

Abstract
Amphiphilic in nature, lipids spontaneously self-assemble into a range of nanostructures in the presence of water. Among lipid self-assembled structures, liposomes and supported lipid bilayers have long held scientific interest for their main applications in drug delivery and plasma membrane models, respectively. In contrast, lipid-based multi-layered membranes on solid supports only recently begun drawing scientists’ attention. New studies on lipid films show that the stacking of multiple bilayers on a solid support yields interestingly complex features to these systems. Namely, multiple layers exhibit cooperative structural and dynamic behavior. In addition, the materials enable compartmentalization, templating, and enhanced release of several molecules of interest. Importantly, supported lipid phases exhibit long-range periodic nano-scale order and orientation that is tunable in response to a changing environment. Herein, we summarize current and pertinent understanding of lipid-based film research focusing on how unique structural characteristics enable the emergence of new applications in biotechnology including label-free biosensors, macroscale drug delivery, and substrate-mediated gene delivery. Our very recent contributions to lipid-based films, focusing on the structural characterization at the meso, nano, and molecular-scale, using Small-Angle X-ray Scattering, Atomic Force Microscopy, Photothermal Induced Resonance, and Solid-State NMR will be also highlighted.
Keywords: lipid films, lipid-polymer hybrids, supported membranes, substrate-mediated delivery, solid-state NMR
1. Introduction
Lipids are the primary components of plasma membranes comprising a hydrophilic polar headgroup and hydrophobic tail(s). The lipid bilayer of plasma membranes separates the interior of cellular components from the external environment, encapsulates membrane proteins, and selectively permeates ions or molecules of interest. Specifically, the capabilities to sense, detect, and transport specific species of plasma membranes have fascinated scientists, leading to active research on functional lipid bilayers.[1–3] Enclosed lipid bilayers in an aqueous solution (termed lipid vesicles or liposomes) have been extensively used as “carriers” for drug and gene delivery based on their ability to encapsulate both hydrophilic and lipophilic molecules into different compartments.[4–7] Lipid bilayers supported onto a solid surface (termed a supported lipid bilayer) also have been exploited largely as plasma membrane models to study basic cellular processes such as lipid-protein interactions.[8–10]
Flat lipid bilayers is the most abundant arrangement of lipids found in nature. However, different types of lipids can self-arrange into various morphologies differing from the zero-curvature bilayer configuration including micellar, hexagonal, and bicontinuous cubic phases.[11–13] Lipid polymorphism has attracted considerable attention because of the unique phase-dependent properties that it entices. For example, lipid cubic phase structures exhibit high internal surface area per volume (≈ 400 m2/g)[14] that enables high loading and fast release of drug molecules or genes.[15,16] In addition, bicontinuous cubic phases have negative Gaussian membrane curvature that promotes endosomal fusion and concomitant efficient small interfering RNA (siRNA) release and gene knockdown.[17,18] Finally the optical properties of isotropic bicontinuous phases and optical transparency can be used to detect biomarkers yielding birefringence upon crystallization.[19] In general, lipid polymorphism has been leveraged for constructing stimuli-responsive self-assembled systems where manipulation of the lipid phases enables “on-demand” release of encapsulated agents.[15,20,21] Examples include leveraging ultrasound-triggered reversible phase transitions between different liquid crystalline lipid phases that regulate the diffusion rates of drug molecules,[20] and pH-sensitive lipid vesicles that transforms from a lamellar phase to an inverted hexagonal phase in acidic conditions, which helps delivering the drug payload of lipid vesicles into the cytoplasm.[22] Despite the exciting properties rendered by lipid polymorphism, most studies exploiting lipid polymorphism have been dedicated to lipid particulate systems.
However, recent studies have shown that lipid polymorphism can be extended to lipid assemblies confined onto surfaces.[23–25] Research on supported lipid films and aspects of surface-mediated phase transitions is still in an early stage of research. This is partly because the use of supported lipid systems has been mostly limited to model membranes where a single bilayer on a solid surface is sufficient to mimic most cellular membranes.
It should be noted that there is a growing interest to develop implantable macroscale drug delivery devices,[26,27] high throughput biosensing systems,[28–30] and substrate-mediated drug/gene delivery.[31,32] Advances in biotechnology such as surface-patterning techniques[33–35], along with microfluidics[36–38] and biodegradable organic electronics[39,40], have enriched research on the aforementioned applications. Following this trend, the need for biointerface membranes adsorbed onto solid substrates, that serve as matrices or scaffolds and that are capable of exerting spatiotemporal control over the release of payloads is rapidly rising.[41,42]
The purpose of this review, is to highlight the potential of lipid films as emerging materials for the development of substrate-mediated biotechnological devices. The first part of review covers aspects of lipid films placed into three categories: supported lipid bilayers, lipid multi-lamellar films, and lipid non-lamellar films with a special focus into functionalities enabled by new lipid multilamellar films. The section of non-lamellar lipid films describes the phase-behavior of supported systems highlighting unanswered questions in phase transformation mechanisms, followed by potential applications enabled by their highly adaptable behavior in response to changing environmental conditions (e.g. humidity). The second part of the review summarizes the research on novel lipid-polymer composite membranes on solid supports that have recently begun to be developed.[43–46] These recent advances on hybrid films composed of self-assembled polymers and lipids are discussed emphasizing new functionalities that are not attainable with non-hybrid systems.
2. Lipid Membranes
2.1 Supported lipid bilayers (SLB)
Since the scope of this paper focuses on lipid multi-layered films, we will briefly discuss the case of a single lipid bilayer supported on a solid substrate. For a comprehensive review see references.[9,10,47,48]
Figure 1 shows a schematic illustration of a supported lipid bilayer. The deposition of a lipid bilayer onto hydrophilic solid substrate leaves a thin water layer (1 nm to 2 nm thick) between the bilayer and the substrate which preserves the fluidity exhibited in membrane native state.[8,49] Locking of a lipid membrane onto the surface is experimentally straightforward in comparison to challenges for producing free-standing bilayer systems. In addition, confined lipid bilayers offer robust and stable platforms that are facile to characterize with a variety of surface-sensitive techniques including quartz crystal microbalance (QCM),[50] atomic force microscopy (AFM),[51] and time-of-flight secondary ion mass spectrometry (SIMS).[52]
Figure 1.
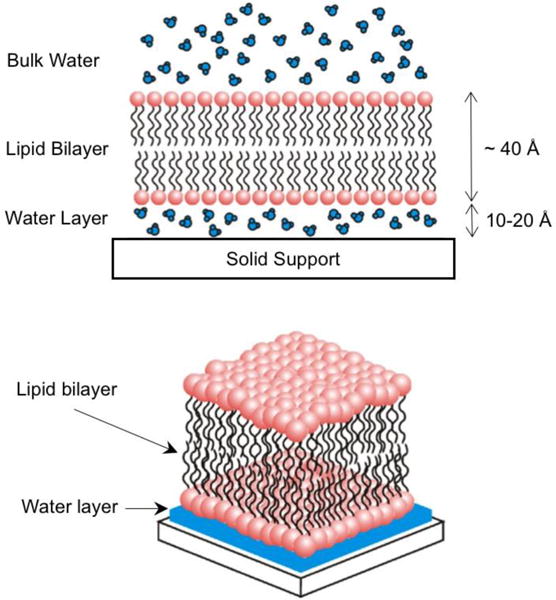
Schematic representation (not to scale) of a supported lipid bilayer. Note the presence of a thin water layer between the substrate and the lipid bilayer. Adapted From [9]. Copyright 2006 Elsevier Ltd.
SLBs can be prepared with different techniques[10]: the Langmuir-Blodgett/Langmuir-Schaefer method in which lipid molecules are spread at the air/water interface and then transferred onto the substrate,[8,53,54] or the adsorption and fusion of lipid vesicles to the substrate,[55–57] or the combination of these two methods.[58] A comprehensive review on the advantages and disadvantages of the SLB preparation methods can be found in Ref.[9,10] There are several applications where SLBs attract intrest. They provide a model membrane platform to study biological processes of the plasma membranes such as protein-lipid interactions, protein-protein interactions, membrane domains, cell adhesion and signaling.[47,59–62] SLBs can be also exploited to realize biological and chemical sensors and on-chip immunoassays in combination with patterning techniques and integrated microfluidic devices.[9,63–65] Recently, SLB-assisted self-assembly of DNA origami was reported, suggesting a new utilization of the SLB as a template for directed self-assembly.[66]
One of the main limitations of the SLBs is that membrane proteins cannot retain 100% of their functionality when reconstituted within the SLBs.[59] To address this problem, advanced SLBs such as tethered-SLBs have been developed.[67] For example, in polymer-cushioned bilayers, a tethered-polymer layer onto the substrate decouples the lipid bilayer and the substrate, thus shielding membrane proteins from the substrate and preserving their functionality.[48,59,67] Since their first discovery, SLBs have been extensively studied and still are an active research field. With the development of nanotechnology, SLBs will continue to find their use in various fields.
2.2 Lipid multilamellar films
Lipid multilamellar films are composed of tens to thousands of lipid bilayers on solid substrates. The extra layers on a supported lipid bilayer add interesting features to the system and inevitable complexity. The structure of lipid multilamellar films has been thoroughly investigated by means of several techniques including X-ray and neutron scattering/reflectivity, and AFM.[68–71] The structure of lipid multilayers under hydration, temperature, and electric field has been also studied in the context of structural stability.[72–76] Despite substantial research on structures, applications of the lipid multilamellar films have remained conceptual until recently. Over the past 5 years, the rapid emergence of macroscale delivery systems that require a supported film format coupled with the developments of new lipid film preparation methods has spurred the integration of this novel technology into various applications. In this section, we will review recent advances on lipid multilamellar film research, with an emphasis on new methodologies to form lipid multilayers and on the recently reported applications.
2.2.1. Lipid supported bilayer VS Lipid multilamellar films
Lipid multilamellar films provide benefits stemming from the “multilayer” architecture that differentiate their applications from those of SLB. Firstly, lipid multilamellar films can provide a practical platform to mimic the stacked membranes found in nature.[78,82–85] Although the plasma membranes consist a bilayer structure, several biological membranes are composed of stacks of bilayer membranes. Examples include thylakoids in the chloroplast of the plant cells[82,83] (Fig. 2A) and myelin sheath in the nerves (Fig. 2B) [84,85]. Figure 2C illustrates multi-layered myelin sheaths wrapping around nerve axons. The stacked membranes in such organelles are thought to play an important role in mediating functionality and adaptability to external conditions in those organelles.[86,87] In this regard, lipid multilamellar films can be utilized as model systems to study the structures and functions of certain organelles.
Figure 2.
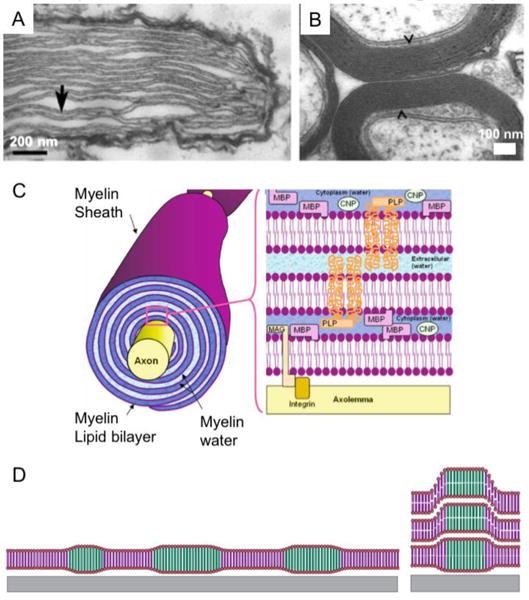
A) Transmission Electron Microcopy (TEM) of thylakoid membrane stacks found in Nematodinium sp. Thylakoids are marked by arrows. Adapted from [77]. Copyright 2015 Macmillan Publishers Ltd. B) TEM of Myelinated axons in rat optic nerve indicated by arrows. Adapted from [78]. Copyright 2015 Association for Research in Vison and Ophthalmology. C) An illustration of multi-layered myelin sheaths wrapping around nerve axons. Adapted from [79]. Copyright 2007 Springer New York. D) An illustration of supported model lipid membranes studied by Tayebi et al.[80] Multicomponent lipid bilayers phase-separate into coexisting domains (left) and domains align across layers showing interlayer alignment (right). Adapted from [81]. Copyright 2012 Macmillan Publishers Ltd.
Secondly, the layering of membranes increases the capacity to store molecules of interest with potential applications in small molecule (drug, genes, and proteins) delivery or sensing. Multilayers could be possibly exploited in concurrent delivery of multiple therapeutic agents in a sequential way or a new way mediated by the substrate.[42]
Additionally, recent findings by Tayebi et al.[80] show interesting structural characteristics that lipid multilamellar films can offer, opening up new possibilities of their applications in photonics and sensing. The authors prepared supported lipid lamellar films from the mixtures of sphingomyelin, cholesterol, and unsaturated phospholipids. Phase separation occurred from the multicomponent mixtures, giving rise to liquid-ordered domains (Lo) and liquid-disordered domains (Ld) in plane.[80] Interestingly, those phase-separated domains in-plane aligned themselves across hundreds of bilayers (along the out-of-plane direction).[80] Figure 2D shows the illustration of stacked lipid multilayers with interlayer smectic ordering of the intralayer domains. Two features of such domains are particularly interesting – serial coupling and compartmentalization – which may lead to unprecedented membrane functions. For example, transport process and/or electrical signals could be amplified through cooperative behavior of the membrane components dictated by the columnar ordering of phase-separated domains.[80,81]
2.2.2. Advances in preparation of lipid multilayers with functionality
Several factors act as a bottleneck for the use of lipid multilamellar films in practical applications. The most significant factor is the mechanical and the chemical instability of the lipid films. The lipid films often cannot withstand perturbation of the buffer solution (delamination and disruption upon hydration)[72] and they are susceptible to oxidation (especially in case of unsaturated lipids).[88,89] Also, the addition of functional components to the lipid films often provides the challenge of retaining the functionalities of those components during the integration process (e.g. drying of lipid amphiphiles in an organic solution is the simple way to form self-assembled lamellar films but the membrane proteins cannot retain their biological activity while going through such process).[90] For practical applications, one should find a way to incorporate functional components to the lipid films without losing the structure and functionality of the assemblies. In addition, to serve as model membranes, the existing lipid multilamellar films should be further developed to better mimic the complexity of nature’s membranes. To this end, new approaches to prepare lipid films have been suggested. In this subsection, we will introduce some of the new methods to prepare lipid lamellar films, highlighting their significance in relation to potential applications.
Overall, the preparation of lipid multilamellar films can be divided into two different approaches. One is bulk-scale direct spreading and the other is a layer-by-layer (LBL) assembly approach. The direct spreading consist of depositing stock solutions of lipids dissolved in an organic solvent to the substrates either by drop-casting or spin-coating.[91] By controlling spin-coating conditions or the density of stock solutions, one can control the thickness of stacked bilayers. This procedure rapidly yields multilamellar lipid films even on large-scale.
