

Abstract
Background
Clostridium spp. can synthesize valuable chemicals and fuels by utilizing diverse waste-stream substrates, including starchy biomass, lignocellulose, and industrial waste gases. However, metabolic engineering in Clostridium spp. is challenging due to the low efficiency of gene transfer and genomic integration of entire biosynthetic pathways.
Results
We have developed a reliable gene transfer and genomic integration system for the syngas-fermenting bacterium Clostridium ljungdahlii based on the conjugal transfer of donor plasmids containing large transgene cassettes (> 5 kb) followed by the inducible activation of Himar1 transposase to promote integration. We established a conjugation protocol for the efficient generation of transconjugants using the Gram-positive origins of replication repL and repH. We also investigated the impact of DNA methylation on conjugation efficiency by testing donor constructs with all possible combinations of Dam and Dcm methylation patterns, and used bisulfite conversion and PacBio sequencing to determine the DNA methylation profile of the C. ljungdahlii genome, resulting in the detection of four sequence motifs with N6-methyladenosine. As proof of concept, we demonstrated the transfer and genomic integration of a heterologous acetone biosynthesis pathway using a Himar1 transposase system regulated by a xylose-inducible promoter. The functionality of the integrated pathway was confirmed by detecting enzyme proteotypic peptides and the formation of acetone and isopropanol by C. ljungdahlii cultures utilizing syngas as a carbon and energy source.
Conclusions
The developed multi-gene delivery system offers a versatile tool to integrate and stably express large biosynthetic pathways in the industrial promising syngas-fermenting microorganism C. ljungdahlii. The simple transfer and stable integration of large gene clusters (like entire biosynthetic pathways) is expanding the range of possible fermentation products of heterologously expressing recombinant strains. We also believe that the developed gene delivery system can be adapted to other clostridial strains as well.
Electronic supplementary material
The online version of this article (10.1186/s13068-019-1448-1) contains supplementary material, which is available to authorized users.
Keywords: Synthetic biology, Metabolic engineering, Acetogen, Conjugation, Inducible promoter, Himar1 transposase, Genomic integration, Syngas fermentation, Isopropanol, Acetone, Bio-fuels
Introduction
The microbial conversion of industrial waste streams into bulk chemicals and fuels is a promising approach to reduce greenhouse gas emissions and offset the effects of climate change [1]. The bacterial genus Clostridium includes several species that can produce valuable chemical building blocks, such as acetone, organic acids, and various short/medium-chain alcohols [2]. For industrial-scale production, the feedstock is often starchy plant material that provides sugars as a carbon and energy source, e.g., C. acetobutylicum can produce acetone using starch as a substrate [3]. However, one drawback is that starch crops cultivated for industrial purposes compete with food and feed crops. A more attractive feedstock is synthesis gas (syngas), a mixture of carbon monoxide, carbon dioxide, and hydrogen, which is emitted in large amounts e.g., by the steel industry and can be produced by the gasification of municipal waste [4]. C. ljungdahlii and C. autoethanogenum among others can utilize syngas as a sole carbon and energy source [4, 5]. These species use the reductive acetyl-CoA pathway, also known as the Wood–Ljungdahl pathway, to reduce CO, CO2, and H2 to the key metabolite acetyl-CoA, which is processed further into biomass and the main fermentation products ethanol and acetate [6–8].
The ability of certain Clostridium species to convert syngas into acetyl-CoA, a central biochemical building block, could be exploited to produce additional chemicals by extending and modifying the corresponding metabolic pathways. However, the genetic manipulation of Gram-positive bacteria remains much more challenging than the amenable processes developed for model Gram-negative species such as Escherichia coli, due to the physiological and biochemical distinctions between these groups of bacteria. Successful metabolic engineering in Gram-positive bacteria requires a strategy to achieve the introduction of multiple transgenes encoding appropriate enzymes and to ensure their stable integration and expression, while overcoming the restriction–methylation system of the host.
Multi-gene transformation in Gram-positive bacteria can be achieved using traditional approaches such as electroporation, but the efficiency of gene transfer tends to be inversely proportional to the plasmid size, as shown in other species such as E. coli [9, 10]. Optimal parameters must be established empirically or systematically for each species [11]. An alternative strategy is conjugation, a natural DNA transfer process that requires only the presence of a dedicated origin of transfer (oriT) on the donor plasmid, with all other functions (tra and mob genes) provided in trans [12]. Conjugation is versatile because it mobilizes large plasmids efficiently and can transfer DNA between species, including transfer between Gram-negative and Gram-positive bacteria [13] and from bacteria to eukaryotes [14]. This method has been used for DNA transfer to C. beijerinckii NCIMB 8052 (formerly Clostridium acetobutylicum NCIB 8052) [15] and C. perfringens [16] and is the standard method for DNA transfer to C. difficile [17]. Although conjugation is an efficient strategy for gene transfer to Clostridium spp., the donor plasmid is susceptible to the host’s restriction–modification system [18]. The decline in conjugation efficiency caused by unmethylated recognition sites within the donor plasmid sequence has been demonstrated for C. difficile [17]. Plasmid instability is also an important issue, especially in an industrial context where the bulk use of antibiotics to maintain selection pressure is too expensive [19, 20]. These issues can be addressed by targeted genomic integration, as has been demonstrated using the ClosTron system based on group II introns [21]. Various transposon systems have also been used to achieve non-targeted integration, including the conjugative transposon Tn916 [22–24] and non-conjugative transposons such as Tn5 [25, 26]. While most approaches use transposon mutagenesis as a means for reverse genetics to disrupt genes [23–25, 27], it has also been applied to integrate entire biosynthetic pathways into several Gram-negative bacteria species [26, 28]. A useful alternative is the mariner-type transposon Himar1 from Haematobia irritans [29], which has the capability to deliver biosynthetic pathways as large as 29.6 and 57.5 kb into the genome [30]. The Himar1 transposase facilitates the random and even multiple integration of DNA sequences flanked by ~ 27-bp inverted terminal repeats (ITRs) without additional cofactors [29], making the system applicable in any host including Clostridium spp. [31]. The integration occurs by a ‘cut and paste’ mechanism in the 2-bp TA target sequence of Himar1, which is duplicated upon insertion [32]. The Staphylococcus xylosus xylose-inducible promoter–repressor system [33] is functional in C. acetobutylicum [34] and can be used to induce the transposase. After conjugal transfer of the donor plasmid, the addition of xylose induces Himar1 transposase expression and promotes the integration of one or more copies of the cassette bounded by the ITRs for increased transgene stability, with multiple integration events enhancing the gene dosage and ultimately the anticipated product yield.
Here we describe the development of an efficient conjugation and genomic integration protocol that allows the transfer of large gene cassettes, e.g. coding for entire metabolic pathways, to the host species C. ljungdahlii and thus facilitates the production of different bulk chemicals and fuels from a syngas-fermenting bacterial strain. The industrial scaling of such a process would allow the recycling of syngas, reducing the environmental harm caused by its release, and channeling this waste stream into the production of valuable chemicals.
Results and discussion
Development and optimization of a conjugation protocol for C. ljungdahlii
The syngas-fermenting bacterium C. ljungdahlii offers the tantalizing prospect of converting an important industrial waste stream into environmentally sustainable platform chemicals and fuels. Although the availability of a complete C. ljungdahlii genome sequence will facilitate this process [35], several technical challenges must be addressed before efficient metabolic engineering is possible, including the low efficiency of transformation with large gene cassettes [36]. The latter reflects a combination of factors including inefficient gene transfer, a hostile host restriction–modification system, and the instability of plasmid DNA.
We addressed these challenges by developing a robust gene delivery system based on conjugation, which efficiently transfers large plasmids, combined with an inducible Himar1 transposase to facilitate cassette integration into the C. ljungdahlii genome. To establish and optimize the conjugation procedure for C. ljungdahlii, the oriT region of the broad host range IncPα plasmid RK2 [37, 38] was transferred to a derivative of pSOS95 vector, which contains the repL (pIM13) origin of replication for Gram-positive bacteria [34, 39], resulting in the vector pSOS-traJ. Several further origins of replication are suitable for C. ljungdahlii [40], and to extend the potential of our conjugation platform we therefore replaced repL with the broad-range origin repH (pCB102) to generate the final vector pSOS-traJ-repH.
Triparental mating was carried out using donor and helper strains of E. coli and the recipient strain of C. ljungdahlii (Fig. 1). We used the E. coli B-strain NEB Express as a donor, since it was reported that B-strains achieve higher C. ljungdahlii transformation efficiencies by electroporation than K-strains [40]. The helper strain Stbl3 carries the vector pRK2013, which supplies the necessary tra and mob functions in trans [41].
Fig. 1.

