

Abstract
Cessation from prolonged use of ∆9-tetrahydrocannabinol (THC), the primary active compound responsible for the cannabimimetic effects of cannabis results in a mild to moderate withdrawal syndrome in humans and laboratory animals. Whereas manipulations of the endogenous cannabinoid system (e.g., cannabinoid receptors and endocannabinoid regulating enzymes) alter nicotine withdrawal, in this study we asked the reciprocal question. Do nicotinic acetylcholine receptors (nAChRs) modulate THC withdrawal? To assess the role of different nAChR subtypes in THC withdrawal, we used transgenic mouse, preclinical pharmacological, and human genetic correlation approaches. Our findings show that selective α3β4* nAChR antagonist, AuIB, and α3β4* nAChR partial agonist, AT-1001, dose-dependently attenuated somatic withdrawal signs in THC-dependent mice that were challenged with the cannabinoid-1 receptor antagonist rimonabant. Additionally, THC-dependent α5 and α6 nAChR knockout (KO) mice displayed decreased rimonabant precipitated somatic withdrawal signs compared with their wild-type counterparts. In contrast, β2 and α7 nAChR KO mice showed no alterations in THC withdrawal signs. Moreover, deletion of β nAChR did not alter the reduced expression of somatic signs by the preferred α6β4* antagonist, BulA[T5A;P60]. Finally, the human genetic association studies indicated that variations in the genes that code for the α5, α3, β4, and α6 nAChRs were associated with cannabis disorder phenotypes. Overall, these findings suggest that α3β4* and α6β4* nAChR subtypes represent viable targets for the development of medications to counteract THC dependence.
Keywords: THC, nicotinic acetylcholine receptors, withdrawal, somatic signs, mice, human genetics
INTRODUCTION
Cannabis consistently remains the most widely used illicit substance in the United States. Although a relatively small percentage of total users develop cannabis use disorder (CUD) (Budney, Roffman, Stephens & Walker 2007), a high prevalence of approximately four million people in the United States in 2016 met the diagnostic criteria for this, which raises concern about dependence to this drug. Indeed, retrospective, prospective, and laboratory studies demonstrate that abrupt discontinuation following chronic exposure to smoked cannabis or oral Δ9-tetrahydrocannabinol (THC), its major psychoactive constituent, elicits a cannabinoid withdrawal syndrome in humans consisting of sleep disturbances, disrupted cognition, decreased appetite, restlessness, irritability, sweating, chills, and nausea (Haney, Ward, Comer, Foltin & Fischman 1999a b). Cannabis withdrawal has been compared to that of tobacco and is reported to increase craving and desire to resume use (Budney, Vandrey, Hughes, Thostenson & Bursac 2008), which presents complications in treating dependence. Currently, no approved pharmacological treatments exist for treating CUD. Although THC effectively ameliorates cannabinoid withdrawal signs in both preclinical (Lichtman, Fisher & Martin 2001) and clinical (Haney, Hart, Vosburg, Nasser, Bennett, Zubaran & Foltin 2004) studies, considering its psychoactive effects, the identification of novel non-THC pharmacotherapies remains an important area of study (Balter, Cooper & Haney 2014).
Nicotinic acetylcholine receptors (nAChRs) represent promising targets for pharmacotherapies to treat CUD. These receptors participate in the addictive properties of other substances of abuse (Muldoon, Jackson, Perez, Harenza, Molas, Rais, Anwar, Zaveri, Maldonado, Maskos, McIntosh, Dierssen, Miles, Chen, De Biasi & Damaj 2014; Srisontiyakul, Kastman, Krstew, Govitrapong & Lawrence 2016) and cannabis users show a high incidence of tobacco use compared to those that smoke only tobacco (Ramo, Delucchi, Hall, Liu & Prochaska 2013). Whereas preclinical rodent studies demonstrate that the endogenous cannabinoid system modulates nicotine reward and dependence (Muldoon, Lichtman, Parsons & Damaj 2013; Gueye, Pryslawsky, Trigo, Poulia, Delis, Antoniou, Loureiro, Laviolette, Vemuri, Makriyannis & Le Foll 2016), the role that nAChRs play in THC dependence remains unexplored. Valjent and colleagues (2002) reported that acute nicotine enhanced the pharmacological effects of THC in the rodent tetrad assay, consisting of in vivo end points indicative of cannabimimetic activity (i.e., antinociception decreased locomotor activity, hypothermia, and catalepsy; Valjent, Mitchell, Besson, Caboche & Maldonado 2002). However, six days of repeated co-administration of low doses of nicotine and THC attenuated THC tolerance in the tetrad assay, but enhanced cannabinoid-1 (CB1) receptor antagonist precipitated somatic withdrawal signs (Valjent, Mitchell, Besson, Caboche & Maldonado 2002).
The α3β4* (asterisk indicates the presence of additional subunits) nAChR subtype is an interesting candidate since the 15q25 gene cluster, which contains the CHRNA5-CHRNA3-CHRNB4 genes, coding for the α5, α3, and β4 nAChR subunits respectively, has shown the most robust findings in human genetic studies as a candidate region contributing to risk of heavy smoking, nicotine dependence, and smoking related diseases in humans (Bierut 2009). In addition, variations in the CHRNA5–CHRNA3–CHRNB4 gene cluster in humans were associated with opioid, alcohol, and cocaine dependence (Wang, Grucza, Cruchaga, Hinrichs, Bertelsen, Budde, Fox, Goldstein, Reyes, Saccone, Saccone, Xuei, Bucholz, Kuperman, Nurnberger, Rice, Schuckit, Tischfield, Hesselbrock, Porjesz, Edenberg, Bierut & Goate 2009). Rodent studies have also confirmed an important role for α5, α3, and β4 nAChR subunits in nicotine and morphine withdrawal (Jackson, Marks, Vann, Chen, Gamage, Warner & Damaj 2010; Jackson, Sanjakdar, Muldoon, McIntosh & Damaj 2013). The α5 nAChR subunit can co-assemble with α3β4* nAChR subtypes to form functional receptors in the peripheral ganglia, as well as centrally, in the medial habenula (MHb) and interpeduncular nucleus (IPN). These brain regions were recently reported to be involved in nicotine withdrawal and intake (Salas, Sturm, Boulter & De Biasi 2009; Fowler & Kenny 2012). Based on these studies, we sought to investigate whether the α3β4* nAChR subtype plays a role in THC dependence.
The α6* nAChR subunit represents another possible modulator of CUD. Expression of α6-containing nAChRs in the brain is largely confined to catecholaminergic nuclei, such as the ventral tegmental area, substantia nigra, and locus coeruleus (LC) (Le Novère, Zoli & Changeux 1996). The α6 nAChR subunit can co-assemble with α4β2* nAChR subtypes and is involved in nicotine-stimulated dopamine release in the striatum (Champtiaux, Gotti, Cordero-Erausquin, David, Przybylski, Léna, Clementi, Moretti, Rossi, Le Novère, McIntosh, Gardier & Changeux 2003). In addition, it can co-assemble with β4-containing nAChRs in other brain regions such as the LC (Léna, de Kerchove D’Exaerde, Cordero-Erausquin, Le Novère, del Mar Arroyo-Jimenez & Changeux 1999) and the hippocampus, where the α6β4* nAChR subtype was shown to modulate norepinephrine release (Azam, Maskos, Changeux, Dowell, Christensen, De Biasi & McIntosh 2010).
