

Abstract
Cell membranes contain a large variety of lipid types and are crowded with proteins, endowing them with the plasticity needed to fulfill their key roles in cell functioning. The compositional complexity of cellular membranes gives rise to a heterogeneous lateral organization, which is still poorly understood. Computational models, in particular molecular dynamics simulations and related techniques, have provided important insight into the organizational principles of cell membranes over the past decades. Now, we are witnessing a transition from simulations of simpler membrane models to multicomponent systems, culminating in realistic models of an increasing variety of cell types and organelles. Here, we review the state of the art in the field of realistic membrane simulations and discuss the current limitations and challenges ahead.
1. Introduction
Membranes are essential components of every cell, providing the cell’s identity as well as defining a large variety of internal compartments. Typical cell membranes may contain hundreds of different lipids, asymmetrically distributed between the two bilayer leaflets and are crowded with proteins covering an estimated membrane area as large as 30%.1−3 The compositional heterogeneity of cellular membranes is now well recognized, leading to a nonuniform lateral distribution of the components.4−6 Together, lipids and proteins form distinct nanodomains with important implications for many cellular processes such as membrane fusion, protein trafficking, and signal transduction. Lipids move proteins, and proteins move lipids in a fascinating protein–lipid interplay.7
Experimental techniques are getting more and more sophisticated to reveal lateral membrane organization and the principles driving it. Experimental advances include improved methods for single-particle tracking, fluorescence correlation spectroscopy, super-resolved imaging, scattering, solid-state NMR, and mass spectrometry, as well as methods to prepare asymmetric model membranes and real cell membrane extracts.8−14 However, the detailed membrane organization proves difficult to probe at the molecular level, despite progress in experimental techniques that can directly probe living cells.15 Computer simulations, in principle, can provide this detail. Techniques such as molecular dynamics (MD) are capable of describing the interactions between all the components in the system at atomic resolution, acting like a “computational microscope”.16,17 Given enough computer power, the behavior of a system can be followed in time long enough to observe the process of interest.
The first MD simulations of surfactants and lipids appeared in the 1980s, shortly after the first published protein simulations,18 at a time when there were only a handful of super computers available for academic research. Complexity in lipid and surfactant systems rapidly increased from simplified ordered decanoate bilayers tethered harmonically to the average position of all headgroup particles19 to a smectic liquid crystal made of decanol, decanoate, water, and sodium ions,20 a micelle,21 and a liquid crystalline DPPC bilayer.22 In the early 1990s several groups published simulation papers on phospholipids with explicit water, including the infamous Berger lipid model23 that, although parametrized on erroneous data, became one of the leading lipid force fields until quite recently. These early papers already targeted a set of diverse problems, including lipid bilayer structure,24−26 transport of small molecules through bilayers,27 effect of cholesterol,28 the hydration force between bilayers,29 and interactions with membrane-active peptides,30 all of which continue to be studied. The first simulations of complete membrane proteins in a lipid environment studied gramicidin A,31 bacteriorhodopsin,32 OmpF porin,33 and phospholipase A.34 An early example of protein-induced bilayer perturbation is found in the work of Tieleman et al.35 Simulations of membrane proteins have since grown immensely in importance and are now widely used. Comprehensive reviews of these pioneering studies are available in the literature.36,37
As computer power grew and became more universally available, lively technical discussions appeared in the literature. Significant matters of debate included the use of cutoffs,39 appropriate boundary conditions for membrane simulations,40 as well as concerns with sampling and questions related to linking experiment and simulation. The latter two are not specific to membrane systems and, not surprisingly, continue to be major topics of both concern and continued research. In addition, during the first decade of the new millennium, we witnessed a growing range of applications of simulations involving collective lipid motion. Key pioneering examples include accessing bilayer undulatory modes,41 spontaneous self-assembly of lipids into a bilayer,42 pore formation by antimicrobial peptides or electrical fields,43−45 lipid flip-flop,46 collective lipid flows,47 domain formation,48 membrane fusion,49 and many more. For an in-depth discussion on these developments, now more than 10 years ago, we refer the reader to a number of earlier reviews.50−52
If we express the scope of a simulation as a combination of system size and simulation length, there has always been a large (maybe even up to 2–3 orders of magnitude) difference between a “typical” simulation and the largest ones in the literature. A typical scope in the early 1990s would be a bilayer model of 72–128 lipids (or 4000–15000 atoms) and simulation times of the order of a hundred picoseconds. For comparison, at the moment, early 2018, a typical simulation study might involve a combination of dozens of simulations on the order of microseconds, where a simulation system might contain 150000 atoms, an increase of at least 5 orders of magnitude. At these time and length scales, many interesting biochemical and biophysical questions can be addressed by simulations on relatively commonly available computer resources. Leadership-category machines allow access to 2–3 orders of magnitude more elaborate studies and coarse-grained models describe similar systems at a computational cost that is 2–3 orders of magnitude lower than a corresponding atomistic model. This massive increase in accessible scope, which now includes a large number of applications, has led to an explosive growth in the use of simulations to study membranes, as well as to the use of simulations in general.53−56
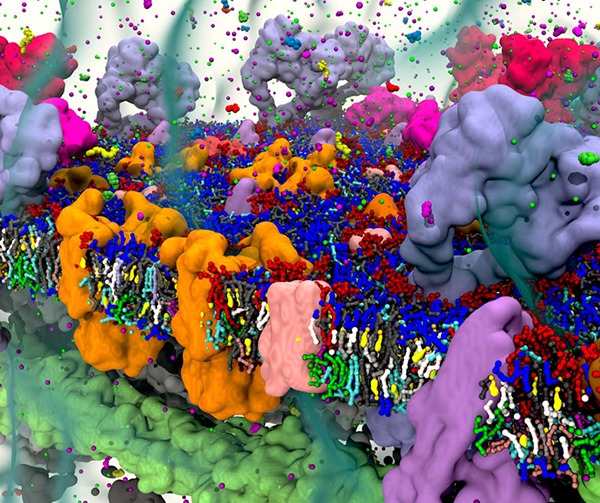
Thanks to the ongoing increase in computer power, sparked by the efficient use of GPUs, together with the development of accurate atomistic and coarse-grain (CG) models and the community-based development of tools to automate setup and analysis of membrane simulations, we are now witnessing a transition from simulations of simplified, model membranes toward multicomponent realistic membranes.57,58 This transition is essential to unravel protein–lipid interplay in the crowded and complex environment of real cell membranes, where experimental detection is difficult and theoretical models fall short. In this review, we focus on this transition, which is becoming apparent during the past five years (Figure 1). We restrict ourselves to particle-based simulation methods, mostly MD, and to simulation studies addressing the lateral and spatial organizational principles of membranes. For a discussion of related topics, not covered in the current review, we refer the reader to a number of other recent reviews, for example, on membrane proteins functioning and activity,59−62 binding of membrane active peptides,63,64 nanoparticle uptake,65−67 drug-membrane interactions,68,69 ionic-liquids and membranes,70 pore formation,71 lipid flip-flop,72 and lipid nanodisks.73
Figure 1.

Growth of complexity of membrane models. From the pioneering stage 30 years ago, basic properties of one and two component membranes were explored around the millennium. From then on, complexity of simulated membrane systems was gradually increased, culminating in the current era of more and more realistic membrane models. POPC, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine; DPPC, 1,2-dipalmitoyl-sn-glycero-3-phosphocholine; POPE, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine; DOPC, 1,2-dioleoyl-sn-glycero-3-phosphocholine; Chol, cholesterol; CLs, cardiolipins; PPPE, 1-palmitoyl-2-palmitoleoyl-phosphatidylethanolamine; PVPG, 1-palmitoyl-2-vacenoyl-phosphatidylglycerol; PVCL2, 1,10-palmitoyl-2,20-vacenoyl cardiolipin; Lps5, E. coli R1 lipopolysaccharide core with repeating units of O6-antigen. From left to right: Reprinted with permission from ref (20). Copyright 1988 AIP Publishing. Adapted from ref (26). Copyright 1993 American Chemical Society. Adapted from ref (42). Copyright 2001 American Chemical Society. Adapted with permission from ref (38). Copyright 2004 American Society for Biochemistry and Molecular Biology. Adapted from ref (311). Copyright 2014 American Chemical Society. Adapted from ref (382). Copyright 2013 American Chemical Society. Adapted from ref (593). Copyright 2014 American Chemical Society. Adapted with permission from ref (643). Copyright 2016 Elsevier.
The rest of this review is organized as follows. We first give an overview of the tools comprising the computational microscope, organized by the level of resolution obtained: from all-atom models via CG models to supra-CG models. Then we provide a comprehensive overview of the current state of the art in modeling membrane systems of increasing complexity, with sections on multicomponent systems, realistic cell membranes, and the current avenues toward full cell models. A short outlook section concludes this review.
