmembrane domains, which could be regulated directly by their interactions with the surrounding bilayer or specific lipids. Receptors are responsible for the recognition and transmission of chemical signals, and their activation upon extracellular ligand binding is key to numerous physiological pathways. Lipid-receptor interactions not only can affect the activation cycle but also the stability and oligomerization of receptors.
MD simulations over the past few decades have successfully characterized structural dynamics and functionally relevant mechanisms for integral membrane proteins of various sizes, shapes and originating organisms.232,233 Many structural and physicochemical aspects of lipid bilayers and specific lipids have been studied computationally, permitting the investigation of interactions between membrane/lipids and important integral proteins. In this section, we provide an overview of the application of MD simulations to the investigation of lipid-protein interactions and lipid-mediated effects on the integral proteins. Some of the most successful applications recently achieved by the combination of hybrid simulation methods or advanced computational techniques, such as free energy calculations or integration of experimentally derived restraints, will be highlighted and discussed in more details.
3.1. Membrane Channels
Membrane channels are transmembrane proteins that facilitate the permeation of various chemical species down their electrochemical gradients across the biological membranes.222,234 Membrane channels are fundamentally important and play key roles in a wide range of cellular and physiological events, such as propagation of electrical signals, neuronal communication, muscle contractions, and apoptosis. Channel gating is regulated by various factors, such as transmembrane voltage, chemical stimuli, and membrane tension. Upon stimulation, channels undergo conformational changes from a closed state to an open state (gating), allowing substrates to flow down their electrochemical gradients. The structures of a membrane-embedded channel and the conformational changes associated with its gating can be directly affected by the surrounding lipids in various ways.
Simulation studies exploring the protein surface hydrophobicity and protein/lipid interfaces have shed light on the importance of membrane structure and thickness on the function of membrane channels. In addition to bulk properties of the membrane, specific interactions with lipids can affect membrane channels. Lipid bilayers not only provide the environment necessary for channels to function properly, but also serve as the medium for small molecules to approach and interact with them. Here, we will review the lipid-protein interactions and lipid-modulated impacts on protein function that have been reported in computational studies of membrane channels. The channels discussed in this section (Figure 11) will be classified into three major categories, based on their gating mechanisms: voltage-gated channels, ligand-gated channels, and mechanosensitive channels. Computational studies on other types of channels such as outer membrane proteins, aquaporins, and phospholipid scramblases will be covered at the end of this section.
Figure 11.
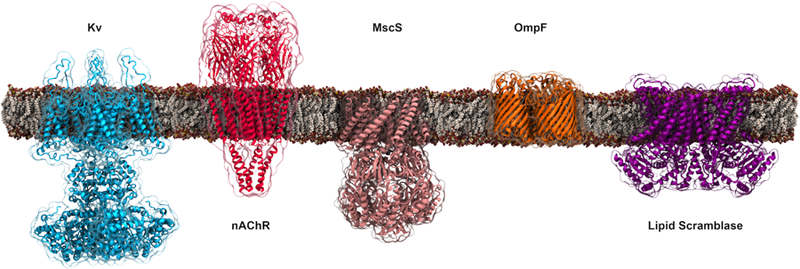
Representative membrane channels covered in Section 3.1. The channels shown from left to right are the voltage-gated potassium channel (Kv, PDB:3LUT) that opens and closes in response to changes in membrane potential; the nicotinic acetylcholine receptor (nAChR, PDB:4BOI), a pentameric ligand-gated ion channel; the mechanosensitive channel of small conductance (MscS, PDB:2VV5) that functions as a pressure relief valve and regulated by membrane tension; the bacterial outer-membrane porin (OmpF, PDB:2OMF) that aids the diffusion of small hydrophilic molecules across the outer membrane of Gram-negative bacteria; and, the fungal phospholipid scramblase (PDB:4WIS) that facilitates the transmembrane movement of phospholipids in an ATP-independent manner.
3.1.1. Voltage-gated Channels
Voltage-gated channels mediate the transmembrane movement of ions in response to the changes in the electrical membrane potential. These channels generally contain four homologous domains/subunits with an ion conduction pore formed along the 4-fold symmetry axis. Each of the four domains/subunits comprises six transmembrane α-helical segments, named S1-S6, with S1-S4 contributing to voltage sensing and S5-S6 forming the pore.235 Voltage-gated channels are generally ion-specific and are involved in the conduction of various cations and anions, such as K+, Na+, Ca2+, and Cl−, crucial for the propagation of electrical signals in excitable cells.222 Computational studies on voltage-gated ion channels have elucidated important aspects of protein dynamics modulated by membrane environments and/or specific lipids.
Voltage-gated K+ (Kv) channels are widely distributed and found in virtually all living organisms and most cell types, where they control a wide variety of cellular functions.234 Several previous computational studies have investigated lipid interactions with the isolated pore domain or voltage sensor domain of Kv channels, providing valuable structural and functional information for more comprehensive studies at a later stage. The structure of bacterial KcsA, which serves as an archetypical pore domain of the Kv members, was used to explore the lipid-protein interactions in early simulations (on the order of tens of nanoseconds).236–238 These simulations revealed not only interactions between surface residues and boundary lipid headgroups, but also specific binding of anionic lipids at the interfacial binding sites between the adjacent subunits. Strong binding of PG lipids to the same sites was also captured in sub-millisecond CG simulations of KcsA in a PC/PG lipid mixture.239 In the KcsA-Kv1.3 chimera, a point-mutation at the corresponding nonannular lipid binding site led to the formation of a salt bridge between its adjacent subunits, which resulted in reduced binding of anionic lipids.239
One of the most exciting features of Kv channels, which attracted a large body of computational studies, is the mechanism of voltage gating. The opening and closing of the pore domain are coupled to the movement of the voltage sensor domain that contains the voltage-sensing basic residues in the S4 segment. Simulations of the Arg-containing short peptides as well as the whole S4 helix within a PC bilayer exhibited a tilting motion relative to the membrane in response to the changes in the external electric field 241 Free energy calculations suggested that membrane insertion of S4 is thermodynamically favorable because the energy gain from shielding the S4 hydrophobic residues from water is larger than the free energy penalty for inserting the charged residues into the hydrophobic core of the membrane.242 The calculations also indicated that the free energy penalty of charge insertion was reduced by membrane deformation that enables the penetration of water molecules into the hydrophobic core to provide a polar micro-environment for the charged residues.242 Membrane thinning and local adaption of the lipid bilayer were also observed for simulations of the complete voltage sensor domain S1-S4 from KvAP, which allows water molecules to hydrate the charged residues and focus the transmembrane electric field 243 Self-assembly CG simulations of various voltage sensor homologs revealed similar interactions with lipid phosphate groups as well as local distortions of the lipid bilayer, providing insight into the molecular basis underlying their stability within the membrane.244
More importantly, AA simulations of the eukaryotic Kv1.2 showed that the interactions between the lipid phosphate groups and the S4 basic residues not only stabilize the channel conformation,245,246 but also play a role in modulating the gating process during the S4 transition under transmembrane potentials240,247 (Figure 12). Another study using a PIP2 (phosphatidyl-4,5-bisphosphate; (PI(4,5)P2) containing PC bilayer demonstrated state-dependent interactions between the anionic phospholipid in the inner leaflet and the basic residues of Kv1.2.248 Even though PIP2 exerts different effects on KCNQ channels compared to Kv1.2, state-dependent interactions between PIP2 and KCNQ channels were also observed in multiple simulations, suggesting a functional role of PIP2 in mediating effective coupling between the voltage sensor domain and the pore domain, and regulating the protein conformational transitions249–251 (Figure 13). CG simulations combined with patch-clamp measurements and site-directed mutagenesis also revealed the presence of a specific phosphatidyl-3,5-bisphosphate (PI(3,5)P2) binding pocket on the PI(3,5)P2-activated two-pore channel hTPC2, which is suggested to mediate coordinated movement during channel gating upon binding of the lipid.252 In addition to phosphoinositides, microsecond AA simulations also identified a putative binding site for a negatively charged polyunsaturated fatty acid (PUFA), an essential component of heart and neuronal cellular membranes, on the open state Shaker Kv channel, providing a structural framework for testing the modulatory role of PUFA on K+ channels.253
Figure 12.

Representative conformations (α, β, γ, δ, and ε) of the Kv1.2 voltage sensor domain revealed during the unbiased and subsequent biased-MD simulations. (A) Molecular views of the five key conformations highlighting the positions of the S4 basic residues (blue sticks) and their binding sites (acidic residues: red sticks, lipid : yellow vdW) during the gating transition. (B) The closest interacting partner with each of the S4 basic residues in the five conformations. Lipid groups were involved in providing counter-charges for the S4 basic residues during the gating process. (C) Positions of the S4 basic residues R1 (black) through R6 (orange) with respect to the membrane midplane (z=0) for each intermediate conformation. Adapted with permission from ref 240. Copyright 2011 Delemonte et al.
Figure 13.

PIP2 molecules access different regions of the KCNQ2 channel depending on protein conformations. (Top) Trajectories of PIP2 illustrating the preferential binding of the lipid molecules to the S4-S5 linker of the open channel (left) and the S2-S3 loop of the closed channel (right), respectively. Reprinted with permission from ref 250. Published by National Academy of Sciences. (Bottom) The schematic model proposed based on the simulations shows that in the closed state, PIP2 is anchored at the S2-S3 loop (right); upon channel activation, PIP2 interacts with the S4-S5 linker and is involved in channel gating (left). Adapted with permission from ref 251. Copyright 2015 Chen et al. Licensed under a Creative Commons Attribution 4.0 International License.
Voltage-gated Na+ (Nav) channels mediate the upstroke of the action potential in most excitable cells and are key targets for numerous anesthetic agents.254 Given significant degrees of structural similarity shared by members of the voltage-gated cation channel (VGCC) family, the gating mechanism of bacterial NavAb channel was investigated computationally taking advantage of the structural information from Kv1.2.255 Biased simulations driven by the template structural models of Kv1.2 provided adequate sampling of NavAb conformations along the activation pathway, and revealed the important role of lipid phosphate groups in coordinating the S4 basic residues during the conformational transitions.255 Moreover, multi-μs unbiased simulations led to the determination of distinct binding sites for lipophilic drug molecules benzocaine and phenytoin to NavAb via two drug-access pathways.256 The lipophilic pathway through the membrane-embedded fenestration was shown to be a low free energy pathway compared to the alternative aqueous route.256 Equilibrium flooding simulations, where a high copy number (concentration) of the ligand is introduced into the simulation system but without applying external biases, also identified putative binding sites and access pathways for general anesthetics isofl and sevoflurane to another archetypical bacterial channel NaChBac, highlighting the importance of the fenestration pathway for drug access.257,258 In addition, AA simulations of six bacterial Nav channels showed that the lipid molecules protruding the fenestrations can displace the side chains of the bottleneck residues and influence the size of the fenestrations.259
Transient receptor potential (TRP) channels are a diverse set of non-selective cation channels that respond to a plethora of physical and chemical stimuli.260 Their overall transmembrane architecture resembles that of the canonical Kv channels.261 AA simulations of the voltage-sensor-like domain of the heat-sensitive TRPV1 captured spontaneous binding of the lipophilic ligand capsaicin from the cytosolic aqueous phase to a membrane-embedded site, implicating the role of the membrane in mediating the effect of the channel-activating ligand.262 Simulations of another heat-sensitive member, TRPV4, showed that the hydrogen bond that secures the protein in a closed state can be counteracted by the interaction of the surrounding lipids, thus increasing the open probability of the channel.263 In addition, an altered pattern of interaction with lipids was captured in simulations of a mutant of TRPV5 compared to the wildtype protein, which may contribute to the experimentally observed disruption of the ion transport in the mutant.264
3.1.2. Ligand-gated Channels
The Cys-loop superfamily of the pentameric ligand-gated ion channels (pLGICs) are anesthetic-sensitive receptors that act in response to release of neurotransmitters from the presynaptic terminal. They are composed of five homologous subunits, with each consisting of a large extracellular domain and four transmembrane segments (M1-M4).265 Tens of nanosecond AA of simulations of the transmembrane domains of nicotinic acetylcholine receptor (nAChR), the prototypical cation channel of this superfamily, captured spontaneous membrane partitioning of the volatile anesthetic halothane from solution into a hydrophobic cavity near the M2-M3 loop, one of the experimentally reported sites for anesthetic binding.266 The binding of halothane was suggested to play a role in channel inhibition by altering the dynamics of the M2-M3 loop, which is implicated in transmitting the effect of the anesthetic to the channel gate.266 In addition, sub-μs AA simulations of intact nAChR and its prokaryotic homolog GLIC (Gloebacter violaceus ligand-gated ion channel) revealed membrane partitioning as well as binding of general anesthetic isoflurane to both the transmembrane and the extracellular domains, in a remarkably similar manner between the two proteins.267 Moreover, high concentrations of desflurane employed in the simulation systems led to the identification of a novel anesthetic binding site in GLIC, accessed via a membrane-embedded tunnel (Figure 14).268,269 Ligand binding at this site inhibited the dissociation of anesthetic from a site previously known, resulting in conformational changes that produce a non-conductive state of the channel.268,269 In addition to serving as the medium for ligand binding, lipids were also observed to specifically interact with the pLGIC channels to potentially influence the allosteric modulation of GLIC270 and the resting state of nAChR.271
Figure 14.

Membrane partitioning and the facilitated binding of anesthetics to the modulation sites of GLIC during the flooding simulations. (A) Time series demonstrating the membrane partitioning of desflurane during the flooding simulations, in which a high copy number of drug molecules were initially placed randomly in solution. (B) Spontaneous binding of desflurane to the membrane domains of GLIC, following its rapid partitioning into the membrane. The desflurane molecule forms several non-specific contacts within the binding site (top inset), which is located near the same region within the membrane where desflurane partitions (bottom inset). Reprinted with permission from ref 269. Copyright 2016 Elsevier.
Inward rectifying K+ (Kir) channels are regulated by the signaling anionic phospholipid PIP2.272,273 In addition to the similar tetrameric architecture observed in other K+ channels, Kir channels also contain a large cytoplasmic domain that not only extends the central ion pore but also plays a role in gating upon ligand binding.274 Early homology modeling and short simulations of mammalian Kir channels allowed for the exploration of general protein dynamics and contacts with the surrounding lipids,275 as well as the docking of PIP2 near the “slide helix” at the cytoplasmic surface of the membrane.276 Spontaneous binding of PIP2 to a similar site was observed in combined CG-AA simulations on three different Kir structures, obtained either from crystallography or by homology modeling.277 Moreover, combined CG-AA simulations starting with apo structures of the eukaryotic Kir2.2 successfully reproduced the binding of PIP2 to the same cluster of basic residues as observed in the PIP2-bound crystal structure.278 The PIP2-bound structure of Kir2.2 was also used to build a homology model for sponge channel AqKir, a distant relative of the vertebrate members that interacts weakly with PIP2 due to the lack of two basic residues in the PIP2 binding site.279 Evaluation of interaction energies showed that restoring the two positive charges by mutations greatly favors the interaction with PIP2 as compared to wildtype AqKir.279 Furthermore, free energy calculations of PIP2-Kir2.2 interactions indicated that neutralizing the binding site residue or PIP2 phosphate charges greatly weakened the interactions, highlighting the role of electrostatics in lipid-protein interactions280 (Figure 15).
Figure 15.

Free energy landscape of PIP2-Kir2.2 interaction. (A) Replica exchange umbrella sampling along a collective variable defined by the distance between the PIP2 headgroup and the center of mass of the interacting Kir subunit. Representative snapshots of the initial configurations in the PIP2-bound and unbound conditions are shown (PIP2: red, Kir2.2 interacting subunit: blue). (B) Two independent sets of simulations, initiated either from the PIP2 bound (blue) or unbound (orange) states, provided convergent results for both the wildtype protein and the R186A mutant (inset) with reduced PIP2-binding affinity. Adapted with permission from ref 280. Copyright 2017 American Chemical Society.
Beyond the binding of PIP2, simulations of the closed state Kir3.1 chimera revealed that the PIP2-driven conformational change of the cytoplasmic domain dilates the G-loop gate in the cytoplasmic pore and results in the formation of an intermediate state between the closed and open states.281 A follow-up study focusing on the cytoplasmic domain of Kir2 channels further identified a loop region involved in the PIP2-induced gating, whose increased flexibility by mutations directly affects its interactions with several important structural elements and thus regulates the gating kinetics of the channels.282 In addition to the cytoplasmic gate, AA simulations of Kir3.2 captured dynamic opening of the helix bundle crossing gate in the transmembrane pore upon a mutation at the PIP2 binding site, inducing tighter interactions with PIP2 compared to the wildtype structure.283 Besides PIP2, molecular docking and AA simulations identified putative binding sites for cholesterol on the transmembrane domain of Kir2.1, providing insight into the mechanism of channel inhibition by cholesterol.284
3.1.3. Mechanosensitive Channels
Mechanosensitive channels are ubiquitous across prokaryotes, archaea, and eukaryotes and implicated in a wide range of biological processes. They sense and gate in response to the mechanical stress of membrane to regulate the flow of solutes in a generally non-selective manner.285 The best studied member of this family is the bacterial mechanosensitive channel of large conductance (MscL), formed by five identical subunits around a central pore that can dilate up to 40 Å in diameter when the channel opens.286 Early multi-nanosecond AA simulations showed that the structure and dynamics of the bacterial Tb-MscL channel are directly affected by changes in lipid headgroups, with decreased number of protein-lipid hydrogen bonds upon the change of lipids from POPE to POPC.287,288 Using an analytical model developed for the bilayer mechanics, a theoretical study on bilayer deformation revealed that the deformation free energy can be on the same order as the free energy differences between the conduction states of the MscL channel, suggesting the involvement of bilayer mechanics in regulating the function of the channel.289 Furthermore, free energy calculations performed using umbrella sampling indicated that tilting the Tb-MscL transmembrane helices results in channel expansion comparable to when an excess surface tension is applied to the membrane.290 Channel opening in response to membrane tension was captured in other AA and CG simulations using either the Tb-MscL X-ray structure or the E. coli channel model, providing insight into the tension-sensing sites on the protein surface.291,292
In addition to the tension-induced gating, AA simulations of the E. coli MscL model structure in a stress-free curved bilayer showed that the asymmetrical addition of the single-tailed lipids can change the bilayer geometry which in turn affects the channel structure within a few nanoseconds.293 Hydrophobic mismatch in a thinner membrane was also shown to widen the transmembrane domains of the E. coli channel.294
The mechanosensitive channel of small conductance (MscS), which is organized as a homoheptamer, acts as a safety valve in bacteria and prevents cell lysis under challenging osmotic conditions.296 Taking advantage of available experimental data, MD simulations with EPR-derived restraints permitted the modeling of MscS in a closed conformation, a key step in determining the molecular mechanism of MscS gating.297 After building side chains to this Cα-only model structure, a follow-up study on the closed state MscS predicted several tension-sensing residues based on interaction energies between the protein and lipids, which successfully guided the experimental identification of lipid-sensing residues near the membrane interface on the extracellular side.298 More importantly, combined CG-AA simulations of the closed and open states of the E. coli MscS captured the migration of lipids into the membrane-exposed protein pockets formed by transmembrane helices, a process accompanied by strong local membrane curvature around the protein (Figure 16).295 Lipids localized in the protein pockets were found to move dynamically in correlation with the conformations of the protein, suggesting a possible mechanism of membrane tension transmission by changes in lipid-protein interactions.
Figure 16.
Lipid exchange between the membrane-exposed pockets of MscS and the bilayer upon gating. (A) Cut away slices showing the packing of lipids in the pockets during the simulations of MscS in the closed (left) and open (right) states. Lipid molecules in the lower pocket reach the pore-sealing residues in the open state, but they are blocked in the closed state (insets). (B) A schematic model derived from the simulations showing that as pressure is applied, the increased lateral tension induces lipid repartitioning (blue arrows, left panel) from the protein pockets to the bilayer, destabilizing the closed state and facilitating the formation of the open state (orange arrow, left panel). Lipid molecules inside the pockets are highlighted with a green headgroup. Adapted with permission from ref 295. Copyright 2015 Springer Nature.
Besides the prokaryotic channels MscL and MscS, computational studies of the eukaryotic mechanosensitive channels TREK-1 and TREK-2 from the two-pore domain K+ (K2P) channel family also provided structural and functional insights into the mechanism of mechanosensitivity. Sub-μs AA simulations of the TREK-1 homology models captured the adsorption of the C-terminal domain onto the membrane surface, highlighting the role of this domain in coupling membrane tension to the gating of the channel.300 A recent extensive simulation study showed that TREK-2 can expand rapidly to switch between the two main conformational states in response to bilayer stretch, a structural transition involving state-dependent changes in lipid-protein interactions (Figure 17).299 In contrast, stretch-induced conformational changes were absent in simulations of the non-mechanosensitive homolog TWIK-1 under the same conditions, suggesting the specificity of the dynamic behavior for mechanosensitive K2P channels. Even though TWIK-1 differs from the mechanosensitive K2Ps in functional and mechanistic terms, both TWIK-1 and TREK-2 simulations captured the penetration of lipid tails into the side fenestrations, which in turn influences the dewetting of the inner pore for ion conduction.299,301
Figure 17.
Bilayer stretch induced TREK-2 conformational change between the two major states. (A) A pressure jump of −80 mbar results in fully reversible currents through the reconstituted TREK-2 channel as recorded at +80 mV. (B) Simulation of membrane stretch by expanding the xy plane of the bilayer (red arrows) induces a protein conformational transition and mechanogating of K2P channels. (C) State-dependent binding of lipids near the protein fenestration. Without stretch, the fenestration is open and lipids (Fen lipids) are bound within the groove between M2 and M4 helices. When the membrane is stretched, the fenestration closes and lipids no longer bind. Adapted with permission from ref 299. Copyright 2017 Elsevier.
3.1.4. Outer Membrane Proteins
The outer membrane of Gram-negative bacteria is asymmetric with lipopolysaccharides (LPS) in the outer leaflet and phospholipids in the inner leaflet 302 The highly anionic nature of LPS deters the penetration of hydrophobic compounds and thus presents an effective barrier to many antibiotics. The outer membrane proteins (OMPs) are featured with a generic transmembrane β-barrel structure and are involved biological processes such as transmembrane transport and cell recognition.303 Inclusion of LPS in the outer leaflet of the simulation systems revealed electrostatics interactions between LPS and the extracellular loop regions of several OMPs, which led to secondary structure variation and loop displacement compared to LPS-free bilayers.304–307 Especially in the case of the trimeric porin OmpF, simulations observed interactions between the charged residues on the protein outer surface and the LPS core sugars, highlighting the importance of LPS in shielding OmpF surface epitopes from antibody recognition.306
In addition to LPS-containing systems, simulations of the OMP β-barrel domains in phospholipid bilayers of various tail lengths revealed hydrophobic mismatch induced lipid sorting or membrane disruption around the β-barrel structures.308,309 Moreover, the tilting motion of the β-barrel from the lipid A acylase PagP was found to facilitate the access of lipid acyl chains into the mouth of the central binding pocket.310 The two-domain homology model of PmOmpA is composed of a transmembrane β-barrel domain as well as a periplasmic α-helical domain.311 Simulations of this multi-domain OMP described structural dynamics of the periplasmic domain, revealing its interaction with the phospholipid headgroups on the periplasmic surface of the membrane.311 Partitioning of the periplasmic domain into the proximal membrane leaflet was also observed for BamA, providing conformations compatible with the binding of the other subunits of the BAM complex.312 This finding suggested the importance of the periplasmic domain in the mechanism of the BAM-facilitated insertion of OMPs.312
Apart from the outer membrane of Gram-negative bacteria, the outer mitochondrial membrane also contains OMPs, such as the voltage-dependent anion channel (VDAC) that allows the flow of ions and metabolites between the cytosol and the mitochondrial intermembrane space.313 Comparative modeling and MD simulations of VDAC in PC or PE bilayers suggested that persistent interactions between acidic residues and PE headgroups may be liable for the enhanced ion selectivity of VDAC observed in PE relative to PC.314 Besides phospholipids, reproducible binding of cholesterol was also observed on multiple surface sites of VDAC, which was suggested to play a role in stabilizing the charged residues inside the channel and localizing the surrounding electrostatic potentials.315
3.1.5. Other Channels
Aquaporins (AQPs) are membrane channels specialized in rapid transport of water across biological membranes.316,317 They are arranged as homotetramers, with each monomer forming a functionally independent water-conducting pore.318,319 In contrast to the previously discussed membrane channels, highly specific lipid-protein interactions were not captured in a set of 1-μs-long CG self-assembling simulations using all structurally known AQPs.320 Although water permeability of AQP4 is reported to strongly depend on the cholesterol content of the enclosing lipid bilayer, both osmotic-gradient experiments and MD simulations using cholesterol-free membrane showed that changes in permeability was caused by the cholesterol-induced changes in membrane thickness, rather than direct cholesterol-AQP4 interactions.321
Aside from the conventional role as water channels, several AQP members have been found to facilitate the conduction of small neutral gas molecules such as O2 and CO2 across the membrane. Although the physiological significance of AQPs in gas transport remains controversial, explicit gas diffusion simulation and implicit ligand sampling of AQP1 showed that the hydrophobic central pore formed at the 4-fold symmetry axis of the tetramer can be used by either O2 or CO2 to cross the membrane.322 The role of the central pore in gas permeation was further confirmed by a study on AQP5, demonstrating that the pore-occluding lipid resolved in the central pore may interfere with gas permeation but leaves the water permeation through the monomeric water pores intact.323 In addition to the central pore, simulations also identified other potential gas pathways that are energetically favorable according to the PMF calculation.322 One such pathway for AQP1 is located between the neighboring monomers near the protein-lipid interface.322
A recently emerging topic involving intimate lipid-protein interactions is the physiologically relevant phospholipid translocation mediated by the lipid scramblases, a family of passive transport proteins whose biochemical identity became known only recently. AA simulations of the fungal phospholipid scramblase nhTMEM16, which is also known to be a non-selective ion channel, captured spontaneous diffusion of lipids between the two leaflets of the bilayer via a surface-exposed hydrophilic aqueduct provided by the protein.325–327 Moreover, both MD simulations and continuum modeling demonstrated significant membrane deformation induced by the protein, which greatly decreases the effective membrane thickness near the lipid-conducting pathway and thus lowers the energy barrier against lipid translocation.325,326,328 In addition, the lipids lining the hydrophilic aqueduct on the surface of the scramblase also play a structural role in forming a ‘proteolipidic’ pore, which is likely to be also used by ions to cross the membrane326 (Figure 18).
Figure 18.
Direct involvement of phospholipids in ion translocation across the membrane, mediated by intimate lipid-protein interactions. Representative snapshots from MD simulations demonstrating: (left) the egress of Na+ through the lateral cytoplasmic fenestrations of the human P2X3 trimer lined by lipid headgroups; (right) the lipids lining the hydrophilic aqueduct on the surface of the nhTMEM16 scramblase play a structural role in forming a ‘proteolipidic’ pore for ion conduction. The lipid headgroups interacting closely with the protein and coordinating the permeating ions are shown in red with the tails drawn in yellow. Reprinted with permission from ref 324. Copyright 2018 Elsevier.
Direct involvement of lipids in ion translocation was also observed in simulations of the human P2X3 receptor, a non-selective cation channel, showing that the increased hydration brought by the lipid headgroups lining the transmembrane fenestrations of the protein can constitute a hydrophilic pathway for ion conduction.329 Besides TMEM16, large-scale ensemble simulations of the class A GPCR opsin also revealed a hydrophilic pathway between two transmembrane helices for lipids translocation, illustrating the unique aspects of this GPCR structure and providing a molecular basis for its scramblase activity.330
3.2. Membrane Transporters
Another major class of membrane transport proteins are transporters. In contrast to channels, which allow simultaneous access of their substrates from both sides of the membrane when they are open, transporters undergo a series of conformational changes during each transport cycle to change the accessibility of the substrate binding site from one side of the membrane to the other.228 This “alternating-access mechanism” prevents the leak of the substrate while allowing the translocation of substrate against its concentration gradient.331 The resulting active transport utilizes diverse sources of energy, including ATP produced in the cell for primary active transporters, or pre-established electrochemical gradients for secondary active transporters. The distinct conformations formed during the transport process are associated with different substrate-binding states that are important for the transporter function.
Both computational and experimental studies have revealed that membrane lipids are not merely forming a passive environment for membrane transporters. They are now recognized to play important roles in regulating membrane transporter function, often through specific interactions, including annular lipid contact or individual lipid binding. In this section, we will review computational studies on membrane transporters that have emphasized the role of membrane/lipid in the regulation of protein structure and function. Major transporters covered here (Figure 19) include the ATP-binding cassette (ABC) transporters, neurotransmitter sodium symporters (NSSs), and H+-coupled transporters. A number of related systems, including protein or adenine nucleotide translocating systems as well as outer membrane transporters are also included in this section.
Figure 19.
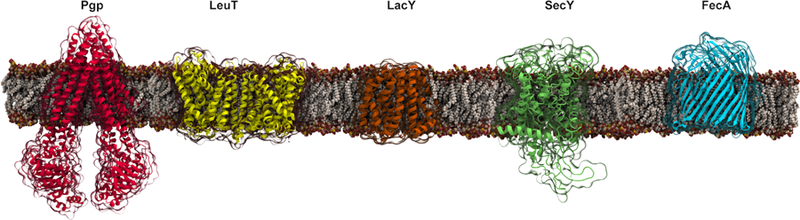
Representative membrane transporters covered in Section 3.2. The transporters shown from left to right are the ATP-dependent multidrug transporter P-glycoprotein (Pgp, PDB:5KPI), a member of the ATP-binding cassette (ABC) transporter family; the bacterial leucine transporter (LeuT, PDB:3MPN), which transports amino acids across the membrane utilizing the electrochemical gradient of Na+; the lactose permease (LacY, PDB:2Y5Y) of the major facilitator superfamily, which catalyzes the translocation of galactopyranoside using the pH gradient; the SecY translocon (PDB:3BO0), which mediates the transmembrane secretion or insertion of nascent proteins; and the β-barrel transporter FecA (PDB:1KMO), which transports ferric citrate across the outer membrane of E. coli.
3.2.1. ATP Binding Cassette Transporters
ATP-binding cassette (ABC) transporters are primary active transporters that harness the energy from ATP hydrolysis to actively transport a broad range of substrates across the membrane either in the import direction or export, depending on their architecture and fold.332 One of the most studied ABC transporters by far is the P-glycoprotein (Pgp), a multidrug exporter that plays a key role in the development of multidrug resistance in cancer cells.333 Pgp has also been proposed to transport lipids and lipid-like substrates from the inner leaflet of the membrane to the outer leaflet.334,335 Before the high-resolution structure of Pgp became available, simulations of the transmembrane domains of the bacterial homolog MsbA revealed marked deformation of the cytosolic leaflet of the membrane near the protein, a preliminary step priming lipid transport.336 Local bilayer deformation was also captured in CG simulations of the MsbA complete structures in its inward-facing, closed, and outward-facing conformations.337 Moreover, preference for anionic lipids in the first annular lipid shell was observed for both Pgp and a bacterial ABC transporter McjD, attributed to the positively charged residues near the headgroup region of the bilayer.338,339 This specific lipid organization in the proximity of the ABC transporters was proposed to be essential for their ATPase activity.
For human Pgp, AA simulations in its inward-facing state demonstrated partial entry of a POPE lipid into the transmembrane lumen, suggesting a novel putative pathway for direct drug recruitment from the membrane.340 A follow-up study using a refined crystal structure and longer simulations captured the entry of two full lipid molecules from the inner leaflet into the central chamber through the openings formed between transmembrane helices (Figure 20).269 Equilibrium simulation of the outward-facing Pgp model, constructed by a combination of structure-based sequence alignment and non-equilibrium simulations, revealed lipid occupancy at the extracellular opening of the transmembrane domain, which influences the dynamics and stability of the outward-facing state and may facilitate substrate exit into the upper leaflet (Figure 20).269 In another study, extensive sampling of lipid diffusion in CG simulations captured multiple simultaneous lipid uptake events for both PC and PE lipids to inward-facing Pgp during a 20-μs simulation.341 Moreover, using μs-long AA simulations, a recent study of bacterial ABC exporter Sav1866 in different membrane environments showed that the outward-facing to inward-facing conformational changes of the transporter is lipid-dependent and only happens in the presence of PE lipids, providing insight into the influence of lipid environment on the alternating access mechanism of ABC exporters.342 In addition to the interaction with phospholipids, 10-μs CG simulations of the apo-Pgp homology model also revealed the binding of cholesterol to the surface crevices between the transmembrane helices, the strength of which was investigated further by calculating the potential of mean force via umbrella sampling.339 Interestingly, the presence of Pgp was found to increase the flip-flop rate of cholesterol.339
Figure 20.
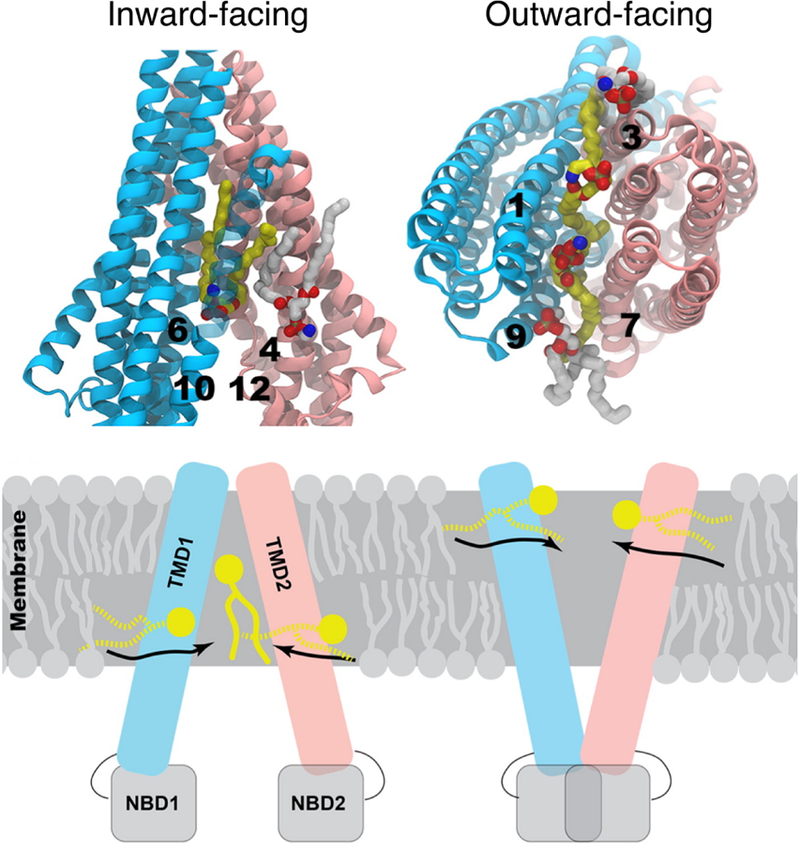
Lipid entry into the lumen of Pgp in its inward-facing and outward-facing states. (Top) Representative simulation snapshots showing the penetration of lipids into the probable drug entry portals from the inner (left) and outer (right) leaflets of the membrane. The location of the protruding lipid suggests a putative pathway for direct drug recruitment from the membrane. The initial and final conformations of the lipid molecules are in white and yellow sticks, respectively. (Bottom) Schematic representation of the inward-facing and outward-facing conformations of Pgp. The half-inserted and fully-inserted lipid molecules are shown in broken and solid yellow lines, respectively. Adapted with permission from ref 269. Copyright 2016 Elsevier.
3.2.2. Neurotransmitter Sodium Symporters
Neurotransmitter sodium symporters (NSSs) mediate the re-uptake of neurotransmitters from the synaptic cleft using the electrochemical gradient of Na+ ions as a driving force.343,344 Their vital roles in neurological pathways make them important drug targets for psychiatric diseases. To gain a better understanding of structural dynamics and functional mechanisms of NSSs, several computational studies on bacterial and mammalian homologs have been conducted since the determination of the first crystal structure of the bacterial homolog, leucine transporter (LeuT) in 2005.345 Short AA simulations of LeuT in three different lipid bilayers revealed a better matching of the DMPC membrane with the hydrophobic transmembrane portion of the protein, compared to the thicker POPE or POPC membranes.346 To quantify membrane deformation around LeuT, driven by the hydrophobic mismatch, the hybrid continuum-molecular dynamics (CTMD) approach was applied to calculate the associated energy cost at the continuum level.347 The study showed that the hydrophobic mismatch is different in distinct conformations (outward-open, occluded, inward-open) of LeuT, and that the differences are connected to the structural elements involved in the conformational transitions during the transport cycle.347,348
Beyond hydrophobic-hydrophilic contacts between the membrane and the embedded protein, specific lipids can regulate the dynamics and conformational transitions of the LeuT-fold NSSs upon direct interactions. Microsecond AA simulations of human dopamine transporter (hDAT) in a PIP2-enriched membrane revealed an inward opening of the transporter triggered by PIP2-mediated electrostatic association of specific structural motifs.349 In addition, CG simulations of the homologous human serotonin transporter (hSERT) demonstrated strong binding of cholesterol to a conserved site, the occupation of which by cholesterol was suggested to modulate the conformational equilibrium of the transporter.350 Besides LeuT-fold NSSs, computational modeling of a structurally distinct aspartate/sodium symporter Gltph showed that lipid or detergent insertion into the domain interface can facilitate the formation of the inward-facing unlocked state, which represents a configuration in the transport cycle that is uniquely suitable for ligand binding and release.351
3.2.3. Proton-coupled Transporters
Lactose permease (LacY), a paradigm for the major facilitator superfamily (MFS), catalyzes the translocation of galactopyranoside using the electrochemical gradient of H+.352 Several MFS transporters, including LacY and XylE, require PE lipids for proper function.353 Multi-nanosecond AA simulations on individual transmembrane helices of LacY showed that the helices need to tilt and/or bend in order to match their hydrophobic surface with the hydrophobic thickness of the POPE bilayer.354 10-ns AA simulations of the complete LacY structure revealed consistent formation of strong salt-bridges between the PE headgroup and functionally important basic residues, which is significantly weak in the presence of PC.355 The highly-conserved residues involved in lipid-protein interactions are crucial for the energy-coupling mechanism of LacY, implying the role of PE lipid in the H+ gradient-sensing mechanism.355 By varying the protonation state of a H+ acceptor residue in LacY, simulations demonstrated a protonation-coupled dynamical interplay between the salt-bridge formations and the global protein conformation when the protein was embedded in a PE membrane.356 The observed structural transition of LacY in the presence of PE was completely absent in a pure PC membrane, suggesting again a lipid-dependent H+-coupling mechanism. A recent study applying MD simulations combined with hydrogendeuterium exchange mass spectrometry (HDX-MS) experiments revealed that direct interactions between the PE headgroup and a conserved cytoplasmic network in XylE can modulate the conformational equilibrium between the OF and IF states.357
In addition, the protonation states of key residues in the H+-coupled multidrug antiporter PfMATE are also associated with protein conformational changes essential for substrate translocation. Using QM/MM simulations combined with classical MD, the potential energy surfaces for H+ transfer reactions between a PC phosphate group and the H+-binding site were obtained, suggesting a role for lipid headgroups as a H+ conductor mediating fast H+ diffusion along the membrane surface (Figure 21).358 Taking advantage of the increased simulation efficiency and longer timescales, CG simulations allowed the observation of association of anionic lipids to the bacterial UraA H+-uracil symporter.359 The preferential interaction of cardiolipin (CDL) mediated by the positively charged residues is likely related to its potential role as a source of buffered protons in the vicinity of the H+-driven symporter.359 Besides the involvement of lipid headgroups, AA simulations with gel- or liquid-phase PC membranes indicated that the physical phase of the lipid bilayer can also alter the structural dynamics of glucose transporter GLUT1, which in turn may affect the substrate translocation pathways within the protein.360
Figure 21.
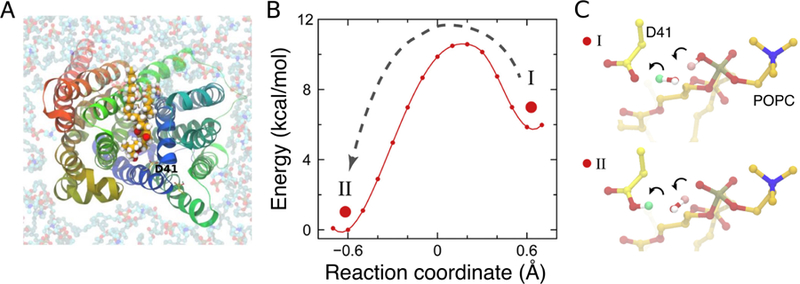
The role of lipids in the H+ transfer reactions of the H+-coupled MATE multidrug transporter. (A) A POPC lipid intruded MATE during the simulation, approaching the H+-binding site D41 with its headgroup. (B) The potential energy surface of H+ transfer from the intruding lipid headgroup to D41 by QM/MM calculations using selected MD snapshots. (C) The optimized structures corresponding to the two local minima in H+ transfer: protonated phosphate group (I) and protonated D41 (II). Adapted with permission from ref 358. Copyright 2016 Elsevier.
3.2.4. Other Transporters
The translocation of proteins out of the bacterial cytoplasm requires two structurally and mechanistically different transporting systems, Sec and Tat.361–363 The Sec translocon complex mediates the transmembrane secretion or insertion of nascent proteins, while the Tat translocase transports proteins in a fully folded form. AA simulations of the bacterial SecY translocon in different states revealed a strong correlation between the conformational transition of SecY and the intercalation of a PC lipid at the lateral gate.364 The pre-open state, which is stabilized by the intercalated lipid molecule, highlighted the importance of lipid-SecY interaction in the early steps of protein translocation through SecY.364 Another computational study showed that the intrusion of lipid acyl chains also affects the water occupancy and dipole alignment within the SecY pore, which may directly relate to the partitioning process of nascent transmembrane helices.365 TatA oligomers constitute the protein-translocating element of the Tat system. Combined CG and AA simulations suggested that the short transmembrane domain of the oligomers can lead to membrane thinning and distortion potentially facilitating the protein transport process.366
The exchange of ADP and ATP across the mitochondrial inner membrane is facilitated by the mitochondrial ADP/ATP carrier (AAC).367 The structure and function of AAC are both dependent on the mitochondrial signature phospholipid CDL.368 CG simulations followed by atomistic refinement identified three conserved CDL binding sites on AACs, characterized by stronger binding of CDL compared to the non-binding regions, along with clear selectivity for CDL over other mitochondrial lipids.369 CG simulations of a large membrane patch containing multiple copies of AAC further suggested a role of CDL in mediating the protein oligomerization process369 (see Section 5.2.4).
AA simulations of the outer membrane autotransporter Hia in an asymmetric membrane model that incorporated the outer membrane unique lipid lipopolysaccharides (LPS) captured the interactions of basic residues with the phosphate and sugar moieties of LPS, which help stabilize the protein within its native membrane environment.370 A study on FecA, an outer membrane transporter, in a LPS-containing membrane showed that extensive interactions between the inner core sugars of LPS and the extracellular residues of the protein significantly affect the dynamics of the loop regions crucial for the protein function.371 Such strong protein-lipid interactions were not observed in the simulation of FecA in a POPC bilayer, highlighting the importance of realistic membrane models in exploring relevant conformational dynamics of membrane proteins.371
3.3. Membrane Receptors
Membrane receptors are proteins that detect chemical signals from outside and transmit them into the cell in the form of various chemical or mechanical signals.372 The action of receptors can be classified as: amplification, relay and integration of signals.373 Amplification increases the effect of signals, while relay directs the onward propagation of signals, and integration allows for the incorporation of signals into other biochemical pathways.373 Some receptors also serve as major drug targets, e.g., G-protein coupled receptors (GPCRs),374 kinase-linked receptors,375 and integrins.376
While numerous receptors are found in the cell, each is linked to a specific biochemical pathway and will only bind to ligands with specific structures and properties. Agonist binding causes activation of the receptor-associated pathway, a process that requires protein conformational changes which can be influenced or even triggered by specific lipid-protein interactions.139,377
In this section, we will cover major classes of membrane receptors studied computationally with regard to protein-lipid interactions, with examples from GPCRs, integrins, and kinase-linked receptors (Figure 22).
Figure 22.
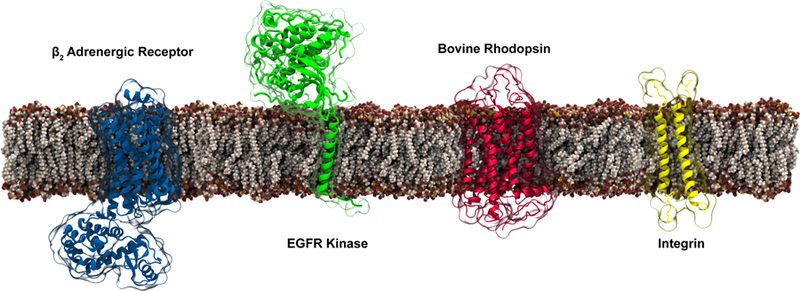
Representative integral membrane receptors covered in Section 3.3. The receptors shown from left to right are β2 adrenergic receptor (PDB:2RH1), a G-protein coupled receptor; the epidermal growth factor receptor (EGFR), (assembled using the transmembrane domain from PDB:5LV6, and the kinase domain from PDB:2JIT), which is associated with diseases such as Alzheimer’s; bovine rhodopsin (PDB:1U19), a photoreceptor required for vision; and the integrin (PDB:2K9J) involved in the key signal transduction pathways in the cell.
3.3.1. G Protein-coupled Receptors (GPCRs)
All GPCRs share a common architecture for their transmembrane domains: a seven-α-helical bundle with three extracellular and three intracellular loops, which are essential for the signal transduction across the cellular membrane.378 The largest phylogenetic class of GPCRs, known as class A, contains only the transmembrane domain with varying lengths and sequence contents of carboxyl and amino termini.379,380 These receptors are able to detect a variety of molecules outside the cell and initiate a wide array of signaling pathways. GPCRs are among the most important targets for currently used drugs.381
GPCRs are known to be functionally regulated by their surrounding lipids.330,382–385 MD simulations have been extensively used to study the regulatory role of lipids. GPCRs known to be regulated by lipids, as captured by MD simulations include rhodopsin,386 β2 adrenergic receptor (β2AR),387 adenosine A2A receptor (A2AR),388 μ-opiod receptor (MOR),389 serotonin 2A receptor (5-HT2AR),390 serotonin 1A receptor (5-HT1AR),391 chemokine receptor (CXCR4),392 and human gonadotropin-releasing hormone receptor (GnRHR).393 MD simulations at various resolutions have been used to not only map the lipid binding sites on GPCRs, but also to explore the role of lipids in the activation, oligomerization, stability, and ligand binding of the receptors.
GPCRs activation dynamics have been studied by AA simulations of rhodopsin, a visual signal transduction protein, in different lipid bilayers, namely DPPC, POPC, DMPC and PLPC, suggesting that charged lipid headgroups, bilayer thickness, length and functionalization of acyl tails induce subtle but significant changes in the protein structure,394 with implications in activation kinetics. Similarly, interaction of ω3 fatty acid docosahexaenoic acid (DHA) with rhodopsin was studied using free-energy calculations. The preferential interaction between rhodopsin and DHA was found to be entropically driven. It was observed that although all acyl chains pay the entropic penalty to interact with rhodopsin, the cost is significantly less for DHA than for other acyl chains.395 A follow-up simulation study highlighted that PE headgroups and DHA stabilized the inactive state of rhodopsin by partial structuring of its intracellular loops.396
AA simulations of β2AR in its active state revealed phospholipid movement to the binding site of the receptor which resulted in a prolonged residence time of the receptor in its active conformation (Figure 23B).387 Similarly, stochastic sidechain fluctuations in the GPCR opsin were shown to open a groove on the protein surface, facilitating rapid bidirectional lipid scrambling between the two leaflets.330 An independent AA simulation of agonist-bound 5-HT2AR showed that protein conformational transition is correlated with associated cholesterol.390 Song et al. reported a CG study of A2AR in a complex membrane model illustrating its specific interactions with GM3, cholesterol and PIP2 as well as lipid stabilization effects on its conformations.397 These specific lipid interactions are believed to play an important role in A2AR activation.
Figure 23.