In the present study, we used converging approaches to determine whether nAChRs modulate CB1 receptor antagonist precipitated withdrawal responses in THC-dependent mice. In particular, using pharmacologic or genetic approaches, we targeted β2*(α5, α6); α7; α3β4*(α5); α6β4*(β2) nAChR subtype, where the * indicates that one or more additional subunit may also be present in that specific nAChR subtype. Accordingly, these studies employed the selective α3β4* nAChR-antagonist AuIB (Luo, Kulak, Cartier, Jacobsen, Yoshikami, Olivera & McIntosh 1998a), the partial α3β4* nAChR agonist AT-1001 (Toll, Zaveri, Polgar, Jiang, Khroyan, Zhou, Xie, Stauber, Costello & Leslie 2012), and the α6β4* nAChR-preferred antagonist BulA[T5A;P6O] (Chi, Kim, Olivera, McIntosh & Han 2006; Azam et al. 2010). Additionally, we used nicotinic α5, α6, α7, and β2 nAChR subunit knock-out (KO) mice.
Finally, in order to ascertain whether variants in nAChRs are associated with cannabis dependence in humans, we conducted association analyses utilizing the Study of Addiction: Gene and Environment (SAGE) European-American and African-American datasets. Specifically, we investigated associations between cannabis dependence and variants in the 15q25 gene cluster and the CHRNA6 gene, which codes for the α6 nAChR subunit.
MATERIALS AND METHODS
Animals
Male C57BL/6J mice were purchased from Jackson Laboratories (Bar Harbor, ME). Mice null for the α5, α6, α7 (Jackson Laboratories) and β2 (Institut Pasteur, Paris, France) nAChR subunits and their wild-type (WT) littermates were bred in an animal care facility at Virginia Commonwealth University. For all experiments, mice (C57BL/6 background) were backcrossed at least 8 to 10 generations. Mutant and WT were obtained from crossing heterozygote mice. This breeding scheme controlled for any irregularities that might occur with crossing solely mutant animals. Animals were 8–10 weeks of age and were group-housed (three to five per cage) under a 12 hr light/dark cycle in a 21°C humidity-controlled Association for Assessment and Accreditation of Laboratory Animal Care -approved animal care facility with ad libitum access to food and water. Experiments were performed during the light cycle and were approved by the Institutional Animal Care and Use Committee of Virginia Commonwealth University.
Drugs
The α3β4* nAChR-selective antagonist AuIB was synthesized as described by Luo et al., (1998). AuIB, purified from the venom of the “court cone”, Conus aulicus, blocks the α3β4* nAChR subtype with > 100 fold higher potency than other receptor combinations, such as α3β2* and α4β4* (Luo et al. 1998a). AT-1001, a partial α3β4* nAChR agonist, and BulA[T5A;P6O], an α6β4* nAChR-selective antagonist, were synthesized as previously described (Chi et al. 2006; Zaveri, Jiang, Olsen, Polgar & Toll 2010). THC and rimonanbant were obtained from the National Institute on Drug Abuse (Baltimore, MD) and were dissolved in a vehicle mixture of ethanol/ emulphor (Rhone-Poulenc, Princeton, NJ)/saline in a ratio of 1:1:18. THC was injected subcutaneous (s.c.) and rimonanbant was injected intraperitoneal (i.p.). AT-1001 was dissolved in 20% DMSO:emulphor:saline solution in a 1:1:18 ratio and the drug was injected i.p. AuIB and BulA[T5A;P6O] were dissolved in physiological saline (0.9% sodium chloride) and 100% saline, respectively, and administered to each animal by intracerebroventricular (i.c.v.) injection. The doses used for AuIB and AT-1001 (1.75, 3.5, and 7 pmol) were based on previous in vivo studies (Jackson et al. 2013; Muldoon et al. 2014), which show a dose-dependent blockade of nicotine and morphine withdrawal in mice. The BulA[T5A;P6O] dose was calculated on the functional ED50 at α6β4* nAChR (Chi et al. 2006).
I.c.v. surgery
The i.c.v. injections have been performed as previously described (Sanjakdar, Maldoon, Marks, Brunzell, Maskos, McIntosh, Bowers & Damaj 2015). Briefly, mice were anesthetized with sodium pentobarbital (45 mg/kg i.p.) on the evening prior to testing, and a scalp incision was made to expose the bregma. Unilateral injection sites were prepared using a 26-gauge needle with a sleeve of polyurethane (PE) tubing to control depth of the needle at a site 2 mm rostral and 2 mm lateral to the bregma at a depth of 2 mm. Animals were sutured in such a way to enable an injection volume of 5 µl using a 26-gauge needle with a sleeve of PE tubing into the lateral ventricle on the morning of testing. The needle was held in place for 20 s to ensure drug delivery.
Repeated THC administration and precipitated withdrawal protocol
Different groups of naïve mice (n=8 for the AuIB and AT-1001 studies; n=10 for the α5 and β2 nAChR WT and KO studies; n=11–12 for the α7 and α6 nAChR WT and KO studies; n=7–8 for the BulA[T5A;P6O] studies) were given two daily s.c. injections of THC (50 mg/kg) or vehicle for 5.5 days, with each injection separated by approximately 12 h. The rationale for the use of THC at 50 mg/kg dose is based on a previous study (Schlosburg, Carlson, Ramesh, Abdullah, Long, Cravatt & Lichtman 2009) showing that this dosing regimen led to robust rimonabant precipitated withdrawal responses in mice. Mice were given an i.p. injection of rimonabant (3 mg/kg) 30 min after THC. For studies using nAChR antagonists, injections were given 10 min prior to rimonabant.
Immediately after rimonabant challenge, mice were individually placed in Plexiglas cages and scored during a 30 min observation period for the manifestation of the following predominant withdrawal signs: 1) front paw tremors that included single-paw twitches or shaking of both paws simultaneously; and 2) head twitches, which generally manifested as rotational shakes of the head, similar to what is described as “wet dog shakes” in rats. Other less common signs such as backing, ptosis, and curling were also recorded. All behaviors were counted as new incidences if either separated by at least 1 s and/or interceded by another distinct behavior. All testing was conducted blind to group assignment.
Human subjects and measurements
Datasets were obtained from the SAGE dataset. SAGE is part of the Gene Environment Association Studies initiative (GENEVA) funded by the National Human Genome Research Institute aiming at understanding the impact of genes and environments on substance dependence and addiction. The SAGE sample consists of three subsamples: the Collaborative Study on the Genetics of Alcoholism (COGA) (Bierut, Saccone, Rice, Goate, Foroud, Edenberg, Almasy, Conneally, Crowe, Hesselbrock, Li, Nurnberger, Porjesz, Schuckit, Tischfield, Begleiter & Reich 2002), the Family Study of Cocaine Dependence (FSCD) (Bierut, Strickland, Thompson, Afful & Cottler 2008), and the Collaborative Genetic Study of Nicotine Dependence (COGEND) (Saccone, Hinrichs, Saccone, Chase, Konvicka, Madden, Breslau, Johnson, Hatsukami, Pomerleau, Swan, Goate, Rutter, Bertelsen, Fox, Fugman, Martin, Montgomery, Wang, Ballinger, Rice & Bierut 2007). Dichotomized variables for tolerance to cannabis, withdrawal, and DSM-IV cannabis use disorder were used in this study. Tolerance to cannabis was defined as either a) a need for markedly increased amounts of the substance to achieve intoxication or desired effect, or b) markedly diminished effect with continued use of the same amount of the substance. Cannabis withdrawal was defined as a) the characteristic withdrawal syndrome for the substance, or b) the same (or closely related) substance is taken to relieve or avoid withdrawal symptoms. The SAGE sample included 2,735 subjects with self-reported European ancestry (SAGE_EA) and 1,317 subjects with self-reported African ancestry (SAGE_AA) with data on cannabis phenotypes.
