

Abstract
Activation of several ADP-ribosylation factors (ARFs) by guanine nucleotide exchange factors (GEFs) regulates recruitment of coat proteins (COPs) on the Golgi complex and is generally assumed to be the target of brefeldin A (BFA). The large ARF-GEFs Golgi-specific BFA resistance factor 1 (GBF1) and BFA-inhibited GEFs (BIGs) localize to this organelle but catalyze exchange preferentially on class II and class I ARFs, respectively. We now demonstrate using quantitative confocal microscopy that these GEFs show a very limited overlap with each other (15 and 23%). In contrast, GBF1 colocalizes with the cis-marker p115 (86%), whereas BIGs overlap extensively with TGN38 (83%). Consistent with these distributions, GBF1, but not BIG1, partially relocalized to peripheral sites after incubation at 15°C. The new GBF1 structures represent peripheral vesicular tubular clusters (VTCs) because 88% of structures analyzed stained for both GBF1 and p115. Furthermore, as expected of VTCs, they rapidly reclustered to the Golgi complex in a microtubule-dependent manner upon warm-up. These observations suggest that GBF1 and BIGs activate distinct subclasses of ARFs in specific locations to regulate different types of reactions. In agreement with this possibility, COPI overlapped to a greater extent with GBF1 (64%) than BIG1 (31%), whereas clathrin showed limited overlap with BIG1, and virtually none with GBF1.
INTRODUCTION
Proper targeting and delivery of both proteins and lipids to the various organelles of eukaryotic cells is ensured by a complex and diverse transport system. Traffic within this system is extremely dynamic and is thought to involve several types of transport carriers generated through the action of the coat protein (COP) I, COPII, and clathrin (Pelham and Rothman, 2000). Cargo initially translocated into the endoplasmic reticulum (ER) is selected into COPII-coated structures for transport from specialized ER regions to form vesicular tubular clusters (VTCs) (Antonny and Schekman, 2001). Preformed COPI complexes are then recruited from the cytosol to nascent VTCs to promote the retrograde movement of escaped ER proteins (Presley et al., 1997; Scales et al., 1997; Stephens et al., 2000). COPI function has also been implicated in movement of VTCs to the Golgi complex (Pepperkok et al., 1993) and transport of cargo across the Golgi stack (Pelham and Rothman, 2000). In agreement with such diverse functions, COPI localizes to several compartments of the secretory pathway, with greatest abundance in VTCs and cis-elements of the Golgi stack (Oprins et al., 1993; Griffiths et al., 1995). In contrast, the distribution of clathrin within the Golgi complex appears more restricted. High- voltage electron microscopy (EM) tomography studies established that the trans-most cisterna produces uniquely and exclusively clathrin-coated buds for traffic to the lysosomal system (Ladinsky et al., 1999). Transport of cargo to the plasma membrane has been proposed to occur from the penultimate cisterna and may involve a yet to be identified lace-like coat first revealed by morphological studies (Ladinsky et al., 1994).
Recruitment of coat proteins on Golgi membranes is regulated by members of the ADP-ribosylation factors (ARFs) family of small GTPases. Sequence comparison of the six mammalian ARFs delineates three classes (Chavrier and Goud, 1999): class I (ARFs 1, 2, and 3), class II (ARFs 4 and 5), and class III (ARF6). With the exception of ARF6 that functions at the plasma membrane (D'Souza-Schorey et al., 1995; Peters et al., 1995), ARFs localize to the Golgi complex. ARFs cycle between inactive cytosolic GDP-bound form and active membrane-associated GTP-bound form. Once activated on the membrane, the most extensively characterized ARF1 was shown to regulate the recruitment of COPI (Donaldson et al., 1992), the clathrin adaptor proteins (AP)-1 and AP-3, as well as the newly identified Golgi-localized, gamma-ear–containing, ARF- binding proteins (GGAs) (Robinson and Bonifacino, 2001). Activated ARF-GTP also stimulates phopholipase D and phosphoinositide 4-kinase activities, leading to membrane remodeling (Brown et al., 1993; Godi et al., 1999).
ARF activation is stimulated by specific guanine nucleotide exchange factors (GEFs) that catalyze the replacement of GDP with GTP. A large number of ARF-GEFs have now been characterized, among which some can be inhibited by brefeldin A (BFA). BFA is a fungal metabolite that reversibly blocks protein secretion and causes a dramatic disappearance of the Golgi complex in many cells (Klausner et al., 1992). The reported mammalian ARF-GEFs can be divided into four families on the basis of size and sequence similarity (Donaldson and Jackson, 2000). Members of the ARNO/cytohesin and EFA6 families all have low molecular weights and contain pleckstrin homology (PH) domains. In contrast, the Gea/GBF/GNOM and Sec7/BIG families contain large GEFs that lack PH domains. A recently reported novel ARF-GEF of intermediate size may define a fifth family (Someya et al., 2001). All ARF-GEFs identified to date possess a central sec7 domain, a module of ∼200 amino acids that is sufficient to catalyze exchange of GDP for GTP in vitro and is the direct target of BFA. Binding of BFA to sensitive GEFs traps the substrate in an abortive ARF-GDP-Sec7 domain-BFA complex (Mansour et al., 1999; Peyroche et al., 1999).
With the exception of EFA6, all mammalian ARF-GEFs are peripheral membrane proteins readily recovered in the high- speed supernatant of cellular homogenates (Frank et al., 1998; Claude et al., 1999; Yamaji et al., 2000; Someya et al., 2001). BFA-inhibited GEF (BIG)1, BIG2, and Golgi-specific BFA resistance factor 1 (GBF1), the three large mammalian GEFs, all localize to the Golgi complex in intact cells, but differ in ARF substrate specificity and BFA sensitivity. BIG1/BIG2 were copurified from bovine brain cytosol as a >670-kDa macromolecular complex based on their BFA- inhibited GEF activity (Morinaga et al., 1996). They show highest sequence similarity to each other and yeast Sec7p. BIG1/BIG2 form a heterodimer (Yamaji et al., 2000) that acts preferentially on class I ARFs (Morinaga et al., 1996). In contrast, GBF1, first identified in our laboratory, is BFA resistant and closely related to the yeast proteins Gea1p/Gea2p. Importantly, GBF1 exhibits GEF activity selectively toward the class II ARF5 (Claude et al., 1999). GBF1 likely exists as a homodimer because it contains a dimerization motif first identified in its plant homolog GNOM (Grebe et al., 2000).
We now report that GBF1 and BIGs can be further distinguished by their predominant localization to cis- and trans-elements of the Golgi, respectively. These observations suggest that ARF-GEFs may participate in coat selection. Furthermore, our observation that BIGs, the only identified BFA-sensitive mammalian ARF- GEFs, localize to the trans-Golgi challenges current thinking that inhibition of an ARF-GEF by BFA is the direct cause for COPI dissociation from membranes of VTCs and the cis-Golgi.
MATERIALS AND METHODS
Tissue Culture and Reagents
Media and culture reagents were purchased
from Invitrogen
(Carlsbad, CA). Disposable plasticware and culture
six-well plates were purchased from Falcon Plastics
(Oxnard, CA). Normal rat kidney (NRK) cells were obtained
from Dr. Kathryn Howell (University of Colorado Health
Science Center, Denver, CO). BHK-21 cells were obtained
from Dr. Thomas Hobman (University of Alberta, Edmonton,
AB, Canada). Monolayers of NRK and BHK-21 cells were
maintained in DMEM supplemented with 10% fetal bovine
serum (Sigma, St. Louis, MO), 100 μg/ml penicillin G, 100
μg/ml streptomycin, and 2 mM glutamine. For incubations
at 15°C, cell monolayers were transferred to DMEM lacking
HCO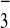 that was supplemented with 20 mM
HEPES pH 7.4. BFA and nocodazole were purchased from Sigma,
dissolved in dimethyl sulfoxide (DMSO), and stored at −
20°C as stock solutions of 10 and 5 mg/ml, respectively.
that was supplemented with 20 mM
HEPES pH 7.4. BFA and nocodazole were purchased from Sigma,
dissolved in dimethyl sulfoxide (DMSO), and stored at −
20°C as stock solutions of 10 and 5 mg/ml, respectively.