Schematic representation of the triparental mating and genomic integration process. A helper strain (E. coli Stbl3) is used to provide the genes necessary for conjugation in trans on the helper plasmid pRK2013. Once the helper plasmid is transferred through the conjugation bridge, the donor strain with the plasmid of interest can transfer its cargo to the recipient Clostridium strain via conjugation. The mechanism of plasmid transfer between Gram-negative and Gram-positive bacteria is currently unknown and is indicated by a dotted arrow. Genomic integration is induced by the addition of 2% xylose to the medium. Plasmid loss is promoted by repeated cultivation in YTF medium with selection on d-cycloserine and clarithromycin only, omitting thiamphenicol
Several conjugation methods were tested with variable results. The most straight-forward method (concentrating, washing, and incubating the mixed strains in liquid medium overnight, as often used with E. coli) was unsuccessful. Switching to solid rich medium achieved better results as suggested by Willets [42], although the frequency of transconjugant C. ljungdahlii colonies varied widely among experimental replicates. We therefore investigated the relationship between the conjugation efficiency and culture volume by conducting parallel triparental mating experiments in liquid medium and on solid medium poured either 2 days or 2 weeks before conjugation, which results in the absorption of different quantities of the concentrated conjugation broth. We also varied the amount of conjugation broth applied to the plates (100 µl or 1 ml).
The lower mating culture volume (100 µl) on a fresh plate gave comparable results to plating the higher volume (1 ml) onto older plates, but the combination of the lower mating culture volume and the well-dried plate for overnight mating gave the best results (Fig. 2a). Placing unsealed fresh plates face down at 37 °C for 1–2 days accelerated the drying process and also increased the number of transconjugants (unpublished observations). The growth of freshly transformed C. ljungdahlii can be poor on the plate or is inhibited by selection pressure, so the harvested cultures were poured into the plates after mating, using low-melting-point agarose to prevent heat shock. The impact of these minor procedural differences may explain the range of conjugation frequencies reported by different research groups using an identical protocol [43].
Fig. 2.
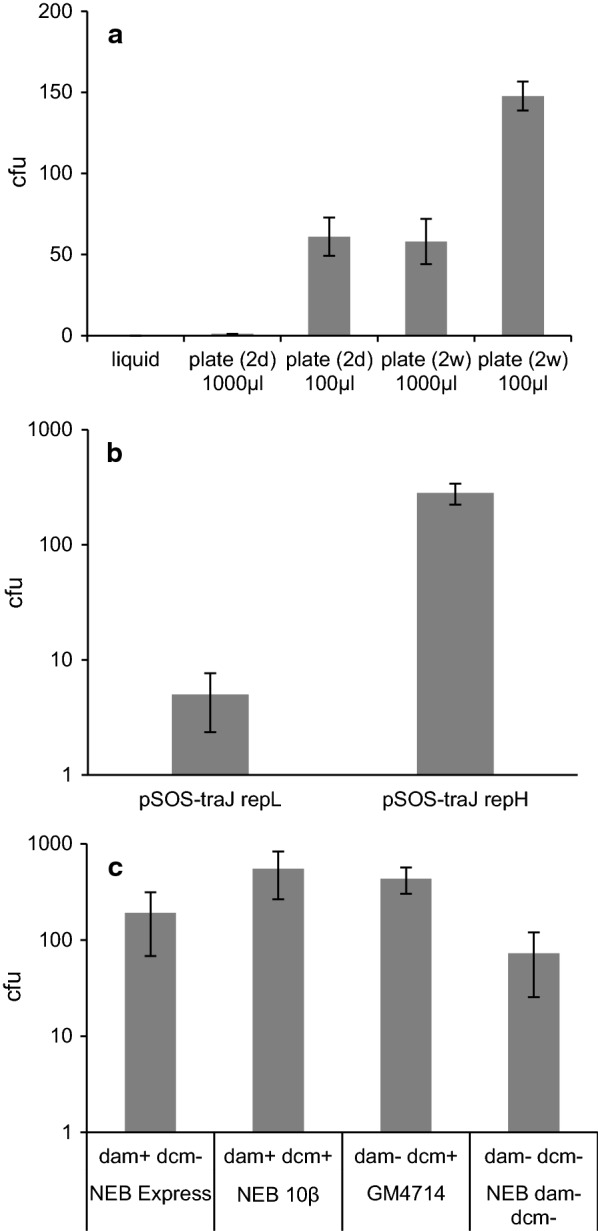
Number of transconjugants per plate when using different mating setups. a The same number of cells was subjected to different mating conditions. Conjugation was performed in liquid medium, applied as 1000 µl or 100 µl onto freshly poured plates (2 days) or as 1000 µl or 100 µl onto 2-week-old well-dried YTF agar plates (2 weeks). b Conjugation was performed using donor plasmids with the Clostridium origin of replication repL and repH. c E. coli strains with different dam and dcm methylation patterns were used as the donor strain containing pSOS-traJ repH. The data represent biological triplicates with standard deviations
To verify the complete transfer of the donor plasmid, 10 transconjugant colonies were picked randomly and the isolated plasmid DNA was reintroduced into E. coli for restriction analysis. Digestion of the plasmids with HindIII revealed the anticipated restriction pattern (unpublished observations).
The impact of different origins of replication
We compared two different origins of replication that are known to function in C. ljungdahlii and other Gram-positive bacteria: repL (replicon pIM13, derived from Bacillus subtilis) and repH (replicon pCB102, derived from C. butyricum). Both origins were used with the same donor plasmid backbone, pSOS-traJ. The repH origin was the most efficient, generating two orders of magnitude more transconjugant C. ljungdahlii colonies than repL (Fig. 2b). This supports the results achieved using the same origins with an electroporation protocol [40] although no firm conclusions can be drawn because the vector backbones and selection procedures were different in this earlier study. It is unclear whether the better performance of the repH origin reflected its impact on mating efficiency (i.e., transfer) or stability (i.e., replication and maintenance). Our experiments indicate the latter is more likely, given the larger number of repH transformants recovered by plasmid rescue and the tendency for repL transformants to undergo a sudden arrest in cell growth after several passages. The instability of repL plasmids in C. ljungdahlii has been reported before [40, 44]. However, the instability of repL plasmids can be of advantage, when limited plasmid stability is desirable.
A typical mating efficiency in our hands was 2.4 × 10−6 transconjugants per recipient cell. This is in the range reported for inter-species transfer especially between Gram-positive and Gram-negative bacteria. For example, Strätz et al. [45] reported a transfer efficiency of ~ 7.3 × 10−6 per recipient when transferring a plasmid from E. coli to Acetobacterium woodii. Efficiencies ranging from 4.7 × 10−5 to less than 3.3 × 10−9 per recipient have been reported for conjugation between E. coli and C. beijerinckii NCIMB 8052 (formerly C. acetobutylicum NCIB 8052) [15]. Although successful conjugation has been reported between E. coli and C. cellulolyticum [46], C. difficile [17], and C. perfringens [16], conjugation has not yet been achieved between E. coli and C. acetobutylicum ATCC 824 [47].
The influence of donor strain dam and dcm methylation on conjugation efficiency
Host DNA is often protected from endogenous restriction endonucleases by modification, which typically involves the methylation of certain bases at the enzyme’s recognition site [18]. The efficiency of C. ljungdahlii transformation by electroporation was reported to be higher when using plasmids isolated from an E. coli B-strain with Dam methylase activity but lacking Dcm methylase activity (dam+dcm−) compared to the same plasmids isolated from a K strain (dam−dcm+) [40]. Furthermore, both Dam and Dcm methylation inhibit transformation and conjugation in C. pasteurianum NRRL B-598 [48], and Dcm methylation reduces the efficiency of transformation by electroporation in C. thermocellum DSM1313 [49]. Dam methylase modifies adenosine residues in the GATC motif, whereas Dcm methylase modifies the second cytidine residue in the CCWGG motif. To investigate the effect of dam and dcm methylation backgrounds on conjugation and colony forming efficiency, E. coli strains with all four possible combinations of dam and dcm methylation were transformed with pSOS-traJ-repH and used as the donor strain for conjugation (Fig. 2c). Although the dam+dcm+ strain generated the most colonies and the dam−dcm− strain generated the fewest, the differences were not extensive and we conclude that (at least under our conditions) Dam and Dcm methylation do not substantially influence the success of conjugation between E. coli and C. ljungdahlii. Our data suggest that C. ljungdahlii has no adverse restriction methylation system or alternatively that conjugation circumvents the restriction system due to its single stranded DNA transfer mechanism and rapid methylation of the newly synthesized double stranded DNA [50]. Nevertheless, we recommend the cloning strain NEB10β (dam+dcm+) not only for its superior performance but also because it has an endA1 and recA genotype, which abolishes nonspecific endonuclease I activity and homologous recombination.
DNA methylation profiling using bisulfite conversion and PacBio sequencing
Another potential drawback of the diverse restriction–modification systems of different bacteria [51] is the unpredictable impact of further host DNA restriction enzymes on heterologous transgenes. We therefore investigated the C. ljungdahlii global DNA methylation profile to identify unique characteristics of this species that might affect conjugation efficiency or transgene expression. Bacterial genomes often contain 5-methylcytidine (m5C), N4-methylcytidine (m4C), and/or N6-methyladenosine (m6A), and various methods are available to map these residues [52]. Putative m5C residues were mapped by sequencing after bisulfite conversion, a deamination reaction that converts unmethylated and therefore unprotected cytidine residues to uridine and thus changes the base-paring properties at that site compared to the reference genome. However, comparison with the reference sequence for C. ljungdahlii DSM 13528 (GenBank CP001666.1) revealed no evidence for the presence of m5C residues. Putative m6A residues were mapped by PacBio sequencing, using a culture harvested in the logarithmic growth phase (Table 1). The most prominent motif was Cm6AGNNNNNGAT (25% of potential sites methylated) together with its reverse complement m6ATCNNNNNCTG (21% of potential sites methylated). Other sites were methylated at lower frequencies: GAATm6AYC (18%), GATAm6AT (13%), and CAAAAm6AR (11%). The data were submitted to REBASE (http://rebase.neb.com) [53].