2. Computational Tools
At the heart of the computational “microscope” lies the simulation algorithm, for which MD is most widely used. MD simulations, in their most basic form, involve numerically solving classical equations of motion for a set of particles over a given time period. The resulting time series, called trajectory, can subsequently be visualized and analyzed in detail. MD simulation algorithms, as well as related algorithms such as Brownian Dynamics, Langevin Dynamics, and Dissipative Particle Dynamics (DPD) have been implemented in a number of simulation software packages; the most widely used in the field of membrane modeling include AMBER,74,75 CHARMM,76 NAMD,77 OpenMM,78 LAMMPS,79 ESPResSo,80 and GROMACS,81,82 as well as the special purpose machine ANTON with the DESMOND software.83 A major limitation of simulations is the limited amount of sampling that can be performed, even when using the largest super computers available today. To more efficiently explore phase space, various enhanced sampling and biasing methods are available, with replica exchange MD (REMD), metadynamics, milestoning, and umbrella sampling (US) among the most popular methods in the field of biomembranes. Noteworthy are recent attempts to adopt these methods specifically in the field of membrane simulations.84−91
Central to the success of an MD simulation is the quality of the force field (FF) (i.e., the set of parameters dictating how the particles interact). In biomolecular simulation in general, there is a variety of FFs, although they fall in a handful of families that continue to be developed and are broadly similar in terms of their potential function and main approximations.92,93 An important distinction between the FFs is the level of resolution considered (Figure 2). Traditionally, full atomistic detail is the highest level of resolution for classical MD simulations (i.e., when quantum degrees of freedom or electronic polarizability are not considered explicitly). However, to increase the spatiotemporal range of simulations, lower resolution level FFs have been developed. These range from CG models that still contain chemical detail to supra-CG models that are more generic in nature and can form a bridge to the continuum level of description. Below, we discuss the current state of the FFs in each of these categories in detail, restricting ourselves to the most popular FFs in lipid membrane simulations.
Figure 2.

Different resolutions in particle-based simulation models of lipid membranes. At the all-atom (AA) level, all atoms are considered explicitly. Upon coarse-graining, small groups of atoms and associated hydrogens are represented by coarse-grain (CG) beads. Moving down in resolution to the supra-CG level, lipids and proteins are represented only qualitative by few-bead models, and solvent is considered implicitly. Further reduction in resolution is achieved by integrating out also the lipid particles by mean-field approaches.
2.1. All-Atom Models
Generally speaking, detailed atomistic lipid parameters have been developed with the same philosophy as protein FFs and in practice in most cases are related to or part of a small number of widely used more general FFs. Although there are many FFs for lipids, and many modifications have been proposed for specific cases, there is only a handful of FFs that aims to be general enough for complex membrane simulations. In the current literature, these can be divided in four families that are still being developed: CHARMM, AMBER, Slipids, and GROMOS. Given the staggering variety of lipid types, developing and testing consistent parameter sets poses significant challenges. Below we describe some of these challenges, followed by a brief description of the most widely used atomistic FFs, setup tools to build complex membrane models, and limitations of atomistic simulations. For an in-depth discussion and comparison of current atomistic FFs, see, for instance, refs (94−97).
2.1.1. Challenge of Atomistic Force Fields
First, the properties of lipid bilayers are determined by the sum of a large number of interactions, some of which are weak but add up to significant contributions. An example is the strong effect of pressure on the structure of lipid bilayers, but pressure has significant contributions from long-range Lennard-Jones interactions. This makes lipid simulations quite sensitive to small variations in parameters, in particular standard schemes used to mitigate cutoff errors routinely used in molecular dynamics simulations and the related treatment of electrostatic interactions.
Second, it has only recently become practical to routinely carry out simulations on a time scale of hundreds of nanoseconds, which is required to get equilibrated properties on a bilayer of ca. 250 lipids of one type of lipid. Thus, any change in parameters requires a large amount of computer time to investigate. For binary mixtures in liquid crystalline phases or their cholesterol-containing analogues (liquid disordered), equilibration times increase to microseconds and much more in the presence of ordered domains. A related problem is that periodic boundary conditions affect the properties of lipids in simulations. Some of the first simulations of bilayers used 32–100 lipids per leaflet, but this amounts to 5–10 lipids in each of the x and y dimension and an artificially constrained length scale compared to the characteristic length scale of lipid interactions in experimental systems.
Third, biological membranes contain a large number of lipid components, which are made of a combination of a limited number of different head groups, linkages, and a limited number of different tails.3 In principle these components should be transferable in FFs, but this requires an additional, large, amount of testing. For mixtures, the number of possible combinations explode. In practice, these components are not reliably transferable and might be considered a reasonable initial model.
Fourth, detailed experimental structural data, primarily from neutron and X-ray scattering and from NMR, have been available for a growing number of lipids, starting with phosphatidylcholine (PC) lipids, but is insufficient to validate models of all biologically interesting lipids. Force field development and detailed experiments these days often go hand-in-hand, as simulations augment the interpretation of experimental results and in some cases drive experiments to parametrize new lipids and more complex systems. Recent reviews on comparing atomistic simulations and experiments include refs (98 and 99). In simulations, PC lipids have generally been the easiest to model, but the resulting parameters have not reliably transferred to other lipid types. More recently, a wider range of model lipids has been studied experimentally, primarily by scattering, including phosphatidylserine (PS), phoshatidylethanolamine (PE), phosphatidylglycerol (PG), and phosphatidylcholine lipids (PC) lipids,100,101 the structure of polyunsaturated lipids,102 and elements of cholesterol.103 These studies provide essential detail for the validation of simulations, but still only span a small subset of all lipids, and have been subject to several reinterpretations, while key elements like sphingomyelins have received less attention. They have also been largely limited to single-component systems, whereas more detailed experimental structural data on mixtures would be very useful for the development of simulation parameters.
Next to scattering, a second major experimental technique is deuterium NMR, which measures the average orientation of C–D bonds in deuterated lipids and can measure dynamics on relevant simulation time scales.104 Since both bond orientations and detailed dynamics can be directly calculated from simulations, they are powerful validation tools.105 By selectively labeling one component in lipid mixtures, details on mixtures can also be obtained. A second major application of deuterium NMR has been the measurement of phase diagrams for simple mixtures.106 Since deuterium, unlike fluorescent probes, barely changes the chemistry of lipids, this is very important data. It remains challenging to calculate phase diagrams for computer models, but this has become feasible for CG simulations (see below) and will soon be more feasible for atomistic simulations.
2.1.2. CHARMM
The most elaborate effort has been put in CHARMM36, an updated lipid FF consistent with the most recent version of the more general CHARMM FF for biomolecular simulation, which includes protein, nucleic acid, and small molecule parameters.107−109 This work is based on extensive parametrization for tails, headgroup components, and specific lipids, and has additional advantages in the large set of parametrized and tested lipids as well as the powerful setup tool CHARMM-GUI (see below).110,111 The CHARMM lipid FF was initially developed for PC lipids but has been massively extended. It includes most common lipids used in biophysical experiments, the main families of lipids found in higher organisms, bacterial lipids specific to extremophiles including ring-containing and branched lipids and hopanoids, a library of LPS from the outer membrane of Gram-negative bacteria, and yeast lipids including sterols. The main repository for lipid parameters is CHARMM-GUI, as no comprehensive review or paper describing the current CHARMM lipidome is available, although individual components have been described in more detail.112−115 The present issue has a detailed review by Leonard et al. with a comprehensive description as of 2018.97 CHARMM lipid parameters are typically used with the CHARMM protein FF, which is implemented in most of the widely used MD programs.
2.1.3. AMBER
AMBER is a widely used FF for proteins, nucleic acids, and small (druglike) molecules, similar to CHARMM. Several groups have attempted to develop AMBER lipid parameters for use with the rest of the AMBER FF, initially based on GAFF, the generalized AMBER FF.116,117 This was tested on a limited set of lipids118 and has not been widely used. The most recent published AMBER-based parameters set is Lipid14.119 Lipid14 appeared in 2014 and has not been widely used yet either. It initially had parameters for six different PC lipids with either saturated or monounsaturated chains. Lipid14 also has updated cholesterol parameters.120 A Lipid17 version with an expanded library is under development and available for testing at the time of writing of this review but has not been formally published yet. Compatible parameters for LPS are also available for AMBER.121 A major advantage of AMBER parameters for simulating complex membranes is the advanced state of the rest of the FF, but a significant amount of development is required to make the FF easily applicable to a variety of lipids and lipid mixtures.
2.1.4. Slipids
Another promising set of FF parameters has been developed by Jämbeck et al., called Slipids (for Stockholm Lipids).122 These have been parametrized to be consistent with AMBER, although this consistency is primarily based on the same charge derivation method as AMBER uses, and the standard for Lennard-Jones parameters is derived from CHARMM.122 The initial paper described DLPC, DMPC, and DPPC, which has been expanded to include monounsaturated PC and PE lipids,123 as well as sphingomyelin, PG, PS, and cholesterol,124 and most recently a set of poly unsaturated PC lipids.125 The protocol for parametrization is sufficiently well-defined that there is a clear path for adding new lipids. This set has not been used as widely as the CHARMM lipids and is still relatively new but so far appears a viable choice that has been used both with AMBER and CHARMM protein FFs. A recent paper derived parameters for a large set of steroids to be consistent with Slipids, which are currently not available for other force fields.126
2.1.5. GROMOS
The GROMOS parameter set is based on the united-atom FF GROMOS 54A7.127 Mark and colleagues developed parameters for a number of lipid types that are consistent with GROMOS 54A7. Computationally, these have an advantage because in most software implementation united-atom lipids are substantially more efficient than all-atom lipids, in contrast to protein FFs where the extra hydrogens have much less impact. As for other FFs, the first lipids to be parametrized were saturated128 and monounsaturated PC lipids.129 In addition, parameters for bacterial lipids with branched fatty acids in their lipid chains,130 with cyclo-propane moieties,131 LPS,132 and for hopanoids and sterols133 are available. The parametrization is consistent in approach and atom types with GROMOS 54A7, which enables lipid–protein simulations, but the number of different lipids that is available and has been tested for this FF is rather limited.