Lipid-modulated structural dynamics of membrane receptors. (A) Normalized probability distribution of cholesterol around inactive (upper panel) and active (lower panel) μ-opioid receptor (MOR) captured in μs-long CG simulations. The colored circles indicate the center of mass of transmembrane (TM) helices: TM1 through 7 are colored in blue, red, grey, orange, yellow, green, and pink, respectively. Inactive and active structures of MOR with residues colored by their probability of being in contact with cholesterol (low to high probability indicates white to blue to green). Reprinted with permission from ref 389. Copyright 2016 Marino et al. Licensed under a Creative Commons Attribution 4.0 International License. (B) Zwitterionic lipid binding to the arginine component of the ionic lock of active, lipid-bound state (upper panel) and inactive, lipid-unbound state (lower panel) of β2AR. Adapted with permission from ref 387. Copyright 2015 Elsevier.
GPCR oligomerization, which is known to be involved in their function and biogenesis,398–400 is strongly modulated by the surrounding lipids, a phenomenon extensively characterized using AA and CG simulations.401 One of the earliest high-resolution crystal structures of human β2AR highlights how the co-crystallized cholesterol molecules at the crystal packing interface can mediate interactions between the receptors.374 Later, MD studies on β2AR characterized specific cholesterol binding sites at the interface of helices I-IV and V,384,402 substantiating experimental results374 and providing direct microscopic mechanism for cholesterol-mediated GPCR dimerization.403,404
Another μs-long CG study on MOR in the presence of an asymmetric lipid bilayer highlighted the role of cholesterol and sphingomyelin on the spatio-temporal organization of the receptor (Figure 23A). The sphingomyelin-rich region around MOR was proposed to induce long-range attractive force on the protomers.389 Similarly, MD simulations have shown that cholesterol can regulate dimerization of CXCR4392,405 and 5-HT1AR,391 with contributions from both direct binding and indirectly influencing bulk properties of the membrane. Furthermore, extensive (μs-long) MD simulations of CXCR4, CCR5, and CCR2 highlighted diverse homo- and heterodimer configurations in a cholesterol rich lipid bilayer.406 Finally, self-assembly simulations and dimerization free energy profiles of epithelial growth factor receptor (EGFR) confirm that along with the favorable protein-protein interactions, non-specific protein-lipid interactions contribute to the dimerization process.407
In addition to activation kinetics and oligomerization, lipids can also modulate the flexibility and stability of GPCRs. A 300-ns MD simulation of A2AR demonstrated higher flexibility and mobility of the protein in a POPC lipid bilayer than POPE. The differential flexibility was shown to stem from different hydrophobic thicknesses and distinct lipid headgroup interactions.388 Palmitoyl modifications at two specific cysteines of rhodopsin resulted in a considerably larger number of contacts with the transmembrane helices, thus stabilizing the protein structure.408 100-ns AA simulations of cannabinoid receptor (CB1) in POPC highlighted a water-mediated H-bond network, aromatic stacking interactions and receptor-lipid interactions contributing to the receptor stability.409 Furthermore, a decrease in phospholipid tail length was found to result in a kink in the transmembrane helices of the receptor to avoid the hydrophobic mismatch. Go-like CG simulations have been employed to study the mechanical stability of the related protein, bacteriorhodopsin, (not a GPCR) in membranes.410 This study qualitatively reproduced the experimentally observed force-extension curves for the mechanical unfolding of the membrane protein and illustrated the decisive role of specific lipids in determining the force patterns. In an other study, homology modeling combined with AA simulations of GnRHR not only revealed its refined structure but also highlighted that the interaction between PC headgroups and polar residues stabilizes the protein structure.393
Cholesterol is also known to alter the dynamics of GPCRs.411 Its binding has been shown to alter the conformational dynamics of β1AR412 and β2AR413 A μs-AA simulation showed that cholesterol binding at the helical interface limits the conformational variability of β2AR,413 thus establishing an allosteric role for cholesterol in modulation of the protein. Similarly, a short MD simulation of a peptide representing one of the transmembrane helices of 5-HT2AR demonstrated that the lipid bilayer with the help of a few water molecules can stabilize the helical elements, even in the presence of helix-disrupting prolines.414 In a simulation study of 5-HT1AR employing homology models constructed upon rhodopsin and β2AR415 in cholesterol-rich and cholesterol-free bilayers, cholesterol was shown to stabilize the receptor.415,416 In comparison, cholesterol binding to 5-HT2AR was shown to stabilize its fluctuations but to increase the overall conformational variability by disrupting H-bond networks.417
The lipid environment of GPCRs is also known to affect their ligand-binding properties. μs-long AA simulations of cannabinoid sn-2-arachidonoylglycerol (2-AG), an endogenous agonist to CB1 highlighted the mechanism of its entry into the receptor.418 The results suggested that 2-AG first partitions into the membrane before entering the binding pocket. This triggers the breaking of the ionic lock between TM helices, allowing a large influx of water. A comparative study of A2AR in different membranes (namely PC, mixed PC/PE, and cholesterol-rich) unambiguously showed that a specific caffeine-binding conformation is stabilized by cholesterol binding to the receptor.419 Furthermore, incorporation of agonists prevented cholesterol binding by disrupting the H-bond interactions on the protein surface. Similarly a 50- μs CG simulation showed that agonist binding to β2AR and A2AR alters their deep cholesterol binding pockets.420
3.3.2. Integrins
Integrins are transmembrane cell adhesion proteins that tie the extracellular matrix to the cell’s cytoskeleton. An integrin consists of non-covalently linked α and β subunits, each comprised of a cytoplasmic tail, a transmembrane helix, and a large ectodomain. Ligand activation of integrin initiates a cascade of signaling pathways and the recruitment of new receptors to the cell surface. Most integrins are expressed by default in their “off” state, which needs to be altered during the activation process to generate a high-affinity ligand binding state (“on” state), thus making the process highly membrane dependent. Integrin activation can occur in response to cytoplasmic and extracellular signals, known as “inside-out” and “outside-in” activations respectively.421 Previous studies have identified talin, a cytoskeletal-associated protein, as a cytoplasmic activator for integrin.376,422–424
MD simulations have highlighted the role of the lipid bilayer in regulating the specific inter-helical interactions between α and β subunits of integrins.426 Leveraging the power of CG simulations, stability dynamics of αLβ2 and αIIβ integrins was studied in model membranes.426 Owing to the inter-helical hydrogen bonding interactions MD simulations predicted optimal packing and orientation for αLβ2.427
CG simulation have successfully captured integrin-lipid binding428 and integrin-talin complex formation429 in symmetric and asymmetric lipid bilayers respectively (Figure 24). The study suggested high residence time of PC lipids around integrins and the importance of PS lipids in stabilizing the F2 domain of talin. Multiscale MD simulations have also shown that binding of the talin head domain to integrin result in its activation.425,430
Figure 24.
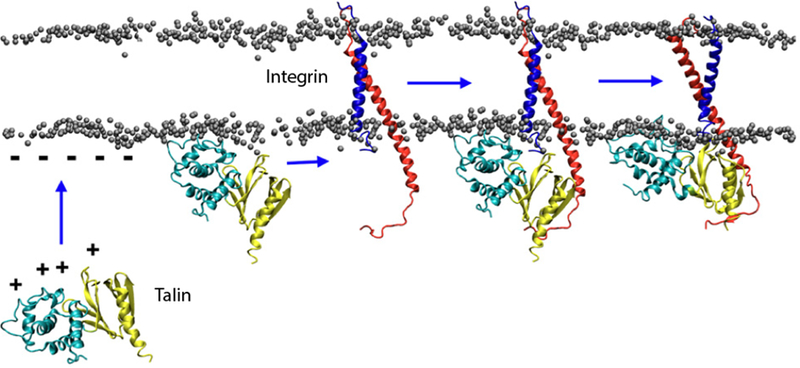
Proposed mechanism of integrin inside-out activation by talin. The figure illustrates the proposed a scissor-like motion of integrin α (red) and β (blue) TM helices that occurs upon the binding of talin (cyan and yellow). Positively charged surface of talin and negatively charged phosphate plane of lipid bilayer are highlighted. Reprinted with permission from ref 425. Copyright 2011 Kalli et al.
3.3.3. Other Receptors
MD simulations were used to study the role of lipids in modulating the structural dynamics and stability of kinase-linked receptors,375 growth factor receptors431 and cluster of differentiation 3 receptor (CD3).432 MD simulations of diacylglycerol kinase highlighted that the protein-lipid interactions ensure proper substrate loading and product release.375 A multiscale MD study of receptor tyrosine kinase (RTK) highlighted the importance of specific interactions between the juxtamembrane part of RTK and PIP2 lipids in modulating the receptor and its nanoscale organization in the cellular membrane.433
The role of charged mutation (valine to glutamate) on Neu receptor, a member of human EGFR family was studied in a PC bilayer by AA simulations. The native receptor was found to be more flexible and to exhibit a tilt to accommodate the membrane thickness,435 thus weakening the TM dimer.436 MD simulations of EGFR and CD3ϵ cytoplasmic tail of T-cell receptor in different lipid bilayers showed TM bending of the EGFR to match the hydrophobic thickness of the bilayer,431 and preferential binding of CD3ϵ to negatively charged lipids in the membrane.432 Similarly, the preferential binding of anionic PS lipid to EGFR results in the autoinhibition of the receptor (Figure 25).434
Figure 25.

Binding of EGFR kinase to the anionic membrane. (A) Electrostatic potential surface of the kinase domain (KD) when in contact with the membrane. The electrostatic potential is from −5 to 5 kB T/e (red to blue). (B) KD interactions with the lipid bilayer and the aggregation of anionic lipids (PS in red) captured from μs-long MD simulations. The KD is attached to the membrane, and the active sites (shown in orange) are sequestered by the membrane except in the active dimer. Reprinted with permission from ref 434. Copyright 2013 Elsevier.
CG simulations have captured the specific binding of fibronectic domain (FN2) of ephrin receptors to anionic PG lipid headgroups.437 In another study, dimerization of the transmembrane domain of the fibroblast growth factor receptor (FGFR) was found to be accompanied by the formation of multiple dimer interfaces whose relative propensities were influenced by the interaction of charged residues with the lipid headgroups.438 Similarly, the interaction of EGFR ectodomain with the membrane was shown to result in conformational changes in the dimer and to stabilize the receptor on the cell surface.439
3.4. Other Integral Membrane Proteins
Lipid interactions also play roles in the structure integrity and function of other integral membrane proteins besides the aforementioned membrane channels, transporters and receptors. Among those, are proteins involved in the aerobic respiration and photosynthesis. The aerobic respiration takes place in the inner mitochondrial membrane, involving a series of proton-coupled electron transfer reactions via enzyme complexes, such as the cytochrome bc1 (bc1) and the cytochrome c oxidase (CcO). As a major lipid of bioenergetic membranes, the interactions of CDL to these enzymes has been explored by MD simulations. Microsecond-long CG simulations along with PMF calculations characterized CDL binding sites that bridge contacts between the bc1 and the CcO, potentially facilitating the electron transfer process.440,441 A CG study of the CcO also observed CDL binding sites proximal to the proton entrances,442 while an AA study of the bc1 observed the occupation of CDLs near a catalytic site of its quinol/quinone substrates.443 These findings suggest that CDLs also take part in the proton uptake, a critical functional process of the bc1 and the CcO.
Photosynthesis is a process in plants and autotrophic bacteria involving proton and electron transfer reactions. Photosystem II (PSII), an enzyme complex located in the thylakoid membrane, which contains high percentages of PG and glycolipids,444 is a main component of the process. A microsecond-long CG simulation of a PSII dimer in a thylakoid-like membrane revealed the flexibility of the complex at the dimer interface.445 In contrast to the dimer, the monomer adopted a tilted orientation which led to membrane buckling.445 A follow-up MD study captured the open state of PSII in thylakoid membranes, which allows free diffusion of the lipids in and out of the membrane.446 AA simulations have been employed to investigate the homodimerization and localization of PufX, which mediates the interactions between protein complexes involved in photosynthetic electron transfer reactions. The presence of tyrosine and tryptophan residues preferentially at the lipid-water interface was shown to contribute to the anchoring of TM helices of PufX to the lipid headgroups.447
Lipid interactions have also been shown to play an important role in modulating the orientation and conformational dynamics of integral membrane proteins. AA simulations have been employed to compare the dynamics of PagP, a bacterial outer membrane protein, in six different lipid bilayers.310 Analysis of lipid-PagP interactions revealed that the N-terminus interacts preferentially with the lipid headgroups to lock the protein configuration in the lipid bilayer. In all tested lipid bilayers, PagP adopted a tilted orientation, facilitating access of lipid tails to the central binding pocket.310 Similarly, AA simulations showed that the orientational dynamics of PilA, an adhesion and mobility factor in bacteria, were dependent on specific H-bonds between protein sidechains and PE headgroups.448 Microsecond-long MD simulations have captured the transmembrane motions of PglB, induced by lipid-linked oligosaccharides, which were found to be coupled with the conformational changes in the loops.449
The activation of integral enzymes and vesicle fusion proteins can also be affected by protein-lipid interactions. Sphingomyelin binding observed in AA simulations was shown to regulate the equilibrium between the active and inactive states of p23 and p24 proteins, which are involved in vesicle biosynthesis.377 In GlpG, a transmembrane protease, an AA study demonstrated that membrane thinning around the protein allows substrate access to the catalytic dyad.450
Sampling of important protein-lipid interactions key to the behabior of the transmembrane proteins may be enhanced by the simulations of individual model TM helices. AA umbrella sampling simulations for translating a TM helix with a protonated arginine across the lipid bilayer, for example, revealed the movement of water and lipid headgroups into the lipid bilayer to interact with the arginine residue.451
A number of MD studies have used experimental restraints, e.g., solid-state NMR chemical shift anisotropy, dipolar coupling and solution NMR residual dipolar coupling, to refine protein structures in explicit lipid bilayers.452 The refined structure of Pf1 coat protein revealed that the hydrophobic mismatch of the TM domain and the lipid bilayer contributes to the domain orientation. Specific protein-lipid interactions between the Pf1 polar residues and the lipid headgroups were shown to stabilize the orientation and the depth of protein insertion in the membrane.452
4. Lipid Dependence of Peripheral Membrane Proteins
Peripheral proteins engage primarily with the surface or interfacial region of one leaflet of the cellular membrane. While some peripheral proteins primarily associate with the membrane through interactions with integral membrane proteins, many others bind primarily through protein-lipid interactions.3 We will mainly focus on latter category, which includes structural proteins such as myelin basic protein, enzymes like cytochrome P450, and proteins involved in blood clotting.3,16,453 Many peripheral proteins are membrane binding domains of larger proteins. Some peripheral proteins are involved in pathophysiological conditions, such as viral membrane binding proteins and bacterial toxins.3
Peripheral proteins use two primary binding modes to associate with the membrane. The first mode requires some combination of strong electrostatic association of charged residues with lipid headgroups and insertion of hydrophobic residues into the membrane core. This association may be mediated through bound ions, as is the case with the binding domain of coagulation factor X (Figure 26).373 In the second mode, the protein may interact only superficially with lipid headgroups but is tethered to the membrane by either an α-helix or a lipid-like anchor such as a covalently-linked farnesyl group (Figure 26).373 In some cases, protein binding to the membrane can be highly dependent on specific lipids. We will discuss such special lipids in Section 5.
Figure 26.
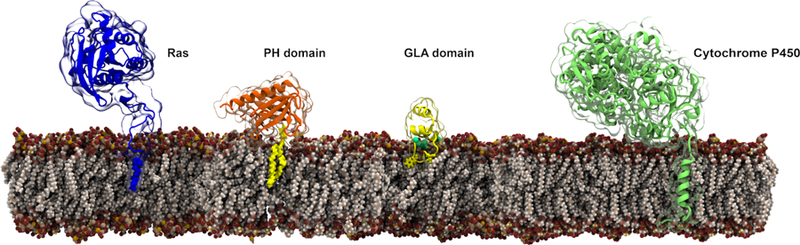
Representative peripheral proteins discussed in Section 4. Ras proteins are key regulators in cell signaling (globular domain (PDB:4OBE), linker was modeled using Rosetta). PH domain (PDB:1UNQ), a PIP binding domain found in signaling proteins. Coagulation factor X GLA domain (PBD:1IOD) is a Ca2+-coordinating domain used by coagulation proteins to bind anionic membranes. Cytochrome P450 enzymes are crucial to metabolism and biosynthesis (globular domain (PDB:1TQN), the transmembrane helix was modeled using Modeller).
For those interested in mechanistic studies of peripheral proteins, information on membrane penetration depth, orientation and interaction sites with the membrane provides invaluable insight. Obtaining this information though simulations may be impeded by the slow pace of lipid diffusion. This constraint can make simulation of spontaneous binding and insertion of peripheral proteins in AA simulations prohibitively difficult. A variety of methods have been used to address this difficulty; these have included initial CG simulations before conversion to AA models, using biases to pull proteins into the membrane, pre-insertion of the protein based on either experimental constraints or initial implicit membrane simulations, and use of specially designed membrane representations such as HMMM.
In this section, we will cover major classes of peripheral proteins for which simulation results have illuminated aspects of lipid-protein interactions. We will first discuss membrane-associated enzymes (Figure 27), an important class of peripheral proteins involved in metabolizing drug molecules, blood coagulation, and phospholipid hydrolysis. We will then turn our attention to proteins directly involved in cell signaling, e.g., Ras GTPases. Next, simulations of “disease-causing” proteins, including toxin proteins, viral proteins, and proteins involved in improper aggregation will be presented. Finally, we will discuss proteins directly involved in membrane curvature and fusion, particularly highlighting BAR domains.
Figure 27.

Results of spontaneous bilayer formation and protein-membrane association from CG simulations of nine monotopic membrane-associated enzymes, and comparison of their binding mode and depth. From right to left, and top column to bottom, these enzymes are: COX-1 (cyclooxygenase 1); ACO (apocarotenoid cleavage oxygenase); fatty acid amide hydrolase (FAAH); COX-2 (cyclooxygenase 1); P450 (cytochrome P450); 11-β-HSD (11-β-hydroxysteroid dehydrogenase); OSC (oxidosqualene cyclase); SHC (squalene hopene cyclase); CrAT (carnitine acyltransferase). Reprinted with permission from ref 454. Copyright 2009 American Chemical Society.
4.1. Membrane-bound Enzymes
Several classes of important membrane-associated enzymes have been studied using MD simulations (Figure 27). These peripheral proteins help to catalyze a variety of biotransformation and biosynthetic reactions.453,454 Here, we will first discuss cytochrome P450, followed by phospholipases and lipases. We will then discuss binding domains required for activity of blood coagulation proteases. The concluding section will detail simulations of additional membrane-associated proteases and enzymes.
4.1.1. Cytochrome P450
Cytochrome P450 (CYP) enzymes are responsible for metabolizing 75% of drugs which undergo biotransformation in the body. Their catalytic domain is globular, and while tethered to the membrane by a transmembrane helix, its direct interaction with the membrane has been shown to arise independently of the tether.178 A number of simulation approaches have been used to characterize the membrane-bound state of CYP3A4, the most important CYP for drug metabolism, including AA,455,456 HMMM178 (Figure 28), multiscale,+ and CG simulations.458 In addition to characterizing binding orientation and depth of the catalytic domain,178,457,458 these studies found that membrane binding affects the ingress and egress channels believed to allow hydrophobic and amphiphilic substrates to move between the catalytic site and the membrane.178,457 Membrane binding was found in HMMM simulations to induce conformational changes in the globular domain at the lipid-protein interface, and to induce opening of the putative ingress channels to the substrate.178 In a multiscale study comparing apo and warfarin-bound catalytic domains, the number of open ingress channels in aqueous solution was found to be greater in apo form than the warfarin-bound state.457 In membrane-bound CYP3A4, however, the same number of open channels was found for both the warfarin-bound and apo forms.457 Microsecond-long AA simulations reported that lipid composition affected the orientation of the catalytic domain and the position of ingress/egress channels relative to the bilayer, with significant differences in orientation between anionic and zwitterionic phospholipids.455
Figure 28.

Spontaneous membrane binding of CYP3A4. (Top) Snapshots taken at different time points in the simulation. (Bottom right) Close-up view of the membrane-bound form of CYP3A4, highlighting residues inserting directly into and interacting with the membrane. (Bottom left) Time evolution of average height of the center of mass for individual membrane anchoring helices (A, F’, G’) in five independent simulations (listed as Mem-1 through Mem-5). The average positions of the phosphorus (PO4) and the nitrogen (choline) atoms of the lipid headgroups are shown as gray and brown dotted lines, respectively.178 Reprinted with permission from ref 178. Copyright 2013 American Chemical Society.
Similar to simulations of CYP3A4, results for other CYPs showed that membrane binding influenced opening and closing of putative substrate ingress/egress channels.459–462 It was reported, in some cases, that ingress channel position in simulations agreed with the height at which the substrate partitions in the bilayer.461,463 Membrane-binding for CYP2C9 was found to stabilize the open conformation of a gate which locks the substrate in the catalytic site.459 HMMM simulations of CYP2J2 demonstrated that mutating the hydrophobic residues that engage with the membrane led to more shallow membrane insertion.194 Some studies have also suggested that CYPs may interact preferentially with specific lipids. A study of CYP2B4, for example, found that membrane binding induced formation of a sphingomyelin (SM)-enriched domain.464 In addition, the orientation of CYP3A4 was found to change upon binding to neutral lipids as compared to negatively charged lipids.455
4.1.2. Phospholipases and Lipases
Lipases and phospholipases are enzymes that catalyze the hydrolysis of various lipid ligands such as triglycerides or phospholipids into fatty acids and other products. These water-soluble proteins feature a flexible lid, which protects the hydrophobic active site in an aqueous surroundings but opens in hydrophobic environments to provide access for lipid substrates. Simulation studies of lipases and phospholipases in the presence of lipid substrates provided insight into the conformational changes of the lid and its role in protein-substrate interactions. AA simulations of Thermomyces lanuginosa lipase revealed the high plasticity of the lid and its role in anchoring lipid aggregate, also suggesting that draining of water from the active site is required for the enzymatic activity after lipid adsorption.465 AA simulations of the cytosolic phospholipase A2 demonstrated selective binding of arachidonyl phospholipids due to the specific shape of the sn-2 tail, providing insight for future design of novel inhibitors of the enzyme.466 Moreover, sub-μs AA simulations of phospholipase A2 under different conditions captured a closed state in the presence of water and an open state upon association with the membrane.467
Simulation of other lipases have also shown conformational changes of the lid following membrane binding.468,469 Multiscale, μs-long simulations of M37 lipase in the presence of a lipid bilayer showed that triglyceride-protein interactions induced large-scale conformational changes, creating a putative substrate entry path.469 The lipase was also shown to bind to anionic phospholipids, in which case no conformational change in the lid was observed. AA simulations of monoacylglycerol lipase (MGL) for 10 ns found conformational changes upon interaction with the membrane as well, a finding consistent with results of mutagenesis and kinetic experiments presented in the same study.468
A number of simulation studies have examined two important phopholipases, phospholipase A2 (PLA2) and C (PLC) (Figure 29). PLA2 breaks down components in dietary fat, releasing fatty acids from the second carbon group of glycerol. Three types of PLA2 isoforms have been studied using MD simulations: Ca2+-independent phospholipase A2 (iPLA2) and cytosolic (cPLA2), which both bind the cytosolic side of the membrane, and secretory (sPLA2) which binds the extracellular side of the membrane. Spontaneous association of pancreatic sPLA2 membrane-binding C2 domain to lipid bilayers was studied using CG and AA simulations, concluding that the protein bound preferentially to anionic lipids472,473 and to lipids with greater fl y (e.g., DOPC as compared to DOPE at the same temperature).473 Specific hydrophobic residues were shown to insert into the membrane core.472,473 AA simulations of the C2 domain of cPLA2, in which the protein was initially pre-embedded in the bilayer based on EPR data, identified residues interacting with PC headgroups.474 Short AA simulations (10 ns) of cPLA2 also suggested that specific binding of ceramide-1-phosphate to the C2 domain changes its tilt relative to the membrane.475 Multiscale simulations of iPLA2 captured spontaneous membrane binding and identified a hydrophobic cleft near the membrane surface potentially involved in lipid extraction from the membrane470 (Figure 29A).
Figure 29.

Membrane bound conformation of phospholipases, and the critical reactions they catalyze. Membrane binding of (A) phospholipase A2 (PLA2). Important membrane-binding residues shown in blue, red, and green. Reprinted with permission from ref 470. Copyright 2013 Bucher et al. Licensed under a Creative Commons Attribution 4.0 International License. The PLA2 reaction is provided underneath the figure. (B) Phospholipase C (PLC) bound to membrane bilayer, higlighting cation-π interactions with PLC anchor residues. Reprinted with permission from ref 471. Copyright 2013 American Chemical Society. The PLC reaction is provided underneath the figure.
The membrane binding of phospholipase C (PLC), which hydrolyzes the phosphodiester bond, (Figure 29B) has also been captured using AA471,476,477 and CG simulations.478 It had been found experimentally that a PLC which specifically hydrolyzes PI bound transiently to PC membranes, hypothetically allowing PLC to enhance its residence on the membrane surface while searching for PI lipids.477 AA simulations confirmed superficial protein binding to the membrane, featuring interacting characteristic of cation-π bonds with tyrosine residues.477 Interactions with lipids were found to dynamically exchange over 100–200 ns (during simulations with total lengths of 500-ns),477 substantiating transient interactions observed experimentally. Another AA simulation of a PLC also found structures characteristic of cation-π interactions between tyrosine residues and PC choline groups. Mutation of the tyrosine residues involved in these interactions was shown experimentally to affect the membrane binding affinity.471
In a CG study of a different PLC, PLCβ2, the effect of lipid composition on activation of the enzyme was studied.478 It had been found experimentally that PC inhibited activation while PE allowed for it, so CG simulations were performed of the PLCβ2 membrane binding domain in the presence of bilayers with varying PE contents.479 It was found that binding depth of the N-terminal residues was greater at higher PE contents; thus, PC headgroups appeared to interfere with deeper membrane penetration.478
4.1.3. Coagulation Proteins
Binding of coagulation proteases to the platelet surface is a highly lipid-regulated process, primarily triggered by increased exposure of anionic lipids to the outer leaflet of the plasma membrane.16 Simulations have been used to study lipid-protein interactions of the membrane binding domains of these proteases and their cofactor proteins, which form complexes with coagulation proteins on the surface of the membrane. Of particular interest are membrane binding domains rich in γ-carboxyglutamate (GLA) residues, which allow for Ca2+-mediated membrane binding of coagulation factors to anionic lipids such as PS, and discoidin domains (C1 and C2), which are involved in binding of several coagulation cofactor proteins.
Simulations have been used to examine PS binding sites of the prothrombin GLA domain (PT-GLA),480 demonstrating a dynamic binding of PS, and identifying more PS binding sites than originally suggested by crystallography.480 The GLA domain of coagulation factor VII (FVII-GLA) has also been studied using AA simulations, with SMD used to assess the rupture force of PS unbinding from wildtype and mutant FVII-GLA domains.481 Ohkubo et al. used SMD to simulate FVII-GLA binding to a 100% PS membrane,177 and later simulated spontaneous binding of the same domain to a PS-HMMM membrane.176 This allowed for demonstration of hydrophobic keel binding to the membrane core, importance of Ca2+ ions in association of FVII-GLA with anionic lipids, and identification of protein residues with significant lipid contacts.176,177 The resulting model allowed in a follow-up study for the fi simulation of full coagulation factor VII in complex with its cofactor, tissue factor, on the surface of the membrane.482 Using the HMMM methodology, Muller et al. were able to extensively sample spontaneous binding of the coagulation factor X GLA (FX-GLA) domain to a 100% PS membrane in search of PS-specific binding sites. Analysis of PS binding during 27 independent 200 ns combined HMMM and full-membrane simulations allowed for characterization of putative PS-specific binding sites.189
For factors V (FV) and VIII (FVIII), membrane binding is mediated by two discoidin domains known as the C1 and C2 domains. Membrane binding of FVIII discoidin C1 and C2 domains186,483 has been studied using both HMMM186 and CG simulations,483 while membrane binding of FV C2 domain was investigated using AA simulations.484 All simulations consistently found that membrane binding was mediated by insertion of residues on hydrophobic “spikes” or loops. HMMM simulation of the FVIII C1 and C2 domains found that both bound through spike residues, but with different orientations.186 In addition, FVIII discoidin C1 and C2 domains were found to induce clustering of anionic lipids during CG simulations.483
4.1.4. Proteases and Other Enzymes
In coagulation proteins, discussed above, the serine protease domain is positioned far above the membrane. In other proteases, the catalytic domain can engage more closely with the membrane. This includes two neutrophil serine proteases involved in destruction of connective tissue in inflammatory diseases such as rheumatoid arthritis, which are shown experimentally to have differential affinity for POPC vesicles.485 These netoutrophil proteases have been studied in extensive AA simulations485,486 in conjunction with surface plasmon resonance experiments.485 The simulations found that the two proteases associated with the membrane using different mechanisms, with one binding by inserting bulky hydrophobic residues into the membrane core,486,487 while the other primarily interacted with the membrane through electrostatic interactions.485 AA simulations have also allowed for characterization of binding orientation and depth for a mitochondrial protease, using the first NMR solution structure of the protein as the starting structure.488 In addition, spontaneous insertion of a hepatitis C protease into PIP2-containing membranes was described using HMMM simulations,489 which allowed for characterization of three PIP2 binding sites.489
AA simulations have also been used to study the membrane binding of two cyclooxygenase enzymes (COX-1 and COX-2),490–492 which are involved in synthesis of prostanoids mediating pain and inflammation. Residues interacting with the hydrophobic core were identified.490,492,493 In addition, membrane lipid order parameters were compared following monoamine oxidase B (MAO-B) dimer and COX-2 membrane binding,493 and it was found that COX-2 binding only affected order parameters for lipids in the cis leaflet while MAO-B binding affected both leaflets. Despite the short length of these simulations (between 1 ns490 and 25 ns491–493)and thus limited sampling of the lipid-protein interactions, differences were observed in the membrane interaction of COX-1 and COX-2. Furthermore, homodimers of COX-1 and COX-2 were found to induce curvature in the trans membrane leaflet.492 More extensive CG simulations compared spontaneous membrane binding of 11 monotopic enzymes including COX-1, COX-2, and seven other oxidoreductases, FAAH (a hydrolase), an isomerase, and a transferase454 (Figure 27). The binding of a fatty acid to FAAH has also been examined with extensive AA simulations in the context of a lipid bilayer.494,495 Considerable differences in membrane penetration were found for COX-1 and COX-2,454 with the latter shown to cause deformation in the bilayer structure.454
CG, AA, and HMMM simulations of other membrane associated enzymes have characterized membrane partitioning,185,496–498 protein-induced perturbation in local membrane curvature,496 protein-induced anionic lipid enrichment,496,499,500 and lipid-mediated dimer stabilization.501 AA simulations of cytotoxic demetallated copper-zinc superoxide dismutase 1 found that the protein was both able to adsorb onto PC bilayers, using its metal binding loops, and to complex with clumps of octanol in solution.502 Another example is PTEN, which hydrolyzes PIP3 to PIP2 and contains both a tyrosine phosphatase domain and a membrane binding C2 domain. PTEN interaction with the membrane has been studied using both CG and AA simulations.500,503–505 Charged reversal mutations of positive residues on the membrane binding surface were shown to reduce interactions of the phosphatase domain with lipids. In addition, the C2 domain induced clustering of anionic lipids500 and was found to bind PS tightly.503
4.2. Cell Signaling Proteins
A variety of peripheral proteins are intimately involved in signaling cascades. Ras proteins and other GTPases, for example, are involved in signaling pathways that promote cell growth, and a number of Ras mutations are known to promote cancer. Here, we will discuss simulation studies of Ras proteins as well as a number of other signaling proteins, such as talin and TIM proteins.
4.2.1. Ras Proteins
Ras (Rat sarcoma) oncoproteins regulate major signaling pathways and key responses to external stimuli in the cell. Ras proteins must associate with the membrane for their signaling activity, as the tight functional coordination of Ras and its effectors is mediated partly by the membrane. There are three isoforms of Ras: H-Ras, N-Ras, and K-Ras, with K-Ras being the most abundant isoform of mutant Ras oncoproteins. The full-length protein is composed of a G-domain, a globular domain which binds and hydrolyzes GTP, and a highly flexible linker. While the G-domain of these isoforms is highly conserved both sequentially and structurally, the main difference between Ras isoforms lies in the highly flexible linker, also called the hypervariable region (HVR). The HVR plays a crucial role in anchoring Ras into the cellular membrane in a lipid-dependent manner, after undergoing post-translational modifications, which include prenylation (e.g., farnesylation) and acylation (e.g., palmitoylation) involving covalent bonding between a fatty acid and a cysteine residue. The anchoring consequently may affect the orientation of the G-domain and its interaction with the membrane.
AA simulations of K-Ras characterized the association of the anchored farnesyl fatty acid with anionic lipids via lysine-PC salt bridges.506,507 K-Ras HVR contains a polylysine sequence which was found to modulate both the interaction of the HVR with the membrane as well as the orientation of the farnesyl fatty acid in the membrane.508 Microsecond-long AA simulations of the K-Ras HVR found evidence of multiple conformational states, which was also supported by free energy calculations using metadynamics.507 In silico mutagenesis of the HVR linker showed that mutating a lysine residue of the polylysine sequence to a glutamine significantly increased the population of ordered conformations.507 Phosphorylation prevented the insertion of the farnesyl tail into the membrane.508 A later study with μs-long simulations found that phosphorylation of K-Ras changed its nanoclustering, resulting in a distinct signaling output and enhanced K-Ras binding affinity to the membrane.509 AA simulations also found that in addition to the G-domain orientation on the membrane, the HVR sequestration, farnesyl insertion and the exchange of GDP to GTP are required to switch between active and inactive forms of K-Ras.510
N-Ras undergoes farnesylation as well as palmitoylation, aiding in membrane anchoring. A detailed simulation analysis of lipidated versus non-lipidated anchors showed that lipidation is essential for N-Ras stability in the membrane. The anchor binding to the membrane was facilitated not only by acyl insertion, but also by interactions of hydrophobic residues of the peptide with the hydrophobic core of the membrane.511 Membrane curvature was found to affect binding; N-Ras preferentially bound DOPC when the bilayer is planar and POPC when curved.512 The structural and conformational flexibility of the N-Ras linker was studied and key residues interacting with the membrane were identified with replica exchange MD in combination with NMR experiments.513–515 CG simulations suggested N-Ras slows the mixing of lipid domains by localizing at their interface.516
H-Ras anchors undergo two palmitoylation events, in addition to farnesylation. Membrane association of H-Ras was studied using AA simulation, which found that both of its palmitoylated cysteine residues contribute to the membrane affinity.511,517,518 Another AA study calculated the transfer free energy of H-Ras from bulk solution into the lipid membrane and found that the GTP-bound form inserted deeper into the membrane than the GDP-bound form.519
The G-domain association to the membrane was also reported in several studies in two distinct ways.511,517,521 AA simulations found differences in orientation of the K-Ras and H-Ras G-domains, potentially resulting in functional implications in terms of downstream effector interactions.522 PIP2 has been found to form long-lived salt bridges with the G-domain, preventing the tumbling or turning motions on the membrane surface.191,523
Formation of dimers and nanoclusters is believed to be necessary for signal activation of Ras proteins. MD simulations identified four sets of possible Ras dimers, whose stability on the membrane surface was evaluated with MM-PBSA.524 Studies of full length and H-Ras linkers revealed that a critical concentration was needed for nanoclustering.525 H-Ras linkers were found to cluster at the boundary of lipid domains because of the respective preference of palmitoyl and farnesyl fatty acids for ordered and disordered membrane domains.526 Key residues influencing signaling have been identified, providing an opportunity for novel drug targets to interfere with signal transduction in oncogenic Ras.527
Cholesterol was also found to enhance the stability of nanoclusters even though it did not appear to be required for their formation.525,528 H-Ras anchors formed reversible nanoclusters in membranes containing more flexible DLiPC lipids, in which the cluster formation depended on both cholesterol and protein concentrations.525,528 Results of sub-μs to μs simulations also suggested that the clusters underwent molecular exchange on the membrane.526 A CG study found a large aggregate of H-Ras molecules formed, independent of the initial orientation of the G-domain. The different orientations influenced the G-domain dynamics during and after H-Ras aggregation, providing insight into the ability of H-Ras to bind downstream effectors520 (Figure 30).
Figure 30.
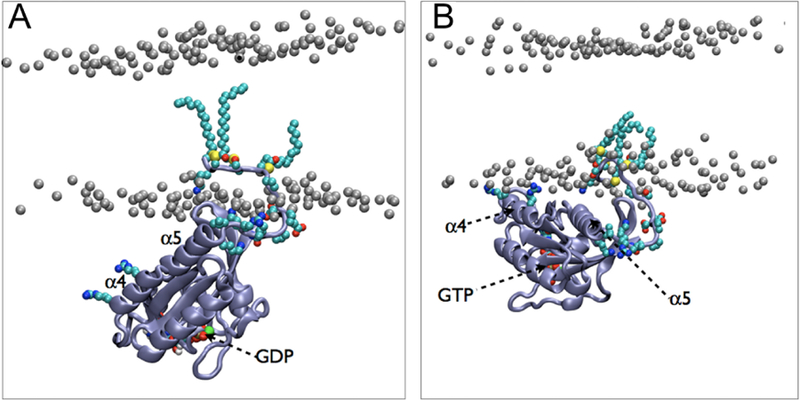
Binding modes of (A) GDP-bound and (B) GTP-bound G-domain of H-Ras observed by CG simulations. The GDP-bound G-domain bound in an approximately perpendicular orientation to the plane of the membrane, while the GTP-bound G-domain bound in a semi-parallel orientation. These distinct orientations can be observed from the positions of α-helix 4 (α4) and α-helix 5 (α5) with respect to the membrane. Reprinted with permission from ref 520. Copyright 2013 Li et al. Licensed under a Creative Commons Attribution 4.0 International License.
The Ras-mediated signaling cascade is dependent on Ras association to its downstream effectors, one of which is Raf (rapidly accelerated fibrosarcoma), whose interactions with Ras have been explored by many AA simulation studies.522,529–531 Simulations of membrane-anchored Raf-1 cysteine rich domain (CRD) to a PS/PC bilayer provided a putative model of K-Ras/Raf-1 complex, in which docking was used to determine the initial configuration of Raf-1 relative to the membrane.529 AA as well as CG simulations in combination with experimental data, showed that the hydrophobic loops of the CRD associated with the membrane, affect the orientation of the Ras-Raf complex, as well as the dimerization of Ras monomers.530 AA simulations also suggested a competition between C-Raf CRD and K-Ras membrane interactions, maintaining the protein complex at the membrane surface, enabling fast signaling.531
4.2.2. Other Signaling Proteins
In addition to Ras, simulation studies have examined lipid-protein interactions in a number of other signaling proteins. CG simulations of the Rab5 HVR showed persistent binding with PIP3, as well as slower diffusion upon enrichment of either cholesterol or PIP3.532 Another AA simulation study found the G-domain of Rab5 to be oriented so that the switch regions of the GDP–bound state are partially buried between the protein and the lipid bilayer, while the switch regions of the GTP–bound state adopt an orientation in which they are fully solvent and effector accessible.533
Other simulations have examined lipid-protein interactions of peripheral proteins which interact with receptors. Multiscale simulations of the Dok-7 protein, which regulates activation of a tryosine kinase, identified PI–specific binding sites.534 Both AA and CG simulations have been used to examine membrane interactions of talin,182,535,536 a key regulator of cell transduction events through its role in activation of cell surface receptor integrin. Simulations studying protein-lipid interactions of integrin were discussed earlier (see Section 3.3.2). Determination of orientation, as well as changes in conformation induced by membrane binding, provided key insight into how talin fulfills its role in activating integrin.182 Talin was found to bind using a hydrophobic anchor, which emerges from the core of the protein following a conformational rearrangement induced by membrane binding.182
Membrane interactions of TIM proteins, which are involved in stimulatory and co-stimulatory signaling of T-cells, have also been studied using simulations. TIM proteins recognize PS specifically, and have been co-crystallized with PS bound to a Ca2+-containing binding site.537 A number of simulations have been used to study PS binding to both TIM1 and TIM4. AA simulations were used to study PS recognition by TIM4. TIM4 was docked to the membrane surface using restraints from X-ray scattering data, and a PS docked into the known crystallographic binding pocket.537 In another AA study, four additional residues which could serve as binding sites for PS were identified.537 An HMMM membrane was used in a study of TIM1, also in conjunction with X-ray reflectivity analysis.538 Two different binding states were identified using HMMM, one likely representative of binding high–PS membranes and the other representative of binding to low–PS membranes.538
4.3. Membrane-bending Proteins
Peripheral proteins can induce and stabilize various degrees of membrane curvature. Membrane-bending effects have been observed for amphiphysin with experimental techniques such as fluorescence microscopy and negative staining electron microscopy.539,540 However, a detailed mechanistic descriptions of how lipid-protein interactions lead to curved membranes are only possible with MD simulations. In this section, we will start with the most extensively studied membrane-bending peripheral protein, the BAR domains, and then continue with proteins containing amphipathic helix such as α synuclein and synaptotagmin, and conclude with other membrane-bending proteins.
4.3.1. BAR Domains
BAR (Bin/Amphiphysin/Rvs) domains are involved in global membrane remodeling process such as vesiculation and tubulation in the cell.541 All BAR domains exist as a crescent-shaped dimer, the formation of which is facilitated by a highly conserved three-helix motif. There are, however, significant sequence/structural variations among BAR domains, giving rise to three subtypes, termed N-BAR, F-BAR, and I-BAR.
As the first attempt to understand the molecular basis of membrane sculpting by the BAR domains, AA MD simulations showed that a single amphiphysin N-BAR domain, which consists of a BAR domain with an N-terminal amphipathic helix, can bind to the lipid bilayer with its positively charged concave surface and induce a positive local curvature with a radius of ~15–25 nm.543 This membrane behavior was validated by a more extensive computational study conducted at four different levels of resolution: AA, residue-based CG and shape-based CG simulations, and a continuum elastic membrane model, finding that different arrangements of N-BAR domains resulted in different membrane bending dynamics (Figure 31).542 It was further demonstrated with sub-millisecond CG simulations that various lattices of amphiphysin generated a wide range of membrane curvatures, with radii ranging from 15 to 100 nm.544 The dominating factor for membrane curvature induced by the N-BAR was concluded to be the scaffolding effect of its concaved surface.545,546 On the other hand, the role of the short N-terminal amphipathic helix, which is believed to be inserted into the membrane headgroup region, has also been evaluated with MD simulations.545,547 The consensus is that in the case of amphiphysin, strong membrane curvature is not generated by the insertion of the amphipathic helix per se. Rather, this amphipathic helix appears to be important for the close N-BAR domain association with the membrane and formation of an optimal lattice to bend the membrane globally.545
Figure 31.

MD simulations revealing membrane curvatures induced by the N-BAR domain. (A) Snapshots from AA simulation of a single amphiphysin N-BAR domain. (B) Snapshots from residue-based CG simulation (RBCG) of a single amphiphysin N-BAR domain. (C) Snapshots from shape-based CG simulation (SBCG) of a single amphiphysin N-BAR domain. (D) Six amphiphysin N-BAR domains in the nonstaggered arrangement. (E) Six amphiphysin N-BAR domains in the staggered arrangement. Upper and middle panels in panels D and E show side- and top-views of the initial setup. Lower panels are snapshots after 20 or 50 ns. The nonstaggered arrangement of BAR domains induces a ripple-shaped membrane while the staggered arrangement results in a uniform curvature. Adapted with permission from ref 542. Copyright 2008 Elsevier.
Endophilin, another N-BAR domain, has also been investigated extensively for its ability to remodel membranes.548–555 Similar to amphiphysin, endophilin needs to form an ordered lattice to cooperatively sculpt the membrane.550,551 Specifically, it was found that when endophilins occupied only 20% or less of the membrane surface, they interacted with each other and formed a linear aggregate.552 In contrast, at 50% or more occupancy, endophilins were found to transform a lipid vesicle into a 3D tubular network.551 In addition, while tensionless membranes promote endophilin association, increasing membrane tension was shown by CG simulations to inhibit their aggregation at the membrane surface and consequently reduce the induced membrane curvature.553
Other members from the BAR domain family that lack the amphipathic helix have also been studied by MD simulation. CG simulations have captured the dynamic process of membrane tubulation by a lattice of F-BAR domains.556 Compared to N-BAR, the F-BAR domains are less curved and induce a smaller membrane curvature individually. When arranged in a lattice, the generated positive membrane curvature was highly dependent on the F-BAR domain density. Lattices with lower protein densities achieved lower curvatures because of the weaker electrostatic interactions between the F-BAR domains and the lipids.556 In contrast, the I-BAR domain has been shown to induce a slight negative membrane curvature to increase the packing of negatively charged DOPS lipids near the positively charged I-BAR surface.557
4.3.2. Amphipathic Helix-containing Proteins
Unlike BAR domains which sculpt the membrane mainly via scaffolding, certain proteins generate membrane curvature by inserting their amphipathic helices into one membrane leaflet.559–563 One representative case is α synuclein, a small neuronal protein regulating synaptic vesicle trafficking but most notable for its association with Parkinson’s disease. It has been hypothesized that α synuclein inhibits membrane fusion through stabilizing a curved structure of the membrane.564 The mechanism by which α synuclein generates curvature with its conformationally flexible amphipathic helix has been studied extensively with MD simulations.558,561,565,566 CG simulations revealed that the α synuclein-lipid complex had an intrinsic positive curvature dictated by the interactions between the protein and nearby lipids, water molecules and ions.561 A follow-up study demonstrated that radially arranged α synuclein proteins could induce budding in a planar membrane (Figure 32).558 Furthermore, CG simulations found that association of α synuclein with a small unilamellar vesicle increased the membrane undulation of the vesicle.565
Figure 32.
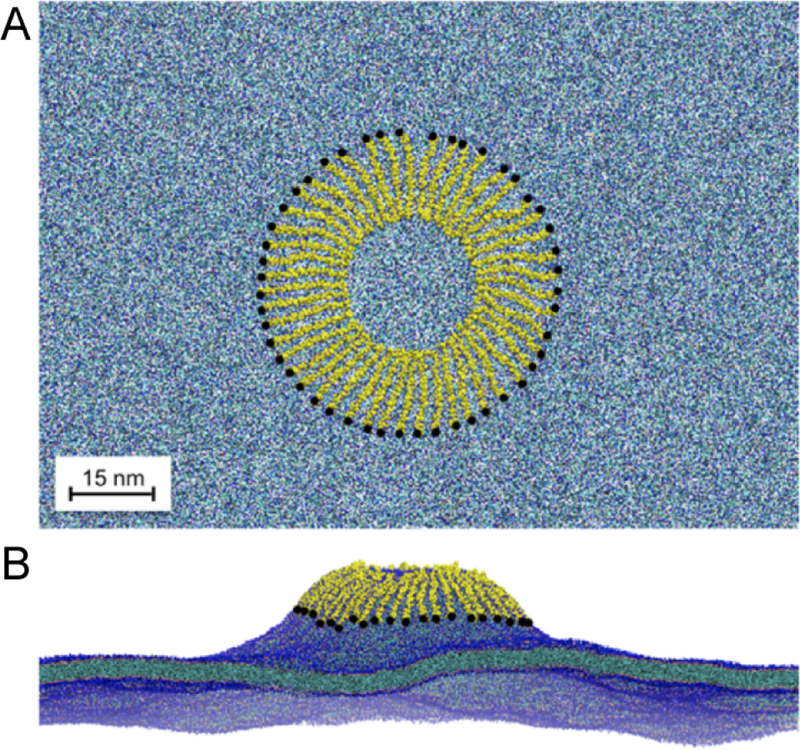
Membrane budding caused by α synuclein. (A) Top-down view of the spoke starting configuration. The system includes 48 α synuclein proteins (yellow) and a pure POPG lipid bilayer (blue). The N-terminus of each protein is marked with a black dot. (B) Snapshot at 300 ns simulation time. The budding tubule extends 25 nm above the bulk lipid bilayer. Reprinted with permission from ref 558. Copyright 2014 American Chemical Society.
The epsin N-terminal homology (ENTH) domain, which is involved in clathrin-mediated endocytosis, contains an amphipathic helix in its compact globular structure. AA simulations revealed that the amphipathic helix of the ENTH domain can wedge into the lipid bilayer with favorable interactions between PIP2 and nearby positively charged protein residues, resulting in a displacement of lipid headgroups and a local membrane curvature.560 Furthermore, CG simulations suggested that large-scale anisotropic membrane remodeling relied on sufficient packing of ENTH domain dimers on the membrane surface.560,567
Similarly, the C2B domain of synaptotagmin, a Ca2+ sensing protein involved in synaptic vesicle fusion, can interact with the membrane via its amphipathic helix. In one MD study, the membrane binding of the C2B domain of synaptotagmin was captured with HMMM.562 After conversion to full membranes, AA simulations revealed how synaptotagmin C2B domain can cooperatively induce a positive membrane curvature by inserting its C-terminal helix into the proximal leaflet inducing a different lipid tail ordering and an imbalance of lateral pressure across the leaflet.562 The membrane insertion and binding orientation of the related C2A domain, which lacks the amphipathic helix, have also been examined with MD simulations.192,568
4.3.3. Other Membrane-bending Proteins
Simulations of annexins, a family of proteins participating in membrane organization and vesicle transport, found both that the convex Ca2+ binding side of annexin was the membrane-interacting site and that annexin can induce a negative membrane curvature in a Ca2+-dependent manner.569,570 In addition, the membrane binding segment of caveolin-1, which drives the formation of caveolae, was also shown by simulations to partition into the membrane and to modulate spontaneous membrane curvature.571,572
Certain peripheral proteins, e.g., Ras proteins (see Section 4.3), bind the membrane via covalently linked lipid moieties including isoprenyl tails, fatty acids, glycosylphosphatidylinositol, and diacylglycerol.573–576 Incorporation of these hydrophobic anchors and nearby protein residues into the lipid bilayer poses a significant perturbation to the membrane structure, potentially inducing membrane curvature. One prominent example is the HVR linkered H-Ras, which undergoes two palmitoylation and one farnesylation post-translational modifications at its C-terminus. The bulky lipid anchor of H-Ras was shown by simulations to change the tail tilting angle of nearby lipids and modulate the membrane thickness.528,573 It has also been demonstrated by MD simulations that full H-Ras aggregates on the membrane and leads to major membrane remodeling, an effect primarily attributed to the area expansion of the proximal leaflet caused by the insertion of lipid anchors.577
4.4. Disease-causing Proteins
MD simulations have been used to characterize membrane-protein interactions critical to the effect of disease-causing proteins, including toxins, prions, and viral proteins. Although some toxins are classified as integral membrane proteins, as they ultimately span the membrane, their initial mode of interaction with host membranes is largely peripheral. We will therefore discuss their simulations here.
4.4.1. Membrane-binding Toxins
It has been suggested that the inhibition of ion channels by certain toxins can be mediated by their membrane binding. For instance, GsMTx4, a cysteine-knot toxin isolated from tarantula venom, can effectively inhibit mechanosensitive ion channels in both enantiomer forms (L- and D-), which is incompatible with an inhibition mechanism involving stereochemistry-specific association with the channels.578 Later, a number of MD studies evaluated membrane binding properties of this group of gating-modifying toxins. Two different membrane insertion modes of GsMTx4 were identified to be dictated by electrostatic interactions between cationic residues and the lipids. Furthermore, the deep insertion of GsMTx4 resulted in significant membrane thinning, which could explain its potentiation effects on mechanosensitive channels.579,580
In the case of VSTx1, another tarantula toxin targeting voltage-gated K+ (Kv) channels, it was first demonstrated with CG simulations that the toxin bound to the interfacial region of the membrane.583 Follow-up CG simulations used umbrella-sampling to quantitatively describe the preference of VSTx1 to partition in the interfacial headgroup region.584,585 PMFs of both CG and AA simulations showed an energy well at the interfacial region and a barrier at the hydrophobic core.585 Furthermore, VSTx1 was simulated along with the archaeal Kv channel KvAP, first using a CG presentation to find the correct association pose and then using a converted AA model to refine the predicted VSTx1/channel interface (Figure 33).581 This multiscale study supported the membrane-mediated inhibition mechanism of gating-modifying toxins.
Figure 33.
Example of a refined voltage sensor (VS)/VsTX1 complex structure, showing a t = 20 ns snapshot from an AA simulation. (A) Complex in a bilayer showing the VS in beige (with the S3b and S4a helices in green and blue, respectively), VsTx1 in red, and the phosphorus atoms of the lipids in purple. (B) View of the complex with the consensus interaction side chains of S4a (consensus between simulation and experiment) in pale blue in a space-filling representation. 581 Reprinted with permission from ref 581. Copyright 2010 Elsevier.
Other MD simulations confirmed similar membrane association modes for related toxins including SGTx1,586 HaTx1,587 and JZTx-III,588 and ProTx-II.582 For instance, it was proposed that ProTx-II binds the membrane via a patch of hydrophobic or positively charged residues, which increases the effective concentration of the toxin in the membrane and enhances the inhibition of Na+ channels (Figure 34). Another study found ProTx-I bound to a model bilayer while another toxin Hd1a did not, suggesting that the membrane interaction was not necessary for all gating-modifying toxins and had to be examined case by case.589
Figure 34.
Model of membrane-mediated binding of ProTx-II to Na+ channels. (A) ProTx-II surface representation, with residues important for binding to the membrane (green) and important for binding to the channel (blue). (B) Putative location of ProTx-II within the lipidic membrane and in the presence of the channel: 1, membrane-binding patch anchors the toxin to the membrane; 2, increased toxin concentration in vicinity of the channel and the toxin orientation facilitate binding to the voltage-sensing domain (VSD) of the channel. Reprinted with permission from ref 582. Copyright 2016 American Society for Biochemistry and Molecular Biology.
One of the most acute components of the snake venom is α-neurotoxin. It consists of 60–62 amino acids forming a rather flat “three-finger” β sheet structure that selectively inhibits the acetylcholine receptor and results in flaccid paralysis.590 Similar to the smaller toxins discussed above, it was found that membrane binding of α-neurotoxin facilitates its delivery to the receptor, in a study combining NMR spectroscopy and MD simulation.591 In particular, specific interactions with the anionic DOPS lipids promoted a specific “standing” orientation of α-neurotoxin on the membrane suitable for receptor inhibition.591 Interestingly, cardiotoxins are structurally homologous to neurotoxins but confer less toxicity and are believed to interact with intracellular components in addition to membrane targets. MD simulations have been used to explore membrane association modes and membrane penetration mechanisms for cobra cardiotoxin CTX A3.592–594
MD simulation has also been employed to study the diphtheria toxin translocation domain (T-domain). Following exposure to the acidic environment of the endosome, the T-domain undergoes a conformational transition which facilitates membrane insertion.595,596 CG simulation of 30 μs allowed for comparison of binding under acidic and neutral pH conditions as well as comparison of binding to bilayers of varying anionic character.596 Two predominant membrane orientations were identified which in a later simulation study were converted to AA and further simulated for several hundred nanoseconds.595
Additionally, MD simulations have proven useful in study of peripheral bacterial toxins which target other bacteria. Simulations have been performed on a bacteriocidal protein produced by lactic acid bacteria, class II bacteriocins, and three immunity proteins, which confer immunity to the host bacteria from their own bacteriocin.597 A set of 30 ns AA simulations were performed, placing each in an environment consistent with NMR experiment, that is, the bacteriocin in the membrane core and the immunity protein in the interfacial region respectively.597 The immunity protein was found to interact with zwitterionic polar headgroups and the bacteriocin.597
4.4.2. Proteins Relevant to Improper Aggregation Diseases
Simulations of proteins involved in diseases of improper aggregation have allowed for identification of modes by which membrane interaction either inhibits or promotes disease. Prion diseases are associated with misfolding of the PrPC protein. When simulated in solution, PrPC showed a tendency to misfold during AA simulations of 10–50 ns. In contrast, when simulated at the surface of the membrane for ~40–80 ns, PrPC remained stable because it tilted toward the membrane surface, rendering the putative sites for misfolding and oligomerization inaccessible.598 Misfolding of Huntingtin protein is related to Huntington’s disease through pathways that likely involve interactions with the membrane. The poly-glutamine region of Huntingtin protein, with a variety of different flanking sequences was found to induce a variety of membrane effects.599 It was suggested that the N-terminus, a 17-residue sequence just before the poly-glutamine region, was important for the binding of Huntingtin protein to membranes. Indeed, the N-terminus peptide was shown to stably bind at the membrane/water interface, forming both favorable hydrophobic interactions and salt bridges with the membrane.600 Apolipoprotein C-II is another known amyloidogenic peptide. Interestingly, the fibrillization of this peptide is rapid in solution but inhibited in the presence of lipids. MD simulations suggested that preferential binding of peptide aromatic residues with lipid hydrophobic tails reduced inter-peptide hydrophobic interactions.601
Amyloid β (Aβ ) peptides are involved in the formation of plaques in Alzheimer’s disease. Extensive simulations have been performed on the two most prevalent forms of Aβ peptides, namely Aβ1−42 and Aβ1−40.602–611 During the simulations, both peptides were shown to bind to lipid bilayers, which influenced their secondary structures603,604,606–608 and perturbed the integrity of the membrane, resulting in a thinner bilayer.606,608–610 It was also found that the N-terminus of Aβ interacted with the membrane at the lipid-water interface while its C-terminus remained mostly buried within the hydrophobic core of the membrane.612
Influence of gangliosides613–617 and sugar molecules618 on Aβ peptides in the context of the membrane has been probed with simulations, and interactions with these moieties were found to induce conformational transitions616,617 and accelerated membrane insertion of the peptides.618 Cholesterol was also reported to affect the conformation of Aβ,619,620 reducing Aβ-induced membrane disruption.621,622 In addition, cholesterol can affect Aβ-membrane interactions through asymmetric distribution within the bilayer.623 Other simulations studying shorter versions of the Aβ peptide624–627 also demonstrated drastic conformational change upon membrane binding625 and assessed lipid composition effects.626–629
To understand how Aβ peptides interact with each other in the context of the membrane, the most intuitive choice is to study the Aβ dimerization in different lipid bilayers. Free energy calculations found that an anionic DOPS bilayer strongly promoted protein-protein interactions and favored Aβ dimerization, while a DPPC bilayer promoted strong protein-lipid interactions.630 Other simulation studies aimed at directly capturing the dimerization process in regular MD simulations.631,632 Interactions between Aβ tetramer and membrane has also been analyzed, and it was shown that the tetramer maintained its β sheet structure in a POPC bilayer and accelerated water permeation across the bilayer.633 On the other hand, the formation of Aβ tetramer was monitored with UA MD simulations, with the resultant tetramer subjected to simulations in the presence of different lipid bilayers. It was observed that the Aβ tetramer significantly perturbed the POPC membrane, but not an ordered-phase (raft) membrane (Figure 35).634
Figure 35.