Table 1.
Results of PacBio sequencing
| Motif and modified position | Modification type | Number of modified motifs detected | Number of motifs in genome | Percentage of modified motifs detected (%) |
|---|---|---|---|---|
| CAGNNNNNGAT | m6A | 581 | 2338 | 24.85 |
| ATCNNNNNCTG | m6A | 494 | 2338 | 21.13 |
| GAATAYC | m6A | 251 | 1375 | 18.25 |
| GATAAT | m6A | 740 | 5769 | 12.83 |
| CAAAAAR | m6A | 504 | 4717 | 10.68 |
Summary of the detected motifs with methylated adenine residues (underlined)
The C. ljungdahlii methylation profile is therefore similar to that of C. autoethanogenum, which features m6A residues in the motifs CAAAAm6AR, GWTAm6AT, and SNNGCAm6AT [54] probably due to its close evolutionary relationship with C. ljungdahlii [55]. However, the above-mentioned motifs showed a methylation frequency of more than 85% in C. autoethanogenum [54]. The low frequency of methylated sites in our sample may reflect the testing of samples in the logarithmic growth phase, which may show limited methylation due to concurrent DNA replication [56]. Interestingly, the Cm6AGNNNNNGAT and m6ATCNNNNNCTG sites that are methylated in C. ljungdahlii are unmethylated in C. autoethanogenum. These motifs match the anticipated target sites for type I restriction–methylation systems, which comprise separate methylation, sequence recognition, and endonuclease domains [57], predicted to be represented by the proteins M.CljII (CLJU_c03310), S.CljII (CLJU_c03320), and CljIIP (CLJU_c03330) in C. ljungdahlii, respectively [53].
Design of a versatile E. coli–C. ljungdahlii shuttle vector for genomic integration
DNA transfer by conjugation must be followed by the stable genomic integration of a transgene cassette in order to maintain the new phenotype in the absence of selection. We therefore developed a new E. coli–C. ljungdahlii shuttle vector for this purpose (Fig. 3a).
Fig. 3.

E. coli-Clostridium shuttle vector for the inducible genomic integration of an acetone biosynthesis gene cluster. a The vector pIM-SLIC contains a multiple cloning site (MCS) and a SwaI restriction site to allow directed and undirected insertion of cassettes. b A cluster comprising the four genes necessary for acetone biosynthesis (adc, thlA, ctfA-ctfB) was inserted by Gibson assembly via the SLIC sites (dark blue triangles) between the ptb promoter (P-ptb, yellow) and the fdx terminator (fdx-T, orange) resulting in pIM-Ace#22. c In C. ljungdahlii acetyl-CoA is derived from syngas (CO, CO2, H2) via the Wood–Ljungdahl pathway. Acetyl-CoA is used as a starting material for acetone biosynthesis by the engineered pathway. Acetone biosynthesis starts with the condensation of two acetyl-CoA molecules to acetoacetyl-CoA and the release of one molecule of CoA by acetyl-CoA acetyltransferase (ThlA). The CoA is then transferred to an acetate molecule by the CoA-transferase (CtfA/CtfB) to yield acetoacetate. Finally, acetone is generated via a decarboxylation reaction catalyzed by acetoacetate decarboxylase (Adc). However, the obtained acetone gets reduced by an endogenous primary secondary alcohol dehydrogenase (psAdh) to the secondary alcohol isopropanol
The shuttle vector includes a pMB1 origin of replication and bla selectable marker for high-copy-number maintenance and selection in E. coli, and a repL origin of replication for maintenance in C. ljungdahlii. Additionally to bla, the catP and ermC selectable markers are included because only they can be used for propagation in both E. coli and C. ljungdahlii (catP is located on the non-integrating vector backbone and ermC is present as part of the transgene cassette and is used for the further selection of integrants). The vector also contains a Himar1 transposase gene controlled by a xylose-inducible promoter–repressor system [33], allowing the induction of transposase activity. The transgene cassette (including ermC) is flanked by ITRs, which act as the substrate for the transposase. The ermC gene is also flanked by flippase recognition target (FRT) sites recognized by FLP recombinase, to allow subsequent marker removal and additional rounds of integration with the same cassette or a different one. An oriT/traJ region allows plasmid transfer by conjugation. SLIC sites are also present to facilitate cloning strategies for the introduction of additional transgenes by Gibson assembly. A variant of the shuttle vector containing repH and a hyperactive Himar1 transposase (H1C9) [58] was generated as well resulting in pIM-SLIC-H1C9-repH. As discussed above, the inclusion of a repH origin of replication instead of repL generated a larger number of transconjugants (Fig. 2b), which may rather reflect the ability of repH to maintain the plasmid as an episome. However, the use of the repL origin is more suitable when rapid vector loss is desirable.
Proof of principle using a Clostridium acetone biosynthesis pathway
The new shuttle vector was tested by introducing a clostridial acetone biosynthesis pathway (Fig. 3b), comprising the genes adc, thlA, and ctfA-ctfB. The C. acetobutylicum thlA and ctfA-ctfB genes were used because this species is a natural producer of acetone [3]. However, the C. acetobutylicum Adc enzyme is inhibited by CO [59, 60], so we used the C. beijerinckii adc gene instead. The three corresponding enzymes catalyze the production of acetone from the intermediate acetyl-CoA (Fig. 3c). The gene cluster was placed under the control of the strong C. acetobutylicum ptb promoter and upstream of the C. pasteurianum DSM525 fdx transcriptional terminator. After verifying the nucleotide sequence of the vector by DNA sequencing, triparental mating was carried out as described above with C. ljungdahlii as the recipient strain. The 12-kb plasmid was successfully transferred to the recipient as shown by comparative restriction analysis after plasmid isolation and reintroduction into E. coli NEB10β (Additional file 1: Figure S1). Even larger constructs of 18, 20, and 25.6 kb were also successfully transferred by conjugation (unpublished observations).
For genomic integration, a single transconjugant containing pIM-Ace#22 was grown repeatedly in YTF medium containing clarithromycin and d-cycloserine for selection and 2% (w/v) xylose to induce transposase activity (Fig. 1). Subsequently, 100 µl of the suspension was spread onto YTF agar plates without xylose or antibiotics, and the resulting colonies were replica plated onto medium containing either clarithromycin or thiamphenicol to identify the integrants. Colonies containing episomal plasmids remain resistant to both antibiotics whereas integrants should be sensitive to thiamphenicol but resistant to clarithromycin. The latter colonies were picked and inoculated into fresh medium to allow the extraction and analysis of genomic DNA. The site of genomic integration was identified by inverse PCR (Additional file 2: Figure S2). Strain Ace#22-24 was investigated in more detail, revealing that the cassette had integrated within a gene encoding a putative membrane protein (CLJU_c32170) at position 3523326. An analytical PCR confirmed the integration at this position and loss of the plasmid backbone (Additional file 3: Figure S3). Whole-genome sequencing was carried out to verify the site of integration, revealing the integration of a single copy of the complete 5-kb cassette comprising the acetone cluster and the ermC selectable marker between two ITRs. Successful genomic integration was also confirmed for an 11.1-kb fragment (unpublished observations) indicating that Himar1 transposase can facilitate the integration of even larger DNA fragments into the C. ljungdahlii genome.
Although Himar1 transposase has mostly been used for the generation of random mutagenesis libraries [31, 61, 62] and also for complementation in a mutant Rickettsia rickettsii strain [63], we have shown for the first time that the same system can be used for the introduction of several genes corresponding to complete metabolic pathways into the C. ljungdahlii genome. The analysis of additional C. ljungdahlii strains possessing Himar1 transposon mediated genomic integrations revealed a random distribution throughout the genome (unpublished observations). The C. ljungdahlii genome is remarkably AT-rich (68.9%) thus offering great variety in terms of the integration site, which allows the exploitation of position effects that could modulate the expression of the integrated transgenes [64]. The xylose-inducible promoter system [33] was useful as a transposase control strategy. Although xylose is metabolized by C. ljungdahlii [5] 2% (w/v) of xylose for induction was sufficient and was not metabolized to completion during cultivation (unpublished observations). Recently, an alternative inducible expression system for C. ljungdahlii was introduced, based on the lac promoter employing lactose, which does not serve as carbon source in C. ljungdahlii [65].
Gene transfer to Clostridium spp. for the purpose of transgene integration has also been achieved using targeted methods, including the ClosTron system based on group II introns [21, 66], allele coupled exchange [67] and the use of counter-selectable markers during screening [68]. The targeting of specific genomic sites allows these methods to be used for gene knockout and knock-in procedures. Although the ClosTron system is a valuable tool to generate deletion mutants, its efficiency declines when introducing heterologous sequences of more than 1 kb [69]. An elaborate but laborious multistep process system has been used to integrate 40 kb of heterologous DNA into the C. acetobutylicum genome [70].