Based on the direct spreading method, Gupta et al.[92] introduced a way to construct lipid-based multilamellar films with improved structural integrity. The authors have prepared composite lipid-silica films by spin-coating the lipids onto a substrate first and then exposing lipid film to vapors of a silica precursor, tetraethyl orthosilicate (TEOS), as schematically shown in Fig. 3A. During the chemical vapor deposition (CVD) process, silica molecules are assumed to penetrate stacks of lipid bilayers and to condense in the water layer within the lipid bilayers, supposedly interacting with lipid hydrophilic headgroups. The final structure of the lipid-silica composite films comprises alternating lipid bilayers and silica-water layer, maintaining long-range alignment of stacked lipid bilayers. The lipid-silica composite films were robust in air for at least 6 months and stable in aqueous solutions (no delamination was observed). More importantly, the fluidity of lipid membranes was retained in the presence of silica, which is important for mimicking biologically relevant conditions. These lipid-silica composite films may serve as robust model systems to study stacked membranes in nature and as durable platforms for device integration. Although composite lipid films have been less explored, they could potentially provide new solutions to overcome the weak integrity of lipid films while preserving native properties of lipid membranes. In such regard, composite lipid-polymer hybrid films will be reviewed in section 3 of this paper.
Figure 3.
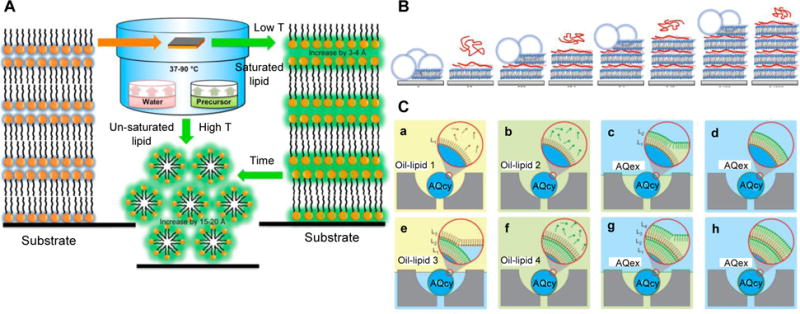
A) Schematic illustration of the CVD process that produces silica encapsulated multilamellar lipid films. When lipids films are exposed to a silica precursor/water vapor environment, silica locate between lipid bilayers (represented in green). Adapted from [92]. Copyright 2013 American Chemical Society. B) Schematic of the buildup process of multilamellar lipid-Poly L-lysine (PLL) films. The lipid vesicle is ruptured and fused onto the substrate followed by addition of PLL (red ribbon) to the lipid bilayer. The steps are repeated to form additional bilayers. Adapted from [93]. Copyright 2015 American Chemical Society. C) Schematic of LBL assembly on microfluidic droplets that enables to build membranes with transbilayer asymmetry. Water-in-oil droplets are trapped in a capture cup (a). Each phase-boundary crossing over the immobilized droplets deposits a new monolayer of lipids on the droplets (c, e, g). Changing the lipid composition in the step (b, f) enables to build asymmetric transbilayers. AQcy stands for aqueous cytoplasmic material and AQex denotes extracellular aqueous phase. Adapted from [94]. Copyright 2012 Macmillan Publishers Ltd.
The LBL method yields to multi-layered lipid films by depositing bilayers one-by-one. The commonly used method to form a base bilayer in contact with the substrate is to induce rupture of giant unilamellar vesicles (GUVs) or large unilamellar vesicles (LUV) at the substrate surface. Multiple interactions to add subsequent bilayers were explored including electrostatic attraction, complementary functional group interactions (e.g. biotin-streptavidin coupling,[95] DNA hybridization[96]), or specific surface chemical interactions (e.g. maleimide-thiol coupling,[61] N-hydroxysuccinimide (NHS)/1-Ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC),[97] amine-sulfhydryl crosslinking[98]).
Heath et al.[93] applied the traditional LBL method in which subsequent layering occurs via electrostatic interactions. The negatively charged lipid bilayer (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine or POPC/1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) or POPG) was formed via vesicle rupture and positively charged poly-L-lysine (PLL) was deposited between lipid bilayers acting as an electrostatic polymeric glue (schematic illustration in Fig. 3B). This approach allows to accommodate a range of proteins in separate bilayers by rupturing different types of proteoliposomes in each bilayer deposition step. In addition, the LBL process utilizing electrostatic interactions is less time consuming and less costly compared to processes that leverage direct chemical interactions.
A novel LBL procedure devised by Matosevic and Paegel allows to build multilamellar lipid membranes with programmable lamellarity and transbilayer asymmetry where lipids are heterogeneously distributed over the two leaflets of the lipid bilayer. [94] As illustrated in Fig. 3C, such layering procedure[94] makes use of monodisperse lipid-stabilized water-in-oil emulsions that are entrapped on microfluidic droplet arrays. The strategy is to mobilize the oil/water phase boundaries over stationary droplets instead of making droplets cross the stationary phase boundaries. For each deposition step, a lipid monolayer is formed on the immobilized droplets crossing the oil/water phase boundary. Because the deposition unit is a monolayer, this method enables to the formation of asymmetric lipid bilayers by varying the chemical composition of the oil-lipid phase. This work provides a systemic route to make model membranes that closely mimic natural cell membranes where the transbilayer asymmetry is ubiquitous.
Furthermore, recent advances in lipid film preparation based on LBL methods would allow for the development of systems that are capable of confining or compartmentalizing molecules of interest in the same way nature does. Not only limited to model membrane systems, such functionality of lipid films could be further applied to engineer advanced drug delivery systems to co-delivery of multiple types of drugs in sequential steps.
In addition to the aforementioned methods other approaches have been developed. Electrospraying has been employed to coat complex surfaces with lipid thin films for the application of complex porous surfaces such as those encountered in food products and pharmaceuticals.[99] Patterning or stamping of stacked lipid bilayers was achieved by polymer stencil lift off (PSLO)[100] or dip-pen nanolithography (DPN)[101] for applications in drug screening and sensing. Building of lipid multilayers from an aqueous dispersion[102] was also tested to facilitate the inclusion of hydrophilic molecules into the lipid films.
2.2.3. Recent applications of lipid multilamellar films
Early application studies on lipid multilamellar films have been confined to the structural studies of reconstituted transmembrane proteins. Recent applications on films, however, show that lipid layers supported on a substrate can have a wide variety of uses. Those applications include artificial cell substrates,[61] bio-electrocatalytic systems for biosensors or photovoltaic cells,[103] matrixes for macroscale drug delivery devices,[104] and substrate-mediated gene delivery.[105] More generally, the use of lipid films to direct the assembly of functional soft and hard materials has been suggested.[106]
Advances on lipid multilamellar films have been driven by the capability of the lipid films to mimic complex multilamellar membranous structures found in nature. A noteworthy direction in lipid film research is their application as a supported film platform for biomaterial-based implantable electronic/photonic devices to realize remotely controlled therapy,[104] which is a rapidly growing field. Figure 4 summarizes recent research directions towards practical applications of lipid multilamellar films.
Figure 4.
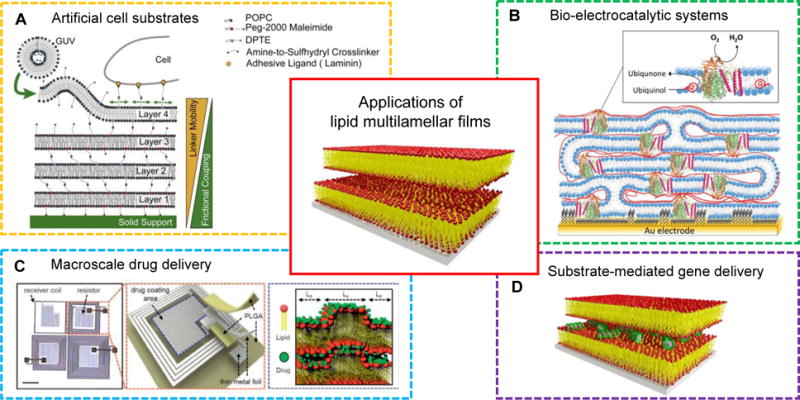
Applications of lipid multilamellar films: artificial cell substrates, bio-electrocatalytic systems, macroscale drug delivery, and substrate-mediated gene delivery. Adapted from [98,103,104]. Copyright 2013 Elsevier Ltd., 2017 WILEY-VCH, 2015 Macmillan Publishers Ltd.
Artificial cell substrates
The extracellular matrix (ECM) provides the physical and biochemical support for surrounding cells. Lipid multilamellar films have been exploited as a cell substrate mimicking the native environment of the ECM.[61,98] Cell behavior in polymer-tethered lipid multilamellar films was studied in response to viscoelastic properties of the films (Fig. 4A).[98] The viscoelastic properties of the cell substrates could be tailored by controlling the number of lipid bilayers or the density of cell adhesion ligands incorporated in the top layer.
Bio-electrocatalytic systems
In nature, electron transport mediated by quinones commonly takes place in the mitochondrial cristae or in the thylakoid stacks in chloroplasts. Quinones are coenzymes that shuttle electrons between membrane enzymes, leading to energy transduction or storage. Heath et al.[103] recently constructed a lipid multilayer matrix with redox-active membrane enzymes for the purpose of mimicking the function of mitochondrial cristae and thylakoid stacks (Fig. 4B). Several lipid-protein bilayers were assembled onto gold electrodes using the LBL method. Lipid membranes containing quinones and quinone-converting enzymes successful provided electron transfer across the membrane layers, as confirmed by means of cyclic voltammetry. Quinones were suggested to diffuse through defect sites within the lipid films where neighboring lipid bilayers are interconnected. This pioneering work could stimulate future studies that model protein interactions in stacked lipid membranes, and advance this field, a step closer to mimic the inherent membrane complexity observed in nature.
Macroscale drug delivery
Macroscale drug delivery (MDD) device, a term defined by Kearney and Mooney,[41] refers to a system delivering bioactive agents such as genes, drugs, and proteins to the desired site by implantation or injection. In order to exert spatiotemporal control over drug storage and release, macroscale biomaterials are often combined into MDD devices in the form of a matrix or reservoir. Hydrogels and polymers have been extensively explored as candidate macroscale biomaterials, but in comparison there is a shortage of MDD studies that leverage lipids as carrier material. Our group recently reported temperature-sensitive lipid multilamellar films that are integrated with electronically programmable and frequency-multiplexed wireless hardware (MDD device).[104] Multiple types of drugs were incorporated into the lipid membranes where the drug transport was actuated by the temperature induced phase transitions of lipid films (Fig. 4C). Below 40 °C, the lipid multilayers comprised two coexisting phases: liquid ordered (Lo) and liquid disordered (Ld). Upon an increase in temperature, the Lo phase transformed into the Ld phase, facilitating diffusion of the hydrophilic drugs out of the films. Such thermotropic phase transition of lipid films would be a useful handle for implantable drug delivery systems with “on/off” switch.
Substrate-mediated gene delivery
One can find another example of utilizing lipid films as a matrix, in the field of gene delivery (Fig. 4D). A new class of matrix-mediated delivery of genes is also referred to as substrate-mediated or surface-based gene delivery. Substrate-mediated gene delivery holds tremendous potential in many biomedical research applications, including medical implant coatings,[107] inductive tissue engineering,[108] and transfected cell microarrays for high-throughput genomic studies.[109,110] To date, carrier materials used in substrate-mediated drug/gene delivery are largely polymers[42] and the use of lipid films as the matrix is scarce. We will point out important aspects of utilizing lipid films in substrate-mediated gene delivery in this section. Our contributions in substrate-mediated siRNA (small or short interfering RNA) delivery are discussed in the section 2.3.
Lipid-DNA films have been successfully prepared by several studies.[111–113] Briefly, lipid-DNA complexes are formed via electrostatic interactions in an aqueous solution and precipitated out as solid film. The complexes are dissolved in 2-propanol or chloroform/ethanol mixtures and then cast as a film. During this process, DNA molecules are intercalated between lipid bilayers. Interestingly, it was shown that DNA undergoes a reversible phase transition between double stranded (active) and single stranded (non-active) conformations at wet and dry conditions, respectively. Concomitant structure changes of lipid (dimethyldidodecylammonium bromide or DDAB) layers were accompanied between bilayer and single layer at wet and dry conditions, respectively. Such phase transition confers greater storage capabilities of lipid-DNA films with humidity-responsive properties, which would be advantageous in MDD applications. Recent studies by Perry et al.[105] tested the in-vitro DNA transfection efficacy of lipid-DNA films. The reported transfection level was very low for the solid films compared to their analogues in particulate form. Although, the gene delivery mechanism from the lipid-DNA films should be further explored, it is postulated that film disassembly plays a significant role in determining the cellular uptake of DNA. When exposed to an aqueous environment, lipid-DNA films can face challenges such as separation of DNA molecules from lipid films leaving DNA unprotected and/or disassembly of the films into large particles that are too big for endocytosis. In this regard, future studies in lipid film-mediated gene delivery should be geared towards understanding cellular uptake mechanisms of genes at a fundamental level with the goal to achieve high DNA transfection efficacies. Also, despite low transfection efficiency, lipid-nucleic acid films are still promising coating materials to deliver genes because lipid molecules can be tuned to quickly respond to external stimuli. We will further discuss the responsive behavior of lipid films and their associated applications in section 2.3.
2.3 Non-lamellar lipid films
Of the two main characteristics of lipids, i.e. excellent biocompatibility and polymorphism, the former has been a main motivation for using lipid films into various systems such as implantable medical devices or cell membrane mimicking structures. The latter, however, has gained considerably less attention in the field of lipid film research. In light of the importance of lipid polymorphism in bulk (herein bulk represents bulk gels or suspensions), the lipid phase behavior—when they are confined to a surface—is also expected to play an important role in the function of the lipid films. To keep up with the increasing need of “film/matrix” materials in various applications and the potential of lipid films for such applications, the structure and properties of non-lamellar lipid films should be better understood. In this section, we will describe the current understanding of lipid polymorphism on confined surfaces, highlighting the applications enabled by their unique capabilities.
2.3.1 Lipid phase behavior on surfaces
It is a general observation that the phase diagram of lipids obtained for bulk systems are translated into films (although there are slight changes which will be discussed later). One can attain non-lamellar phases of lipid films using the same composition and temperature as determined to bulk systems. This includes inverted hexagonal phases(HII),[16,24] inverse discrete micellar cubic (Q227, Fd3m),[23,24] and inverse bicontinuous cubic (QII) phases of two distinct symmetries (diamond Pn3m and gyroid Ia3d).[16,25,114,115] In the category of “non-lamellar” films, the preparation of lipid bicontinuous cubic phase films is of particular interest. These structures comprise a continuous lipid bilayer where the mid-planes conform to periodic minimal surface with the negative Gaussian curvature. The bilayer is in contact with two interwoven yet unconnected networks of water channels (Fig. 5A).[114,116] Such inherent structure of the lipid cubic phase yields large surface area to volume ratios with uniform water channels and 3D isotropic diffusion.[14] Bicontinuous lipid cubic phases in bulk have been utilized for various applications including membrane protein crystallization and drug/gene delivery.[14,17,117–119]
Figure 5.