2.1.6. Polarizable Models
Although the further improvement of standard atomistic FFs has arguably been the most important recent development, together with increased time scales accessible with newer computers and GPUs, in the slightly longer term recent work on polarizable lipid FFs may become very important. In standard atomistic FFs, we assume that the details of electronic motion are averaged out. The main consequence of this is that the partial charge of atoms cannot respond to the environment, although this is an important effect in some cases. Classical FFs that address this are called polarizable or nonadditive FFs, essentially with charges that will respond to the environment.134 Such FFs were routinely forecast as the next step even more than 30 years ago, but in practice their cost and the effort required to develop consistent FFs has made progress slow. In the past few years, two different approaches have been applied to membrane simulations, while a third, more detailed and expensive method has been used in other biomolecular systems but not yet on membranes to our knowledge. In the Drude oscillator model,135 small charges on springs attached to the nucleus (the standard atomistic atom) are able to move around in response to the local electric field, thus changing the charge distribution. In the FlexQ method,136 charges equilibrate locally. Both methods have been applied to model systems, including PC lipids, peptides, and nucleic acids.137−141 Simulations of mixed polarizable/standard systems have also been used, as in principle the most polarizable atoms could be treated as polarizable. Examples are systems with the lipid chains as polarizable142 or simulations with a permeating molecule as polarizable.143 A third model, AMOEBA, is considerably more complicated but is now used in biomolecular simulation144,145 and would be interesting to test in membranes.
At the current state of the art, it is clear that there are viable polarizable models for membranes. They have been tested on relatively limited cases so far, primarily PC lipids. Probably the most striking difference between standard atomistic and polarizable models is a large difference in the dipole potential across the water/lipid interface. Unfortunately, this property is not easy to measure or interpret. Other properties appear less critical, and it remains to be seen in more detail where the strengths and weaknesses of these more complicated models lie.
2.1.7. Limitations/Developments of AA Models
Lipid FFs do not divide readily into neat categories, but broadly speaking, there are recognizable families in addition to a large number of more ad-hoc modifications with generally more limited reach. Such modifications allow optimizations for a specific purpose, but in the context of complex membranes, they do not generalize sufficiently to be useful. For complex membranes, a consistent set of lipid parameters, including all relevant types for the problem at hand, which may include sterols or unusual bacterial, mitochondrial, or endosomal lipids, and a consistent set of protein parameters is essential. We argue that this requirement is currently not met by any set of parameters, although CHARMM comes closest.
An additional complexity is the reliance of all FFs on very specific cutoff values for Lennard-Jones interactions and corresponding shift functions to deal with cutoff artifacts. One consequence of this is that it is not trivial to exactly match the results of simulations with the CHARMM FF in NAMD, AMBER, or GROMACS when attempting to match the original parametrization conditions in the CHARMM simulation software. Anecdotally, results have been dramatically different as lipids undergo phase transitions to the gel phase at the wrong temperatures, although recent updates to simulation algorithms in different software packages offer significant improvements, tested in, for example, ref (146). One thorough solution for this would be to reparametrize entire FFs to not use cut-offs at all, which has become more realistic in recent years with the development of efficient lattice sum methods. Unfortunately, it is hard to see where the resources for the effort would come from to reparametrize the most widely used, and most complex, FFs. This is an effort that would have a wide impact on the field, making lipid force fields more transferrable and, therefore, ought to be funded. An interesting initiative uses a form of crowdsourcing to collect validation data on a variety of lipids in an open science format. The project identified a number of issues with the headgroups and glycerol backbones of PC lipids and provides an important database of simulation data.98,147 A more technical consideration is that changes in algorithms, often coupled to changes in computer hardware that favor one type of optimization over another, do affect simulation results.148 This will continue to be a concern and require simple test systems for regression testing as actual research systems become increasingly complex.
In addition, there are intrinsic limitations in the use of finite systems with periodic boundaries. This has been documented for the calculation of electrostatic properties, but more recently it was shown by Camley et al. that the diffusion coefficients of membrane-embedded objects have a nontrivial dependence on both the box shape and box size, and in particular show a strong dependence on the normal direction to the membrane.149 This is perhaps counterintuitive, but the water layer surrounding the membrane couples hydrodynamically to the membrane and diffusion coefficients do not converge with increasing size of the membrane patch. Subsequent large-scale simulations confirmed this behavior, and analytical expressions to correct for these artifacts have recently been introduced.150−152 Such considerations become increasingly important as simulations model larger and increasingly complex systems and begin to overlap with direct measurements of diffusion of membrane proteins by spectroscopic methods.
One additional use of deuterium NMR that could be expanded is the measurement of order parameters of a “reporter” lipid like DMPC or POPC, which are readily available in deuterated form, as a function of concentration in mixtures. More generally, deuterium NMR has not been widely applied to mixtures, except for investigations involving cholesterol, and it is challenging to obtain funding for this, but this would be important data to validate simulations of lipid mixtures.
In addition to lipids, sterols play an important biological role and require careful parametrization. Lipid–protein interactions introduce additional complexities. A lack of useful experimental data to validate simulations is a limiting factor in model improvement in many cases. Finally, improved parameters for ions, in particular their tendency to adsorb to the membrane/water interface, remains an ongoing and important area of research.153−156
2.1.8. Setup Tools
Historically, great effort was spent on creating starting structures for simulations that were as close as possible to equilibrium, because limited simulation time scales (nanoseconds) compared to phospholipid diffusion and other motions (tens of nanoseconds or more) meant that poor starting structures completely biased the simulation results.157−161 As computers became faster, starting structures for relatively simple systems became less problematic, as even starting from random mixtures in solution resulted in equilibrated bilayers.42,162 However, for complex membranes of the type described here, or even basic mixtures or membrane proteins in basic mixtures, we are now in a situation again that it takes microseconds or much longer to equilibrate starting structures, a key prerequisite for useful simulations. A second problem is that finding errors in initial structures is almost impossible in very large simulations, which puts stringent demands on useful setup methods. This will continue to be an area of development for the foreseeable future. Here we will discuss some widely used tools.
Perhaps the most widely used tool is CHARMM-GUI, a graphical interface developed by Im and co-workers to set up a broad range of biomolecular simulations, for most of the major molecular dynamics packages. One of its uses is the conversion of CHARMM FFs to input formats that can be used in GROMACS, NAMD, OpenMM, and other software.163 For membranes, it can build structures based on a desired composition using an extensive library of lipids, including bacterial lipids, a large library of lipopolysaccharides for outer membranes from Gram-negative bacteria, and a library of yeast-specific lipids. One major problem with these systems is the slow equilibration time. A related tool has recently been developed by de Fabritis and co-workers, coined HTMD (High Throughput MD).164 HTMD offers a platform for preparation of MD simulations in general, including membrane/protein systems. Starting from PDB structures, the platform assists in building the system using well-known force fields, and in applying standardized protocols for running the simulations.
Two other methods try to use simpler model descriptions to initially equilibrate a system, after which the systems are converted to atomistic detail. The insane (INSert membrANE) method uses the Martini FF and command-line tools to create arbitrary membranes at the coarse-grained level, which can be equilibrated and then converted to atomistic simulations.165,166 This is a potentially powerful approach, but there is no guarantee at the moment that Martini and atomistic FFs (or indeed different atomistic FFs) give the same equilibrium distribution of lipids in a mixture, insane is specific to GROMACS,167 and backmapping of very complex systems from Martini to atomistic is not always straightforward.