Expression of the acetone cluster genes and product formation
Finally, having confirmed the integration of an intact cassette in strain Ace#22-24, we used targeted proteomics to confirm the presence of the corresponding heterologous enzymes. Accordingly, in addition to the internal control peptides, we also detected the proteotypic peptides for ThlA, CtfA, CtfB, and Adc, with Adc producing the weakest ion counts (Additional file 4: Figure S4).
To verify the activity of the transgene cluster, we measured the amount of acetone and alcohol produced by strain Ace#22-24. Therefore, a serum bottle containing modified ATCC medium was inoculated with the genomic integrant of C. ljungdahlii and gassed for 15 s with artificial syngas (comprising an equimolar mixture of CO, CO2, and H2). After incubation for 2 weeks in a sealed bottle in the syngas atmosphere, the acetone concentration was 0.6 mM, the ethanol concentration was 9 mM, and we also detected 2.4 mM isopropanol (Fig. 4).
Fig. 4.

Chromatogram of fermentative products of C. ljungdahlii wild type and integration strain Ace#22-24. C. ljungdahlii wild type (black) and integration strain Ace#22-24 containing the acetone biosynthesis cluster (red) were grown in modified medium on syngas (33% CO, 33% CO2, 33% H2) for 2 weeks. The supernatant was analyzed by GC–MS
In a previous report, the expression of heterologous acetone biosynthesis genes resulted in the production of 13 mM acetone in C. ljungdahlii using syngas as the carbon source with an inducible expression system [65]. The difference in final product concentration and profile may reflect differences in the detection methods used or a gene dosage effect, given that our strain contained a single copy of the transgene cassette. The ability of C. ljungdahlii to reduce acetone to isopropanol reflects the activity of a secondary alcohol dehydrogenase—such activity has already been detected in C. ragsdahlii but not in C. carboxidivorans [71]. The corresponding C. autoethanogenum and C. ljungdahlii enzymes have been characterized more recently [72]. The production of 0.6 mM acetone and 2.4 mM isopropanol by C. ljungdahlii strain Ace#22-24 in this study suggests that up to 3 mM acetone could be generated if the alcohol dehydrogenase was inactivated, e.g., by gene knockout. However, isopropanol itself is a valuable substance, since it can be used as a drop-in fuel in order to reduce crystallization temperature of biodiesel [73].
A steadily growing toolbox of strategies becomes available to increase product yields of the desired products in the genus Clostridium. A recent study in A. woodii showed the impact of different promoters and different origins of replication (resulting in different plasmid copy numbers) on the heterologous expression of the C. acetobutylicum acetone gene cluster, resulting in the accumulation of up to 15.2 mM acetone in sealed serum bottles [74]. Hence, further improvements in the yield of this or other heterologous metabolic pathways might be achieved by selecting strong native, heterologous or even synthetic promoters [75], screening for mutants with different integration sites or multiple copies of the cassette, knocking out competing pathways using CRISPR/Cas9 and related technologies [76], in addition to standard approaches such as medium and process optimization.
Conclusion
The difficulty of transferring large DNA fragments into the genome of C. ljungdahlii represented a major obstacle, which severely limited the metabolic engineering of this industrial relevant organism. In order to obtain products such as acetone/isopropanol or other bio-chemicals and bio-fuels from syngas, a method for transferring metabolic pathways comprising several genes is required. Furthermore, for the purpose of obtaining a stable industrial fermentation strain, genomic integration of heterologous genes is desirable. We were able to transfer plasmids of up to 25.6 kb by conjugation and integrate clusters of up to 11.1 kb into the genome of C. ljungdahlii. This method shows the clear advantage over other systems based e.g., on homologous recombination, in that it does not require additional cloning of homology arms or preparation of a target site within the genome of the host strain [68, 77]. In this example, we have demonstrated proof of principle for the construction, conjugal transfer and genomic integration of an acetone biosynthesis pathway in C. ljungdahlii. Functionality was confirmed by the formation of acetone and isopropanol from syngas. Our straight-forward strategy for gene delivery and integration provides a basis for the bio-based production of chemicals and fuels using industrial waste streams not only in syngas-fermenting strains but our approach can be easily adapted to other clostridial strains as well.
Materials and methods
Bacterial strains and cultivation
A detailed list of bacterial strains used in this study is provided in Additional file 5: Table S1. E. coli strains NEB10β and NEB Express (New England Biolabs, Ipswich, MA, USA) were used as conjugation donors. NEB10β was also used for general plasmid cloning procedures. E. coli conjugation helper strain Stbl3 was used as part of the GeneArt® Seamless PLUS Cloning and Assembly Kit (Thermo Fisher Scientific, Waltham, MA, USA). This strain contains pRK2013, which provides the mob (mobility) and tra (transfer) gene functions necessary for efficient conjugation [78]. All E. coli strains were cultivated aerobically at 37 °C in LB medium according to Lennox (Carl Roth, Karlsruhe, Germany) on a rotary shaker at 160 rpm. For single-reagent selection, the final antibiotic concentrations were 100 µg/ml ampicillin, 250 µg/ml erythromycin, 34 µg/ml chloramphenicol, and 100 µg/ml kanamycin. For double or triple selection, the antibiotic concentrations were reduced to 50 µg/ml ampicillin, 50 µg/ml erythromycin, and (where applicable) 3.4 µg/ml chloramphenicol. C. ljungdahlii DSM-13528 (Deutsche Sammlung von Mikro-organismen und Zellkulturen GmbH, Leibniz, Germany) was cultivated in a Bactron-IV anaerobic workbench (Shellab, Cornelius, OR, USA) under strict anaerobic conditions (5% H2, 10% CO2, 85% N2; Westfalen AG, Münster, Germany). The cells were cultivated without shaking in yeast tryptone fructose (YTF) medium (16 g/l tryptone, 10 g/l yeast extract, 4 g/l sodium chloride (pH 6.0) supplemented with 5 g/l fructose, 0.75 g/l l-cysteine and optionally 2 mg/l resazurin as a redox indicator). Alternatively, modified Protein Expression Technology Center (PETC) 1754 medium (ATCC, Manassas, VA, USA) was used as a defined medium by replacing sodium bicarbonate with 20 mM Bis–Tris. For cultivation on solid plates, we added 1.5% (w/v) BD Difco agar (Becton–Dickinson, Franklin Lakes, NJ, USA). When pouring the liquid cultures into plates, we added 1.5% (w/v) low-melting-point agarose (Carl Roth). Low-level oxygen contamination was detected on agar plates prepared as above (omitting l-cysteine but including resazurin) by incubating them in an anaerobic atmosphere. Where applicable, antibiotics were used at the following concentrations: 4 µg/ml clarithromycin, 4 µg/ml thiamphenicol, and 200 µg/ml d-cycloserine (the latter to counter-select against E. coli).
Vector construction
Shuttle vector pSOS-MCS is based on pSOS95 (GenBank AY187686.1; [79]). A BamHI-NarI fragment containing the ctfA-ctfB-adc cassette was replaced with the multiple cloning site from pYUBDuet, prepared with the same enzymes. A cluster containing the acetone biosynthesis pathway was designed, encompassing the genes for acetoacetate decarboxylase (adc, CA_P0165, Clostridium beijerinckii), acetyl-CoA acetyltransferase (thlA CA_C2873, C. acetobutylicum), CoA-transferase subunit A (ctfA, CA_P0163, C. acetobutylicum), and CoA-transferase subunit B (ctfB, CA_P0164, C. acetobutylicum). The genes were synthesized by GenScript (Piscataway, NJ, USA) and integrated into pSOS-MCS by Gibson assembly [80] to generate vector pSOS-MCS-acetone. An oriT region including the adjacent traJ gene was integrated in the same manner to generate vector pSOS-MCS-traJ. The origin of replication repL in the vector was replaced with repH [40]. The pSOS-MCS-traJ and repH sequences were amplified by PCR using primer pairs pSOS-traJ del repL for/rev and repH-pSOS-SLIC for/rev, respectively, and the products were joined by isothermal in vitro recombination [80] to produce the final vector pSOS-traJ-repH.
The pIM vector was assembled from four synthetic fragments (GenScript) and an oriT region with the adjacent traJ gene were inserted by Gibson assembly resulting in pIM-traJ. The shuttle vector pIM-SLIC was derived from vector pIM-traJ to support sequence and ligation independent cloning (SLIC) and Gibson assembly [80]. Therefore, a SacII fragment of pET41a containing the multiple cloning sites was prepared and inserted into pIM-traJ, which had been linearized with SacII. Plasmid pIM-SLIC was used to introduce the amino acid exchanges for the hyperactive Himar1 C9 transposase version [58] by Quick Change PCR with primers QC_himar_for1 and QC_himar_rev1. Subsequently, the repH origin of replication was introduced by Gibson assembly using fragments amplified by primer pairs pIM-repH SLIC for1/rev2 for repH and pIM-repH SLIC for2/rev1 for the remaining backbone resulting in pIM-SLIC-H1C9-repH.