Antibodies
Antibodies against BIG1 were raised in rabbits according to standard procedures (Harlow and Lane, 1988) by using a hexa-histidine-tagged form of the sec7 domain of human BIG1 (termed M1) encompassing residues 560–890. The recombinant protein was expressed and purified as described (Mansour et al., 1999). Antibodies to BIG1 were affinity purified against strips of nitrocellulose onto which the recombinant M1 had been transferred after SDS- PAGE. Bound antibody was recovered by acid elution as described (Harlow and Lane, 1988) and used at 1:50 dilution for immunofluorescence (IF) study. Alexa488-labeled anti- BIG1 antibodies were prepared according to manufacturer's instructions by using Alexa Fluor 488 protein labeling kit (A-10235; Molecular Probes, Eugene, OR) and used at 1:10 dilution for IF. IgG fractions purified from crude serum by chromatography on protein A-Sepharose after ammonium sulfate precipitation were used for this purpose. The specificity of this antibody was confirmed by immunoblot and IF. Rabbit polyclonal antibody against GBF1 (H154, 1:500 dilution) has been described previously (Claude et al., 1999). For IF, the following monoclonal antibodies were used: anti- hemagglutinin (HA) (clone 3F10; Roche Molecular Biochemicals, Indianapolis, IN) at 1:50; anti-p115 (clone 3A10; Waters et al., 1992; a kind gift from Dr. G. Waters, Princeton University, Princeton, NJ) at 1:1000; anti-p58 (clone 7DB2; a kind gift from Dr. L. Hendricks, Centocor, Philadelphia, PA) at 1:150; anti- mannosidase II (Man II) (clone 53FC3; Burke et al., 1982; a kind gift from Dr. B. Burke, University of Calgary, Calgary, AB, Canada) at 1:50; anti-α-tubulin (clone no. B-5-1-2; Sigma) at 1:4000; anti-β-COP (clone M3A5; Allan and Kreis, 1986; a kind gift from Dr. T. Kreis, University of Geneva, Geneva, Switzerland) at 1:300; and anti-clathrin (clone X22; a kind gift from Dr. S. Sorkin, University of Colorado Health Science Center) at 1:200. The following rabbit polyclonal antibodies were used: anti- TGN38 (a kind gift from Dr. J. Barasch, Columbia University, New York, NY) at 1:2000; anti-p58 (Molly 6; Saraste et al., 1987; a kind gift from Dr. J. Saraste, University of Bergen, Bergen, Norway) at 1:100; and anti-Man II (a kind gift from Dr. K. Moremen, University of Georgia, Athens, GA) at 1:2000. The following fluorescent secondary antibodies were used: fluorescein isothiocyanate-conjugated goat anti-rat antibody (Jackson Immunoresearch Laboratories) at 1:100; and Alexa594- conjugated goat anti-rabbit and Alexa488-conjugated goat anti-mouse antibodies (Molecular Probes) at 1:600.
Construction of HA-tagged BIG2 N-Terminus and Transient Expression
A truncated cDNA encoding a tagged-form of the N terminus of BIG2 was generated by polymerase chain reaction (PCR) from a yeast two-hybrid human pancreatic library (CLONTECH, Palo Alto, CA) by using the Expand high fidelity PCR kit (Roche Molecular Biochemicals) and primers F1 5′- CGAGATCTTCTAGACAGGAGAGCCAGACCAAGAGCATGTTCG-3′ and R1A 5′-CTTCCTCGAGTCATGTCATTCCCAGCTCATGTCCACTTCTTCC-3′. This fragment, which encodes residues 2–552 of hBIG2, was directionally cloned into a pCEP4 derivative, pMCL, that yields fusion proteins with an HA tag (MAYPYDVPDYASGT; underlined residues) at the N terminus (Mansour et al., 1999). Briefly, the insert in pMCL-HA-N2 was excised with NheI and XhoI and replaced with the PCR fragment by using the XbaI and XhoI sites engineered into F1 and R1A, respectively. Several clones from two distinct PCR reactions were sequenced on both strands to ensure construct fidelity. This analysis revealed a two-nucleotide difference (619GA instead of 619AG) from the sequence of BIG2 published by Togawa et al. (1999), leading to a change from Arg to Glu at position 207. This corrected sequence matches the published human genomic sequence on the National Center for Biotechnology Information site (accession no. NT_011361.3). For transient expression from this and other plasmids, BHK-21 cells grown on coverslips were transfected with designated plasmids by using FuGENE 6 transfection reagent according to manufacturer's instructions (Roche Molecular Biochemicals) and cultured for 20–24 h before fixation.
Immunofluorescence
Cells grown on glass coverslips were washed once in phosphate-buffered saline and fixed with either methanol (−20°C, 6 min) or 3% paraformaldehyde in phosphate-buffered saline (room temperature, 20 min). Double labeling with mouse and rabbit antibodies was processed as described previously (Mansour et al., 1999). For double labeling with two rabbit primary antibodies, cells were first decorated with the GBF1 or TGN38 antisera for 90 min, followed by Alexa594- labeled anti-rabbit IgG for 60 min. Cells were then incubated a second time for 60 min with GBF1 or TGN38 antisera, before final labeling for 60 min with Alexa488- labeled BIG1 antiserum (9D3). This precaution minimized potential labeling of the 9D3 antibodies with Alexa594 anti- rabbit IgG remaining in the sample. Several control experiments confirmed the lack of cross-reaction. For Figures 1C and 7, mounted coverslips were viewed with an Axioskop microscope (Carl Zeiss, Thornwood, NY) equipped with a Spot 1.1 digital camera (Diagnostic Products, Los Angeles, CA). To avoid artifacts due to shifts between measurements, merged images were acquired using a dichroic filter that simultaneously captures the signal from both the red and green fluorophore.
Figure 1.

Endogenous BIG1 localizes to the Golgi complex through sequences present in the N-terminal third of the protein. (A) Both crude and affinity-purified 9D3 serum (anti-BIG1) label a protein of 208 kDa, the size predicted from the cDNA of hBIG1. NRK cell lysates (400 μg) was loaded into the wide lane (7.3 cm) of an 8% gel, separated by SDS-PAGE, and then transferred to a nitrocellulose membrane. Membrane strips (0.5 cm in width) were incubated with the indicated sera at equivalent dilutions and processed for enhanced chemiluminescence. One strip was incubated with antibody in the presence of 2 μg antigen, as indicated. The position of molecular weight standards run in parallel is shown on the right. Shown are representative data from two experiments with similar results. (B) Both crude and affinity-purified 9D3 serum specifically localize endogenous BIG1 to a perinuclear structure by IF. NRK cells were fixed and processed for IF by incubation with the indicated 9D3 sera (crude serum or purified antibody) in the absence or presence of the immunizing antigen (1 μg). Shown are single slice confocal images. (C) N-Terminal third fragment of BIG1 (N1- BIG1) is recruited to the same subcompartment within the Golgi complex as full-length endogenous BIG1. NRK cells were transiently transfected with constructs encoding HA- tagged N1 fragment of BIG1 and processed for IF by incubation with rabbit serum 9D3 and monoclonal anti-HA 3F10. Cell that has been transfected is indicated by arrowhead. Images were obtained by standard epifluorescence microscopy because the limited number of transfectants made confocal microscopy impractical in this case. The middle image was taken using a dichroic filter to simultaneously capture the signal from both the red and green fluorophores. Bars (B and C), 10 μm.
Confocal Microscopy and Image Quantitation
Confocal images were acquired using an LSM 510 (Carl Zeiss) equipped with a 63× objective (numerical aperture = 1.4). When two markers were imaged in the same cells, each fluorophore was excited and detected sequentially (multitrack mode) to avoid channel bleed- through. Laser intensity and filters were adjusted to give maximum signal (grayscale intensity of 255). Before final scanning, both channels were checked to ensure that no pixels were oversaturated. Tests confirmed that under our detection conditions, images obtained in the red and green channels were in register to within 60 nm. Unless otherwise indicated, a single focal plane (0.8–1 μm) was analyzed. For Figure 2, a series of z sections were collected and displayed using the ortho mode in LSM510 software provided with the microscope. For Figure 6, a series of z sections were collected at 0.2- and 0.3- μm intervals and used to generate projections that better reveal peripheral structures.
Figure 2.

GBF1 and BIG1 are recruited to different compartments of the Golgi complex. (a) BHK-21 cells were transiently transfected with a plasmid encoding HA-tagged full-length BIG1 and processed for IF by using antibodies against the HA epitope (3F10/fluorescein) and GBF1 (H154/Texas Red). (b) NRK cells were fixed and processed for IF by using antibodies against GBF1 (H154/Alexa594) and BIG1 (Alexa488-conjugated 9D3). Both images (a and b) were obtained by collecting stacks of optical sections (3.2 μm in thickness for BHK-21 cell [a] and 2.0 μm for NRK cell [b]) by using a confocal microscope. Shown in the center is an X-Y slice. Above and to the right are slices through the stack that reveal the distribution in the X-Z and Y-Z planes at the position indicated by the magenta and green lines, respectively. Bars, 10 μm. Similar results were obtained from at least three independent experiments.
Figure 6.