Representative conformations of Aβ tetramer and tetramer-membrane interactions. The images represent the central structure of the largest cluster from the last 250 ns of each simulation, with percentages representing the cluster size (percentage of frames belonging to the cluster). Aβ tetramer binding significantly perturbed POPC membranes, whereas the cholesterol-rich membrane remained relatively unperturbed. Reprinted with permission from ref 634. Copyright 2016 Elsevier.
The interactions between a variety of protofi ar Aβ oligomers and the membrane have been studied with MD simulations.635–640 Though the protofibrillar oligomeric structures were perturbed to different degrees across the different monomers upon binding to the membrane, the β-sheet content was well preserved during the simulations,635–639 which ranged from 150 ns to 500 ns. The main driving force for membrane binding was found to be the interactions between the N-terminal charged residues of Aβ and the lipid headgroups,635,636,638 and the membrane association was found to be stronger with anionic lipids.637,638 The binding of oligomeric protofibrillar Aβ caused thinning of the membrane, most notably at the interacting leaflet. 636 The free energy of embedding the protofibrillar Aβ trimers into a DPPC bilayer was determined to be around −70 kcal/mol, clearly indicating the favorable interactions (electrostatic and hydrophobic) between the Aβ peptides and the lipid bilayer.641
MD simulations found that oligomers of hIAPP (human islet amyloid polypeptide) fragments could disrupt the membrane, an effect which was alleviated by cholesterol.643,644 In contrast, its full-length monomer had little effect on the membrane integrity.645–647 Interestingly, changing the protonation state of a histidine residue modulated the membrane interaction strength as well as the conformational flexibility of the C-terminal portion of hI-APP.196 Transition from membrane-bound α-helical hIAPP to β-strand containing oligomers was captured in a later study, with the self-assembly process requiring a neutral histidine at position 18.197
The ion channel form648 as well as the protofibrillar oligomers649 of hIAPP were also studied using molecular modeling and MD simulations. To probe interactions between different amyloid peptides, CG simulations were used to study the cross-seeding of Aβ and hIAPP on various membranes.650 A specific orientation was preferred by the Aβ-hIAPP cross-seeding assembly and was associated strongly with lipid bilayers composed of either PC or PC/PG, through the N-terminus of Aβ.650 This explained the observation that pure hIAPP fibrils and hybrid Aβ-hIAPP fibrils are morphologically similar. 650
Simulations of α-synuclein discussed earlier (see Section 4.4) have examined its curvature effects.558,561,565,566 The aggregation of α-synuclein is believed to be highly dependent on its membrane interactions. MD simulation suggested that α-synuclein changed conformation upon binding to membrane and can readily penetrate the membrane.651 Interactions between different portions of α-synuclein and the membrane were also studied computationally.652,653 CG simulations were used to model interactions of α-synuclein in bilayers of mixed zwitterionic and anionic lipids,642 finding that anionic lipids draw α-synuclein into the lipid-disordered phase.642 Remarkably, α-synuclein also showed a preference for PUFA (polyunsaturated fatty acid) chains (Figure 36).642 AA simulations comparing dynamics and lipid-protein interactions of α-synuclein with micelles and bicelles654 suggested the protein was less dynamic in bicelles.654 Using twenty independent simulations, it was shown that α-synuclein can have a highly variable insertion depth into the bilayer at equilibrium. 184
Figure 36.
(A) Distribution of lipid-protein distances between lipids with polyunsaturated fatty acid (PUFA) and all-saturated lipids. The black plot shows the distances between the PUFA chain and the protein. The red plot shows the distances between the saturated chain and the protein. This data was sampled using 0.1 nm radial bins and averaged over the last 500 ns of a 10.5 μs CG simulation. (B) Snapshot of a single asymmetric lipid near the α-synuclein helix which is perpendicular to the page. Reprinted with permission from ref 642. Copyright 2017 Elsevier.
4.4.3. Viral Proteins
Viruses hijack the normal function of cells and cause a range of diseases from the common cold to AIDS. Remarkably, they accomplish the invasion with only a handful of viral proteins, a large fraction of which are known to function by interacting with membranes.
To enter the cell, many viruses need to fuse their envelope with the host membrane, a process usually facilitated by surface viral proteins. One of the most prominent cases is the hairpin-like N-terminal fusion peptide of hemagglutinin from the influenza virus. Early AA simulations captured insertion of the fusion peptide into a PC bilayer, resulting in membrane thinning near the peptide.655,656 Later simulations revealed consistent peptideinduced membrane perturbation and provided further details of pH-dependence of its membrane association, 657 aggregation behavior,658 conformational dynamics,659,660 and mutation effect.661,662 Notably, HMMM simulations captured its spontaneous membrane insertion179 and CG simulations demonstrated how a bundle of hemagglutinin fusion peptides could stabilize a hypothetical pre-fusion structure. 663
Another notable viral protein related to viral entry is the glycoprotein 41 (gp41) of HIV. The N-terminal part of gp41 was found in UA simulations to penetrate into the membrane regardless of its initial orientation, which affected both structure and dynamics of the nearby lipids.664 The membrane-spanning domain of gp41, on the other hand, assumed a tilted α-helical conformation with its central arginine residue “snorkeling” to either side of the membrane.665,666 Additionally, the membrane-proximal external region of gp41 induced phospholipid protrusion in the cholesterol-enriched rigid envelope during AA simulations.667 Interestingly, synthetic peptides mimicking the C-terminal portion of gp41 that were used in clinical trials as HIV inhibitor candidates, were found to interact weaker with cholesterol-containing ordered lipid domains.668,669
Other simulated fusogenic viral proteins include glycoprotein gH of the herpes simplex virus,670 envelope protein of the Dengue virus,671 fusogenic protein F of the parainfluenza virus,672 as well as lytic peptide C from the non-enveloped flock house virus.673 They were all found to insert into the membrane and cause disordering in the nearby lipids during simulations, consistent with their fusogenic role. Notably, fusion protein Gc from Rift Valley fever virus was shown in MD simulations to bind to membranes by accommodating the headgroup of a PC lipid, which initiates the membrane reorganization process.674
In addition to viral entry, proteins are required to mediate the exit of replicated viruses from the cell. VP40 is a membrane-associated protein thought to be necessary for viral budding in Ebola and Marburg viruses. Multiple studies have employed MD simulation to study VP40 membrane association. 675–679 Particularly, CG simulations showed how Ebola VP40 hexamer induced negative curvature and promoted clustering of PIP2 lipids.676 Another VP40 simulation showed that its dimer would not bind the membrane in the absence of anionic lipids.678 In accordance with this conclusion, AA simulations of the Marburg VP40 dimer revealed favorable interactions between a lysine residue and anionic lipid headgroups (Figure 37).675,679
Figure 37.

Marburg VP40 undergoing substantial conformational rearrangements upon binding to the membrane. (A-C) Snapshots of the VP40 dimer association with the plasma membrane at different time points. (D) Various lipid types interacting with the basic loop 1 and basic loop 2 residues at 300 ns. The lipids are colored as: POPS-cyan, POPI-green, POPC-gray, POPE-purple. Adapted with permission from ref 675. Copyright 2017 The Royal Society of Chemistry.
Some viral proteins interact with membranes during secretion and endocytosis. HIV Tat protein, a regulatory protein that enhances viral transcription, is known to interact with the endosomal membrane. Strikingly, a small region of Tat protein can translocate cargoes of different molecular sizes across the membrane independent of ATP and has received much attention from the biophysical community.680–684 Pore nucleation was demonstrated to be caused by the insertion of charged side chains of Tat,680 and AA simulations suggested that Tat binding induced both membrane curvature 681 and bilayer thinning.682 Other simulations found that Tat inserted into the hydrophobic core more readily in mixed PC:PE bilayers than in pure PC bilayers.683 Cholesterol was shown to hinder pore formation while anionic lipids were found to reduce the free energy barrier across the membrane for Tat peptide.684
4.5. Lipid Transfer Systems
Lipoproteins are complex lipid-protein particles involved in transport of fat molecules, such as cholesteryl ester (CE) and triacylglycerol (TG) in blood or extracellular fluid .685–687 The computationally most studied lipoproteins are HDL (high density lipoprotein) and LDL (low density lipoprotein), which differ in the fat/protein ratio of the particles. HDL helps the removal of excess cholesterol from plasma, while LDL has been implicated in the development of atherosclerosis.685,686
Due to the heterogeneity of shape and size, the details of how lipoproteins form and the structure they assume in the lipid-associated states are difficult to characterize experimentally. CG simulations starting from disordered protein-lipid complexes revealed the assembly of discoidal HDL structures on the μs timescale.688 The final structure included a lipid bilayer of 160 DPPC lipids wrapped by two apoA-I scaffold proteins, which is in great agreement with the double-belt discoidal model deduced from experimental data (Figure 38).688 CG simulations also showed that truncation of the bilayer section in the discoidal structure led to various HDL configurations, providing insight on the early steps involved in the formation of HDL.689 Moreover, replacing an adequate number of phospholipids in the discoidal HDL with a cluster of CE resulted in structural transition of HDL into prolate ellipsoidal shapes, with CE shielded inside the particles.690
Figure 38.

Snapshots from a 4 μs CG simulation of the assembly of an HDL particle with lipids initially randomly scattered. At the end of 4 μs, the simulation captured a discoidal particle with beltlike arrangement of the scaffold proteins. The two scaffold proteins (backbone only), are shown in blue and red, and the DPPC lipids are shown in dark and light tan. Reprinted with permission from ref 688. Copyright 2007 Elsevier.
CG simulations with a lipid composition matching the human plasma HDL revealed that the interfacial region of the spheroidal HDL is largely composed of phospholipids, lysolipids and cholesterols, while CE and TG are mainly confined to the core of the particles.691 The simulations also showed that apoA-I proteins interact strongest with cholesterol, which may be relevant to the cholesterol transport processes of HDL.691 LDL is larger and less dense compared to HDL, consisting of an apoB-100 protein wrapping around a lipid droplet of 3000 to 6000 lipids.692 Multi-μs CG simulations of formation of LDL demonstrated loose packing of lipids on the surface of the large lipid-protein complex, revealing a feature likely to foster the transfer of lipid between lipoproteins.693
In plasma, the transfer of CE from HDL to LDL and VLDL (very low density lipoprotein) is catalyzed by cholesteryl ester transfer protein (CETP).694 Sub-μs AA simulations of CETP-HDL interactions revealed upright penetration of CETP into the HDL surface, and the migration of CE from the core of HDL into the opening of CETP.695 AA and CG simulations also showed that CETP binding to the HDL-like lipid droplet induces formation of a small hydrophobic patch on the surface of the droplet, opening a route for the core CEs to access the binding pocket in CETP.696 Microsecond-long AA simulations of the CE-bound CETP showed that the structural flexibility of CETP affects the conformations of the two bound CEs and results in a continuous tunnel traversing the protein.697 While retaining both CEs inside the CETP core tunnel, the binding of two charged phospholipids in AA simulations was shown to maintain the protein conformation upholding a wide tunnel, which may facilitate the transfer of CEs between the lipoproteins.698 To speed up CE transfer, which happens approximately on a second timescale, SMD simulations were applied to drive CE through the entire 60– Å –long central tunnel of CETP.699 The predicted transfer rate estimated from the simulations was comparable with those deduced from physiological measurements.699
In addition to CETP, structural details of sterol transport proteins, Osh4 and Niemann-Pick (NP) proteins, and their interactions with sterol ligands have been characterized computationally, providing atomistic information on the binding and releasing mechanism of cholesterol700,701 as well as sterol ligand specificity.702 Cholesterol transport between Niemann-Pick protein C2 (NPC2) and Niemann-Pick protein C1 (NPC1) has been studied by MM/GBSA (molecular mechanics/generalized born surface area) calculations 703 and QM/MM (quantum mechanical/molecular mechanical).704 The MM/GBSA study identified key protein residues for cholesterol binding,703 while the QM/MM study illustrated cholesterol conformational changes during the transport process.704
Fatty acid binding proteins (FABPs) are intracellular carriers that transfer free fatty acids and other detergent-like compounds between cellular compartments. AA simulations revealed that the association of FABPs onto anionic membranes is driven mainly by electrostatic interactions, which determine not only the preferred binding orientation, but also the conformational changes of the proteins.705,706 In addition, simulations in the absence of lipid bilayer suggested that the surface of an FABP can sequester free palmitate from solution, a step priming the migration of the ligand into a more specific binding site.707 FABP was also shown to form a stable complex with palmitate mini-micelle, implying its high efficiency in clearing fatty acid from the cytoplasmic matrix.708 Besides this study of an FABP, palmitate penetration into another fatty acid carrier β-lactoglobulin was found to be driven mainly by hydrophobic interactions, according to the free energy calculation and decomposition analysis.709 Using distance restraints from NMR, AA simulations of a plant lipid transfer protein captured two stable binding modes of palmitate inside an internal cavity, characterized by specific hydrogen-bonding patterns with the protein.710
Another class of proteins that mediate lipid exchange are the phosphatidylinositol-transfer proteins (PITPs), which transfer PI (phosphatidylinositol) and PC (phosphatidylcholine) between different cellular membranes.711 Microsecond-long AA simulations of a mammalian PITP captured not only spontaneous association of the protein with the membrane but also partial loading of a single PC lipid into the binding pocket.712 Moreover, umbrella sampling simulations showed that the free energy of desorption of PI or PC from membrane is dramatically reduced in the presence of PITP, emphasizing the remarkable role of PITP in facilitating lipid loading and unloading processes.712
4.6. Other Peripheral Proteins
Some peripheral proteins interact with the membrane and form essential cellular structures. One case studied with MD simulations is myelin basic protein (MBP), a crucial component of the myelin sheath wrapped around the neuronal axons in the central nervous system. AA simulations used to characterize the membrane binding of a conserved central segment of MBP found that phosphorylation changed the orientation and reduced the penetration depth of MBP into the membrane.713 A follow-up simulation study showed that phosphorylation reduced the α-helical content of the central segment, which could inhibit the MBP-membrane association. 714
Another example of a structural peripheral protein is lung surfactant proteins, an important part of lung surfactant located at the air-water interface of the lung alveoli. Lung surfactant is a complex mixture of lipid monolayer and protein components; it can reduce the surface tension of the air-water interface during the breathing cycle by rapid exchange of the lipids with other lipid reservoirs. CG simulations of a mammalian lung surfactant with the surfactant protein C (SP-C) revealed a reversible transformation between the monolayer and the associated bilayer reservoirs under membrane compression and expansion. 715 Simulations also showed that surfactant protein B (SP-B) can mediate the association of the bilayer reservoirs with the protein-free monolayer and induce lipid flow in between under surface tension.716 In addition to the monolayer-bilayer association, μs-scale CG simulations showed that SP-B monomer can also trigger vesicle fusion by facilitating the formation of a hemifusion intermediate.717 Vesicle fusion was not observed in the absence of SP-B in either unbiased simulations or upon application of a restraining potential to keep the vesicles in close proximity.717 Moveover, AA simulations also identified specific LPS binding sites on lung surfactant protein A (SP-A), which may play additional roles in pathogen recognition and host defense.718 Besides known surfactant proteins, AA simulations of two putative surfactant proteins with both DPPC monolayer and bilayer captured strong lipid-protein interactions, supporting their potential role as surfactant proteins.719
Certain well-known “soluble” proteins can also bind biological membranes. For instance tubulin, a major component in eukaryotic microtubules, is found to be bound with the mitochondrial outer membrane. MD simulations of tubulin with mimetic mitochondrial membranes suggested that its amphipathic helix can bind to the membrane, preferably to the less bulky PE lipids compared to PC.720 Microtubule-associated protein light chain 3 (LC3) can be reversibly conjugated with a PE lipid, which is important for its association with the autophagosome membrane. CG simulations of PE-anchored LC3 found multiple conformational rearrangements during its recruitment and insertion into membranes.721
CD1 (cluster of differentiation 1) is a family of glycoproteins expressed on the surface of various human antigen-presenting cells. They are involved in the presentation of lipid antigens for the recognition by T cell receptors. Although this lipid presentation does not occur in the context of membranes, it involves many relevant protein-lipid interactions which have been studied with MD simulations. Using a position-specific homology modeling protocol that utilizes the structural information available from other CD1 isoforms, an atomic structure model of CD1c was constructed and used in the characterization of specific features of the binding domain.722 A follow-up study on five human CD1 isoforms in the lipid-bound and lipid-free states showed that the hydrophobic binding grooves of CD1b-e, but not CD1a, collapse irreversibly in the absence of a lipid antigen, suggesting dependence on helper proteins such as lipid transfer proteins for lipid reloading.723 Endothelial protein C receptor (EPCR), a CD1 homolog, is important in regulating protein C functions upon its binding to the protein C GLA domain. Compared to the PE bound simulation, the unbound EPCR structure presented a reduced interaction surface for the GLA domain, confirming the role of PE in establishing the proper EPCR conformation for interaction with its partner protein.724
5. Special Lipids
Structural and functional modulation of proteins by lipids can be highly specific, requiring particular species of lipids.16,17 This section discusses the simulation results that either demonstrate specificity to a particular kind of lipid, or, shed light on the mechanism of such interactions. While specific binding to zwitterionic glycerophospholipids like phosphatidylcholine (PC) and phosphatidylethanolamine (PE) with functional ramifications can occur,674 these lipids are generally used in MD simulations to represent the bulk of the lipid bilayer. In fact, the majority of the MD simulations of membrane proteins in the past have used single-lipid membranes of PC or PE, to represent mammalian or bacterial membranes, respectively. We have therefore discussed studies demonstrating specific binding sites for these zwitterionic glycerophospholipids in earlier sections. In this section, we focus on key lipids for which a regulatory role has been well established. We also discuss simulations with sphingomyelin (SM) and lipopolysaccharides (LPSs), which have been parameterized more recently.
We will begin with cholesterol, a unique steroid lipid which can regulate protein function through both specific binding interactions and cholesterol-induced effects on bulk properties of the membrane (Figure 39).725 Simulations have been widely used to identify cholesterol binding sites and capture direct modulation of such binding to protein structure and dynamics. The membrane thickening and ordering effects observed in cholesterol-rich bilayer simulations also provide insight into the influence of lipid micro-environment on protein function.
Figure 39.
Special lipids modulating protein structure and function. Sphingomyelin, phosphatidylinositol 4,5-bisphosphate (PIP2), phosphatidylglycerol (PG), cholesterol, phosphatidic acid (PA), lipopolysaccharides (LPS), phosphatidylserine (PS), and cardiolipin.
Anionic lipids will be discussed next (Figure 39). Asymmetric distribution of anionic lipids between the inner and outer leaflets is actively maintained by active transporters,6 keeping a higher concentration of anionic lipids in the inner leaflet. Anionic lipid expression on the outer leaflet can occur during cell signaling events and in a number of pathological conditions. 16,726,727 PIP lipids, in particular, have important signaling properties, and we will detail simulations studying PIP-specific binding domains. We will discuss charge-driven interaction as well as interactions specific to particular species of phospholipids. We will also review simulations studying specific interactions of cardiolipin (CDL) with proteins, a lipid with a particularly important role in mitochondria.
Finally, we will discuss simulations probing specific interactions with SM and LPS, lipids which have been parameterized relatively recently. The composition of lipids exposed to the extracellular environment of mammalian and bacterial cells is drastically different. For mammalian cells, the outer leaflet is primarily composed of PC, along with small concentrations of PE and SM. The relatively small concentration of SM, which is critical in the formation of lipid microdomains by tightly intercalating cholesterol molecules and plays a critical role in membrane trafficking (Figure 39).728,729 On the other hand, Gram-negative bacteria have a double-membrane envelope that enables them to survive harsh environmental conditions and prevents the entry of large polar and small hydrophobic molecules into the cell. A major component of the bacterial outer cell membrane is LPS, a glycolipid with a number of sugar moieties and multiple fatty acyl chains.
5.1. Cholesterol
Cholesterol, an abundant component in mammalian plasma membranes, plays an essential role in regulating membrane proteins.730 Cholesterol concentration significantly alters membrane thickness, fluidity, and curvature, and induces the formation of lipid domains or cholesterol-enriched regions with saturated phospholipids and sphingolipids.731–733 Such membrane alteration has been attributed to the indirect cholesterol modulation of protein structure and dynamics. With recent advancements in structural biology, putative cholesterol binding sites have been identified in crystal and cryo-EM structures, indicating the possibility of direct cholesterol modulation.725,734 Based on protein sequence analysis and structural studies, several cholesterol binding motifs have been proposed, including two sequence-based motifs, CRAC (Cholesterol Recognition Amino acid Consensus) and CARC (reversed CRAC), and a structure-based motif in the GPCR family, CCM (Cholesterol Consensus Motif).735,736 In all these motifs, basic residues (R/K), aromatic residues (F/Y/W) and aliphatic residues (V/L/I) are suggested to form direct interactions with the cholesterol hydroxyl group, fused rings, and the hydrocarbon tail, respectively. To characterize and validate these cholesterol consensus motifs, AA and CG simulations have been extensively performed to sample protein surface and identify sites with high cholesterol occupancy. In this section, we will focus on MD simulations studying specific cholesterol interactions as well as its direct and indirect effects on membrane protein function.
GPCRs are a major protein family functionally regulated by cholesterol, and thus the most studied systems with regard to cholesterol-protein interaction.737 Lipid interactions with GPCRs have been discussed at length earlier in this review (see Section 3.3.1). Here, we will focus on the use of MD simulations to characterize the effects of cholesterol on GPCR structure and dynamics.
One of the earliest AA simulations showed that the stabilizing effect of cholesterol on the native state of rhodopsin was independent from direct cholesterol binding to the protein.386 A later study further supported that cholesterol affected the structural stability of rhodopsin via membrane-mediated modulation rather than direct specific interaction.738 Direct modulating effects of cholesterol packing had not been observed in detail until μs-long simulations revealed specific cholesterol binding and its dynamic perturbation to the rhodopsin structure. 411 The study was subsequently followed up by a CG study to further validate the predicted cholesterol binding sites with higher statistical certainty, owing to the longer timescale achievable by CG simulations.739 Altogether, these studies of rhodopsin determined common cholesterol-GPCR interactions and cholesterol modulation effects at various spatial resolutions and timescales.
Experimentally, cholesterol has been frequently used in crystallography to stabilize GPCR structures, e.g., the human β2AR structure with co-crystallized cholesterol molecules resolved at the crystal packing interface.374 The first establishment of a specific cholesterol binding site, namely the CCM site, was reported in a later structural study of class A GPCR β2AR.740 Following these structural studies, cholesterol binding sites were identified in class A GPCRs including β1AR,402 β2AR,384,402,403,420,741 A2AR,420,742–744 5-HT1AR,415,745 and a class F GPCR Smoothened746 by AA and CG simulations as well as a recent study based on alchemical free energy perturbation. 741
Particularly, in a systematic mapping of cholesterol binding sites on β2AR using μs AA simulations (Figure 40A), a cholesterol binding site was found at the same location as the co-crystallized cholesterol at the crystal packing interface, suggesting the role of cholesterol in mediating GPCR dimerization.403 Cholesterol occupancy was then demonstrated in a CG simulation study to restrict certain β2AR dimer formation and thus alter the distribution of dimer interfaces (Figure 40B).404 Using extensive CG simulations, the diverse roles of cholesterol in regulating GPCR oligomerization have also been illustrated in other class A GPCRs, e.g., 5-HT1AR,391 chemokine receptors (CXCR4, CCR5, CCR2),392,406 and MOR,389 showing the importance of both direct, binding-induced and indirect, membrane-mediated effects.
Figure 40.
Cholesterol modulation of human β2AR characterized by MD simulations. (A) Cholesterol binding sites are shown in orange, and POPC binding sites shown in blue for comparison. Reprinted with permission from ref 403. Copyright 2013 American Chemical Society. (B) β2AR dimer interface formed by helices IV/V and I/II at 0% and 50% cholesterol (Chol) concentration, respectively. Adapted with permission from ref 404. Copyright 2014 Elsevier. (C) β2AR conformational dynamics restricted by cholesterol binding. LL and LG denote the distances between the Cα atoms of D113-S207 at the extracellular ligand-binding site and R131-E268 at the intracellular G-protein interface, respectively. Reprinted with permission from ref 413. Copyright 2016 Manna et al. Licensed under a Creative Commons Attribution 4.0 International License.
Cholesterol binding has been shown by AA simulations to affect ligand binding in GPCRs, especially in A2AR.419,747,748 A μs simulation captured the disruption of cholesterol binding at the A2AR CCM site due to changes of protein side-chain dynamics triggered by agonist binding.747 Interestingly, another study reported a stabilizing effect of cholesterol on caffeine binding via inducing conformational rearrangement of protein side chains.419 In a later study, a pathway for cholesterol access to the A2AR ligand binding pocket was discovered, indicating a distinct mechanism of cholesterol action by directly modulating the orthosteric ligand binding site.748 Apart from AA simulations, two CG simulations have elucidated how cholesterol molecules may facilitate ligand binding in 5-HT1AR415 and how agonists affect cholesterol binding to β2AR and A2AR.420
Additionally, long-timescale AA simulations have shown the influence of cholesterol on the conformational dynamics of several GPCRs, including β1AR,412 β2AR,413 5-HT1AR416 and 5-HT2AR.390,417 As an example, μs simulations unveiled reduced conformational variability of β2AR caused by cholesterol binding (Figure 40C), suggesting an allosteric modulation of β2AR.413 Long-timescale simulations have also characterized a special modulatory effect of cholesterol on stabilizing the secondary structure of an amphipathic juxtamembrane helix in CB1,749 a class A GPCR, and mGluR2,750 a class C GPCR. The observed stabilization was attributed either to a cooperation of cholesterol binding near the CCM site and palmitoylation of a cysteine residue,749 or to a combination of direct interaction and indirect membrane thickening effects of cholesterol.750
Besides GPCRs, MD simulations have identified specific cholesterol binding sites in other integral membrane proteins, some of which contain CRAC or CARC motifs.350,751–756 A variety of cholesterol binding sites and modes have been characterized in ion channels including Kir,757,758 BK751 and VDACs.315 In pentameric ligand-gated ion channels such as nAChR752,759,760 and GABAAR,761 cholesterol molecules have been found to bind preferentially to the non-annular sites located at the inter-subunit interfaces. Similar non-annular cholesterol sites have been confirmed by various simulation techniques as well as free energy calculations in ABC transporters, including Pgp339,762 and ABCG1.753 Likewise, conserved cholesterol interaction sites have been uncovered in neurotransmitter sodium symporters (NSSs).350,754,755 In all proteins mentioned above, as well as in several other integral membrane proteins, including opioid peptide dynorphin A,763 aquaporin,764 and integrin,429 specific hydrogen-bonding, π-stacking or hydrophobic interactions have been characterized as key components for cholesterol association.
Cholesterol interactions have been indicated in simulations to directly stabilize transmembrane helices and thus modulate integral membrane protein conformations. In one of the NSSs, serotonin transporter, cholesterol binding has been shown to affect ion coordination and transporter activity.754 Meanwhile, evidence from MD simulations has illustrated a common regulatory mechanism among NSSs that involves cholesterol-induced stabilization of the transporter in a particular state, thereby inhibiting its transport cycle.350,755 Modulation of protein conformational dynamics by direct cholesterol contact has been also found in phospholamban,765 MHC-II protein,756 tetraspanin CD81766 and the C-terminal transmembrane helix of phospholipid scramblase 1.767 One special case for such cholesterol modulation was manifested in the helix-helix dimer formation of an epidermal growth factor receptor, ErbB2.768 In contrast, indirect cholesterol regulation has been described for Ca2+-ATPase,769 NavAb channel770 and endoplasmic reticulum stress sensor Ire1,771 suggesting an alternative effect of cholesterol by enhancing lipid packing instead of specific, direct association.
For peripheral membrane proteins, MD simulations have shed light on both direct and indirect effects of cholesterol on their membrane association. One such system of particular interest comprises of amyloidogenic proteins implicated in Alzheimer’s disease, including amyloid beta (Aβ) and amyloid precursor proteins (APPs).772,773 Since lipid modulation of Aβ peptides has been discussed in detail earlier (see Section 4.5.2), we will only highlight key modulatory mechanisms of cholesterol here. Multiple cholesterol interaction sites were characterized in the APP C99 domain,774 where the protonation states of two negatively charged residues were identified as critical in direct cholesterol interactions from constant pH simulations.775 Meanwhile, cholesterol-induced membrane effects, rather than direct binding, were shown in another CG simulation to result in the inhibition of APP C99 domain dimerization.776 Such dual effects of cholesterol have also been illustrated in Aβ membrane association, 622,623,627,777 conformational transition,619,778,779 as well as oligomerization and pore formation. 780,781 Moreover, the interplay between cholesterol and ganglioside GM1, another lipid associated with membrane effects, has been found to enhance the membrane assembly of Aβ peptides. 617,629 A variety of cholesterol-triggered effects have been observed in other peripheral protein systems, including α-synuclein,652 leukotoxin,782 stromal interaction molecule 1,783 caveolin-1,571,784 cytochrome P450,785 viral proteins,665–667,670,674,786 H-Ras,520,525,526,528 Niemann-Pick type C2 protein,787 and the complex of thrombospondin-1 and calreticulin. 788
5.2. Anionic Phospholipids
While constituting a minor component of the cell membrane (around 30% of the total phospholipids present) anionic phospholipids have a considerable impact on function and regulation of the cell and of membrane proteins.3,6,16,789 The outer leaflet of mammalian plasma membranes is primarily composed of neutral lipids such as PC and SM along with a small fraction of PE. The inner leaflet, in comparison, contains the majority of PE in the cell and a variety of anionic phospholipids including phosphatidylserine (PS), phosphatidylglcerol (PG), phosphatidic acid (PA), and phosphoinositides (PIs).3,7 Bilayer asymmetry is both actively maintained and dissipated by membrane proteins,6–8 and we have discussed simulations of proteins, such as scramblases, involved in the dissipation of asymmetry (see Section 3.1.5). Asymmetry is lost and large numbers of anionic lipids are flipped to the surface of the membrane during normal initiation of blood coagulation and apoptosis.16,726,727 A number of pathological conditions, including cancer, also result in heightened concentrations of negatively charged lipids on the surface of cells.727,790 Some anionic phospholipids regulate membrane protein functions in a highly specific way. PS and PA bind specifically to a number of different proteins, often as elements of complex signaling pathways.17 Cardiolipin (CDL), found in bacterial and mitochondrial membranes, is required for optimal function of mitochondrial respiratory and bioengergetic enzymes. In plant membranes, PG has been shown to bind tightly in photosynthetic complexes.
Here we discuss use of simulations in characterizing protein interactions with anionic lipids. We will first discuss simulations where anionic lipids are demonstrated either to be required for protein binding or to form clusters around the protein after binding. We will then detail simulations in which binding of anionic lipids has been shown to induce changes in conformation or orientation of membrane binding proteins. Further, we will discuss simulations in which differential protein-lipid interactions are characterized, first detailing simulations of PS, PG and PA. Additional sections will be dedicated to simulations of CDL and PI specific interactions.
5.2.1. Preferential Interactions with Anionic Lipids
Simulations of bilayers containing both PS and PG have been used to demonstrate specifity to anionic membranes. While many of the systems highlighted here are peripheral proteins, given the prevalence and involvement of anionic lipids in drawing peripheral proteins to the membrane, many simulations have also been performed with PG or PS in the membrane for channels15,295,306,307,310,330,347,355,375,387,452,791–802 and receptors.387,437,803 Preference of proteins to bind anionic membranes has been frequently shown by demonstrating clustering of anionic lipids around a protein using both CG483,496,676,804,805 and AA simulations.499,506,675 Proteins with positive residues interacting at the interfacial region have been shown to generate clusters of PG or PS,246,338,359,437,469,483,506,535,803,804 and simulation of a bacterial monoglucosyldiacylglycerol synthase (Figure 41) demonstrated local enrichment of CDL and PG in the immediate area of the protein.496 A study of the tumor-suppressor protein PTEN demonstrated anionic lipid clustering, along with loss of the clustering effect upon charge reversal mutations of positively charged residues.500 Lipid clustering demonstrated in these studies may be driven purely by electrostatics. Simulations of proteins binding to a membrane containing multiple anionic lipids and inducing clustering of a single species, such as simulations of Ebola VP40 clustering PIP2676 and Marburg VP40 clustering PS,675 however, suggest involvement of more specific lipid-protein interactions.
Figure 41.

Lipid headgroup density profiles around the protein for (A) all phospholipids, (B) PE, (C) PG, and (D) cardiolipin (CL in figure). (C) and (D) show the density profiles of the three simulations for PG and cardiolipin, respectively. The values in the color chart show the relative lipid enrichment around the protein, defined as the ratio between the local lipid density (δlocal) and the average lipid density (δrandom). The yellow line indicates the protein footprint, and the yellow arrow connects the N and the C domains, indicating the orientation of the protein. The protein is not shown for clarity. Some protein residues interacting directly with the membrane are shown as colored symbols: aromatic residues in green, hydrophobic residues in red and positively charged residues in blue, as detailed in (A). (E and F) Monoglucosyldiacylglycerol synthase bound to the membrane (E), and monoglucosyldiacylglycerol synthase outside of the membrane with potential membrane binding segments (marked S) highlighted in color (F). Adapted with permission from ref 496. Copyright 2014 John Wiley and Sons.
An alternate method of demonstrating anionic lipid preference for peripheral proteins is to compare spontaneous binding to neutral and anionic membranes. Simulation studies of Ebola VP40,677 and the Aβ-hIAPP assembly650 showed spontaneous binding to both neutral and anionic membranes, but more protein-lipid contacts and thus stronger binding to the anionic membranes were found in both studies. 650,677 In an integrated experimental/simulation study of the bovine α-lactalbumin oleic acid complex, which is cytotoxic to tumor cells, CG simulations demonstrated that the complex bound in a sustained fashion to a PG-containing but only intermittently to a PC-only membrane.806 Similarly, CG simulations of M37 lipase,469 the auxiliary subunits of voltage-gated Ca2+ channels,807 and diptheria toxin T-domain, 596 as well as AA simulations of Osh4808,809 and a bile acid-binding protein,706 found only transient interactions between the protein and zwitterionic membranes, but stable binding to anionic membranes.469,596,706,807,808
5.2.2. Conformational Changes Induced by Anionic Lipids
Proteins have been demonstrated to undergo conformational transitions or adopt differential orientations upon binding to anionic membranes. In comparative simulations of a β-amyloid dimer binding to PS/PC and PC membranes, it was found that the protein bound to a PS-containing membrane was more folded, aggregated, and more tilted on the surface of the bilayer.810 Protein kinase C1 binding to a PS membrane resulted in its conformation changes, and a change in the PS distribution in the immediate area of its binding.499 In PTEN, it was demonstrated that anionic lipids changed positioning of the anionic C-terminal tail, which in solution obstructed the membrane-binding interface.503 An HMMM study of α-synuclein showed binding to anionic lipids changed conformational state of the protein,184 with PS interactions promoting a transition from a bent conformation to a more extended one.180 CD3ϵ receptor secondary structure was also found to be affected by PG in CG simulations, with the protein adopting stretches of α-helices when bound to DOPG and DOPC, but not in the presence of DPPC.432 Anionic-lipid interactions were also found to be important for stabilizing the structure of the epidermal growth factor receptor dimer.434 In talin, a large interdomain conformational change allowing for F3 subdomain interaction with the membrane surface has been shown to result from binding to anionic lipids.182
In some cases, a drastic conformational change is not observed but the orientation and/or depth of binding are affected by the presence of anionic lipids. Orientation of the diptheria toxin T2 domain was found to stabilize with an increase of the anionic lipid content, with a larger number of protein-lipid contacts at higher PG contents observed at both pH levels studied. 595,596 A simulation study of cytochrome P450 found that the enzyme bound at a more tilted orientation and at a greater depth to membranes containing PG or PS.455
HMMM simulation allowed atomic-level differences in binding of RecA to be captured for membranes containing three different anionic lipids, namely PS, PG, and CDL.195 Fluorescence experiments had previously shown that the protein behaved differently when bound to liposomes containing PG and CDL. Binding to PG and CDL was similar, with insertion of key motifs being different in CDL than in either PS or PG.195 In addition, PS appeared to drive a different binding mode than the two other anionic lipids.195
5.2.3. Characterization of Anionic Lipid Binding Sites
Simulations have also been used to characterize specific binding sites for anionic lipids on membrane proteins affected by these lipids. In the simplest cases, a single anionic lipid was either docked to a protein or adopted from an x-ray structure, whose binding stability was assessed by MD simulations.481,811 PG binding sites have been characterized for receptors as well as channels. Using biased and unbiased simulations, it was demonstrated that PG preferentially binds to the CD3ϵ receptor tail.432 These simulations demonstrated a reduced membrane association in PC and that it was less difficult to detach the tail from a PC membrane as opposed to a PG-containing membrane.432 Extensive (0.25 ms) CG simulations demonstrated that anionic lipids entered the empty G-protein binding site of β2AR.387 The lipid-protein interaction stabilized the active state, preventing ionic interactions required for the inactive state to form.387 AA simulations as short as 10 ns performed on bacterial K+ channel KcsA in a mixed bilayer were able to capture the binding of PG lipids at an interfacial site between the neighboring subunits, 237,238 where they appeared to stabilize the dimer. Sub-ms CG simulations allowed the evaluation of PG binding lifetime at these functionally important binding sites.239
A large data set produced using HMMM simulations was used to characterize putative PS specific binding sites in the coagulation factor X GLA domain. Detailed analysis identified where each PS functional group bound during each of the 27 independent 100-ns replicates, allowing for identification of sites most likely to be highly PS–specific (Figure 42).189 A PG binding site on talin was found in simulations to be key to protein regulation, with mutations at these residues leading to talin binding with a perturbed orientation, likely changing the binding interface with integrin.535 These residues had previously been mutated experimentally and determined crucial for talin-membrane association. 535 HMMM simulations of spontaneous talin binding to a PS-bilayer confirmed the importance of these same basic residues in the association of talin to the membrane.182 While one PS binding site was identified using x-ray crystallography for Tim4, AA simulations identified four additional basic residues which could serve as binding sites for PS.812
Figure 42.
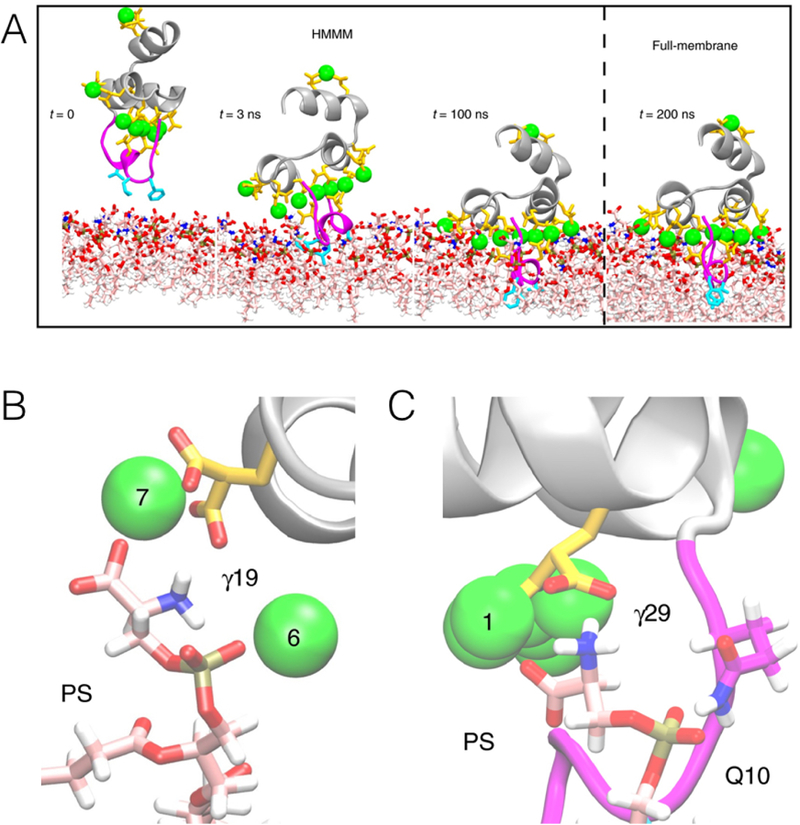
Putative PS binding sites for coagulation factor X GLA domain (FX-GLA). Ca2+ ions are shown in green, and specialized GLA residues (marked as γ) are shown in gold. The GLA domain membrane binding loop is shown in magenta, hydrophobic residues which insert into the core of the membrane are shown in cyan, and the rest of the protein is shown in silver. Lipids are shown with carbon atoms in light pink, nitrogen in blue, and oxygen in red. (A) Spontaneous binding of FX-GLA domain to an HMMM bilayer. 189 (B-C) Putative PS–specific binding sites. Sites identified as most likely to be PS specific because all three charged groups of the PS lipid interact with the protein simultaneously. 189 Adapted with permission from ref 189. Copyright 2017 John Wiley and Sons.
PA has been identified as an important signaling lipid, but thus far few simulations have been performed assessing PA binding to proteins. 31P NMR and MD simulation were used in conjunction to study PA interactions with charged residues. Basic amino acids were found to increase the charge of PA by forming hydrogen bonds with the phosphate group, thereby stabilizing protein-lipid interactions.813 The results showed that this electrostatic/hydrogen bond switch was the basis for preferential interaction of LYS and ARG residues with PA.813
Simulations of entire proteins interacting with PA have included Dvl2 DEP814 and MIT domain,815 both of which used 100% PA membranes. MD simulations, in conjunction with NMR, were used to study binding of Dvl2 DEP to PA,814 allowing for identification of specific ARG and LYS residues on a basic helix important for binding to PA.814 Simulations of the MIT domain compared binding to pure PA and pure PC membranes.815 The domain was found be absorbed with higher affinity onto the PA membranes through interactions of its basic residues, and a particular helix was identified as a potential PA binding hotspot.815
5.2.4. Cardiolipin
Cardiolipin (CDL), an anionic phospholipid containing four acyl chains and two phosphate groups, is the signature phospholipid of the inner bacterial membrane and the inner mitochondrial membrane in eukaryotic cells. Its presence is important not only for maintaining structure, but also for proper functioning of various proteins, many of which play diverse roles ranging from energy metabolism to apoptosis. Co-crystallized structures of protein-CDL complexes together with molecular docking have provided insight into structural implications of CDL binding.816 In this section, a collection of representative CDL-interacting proteins investigated using simulation approaches is presented.
In the mitochondrial inner membrane, CDL plays an important role in the function and supramolecular organization of the respiratory chain complexes, which largely contribute to the biosynthesis of ATP. Sub-μs CG simulations of cytochrome c oxidase (CcO), the terminal oxidase of the aerobic respiratory chain, in a mitochondrial membrane mimetic have successfully identified high-affinity binding sites of CDL.442 AA and CG simulations of cytochrome bc1 (bc1), which transfers electrons to CcO, also captured spontaneous binding of CDL to preferential interacting sites (Figure 43).440,443 In the self-assembly CG simulations of bc1 and CcO, CDL was shown to play a structural role in bridging the respiratory chain complexes into supercomplexes (Figure 44).440,441 Free energy calculations demonstrated that CDLs have a stronger binding affinity compared to other mitochondrial lipids, providing a key example of lipid-mediated protein-protein interactions.441 The formation of the supercomplex could facilitate rapid electron transfer between proteins, thereby maintaining efficient energy transduction.
Figure 43.
CG simulations describe the diffusion of CDL in a mixed POPC/CDL bilayer and enable the detection of stable binding sites of CDL on the surface of the cytochrome bc1 complex. The CDL binding sites shown in yellow volume are mapped at an isovalue corresponding to at least 5 times the average bulk density. Reprinted with permission from ref 440. Copyright 2013 American Chemical Society.
Figure 44.
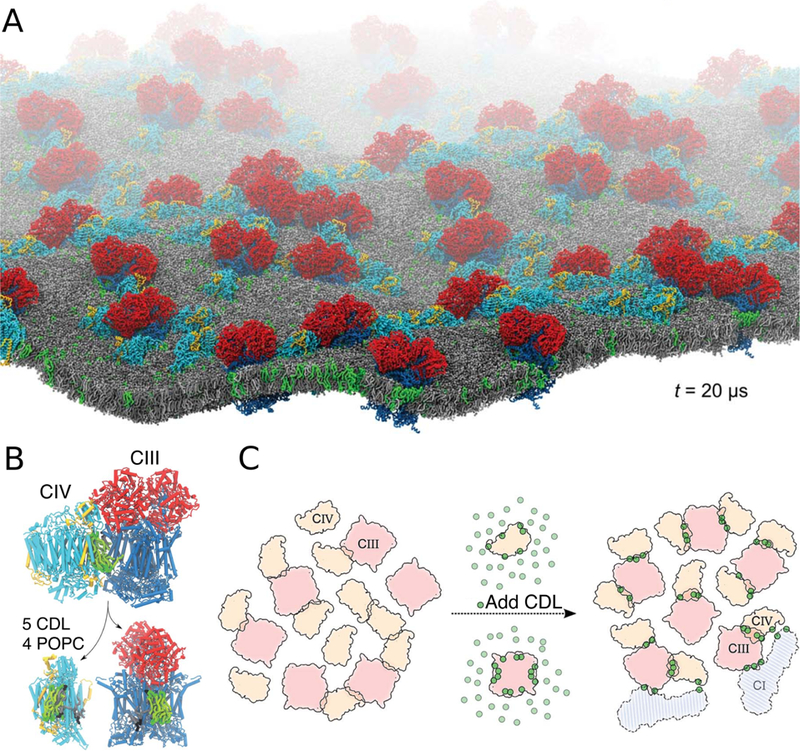
CDL mediated formation of respiratory supercomplexes. (A) View of the CDL-containing system after 20 μs self-assembly CG simulation. CDL and POPC are shown in green and grey, respectively. Cytochrome bc1 (complex III, CIII) and Cytochrome CcO (complex IV, CIV) are colored as in (B). (B) A snapshot at the end of the simulation showing the lipid content at the bc1/CcO interface. (C) Schematic model of CDL implication in the formation of the supercomplexes. The presence of CDL (green dots) increases the contacts between bc1 and CcO. Two copies of NADH dehydrogenase (complex I, CI) are shown to illustrate its possible integration to the bc1/CcO supercomplexes formed during the simulations. Adapted with permission from ref 441. Copyright 2016 The Royal Society of Chemistry.
The mitochondrial adenine nucleotide translocase, also known as the ADP/ATP carrier (AAC), is one of the best characterized members of the mitochondrial carrier family, the optimal activity of which relies on the presence of CDL. High-affinity CDL binding sites identified using CG and AA simulations on the bovine and yeast AACs were shown to agree well with those inferred from crystal structures and NMR measurements.369,817 Free energy calculation showed that CDL not only is selectively favored over zwitterionic lipids at the binding sites, but also binds tighter compared to the non-binding regions of the protein (Figure 45).280,369 Moreover, CG simulations of a large membrane patch containing multiple copies of AAC showed that CDL binding promotes the AAC oligomerization.369
Figure 45.
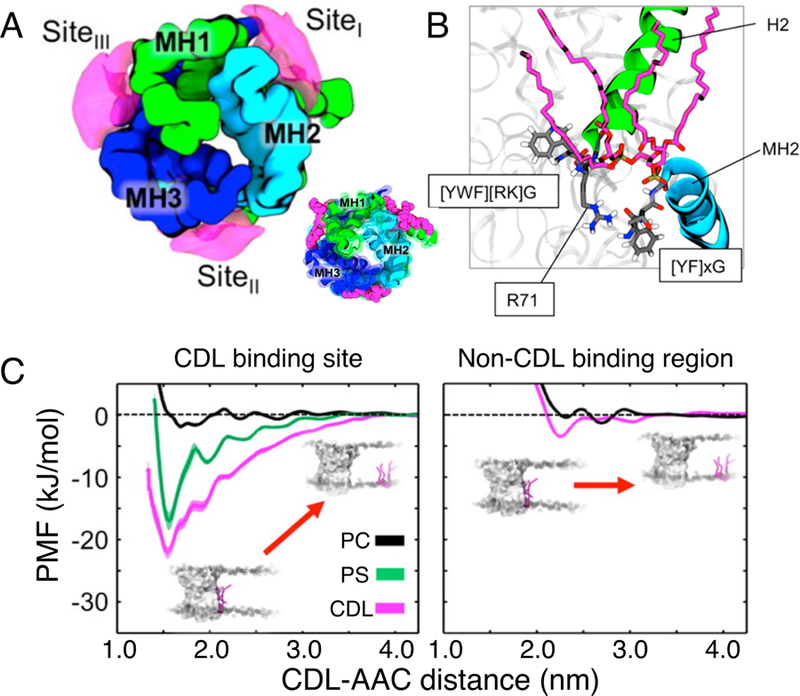
CDL interaction with the mitochondrial ADP/ATP carrier (AAC). (A) The time-averaged probability density surface of the CDL binding sites (magenta) revealed by CG simulations. Inset illustrates CDL binding sites observed in the X-ray structure (PDB: 1OKC). (B) The arrangement of the conserved motifs (grey) around the bound CDL (magenta) after refinement with AA simulations. (C) Potential of mean force profiles for the interaction of various lipids (CDL, PC and PS) at the X-ray observed CDL binding site (left) and a control non-CDL binding region (right). At the X-ray binding site, CDL binds more favorably than PS and PC. Adapted with permission from ref 369. Copyright 2016 American Chemical Society.
In contrast to the stable association of CDLs on the surface grooves of bc1, CcO, and AAC, the binding of CDL to the convex surface of the Fo domain of FoF1-ATP synthase was shown to be highly specific, although transient.818 Since the Fo domain is involved in proton translocation, its transient but repeated contacts with CDL suggest a role of CDL in stabilizing and “lubricating” the rotation of the domain, or aiding in proton transfer through this protein complex.
In addition to mitochondrial membrane proteins, computational studies also captured the interactions of CDL with plasma membrane proteins. For example, lipid organization in the proximity of the ABC transporter McjD demonstrated the preferential association of anionic lipids CDL and PG over zwitterionic lipid PE during the simulations, which is essential for the protein function. 338 Self-assembly simulations of lipids around the bacterial UraA H+-uracil symporter revealed several potential binding sites of CDL, which were further validated by in silico mutations that abolished the binding.359 Since CDL may act as a proton reservoir, its interaction with UraA implies a role of CDL in the H+-driven transport function. Beyond that, CDL was also found to bind at the dimer interface of LeuT and NhaA transporters, suggesting a role in their oligomerization.802
Another functionally important aspect of CDL that has been characterized computationally is its role in modulating the association of peripheral proteins with membranes. Using distinct membrane models, simulations have elucidated the enhancement of membrane anchoring by the electrostatic interactions between the CDL headgroups and proteins.195,496,819–821 In one example, CDL was found to promote the association of the membrane-bound soluble receptor domain of Tim50 to the transmembrane channel Tim23, highlighting the importance of CDL in the mechanism of the mitochondrial transport complex. 820 Another example is cytochrome c, a soluble protein located in the mitochondrial inter-membrane space, in which binding to CDL-rich membranes was found to result in the clustering of CDL lipids, thereby inducing a negative membrane curvature.821
5.2.5. Phosphoinositides
Phosphoinositides or phosphatidylinositol phosphate (PIP) molecules constitute a special class of phospholipids responsible for mediating signaling processes and membrane dynamics.822–824 PIP-protein interactions are essential in the membrane association of many proteins including phosphatases and kinases that control PIP concentrations,825,826 as well as PIP transfer proteins that regulate PIP cellullar localization.827,828 The increase of the PIP level enhances the recruitment of peripheral PIP effector proteins to membrane and further triggers the activation of their downstream pathways.826,829 Multiple PIP binding domains have been structurally characterized in these PIP binding proteins, showing various PIP specificities in terms of their different phosphorylation at the inositol ring.9,830 PIP lipids also act as regulators of transmembrane proteins, in particular ion channels.831,832 Together with free energy methods, AA and CG simulations have illuminated diverse binding modes in PIP binding targets. Here, we will review simulation studies on specific PIP-protein interactions, with some emphasis on phosphatidylinositol 4,5-bisphosphate (PIP2), one of the most abundant and well-studied signaling PIP lipids.
Pleckstrin homology (PH) domain is among the most representative PIP binding domains commonly found in cellular signaling proteins.830,833 Despite similar folds, PH domains exhibit various binding specifi for PIP lipids and their soluble headgroups, inositol phosphates (IPs).9,830 Driven by the determination of PH domain structures with co-crystalized PIP/IPs, AA simulation studies have provided atomistic details on how PIP/IP binding affinities are determined by the structural characteristics of PH domain from multiple proteins, including enzymes such as phospholipase C δ1,834 protein kinase B,835 and Bruton’s tyrosine kinase,836 and cell adhesion protein kindlin-1.837 Several simulation techniques, including nonequilibrium MD and Brownian dynamics simulations, have been used to capture spontaneous binding of the general receptor for phosphoinositides isoform 1 (GRP1) PH domain to membranes containing phosphatidylinositol 3,4,5-trisphosphate (PIP3).838–840 CG simulations have also proven powerful in sampling PH domain membrane targeting as well as exploring various PH domain membrane binding modes in nucleotide exchange factor Brag2,841 exocyst protein Sec3,842,843 and adaptor proteins Dok7534 and DAPP1.844,845 Particularly, in a modified version of CG MARTINI protein model with improved sampling of protein side-chain dynamics, the translational and orientational motions of the PLCδ1 PH domain were greatly enhanced compared to AA simulations during the association to PIP2-containing membranes.846
Free energy methods, including metadynamics and umbrella sampling, have been recently utilized to quantitatively study lipid specificities in PIP-PH domain interaction.847–849 Notably, two different PIP2 binding pockets were identified at the PH domain of ACAP1BAR−PH protein with extensive AA and umbrella sampling simulations (Figure 46A).850 Different binding modes were also characterized in two other studies adopting a systematic multiscale approach, which brought together detailed PIP-PH domain interaction resolved in crystal structures and thermodynamic information derived from simulations (Figure 46B).851,852 Altogether, these simulations have depicted a well-rounded view of PIP-PH domain interactions.
Figure 46.