The pIM-Ace#22 vector was prepared by amplifying the complete acetone gene cluster in pSOS-MCS-acetone using Herculase II DNA polymerase (Agilent Technologies, Santa Clara, CA, USA) and primers Ace_096 and Ace_087, resulting in a 3.3-kb product. Vector pIM-SLIC was amplified as above using primers Gn_026 and Gn_027. The products were combined by isothermal in vitro recombination at 50 °C. The primers used in this study are listed in Additional file 6: Table S2.
Conjugation (triparental mating)
A pre-culture of C. ljungdahlii inoculated from an exponentially growing culture was cultivated overnight in YTF medium and diluted the following morning to an OD600 of 0.05. The diluted culture was incubated anaerobically at 37 °C and split successively into several tubes depending on the number of planned conjugations (40 ml each) and was grown until the OD600 reached 0.2–0.3.
In the meantime, an inoculation loop full of E. coli helper strain Stbl3 with pRK2013 and donor strain NEB Express or NEB 10β containing the plasmid to be conjugated were transferred from overnight agar plates into separate shake flasks containing 100 ml and 50 ml LB medium, respectively, supplemented with the appropriate antibiotics. The cultures were incubated until the OD600 reached 0.2–0.5, then, for each conjugation 10 ml of the helper strain and 40 ml of the donor strain were harvested by centrifugation (4000g, 10 min, room temperature), washed with fresh LB medium to remove residual antibiotics and centrifuged at 12,000g for 1 min at room temperature. The cells were then washed with anaerobic YTF medium to remove residual oxygen, centrifuged and resuspended in 100–200 µl fresh YTF medium.
The E. coli donor and helper strains were mixed and incubated at 37 °C anaerobically, and meanwhile the C. ljungdahlii cells were recovered by centrifugation (4000g, 15 min, room temperature) in tightly closed 50-ml Corning tubes. The supernatant was carefully discarded and the pellet resuspended in the residual medium and pooled. Aliquots were added to the E. coli mixed culture to initiate triparental mating. The relative proportions of the three strains were equivalent to OD600 values (in total) of ~ 5 for the E. coli helper strain, 2–20 for the E. coli donor strain and 4.5–12 for the recipient C. ljungdahlii strain. The cell suspension was spread onto YTF agar plates without selection and allowed to dry completely at 37 °C for 12–20 h. The cells were resuspended by adding 2–3 ml fresh medium to the agar surface and dislodging the cells with an inoculation loop. The suspension was transferred to Petri dishes and mixed with YTF plus 1.5% (w/v) low-melting-point agarose at ~ 37 °C, which was allowed to solidify at room temperature for 1 h before transfer to 37 °C (Bactron-IV). The first colonies became visible after 3–7 days, depending on the origin of replication and concentration of antibiotics.
Genomic integration
Single colonies were inoculated into YTF medium containing 4 µg/ml clarithromycin, 4 µg/ml thiamphenicol, and 200 µg/ml d-cycloserin. After verification by plasmid rescue and PCR sequencing using primers 16S_fD1 and 16S_rD1 [81], gene cluster integration was induced by transferring the cells to YTF medium as above but lacking thiamphenicol and containing 2% (w/v) xylose. The cells were subcultured at least three times in Hungate tubes. Single colonies were obtained by streaking on YTF agar plates or by spreading 2-µl aliquots into YTF agar plates without clarithromycin, thiamphenicol, and xylose. Plasmid loss was confirmed by replica plating onto agar plates without antibiotics, or supplemented with clarithromycin alone, or supplemented with clarithromycin and thiamphenicol.
Inverse PCR
To confirm integration, genomic DNA was isolated using the Genomic DNA from Tissue Kit (Macherey–Nagel, Düren, Germany) according to the manufacturer’s recommendations for difficult-to-lyse bacteria, with one exception: after treatment with lysozyme (20 mg/ml) in lysis buffer (20 mM Tris/HCl, 2 mM EDTA, 1% Triton X-100, pH 8.0) the cells were resuspended in fresh lysis buffer (without lysozyme) before adding proteinase K. From the isolated genomic DNA 500 ng was digested with different restriction enzymes (1 µl of an appropriate restriction enzyme, e.g., AseI, BsrGI, HindIII, or PstI (New England Biolabs) in 30 µl buffer for 6 h) and 5 µl of the heat-inactivated reaction mixture was used for ligation in a reaction volume of 20 µl containing one unit of T4 DNA ligase (Thermo Fisher Scientific) at 15 °C overnight. The products were amplified from 1 µl of a 1:20 dilution using Herculase II DNA polymerase and primers iPCR_mlsR_for01 and iPCR_mlsR_rev01 for PCR (initial denaturation at 95 °C for 5 min, 40 cycles of denaturation at 95 °C for 30 s, annealing at 53 °C for 30 s, and elongation at 72 °C for 3 min 30 s, then a final elongation step at 72 °C for 5 min before holding at 4 °C). The PCR products were verified by in-house sequencing using an ABI 3730 DNA Analyser (Applied Biosystems, Thermo Fisher Scientific).
Methylation analysis and whole-genome sequencing
The cytosine methylation status of the C. ljungdahlii genome was characterized by bisulfite sequencing [82] using a 10-µg aliquot of genomic DNA obtained from stationary cell cultures. Bisulfite conversion and sequencing was carried out by Zymo Research (Irvine, CA, USA). Methylated adenines were detected by PacBio single-molecule, real-time DNA sequencing [83]. Genomic DNA was prepared from wild-type cultures in the logarithmic growth phase (OD600 = 0.6) and sequenced by Microsynth (Balgach, Switzerland). A SMRTbell™ library was prepared according to the 10-kb template protocol using the DNA Template Kit 2.0, and was sequenced using P4-C2 chemistry on an RSII instrument (all from Pacific Biosciences, Menlo Park, CA, USA). The whole-genome sequencing of integrant Ace#22-24 was carried out by Seq-IT GmbH & Co KG (Kaiserslautern, Germany) using 2 µg of genomic DNA. The library was prepared using the Nextera XT DNA Sample Preparation Kit (Illumina, Inc., San Diego, CA, USA) and was sequenced using a MiSeq Benchtop Sequencer (Illumina) generating paired-end reads of 2 × 250 bp with a coverage of ~ 20–100×.
Protein extraction and LC–MS/MS analysis
Protein samples were prepared as previously described [84] with minor modifications. Briefly, 35 ml of C. ljungdahlii culture grown in modified ATCC 1754 medium with 0.5% (w/v) fructose was harvested by centrifugation (4000g, 15 min, 4 °C) and the pellet was resuspended in 1 ml freshly prepared 50 mM ammonium bicarbonate (pH 7.8). The cells were disrupted by milling with 500 mg 0.1-mm zirconia/silica beads (Carl Roth). After centrifugation (13,000g, 2 min, 4 °C) the soluble protein fraction was transferred into a fresh Eppendorf tube. The protein concentration was determined using the Bradford assay, and an aliquot containing 200 µg total protein was then processed, digested with trypsin and desalted as previously described [84]. The digested samples were analyzed by LC–MS/MS using a Shimadzu UFLC (Shimadzu Corp., Kyoto, Japan) fitted with a Supelco Ascentis® Express Peptide ES-C18 reversed-phase column (50 × 2.1 mm, 2.7 µm particle size; Sigma-Aldrich, Munich, Germany). Fractionated samples were injected into a QTRAP6500 triple-quadrupole mass spectrometer (AB Sciex, Framingham, MA, USA) managed with Analyst software v1.6.2. The following proteotypic peptides were used to confirm the presence of the acetone cluster: APYLANNAR (ThlA), TGLGTLIEK (CtfA), NTTIDEIR (CtfB), and EYLNIIYR (Adc). The following proteotypic peptides were used as C. ljungdahlii internal controls: DFEPVLER (CODH/ACS, CO dehydrogenase/acetyl-CoA synthase complex beta subunit, CLJU_c37550), AIEAGLK (MeTr, methyltetrahydrofolate:corrinoid/iron-sulfur protein, CLJU_c37560), and IVVGYTR (F-THFL, formate-tetrahydrofolate ligase, CLJU_c37650). Peptide FDGTVEVK (glyceraldehyde 3-phosphate dehydrogenase) was used to exclude potential E. coli contamination.
GC–MS analysis
The concentrations of acetone, isopropanol, ethanol, and acetate were determined by GC–MS analysis using a Shimadzu GCMS-QP2010S instrument. Supernatants from bacterial cultures were diluted 1:10 in 100% methanol spiked with 5.5 mM 1,3-propanediol as an internal standard, and were fractionated on an InertCap FFAP capillary column (0.25 mm × 30 m, 0.25 µm film thickness; GL Sciences, Torrance, CA, USA). A 1-µl sample was evaporated at 200 °C, and the following temperature profile was then applied: initial 3 min hold at 50 °C, temperature gradient (35 °C/min) to 220 °C, and final hold at 220 °C for 2 min. The eluate was ionized at 1 kV and the total ion count was monitored. Absolute concentrations of analytes were calculated using corresponding calibration curves.