GBF1, but not BIG1, redistributes from the Golgi complex to peripheral VTCs at 15°C. NRK cells incubated for 2 h in a 15°C water bath before fixation were processed for double-label IF by using monoclonal antibodies against p115 and polyclonal antibodies that recognize either GBF1 (a–c) or BIG1 (d–f). To better reveal small peripheral structures, projections of several confocal slices are shown. Middle panels (b and e) show superimposed left and right images. As indicated by arrowheads in images a–c, the peripheral GBF1 and p115 staining patterns are almost identical. Peripheral structures appear as either yellow or orange dots depending on the relative intensity of red and green signals. The apparent partial overlap between BIG1 and p115 in perinuclear area shown in image e is due to the overexposure of perinuclear signals to better display small peripheral structures. Bars, 10 μm. The distinct response to temperature shift for GBF1 and BIG1 was observed in each of at least three separate experiments.
Quantitation of IF images was performed using either NIH Image (version 1.62, downloaded from http://rsb.info.nih.gov/nih-image) or MetaMorph (version 4.5r5; Universal Imaging, Downington, PA). Our approach to quantify the degree of overlap of GBF1 and p115 in peripheral structures (Figure 6) was based on that described by Hammond and Glick (2000). Briefly, NRK cells incubated at 15°C for 2 h were costained with H154 (anti-GBF1) and 3A10 (anti-p115). NIH Image was then used to generate separate masks for the green and red signal by using a range of threshold values that retained all discernible peripheral structures. For practical reasons, analysis was confined to smaller peripheral structures (<0.7 μM) with clearly identifiable centers; the number of structures eliminated from analysis was <5% of total. Two peripheral structures were defined as colocalized if their geometric centers (ultimate points in NIH Image) were within three pixels (0.2 μm) from each other. Results are expressed as percentage of total spots chosen for analysis in the green mask that were concentric with spots in the red mask. A total of seven cells (521 peripheral structures) was analyzed. Comparison of masks generated with various threshold values established that the choice of parameter did not significantly alter the outcome.
The extent of overlap between GBF1, BIG1, and several other markers in the perinuclear Golgi area (Figure 9) was quantitated as follows. Single focal plane images were used for this purpose. Pixels of interest were first identified by generating a mask for each channel to eliminate background signal resulting from nonspecific binding or out- of-focus signal. The perinuclear region is the thickest portion of the cell where intensity measurement errors due to out-of-focal plane fluorescence is most pronounced. Using MetaMorph software, we overcame this problem by defining a threshold value in red and green channels separately that includes only the brighter labeled structures. Image capture conditions had been optimized to yield intensity values near the maximum 255, while avoiding oversaturation. For such images, preliminary studies established that threshold values near 100 were necessary to retain fine structure within the Golgi complex. For a few weaker samples, the maximum value was <255 and in those cases, an equivalent threshold value was defined as 40% of the maximum intensity. This procedure ultimately yielded binary “masks” that had values of 1 for the pixels with intensity above threshold value and 0 for all others. Shared pixels between the red and green masks were then identified with the AND function.
Figure 9.

Quantitative analysis of the distribution of ARF-GEFs relative to Golgi markers and coat proteins. The extent of overlap observed in the perinuclear area by using antibodies to the indicated proteins was measured as described in MATERIALS AND METHODS. The percentage of IF signal intensity present in shared pixels (yellow) relative to total signal was measured for both the red (top) and green (bottom) channels. Red and green only values were defined as the difference between 100% and the overlap measured for the corresponding channel. Red signal was obtained by staining with polyclonal anti-GBF1 or anti- BIG1, whereas green signal was obtained with monoclonal anti-p115, anti-β-COP, or Alexa488-conjugated polyclonal anti-BIG1 (see legends of Figures 2, 3, and 8 for details). The number of separate images used for quantitation was five for GBF1/p115, four for TGN38/BIG1, five for GBF1/BIG1, six for GBF1/β-COP, and six for BIG1/β-COP. Error bars correspond to the SD of the mean.
Rather than simply comparing the “number” of shared pixels to the total, overlap was defined as the percentage of total signal “intensity” present in shared pixels. This approach yields a more accurate estimate of overlap because it weighs preferentially those pixels with greater intensity. To do this, we first had to recover the pixel intensity value information lost during processing. The binarized masks described above were modified using a combination of subtract and multiply functions in MetaMorph to regain the green and/or red intensity values above threshold. To calculate overlap for the green signal, we then used the ratio of the average intensity of the green pixels in the AND mask over that of the green mask. The converse calculation was performed for the red signal by using the red mask and the AND mask modified with red pixel values. To eliminate contribution from peripheral staining, the analysis was restricted to the perinuclear Golgi structure by using MetaMorph to select identical measurement areas in all three images (red, green, AND). For any given pair of markers, several cells were analyzed.
The validity of our quantitation method was established using two pairs of markers known to overlap either well or poorly. Images taken from BHK-21 cells that were transfected with HA-tagged full-length BIG1 and doubly stained with anti-BIG1 and anti-HA antibodies were used as positive control (our unpublished data). By using the above- mentioned quantitation method, 85% of BIG1 signal overlaps with HA-positive structures, whereas 90% of HA signal overlaps with BIG1-positive structures. Images taken from NRK cells doubly stained for TGN38 and p115 were used as negative control (Figure 3, a–c). We found that 29% of the TGN38 signal overlaps with p115 and 15% of p115 signal overlaps with TGN38. The small overlap between p115 and TGN38 likely results from the limitation of the IF confocal microscopy; however, we cannot exclude a partial colocalization of these two proteins.
Figure 3.

GBF1 and BIG1 localize to cis- and trans-compartments of the Golgi complex, respectively. NRK cells were fixed and processed for double-label IF by using either polyclonal anti-TGN38 (a), anti-GBF1 (d), or anti-BIG1 (g), and monoclonal anti-p115 (c, f, and i), or the two polyclonal antibodies Alexa488-conjugated 9D3 (anti-BIG1, j) and anti-TGN38 (l). Shown are single slice confocal images. Middle panels (b, e, h, and k) show merged left and right images. The inset shows a threefold magnification of the area indicated by an arrowhead. Bar, 10 μm. Shown are representative data from at least three experiments with similar results.
Nocodazole Treatment
We used a modified procedure to examine the microtubule requirement for VTC movement. Preliminary experiments established that we could not use the standard approach of brief incubation at 4°C with nocodazole before warm-up. Under these conditions, within 1 min of warm-up, depolymerization was incomplete and sufficient microtubules remained to facilitate fast movement of GBF1 from peripheral structures to the perinuclear area. To solve this problem, we included 5 μg/ml nocodazole during the 2 h at 15°C. Staining with anti-α-tubulin antibody confirmed that this method caused a complete loss of the microtubule array. The presence of nocodazole during the 15°C incubation neither prevented redistribution of p115 nor caused redistribution of Golgi resident enzymes such as Man II (our unpublished data).
RESULTS
Endogenous BIG1 Localizes to Golgi Complex through Sequences Present in N-Terminal Third of Protein
Our previous localization studies of BIG1 revealed that overexpressed HA-tagged BIG1 overlaps to a significant extent with a well-characterized Golgi marker, Man II (Mansour et al., 1999). To examine the localization of endogenous BIG1, antibodies were raised by immunizing rabbits with recombinantly produced fragments of BIG1 (see MATERIALS AND METHODS). One serum, 9D3, that recognizes a truncation containing the Sec7 domain termed M1 (Mansour et al., 1999) worked best for IF and was therefore chosen for further analysis. When assayed by immunoblotting, affinity-purified 9D3 showed greater specificity than crude serum and labeled specifically a protein of 208 kDa, the size predicted from the cDNA (Figure 1A). When used for IF, both 9D3 crude serum and affinity-purified 9D3 gave nearly identical perinuclear signals with little cytoplasmic staining (Figure 1B). Addition of excess immunizing protein (M1) eliminated perinuclear staining (Figure 1B). The neutralization of perinuclear staining by excess antigen was specific to BIG1 because it had no effect on the costaining with monoclonal antibodies against p115 (our unpublished data).
Previously, we established that the N-terminal third fragment of BIG1 (N1-BIG1) contains targeting information but could not determine whether N1-BIG1 was recruited to the same subcompartment within the Golgi complex as full-length endogenous BIG1 (Mansour et al., 1999). Our new serum, 9D3, which specifically recognizes the endogenous BIG1, but not N1-BIG1 that lacks the Sec7 domain, allowed us to address this question. NRK cells were transfected with a vector encoding HA-N1-BIG1 and then double stained with anti- HA and 9D3 (Figure 1C). The representative images presented in Figure 1C reveal that overexpressed exogenous N1-BIG1 colocalizes extensively with endogenous BIG1. These observations demonstrate that the targeting information in the N-terminal third of the protein is sufficient to localize the protein to the correct Golgi subcompartment. Furthermore, these results not only confirm the perinuclear Golgi localization obtained with overexpressed HA-tagged forms (Mansour et al., 1999) but also provide additional evidence for the specificity of our new 9D3 serum.