A second way of speeding up the equilibration of membrane simulations has been put forward by the Tajkhorshid group, called the Highly Mobile Membrane Mimetic (HMMM) approach.168 In this approach, the aim is to speed up lipid diffusion as it is often found to be the rate-limiting factor in membrane dynamics. Increased lipid mobility is achieved by separating the lipid heads from the tails; in fact, the HMMM bilayer consists of two monolayers of very short tail lipids with a bulk organic (or imaginary, as it does not have to actually exist as chemical) solvent in between to represent the membrane interior. The performance of the model was tested by comparing side chain free energy profiles between HMMM and full lipid representations, showing very good agreement in the interfacial part but less accuracy in the membrane interior.169 So far, the model has been mainly applied to study binding of peripheral proteins and has been shown to be an efficient tool to predict their membrane bound state.170
2.2. CG Models
The large time and length scales over which cellular processes operate has spurred the development of a large number of CG lipid FFs, following the pioneering work of Smit et al.171 and Goetz and Lipowsky172 in the 90s. Today, CG lipid models span all the way from a generic, supra-CG level of resolution to near-atomistic models. Here we focus on models that retain chemical specificity and are therefore able to distinguish specific lipid types. These kinds of models usually group 3–6 heavy atoms per CG bead, reducing a typical lipid to around 8–14 beads. Below we discuss the overall parametrization strategy for CG models (top down versus bottom up) and describe recent progress in some of the more popular CG lipid models used for cell membranes, namely the Martini, Shinoda/Devane/Klein (SDK), the SIRAH, and ELBA FFs, as well as a number of solvent-free models. The growing number of tools to automate the simulation workflow and the limitations inherent to CGing are also discussed. For a broader overview, we direct the reader to a number of other reviews on CG membrane simulations.173−176
2.2.1. Top Down versus Bottom Up
Parameterization of CG models may follow either a bottom-up strategy (also denoted structure-based coarse-graining) or a top-down strategy (thermodynamic-based coarse-graining). In the bottom-up approach, effective CG interactions are extracted from reference data, such as atomistically detailed simulations or structural databanks, aiming at a faithful reproduction of the structural features of the reference data. In the top-down approach, the focus lies on reproducing experimental data, especially thermodynamic properties such as density, heat of vaporization, and partitioning data. Both approaches have their own advantages and disadvantages. Focusing on reproducing structural details often leads to highly accurate CG models; however, the accuracy is usually limited to the state point at which the parameters were derived. Besides, the resulting CG potentials typically contain detailed features that limit the integration time step and are not always straightforward to interpret from a physicochemical point of view. Relying on thermodynamic data comes at the price of limited structural accuracy but with the benefit of reproducing global partitioning of the CG molecules over a wider range of state points. In practice, many CG FFs use a combination of these two approaches to maximize accuracy on the one hand and transferability on the other. Note that, inherent to the nature of coarse graining, it is impossible to obtain fully transferable models nor to represent all features of the underlying compound at the same time (the “representability problem”177,178). There is no unique method to construct CG potentials from higher resolution data. A full representation of higher-order correlations requires multibody potentials, which are impractical and computationally expensive, thereby defeating the purpose of coarse graining. Even when the pair correlations are well-described, other system properties such as the pressure or energy cannot be matched at the same time unless higher-order terms are included in the force field. The art of coarse graining is in the compromise of assessing which level of detail needs to be included. The best choice of CG model, in the end, will depend on the application at hand. For in depth reviews on this topic, see, for example, Brini et al.,179 Ingólfsson et al.,180 and Noid.181
2.2.2. Martini Model
The Martini FF,182,183 developed jointly in the laboratories of Marrink and Tieleman, is currently the most widely applied CG FF for biomembranes. The philosophy behind Martini is to present an extendable CG model based on simple modular building blocks, using few parameters and standard interaction potentials to maximize applicability and transferability. Martini uses an approximate 4:1 mapping and combines top-down and bottom-up parametrization strategies. Due to the modularity of Martini, a large set of different lipid types have been parametrized, covering all common lipid heads that can be straightforwardly combined with tails of varying length and degree of saturation.165 More specialized lipids, such as glycolipids,184,185 PEGylated lipids,186,187 cardiolipins,188,189,114 tetraether lipids,190 lipopolysaccharides (LPS),191−194 and a variety of sterols and sterol-like compounds (cholesterol, ergosterol, hopanoids)195 are available as well, enabling simulation of complex membranes with realistic lipid compositions (see section 3.2). The Martini model is implemented in a number of major simulation packages, including GROMACS NAMD, LAMMPS, as well as in the Materials Science Suite.196
In addition to lipids, Martini has been extended to the most important classes of biomolecules (proteins,197,198 carbohydrates,199 nucleotides200,201), as well as a large variety of polymers202 and nanoparticles.203 This variety makes the Martini model ideally suited to study a wide range of membrane-related processes, including interaction with nonbiological particles such as polymer-induced formation of nanodisks204 or penetration of gold particles.205 For processes for which long-range electrostatic interactions are deemed important, polarizable water and ion models have been developed.206−208 A major limitation of the Martini FF is the inability to model protein folding events. The use of isotropic interaction potentials cannot capture the directionality of hydrogen-bonding patterns that underlie protein conformational stability. Instead, an elastic network is used to constrain proteins, as well as nucleotides, to a reference (e.g., X-ray) structure.209 A recently introduced combination of Martini with Go models allows sampling also of unfolded protein states and is a promising method to further extend the range of applications.210 Another limitation, that also affects all-atom FFs, is the stickiness of larger biomolecules including proteins. Although this problem can be alleviated by ad-hoc approaches, for example, by downscaling protein–protein interactions or increasing protein hydration strength,211−213 the origin of the problem appears to reside in the different CG mapping densities of these biomolecules compared to the surrounding solvent. In the forthcoming new version of the model (Martini 3.0), these interactions have been balanced more carefully, resolving this issue. More background on Martini is provided in a perspective paper by the main developers214 and on the Martini webportal http://cgmartini.nl.
2.2.3. SDK Model
Klein and co-workers are among the pioneers in developing CG lipid models. Their model is based on a 3:1 mapping and therefore somewhat more detailed than the Martini model. Besides, the model uses softer interaction potentials, allowing for a better reproduction of heats of vaporization and surface tensions. The latest version of the model, the SDK FF (Shinoda, Devane, Klein215) also combines bottom-up and top-down parametrization and has resulted in improved transferability. Applications of the SDK model include studies of the phase behavior of lipid monolayers, vesicle fusion, and membrane partitioning of fullerenes (reviewed in Shinoda et al.,216). Recently the model has been extended to include triglycerides, allowing the study of formation of lipid droplets.217 A drawback of the SDK model is that only a limited number of lipid parameters are available currently, and no compatible protein model has been developed. Furthermore, the SDK model is only implemented in the LAMMPS software package, and no active development site is maintained. A recent extension of the SDK model, called the SPICA (Surface Property fitting Coarse graining) force field, includes improved parameters for cholesterol and different lipid types allowing realistic simulations of domain formation.218
2.2.4. ELBA Model
The ELBA (electrostatics-based) CG lipid FF developed by Orsi and co-workers,219 focuses on modeling lipid–water interactions and capturing important electrostatic contributions. The model uses a 3:1 mapping but represents each water molecule individually using soft sticky dipole potentials and incorporates electrostatics in the CG lipid beads as point charges or point dipoles. A few lipid types have been parametrized by matching lipid properties, such as volume and area per lipid, average segmental tail order parameter, spontaneous curvature, and dipole potential. Most recently, an ELBA model for cholesterol has been developed that matches experimental phase behavior for binary DPPC/cholesterol mixtures.220 Applications of the ELBA FF have thus far been focused on permeation of drugs and other compounds across bilayers but only using some standard lipid types. Compared to Martini, the major advantage of the ELBA models lies in the more accurate description of the electrostatic interactions. As with the SDK model, however, only few lipid types have been parametrized, and the model is only available within LAMMPS. More information is available on the Web site http://www.orsi.sems.qmul.ac.uk/elba/.
2.2.5. SIRAH Force Field
SIRAH (South-American initiative for a rapid and accurate Hamiltonian) is a top-down CG FF developed by Pantano and co-workers to model proteins and DNA.221,222 The SIRAH model has a similar mapping as the Martini model and also treats solvent and ions explicitly. Interestingly, the SIRAH FF has recently been extended to include lipids.223 So far, only parameters for DMPC lipids have been published, but the ability to model lipids opens the way to a broad range of applications involving cell membranes in the future. The FF is available for both GROMACS and AMBER. An important aspect of SIRAH is that it allows sampling of conformational changes of proteins, due to a higher resolution of the peptide backbone. More details of the FF can be found at the Web site http://www.sirahff.com/.
2.2.6. Solvent-Free Models
A number of other models should be mentioned, in particular, recent attempts to parametrize solvent-free lipid models that retain chemical detail. Implicit solvent models considerably reduce computational cost but do need to incorporate the excluded solvent interactions into the effective potentials between the CG beads. In the pioneering work of the Voth group,224,225 a bottom-up strategy based on force matching between CG and AA systems is used to derive detailed solvent-free models for a number of different lipid mixtures. Hills and co-workers used this strategy also for development of a solvent-free protein model, CgProt,226 which was recently combined with a lipid FF parametrized using the same strategy.227 Lyubartsev and co-workers228 used another bottom-up strategy, the Newton inversion method, to capture the fine details of the AA lipid models into CG potentials. Wang and Deserno229 and Sodt and Head-Gordon230 followed a more pragmatic top-down approach, adding long-range attractive interactions in the lipid tails to mimic the hydrophobic effect, tuned to fit experimental data. The model of Wang and Deserno has also been successfully combined with a CG protein model and coined the PLUM model.231,232 Curtis and Hall,233 in their LIME (lipid intermediate resolution model) FF, use hard-sphere and square-well potentials in order to use discontinuous molecular dynamics and gain even greater speedup. An implicit solvent version of the Martini FF has also been developed by the Marrink group, coined Dry Martini,234 using a rescaled interaction matrix that accounts for the hydrophobic and solvation effects. The Dry Martini model can also be combined with stochastic rotational dynamics to incorporate hydrodynamics (denoted STRD Martini).235 Wan, Gao, and Fang developed a DPD model based on Martini type mapping that can be used for both lipids and peptides.236 In a recent extension of the popular CG protein model PRIMO, developed by Feig and co-workers, an implicit membrane environment has been added to study membrane protein folding and aggregation.237
2.2.7. Limitations/Developments of CG Models
As discussed above, parametrization and validation of CG models relies either on experimental data (top-down) or higher resolution data (bottom-up). Experimental data on suitable reference systems, however, is not always available or not easy to interpret. For instance, dimerization free energies of TM peptides in model lipid membranes form a perfect test system to validate CG simulations. The free energy of this process can be easily obtained from CG simulations with the help of advanced sampling and biasing techniques. In principle, this allows comparing to the same quantity derived from association constants measured using FRET assays. However, the bound and unbound states are ill-defined, hampering a straightforward comparison. Relying on all-atom reference simulations, on the other hand, is also problematic, for two reasons. First, sampling issues at the all-atom level prevent careful validation of most processes involving protein–lipid or protein–protein binding. Second, shortcomings of the all-atom models are inherited by the CG models. In this regard, it is helpful to calibrate CG models not on a single reference FF but to use multiple ones in the absence of clearly validated targets.