Statistical analysis
For all mouse data, statistical analyses were performed using the computer program GraphPad Prism version 6.0 (GraphPad Softwer, Inc., San Diego, CA). Data were analyzed with one-way analysis of variance with treatment as the between subject factor or two-way analysis of variance with treatment and genotype as between subject factors. A P value of <0.05 was considered statistically significant. Significant results were further analyzed using the Neuman-Keuls or Holm-Sidak’s post-hoc test. All data are expressed as the mean ± SEM.
For the human data, statistical analyses of the SAGE_EA and SAGE_AA samples for the intervals covering the 15q25 gene cluster (CHRNA5/CHRNA3/CHRNB4), and the CHRNA6 gene were conducted using the PLINK program (Purcell, Neale, Todd-Brown, Thomas, Ferreira, Bender, Maller, Sklar, de Bakker, Daly & Sham 2007). In these analyses, cannabis phenotypes were treated as case control and analyzed by logistic regression. In all analyses, sex and age at interview were used as covariates. Race was included as an additional covariate in the combined sample (n=4,025). Correction for multiple comparisons was applied to all significant results; however, the P-values reported in the paper are the uncorrected values.
RESULTS
AuIB and AT-1001 dose-dependently attenuate rimonabant-precipitated THC withdrawal signs
To evaluate the effects of α3β4* nAChR receptor inhibition on rimonabant precipitated THC withdrawal, mice received repeated injections of THC (50 mg/kg) and were then administered AuIB (1.75, 3.5, or 7 pmol/mouse, i.c.v.) or vehicle before rimonabant (3 mg/kg, i.p.) challenge. As previously demonstrated, rimonabant precipitated a significant increase in total somatic signs in THC-treated mice (Schlosburg et al. 2009). AuIB dose-dependently reduced rimonabant-precipitated somatic signs [F (6, 48) = 49.24, P <0.0001; Figure 1A]. In the absence of rimonabant, the highest dose of AuIB (7 pmol) had no significant effects in mice given repeated injections of THC or vehicle. Rimonabant administered to mice given repeated administration of vehicle did not elicit somatic withdrawal signs. Additionally, the partial α3β4* nAChR agonist AT-1001 (0.3, 3 mg/kg, i.p.) dose-dependently decreased the magnitude of rimonabant-precipitated withdrawal signs in THC-dependent mice [F (4, 35) = 11.93, P <0.0001; Figure 1B], but did not produce significant effects when its highest dose was administered in vehicle- or THC-treated mice in the absence of rimonabant challenge. The reductions in withdrawal were comparable across observed withdrawal signs. An injection of rimonabant in non-dependent mice did not alter the number of somatic signs compared with the vehicle-treated group.
Figure 1. Functional antagonism of α3β4* nAChRs reduces THC withdrawal signs.
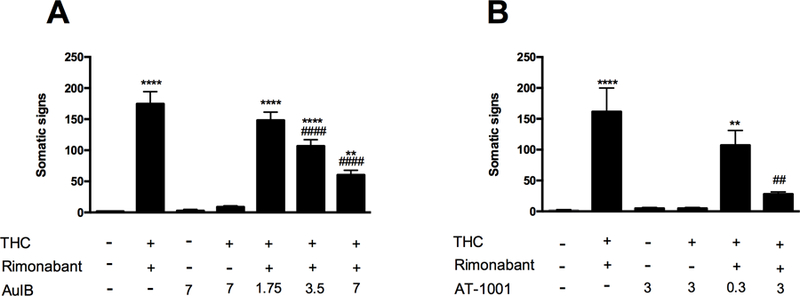
(A) THC-dependent mice pretreated with the selective α3β4* nAChR antagonist, AuIB (1.75, 3.5, 7 pmol, i.c.v.), show a decrease in somatic signs compared with the group of mice injected with THC and rimonabant. The injection of AuIB (7 pmol/mouse, i.c.v.) by itself or in combination with THC does not affect the total somatic signs compared to vehicle-treated group. ****P < 0.0001, **P < 0.01 versus vehicle+vehicle+vehicle group; ####P < 0.0001 versus THC+rimonabant+AuIB (1.75 pmol) group. (B) The partial agonist, AT-1001 (0.3, 3 mg/kg, i.p.), dose-dependently decrease the total somatic signs compared to the group of mice injected with THC and rimanabant. The injection of AT-1001 (3 mg/kg, i.p.) by itself or in combination with THC does not affect the total somatic signs compared to vehicle-treated group. **** P < 0.0001, **P < 0.01 versus vehicle+vehicle+vehicle group; ##P < 0.01 versus THC+rimonabant+AT-1001 (0.3 mg/kg) group. Data reflect mean ± SEM, n=8 mice per group.
THC-dependent α5* nAChR KO mice show reduced rimonabant-precipitated withdrawal signs
Given that the α5* nAChR subunit often associates with α3β4* nAChR subtypes, here we tested the involvement of α5*-containing nAChRs in THC withdrawal using a genetic approach. Rimonabant challenge elicited significant decrease of somatic withdrawal signs in THC-dependent α5 KO compared to the WT counterpart [Finteraction (6, 48) = 49.24, P <0.0001; Figure 2]. The reductions in withdrawal responses were comparable across observed withdrawal signs. The injection of rimonabant alone did not affect the number of somatic signs in either vehicle-treated WT or KO mice (Figure 2).
Figure 2. Deletion of α5 nAChRs produces reduced rimonabant precipitated withdrawal responses in THC-dependent mice.
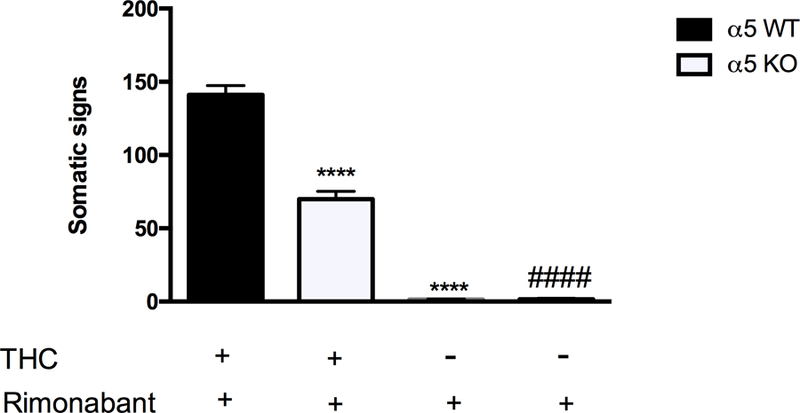
Rimonabant produces decreased withdrawal signs in α5 KO mice given repeated doses of THC compared with WT controls. The injection of rimonabant by itself did not affect the physical THC withdrawal signs in both α5 WT and KO mice. ****P < 0.0001 versus α5 WT (THC+rimonabant) group; ####P < 0.0001 versus α5 KO (THC+rimonabant). Data reflect mean ± SEM, n=10 mice per group.
α6 nAChR subunits are involved in THC withdrawal
Figure 3 depicts the consequences of deletion of β2, α7, and α6 nicotinic subunits on rimonabant-precipitated withdrawal responses in THC-dependent mice. Neither β2 (Figure 3A) nor α7 (Figure 3B) nAChR KO mice displayed altered magnitudes of withdrawal compared with WT mice. In contrast, α6 nAChR KO mice showed significant reductions in somatic withdrawal signs compared to their WT counterparts [Finteraction (1, 43) = 13.13, P = 0.0008; Figure 3C]. The reductions in withdrawal were comparable across observed withdrawal signs. Rimonabant did not affect this measure in control vehicle-treated WT or KO mice.
Figure 3. THC withdrawal signs require α6, but not α7 or β2, nAChR subunits.