GBF1 and BIG1 Are Recruited to Different Compartments of Golgi Complex
Experiments with BHK cells overexpressing HA-tagged BIG1 first revealed that both GBF1 and overexpressed tagged BIG1 localized primarily to perinuclear structures but overlapped with each other to a very limited extent (Figure 2a). Analysis of red and green signal along the z-axis confirmed the extent of signal separation (Figure 2a, side panels). To establish that the lack of overlap did not result from the use of a tagged protein, we further compared the localization of endogenous BIG1 and GBF1. Because the only antibodies available against these two GEFs were polyclonal, one of them (anti-BIG1 rabbit serum, 9D3) was conjugated with the fluorophore Alexa488. Double staining of NRK cells with this antibody and a previously characterized anti-GBF1 polyclonal serum (H154) confirmed that GBF1 and BIG1 do not overlap (Figure 2b). Although usually in proximity, the BIG1 and GBF1 signals not only remained clearly separate from each other but also often took on very different patterns. Quantitative analysis discussed in more detail in Figure 9 confirmed this interpretation.
GBF1 and BIG1 Localize to cis- and trans-Compartments of Golgi Complex, Respectively
Double staining of NRK cells with antibodies for two well-characterized markers of the cis-Golgi and trans-Golgi network (TGN), p115 and TGN38, respectively, confirmed the feasibility of distinguishing cis-Golgi and trans-Golgi proteins by IF at light microscopy level in these cells (Figure 3, a–c). NRK cells decorated separately with antibodies raised against either GEFs and each costained for the common cis-marker p115 revealed that GBF1 largely colocalized with p115 (Figure 3, d–f). In contrast, the staining for BIG1 appeared largely distinct from that observed for p115 (Figure 3, g–i). Identical results were obtained with another cis-Golgi marker, p58 (our unpublished data). These results indicated that GBF1 associates primarily with early compartments of the Golgi complex and confirmed our initial observation of distinct localization for GBF1 and BIG1 (Figure 2). In addition, they suggested that BIG1 might localize to trans-elements of the Golgi complex. This possibility was tested directly by comparing the localization of BIG1 and TGN38 by using Alexa488-labeled BIG1 antibody and a polyclonal serum to TGN38. These experiments established that BIG1 largely overlapped with TGN38 (Figure 3, j–l). Similar results were obtained by costaining transfected NRK cells expressing HA-tagged BIG1 with antibodies to HA and TGN38 (our unpublished data). Higher magnifications of selected regions of merged images are presented in boxed insets to better illustrate the relative localization of the GEFs. We conclude that GBF1 and BIG1 localize to cis- and trans- elements of the Golgi complex, respectively. Quantitative analysis described in more detail in Figure 9 clearly supported this interpretation.
Previous work by Vaughan and colleagues predict that BIG2 should have a distribution nearly identical to that of BIG1 and thus be distinct from that of GBF1: Coimmune- precipitation studies established that the bulk of BIG1 and BIG2 exists as a complex, and initial IF studies suggested extensive colocalization of these two GEFs (Yamaji et al., 2000). Because our attempts to generate antibodies that recognize endogenous BIG2 were unsuccessful, we chose to investigate the relative distribution of BIG2 and GBF1 by constructing a series of HA-tagged fragments containing the N-terminal domain of BIG2 (see MATERIALS AND METHODS). As was the case for BIG1, a fragment containing the N-terminal third of BIG2 (HA-N-BIG2) localized to a perinuclear structure, which resembled that observed with several Golgi markers (our unpublished data). More detailed analysis demonstrated that the staining pattern of BIG2, although perinuclear, remained quite distinct from that of the cis-Golgi marker p58 (Figure 4, a–c). More importantly, double staining of BHK cells overexpressing HA-N-BIG2 for HA and GBF1 confirmed that there is little overlap between GBF1 and BIG2 (Figure 4, d–f). Higher magnifications of selected regions of merged images are presented in boxed insets to better illustrate the relative localization of exogenously expressed HA-N-BIG2. We conclude that the two ARF-GEF families localize and function in distinct regions of the Golgi complex.
Figure 4.

N terminal fragment of BIG2, like BIG1, does not colocalize with GBF1 and the cis-Golgi marker p58. BHK-21 cells were transiently transfected with a plasmid encoding HA-tagged N- BIG2 and processed for double-label IF by using monoclonal anti-HA and polyclonal antibodies against either p58 (a–c) or GBF1 (d–f). Shown are single slice confocal images obtained with the indicated antibody. Middle panels (b and e) show superimposed left and right images. The inset shows a threefold magnification of the area indicated by an arrowhead. Bars, 5 μm. Distinct staining for HA-BIG2 and GBF1 was observed in each of four experiments.
GBF1 and BIG1 Redistribute to Distinct Structures in Response to BFA
To further test the behavior of GBF1 and BIG1 as proteins of cis- and trans-Golgi compartments, we examined the effect of the fungal metabolite BFA on their distribution (Figure 5). In most animal cells, BFA treatment will induce extensive tubulation of both the TGN and central stack of the Golgi complex, but cause their resident enzymes to redistribute to distinct compartments. BFA-induced tubules from the main Golgi stack eventually fuse with the ER. In contrast, tubulation of trans-cisternae leads to formation of a hybrid organelle with the endosomal system that clusters near the microtubule-organizing center (Klausner et al., 1992b). Double staining of NRK cells treated with 10 μg/ml BFA for 30 min at 37°C before fixation established that the two GEFs moved to distinct structures after BFA treatment (Figure 5, c and d). Whereas GBF1 redistributed to a diffuse ER pattern similar to that observed with Man II (Figure 5, c and e), BIG1 appeared in a dense collection of fine punctate structures near the nucleus, reminiscent of that observed with TGN38 (Figure 5, d and f).
Figure 5.

GBF1 and BIG1 redistribute to distinct structures in response to BFA. NRK cells were treated with either 0.1% DMSO (control) or 10 μg/ml BFA for 30 min at 37°C before fixation and processing for IF. Cells were either double stained using two polyclonal antibodies (a–d) against GBF1 (H154/Alexa594; a and c) and BIG1 (Alexa488-conjugated 9D3; b and d) or singly stained using polyclonal antibodies against Man II (e) or TGN38 (f). Shown are single-slice confocal images. Bar, 10 μm. Similar results were obtained from at least two independent experiments.
GBF1, but not BIG1, Redistributes from Golgi Complex to Peripheral VTGs at 15°C
The cis-Golgi localization of GBF1 suggests that it regulates events at the interface between the ER and Golgi complex. To test this possibility, we examined the effect of incubation at lower temperature on the subcellular localization of GBF1. Protein traffic between the ER and Golgi stack in animal cells is temperature sensitive and blocked at temperatures <15°C. Under these conditions, cargo accumulates in small punctate peripheral structures called VTCs (Saraste et al., 1986). Proteins that shuttle between the cis-Golgi and VTCs, p115 and KDEL receptor, for example, also accumulate in peripheral VTC structures during prolonged incubation at 15°C (Alvarez et al., 1999). To examine how GBF1 and BIG1 react during treatment at lower temperature, NRK cells either kept at 37°C or incubated at 15°C for 2 h before fixation were processed for IF by using antibodies that recognize p115, endogenous GBF1, or endogenous BIG1. As shown in Figure 6, GBF1, like p115, largely redistributed to peripheral punctate structures during the 15°C block (Figure 6, a–c). In contrast, BIG1 remained in a tight ribbon-like perinuclear structure under conditions where p115 clearly redistributed throughout the cell (Figure 6, d–f).
The appearance of GBF1 in peripheral structures suggested that GBF1 might function to regulate ARF activation during formation and/or maturation of VTCs. Extensive colocalization of p115 and GBF1 in merged images indeed suggests that GBF1 associates with p115-positive structures at reduced temperatures (Figure 6b). Additional experiments confirmed the expected presence of COPI in those peripheral structures (our unpublished data). Quantitative analysis of Figure 6b and several similar images established that 88 ± 2% of peripheral structures examined (7 cells/521 structures) stain for both GBF1 and p115 (see MATERIALS AND METHODS). As predicted from the range of color (orange-yellow-lime) in the merged images, the ratio of green-to-red signal within these overlapping peripheral structures was variable. In spite of this unexplained variation, the presence of p115 in GBF1-positive peripheral structures clearly suggests a function for GBF1 at early stages of exocytosis.