Naturally, limitations of CG models arise from the reduced level of resolution. As discussed above, most CG models face limitations in the extent to which protein structural transitions can be captured, owing to the absence of directional hydrogen bonds or alternative potentials that introduce directionality. One avenue to improve the accuracy of CG models is through multiscaling, combining the sampling speed of CG models with the accuracy of atomistic models. This can be achieved in a static way, in which part of the system is modeled at high resolution and surrounded by a CG environment or in a dynamic way in which molecules can change their resolution on the fly. Despite the progress in multiscale method development, applications of such methods to lipid membranes have been very limited. In a proof of principle application,238 a multiscale method was used to simulate an atomistic protein channel in a CG Martini bilayer. Proper coupling of the electrostatic interactions between the two levels of resolution, however, remained problematic due to the poor short-range screening behavior of the CG solvent. To achieve a quantitatively more accurate method, cross optimization of the interactions between CG and the atomistic FF is probably necessary as has been attempted in the PACE FF in which Martini lipids are combined with a near-atomistic protein model.239 The ELBA FF has also been used in a multiscale setup, in particular to study permeation of AA drugs across CG membranes.240 The level of detail retained in the ELBA model is high enough that the AA-CG cross interactions can be based on standard combination rules. Multiscale simulations with the SIRAH FF have also been reported241 but not (yet) involving lipid membranes. In an implicit membrane environment, the PRIMO FF can be combined with CHARMM.242
At the moment, more powerful are so-called serial multiscaling schemes that are used to reconstruct all-atom detail from a given CG configuration (“backmapping”). Most commonly applied backmapping tools for lipid systems include fragment-based approaches,243,244 simulated annealing,245 and usage of geometrical rules.166,246,247 There is also a promising new multiscale tool GADDLE maps which is based on a Monte Carlo sampling algorithm.248 Typically, backmapping is used either to validate specific interactions observed in CG simulations or to focus on some atomic details of the system of interest. Note, however, that the amount of sampling that can be performed at the atomistic level is usually limited. Therefore, finding that a CG configuration is also stable at the atomistic level, albeit encouraging, is not a proof of the validity of the CG model. The opposite, for example, observing that the CG configuration is unstable at the all-atom level, may however point to a limitation of the CG model.
Milano and co-workers have developed an interesting hybrid particle-field scheme, combining molecular dynamics with self-consistent field theory (the hybrid MD-SCF).249,250 The main difference of the hybrid MD-SCF method in relation to other CG approaches is that the calculation of the nonbonded interactions between the CG particles is replaced by an evaluation of an external potential on the local density. With this scheme, the hybrid MD-SCF method allows the usage of mapping and bonded parameters commonly used in other CG approaches in combination with an efficient parallelization for the calculation of interaction forces, obtained via an average density field.251 Lipid applications are still limited, which includes simulations of phospholipids in bilayer and nonlamellar phases, with lipids mapping and bonded parameters based in the Martini scheme.252,253 More recently, a flexible CG model for protein has been introduced, allowing studies of conformational changes, even in a lipid environment.254 The hybrid SCF-MD is available in a dedicated software package called OCCAM. More details of the method are available at the Web site http://www.occammd.org/.
2.2.8. High-Throughput Tools
One of the advantages of CG models is that they provide easy access to high-throughput applications. Hundreds or thousands of simulations can be performed, systematically exploring, for example, lipid membrane composition or protein mutant libraries. A nice example is the membrane protein database MemProtMD, developed by Sansom and co-workers: based on self-assembly simulations, configurations of all classes of membrane proteins embedded in a natural lipid environment are provided.255,256 To facilitate high-throughput applications, many new and improved methods have been developed to help set up initial simulation configurations. A key example is the CHARMM-GUI framework (see also discussion above), which currently supports also the CG Martini FF.257,258 A drawback of CHARMM-GUI is that it is not command-line-based and therefore cannot be integrated into automated workflows. An example of a command-line-based tool is Moltemplate (http://www.moltemplate.org/), a generic molecular builder for LAMMPS, with support for the CG models Martini and SDK. Another command-line based tool called insane is a popular membrane-building tool associated with the Martini FF and allows for on the fly generation of new lipid templates.165 A number of programs have also been developed that automatically setup and run CG simulations for high-throughput screening of protein–protein interactions, such as Sidekick259 and Docking Assay For Transmembrane components (DAFT).260 To further automize the simulation workflow, current efforts are also being directed toward automated CG topology builders.261−264 Here, one of the main challenges is to automate the mapping of the underlying atomistic structure to the CG representation, a nontrivial problem. The power of such a tool is illustrated in a recent paper from Bereau and co-workers,265 who established linear relations between bulk membrane partitioning and the potential of mean force covering more than 400000 drug compounds.
2.3. Supra-CG Models
A longer-term aim of simulation of complex biological membranes is to enable us to relate molecular structures of their lipids and protein components to cellular phenotypes. This requires us to be able to compare the behavior of membrane simulations more directly to experiments at the cellular level, for example, via various super-resolution imaging modalities. The CG models described above all have a similar level of granularity, whereby each CG particle corresponds to 3–4 heavy (i.e., not hydrogen) atoms, such that, for example, a phospholipid molecule is represented by 10–15 CG beads. The advantage of this level of granularity is that it allows retention of chemical specificity of, for example, lipid headgroups in their interactions with proteins. The disadvantage is that it restricts practical applications to systems of ∼2 M particles (i.e., ∼8 M heavy atoms), equivalent to a length scale of <100 nm, on time scales up to the millisecond range. We need to move beyond these limitations in order to address dynamic events in membrane cell biology. For example, at the lower scale of cell membrane events, a clathrin-coated vesicle has a diameter of 100 nm and is formed by budding on a time scale of 20 s.266 Here we discuss current approaches to simulate such large-scale collective phenomena, requiring a further reduction in resolution denoted supra-CGing. For other reviews in this field, see, for example, refs (267 and 268).
2.3.1. Supra CGing Approaches
In order to address events on these larger scales, supra-CGing approaches are needed. A number of approaches may be adopted in order to reach the desired meso and micro scales. At a simple level, one can employ CG models with fewer particles, for example, just a few particles per lipid molecule (e.g., the model by Ayton and Voth269) or even a few particles to represent a protein molecule or domain (e.g., models by Zhang et al.270,271). Alternatively, one may both reduce the number of particles and use modified interactions that smoothen the energy surface (as in DPD models, e.g., Venturoli et al.272). A more radical level of simplification (to reach even larger scales) may be to integrate out lipids (and water) altogether, such that proteins are represented as particles interacting in a continuum membrane environment. For all of these approaches, parametrization is a challenge, especially if one wishes to retain a degree of chemical specificity in these higher-level models, which is essential if they are to be used to address genuinely biological questions. Voth and co-workers have developed a theoretical framework for obtaining and interpreting such supra-CG models.273,274
2.3.2. Few-Bead Lipids
A number of groups have explored CG models in which only a small number of particles are used to represent each lipid molecule.275,276 For example, Voth and colleagues have developed a framework for “aggressive” CGing of lipids in which, for example, two or three particles can represent each lipid molecule in a (solvent free) model. This can be used to simulate, for example, 200 nm diameter lipid vesicles containing ∼500000 lipids.269 A related model has also been developed for charged lipids to capture the electrostatic interactions of their headgroups in a “broad brush” fashion which has been used to model both mixed lipid vesicles and (peripheral) protein/charged lipid bilayer interactions.277 A key feature of these models is to combine analytical potentials (e.g., Gay-Berne models) to describe the generic anisotropic behavior of the lipids with more detailed force-matched potentials that provide an element of chemical specificity.
A similar level of granularity to that in “standard” CG representations is employed in DPD models,272 which smooth the energy surface for interactions between lipid molecules. The advantage of the soft potential employed is to enhance diffusion, although it may result in, for example, unphysical lipid overlaps. DPD models have been used, for example, to examine the effects of cholesterol on lipid bilayer structure.278 Comparable models have also been applied to examine mechanisms of fusion between lipid bilayer and vesicles.279 A supra CG model for lipids based on soft interactions has also been developed by Laradji and co-workers and applied to study a variety of phase transitions and membrane remodeling processes.280
2.3.3. Reduced Protein Models
There are two broad approaches to the representation of proteins in supraCG simulations. One approach relies on idealizing/simplifying the representation of proteins in a fashion which (it is hoped) will retain the essence (but not the specific chemical details) of protein/protein and protein/lipid interactions. The other relies on simplified models of proteins comprising just a few particles, the interactions between which are parametrized on the basis of more detailed CG and/or atomistic simulations. The former approach has been used extensively within a DPD framework to study protein/lipid and protein/protein interactions in simplified models of biomembranes. The latter approach has been applied to much larger and more complex biological membrane systems in order to capture their emergent behavior on a meso scale.
In a series of DPD simulation studies, Smit et al. have modeled membrane proteins as, for example, rodlike structures with hydrophobic cores and polar caps and have used these to explore protein/membrane interactions and also the free energy landscapes of protein–protein interactions within membranes.281,282 Protein/protein interaction potentials of mean force (PMFs) computed from DPD simulations have been used to develop larger scale 2D models in which proteins are treated as disks interacting through those PMFs.283 This provides an interesting route to capturing protein/protein interactions in very large-scale simulations. Weiss and colleagues have also used comparable DPD models of membrane proteins to explore, in a generalized fashion, the influence of membrane protein structure on, for example, diffusion.284−286 DPD simulations in which membrane binding proteins were represented as highly simplified Janus-like particles have been used to propose models of large-scale dynamic events such as membrane vesiculation.287 Again, this provides an interesting supraCG route to large-scale biomembrane behavior, but parametrization will be challenging if biologically realistic specificity and complexity is to be preserved in such models.
A promising route to supraCG models of membrane proteins that retain a degree of specificity, in terms of the irregular shapes and dynamics of those proteins, is provided by the work of, for example, Voth and colleagues in which protein domains are represented by a small number of particles.270,271 The supraCG mapping in these ED-CG models is achieved by matching the dynamics of the CG model to a more detailed essential dynamics (ED) description derived either from atomistic simulations combined with PCA270 or by an elastic network model (ENM) of the protein.271 These models have been used in, for example, studies of membrane remodelling288 (see below).
An even coarser level of granularity, in terms of representation of membrane proteins, has been explored in a DPD study of the organization of membrane protein complexes in simple models of photosynthetic membranes. In this study, the protein complexes were represented by a model in which a protein (or protein oligomer) is represented by a single particle, combined with a two particle per lipid molecule model. The protein particles were parametrized phenomenologically on the basis of experimental (electron microscopy) data for their supercomplexes.289 Such a model allows large-scale (500 nm) organization of membranes to be explored, although the parametrization does rely on appropriate experimental data being available.