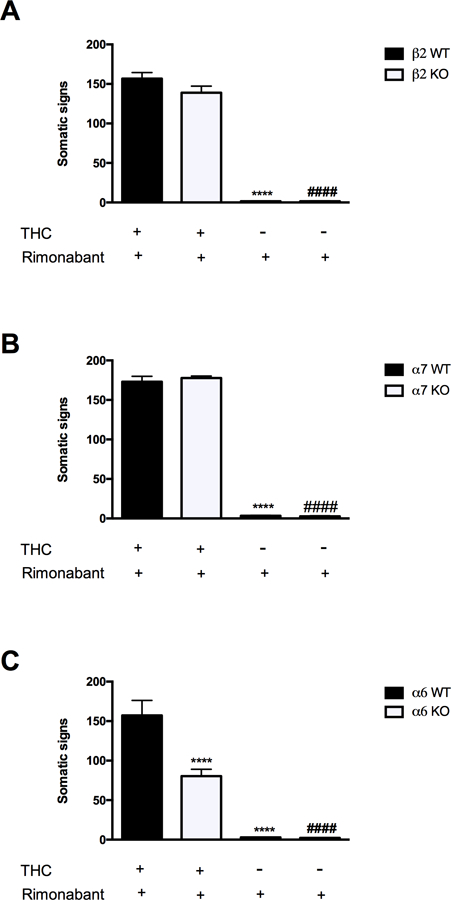
(A) Both THC-dependent β2 nAChR WT and KO mice show an increase in total somatic signs when challenged with rimonabant. ****P < 0.0001 versus β2 WT (THC+rimonabant) group; ####P < 0.0001 versus β2 KO (THC+rimonabant). Data reflect mean ± SEM, n=10 mice per group. (B) α7 nAChR WT and KO mice display the same increase in somatic sign after THC and rimonabant injection. **** p < 0.0001 versus α7 WT (THC+rimonabant) group; ####P < 0.0001 versus α7 KO (THC+rimonabant). Data reflect mean ± SEM, n=11–12 mice per group. (C) Compared to α6 WT mice, α6 nAChR KO mice show a significant reduction in THC withdrawal. The injection of rimonabant alone in β2, α7, and α6 WT and KO mice does not alter the physical THC withdrawal signs compared to the respective vehicle-treated groups. ****P < 0.0001 versus α6 WT (THC+rimonabant) group; ####P < 0.0001 versus α6 KO (THC+rimonabant). Data reflect mean ± SEM, n=11–12 mice per group.
BulA[T5A;P6O] attenuates somatic THC withdrawal signs in a β4-dependent and β2-independent mechanism
As shown in Figure 4A, i.c.v. administration of the α6β4* nAChR-preferred antagonist BulA[T5A;P6O] dose-dependently reduced the expression of rimonabant-precipitated somatic signs in THC-dependent C57BL/6J mice [F (6, 49) = 106.9, P <0.0001]. The reductions in withdrawal were comparable across observed withdrawal signs. We next tested high dose BulA[T5A;P6O] (200 pmol/mouse; i.c.v.) in β2 nAChR KO and WT mice. This high dose of BulA[T5A;P60] evoked ameliorated withdrawal responses to a similar degree in each genotype [F (2, 41) = 195.7, P <0.0001; Figure 4B]. No significant main effects of genotype were detected (Figure 4B). As before, rimonabant precipitated a similar magnitude of withdrawal responses in β2 nAChR KO and WT and THC-dependent mice but was without effect in non-dependent mice.
Figure 4. BulA[T5A;P6O] blockade of physical THC withdrawal responses is not altered by deletion of β2 nAChRs.
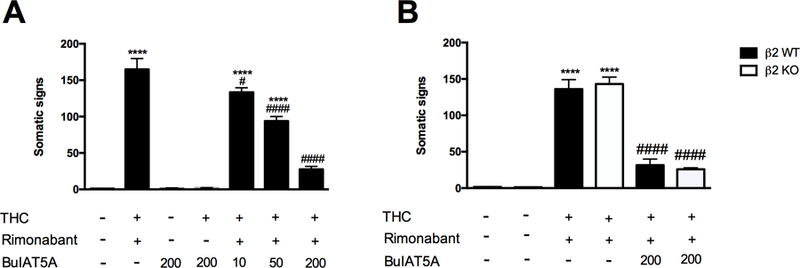
(A) BulA[T5A;P6O] (10, 50, 200 pmol/mouse, i.c.v.), a preferred α6β4* nAChR antagonist, dose-dependently reverses the increase of total somatic signs in THC-dependent mice challenged with rimonabant. The injection of BulA[T5A;P6O] (200 pmol/mouse, i.c.v.) by itself or in combination with THC does not affect the total somatic signs compared to vehicle-treated group. ****P < 0.0001 versus vehicle+vehicle+vehicle group; ####P < 0.0001, #P < 0.05 versus THC+rimonabant group. Data reflect mean ± SEM, n=8 mice per group. (B) The effects evoked by the high dose of Bul[T5A;P6O] are not altered in β2 nAChR KO mice. ****P < 0.0001 versus vehicle+vehicle+vehicle respective groups; ####P < 0.0001 versus THC+rimonabant respective groups. Data reflect mean ± SEM, n=7–8 mice per group.
Variants in the 15q25 gene clusters are associated with CUD
The interval spanning the 15q25 cluster was analyzed in the SAGE sample. Results showed that variants in the CHRNA3 (rs615470) and CHRNA5 (rs190004177) genes were associated with increased risk for cannabis withdrawal symptoms in the SAGE_AA and combined samples (Table 1). The variant rs190004177 was also associated with increased risk for DSM-IV CUD in the SAGE_EA and combined samples. Alternatively, three protective variants for cannabis tolerance in the CHRNA3 gene (rs190825809, rs112712252l, rs116932868), and two protective variants for DSM-IV criteria (CHRNA3-rs190245674, CHRNA5-rs115472979) were identified in the SAGE_EA, SAGE_AA, and combined samples. No markers survived corrections for multiple testing.
Table 1. Variants in the 15q25 gene cluster and marijuana dependence.
Association analysis conducted in the SAGE European American (EA) and African-American (AA) datasets. Risk variants for marijuana withdrawal, and protective variants for marijuana tolerance and DSM-IV criteria were identified. Significant results are bolded and underlined. No values survived correction for multiple testing after the meta-analysis [p-value x no. or markers (293) x no. of phenotypes (3) x no. of datasets (2)]. Significant Cochran’s Q statistic p-values (Q_P) are italicized.