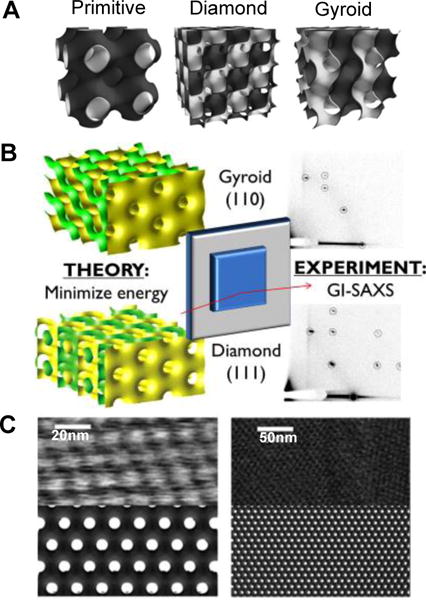
A) Schematic representation of three different bicontinuous cubic phases. The minimal surface represents the mid-plane of a lipid bilayer. Each side of the bilayer has a water domain (represented in dark grey and white) and these domains don’t penetrate. Adapted from [114]. Copyright 2010 The Royal Society of Chemistry B) The crystallographic orientation that cubic phase film adopts with respect to the surface can be predicted from theoretical considerations of surface energy minimization. The predictions are in good agreement with experimental observation. Adapted from [115]. Copyright 2014 American Chemical Society C) AFM images of the diamond cubic phase in water (top) and simulated surface of (111) plane of the cubic diamond phase. The cubic phase films can be directly imaged using AFM. Adapted from [114]. Copyright 2010 The Royal Society of Chemistry.
Monoolein (MO) and phytantriol, well known as cubic-phase forming lipids in bulk, form cubic-phases in supported thin films.[25,115] We recently prepared a positively charged gyroid phase (QIIG) to incorporate negatively charged siRNA in the lipid films, by including a cationic lipid, 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP) to the MO mixtures.[16] Unlike lipid multilamellar films, the lipid cubic phase films can be prepared exclusively by the direct spreading method via drop casting or spin coating from organic solutions including lipids.
The structure of the lipid cubic phase films has been mainly investigated by grazing-incidence X-ray scattering (GISAXS) and AFM. GISAXS in reflection geometry, providing structural information in both parallel and perpendicular directions to the thin film surface. Averaged information on the lipid cubic phase films regarding the symmetry (space group), unit cell size, alignment on surfaces can be readily obtained from the GISAXS measurements. Figure 5B shows an example of GISAXS data obtained for the lipid bicontinuous cubic diamond phase with the (111) plane and gyroid phase with the (110) plane oriented parallel to the substrate.[115] While time-resolved synchrotron experiments would enable the investigation of phase transition kinetics, GISAXS as an average technique cannot provide information on the boundary region of adjacent domains or on the interface between the films and air or water. In this regard, AFM measurements can provide complementary information on the structure of the lipid films because AFM allows direct visualization of domain size, individual water channels, and epitaxy in the lipid films.[114] Figure 5C presents AFM images of supported QII phases in excess water where the nanostructure and film epitaxy is clearly seen.[114]
From the characterization of the lipid cubic phase films, one prominent feature was observed. Lipids align themselves in a highly-ordered manner exhibiting preferential orientation with respect to the substrate. Such preferential orientation can be explained in terms of the interfacial energy between the lipid phase and the substrate. When the lipid bilayers of the cubic phase are in contact with the surface, they form a closed surface in a way that free edges of the lipid bilayer are shielded due to the hydrophobic effect. The closure of the bilayers of the cubic periodic surface results in the formation of both positive and negative mean curvatures, which generates the bending energy.[115,120] Thus, when the lipid cubic phase encounters the substrate interface, it aligns with respect to the substrate in a way that minimize the interfacial energy.[114,119 The preferential orientation adopted by the cubic phase films can be predicted from thermodynamic interfacial energy calculations, and is well corroborated by experimental observations.[115] Those oriented films could provide a good model system to study the pathways of lipid phase transitions, unveiling the epitaxial relationships during transformations.[121–123]
Nylander et al.[24] recently investigated the layers dynamics in the cubic (Fd3m) phase and hexagonal (HII) phase at a Si substrate surface. The cubic phase layers appeared more rigid at the substrate interface compared to the hexagonal phase layers based on neutron reflectometry and grazing incidence neutron spin echo spectroscopy (GINESES) experiments. The rigidity of the Fd3m phase is attributed to the suppressed undulations at the interface whereas the HII phase experiences undulations coming from the hydrodynamic interactions between the HII phase cylinders and the substrate. It is notable that the distance from the substrate to the first HII layer seemed to affect the length of the hexagonally ordered cylinders with their long axis parallel to the surface. Further studies on the surface effect could provide a valuable design handle in controlling the domain size of lipid HII phase films.
Very little work has been carried out towards the effect of different substrates on the cubic structure and its dynamics in non-lamellar lipid films. It would be interesting to understand the role of hydrophilic/hydrophobic functionalization of the surface in determining the structure of the lipid non-lamellar phase on solid supports (e.g. the domain alignment, flexibility of the layers, and shift in phase boundaries). Understanding the self-assembly, thermodynamics and dynamics of lipid films onto solid supports will allow for their rational design of functional films with tunable polymorphic properties.
2.3.2 Applications of non-lamellar lipid films
Applications of non-lamellar lipid films are almost unexplored at this point. Nanostructured lipid thin films have been utilized as matrices for substrate-mediated gene delivery,[16] as templates for in-situ metal growth into periodic nanostructures,[124,125] and as host materials to incorporate nanoparticles with macroscopic alignment.[106] Since we limit the scope of this paper to biomedical-related applications, we will introduce the previous application of non-lamellar lipid films focusing on substrate-mediated gene delivery.
We have recently prepared three different lipid films adopting a 1D lamellar, 2D HII and 3D Q GII or mixture of two phases with proper control of lipid compositions, temperature, and relative air humidity.[16] Those lipid films were shown to experience reversible phase transitions from one to another phase upon a temperature or humidity change.
When siRNA molecules were incorporated into the lipid films via electrostatic interactions, we observed slight changes in the phase behavior (Table 1) compared to neat lipid films. At 70/30 and 60/40 GMO/DOTAP (mol %/mol %) compositions, the neat lipid films exhibited a QIIG phase while lipid-siRNA films showed the coexistence of QIIG and HII phases. This implies that the incorporation of siRNA molecules into the QIIG phase directs the distortion of the water channels leading to a phase transformation into the HII phase. This behavior has been observed for bulk systems too.[126] It is also noteworthy from Table 1 that the lipid-siRNA films exhibit shifted phase boundaries compared to the lipid-siRNA particulates in solution. The QIIG and HII phases are preserved at higher DOTAP molar percent in films compared to bulk. The shift in phase boundaries may come from different responsiveness between films and bulk with respect to the introduction of siRNA negative charges to the assemblies. The lipid films are interfaced with two parts—the substrate and water layer—whereas the particulates are only exposed to bulk water. The changes in the net charge of the lipid head-groups due to siRNA pinning, affect the effective area per lipid head-group and hence the lipid molecule geometry. In turn, this concomitantly alters the interfacial energy, which is a function of the mean curvature of the membranes in the lipid constructs. Such changes in interfacial energy accompanied by membrane charge density overall may affect the epitaxial (orientational) relationships during phase transformations (QIIG to HII and HII to Lα), resulting in the shifts of the phase boundaries.
Table 1.
Phase diagram of MO/DOTAP lipid films with and without siRNA. RH stands for relative air humidity. , HII, Lα symbols represent a bicontinuous gyroid cubic, an inverted hexagonal, and a lamellar phase, respectively. Adapted from [16]. Copyright 2016 WILEY-VCH.
| Molar Ratio (mol%/mol%) | Dry Film (25°C, 20~40% RH) | Wet Film (37°C, 95±5% RH) | Bulk Solution (37°C) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||
| GMO/DOTAP | − siRNA | + siRNA | − siRNA | + siRNA | − siRNA | + siRNA | ||||
| 85/15 | Lα | Lα |
|
|
|
|
||||
| 75/25 | Lα/HII | Lα/HII |
|
|
|
|
||||
| 70/30 | HII | HII |
|
+HII |
|
HII | ||||
| 60/40 | HII | HII |
|
+HII |
|
HII+Lα | ||||
| 50/50 | HII | HII | Lα | Lα | Lα | Lα | ||||
| 25/75 | HII | HII | Lα | Lα | Lα | Lα | ||||
Figure 6 shows GISAXS diffraction patterns of a QIIG phase before and after siRNA incorporation. The elongated diffusion spots after siRNA inclusion indicates that some degree of order is lost with a distribution of domain orientations, but the preferred uniaxial orientation is still preserved. Interestingly, the composite lipid films not always show out-of-plane alignment with respect to the substrate. When platinum was electrodeposited into one of the two water channels of the lipid cubic phase template (QIID), a net crystallographic orientation of the Pt film relative to the substrate was dependent on the thickness of the Pt film.[124,125] When the thickness of Pt film was 1 μm to 2 μm thick, the nanostructured Pt films having Fd3m morphology showed out-of-plane alignment with the (111) plane parallel to the substrate, whereas for thicker Pt films (> 2.8 μm) the orientational order was lost.[125] In our work, we observe that the uniaxial orientation of the lipid-siRNA film is maintained for the whole film thickness (≈ 30 μm). We expect further studies would disclose the relationship between the film thickness and the structure alignment in terms of interfacial energy.
Figure 6.
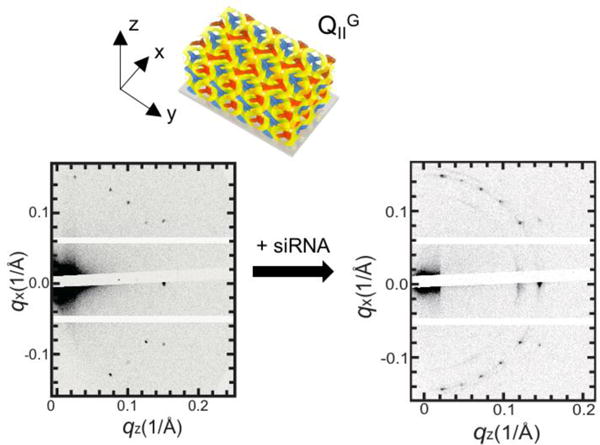
GISAXS data of 70/30 (mol %/mol %) GMO/DOTAP films before and after siRNA incorporation equilibrated at air humidity. Upon siRNA addition, the QIIG phase transformed into a mixture of the QIIG and HII phases. Note that lipid-siRNA films are more disordered compared to the lipid-only films. Adapted from [16]. Copyright 2016 WILEY-VCH.
When three different nanostructured lipid films were applied in siRNA knockdown experiments, the QIIG phase film showed the superior siRNA silencing efficacy compared to other films. This is attributed to the inherent structural properties of the QIIG phases that possess positive Gaussian modulus leading to efficient endosomal escape.[16,17] The significance of this work is the exploitation of nanostructures for the first time to regulate the efficacy in substrate-mediated gene delivery applications. Also, it is worth noting that the responsive phase behavior of nanostructured lipid thin films was controlled with changes in relative humidity. Lipid assemblies, especially liposomes, have received tremendous attention as stimuli-responsive materials in various fields. They can be prepared to be responsive to external stimuli such as water content, temperature, pressure, or ultrasound,[20] mimicking the responsiveness of living organisms. Different lipid phases show distinct properties of molecular diffusion, cell adhesion, and permeation, etc.[14,16,118,127] Thus, the use of lipid nanostructured films that take advantage of stimuli-responsive properties would give rise to tunable diffusion and adhesion/permeation properties for encapsulated molecules of interest.
On a final note, recent studies of lipid cubic phases in bulk are widening their functionality, which could further extend future applications of lipid cubic phase films. For example, Mezzenga and collaborators demonstrated that lipid cubic phases in bulk can serve as matrices for the detection of a vast class of analytes including disease biomarkers, viruses, bacteria, and parasites.[19] In this work, a new assay principle was introduced based on the optical properties of the bicontinuous cubic phase. Optically isotropic lipid cubic phases become birefringent upon enzymatic reactions, hence birefringence signal can be used as an optical output.[19] Such changes in optical properties of the lipid cubic phase could be further applied to the cubic phase thin films for high throughput biosensing and biocatalytic fuel cell devices.
3. Lipid-Polymer Hybrid Membranes
Block copolymers composed of hydrophilic and hydrophobic groups can self-assemble into various structures in a similar manner to lipids such as micelles, vesicles, and tubes.[128,129] Despite the similarities between lipids and block copolymers in terms of their amphiphilic nature and the ability to self-assemble into various morphologies, research on lipid assemblies and block copolymer assemblies have followed separate routes.
Polymer assemblies and lipid assemblies show different physicochemical properties. Polymersome membranes are usually thicker (≈ 8 nm to 50 nm per single bilayer) than that of liposomes (≈ 5 nm per single bilayer) because of the higher molecular weight of the polymer blocks, leading to better stability and higher mechanical strength compared to liposomes.[43,44,130,131] However, thick membranes also yield poor membrane permeability which can limit the diffusion of small molecules that are encapsulated inside polymersomes, [130] as well as poor membrane-fusion capabilities. The synthetic nature of block copolymers allows versatility to modulate chemical functionality but simultaneously results in lack of biocompatibility. In contrast, lipid membranes found in all cell membranes are biocompatible.[43,44]
In an effort to combine the benefits of the two materials, hybrid systems, composed by lipid-polymer mixtures have been recently explored[132–134,44,135–137]. By mixing lipids and block copolymers in the same membrane, one can expect to obtain hybrid membranes with tunable structural properties and good biocompatibility. A broader range of chemical compositions, molecular weight, hydrophobicity, and surface charge of assembling building blocks (i.e. block copolymer and lipids) would enable modulation of the hybrid membranes physicochemical propertiesincluding permeability, mechanical stability, and solubility of encapsulated molecules. Moreover, from a materials science perspective, hybrid systems of two different materials are exciting as they may give rise to new structures and properties otherwise not attainable with single component systems.
Research on lipid-polymer hybrid membranes is at a very early stage of development with just a few studies reported so far. However, previous work already alludes to numerous potential applications of hybrid membranes. This includes model systems mimicking plasma membranes, sensors, and small molecule (drug, genes, and proteins) delivery platforms. Hybrid vesicles, composed of mixtures of phospholipids and block copolymers, have been shown to form various membrane phases with different miscibility.[43,135] Molecular and macroscopic parameters such as copolymer architecture, lipid fluidity, hydrophobic mismatch or chemical compatibility between lipids and polymers determine the final phase of the hybrid vesicles. Both lateral phase separation at the nanoscale and micrometer scale or complete fission into separate vesicles have been reported.[43,44] Careful selection of lipid and copolymer molecules with engineering parameters such as temperature, cooling rate, and osmotic control enables the modulation of phase separation at the micro- and nano-scale.[45,136–138] Such complicated but diverse phase behavior of lipid-polymer hybrid systems may offer new insights to understand complex plasma membrane systems where the presence of phase separated domains goes beyond classical liquid-ordered and -disordered lipid-only phases. Also, the compositional variety of hybrid membranes could possibly confer advanced functionalities such as control over membrane compartmentalization, diffusion rates of membrane components, and mechanical stability.