2.3.4. Meso Models
One approach to developing very large-scale models of, for example, membrane protein cluserting in a bilayer environment is to ignore the lipids and model protein/protein interactions using data derived from CG simulations of protein interactions in a bilayer model. A possible approach to this is sketched out by, for example, Yiannourakou et al.283,290 using PMFs from DPD simulations of simple model membrane protein–protein interactions (see above) as parameters for 2D MC simulations of clustering of proteins. More recently, a comparable approach has been employed in which membrane proteins were modeled as 2D disks with “sticky patches” for interactions based on analysis of protein–protein contacts in large-scale CG-MD simulations291 (see below). It is also feasible to derive knowledge-based potentials from protein databases to represent the interactions of proteins with the implicit membrane environment, as in the recent work of Wang et al.292 All of these models currently ignore the complexities of the lipid bilayer, but in the longer term, it may be possible to combine them with continuum representations of multicomponent lipid bilayers, for example, Hu et al.293 and Nepal et al.294
An alternative approach is provided by the MesM-P (mesoscopic membrane with proteins) model of Voth and colleagues which allows, for example, membrane vesicle geometry to be explored as a function of the protein density and properties.295 There are also various mesoscale cell models representing the membrane by a triangulated surface296−299 and mesoscale models of proteins based on finite element models.300 Indeed, it would be timely for a systematic exploration of the “zoo” of existing and potential mesoscopic models301,302 to establish which classes of these may be successfully linked to underlying CG models of biological specificity in order to successfully enable quantitative predictive cellular level membrane dynamics.
3. Increasing Complexity
The complexity of cellular membrane is really staggering. There exists more than a thousand different lipid types that are found in biological membranes, with, in some cases, hundreds present in the same membrane.3 Embedded in this complex lipid mixture is a plethora of membrane proteins, either transmembrane or peripherally bound. On top of this, many cell membranes are highly curved, and interact with components of the surrounding medium such as the cytoskeleton or neighboring organelles or cells. And all of this happens under, constantly changing, nonequilibrium conditions. To capture this complexity, it is evident that we need to move beyond modeling highly simplified model membrane systems containing one or two lipid components only and being surrounded by excess aqueous solvent, notwithstanding the continued usefulness of studying simplified systems.
Below, we describe our current ability to increase the level of complexity, making use of the improvements of lipid FFs as discussed in the preceding section. We first describe simulation studies that thoroughly explore the basic behavior of multicomponent lipid and lipid protein mixtures (section 3.1), followed by the ongoing efforts to model specific membranes in realistic detail (section 3.2), on our way to full cell models (section 3.3).
3.1. Multicomponent Membranes
In this section. we provide an overview of the growing body of simulation studies that consider multicomponent membranes to understand the organizational principles of membranes at a fundamental level, including formation and structure of lipid domains, binding of specific lipids to membrane proteins, membrane-mediated protein–protein interactions, and lipid- or protein-induced membrane curvature.
3.1.1. Lipid Domains
Given the diversity of lipid types in cell membranes, a nonuniform distribution of lipids in the lateral plane of the membrane is rather likely. In fact, the heterogeneous nature of the cell membrane is underlying the raft concept,4,5 which, in its current form, states that specific lipids together with proteins can cluster into nanodomains. These nanodomains may be transient and too small to be detected by experimental means but may also grow into more stable functional platforms when needed.303,304 The propensity to form distinct phases is already found in model membranes composed of ternary mixtures of saturated lipids, unsaturated lipids, and cholesterol, capable of forming coexisting liquid-ordered (Lo) and liquid-disordered (Ld) domains. In fact, ternary and quaternary mixtures display a rich behavior of domain formation processes ranging from critical fluctuations, modulated phases, all the way to macroscopic phase separation. Interestingly, extracts from real cells show similar phase behavior,305,306 pointing to the possible biological relevance of this fundamental aspect of multicomponent lipid membranes.
Although mean field theories describe these phenomena in a qualitative way,307−309 MD simulations prove essential in providing the molecular details of both the structural and kinetic aspects of lipid nanodomains. Important insight into the structure of the Lo phase has been obtained by recent all-atom models of the groups of Vattulainen310 and Lyman,311,312 revealing the presence of substructures within these domains. Still, with all-atom models, spontaneous segregation into coexisting Lo/Ld domains is proving difficult to observe, likely hampered by the slow kinetics of phase separation. Only the onset of the process has thus far been captured with atomistic models.313,314
Here, CG models have proven very valuable. An important breakthrough was reported by Risselada and Marrink,48 who simulated the spontaneous formation of Lo and Ld domains in ternary mixtures of saturated and unsaturated lipids together with cholesterol based on the Martini model. Follow up studies have further explored the properties of these domains as a function of lipid composition,315−322 including the effect of hybrid lipids (lipids with one saturated and one unsaturated tail) that act as linactants (i.e., decrease the line tension between the domains).323−327 Making use of high-throughput simulation strategies, complete lipid phase diagrams can nowadays be established, for example, for binary lipid/cholesterol systems as a function of temperature,328,329 as well as ternary and even quaternary mixtures.330−334Figure 3 provides an example of a ternary phase diagram from Carpenter et al.335 based on the Martini model, in comparison to the experimental phase diagram.336 In the study of Ackerman and Feigenson,330 concentrations of DPPC and cholesterol are fixed, whereas the nanodomain-inducing lipid 16:0,18:2-PC (PUPC) is incrementally replaced by the macrodomain-inducing lipid 18:2,18:2-PC (DUPC). Extensive simulations of this four-component system reveal that lipid demixing increases as the amount of DUPC increases, in agreement with the experimental phase diagram. Furthermore, domain size and interleaflet alignment change sharply over a narrow range of replacement of PUPC by DUPC, indicating that intraleaflet and interleaflet behaviors are coupled. It turns out to be not always trivial to assign phases in these mixtures, due to the challenge of identifying a phase based on local physical properties, in addition to challenges due to finite size and hysteresis effects.328,331,337
Figure 3.

Example of a multicomponent membrane phase diagram. The ternary lipid mixture dioleoyl phosphocholine (DOPC), DPPC, and cholesterol exhibits a range of interesting phase behavior. Experimental phase diagram is shown on the left, and a simulated diagram using further optimized Martini parameters for DOPC and DPPC on the right. Inserts show snapshots of four of the different simulations illustrating the phase separation. The lipids are colored red, blue, and green for DOPC, DPPC, and cholesterol, respectivly. Adapted from ref (335). Copyright 2018 American Chemical Society.
An interesting and ongoing topic of discussion is the extent to which the domains in opposing leaflets are registered (i.e., occupy the same lateral position as a result of an inter leaflet coupling mechanism). In principle, domains can be both registered or antiregistered, or anything in between. Theoretically, driving forces that govern the extent of registration include minimization of the line tension at the domain boundaries, minimization of the interleaflet surface tension, release of curvature frustration, electrostatic coupling, lipid or cholesterol tail interdigitation, and flip-flopping of additives or cholesterol.338−340 Currently, simulation studies in support of each of these mechanisms can be found,48,341−349 probably pointing to a subtle interplay of all of these effects occurring in realistic membranes.
To add another layer of complexity, the organization of the nanodomains can be tuned by a number of additional factors, as evidenced by simulation studies on the effect of stress350 and immobilization,351,352 as well as the addition of other additives such as hydrophobic compounds,353,354 sugars,355,356 or ions.357 However, predictions from simulations are not always in agreement with experiments,358 pointing to differences between experimental and computational time and length scales (notably, the contribution of the domain boundaries is much more dominant in simulation studies359) and/or deficiencies in the nature of the FFs, and/or challenges in the interpretation of experimental data.
Due to the different nature of the Lo and Ld domains, the former being enriched in cholesterol and saturated lipids and more densely packed, one naturally expects a nonequal distribution of membrane proteins between these phases. Experiments confirm this expectation, showing a common preference for proteins to reside in the Ld domains, unless specific lipid anchors, commonly post-translationally attached to membrane proteins in vivo, are present.360 A number of recent MD studies have addressed the driving forces underlying this sorting process. A pioneering study was performed by Schäfer et al.,361 revealing the molecular mechanism behind the generic preference for peptide and proteins to reside in Ld domains. In accordance with the authors, inclusions disturb the tight packing of saturated lipids and cholesterol in the Lo domain, providing an enthalpic driving force for sorting into Ld domains. In a subsequent work, De Jong et al.362 demonstrated that lipid anchors can indeed provide a counter-force to steer proteins toward the more ordered Lo domains. The importance of lipid anchors in dictating sorting behavior is also clearly demonstrated in the work of Gorfe and co-workers363−365,359 as well as others.362,366 One highlight is the study showing that different Ras variants (H-Ras, N-Ras, K-Ras) have different propensities to segregate into Lo or Ld domains, driven by the opposite preference of palmitoyl and farnesyl anchors for ordered and disordered membrane domains.363 Localization of Ras clusters at the domain boundaries may further lead to a reduction in line tension and destabilization of the domains. Parton et al.367 show that influenza hemagglutinin, a TM protein containing a number of palmitoyl anchors, also resides in proximity of Lo regions.
Apart from lipid anchors, simulation studies have revealed a number of other mechanisms that effect sorting. Restriction of tilt, for instance as a consequence of protein anchoring to the cytoskeleton, is important as it prevents release of lipid mismatch through sorting.368 Likewise, fixed membrane curvature may lead to sorting of TM peptides.369 In addition, specific lipids can mediate the sorting behavior. A striking example is the sorting of peptides into Lo domains under the influence of gangliosides.362,370 Another example is the observation of cholesterol mediated sorting in a joint experimental-computational framework.371 Here it is shown that cholesterol may constrain the structural adaptations at the peptide-lipid interface under mismatch, resulting in a sorting potential.