| SAGE EA, n= 2735 | SAGE AA, n= 1317 | Combined, n= 4052 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Phenotype | SNP | Gene | Allele | OR | P | OR | P | OR | P | Q_P |
| rs190825809 | CHRNA3 | C | 0.49 | 0.01 | 0.49 | 0.07 | 0.49 | 0.002 | 0.99 | |
| Tolerance | rs112712252 | CHRNA3 | G | 0.46 | 0.006 | 0.65 | 0.26 | 0.52 | 0.004 | 0.45 |
| rs116932868 | CHRNA3 | G | 0.47 | 0.01 | 0.63 | 0.31 | 0.52 | 0.009 | 0.60 | |
| rs16969968 | CHRNA5 | A | 1.08 | 0.34 | 1.07 | 0.77 | 1.08 | 0.32 | 0.98 | |
| rs615470 | CHRNA3 | C | 1.05 | 0.61 | 1.45 | 0.002 | 1.18 | 0.02 | 0.03 | |
| Withdrawal | rs190004177 | CHRNA5 | C | 3.17 | 0.05 | 1.38 | 0.008 | 2.87 | 0.03 | 0.74 |
| rs117349742 | CHRNA5 | A | 1.01 | 0.91 | 2.18 | 0.42 | 1.14 | 0.08 | 0.04 | |
| rs16969968 | CHRNA5 | A | 1.03 | 0.76 | 0.93 | 0.78 | 1.02 | 0.85 | 0.72 | |
| rs190004177 | CHRNA5 | C | 2.69 | 0.03 | 5.17 | 0.08 | 3.07 | 0.008 | 0.49 | |
| DSM-IV | rs190245674 | CHRNA3 | T | 0.70 | 0.08 | 0.58 | 0.04 | 0.66 | 0.008 | 0.55 |
| rs115472979 | CHNRA5 | A | 0.63 | 0.11 | 0.58 | 0.08 | 0.60 | 0.02 | 0.84 | |
| rs16969968 | CHRNA5 | A | 1.01 | 0.90 | 0.93 | 0.75 | 1.00 | 0.99 | 0.73 | |
| SAGE EA, n= 2735 | SAGE AA, n= 1317 | Combined, n= 4052 | ||||||||
| Phenotype | SNP | Gene | Allele | OR | P | OR | P | OR | P | Q_P |
| rs190825809 | CHRNA3 | C | 0.49 | 0.01 | 0.49 | 0.07 | 0.49 | 0.002 | 0.99 | |
| Tolerance | rs112712252 | CHRNA3 | G | 0.46 | 0.006 | 0.65 | 0.26 | 0.52 | 0.004 | 0.45 |
| rs116932868 | CHRNA3 | G | 0.47 | 0.01 | 0.63 | 0.31 | 0.52 | 0.009 | 0.60 | |
| rs16969968 | CHRNA5 | A | 1.08 | 0.34 | 1.07 | 0.77 | 1.08 | 0.32 | 0.98 | |
| rs615470 | CHRNA3 | C | 1.05 | 0.61 | 1.45 | 0.002 | 1.18 | 0.02 | 0.03 | |
| Withdrawal | rs190004177 | CHRNA5 | C | 3.17 | 0.05 | 1.38 | 0.008 | 2.87 | 0.03 | 0.74 |
| rs117349742 | CHRNA5 | A | 1.01 | 0.91 | 2.18 | 0.42 | 1.14 | 0.08 | 0.04 | |
| rs16969968 | CHRNA5 | A | 1.03 | 0.76 | 0.93 | 0.78 | 1.02 | 0.85 | 0.72 | |
| rs190004177 | CHRNA5 | C | 2.69 | 0.03 | 5.17 | 0.08 | 3.07 | 0.008 | 0.49 | |
| DSM-IV | rs190245674 | CHRNA3 | T | 0.70 | 0.08 | 0.58 | 0.04 | 0.66 | 0.008 | 0.55 |
| rs115472979 | CHNRA5 | A | 0.63 | 0.11 | 0.58 | 0.08 | 0.60 | 0.02 | 0.84 | |
| rs16969968 | CHRNA5 | A | 1.01 | 0.90 | 0.93 | 0.75 | 1.00 | 0.99 | 0.73 | |
Variants in the CHRNA6 gene cluster are associated with a protective effect against CUD
Analyses of the SAGE sample identified variants protective against cannabis tolerance and withdrawal in the SAGE_EA and combined samples (Table 2). These findings suggest that subjects carrying the minor alleles for these variants were less likely to experience cannabis tolerance (rs79010274, rs150379145, rs145060765) and withdrawal (rs183424710, rs6982753, rs80215470-SAGE_EA only), than those carrying the major allele. No significant association was identified for DSM-IV criteria. Markers did not survive correction for multiple testing.
Table 2. Variants in the CHRNA6 gene and marijuana dependence.
Association analysis conducted in the SAGE European American (EA) and African-American (AA) datasets show that variants in the CHRNA6 gene cluster are significantly associated with a protective effect against marijuana tolerance and withdrawal. Uncorrected p-values are shown for individual datasets. Significant results are bolded and underlined. No markers survived correction for multiple testing [p-value x no. of markers (127) x no. of phenotypes (3) x no. of datasets (2)]. Significant Cochran’s Q statistic p-values (Q_P) are italicized.
| SAGE EA, n= 2735 | SAGE AA, n= 1317 | Combined, n= 4052 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Phenotype | SNP | Allele | OR | P | OR | P | OR | P | Q_P |
| Tolerance | rs79010274 | G | 0.54 | 0.09 | 0.42 | 0.12 | 0.50 | 0.02 | 0.70 |
| rs150379145 | C | 0.67 | 0.11 | 0.61 | 0.17 | 0.65 | 0.03 | 0.83 | |
| rs145060765 | G | 0.67 | 0.12 | 0.60 | 0.17 | 0.65 | 0.04 | 0.81 | |
| Withdrawal | rs183424710 | A | 0.48 | 0.003 | 0.86 | 0.27 | 0.75 | 0.02 | 0.04 |
| rs6982753 | G | 0.50 | 0.005 | 0.89 | 0.39 | 0.78 | 0.03 | 0.04 | |
| rs80215470 | A | 0.50 | 0.006 | 0.91 | 0.46 | 0.80 | 0.05 | 0.04 | |
| DSM-IV | rs183424710 | A | 0.72 | 0.20 | 0.85 | 0.18 | 0.83 | 0.09 | 0.67 |
| rs6982753 | G | 0.75 | 0.25 | 0.85 | 0.20 | 0.84 | 0.11 | 0.76 | |
| rs145060765 | G | 0.79 | 0.32 | 0.82 | 0.59 | 0.75 | 0.17 | 0.78 | |
DISCUSSION
Using mouse genetics, behavioral pharmacology, and human genetic association approaches, we make the unique observation that multiple nAChR subtypes (i.e., α3β4*(α5), α5*, α6β4*(β2), and α6*) play important roles in THC withdrawal. In contrast, the genetic data suggest that β2* and α7* subunits appear dispensible in the development of THC dependence. Specifically, central administration of the α3β4* receptor antagonist AuIB dose-dependently blocked the expression of THC withdrawal signs. Additionally, systemic administration of AT-1001 significantly reduced the magnitude of somatic signs in THC-dependent mice. AuIB is 100-fold more potent at α3β4* nAChRs compared to other heteromeric nAChR combinations, and 10-fold more potent at α3β4* than at the α7 homomeric nAChR subtype (Luo, Kulak, Cartier, Jacobsen, Yoshikami, Olivera & McIntosh 1998b). In vitro electrophysiological studies have shown that AT-1001 has partial agonist activity at the expressed α3β4* nAChR subtype, evoking 35% of maximum acetylcholine (Ach) response. However, at the same concentrations, AT-1001 produced desensitization of the ACh response in vitro, suggesting that it acts as a functional antagonist at the α3β4* nAChR (Zaveri, Bertrand, Yasuda & Bertrand 2015). Recently, it has been shown that AT-1001 administration to nicotine-dependent rats induces minimal withdrawal signs, compared to those produced by the non-selective nicotine antagonist, mecamylamine, after seven days of nicotine exposure (Yuan, Malagon, Yasuda, Belluzzi, Leslie & Zaveri 2017). The reduction of somatic withdrawal signs in THC-dependent mice by AuIB and AT-1001 suggests that α3β4* nAChR subtypes represent a promising target to treat THC dependence.