The original studies on lipid-polymer hybrid membranes were performed on GUVs because of the ease of optical characterization. Besides GUVs, recent work has explored other systems including large unilamellar vesicles (LUVs),[138] tubular vesicles,[139] planar membranes,[140,141] and most recently from our group, multi-layered films.[142] In this section, we will review the lipid-polymer hybrid systems focusing on planar membranes in the form of suspended monolayers, solid-supported bilayers, or multi-layered films on solid supports. We will highlight the approaches to construct hybrid membranes, followed by the structure, function, and possible applications of the newly assembled structures.
3.1 Suspended lipid-polymer monolayer
A mixed lipid-polymer monolayer at the air/water interface has been studied as a platform for directed membrane protein insertion and for controlled localization of polymer-functionalized nanoparticles in the membranes. Two important features of the mixed lipid-polymer monolayer give rise to controlled distribution of molecules embedded into the hydrophobic region: surface hydrophobicity and heterogeneity (phase-separated domains).[141,143]
Kowal et al.[141] combined amphiphilic copolymer poly(dimethyliloxane)-block-poly(2-methyl-2-oxazoline) or PDMS-b-PMOXA with various phospholipids to investigate the tunability of membrane protein incorporation into the monolayer. The mixtures of lipids and polymers resulted in phase separation of two components into different domains, of which the size and shape were affected by the type of lipids and lipid/polymer compositions. Interestingly, membrane proteins were preferentially located into the more fluid regions of the monolayer. When saturated lipids (1,2-dipalmitoyl-sn-glycero-3-phophocholine (DPPC), DPPE) were mixed with PDMS-b-PMOXA, proteins favored the polymer-rich domains over lipid-rich domains while the opposite trend was observed for unsaturated lipid (DOPC)-polymer mixtures. This work raises a possibility of achieving model membranes that are biologically relevant by introducing controlled distribution of membrane proteins at the desired sites.
The work by Olubummo et al.[143] also demonstrates the localization of molecules of interest (polymer-coated nanoparticles) into mixed lipid/polymer monolayers. The mixture of block copolymer poly(isobutylene)-block-poly(ethylene oxide) or PIB-b-PEO and phospholipid DPPC formed a monolayer at the air/water interface. It was observed that the presence of PIB-b-PEO in the hybrid layer disturbs the lipid packing, inducing rearrangement of lipid molecules followed by changes in the LE (liquid-expanded)/LC (liquid-condensed) transition plateau. In addition, the lipid-polymer system phase separated into different domains with the size of the polymer domains increasing with the polymer content. Surface functionalization of CdSe nanoparticles played a pivotal role in controlling the location of those nanoparticles in the monolayer. The particles coated with PIB were homogeneously distributed whereas the ones coated with PIB-b-PEO showed heterogeneous distribution (preferentially embedded into the polymer domains).
Partitioning molecules into selective membrane regions is an interesting outcome enabled by the mixing of different self-assembling building blocks: lipids and block-co-polymers. The factors driving phase-separation of lipid-polymer mixtures should be further explored to fine-control the layer structure and the distribution of molecules of interest. Hybrid monolayer systems may be useful to the development of membranes where spatial control of embedded components is required.
3.2 Supported lipid-polymer bilayer (SLPB)
Supported lipid-polymer bilayers (SLPB) are analogues to supported lipid bilayers (SLB) but differing with respect to composition, comprising block copolymers in addition to lipids. It should be noted that SLPBs are different systems than polymer cushioned lipid bilayers where a hydrophilic polymer brush is located between a (polymerized) lipid bilayer and the solid support. While polymer brush-SLB systems utilize polymers to fill the space between lipid bilayers and the substrates, polymers in SLPBs interact with the lipids within the same bilayer thus providing new biophysical/biochemical properties to the membrane. The concept of introducing heterogeneity into the membrane is inspired by nature. Cell membranes comprise a vast class of lipids and proteins and heterogeneities are known to mediate various cellular processes.
The work by Gettel et al.[140] demonstrates the construction of SLPBs and their use as model membranes for studying obstructed diffusion. The main findings of this work are depicted in Fig. 7. They have exploited UV photochemical techniques to pattern surfaces. The substrates were patterned by exposing n-octadecyltrichlorosilane (OTS) covered substrates with a photomask to ozone-generating, short-wavelength UV light (187 nm - 254 nm). This is schematically represented in Fig. 7A. Two different approaches were developed to form the SLPBs: 1) mixtures of lipid-polymer hybrid vesicles were adsorbed and fused into the patterned substrates, resulting in the formation of a monolayer or a bilayer on the hydrophobic and hydrophilic regions, respectively; and 2) designated regions of surfaces all filled with polymer bilayers were selectively removed and backfilled with lipid bilayers through patterning.
Figure 7.
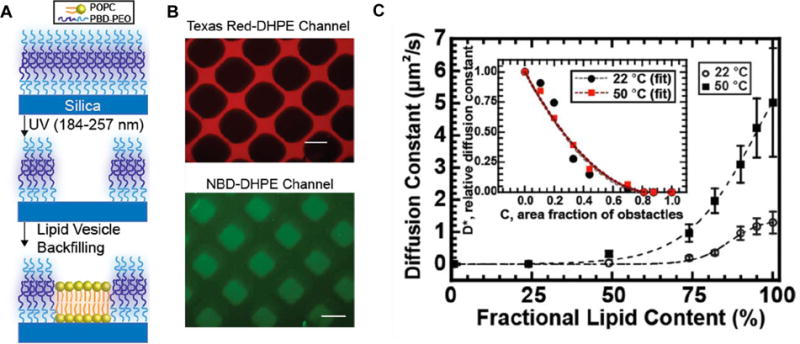
A) The process of lipid-polymer supported membrane preparation: deposition of polymer PBDPEO bilayer followed by selective removal and backfilling with lipid POPC bilayer. B) Epifluorescence images of polymer (doped with Texas Red-DHPE) and lipid (doped with NBD-DHPE) bilayers. Scale bar = 100 μm. C) Diffusion constants of probe lipids in hybrid lipid-polymer membranes at different molar ratios. The inset shows the relative diffusion constant of the probe lipids per area fraction of polymers. Adapted from [140]. Copyright 2014 American Chemical Society.
Interestingly, the mixtures of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and poly(butadiene-b-ethylene oxide) (PBDPEO) did not show lateral phase-separation at the macroscopic level when adsorbed onto amphiphilic surfaces as observed by epifluorescence images (Fig. 7B). Such seemingly homogenous distribution of lipid and polymer was unexpected because of the height mismatch between the POPC (≈ 5 nm thick) bilayer) and the PBDPEO (10 nm to 12 nm thick) bilayer. Indeed, phase-separation was observed for GUVs made of comparable mixtures. The observed membrane homogeneity implies an irreversible adsorption of components which was investigated by measuring the lateral diffusion coefficient of POPC in the SLPB (Fig. 7C). The POPC lateral diffusion was clearly hindered by the presence of PBDPEO on solid supports. The diffusion behavior of POPC in the SLPB clearly differs from the one in freely floating membranes (e.g. GUVs). The mechanism of substrate-mediated fusion of PBDPEO is thought to be responsible for bringing irreversibility in the SLPB, which needs to be further investigated. The implication of this study on SLPBs is that it allows the investigation of obstructed diffusion behavior of transmembrane proteins and lipids in plasma membranes,[144] a process that is still not well understood. Besides block copolymers, another type of synthetic polymer (dendrimer) was employed to form lipid-dendrimer co-assembly structures on solid supports.[145] The hybrid dendrimer/POPC vesicles were exposed to the hydrophilic substrates and incubated in an aqueous environment to allow them to form a well-defined supported bilayer. The fluidity and stability of the membrane could be modulated by controlling the generation (the number of repeated branching cycles) and concentration of the dendrimer. Also, the versatility of the functional end groups in the dendrimers eased the conjugation of biological recognition ligands to the membrane, offering new opportunities to develop powerful sensors.
3.3 Multi-layered phase-separated films
In this section, we discuss our recent work on multi-layered lipid-polymer hybrid films systems with nanostructures that enable synergistic and controlled delivery of paclitaxel – a powerful drug that is often challenging to encapsulate and release.[142] We also include very recent (unpublished) structural and chemical composition characterization obtained by photothermal induced resonance (PTIR, Fig. 10) and dynamical characterization obtained by solid-state NMR (Fig. 11).
Figure 10.
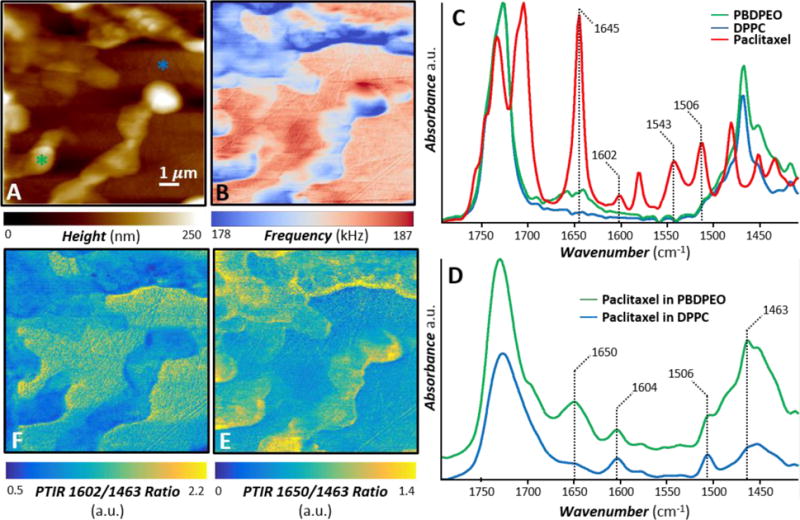
PTIR nanoscale chemical imaging. A) AFM topography image and B) AFM contact frequency image of a paclitaxel loaded (0.05 molar fraction) hybrid 1:1 molar ratio DPPC/PBDPEO film. The features with higher topography and lower contact frequency identify the polymer reach domains. C) Normalized FTIR spectra of PBDPEO (green), DPPC (blue) and paclitaxel (red). D) Characteristic PTIR spectra (displayed with a common intensity scale) obtained in the color-coded positions identified in panel A. In addition to the polymer and lipid characteristic bands (1463 cm−1, 1730 cm−1), a few distinct and characteristic paclitaxel bands indicate that the drug is partitioned in both the polymer and lipid phases. E) PTIR ratio map obtained by dividing the intensity of the 1650 cm−1 PTIR map (paclitaxel amide I band) over the 1463 cm−1 PTIR map (polymer and lipid band) reveals the heterogeneous distribution of paclitaxel in the polymer rich phase. F) PTIR ratio map obtained by dividing the intensity of the 1602 cm−1 PTIR map (paclitaxel C=C stretching band) over the 1463 cm−1 PTIR map (polymer and lipid band).
Figure 11.
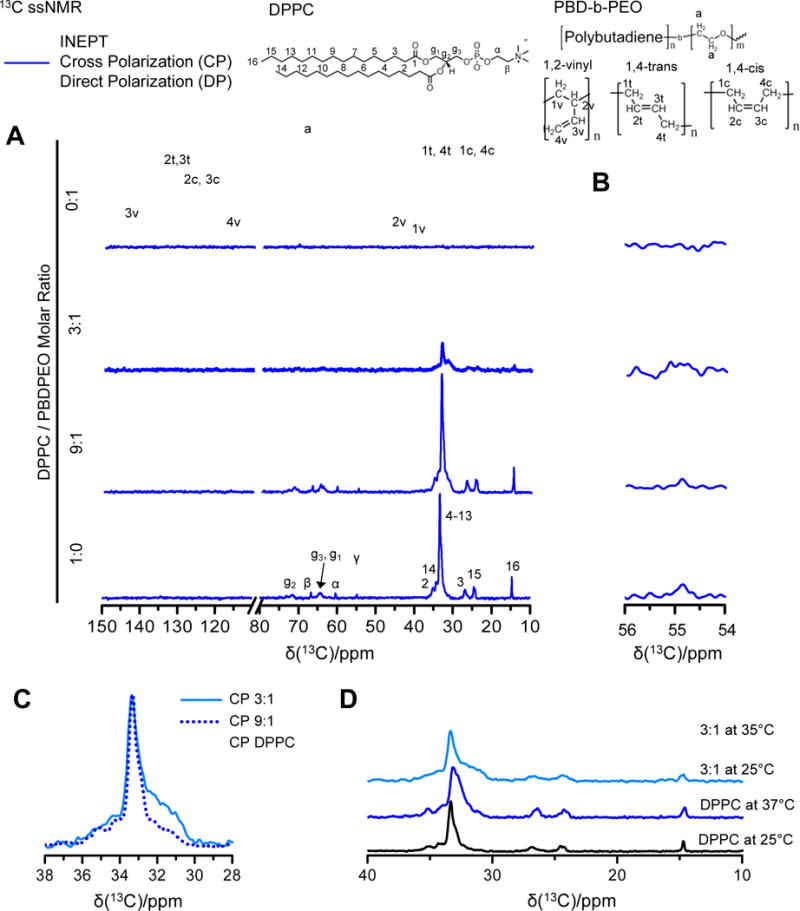
13C solid state NMR spectra of lipids, polymers, and lipid-polymer hybrids. Top: Molecular structure of lipid DPPC and polymer PBDPEO with the carbon atom labeling. A) The combined DP (gray lines)-CP (blue lines)-INEPT (red lines) sets of DPPC (bottom), 9:1 molar ratio DPPC/PBDPEO, 3:1 molar ratio DPPC/PBDPEO (middle), and PBDPEO (top). All samples were measured at 25 °C. The spectrum of DPPC exhibits dominant CP signals compared to INEPT while that of PBDPEO presents the opposite trend: strong INEPT signals without CP. The peaks at 54.8 ppm (denoted by dashed lines) are enlarged in B) to emphasize the relative signal amplitudes of DP, CP, and INEPT. Resonances at 54.8 ppm corresponds to the DPPC headgroup moiety “γ”. As PBDPEO is incorporated into DPPC membranes, CP of headgroup “γ” decreases along with increased INEPT to DP ratios, indicating that the lipid headgroup becomes fluidic upon polymer incorporation. C) The CP spectra of DPPC (black, solid) and 9:1 molar ratio DPPC/PBDPEO (blue, dotted) and 3:1 molar ratio DPPC/PBDPEO (skyblue, solid). The spectra are normalized to equal intensity of the 33 ppm shift which is characteristic of the acyl chains adapting all-trans conformations. The significant line broadening is observed in the regions of 30-33 ppm for DPPC/PBDPEO hybrids, which can be attributed to the perturbed lipid acyl chain packing in the presence of polymer yielding a distribution of different conformations. D) The CP spectra of DPPC and 3:1 molar ratio DPPC/PBDPEO are compared at two different temperatures, 25 °C and 37 °C. Spectra are labelled by compound name. The broad peak of DPPC/PBDPEO differs from that of DPPC even at elevated temperature. The appearance of carbon peak at 31 ppm from 3:1 molar ratio DPPC/PBDPEO at 37 °C reveals that some portions of acyl chains in the hybrids already transform into liquid-like conformations below the Tm of DPPC (41 °C).