For more in depth discussion on the topic of lipid domain simulations, please consult recent reviews from Bennett and Tieleman372 and Róg and Vattulainen,373 as well as the comprehensive review of Hof and co-workers covering both experimental and computational studies on membrane nanodomains.374
3.1.2. Protein–Lipid Binding Sites
Identifying lipid binding sites on membrane proteins is a rapidly growing area. Experimentally more and more lipid binding sites are being discovered,375 thanks to an increasing number of techniques. Traditionally, X-ray techniques may reveal tightly bound, cocrystallized lipids, but advanced mutagenesis studies or chemical cross-linking techniques are used to probe also weaker bound lipids that are washed away under the harsh crystallization conditions. New techniques such as the use of lipid nanodisks to isolate membrane proteins with their native lipid environment,376 as well as mass-spectrometry (MS),377 hold a lot of promise to further this development. On the basis of the strength of binding, two classes of lipid binding sites can be differentiated, namely specific and nonspecific binding sites. The former involves tightly bound lipids that occupy specific sites inside or at the protein surface. The latter refers to lipids only showing a weak protein affinity, occupying the annular shell around the protein. The ability of membrane proteins to recruit and bind specific lipid types is of functional importance. For instance, lipid binding may dictate the sorting behavior of proteins between different membrane domains and may facilitate protein insertion by lowering the cost of hydrophobic mismatch;378 bound lipids may either protect proteins against aggregation (locking mechanism) or bridge proteins together into functional supercomplexes (bridging mechanism); specific binding sites may be involved in enzymatic reactions (e.g., donating protons or electrons) or more generally provide structural stability and stabilize specific protein conformations.
Computational studies are entering this field at a rapid pace. Both all-atom and CG simulations have proven useful to look at both specific and nonspecific lipid binding. In a typical setup, a membrane protein is embedded in a bilayer composed of two or three components, including the putative binding lipids. Binding sites are then identified by constructing density maps (“heat” maps) and some user defined density threshold. In principle, MD simulations also allow for quantification of the strength of lipid binding, through computation of a PMF.379,401,85,380 In AA simulations, strongly bound lipids can usually be distinguished from weak or nonbinding lipids, but equilibration remains problematic. In particular, lipid exchange rates may become prohibitively large. Even nonbinding lipids can occupy a given site for hundreds of nanoseconds. Currently, the best strategy is to use multiple runs from different starting configurations to get a handle on the reproducibility of the results. For instance, Rogaski and Klauda381 generated five different orientations of a peripheral membrane protein to study its binding to a lipid bilayer and found consistent interaction modes requiring the presence of anionic lipids. Alternatively, CG models can be used. At CG resolution, reversible lipid–protein binding events can be observed on time scales of 10–100s of microseconds. In a state-of-the-art example, Arnarez et al.382 identified six cardiolipin (CL) binding sites on the respiratory chain complex cytochrome bc1. To provide a fully atomistic view of these binding sites, the CG configurations were backmapped to AA resolution, a procedure also frequently used by the group of Sansom.383,384 AA simulations can also serve to refine X-ray data, as shown by Aponte-Santamaria et al.385,386 in the case of cocrystallized DMPC lipids around aquaporin-0.
The full power of simulation models in this area is best demonstrated by a fast-growing number of studies that can reproduce experimental binding sites. An impressive example is the specific binding of C18-SM to a binding pocket in the TM domain of the COPI machinery protein.387 In this joint computational-experimental study, MD simulations reveal a close interaction between C18-SM and the transmembrane domain, as suggested by mutagenesis data. Interestingly, the interaction is found to be very specific, depending on both the headgroup and the backbone of the sphingolipid, as well as on a signature sequence of the protein. The verification of experimental cholesterol binding sites of GPCRs is another hot topic. Successful validations have now been made in case of the human A(2A) adenosine receptor,388−390 the β2-adrenergic receptor,391,392 and rhodopsin,393 recently reviewed by Sengupta et al.394 (Figure 4). Simulations of sterol binding to the known sterol binding sites of the oxysterol binding protein osh4395 and the mitochondrial voltage gated anion channel VDAC1,396 cholesterol binding to the family of monoamine transporters,397 as well as a study of binding of cholesteryl esters to their binding pocket in the cholesteryl ester transfer protein398 are also worth mentioning. An increasing number of examples exist also for anionic lipids, which are key regulators of membrane protein behavior. For instance, simulations of the Sansom group identify phosphatidylinositol-triphosphate (PIP3) binding sites on the pleckstrin homology domain399 and PIP2 binding sites on the inwardly rectifying potassium (Kir) channels400,383 in line with X-ray and mutagenesis data. Other examples include binding sites for CL on the respiratory chain complexes cytochrome c oxidase and cytochrome bc1,382,401 reproducing sites known from earlier structural studies and buried into protein cavities, as well as validation of CL binding sites on the mitochondrial ADP/ATP carrier402,403 and reproduction of known DPPG binding sites of potassium channels KcsA and chimeric KcsA-Kv1.3.404
Figure 4.
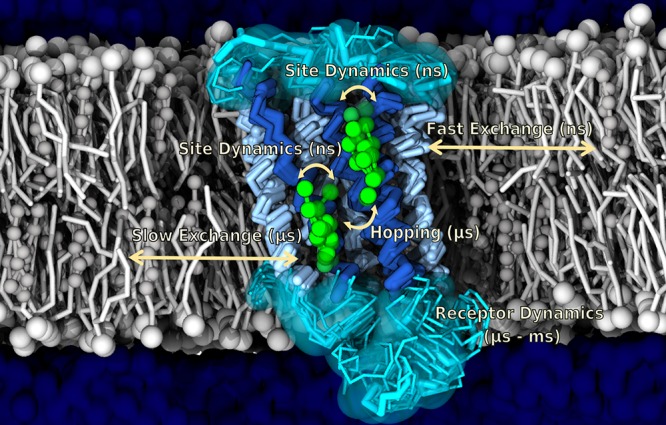
Example of protein–lipid binding modes. Cholesterol binding to a GPCR with indications of fast and slow exchange dynamics, obtained from MD simulations by Sengupta and co-workers.394
Given the ability of current simulation studies to reproduce known lipid binding sites, the prediction of novel binding sites becomes interesting. A number of studies on respiratory chain complexes reveal hitherto unknown CL binding sites on the membrane-exposed surfaces of these proteins.382,401,405−408 Surface-bound CLs could play an important role in, for example, proton uptake or by providing structural integrity of the complexes and supercomplexes. CL binding sites are also predicted for a number of other bacterial proteins.409−412 Likewise, simulation studies on GPCRs are pointing at novel cholesterol binding sites, for instance, in case of the serotonin(1A) receptor413 the A(2A) adenosine receptor,414 and the Smoothened receptor,380 and on the importance of PIP2 in regulating conformational states.415 Cholesterol binding sites are also found on the Kir2.2 channel, depending on the open/close state of the channel.416 In a study on nicotinic acetylcholine receptors (nAChR), cholesterol competes with PUFA containing lipids to occupy binding sites.417 The list of predicted lipid binding sites keeps growing. Other examples are the discovery of a PE binding site, stabilized by a lipid-mediated salt bridge, on lactose permease,418 and of numerous PG binding sites on the ammonium transporter AmtB.419 Binding sites for PI lipids were found in a combined experimental and computational study of the eukaryotic purine symporter UapA.420 Their presence at the dimer interface suggests a role in structural stability of the complex. A similar study revealed PIP2 binding to mammalian two pore channels, forming a cross-link between two parts of the channel and enabling their coordinated movement during channel gating.421 The power of combining modeling and experimental studies is further illustrated by the discovery of ceramide binding sites on one of the TM helices of Late Endosomal Protein LAPTM4B.422 Although quite often overlooked, the absence of lipid binding sites is also useful information that can be extracted from simulations. For instance, in a multiscale study, Stansfeld et al. report no specific DPPC binding sites for various members of the aquaporin family.384
Noteworthy are also an increasing number of studies that reveal pathways for protein-mediated lipid flip-flop, a mechanism that has been hypothesized already a while ago but not been observed directly (Figure 5). Khelashvili et al. identified the existence of a series of weak lipid binding spots along the surface of Opsin, allowing flip-flopping of POPC lipids423 (Figure 5A). Spontaneous penetration of lipid head groups into the membrane interior along the aqueduct nhTMEM16, a fungal scramblase, was observed during a simulation of Tajkhorshid and co-workers424 (also noted earlier244), resulting in a continuous file of lipids connecting the outer and inner leaflets (Figure 5C). Full permeation of a POPC lipid from the inner leaflet to the outer leaflet of the membrane was captured during the simulation. Additional full translocations of lipids, including the charged lipid POPS, took place when a transmembrane voltage was applied. Further details of the translocation pathway of nhTMEM16 were revealed in a similar study by Lee et al.425 and in a study on the TMEM16K member of the family.426 In another study, Koch et al.427 discovered a potential flippase activity of the SecYEG channel. They found the presence of a single binding site for DOPG lipids that can be reached from both membrane leaflets and thus allows spontaneous flip-flop of an anionic lipid (Figure 5B). Transfer of lipids between different protein complexes is observed in the study of Huber et al.428 Here, extensive simulations were used to show a downhill pathway of transfer of lipid A from bacterial outer membrane models through CD14 to the terminal TLR4/MD-2 complex. Aksimentiev and co-workers429 reported a synthetic DNA nanopore specialized in flipping lipids, by stabilizing a toroidal (i.e., lipid lined) pore. MD simulations show indeed spontaneous lipid scrambling to occur on a submicrosecond time scale.
Figure 5.