Other in vivo studies have also implicated an important role of α5, α3, and β4 nAChR subunits in somatic and affective nicotine withdrawal (Salas, Cook, Bassetto & De Biasi 2004; Jackson et al. 2010, 2013). The present findings that α5 nAChR KO mice treated repeatedly with THC show reduced rimonabant-precipitated somatic withdrawal signs compared with their WT counterparts, suggest the involvement of this subunit in THC withdrawal signs. Although the brain regions that mediate the effects of α3β4* and α5 nAChRs on THC dependence were not investigated in the present study, previous evidence shows that the association of α5 nAChR subunit with α3β4* nAChR subtypes occurs in the peripheral ganglia, MHb and IPN (Quick, Ceballos, Kasten, McIntosh & Lester 1999; Whiteaker, Peterson, Xu, McIntosh, Paylor, Beaudet, Collins & Marks 2002), which are reported to be involved in nicotine withdrawal and intake (Salas et al. 2009; Fowler & Kenny 2012). As such, nAChRs in the MHb-IPN pathway may contribute to the mediation of THC withdrawal responses. Additionally, Görlich and colleagues have reported that elevated sensitivity to nicotine in mice undergoing withdrawal occurs from an increase in the firing and pace-making activity of the cholinergic MHb neurons and involves activation of only the α3β4* nAChR, but not α4β2*, α4β4*, α3β2*, α7, and β3* nAChRs (Görlich, Antolin-Fontes, Ables, Frahm, Slimak, Dougherty & Ibañez-Tallon 2013). Our findings that α7* KO mice and β2* KO given repeated THC display similar rimonabant-precipitated withdrawal responses as WT mice agree with results of the previous studies. Thus, it appears that α3β4* and α5* nAChRs play important roles in modulating THC withdrawal, and these actions may be mediated through the MHb-IPN pathway. However, it should also be noted that deletions of these subunits may also alter the acute pharmacological effects of THC, which could have implications on consequences of repeated THC administration.
While the exact concentrations of the α6β4* nAChR antagonist BulA[T5A;P6O] at the sites of action were not determined and the in vivo α6β4/α3β4 selectivity of this compound is not known, our collective data suggest that α6*-containing nAChRs also play an important role in mediating THC withdrawal signs. Indeed, α6 KO mice show a significant decrease in somatic signs compared to their WT counterparts. In line with these findings, varenicline, a nicotinic receptor-based therapeutics available in clinic that has been reported to have activity at α6 nAChR subunit (Bordia, Hrachova, Chin, McIntosh & Quik 2012), has been shown to be well-tolerated for the treatment of co-occurring cannabis and tobacco use in humans (Adams, Arnsten, Ning & Nahvi 2018). Furthermore, this study shows that BulA[T5A;P6O] reduced total somatic signs in both WT and β2 nAChR KO mice, suggesting that α6β4*, not α6β2*, nAChRs mediate these effects. Interestingly, α6-containing nAChRs play a necessary role for almost all nicotine-stimulated norepinephrine release in mice. In particular, a saturated concentration of BulA[T5A;P6O] evoked a 32% blockade of nicotine-release of norepinephrine in the hippocampus of mice, indicating that approximately one-third of this release is due to nAChRs containing an α6/β4 interface (Azam et al. 2010). Therefore, it is possible that α6β4* nAChR subtypes may mediate nicotine- as well THC-induced release of norepinephrine. In agreement, preclinical data have shown that cannabinoid withdrawal results in noradrenergic hyperactivity (Hart 2005), and α2-receptor agonists decrease noradrenergic cell firing and release (Carter 1997). However, despite the fact that combination of dronabinol and lofexidine, an α2 noradrenergic agonist, failed to reduce cannabis withdrawal in humans (Levin, Mariani, Pavlicova, Brooks, Glass, Mahony, Nunes, Bisaga, Dakwar, Carpenter, Sullivan & Choi 2016), it remains possible that norepinephrine blockade with a more well-tolerated dose of lofexidine in humans would result in the reduction of THC withdrawal, as observed in the present study. Nevertheless, future studies are needed to test this hypothesis as well as investigate emotional and cognitive aspects of THC dependence and withdrawal.
The individual and combined reward effects of cannabis and tobacco have been deeply investigated in humans from a physiologic and psychologic point of view (Meier & Hatsukami 2016; Hindocha, Freeman, Xia, Shaban & Curran 2017a; Hindocha, Lawn, Freeman & Curran 2017b). From a genetic prospective, it has been shown that polymorphisms in the CHRNA5–CHRNA3–CHRNB4 gene cluster in humans are associated with alcohol, opioid, and cocaine dependence. These findings prompted us to investigate the possible correlation between genetic variation in this gene cluster and THC withdrawal. Findings from the human genetic association studies indicate that variations in the genes that code for the α5, α3, β4, and α6 nAChR subtypes are associated with CUD phenotypes, specifically tolerance and withdrawal. The association with protective effects in the CHRNA6, suggesting reduced likelihood of minor allele carriers to experience cannabis tolerance and withdrawal, coincide with the reduced somatic signs observed in the α6 genetic and pharmacological data in mice. Similarly, findings of protective variants in the CHRNA3 and CHRNA5 genes are supported by animal data. It is noted that risk variants for withdrawal were also identified in the CHRNA3 (rs615470) and CHRNA5 (rs190004177) genes. However, it is likely that specific haplotypes are associated with protective effects, and individuals with the protective phenotype may possess the major allele (i.e., the non-risk allele) at these positions. Haplotype analyses would be necessary to confirm this possibility.
In addition to the present findings, other studies suggest the involvement of other nAChR subunits in CUD. A genome-wide association study recently identified a significant correlation between the expression of CHRNA2, coding for the neuronal α2 nAChR subunit, and CUD. Specifically, individuals diagnosed with CUD showed decreased expression of CHRNA2 in the cerebellum and other brain regions, raising the possibility that CHRNA2 may represent a potential therapeutic target to counteract THC dependence (Demontis, Rajagopal, Als, Grove, Pallesen, Hjorthoj, Qvist, Christensen, Bybjerg-Grauholm, Baekvad-Hansen, Huckins, Stahl, Timmermann, Agerbo, Hougaard, Werge, Mors, Mortensen, Nordentoft, Daly, Nyegaard & Borglum 2018). However, as the present study did not examine the role of α2 nAChR in THC dependence, future preclinical studies are needed to test this hypothesis. Our results with genes coding for nAChR subtypes add to the list of genes recently reported by the largest genome-wide association study meta-analysis for lifetime cannabis use that identified 29 genome-wide significant genes in four independent loci containing significant SNP associations. Among others, the most significant associated gene was CADM2, coding for a synaptic cell adhesion molecule, which has previously been associated with substance use and risk-taking phenotypes (Mounteney, Griffiths, Sedefov, Noor, Vicente & Simon 2016; Charilaou, Agnihotri, Garcia, Badheka, Frenia & Yegneswaran 2017).
The present animal and human findings with THC along with previous laboratory animal studies that implicate the involvement of α3β4* nAChRs in opioid (Taraschenko, Panchal, Maisonneuve & Glick 2005), suggest a critical role for this nicotinic subtype in regulating drug dependence.
In summary, the present findings suggest that multiple nAChRs contribute to the development and/or expression of THC withdrawal. Interestingly, these results suggest bidirectional modulation of the endogenous cannabinoid and endogneous nicotinic systems in tobacco and cud. More specifically, the α3β4* and/or α3β4α5* and α6β4* nAChRs subtypes represent viable targets for the development of medications to alleviate the somatic signs associated with cannabis withdrawal signs.
Acknowledgements
The authors would like to thank Tie Han for his technical assistance with the withdrawal studies. This research was supported by National Institute on Drug Abuse (grant DA032246) to M. I. D. and X.C. and National Institute of General Medical Sciences (grants GM103801 and GM48677) to JMM. Funding support for SAGE was provided through the NIH Genes, Environment and Health Initiative [GEI] (U01 HG004422). SAGE is one of the genome-wide association studies funded as part of the GENEVA under GEI. Assistance with phenotype harmonization and genotype cleaning, as well as with general study coordination, was provided by the GENEVA Coordinating Center (U01 HG004446). Assistance with data cleaning was provided by the National Center for Biotechnology Information. Support for collection of datasets and samples was provided by COGA (U10 AA008401), COGEND (P01 CA089392), and FSCD (R01 DA013423). Funding support for genotyping, which was performed at the Johns Hopkins University Center for Inherited Disease Research, was provided by the NIH GEI (U01HG004438), the National Institute on Alcohol Abuse and Alcoholism, the National Institute on Drug Abuse, and the NIH contract “High throughput genotyping for studying the genetic contributions to human disease” (HHSN268200782096C). The datasets used for the analyses described in this manuscript were obtained from dbGaP at http://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000092.v1.p1 through dbGaP accession number phs000092.v1.p.