PIP-PH domain interaction examined by free energy and multiscale methods. (A) Free energy profiles of PIP2/POPS binding to two different pockets (1&2) in a PH domain calculated from umbrella sampling. Reprinted with permission from ref 850. Copyright 2017 American Chemical Society. (B) A multiscale approach combining crystallographic data and MD simulations to characterize PH domain binding to a PIP-containing membrane. Reprinted with permission from ref 852. Copyright 2018 Elsevier.
In addition to simulations of the PH domain, MD simulations have been widely used to study PIP interactions with other proteins containing PIP binding domains. For instance, AA simulations of protein kinase Cα C2 domain have provided insights into PIP2/PIP3 specifity,853 PIP2 stoichiometry,854,855 C2 domain docking geometry,854,855 and the weakening effect of membrane diacylglycerol on PIP2-C2 domain interaction.855 The critical role of Ca2+ has also been illustrated in promoting PIP2 binding at the C2 domain of enzyme phospholipase D856 and exocytosis-associated protein double C2 domain protein B.857 Likewise, MD simulations have elucidated how PIP2 influences membrane dynamics by its interaction with the FERM domain of cell adhesion proteins including talin,858 moesin859,860 and focal adhesion kinases.861–863 Combining CG with AA simulations, PIP interactions with PTEN domains and PTEN-like domain have been investigated in phosphatase PTEN proteins500,504 and endocytosis-associated protein auxilin-1,864 suggesting a synergy between specific and non-specific protein-lipid interactions in the membrane targeting process. Such a synergy has also been described in the association of two phosphatidylinositol 3-phosphate (PI3P) specific binding domains, FYVE and PX domains, with PI3P-containing membranes.865,866 Besides, specific PIP interactions have been revealed for other PIP binding domains such as the TH domain in the tumor necrosis factor-α-induced protein 8-like (TIPE) family867 and the PROPPIN domain in β-propellers.868
Similar to other anionic lipids, PIP interactions with peripheral proteins are often accompanied with PIP lipid clustering, as observed in the MD studies of viral proteins675,676,678,805 and curvature-inducing domains including amphiphysin N-BAR domain,869 AP180 ANTH domain869 and epsin ENTH domain.560,869 Futhermore, PIP-containing membrane association and specific PIP interactions have been characterized in simulations of peripheral proteins without specific PIP binding domains, including Osh4,808 HCV protein,489 Rab5,532 K-Ras,191,523 fibroblast growth factor 2,870 MARKCS-ED peptide,190 gelsolin,871 cofilin, 859 L-selectin,860 syntaxin,872,873 and other fusion-associated proteins.842,843
Upon its association with integral membrane proteins, PIP2 has been found in MD simulations to cluster around proteins due to its highly negatively charged nature.874–877 Moreover, PIP2 can regulate integral membrane proteins in a unique way because of its larger head-group compared to other anionic phospholipids, in that it is able to interact with proteins at both their transmembrane (TM) domain and their cytosolic linker/domain or juxtamembrane (JM) domain. For example, conserved PIP2 binding sites have been identified at the TM subunit interfaces in Kir channels.276,277,280 In addition, basic residues at the cytosolic C-terminal linker/tether and loops of Kir channels have also been characterized as PIP2 binding targets.278,279,281–283 Similarly, direct PIP2 interactions with cytosolic linkers have been illustrated to be involved in gating of Kv channels248–251 and the human two-pore channel 2.252 In several other channels and transporters, e.g., the TRPV4 channel,878 the glutamate transporter,879 and the dopamine transporter,349 PIP2 modulation has been observed at their cytoplasmic N-terminal or C-terminal region. Another typical case of interest is PIP2-JM domain association in receptors such as receptor tyrosine kinases433 and the epidermal growth factor receptor.880,881 Multiscale approaches, umbrella sampling, and large-scale CG simulations have shed light on conserved PIP2 binding sites,433 free energy landscapes governing PIP2-JM domain interaction,880 and the interplay between protein organization and PIP2 clustering in complex plasma membranes,881 respectively. A recent CG study also demonstrated a dual role of PIP2 in inducing the activation of GPCR A2AR by stabilizing a TM helix and associating A2AR with an engineered G protein.397
5.3. Sphingomyelin and LPS
Recent advancements in force field parametrization882 and computational modeling883 have made it feasible to perform biomolecular simulations with more realistic and complex membrane compositions. In this section we will focus on the MD simulations which have lever-aged these recent force field advancements to study the behavior of sphingomyelin (SM) and lipopolysaccharide (LPS).
5.3.1. Sphingolipids
SM is a sphingolipid ubiquitously found in plants, fungi, animals and prokaryotic membranes. It consists of phosphoethanolamine and ceramide or phosphocholine head group, thus classified as sphingophospholipids. 884 In mammals, SM forms 85% of all sphingophospholipids, majority of which is located in the outer leaflet of the plasma membrane885 and indispensable for the viability of mammalian cells.886 Along with cholesterol and other phospholipids, SM is implicated in the formation of microdomains in biological membranes.728,729 These microdomains work as a platform for cellular processes such as signal transduction, protein sorting and membrane trafficking.887,888 Many signaling proteins, such as kinases, GPCRs, growth factor receptors, and PKC, are primarily found to localize in the cholesterol and sphingomyelin rich domains, where sphingomyelin allows for tight intercalation of cholesterol.888 Thus, sphingolipids play an important role in cell growth, death, migration, adhesion and inflammation. 889 Abnormally high levels of SM in Neimann-Pick diseases are believed to modulate the cellular behavior. Binding of Niemann-Pick protein C2 (NPC2) to endosomal/lysosomal membrane was studied using MD simulations,787 capturing two competitively favorable membrane binding modes with a low energy barrier for their interconversion. The first mode was shown to be disrupted by the incorporation of SM while the second binding mode was invariant to SM concentration. Apart from modulating the cell behavior, SM was found to play an important role in the activation of GPCRs890 and orientational dynamics of CD2 ectodomain.891
Besides transmembrane proteins, GPI-anchored proteins, in which a glycolipid is attached to the C-terminus of a protein, show a strong affinity to the highly ordered lipid bilayers rich in cholesterol and SM.892 A combination of biophysical experiments and MD simulations highlighted SM binding sites on CYP2B4.464 The stability of the protein was enhanced by the formation of SM-rich domains, which drastically altered the binding of a hydrophilic ligand. SM and ganglioside (GM-1), another sphingolipid, were shown to promote structural changes and peptide aggregation in Aβ,617,619 aquaporin (AQP1),893 WALP23,893 pore-forming toxins894 and cholera toxin.895,896 The oligosaccharide group on GM1 was found to act as a scaffold for Aβ binding. Starting from an α-helical structure, the bound Aβ monomer formed a β-hairpin motif, and the β structure was further enhanced due to peptide-peptide interaction in the dimer state.
5.3.2. Lipopolysaccharides
The outer membrane (OM) of Gram-negative bacteria is a highly asymmetric lipid bilayer. The inner leaflet is exclusively composed of PE, PG, and CDL, while the outer leaflet is composed of only LPS. An LPS molecule consists of a hydrophobic lipid A moiety embedded in the OM, a hydrophilic core of oligosaccharides and repeating chains of O-antigen polysaccharide. Lipid A, which consists of a phylogenetically conserved core and highly variable O- and N-acylated β-(1→6)-linked, forms the basis for serogroup determination in bacteria. Given its highly charged nature, LPS makes the bacterial surface strongly polar thereby preventing the diffusion of hydrophobic drugs, dyes and antibodies. LPS is also known to modulate the structural dynamics of the outer membrane porins.
As compared to a pure phospholipid bilayer, simulation studies of the bacterial outer membrane porins such as BAM,312 FecA,371 OprH,305 OmpF306 and OprF304 in LPS-containing bilayers have revealed differential dynamics in their extracellular loops. The loop dynamics were shown to be dependent on LPS composition and were greatly reduced by the incorporation of O-antigen subunits (Figure 47). As it is difficult to obtain a defined orientation of the loops from solution NMR, the advancements in LPS modeling may provide an opportunity to refine the structure of outer membrane proteins in their natural environment. In addition to the loop dynamics, conformational flexibility of lipid A modulates the dimerization and activation of myeloid differentiation factor which is involved in the control of bacterial infections.897 MD simulations have highlighted the critical binding features of surfactant protein A (SP-A), involved in DPPC and lipid A binding.718 Along with the three-walled tyrosine cage on SP-A, which was shown to form cation-π interactions with the lipid headgroups, a positively charged cluster on the protein surface was also shown to be critical for lipid A binding. Furthermore, steered MD simulations have suggested that SP-A binds lipid A more tighly than DPPC.
Figure 47.