Analogous to stacked bilayers of ternary lipid mixtures by Tayebi et al.,[80] binary mixtures of lipid/polymer exhibit a peculiar phase behavior. Lipids and polymers phase-separate into lipid-rich and polymer-rich domains and those domains are in registry across micrometer-thick films, therefore yielding a three- dimensional phase separation. Such 3D segregation imposes synergistic permeability of encapsulated hydrophobic drug molecules (Paclitaxel) through the hybrid membranes.
Mixtures of lipids (DPPC) and polymers (PBDPEO) in an organic solution were dried onto a solid surface, yielding self-assembled hybrid films. Figure 8 shows the structural characterization of the self-assembled lipid-polymer films by Confocal Laser Scanning Microscopy (CLSM), AFM, and GISAXS. Co-existence of polymer-rich and lipid-rich domains in the membrane and their out-of-plane alignment were confirmed by CLSM (Fig. 8A) and GISAXS (Fig. 8B), respectively. As in multilamellar lipid films,[80] like-domains stacked up across multiple membrane layers aligning themselves parallel to the substrate. The large height mismatch between lipid and polymer individual layers leads to formation of extensive phase boundaries, as measured by AFM topography & phase images (Fig. 8C).
Figure 8.
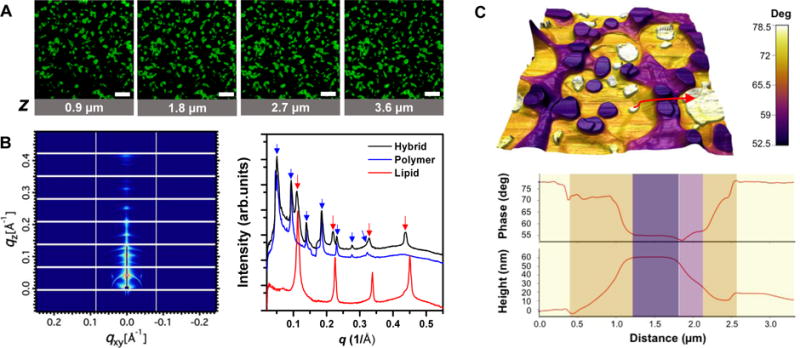
A) Confocal fluorescence microscopy images obtained at different depths (z) inside lipid-polymer hybrid membranes (1:1 molar ratio DPPC/PBDPEO) in bulk water doped with NBD-DPPE (0.001 molar fraction). Binary spatial patterns continue across the membrane normal, suggesting domain alignment across multilamellar membranes. Scale bars = 50 μm. B) GISAXS two-dimensional raw data of hybrid membranes (left) and one-dimensional I(q) profiles of hybrid, lipid, and polymer membranes (right) obtained at > 95% relative humidity. C) AFM phase contrast image overlaid onto pseudo-3D topography of the hybrid membranes (top). Cross-sectional profiles of the phase and topography along the arrow marked in the image (bottom). Adapted from [142].
Figure 9 shows in-vitro cumulative drug paclitaxel release profiles from neat lipid, neat polymer, and lipid-polymer hybrid films each loaded with paclitaxel (0.02 molar fraction). Interestingly, hybrid membranes showed a synergistic permeability compared to single-component films that was attributed to the presence of the extensive domain interfaces in the hybrid films which impede paclitaxel crystallization and provide areas for enhanced diffusion (i.e. leakiness).
Figure 9.
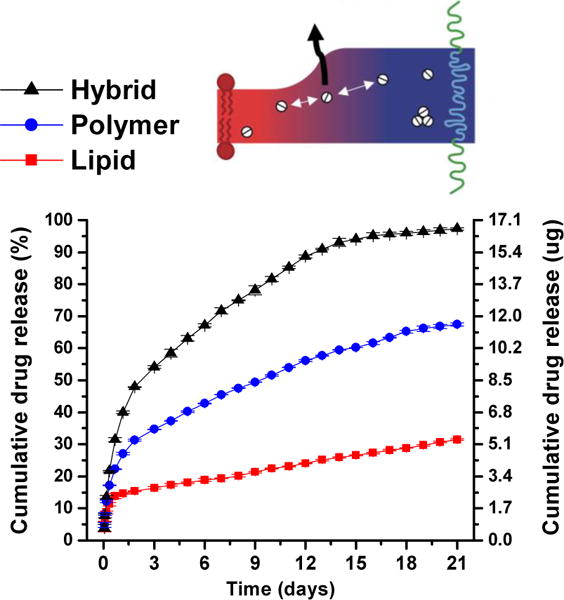
In-vitro cumulative release profiles of paclitaxel from 1:1 molar ratio DPPC/PBDPEO hybrid (black triangles), PBD-b-PEO (blue circles), and DPPC (red squares) membranes with 0.02 molar fraction paclitaxel incorporated. Adapted from [142].
Here, we leverage PTIR experiments to study the distribution of paclitaxel in a paclitaxel loaded (0.05 molar fraction) hybrid DPPC/PBDPEO (1:1 molar ratio) membrane with nanoscale resolution (Fig. 10). PTIR, also known as AFM-IR, is an emergent technique that combines the high spatial resolution of AFM with the composition specificity of infrared (IR) spectroscopy.[146,147] The proportionality between the PTIR signal and the energy absorbed locally by the sample,[148,149] as in conventional IR spectroscopy, allows material identification at the nanoscale by comparison with far-field IR spectral databases.[147] Recent reviews[146,147] discuss the PTIR working principles and an ever growing list of applications spanning from biology[150,151] to materials science,[152–155] and includes studies on the nanoscale distribution of drug-polymer blends.[156,157] Recently, PTIR has been extended to the visible range,[158] enabling measurement of semiconductor bandgap at the nanoscale.[159,160] Furthermore, the development of nano-sized picogram-scale probes capable of capturing the sample thermalization dynamics in PTIR experiments, has added the ability to measure local thermal conductivity of the sample.[161]
The AFM topography (Fig. 10A) and contact resonance frequency (Fig. 10B) images of the paclitaxel loaded hybrid film show the phase separated polymer rich (higher topography, lower frequency) and lipid rich (lower topography, higher frequency) domains. Fig. 10B suggests that the polymer rich domains are softer (lower frequency) than the lipid domains because the AFM contact resonance frequency is proportional to the local sample stiffness.[162] This observation corroborates Fig. 8C, where the thicker polymer domains display a lower phase value than the lipids due to the lower Young’s modulus of the polymer.[163] The FTIR spectra of pure paclitaxel, DPPC, and PBDPEO (Fig. 10C) show that the lipid and the polymer have very similar IR spectra (i.e. it is difficult to spectroscopically differentiate them). However, the paclitaxel spectrum shows a few distinct bands that do not overlap with the polymer or the lipid bands, and these peaks: 1645 cm−1 (Amide I), 1602 cm−1 (C=C stretching), 1543 cm−1 (amide II), and 1506 cm−1 (C=C stretching)[164] were leveraged for the subsequent PTIR experiments. In addition to prominent polymer and lipid bands (≈ 1730 cm−1 and ≈ 1463 cm−1), representative PTIR spectra (Fig. 10D) reveal the presence of paclitaxel (see bands at 1650 cm−1, 1604 cm−1 and 1506 cm−1) in both the polymer and the lipid domains. Notably the amide I peak of paclitaxel at 1650 cm−1 broadens upon the drug’s inclusion in the polymer, and broadens and weakens considerably upon the inclusion in the lipid (Fig. 10D), perhaps suggesting a stronger interaction between the amide group in the drug and the lipid. In contrast the C=C stretching bands at 1604 cm−1 and 1506 cm−1 are relatively stronger for the paclitaxel incorporated in the lipid phase than in the polymer phase, suggesting a stronger interaction of aromatic groups in paclitaxel with the PBD block in the polymer than with the lipid phase. PTIR chemical maps are obtained by illuminating the sample at a given wavelength while scanning the AFM probe on the sample to enable the visualization of different components. However, it is known that sample locations with higher stiffness, characterized by higher contact resonant frequency (Fig. 10B), can provide a little stronger PTIR signal amplitude.[165] Consequently, it is a common practice[165] to analyze a ratio of PTIR maps obtained at two wavelengths (Fig. 10 E, F) to cancel out the effect of the sample stiffness variability on the PTIR signal intensity because such effect at each location is wavelength independent. The PTIR ratio map (Fig 10E) of the 1650 cm−1 band (amide I of paclitaxel) over the 1463 cm−1 band (polymer and lipid) highlights the spatial distribution of paclitaxel in the film and, particularly, the heterogeneous distributions in the polymer rich phase. Interestingly, it appears that the paclitaxel concentration is somewhat enhanced along many boundaries between the polymer rich and lipid rich domains, thus corroborating our previous hypothesis.[142] The PTIR ratio map (Fig 10F) of the 1602 cm−1 band (C=C stretching of paclitaxel) over the 1463 cm−1 band (polymer and lipid band) shows that the overall distribution of paclitaxel in the lipid rich phase is homogeneous, however a slightly stronger intensity is observed along some lipid-polymer interfaces, perhaps suggesting again a slightly higher concentration of the drug in these regions.
To investigate the changes in molecular configuration that lipid molecules adopts upon polymer incorporation, we performed 13C ssNMR experiments which can provide atomically resolved information about molecular conformations and reorientational dynamics. The results are presented in Figure 11. Three different 13C NMR measurements were combined: DP (direct-polarization), CP (cross-polarization),[166] and refocused INEPT (insensitive nuclei enhanced by polarization transfer).[167] The two different methods of 1H-13C polarization transfer used for enhancing the 13C signals (CP and INEPT) complement each other as they respond differently to the reorientational dynamics of the C-H bonds (which can be quantified with the correlation time τC and order parameter SCH[168]). Solids (τC > 0.1 μs and/or |SCH| > 0.5) yield intense CP signals without INEPT while anisotropic liquids (τC < 10 ns and 0.05 < |SCH| <0.2) give comparable CP and INEPT signals.[169,170] For isotropic liquids (τC < 10 ns and |SCH| < 0.01), strong INEPT with vanishing CP is observed.[169,170] Thus from the relative ratios of INEPT and CP signals, one can readily obtain qualitative information about molecular segment mobility.
Figure 11A shows the DP-CP-INEPT set of 13C spectra of DPPC (bottom), DPPC/PBDPEO hybrids (middle), and PBDPEO (top). The DP, CP, and INEPT spectra were superimposed and color-coded in gray, blue, and red, respectively. The peaks were assigned based on single-component control samples of DPPC and PBDPEO, of which values agree with the previous references.[171–173] For DPPC measured at 25 °C, the polarization transfer efficiency ICP > IDP≫IINEPT ≈ 0 was observed as expected for the solid gel phase[174] (Tm, DPPC = 41°C). An opposite trend was found for PBDPEO, suggesting that polymer carbon chains are liquid-like and mobile. DPPC/PBDPEO hybrids display peaks associated with DPPC and PBDPEO which seem to be the simple overlap of two phases at first glance. A closer look, however, reveals two characteristic differences between hybrids and single-component samples. Figure 11B shows the magnified region of 54 ppm – 56 ppm corresponding to the lipid headgroup moiety (γ). The relative intensities of DP, CP, and INEPT of the carbon γ clearly show the changes in lipid headgroup dynamics when polymer is present. For DPPC, the polarization transfer efficiency follows the order IINEPT ≈ IDP ≫ ICP > 0 but as the PBDPEO content increases, the relative intensities become IINEPT > IDP ≫ ICP ≈ 0. Such increase in INEPT/DP ratios and absence of CP signals[169,170] indicate that the segment γ in DPPC which has some degree of anisotropy in its pure phase undergoes complete isotropic reorientation when PBDPEO co-exists. This is a clear indication that the system assembles as a hybrid polymer-lipid membrane and not as two completely phase-separated sets of polymers and lipids.
Other notable differences between pure and hybrid phases are shown in Fig. 11C and 11D. Figure 11C compares normalized CP scans of DPPC and DPPC/PBDPEO hybrids in a range of 28 ppm -38 ppm. One can easily distinguish whether the lipid tail hydrocarbon chains are in an all-trans conformation or chain configurations containing gauche forms by looking at the acyl chain peak positions. The broad peak centered at 33 ppm originates from the central segment of the acyl chains (C4-13) in all-trans conformation[174] whereas liquid acyl chains with a distribution of trans- and gauche conformations show the peak at 31 ppm.[175] Interestingly, DPPC/PBDPEO hybrids exhibit the acyl chain peak with a broad shoulder in comparison to DPPC, indicating that some portions of the acyl chains are in not only all-trans but acquires a distribution of trans- and gauche conformations. Such behavior on the conformational distribution of the acyl chains can only be understood as a perturbation of lipid chain packing due to the coexistence of polymer molecules within the membrane.
To further investigate the characteristics of different chain conformations in DPPC/PBDPEO hybrids, we compared DPPC and DPPC/PBDPEO hybrids at 37 °C at which DPPC is in the ripple phase . The ripple phase is characterized by periodically undulated bilayers with troughs and ridges.[176] Figure 10D shows the 37 °C data obtained for DPPC where an upfield shift of the C4-13 peak with a broadened shoulder can be observed. Notably, the peak shape clearly differs from that obtained for DPPC/PBDPEO at 25 °C, indicating that the broadened C4-13 peak of DPPC/PBDPEO is not just a simple result of the DPPC phase transition from the gel to ripple ( to ). Continuous resonances of DPPC/PBDPEO in the 31-33 ppm region rather reflect a wide distribution of chain conformations in the DPPC acyl chains. At 37 °C, the shoulder of the main acyl chain peak at 33 ppm of DPPC/PBDPEO separates out to another peak centered at 31 ppm, indicating that an elevated temperature causes some of the hydrocarbon chains to acquire a liquid-like distribution of conformations. Taken all together, the DPPC acyl chains go through conformational changes when PBDPEO molecules are incorporated and such distribution of different conformations in DPPC/PBDPEO differs from the ones seen in pure DPPC phases ( or ). In AFM phase imaging (Fig. 8C), we observed gradual AFM topography and phase changes between lipid domains and polymer domains, likely indicating the presence of mixed domains where lipid and polymer molecules co-exist. In line with the AFM phase data, the continuous resonances at 31ppm - 33 ppm of DPPC/PBDPEO may come from the wide range of (or gradual conformational changes in) the molecular states that DPPC adapt in the presence of PBDPEO molecules.