Footnotes
Conflict of interest
The authors do not have any conflicts of interest or financial disclosures to make
References
- Adams TR, Arnsten JH, Ning Y & Nahvi S (2018) Feasibility and Preliminary Effectiveness of Varenicline for Treating Co-Occurring Cannabis and Tobacco Use. J Psychoactive Drugs 50:12–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azam L, Maskos U, Changeux J-P, Dowell CD, Christensen S, De Biasi M & McIntosh JM (2010) α-Conotoxin BuIA[T5A;P6O]: a novel ligand that discriminates between α6ß4 and α6ß2 nicotinic acetylcholine receptors and blocks nicotine-stimulated norepinephrine release. FASEB J 24:5113–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balter RE, Cooper ZD & Haney M (2014) Novel Pharmacologic Approaches to Treating Cannabis Use Disorder. Curr Addict reports 1:137–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierut LJ (2009) Nicotine dependence and genetic variation in the nicotinic receptors. Drug Alcohol Depend 104:S64–S69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierut LJ, Saccone NL, Rice JP, Goate A, Foroud T, Edenberg H, Almasy L, Conneally PM, Crowe R, Hesselbrock V, Li TK, Nurnberger J, Porjesz B, Schuckit MA, Tischfield J, Begleiter H & Reich T (2002) Defining alcohol-related phenotypes in humans. The Collaborative Study on the Genetics of Alcoholism. Alcohol Res Health 26:208–13. [PMC free article] [PubMed] [Google Scholar]
- Bierut LJ, Strickland JR, Thompson JR, Afful SE & Cottler LB (2008) Drug use and dependence in cocaine dependent subjects, community-based individuals, and their siblings. Drug Alcohol Depend 95:14–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordia T, Hrachova M, Chin M, McIntosh JM & Quik M (2012) Varenicline is a potent partial agonist at α6β2* nicotinic acetylcholine receptors in rat and monkey striatum. J Pharmacol Exp Ther 342:327–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budney AJ, Roffman R, Stephens RS & Walker D (2007) Marijuana dependence and its treatment. Addict Sci Clin Pract 4:4–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budney AJ, Vandrey RG, Hughes JR, Thostenson JD & Bursac Z (2008) Comparison of cannabis and tobacco withdrawal: Severity and contribution to relapse. J Subst Abuse Treat 35:362–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter AJ (1997) Hippocampal noradrenaline release in awake, freely moving rats is regulated by alpha-2 adrenoceptors but not by adenosine receptors. J Pharmacol Exp Ther 281:648–54. [PubMed] [Google Scholar]
- Champtiaux N, Gotti C, Cordero-Erausquin M, David DJ, Przybylski C, Léna C, Clementi F, Moretti M, Rossi FM, Le Novère N, McIntosh JM, Gardier AM & Changeux J-P (2003) Subunit composition of functional nicotinic receptors in dopaminergic neurons investigated with knock-out mice. J Neurosci 23:7820–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charilaou P, Agnihotri K, Garcia P, Badheka A, Frenia D & Yegneswaran B (2017) Trends of Cannabis Use Disorder in the Inpatient: 2002 to 2011. Am J Med 130:678–687.e7. [DOI] [PubMed] [Google Scholar]
- Chi S-W, Kim D-H, Olivera BM, McIntosh JM & Han K-H (2006) NMR structure determination of α-conotoxin BuIA, a novel neuronal nicotinic acetylcholine receptor antagonist with an unusual 4/4 disulfide scaffold. Biochem Biophys Res Commun 349:1228–1234. [DOI] [PubMed] [Google Scholar]
- Demontis D, Rajagopal VM, Als TD, Grove J, Pallesen J, Hjorthoj C, Qvist P, Christensen JH, Bybjerg-Grauholm J, Baekvad-Hansen M, Huckins LM, Stahl EA, Timmermann A, Agerbo E, Hougaard DM, Werge T, Mors O, Mortensen PB, Nordentoft M, Daly M, Nyegaard M & Borglum AD (2018) Genome-wide association study implicates CHRNA2 in cannabis use disorder. bioRxiv:237321. [DOI] [PMC free article] [PubMed]
- Fowler CD & Kenny PJ (2012) Habenular Signaling in Nicotine Reinforcement. Neuropsychopharmacology 37:306–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Görlich A, Antolin-Fontes B, Ables JL, Frahm S, Slimak MA, Dougherty JD & Ibañez-Tallon I (2013) Reexposure to nicotine during withdrawal increases the pacemaking activity of cholinergic habenular neurons. Proc Natl Acad Sci U S A 110:17077–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gueye AB, Pryslawsky Y, Trigo JM, Poulia N, Delis F, Antoniou K, Loureiro M, Laviolette SR, Vemuri K, Makriyannis A & Le Foll B (2016) The CB1 Neutral Antagonist AM4113 Retains the Therapeutic Efficacy of the Inverse Agonist Rimonabant for Nicotine Dependence and Weight Loss with Better Psychiatric Tolerability. Int J Neuropsychopharmacol 19:pyw068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haney M, Hart CL, Vosburg SK, Nasser J, Bennett A, Zubaran C & Foltin RW (2004) Marijuana withdrawal in humans: effects of oral THC or divalproex. Neuropsychopharmacology 29:158–70. [DOI] [PubMed] [Google Scholar]
- Haney M, Ward AS, Comer SD, Foltin RW & Fischman MW (1999a) Abstinence symptoms following oral THC administration to humans. Psychopharmacology (Berl) 141:385–94. [DOI] [PubMed] [Google Scholar]
- Haney M, Ward AS, Comer SD, Foltin RW & Fischman MW (1999b) Abstinence symptoms following smoked marijuana in humans. Psychopharmacology (Berl) 141:395–404. [DOI] [PubMed] [Google Scholar]
- Hart CL (2005) Increasing treatment options for cannabis dependence: A review of potential pharmacotherapies. Drug Alcohol Depend 80:147–159. [DOI] [PubMed] [Google Scholar]
- Hindocha C, Freeman TP, Xia JX, Shaban NDC & Curran HV (2017a) Acute memory and psychotomimetic effects of cannabis and tobacco both ‘joint’ and individually: a placebo-controlled trial. Psychol Med 47:2708–2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindocha C, Lawn W, Freeman TP & Curran HV (2017b) Individual and combined effects of cannabis and tobacco on drug reward processing in non-dependent users. Psychopharmacology (Berl) 234:3153–3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson KJ, Marks MJ, Vann RE, Chen X, Gamage TF, Warner JA & Damaj MI (2010) Role of 5 Nicotinic Acetylcholine Receptors in Pharmacological and Behavioral Effects of Nicotine in Mice. J Pharmacol Exp Ther 334:137–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson KJ, Sanjakdar SS, Muldoon PP, McIntosh JM & Damaj MI (2013) The α3β4* nicotinic acetylcholine receptor subtype mediates nicotine reward and physical nicotine withdrawal signs independently of the α5 subunit in the mouse. Neuropharmacology 70:228–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Léna C, de Kerchove D’Exaerde A, Cordero-Erausquin M, Le Novère N, del Mar Arroyo-Jimenez M & Changeux JP (1999) Diversity and distribution of nicotinic acetylcholine receptors in the locus ceruleus neurons. Proc Natl Acad Sci U S A 96:12126–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin FR, Mariani JJ, Pavlicova M, Brooks D, Glass A, Mahony A, Nunes EV, Bisaga A, Dakwar E, Carpenter KM, Sullivan MA & Choi JC (2016) Dronabinol and lofexidine for cannabis use disorder: A randomized, double-blind, placebo-controlled trial. Drug Alcohol Depend 159:53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtman AH, Fisher J & Martin BR (2001) Precipitated cannabinoid withdrawal is reversed by Delta(9)-tetrahydrocannabinol or clonidine. Pharmacol Biochem Behav 69:181–8. [DOI] [PubMed] [Google Scholar]