Additional files
Additional file 1: Figure S1. Restriction digest analysis of re-isolated plasmid pIM-Ace#22. To verify the complete transfer of pIM-Ace#22 into C. ljungdahlii by conjugation the plasmid was isolated and retransformed into E. coli. An agarose gel from pIM Ace#22 and the reisolated plasmid is shown after restriction digest with EcoRV, ScaI and XbaI.
Additional file 2: Figure S2. Inverse PCR on genomic integrant strain Ace#22-24. To identify the site of integration of the acetone biosynthesis pathway including the resistance cassette into the genome of strain Ace#22-24 an inverse PCR was performed after digestion with the indicated restriction endonucleases and re-ligation. The PCR products were separated and visualized in a 1% agarose gel stained with redsafe.
Additional file 3: Figure S3. Analytical PCR for verification of the genomic integrant strain Ace#22-24. Several PCRs were performed on C. ljungdahlii wild type strain (wt), integration strain Ace#22-14 (gInt) and controls (K−/K+). Primer pairs used were binding close to the site of integration, on the catP resistance gene encoded on the plasmid backbone and the ermC resistance gene encoded on the integration cassette between the two ITR sites.
Additional file 4: Figure S4. Chromatograms of proteotypic peptides of selected proteins of the Wood–Ljungdahl pathway and the acetone biosynthesis pathway. Protein raw extract was prepared from C. ljungdahlii wild type and strain Ace#22-24 with integrated acetone biosynthesis cluster and was subjected to tryptic digest for LC–MS/MS analysis. Targeted proteomics was performed analyzing proteotypic peptides of the Wood–Ljungdahl pathway (MeTr, F-THFL and CODH/ACS) as internal controls and the heterologously expressed acetone biosynthesis pathway (ThlA, CtfA, CtfB and Adc) with at least two transitions (y) per peptide.
Additional file 5: Table S1. Organisms used in this study.
Additional file 6: Table S2. Oligonucleotides used in this study.
Authors’ contributions
GP, SdV and SJ designed the experiments. SdV conducted most of the cloning work and GC–MS measurements. GP generated and analyzed the strains, conducted part of the targeted proteomics analysis and experiments related to determination of the methylation pattern. SJ conceived the project. GP drafted and wrote the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We wish to thank Daniel Volke for establishing the QTRAP6500 triple-quadrupole mass spectrometer for targeted proteomics analysis and Jens Lennartz for technical assistance in sample preparation. We are grateful to Elisabeth Raleigh for providing E. coli strain GM4714. We would like to express our gratitude to Richard Twyman, Stefan Gaida and Christopher McElroy for critical reading of the manuscript.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
All data generated or analyzed during this study are included in this published article (and its additional files) or are available from the corresponding author on reasonable request.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
This work was supported by the internal Fraunhofer program MAVO SyngasFerm (Grant Number MAVO825 794).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Tollefson J, Weiss KR. Nations approve historic global climate accord. Nature. 2015;528:315–316. doi: 10.1038/528315a. [DOI] [PubMed] [Google Scholar]
- 2.Tracy BP, Jones SW, Fast AG, Indurthi DC, Papoutsakis ET. Clostridia: the importance of their exceptional substrate and metabolite diversity for biofuel and biorefinery applications. Curr Opin Biotechnol. 2012;23:364–381. doi: 10.1016/j.copbio.2011.10.008. [DOI] [PubMed] [Google Scholar]
- 3.Jones DT, Woods DR. Acetone-butanol fermentation revisited. Microbiol Rev. 1986;50:484–524. doi: 10.1128/mr.50.4.484-524.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liew FM, Köpke M, Simpson SD. Gas fermentation for commercial biofuels production. London: INTECH Open Access Publisher; 2013. [Google Scholar]
- 5.Tanner RS, Miller LM, Yang D. Clostridium ljungdahlii sp. nov., an acetogenic species in clostridial rRNA homology group I. Int J Syst Bacteriol. 1993;43:232–236. doi: 10.1099/00207713-43-2-232. [DOI] [PubMed] [Google Scholar]
- 6.Ljungdahl LG. Total synthesis of acetate from CO2 by heterotrophic bacteria. Annu Rev Microbiol. 1969;23:515–538. doi: 10.1146/annurev.mi.23.100169.002503. [DOI] [PubMed] [Google Scholar]
- 7.Ragsdale SW. The eastern and western branches of the Wood/Ljungdahl pathway: how the east and west were won. BioFactors. 1997;6:3–11. doi: 10.1002/biof.5520060102. [DOI] [PubMed] [Google Scholar]
- 8.Ragsdale SW, Pierce E. Acetogenesis and the Wood–Ljungdahl pathway of CO2 fixation. Biochim Biophys Acta. 2008;1784:1873–1898. doi: 10.1016/j.bbapap.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sheng Y, Mancino V, Birren B. Transformation of Escherichia coli with large DNA molecules by electroporation. Nucleic Acids Res. 1995;23:1990–1996. doi: 10.1093/nar/23.11.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Szostkova M, Horakova D. The effect of plasmid DNA sizes and other factors on electrotransformation of Escherichia coli JM109. Bioelectrochem Bioenergy. 1998;47:319–323. doi: 10.1016/S0302-4598(98)00203-7. [DOI] [Google Scholar]
- 11.Pyne ME, Bruder M, Moo-Young M, Chung DA, Chou CP. Technical guide for genetic advancement of underdeveloped and intractable Clostridium. Biotechnol Adv. 2014;32:623–641. doi: 10.1016/j.biotechadv.2014.04.003. [DOI] [PubMed] [Google Scholar]
- 12.Schroder G, Lanka E. The mating pair formation system of conjugative plasmids-A versatile secretion machinery for transfer of proteins and DNA. Plasmid. 2005;54:1–25. doi: 10.1016/j.plasmid.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 13.Trieu-Cuot P, Arthur M, Courvalin P. Origin, evolution and dissemination of antibiotic resistance genes. Microbiol Sci. 1987;4:263–266. [PubMed] [Google Scholar]
- 14.Waters VL. Conjugation between bacterial and mammalian cells. Nat Genet. 2001;29:375–376. doi: 10.1038/ng779. [DOI] [PubMed] [Google Scholar]
- 15.Williams DR, Young DI, Young M. Conjugative plasmid transfer from Escherichia coli to Clostridium acetobutylicum. J Gen Microbiol. 1990;136:819–826. doi: 10.1099/00221287-136-5-819. [DOI] [PubMed] [Google Scholar]
- 16.Lyras D, Rood JI. Conjugative transfer of RP4-oriT shuttle vectors from Escherichia coli to Clostridium perfringens. Plasmid. 1998;39:160–164. doi: 10.1006/plas.1997.1325. [DOI] [PubMed] [Google Scholar]
- 17.Purdy D, O’Keeffe TA, Elmore M, Herbert M, McLeod A, Bokori-Brown M, Ostrowski A, Minton NP. Conjugative transfer of clostridial shuttle vectors from Escherichia coli to Clostridium difficile through circumvention of the restriction barrier. Mol Microbiol. 2002;46:439–452. doi: 10.1046/j.1365-2958.2002.03134.x. [DOI] [PubMed] [Google Scholar]
- 18.Suzuki H. Host-mimicking strategies in DNA methylation for improved bacterial transformation. London: INTECH Open Access Publisher; 2012. [Google Scholar]
- 19.Bron S, Luxen E. Segregational instability of pUB110-derived recombinant plasmids in Bacillus subtilis. Plasmid. 1985;14:235–244. doi: 10.1016/0147-619X(85)90007-1. [DOI] [PubMed] [Google Scholar]
- 20.Friehs K. Plasmid copy number and plasmid stability. Adv Biochem Eng Biotechnol. 2004;86:47–82. doi: 10.1007/b12440. [DOI] [PubMed] [Google Scholar]
- 21.Heap JT, Kuehne SA, Ehsaan M, Cartman ST, Cooksley CM, Scott JC, Minton NP. The ClosTron: mutagenesis in Clostridium refined and streamlined. J Microbiol Methods. 2010;80:49–55. doi: 10.1016/j.mimet.2009.10.018. [DOI] [PubMed] [Google Scholar]
- 22.Franke AE, Clewell DB. Evidence for a chromosome-borne resistance transposon (Tn916) in Streptococcus faecalis that is capable of “conjugal” transfer in the absence of a conjugative plasmid. J Bacteriol. 1981;145:494–502. doi: 10.1128/jb.145.1.494-502.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin WJ, Johnson EA. Transposon Tn916 mutagenesis in Clostridium botulinum. Appl Environ Microbiol. 1991;57:2946–2950. doi: 10.1128/aem.57.10.2946-2950.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lyristis M, Bryant AE, Sloan J, Awad MM, Nisbet IT, Stevens DL, Rood JI. Identification and molecular analysis of a locus that regulates extracellular toxin production in Clostridium perfringens. Mol Microbiol. 1994;12:761–777. doi: 10.1111/j.1365-2958.1994.tb01063.x. [DOI] [PubMed] [Google Scholar]