In this work, we have demonstrated the use of multilayered lipid-polymer hybrid films for substrate-mediated drug delivery applications. Intralayer and interlayer domain ordering of hybrid films has implications for accurate control of the permeation behavior of embedded solutes through the films.
4. Summary and Outlook
Until recently, lipid films have been mostly used as model systems to mimic cell membranes. The aim of this review is to show that supported lipid-based materials have the potential to catalyze many substrate-mediated applications such as drug/gene delivery, biomimetic energy conversion, and sensing.
Lipids are biocompatible and possess the capability to self-assemble into various phases both in nature and in artificial systems. Lipid molecules are the structural motifs that comprise cell membranes, indicating that lipid self-assembled structures are versatile in terms of adapting to external stimuli/environmental changes. These characteristics should be very compelling arguments to investigate and exploit lipid materials beyond cell membrane models. One can envisage biocompatible lipid films becoming instrumental to the development of new medical devices requiring susceptibility in response to specific stimuli. Importantly, when lipids self-assemble onto a support, interesting structural features are observed such as specific orientation and stacking. Multicomponent lipid mixtures phase-separate into co-existing domains and those domains stack up in registry, resulting in columnar alignment across lipid multilayers.[80]
Lipid polymorphism as seen in the bulk is mostly retained for lipids prepared as supported films with the added complexity of the preferred orientation of the polymorphic phases. A theoretical framework to understand the orientation of lipid films has been established by Latypova et al.[120] and Richardson et al.[115] in the context of thermodynamic minimization for surface energy. Further understanding on substrate effects will allow the engineering of on-demand structures and orientation. Substrate modification based on recent surface functionalization technologies is expected to play a key role in dictating the structure of lipid films (from a few layers adjacent to the substrate propagating to the entire film thickness).
Artificial transplants, stents, scaffolds for tissue engineering, surface-based drug/gene delivery, and macro-scale drug delivery devices all necessitate biocompatible and functional coatings. A few, original studies that employ lipid materials in those applications have already demonstrated their great utility as reservoirs of several therapeutic cargos and their ability to release active species in response to stimuli. It should be noted that phase transformations in lipid films are fast and involve the conversion between systems that have dramatically distinct nanostructures, permeability, and diffusion behaviors, a feature that is less prominent in polymer systems.
The fact that lipids are so adaptable, however, also constitutes a bottleneck for their application. One important aspect is the lack of mechanical robustness and stability when interfaced with hard materials or harsh environments. A clear future direction in this field will be the development of mechanically robust lipid-based materials capable of retaining the biocompatibility and the capacity to quickly transform into different structures as a function of specific environmental cues. There have been a few efforts to address this challenge by incorporating additional components such as lipid-silica or copolymers. The key in those processes is to maintain the functionality of the systems, in particular of the bioactive agents incorporated into the lipid-based films. We argue that one of the most promising developments in the field will revolve around composite lipid-polymer hybrid materials. The literature cited in this review demonstrates that this is a rapidly emerging research field over the past 5 years. Concurrent self-assembly of lipids and polymers into the same membrane results in interesting hybrid membranes showing a diverse phase behavior spanning from homogeneous mixing to micro- and macro phase separation. Inherent advantages provided by polymer systems include ease composition engineering, tunable mechanical stability, and membrane permeability that are expected to synergistically cooperate with the advantages provided by lipid systems such as biocompatibility and responsivness towards external stimuli. Incorporation of distinct functional species (hydrophobic/hydrophilic drugs, nanoparticles, or proteins) could be carefully directed to be co-assembled in certain membrane domains and released at different time points. Such location selectivity of functional components is one advantage conferred by introducing in-plane heterogeneities into the lipid membranes. The fact that those heterogeneities align in registry across a wide space field, offers an opportunity to control active species concentration by stacking layers at different thicknesses. Although the nanostructure and the orientation of the phases can be somewhat modulated and predicted there is still an enduring lack of control over the size, shape, and distribution of in-plane phase-separated membrane domains which should be addressed in future research. With the exception of a few studies conducted in our laboratory shown here, multi-layered hybrid lipid-polymer films are essentially an unexplored material system which deserves further exploration. In addition, phase behavior studies of hybrid films have been limited to planar membrane systems although a rich polymorphism akin to lipid-only or polymer-only systems should be expected. Structural and chemical diversities brought by the lipid-polymer hybrid films would broaden the application space of lipid-based films. To meet the rapidly rising demand for coatings, matrices, and scaffold materials in biotechnology applications we expect to anticipate that more research efforts will be devoted to lipid-based films.
5. Experimental Section
5.1 Materials
1,2-dipalmitoyl-sn-glycero-3-phosphocholine (16:0 PC or DPPC) was purchased from Avanti Polar Lipids† (Alabaster; AL, USA). Amphiphilic diblock copolymer, poly(butadiene-b-ethylene oxide) (PBD-b-PEO), was purchased from Polymer Source, Inc.† (Quebec, Canada). The catalog number is P19015-BdEO. Its average molecular weight (Mn) was reported to be 4000 with PBD block (rich in 1,4 microstructure) 2500 and PEO block 1500, respectively. The reported polydispersity was 1.06. Paclitaxel was purchased from Thermo Fisher Scientific (Waltham; MA, USA). All solvents used were of high performance liquid chromatography (HPLC) grade and purchased from Sigma-Aldrich (St Louis; MO, USA). All chemicals and materials were used as received.
5.2 Sample Preparation
Samples for NMR experiments were prepared using thin film hydration method.[177] The stock solutions of phospholipids and/or block copolymers (25 mg/mL) were dissolved in chloroform in the desired molar ratio. The solutions were prepared in glass vials and the solvent was slowly dried with a nitrogen stream. For complete solvent evaporation, samples were put into the vacuum desiccator overnight. The films were hydrated with deionized water and incubated at 45 °C for 5h -12 h. The final concentration of ≈ 200 mM with the volume of ≈ 20 μL was transferred into NMR rotor inserts.
Samples for PTIR experiments were prepared using spin-coating method. Lipids and polymers were dissolved in chloroform and paclitaxel drugs were dissolved in ethanol. A stock solution was prepared with the desired ratio of each component in mixtures (concentration: 10 mg/mL - 25 mg/mL in solvent: chloroform/ethanol 4:1 volume ratio). The stock solution was either dropped onto the substrate followed by a solvent evaporation under a fume hood or spin-coated at 3000 rpm for 30 s.
5.3 PTIR experiments
PTIR spectra and images were obtained with a commercial PTIR instrument interfaced with a quantum cascade laser tunable from 1934 cm−1 (5.17 μm) to 1136 cm−1 (8.80 μm). The PTIR laser illuminates the sample from the top at ≈ 20° angle from the sample plane. Commercially available 450 μm long and 50 μm wide gold-coated silicon AFM probes with a nominal spring constant between 0.07 and 0.4 N/m were used for all the PTIR experiments. A paclitaxel loaded (0.05 molar fraction) hybrid lipid-polymer film was spin coated onto ZnS flat substrate to minimize the background absorption contribution of the substrate. PTIR spectra and images were obtained by tuning the laser repetition rate to resonantly excite[178] the AFM cantilever second bending oscillation mode. In contrast to the original implementation of the resonance enhanced PTIR method that used lock-in detection at the cantilever resonant frequency,[178] we leveraged a phase lock-in loop to better account for resonant frequency shifts as a function of location (or time) due variations in the sample-probe interactions. PTIR spectra were obtained by tuning the laser at intervals of 2 cm−1. Up to six spectra were acquired and averaged for each tip location, and smoothed by considering two adjacent points. AFM topography and PTIR maps were acquired simultaneously illuminating the sample at constant wavelength. Because the sample mechanical properties are known to influence the amplitude of the PTIR response,[165] throughout the manuscript we use PTIR absorption map ratios, to cancel out the effect of the local mechanical properties of the sample on the data.
5.4 FTIR experiments
FTIR spectra were acquired with a commercial in total internal reflection geometry (4 cm−1 spectral resolution). Each spectrum is the average of 128 consecutive scans.
5.5 Polarization Transfer Solid-state Nuclear Magnetic Resonance (PT ssNMR)
NMR experiments were performed on a Bruker Avance-II 500 spectrometer† (Karlsruhe, Germany) equipped with a 4 mm 13C/31P/1H Efree probe and a 11.7 T magnet, giving resonance frequencies of 500 MHz for 1H and 125 MHz for 13C. 1D 1H direct-polarization (DP) spectra and 13C DP, cross-polarization (CP),[166] and insensitive nuclei enhanced by polarization transfer (INEPT)[167] spectra were measured at 5 kHz magic-angle spinning (MAS) and temperatures from 298 to 316 K. The samples were equilibrated for 1 h at each temperature before measurements. The 13C spectra were acquired with 31.25 kHz spectral width and 100 ms acquisition time under 48 kHz TPPM 1H decoupling.[179] The 13C chemical shifts were calibrated by referring to the signal of solid α-glycine at 43.67 ppm.[180] The CP acquisition parameters were contact time 1 ms, 80 kHz 13C nutation frequency, and linear ramp of 1H nutation frequency from 72 to 88 kHz, while the delays in the INEPT sequence were t= 1.2 ms and t′ = 1.8 ms. All 90° and 180° pulses were applied at 80 kHz nutation frequency. Accumulation of 1280 transients at a recycle delay of 5 s yielded a measurement time of 107 min per spectrum. 1D NMR spectra were processed with a commercial data analysis package with the following processing parameters; the line broadening factor (LB) was set to be 10 Hz, and the size of real spectrum (SI) was 8192 and the size of fid (TD) was 3120.
Acknowledgments
This work was supported by the National Science Foundation under grant no. DMR-1554435 (phase behavior) and the National Institutes of Health under grant no. 1DP2EB024377-01 (drug-release). M.T. acknowledges support under the Cooperative Research Agreement between the University of Maryland, and the National Institute of Standards and Technology Center for Nanoscale Science and Technology, Award 70NANB14H209, through the University of Maryland.
Biographies

Minjee Kang is in the final year of the Ph.D. program in Materials Science and Engineering at the University of Illinois, Urbana-Champaign (Prof. Cecilia Leal’s Lab). She received her bachelor’s degree in Pohang University of Science and Technology, South Korea (2012). Her research focus on exploring the self-assembly behavior of lipids and amphiphilic block co-polymers. She is interested in understanding the structures and interactions of these materials leading to their application in medicine and biotechnology.

Daniel Topgaard is a Professor of Physical Chemistry at Lund University, Sweden. His primary research interest is development of magnetic resonance methods for investigating structure and dynamics of soft matter. He received his Ph.D. from Lund University and was a postdoctoral fellow at the Materials Sciences Division, Ernest Orlando Lawrence Berkeley National Laboratory and the Department of Chemistry, University of California,Berkeley. His innovations in the field of magnetic resonance imaging are commercialized by CR Development AB, Sweden.

Cecilia Leal is an Assistant Professor of Materials Science and Engineering at the University of Illinois, Urbana-Champaign since 2012. Her research interests focus on structures and interactions of soft matter that can be functionalized to operate as therapeutic or biotechnological devices. Cecilia received her Ph.D. in Physical Chemistry from the University of Lund, Sweden and was a postdoctoral fellow for three years at the group of Prof. Safinya at the University of California, Santa Barbara.
Footnotes
The materials used in this paper require the identification of a commercial product and its supplier. The inclusion of such information should in no way be constructed as indicating that such product or supplier is endorsed by NIST or is recommended by NIST or that is necessarily the best material for the purpose described.
The equipment used in this paper requires the identification of a commercial product and its supplier. The inclusion of such information should in no way be constructed as indicating that such product or supplier is endorsed by NIST or is recommended by NIST or that is necessarily the best material for the purpose described.