- Luo S, Kulak JM, Cartier GE, Jacobsen RB, Yoshikami D, Olivera BM & McIntosh JM (1998a) alpha-conotoxin AuIB selectively blocks alpha3 beta4 nicotinic acetylcholine receptors and nicotine-evoked norepinephrine release. J Neurosci 18:8571–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo S, Kulak JM, Cartier GE, Jacobsen RB, Yoshikami D, Olivera BM & McIntosh JM (1998b) alpha-conotoxin AuIB selectively blocks alpha3 beta4 nicotinic acetylcholine receptors and nicotine-evoked norepinephrine release. J Neurosci 18:8571–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier E & Hatsukami DK (2016) A review of the additive health risk of cannabis and tobacco co-use. Drug Alcohol Depend 166:6–12. [DOI] [PubMed] [Google Scholar]
- Mounteney J, Griffiths P, Sedefov R, Noor A, Vicente J & Simon R (2016) The drug situation in Europe: an overview of data available on illicit drugs and new psychoactive substances from European monitoring in 2015. Addiction 111:34–48. [DOI] [PubMed] [Google Scholar]
- Muldoon PP, Jackson KJ, Perez E, Harenza JL, Molas S, Rais B, Anwar H, Zaveri NT, Maldonado R, Maskos U, McIntosh JM, Dierssen M, Miles MF, Chen X, De Biasi M & Damaj MI (2014) The α3β4* nicotinic ACh receptor subtype mediates physical dependence to morphine: mouse and human studies. Br J Pharmacol 171:3845–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muldoon PP, Lichtman AH, Parsons LH & Damaj MI (2013) The role of fatty acid amide hydrolase inhibition in nicotine reward and dependence. Life Sci 92:458–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Novère N, Zoli M & Changeux JP (1996) Neuronal nicotinic receptor alpha 6 subunit mRNA is selectively concentrated in catecholaminergic nuclei of the rat brain. Eur J Neurosci 8:2428–39. [DOI] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ & Sham PC (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81:559–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick MW, Ceballos RM, Kasten M, McIntosh JM & Lester RA (1999) Alpha3beta4 subunit-containing nicotinic receptors dominate function in rat medial habenula neurons. Neuropharmacology 38:769–83. [DOI] [PubMed] [Google Scholar]
- Ramo DE, Delucchi KL, Hall SM, Liu H & Prochaska JJ (2013) Marijuana and tobacco co-use in young adults: patterns and thoughts about use. J Stud Alcohol Drugs 74:301–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saccone SF, Hinrichs AL, Saccone NL, Chase GA, Konvicka K, Madden PAF, Breslau N, Johnson EO, Hatsukami D, Pomerleau O, Swan GE, Goate AM, Rutter J, Bertelsen S, Fox L, Fugman D, Martin NG, Montgomery GW, Wang JC, Ballinger DG, Rice JP & Bierut LJ (2007) Cholinergic nicotinic receptor genes implicated in a nicotine dependence association study targeting 348 candidate genes with 3713 SNPs. Hum Mol Genet 16:36–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salas R, Cook KD, Bassetto L & De Biasi M (2004) The alpha3 and beta4 nicotinic acetylcholine receptor subunits are necessary for nicotine-induced seizures and hypolocomotion in mice. Neuropharmacology 47:401–7. [DOI] [PubMed] [Google Scholar]
- Salas R, Sturm R, Boulter J & De Biasi M (2009) Nicotinic receptors in the habenulo-interpeduncular system are necessary for nicotine withdrawal in mice. J Neurosci 29:3014–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjakdar SS, Maldoon PP, Marks MJ, Brunzell DH, Maskos U, McIntosh JM, Bowers MS & Damaj MI (2015) Differential roles of α6β2* and α4β2* neuronal nicotinic receptors in nicotine- and cocaine-conditioned reward in mice. Neuropsychopharmacology 40:350–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlosburg JE, Carlson BLA, Ramesh D, Abdullah RA, Long JZ, Cravatt BF & Lichtman AH (2009) Inhibitors of endocannabinoid-metabolizing enzymes reduce precipitated withdrawal responses in THC-dependent mice. AAPS J 11:342–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srisontiyakul J, Kastman HE, Krstew EV., Govitrapong P & Lawrence AJ (2016) The Nicotinic α6-Subunit Selective Antagonist bPiDI Reduces Alcohol Self-Administration in Alcohol-Preferring Rats. Neurochem Res 41:3206–3214. [DOI] [PubMed] [Google Scholar]
- Taraschenko OD, Panchal V, Maisonneuve IM & Glick SD (2005) Is antagonism of alpha3beta4 nicotinic receptors a strategy to reduce morphine dependence? Eur J Pharmacol 513:207–18. [DOI] [PubMed] [Google Scholar]
- Toll L, Zaveri NT, Polgar WE, Jiang F, Khroyan TV, Zhou W, Xie X, Stauber GB, Costello MR & Leslie FM (2012) AT-1001: A High Affinity and Selective α3β4 Nicotinic Acetylcholine Receptor Antagonist Blocks Nicotine Self-Administration in Rats. Neuropsychopharmacology 37:1367–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valjent E, Mitchell JM, Besson M-J, Caboche J & Maldonado R (2002) Behavioural and biochemical evidence for interactions between Δ9-tetrahydrocannabinol and nicotine. Br J Pharmacol 135:564–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JC, Grucza R, Cruchaga C, Hinrichs AL, Bertelsen S, Budde JP, Fox L, Goldstein E, Reyes O, Saccone N, Saccone S, Xuei X, Bucholz K, Kuperman S, Nurnberger J, Rice JP, Schuckit M, Tischfield J, Hesselbrock V, Porjesz B, Edenberg HJ, Bierut LJ & Goate AM (2009) Genetic variation in the CHRNA5 gene affects mRNA levels and is associated with risk for alcohol dependence. Mol Psychiatry 14:501–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteaker P, Peterson CG, Xu W, McIntosh JM, Paylor R, Beaudet AL, Collins AC & Marks MJ (2002) Involvement of the alpha3 subunit in central nicotinic binding populations. J Neurosci 22:2522–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan M, Malagon AM, Yasuda D, Belluzzi JD, Leslie FM & Zaveri NT (2017) The α3β4 nAChR partial agonist AT-1001 attenuates stress-induced reinstatement of nicotine seeking in a rat model of relapse and induces minimal withdrawal in dependent rats. Behav Brain Res 333:251–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaveri N, Jiang F, Olsen C, Polgar W & Toll L (2010) Novel α3β4 nicotinic acetylcholine receptor-selective ligands. Discovery, structure-activity studies, and pharmacological evaluation. J Med Chem 53:8187–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaveri NT, Bertrand S, Yasuda D & Bertrand D (2015) Functional characterization of AT-1001, an α3β4 nicotinic acetylcholine receptor ligand, at human α3β4 and α4β2 nAChR. Nicotine Tob Res 17:361–7. [DOI] [PMC free article] [PubMed] [Google Scholar]