To further test the functional significance of GBF1 in peripheral structures, we determined whether this redistribution was readily reversible upon warm-up. Previous studies established that cargo accumulated in peripheral structures at 15°C migrates toward the Golgi stack at speeds ∼1 μm/s and does so in a microtubule-dependent manner (Presley et al., 1997; Scales et al., 1997). We found that GBF1 accumulated in peripheral structures at 15°C does return to perinuclear localization upon warm-up to 37°C. This redistribution was extremely rapid and complete in less than 1 min (Figure 7, left, DMSO). This result is consistent with previous estimates of migration rates for ER- Golgi transport intermediates mentioned above. In contrast, GBF1 remained in peripheral structures when incubations were performed under conditions where microtubules have been disrupted with nocodazole (Figure 7, right, NOZ). Significant amounts of GBF1 remained in peripheral structures even after a 10-min incubation in the absence of microtubules (Figure 7j). Under these conditions, Man II, a Golgi-resident enzyme of medial cisternae, clearly did not redistribute to peripheral structures (Figure 7l). The observation that GBF1 associates with peripheral structures clearly distinct from the perinuclear structures positive for Man II further strengthens our conclusion that GBF1 is involved in traffic between the ER and Golgi complex.
Figure 7.

Redistribution of GBF1 from the 15°C peripheral compartments upon warm-up to 37°C is microtubule dependent. NRK cells were transferred to DMEM containing HEPES pH 7.4 and either 0.1% DMSO vehicle control or 5 μg/ml nocodazole (NOZ) and then incubated in a 15°C water bath for 2 h. Cells were either immediately fixed (0′) or quickly transferred to 37°C water bath and incubated for additional 1 min (1′), 3 min (3′), or 10 min (10′) before fixation. Coverslips were processed for IF by using polyclonal anti-GBF1 (e–h) or double-label IF (a–d and i–l) by using polyclonal anti- GBF1 (c and d, and I and j) and monoclonal anti-Man II (53FC3) (a and b, and k and l). Images obtained by standard epifluorescence microscopy are presented. Bar, 10 μm. Similar results were obtained in at least two independent experiments.
GBF1, but not BIG1, Overlaps Significantly with COPI
Several studies established that COPI components associate primarily with VTCs and cis-compartment of the Golgi complex (Oprins et al., 1993; Griffiths et al., 1995). Furthermore, the association of COPI with Golgi membranes has been shown in several studies to be very sensitive to BFA (Donaldson et al., 1990; Oprins et al., 1993). The demonstration that a BFA-resistant GEF (GBF1) rather than the BFA-sensitive ones (BIGs) are present in cis- regions of the Golgi prompted us to examine the relative distributions of the GEFs with known coat components. Double labeling of NRK cells show that GBF1, but not BIG1, overlaps significantly with the COPI coat (Figure 8, a–c). Although a few punctate peripheral structures positive for β-COP appear to lack GBF1 (Figure 8, b and c, arrows), the majority of the perinuclear structures show extensive overlap of these two markers (Figure 8b). Close examination reveals that the majority of bright β-COP-positive structures also stain for GBF1. However, as illustrated in the inset, a significant amount of β-COP signal often surrounds structures containing both markers. In contrast, BIG1- positive structures although in close apposition, remain largely distinct from those labeled with β-COP antibody (Figure 8e, inset).
Figure 8.

GBF1 and BIG1 overlap with distinct sets of coat proteins: GBF1 significantly with β-COP and BIG1 preferentially with clathrin. NRK cells were fixed and processed for double-label IF by using anti-β-COP and anti-GBF1 (a–c), anti-β-COP and anti-BIG1 (d–f), anti- clathrin and anti-GBF1 (g–i), or anti-clathrin and anti- BIG1 (j–l). Shown are single-slice confocal images taken in the indicated channel. Middle panels (b, e, h, and k) show superimposed left and right images. The inset shows a fourfold magnification of the area indicated by arrowhead. Arrows in b, c, e, and f point to some peripheral structures stained by anti-β-COP only. Bars, 5 μm. Shown are representative data from at least two experiments.
These results suggest that the primary function of BIGs may be to regulate activation of ARFs for recruitment of other coat proteins such as APs and/or GGAs in trans- elements of the Golgi. In agreement with this possibility, double staining with antibodies against clathrin and either of the two ARF-GEFs reveals closer association of clathrin with BIG1 than with GBF1 (Figure 8, g–l). The overlap between clathrin and BIG1 remains partial and appears limited to the perinuclear regions. The insets in Figure 8, h and k, illustrate the difference in the degree of overlap of the two GEFs with clathrin.
To better estimate the extent of overlap between GEFs and other proteins, we developed and tested a quantitative approach that yields the extent of signal intensity from each fluorophore that is present in shared pixels (see MATERIALS AND METHODS). Application of this method to images similar to those presented in Figures 2 and 3 confirmed our previous conclusion that GBF1 and BIG1 showed poor overlap with each other but did colocalize with markers of the cis- and trans- elements of the Golgi complex, respectively (Figure 9). For example, only 15% of the GBF1 signal is present in BIG1-positive structures, whereas the converse analysis shows only 23% of the BIG1 signal present in GBF1-positive structures. In contrast, the GBF1 and p115 signal overlapped significantly with each other (81 and 86%), whereas the BIG1 and TGN38 signal showed similarly high degree of overlap (83 and 92%). The small amount of overlap observed between GBF1 and BIG1 (15 and 23%) was similar to that measured for TGN38 and p115 (15 and 29%; see MATERIALS AND METHODS). Using this method on cells double stained for β-COP and GBF1, we found that nearly 85% of the GBF1 signal in the perinuclear area was present in β-COP-positive pixels. Consistent with images such as shown in Figure 8b, inset, a lower percentage (64%) of the β-COP signal in the perinuclear area overlapped with GBF1. In contrast, the extent of overlap measured with cells stained for BIG1 and β-COP was much lower, in the range of 31–37%. The extensive labeling of clathrin in peripheral structures prevented a meaningful quantitative analysis. The impact of these observation on the mechanism responsible for release of COPI from Golgi membranes by BFA will be discussed below.
DISCUSSION
The distinct substrate specificities and BFA sensitivities of members of the GBF1 and BIG families led us to explore whether these two classes of ARF-GEFs associate with distinct subcompartments of the Golgi complex. Examination of the steady-state localization and dynamics upon low-temperature or BFA treatment revealed dramatic differences. Using p115 and TGN38 as cis- and trans-Golgi markers, we found that GBF1 and BIG1 associated preferentially with cis- and trans-compartments, respectively. Consistent with these observations, GBF1 and BIG1 displayed different responses to external treatment. These observations suggest that GBF1 and BIGs may activate distinct subclasses of ARFs in unique locations to regulate different types of reactions. In agreement with this possibility, we found that the COPI coat overlapped to a greater extent with GBF1 than BIG1, whereas clathrin showed limited overlap with BIG1, and virtually none with GBF1.
Localization of Large ARF-GEFs
Large ARF-GEFs of the GBF1 and BIG subfamilies were localized using confocal microscopy. Despite multiple attempts, we were not able to use available antibodies to examine the relative localization of endogenous ARF-GEFs by using immuno-electron microscopy. Fortunately, the significant differences in localization of the two GEF families made assignment to distinct subcompartments of the Golgi complex possible even at the light microscope level. Costaining for endogenous GBF1 and either HA-tagged or endogenous BIG1 revealed very clear separation in staining pattern for these two ARF-GEF classes. Such separation between GBF1 and BIGs was observed in at least two cell types and with various combinations of endogenous or HA- tagged forms of BIGs. Quantitative analysis of confocal images confirmed this separation and revealed an extent of overlap as low as that measured with the well-characterized cis- and trans-Golgi markers p115 and TGN38.
Our localization of GBF1 to early compartments of the secretory pathway was based on both its distribution and response to various treatments. At steady state, GBF1 overlapped to a large extent with both p115 and p58. In addition, as expected of a cis-Golgi protein, GBF1 partially redistributed from the Golgi complex to peripheral sites after incubation at 15°C, whereas it appeared in a diffuse reticular ER pattern in the presence of BFA. These observations are consistent with our previous EM study (Claude et al., 1999). In rat hepatocytes, although GBF1 was found at the highest concentration in stacked regions, a significant fraction of the protein also localized to smooth tubules corresponding to VTCs in these cells (Dahan et al., 1994; Lavoie et al., 1999). Several observations similarly established that BIGs reside primarily in trans- compartments: they overlapped with TGN38 and their perinuclear localization was not altered at 15°C. Interestingly, the BFA-sensitive BIG1, like GBF1, was clearly not released from membranes upon BFA treatment. In this case, it redistributed like TGN38 to a dense collection of fine punctate structures in the perinuclear area upon BFA treatment. These results significantly extend the previous demonstration by Vaughan and colleagues that BIG1 and BIG2 exist in a large complex that localizes in the perinuclear region (Yamaji et al., 2000). These observations are also consistent with our previous report that BIG1 overlaps better with Man II than with ERGIC-53 (Mansour et al., 1999). The apparent discrepancy with the conclusion of Vaughan and colleagues that BFA causes little change in BIGs' distribution (Yamaji et al., 2000) most likely reflects the slow kinetics of BFA's effect on the TGN and the short 10- min treatment used by those authors (our unpublished data).