References
- 1.Simons K, Vaz WLC. Annu Rev Biophys Biomol Struct. 2004;33:269. doi: 10.1146/annurev.biophys.32.110601.141803. [DOI] [PubMed] [Google Scholar]
- 2.van Meer G, Voelker DR, Feigenson GW. Nat Rev Mol Cell Biol. 2008;9:112. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tu Y, Peng F, Adawy A, Men Y, Abdelmohsen LKEA, Wilson DA. Chem Rev. 2016;116:2023. doi: 10.1021/acs.chemrev.5b00344. [DOI] [PubMed] [Google Scholar]
- 4.Gregoriadis G, Ryman BE. Biochem J. 1971;124:58P. doi: 10.1042/bj1240058p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gregoriadis G. FEBS Lett. 1973;36:292. doi: 10.1016/0014-5793(73)80394-1. [DOI] [PubMed] [Google Scholar]
- 6.Allen TM, Cullis PR. Adv Drug Deliv Rev. 2013;65:36. doi: 10.1016/j.addr.2012.09.037. [DOI] [PubMed] [Google Scholar]
- 7.Kraft JC, Freeling JP, Wang Z, Ho RJY. J Pharm Sci. 2014;103:29. doi: 10.1002/jps.23773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tamm LK, McConnell HM. Biophys J. 1985;47:105. doi: 10.1016/S0006-3495(85)83882-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Castellana ET, Cremer PS. Surf Sci Rep. 2006;61:429. doi: 10.1016/j.surfrep.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Richter RP, Bérat R, Brisson AR. Langmuir. 2006;22:3497. doi: 10.1021/la052687c. [DOI] [PubMed] [Google Scholar]
- 11.Cullis PR, de Kruijff B. Biochim Biophys Acta. 1979;559:399. doi: 10.1016/0304-4157(79)90012-1. [DOI] [PubMed] [Google Scholar]
- 12.Cullis PR, Hope MJ, Tilcock CP. Chem Phys Lipids. 1986;40:127. doi: 10.1016/0009-3084(86)90067-8. [DOI] [PubMed] [Google Scholar]
- 13.Seddon JM, Templer RH. Handb Biol Phys. Elsevier; 1995. pp. 97–160. [Google Scholar]
- 14.Ericsson B, Eriksson PO, Löfroth JE, Engström S. In: Polym Drugs Drug Deliv Syst. Dunn RL, Ottenbrite RM, editors. American Chemical Society; Washington, DC: 1991. pp. 251–265. [Google Scholar]
- 15.Fong WK, Hanley T, Boyd BJ. J Controlled Release. 2009;135:218. doi: 10.1016/j.jconrel.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 16.Kang M, Leal C. Adv Funct Mater. 2016;26:5610. doi: 10.1002/adfm.201704356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leal C, Bouxsein NF, Ewert KK, Safinya CR. J Am Chem Soc. 2010;132:16841. doi: 10.1021/ja1059763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim H, Leal C. ACS Nano. 2015;9:10214. doi: 10.1021/acsnano.5b03902. [DOI] [PubMed] [Google Scholar]
- 19.Vallooran JJ, Handschin S, Pillai SM, Vetter BN, Rusch S, Beck HP, Mezzenga R. Adv Funct Mater. 2016;26:181. [Google Scholar]
- 20.Kang M, Huang G, Leal C. Soft Matter. 2014;10:8846. doi: 10.1039/c4sm01431f. [DOI] [PubMed] [Google Scholar]
- 21.Boyd BJ, Fong W-K. In: Self-Assem Supramol Archit. Garti N, Somasundaran P, Mezzenga R, editors. John Wiley & Sons, Inc.; Hoboken, NJ, USA: 2012. pp. 257–288. [Google Scholar]
- 22.Guo X, Andrew MacKay J, Szoka FC. Biophys J. 2003;84:1784. doi: 10.1016/s0006-3495(03)74986-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wadsäter M, Barauskas J, Nylander T, Tiberg F. Soft Matter. 2013;9:8815. doi: 10.1039/c4sm02296c. [DOI] [PubMed] [Google Scholar]
- 24.Nylander T, Soltwedel O, Ganeva M, Hirst C, Holdaway J, Arteta MY, Wadsäter M, Barauskas J, Frielinghaus H, Holderer O. J Phys Chem B. 2017;121:2705. doi: 10.1021/acs.jpcb.6b11038. [DOI] [PubMed] [Google Scholar]
- 25.Rittman M, Amenitsch H, Rappolt M, Sartori B, O’Driscoll BMD, Squires AM. Langmuir ACS J Surf Colloids. 2013;29:9874. doi: 10.1021/la401580y. [DOI] [PubMed] [Google Scholar]
- 26.Grayson ACR, Shawgo RS, Johnson AM, Flynn NT, Li Y, Cima MJ, Langer R. Proc IEEE. 2004;92:6. [Google Scholar]
- 27.Joung YH. Int Neurourol J. 2013;17:98. doi: 10.5213/inj.2013.17.3.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.El-Ali J, Sorger PK, Jensen KF. Nature. 2006;442:403. doi: 10.1038/nature05063. [DOI] [PubMed] [Google Scholar]
- 29.Cheng M, Cuda G, Bunimovich Y, Gaspari M, Heath J, Hill H, Mirkin C, Nijdam A, Terracciano R, Thundat T. Curr Opin Chem Biol. 2006;10:11. doi: 10.1016/j.cbpa.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 30.LaFratta CN, Walt DR. Chem Rev. 2008;108:614. doi: 10.1021/cr0681142. [DOI] [PubMed] [Google Scholar]
- 31.Shmueli RB, Anderson DG, Green JJ. Expert Opin Drug Deliv. 2010;7:535. doi: 10.1517/17425241003603653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang CHK, Pun SH. Trends Biotechnol. 2011;29:119. doi: 10.1016/j.tibtech.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 33.Jung DR, Kapur R, Adams T, Giuliano KA, Mrksich M, Craighead HG, Taylor DL. Crit Rev Biotechnol. 2001;21:111. doi: 10.1080/20013891081700. [DOI] [PubMed] [Google Scholar]
- 34.Ogaki R, Alexander M, Kingshott P. Mater Today. 2010;13:22. [Google Scholar]
- 35.Lei Y, Yang S, Wu M, Wilde G. Chem Soc Rev. 2011;40:1247. doi: 10.1039/b924854b. [DOI] [PubMed] [Google Scholar]
- 36.Berthier J, Silberzan P. Microfluidics for Biotechnology. Artech House; Boston: 2006. [Google Scholar]
- 37.Nge PN, Rogers CI, Woolley AT. Chem Rev. 2013;113:2550. doi: 10.1021/cr300337x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oliveira AF, Pessoa ACSN, Bastos RG, de la Torre LG. Biotechnol Prog. 2016;32:1372. doi: 10.1002/btpr.2350. [DOI] [PubMed] [Google Scholar]
- 39.Bettinger CJ, Bao Z. Polym Int. 2010;59:563. doi: 10.1002/pi.2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Irimia-Vladu M. Chem Soc Rev. 2014;43:588. doi: 10.1039/c3cs60235d. [DOI] [PubMed] [Google Scholar]
- 41.Kearney CJ, Mooney DJ. Nat Mater. 2013;12:1004. doi: 10.1038/nmat3758. [DOI] [PubMed] [Google Scholar]
- 42.Zelikin AN. ACS Nano. 2010;4:2494. doi: 10.1021/nn100634r. [DOI] [PubMed] [Google Scholar]
- 43.Le Meins JF, Schatz C, Lecommandoux S, Sandre O. Mater Today. 2013;16:397. [Google Scholar]
- 44.Schulz M, Olubummo A, Binder WH. Soft Matter. 2012;8:4849. doi: 10.1039/c3sm52040d. [DOI] [PubMed] [Google Scholar]
- 45.Schulz M, Binder WH. Macromol Rapid Commun. 2015;36:2031. doi: 10.1002/marc.201500344. [DOI] [PubMed] [Google Scholar]
- 46.Liu Y, Liu B, Nie Z. Nano Today. 2015;10:278. [Google Scholar]
- 47.Kiessling V, Domanska MK, Murray D, Wan C, Tamm LK. Wiley Encycl Chem Biol. John Wiley & Sons, Inc.; Hoboken, NJ, USA: 2008. [Google Scholar]
- 48.Jackman J, Knoll W, Cho NJ. Materials. 2012;5:2637. [Google Scholar]
- 49.Johnson SJ, Bayerl TM, McDermott DC, Adam GW, Rennie AR, Thomas RK, Sackmann E. Biophys J. 1991;59:289. doi: 10.1016/S0006-3495(91)82222-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Keller CA, Kasemo B. Biophys J. 1998;75:1397. doi: 10.1016/S0006-3495(98)74057-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mingeot-Leclercq MP, Deleu M, Brasseur R, Dufrêne YF. Nat Protoc. 2008;3:1654. doi: 10.1038/nprot.2008.149. [DOI] [PubMed] [Google Scholar]
- 52.Kraft ML. Science. 2006;313:1948. doi: 10.1126/science.1130279. [DOI] [PubMed] [Google Scholar]
- 53.Blodgett KB. J Am Chem Soc. 1935;57:1007. [Google Scholar]
- 54.Ulman A. An Introduction to Ultrathin Organic Films: From Langmuir-Blodgett to Self-Assembly. Academic Press; Boston: 1991. [Google Scholar]
- 55.Cremer PS, Boxer SG. J Phys Chem B. 1999;103:2554. [Google Scholar]
- 56.Richter R, Mukhopadhyay A, Brisson A. Biophys J. 2003;85:3035. doi: 10.1016/S0006-3495(03)74722-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lind TK, Cárdenas M, Wacklin HP. Langmuir. 2014;30:7259. doi: 10.1021/la500897x. [DOI] [PubMed] [Google Scholar]
- 58.Kalb E, Frey S, Tamm LK. Biochim Biophys Acta. 1992;1103:307. doi: 10.1016/0005-2736(92)90101-q. [DOI] [PubMed] [Google Scholar]
- 59.Wagner ML, Tamm LK. Biophys J. 2000;79:1400. doi: 10.1016/S0006-3495(00)76392-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kam L, Boxer SG. J Biomed Mater Res. 2001;55:487. doi: 10.1002/1097-4636(20010615)55:4<487::aid-jbm1041>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 61.Minner DE, Rauch P, Käs J, Naumann CA. Soft Matter. 2014;10:1189. doi: 10.1039/c3sm52298a. [DOI] [PubMed] [Google Scholar]
- 62.Vafaei S, Tabaei SR, Biswas KH, Groves JT, Cho NJ. Adv Healthc Mater. 2017;6:1700243. doi: 10.1002/adhm.201700243. [DOI] [PubMed] [Google Scholar]
- 63.Groves JT. Science. 1997;275:651. doi: 10.1126/science.275.5300.651. [DOI] [PubMed] [Google Scholar]
- 64.Phillips KS, Cheng Q. Anal Chem. 2005;77:327. doi: 10.1021/ac049356+. [DOI] [PubMed] [Google Scholar]
- 65.Moran-Mirabal JM, Edel JB, Meyer GD, Throckmorton D, Singh AK, Craighead HG. Biophys J. 2005;89:296. doi: 10.1529/biophysj.104.054346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Suzuki Y, Endo M, Sugiyama H. Nat Commun. 2015;6:8052. doi: 10.1038/ncomms9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Knoll W, Frank CW, Heibel C, Naumann R, Offenhäusser A, Rühe J, Schmidt EK, Shen WW, Sinner A. Rev Mol Biotechnol. 2000;74:137. doi: 10.1016/s1389-0352(00)00012-x. [DOI] [PubMed] [Google Scholar]
- 68.Salditt T. J Phys Condens Matter. 2005;17:R287. [Google Scholar]
- 69.Tristram-Nagle S, Nagle JF. Chem Phys Lipids. 2004;127:3. doi: 10.1016/j.chemphyslip.2003.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pompeo G, Girasole M, Cricenti A, Cattaruzza F, Flamini A, Prosperi T, Generosi J, Congiu Castellano A. Biochim Biophys Acta BBA - Biomembr. 2005;1712:29. doi: 10.1016/j.bbamem.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 71.Nagle JF, Tristram-Nagle S. Biochim Biophys Acta. 2000;1469:159. doi: 10.1016/s0304-4157(00)00016-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Perino-Gallice L, Fragneto G, Mennicke U, Salditt T, Rieutord F. Eur Phys J E - Soft Matter. 2002;8:275. doi: 10.1140/epje/i2001-10101-0. [DOI] [PubMed] [Google Scholar]
- 73.Nowak B, Paulus M, Nase J, Salmen P, Degen P, Wirkert FJ, Honkimäki V, Tolan M. Langmuir. 2016;32:2638. doi: 10.1021/acs.langmuir.5b04554. [DOI] [PubMed] [Google Scholar]
- 74.Hønger T, Mortensen K, Ipsen JH, Lemmich J, Bauer R, Mouritsen OG. Phys Rev Lett. 1994;72:3911. doi: 10.1103/PhysRevLett.72.3911. [DOI] [PubMed] [Google Scholar]
- 75.Ma Y, Ghosh SK, Bera S, Jiang Z, Schlepütz CM, Karapetrova E, Lurio LB, Sinha SK. Phys Chem Chem Phys. 2016;18:1225. doi: 10.1039/c5cp04703j. [DOI] [PubMed] [Google Scholar]
- 76.Constantin D, Ollinger C, Vogel M, Salditt T. Eur Phys J E. 2005;18:273. doi: 10.1140/epje/e2005-00028-7. [DOI] [PubMed] [Google Scholar]
- 77.Gavelis GS, Hayakawa S, White RA, III, Gojobori T, Suttle CA, Keeling PJ, Leander BS. Nature. 2015;523:204. doi: 10.1038/nature14593. [DOI] [PubMed] [Google Scholar]
- 78.Payne SC, Bartlett CA, Harvey AR, Dunlop SA, Fitzgerald M. Investig Opthalmology Vis Sci. 2012;53:6093. doi: 10.1167/iovs.12-10080. [DOI] [PubMed] [Google Scholar]
- 79.Laule C, Vavasour IM, Kolind SH, Li DKB, Traboulsee TL, Moore GRW, MacKay AL. Neurotherapeutics. 2007;4:460. doi: 10.1016/j.nurt.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tayebi L, Ma Y, Vashaee D, Chen G, Sinha SK, Parikh AN. Nat Mater. 2012;11:1074. doi: 10.1038/nmat3451. [DOI] [PubMed] [Google Scholar]
- 81.Bechinger B. Nat Mater. 2012;11:1005. doi: 10.1038/nmat3507. [DOI] [PubMed] [Google Scholar]
- 82.Bald D, Kruip J, Rögner M. Photosynth Res. 1996;49:103. doi: 10.1007/BF00117661. [DOI] [PubMed] [Google Scholar]
- 83.Pribil M, Labs M, Leister D. J Exp Bot. 2014;65:1955. doi: 10.1093/jxb/eru090. [DOI] [PubMed] [Google Scholar]
- 84.Schmitt FO, Bear RS, Palmer KJ. J Cell Comp Physiol. 1941;18:31. [Google Scholar]
- 85.Geren BB. Exp Cell Res. 1954;7:558. doi: 10.1016/s0014-4827(54)80098-x. [DOI] [PubMed] [Google Scholar]
- 86.Trissl HW, Wilhelm C. Trends Biochem Sci. 1993;18:415. doi: 10.1016/0968-0004(93)90136-b. [DOI] [PubMed] [Google Scholar]
- 87.Hartline DK. Neuron Glia Biol. 2008;4:153. doi: 10.1017/S1740925X09990263. [DOI] [PubMed] [Google Scholar]
- 88.Brodnitz MH. J Agric Food Chem. 1968;16:994. [Google Scholar]
- 89.Reis A, Spickett CM. Biochim Biophys Acta BBA - Biomembr. 2012;1818:2374. doi: 10.1016/j.bbamem.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 90.Crowe JH, Crowe LM, Carpenter JF, Rudolph AS, Wistrom CA, Spargo BJ, Anchordoguy TJ. Biochim Biophys Acta BBA - Rev Biomembr. 1988;947:367. doi: 10.1016/0304-4157(88)90015-9. [DOI] [PubMed] [Google Scholar]
- 91.Mennicke U, Salditt T. Langmuir. 2002;18:8172. [Google Scholar]
- 92.Gupta G, Iyer S, Leasure K, Virdone N, Dattelbaum AM, Atanassov PB, López GP. ACS Nano. 2013;7:5300. doi: 10.1021/nn401123p. [DOI] [PubMed] [Google Scholar]