Computational modeling of OprH in an LPS bilayer. Chemical structures of lipid A, LPS core sequences and O-antigen of outer membrane (OMs) with the corresponding simulation box are shown in (A-F). The color representations are as follows: pink spheres, lipid A; orange sticks, O10-antigen polysaccharides; gray sticks, core sugars; blue spheres, PPPE; orange spheres, PVPG; magenta spheres, PVCL2; small cyan spheres, Ca2+ ions; small magenta spheres, K+ ions; small green spheres, Cl ions. 305 Reprinted with permission from ref 305. Copyright 2017 Elsevier.
6. Protein-lipid Interactions and Membrane Structure
The biological membrane structure undergoes drastic remodeling during cell division, vesicle trafficking, viral entry, and other membrane-mediated cellular events.898–901 Being a liquid crystalline mesophase, the biological membrane structure is a unique combination of solid crystals and conventional liquids.902 Correspondingly, it can be studied from two complementary perspectives: mechanically, as a thin elastic solid sheet with specific moduli of bending and compressing, 903 or, dynamically, as a 2D array of freely diffusing lipids.904 Experimental results have provided phenomenological evidence of proteins modulating membrane structure from both perspectives. On one hand, high-resolution structures of membrane-associated protein assemblies suggest their ability to sculpt membranes, e.g., the cage-shaped structure of clathrin-coated vesicles during endocytosis,905 and the V-shaped ATP synthase dimer from yeast mitochondria.906 On the other hand, NMR and EPR measurements of isotope or nitroxide radical labelled lipids show evident perturbation in lipid dynamics when integral proteins are added to membranes.907
Microscopically, most membrane proteins introduce local perturbations to lipid bilayers via hydrophobic mismatches and electrostatic interactions. These effects can translate into a larger-scale global membrane curvature under certain scenarios which include, but are not limited to, helix insertion into one leaflet incorporation of an irregularly-shaped integral membrane protein, or scaffolding by a peripheral membrane protein.908 Although these mechanisms are difficult to be quantitatively described experimentally, they can be elucidated by MD simulations which capture molecular events with both high temporal and spatial precisions. This part of the review focuses on simulation studies on how proteins modulate the mechanical and dynamical properties of the lipid bilayers. It also covers a related subject that proteins change conformation or redistribute along the lipid bilayer upon sensing the membrane structure.
6.1. Membrane Mechanical Properties
Each defined membrane structure has a specific set of mechanical properties,909–911 which dictate its response to forces resulting from the binding of peripheral proteins or the incorporation of integral membrane proteins, exerting distinct stresses to the membrane and eliciting different membrane effects.912
Asymmetrically bound peripheral proteins are well-suited to produce a curved membrane because they can accommodate divergent stresses experienced by the two leaflet stretching for one leaflet and compression for the other. Examples of membrane-bending peripheral proteins that have been computationally well-characterized include BAR domains which scaffold the membrane mainly with their curved faces,542,543,548,556 and synaptotagmin C2B domain which achieves membrane bending via insertion of its amphiphatic helix into the membrane.562 These membrane-bending peripheral proteins have been discussed in Section 4.4.
Integral membrane proteins can also result in membrane curvature. In AA simulations of three different protein assemblies from the chromatophore of purple bacteria, it was observed that the light harvesting complex II (LHII) bent the membrane patch within 10 ns.793,794 This effect was attributed to a combination of the intrinsic shape of LHII and the electrostatic interactions between conserved charged residues and nearby lipids on the cytoplasmic side. CG simulations of the ATP synthase dimer from mitochondria also revealed an anisotropic membrane curvature induced by the dimer.906 Furthermore, when multiple dimers were arranged side by side, they generated a highly curved membrane ridge reminiscent of the boundary of mitochondria cristae.906
In most cases, integral membrane proteins cause only local deformations in membrane thickness and curvature. Nevertheless, these membrane perturbation profiles can reflect specific modulations by proteins, and have recently been proposed in a CG simulation study to serve as the fingerprints of membrane proteins.913 In this series of simulations, four copies of each protein from ten classes were placed in the same lipid bilayers to sample their membrane perturbation profiles, which turned out to be intriguingly complex yet distinct across these studied membrane protein categories (Figure 48).913
Figure 48.
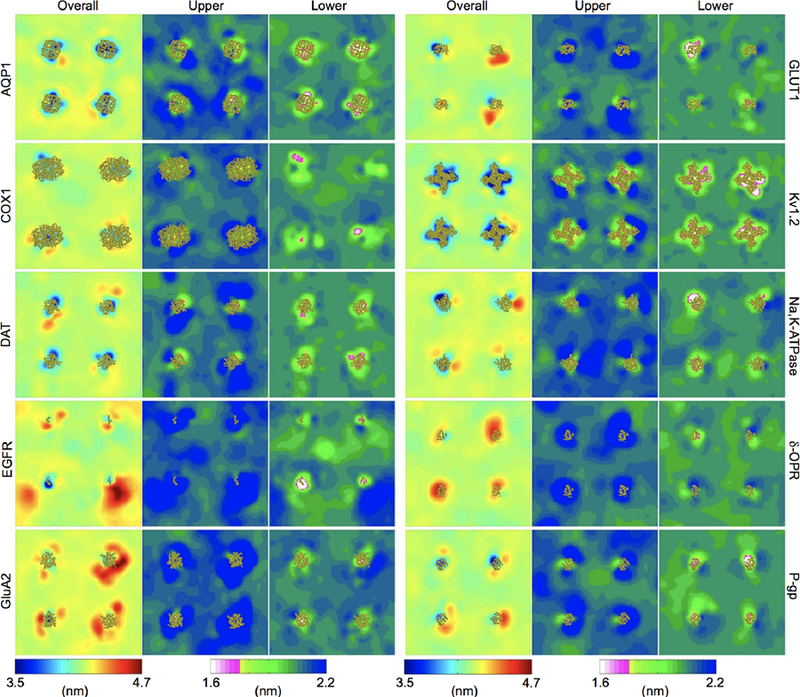
Membrane thickness profiles near ten different membrane proteins from CG simulations. For each of the simulation systems, membrane thickness is shown as 2D maps, averaged over the simulations from 25 to 30 μs. Overall thickness, the distance calculated between the upper and lower surfaces, is shown color-coded according to a 3.5–4.7 nm scale. The thickness for the upper leaflet (as distance between the upper and the middle plane) and for the lower leaflet (as distance between the lower and the middle plane) is shown on a different color scale, ranging from 1.6 to 2.2 nm. The position of the four protein copies in each simulation box is indicated by drawing the proteins in yellow ribbons. The studied membrane proteins are aquaporin AQP1, cyclooxyge-nase COX1, dopamine transporter DAT, epidermal growth factor receptor EGFR, AMPA receptor GluA2, glucose transporter GLUT1, K+ channel Kv1.2, Na+-K+ pump Na,K-ATPase, opioid receptor δ-OPR, and P-glycoprotein P-gp. Reproduced with permission from ref 913. Copyright 2018 American Chemical Society.
The membrane perturbations caused by integral membrane proteins have been also extensively studied with AA simulations, which can yield more details than CG simulations. It was shown that the sensing domain of archaeal K+ channel KvAP induced a significant membrane deformation along its S4 helix; such deformation was mostly observed around the positively charged half-helix, with a much weaker effect around the other hydrophobic half-helix.792,914 Intra-membrane protease GlpG, which has a non-cylindrical shape and a short hydrophobic thickness, rearranged the nearby lipids and caused non-uniform thinning of membrane around the protein.450 With a combination of AA simulations and elastic membrane model, it has been demonstrated that helical transmembrane proteins such as rhodopsin915 and LeuT347 induce local membrane deformations to establish optimal hydrophobic-polar interactions between the lipids and the protein. Similarly, AA simulations have captured membrane thinning in the vicinity of β-barrel proteins including BamA and the outer membrane phospholipase A (OmpLA).307,312 Furthermore, by analyzing membrane thickness, area per lipid, and lipid tilt angle, AA simulations showed that the influence of integral proteins on the membrane structure extended up to 3 nm away from the protein boundary, much farther beyond the first shell of lipids.801,916
In addition, the lipid bilayer itself has been found to undergo thermal fluctuations that are inversely correlated to the membrane stiffness, and the association of densely packed membrane receptors was shown to reduce such membrane fluctuations, rendering the membrane deformation more dispersed and with a smaller amplitude.876 In a follow-up study, it was demonstrated that integral proteins altered the bending rigidity of the membrane in a protein-concentration and lipid-composition dependent manner, while peripheral proteins had little effect on membrane stiffness.799
Furthermore, dynamics and structural changes of integral proteins can result in changes in mechanical properties of the membrane, and/or be affected by them. For example, the function of membrane transporters usually call for major conformational changes, which are accommodated by the surrounding membrane and result in different stresses to the membrane. Two different conformations of Ca2+ translocating ATPase SERCA embedded in lipid bilayers, for example, caused different local deformation profiles of the membrane, due to differntial sidechain conformations and helix tilts.917
6.2. Membrane Dynamical Properties
Membrane structure is far from homogeneous and static.918,919 Different membrane phases/domains form, migrate, and disintegrate as individual lipid molecules continuously diffuse within the membrane. Aside from their intrinsic properties,920 lipid motions, including translation and rotation, are heavily influenced by membrane proteins.
A single transmembrane helix was shown to significantly reduce diffusion of lipids in its vicinity; in particular, the positively charged lysine and arginine residues trapped the negatively charged POPS lipids by electrostatic interactions.921 Later CG simulations also found a reduced diffusion of phospholipids around larger transmembrane proteins up to 3 nm from the protein boundary.796 An asymmetry of lipid diffusion within the inner and outer leaflet was observed in the OmpF porin system (Figure 49) and attributed to the asymmetry in charged protein residues interacting with lipid headgroups.796,801 Moreover, it was found that clusters of influenza hemagglutinin tuned the diffusion of nearby lipids differently, leading to enrichment of ordered lipids within the protein cluster.922 In extreme cases, lipid diffusion is reduced by proteins to such a degree that they appear to be bound in a fixed pose. For instance, annular lipids were resolved in 2D crystals of aquaporin-0.923 Intriguingly, when the aquaporin-0 tetramer was simulated in a DMPC bilayer, certain lipids were found to be trapped near the protein surface, adopting similar conformations to the ones observed in the crystal structure. 924 Later, it was demonstrated with MD simulations that localization of these annular lipids was more critically dependent on the mobility of protein surface residues than the mobility of lipids themselves.925
Figure 49.
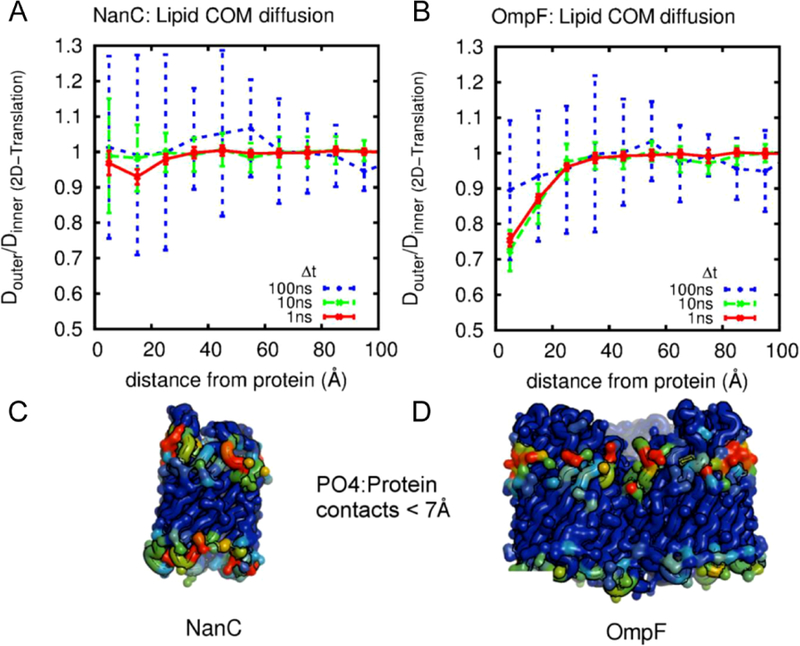
Leaflet asymmetry of lipid mobility near NanC and OmpF. Ratio of diffusion coefficients between inner and outer leaflet for (A) NanC and (B) OmpF as a function of distance from the protein at different times. Error bars are the standard errors from 6 sub-trajectories. Asymmetry can be seen in the OmpF simulations for distances from the protein of less than 20 Å. (C,D) The corresponding proteins colored based on time averaged number of contacts (cutoff 7 Å) to lipid phosphates on a blue (0%) to red (100%) scale. Reprinted with permission from ref 796. Copyright 2013 Goose, Sansom. Licensed under a Creative Commons Attribution 4.0 International License.
In sharp contrast to lateral lipid diffusion, spontaneous lipid flip-flop across leaflets occurs more rarely due to the high energy cost associated with embedding the lipid headgroups into the hydrophobic interior of the membrane.926 In the cell, translayer diffusion of lipids is usually facilitated by scramblases or ATP-dependent flippases/floppases, and sometimes by certain lipid components such as ceramide. 927 In an umbrella sampling study based on AA simulations, the free energy barrier for PE and PG flip-flop was shown to be decreased by a few kJ/mol when a model transmembrane peptide was present in the membrane, while the barrier for PC flipping was not affected.928 However, the molecular basis for protein-facilitated lipid scrambling was not revealed until recent computational studies on scramblase,325–327 where simulations unequivocally captured events of lipid translayer diffusion explicitly (see Section 3.1.5).
6.3. Membrane Structure Sensing
Interactions between lipids and proteins are reciprocal. While proteins influence the membrane structure, they can also sense the surrounding membrane structure and change their physical behaviors correspondingly. These responses include diffusion/distribution within the membrane, oligomerization, and structural rearrangement. Though membrane structure sensing is discussed throughout this review, we believe presenting a consolidated selection of representative cases studied with MD simulations is useful.
Firstly, protein diffusion can be affected by membrane curvature. A fraction of ENTH domains have been shown to diff faster than the lipids, indicating that the free energy gradient generated by the membrane curvature field is sensed by the proteins.567 Secondly, distribution of proteins on/in the bilayer can be affected by the membrane structure. With CG simulations, it has been shown that the transmembrane helical model peptide WALP23 accumulates in curved membranes, regardless of the lipid composition.929 Similarly, the amphipathic helix of the influenza virus M2 channel binds predominately to the positively curved surface of a buckled membrane.930 Protein partitioning is also altered by the membrane phase. For instance, glycophorin A dimer favored the lipid-disordered phase over the lipid-ordered one.931 Thirdly, protein oligomerization can be modulated by the membrane thickness.932 Among four different lipid bilayers simulated, rhodopsin self-assembly was more prominent in thinner bilayers due to a greater hydrophobic mismatch.932 Lastly, proteins may change their tertiary structure in response to the membrane curvature.933 For instance, it has been shown for the N-terminal helix of endophilin that lipid packing defects of a convex membrane promoted helical folding by several kcal/mol while flat and concave membrane surfaces inhibited folding.933
MD simulation is indispensable and widely adopted to provide insight into molecular mechanisms for the crosstalk between protein and membrane structure with high spatial and temporal precisions. It should be noted that another class of computational methods can also evaluate the bilayer deformation and membrane curvature caused by protein-lipid interactions.328 These methods supplement the Helfrich’s elastic membrane model934 with either boundary condition from the transmembrane protein structure or Hamiltonian terms describing protein-membrane interactions.935–944 Many authors found their results from the elastic model to agree well with those from AA or CG simulations.944–947 Elastic membrane models are computationally less expensive, at the cost of losing the molecular detail and temporal relevance, but have the potential to qualitatively capture slow membrane remodeling events.
7. Future Directions and Concluding Remarks
The importance of lipids in membrane protein function is now well established, both through a large body of experimental studies employing a variety of biochemical and biophysical techniques, and also by numerous simulation studies, as reviewed in this article, offering complementary high spatial and temporal resolutions and thus providing a more detailed picture of the underlying molecular phenomena. Lipids exert their effects either through modulating bulk properties of the membrane which in turn can impact conformational dynamics and equilibria of membrane proteins, or via specific, direct interactions with membrane proteins. Specific lipid types have been shown to be directly involved in key signaling pathways, and the cell often relies on modulating their concentration and/or localization within the membrane to activate or shut down such pathways. Biological membranes and their lipid constituents, therefore, can no longer be viewed as passive hydrophobic barriers, merely forming boundaries around the cell and its inner compartments. We have only begun to discover the many ways by which the dynamically controlled heterogeneity of lipid bilayers in biological membranes can modulate the function of membrane proteins. With the emergence of more powerful hardware and advanced simulation techniques and algorithms, we can expect an even larger impact of simulations on our understanding of biological membranes and the role of lipids.
A major aspect in recent simulation studies of biological membranes has been the introduction of increasingly more realistic lipid compositions, a feature that has experienced substantial improvement over the last decade. Recent development of various AA371,948 and CG949,950 models have greatly expanded our ability to model increasingly complex membranes, such as the bacterial outer membrane (OM), an asymmetric membrane composed exclusively of lipopolysaccharides (LPS) in the outer leaflet and of a mixture of lipids including anionic ones in the inner leaflet (in E. coli : 90% PE, 5% PG, and 5% CDL).951 With these advancements we can now start to ask questions about, e.g., mechanical properties of the bacterial OM, and how the LPS composition of the outer leaflet may interfere with OM proteins. The library of lipids and their derivatives available for simulation studies will continue to expand as more information becomes available on the lipid composition of membranes in various cells and organelles and as novel roles of lipids are discovered experimentally.
Similar to other areas of biomolecular simulation, computational studies of membranes will continue to benefit from more accurate descriptions offered by better treatments of the interactions. Hybrid QM/MM simulations, which have been widely used in studies of proteins and enzymes, are expected to play a more visible role in simulation studies of lipids and membranes. As an example, one can easily imagine that the interaction between the ring system of cholesterol704 and the protein environment is affected by electronic processes such as charge delocalization and transfer, which cannot be handled sufficiently accurate with classical force fields. Other examples include cation-π interactions, which can easily arise in interactions between positively charged moieties of lipids and aromatic sidechains of the embedded protein. Yet another example regards a challenging aspect in setting up MD studies of lipids related to the prediction of the titration states of protonatable moieties, a feature that can be dynamically changed not only by mixing of lipids within the bilayer, but also by binding of peptides and proteins to the membrane. “Constant pH” MD simulations, though still classical, offer reasonably affordable methods with good accuracy to take into account such effects. Protein binding to anionic lipids such as PA is a good example in this context. While PA in its isolated form carries a charge of −1, the pKa of the second protonation is close to physiological pH, and PA binding to some proteins is known to be pH dependent.814 Such effects could also be relevant to PIP lipids. Either QM or constant pH simulations could credibly be used to investigate these phenomena. Polarizable force fields, as their computational cost decreases, will also allow for greater accuracy in modeling proteins and their interaction with lipid phases, and thus enable researchers to more accurately characterize membrane-associated systems and processes by MD simulation. In addition, changes in charge distribution could be of interest in systems where protein-lipid interactions are particular to a specific lipid species, potentially giving insight into the source of the specificity.
Description of protein-lipid interactions will continue to improve with the advanced enhanced sampling techniques and free energy methods. While a number of studies have employed these techniques in simulation studies of membranes and membrane proteins, the main objective of these studies has been largely on aspects other than lipid-protein interactions, e.g., conformational landscape of membrane transporters, or accelerated mixing simulations of pure lipid bilayers. Given the slow diff of lipids, achieving converged free energies of lipid-protein interactions continues to be among the more challenging tasks, but we have already started to explore such processes at a more quantitative level with advanced simulation techniques.
Finally, we should expect more simulation studies in the future where realistic non-planar geometries of membranes as relevant to many cellular structures and processes, e.g., vesicle formation and curved membranes of organelles, will be included in the simulation system. One such example is the SNARE-mediated membrane fusion,952 which is modulated by SM (Sec1/Munc18-like) proteins, complexins, and synaptotagmins. Such mechanical aspects are known to be coupled to lipid distribution and protein localization. To bridge the gap between simulation and physiology, future membrane simulations need to incorporate richer biological contexts. In addition, to capture larger and slower membrane remodeling events by proteins with molecular accuracy, new approaches that can leverage the precision of AA as well as the efficiency of CG needs to be developed. Equally importantly, there is a need for methodology allowing to construct such large, cell-scale systems, which can easily add up to many billions of atoms/particles, with reasonable ease and efficiency.
The future of simulation studies of membranes and membrane proteins is very promising. The field can expect to see more examples in which molecular simulations of increasing accuracy, realism, and quantification will provide novel insight into the molecular mechanisms underlying biological observables. Combined with novel experimental techniques and studies unraveling new biological roles for lipids, MD simulations will allow us to understand why lipids were selected by evolution to play such well-tuned regulatory roles in membrane biology and protein function.
9. Acknowledgements
The authors would like to acknowledge support from the National Institute of General Medical Sciences of the National Institutes of Health under awards P41-GM104601, U54-GM087519, R01-GM101048, and R01-GM123455. MPM would like to acknowledge support of the National Heart, Lung and Blood Institute of the National Institutes of Health under award F31-HL136155. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
8. Biographies
Melanie P. Muller earned a B.S. in Mathematics and a B.S. in Chemistry in 2014 from the University of Massachusetts Amherst, after earning an AGS from Middlesex Community College in Connecticut. She is currently a duel degree Medical and Doctoral student at the University of Illinois at Urbana-Champaign, working toward a Ph.D. in Biophysics and Quantitative Biology under the supervision of Prof. Emad Tajkhorshid. Her research interests include application of computational modeling and advanced simulation techniques to coagulation protease binding to and complex formation on the cellular membrane.
Tao Jiang received her Ph.D. degree in Biophysics and Computational Biology at University of Illinois at Urbana-Champaign in 2017 under the supervision of Prof. Emad Tajkhorshid for her study of membrane transport proteins using molecular dynamics simulations. She is currently a postdoctoral research associate in the Department of Biochemistry and the NIH Center for Macromolecular Modeling & Bioinformatics. Her research interests include the application of computational modeling and advanced simulation techniques to understand the dynamical properties and functional mechanisms of membrane transport machineries, such as channels, transporters, and phospholipid scramblases.
Chang Sun received his B.S. in Pharmacy from Fudan University in 2010, where he became interested in membrane proteins. He then went to the University of Illinois at Urbana-Champaign to study bioenergetic enzymes with optical and electron paramagnetic resonance spectroscopy under the supervision of Profs. Colin A. Wraight and Robert B. Gennis. After receiving his Ph.D. in Biochemistry in 2016, he started to collaborate with John Rubinstein’s group to determine membrane protein structures using cryo-EM. In 2017, he joined Emad Tajkhorshid’s group as a postdoctoral research associate and had been focusing on modeling atomic structure from cryo-EM density maps. His research interests include cryo-EM, computational biology, bioenergetics, and neuroscience.
Muyun Lihan received his B.Sc. in Biological Sciences from Peking University in 2014 and is currently Ph.D. student of Biophysics and Quantitative Biology at the University of Illinois at Urbana-Champaign under the supervision of Prof. Emad Tajkhorshid. Before he joined Prof. Tajkhorshid’s group, he worked on computational antibody design in the late Prof. Klaus Schulten’s group from 2014 to 2016. His current research interest is focused on understanding protein-cholesterol interactions using molecular dynamics simulations and machine learning techniques.
Shashank Pant graduated from the Indian Institute of Science Education and Research, Kolkata, India in 2015 with a B.S./M.S. in chemical sciences. He is currently a Ph.D. student of Biophysics and Quantitative biology at University of Illinois at Urbana-Champaign. Under the supervision of Prof. Emad Tajkhorshid, he is using enhanced sampling molecular dynamics simulations to investigate the conformational transition of various membrane transporters and the role of lipids in modulating their activity.
Paween Mahinthichaichan received his Ph.D. in Biochemistry with training in molecular biophysics from the University of Illinois at Urbana-Champaign in 2016 under the guidance of Profs. Emad Tajkhorshid and Robert Gennis. He is continuing his postdoctoral research with Prof. Tajkhorshid. His research primarily focuses on elucidating the delivery of gases and small molecules to biological macromolecules, and the effects of membrane compositions on the transport of those molecules across biological membranes.
Anda Trifan obtained her B.Sc in Chemistry from DePaul University in 2012. She is currently pursuing her Ph.D. in Biophysics from the University of Illinois at Urbana-Champaign. Under the guidance of Prof. Emad Tajkhorshid, her current research focuses on Ras protein-membrane interactions using classical molecular dynamics and QM/MM simulations.
Emad Tajkhorshid, PhD, is a Professor of Biochemistry, Biophysics and Quantitative Biology, Chemistry, Bioengineering, and Pharmacology at the University of Illinois at Urbana-Champaign. After receiving his initial training in Pharmacy and in Medicinal Chemistry and Pharmacology at Tehran University, and a PhD in Molecular Biophysics at the University of Heidelberg, he moved to UIUC initially as a postdoctoral associate with Prof. Klaus Schulten before joining the faculty in 2007 as an Assistant Professor, fast tracking to Associate Professor in 2010 and to Full Professor in 2013. He is currently a University of Illinois Scholar and an Endowed Chair in Biochemistry, and the Director of the NIH Biotechnology Resource Center for Macro-molecular Modeling and Bioinformatics. Prof. Tajkhorshids research focuses on developing computational methods and tools for biomolecular modeling and simulation, and their application to complex biological systems, most prominently in the area of biological membranes, membrane proteins, and membrane-associated phenomena with a strong emphasis on lipid-protein interactions. Examples include mechanistic studies of membrane transport proteins, principles of energy transduction and coupling in bioenergetic proteins, and lipid modulation of protein function, e.g., in signaling proteins associated with the cellular membrane. He has authored over 200 research articles (H-index 64) with over 24,000 citations.
References
- (1).Phillips R; Theriot J; Kondev J; Garcia H Physical biology of the cell ; Garland Science, 2012.
- (2).Edidin M Lipids on the frontier: A century of cell–membrane bilayers. Nat. Rev. Mol. Cell Biol 2003, 4, 414–418. [DOI] [PubMed] [Google Scholar]
- (3).Gennis RB Biomembranes: Molecular Structure and Function; Springer-Verlag: New York, 1989. [Google Scholar]
- (4).van Meer G; Voelker DR; Feigenson GW Membrane lipids: where they are and how they behave. Nat. Rev. Mol. Cell Biol 2008, 9, 112–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Spector AA; Yorek MA Membrane lipid composition and cellular function. J. Lipid Res 1985, 26, 1015–1035. [PubMed] [Google Scholar]
- (6).Devaux PF; Zachowski A Maintenance and consequences of membrane phospholipid asymmetry. Chem. Phys. of Lipids 1994, 73, 107–120. [Google Scholar]
- (7).Dowhan W Molecular basis for membrane phospholipid diversity: Why are there so many lipids? Annu. Rev. Biochem 1997, 66, 199–232. [DOI] [PubMed] [Google Scholar]
- (8).Bevers EM; Comfurius P; Dekkers DW; Zwaal RF Lipid translocation across the plasma membrane of mammalian cells. Biochim. Biophys. Acta 1999, 1439, 317–330. [DOI] [PubMed] [Google Scholar]
- (9).Lemmon MA Membrane recognition by phospholipid-binding domains. Nat. Rev. Mol. Cell Biol 2008, 9, 99–111. [DOI] [PubMed] [Google Scholar]
- (10).Stahelin RV Lipid binding domains: more than simple lipid effectors. J. Lipid Res 2009, 50, S299–S304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Wallin E; von Heijne G Genome-wide analysis of integral membrane proteins from eubacterial, archaen, and eukaryotic organisms. Prot. Sci 1998, 7, 1029–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).von Heijne G; Manoil C Membrane proteins: from sequence to structure. Protein Eng 1990, 4, 109. [DOI] [PubMed] [Google Scholar]
- (13).Shibata Y; Hu J; Kozlov MM; Rapoport TA Mechanisms shaping the membranes of cellular organelles. Annu. Rev. Cell. Dev. Biol 2009, 25, 329–354. [DOI] [PubMed] [Google Scholar]
- (14).Bevers EM; Williamson PL Getting to the outer leaflet: Physiology of phosphatidylserine exposure at the plasma membrane. Physiol. Rev 2016, 96, 605–645. [DOI] [PubMed] [Google Scholar]
- (15).Laganowsky A; Reading E; Allison TM; Ulmschneider MB; Degiacomi MT; Baldwin AJ; Robinson CV Membrane proteins bind lipids selectively to modulate their structure and function. Nature 2014, 510, 172–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Zwaal RFA; Comfurius P; Bevers EM Lipid-protein interactions in blood coagulation. Biochim. Biophys. Acta 1998, 1376, 433–453. [DOI] [PubMed] [Google Scholar]
- (17).Stace CL; Ktistakis NT Phosphatidic acid- and phosphatidylserine-binding proteins. Biochim. Biophys. Acta 2006, 1761, 913–926. [DOI] [PubMed] [Google Scholar]
- (18).Comfurious P; Smeets E; Willems G; Bevers E; Zwaal R Assembly of the prothrombinase complex on lipid vesicles depends on the stereochemical configuration of the polar headgroup of phosphatidylserine. Biochemistry 1994, 33, 10319–10324. [DOI] [PubMed] [Google Scholar]
- (19).Wymann MP; Schneiter R Lipid signalling in disease. Nat. Rev. Mol. Cell Biol 2008, 9, 162–176. [DOI] [PubMed] [Google Scholar]
- (20).Straub JE; Thirumalai D Membrane-protein interactions are key to understanding amyloid formation. J. Phys. Chem. Lett 2014, 5, 633–635. [DOI] [PubMed] [Google Scholar]
- (21).Schmidt D; MacKinnon R Voltage-dependent K+ channel gating and voltage sensor toxin sensitivity depend on the mechanical state of the lipid membrane. Proc. Natl. Acad. Sci. USA 2008, 105, 19276–19281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Rojko N; Anderluh G How lipid membranes affect pore forming toxin activity. Acc. Chem. Res 2015, 48, 3073–3079. [DOI] [PubMed] [Google Scholar]
- (23).Harrison SC Viral membrane fusion. Virology 2015, 479–480, 498–507. [DOI] [PMC free article] [PubMed]
- (24).Altan-Bonnet N Lipid tales of viral replication and transmission. Trends Cell. Biol 2017, 27, 201–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Wisedchaisri G; Gonen T Fragment-based phase extension for three-dimensional structure determination of membrane proteins by electron crystallography. Structure 2011, 19, 976–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Norimatsu Y; Hasegawa K; Shimizu N; Toyoshima C Protein-phospholipid interplay revealed with crystals of a calcium pump. Nature 2017, 545, 193–198. [DOI] [PubMed] [Google Scholar]
- (27).Gao Y; Cao E; Julius D; Cheng Y TRPV1 structures in nanodiscs reveal mechanisms of ligand and lipid action. Nature 2016, 534, 347–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Yamamoto K; Caporini MA; Im S-C; Waskell L; Ramamoorthy A Transmembrane interactions of full-length mammalian bitopic cytochrome-P450-cytochrome-b5 complex in lipid bilayers revealed by sensitivity-enhanced dynamic nuclear polarization solid-state nmr spectroscopy. Sci. Rep 2017, 7 . [DOI] [PMC free article] [PubMed]
- (29).Landgraf KE; Malmberg NJ; Falke JJ Effect of PIP2 binding on the membrane docking geometry of PKCα C2 domain: An EPR site-directed spin-labeling and relaxation study. Biochemistry 2008, 47, 8301–8316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Stevenson P; Tokmakoff A Time-resolved measurements of an ion channel conformational change driven by a membrane phase transition. Proc. Natl. Acad. Sci. USA 2017, 10840–10845. [DOI] [PMC free article] [PubMed]
- (31).Arauz E; Aggarwal V; Jain A; Ha T; Chen J Single-molecule analysis of lipid-protein interactions in crude cell lysates. Anal. Chem 2016, 88, 4269–4276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Tavoosi N; Davis-Harrison RL; Pogorelov TV; Ohkubo YZ; Arcario MJ; Clay MC; Rienstra CM; Tajkhorshid E; Morrissey JH Molecular determinants of phospholipid synergy in blood clotting. J. Biol. Chem 2011, 286, 23247–23253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Chlanda P; Zimmerberg J Protein-lipid interactions critical to replication of the influenza A virus. FEBS Lett 2016, 590, 1940–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Papahadjopoulos D; Kimelberg HK Phospholipid vesicles (liposomes) as models for biological membranes: Their properties and interactions with cholesterol and proteins. Proc. Surf. Sci 1974, 4, 141–232. [Google Scholar]
- (35).Bayburt TH; Sligar SG Membrane protein assembly into Nanodiscs. FEBS Lett 2010, 584, 1721–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Castellana ET; Cremer PS Solid supported lipid bilayers: From biophysical studies to sensor design. Surf. Sci. Rep 2006, 61, 429–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Verger R; Pattus F Lipid-protein interactions in monolayers. Chem. Phys. of Lipids 1982, 30, 189–227. [Google Scholar]
- (38).Watanabe R; Soga N; Fujita D; Tabata KV; Yamauchi L; Hyeon Kim S; Asanuma D; Kamiya M; Urano Y; Suga H et al. Arrayed lipid bilayer chambers allow single-molecule analysis of membrane transporter activity. Nat. Commun 2014, 5, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Sun C; Carey A-M; Gao B-R; Wraight CA; Woodbury NW; Lin S Ultrafast electron transfer kinetics in the LM dimer of bacterial photosynthetic reaction center from Rhodobacter sphaeroides. J. Phys. Chem. B 2016, 120, 5395–5404. [DOI] [PubMed] [Google Scholar]
- (40).Alonso A; Restall CJ; Turner M; Gomez-Fernandez JC; Goñi FM; Chapman D Protein-lipid interactions and differential scanning calorimetric studies of bacteriorhodopsin reconstituted lipid-water systems. Biochim. Biophys. Acta, Biomembr 1982, 689, 283–289. [Google Scholar]
- (41).Anbazhagan V; Sankhala RS; Singh BP; Swamy MJ Isothermal titration calorimetric studies on the interaction of the major bovine seminal plasma protein, PDC-109 with phospholipid membranes. PLoS One 2011, 6 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Garcia-Celma J; Szydelko A; Dutzler R Functional characterization of a ClC transporter by solid-supported membrane electrophysiology. J. Gen. Physiol 2013, 141, 479–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Gonzalez-Gutierrez G; Wang Y; Cymes G; Tajkhorshid E; Grosman C Chasing the open-state structure of pentameric ligand-gated ion channels. J. Gen. Physiol 2017, 149, 1119–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Barrera NP; Zhou M; Robinson CV The role of lipids in defining membrane protein interactions: Insights from mass spectrometry. Trends Cell. Biol 2013, 23, 1–8. [DOI] [PubMed] [Google Scholar]
- (45).Cong X; Liu Y; Liu W; Liang X; Russell DH; Laganowsky A Determining membrane protein-lipid binding thermodynamics using native mass spectrometry. J. Am. Chem. Soc 2016, 138, 4346–4349. [DOI] [PubMed] [Google Scholar]
- (46).Marsh D; Horváth LI Structure, dynamics and composition of the lipid-protein interface. Perspectives from spin-labelling. Biochim. Biophys. Acta, Rev. Biomembr 1998, 1376, 267–296. [DOI] [PubMed] [Google Scholar]
- (47).Kaplan M; Pinto C; Houben K; Baldus M Nuclear magnetic resonance (NMR) applied to membrane-protein complexes. Quart. Rev. Biophys 2016, 49, e15. [DOI] [PubMed] [Google Scholar]
- (48).Puchner EM; Walter JM; Kasper R; Huang B; Lim WA Counting molecules in single organelles with superresolution microscopy allows tracking of the endosome maturation trajectory. Proc. Natl. Acad. Sci. USA 2013, 110, 16015–16020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Jakobs S; Wurm CA Super-resolution microscopy of mitochondria. Curr. Opin. Chem. Biol 2014, 20, 9–15. [DOI] [PubMed] [Google Scholar]
- (50).Huang F; Sirinakis G; Allgeyer ES; Schroeder LK; Duim WC; Kromann EB; Phan T; Rivera-Molina FE; Myers JR; Irnov I et al. Ultra-high resolution 3D imaging of whole cells. Cell 2016, 166, 1028–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Galiani S; Waithe D; Reglinski K; Cruz-Zaragoza LD; Garcia E; Clausen MP; Schliebs W; Erdmann R; Eggeling C Super-resolution microscopy reveals compartmentalization of peroxisomal membrane proteins. J. Cell Biol 2016, 291, 16948–16962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Akyuz N; Altman RB; Blanchard SC; Boudker O Transport dynamics in a glutamate transporter homologue. Nature 2013, 502, 114–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).McHaourab HS; Steed PR; Kazmier K Toward the fourth dimension of membrane protein structure: Insight into dynamics from spin-labeling EPR spectroscopy. Structure 2011, 19, 1549–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Khantwal CM; Abraham SJ; Han W; Jiang T; Chavan TS; Cheng RC; Elvington SM; Liu CW; Mathews II; Stein RA et al. Revealing an outward-facing open conformational state in a CLC Cl−/H+ exchange transporter. eLife 2016, 5, e11189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).King GI; White SH Determining bilayer hydrocarbon thickness from neutron diffraction measurements using strip-function models. Biophys. J 1986, 49, 1047–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Mitra K; Ubarretxena-Belandia I; Taguchi T; Warren G; Engelman DM Modulation of the bilayer thickness of exocytic pathway membranes by membrane proteins rather than cholesterol. Proc. Natl. Acad. Sci. USA 2004, 101, 4083–4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Suresh S; Edwardson JM The endophilin N-BAR domain perturbs the structure of lipid bilayers. Biochemistry 2010, 49, 5766–5771. [DOI] [PubMed] [Google Scholar]
- (58).Chaibva M; Burke KA; Legleiter J Curvature enhances binding and aggregation of Huntingtin at lipid membranes. Biochemistry 2014, 53, 2355–2365. [DOI] [PubMed] [Google Scholar]
- (59).Lee AG Lipid-protein interactions in biological membranes: a structural perspective. Biochim. Biophys. Acta 2003, 1612, 1–40. [DOI] [PubMed] [Google Scholar]
- (60).Opella SJ; Marassi FM Applications of NMR to membrane proteins. Arch. Biochem. Biophys 2017, 628, 92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Jin P; Bulkley D; Guo Y; Zhang W; Guo Z; Huynh W; Wu S; Meltzer S; Cheng T; Jan LY et al. Electron cryo-microscopy structure of the mechanotransduction channel NOMPC. Nature 2017, 547, 118–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Dror RO; Dirks RM; Grossman J; Xu H; Shaw DE Biomolecular simulation: A computational microscope for molecular biology. Annu. Rev. Biophys 2012, 41, 429–452. [DOI] [PubMed] [Google Scholar]
- (63).Lee EH; Hsin J; Sotomayor M; Comellas G; Schulten K Discovery through the computational microscope. Structure 2009, 17, 1295–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Nelson M; Humphrey W; Gursoy A; Dalke A; Kalé L; Skeel RD; Schulten K NAMD – A parallel, object-oriented molecular dynamics program. Int. J. Supercomp. Appl. High Perform. Comp 1996, 10, 251–268. [Google Scholar]
- (65).Bekker H; Berendsen HJC; Dijkstra EJ; Achterop S; van Drunen R GROMACS: A parallel computer for molecular dynamics simulations. Proceedings of the 4th Intl. Conference Physics Computing ‘92. Singapore, 1993; pp 252–256. [Google Scholar]
- (66).Pearlman DA; Case DA; Caldwell JW; Ross WS; Cheatham T; Debolt S; Ferguson D; Seibel G; Kollman P AMBER, a package of computer-programs for applying molecular mechanics, normal-mode analysis, molecular-dynamics and free-energy calculations to simulate the structural and energetic properties of molecules. Comput. Phys. Commun 1995, 91, 1–41. [Google Scholar]
- (67).Letts JA; Fiedorczuk K; Sazanov LA The architecture of respiratory supercomplexes. Nature 2016, 537, 644–648. [DOI] [PubMed] [Google Scholar]
- (68).Berendsen HJC; van Gunsteren WF; Egberts E; de Vlieg J Supercomputer Research in Chemistry and Chemical Engineering ; Chapter 7, pp 106–122. [Google Scholar]
- (69).van der Ploeg P; Berendsen HJC Molecular dynamics simulation of a bilayer membrane. J. Chem. Phys 1982, 76, 3271–3276. [Google Scholar]
- (70).van der Ploeg P; Berendsen HJC Molecular dynamics of a bilayer membrane. Mol. Phys 1983, 49, 233–248. [Google Scholar]
- (71).Heller H; Schaefer M; Schulten K Molecular dynamics simulation of a bilayer of 200 lipids in the gel and in the liquid crystal-phases. J. Phys. Chem 1993, 97, 8343–8360. [Google Scholar]
- (72).Woolf T; Roux B Molecular dynamics simulation of the gramicidin channel in a phospholipid bilayer. Proc. Natl. Acad. Sci. USA 1994, 91, 11631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Shelley JC; Shelley MY; Reeder RC; Bandyopadhyay S; Klein ML A coarse grain model for phospholipid simulations. J. Phys. Chem. B 2001, 105, 4464–4470. [Google Scholar]
- (74).Egberts E; Berendsen HJC Molecular dynamics simulation of a smectic liquid crystal with atomic detail. J. Chem. Phys 1988, 89, 3718–3732. [Google Scholar]
- (75).Damodaran KV; Merz KM Jr.; Gaber BP Structure and dynamics of the dilauroylphos-phatidylethanolamine lipid bilayer. Biochemistry 1992, 31, 7656–7664. [DOI] [PubMed] [Google Scholar]
- (76).Damodaran K; Merz K Jr. Head group–water interactions in lipid bilayers: A comparison between DMPC- and DLPE-based lipid bilayers. Langmuir 1993, 9, 1179–1183. [Google Scholar]
- (77).Venable RM; Zhang Y; Hardy BJ; Pastor RW Molecular dynamics simulations of a lipid bilayer and of hexadecane: An investigative of membrane fluidity. Science 1993, 262, 223–226. [DOI] [PubMed] [Google Scholar]
- (78).Edholm O; Johansson J Lipid bilayer polypeptide interactions studied by molecular dynamics simulation. Eur. Biophys. J 1987, 14, 203–209. [DOI] [PubMed] [Google Scholar]
- (79).Woolf TB; Roux B Structure, energetics, and dynamics of lipid-protein interactions: a molecular dynamics study of the gramicidin A channel in a DMPC bilayer. Proteins: Struct., Func., Gen 1996, 24, 92–114. [DOI] [PubMed] [Google Scholar]
- (80).Woolf TB; Roux B The binding site of sodium in the gramacidin A channel: comparison of molecular dynamics with solid-state NMR data. Biophys. J 1997, 72, 1930–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Zhou F; Schulten K Molecular dynamics study of the activation of phospholipase A2 on a membrane surface. Proteins: Struct., Func., Gen 1996, 25, 12–27. [DOI] [PubMed] [Google Scholar]
- (82).Roux B; Prod’hom B; Karplus M Ion transport in the gramicidin channel: molecular dynamics study of single and double occupancy. Biophys. J 1995, 68, 876–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Zhong Q; Jiang Q; Moore PB; Newns DM; Klein ML Molecular dynamics simulation of a synthetic ion channel. Biophys. J 1998, 74, 3–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Arcario MJ; Ohkubo YZ; Tajkhorshid E Capturing spontaneous partitioning of peripheral proteins using a biphasic membrane-mimetic model. J. Phys. Chem. B 2011, 115, 7029–7037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Shelley JC; Shelley MY; Reeder RC; Bandyopadhyay S; Moore PB; Klein ML Simulations of phospholipids using a coarse grain model. J. Phys. Chem. B 2001, 105, 9785–9792. [Google Scholar]
- (86).Ponder JW; Case DA Protein Simulations; Advances in Protein Chemistry Supplement C; Academic Press, 2003; Vol. 66; pp 27–85. [DOI] [PubMed] [Google Scholar]
- (87).Mackerell AD Empirical force fields for biological macromolecules: Overview and issues. J. Comp. Chem 2004, 25, 1584–1604. [DOI] [PubMed] [Google Scholar]
- (88).Monticelli L; Tieleman DP In Biomolecular Simulations: Methods and Protocols; Monticelli L, Salonen E, Eds.; Humana Press: Totowa, NJ, 2013; Chapter Force Fields for Classical Molecular Dynamics, pp 197–213. [DOI] [PubMed] [Google Scholar]
- (89).Brooks BR; Bruccoleri RE; Olafson BD; States DJ; Swaminathan S; Karplus M CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comp. Chem 1983, 4, 187–217. [Google Scholar]
- (90).MacKerell AD Jr.; Bashford D; Bellott M; Dunbrack JRL; Evanseck J; Field MJ; Fischer S; Gao J; Guo H; Ha S et al. Self-consistent parameterization of biomolecules for molecular modeling and condensed phase simulations. FASEB J 1992, 6, A143–A143. [Google Scholar]
- (91).MacKerell AD Jr.; Bashford D; Bellott M; Dunbrack RL Jr.; Evanseck JD; Field MJ; Fischer S; Gao J; Guo H; Ha S et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- (92).Weinmann H-J; Brasch RC; Press W-R; Wesbey GE Characteristics of Gd-DTPA complex: A potential NMR contrast agent. Am. J. Roentgenology 1984, 142, 619–624. [DOI] [PubMed] [Google Scholar]
- (93).Cornell WD; Cieplak P; Bayly CI; Gould IR; Merz KM Jr.; Ferguson DM; Spellmeyer DC; Fox T; Caldwell JW; Kollman PA A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc 1995, 117, 5179–5197. [Google Scholar]
- (94).<ball-atom additive force field for sphingomyelin: Elucidation of hydrogen bonding and of positive all-atom additive force field for sphingomyelin: Elucidation of hydrogen bonding and of positive />van Gunsteren WF; Berendsen HJC Gromos manual. BIOMOS b. v.: Lab. of Phys. Chem., Univ. of Groningen, 1987. [Google Scholar]
- (95).Jorgensen W; Tirado-Rives J The OPLS potential functions for protein energy minimization for crystals of cyclic peptides and crambin. J. Am. Chem. Soc 1988, 110, 3469. [DOI] [PubMed] [Google Scholar]
- (96).Kamisnki GA; Friesner RA; Tirado-Rives J; Jorgensen WL Evaluation and reparameterization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. B 2001, 105, 6476–6487. [Google Scholar]
- (97).Schlenkrich M; Brickmann J; MacKerell AD Jr.; Karplus M In Biological Membranes: A Molecular Perspective from Computation and Experiment ; Merz KM, Roux B, Eds.; Birkhauser: Boston, 1996; pp 31–81. [Google Scholar]
- (98).Pastor RW; MacKerell AD Development of the CHARMM force field for lipids. J. Phys. Chem. Lett 2011, 2, 1526–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Berger O; Edholm O; Jähnig F Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys. J 1997, 72, 2002–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Feller SE; MacKerell AD Jr. An improved empirical potential energy function for molecular simulations of phospholipids. J. Phys. Chem. B 2000, 104, 7510–7515. [Google Scholar]
- (101).Klauda JB; Brooks BR; MacKerell AD Jr.; Venable RM; Pastor RW An ab initio study on the torsional surface of alkanes and its effect on molecular simulations of alkanes and a DPPC bilayer. J. Phys. Chem. B 2005, 109, 5300–5311. [DOI] [PubMed] [Google Scholar]
- (102).Klauda JB; Venable RM; Freites JA; O’Connor JW; Tobias DJ; Mondragon-Ramirez C; Vorobyov I; MacKerell AD Jr.; Pastor RW Update of the CHARMM all-atom additive force field for lipids: Validation on six lipid types. J. Phys. Chem. B 2010, 114, 7830–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Venable RM; Sodt AJ; Rogaski B; Rui H; Hatcher E; A. D. M. Jr.; Pastor RW; Klauda JB CHARMM all-atom additive force field for sphingomyelin: Elucidation of hydrogen bonding and of positive curvature. Biophys. J 2014, 107, 134–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Tieleman DP; Marrink S-J Lipids out of equilibrium: Energetics of desorption and pore mediated flip-flop. J. Am. Chem. Soc 2006, 128, 12462–12467. [DOI] [PubMed] [Google Scholar]
- (105).Jämbeck JPM; Lyubartsev AP Derivation and systematic validation of a refined all-atom force field for phosphatidylcholine lipids. J. Phys. Chem. B 2012, 116, 3164–3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Jämbeck JPM; Lyubartsev AP Another piece of the membrane puzzle: Extending Slipids further. J. Chem. Theory Comput 2013, 9, 774–784. [DOI] [PubMed] [Google Scholar]
- (107).Tessier M; DeMarco M; Yongye A; Woods R Extension of the GLYCAM06 biomolecular force field to lipids, lipid bilayers and glycolipids. Mol. Sim 2008, 34, 349–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Sapay N; Tieleman DP Combination of the CHARMM27 force field with united-atom lipid force fields. J. Comp. Chem 2011, 32, 1400–1410. [DOI] [PubMed] [Google Scholar]
- (109).Cordomí A; Caltabiano G; Pardo L Membrane protein simulations using amber force field and berger lipid parameters. J. Chem. Theory Comput 2012, 8, 948–958. [DOI] [PubMed] [Google Scholar]
- (110).Bennett WFD; Hong CK; Wang Y; Tieleman DP Antimicrobial peptide simulations and the influence of force field on the free energy for pore formation in lipid bilayers. J. Chem. Theory Comput 2016, 12, 4524–4533. [DOI] [PubMed] [Google Scholar]
- (111).Skjevik ÅA; Made BD; Walker RC; Teigen K LIPID11: A modular framework for lipid simulations using amber. J. Phys. Chem. B 2012, 116, 11124–11136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (112).Wang J; Wolf RM; Caldwell JW; Kollman PA; Case DA Developing and testing of a general amber force field. J. Comp. Chem 2004, 25, 1157–1174. [DOI] [PubMed] [Google Scholar]
- (113).Dickson CJ; Madej BD; Skjevik ÅA; Betz RM; Teigen K; Gould IR; Walker RC Lipid14: The amber lipid force field. J. Chem. Theory Comput 2014, 10, 865–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Madej BD; Gould IR; Walker RC A parameterization of cholesterol for mixed lipid bilayer simulation within the amber lipid14 force field. J. Phys. Chem. B 2015, 119, 12424–12435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Kulig W; Pasenkiewicz-Gierula M; Róg T Topologies, structures and parameter files for lipid simulations in GROMACS with the OPLS-aa force field: DPPC, POPC, DOPC, PEPC, and cholesterol. Dat. Brief 2015, 5, 333–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (116).Maciejewski A; Pasenkiewicz-Gierula M; Cramariuc O; Vattulainen I; Ròg T Refined OPLS all-atom force field for saturated phosphatidylcholine bilayers at full hydration. J. Phys. Chem. B 2014, 118, 4571–4581. [DOI] [PubMed] [Google Scholar]
- (117).Halgren TA; Damm W Polarizable force fields. Curr. Opin. Struct. Biol 2001, 11, 236–242. [DOI] [PubMed] [Google Scholar]
- (118).Warshel A; Levitt M Theoretical studies of enzymatic reactions: Dielectric, electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme. J. Mol. Biol 1976, 103, 227–249. [DOI] [PubMed] [Google Scholar]
- (119).Stillinger FH; David CW Polarization model for water and its ionic dissociation products. J. Chem. Phys 1978, 69, 1473–1484. [Google Scholar]
- (120).Ponder JW; Wu C; Ren P; Pande VS; Chodera JD; Schnieders MJ; Haque I; Mobley DL; Lambrecht DS; DiStasio RA et al. Current status of the AMOEBA polarizable force field. J. Phys. Chem. B 2010, 114, 2549–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Shi Y; Xia Z; Zhang J; Best R; Wu C; Ponder JW; Ren P Polarizable atomic multipole-based AMOEBA force field for proteins. J. Chem. Theory Comput 2013, 9, 4046–4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (122).Lopes PEM; Huang J; Shim J; Luo Y; Li H; Roux B; MacKerell AD Jr. Polarizable force field for peptides and proteins based on the classical Drude oscillator. J. Chem. Theory Comput 2013, 9, 5430–5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (123).Chowdhary J; Harder E; Lopes PEM; Huang L; MacKerell AD Jr.; Roux B A polarizable force field of dipalmitoylphosphatidylcholine based on the classical Drude model for molecular dynamics simulations of lipids. J. Phys. Chem. B 2013, 117, 9142–9160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (124).Li H; Chowdhary J; Huang L; He X; MacKerell AD; Roux B Drude polarizable force field for molecular dynamics simulations of saturated and unsaturated zwitterionic lipids. J. Chem. Theory Comput 2017, 13, 4535–4552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (125).Huang J; Lopes PE; Roux B; MacKerell AD Jr Recent advances in polarizable force fields for macromolecules: microsecond simulations of proteins using the classical drude oscillator model. J. Phys. Chem. Lett 2014, 5, 3144–3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (126).Vorobyov I; Li L; Allen TW Assessing atomistic and coarse-grained force fields for protein-lipid interactions: the formidable challenge of an ionizable side chain in a membrane. J. Phys. Chem. B 2008, 112, 9588–9602. [DOI] [PubMed] [Google Scholar]
- (127).Grubmüller H; Heymann B; Tavan P Ligand binding and molecular mechanics calculation of the streptavidin-biotin rupture force. Science 1996, 271, 997–999. [DOI] [PubMed] [Google Scholar]
- (128).Isralewitz B; Gao M; Schulten K Steered molecular dynamics and mechanical functions of proteins. Curr. Opin. Struct. Biol 2001, 11, 224–230. [DOI] [PubMed] [Google Scholar]
- (129).Vendruscolo M; Dobson CM Protein dynamics: Moore’s law in molecular biology. Curr. Biol 2011, 21, R68–R70. [DOI] [PubMed] [Google Scholar]
- (130).Kolinski A; Skolnick J Reduced models of proteins and their applications. Polymer 2004, 45, 511–524. [Google Scholar]
- (131).Tozzini V Coarse-grained models for proteins. Curr. Opin. Struct. Biol 2005, 15, 144–150. [DOI] [PubMed] [Google Scholar]
- (132).Rader AJ Coarse-grained models: Getting more with less. Curr. Opin. Pharmacol 2010, 10, 753–759. [DOI] [PubMed] [Google Scholar]
- (133).Wu C; Shea JE Coarse-grained models for protein aggregation. Curr. Opin. Struct. Biol 2011, 21, 209–220. [DOI] [PubMed] [Google Scholar]
- (134).Riniker S; Allison JR; Van Gunsteren WF On developing coarse-grained models for biomolecular simulation: A review. Phys. Chem. Chem. Phys 2012, 14, 12423–12430. [DOI] [PubMed] [Google Scholar]
- (135).Takada S Coarse-grained molecular simulations of large biomolecules. Curr. Opin. Struct. Biol 2012, 22, 130–137. [DOI] [PubMed] [Google Scholar]
- (136).Saunders MG; Voth GA Coarse-graining methods for computational biology. Annu. Rev. Biophys 2013, 42, 73–93. [DOI] [PubMed] [Google Scholar]
- (137).Baaden M; Marrink SJ Coarse-grain modelling of protein-protein interactions. Curr. Opin. Struct. Biol 2013, 23, 878–886. [DOI] [PubMed] [Google Scholar]
- (138).Ingólfsson HI; Lopez CA; Uusitalo JJ; de Jong DH; Gopal SM; Periole X; Marrink SJ The power of coarse graining in biomolecular simulations. WIREs Comput. Mol. Sci 2014, 4, 225–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (139).Sorenson JM; Head-Gordon T The importance of hydration for the kinetics and thermodynamics of protein folding: Simplified lattice models. Fold. Des 1998, 3, 523–534. [DOI] [PubMed] [Google Scholar]
- (140).Shakhnovich E Protein folding thermodynamics and dynamics: Where physics, chemistry, and biology meet. Chem. Rev 2006, 106, 1559–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (141).Kolinski A; Jaroszewski L; Rotkiewicz P; Skolnick J An efficient Monte Carlo model of protein chains. Modeling the short-range correlations between side group centers of mass. J. Phys. Chem. B 1998, 102, 4628–4637. [Google Scholar]
- (142).Kolinski A Protein modeling and structure prediction with a reduced representation. Rev. Lit. Arts Am 2004, 51, 349–371. [PubMed] [Google Scholar]
- (143).Liwo A; Baranowski M; Czaplewski C; Go-lás E; He Y; Jagiel-a D; Krupa P; Maciejczyk M; Makowski M; Mozolewska MA et al. A unified coarse-grained model of biological macromolecules based on mean-field multipole-multipole interactions. J. Mol. Mod 2014, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (144).Gopal SM; Mukherjee S; Cheng YM; Feig M PRIMO/PRIMONA: A coarse-grained model for proteins and nucleic acids that preserves near-atomistic accuracy. Proteins: Struct., Func., Bioinf 2010, 78, 1266–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (145).Kar P; Gopal SM; Cheng YM; Predeus A; Feig M PRIMO: A transferable coarse-grained force field for proteins. J. Chem. Theory Comput 2013, 9, 3769–3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (146).Kar P; Gopal SM; Cheng YM; Panahi A; Feig M Transferring the PRIMO coarse-grained force field to the membrane environment: Simulations of membrane proteins and helix-helix association. J. Chem. Theory Comput 2014, 10, 3459–3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (147).Das R; Baker D Macromolecular modeling with rosetta. Annu. Rev. Biochem 2008, 77, 363–382. [DOI] [PubMed] [Google Scholar]
- (148).De Jong DH; Singh G; Bennett WF; Arnarez C; Wassenaar TA; Schäfer LV; Periole X; Tieleman DP; Marrink SJ Improved parameters for the martini coarse-grained protein force field. J. Chem. Theory Comput 2013, 9, 687–697. [DOI] [PubMed] [Google Scholar]
- (149).Kmiecik S; Gront D; Kolinski M; Wieteska L; Dawid AE; Kolinski A Coarse-grained protein models and their applications. Chem. Rev 2016, 116, 7898–7936. [DOI] [PubMed] [Google Scholar]
- (150).Marrink SJ; de Vries AH; Mark AE Coarse grained model for semiquantitative lipid simulations. J. Phys. Chem. B 2004, 108, 750–760. [Google Scholar]
- (151).Marrink SJ; Risselada HJ; Yefimov S; Tieleman DP; de Vries AH The MARTINI force field: coarse grained model for biomolecular simulations. J. Phys. Chem. B 2007, 111, 7812–7824. [DOI] [PubMed] [Google Scholar]
- (152).Monticelli L; Kandasamy SK; Periole X; Larson RG; Tieleman DP; Marrink SJ The MARTINI coarse grained forcefield: Extension to proteins. J. Chem. Theory Comput 2008, 4, 819–834. [DOI] [PubMed] [Google Scholar]
- (153).Shih AY; Arkhipov A; Freddolino PL; Schulten K Coarse grained protein-lipid model with application to lipoprotein particles. J. Phys. Chem. B 2006, 110, 3674–3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (154).Bond PJ; Holyoake J; Ivetac A; Khalid S; Sansom M Coarse-grained molecular dynamics simulations of membrane proteins and peptides. J. Struct. Biol 2007, 157, 593–605. [DOI] [PubMed] [Google Scholar]
- (155).Yesylevskyy SO; Schäfer LV; Sengupta D; Marrink SJ Polarizable water model for the coarse-grained MARTINI force field. PLoS Comput. Biol 2010, 6, e1000810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (156).Arnarez C; Uusitalo JJ; Masman MF; Ingólfsson HI; De Jong DH; Melo MN; Periole X; De Vries AH; Marrink SJ Dry martini, a coarse-grained force field for lipid membrane simulations with implicit solvent. J. Chem. Theory Comput 2015, 11, 260–275. [DOI] [PubMed] [Google Scholar]
- (157).Marrink SJ; Tieleman DP Perspective on the MARTINI model. Chem. Soc. Rev 2013, 42, 6801–6822. [DOI] [PubMed] [Google Scholar]
- (158).Fritz D; Koschke K; Harmandaris VA; van der Vegt NFA; Kremer K Multiscale modeling of soft matter: scaling of dynamics. Phys. Chem. Chem. Phys 2011, 13, 10412–10420. [DOI] [PubMed] [Google Scholar]
- (159).Vieth M; Kolinski A; Brooks CL; Skolnick J Prediction of the folding pathways and structure of the gcn4 leucine zipper. J. Mol. Biol 1994, 237, 361–367. [DOI] [PubMed] [Google Scholar]
- (160).Fan ZZ; Hwang J-K; Warshel a. Using simplified protein representation as a reference potential for all-atom calculations of folding free energy. Theor. Chim. Acta 1999, 103, 77–80. [Google Scholar]
- (161).Feig M; Karanicolas J; Brooks CL MMTSB Tool Set: Enhanced sampling and multiscale modeling methods for applications in structural biology. J. Mol. Graph. Model 2004, 22, 377–395. [DOI] [PubMed] [Google Scholar]