Mechanism of GEF Recruitment to Specific Membranes
Greater than 90% of GBF1 and BIGs are recovered in the soluble fraction of cellular homogenates (Claude et al., 1999; Yamaji et al., 2000). Our localization of these proteins to distinct Golgi subcompartment in intact cells therefore predicts that the interaction of ARF-GEFs with membranes is readily reversible and therefore regulated. Furthermore, in contrast to members of the ARNO and EFA6 group, these GEFs lack PH domains and likely require receptor-like proteins to mediate membrane recruitment.
Our work provides interesting starting points to analyze the regulation of recruitment of BIGs to the TGN. We established that HA-tagged forms containing the N-terminal third of either BIG1 (Mansour et al., 1999) or BIG2 (Figure 5) localized to the Golgi complex. Further analysis established, in the case of BIG1, that HA-tagged full-length protein and truncations colocalized with the endogenous protein (Figure 1C). The information present within the N-terminal domain is therefore sufficient to direct the protein to the correct Golgi subcompartment. We cannot at this point determine whether the N-terminal third contains targeting information or is responsible for interaction with a partner in a hetero-oligomeric complex that contains such targeting information.
The appearance of GBF1 in peripheral structures at 15°C suggests that VTCs produced de novo from the ER represent a major site for GBF1 recruitment. Indeed, one of our anti- GBF1 sera yields a more peripheral staining pattern for GBF1 (our unpublished data). As shown in Figure 7, these GBF1- labeled peripheral structures cluster very rapidly in the perinuclear area upon warm-up from 15°C and this movement requires microtubules. Therefore, the fact that at steady state most GBF1 localizes to perinuclear stacks may simply result from the rapid migration of transport complexes along microtubules from the cell periphery to the Golgi area. GBF1 may remain associated with these structures during transport and after their fusion to produce cis- compartments of the Golgi complex. We cannot exclude the possibility that GBF1 is in constant dynamic equilibrium with a cytoplasmic pool during transport and/or that additional GBF1 is recruited directly on the Golgi stacks. Clearly, the mechanism responsible for the recruitment of GBF1 to nascent VTCs, or its release from late Golgi compartments, is of great interest. Identification of interaction partners for GBF1 and the N-terminal domain of BIGs will likely be required to elucidate the mechanism of ARF-GEFs recruitment to the Golgi complex.
Inhibition of ER-Golgi Traffic by BFA
The available evidence suggests that BFA blocks ER- Golgi transport by causing dissociation of COPI from VTCs. The initial reports of COPI association with VTCs (Oprins et al., 1993; Griffiths et al., 1995) have been extended by several live-cell imaging studies, which confirm that recruitment of COPI to VTCs occurs early as cargo emerges from ER exit sites (Presley et al., 1997; Scales et al., 1997; Stephens et al., 2000). Although no published report specifically examined loss of COPI from peripheral VTCs at short time (<1 min) upon BFA addition, live-cell imaging studies with green fluorescent protein- tagged forms of COPI subunits recently confirmed that membrane association of COPI to peripheral VTCs is indeed sensitive to BFA (Presley, personal communication). Such loss of COPI from VTCs should block ER-Golgi transport because COPI function has clearly been shown to be required for maturation and/or movement of VTCs to the Golgi complex. In vitro studies with an assay that reconstitutes formation of VTCs from transitional ER confirmed that VTC formation required COPI components and was BFA sensitive (Lavoie et al., 1999).
It is generally assumed that the effect of BFA on the formation of VTCs results from inhibition of an ARF-GEF. However, in this study we have convincingly localized BIGs, the only mammalian ARF-GEF clearly inhibited by BFA, to trans-element of the Golgi. How then can one explain the effect of BFA on early steps in protein traffic? Although the precise mechanism remains unclear, several possibilities come to mind. For example, in vitro assays established that 50-kDa BFA-ADP–ribosylated substrate promotes scission of COPI vesicles (Spano et al., 1999; Weigert et al., 1999), and its inactivation by ADP ribosylation in response to BFA could prevent release of VTCs from ER exit sites. Furthermore, 50- kDa BFA-ADP–ribosylated substrate is a lyso-phosphatidate acyl transferase that produces phosphatidic acid (Weigert et al., 1999) and its inactivation by BFA could alter membrane composition and interfere either directly or indirectly with COPI recruitment. Alternatively, we cannot exclude the possibility that other ARF-GEFs are present in early Golgi compartments and/or that GBF1 is BFA sensitive under physiological conditions.
ARF-GEFs May Determine Specificity of Coat Recruitment
Our hypothesis that GBF1 acts as a regulator for COPI recruitment in cis-compartments of the Golgi complex is supported by several lines of evidence. First, at steady state 85% of the GBF1-positive perinuclear structures also stained with β-COP. Second, the recruitment of GBF1 to peripheral ERGIC at 15°C further supports its involvement in regulation of COPI for maturation of those structures. Under normal conditions, formation of VTCs from ER exit sites involves the sequential recruitment of COPII and COP1 components onto ER-derived membranes. It seems reasonable to assume that an ARF-GEF would have to be recruited from a cytoplasmic pool de novo to initiate COPI recruitment on the nascent structures. The appearance of GBF1 on peripheral structures at 15°C indicates that GBF1 could have that function.
The cis-Golgi localized COPI (Oprins et al., 1993) has been implicated in bidirectional traffic between ER and Golgi (Orci et al., 1997), and our studies to date cannot resolve whether GBF1 participates primarily in one or in both directions. Among mammalian ARFs- GEFs, GBF1 is unique in catalyzing exchange preferentially on class II ARFs (Claude et al., 1999). Class II ARFs are found at significantly lower levels than class I ARFs (Berger et al., 1995) and could simply have a redundant function. After all, in vitro studies showed that ARF1, 3, and 5 facilitate with similar efficiency the recruitment of COPI and AP-1 onto Golgi-enriched membranes (Liang and Kornfeld, 1997). On the other hand, the substrate specificity of GBF1 (Claude et al., 1999) and the identification of arfophilin, an ARF effector that specifically recognizes ARF5 and ARF6 (Shin et al., 1999; Shin et al, 2001), suggest otherwise. A recent analysis of the retrograde traffic of cholera toxin provides further evidence that ARF1 and ARF5 may play different functions in vivo (Morinaga et al., 2001). Of course, we cannot rule out other functions for ARF5 and GBF1 such as activation of lipid-modifying enzymes. Further characterization of arfophilin or other ARF5- specific effectors may be required to resolve this issue.
Several proteins implicated in the assembly of clathrin have been identified in trans-elements of the Golgi complex. These include the adaptins AP-1 and AP-3, as well as a new class of proteins termed GGAs that appear to function in sorting cargo into nascent clathrin-coated vesicles (Puertollano et al., 2001; Zhu et al., 2001). Interestingly, the recruitment of these proteins to Golgi membranes is sensitive to BFA (Robinson and Bonifacino, 2001) and is most likely regulated by the BFA-sensitive and TGN-localized BIGs. As expected, we observed clear but limited overlap in the distribution of clathrin and BIG1. Our observation that many clathrin- positive structures lacked BIGs was not surprising because much clathrin is involved in endocytosis.
Our hypothesis that GBF1 and BIGs perform distinct functions in different environments is consistent with several morphological and functional studies with their yeast homologues, Gea1p/Gea2p and Sec7p. These proteins, like their mammalian homologues, associate preferentially with early and late Golgi cisternae, respectively (Franzusoff et al., 1991; Spang et al., 2001). Furthermore, Gea1p and Gea2p have nonredundant but overlapping functions in retrograde traffic between the Golgi complex and the ER, while Sec7 mutants do not show defects in this step of traffic (Spang et al., 2001). Sec7p, like BIGs, probably works at the trans-Golgi, and its reported involvement in ER-golgi traffic (Deitz et al., 2000) may result from indirect effects.
How each class of ARF-GEF regulates the recruitment of different types of coat components on their respective compartments remains unclear. It seems unlikely that activation of distinct classes of ARFs is sufficient to account for coat selectivity. Although both ARF1 and ARF5 have been found associated with Golgi membranes (Tsai et al., 1992; Cavenagh et al., 1996; Morinaga et al., 2001), to date neither has been localized to a specific subcompartment at either the light or EM level. ARNOs catalyze exchange on all three classes of ARFs (Donaldson and Jackson, 2000), and recent reports localize these small GEFs to the Golgi complex in some cells (Lee et al., 2000); class I and class II ARFs may therefore not show a polarized distribution that match coat proteins. On the other hand, the striking similarity between the polarized distribution of the two large ARF-GEF classes and the two main type of protein coats on the Golgi complex suggest that the GEFs may actually participate directly in effector and/or coat selection. By acting as transient molecular scaffolds, the large ARF-GEFs could provide some of the required specificity to direct, specific interactions between GEFs and downstream effectors in a spatially and temporally regulated manner. Ongoing efforts in several laboratories to identify partners for ARFs and their regulators will, in time, test this hypothesis.