- 25.Vidal JE, Chen J, Li J, McClane BA. Use of an EZ-Tn5-based random mutagenesis system to identify a novel toxin regulatory locus in Clostridium perfringens strain 13. PLoS ONE. 2009;4:e6232. doi: 10.1371/journal.pone.0006232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Loeschcke A, Markert A, Wilhelm S, Wirtz A, Rosenau F, Jaeger KE, Drepper T. TREX: a universal tool for the transfer and expression of biosynthetic pathways in bacteria. ACS Synth Biol. 2013;2:22–33. doi: 10.1021/sb3000657. [DOI] [PubMed] [Google Scholar]
- 27.Le Breton Y, Mohapatra NP, Haldenwang WG. In vivo random mutagenesis of Bacillus subtilis by use of TnYLB-1, a mariner-based transposon. Appl Environ Microbiol. 2006;72:327–333. doi: 10.1128/AEM.72.1.327-333.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang X, Wang T, Zhou W, Jia X, Wang H. Use of a Tn5-based transposon system to create a cost-effective Zymomonas mobilis for ethanol production from lignocelluloses. Microb Cell Fact. 2013;12:41. doi: 10.1186/1475-2859-12-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lampe DJ, Churchill ME, Robertson HM. A purified mariner transposase is sufficient to mediate transposition in vitro. EMBO J. 1996;15:5470–5479. doi: 10.1002/j.1460-2075.1996.tb00930.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fu J, Wenzel SC, Perlova O, Wang J, Gross F, Tang Z, Yin Y, Stewart AF, Muller R, Zhang Y. Efficient transfer of two large secondary metabolite pathway gene clusters into heterologous hosts by transposition. Nucleic Acids Res. 2008;36:e113. doi: 10.1093/nar/gkn499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cartman ST, Minton NP. A mariner-based transposon system for in vivo random mutagenesis of Clostridium difficile. Appl Environ Microbiol. 2010;76:1103–1109. doi: 10.1128/AEM.02525-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Choi KH, Kim KJ. Applications of transposon-based gene delivery system in bacteria. J Microbiol Biotechnol. 2009;19:217–228. doi: 10.4014/jmb.0811.669. [DOI] [PubMed] [Google Scholar]
- 33.Sizemore C, Buchner E, Rygus T, Witke C, Gotz F, Hillen W. Organization, promoter analysis and transcriptional regulation of the Staphylococcus xylosus xylose utilization operon. Mol Gen Genet. 1991;227:377–384. doi: 10.1007/BF00273926. [DOI] [PubMed] [Google Scholar]
- 34.Girbal L, Mortier-Barriere I, Raynaud F, Rouanet C, Croux C, Soucaille P. Development of a sensitive gene expression reporter system and an inducible promoter-repressor system for Clostridium acetobutylicum. Appl Environ Microbiol. 2003;69:4985–4988. doi: 10.1128/AEM.69.8.4985-4988.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Köpke M, Held C, Hujer S, Liesegang H, Wiezer A, Wollherr A, Ehrenreich A, Liebl W, Gottschalk G, Durre P. Clostridium ljungdahlii represents a microbial production platform based on syngas. Proc Natl Acad Sci USA. 2010;107:13087–13092. doi: 10.1073/pnas.1004716107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fisher AK, Freedman BG, Bevan DR, Senger RS. A review of metabolic and enzymatic engineering strategies for designing and optimizing performance of microbial cell factories. Comput Struct Biotechnol J. 2014;11:91–99. doi: 10.1016/j.csbj.2014.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Heap JT, Pennington OJ, Cartman ST, Minton NP. A modular system for Clostridium shuttle plasmids. J Microbiol Methods. 2009;78:79–85. doi: 10.1016/j.mimet.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 38.Pansegrau W, Lanka E, Barth PT, Figurski DH, Guiney DG, Haas D, Helinski DR, Schwab H, Stanisich VA, Thomas CM. Complete nucleotide sequence of Birmingham IncP alpha plasmids. Compilation and comparative analysis. J Mol Biol. 1994;239:623–663. doi: 10.1006/jmbi.1994.1404. [DOI] [PubMed] [Google Scholar]
- 39.Mermelstein LD, Welker NE, Bennett GN, Papoutsakis ET. Expression of cloned homologous fermentative genes in Clostridium acetobutylicum ATCC 824. Biotechnology. 1992;10:190–195. doi: 10.1038/nbt0292-190. [DOI] [PubMed] [Google Scholar]
- 40.Leang C, Ueki T, Nevin KP, Lovley DR. A genetic system for Clostridium ljungdahlii: a chassis for autotrophic production of biocommodities and a model homoacetogen. Appl Environ Microbiol. 2013;79:1102–1109. doi: 10.1128/AEM.02891-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Friedman AM, Long SR, Brown SE, Buikema WJ, Ausubel FM. Construction of a broad host range cosmid cloning vector and its use in the genetic analysis of Rhizobium mutants. Gene. 1982;18:289–296. doi: 10.1016/0378-1119(82)90167-6. [DOI] [PubMed] [Google Scholar]
- 42.Willetts N. Conjugation. Methods Microbiol. 1988;21:49–77. doi: 10.1016/S0580-9517(08)70070-9. [DOI] [Google Scholar]
- 43.Lampkowska J, Feld L, Monaghan A, Toomey N, Schjorring S, Jacobsen B, van der Voet H, Andersen SR, Bolton D, Aarts H, et al. A standardized conjugation protocol to asses antibiotic resistance transfer between lactococcal species. Int J Food Microbiol. 2008;127:172–175. doi: 10.1016/j.ijfoodmicro.2008.06.017. [DOI] [PubMed] [Google Scholar]
- 44.Held CM. Funktionelle Genomanalyse von Clostridium ljungdahlii. Doctoral dissertation, TU München. 2013.
- 45.Stratz M, Sauer U, Kuhn A, Durre P. Plasmid transfer into the homoacetogen Acetobacterium woodii by electroporation and conjugation. Appl Environ Microbiol. 1994;60:1033–1037. doi: 10.1128/aem.60.3.1033-1037.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jennert KC, Tardif C, Young DI, Young M. Gene transfer to Clostridium cellulolyticum ATCC 35319. Microbiology. 2000;146:3071–3080. doi: 10.1099/00221287-146-12-3071. [DOI] [PubMed] [Google Scholar]
- 47.Cartman ST, Heap JT, Kuehne SA, Cooksley CM, Ehsaan M, Winzer K, Minton NP. Clostridial gene tools. In: Dürre P, editor. Systems biology of Clostridium. Imperial College Press; 2014. p. 21–55.
- 48.Kolek J, Sedlar K, Provaznik I, Patakova P. Dam and Dcm methylations prevent gene transfer into Clostridium pasteurianum NRRL B-598: development of methods for electrotransformation, conjugation, and sonoporation. Biotechnol Biofuels. 2016;9:14. doi: 10.1186/s13068-016-0436-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guss AM, Olson DG, Caiazza NC, Lynd LR. Dcm methylation is detrimental to plasmid transformation in Clostridium thermocellum. Biotechnol Biofuels. 2012;5:30. doi: 10.1186/1754-6834-5-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stein DC, Gregoire S, Piekarowicz A. Restriction of plasmid DNA during transformation but not conjugation in Neisseria gonorrhoeae. Infect Immun. 1988;56:112–116. doi: 10.1128/iai.56.1.112-116.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Murray IA, Clark TA, Morgan RD, Boitano M, Anton BP, Luong K, Fomenkov A, Turner SW, Korlach J, Roberts RJ. The methylomes of six bacteria. Nucleic Acids Res. 2012;40:11450–11462. doi: 10.1093/nar/gks891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shanmuganathan R, Basheer NB, Amirthalingam L, Muthukumar H, Kaliaperumal R, Shanmugam K. Conventional and nanotechniques for DNA methylation profiling. J Mol Diagn. 2013;15:17–26. doi: 10.1016/j.jmoldx.2012.06.007. [DOI] [PubMed] [Google Scholar]
- 53.Roberts RJ, Vincze T, Posfai J, Macelis D. REBASE—a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 2015;43:D298–D299. doi: 10.1093/nar/gku1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Utturkar SM, Klingeman DM, Bruno-Barcena JM, Chinn MS, Grunden AM, Köpke M, Brown SD. Sequence data for Clostridium autoethanogenum using three generations of sequencing technologies. Sci Data. 2015;2:150014. doi: 10.1038/sdata.2015.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stackebrandt E, Kramer I, Swiderski J, Hippe H. Phylogenetic basis for a taxonomic dissection of the genus Clostridium. FEMS Immunol Med Microbiol. 1999;24:253–258. doi: 10.1111/j.1574-695X.1999.tb01291.x. [DOI] [PubMed] [Google Scholar]
- 56.Kozdon JB, Melfi MD, Luong K, Clark TA, Boitano M, Wang S, Zhou B, Gonzalez D, Collier J, Turner SW, et al. Global methylation state at base-pair resolution of the Caulobacter genome throughout the cell cycle. Proc Natl Acad Sci USA. 2013;110:E4658–E4667. doi: 10.1073/pnas.1319315110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Loenen WA, Dryden DT, Raleigh EA, Wilson GG. Type I restriction enzymes and their relatives. Nucleic Acids Res. 2014;42:20–44. doi: 10.1093/nar/gkt847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lampe DJ, Akerley BJ, Rubin EJ, Mekalanos JJ, Robertson HM. Hyperactive transposase mutants of the Himar1 mariner transposon. Proc Natl Acad Sci USA. 1999;96:11428–11433. doi: 10.1073/pnas.96.20.11428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hüsemann MH, Papoutsakis ET. Enzymes limiting butanol and acetone formation in continuous and batch cultures of Clostridium acetobutylicum. Appl Microbiol Biotechnol. 1989;31:435–444. doi: 10.1007/BF00270772. [DOI] [Google Scholar]