- (162).Villa E; Balaeff A; Schulten K Structural dynamics of the Lac repressor-DNA complex revealed by a multiscale simulation. Proc. Natl. Acad. Sci. USA 2005, 102, 6783–6788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (163).Shi Q; Izvekov S; Voth GA Mixed atomistic and coarse-grained molecular dynamics: Simulation of a membrane-bound ion channel. J. Phys. Chem. B 2006, 110, 15045–15048. [DOI] [PubMed] [Google Scholar]
- (164).Lyman E; Ytreberg FM; Zuckerman DM Resolution exchange simulation. Phys. Rev. Lett 2006, 96, 028105. [DOI] [PubMed] [Google Scholar]
- (165).Ayton GS; Blood PD; Voth GA Membrane remodeling from N-BAR domain interactions: Insights from multi-scale simulation. Biophys. J 2007, 92, 3595–3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (166).Kamerlin SC; Vicatos S; Dryga A; Warshel A Coarse-Grained (Multiscale) Simulations in Studies of Biophysical and Chemical Systems. Annu. Rev. Phys. Chem 2011, 62, 41–64. [DOI] [PubMed] [Google Scholar]
- (167).Han W; Wan C-K; Jiang F; Wu Y-D PACE force field for protein simulations. 1. full parameterization of version 1 and verification. J. Chem. Theory Comput 2010, 6, 3373–3389. [DOI] [PubMed] [Google Scholar]
- (168).Han W; Wan C-K; Wu Y-D PACE force field for protein simulations. 2. folding simulations of peptides. J. Chem. Theory Comput 2010, 6, 3390–3402. [DOI] [PubMed] [Google Scholar]
- (169).Wan C-K; Han W; Wu Y-D Parameterization of PACE force field for membrane environment and simulation of helical peptides and helix-helix association. J. Chem. Theory Comput 2012, 8, 300–313. [DOI] [PubMed] [Google Scholar]
- (170).Kerdcharoen T; Morokuma K ONIOM-XS: An extension of the ONIOM method for molecular simulation in condensed phase. Chem. Phys. Lett 2002, 355, 257–262. [Google Scholar]
- (171).Hofer TS; Pribil AB; Randolf BR; Rode BM Structure and dynamics of solvated Sn(II) in aqueous solution: An ab initio QM/MM MD approach. J. Am. Chem. Soc 2005, 127, 14231–14238. [DOI] [PubMed] [Google Scholar]
- (172).Heyden A; Lin H; Truhlar DG Adaptive partitioning in combined quantum mechanical and molecular mechanical calculations of potential energy functions for multiscale simulations. J. Phys. Chem. B 2007, 111, 2231–2241. [DOI] [PubMed] [Google Scholar]
- (173).Praprotnik M; Site LD; Kremer K Multiscale simulation of soft matter: From scale bridging to adaptive resolution. Annu. Rev. Phys. Chem 2008, 59, 545–571. [DOI] [PubMed] [Google Scholar]
- (174).Nielsen SO; Bulo RE; Moore PB; Ensing B Recent progress in adaptive multiscale molecular dynamics simualtions of soft matter. Phys. Chem. Chem. Phys 2010, 12, 12401–12414. [DOI] [PubMed] [Google Scholar]
- (175).Potestio R; Fritsch S; Español P; Delgado-Buscalioni R; Kremer K; Everaers R; Donadio D Hamiltonian adaptive resolution simulation for molecular liquids. Phys. Rev. Lett 2013, 110, 1–11. [DOI] [PubMed] [Google Scholar]
- (176).Ohkubo YZ; Pogorelov TV; Arcario MJ; Christensen GA; Tajkhorshid E Accelerating membrane insertion of peripheral proteins with a novel membrane mimetic model. Biophys. J 2012, 102, 2130–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (177).Ohkubo YZ; Tajkhorshid E Distinct structural and adhesive roles of Ca2+ in membrane binding of blood coagulation factors. Structure 2008, 16, 72–81. [DOI] [PubMed] [Google Scholar]
- (178).Baylon JL; Lenov IL; Sligar SG; Tajkhorshid E Characterizing the membrane-bound state of cytochrome P450 3A4: Structure, depth of insertion, and orientation. J. Am. Chem. Soc 2013, 135, 8542–8551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (179).Baylon JL; Tajkhorshid E Capturing spontaneous membrane insertion of the influenza virus hemagglutinin fusion peptide. J. Phys. Chem. B 2015, 119, 7882–7893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (180).Vermaas JV; Tajkhorshid E A microscopic view of phospholipid insertion into biological membranes. J. Phys. Chem. B 2014, 118, 1754–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (181).Pogorelov TV; Vermaas JV; Arcario MJ; Tajkhorshid E Partitioning of amino acids into a model membrane: Capturing the interface. J. Phys. Chem. B 2014, 118, 1481–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (182).Arcario MJ; Tajkhorshid E Membrane-induced structural rearrangement and identification of a novel membrane anchor in Talin F2F3. Biophys. J 2014, 107, 2059–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (183).Blanchard AE; Arcario MJ; Schulten K; Tajkhorshid E A highly tilted membrane configuration for the pre-fusion state of synaptobrevin. Biophys. J 2014, 107, 2112–2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (184).Vermaas JV; Tajkhorshid E Conformational heterogeneity of α-synuclein in membrane. Biochim. Biophys. Acta, Biomembr 2014, 1838, 3107–3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (185).Rhéault J-F; Gagné E; Guertin M; Lamoureux G; Auger M; Lagüe P Molecular model of hemoglobin N from Mycobacterium tuberculosis bound to lipid bilayers: A combined spectroscopic and computational study. Biochemistry 2015, 54, 2073–2084. [DOI] [PubMed] [Google Scholar]
- (186).Madsen JJ; Ohkubo YZ; Peters GH; Faber JH; Tajkhorshid E; Olsen OH Membrane interaction of the factor VIIIa discoidin domains in atomistic detail. Biochemistry 2015, 54, 6123–6131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (187).Vermaas JV; Baylon JL; Arcario MJ; Muller MP; Wu Z; Pogorelov TV; Tajkhorshid E Efficient exploration of membrane-associated phenomena at atomic resolution. J. Membr. Biol 2015, 248, 563–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (188).Baylon JL; Vermaas JV; Muller MP; Arcario MJ; Pogorelov TV; Tajkhorshid E Atomic-level description of protein-lipid interactions using an accelerated membrane model. Biochim. Biophys. Acta, Biomembr 2016, 1858, 1573–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (189).Muller MP; Wang Y; Morrissey JH; Tajkhorshid E Lipid specificity of the membrane binding domain of coagulation factor X. J. Thromb. Haem 2017, 15, 2005–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (190).Qi Y; Cheng X; Lee J; Vermaas JV; Pogorelov TV; Tajkhorshid E; Park S; Klauda JB; Im W CHARMM-GUI HMMM builder for membrane simulations with the highly mobile membrane-mimetic model. Biophys. J 2015, 109, 2012–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (191).Gregory MC; McLean MA; Sligar SG Interaction of KRas4b with anionic membranes: A special role for PIP2. Biochem. Biophys. Res. Commun 2017, 487, 351–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (192).Vermaas JV; Tajkhorshid E Differential membrane binding mechanics of synaptotagmin isoforms observed at atomic detail. Biochemistry 2017, 56, 281–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (193).Vermaas JV; Pogorelov TV; Tajkhorshid E Extension of the highly mobile membrane mimetic to transmembrane systems through customized in silico solvents. J. Phys. Chem. B 2017, 121, 3764–3776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (194).McDougle DR; Baylon JL; Meling DD; Kambalyal A; Grinkova YV; Hammernik J; Tajkhorshid E; Das A Incorporation of charged residues in the CYP2J2 FG loop disrupts CYP2J2–lipid bilayer interactions. Biochim. Biophys. Acta, Biomembr 2015, 1848, 2460–2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (195).Zhang L; Rajendram M; Weibel DB; Yethiraj A; Cui Q Ionic hydrogen bonds and lipid packing defects determine the binding orientation and insertion depth of reca on multicomponent lipid bilayers. J. Phys. Chem. B 2016, 120, 8424–8437. [DOI] [PubMed] [Google Scholar]
- (196).Skeby KK; Andersen OJ; Pogorelov TV; Tajkhorshid E; Schiott B Conformational dynamics of the human islet amyloid polypeptide in a membrane environment: Toward the aggregation prone form. Biochemistry 2016, 55, 2031–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (197).Christensen M; Skeby KK; Schiøtt B Identification of key interactions in the initial self-assembly of amylin in a membrane environment. Biochemistry 2017, 56, 4884–4894. [DOI] [PubMed] [Google Scholar]
- (198).Aksimentiev A; Schulten K Extending the molecular modeling methodology to study insertion of membrane nanopores. Proc. Natl. Acad. Sci. USA 2004, 101, 4337–4338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (199).Kandt C; Ash WL; Tieleman DP Setting up and running molecular dynamics simulations of membrane proteins. Methods 2007, 41, 475–488. [DOI] [PubMed] [Google Scholar]
- (200).Vermaas JV; Trebesch N; Mayne C; Thangapandian S; Shekhar M; Mahinthichaichan P; Baylon JL; Jiang T; Wang Y; Muller MP et al. In Computational Approaches for Studying Enzyme Mechanism Part B ; Voth GA, Ed.; Academic Press, 2016; Vol. 578; Chapter 16, pp 373–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (201).Wolf MG.; Hoefling M.; Aponte-Santamaría C.; Grubmüller H; Groenhof G g_membed: Efficient insertion of a membrane protein into an equilibrated lipid bilayer with minimal perturbation. J. Comp. Chem 2010, 31, 2169–2174. [DOI] [PubMed] [Google Scholar]
- (202).Staritzbichler R; Anselmi C; Forrest LR; Faraldo-Gómez JD GRIFFIN: A versatile methodology for optimization of protein-lipid interfaces for membrane protein simulations. J. Chem. Theory Comput 2011, 7, 1167–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (203).Javanainen M Universal method for embedding proteins into complex lipid bilayers for molecular dynamics simulations. J. Chem. Theory Comput 2014, 10, 2577–2587. [DOI] [PubMed] [Google Scholar]
- (204).Lomize MA; Lomize AL; Pogozheva LD; Mosberg HI OPM: Orientations of proteins in membranes database. Bioinformatics 2006, 22, 623–625. [DOI] [PubMed] [Google Scholar]
- (205).Lomize AL; Pogozheva ID; Lomize MA; Mosberg HI Positioning of proteins in membranes: A computational approach. Prot. Sci 2006, 15, 1318–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (206).Lomize AL; Pogozheva ID; Lomize MA; Mosberg HI The role of hydrophobic interactions in positioning of peripheral proteins in membranes. BMC Struct. Biol 2007, 7, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (207).Basyn F; Charloteaux B; Thomas A; Brasseur R Prediction of membrane protein orientation in lipid bilayers: a theoretical approach. J. Mol. Graph. Model 2001, 20, 235–244. [DOI] [PubMed] [Google Scholar]
- (208).Lomize MA; Pogozheva ID; Joo H; Mosberg HI; Lomize AL OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res 2012, 40, D370–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (209).Ducarme P; Rahman M; Brasseur R IMPALA: A simple restraint field to simulate the biological membrane in molecular structure studies. Proteins: Struct., Func., Bioinf 1998, 30, 357–371. [PubMed] [Google Scholar]
- (210).Nugent T; Jones DT Membrane protein orientation and refinement using knowledge-based statistical potential. BMC Bioinform 2013, 14, 276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (211).Schmidt TH; Kandt C LAMBADA and InflateGRO2: Efficient membrane alignment and insertion of membrane proteins for molecular dynamics simulations. J. Chem. Inf. Model 2012, 52, 2657–2669. [DOI] [PubMed] [Google Scholar]
- (212).Stansfeld PJ; Goose JE; Caffrey M; Carpenter EP; Parker JL; Newstead S; Sansom MS MemProtMD: automated insertion of membrane protein structures into explicit lipid membranes. Structure 2015, 23, 1350–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (213).Jo S; Kim T; Im W Automated builder and database of protein/membrane complexes for molecular dynamics simulations. PLoS One 2007, 2, e880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (214).Humphrey W; Dalke A; Schulten K VMD – Visual Molecular Dynamics. J. Mol. Graphics 1996, 14, 33–38. [DOI] [PubMed] [Google Scholar]
- (215).Pettersen EF; Goddard TD; Huang CC; Couch GS; Greenblatt DM; Meng EC; Ferrin TE UCSF Chimera - A visualization system for exploratory research and analysis. J. Comp. Chem 2004, 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- (216).Park S; Beaven AH; Klauda JB; Im W How tolerant are membrane simulations with mismatch in area per lipid between leaflets? J. Chem. Theory Comput 2015, 11, 3466–3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (217).Jefferys E; Sands ZA; Shi J; Sansom MS; Fowler PW Alchembed: A computational method for incorporating multiple proteins into complex lipid geometries. J. Chem. Theory Comput 2015, 11, 2743–2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (218).Jo S; Kim T; Iyer VG; Im W CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comp. Chem 2008, 29, 1859–1865. [DOI] [PubMed] [Google Scholar]
- (219).Cheng X; Jo S; Lee HS; Klauda JB; Im W CHARMM-GUI micelle builder for pure/mixed micelle and protein/micelle complex systems. J. Chem. Inf. Model 2013, 53, 2171–2180. [DOI] [PubMed] [Google Scholar]
- (220).Jo S; Cheng X; Lee J; Kim S; Park S-J; Patel DS; Beaven AH; Lee KI; Rui H; Parks S et al. CHARMM-GUI 10 years for biomolecular modeling and simulation. J. Comp. Chem 2017, 38, 1114–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (221).Hsu P-C; Bruininks BMH; Jefferies D; Cesar Telles de Souza P; Lee J; Patel DS; Marrink SJ; Qi Y; Khalid S; Im W CHARMM-GUI Martini Maker for modeling and simulation of complex bacterial membranes with lipopolysaccharides. J. Comp. Chem 2017, 38, 2354–2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (222).Alberts B; Johnson A; Lewis J; Raff M; Roberts K; Walter P Molecular Biology of The Cell, 5th ed.; Garland Science: New York, 2008. [Google Scholar]
- (223).Hille B Ionic channels of excitable membranes, 2nd ed.; Sinauer Associates: Sunderland, MA, 1992. [Google Scholar]
- (224).Overington JP; Al-Lazikani B; Hopkins AL How many drug targets are there? Nat. Rev. Drug Disc 2006, 5, 993–996. [DOI] [PubMed] [Google Scholar]
- (225).Yildirim MA; Goh K-I; Cusick ME; Barabasi A-L; Vidal M Drug–target network. Nat. Biotechnol 2007, 25, 1119–1126. [DOI] [PubMed] [Google Scholar]
- (226).Cooper K; Jakobsson E; Wolynes P The theory of ion transport through membrane channels. Prog. Biophys. molec. Biol 1985, 46, 51–96. [DOI] [PubMed] [Google Scholar]
- (227).Gouaux E; Mackinnon R Principles of selective ion transport in channels and pumps. Science 2005, 310, 1461–1465. [DOI] [PubMed] [Google Scholar]
- (228).Gadsby DC Ion channels versus ion pumps: the principal difference, in principle. Nat. Rev. Mol. Cell Biol 2009, 10, 344–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (229).Enkavi G; Li J; Mahinthichaichan P; Wen P-C; Huang Z; Shaikh SA; Tajkhorshid E In Biomolecular Simulations; Monticelli L, Salonen E, Eds.; Methods in Molecular Biology; Humana Press, 2013; Vol. 924; pp 361–405. [DOI] [PubMed] [Google Scholar]
- (230).Enkavi G; Li J; Wen P; Thangapandian S; Moradi M; Jiang T; Han W; Tajkhorshid E Annual Reports in Computational Chemistry ; Elsevier, 2014; Vol. 10; Chapter 4, pp 77–125. [Google Scholar]
- (231).Jiang T; Han W; Maduke M; Tajkhorshid E Molecular basis for differential anion binding and proton coupling in the Cl−/H+ exchanger ClC-ec1. J. Am. Chem. Soc 2016, 138, 3066–3075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (232).Ash WL; Zlomislic MR; Oloo EO; Tieleman DP Computer simulations of membrane proteins. Biochim. Biophys. Acta, Biomembr 2004, 1666, 158–189. [DOI] [PubMed] [Google Scholar]
- (233).Stansfeld PJ Computational studies of membrane proteins: from sequence to structure to simulation. Curr. Opin. Struct. Biol 2017, 45, 133–141. [DOI] [PubMed] [Google Scholar]
- (234).Hille B Ionic channels of excitable membranes, 3rd ed.; Sinauer Associates: Sunderland, MA, 2001. [Google Scholar]
- (235).Bezanilla F The voltage sensor in voltage-dependent ion channels. Physiol. Rev 2000, 80, 555–592. [DOI] [PubMed] [Google Scholar]
- (236).Deol SS; Bond PJ; Domene C; Sansom MS Lipid-protein interactions of integral membrane proteins: A comparative study. Biophys. J 2004, 87, 3737–3749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (237).Deol SS; Domene C; Bond PJ; Sansom MS Anionic phospholipid interactions with the potassium channel KcsA: Simulation studies. Biophys. J 2006, 90, 822–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (238).Sansom M; Bond P; Deol S; Grottesi A; Haider S; Sands Z Molecular simulations and lipid-protein interactions: potassium channels and other membrane proteins. Biochem. Soc. Trans 2005, 33, 916–920. [DOI] [PubMed] [Google Scholar]
- (239).Weingarth M; Prokofyev A; Van Der Cruijsen EAW; Nand D; Bonvin AMJJ; Pongs O; Baldus M Structural determinants of specific lipid binding to potassium channels. J. Am. Chem. Soc 2013, 135, 3983–3988. [DOI] [PubMed] [Google Scholar]
- (240).Delemotte L; Tarek M; Klein ML; Amaral C; Treptow W Intermediate states of the Kv1.2 voltage sensor from atomistic molecular dynamics simulations. Proc. Natl. Acad. Sci. USA 2011, 108, 6109–6114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (241).Nishizawa M; Nishizawa K Molecular dynamics simulation of Kv channel voltage sensor helix in a lipid membrane with applied voltage. Biophys. J 2008, 95, 1729–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (242).Wee CL; Chetwynd A; Sansom MS Membrane insertion of a voltage sensor helix. Biophys. J 2011, 100, 410–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (243).Krepkiy D; Mihailescu M; Freites J; Schow E; Worcester D; Gawrisch K; Tobias D; White S; Swartz K Structure and hydration of membranes embedded with voltage-sensing domains. Nature 2009, 462, 473–U168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (244).Mokrab Y; Sansom MS Interaction of diverse voltage sensor homologs with lipid bilayers revealed by self-assembly simulations. Biophys. J 2011, 100, 875–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (245).Jogini V; Roux B Dynamics of the Kv1.2 voltage-gated K+ channel in a membrane environment. Biophys. J 2007, 93, 3070–3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (246).Bjelkmar P; Niemelä P; Vattulainen I; Lindahl E Conformational changes and slow dynamics through microsecond polarized atomistic molecular simulation of an integral Kv1.2 ion channel. PLoS Comput. Biol 2009, 5, e1000289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (247).Delemotte L; Kasimova MA; Klein ML; Tarek M; Carnevale V Free-energy landscape of ion-channel voltage-sensor–domain activation. Proc. Natl. Acad. Sci. USA 2015, 112, 124–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (248).Kasimova MA; Tarek M; Shaytan AK; Shaitan KV; Delemotte L Voltage-gated ion channel modulation by lipids: Insights from molecular dynamics simulations. Biochim. Biophys. Acta, Biomembr 2014, 1838, 1322–1331. [DOI] [PubMed] [Google Scholar]
- (249).Kasimova MA; Zaydman MA; Cui J; Tarek M PIP2-dependent coupling is prominent in Kv7.1 due to weakened interactions between S4-S5 and S6. Sci. Rep 2015, 5 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- (250).Zhang Q; Zhou P; Chen Z; Li M; Jiang H; Gao Z; Yang H Dynamic PIP2 interactions with voltage sensor elements contribute to KCNQ2 channel gating. Proc. Natl. Acad. Sci. USA 2013, 110, 20093–20098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (251).Chen L; Zhang Q; Qiu Y; Li Z; Chen Z; Jiang H; Li Y; Yang H Migration of PIP2 lipids on voltage-gated potassium channel surface influences channel deactivation. Sci. Rep 2015, 5 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- (252).Kirsch SA; Kugemann A; Carpaneto A; Böckmann RA; Dietrich P Phosphatidylinositol-3,5-bisphosphate lipid-binding-induced activation of the human two-pore channel 2. Cell. Mol. Life Sci 2018, 75, 3803–3815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (253).Yazdi S; Stein M; Elinder F; Andersson M; Lindahl E The molecular basis of polyunsaturated fatty acid interactions with the Shaker voltage-gated potassium channel. PLoS Comput. Biol 2016, 12, e1004704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (254).Andavan G; Lemmens-Gruber R Voltage-gated sodium channels: Mutations, channelopathies and targets. Curr. Med. Chem 2011, 18, 377–397. [DOI] [PubMed] [Google Scholar]
- (255).Amaral C; Carnevale V; Klein ML; Treptow W Exploring conformational states of the bacterial voltage-gated sodium channel NavAb via molecular dynamics simulations. Proc. Natl. Acad. Sci. USA 2012, 109, 21336–21341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (256).Boiteux C; Vorobyov I; French RJ; French C; Yarov-Yarovoy V; Allen TW Local anesthetic and antiepileptic drug access and binding to a bacterial voltage-gated sodium channel. Proc. Natl. Acad. Sci. USA 2014, 111, 13057–13062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (257).Raju SG; Barber AF; LeBard DN; Klein ML; Carnevale V Exploring volatile general anes-thetic binding to a closed membrane–bound bacterial voltage–gated sodium channel via computation. PLoS Comput. Biol 2013, 9, e1003090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (258).Barber AF; Carnevale V; Klein ML; Eckenhoff RG; Covarrubias M Modulation of a voltage-gated Na+ channel by sevoflurane involves multiple sites and distinct mechanisms. Proc. Natl. Acad. Sci. USA 2014, 111, 6726–6731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (259).Kaczmarski JA; Corry B Investigating the size and dynamics of voltage-gated sodium channel fenestrations: A molecular dynamics study. CHANNELS 2014, 8 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- (260).Clapham DE TRP channels as cellular sensors. Nature 2003, 426, 517–524. [DOI] [PubMed] [Google Scholar]
- (261).Liao M; Cao E; Julius D; Cheng Y Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 2013, 504, 107–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (262).Hanson SM; Newstead S; Swartz KJ; Sansom MS Capsaicin interaction with TRPV1 channels in a lipid bilayer: Molecular dynamics simulation. Biophys. J 2015, 108, 1425–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (263).Teng J; Loukin SH; Anishkin A; Kung C A competing hydrophobic tug on L596 to the membrane core unlatches S4-S5 linker elbow from TRP helix and allows TRPV4 channel to open. Proc. Natl. Acad. Sci. USA 2016, 113, 11847–11852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (264).Wang M; Quinn CM; Perilla JR; Zhang H; R. S. Jr.; Hou G; Byeon I-J; Suiter CL; Ablan S; Urano E et al. Quenching protein dynamics interferes with HIV capsid maturation. Nat. Commun 2017, 8, 1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (265).Unwin N Refined structure of the nicotinic acetylcholine receptor at 4 Å resoultion. J. Mol. Biol 2005, 346, 967–989. [DOI] [PubMed] [Google Scholar]
- (266).Vemparala S; Saiz L; Eckenhoff RG; Klein ML Partitioning of anesthetics into a lipid bilayer and their interaction with membrane-bound peptide bundles. Biophys. J 2006, 91, 2815–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (267).Brannigan G; LeBard DN; Hénin J; Eckenhoff RG; Klein ML Multiple binding sites for the general anesthetic isoflurane identified in the nicotinic acetylcholine receptor transmembrane domain. Proc. Natl. Acad. Sci. USA 2010, 107, 14122–14127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (268).Arcario MJ; Mayne CG; Tajkhorshid E A membrane-embedded pathway delivers general anesthetics to two interacting binding sites in the Gloeobacter violaceus ion channel. J. Biol. Chem 2017, 292, 9480–9492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (269).Mayne CG; Arcario MJ; Mahinthichaichan P; Baylon JL; Vermaas JV; Navidpour L; Wen P-C; Thangapandian S; Tajkhorshid E The cellular membrane as a mediator for small molecule interaction with membrane proteins. Biochim. Biophys. Acta, Biomembr 2016, 1858, 2290–2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (270).Carson S; Wanunu M Challenges in DNA motion control and sequence readout using nanopore devices. Nanotechnology 2015, 26, 074004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (271).Dickey AN; Faller R Behavioral differences between phosphatidic acid and phosphatidylcholine in the presence of the nicotinic acetylcholine receptor. Biophys. J 2008, 95, 5637–5647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (272).Logothetis DE; Petrou VI; Adney SK; Mahajan R Channelopathies linked to plasma membrane phosphoinositides. Pflug. Arch. Eur. J. Physiol 2010, 460, 321–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (273).Fürst O; Mondou B; D’Avanzo N Phosphoinositide regulation of inward rectifier potassium (Kir) channels. Front. Physiol 2014, 4, 404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (274).Tao X; Avalos JL; Chen J; MacKinnon R Crystal structure of the eukaryotic strong inward-rectifier K+ channel Kir2.2 at 3.1 Å resolution. Science 2009, 326, 1668–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (275).Haider S; Khalid S; Tucker SJ; Ashcroft FM; Sansom MS Molecular dynamics simulations of inwardly rectifying (Kir) potassium channels: A comparative study. Biochemistry 2007, 46, 3643–3652. [DOI] [PubMed] [Google Scholar]
- (276).Haider S; Tarasov AI; Craig TJ; Sansom MS; Ashcroft FM Identification of the PIP2-binding site on Kir6.2 by molecular modelling and functional analysis. EMBO J 2007, 26, 3749–3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (277).Stansfeld PJ; Hopkinson R; Ashcroft FM; Sansom MSP PIP2-binding site in Kir channels: Definition by multiscale biomolecular simulations. Biochemistry 2009, 48, 10926–10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (278).Schmidt MR; Stansfeld PJ; Tucker SJ; Sansom MS Simulation-based prediction of phosphatidylinositol 4,5-bisphosphate binding to an ion channel. Biochemistry 2013, 52, 279–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (279).Tang QY; Larry T; Hendra K; Yamamoto E; Bell J; Cui M; Logothetis DE; Boland LM Mutations in nature conferred a high affinity phosphatidylinositol 4,5-bisphosphate-binding site in vertebrate inwardly rectifying potassium channels. J. Biol. Chem 2015, 290, 16517–16529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (280).Domanski J; Hedger G; Best RB; Stansfeld PJ; Sansom MSP Convergence and sampling in determining free energy landscapes for membrane protein association. J. Phys. Chem. B 2017, 121, 3364–3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (281).Meng XY; Zhang HX; Logothetis DE; Cui M The molecular mechanism by which PIP2 opens the intracellular G-loop gate of a Kir3.1 channel. Biophys. J 2012, 102, 2049–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (282).An HL; Lü SQ; Li JW; Meng XY; Zhan Y; Cui M; Long M; Zhang HL; Logothetis DE The cytosolic GH loop regulates the phosphatidylinositol 4,5-bisphosphate-induced gating kinetics of Kir2 channels. J. Biol. Chem 2012, 287, 42278–42287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (283).Lacin E; Aryal P; Glaaser IW; Bodhinathan K; Tsai E; Marsh N; Tucker SJ; Sansom MS; Slesinger PA Dynamic role of the tether helix in PIP2-dependent gating of a G protein-gated potassium channel. J. Gen. Physiol 2017, 149, 799–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (284).Rosenhouse-Dantsker A; Noskov S; Durdagi S; Logothetis DE; Levitan I Identification of novel cholesterol-binding regions in Kir2 channels. J. Biol. Chem 2013, 288, 31154–31164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (285).Hamill OP; Martinac B Molecular basis of mechanotransduction in living cells. Physiol. Rev 2001, 81, 685–740. [DOI] [PubMed] [Google Scholar]
- (286).Spencer RH; Chang G; Rees DC ‘Feeling the pressure’: structural insights into a gated mechanosensitive channel. Curr. Opin. Struct. Biol 1999, 9, 448–454. [DOI] [PubMed] [Google Scholar]
- (287).Elmore DE; Dougherty DA Molecular dynamics simulations of wild-type and mutant forms of the Mycobacterium tuberculosis MscL channel. Biophys. J 2001, 81, 1345–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (288).Elmore DE; Dougherty DA Investigating lipid composition effects on the mechanosensitive channel of large conductance (MscL) using molecular dynamics simulations. Biophys. J 2003, 85, 1512–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (289).Wiggins P; Phillips R Membrane-protein interactions in mechanosensitive channels. Biophys. J 2005, 88, 880–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (290).Rui H; Kumar R; Im W Membrane tension, lipid adaptation, conformational changes, and ener-getics in MscL gating. Biophys. J 2011, 101, 671–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (291).Sawada Y; Murase M; Sokabe M The gating mechanism of the bacterial mechanosensitive channel mscl revealed by molecular dynamics simulations: from tension sensing to channel opening. Channels (Austin) 2012, 6, 317–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (292).Mukherjee N; Jose MD; Birkner JP; Walko M; Ingólfsson HI; Dimitrova A; Arnarez C; Marrink SJ; Koçer A The activation mode of the mechanosensitive ion channel, MscL, by lysophos-phatidylcholine differs from tension-induced gating. FASEB J 2014, 28, 4292–4302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (293).Meyer GR; Gullingsrud J; Schulten K; Martinac B Molecular dynamics study of MscL interactions with a curved lipid bilayer. Biophys. J 2006, 91, 1630–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (294).Debret G; Valadié H; Stadler AM; Etchebest C New insights of membrane environment effects on MscL channel mechanics from theoretical approaches. Proteins: Struct., Func., Gen 2008, 71, 1183–1196. [DOI] [PubMed] [Google Scholar]
- (295).Pliotas C; Dahl ACE; Rasmussen T; Mahendran KR; Smith TK; Marius P; Gault J; Banda T; Rasmussen A; Miller S et al. The role of lipids in mechanosensation. Nat. Struct. Mol. Biol 2015, 22, 991–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (296).Bass RB; Strop P; Barclay M; Rees DC Crystal structure of Escherichia coli MscS, a voltage-modulated and mechanosensitive channel. Science 2002, 298, 1582–1587. [DOI] [PubMed] [Google Scholar]
- (297).Vasquez V; Sotomayor M; Cortes DM; Roux B; Schulten K; Perozo E Three dimensional architecture of membrane-embedded MscS in the closed conformation. J. Mol. Biol 2008, 378, 55–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (298).Malcolm HR; Heo YY; Elmore DE; Maurer JA Defining the role of the tension sensor in the mechanosensitive channel of small conductance. Biophys. J 2011, 101, 345–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (299).Aryal P; Jarerattanachat V; Clausen MV; Schewe M; McClenaghan C; Argent L; Conrad LJ; Dong YY; Pike AC; Carpenter EP et al. Bilayer-mediated structural transitions control mechanosensitivity of the TREK-2 K2P channel. Structure 2017, 25, 708–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (300).Treptow W; Klein ML The membrane-bound state of K2P potassium channels. J. Am. Chem. Soc 2010, 132, 8145–8151. [DOI] [PubMed] [Google Scholar]
- (301).Aryal P; Abd-Wahab F; Bucci G; Sansom MSP; Tucker SJ Influence of lipids on the hydrophobic barrier within the pore of the TWIK-1 K2P channel. Channels (Austin) 2015, 9, 44–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (302).Bos MP; Robert V; Tommassen J Biogenesis of the gram-negative bacterial outer membrane. Annu. Rev. Microbiol 2007, 61, 191–214. [DOI] [PubMed] [Google Scholar]
- (303).Koebnik R; Locher KP; Van Gelder P Structure and function of bacterial outer membrane proteins: barrels in a nutshell. Mol. Microbiol 37, 239–253. [DOI] [PubMed] [Google Scholar]
- (304).Straatsma TP; Soares TA Characterization of the outer membrane protein oprf of Pseu-domonas aeruginosa in a lipopolysaccharide membrane by computer simulation. Proteins: Struct., Func., Bioinf 2009, 74, 475–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (305).Lee J; Patel DS; Kucharska I; Tamm LK; Im W Refinement of OprH-LPS interactions by molecular simulations. Biophys. J 2017, 112, 346–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (306).Patel DS; Re S; Wu EL; Qi Y; Klebba PE; Widmalm G; Yeom MS; Sugita Y; Im W Dynamics and interactions of OmpF and LPS: Influence on pore accessibility and ion permeability. Biophys. J 2016, 930–938. [DOI] [PMC free article] [PubMed]
- (307).Wu EL; Fleming PJ; Yeom MS; Widmalm G; Klauda JB; Fleming KG; Im WE coli outer membrane and interactions with OmpLA. Biophys. J 2014, 106, 2493–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (308).Yin F; Kindt JT Hydrophobic mismatch and lipid sorting near ompa in mixed bilayers: Atomistic and coarse-grained simulations. Biophys. J 2012, 102, 2279–2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (309).Schiffrin B; Calabrese AN; Higgins AJ; Humes JR; Ashcroft AE; Kalli AC; J.Brockwell D; Radford SE Effects of periplasmic chaperones and membrane thickness on BamA-catalyzed outer-membrane protein folding. J. Mol. Biol 2017, 429, 3776–3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (310).Cox K; Sansom MSP One membrane protein, two structures and six environments: A comparative molecular dynamics simulation study of the bacterial outer membrane protein PagP. Mol. Membr. Biol 2009, 26, 205–214. [DOI] [PubMed] [Google Scholar]
- (311).Carpenter T; Khalid S; Sansom MS A multidomain outer membrane protein from Pasteurella multocida: Modelling and simulation studies of PmOmpA. Biochim. Biophys. Acta, Biomembr 2007, 1768, 2831–2840. [DOI] [PubMed] [Google Scholar]
- (312).Fleming PJ; Patel DS; Wu EL; Qi Y; Yeom MS; Sousa MC; Fleming KG; Im W BamA POTRA domain interacts with a native lipid membrane surface. Biophys. J 2016, 110, 2698–2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (313).Hodge T; Colombini M Regulation of metabolite flux through voltage-gating of VDAC channels. J. Membr. Biol 1997, 157, 271–279. [DOI] [PubMed] [Google Scholar]
- (314).Mlayeh L; Krammer EM; Léonetti M; Prévost M; Homblé F The mitochondrial VDAC of bean seeds recruits phosphatidylethanolamine lipids for its proper functioning. Biochim. Biophys. Acta, Bioener 2017, 1858, 786–794. [DOI] [PubMed] [Google Scholar]
- (315).Weiser BP; Salari R; Eckenhoff RG; Brannigan G Computational investigation of cholesterol binding sites on mitochondrial VDAC. J. Phys. Chem. B 2014, 118, 9852–9860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (316).Agre P; Kozono D Aquaporin water channels: molecular mechanisms for human diseases. FEBS Lett 2003, 555, 72–78. [DOI] [PubMed] [Google Scholar]
- (317).Agre P The aquaporin water channels. Proc Am Thorac Soc 2006, 3, 5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (318).Tajkhorshid E; Nollert P; Jensen MO; Miercke LJW; O’Connell J; Stroud RM; Schulten K Control of the selectivity of the aquaporin water channel family by global orientational tuning. Science 2002, 296, 525–530. [DOI] [PubMed] [Google Scholar]
- (319).de Groot BL; Grubmüller H Water permeation across biological membranes: Mechanism and dynamics of aquaporin-1 and GlpF. Science 2001, 294, 2353–2357. [DOI] [PubMed] [Google Scholar]
- (320).Stansfeld P; Jefferys E; Sansom M Multiscale simulations reveal conserved patterns of lipid interactions with aquaporins. Structure 2013, 21, 810–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (321).Tong J; Wu Z; Briggs MM; Schulten K; Mclntosh TJ The water permeability and pore entrance structure of aquaporin-4 channels depend on lipid bilayer thickness. Biophys. J 2016, 111, 90–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (322).Wang Y; Cohen J; Boron WF; Schulten K; Tajkhorshid E Exploring gas permeability of cellular membranes and membrane channels with molecular dynamics. J. Struct. Biol 2007, 157, 534–544. [DOI] [PubMed] [Google Scholar]
- (323).Zhang YB; Chen LY In silico study of Aquaporin V: Effects and affinity of the central pore-occluding lipid. Biophys. Chem 2013, 171, 24–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (324).Wen P-C; Mahinthichaichan P; Trebesch N; Jiang T; Zhao Z; Shinn E; Yuhang Wang MS; Kapoor K; Chan CK; Tajkhorshid E Microscopic view of lipids and their diverse biological functions. Curr. Opin. Struct. Biol 2018, 51, 177–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (325).Bethel NP; Grabe M Atomistic insight into lipid translocation by a TMEM16 scramblase. Proc. Natl. Acad. Sci. USA 2016, 113, 14049–14054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (326).Jiang T; Yu K; Hartzell HC; Tajkhorshid E Lipids and ions traverse the membrane by the same physical pathway in the nhTMEM16 scramblase. eLife 2017, 6, e28671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (327).Lee B-C; Khelashvili G; Falzone M; Menon AK; Weinstein H; Accardi A Gating mechanism of the extracellular entry to the lipid pathway in a TMEM16 scramblase. Nat. Commun 2018, 9, 3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (328).Argudo D; Bethel NP; Marcoline FV; Wolgemuth CW; Grabe M New continuum approaches for determining protein-induced membrane deformations. Biophys. J 2017, 112, 2159–2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (329).Mansoor SE; Lüa W; Oosterheert W; Shekhar M; Tajkhorshid E; Gouaux E X-ray structures of human P2X3 receptor define complete gating cycle and antagonist action. Nature 2016, 538, 66–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (330).Morra G; Razavi AM; Pandey K; Weinstein H; Menon AK; Khelashvili G Mechanisms of lipid scrambling by the G protein-coupled receptor opsin. Structure 2018, 26, 356–367.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (331).Jardetzky O Simple allosteric model for membrane pumps. Nature 1966, 211, 969–970. [DOI] [PubMed] [Google Scholar]
- (332).Higgins CF ABC transporters: From microorganisms to man. Annu. Rev. Cell Biol 1992, 8, 67–113. [DOI] [PubMed] [Google Scholar]
- (333).Szakács G; Paterson JK; Ludwig JA; Booth-Genthe C; Gottesman MM Targeting multidrug resistance in cancer. Nat. Rev. Drug Disc 2006, 5, 219–234. [DOI] [PubMed] [Google Scholar]
- (334).Romsicki Y; Sharom FJ Phospholipid flippase activity of the reconstituted P-glycoprotein multidrug transporter. Biochemistry 2001, 40, 6937–6947. [DOI] [PubMed] [Google Scholar]
- (335).Eckford PDW; Sharom FJ The reconstituted P-glycoprotein multidrug transporter is a flippase for glucosylceramide and other simple glycosphingolipids. Biochem. J 2005, 389, 517–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (336).Haubertin DY; Madaoui H; Sanson A; Guérois R; Orlowski S Molecular dynamics simulations of E. coli MsbA transmembrane domain: Formation of a semipore structure. Biophys. J 2006, 91, 2517–2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (337).Ward AB; Guvench O; R. D. H. Jr Coarse grain lipid-protein molecular interactions and diffusion with MsbA flippase. Proteins: Struct., Func., Bioinf 2012, 80, 2178–2190. [DOI] [PubMed] [Google Scholar]
- (338).Mehmood S; Corradi V; Choudhury HG; Hussain R; Becker P; Axford D; Zirah S; Rebuffat S; Tieleman DP; Robinson CV et al. Structural and functional basis for lipid synergy on the activity of the antibacterial peptide ABC transporter McjD. J. Biol. Chem 2016, 291, 21656–21668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (339).Domicevica L; Koldsø H; Biggin PC Multiscale molecular dynamics simulations of lipid interactions with P-glycoprotein in a complex membrane. J. Mol. Graph. Model 2018, 80, 147–156. [DOI] [PubMed] [Google Scholar]
- (340).Wen P-C; Verhalen B; Wilkens S; Mchaourab HS; Tajkhorshid E On the origin of large flexibility of p-glycoprotein in the inward-facing state. J. Biol. Chem 2013, 288, 19211–19220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (341).Barreto-Ojeda E; Corradi V; Gu R-X; Tieleman DP Coarse-grained molecular dynamics simulations reveal lipid access pathways in P-glycoprotein. J. Gen. Physiol 2018, 150, 417–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (342).Immadisetty K; Hettige J; Moradi M Lipid-dependent alternating access mechanism of a bacterial multidrug abc exporter. ACSCS 2019, 5, 43–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (343).Kanner BI; Zomot E Sodium-coupled neurotransmitter transporters. Chem. Rev 2008, 108, 1654–1668. [DOI] [PubMed] [Google Scholar]
- (344).Focke PJ; Wang X; Larsson HP Neutrotransmitter transporters: Structure meets function. Structure 2013, 21, 694–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (345).Yamashita A; Singh SK; Kawate T; Jin Y; Gouaux E Crystal structure of a bacterial homologue of Na+/Cl−-dependent neurotransmitter transporters. Nature 2005, 437, 215–223. [DOI] [PubMed] [Google Scholar]
- (346).Pantano DA; Klein ML Characterization of membrane-protein interactions for the leucine transporter from Aquifex aeolicus by molecular dynamics calculations. J. Phys. Chem. B 2009, 113, 13715–13722. [DOI] [PubMed] [Google Scholar]
- (347).Mondal S; Khelashvili G; Shi L; Weinstein H The cost of living in the membrane: a case study of hydrophobic mismatch for the multi-segment protein LeuT. Chem. Phys. of Lipids 2013, 169, 27–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (348).Grouleff J; Søndergaard S; Koldsø H; Schiøtt B Properties of an inward-facing state of LeuT: Conformational stability and substrate release. Biophys. J 2015, 108, 1390–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (349).Khelashvili G; Stanley N; Sahai MA; Medina J; LeVine MV; Shi L; Fabritiis GD; Weinstein H Spontaneous inward opening of the dopamine transporter is triggered by PIP2-regulated dynamics of the N-terminus. ACS Chem. Neurosci 2015, 6, 1825–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (350).Laursen L; Severinsen K; Kristensen KB; Periole X; Overby M; Müller HK; Schiøtt B; Sinning S Cholesterol binding to a conserved site modulates conformation, pharmacology and transport kinetics of the human serotonin transporter. J. Biol. Chem 2018, 293, 3510–3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (351).Akyuz N; Georgieva ER; Zhou Z; Stolzenberg S; Cuendet MA; Khelashvili G; Altman RB; Terry DS; Freed JH; Weinstein H et al. Transport domain unlocking sets the uptake rate of an aspartate transporter. Nature 2015, 518, 68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (352).Kaback HR Structure and mechanism of the lactose permease. C. R. Biol 2005, 328, 557–567. [DOI] [PubMed] [Google Scholar]
- (353).Dowhan W; Bogdanov M Lipid-dependent membrane protein topogensis. Annu. Rev. Biochem 2009, 78, 515–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (354).Yeagle PL; Bennett M; Lemaître V; Watts A Transmembrane helices of membrane proteins may flex to satisfy hydrophobic mismatch. Biochim. Biophys. Acta, Biomembr 2007, 1768, 530–537. [DOI] [PubMed] [Google Scholar]
- (355).Lensink MF; Govaerts C; Ruysschaert JM Identification of specific lipid-binding sites in integral membrane proteins. J. Biol. Chem 2010, 285, 10519–10526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (356).Andersson M; Bondar AN; Freites JA; Tobias DJ; Kaback HR; White SH Proton-coupled dynamics in lactose permease. Structure 2012, 20, 1893–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (357).Martens C; Shekhar M; Borysik A; Lau A; Reading E; Tajkhorshid E; Booth PJ; Politis A Direct protein-lipid interactions shape the conformational landscape of secondary transporters. Nat. Commun 2018, 9, 4151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (358).Nishima W; Mizukami W; Tanaka Y; Ishitani R; Nureki O; Sugita Y Mechanisms for two-step proton transfer reactions in the outward-facing form of MATE transporter. Biophys. J 2016, 110, 1346–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (359).Kalli AC; Sansom MS; Reithmeier RA Molecular dynamics simulations of the bacterial UraA+-uracil symporter in lipid bilayers reveal a closed state and a selective interaction with cardiolipin. PLoS Comput. Biol 2015, 11, 1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (360).Iglesias-Fernandez J; Quinn PJ; Naftalin RJ; Domene C Membrane phase-dependent occlusion of intramolecular GLUT1 cavities demonstrated by simulations. Biophys. J 2017, 112, 1176–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (361).Park E; Rapoport TA Mechanisms of Sec61/SecY-mediated protein translocation across membranes. Annu. Rev. Biophys 2012, 41, 21–40. [DOI] [PubMed] [Google Scholar]
- (362).Fröbel J; Rose P; Müller M Twin-arginine-dependent translocation of folded proteins. Phil. Trans. R. Soc. Lond. B 2012, 367, 1029–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (363).Palmeira A; Vasconcelos MH; Paiva A; Fernandes MX; Pinto M; Sousa E Dual inhibitors of P-glycoprotein and tumor cell growth: (re)discovering thioxanthones. Biochem. Pharmacol 2012, 83, 57–68. [DOI] [PubMed] [Google Scholar]
- (364).Mori T; Ishitani R; Tsukazaki T; Nureki O; Sugita Y Molecular mechanisms underlying the early stage of protein translocation through the Sec translocon. Biochemistry 2010, 49, 945–950. [DOI] [PubMed] [Google Scholar]
- (365).Capponi S; Heyden M; Bondar A-N; Tobias DJ; White SH Anomalous behavior of water inside the SecY translocon. Proc. Natl. Acad. Sci. USA 2015, 112, 9016–9021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (366).Rodriguez F; Rouse SL; Tait CE; Harmer J; De Riso A; Timmel CR; Sansom MS; Berks BC; Schnell JR Structural model for the protein-translocating element of the twin-arginine transport system. Proc. Natl. Acad. Sci. USA 2013, 110, E1092–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (367).Klingenberg M The ADP and ATP transport in mitochondria and its carrier. Biochim. Biophys. Acta, Biomembr 2008, 1778, 1978–2021. [DOI] [PubMed] [Google Scholar]
- (368).Klingenberg M Cardiolipin and mitochondrial carriers. Biochim. Biophys. Acta 2009, 1788, 2048–2058. [DOI] [PubMed] [Google Scholar]
- (369).Hedger G; Rouse SL; Domanski J; Chavent M; Koldso H; Sansom MSP Lipid-loving ANTs: Molecular simulations of cardiolipin interactions and the organization of the adenne nucleotide translocase in model mitochondrial membranes. Biochemistry 2016, 55, 6238–6249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (370).Holdbrook DA; Piggot TJ; Sansom MSP; Khalid S Stability and membrane interactions of an autotransport protein: MD simulations of the Hia translocator domain in a complex membrane environment. Biochim. Biophys. Acta, Biomembr 2013, 1828, 715–723. [DOI] [PubMed] [Google Scholar]
- (371).Piggot TJ; Holdbrook DA; Khalid S Conformational dynamics and membrane interactions of the E. coli outer membrane protein FecA: A molecular dynamics simulation study. Biochim. Biophys. Acta, Biomembr 2013, 1828, 284–293. [DOI] [PubMed] [Google Scholar]
- (372).Hall J Guyton and Hall Textbook of Medical Physiology ; Elsevier Saunders: Philadelphia, 2016. [Google Scholar]
- (373).Alberts B; Bray D; Hopkin K; Johnson A; Lewis J; Raff M; Roberts K; Walter P Essential Cell Biology ; Garland Science: New York, 2014. [Google Scholar]
- (374).Cherezov V; Rosenbaum D; Hanson M; Rasmussen S; Thian F; Kobilka T; Choi H; Kuhn P; Weis W; Kobilka B et al. High-resolution crystal structure of an engineered human β2-adrenergic G protein-coupled receptor. Science 2007, 318, 1258–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (375).Li D; Stansfeld PJ; Sansom MSP; Keogh A; Vogeley L; Howe N; Lyons JA; Aragao D; Fromme P; Fromme R et al. Ternary structure reveals mechanism of a membrane diacylglycerol kinase. Nat. Commun 2015, 6, 10140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (376).Campbell ID; Ginsberg MH The talin-tail interaction places integrin activation on FERM ground. Trends Biochem. Sci 2004, 29, 429–435. [DOI] [PubMed] [Google Scholar]
- (377).Contreras F-X; Ernst AM; Haberkant P; Björkholm P; Lindahl E; Gönen B; Tischer C; Elofsson A; von Heijne G; Thiele C et al. Molecular recognition of a single sphingolipid species by a protein’s transmembrane domain. Nature 2012, 481, 525–529. [DOI] [PubMed] [Google Scholar]
- (378).Rosenbaum DM; Rasmussen SG; Kobilka BK The structure and function of G-protein-coupled receptors. Nature 2009, 459, 356–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (379).Fredriksson R; Lagerström MC; Lundin L-G; Schiöth LB The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol 2003, 63, 1256–1272. [DOI] [PubMed] [Google Scholar]
- (380).Lagerström MC; Schiöth LB Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat. Rev. Drug Disc 2008, 7, 339–357. [DOI] [PubMed] [Google Scholar]
- (381).Hill SJ G-protein-coupled receptors: past, present and future. Br. J. Pharmacol 2009, 147, S27–S37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (382).Paila YD; Chattopadhyay A In Sub-cellular biochemistry ; Harris JR, Ed.; Subcellular Biochemistry; Springer Netherlands, 2010; Vol. 51; pp 439–466. [DOI] [PubMed] [Google Scholar]
- (383).Oates J; Watts A Uncovering the intimate relationship between lipids, cholesterol and GPCR activation. Curr. Opin. Struct. Biol 2011, 21, 802–7. [DOI] [PubMed] [Google Scholar]
- (384).Mahmood MI; Liu X; Neya S; Hoshino T Influence of lipid composition on the structural stability of G-protein coupled receptor. Chem. Pharm. Bull 2013, 61, 426–437. [DOI] [PubMed] [Google Scholar]
- (385).Zhang D; Zhao Q; Wu B Structural studies of G protein-coupled receptors. Mol. Cell 2015, 38, 836–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (386).Pitman MC; Grossfield A; Suits F; Feller SE Role of cholesterol and polyunsaturated chains in lipid-protein interactions: Molecular dynamics simulation of rhodopsin in a realistic membrane environment. J. Am. Chem. Soc 2005, 127, 4576–4577. [DOI] [PubMed] [Google Scholar]
- (387).Neale C; Herce HD; Pomés R; Garćıa AE Can specific protein-lipid interactions stabilize an active state of the β2 adrenergic receptor? Biophys. J 2015, 109, 1652–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (388).Ng HW; Laughton CA; Doughty SW Molecular dynamics simulations of the adenosine A2a receptor in POPC and POPE lipid bilayers: Effects of membrane on protein behavior. J. Chem. Inf. Model 2014, 54, 573–581. [DOI] [PubMed] [Google Scholar]
- (389).Marino KA; Prada-Gracia D; Provasi D; Filizola M Impact of lipid composition and receptor conformation on the spatio-temporal organization of μ-Opioid receptors in a multicomponent plasma membrane model. PLoS Comput. Biol 2016, 12, e1005240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (390).Shan J; Khelashvili G; Mondal S; Mehler EL; Weinstein H Ligand-dependent conformations and dynamics of the serotonin 5-HT2A receptor determine its activation and membrane-driven oligomerization properties. PLoS Comput. Biol 2012, 8 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- (391).Prasanna X; Sengupta D; Chattopadhyay A Cholesterol-dependent conformational plasticity in GPCR dimers. Sci. Rep 2016, 6 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- (392).Pluhackova K; Gahbauer S; Kranz F; Wassenaar TA; Böckmann RA Dynamic cholesterol-conditioned dimerization of the G protein coupled chemokine receptor type 4. PLoS Comput. Biol 2016, 12, 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (393).Jardón-Valadez E; Ulloa-Aguirre A; Piñeiro A Modeling and molecular dynamics simulation of the human gonadotropin-releasing hormone receptor in a lipid bilayer. J. Phys. Chem. B 2008, 112, 10704–10713. [DOI] [PubMed] [Google Scholar]
- (394).Cordomí A; Perez JJ Molecular dynamics simulations of rhodopsin in different one-component lipid bilayers. J. Phys. Chem. B 2007, 111, 7052–7063. [DOI] [PubMed] [Google Scholar]
- (395).Grossfield A; Feller SE; Pitman MC Contribution of omega-3 fatty acids to the thermodynamics of membrane protein solvation. J. Phys. Chem. B 2006, 110, 8907–8909. [DOI] [PubMed] [Google Scholar]
- (396).Salas-Estrada LA; Leioatts N; Romo TD; Grossfield A Lipids alter rhodopsin function via ligand-like and solvent-like interactions. Biophys. J 2018, 114, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (397).Song W; Yen H-Y; Robinson CV; Sansom M State-dependent lipid interactions with the A2a receptor revealed by MD simulations using in vivo-mimetic membranes. Structure 2019, 27, 392–403.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (398).Park PS-H; Filipek S; Wells JW; Palczewski K Oligomerization of G protein-coupled receptors: Past, Present, and Future. Biochemistry 2004, 43, 15643–15656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (399).Gomes I; Jordan BA; Gupta A; Rios C; Trapaidze N; Devi LA G protein coupled receptor dimerization: Implications in modulating receptor function. J. Mol. Med 2001, 79, 226–242. [DOI] [PubMed] [Google Scholar]
- (400).George SR; O’Dowd BF; Lee SP G-protein-coupled receptor oligomerization and its potential for drug discovery. Nat. Rev. Drug Disc 2002, 1, 808–820. [DOI] [PubMed] [Google Scholar]
- (401).Khelashvili G; Albornoz PBC; Johner N; Mondal S; Caffrey M; Weinstein H Why GPCRs behave differently in cubic and lamellar lipidic mesophases. J. Am. Chem. Soc 2012, 134, 15858–15868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (402).Cang X; Yang L; Yang J; Luo C; Zheng M; Yu K; Yang H; Jiang H Cholesterol-β1AR interaction versus cholesterol-β2AR interaction. Proteins: Struct., Func., Bioinf 2014, 82, 760–770. [DOI] [PubMed] [Google Scholar]
- (403).Cang X; Du Y; Mao Y; Wang Y; Yang H; Jiang H Mapping the functional binding sites of cholesterol in β2-adrenergic receptor by long-time molecular dynamics simulations. J. Phys. Chem. B 2013, 117, 1085–1094. [DOI] [PubMed] [Google Scholar]
- (404).Prasanna X; Chattopadhyay A; Sengupta D Cholesterol modulates the dimer interface of the β2-adrenergic receptor via cholesterol occupancy sites. Biophys. J 2014, 106, 1290–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (405).Yue T; Sun M; Zhang S; Ren H; Ge B; Huang F How transmembrane peptides insert and orientate in biomembranes: A combined experimental and simulation study. Phys. Chem. Chem. Phys 2016, 18, 17483–17494. [DOI] [PubMed] [Google Scholar]
- (406).Gahbauer S; Pluhackova K; Böckmann RA Closely related, yet unique: Distinct homo-and heterodimerization patterns of G protein coupled chemokine receptors and their fine-tuning by cholesterol. PLoS Comput. Biol 2018, 14, e1006062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (407).Prasanna X; Praveen PJ; Sengupta D Sequence dependent lipid-mediated effects modulate the dimerization of ErbB2 and its associative mutants. Phys. Chem. Chem. Phys 2013, 15, 19031. [DOI] [PubMed] [Google Scholar]
- (408).Olausson BES; Grossfield A; Pitman MC; Brown MF; Feller SE; Vogel A Molecular dynamics simulations reveal specific interactions of post-translational palmitoyl modifications with rhodopsin in membranes. J. Am. Chem. Soc 2012, 134, 4324–4331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (409).Shim JY Transmembrane helical domain of the cannabinoid CB1 receptor. Biophys. J 2009, 96, 3251–3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (410).Cieplak M; Filipek S; Janovjak H; Krzyśko K A. Pulling single bacteriorhodopsin out of a membrane: Comparison of simulation and experiment. Biochim. Biophys. Acta, Biomembr 2006, 1758, 537–544. [DOI] [PubMed] [Google Scholar]
- (411).Khelashvili G; Grossfield A; Feller SE; Pitman MC; Weinstein H Structural and dynamic effects of cholesterol at preferred sites of interaction with rhodopsin identified from microsecond length molecular dynamics simulations. Proteins: Struct., Func., Bioinf 2009, 76, 403–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (412).Ghumman A; Robinson D Modulation of the turkey β1-adrenergic receptor by membrane rafts - insight from molecular dynamics. Chem. Sel 2016, 1, 4274–4276. [Google Scholar]
- (413).Manna M; Niemelä M; Tynkkynen J; Javanainen M; Kulig W; Müller DJ; Rog T; Vattulainen I Mechanism of allosteric regulation of β2-adrenergic receptor by cholesterol. eLife 2016, 5, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (414).Duong TH; Mehler EL; Weinstein H Molecular dynamics simulation of membranes and a transmembrane helix. J. Chem. Phys 1999, 151, 358–387. [Google Scholar]
- (415).Paila YD; Tiwari S; Sengupta D; Chattopadhyay, A. Molecular modeling of the human serotonin 1A receptor: role of membrane cholesterol in ligand binding of the receptor. Mol. BioSyst 2011, 7, 224–234. [DOI] [PubMed] [Google Scholar]
- (416).Patra SM; Chakraborty S; Shahane G; Prasanna X; Sengupta D; Maiti PK; Chattopadhyay A Differential dynamics of the serotonin(1A) receptor in membrane bilayers of varying cholesterol content revealed by all atom molecular dynamics simulation. Mol. Membr. Biol 2015, 32, 127–137. [DOI] [PubMed] [Google Scholar]
- (417).Ramírez-Anguita JM; Rodríguez-Espigares I; Guixà-González R; Bruno A; Torrens-Fontanals M; Varela-Rial A; Selent J Membrane cholesterol effect on the 5-HT2A receptor: Insights into the lipid-induced modulation of an antipsychotic drug target. Biotechnol. Appl. Biochem 2017, 29–37. [DOI] [PubMed]
- (418).Hurst DP; Grossfield A; Lynch DL; Feller S; Romo TD; Gawrisch K; Pitman MC; Reggio PH A lipid pathway for ligand binding is necessary for a cannabinoid G protein-coupled receptor. J. Biol. Chem 2010, 285, 17954–17964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (419).Cao R; Rossetti G; Bauer A; CarIoni P Binding of the antagonist caffeine to the human adenosine receptor hA2AR in nearly physiological conditions. PLoS One 2015, 10 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- (420).Genheden S; Essex JW; Lee AG G protein coupled receptor interactions with cholesterol deep in the membrane. Biochim. Biophys. Acta, Biomembr 2017, 1859, 268–281. [DOI] [PubMed] [Google Scholar]
- (421).Hynes RO Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [DOI] [PubMed] [Google Scholar]
- (422).Tadokoro S; Shattil SJ; Eto K; Tai V; Liddington RC; de Pereda JM; Ginsberg MH; Calderwood DA Talin binding to integrin β tails: A final common step in integrin activation. Science 2003, 302, 103–106. [DOI] [PubMed] [Google Scholar]
- (423).Shattil SJ; Kim C; Ginsberg MH The final steps of integrin activation: The end game. Nat. Rev. Mol. Cell Biol 2010, 11, 288–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (424).Moser M; Legate KR; Zent R; Fässler R The tail of integrins, talin, and kindlins. Science 2009, 324, 895–899. [DOI] [PubMed] [Google Scholar]
- (425).Kalli AC; Campbell ID; Sansom MS Multiscale simulations suggest a mechanism for integrin inside-out activation. Proc. Natl. Acad. Sci. USA 2011, 108, 11890–11895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (426).Chng C-P; Tan S-M Leukocyte integrin αLβ2 transmembrane association dynamics revealed by coarse-grained molecular dynamics simulations. Proteins: Struct., Func., Bioinf 2011, 79, 2203–2213. [DOI] [PubMed] [Google Scholar]
- (427).Vararattanavech A; Chng C-P; Parthasarathy K; Tang X-Y; Torres J; Tan S-M A transmembrane polar interaction is involved in the functional regulation of integrin αLβ2. J. Mol. Biol 2010, 398, 569–583. [DOI] [PubMed] [Google Scholar]
- (428).Schmidt T; Suk JE; Ye F; Situ AJ; Mazumder P; Ginsberg MH; Ulmer TS Annular anionic lipids stabilize the integrin αIIbβ3 transmembrane complex. J. Biol. Chem 2015, 290, 8283–8293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (429).Kalli AC; Rog T; Vattulainen I; Campbell ID; Sansom MSP The integrin receptor in biologically relevant bilayers: Insights from molecular dynamics simulations. J. Membr. Biol 2017, 250, 337–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (430).Mehrbod M; Mofrad MR Localized lipid packing of transmembrane domains impedes integrin clustering. PLoS Comput. Biol 2013, 9, e1002948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (431).Rg T; Murzyn K; Karttunen M; Pasenkiewicz-Gierula M Nonpolar interactions between transmembrane helical EGF peptide and phosphatidylcholines, sphingomyelins and cholesterol. molecular dynamics simulation studies. J. Pharmaceut. Sci 2008, 14, 374–382. [DOI] [PubMed] [Google Scholar]
- (432).López CA; Sethi A; Goldstein B; Wilson BS; Gnanakaran S Membrane-mediated regulation of the intrinsically disordered CD3E cytoplasmic tail of the TCR. Biophys. J 2015, 108, 2481–2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (433).Hedger G; Sansom MS; Koldso H The juxtamembrane regions of human receptor tyrosine kinases exhibit conserved interaction sites with anionic lipids. Nat. Sci. Rep 2015, 5, 9198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (434).Arkhipov A; Shan Y; Das R; Endres NF; Eastwood MP; Wemmer DE; Kuriyan J; Shaw DE Architecture and membrane interactions of the EGF receptor. Cell 2013, 152, 557–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (435).Soumana OS; Aller P; Garnier N; Genest M Transmembrane peptides from tyrosine kinase receptor. Mutation-related behavior in a lipid bilayer investigated by molecular dynamics simulations. J. Biomol. Struct. Dyn 2005, 23, 91–100. [DOI] [PubMed] [Google Scholar]