ACKNOWLEDGMENTS
We thank B.P. Zhao for the construction, expression, and preparation of recombinant proteins used for generation of BIG1-specific antisera; L. Ho for the cloning and sequencing of HA-BIG2; and M. Hughes and H. Vandertol-Vanier for maintenance of cultured cells. We are particularly grateful to Dr. X. Sun and H. Chan for insights and helpful advice in developing the procedures for quantitation of confocal images presented in Figure 9. Finally, we thank Drs. A. Claude and J.J. Bergeron, as well as Ms. M. Schneider for helpful comments on the manuscript. This study was supported by a grant (to P.M.) and predoctoral studentship (to X.Z.) from the Canadian Institutes of Health Research.
Abbreviations used:
- AP
adaptor protein
- ARF
ADP- ribosylation factor
- BFA
brefeldin A
- BIG
BFA-inhibited GEF
- COP
coat protein
- GBF1
Golgi-specific BFA resistance factor 1
- GEF
guanine nucleotide exchange factor
- HA
hemagglutinin
- IF
immunofluorescence
- Man II
mannosidase II
- NRK
normal rat kidney
- VTC
vesicular-tubular cluster
Footnotes
DOI:10.1091/mbc.01–08-0420.
REFERENCES
- Allan VJ, Kreis TE. A microtubule- binding protein associated with membranes of the Golgi apparatus. J Cell Biol. 1986;103:2229–2239. doi: 10.1083/jcb.103.6.2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez C, Fujita H, Hubbard A, Sztul E. ER to Golgi transport: requirement for p115 at a pre-Golgi VTC stage. J Cell Biol. 1999;147:1205–1222. doi: 10.1083/jcb.147.6.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonny B, Schekman R. ER export: public transportation by the COPII coach. Curr Opin Cell Biol. 2001;13:438–443. doi: 10.1016/s0955-0674(00)00234-9. [DOI] [PubMed] [Google Scholar]
- Berger SJ, Resing KA, Taylor TC, Melancon P. Mass-spectrometric analysis of ADP-ribosylation factors from bovine brain: identification and evidence for homogeneous acylation with the C14:0 fatty acid (myristate) Biochem J. 1995;311:125–132. doi: 10.1042/bj3110125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown HA, Gutowsk S, Moomaw CR, Slaughter C, Sternweis PC. ADP- ribosylation factor, a small GTP-dependent regulatory protein, stimulates phospholipase D activity. Cell. 1993;75:1137–1144. doi: 10.1016/0092-8674(93)90323-i. [DOI] [PubMed] [Google Scholar]
- Burke B, Griffiths G, Reggio H, Louvard D, Warren G. A monoclonal antibody against a 135-K Golgi membrane protein. EMBO J. 1982;1:1621–1628. doi: 10.1002/j.1460-2075.1982.tb01364.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavenagh MM, Whitney JA, Caroll K, Zhang C, Boman AL, Rosenwald AG, Melman I, Kahn RA. Intracellular distribution of ARF proteins in mammalian cells. ARF6 is uniquely localized to the plasma membrane. J Biol Chem. 1996;271:21767–21774. doi: 10.1074/jbc.271.36.21767. [DOI] [PubMed] [Google Scholar]
- Chavrier P, Goud B. The role of ARF and Rab GTPases in membrane transport. Curr Opin Cell Biol. 1999;11:466–475. doi: 10.1016/S0955-0674(99)80067-2. [DOI] [PubMed] [Google Scholar]
- Claude A, Zhao BP, Kuziemsky CE, Dahan S, Berger SJ, Yan JP, Armold AD, Sullivan EM, Melancon P. GBF1: a novel Golgi-associated BFA-resistant guanine nucleotide exchange factor that displays specificity for ADP-ribosylation factor 5. J Cell Biol. 1999;146:71–84. [PMC free article] [PubMed] [Google Scholar]
- D'Souza-Schorey C, Li G, Colombo MI, Stahl PD. A regulatory role for ARF6 in receptor-mediated endocytosis. Science. 1995;267:1175–1178. doi: 10.1126/science.7855600. [DOI] [PubMed] [Google Scholar]
- Dahan S, Ahluwalia JP, Wong L, Posner BI, Bergeron JJ. Concentration of intracellular hepatic apolipoprotein E in Golgi apparatus saccular distensions and endosomes. J Cell Biol. 1994;127:1859–1869. doi: 10.1083/jcb.127.6.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deitz SB, Rambourg A, Kepes F, Franzusoff A. Sec7p directs the transitions required for yeast Golgi biogenesis. Traffic. 2000;1:172–183. doi: 10.1034/j.1600-0854.2000.010209.x. [DOI] [PubMed] [Google Scholar]
- Donaldson JG, Cassel D, Kahn RA, Klausner RD. ADP-ribosylation factor, a small GTP-binding protein, is required for binding of the coatomer protein beta-COP to Golgi membranes. Proc Natl Acad Sci USA. 1992;89:6408–6412. doi: 10.1073/pnas.89.14.6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson JG, Jackson CL. Regulators and effectors of the ARF GTPases. Curr Opin Cell Biol. 2000;12:475–482. doi: 10.1016/s0955-0674(00)00119-8. [DOI] [PubMed] [Google Scholar]
- Donaldson JG, Lippincott-Schwartz J, Bloom GS, Kreis TE, Klausner RD. Dissociation of a 110-kD peripheral membrane protein from the Golgi apparatus is an early event in brefeldin A action. J Cell Biol. 1990;111:2295–2306. doi: 10.1083/jcb.111.6.2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank S, Upender S, Hansen SH, Casanova JE. ARNO is a guanine nucleotide exchange factor for ADP- ribosylation factor 6. J Biol Chem. 1998;273:23–27. doi: 10.1074/jbc.273.1.23. [DOI] [PubMed] [Google Scholar]
- Franzusoff A, Redding K, Crosby J, Fuller RS, Schekman R. Localization of components involved in protein transport and processing through the yeast Golgi apparatus. Cell Biol. 1991;112:27–37. doi: 10.1083/jcb.112.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godi A, Pertile P, Meyers R, Marra P, Di Tullio G, Iurisci C, Luini A, Corda D, De Matteis MA. A.R.F. mediates recruitment of PtdIns-4-O.H. kinase- beta and stimulates synthesis of PtdIns(4,5)P2 on the Golgi complex. Nat Cell Biol. 1999;1:280–287. doi: 10.1038/12993. [DOI] [PubMed] [Google Scholar]
- Grebe M, Gadea J, Steinmann T, Kientz M, Rahfeld JU, Salchert K, Koncz C, Jurgens G. A conserved domain of the arabidopsis GNOM protein mediates subunit interaction and cyclophilin 5 binding. Plant Cell. 2000;12:343–356. doi: 10.1105/tpc.12.3.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths G, Pepperkok R, Locker JK, Kreis TE. Immunocytochemical localization of beta-COP to the ER-Golgi boundary and the TGN. J Cell Sci. 1995;108:2839–2856. doi: 10.1242/jcs.108.8.2839. [DOI] [PubMed] [Google Scholar]
- Hammond AT, Glick BS. Dynamics of transitional endoplasmic reticulum sites in vertebrate cells. Mol Biol Cell. 2000;11:3013–3030. doi: 10.1091/mbc.11.9.3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlow E, Lane D. Antibodies: A Laboratory Manual. Cold Spring Harbor: NY, Cold Spring Harbor Laboratory Press; 1988. [Google Scholar]
- Klausner RD, Donaldson JG, Lippincott SJ. Brefeldin A: insights into the control of membrane traffic and organelle structure. J Cell Biol. 1992a;116:1071–1080. doi: 10.1083/jcb.116.5.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladinsky MS, Kremer JR, Furcinitti PS, MaIntosh RJ, Howell KE. HVEM tomograpgy of the trans-Golgi network: structural insights and identification of a lace- like vesicle coat. J Cell Biol. 1994;127:29–38. doi: 10.1083/jcb.127.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladinsky MS, Mastronarde DN, McIntosh JR, Howell KE, Staehelin LA. Golgi structure in three dimensions: functional insights from the normal rat kidney cell. J Cell Biol. 1999;144:1135–1149. doi: 10.1083/jcb.144.6.1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie C, Paiement J, Dominguez M, Roy L, Dahan S, Gushue JN, Bergeron JJ. Roles for alpha(2)p24 and COPI in endoplasmic reticulum cargo exit site formation. J Cell Biol. 1999;146:285–299. doi: 10.1083/jcb.146.2.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SY, Mansour M, Pohajdak B. B2-1, a Sec7- and pleckstrin homology domain-containing protein, localizes to the Golgi complex. Exp Cell Res. 2000;256:515–521. doi: 10.1006/excr.2000.4845. [DOI] [PubMed] [Google Scholar]