- 93.Heath GR, Li M, Polignano IL, Richens JL, Catucci G, O’Shea P, Sadeghi SJ, Gilardi G, Butt JN, Jeuken LJC. Biomacromolecules. 2016;17:324. doi: 10.1021/acs.biomac.5b01434. [DOI] [PubMed] [Google Scholar]
- 94.Matosevic S, Paegel BM. Nat Chem. 2013;5:958. doi: 10.1038/nchem.1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Murray DH, Tamm LK, Kiessling V. J Struct Biol. 2009;168:183. doi: 10.1016/j.jsb.2009.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chung M, Lowe RD, Chan YHM, Ganesan PV, Boxer SG. J Struct Biol. 2009;168:190. doi: 10.1016/j.jsb.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Han X, Achalkumar AS, Cheetham MR, Connell SDA, Johnson BRG, Bushby RJ, Evans SD. ChemPhysChem. 2010;11:569. doi: 10.1002/cphc.200900798. [DOI] [PubMed] [Google Scholar]
- 98.Lautscham LA, Lin CY, Auernheimer V, Naumann CA, Goldmann WH, Fabry B. Biomaterials. 2014;35:3198. doi: 10.1016/j.biomaterials.2013.12.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Khan MKI, Mujawar LH, Schutyser MAI, Schroën K, Boom R. Food Bioprocess Technol. 2013;6:3047. [Google Scholar]
- 100.Zhu Y, Negmi A, Moran-Mirabal J. Membranes. 2015;5:385. doi: 10.3390/membranes5030385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nafday OA, Lowry TW, Lenhert S. Small. 2012;8:1021. doi: 10.1002/smll.201102096. [DOI] [PubMed] [Google Scholar]
- 102.Sironi B, Snow T, Redeker C, Slastanova A, Bikondoa O, Arnold T, Klein J, Briscoe WH. Soft Matter. 2016;12:3877. doi: 10.1039/c6sm00369a. [DOI] [PubMed] [Google Scholar]
- 103.Heath GR, Li M, Rong H, Radu V, Frielingsdorf S, Lenz O, Butt JN, Jeuken LJC. Adv Funct Mater. 2017:1606265. [Google Scholar]
- 104.Lee CH, Kim H, Harburg DV, Park G, Ma Y, Pan T, Kim JS, Lee NY, Kim BH, Jang KI, Kang SK, Huang Y, Kim J, Lee KM, Leal C, Rogers JA. NPG Asia Mater. 2015;7:e227. doi: 10.1038/am.2015.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Perry SL, Neumann SG, Neumann T, Cheng K, Ni J, Weinstein JR, Schaffer DV, Tirrell M. AIChE J. 2013;59:3203. [Google Scholar]
- 106.Steer D, Kang M, Leal C. Nanotechnology. 2017;28:142001. doi: 10.1088/1361-6528/aa5d77. [DOI] [PubMed] [Google Scholar]
- 107.Shah NJ, Hyder MN, Moskowitz JS, Quadir MA, Morton SW, Seeherman HJ, Padera RF, Spector M, Hammond PT. Sci Transl Med. 2013;5:191ra83. doi: 10.1126/scitranslmed.3005576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Solanki A, Shah S, Yin PT, Lee KB. Sci Rep. 2013;3:1553. doi: 10.1038/srep01553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bailey SN, Wu RZ, Sabatini DM. Drug Discov Today. 2002;7:S113. doi: 10.1016/s1359-6446(02)02386-3. [DOI] [PubMed] [Google Scholar]
- 110.Wheeler DB, Carpenter AE, Sabatini DM. Nat Genet. 2005;37(Suppl):S25. doi: 10.1038/ng1560. [DOI] [PubMed] [Google Scholar]
- 111.Neumann T, Gajria S, Tirrell M, Jaeger L. J Am Chem Soc. 2009;131:3440. doi: 10.1021/ja809349m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Neumann T, Gajria S, Bouxsein NF, Jaeger L, Tirrell M. J Am Chem Soc. 2010;132:7025. doi: 10.1021/ja909514j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Fukushima T. Biomaterials. 2004;25:5491. doi: 10.1016/j.biomaterials.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 114.Rittman M, Frischherz M, Burgmann F, Hartley PG, Squires A. Soft Matter. 2010;6:4058. [Google Scholar]
- 115.Richardson SJ, Staniec PA, Newby GE, Terrill NJ, Elliott JM, Squires AM, Góźdź WT. Langmuir. 2014;30:13510. doi: 10.1021/la503313n. [DOI] [PubMed] [Google Scholar]
- 116.Templer RH, Seddon JM, Warrender NA, Syrykh A, Huang Z, Winter R, Erbes J. J Phys Chem B. 1998;102:7251. [Google Scholar]
- 117.Landau EM, Rosenbusch JP. Proc Natl Acad Sci U S A. 1996;93:14532. doi: 10.1073/pnas.93.25.14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bender J, Ericson MB, Merclin N, Iani V, Rosén A, Engström S, Moan J. J Controlled Release. 2005;106:350. doi: 10.1016/j.jconrel.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 119.Kang M, Kim H, Leal C. Curr Opin Colloid Interface Sci. 2016;26:58. doi: 10.1016/j.cocis.2016.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Latypova L, Góźdź WT, Pierański P. Langmuir. 2014;30:488. doi: 10.1021/la4040744. [DOI] [PubMed] [Google Scholar]
- 121.Rancon Y, Charvolin J. J Phys Chem. 1988;92:2646. [Google Scholar]
- 122.Vigild ME, Almdal K, Mortensen K, Hamley IW, Fairclough JPA, Ryan AJ. Macromolecules. 1998;31:5702. [Google Scholar]
- 123.Squires AM, Akbar S, Tousley ME, Rokhlenko Y, Singer JP, Osuji CO. Langmuir. 2015;31:7707. doi: 10.1021/acs.langmuir.5b01676. [DOI] [PubMed] [Google Scholar]
- 124.Akbar S, Elliott JM, Rittman M, Squires AM. Adv Mater. 2013;25:1160. doi: 10.1002/adma.201203395. [DOI] [PubMed] [Google Scholar]
- 125.Richardson SJ, Burton MR, Staniec PA, Nandhakumar IS, Terrill NJ, Elliott JM, Squires AM. Nanoscale. 2016;8:2850. doi: 10.1039/c5nr06691c. [DOI] [PubMed] [Google Scholar]
- 126.Leal C, Ewert KK, Bouxsein NF, Shirazi RS, Li Y, Safinya CR. Soft Matter. 2013;9:795. doi: 10.1039/C2SM27018H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sun W, Vallooran JJ, Fong WK, Mezzenga R. J Phys Chem Lett. 2016;7:1507. doi: 10.1021/acs.jpclett.6b00416. [DOI] [PubMed] [Google Scholar]
- 128.Lindman B, Alexandridis P, editors. Amphiphilic Block Copolymers: Self-Assembly and Applications. Elsevier; Amsterdam; New York: 2000. [Google Scholar]
- 129.Blanazs A, Armes SP, Ryan AJ. Macromol Rapid Commun. 2009;30:267. doi: 10.1002/marc.200800713. [DOI] [PubMed] [Google Scholar]
- 130.Bermúdez H, Hammer DA, Discher DE. Langmuir. 2004;20:540. doi: 10.1021/la035497f. [DOI] [PubMed] [Google Scholar]
- 131.Song Z, Kim H, Ba X, Baumgartner R, Lee JS, Tang H, Leal C, Cheng J. Soft Matter. 2015;11:4091. doi: 10.1039/c5sm00820d. [DOI] [PubMed] [Google Scholar]
- 132.Ruysschaert T, Sonnen AFP, Haefele T, Meier W, Winterhalter M, Fournier D. J Am Chem Soc. 2005;127:6242. doi: 10.1021/ja043600x. [DOI] [PubMed] [Google Scholar]
- 133.Nam J, Beales PA, Vanderlick TK. Langmuir. 2011;27:1. doi: 10.1021/la103428g. [DOI] [PubMed] [Google Scholar]
- 134.Nam J, Vanderlick TK, Beales PA. Soft Matter. 2012;8:7982. [Google Scholar]
- 135.Chemin M, Brun PM, Lecommandoux S, Sandre O, Le Meins JF. Soft Matter. 2012;8:2867. [Google Scholar]
- 136.Chen D, Santore MM. Soft Matter. 2015;11:2617. doi: 10.1039/c4sm02502d. [DOI] [PubMed] [Google Scholar]
- 137.Dao TPT, Fernandes F, Ibarboure E, Ferji K, Prieto M, Sandre O, Le Meins JF. Soft Matter. 2017;13:627. doi: 10.1039/c6sm01625a. [DOI] [PubMed] [Google Scholar]
- 138.Dao TPT, Brûlet A, Fernandes F, Er-Rafik M, Ferji K, Schweins R, Chapel JP, Fedorov A, Schmutz M, Prieto M, Sandre O, Le Meins JF. Langmuir. 2017;33:1705. doi: 10.1021/acs.langmuir.6b04478. [DOI] [PubMed] [Google Scholar]
- 139.Lim SK, Wong A, De Hoog HPM, Rangamani P, Parikh A, Nallani M, Sandin S, Liedberg B. Soft Matter. 2017;13:1107. doi: 10.1039/c6sm01753c. [DOI] [PubMed] [Google Scholar]
- 140.Gettel DL, Sanborn J, Patel MA, de Hoog HP, Liedberg B, Nallani M, Parikh AN. J Am Chem Soc. 2014;136:10186. doi: 10.1021/ja5037308. [DOI] [PubMed] [Google Scholar]
- 141.Kowal J, Wu D, Mikhalevich V, Palivan CG, Meier W. Langmuir. 2015;31:4868. doi: 10.1021/acs.langmuir.5b00388. [DOI] [PubMed] [Google Scholar]
- 142.Kang M, Lee B, Leal C. submitted. 2017 [Google Scholar]
- 143.Olubummo A, Schulz M, Lechner BD, Scholtysek P, Bacia K, Blume A, Kressler J, Binder WH. ACS Nano. 2012;6:8713. doi: 10.1021/nn3023602. [DOI] [PubMed] [Google Scholar]
- 144.Fujiwara TK, Iwasawa K, Kalay Z, Tsunoyama TA, Watanabe Y, Umemura YM, Murakoshi H, Suzuki KGN, Nemoto YL, Morone N, Kusumi A. Mol Biol Cell. 2016;27:1101. doi: 10.1091/mbc.E15-04-0186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hinman SS, Ruiz CJ, Cao Y, Ma MC, Tang J, Laurini E, Posocco P, Giorgio S, Pricl S, Peng L, Cheng Q. ACS Appl Mater Interfaces. 2017;9:1029. doi: 10.1021/acsami.6b11556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Centrone A. Annu Rev Anal Chem. 2015;8:101. doi: 10.1146/annurev-anchem-071114-040435. [DOI] [PubMed] [Google Scholar]
- 147.Dazzi A, Prater CB. Chem Rev. 2017;117:5146. doi: 10.1021/acs.chemrev.6b00448. [DOI] [PubMed] [Google Scholar]
- 148.Dazzi A, Glotin F, Carminati R. J Appl Phys. 2010;107:124519. [Google Scholar]
- 149.Lahiri B, Holland G, Centrone A. Small. 2013;9:439. doi: 10.1002/smll.201200788. [DOI] [PubMed] [Google Scholar]
- 150.Ruggeri FS, Longo G, Faggiano S, Lipiec E, Pastore A, Dietler G. Nat Commun. 2015;6:7831. doi: 10.1038/ncomms8831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Dazzi A, Prazeres R, Glotin F, Ortega JM, Al-Sawaftah M, de Frutos M. Ultramicroscopy. 2008;108:635. doi: 10.1016/j.ultramic.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 152.Katzenmeyer AM, Canivet J, Holland G, Farrusseng D, Centrone A. Angew Chem Int Ed. 2014;53:2852. doi: 10.1002/anie.201309295. [DOI] [PubMed] [Google Scholar]
- 153.Chae J, Lahiri B, Centrone A. ACS Photonics. 2016;3:87. doi: 10.1021/acsphotonics.5b00466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Gong L, Chase DB, Noda I, Liu J, Martin DC, Ni C, Rabolt JF. Macromolecules. 2015;48:6197. [Google Scholar]
- 155.Strelcov E, Dong Q, Li T, Chae J, Shao Y, Deng Y, Gruverman A, Huang J, Centrone A. Sci Adv. 2017;3:e1602165. doi: 10.1126/sciadv.1602165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Purohit HS, Taylor LS. Mol Pharm. 2015;12:1623. doi: 10.1021/acs.molpharmaceut.5b00041. [DOI] [PubMed] [Google Scholar]
- 157.Van Eerdenbrugh B, Lo M, Kjoller K, Marcott C, Taylor LS. Mol Pharm. 2012;9:1459. doi: 10.1021/mp300059z. [DOI] [PubMed] [Google Scholar]
- 158.Katzenmeyer AM, Holland G, Kjoller K, Centrone A. Anal Chem. 2015;87:3154. doi: 10.1021/ac504672t. [DOI] [PubMed] [Google Scholar]
- 159.Chae J, Dong Q, Huang J, Centrone A. Nano Lett. 2015;15:8114. doi: 10.1021/acs.nanolett.5b03556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Yoon Y, Chae J, Katzenmeyer AM, Yoon HP, Schumacher J, An S, Centrone A, Zhitenev N. Nanoscale. 2017;9:7771. doi: 10.1039/c7nr01480e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Chae J, Ramer G, Stravila V, Holland G, Yoon Y, Talin AA, Allendorf MD, Aksyuk V, Centrone A. submitted. 2017 doi: 10.1021/acs.nanolett.7b02404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Yablon DG, Gannepalli A, Proksch R, Killgore J, Hurley DC, Grabowski J, Tsou AH. Macromolecules. 2012;45:4363. [Google Scholar]
- 163.Magonov SN, Elings V, Whangbo MH. Surf Sci. 1997;375:L385. [Google Scholar]
- 164.Socrates G. J Chem Educ. 1995;72:A93. [Google Scholar]
- 165.Barlow DE, Biffinger JC, Cockrell-Zugell AL, Lo M, Kjoller K, Cook D, Lee WK, Pehrsson PE, Crookes-Goodson WJ, Hung CS, Nadeau LJ, Russell JN. The Analyst. 2016;141:4848. doi: 10.1039/c6an00940a. [DOI] [PubMed] [Google Scholar]
- 166.Pines A, Gibby MG, Waugh JS. Chem Phys Lett. 1972;15:373. [Google Scholar]
- 167.Morris GA, Freeman R. J Am Chem Soc. 1979;101:760. [Google Scholar]
- 168.Halle B, Wennerström H. J Chem Phys. 1981;75:1928. [Google Scholar]
- 169.Nowacka A, Bongartz NA, Ollila OHS, Nylander T, Topgaard D. J Magn Reson. 2013;230:165. doi: 10.1016/j.jmr.2013.02.016. [DOI] [PubMed] [Google Scholar]
- 170.Nowacka A, Mohr PC, Norrman J, Martin RW, Topgaard D. Langmuir. 2010;26:16848. doi: 10.1021/la102935t. [DOI] [PubMed] [Google Scholar]
- 171.Mavromoustakos T, Theodoropoulou E, Yang DP. Biochim Biophys Acta BBA - Biomembr. 1997;1328:65. doi: 10.1016/s0005-2736(97)00078-3. [DOI] [PubMed] [Google Scholar]
- 172.Canto LB, Mantovani GL, deAzevedo ER, Bonagamba TJ, Hage E, Pessan LA. Polym Bull. 2006;57:513. [Google Scholar]
- 173.Mahou R, Wandrey C. Polymers. 2012;4:561. [Google Scholar]
- 174.Jagalski V, Barker R, Topgaard D, Günther-Pomorski T, Hamberger B, Cárdenas M. Biochim Biophys Acta BBA - Biomembr. 2016;1858:2827. doi: 10.1016/j.bbamem.2016.08.008. [DOI] [PubMed] [Google Scholar]
- 175.Earl WL, VanderHart DL. Macromolecules. 1979;12:762. [Google Scholar]
- 176.Rappolt M, Rapp G. Eur Biophys J. 1996;24:381. [Google Scholar]
- 177.Lasic DD. Liposomes: From Physics to Applications. Elsevier; Amsterdam; New York: 1993. [Google Scholar]
- 178.Lu F, Jin M, Belkin MA. Nat Photonics. 2014;8:307. [Google Scholar]
- 179.Bennett AE, Rienstra CM, Auger M, Lakshmi KV, Griffin RG. J Chem Phys. 1995;103:6951. [Google Scholar]
- 180.Hayashi S, Hayamizu K. Bull Chem Soc Jpn. 1991;64:685. [Google Scholar]