- (436).Beevers AJ; Nash A; Salazar-Cancino M; Scott DJ; Notman R; Dixon AM Effects of the oncogenic V664E mutation on membrane insertion, structure, and sequence-dependent interactions of the Neu transmembrane domain in micelles and model membranes: An integrated biophysical and simulation study. Biochemistry 2012, 51, 2558–2568. [DOI] [PubMed] [Google Scholar]
- (437).Chavent M; Seiradake E; Jones EY; Sansom MS Structures of the EphA2 receptor at the membrane: Role of lipid interactions. Structure 2016, 24, 337–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (438).Reddy T; Manrique S; Buyan A; Hall BA; Chetwynd A; Sansom MSP Primary and secondary dimer interfaces of the fibroblast growth factor receptor 3 transmembrane domain: Characterization via multiscale molecular dynamics simulations. Biochemistry 2014, 53, 323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (439).Kästner J; Loeffler HH; Roberts SK; Martin-Fernandez ML; Winn MD Ectodomain orientation, conformational plasticity and oligomerization of ErbB1 receptors investigated by molecular dynamics. J. Struct. Biol 2009, 167, 117–128. [DOI] [PubMed] [Google Scholar]
- (440).Arnarez C; Mazat J-P; Elezgaray J; Marrink S-J; Periole X Evidence for cardiolipin binding sites on the membrane-exposed surface of the cytochrome bc1. J. Am. Chem. Soc 2013, 135, 3112–20. [DOI] [PubMed] [Google Scholar]
- (441).Arnarez C; Marrink SJ; Periole X Molecular mechanism of cardiolipin-mediated assembly of respiratory chain supercomplexes. Chem. Sci 2016, 7, 4435–4443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (442).Arnarez C; Marrink SJ; Periole X Identification of cardiolipin binding sites on cytochrome c oxidase at the entrance of proton channels. Sci. Rep 2013, 3, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (443).Pöyry S; Cramariuc O; Postila PA; Kaszuba K; Sarewicz M; Osyczka A; Vattulainen I; Rog T Atomistic simulations indicate cardiolipin to have an integral role in the structure of the cytochrome bc1 complex. Biochim. Biophys. Acta, Biomembr 2013, 1827, 769–778. [DOI] [PubMed] [Google Scholar]
- (444).Kobayashi K; Endo K; Wada H Specific distribution of phosphatidylglycerol to photosystem complexes in the thylakoid membrane. Front. Plant Sci 2017, 8, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (445).Van Eerden FJ; Van Den Berg T; Frederix PW; De Jong DH; Periole X; Marrink SJ Molecular dynamics of photosystem II embedded in the thylakoid membrane. J. Phys. Chem. B 2017, 121, 3237–3249. [DOI] [PubMed] [Google Scholar]
- (446).Van Eerden FJ; Melo MN; Frederix PW; Marrink SJ Prediction of thylakoid lipid binding sites on photosystem II. Biophys. J 2017, 113, 2669–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (447).Hsin J; LaPointe L; Kazy A; Chipot C; Senes A; Schulten K Oligomerization state of photo-synthetic core complexes is correlated with the dimerization affinity of a transmembrane helix. J. Am. Chem. Soc 2011, 133, 14071–14081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (448).Lemkul JA; Bevan DR Characterization of interactions between PilA from Pseudomonas aeruginosa strain K and a model membrane. J. Phys. Chem. B 2011, 115, 8004–8008. [DOI] [PubMed] [Google Scholar]
- (449).Lee HS; Im W Transmembrane motions of PglB induced by LLO are coupled with EL5 loop conformational changes necessary for OST activity. Glycobiology 2017, 27, 734–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (450).Bondar AN; del Val C; White SH Rhomboid protease dynamics and lipid interactions. Structure 2009, 17, 395–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (451).Dorairaj S; Allen TW On the thermodynamic stability of a charged arginine side chain in a transmembrane helix. Proc. Natl. Acad. Sci. USA 2007, 104, 4943–4948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (452).Cheng X; Jo S; Marassi FM; Im W NMR-based simulation studies of Pf1 coat protein in explicit membranes. Biophys. J 2013, 105, 691–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (453).Bracey MH; Cravatt BF; Stevens RC Structural commonalities among integral membrane enzymes. FEBS Lett 2004, 567, 159–165. [DOI] [PubMed] [Google Scholar]
- (454).Balali-Mood K; Bond PJ; Sansom MS Interaction of monotopic membrane enzymes with a lipid bilayer: A coarse-grained MD simulation study. Biochemistry 2009, 48, 2135–2145. [DOI] [PubMed] [Google Scholar]
- (455).Navrátilová V; Paloncýová M; Berka K; Otyepka M Effect of lipid charge on membrane immersion of cytochrome P450 3A4. J. Phys. Chem. B 2016, 120, 11205–11213. [DOI] [PubMed] [Google Scholar]
- (456).Denisov IG; Shih AY; Sligar SG Structural differences between soluble and membrane bound cytochrome P450s. J. Inorg. Biochem 2012, 108, 150–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (457).Lonsdale R; Rouse SL; Sansom MSP; Mulholland AJ A multiscale approach to modelling drug metabolism by membrane-bound cytochrome P450 enzymes. PLoS Comput. Biol 2014, 10, e1003714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (458).Mustafa G; Nandekar PP; Yu X; Wade RC On the application of the MARTINI coarse-grained model to immersion of a protein in a phospholipid bilayer. J. Chem. Phys 2015, 143, 243139. [DOI] [PubMed] [Google Scholar]
- (459).Cojocaru V; Balali-Mood K; Sansom MSP; Wade RC Structure and dynamics of the membrane-bound cytochrome P450 2C9. PLoS Comput. Biol 2011, 7, e1002152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (460).Di Nardo G; Cimicata G; Baravalle R; Dell’Angelo V; Ciaramella A; Catucci G; Ugliengo P; Gilardi G Working at the membrane interface: Ligand-induced changes in dynamic conformation and oligomeric structure in human aromatase. Biotechnol. Appl. Biochem 2018, 65, 46–53. [DOI] [PubMed] [Google Scholar]
- (461).Berka K; Hendrychová T; Anzenbacher P; Otyepka M Membrane position of ibuprofen agrees with suggested access path entrance to cytochrome P450 2C9 active site. J. Phys. Chem. A 2011, 115, 11248–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (462).Jeřábek P; Florián J; Martínek V Lipid molecules can induce an opening of membrane-facing tunnels in cytochrome P450 1A2. Phys. Chem. Chem. Phys 2016, 18, 30344–30356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (463).Berka K; Paloncyóvá M; Anzenbacher P; Otyepka M Behavior of human cytochromes P450 on lipid membranes. J. Phys. Chem. B 2013, 117, 11556–11564. [DOI] [PubMed] [Google Scholar]
- (464).Carlo B; Ranjan SB; Thirupathi R; G. M-MI; Sang-Choul I; M. AG; Lucy W; Ayyalusamy R Cytochrome-p450-induced ordering of microsomal membranes modulates affinity for drugs. Angew. Chem. Int. Ed. Engl 2018, 57, 3391–3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (465).Santini S; Crowet JM; Thomas A; Paquot M; Vandenbol M; Thonart P; Wathelet JP; Blecker C; Lognay G; Brasseur R et al. Study of thermomyces lanuginosa lipase in the presence of tributyrylglycerol and water. Biophys. J 2009, 96, 4814–4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (466).Grandits M; Oostenbrink C Selectivity of cytosolic phospholipase A2 type IV toward arachidonyl phospholipids. J. Mol. Recognit 2015, 28, 447–457. [DOI] [PubMed] [Google Scholar]
- (467).Mouchlis VD; Bucher D; McCammon JA; Dennis EA Membranes serve as allosteric activators of phospholipase A2, enabling it to extract, bind, and hydrolyze phospholipid substrates. Proc. Natl. Acad. Sci. USA 2015, 112, E516–E525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (468).Nasr ML; Shi X; Bowman AL; Johnson M; Zvonok N; Janero DR; Vemuri VK; Wales TE; Engen JR; Makriyannis A Membrane phospholipid bilayer as a determinant of monoacylglycerol lipase kinetic profile and conformational repertoire. Prot. Sci 2013, 22, 774–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (469).Willems N; Lelimousin M; Koldsø H; Sansom MS Interfacial activation of M37 lipase: A multi-scale simulation study. Biochim. Biophys. Acta, Biomembr 2017, 1859, 340–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (470).Bucher D; Hsu YH; Mouchlis VD; Dennis EA; McCammon JA Insertion of the Ca2+-independent phospholipase A2 into a phospholipid bilayer via coarse-grained and atomistic molecular dynamics simulations. PLoS Comput. Biol 2013, 9 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- (471).Grauffel C; Yang B; He T; Roberts MF; Gershenson A; Reuter N Cation-π interactions as lipid-specific anchors for phosphatidylinositol-specific phospholipase C. J. Am. Chem. Soc 2013, 135, 5740–5750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (472).Chze LW; Balali-Mood K; Gavaghan D; Sansom MS The interaction of phospholipase A2 with a phospholipid bilayer: Coarse-grained molecular dynamics simulations. Biophys. J 2008, 95, 1649–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (473).Qin SS; Yu YX; Li QK; Yu ZW Interaction of human synovial phospholipase A2 with mixed lipid bilayers: A coarse-grain and all-atom molecular dynamics simulation study. Biochemistry 2013, 52, 1477–1489. [DOI] [PubMed] [Google Scholar]
- (474).Jaud S; Tobias DJ; Falke JJ; White SH Self-induced docking site of a deeply embedded peripheral membrane protein. Biophys. J 2007, 92, 517–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (475).Ward KKE; Bhardwaj N; Vora M; Chalfant CEC; Lu H; Stahelin RV The molecular basis of ceramide-1-phosphate recognition by C2 domains. J. Lipid Res 2013, 54, 636–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (476).Cheng J; Karri S; Grauffel C; Wang F; Reuter N; Roberts MF; Wintrode PL; Gershenson A Does changing the predicted dynamics of a phospholipase C alter activity and membrane binding? Biophys. J 2013, 104, 185–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (477).Yang B; Pu M; Khan HM; Friedman L; Reuter N; Roberts MF; Gershenson A Quantifying transient interactions between bacillus phosphatidylinositol-specific phospholipase-C and phosphatidylcholine-rich vesicles. J. Am. Chem. Soc 2015, 137, 14–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (478).Han DS; Golebiewska U; Stolzenberg S; Scarlata SF; Weinstein H A dynamic model of membrane-bound phospholipase C2 activation by G subunits. Mol. Pharmacol 2011, 80, 434–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (479).Han D; Pal S; Nangreave J; Deng Z; Liu Y; Yan H DNA origami with complex curvatures in three-dimensional space. Science 2011, 332, 342–6. [DOI] [PubMed] [Google Scholar]
- (480).Rodríguez Y; Mezei M; Osman R The PT1-Ca2+ Gla domain binds to a membrane through two dipalmitoylphosphatidylserines. a computational study. Biochemistry 2008, 47, 13267–13278. [DOI] [PubMed] [Google Scholar]
- (481).Taboureau O; Olsen OH Computational study of coagulation factor VIIa’s affinity for phospholipid membranes. Eur. Biophys. J 2007, 36, 133–144. [DOI] [PubMed] [Google Scholar]
- (482).Ohkubo YZ; Morrissey JH; Tajkhorshid E Dynamical view of membrane binding and complex formation of human factor VIIa and tissue factor. J. Thromb. Haem 2010, 8, 1044–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (483).Du J; Wichapong K; Hackeng TM; Nicolaes GAF Molecular simulation studies of human coagulation factor VIII C domain-mediated membrane binding. Thromb. Haemost 2015, 113, 373–384. [DOI] [PubMed] [Google Scholar]
- (484).Mollica L; Franternali F; Musco G Interactions of the C2 domain of human factor V with a model membrane. Proteins: Struct., Func., Bioinf 2006, 64, 363–375. [DOI] [PubMed] [Google Scholar]
- (485).Schillinger AS; Grauffel C; Khan HM; Halskau Ø; Reuter N Two homologous neutrophil serine proteases bind to POPC vesicles with different affinities: When aromatic amino acids matter. Biochim. Biophys. Acta, Biomembr 2014, 1838, 3191–3202. [DOI] [PubMed] [Google Scholar]
- (486).Broemstrup T; Reuter N How does proteinase 3 interact with lipid bilayers? Phys. Chem. Chem. Phys 2010, 12, 7487–96. [DOI] [PubMed] [Google Scholar]
- (487).Schicht M; Rausch F; Finotto S; Mathews M; Mattil A; Schubert M; Koch B; Traxdorf M; Bohr C; Worlitzsch D et al. SFTA3, a novel protein of the lung: three-dimensional structure, characterisation and immune activation. Eur. Respir J 2014, 44, 447–456. [DOI] [PubMed] [Google Scholar]
- (488).Ramelot TA; Yang Y; Sahu ID; Lee HW; Xiao R; Lorigan GA; Montelione GT; Kennedy MA NMR structure and MD simulations of the AAA protease intermembrane space domain indicates peripheral membrane localization within the hexaoligomer. FEBS Lett 2013, 587, 3522–3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (489).Hung HM; Hang TD; Nguyen MT Molecular details of spontaneous insertion and interaction of HCV non-structure 3 protease protein domain with PIP2-containing membrane. Proteins: Struct., Func., Bioinf 2018, 1–33. [DOI] [PubMed]
- (490).Nina M; Bernéche S; Roux B Anchoring of monotopic membrane protein: The binding of prostaglandin H2 synthase-1 to the surface of a phospholipid bilayer. Eur. Biophys. J 2000, 29, 439–454. [DOI] [PubMed] [Google Scholar]
- (491).Fowler PW; Coveney PV A computational protocol for the integration of the monotopic protein prostaglandin H2 synthase into a phospholipid bilayer. Biophys. J 2006, 91, 401–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (492).Wan S; Coveney PV A comparative study of the COX-1 and COX-2 isozymes bound to lipid membranes. J. Comp. Chem 2009, 30, 1038–1050. [DOI] [PubMed] [Google Scholar]
- (493).Fowler PW; Balali-Mood K; Deol S; Coveney PV; Sansom MS Monotopic enzymes and lipid bilayers: A comparative study. Biochemistry 2007, 46, 3108–3115. [DOI] [PubMed] [Google Scholar]
- (494).Palermo G; Bauer I; Campomanes P; Cavalli A; Armirotti A; Girotto S; Rothlisberger U; De Vivo M Keys to lipid selection in fatty acid amide hydrolase catalysis: Structural flexibility, gating residues and multiple binding pockets. PLoS Comput. Biol 2015, 11, 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (495).Palermo G; Campomanes P; Neri M; Piomelli D; Cavalli A; Rothlisberger U; De Vivo M, Wagging the tail: Essential role of substrate flexibility in FAAH catalysis. J. Chem. Theory Comput 2013, 9, 1202–1213. [DOI] [PubMed] [Google Scholar]
- (496).Ge C; Gõmez-Llobregat J; Skwark MJ; Ruysschaert JM; Wieslander Å; Lindén M Membrane remodeling capacity of a vesicle-inducing glycosyltransferase. FEBS J 2014, 281, 3667–3684. [DOI] [PubMed] [Google Scholar]
- (497).Rodrigues CI; Hardy DJ; Stone JE; Schulten K; Hwu WW GPU acceleration of cutoff pair potentials for molecular modeling applications. CF’08: Proceedings of the 2008 conference on Computing Frontiers. New York, NY, USA, 2008; pp 273–282. [Google Scholar]
- (498).Zhou Q; Li J; Yu H; Zhai Y; Gao Z; Liu Y; Pang X; Zhang L; Schulten K; Sun F et al. Molecular insights into the membrane-associated phosphatidylinositol 4-kinase IIα. Nat. Commun 2014, 5, 3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (499).Li J; Ziemba BP; Falke JJ; Voth GA Interactions of protein kinase C-α C1A and C1B domains with membranes: A combined computational and experimental study. J. Am. Chem. Soc 2014, 136, 11757–11766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (500).Lumb CN; Sansom MS Defining the membrane-associated state of the PTEN tumor suppressor protein. Biophys. J 2013, 104, 613–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (501).Rodrigues ML; Scott KA; Sansom MS; Pereira IA; Archer M Quinol oxidation by c-type cytochromes: Structural characterization of the menaquinol binding site of NrfHA. J. Mol. Biol 2008, 381, 341–350. [DOI] [PubMed] [Google Scholar]
- (502).Chng CP; Strange RW Lipid-associated aggregate formation of superoxide dismutase-1 is initiated by membrane-targeting loops. Proteins Struct. Funct. Bioinforma 2014, 82, 3194–3209. [DOI] [PubMed] [Google Scholar]
- (503).Shenoy SS; Nanda H; Lösche M Membrane association of the PTEN tumor suppressor: Electro-static interaction with phosphatidylserine-containing bilayers and regulatory role of the C-terminal tail. J. Struct. Biol 394–408. [DOI] [PMC free article] [PubMed]
- (504).Kalli AC; Devaney I; Sansom MS Interactions of phosphatase and tensin homologue (PTEN) proteins with phosphatidylinositol phosphates: Insights from molecular dynamics simulations of PTEN and voltage sensitive phosphatase. Biochemistry 2014, 53, 1724–1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (505).Nanda H; Heinrich F; Lösche M Membrane association of the PTEN tumor suppressor: Neutron scattering and MD simulations reveal the structure of protein-membrane complexes. Methods 2015, 77, 136–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (506).Janosi L; Gorfe AA Segregation of negatively chagred phospholipids by the polycationic and farnesylated membrane anchor of Kras. Biophys. J 2010, 99, 3666–3674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (507).Zhou Y; Prakash P; Liang H; Cho K-J; Gorfe AA; Hancock JF Lipid-sorting specificity encoded in K-Ras membrane anchor regulates signal output. Cell 2017, 168, 239–251.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (508).Jang H; Abraham SJ; Chavan TS; Hitchinson B; Khavrutskii L; Tarasova NI; Nussinov R; Gaponenko V Mechanisms of membrane binding of small GTPase K-ras4B farnesylated hypervariable region. J. Biol. Chem 2015, 290, 9465–9477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (509).Cho K; Casteelb DE; Prakasha P; Tana L; van der Hoevenc D; Salimd AA; Kime C; Capond RJ; Laceyf E; Cunhaa SR et al. AMPK and endothelial nitric oxide synthase signaling regulates K-Ras plasma membrane interactions via cyclic GMP-dependent protein kinase 2. Mol. Cell. Biol 2016, 36, 3086–3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (510).Jang H; Banerjee A; Chavan TS; Lu S; Zhang J; Gaponenko V; Nussinov R The higher level of complexity of K-Ras4B activation at the membrane. FASEB J 2016, 30, 1643–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (511).Gorfe AA; Pellarin R; Caflisch A Membrane localization and flexibility of a lipidated peptide studied by molecular dynamics simulations. J. Am. Chem. Soc 2004, 126, 15277–15286. [DOI] [PubMed] [Google Scholar]
- (512).Larsen JB; Kennard C; Pedersen SL; Jensen KJ; Uline MJ; Hatzakis NS; Stamous D Membrane curvature and lipid composition synergize to regulate N-Ras anchor recruitment. Biophys. J 2017, 113, 1269–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (513).Vogel A; Tan KT; Waldmann H; Feller SE; Brown MF; Huster D Flexibility of ras lipid modifications studied by 2H solid-state NMR and molecular dynamics simulations. Biophys. J 2007, 93, 2697–2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (514).Vogel A; Reuther G; Roark MB; Tan K-T; Waldmann H; Feller SE; Huster D Backbone conformational flexibility of the lipid modified membrane anchor of the human N-Ras protein investigated by solid-state NMR and molecular dynamics simulation. Biochim. Biophys. Acta, Biomembr 2010, 1798, 275–285. [DOI] [PubMed] [Google Scholar]
- (515).Vogel A; Roark M; Feller SE A reinterpretation of neutron scattering experiments on a lipdated Ras peptide using replica exchange molecular dynamics. Biochim. Biophys. Acta, Biomembr 2012, 1818, 219–224. [DOI] [PubMed] [Google Scholar]
- (516).Jefferys E; Sansom MSP; Fowler PW NRas slows the rate at which a model lipid bilayer phase separates. Faraday Discuss 2014, 169, 209–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (517).Gorfe AA; Hanzal-Bayer M; Abankwa D; Hancock JF; McCammon JA Structure and dynamics of the full-length lipid-modified H-Ras protein in a 1,2-dimyristoylglycero-3-phosphocholine bilayer. J. Med. Chem 2007, 50, 674–684. [DOI] [PubMed] [Google Scholar]
- (518).Gorfe AA; Babakhani A; McCammon JA H-ras protein in a bilayer: Interaction and structure perturbation. J. Am. Chem. Soc 2007, 129, 12280–12286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (519).Gorfe AA; Babakhani A; McCammon JA Membrane insertion free energy profile of H-Ras membrane anchor. Angew. Chem. Int. Ed. Engl 2007, 46, 8234–8237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (520).Li H; Gorfe AA Aggregation of lipid-anchored full-length H-Ras in lipid bilayers: Simulations with the MARTINI force field. PLoS One 2013, 8, 2467–2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (521).Prakash P; Zhou Y; Liang H; Hancock JF; Gorfe AA Oncogenic K-Ras binds to an anionic membrane in two distinct orientations: A molecular dynamics analysis. Biophys. J 2016, 110, 1125–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (522).Abankwa D; Gorfe AA; Inder K; Hancock JF Ras membrane orientation and nanodomain localization generate isoform diversity. Proc. Natl. Acad. Sci. USA 2010, 107, 1130–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (523).Li Z-L; Buck M Computational modeling reveals that signaling lipids modulate the orientation of K-Ras4A at the membrane reflecting protein topology. Structure 2017, 25, 679–689.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (524).Jang H; Muratcioglu S; Gursoy A; Keskin O; Nussinov R Membrane-associated Ras dimers are isoform-specific: K-Ras dimers differ from H-Ras dimers. Biochem. J 2016, 473, 1719–1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (525).Li Z; Janosi L; Gorfe AA Formation and domain partitioning of H-ras peptide nanoclusters: Effects of peptide concentration and lipid composition. J. Am. Chem. Soc 2012, 134, 17278–17285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (526).Janosi L; Li Z; Hancock JF; Gorfe AA Organization, dynamics, and segregation of Ras nanoclusters in membrane domains. Proc. Natl. Acad. Sci. USA 2012, 109, 8097–8102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (527).Guldenhaupt J; Rudack T; Bachler P; Mann D; Triola G; Waldmann H; Kotting C; Gerwert K N-Ras forms dimers at POPC membranes. Biophys. J 2012, 103, 1585–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (528).Lin X; Li Z; Gorfe A Reversible effects of peptide concentration and lipid composition on H-Ras lipid anchor clustering. Biophys. J 2015, 109, 2467–2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (529).Li S; Jang H; Zhang J; Nussinov R Raf-1 cysteine-rich domain increases the affinity of K-Ras/Raf at the membrane, promoting mapk signaling. Structure 2018, 26, 513–525.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (530).Travers T; Lòpez CA; Van QN; Neale C; Tonelli M; Stephen AG; Gnanakaran S Molecular recognition of RAS/RAF complex at the membrane: Role of RAF cysteine-rich domain. Sci. Rep 2018, 1–15. [DOI] [PMC free article] [PubMed]
- (531).Li J; Liu X; Tian F; Yue T; Zhang X; Cao D Spontaneous insertion of GPI anchors into cholesterol-rich membrane domains. AIP Adv 2018, 8 . [Google Scholar]
- (532).Edler E; Schulze E; Stein M Membrane localization and dynamics of geranylgeranylated Rab5 hypervariable region. Biochim. Biophys. Acta, Biomembr 2017, 1859, 1335–1349. [DOI] [PubMed] [Google Scholar]
- (533).Edler E; Stein M Probing the druggability of membrane-bound Rab5 by molecular dynamics simulations. J. Enzyme Inhib. Med. Chem 2017, 32, 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (534).Buyan A; Kalli AC; Sansom MSP Multiscale simulations suggest a mechanism for the association of the Dok7 PH domain with PIP-containing membranes. PLoS Comput. Biol e1005028. [DOI] [PMC free article] [PubMed]
- (535).Kalli AC; Wegener KL; Goult BT; Anthis NJ; Campbell ID; Sansom MS The structure of the talin/integrin complex at a lipid bilayer: An NMR and MD simulation study. Structure 2010, 18, 1280–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (536).Kalli AC; Campbell ID; Sansom MSP Conformational changes in talin on binding to anionic phospholipid membranes facilitate signaling by integrin transmembrane helices. PLoS Comput. Biol 2013, 9, e1003316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (537).Tietjen GT; Gong Z; Chen C-H; Vargas E; Crooks JE; Cao KD; Heffern CTR; Henderson JM; Meron M; Lin B et al. Molecular mechanism for differential recognition of membrane phosphatidylserine by the immune regulatory receptor Tim4. Proc. Natl. Acad. Sci. USA 2014, 111, E1463–E1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (538).Tietjen GT; Baylon JL; Kerr D; Gong Z; Henderson JM; Heffern CT; Meron M; Lin B; Schlossman ML; Adams EJ et al. Coupling X-ray reflectivity and in silico binding to yield dynamics of membrane recognition by Tim1. Biophys. J 2017, 113, 1505–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (539).A. Leventis P; M. Chow B; Stewart BA; Iyengar B; Campos AR; Boulianne GL Drosophila amphiphysin is a post-synaptic protein required for normal locomotion but not endocytosis. Traffic 2011, 2, 839–850. [DOI] [PubMed] [Google Scholar]
- (540).Razzaq A; Robinson IM; McMahon HT; Skepper JN; Su Y; Zelhof AC; Jackson AP; Gay NJ; O’Kane CJ Amphiphysin is necessary for organization of the excitation-contraction coupling machinery of muscles, but not for synaptic vesicle endocytosis in Drosophila. Genes and Devel 2001, 15, 2967–2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (541).Suetsugu S Higher-order assemblies of bar domain proteins for shaping membranes. Microscopy 2016, 65, 201–210. [DOI] [PubMed] [Google Scholar]
- (542).Arkhipov A; Yin Y; Schulten K Four-scale description of membrane sculpting by BAR domains. Biophys. J 2008, 95, 2806–2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (543).Blood PD; Voth GA Direct observation of Bin/amphiphysin/Rvs (BAR) domain-induced membrane curvature by means of molecular dynamics simulations. Proc. Natl. Acad. Sci. USA 2006, 103, 15068–15072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (544).Yin Y; Arkhipov A; Schulten K Simulations of membrane tubulation by lattices of amphiphysin N-BAR domains. Structure 2009, 17, 882–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (545).Arkhipov A; Yin Y; Schulten K Membrane-bending mechanism of amphiphysin N-BAR domains. Biophys. J 2009, 97, 2727–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (546).Lyman E; Cui H; Voth GA Water under the BAR. Biophys. J 2010, 99, 1783–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (547).Blood PD; Swenson RD; Voth GA Factors influencing local membrane curvature induction by N-BAR domains as revealed by molecular dynamics simulations. Biophys. J 2008, 95, 1866–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (548).Cui H; Ayton GS; Voth GA Membrane binding by the endophilin N-BAR domain. Biophys. J 2009, 97, 2746–2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (549).Mim C; Cui H; Gawronski-Salerno JA; Frost A; Lyman E; Voth GA; Unger VM Structural basis of membrane bending by the N-BAR protein endophilin. Cell 2012, 149, 137–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (550).Cui H; Mim C; Vázquez FX; Lyman E; Unger VM; Voth GA Understanding the role of amphipathic helices in N-BAR domain driven membrane remodeling. Biophys. J 2013, 104, 404–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (551).Simunovic M; Mim C; Marlovits TC; Resch G; Unger VM; Voth GA Protein-mediated transformation of lipid vesicles into tubular networks. Biophys. J 2013, 105, 711–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (552).Simunovic M; Srivastava A; Voth GA Linear aggregation of proteins on the membrane as a prelude to membrane remodeling. Proc. Natl. Acad. Sci. USA 2013, 110, 20396–20401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (553).Simunovic M; Voth GA Membrane tension controls the assembly of curvature-generating proteins. Nat. Commun 2015, 6, 7219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (554).Simunovic M; Evergren E; Golushko I; Prévost C; Renard H-F; Johannes L; McMahon HT; Lorman V; Voth GA; Bassereau P How curvature-generating proteins build scaffolds on membrane nanotubes. Proc. Natl. Acad. Sci. USA 2016, 113, 11226–11231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (555).Simunovic M; Šaríc A; Henderson JM; Lee KYC; Voth GA Long-range organization of membrane-curving proteins. ACS Cent. Sci 2017, 3, 1246–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (556).Yu H; Schulten K Membrane sculpting by F-BAR domains studied by molecular dynamics simulations. PLoS Comput. Biol 2013, 9:e1002892 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- (557).Takemura K; Hanawa-Suetsugu K; Suetsugu S; Kitao A Salt bridge formation between the I-BAR domain and lipids increases lipid density and membrane curvature. Sci. Rep 2017, 7, 6808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (558).Braun AR; Lacy MM; Ducas VC; Rhoades E; Sachs JN α-Synuclein-induced membrane remodeling is driven by binding affinity, partition depth, and interleaflet order asymmetry. J. Am. Chem. Soc 2014, 136, 9962–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (559).González-Rubio P; Gautier R; Etchebest C; Fuchs PF Amphipathic-Lipid-Packing-Sensor interactions with lipids assessed by atomistic molecular dynamics. Biochim. Biophys. Acta, Biomembr 2011, 1808, 2119–2127. [DOI] [PubMed] [Google Scholar]
- (560).Lai C-L; Jao CC; Lyman E; Gallop JL; Peter BJ; McMahon HT; Langen R; Voth GA Membrane binding and self-association of the epsin n-terminal homology domain. J. Mol. Biol 2012, 423, 800–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (561).Braun AR; Sevcsik E; Chin P; Rhoades E; Tristram-Nagle S; Sachs JN α-Synuclein induces both positive mean curvature and negative gaussian curvature in membranes. J. Am. Chem. Soc 2012, 134, 2613–2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (562).Wu Z; Schulten K Synaptotagmin’s role in neurotransmitter release likely involves Ca2+-induced conformational transition. Biophys. J 2014, 107, 1156–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (563).Gill RL; Castaing J-P; Hsin J; Tan IS; Wang X; Huang KC; Tian F; Ramamurthi KS Structural basis for the geometry-driven localization of a small protein. Proc. Natl. Acad. Sci 2015, 112, E1908–E1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (564).Choi B-K; Choi M-G; Kim J-Y; Yang Y; Lai Y; Kweon D-H; Lee NK; Shin Y-K Large α-synuclein oligomers inhibit neuronal snare-mediated vesicle docking. Proc. Natl. Acad. Sci. USA 2013, 110, 4087–4092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (565).Braun AR; Sachs JN α-Synuclein reduces tension and increases undulations in simulations of small unilamella vesicles. Biophys. J 2015, 108, 1848–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (566).Braun AR; Lacy MM; Ducas VC; Rhoades E; Sachs JN α-Synuclein’s uniquely long amphipathic helix enhances its membrane binding and remodeling capacity. J. Membr. Biol 2017, 250, 183–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (567).Bradley RP; Radhakrishnan R Curvature-undulation coupling as a basis for curvature sensing and generation in bilayer membranes. Proc. Natl. Acad. Sci. USA 2016, 113, E5117–E5124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (568).Chon NL; Osterberg JR; Henderson J; Khan HM; Reuter N; Knight JD; Lin H Membrane docking of the synaptotagmin 7 C2A domain: Computation reveals interplay between electrostatic and hydrophobic contributions. Biochemistry 2015, 54, 5696–5711. [DOI] [PubMed] [Google Scholar]
- (569).Zhuxi C; Yanyan M; Jing Y; Tao Z; Lifen Z; Kunqian Y; Mingyue Z; Hualiang J; Huaiyu Y Characterizing the binding of annexin V to a lipid bilayer using molecular dynamics simulations. Proteins: Struct., Func., Bioinf 2014, 82, 312–322. [DOI] [PubMed] [Google Scholar]
- (570).Hakobyan D; Gerke V; Heuer A Modeling of annexin A2-membrane interactions by molecular dynamics simulations. PLoS One 2017, 12, e0185440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (571).Sengupta D Cholesterol modulates the structure, binding modes, and energetics of caveolin-membrane interactions. J. Phys. Chem. B 2012, 116, 14556–14564. [DOI] [PubMed] [Google Scholar]
- (572).Rui H; Root KT; Lee J; Glover KJ; Im W Probing the U-shaped conformation of caveolin-1 in a bilayer. Biophys. J 2014, 106, 1371–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (573).Li Z; Gorfe AA Deformation of a two-domain lipid bilayer due to asymmetric insertion of lipid-modified Ras peptides. Soft Mat 2013, 9, 11249–11256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (574).Weng J; Yang Y; Wang W Lipid regulated conformational dynamics of the longin SNARE protein Ykt6 revealed by molecular dynamics simulations. J. Phys. Chem. A 2015, 119, 1554–1562. [DOI] [PubMed] [Google Scholar]
- (575).Ray A; Jatana N; Thukral L Lipidated proteins: Spotlight on protein-membrane binding interfaces. Prog. Biophys. Mol. Biol 2017, 128, 74–84. [DOI] [PubMed] [Google Scholar]
- (576).Sun C; Benlekbir S; Venkatakrishnan P; Wang Y; Tajkhorshid E; Rubinstein JL; Gennis RB Structure of the alternative complex III in a supercomplex with cytochrome oxidase. Nature 2018, 557, 123–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (577).Li H; Gorfe AA Membrane remodeling by surface-bound protein aggregates: Insights from coarsegrained molecular dynamics simulation. J. Phys. Chem. Lett 2014, 5, 1457–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (578).Suchyna T; Tape S; Koeppe R; Andersen O; Sachs F; Gottlieb P Bilayer-dependent inhibition of mechanosensitive channels by neuroactive peptide enantiomers. Nature 2004, 430, 235–240. [DOI] [PubMed] [Google Scholar]
- (579).Nishizawa M; Nishizawa K Molecular dynamics simulations of a stretch-activated channel inhibitor GsMTx4 with lipid membranes: Two binding modes and effects of lipid structure. Biophys. J 4233–4243. [DOI] [PMC free article] [PubMed]
- (580).Chen R; Chung S-H Effect of gating modifier toxins on membrane thickness: Implications for toxin effect on gramicidin and mechanosensitive channels. Toxins 2013, 5, 456–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (581).Wee CL; Gavaghan D; Sansom MS Interactions between a voltage sensor and a toxin via multiscale simulations. Biophys. J 2010, 98, 1558–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (582).Henriques ST; Deplazes E; Lawrence N; Cheneval O; Chaousis S; Inserra M; Thongyoo P; King GF; Mark AE; Vetter I et al. Interaction of tarantula venom peptide ProTx-II with lipid membranes is a prerequisite for its inhibition of human voltage-gated sodium channel NaV1.7. J. Biol. Chem 2016, 291, 17049–17065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (583).Bemporad D; Sands ZA; Wee CL; Grottesi A; Sansom MS Vstx1, a modifier of Kv channel gating, localizes to the interfacial region of lipid bilayers. Biochemistry 2006, 45, 11844–11855. [DOI] [PubMed] [Google Scholar]
- (584).Chze LW; Gavaghan D; Sansom MS Lipid bilayer deformation and the free energy of interaction of a Kv channel gating-modifier toxin. Biophys. J 2008, 95, 3816–3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (585).Wee CL; Ulmschneider MB; Sansom MS Membrane/toxin interaction energetics via serial multiscale molecular dynamics simulations. J. Chem. Theory Comput 2010, 6, 966–976. [DOI] [PubMed] [Google Scholar]
- (586).Chze LW; Bemporad D; Sands ZA; Gavaghan D; Sansom MSP SGTx1, a Kv channel gating-modifier toxin, binds to the interfacial region of lipid bilayers. Biophys. J 2007, 92, L7–L9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (587).Nishizawa M; Nishizawa K Interaction between K+ channel gate modifier hanatoxin and lipid bilayer membranes analyzed by molecular dynamics simulation. Eur. Biophys. J 2006, 35, 373–381. [DOI] [PubMed] [Google Scholar]
- (588).Wu L; Xie SS; Meng E; Li WY; Liu L; Zhang DY Molecular dynamics simulation reveals unique interplays between a tarantula toxin and lipid membranes. J. Membr. Biol 2017, 250, 315–325. [DOI] [PubMed] [Google Scholar]
- (589).Deplazes E; Henriques ST; Smith JJ; King GF; Craik DJ; Mark AE; Schroeder CI Membrane-binding properties of gating modifier and pore-blocking toxins: Membrane interaction is not a prerequisite for modification of channel gating. Biochim. Biophys. Acta, Biomembr 2016, 1858, 872–882. [DOI] [PubMed] [Google Scholar]
- (590).Rees B; Bilwes A Three-dimensional structures of neurotoxins and cardiotoxins. Chem. Res. Toxicol 1993, 6, 385–406. [DOI] [PubMed] [Google Scholar]
- (591).Lesovoy DM; Bocharov EV; Lyukmanova EN; Kosinsky YA; Shulepko MA; Dolgikh DA; Kirpichnikov MP; Efremov RG; Arseniev AS Specific membrane binding of neurotoxin II can facilitate its delivery to acetylcholine receptor. Biophys. J 2009, 97, 2089–2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (592).Huang W-N; Sue S-C; Wang D-S; Wu P-L; Wu W-G Peripheral binding mode and penetration depth of cobra cardiotoxin on phospholipid membranes as studied by a combined ftir and computer simulation approach. Biochemistry 2003, 42, 7457–7466. [DOI] [PubMed] [Google Scholar]
- (593).Levtsova OV; Antonov MY; Mordvintsev DY; Utkin YN; Shaitan KV; Kirpichnikov MP Steered molecular dynamics simulations of cobra cytotoxin interaction with zwitterionic lipid bilayer: No penetration of loop tips into membranes. Comp. Biol. Chem 2009, 33, 29–32. [DOI] [PubMed] [Google Scholar]
- (594).Su ZY; Wang YT Coarse-grained molecular dynamics simulations of cobra cytotoxin A3 interactions with a lipid bilayer: Penetration of loops into membranes. J. Phys. Chem. B 2011, 115, 796–802. [DOI] [PubMed] [Google Scholar]
- (595).Flores-Canales JC; Kurnikova M Microsecond simulations of the diphtheria toxin translocation domain in association with anionic lipid bilayers. J. Phys. Chem. B 2015, 119, 12074–12085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (596).Flores-Canales JC; Vargas-Uribe M; Ladokhin AS; Kurnikova M Membrane association of the diphtheria toxin translocation domain studied by coarse-grained simulations and experiment. J. Membr. Biol 2015, 248, 529–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (597).Soliman W; Bhattacharjee S; Kaur K Molecular dynamics simulation study of interaction between a class IIa bacteriocin and its immunity protein. Biochim. Biophys. Acta, Proteins Proteomics 2007, 1774, 1002–1013. [DOI] [PubMed] [Google Scholar]
- (598).Cheng CJ; Koldsø H; Van der Kamp MW; Schiøtt B; Daggett V Simulations of membrane-bound diglycosylated human prion protein reveal potential protective mechanisms against misfolding. Neurochem 2017, [DOI] [PMC free article] [PubMed]
- (599).Nagarajan A; Jawahery S; Matysiak S The effects of flanking sequences in the interaction of polyglutamine peptides with a membrane bilayer. J. Phys. Chem. B 2014, 118, 6368–6379. [DOI] [PubMed] [Google Scholar]
- (600).Côté S; Wei G; Mousseau N Atomistic mechanisms of Huntingtin N-terminal fragment insertion on a phospholipid bilayer revealed by molecular dynamics simulations. Proteins: Struct., Func., Bioinf 2014, 82, 1409–1427. [DOI] [PubMed] [Google Scholar]
- (601).Hung A; Yarovsky I Inhibition of peptide aggregation by lipids: Insights from coarse-grained molecular simulations. J. Mol. Graph. Model 2011, 29, 597–607. [DOI] [PubMed] [Google Scholar]
- (602).Davis CH; Berkowitz ML Interaction between amyloid-β (1–42) peptide and phospholipid bilayers: A molecular dynamics study. Biophys. J 2009, 96, 785–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (603).Lemkul JA; Bevan DR A comparative molecular dynamics analysis of the amyloid β-peptide in a lipid bilayer. Arch. Biochem. Biophys 2008, 470, 54–63. [DOI] [PubMed] [Google Scholar]
- (604).Davis CH; Berkowitz ML Structure of the amyloid-β (1–42) monomer absorbed to model phospholipid bilayers: A molecular dynamics study. J. Phys. Chem. B 2009, 113, 14480–14486. [DOI] [PubMed] [Google Scholar]
- (605).Yinon S; Stewart D; Nelson A; Robert GH Models of membrane-bound alzheimer’s abeta peptide assemblies. Proteins: Struct., Func., Bioinf 2010, 78, 3473–3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (606).Ahyayauch H; Raab M; Busto JV; Andraka N; Arrondo J-LR; Masserini M; Tvaroska I; Goñi FM Binding of β-Amyloid (1–42) peptide to negatively charged phospholipid membranes in the liquid-ordered state: Modeling and experimental studies. Biophys. J 2012, 103, 453–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (607).Brown AM; Bevan DR Influence of sequence and lipid type on membrane perturbation by human and rat amyloid β-peptide (142). Arch. Biochem. Biophys 2017, 614, 1–13. [DOI] [PubMed] [Google Scholar]
- (608).Kargar F; Emadi S; Fazli H The molecular behavior of a single β-amyloid inside a dipalmitoylphos-phatidylcholine bilayer at three different temperatures: An atomistic simulation study: Aβ interaction with DPPC: Atomistic simulation. Proteins: Struct., Func., Bioinf 2017, 85, 1298–1310. [DOI] [PubMed] [Google Scholar]
- (609).Pobandt T; Knecht V Free energy of lipid bilayer defects affected by Alzheimer’s disease-associated amyloid-β42 monomers. J. Phys. Chem. B 2014, 118, 3507–3516. [DOI] [PubMed] [Google Scholar]
- (610).Lockhart C; Kilmov DK Binding of Aβ peptide creates lipid density depression in DMPC bilayer. Biochim. Biophys. Acta, Biomembr 2014, 1838, 2678–2688. [DOI] [PubMed] [Google Scholar]
- (611).Bera S; Korshavn KJ; Kar RK; Lim MH; Ramamoorthy A; Bhunia A Biophysical insights into the membrane interaction of the core amyloid-forming Aβ(40) fragment K16-K28 and its role in the pathogenesis of Alzheimer’s disease. Phys. Chem. Chem. Phys 2016, 18, 16890–16901. [DOI] [PubMed] [Google Scholar]
- (612).Zhao LN; Chiu SW; Benoit J; Chew LY; Mu Y Amyloid β peptides aggregation in a mixed membrane bilayer: A molecular dynamics study. J. Phys. Chem. B 2011, 115, 12247–12256. [DOI] [PubMed] [Google Scholar]
- (613).Lemkul JA; Bevan DR Lipid composition influences the release of Alzheimer’s amyloid β-peptide from membranes. Prot. Sci 2011, 20, 1530–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (614).Yahi N; Aulas A; Fantini J How cholesterol constrains glycolipid conformation for optimal recognition of Alzheimer’s β amyloid peptide (Aβ1–40). PLoS One 2010, 5, e9079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (615).Hoshino T; Mahmood MI; Mori K; Matsuzaki K Binding and aggregation mechanism of amyloid β-peptides onto the GM1 ganglioside-containing lipid membrane. J. Phys. Chem. B 2013, 117, 8085–8094. [DOI] [PubMed] [Google Scholar]
- (616).Devarajan S; Sharmila JS Molecular dynamics study of GM1 ganglioside complex with amyloid β peptide (Aβ42) in lipid membrane. J. Mol. Liq 2014, 195, 59–64. [Google Scholar]
- (617).Manna M; Mukhopadhyay C Binding, conformational transition and dimerization of amyloid-β peptide on GM1-containing ternary membrane: Insights from molecular dynamics simulation. PLoS One 2013, 8 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- (618).Izmitli A; Schebor C; McGovern MP; Reddy AS; Abbott NL; De Pablo JJ Effect of trehalose on the interaction of Alzheimer’s Aβ-peptide and anionic lipid monolayers. Biochim. Biophys. Acta, Biomembr 2011, 1808, 26–33. [DOI] [PubMed] [Google Scholar]
- (619).Zhou X; Xu J Free cholesterol induces higher β-sheet content in Aβ peptide oligomers by aromatic interaction with Phe19. PLoS One 2012, 7 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- (620).Qiu L; Buie C; Reay A; Vaughn MW; Cheng KH Molecular dynamics simulations reveal the protective role of cholesterol in β amyloid protein-induced membrane disruptions in neuronal membrane mimics. J. Phys. Chem. B 2011, 115, 9795–9812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (621).Qiu H; Sarathy A; Leburton J-P; Schulten K Intrinsic stepwise translocation of stretched ssDNA in graphene nanopores. Nano Lett 2015, 15, 8322–8330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (622).Yu X; Zheng J Cholesterol promotes the interaction of Alzheimer β-amyloid monomer with lipid bilayer. J. Mol. Biol 2012, 421, 561–571. [DOI] [PubMed] [Google Scholar]
- (623).Liguori N; Nerenberg PS; Head-Gordon T Embedding Aβ42 in heterogeneous membranes depends on cholesterol asymmetries. Biophys. J 2013, 105, 899–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (624).Lu Y; Shi X-F; Salsbury FR; Derreumaux P Influence of electric field on the amyloid-β (29–42) peptides embedded in a membrane bilayer. J. Chem. Phys 2018, 148, 045105. [DOI] [PubMed] [Google Scholar]
- (625).Kim S; Klimov DK Binding to the lipid monolayer induces conformational transition in Aβ monomer. J. Mol. Mod 2013, 19, 737–750. [DOI] [PubMed] [Google Scholar]
- (626).Lockhart C; Klimov DK The Alzheimer’s disease Aβ peptide binds to the anionic DMPS lipid bilayer. Biochim. Biophys. Acta, Biomembr 2016, 1858, 1118–1128. [DOI] [PubMed] [Google Scholar]
- (627).Lockhart C; Klimov DK Cholesterol changes the mechanism of Aβ peptide binding to the DMPC bilayer. J. Chem. Inf. Model 2017, 2554–2565. [DOI] [PubMed]
- (628).Wang Y; Kraut R; Mu Y Aβ1–25-derived sphingolipid-domain tracer peptide SBD interacts with membrane ganglioside clusters via a coil-helix-coil motif. Int. J. Mol. Sci 2015, 16, 26318–26322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (629).Fantini J; Yahi N; Garmy N Cholesterol accelerates the binding of Alzheimer’s β-amyloid peptide to ganglioside GM1 through a universal hydrogen-bond-dependent sterol tuning of glycolipid conformation. Front. Physiol 2013, 4, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (630).Davis CH; Berkowitz ML A molecular dynamics study of the early stages of amyloid-β(1–42) oligomerization: The role of lipid membranes. Proteins: Struct., Func., Bioinf 2010, 78, 2533–2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (631).Lemkul JA; Bevan DR Aggregation of Alzheimer’s amyloid β-peptide in biological membranes: A molecular dynamics study. Biochemistry 2013, 52, 4971–4980. [DOI] [PubMed] [Google Scholar]
- (632).Vahed M; Neya S; Matsuzaki K; Hoshino T Simulation study on complex conformations of Aβ 42 peptides on a GM1 ganglioside-containing lipid membrane. Chem. Pharm. Bull 2018, 66, 170–177. [DOI] [PubMed] [Google Scholar]
- (633).Poojari C; Kukol A; Strodel B How the amyloid-β peptide and membranes affect each other: An extensive simulation study. Biochim. Biophys. Acta, Biomembr 2013, 1828, 327–339. [DOI] [PubMed] [Google Scholar]
- (634).Brown AM; Bevan DR Molecular dynamics simulations of amyloid β-peptide (1–42): Tetramer formation and membrane interactions. Biophys. J 2016, 111, 937–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (635).Tofoleanu F; Buchete NV Molecular interactions of Alzheimer’s Aβ protofilaments with lipid membranes. J. Mol. Biol 2012, 421, 572–586. [DOI] [PubMed] [Google Scholar]
- (636).Jang H; Connelly L; Teran Arce F; Ramachandran S; Kagan BL; Lal R; Nussinov R Mechanisms for the insertion of toxic, fibril-like β-amyloid oligomers into the membrane. J. Chem. Theory Comput 2013, 9, 822–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (637).Yu X; Wang Q; Pan Q; Zhou F; Zheng J Molecular interactions of Alzheimer amyloid-β oligomers with neutral and negatively charged lipid bilayers. Phys. Chem. Chem. Phys 2013, 15, 8878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (638).Tofoleanu F; Brooks BR; Buchete NV Modulation of Alzheimer’s Aβ protofilament-membrane interactions by lipid headgroups. ACS Chem. Neurosci 2015, 6, 446–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (639).Dong X; Sun Y; Wei G; Nussinov R; Ma B Binding of protofibrillar Aβ trimers to lipid bilayer surface enhances Aβ structural stability and causes membrane thinning. Phys. Chem. Chem. Phys 2017, 19, 27556–27569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (640).Zhang M; Ren B; Liu Y; Liang G; Sun Y; Xu L; Zheng J Membrane interactions of hiapp monomer and oligomer with lipid membranes by molecular dynamics simulations. ACS Chem. Neurosci 2017, 8, 1789–1800. [DOI] [PubMed] [Google Scholar]
- (641).Ngo ST; Hung HM; Tran KN; Nguyen MT Replica exchange molecular dynamics study of the amyloid beta (11–40) trimer penetrating a membrane. RSC Adv 2017, 7, 7346–7357. [Google Scholar]
- (642).Brummel BE; Braun AR; Sachs JN Polyunsaturated chains in asymmetric lipids disorder raft mixtures and preferentially assoiciate with α-Synuclein. Biochim. Biophys. Acta, Biomembr 2017, 1859, 529–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (643).Jiang P; Xu W; Mu Y Amyloidogenesis abolished by proline substitutions but enhanced by lipid binding. PLoS Comput. Biol 2009, 5, e1000357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (644).Xu W; Wei G; Su H; Nordenskiöld L; Mu Y Effects of cholesterol on pore formation in lipid bilayers induced by human islet amyloid polypeptide fragments: A coarse-grained molecular dynamics study. Phys. Rev. E 2011, 84, 1–8. [DOI] [PubMed] [Google Scholar]
- (645).Jia Y; Qian Z; Zhang Y; Wei G Adsorption and orientation of human islet amyloid polypeptide (hIAPP) monomer at anionic lipid bilayers: Implications for membrane-mediated aggregation. Int. J. Mol. Sci 2013, 14, 6241–6258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (646).Guo C; Cote S; Mousseau N; Wei G Distinct helix propensities and membrane interactions of human and rat IAPP(1–19) monomers in anionic lipid bilayers. J. Phys. Chem. B 2015, 119, 3366–3376. [DOI] [PubMed] [Google Scholar]
- (647).Qian Z; Jia Y; Wei G Binding orientations and lipid interactions of human amylin at zwitterionic and anionic lipid bilayers. J. Diabetes Res 2016, 2016, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (648).Zhao J; Luo Y; Jang H; Yu X; Wei G; Nussinov R; Zheng J Probing ion channel activity of human islet amyloid polypeptide (amylin). Biochim. Biophys. Acta, Biomembr 2012, 1818, 3121–3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (649).Qian Z; Zou Y; Zhang Q; Chen P; Ma B; Wei G; Nussinov R Atomistic-level study of the interactions between hIAPP protofibrils and membranes: Influence of pH and lipid composition. Biochim. Biophys. Acta, Biomembr 2018, 1860, 1818–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (650).Zhang M; Hu R; Ren B; Chen H; Jiang B; Ma J; Zheng J Molecular understanding of Aβ-hIAPP cross-seeding assemblies on lipid membranes. ACS Chem. Neurosci 2017, 8, 524–537. [DOI] [PubMed] [Google Scholar]
- (651).Tsigelny IF; Sharikov Y; Wrasidlo W; Gonzalez T; Desplats PA; Crews L; Spencer B; Masliah E Role of α-synuclein penetration into the membrane in the mechanisms of oligomer pore formation. FEBS J 2012, 279, 1000–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (652).Fantini J; Carlus D; Yahi N The fusogenic tilted peptide (67–78) of α-synuclein is a cholesterol binding domain. Biochim. Biophys. Acta, Biomembr 2011, 1808, 2343–2351. [DOI] [PubMed] [Google Scholar]
- (653).Pfefferkorn CM; Heinrich F; Sodt AJ; Maltsev AS; Pastor RW; Lee JC Depth of α-synuclein in a bilayer determined by fluoresence, neutron reflectometry, and computation. Biophys. J 2012, 102, 613–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (654).Perlmutter JD; Braun AR; Sachs JN Curvature dynamics of α-synuclein familial Parkinson disease mutants. J. Biol. Chem 2009, 284, 7177–7189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (655).Lagüe P; Roux B; Pastor RW Molecular dynamics simulations of the influenza hemagglutinin fusion peptide in micelles and bilayers: Conformational analysis of peptide and lipids. J. Mol. Biol 2005, 354, 1129–1141. [DOI] [PubMed] [Google Scholar]
- (656).Vaccaro L; Cross KJ; Kleinjung J; Straus SK; Thomas DJ; Wharton SA; Skehel JJ; Franternali F Plasticity of influenza haemagglutinin fusion peptides and their interaction with lipid bilayers. Biophys. J 2005, 88, 25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (657).Jang H; Michaud-Agrawal N; Johnston JM; Woolf TB How to lose a kinh and gain a helix: pH independent conformational changes of the fusion domains from influenza hemagglutinin in heterogeneous lipid bilayers. Proteins 2008, 72, 299–312. [DOI] [PubMed] [Google Scholar]
- (658).Vaidya NK; Huang H; Takagi S Coarse grained molecular dynamics simulation of interaction between hemagglutinin fusion peptides and lipid bilayer membranes. Adv. Appl. Math. Mech 2010, 2, 430–450. [Google Scholar]
- (659).Brice AR; Lazaridis T Structure and dynamics of a fusion peptide helical hairpin on the membrane surface: Comparison of molecular simulations and NMR. J. Phys. Chem. B 2014, 118, 4461–4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (660).Haria NR; Monticelli L; Fraternali F; Lorenz CD Plasticity and conformational equilibria of influenza fusion peptides in model lipid bilayers. Biochim. Biophys. Acta, Biomembr 2014, 1838, 1169–1179. [DOI] [PubMed] [Google Scholar]
- (661).Li J; Das P; Zhou R Single mutation effects on conformational change and membrane deformation of influenza hemagglutinin fusion peptides. J. Phys. Chem. B 2010, 114, 8799–8806. [DOI] [PubMed] [Google Scholar]
- (662).Légaré S; Lagüe P The influenza fusion peptide promotes lipid polar head intrusion through hydrogen bonding with phosphates and N-terminal membrane insertion depth. Proteins: Struct., Func., Bioinf 2014, 82, 2118–2127. [DOI] [PubMed] [Google Scholar]
- (663).Risselada HJ; Marelli G; Fuhrmans M; Smirnova YG; Grubmüller H; Marrink SJ; Müller M Line-tension controlled mechanism for influenza fusion. PLoS One 2012, 7 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- (664).Promsri S; Ullmann MG; Hannongbua S Molecular dynamics simulation of HIV-1 fusion domain-membrane complexes: Insight into the N-terminal gp41 fusion mechanism. Biophys. Chem 2012, 170, 9–16. [DOI] [PubMed] [Google Scholar]
- (665).Gangupomu VK; Abrams CF All-atom models of the membrane-spanning domain of HIV-1 gp41 from metadynamics. Biophys. J 2010, 99, 3438–3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (666).Baker MK; Gangupomu VK; Abrams CF Characterization of the water defect at the HIV-1 gp41 membrane spanning domain in bilayers with and without cholesterol using molecular simulations. Biochim. Biophys. Acta - Biomembr 2014, 1838, 1396–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (667).Apellaniz B; Rujas E; Carravilla P; Requejo-Isidro J; Huarte N; Domene C; Nieva JL Cholesterol-dependent membrane fusion induced by the gp41 membrane-proximal external region-transmembrane domain connection suggests a mechanism for broad HIV-1 neutralization. J. Virol 2014, 88, 13367–13377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (668).Martins do Canto AM; Palace Carvalho AJ; Prates Ramalho JP; Loura LM Structure and conformation of HIV fusion inhibitor peptide T-1249 in presence of model membranes: A molecular dynamics study. J. Mol. Struc 2010, 946, 119–124. [Google Scholar]
- (669).Martins do Canto AM; Palace Carvalho AJ; Prates Ramalho JP; Loura LM Molecular dynamics simulation of HIV fusion inhibitor T-1249: Insights on peptide-lipid interaction. Comput. Math. Methods Med 2012, 2012 . [DOI] [PMC free article] [PubMed]
- (670).Vitiello G; Falanga A; Petruk AA; Merlino A; Fragneto G; Paduano L; Galdiero S; D’Errico G Fusion of raft-like lipid bilayers operated by a membranotropic domain of the HSV-type I glycoprotein gH occurs through a cholesterol-dependent mechanism. Soft Mat 2015, 11, 3003–3016. [DOI] [PubMed] [Google Scholar]
- (671).Rogers DM; Kent MS; Rempe SB Molecular basis of endosomal-membrane association for the dengue virus envelope protein. Biochim. Biophys. Acta, Biomembr 2015, 1848, 1041–1052. [DOI] [PubMed] [Google Scholar]
- (672).Donald JE; Zhang Y; Fiorin G; Carnevale V; Slochower DR; Gai F; Klein ML; De-Grado WF Transmembrane orientation and possible role of the fusogenic peptide from parainfluenza virus 5 (PIV5) in promoting fusion. Proc. Natl. Acad. Sci. USA 2011, 108, 3958–3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (673).Bajaj S; Dey D; Bhukar R; Kumar M; Banerjee M Non-enveloped virus entry: Structural determinants and mechanism of functioning of a viral lytic peptide. J. Mol. Biol 2016, 428, 3540–3556. [DOI] [PubMed] [Google Scholar]
- (674).Guardado-Calvo P; Atkovska K; Jeffers SA; Grau N; Backovic M; Pérez-Vargas J; De Boer SM; Tortorici MA; Pehau-Arnaudet G; Lepault J et al. A glycerophospholipid-specific pocket in the RVFV class II fusion protein drives target membrane insertion. Science 2017, 358, 663–667. [DOI] [PubMed] [Google Scholar]
- (675).Bhattarai N; GC JB; Gerstman BS; Stahelin RV; Chapagain PP Plasma membrane association facilitates conformational changes in the Marburg virus protein VP40 dimer. RSC Adv 2017, 7, 22741–22748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (676).GC JB; Gerstman BS; Stahelin RV; Chapagain PP The Ebola virus protein VP40 hexamer enhances the clustering of PI(4,5)P2 lipids in the plasma membrane. Phys. Chem. Chem. Phys 2016, 18, 28409–28417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (677).Del Vecchio K; Frick CT; Gc JB; Oda S.-i.; Gerstman BS; Saphire EO; Chapagain PP; Stahelin RV A cationic, C-terminal patch and structural rearrangements in Ebola virus matrix VP40 protein control its interactions with phosphatidylserine. J. Biol. Chem 2018, 293, 3335–3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (678).GC JB; Gerstman BS; Chapagain PP Membrane association and localization dynamics of the Ebola virus matrix protein VP40. Biochim. Biophys. Acta, Biomembr 2017, 1859, 2012–2020. [DOI] [PubMed] [Google Scholar]
- (679).Wijesinghe KJ; Urata S; Bhattarai N; Kooijman EE; Gerstman BS; Chapagain PP; Li S; Stahelin RV Detection of lipid-induced structural changes of the Marburg virus matrix protein VP40 using hydrogen/deuterium exchange-mass spectrometry. J. Biol. Chem 2017, 292, 6108–6122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (680).Herce HD; García AE Molecular dynamics simulations suggest a mechanism for translocation of the HIV-1 TAT peptide across lipid membranes. Proc. Natl. Acad. Sci. USA 2007, 104, 20805–20810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (681).Yesylevskyy S; Marrink S-J; Mark AE Alternative mechanisms for the interaction of the cell-penetrating peptides penetratin and the TAT peptide with lipid bilayers. Biophys. J 2009, 97, 40–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (682).Akabori K; Huang K; Treece BW; Jablin MS; Maranville B; Woll A; Nagle JF; Garcia AE; Tristram-Nagle S HIV-1 Tat membrane interactions probed using X-ray and neutron scattering, CD spectroscopy and MD simulations. Biochim. Biophys. Acta, Biomembr 2014, 1838, 3078–3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (683).Neale C; Huang K; García A; Tristram-Nagle S Penetration of HIV-1 Tat47–57 into PC/PE bilayers assessed by MD simulation and X-ray scattering. Membranes (Basel) 2015, 5, 473–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (684).Hu Y; Patel S Thermodynamics of cell-penetrating HIV1 TAT peptide insertion into PC/PS/CHOL model bilayers through transmembrane pores: the roles of cholesterol and anionic lipids. Soft Mat 2016, 12, 6716–6727. [DOI] [PubMed] [Google Scholar]
- (685).WP C; RJ G; PF W; RD A; S, K.; WB, K. Incidence of coronary heart disease and lipoprotein cholesterol levels: The framingham study. J. Am. Med. Assoc 1986, 256, 2835–2838. [PubMed] [Google Scholar]
- (686).Hevonoja T; Pentikäinen MO; Hyvönen MT; Kovanen PT; Ala-Korpela M Structure of low density lipoprotein (LDL) particles: Basis for understanding molecular changes in modified LDL. Biochim. Biophys. Acta 2000, 1488, 189–210. [DOI] [PubMed] [Google Scholar]
- (687).Linsel-Nitschke P; Tall AR HDL as a target in the treatment of atherosclerotic cardiovascular disease. Nat. Rev. Drug Disc 2005, 4, 193–205. [DOI] [PubMed] [Google Scholar]
- (688).Shih AY; Freddolino PL; Arkhipov A; Schulten K Assembly of lipoprotein particles revealed by coarse-grained molecular dynamics simulations. J. Struct. Biol 2007, 157, 579–592. [DOI] [PubMed] [Google Scholar]
- (689).Damirchi B; Saidi M; Rismanian M; Firoozabadi B; Amininasab M An alternative mechanism for the formation of high density lipoprotein in peripheral tissue. Sci. Iran 2016, 23, 600–608. [Google Scholar]
- (690).Catte A; Patterson JC; Vashtovyy D; Jones MK; Gu F; Li L; Rampioni A; Sengupta D; Vuorela T; Niemela P et al. Structure of spheroidal HDL particles revealed by combined atomistic and coarse grained simulations. Biophys. J 2008, 94, 2306–2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (691).Vuorela T; Catte A; Niemelä PS; Hall A; Hyvönen MT; Marrink S-J; Karttunen M; Vattulainen I Role of lipids in spheroidal high density lipoproteins. PLoS Comput. Biol 2010, 6, e1000964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (692).Segrest JP; Jones MK; De Loof H; Dashti N Structure of apolipoprotein B-100 in low density lipoproteins. J. Lipid Res 2001, 42, 1346–1367. [PubMed] [Google Scholar]