- Liang JO, Kornfeld S. Comparative activity of ADP-ribosylation factor family members in the early steps of coated vesicle formation on rat liver Golgi membranes. J Biol Chem. 1997;272:4141–4148. doi: 10.1074/jbc.272.7.4141. [DOI] [PubMed] [Google Scholar]
- Mansour SJ, Skaug J, Zhao XH, Giordano J, Scherer SW, Melancon P. p200 ARF-GEP1: a Golgi-localized guanine nucleotide exchange protein whose Sec7 domain is targeted by the drug brefeldin A. Proc Natl Acad Sci USA. 1999;96:7968–7973. doi: 10.1073/pnas.96.14.7968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morinaga N, Kaihou Y, Vitale N, Moss J, Noda M. Involvement of ADP-ribosylation factor 1 in cholera toxin-induced morphological changes of Chinese hamster ovary cells. J Biol Chem. 2001;276:22838–22843. doi: 10.1074/jbc.M101184200. [DOI] [PubMed] [Google Scholar]
- Morinaga N, Tsai SC, Moss J, Vaughan M. Isolation of a brefeldin A-inhibited guanine nucleotide-exchange protein for ADP ribosylation factor (ARF) 1 and ARF3 that contains a Sec7-like domain. Proc Natl Acad Sci USA. 1996;93:12856–12860. doi: 10.1073/pnas.93.23.12856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oprins A, Duden R, Kreis TE, Geuze HJ, Slot JW. Beta-COP localizes mainly to the cis-Golgi side in exocrine pancreas. J Cell Biol. 1993;121:49–59. doi: 10.1083/jcb.121.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orci L, Stamnes M, Ravazzola M, Amherdt M, Perrelet A, Sollner TH, Rothman JE. Bidirectional transport by distinct populations of COPI-coated vesicles. Cell. 1997;90:335–349. doi: 10.1016/s0092-8674(00)80341-4. [DOI] [PubMed] [Google Scholar]
- Pelham HR, Rothman JE. The debate about transport in the Golgi—two sides of the same coin? Cell. 2000;102:713–719. doi: 10.1016/s0092-8674(00)00060-x. [DOI] [PubMed] [Google Scholar]
- Pepperkok R, Scheel J, Horstmann H, Hauri HP, Griffiths G, Kreis TE. Beta-COP is essential for biosynthetic membrane transport from the endoplasmic reticulum to the Golgi complex in vivo. Cell. 1993;74:71–82. doi: 10.1016/0092-8674(93)90295-2. [DOI] [PubMed] [Google Scholar]
- Peters PJ, Hsu VW, Ooi CE, Finazzi D, Teal SB, Oorschot V, Donaldson JG, Klausner RD. Overexpression of wild-type and mutant ARF1 and ARF6: distinct perturbations of nonoverlapping membrane compartments. J Cell Biol. 1995;128:1003–1017. doi: 10.1083/jcb.128.6.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyroche A, Antonny B, Robineau S, Acker J, Cherfils J, Jackson CL. Brefeldin A acts to stabilize an abortive ARF-GDP-Sec7 domain protein complex: involvement of specific residues of the Sec7 domain. Mol Cell. 1999;3:275–285. doi: 10.1016/s1097-2765(00)80455-4. [DOI] [PubMed] [Google Scholar]
- Presley JF, Cole NB, Schroer TA, Hirschberg K, Zaal KJ, Lippincott-Schwartz J. ER-to-Golgi transport visualized in living cells [see comments] Nature. 1997;389:81–85. doi: 10.1038/38001. [DOI] [PubMed] [Google Scholar]
- Puertollano R, Aguilar RC, Gorshkova I, Crouch RJ, Bonifacino JS. Sorting of mannose 6-phosphate receptors mediated by the GGAs. Science. 2001;292:1712–1716. doi: 10.1126/science.1060750. [DOI] [PubMed] [Google Scholar]
- Robinson MS, Bonifacino JS. Adaptor- related proteins. Curr Opin Cell Biol. 2001;13:444–453. doi: 10.1016/s0955-0674(00)00235-0. [DOI] [PubMed] [Google Scholar]
- Saraste J, Palade GE, Farquhar MG. Temperature-sensitive steps in the transport of secretory proteins through the Golgi complex in exocrine pancreatic cells. Proc Natl Acad Sci USA. 1986;83:6425–6429. doi: 10.1073/pnas.83.17.6425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraste J, Palade GE, Farquhar MG. Antibodies to rat pancreas Golgi subfractions: identification of a 58-kD cis-Golgi protein. J Cell Biol. 1987;105:2021–2029. doi: 10.1083/jcb.105.5.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scales SJ, Pepperkok R, Kreis TE. Visualization of ER-to-Golgi transport in living cells reveals a sequential mode of action for COPII and COPI. Cell. 1997;90:1137–1148. doi: 10.1016/s0092-8674(00)80379-7. [DOI] [PubMed] [Google Scholar]
- Shin OH, Couvillon AD, Exton JH. Arfophilin is a common target of both class II and class III ADP-ribosylation factors. Biochemistry. 2001;40:10846–10852. doi: 10.1021/bi0107391. [DOI] [PubMed] [Google Scholar]
- Shin OH, Ross AH, Mihai I, Exton JH. Identification of arfophilin, a target protein for GTP-bound class II ADP-ribosylation factors. J Biol Chem. 1999;274:36609–36615. doi: 10.1074/jbc.274.51.36609. [DOI] [PubMed] [Google Scholar]
- Someya A, Sata M, Takeda K, Pacheco-Rodriguez G, Ferrans VJ, Moss J, Vaughan M. ARF-GEP100, a guanine nucleotide-exchange protein for ADP-ribosylation factor 6. Proc Natl Acad Sci USA. 2001;98:2413–2418. doi: 10.1073/pnas.051634798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spang A, Herrmann JM, Hamamoto S, Schekman R. The ADP ribosylation factor-nucleotide exchange factors Gea1p and Gea2p have overlapping, but not redundant functions in retrograde transport from the Golgi to the endoplasmic reticulum. Mol Biol Cell. 2001;12:1035–1045. doi: 10.1091/mbc.12.4.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spano S, Silletta MG, Colanzi A, Alberti S, Fiucci G, Valente C, Fusella A, Salmona M, Mironov A, Luini A, et al. Molecular cloning and functional characterization of brefeldin A-ADP- ribosylated substrate. A novel protein involved in the maintenance of the Golgi structure. J Biol Chem. 1999;274:17705–17710. doi: 10.1074/jbc.274.25.17705. [DOI] [PubMed] [Google Scholar]
- Stephens DJ, Lin-Marq N, Pagano A, Pepperkok R, Paccaud JP. COPI-coated ER-to-Golgi transport complexes segregate from COPII in close proximity to ER exit sites. J Cell Sci. 2000;113:2177–2185. doi: 10.1242/jcs.113.12.2177. [DOI] [PubMed] [Google Scholar]
- Togawa A, Morinaga N, Ogasawara M, Moss J, Vaughan M. Purification and cloning of a brefeldin A-inhibited guanine nucleotide-exchange protein for ADP- ribosylation factors. J Biol Chem. 1999;274:12308–12315. doi: 10.1074/jbc.274.18.12308. [DOI] [PubMed] [Google Scholar]
- Tsai SC, Adamik R, Haun RS, Moss J, Vaughan M. Differential interaction of ADP-ribosylation factors 1, 3, and 5 with rat brain Golgi membranes. Proc Natl Acad Sci USA. 1992;89:9272–9276. doi: 10.1073/pnas.89.19.9272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters MG, Clary DO, Rothman JE. A novel 115-kD peripheral membrane protein is required for intercisternal transport in the Golgi stack. J Cell Biol. 1992;118:1015–1026. doi: 10.1083/jcb.118.5.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigert R, Silletta MG, Spano S, Turacchio G, Cericola C, Colanzi A, Senatore S, Mancini R, Polishchuk EV, Salmona M, et al. CtBP/BARS induces fission of Golgi membranes by acylating lysophosphatidic acid. Nature. 1999;402:429–433. doi: 10.1038/46587. [DOI] [PubMed] [Google Scholar]
- Yamaji R, Adamik R, Takeda K, Togawa A, Pacheco-Rodriguez G, Ferrans VJ, Moss J, Vaughan M. Identification and localization of two brefeldin A-inhibited guanine nucleotide-exchange proteins for ADP-ribosylation factors in a macromolecular complex. Proc Natl Acad Sci USA. 2000;97:2567–2572. doi: 10.1073/pnas.97.6.2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Doray B, Poussu A, Lehto VP, Kornfeld S. Binding of GGA2 to the lysosomal enzyme sorting motif of the mannose 6-phosphate receptor. Science. 2001;292:1716–1718. doi: 10.1126/science.1060896. [DOI] [PubMed] [Google Scholar]