- 60.Hüsemann MH, Papoutsakis ET. Comparison between in vivo and in vitro enzyme activities in continuous and batch fermentations of Clostridium acetobutylicum. Appl Microbiol Biotechnol. 1989;30:585–595. doi: 10.1007/BF00255364. [DOI] [Google Scholar]
- 61.Liu H, Bouillaut L, Sonenshein AL, Melville SB. Use of a mariner-based transposon mutagenesis system to isolate Clostridium perfringens mutants deficient in gliding motility. J Bacteriol. 2013;195:629–636. doi: 10.1128/JB.01288-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang Y, Grosse-Honebrink A, Minton NP. A universal mariner transposon system for forward genetic studies in the genus Clostridium. PLoS ONE. 2015;10:e0122411. doi: 10.1371/journal.pone.0122411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Clark TR, Ellison DW, Kleba B, Hackstadt T. Complementation of Rickettsia rickettsii RelA/SpoT restores a nonlytic plaque phenotype. Infect Immun. 2011;79:1631–1637. doi: 10.1128/IAI.00048-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bryant JA, Sellars LE, Busby SJ, Lee DJ. Chromosome position effects on gene expression in Escherichia coli K-12. Nucleic Acids Res. 2014;42:11383–11392. doi: 10.1093/nar/gku828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Banerjee A, Leang C, Ueki T, Nevin KP, Lovley DR. Lactose-inducible system for metabolic engineering of Clostridium ljungdahlii. Appl Environ Microbiol. 2014;80:2410–2416. doi: 10.1128/AEM.03666-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kuehne SA, Minton NP. ClosTron-mediated engineering of Clostridium. Bioengineered. 2012;3:247–254. doi: 10.4161/bioe.21004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ng YK, Ehsaan M, Philip S, Collery MM, Janoir C, Collignon A, Cartman ST, Minton NP. Expanding the repertoire of gene tools for precise manipulation of the Clostridium difficile genome: allelic exchange using pyrE alleles. PLoS ONE. 2013;8:e56051. doi: 10.1371/journal.pone.0056051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Al-Hinai MA, Fast AG, Papoutsakis ET. Novel system for efficient isolation of Clostridium double-crossover allelic exchange mutants enabling markerless chromosomal gene deletions and DNA integration. Appl Environ Microbiol. 2012;78:8112–8121. doi: 10.1128/AEM.02214-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Plante I, Cousineau B. Restriction for gene insertion within the Lactococcus lactis Ll.LtrB group II intron. RNA. 2006;12:1980–1992. doi: 10.1261/rna.193306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Heap JT, Ehsaan M, Cooksley CM, Ng YK, Cartman ST, Winzer K, Minton NP. Integration of DNA into bacterial chromosomes from plasmids without a counter-selection marker. Nucleic Acids Res. 2012;40:e59. doi: 10.1093/nar/gkr1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ramachandriya KD, Wilkins MR, Delorme MJ, Zhu X, Kundiyana DK, Atiyeh HK, Huhnke RL. Reduction of acetone to isopropanol using producer gas fermenting microbes. Biotechnol Bioeng. 2011;108:2330–2338. doi: 10.1002/bit.23203. [DOI] [PubMed] [Google Scholar]
- 72.Köpke M, Gerth ML, Maddock DJ, Mueller AP, Liew F, Simpson SD, Patrick WM. Reconstruction of an acetogenic 2,3-butanediol pathway involving a novel NADPH-dependent primary–secondary alcohol dehydrogenase. Appl Environ Microbiol. 2014;80:3394–3403. doi: 10.1128/AEM.00301-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lee I, Johnson LA, Hammond EG. Use of branched-chain esters to reduce the crystallization temperature of biodiesel. J Am Oil Chem Soc. 1995;72:1155–1160. doi: 10.1007/BF02540982. [DOI] [Google Scholar]
- 74.Hoffmeister S, Gerdom M, Bengelsdorf FR, Linder S, Fluchter S, Ozturk H, Blumke W, May A, Fischer RJ, Bahl H, Durre P. Acetone production with metabolically engineered strains of Acetobacterium woodii. Metab Eng. 2016;36:37–47. doi: 10.1016/j.ymben.2016.03.001. [DOI] [PubMed] [Google Scholar]
- 75.Yang G, Jia D, Jin L, Jiang Y, Wang Y, Jiang W, Gu Y. Rapid generation of universal synthetic promoters for controlled gene expression in both gas-fermenting and saccharolytic Clostridium species. ACS Synth Biol. 2017;6:1672–1678. doi: 10.1021/acssynbio.7b00155. [DOI] [PubMed] [Google Scholar]
- 76.Huang H, Chai C, Li N, Rowe P, Minton NP, Yang S, Jiang W, Gu Y. CRISPR/Cas9-based efficient genome editing in Clostridium ljungdahlii, an autotrophic gas-fermenting bacterium. ACS Synth Biol. 2016;5:1355–1361. doi: 10.1021/acssynbio.6b00044. [DOI] [PubMed] [Google Scholar]
- 77.Huang H, Chai C, Yang S, Jiang W, Gu Y. Phage serine integrase-mediated genome engineering for efficient expression of chemical biosynthetic pathway in gas-fermenting Clostridium ljungdahlii. Metab Eng. 2019;52:293–302. doi: 10.1016/j.ymben.2019.01.005. [DOI] [PubMed] [Google Scholar]
- 78.Figurski DH, Helinski DR. Replication of an origin-containing derivative of plasmid RK2 dependent on a plasmid function provided in trans. Proc Natl Acad Sci USA. 1979;76:1648–1652. doi: 10.1073/pnas.76.4.1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Desai RP, Papoutsakis ET. Antisense RNA strategies for metabolic engineering of Clostridium acetobutylicum. Appl Environ Microbiol. 1999;65:936–945. doi: 10.1128/aem.65.3.936-945.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, 3rd, Smith HO. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009;6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 81.Weisburg WG, Barns SM, Pelletier DA, Lane DJ. 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol. 1991;173:697–703. doi: 10.1128/jb.173.2.697-703.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Frommer M, McDonald LE, Millar DS, Collis CM, Watt F, Grigg GW, Molloy PL, Paul CL. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc Natl Acad Sci USA. 1992;89:1827–1831. doi: 10.1073/pnas.89.5.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Flusberg BA, Webster DR, Lee JH, Travers KJ, Olivares EC, Clark TA, Korlach J, Turner SW. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat Methods. 2010;7:461–465. doi: 10.1038/nmeth.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gaida SM, Liedtke A, Jentges AH, Engels B, Jennewein S. Metabolic engineering of Clostridium cellulolyticum for the production of n-butanol from crystalline cellulose. Microb Cell Fact. 2016;15:6. doi: 10.1186/s12934-015-0406-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Figure S1. Restriction digest analysis of re-isolated plasmid pIM-Ace#22. To verify the complete transfer of pIM-Ace#22 into C. ljungdahlii by conjugation the plasmid was isolated and retransformed into E. coli. An agarose gel from pIM Ace#22 and the reisolated plasmid is shown after restriction digest with EcoRV, ScaI and XbaI.
Additional file 2: Figure S2. Inverse PCR on genomic integrant strain Ace#22-24. To identify the site of integration of the acetone biosynthesis pathway including the resistance cassette into the genome of strain Ace#22-24 an inverse PCR was performed after digestion with the indicated restriction endonucleases and re-ligation. The PCR products were separated and visualized in a 1% agarose gel stained with redsafe.
Additional file 3: Figure S3. Analytical PCR for verification of the genomic integrant strain Ace#22-24. Several PCRs were performed on C. ljungdahlii wild type strain (wt), integration strain Ace#22-14 (gInt) and controls (K−/K+). Primer pairs used were binding close to the site of integration, on the catP resistance gene encoded on the plasmid backbone and the ermC resistance gene encoded on the integration cassette between the two ITR sites.
Additional file 4: Figure S4. Chromatograms of proteotypic peptides of selected proteins of the Wood–Ljungdahl pathway and the acetone biosynthesis pathway. Protein raw extract was prepared from C. ljungdahlii wild type and strain Ace#22-24 with integrated acetone biosynthesis cluster and was subjected to tryptic digest for LC–MS/MS analysis. Targeted proteomics was performed analyzing proteotypic peptides of the Wood–Ljungdahl pathway (MeTr, F-THFL and CODH/ACS) as internal controls and the heterologously expressed acetone biosynthesis pathway (ThlA, CtfA, CtfB and Adc) with at least two transitions (y) per peptide.
Additional file 5: Table S1. Organisms used in this study.
Additional file 6: Table S2. Oligonucleotides used in this study.
Data Availability Statement
All data generated or analyzed during this study are included in this published article (and its additional files) or are available from the corresponding author on reasonable request.