- (693).Murtola T; Vuorela TA; Hyvönen MT; Marrink S-J; Karttunen M; Vattulainen I Low density lipoprotein: structure, dynamics, and interactions of apoB-100 with lipids. Soft Mat 2011, 7, 8135. [Google Scholar]
- (694).Barter PJ; Brewer HB; Chapman MJ; Hennekens CH; Rader DJ; Tall AR Cholesteryl ester transfer protein: a novel target for raising HDL and inhibiting atherosclerosis. Arter. Thromb. Vasc. Biol 2003, 23, 160–1677. [DOI] [PubMed] [Google Scholar]
- (695).Cilpa-Karhu G; Jauhiainen M; Riekkola M-L Atomistic MD simulation reveals the mechanism by which CETP penetrates into HDL enabling lipid transfer from HDL to CETP. J. Lipid Res 2015, 56, 98–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (696).Koivuniemi A; Vuorela T; Kovanen PT; Vattulainen I; Hyvönen MT Lipid exchange mechanism of the cholesteryl ester transfer protein clarified by atomistic and coarse-grained simulations. PLoS Comput. Biol 2012, 8 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- (697).Chirasani VR; Revanasiddappa PD; Senapati S Structural plasticity of cholesteryl ester transfer protein assists the lipid transfer activity. J. Biol. Chem 2016, 291, 19462–19473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (698).Revanasiddappa PD; Sankar R; Senapati S Role of the bound phospholipids in the structural stability of cholesteryl ester transfer protein. J. Phys. Chem. B 2018, 122, 4239–4248. [DOI] [PubMed] [Google Scholar]
- (699).Lei D; Rames M; Zhang X; Zhang L; Zhang S; Ren G Insights into the tunnel mechanism of cholesteryl ester transfer protein through all-atom molecular dynamics simulations. J. Biol. Chem 2016, 291, 14034–14044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (700).Singh RP; Brooks BR; Klauda JB Binding and release of cholesterol in the Osh4 protein of yeast. Proteins: Struct., Func., Bioinf 2009, 75, 468–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (701).Canagarajah BJ; Hummer G; Prinz WA; Hurley JH Dynamics of cholesterol exchange in the oxysterol binding protein family. J. Mol. Biol 2008, 378, 737–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (702).Poongavanam V; Kongsted J; Wüstner D Computational analysis of sterol ligand specificity of the Niemann Pick C2 protein. Biochemistry 2016, 55, 5165–5179. [DOI] [PubMed] [Google Scholar]
- (703).Estiu G; Khatri N; Wiest O Computational studies of the cholesterol transport between NPC2 and the N-terminal domain of NPC1 (NPC1(NTD)). Biochemistry 6879–6891. [DOI] [PubMed]
- (704).Elghobashi-Meinhardt N Niemann-pick type C disease: A QM/MM study of conformational changes in cholesterol in the NPC1(NTD) and NPC2 binding pockets. Biochemistry 2014, 53, 6603–6614. [DOI] [PubMed] [Google Scholar]
- (705).Galassi VV; Villarreal MA; Posada V; Montich GG Interactions of the fatty acid-binding protein ReP1-NCXSQ with lipid membranes. Influence of the membrane electric field on binding and orientation. Biochim. Biophys. Acta, Biomembr 2014, 1838, 910–920. [DOI] [PubMed] [Google Scholar]
- (706).Villarreal MA; Perduca M; Monaco HL; Montich GG Binding and interactions of L-BABP to lipid membranes studied by molecular dynamic simulations. Biochim. Biophys. Acta, Biomembr 2008, 1778, 1390–1397. [DOI] [PubMed] [Google Scholar]
- (707).Levin LB-A; Ganoth A; Amram S; Nachliel E; Gutman M; Tsfadia Y Insight into the interaction sites between fatty acid binding proteins and their ligands. J. Mol. Mod 2010, 16, 929–938. [DOI] [PubMed] [Google Scholar]
- (708).Levin LB-A; Nachliel E; Gutman M; Tsfadia Y Molecular dynamics study of the interaction between fatty acid binding proteins with palmitate mini-micelles. Mol. Cell. Biol 2009, 326, 29–33. [DOI] [PubMed] [Google Scholar]
- (709).Bello M; García-Hernández E Ligand entry into the calyx of β-lactoglobulin. Biopolymers 2014, 101, 744–757. [DOI] [PubMed] [Google Scholar]
- (710).Smith LJ; Van Gunsteren WF; Allison JR Multiple binding modes for palmitate to barley lipid transfer protein facilitated by the presence of proline 12. Prot. Sci 2013, 22, 56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (711).Wirtz KWA Phospholipid transfer proteins. Annu. Rev. Biochem 1991, 60, 73–99. [DOI] [PubMed] [Google Scholar]
- (712).Grabon A; Orl-owski A; Tripathi A; Vuorio J; Javanainen M; Ròg T; Lönnfors M; Mc-Dermott MI; Siebert G; Somerharju P et al. Dynamics and energetics of the mammalian phos-phatidylinositol transfer protein phospholipid exchange cycle. J. Biol. Chem 2017, 292, 14438–14455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (713).Polverini E; Coll EP; Tieleman DP; Harauz G Conformational choreography of a molecular switch region in myelin basic protein-molecular dynamics shows induced folding and secondary structure type conversion upon threonyl phosphorylation in both aqueous and membrane-associated environments. Biochim. Biophys. Acta, Biomembr 2011, 1808, 674–683. [DOI] [PubMed] [Google Scholar]
- (714).Vassall KA; Bessonov K; De Avila M; Polverini E; Harauz G The effects of threonine phos-phorylation on the stability and dynamics of the central molecular switch region of 18.5-kDa myelin basic protein. PLoS One 2013, 8, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (715).Baoukina S; Monticelli L; Amrein M; Tieleman DP The molecular mechanism of monolayer-bilayer transformations of lung surfactant from molecular dynamics simulations. Biophys. J 2007, 93, 3775–3782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (716).Baoukina S; Tieleman DP Lung surfactant protein SP-B promotes formation of bilayer reservoirs from monolayer and lipid transfer between the interface and subphase. Biophys. J 2011, 100, 1678–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (717).Baoukina S; Tieleman DP Direct simulation of protein-mediated vesicle fusion: Lung surfactant protein. Biophys. J 2010, 99, 2134–2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (718).Goh BC; Wu H; Rynkiewicz MJ; Schulten K; Seaton BA; McCormack FX Elucidation of lipid binding sites on lung surfactant protein A using X-ray crystallography, mutagenesis and molecular dynamics simulations. Biochemistry 2016, 55, 3692–3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (719).Rausch F; Schicht M; Bräuer L; Paulsen F; Brandt W Protein modeling and molecular dynamics simulation of the two novel surfactant proteins SP-G and SP-H. J. Mol. Mod 2014, 20, 2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (720).Hoogerheide DP; Noskov SY; Jacobs D; Bergdoll L; Silin V; Worcester DL; Abramson J; Nanda H; Rostovtseva TK; Bezrukov SM Structural features and lipid binding domain of tubulin on biomimetic mitochondrial membranes. Proc. Natl. Acad. Sci. USA 2017, 114, E3622–E3631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (721).Thukral L; Sengupta D; Ramkumar A; Murthy D; Agrawal N; Gokhale RS The molecular mechanism underlying recruitment and insertion of lipid-anchored LC3 protein into membranes. Biophys. J 2015, 109, 2067–2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (722).Garzón D; Bond PJ; Faraldo-Gómez JD Predicted structural basis for CD1c presentation of mycobacterial branched polyketides and long lipopeptide antigens. Mol. Immunol 2009, 47, 253–260. [DOI] [PubMed] [Google Scholar]
- (723).Garzón D; Anselmi C; Bond PJ; Faraldo-Gómez JD Dynamics of the antigen-binding grooves in CD1 proteins: Reversible hydrophobic collapse in the lipid-free state. J. Biol. Chem 2013, 288, 19528–19536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (724).Chiappori F; Merelli I; Milanesi L; Rovida E Exploring the role of the phospholipid ligand in endothelial protein C receptor: A molecular dynamics study. Proteins: Struct., Func., Bioinf 2010, 78, 2679–2690. [DOI] [PubMed] [Google Scholar]
- (725).Contreras FX; Ernst AM; Wieland F; Brügger B Specificity of intramembrane protein-lipid interactions. Cold Spring Harbor Perspect. Biol 2011, 3, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (726).Fadok VA; Voelker DR; Campbell PA; Cohen JJ; Bratton DL; Henson PM Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J. Immunol 1992, 148, 2207–2216. [PubMed] [Google Scholar]
- (727).Zwaal RFA; Comfurius P; Bevers EM Surface exposure of phosphatidylserine in pathological cells. Cell. Mol. Life Sci 2005, 62, 971–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (728).Sonnino S; Prinetti A; Nakayama H; Yangida M; Ogawa H; Iwabuchi K Role of very long fatty acid-containing glycosphingolipids in membrane organization and cell signaling: the model of lactosylceramide in neutrophils. Glycoconj. J 2009, 26, 615–621. [DOI] [PubMed] [Google Scholar]
- (729).Sonnino S; Prinetti A Lipids and membrane lateral organization. Front. Physiol 2010, 1, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (730).Grouleff J; Irudayam SJ; Skeby KK; Schiøtt B The influence of cholesterol on membrane protein structure, function, and dynamics studied by molecular dynamics simulations. Biochim. Biophys. Acta 2015, 1848, 1783–1795. [DOI] [PubMed] [Google Scholar]
- (731).Rheinstädter MC; Mouritsen OG Small-scale structure in fluid cholesterol-lipid bilayers. Curr. Opin. Colloid Interface Sci 2013, 18, 440–447. [Google Scholar]
- (732).Lu SM; Fairn GD Mesoscale organization of domains in the plasma membrane - beyond the lipid raft. Crit. Rev. Biochem. Mol. Biol 2018, 53, 192–207. [DOI] [PubMed] [Google Scholar]
- (733).Bieberich E Sphingolipids and lipid rafts : Novel concepts and methods of analysis. Chem. Phys. of Lipids 2018, 216, 114–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (734).Paila YD; Tiwari S; Chattopadhyay A Are specific nonannular cholesterol binding sites present in G-protein coupled receptors? Biochim. Biophys. Acta, Biomembr 2009, 1788, 295–302. [DOI] [PubMed] [Google Scholar]
- (735).Fantini J; Barrantes F How cholesterol interacts with membrane proteins: an exploration of cholesterol-binding sites including CRAC, CARC, and tilted domains. Front. Physiol 2013, 4, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (736).Di Scala C; Baier CJ; Evans LS; Williamson PT; Fantini J; Barrantes FJ Curr. Top. Membr, 1st ed.; Elsevier Inc., 2017; Vol. 80; pp 3–23. [DOI] [PubMed] [Google Scholar]
- (737).Sengupta D; Chattopadhyay A Molecular dynamics simulations of GPCR-cholesterol interaction: An emerging paradigm. Biochim. Biophys. Acta, Biomembr 2015, 1848, 1775–1782. [DOI] [PubMed] [Google Scholar]
- (738).Grossfield A; Feller SE; Pitman MC A role for direct interactions in the modulation of rhodopsin by ω−3 polyunsaturated lipids. Proc. Natl. Acad. Sci. USA 2006, 103, 4888–4893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (739).Horn JN; Kao T-C; Grossfield A In Advances in Experimental Medicine and Biology ; Filizola M, Ed.; Advances in Experimental Medicine and Biology; Springer Netherlands: Dordrecht, 2014; Vol. 796; pp 75–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (740).Hanson MA; Cherezov V; Griffith MT; Roth CB; Jaakola VP; Chien EY; Velasquez J; Kuhn P; Stevens RC A specific cholesterol binding site is established by the 2.8 Å structure of the human β2-adrenergic receptor. Structure 2008, 16, 897–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (741).Salari R; Joseph T; Lohia R; Henin J; Brannigan G A streamlined, general approach for computing ligand binding free energies and its application to GPCR-bound cholesterol 2018, 1–41. [DOI] [PMC free article] [PubMed]
- (742).Lyman E; Higgs C; Kim B; Lupyan D; Shelley JC; Farid R; Voth GA A role for a specific cholesterol interaction in stabilizing the apo configuration of the human A2A adenosine receptor. Structure 2009, 17, 1660–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (743).Lee JY; Lyman E Predictions for cholesterol interaction sites on the A2A adenosine receptor. J. Am. Chem. Soc 2012, 134, 16512–16515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (744).Rouviere E; Arnarez C; Yang L; Lyman E Identification of two new cholesterol interaction sites on the A2A adenosine receptor. Biophys. J 2017, 113, 2415–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (745).Sengupta D; Chattopadhyay A Identification of cholesterol binding sites in the serotonin. J. Phys. Chem. B 2012, 116, 12991–12996. [DOI] [PubMed] [Google Scholar]
- (746).Hedger G; Koldsø H; Chavent M; Siebold C; Rohatgi R; Sansom MS Cholesterol interaction sites on the transmembrane domain of the hedgehog signal transducer and class F G protein-coupled receptor Smoothened. Structure [DOI] [PMC free article] [PubMed]
- (747).Lee JY; Patel R; Lyman E Ligand-dependent cholesterol interactions with the human A2A adenosine receptor. Chem. Phys. of Lipids 2013, 169, 39–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (748).Guixà-González R; Albasanz JL; Rodriguez-Espigares I; Pastor M; Sanz F; Martí-Solano M; Manna M; Martinez-Seara H; Hildebrand PW; Martín M et al. Membrane cholesterol access into a G-protein-coupled receptor. Nat. Commun 2017, 8, 14505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (749).Oddi S; Stepniewski TM; Totaro A; Selent J; Scipioni L; Dufrusine B; Fezza F; Dainese E; Maccarrone M Palmitoylation of cysteine 415 of CB1 receptor affects ligand-stimulated internalization and selective interaction with membrane cholesterol and caveolin 1. Biochim. Biophys. Acta, Mol. Cell Biol. of Lipids 2017, 1862, 523–532. [DOI] [PubMed] [Google Scholar]
- (750).Bruno A; Costantino G; de Fabritiis G; Pastor M; Selent J Membrane-sensitive conformational states of helix 8 in the metabotropic Glu2 receptor, a class C GPCR. PLoS One 2012, 7 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- (751).Singh AK; McMillan J; Bukiya AN; Burton B; Parrill AL; Dopico AM Multiple cholesterol recognition/interaction amino acid consensus (CRAC) motifs in cytosolic C tail of Slo1 subunit determine cholesterol sensitivity of Ca2+- and voltage-gated K+ (BK) channels. J. Biol. Chem 2012, 287, 20509–20521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (752).Baier CJ; Fantini J; Barrantes FJ Disclosure of cholesterol recognition motifs in transmembrane domains of the human nicotinic acetylcholine receptor. Sci. Rep 2011, 1, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (753).Sharpe LJ; Rao G; Jones PM; Glancey E; Aleidi SM; George AM; Brown AJ; Gelissen IC Cholesterol sensing by the ABCG1 lipid transporter: Requirement of a CRAC motif in the final transmembrane domain. Biochim. Biophys. Acta, Mol. Cell Biol. of Lipids 2015, 1851, 956–964. [DOI] [PubMed] [Google Scholar]
- (754).Ferraro M; Masetti M; Recanatini M; Cavalli A; Bottegoni G Mapping cholesterol interaction sites on serotonin transporter through coarse-grained molecular dynamics. PLoS One 2016, 11, 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (755).Zeppelin T; Ladefoged LK; Sinning S; Periole X; Schiøtt B; Schäfer L A direct interaction of cholesterol with the dopamine transporter prevents its out-to-inward transition. PLoS Comput. Biol 2018, 14, e1005907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (756).Roy K; Mandloi S; Chakrabarti S; Roy S Cholesterol corrects altered conformation of MHC-II protein in Leishmania donovani infected macrophages: Implication in therapy. PLoS Negl. Trop. Dis 2016, 10, 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (757).Rosenhouse-Dantsker A; Noskov S; Durdagi S; Logothetis DE; Levitan I Identification of novel cholesterol-binding regions in Kir2 channels. J. Biol. Chem 2013, 288, 31154–31164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (758).Fürst O; Nichols CG; Lamoureux G; D’Avanzo N Identification of a cholesterol-binding pocket in inward rectifier K+ (Kir) channels. Biophys. J 2014, 107, 2786–2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (759).Brannigan G; Hénin J; Law R; Eckenhoff R; Klein ML Embedded cholesterol in the nicotinic acetylcholine receptor. Proc. Natl. Acad. Sci. USA 2008, 105, 14418–14423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (760).Cheng MH; Xu Y; Tang P Anionic lipid and cholesterol interactions with α4β2 nAChR: Insights from MD simulations. J. Phys. Chem. B 2009, 113, 6964–6970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (761).Hénin J; Salari R; Murlidaran S; Brannigan G A predicted binding site for cholesterol on the GABAA receptor. Biophys. J 2014, 106, 1938–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (762).O’Mara ML; Mark AE The effect of environment on the structure of a membrane protein: P-glycoprotein under physiological conditions. J. Chem. Theory Comput 2012, 8, 3964–3976. [DOI] [PubMed] [Google Scholar]
- (763).Khelashvili G; Mondal S; Andersen OS; Weinstein H Cholesterol modulates the membrane effects and spatial organization of membrane-penetrating ligands for G-protein coupled receptors. J. Phys. Chem. B 2010, 114, 12046–12057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (764).O’Conner JW; Klauda JB Lipid membranes with a majority of cholesterol: Applications to the ocular lens and aquaporin 0. J. Phys. Chem. B 2011, 115, 6455–6464. [DOI] [PubMed] [Google Scholar]
- (765).Manna M; Mukhopadhyay C Cholesterol driven alteration of the conformation and dynamics of phospholamban in model membranes. Phys. Chem. Chem. Phys 2011, 13, 20188. [DOI] [PubMed] [Google Scholar]
- (766).Zimmerman B; Kelly B; McMillan BJ; Seegar TCM; Dror RO; Kruse AC; Blacklow SC Crystal structure of a full-length human tetraspanin reveals a cholesterol-binding pocket. Cell 2016, 167, 1041–1051.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (767).Venken T; Schillinger AS; Fuglebakk E; Reuter N Interactions stabilizing the C-terminal helix of human phospholipid scramblase 1 in lipid bilayers: A computational study. Biochim. Biophys. Acta, Biomembr 2017, 1859, 1200–1210. [DOI] [PubMed] [Google Scholar]
- (768).Prakash A; Janosi L; Doxastakis M GxxxG motifs, phenylalanine, and cholesterol guide the self-association of transmembrane domains of ErbB2 receptors. Biophys. J 2011, 101, 1949–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (769).Autzen HE; Siuda I; Sonntag Y; Nissen P; Møller JV; Thøgersen L Regulation of the Ca2+-ATPase by cholesterol: A specific or non-specific effect? Mol. Membr. Biol 2015, 32, 75–87. [DOI] [PubMed] [Google Scholar]
- (770).Suwattanasophon C; Wolschann P; Faller R Molecular dynamics simulations on the interaction of the transmembrane Na+ Ab channel with cholesterol and lipids in the membrane. J. Biomol. Struct. Dyn 2016, 34, 318–326. [DOI] [PubMed] [Google Scholar]
- (771).Halbleib K; Pesek K; Covino R; Hofbauer HF; Wunnicke D; I. Hänelt GH; Ernst R Activation of the unfolded protein response by lipid bilayer stress. Mol. Cell 2017, 67, 673–684. [DOI] [PubMed] [Google Scholar]
- (772).Di Scala C; Chahinian H; Yahi N; Garmy N; Fantini J Interaction of Alzheimer’s β-amyloid peptides with cholesterol: Mechanistic insights into amyloid pore formation. Biochemistry 2014, 53, 4489–4502. [DOI] [PubMed] [Google Scholar]
- (773).Song Y; Kenworthy AK; Sanders CR Cholesterol as a co-solvent and a ligand for membrane proteins. Prot. Sci 2014, 23, 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (774).Li C-D; Xu Q; Gu R-X; Qu J; Wei D-Q The dynamic binding of cholesterol to the multiple sites of C99: as revealed by coarse-grained and all-atom simulations. Phys. Chem. Chem. Phys 2017, 19, 3845–3856. [DOI] [PubMed] [Google Scholar]
- (775).Panahi A; Bandara A; Pantelopulos GA; Dominguez L; Straub JE Specific binding of cholesterol to C99 domain of amyloid precursor protein depends critically on charge state of protein. J. Phys. Chem. Lett 2016, 7, 3535–3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (776).Sun F; Chen L; Wei P; Chai M; Ding X; Xu L; Luo SZ Dimerization and structural stability of amyloid precursor proteins affected by the membrane microenvironments. J. Chem. Inf. Model 2017, 57, 1375–1387. [DOI] [PubMed] [Google Scholar]
- (777).Qiu X; Rau DC; Parsegian VA; Fang LT; Knobler CM; Gelbart WM Salt-dependent DNA-DNA spacings in intact bacteriophage λ reflect relative importance of DNA self-repulsion and bending energies. Phys. Rev. Lett 2011, 106, 028102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (778).Di Scala C; Yahi N; Leliévre C; Garmy N; Chahinian H; Fantini J Biochemical identification of a linear cholesterol-binding domain within Alzheimer’s β amyloid peptide. ACS Chem. Neurosci 2013, 4, 509–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (779).Cheng KH; Qiu L; Cheng SY; Vaughn MW Lipid insertion domain unfolding regulates protein orientational transition behavior in a lipid bilayer. Biophys. Chem 2015, 206, 22–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (780).Di Scala C; Troadec JD; Leliévre C; Garmy N; Fantini J; Chahinian H Mechanism of cholesterol-assisted oligomeric channel formation by a short Alzheimer β-amyloid peptide. J. Neurochem 2014, 128, 186–195. [DOI] [PubMed] [Google Scholar]
- (781).Pannuzzo M On the physiological/pathological link between Aβ peptide, cholesterol, calcium ions and membrane deformation: A molecular dynamics study. Biochim. Biophys. Acta, Biomembr 2016, 1858, 1380–1389. [DOI] [PubMed] [Google Scholar]
- (782).Miller CM; Brown AC; Mittal J Disorder in cholesterol-binding functionality of CRAC peptides: A molecular dynamics study. J. Phys. Chem. B 2014, 118, 13169–13174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (783).Pacheco J; Dominguez L; Bohórquez-Hernández A; Asanov A; Vaca L A cholesterol-binding domain in STIM1 modulates STIM1-Orai1 physical and functional interactions. Sci. Rep 2016, 6, 29634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (784).Liu H; Yang L; Zhang Q; Mao L; Jiang H; Yang H Probing the structure and dynamics of caveolin-1 in a caveolae-mimicking asymmetric lipid bilayer model. Eur. Biophys. J 2016, 45, 511–521. [DOI] [PubMed] [Google Scholar]
- (785).Navrátilová V; Paloncýová M; Kajšová M; Berka K; Otyepka M Effect of cholesterol on the structure of membrane-attached cytochrome P450 3A4. J. Chem. Inf. Model 2015, 55, 628–635. [DOI] [PubMed] [Google Scholar]
- (786).Vitiello G; Fragneto G; Petruk AA; Falanga A; Galdiero S; D’Ursi AM; Merlino A; D’Errico G Cholesterol modulates the fusogenic activity of a membranotropic domain of the FIV glycoprotein gp36. Soft Mat 2013, 9, 6442. [Google Scholar]
- (787).Enkavi G; Mikkolainen H; Güngör B; Ikonen E; Vattulainen I Concerted regulation of npc2 binding to endosomal/lysosomal membranes by bis(monoacylglycero)phosphate and sphingomyelin. PLoS Comput. Biol 2017, 13 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- (788).Wang L; Murphy-Ullrich JE; Song Y Molecular insight into the effect of lipid bilayer environments on thrombospondin-1 and calreticulin interactions. Biochemistry 2014, 53, 6309–6322. [DOI] [PubMed] [Google Scholar]
- (789).Buckland AG; Wilton DC Anionic phospholipids, interfacial binding and the regulation of cell functions. Biochim. Biophys. Acta 2000, 1483, 199–216. [DOI] [PubMed] [Google Scholar]
- (790).Ran S; Downes A; Thorpe PE Increased exposure of anionic phospholipids on the surface of tumor blood vessels. Cancer Res 2002, 62, 6132–6140. [PubMed] [Google Scholar]
- (791).Baudry J; Tajkhorshid E; Molnar F; Phillips J; Schulten K Molecular dynamics study of bacteriorhodopsin and the purple membrane. J. Phys. Chem. B 2001, 105, 905–918. [Google Scholar]
- (792).Sands ZA; Sansom MS How does a voltage sensor interact with a lipid bilayer? Simulations of a potassium channel domain. Structure 2007, 15, 235–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (793).Chandler D; Hsin J; Harrison CB; Gumbart J; Schulten K Intrinsic curvature properties of photosynthetic proteins in chromatophores. Biophys. J 2008, 95, 2822–2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (794).Chandler DE; Gumbart J; Stack JD; Chipot C; Schulten K Membrane curvature induced by aggregates of LH2s and monomeric LH1s. Biophys. J 2009, 97, 2978–2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (795).Javanainen M; Hammaren H; Monticelli L; Jeon J-H; Miettinen MS; Martinez-Seara H; Metzler R; Vattulainen I Anomalous and normal diffusion of proteins and lipids in crowded lipid membranes. Faraday Discuss 2013, 161, 397–417. [DOI] [PubMed] [Google Scholar]
- (796).Goose JE; Sansom MSP Reduced lateral mobility of lipids and proteins in crowded membranes. PLoS Comput. Biol 2013, 9, e1003033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (797).Eggensperger S; Fisette O; Parcej D; Schäfer LV; Tampé R An annular lipid belt is essential for allosteric coupling and viral inhibition of the antigen translocation complex TAP (Transporter Associated with Antigen Processing). J. Biol. Chem 2014, 289, 33098–33108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (798).Wickles S; Singharoy A; Andreani J; Seemayer S; Bischoff L; Berninghausen O; Soeding J; Schulten K; van der Sluis E; Beckmann R A structural model of the active ribosome-bound membrane protein insertase YidC. eLife 2014, 3:e03035 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- (799).Fowler PW; Hélie J; Duncan A; Chavent M; Koldsø H; Sansom MSP Membrane stiffness is modified by integral membrane proteins. Soft Mat 2016, 12, 7792–7803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (800).Terasaka E; Yamada K; Wang P; Hosokawa K; Yamagiwa R; Matsumoto K; Ishii S; Mori T; Yagi K; Sawai H et al. Dynamics of nitric oxide controlled by protein complex in bacterial system. Proc. Natl. Acad. Sci. USA 2017, 114, 9888–9893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (801).Duneau JP; Khao J; Sturgis JN Lipid perturbation by membrane proteins and the lipophobic effect. Biochim. Biophys. Acta, Biomembr 2017, 1859, 1–5. [DOI] [PubMed] [Google Scholar]
- (802).Gupta K; Donlan JAC; Hopper JTS; Uzdavinys P; Landreh M; Struwe WB; Drew D; Baldwin AJ; Stansfeld PJ; Robinson CV The role of interfacial lipids in stabilizing membrane protein oligomers. Nature 2017, 541, 421–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (803).Bolivar JH; Muñoz-García JC; Castro-Dopico T; Dijkman PM; Stansfeld PJ; Watts A Interaction of lipids with the neurotensin receptor 1. Biochim. Biophys. Acta, Biomembr 2016, 1858, 1278–1287. [DOI] [PubMed] [Google Scholar]
- (804).Ermakova E; Zuev Y Interaction of scots pine defensin with model membrane by coarse-grained molecular dynamics. J. Membr. Biol 2017, 250, 205–216. [DOI] [PubMed] [Google Scholar]
- (805).Ashworth Briggs EL; Gomes RG; Elhussein M; Collier W; Stuart Findlow I; Khalid S; McCormick CJ; Williamson PT Interaction between the NS4B amphipathic helix, AH2, and charged lipid headgroups alters membrane morphology and AH2 oligomeric state - Implications for the Hepatitis C virus life cycle. Biochim. Biophys. Acta, Biomembr 2015, 1848, 1671–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (806).Chaudhuri A; Prasanna X; Agiru P; Chakraborty H; Rydström A; Ho JC; Svanborg C; Sengupta D; Chattopadhyay A Protein-dependent membrane interaction of a partially disordered protein complex with oleic acid: Implications for cancer lipidomics. Sci. Rep 2016, 6, 35015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (807).Kim DI; Kang M; Kim S; Lee J; Park Y; Chang I; Suh BC Molecular basis of the membrane interaction of the β2e subunit of voltage-gated Ca2+ channel. Biophys. J 2015, 109, 922–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (808).Rogaski B; Klauda JB Membrane-binding mechanism of a peripheral membrane protein through microsecond molecular dynamics simulations. J. Mol. Biol 2012, 423, 847–861. [DOI] [PubMed] [Google Scholar]
- (809).Rogaski B; Lim JB; Klauda JB Sterol binding and membrane lipid attachment to the Osh4 protein of yeast. J. Phys. Chem. B 2010, 114, 13562–13573. [DOI] [PubMed] [Google Scholar]
- (810).Cheng SY; Chou G; Buie C; Vaughn MW; Compton C; Cheng KH Maximally asymmetric transbilayer distribution of anionic lipids alters the structure and interaction with lipids of an amyloidogenic protein dimer bound to the membrane surface. Chem. Phys. of Lipids 2016, 196, 33–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (811).Konshina AG; Boldyrev IA; Utkin YN; Omel’kov AV; Efremov RG Snake cytotoxins bind to membranes via interactions with phosphatidylserine head groups of lipids. PLoS One 2011, 6 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- (812).Tietjen GT; Gong Z; Chen C-H; Vargas E; Crooks JE; Cao KD; Heffern CTR; Henderson JM; Merone M; Lin B et al. Molecular mechanism for differential recognition of membrane phosphatidylserine by the immune regulatory receptor Tim4. Proc. Natl. Acad. Sci. USA 2014, 111 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- (813).Kooijman EE; Tieleman DP; Testerink C; Munnik T; Rijkers DTS; Burger KNJ; de Kruijff B An electrostatic/hydrogen bond switch as the basis for the specific interaction of phosphatidic acid with proteins. J. Biol. Chem 2007, 282, 11356–11364. [DOI] [PubMed] [Google Scholar]
- (814).Capelluto DG; Zhao X; Lucas A; Lemkul JA; Xiao S; Fu X; Sun F; Bevan DR; Finkielstein CV Biophysical and molecular-dynamics studies of phosphatidic acid binding by the Dvl-2 DEP domain. Biophys. J 2014, 106, 1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (815).Kuang G; Liang L; Brown C; Wang Q; Bulone V; Tu Y Insight into the adsorption profiles of the Saprolegnia monoica chitin synthase MIT domain on POPA and POPC membranes by molecular dynamics simulation studies. Phys. Chem. Chem. Phys 2016, 18, 5281–90. [DOI] [PubMed] [Google Scholar]
- (816).Planas-Iglesias J; Dwarakanath H; Mohammadyani D; Yanamala N; Kagan VE; Klein-Seetharaman J Cardiolipin interactions with proteins. Biophys. J 2015, 109, 1282–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (817).Duncan AL; Ruprecht JJ; Kunji ER; Robinson AJ Cardiolipin dynamics and binding to conserved residues in the mitochondrial ADP/ATP carrier. Biochim. Biophys. Acta, Biomembr 2018, [DOI] [PMC free article] [PubMed]
- (818).Duncan AL; Robinson AJ; Walker JE Cardiolipin binds selectively but transiently to conserved lysine residues in the rotor of metazoan ATP synthases. Proc. Natl. Acad. Sci. USA 2016, 113, 8687–8692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (819).Karo J; Peterson P; Vendelin M Molecular dynamics simulations of creatine kinase and adenine nucleotide translocase in mitochondrial membrane patch. J. Biol. Chem 2012, 287, 7467–7476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (820).Malhotra K; Modak A; Nangia S; Daman TH; Gunsel U; Robinson VL; Mokranjac D; May ER; Alder NN Cardiolipin mediates membrane and channel interactions of the mitochondrial TIM23 protein import complex receptor Tim50. Sci. Adv 2017, 3 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- (821).Mohammadyani D; Yanamala N; Samhan Arias AK; Kapralov AA; Stepanov G; Nuar N; Planas-Iglesias J; Sanghera N; Kagan VE; Klein-Seetharaman J Structural characterization of cardiolipin-driven activation of cytochrome c into a peroxidase and membrane perturbation. Biochim. Biophys. Acta, Biomembr 2018, 1860, 1057–1068. [DOI] [PubMed] [Google Scholar]
- (822).Di Paolo G; De Camilli P Phosphoinositides in cell regulation and membrane dynamics. Nature 2006, 443, 651–657. [DOI] [PubMed] [Google Scholar]
- (823).Balla T Phosphoinositides: Tiny lipids with giant impact on cell regulation. Physiol. Rev 2013, 93, 1019–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (824).Schink KO; Tan K-W; Stenmark H Phosphoinositides in control of membrane dynamics. Annu. Rev. Cell Dev. Biol 2016, 32, 143–171. [DOI] [PubMed] [Google Scholar]
- (825).Hsu FS; Mao Y The structure of phosphoinositide phosphatases: Insights into substrate specificity and catalysis. Biochim. Biophys. Acta, Mol. Cell Biol. of Lipids 2015, 1851, 698–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (826).Choi S; Thapa N; Tan X; Hedman AC; Anderson RA PIP kinases define PI4,5P2 signaling specificity by association with effectors. Biochim. Biophys. Acta, Mol. Cell Biol. of Lipids 2015, 1851, 711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (827).Kim YJ; Hernandez MLG; Balla T Inositol lipid regulation of lipid transfer in specialized membrane domains. Trends Cell. Biol 2013, 23, 270–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (828).Grabon A; Khan D; Bankaitis VA Phosphatidylinositol transfer proteins and instructive regulation of lipid kinase biology. Biochim. Biophys. Acta, Mol. Cell Biol. of Lipids 2015, 1851, 724–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (829).Martin TF PI(4,5)P2-binding effector proteins for vesicle exocytosis. Biochim. Biophys. Acta, Mol. Cell Biol. of Lipids 2015, 1851, 785–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (830).Hammond GRV; Balla T Polyphosphoinositide binding domains: Key to inositol lipid biology. Biochim. Biophys. Acta, Mol. Cell Biol. of Lipids 2015, 1851, 746–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (831).Hansen SB Lipid agonism: The PIP2 paradigm of ligand-gated ion channels. Biochim. Biophys. Acta, Mol. Cell Biol. L 2015, 1851, 620–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (832).Hille B; Dickson EJ; Kruse M; Vivas O; Suh BC Phosphoinositides regulate ion channels. Biochim. Biophys. Acta, Mol. Cell Biol. of Lipids 2015, 1851, 844–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (833).Lenoir M; Kufareva I; Abagyan R; Overduin M Membrane and protein interactions of the pleck-strin homology domain superfamily. Membranes 2015, 5, 646–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (834).Psachoulia E; sansom MSP Interactions of the pleckstrin homology domain with phosphatidyli-nositol phosphate and membranes: Characterization via molecular dynamics simulations. Biochemistry 2008, 47, 4211–4220. [DOI] [PubMed] [Google Scholar]
- (835).Rosen SAJ; Gaffney PRJ; Spiess B; Gould IR Understanding the relative affinity and specificity of the pleckstrin homology domain of protein kinase B for inositol phosphates. Phys. Chem. Chem. Phys 2012, 14, 929–936. [DOI] [PubMed] [Google Scholar]
- (836).Lu D; Jiang J; Liang Z; Sun M; Luo C; Shen B; Hu G Molecular dynamic simulation to explore the molecular basis of Btk-PH domain interaction with Ins(1,3,4,5)P4. Sci. World J 2013, 2013 . [DOI] [PMC free article] [PubMed]
- (837).Yates LA; Lumb CN; Brahme NN; Zalyte R; Bird LE; De Colibus L; Owens RJ; Calderwood DA; Sansom MS; Gilbert RJ Structural and functional characterization of the kindlin-1 pleckstrin homology domain. J. Biol. Chem 2012, 287, 43246–43261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (838).Lumb CN; He J; Xue Y; Stansfeld PJ; Stahelin RV; Kutateladze TG; Sansom MSP Biophysical and computational studies of membrane penetration by the GRP1 pleckstrin homology domain. Structure 2011, 19, 1338–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (839).Lumb CN; Sansom MS Finding a needle in a haystack: The role of electrostatics in target lipid recognition by PH domains. PLoS Comput. Biol 2012, 8, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (840).Lai C-L; Srivastava A; Pilling C; Chase AR; Falke JJ; Voth GA Molecular mechanism of membrane binidng of the GRP1 PH domain. J. Mol. Biol 2013, 425, 3073–3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (841).Karandur D; Nawrotek A; Kuriyan J; Cherfils J Multiple interactions between an Arf/GEF complex and charged lipids determine activation kinetics on the membrane. Proc. Natl. Acad. Sci. USA 2017, 114, 201707970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (842).Pleskot R; Cwiklik L; Jungwirth P; Žárský V; Potocký M Membrane targeting of the yeast exocyst complex. Biochim. Biophys. Acta, Biomembr 2015, 1848, 1481–1489. [DOI] [PubMed] [Google Scholar]
- (843).Bloch D; Pleskot R; Pejchar P; Potocký M; Trpkošová P; Cwiklik L; Vukašinović N; Sternberg H; Yalovsky S; Žárský V Exocyst SEC3 and phosphoinositides define sites of exocytosis in pollen tube initiation and growth. Plant Physiol 2016, 172, pp.00690.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (844).Yamamoto E; Kalli AC; Akimoto T; Yasuoka K; Sansom MS Anomalous dynamics of a lipid recognition protein on a membrane surface. Sci. Rep 2015, 5, 18245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (845).Yamamoto E; Akimoto T; Kalli AC; Yasuoka K; Sansom MS Dynamic interactions between a membrane binding protein and lipids induce fluctuating diffusivity. Sci. Adv 2017, 3, e1601871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (846).Herzog FA; Braun L; Schoen I; Vogel V Improved side chain dynamics in MARTINI simulations of protein-lipid interfaces. J. Chem. Theory Comput 2016, 12, 2446–2458. [DOI] [PubMed] [Google Scholar]
- (847).Kuang G; Bulone V; Tu Y Computational studies of the binding profile of phosphoinositide PtdIns(3,4,5)P3 with the pleckstrin homology domain of an oomycete cellulose synthase. Sci. Rep 2016, 6, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (848).Naughton FB; Kalli AC; Sansom MS Association of peripheral membrane proteins with membranes: Free energy of binding of GRP1 PH domain with phosphatidylinositol phosphate-containing model bilayers. J. Phys. Chem. Lett 2016, 7, 1219–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (849).Ni T; Kalli AC; Naughton FB; Yates LA; Naneh O; Kozorog M; Anderluh G; Sansom MS; Gilbert RJ Structure and lipid-binding properties of the kindlin-3 pleckstrin homology domain. Biochem. J 2017, 474, 539–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (850).Chan KC; Lu L; Sun F; Fan J Molecular details of the PH domain of ACAP1BAR−P H protein binding to PIP-containing membrane. J. Phys. Chem. B 2017, 121, 3586–3596. [DOI] [PubMed] [Google Scholar]
- (851).Yamamoto E; Kalli AC; Yasuoka K; Sansom MSP Interactions of pleckstrin homology domains with membranes: Adding back the bilayer via high-throughput molecular dynamics. Structure 2016, 24, 1421–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (852).Naughton FB; Kalli AC; Sansom MS Modes of interaction of pleckstrin homology domains with membranes: Toward a computational biochemistry of membrane recognition. J. Mol. Biol 2018, 430, 372–388. [DOI] [PubMed] [Google Scholar]
- (853).Manna D; Bhardwaj N; Vora MS; Stahelin RV; Lu H; Cho W Differential roles of phosphatidylserine, PtdIns(4,5)P2, and PtdIns(3,4,5)P3 in plasma membrane targeting of C2 domains. J. Biol. Chem 2008, 283, 26047–26058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (854).Lai C-L; Landgraf KE; Voth GA; Falke JJ Membrane docking geometry and target lipid stoichiometry of membrane-bound PKC C2 domain: A combined molecular dynamics and experimental study. J. Mol. Biol 2010, 402, 301–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (855).Alwarawrah M; Wereszczynski J Investigation of the effect of bilayer composition on PKCα-C2 domain docking using molecular dynamics simulations. J. Phys. Chem. B 2017, 121, 78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (856).Dreßler L; Michel F; Thondorf I; Mansfeld J; Golbik R; Ulbrich-Hofmann R Metal ions and phosphatidylinositol 4,5-bisphosphate as interacting effectors of α-type plant phospholipase D. Phytochemistry 2017, 138, 57–64. [DOI] [PubMed] [Google Scholar]
- (857).Michaeli L; Gottfried I; Bykhovskaia M; Ashery U Phosphatidylinositol (4, 5)-bisphosphate targets double C2 domain protein B to the plasma membrane. Traffic 825–839. [DOI] [PMC free article] [PubMed]
- (858).Or-lowski A; Kukkurainen S; Pöyry A; Rissanen S; Vattulainen I; Hytönen VP; Ròg T PIP2 and talin join forces to activate integrin. J. Phys. Chem. B 2015, 119, 12381–12389. [DOI] [PubMed] [Google Scholar]
- (859).Senju Y; Kalimeri M; Koskela EV; Somerharju P; Zhao H; Vattulainen I; Lappalainen P Mechanistic principles underlying regulation of the actin cytoskeleton by phosphoinositides. Proc. Natl. Acad. Sci. USA 2017, 114, 201705032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (860).Sun F; Schroer CF; Xu L; Yin H; Marrink SJ; Luo SZ Molecular dynamics of the association of L-selectin and FERM regulated by PIP2. Biophys. J 2018, 114, 1858–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (861).Zhou J; Bronowska A; Le Coq J; Lietha D; Gräter F Allosteric regulation of focal adhesion kinase by PIP2 and ATP. Biophys. J 2015, 108, 698–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (862).Feng J; Mertz B Novel phosphotidylinositol 4,5-bisphosphate binding sites on focal adhesion kinase. PLoS One 2015, 10, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (863).Herzog FA; Braun L; Schoen I; Vogel V Structural insights how PIP2 imposes preferred binding orientations of FAK at lipid membranes. J. Phys. Chem. B 2017, 121, 3523–3535. [DOI] [PubMed] [Google Scholar]
- (864).Kalli AC; Morgan G; Sansom MS Interactions of the auxilin-1 pten-like domain with model membranes result in nanoclustering of phosphatidyl inositol phosphates. Biophys. J 2013, 105, 137–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (865).Psachoulia E; Sansom MSP PX- and FYVE-mediated interactions with membranes: Simulation studies. Biochemistry 2009, 48, 5090–5095. [DOI] [PubMed] [Google Scholar]
- (866).Jia Z; Ghai R; Collins BM; Mark AE The recognition of membrane-bound PtdIns3P by PX domains. Proteins: Struct., Func., Bioinf 2014, 82, 2332–2342. [DOI] [PubMed] [Google Scholar]
- (867).Antony P; Baby B; Vijayan R Molecular insights into the binding of phosphoinositides to the TH domain region of TIPE proteins. J. Mol. Mod 2016, 22 . [DOI] [PubMed] [Google Scholar]
- (868).Busse RA; Scacioc A; Krick R; Pérez-Lara Á; Thumm M; Kühnel K Characterization of PROPPIN-phosphoinositide binding and role of loop 6CD in PROPPIN-membrane binding. Biophys. J 2015, 108, 2223–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (869).Picas L; Viaud J; Schauer K; Vanni S; Hnia K; Fraisier V; Roux A; Bassereau P; Gaits-Iacovoni F; Payrastre B et al. BIN1/M-Amphiphysin2 induces clustering of phosphoinositides to recruit its downstream partner dynamin. Nat. Commun 2014, 5 . [DOI] [PubMed] [Google Scholar]
- (870).Steringer JP; Lange S; Č ujová S; Šachl R; Poojari C; Lolicato F; Beutel O; Müller HM; Unger S; Coskun Ü . et al. Key steps in unconventional secretion of fibroblast growth factor 2 reconstituted with purified components. eLife 2017, 6 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- (871).Mondal S; Chandra A; Venkatramani R; Datta A Optically sensing phospholipid induced coilhelix transitions in the phosphoinositide-binding motif of gelsolin. Faraday Discuss 2018, 207, 437–458. [DOI] [PubMed] [Google Scholar]
- (872).Khelashvili G; Galli A; Weinstein H Phosphatidylinositol 4,5-biphosphate (PIP2) lipids regulate the phosphorylation of syntaxin N-terminus by modulating both its position and local structure. Biochemistry 2012, 51, 7685–7698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (873).Sharma S; Kim BN; Stansfeld PJ; Sansom MS; Lindau M A coarse grained model for a lipid membrane with physiological composition and leaflet asymmetry. PLoS One 2015, 10, e0144814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (874).Holdbrook DA; Leung YM; Piggot TJ; Marius P; Williamson PT; Khalid S Stability and membrane orientation of the fukutin transmembrane domain: A combined multiscale molecular dynamics and circular dichroism study. Biochemistry 2010, 49, 10796–10802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (875).Koldsø H; Shorthouse D; Hélie J; Sansom MSP Lipid clustering correlates with membrane curvature as revealed by molecular simulations of complex lipid bilayers. PLoS Comput. Biol 2014, 10, e1003911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (876).Koldsø H; Sansom MSP Organization and dynamics of receptor proteins in a plasma membrane. J. Am. Chem. Soc 2015, 137, 14694–14704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (877).Duncan AL; Reddy T; Koldso H; Helie J; Fowler PW; Chavent M; Sansom MSP Protein crowding and lipid complexity influence the nanoscale dynamic organization of ion channels in cell membranes. Sci. Rep 2017, 7, 16647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (878).Takahashi N; Hamada-Nakahara S; Itoh Y; Takemura K; Shimada A; Ueda Y; Kitamata M; Matsuoka R; Hanawa-Suetsugu K; Senju Y et al. TRPV4 channel activity is modulated by direct interaction of the ankyrin domain to PI(4,5)P2. Nat. Commun 2014, 5, 4994. [DOI] [PubMed] [Google Scholar]
- (879).Seebohm G; Wrobel E; Pusch M; Dicks M; Terhag J; Matschke V; Rothenberg I; Ursu ON; Hertel F; Pott L et al. Structural basis of PI(4,5)P2-dependent regulation of GluA1 by phosphatidylinositol-5-phosphate 4-kinase, type II, alpha (PIP5K2A). Pflug. Arch. Eur. J. Physiol 2014, 466, 1885–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (880).Hedger G; Shorthouse D; Koldso H; Sansom MSP Free energy landscape of lipid interactions with regulatory binding sites on the transmembrane domain of the EGF receptor. J. Phys. Chem. B 2016, 120, 8154–8163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (881).Halim KBA; Koldsø H; Sansom MSP Interactions of the EGFR juxtamembrane domain with PIP2-containing lipid bilayers: Insights from multiscale molecular dynamics simulations. Biochim. Biophys. Acta, Gen. Subj 2015, 1850, 1017–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (882).Ma H; Cummins DD; Edelstein NB; Kahn A; Llewellyn MD; Picudella T; Willsey SR; Nangia S Modeling diversity in structures of bacterial outer membrane lipids. J. Chem. Theory Comput 2017, 13, 811–824. [DOI] [PubMed] [Google Scholar]
- (883).Patel DS; Qi Y; Im W Modeling and simulation of bacterial outer membranes and interactions with membrane proteins. Curr. Opin. Struct. Biol 2017, 43, 131–140. [DOI] [PubMed] [Google Scholar]
- (884).Voet DJ; Voet JG; Pratt CW Principles of Biochemistry ; Wiley, 2008. [Google Scholar]
- (885).RN K Sphingomyelin and derivatives as cellular signals. Prog. Lipid Res 1991, 30, 1–38. [DOI] [PubMed] [Google Scholar]
- (886).FG T; P, T.; JC, H. The multigenic sphingomyelin synthase family. J. Biol. Chem 2006, 281, 29421–29425. [DOI] [PubMed] [Google Scholar]
- (887).DA B; E, L. Structure and function of sphingolipid- and cholesterol-rich membrane rafts. J. Biol. Chem 2000, 275, 17221–17224. [DOI] [PubMed] [Google Scholar]
- (888).Simons K; Toomre D Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol 2000, 1, 31–39. [DOI] [PubMed] [Google Scholar]
- (889).Hannun YA; Obeid LM Principles of bioactive lipid signalling: lessons from sphingolipids. Nat. Rev. Mol. Cell Biol 2008, 9, 139–150. [DOI] [PubMed] [Google Scholar]
- (890).Troupiotis-Tsäılaki A; Zachmann J; González-Gil I; Gonzalez A; Ortega-Gutiérrez S; Lòpez-Rodríguez ML; Pardo L; Govaerts C Ligand chain length drives activation of lipid G protein-coupled receptors. Sci. Rep 2017, 7, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (891).Polley A; Or-lowski A; Danne R; Gurtovenko AA; Bernardino de La Serna J; Eggeling C; Davis SJ; Ròg T; Vattulainen I Glycosylation and lipids working in concert direct CD2 ectodomain orientation and presentation. J. Phys. Chem. Lett 2017, 8, 1060–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (892).Wu EL; Qi Y; Park S; Mallajosyula SS; Mackerell AD; Klauda JB; Im W Insight into early-stage unfolding of GPI-anchored human prion protein. Biophys. J 2015, 109, 2090–2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (893).Gu RX; Ingólfsson HI; De Vries AH; Marrink SJ; Tieleman DP Ganglioside-lipid and ganglioside-protein interactions revealed by coarse-grained and atomistic molecular dynamics simulations. J. Phys. Chem. B 2017, 121, 3262–3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (894).Soto C; del Valle A; Valiente PA; Ros U; Lanio ME; Hernández AM; Alvarez C Differential binding and activity of the pore-forming toxin sticholysin II in model membranes containing diverse ceramide-derived lipids. Biochimie 2017, 138, 20–31. [DOI] [PubMed] [Google Scholar]
- (895).Sridhar A; Kumar A; Dasmahapatra AK Multi-scale molecular dynamics study of cholera pentamer binding to a GM1-phospholipid membrane. J. Mol. Graph. Model 2016, 68, 236–251. [DOI] [PubMed] [Google Scholar]
- (896).Basu I; Mukhopadhyay C Insights into binding of cholera toxin to GM1 containing membrane. Langmuir 2014, 30, 15244–15252. [DOI] [PubMed] [Google Scholar]
- (897).Artner D; Oblak A; Ittig S; Garate JA; Horvat S; Arrieumerlou C; Hofinger A; Ootenbrink C; Jerala R; Kosma P et al. Conformationally constrained lipid A mimetics for exploration of structural basis of TLR4/MD-2 activation by lipopolysaccharide. ACS Chem. Biol 2013, 8, 2423–2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (898).Rust MJ; Lakadamyali M; Zhang F; Zhuang X Assembly of endocytic machinery around individual influenza viruses during viral entry. Nat. Struct. Mol. Biol 2004, 11, 567–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (899).Dowhan W; Mileykovskaya E; Bogdanov M Diversity and versatility of lipid-protein interactions revealed by molecular genetic approaches. Biochim. Biophys. Acta, Biomembr 2004, 1666, 19–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (900).Martens S; McMahon HT Mechanisms of membrane fusion: Disparate players and common principles. Nat. Rev. Mol. Cell Biol 2008, 9, 543–556. [DOI] [PubMed] [Google Scholar]
- (901).Pomorski TG; Nylander T; Cárdenas M Model cell membranes: Discerning lipid and protein contributions in shaping the cell. Adv. Colloid Interface Sci 2014, 205, 207–220. [DOI] [PubMed] [Google Scholar]
- (902).Brown MF Soft matter in lipid-protein interactions. Annu. Rev. Biophys 2017, 46, 379–410. [DOI] [PubMed] [Google Scholar]
- (903).Hianik T Advances in Planar Lipid Bilayers and Liposomes; 2011; Vol. 13; pp 33–72. [Google Scholar]
- (904).Wohlert J; Edholm O Dynamics in atomistic simulations of phospholipid membranes: Nuclear magnetic resonance relaxation rates and lateral diffusion. J. Chem. Phys 2006, 125, 204703. [DOI] [PubMed] [Google Scholar]
- (905).Fotin A; Kirchhausen T; Grigorieff N; Harrison SC; Walz T; Cheng Y Structure determination of clathrin coats to subnanometer resolution by single particle cryo-electron microscopy. J. Struct. Biol 2006, 156, 453–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (906).Davies KM; Anselmi C; Wittig I; Faraldo-Gomez JD; Kuhlbrant W Structure of the yeast F1Fo-ATP synthase dimer and its role in shaping the mitochondrial cristae. Proc. Natl. Acad. Sci. USA 2012, 109, 13602–13607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (907).Davis JH The influence of membrane proteins on lipid dynamics. Chem. Phys. of Lipids 1986, 40, 223–258. [DOI] [PubMed] [Google Scholar]
- (908).McMahon HT; Gallop JL Membrane curvature and mechanisms of dynamic cell membrane remodeling. Nature 2005, 438, 590–596. [DOI] [PubMed] [Google Scholar]
- (909).Tieleman DP; Leontiadou H; Mark AE; Marrink SJ Simulation of pore formation in lipid bilayers by mechanical stress and electric fields. J. Am. Chem. Soc 2003, 125, 6382–6383. [DOI] [PubMed] [Google Scholar]
- (910).Zhu C; Bao G; Wang N Cell mechanics: mechanical response, cell adhesion, and molecular deformation. Annu. Rev. Biomed. Eng 2000, 2, 189–226. [DOI] [PubMed] [Google Scholar]
- (911).Maftouni N; Amininasab M; Ejtehadi MR; Kowsari F; Dastvan R Nanomechanical properties of lipid bilayer: Asymmetric modulation of lateral pressure and surface tension due to protein insertion in one leaflet of a bilayer. J. Chem. Phys 2013, 138 . [DOI] [PubMed]
- (912).Nicolson GL The fluid-mosaic model of membrane structure: Still relevant to understanding the structure, function and dynamics of biological membranes after more than 40 years. Biochim. Biophys. Acta, Biomembr 2014, 1838, 1451–1466. [DOI] [PubMed] [Google Scholar]
- (913).Corradi V; Mendez-Villuendas E; Inglfsson HI; Gu R-X; Siuda I; Melo MN; Moussatova A; DeGagn LJ; Sejdiu BI; Singh G et al. Lipid-protein interactions are unique fingerprints for membrane proteins. ACS Cent. Sci 2018, 4, 709–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (914).Bond PJ; Sansom M Bilayer deformation by the Kv channel voltage sensor domain revealed by self-assembly simulations. Proc. Natl. Acad. Sci. USA 2007, 104, 2631–2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (915).Mondal S; Khelashvili G; Shan J; Andersen OS; Weinstein H Quantitative modeling of membrane deformations by multihelical membrane proteins: application to G-protein coupled receptors. Biophys. J 2011, 101, 2092–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (916).Lervik A; Bresme F; Kjelstrup S Molecular dynamics simulations of the Ca2+-pump: a structural analysis. Phys. Chem. Chem. Phys 2012, 14, 3543. [DOI] [PubMed] [Google Scholar]
- (917).Sonntag Y; Musgaard M; Olesen C; Schiøtt B; Møller JV; Nissen P; Thøgersen L Mutual adaptation of a membrane protein and its lipid bilayer during conformational changes. Nat. Commun 2011, 2, 304. [DOI] [PubMed] [Google Scholar]
- (918).Ramstedt B; Slotte JP Sphingolipids and the formation of sterol-enriched ordered membrane domains. Biochim. Biophys. Acta 2006, 1758, 1945–1956. [DOI] [PubMed] [Google Scholar]
- (919).Mileykovskaya E; Dowhan W Cardiolipin membrane domains in prokaryotes and eukaryotes. Biochim. Biophys. Acta, Biomembr 2009, 1788, 2084–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (920).Lindbolm G; Orädd G Lipid lateral diffusion and membrane heterogeneity. Biochim. Biophys. Acta 2009, 1788, 234–244. [DOI] [PubMed] [Google Scholar]
- (921).Koldsø H; Sansom MSP Local lipid reorganization by a transmembrane protein domain. J. Phys. Chem. Lett 2012, 3, 3498–3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (922).Parton DL; Tek A; Baaden M; Sansom MSP Formation of raft-like assemblies within clusters of influenza hemagglutinin observed by MD simulations. PLoS Comput. Biol 2013, 9, e1003034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (923).Gonen T; Cheng Y; Sliz P; Hiroaki Y; Fujiyoshi Y; Harrison SC; Walz T Lipid-protein interactions in double-layered two-dimensional AQP0 crystals. Nature 2005, 438, 633–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (924).Aponte-Santamaria C; Briones R; Schenk AD; Walz T; de Groot BL Molecular driving forces defining lipid positions around aquaporin-0. Proc. Natl. Acad. Sci. USA 2012, 109, 9887–9892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (925).Briones R; Aponte-Santamaría C; de Groot BL Localization and ordering of lipids around aquaporin-0: Protein and lipid mobility effects. Front. Physiol 2017, 8, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (926).Pomorski TG; Meno AK Lipid somersaults: Uncovering the mechanisms of protein-mediated lipid flipping. Prog. Lipid Res 2016, 64, 69–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (927).Contreras F-X; Villar A-V; Alonso A; Kolesnick RN; Goñi e. M. Sphingomyelinase activity causes transbilayer lipid translocation in model and cell membranes. J. Biol. Chem 2003, 278, 37169–37174. [DOI] [PubMed] [Google Scholar]
- (928).Sapay N; Bennett WF; Tieleman DP Molecular simulations of lipid flip-flop in the presence of model transmembrane helices. Biochemistry 2010, 49, 7665–7673. [DOI] [PubMed] [Google Scholar]
- (929).Wu Q-Y; Liang Q Interplay between curvature and lateral organization of lipids and peptides/proteins in model membranes. Langmuir 2014, 30, 1116–1122. [DOI] [PubMed] [Google Scholar]
- (930).Martyna A; Gómez-Llobregat J; Lindén M; Rossman JS Curvature sensing by a viral scission protein. Biochemistry 2016, 55, 3493–3496. [DOI] [PubMed] [Google Scholar]
- (931).Flinner N; Schleiff E Dynamics of the glycophorin a dimer in membranes of native-like composition uncovered by coarse-grained molecular dynamics simulations. PLoS One 2015, 10 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- (932).Periole X; Huber T; Marrink S-J; Sakmar TP G protein-coupled receptors self-assemble in dynamics simulations of model bilayers. J. Am. Chem. Soc 2007, 129, 10126–10132. [DOI] [PubMed] [Google Scholar]
- (933).Cui H; Lyman E; Voth GA Mechanism of membrane curvature sensing by amphipathic helix containing proteins. Biophys. J 2011, 100, 1271–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (934).Helfrich W Elastic properties of lipid bilayers: theory and possible experiments. Z. Naturforsch 1973, 28, 693–703. [DOI] [PubMed] [Google Scholar]
- (935).Naji A; Atzberger PJ; Brown FLH Hybrid elastic and discrete-particle approach to biomembrane dynamics with application to the mobility of curved integral membrane proteins. Phys. Rev. Lett 2009, 102, 138102. [DOI] [PubMed] [Google Scholar]
- (936).Lee KI; Pastor RW; Andersen OS; Im W Assessing smectic liquid-crystal continuum models for elastic bilayer deformations. Chem. Phys. of Lipids 2013, 169, 19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (937).Mondal S; Johnston JM; Wang H; Khelashvili G; Filizola M; Weinstein H Membrane driven spatial organization of GPCRs. Sci. Rep 2013, 3, 2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (938).Khelashvili G; Harries D; Weinstein H Modeling membrane deformations and lipid demixing upon protein-membrane interaction: The BAR dimer adsorption. Biophys. J 2009, 97, 1626–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (939).Liu J; Tourdot R; Ramanan V; Agrawal NJ; Radhakrishanan R Mesoscale simulations of curvature-inducing protein partitioning on lipid bilayer membranes in the presence of mean curvature fields. Mol. Phys 2012, 110, 1127–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (940).Tourdot RW; Ramakrishnan N; Baumgart T; Radhakrishnan R Application of a free-energy-landscape approach to study tension-dependent bilayer tubulation mediated by curvature-inducing proteins. Phys. Rev. E 2015, 92, 042715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (941).Noguchi H Formation of polyhedral vesicles and polygonal membrane tubes induced by banana-shaped proteins. J. Chem. Phys 2015, 143, 243109. [DOI] [PubMed] [Google Scholar]
- (942).Sapp K; Maibaum L Suppressing membrane height fluctuations leads to a membrane-mediated interaction among proteins. Phys. Rev. E 2016, 94, 052414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (943).Davtyan A; Simunovic M; Voth GA Multiscale simulations of protein-facilitated membrane remodeling. J. Struct. Biol 2016, 196, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (944).Davtyan A; Simunovic M; Voth GA The mesoscopic membrane with proteins (MesM-P) model. J. Chem. Phys 2017, 147, 044101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (945).Yoo J; Jackson MB; Cui Q A comparison of coarse-grained and continuum models for membrane bending in lipid bilayer fusion pores. Biophys. J 2013, 104, 841–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (946).Sodt AJ; Beaven AH; Andersen OS; Im W; Pastor RW Gramicidin A channel formation induces local lipid redistribution II: A 3D continuum elastic model. Biophys. J 2017, 112, 1198–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (947).Argudo D; Bethel NP; Marcoline FV; Grabe M Continuum descriptions of membranes and their interaction with proteins: Towards chemically accurate models. Biochim. Biophys. Acta, Biomembr 2016, 1858, 1619–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (948).Kirschner K; Lins R; Maass A; Soares T A Glycam-based force field for simulations of lipopolysac-charide membranes: Parametrization and validation. J. Chem. Theory Comput 2012, 8, 4719–4731. [DOI] [PubMed] [Google Scholar]
- (949).Ma W; Schulten K Mechanism of substrate translocation by a ring-shaped ATPase motor at millisecond resolution. J. Am. Chem. Soc 2015, 137, 3031–3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (950).Jefferies D; Hsu P-C; Khalid S Through the lipopolysaccharide glass: A potent antimicrobial peptide induces phase changes in membranes. Biochemistry 2017, 56, 1672–1679. [DOI] [PubMed] [Google Scholar]
- (951).Lugtenberg E; Peters R Distribution of lipids in cytoplasmic and outer membranes of Escherichia Coli K12. Biochim. Biophys. Acta, Lipids Lipid Metab 1976, 441, 38–47. [DOI] [PubMed] [Google Scholar]
- (952).Risselada H; Kutzner C; Grubmuller H Caught in the act: Visualization of SNARE-mediated fusion events in molecular detail. Chem. Bio. Chem 2011, 12, 1049–1055. [DOI] [PubMed] [Google Scholar]