Protein-induced lipid flip-flop. (A) Overlay of POPC lipids bound to Opsin at intermediate stages of the flip-flop pathway.423 (B) CG trajectory of a DOPG lipid during a (partial) flip-flop mediated by the SecYEG complex.427 (C) Overlay of POPC configurations bound to a fungal scamblase, defining a flip-flop pathway.424 In all snapshots, the lipid phosphate groups are represented by red spheres. The tails are colored from yellow to green to visualize the flip-flop pathway.
Furthermore, MD simulations are frequently used to study nonspecific lipid binding. Key papers in this area are from Vattulainen and co-workers,430,431 in which they show strong correlations between the lateral diffusion of membrane proteins and a shell of 50–100 annular lipids. This kind of protein–lipid binding is observed even in lipid membranes composed of a single lipid type and therefore clearly nonspecific. On the basis of CG simulations of a large variety of membrane proteins, Sturgis and collaborators show that even simple mixtures of nonspecifically bound lipids already give rise to a complex perturbation pattern around the protein that is not easily described by elastic membrane deformations and can be long-range in nature.432 In multicomponent membranes, nonspecific lipid binding will lead to an inhomogeneous distribution of lipids around the protein in general. One of the first examples in this respect is the study of Grossfield et al. on the enrichment of polyunsaturated lipid chains around rhodopsin433 in mixed SDPC/SDPE/Chol bilayers. A recent extension of this work points at a dependence of these interactions on the conformational state of the protein.434 Other examples of nonspecific lipid binding include the recruitment of short-tail lipids around OmpA,435 of anionic (POPA) lipids at the gap junction hemichannel connexion-26,436 the nonspecific binding of POPS to TM and juxtamembrane domains of cytokines437 and to integrin,438 weak binding of annular PC/PE lipids to MsbA flippase,439 accumulation of cholesterol at the C-terminal helix of a phospholipase scramblase,440 accumulation of GM1 around WALP peptides362 and aquaporin,185 preferential binding of PE over PC lipids in secondary transporters441 and VDAC,442 and redistribution of PC lipids around the gramicidin A channel dependent on tail length and unsaturation.443
A related topic is the membrane binding of peripheral proteins, which often requires the presence of specific lipids that provide the necessary driving force for stable protein–membrane interactions. Simulation studies in this category reveal the CL mediated membrane binding of creatine kinase (MtCK),444 the enrichment of phosphatidic acid (PA) and PIP2 at the membrane binding spot of actin capping protein (CP),445 the increased propensity of negative lipids to interact with a glycosyltransferase,446 and PAs clustering around the acylated pleckstrin homology domain-containing protein (APH).447 The signaling lipids PI and PIPs seem to play a particularly important role. Many examples can be found in the recent simulation literature in which PIs or PIPs drive the protein binding and orientation, for example, the phox-homology domain (PX),448 auxilin,449 BIN1/M-Amphiphysin2,450 MIM I-BAR,451 GTPases,452 KIdney- and BRAin-expressed protein (KIBRA),453 the HIV-1 matrix protein,454 as well as a variety of actin binding proteins.455−457 Other examples of lipid-mediated membrane binding are the ganglioside mediated binding of cholera toxin,458 competitive binding of bis(monoacylglycero)phosphate and SM to Niemann-Pick Protein C2,459 the role of cation-pi interactions in stabilizing the binding of phospholipases,460 and the modulating effect of cholesterol on the depth, orientation, and conformation of the membrane binding fragment caveolin-1.461
Nonspecific clustering as well as specific binding of lipids by membrane active peptides is another area where simulation studies are increasingly being used.462−464 Simulations addressing lipid-peptide interplay in case of amyloid peptides also remains a hot topic, in particular with respect to membrane-mediated fiber formation.465−468 For a more elaborate description of modeling of protein and peptide-lipid interactions, we refer to previous reviews by Hedger and Sansom,469 Grouleff et al.,470 Wen et al.,471 as well as to Corradi et al. in this issue.472
3.1.3. Lipid-Mediated Protein Oligomerization
Lipid-mediated interactions are of key importance in driving the clustering of membrane proteins. Clustering forces of this kind include release of membrane curvature stress, capillary condensation, lipid depletion effects, and Casimir type forces (arising from perturbed fluctuations in, for example, lipid density or thickness). Depending on the system details, such lipid-mediated effects can dominate direct protein–protein interactions and can be long-range in nature.473−475 Understanding the molecular driving forces that are ultimately responsible for the sorting and clustering of membrane proteins is currently an active field of research in which simulation studies play a key role.476
To systematically study clustering of membrane proteins, basically two approaches are followed: spontaneous self-assembly, which allows also for the formation of higher-order oligomers, or biased simulations to determine the protein–protein dimerization free energy. In self-assembly simulations, the complexation of proteins is simply followed over time. Self-assembly simulations are typically performed using many independent replicas to probe the reproducibility of the interfaces formed. The behavior of WT and mutant proteins can be compared, providing additional insights into the packing motifs. In the case of single TM helices, a growing number of studies demonstrate that the experimentally determined interfaces can be reproduced, even when using CG models to speed up the sampling (reviewed by Psachoulia et al.477). Such approaches require a high-throughput approach to obtain statistically relevant results.478,479 The ability to predict packing of TM helices has paved the way for CG modeling studies of self-assembly of larger protein complexes, in particular, protein complexes in which the interface is formed by single TM helices.480−482 For instance, in a joint experimental/modeling effort, van den Boogaart et al.483 revealed the molecular organization of syntaxin clusters and showed that syntaxin clustering is mediated by electrostatic interactions with the strongly anionic lipid phosphatidylinositol-4,5-bisphosphate (PIP2). The mediating role of PIP2 is also apparent from a simulation study of the binding of FERM to L-selectin.457 Not only is PIP2 required for efficient binding of FERM to the plasma membrane but also PIP2 induces a conformational change of L-selectin which allows the formation of the heterocomplex. Computational evidence for the role of PIP2 in activating integrin, by stabilizing an integrin-talin heterocomplex, has also been obtained,484 as well as revealing the role of PIPs in modulating EphA2, a receptor tyrosine kinase.485 As another example of lipid-mediated protein complex formation, PS lipids are found to steer the formation of the RAS/RAF complex in a multiscale study by Travers et al.486
Self-assembly studies of polytopic membrane proteins are still hampered by slow kinetics, but the onset of protein oligomer formation can be simulated. Following the pioneering studies of Periole et al.,487 the oligomerization tendency of GPCRs as well as other membrane proteins has been simulated through self-assembly by a number of groups.488−492 These studies, mostly based on the Martini FF, often indicate formation of stringlike clusters of proteins with preferred protein–protein interfaces. A clear effect of cholesterol was reported in steering the dimer interface formation in case of the beta2-adrenergic receptor493 and chemokine receptors.494,495 Similarly, both cholesterol and PIP2 lipids were found to affect the interfaces formed in large-scale self-assembly simulations of human serotonin transporters.496 Self-assembly simulations of respiratory chain complexes by Arnarez et al.382 show complexation between cytochrome bc1 and cytochrome c oxidase and point toward a specific role for CL in bridging the proteins together. Clustering of mitochondrial translocases was also shown to depend on the presence of CL in large-scale CG MD simulations.402 In another recent example, gangliosides were observed to bridge tetraspanin CD81 proteins into higher-order aggregates497 (Figure 6). However, on the multimicrosecond time scale accessible with current simulations, equilibration of the protein–protein interfaces of polytopic membrane proteins has not yet been achieved. Reversible sampling of protein–protein binding/unbinding events is extremely challenging for models that retain chemical specificity, the more so in the crowded environment of real cells (see below).
Figure 6.
Example of lipid-mediated protein–protein oligomerization. The snapshots show how gangliosides mediate cluster formation of tetraspanin CD81 proteins. Reproduced from ref (497). Copyright 2014 American Chemical Society.
In addition to affecting the kinetics, protein crowding can also impact the thermodynamic behavior of the system. Domanski et al.498 found that, at lipid/protein ratios characteristic of real membranes, TM peptides can induce coalescence of their annular lipid shells triggering large-scale domain segregation. This is reminiscent of capillary condensation, predicted by Mouritsen and co-workers already in the late nineties based on MC simulations using a highly simplified membrane model.499 Likewise, Ackerman and Feigenson500 observed growth of nanodomains induced by WALP TM peptides, in agreement with the experiment. They furthermore showed that WALPs can induce registration of domains. Lipid-mediated protein crowding was also observed in the study of Guixà-González et al.501 Here, the presence of lipids containing omega-3 polyunsaturated fatty acids (PUFA) drives the oligomerization of adenosine A2A and dopamine D2 receptors, again via coalescence of the annular protein shells enriched in the PUFA containing lipids (Figure 7).
Figure 7.
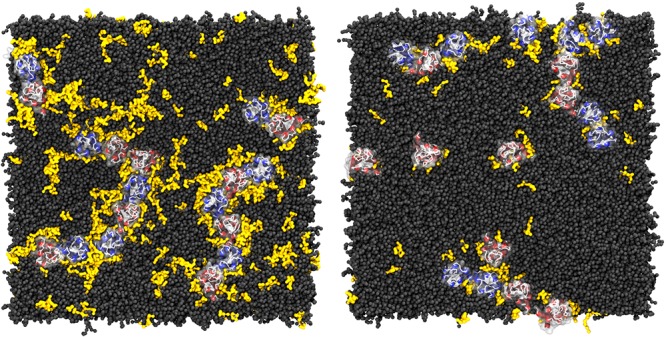
Example of lipid-mediated protein clustering. Snapshots of clustering of GPCRs in a healthy membrane (left), containing DHA, and a nonhealthy membrane (right) depleted of DHA. In the healthy case, higher order oligomers, stabilized by DHA, are more present. Reproduced with permission from ref (501). Copyright 2016 Nature (http://creativecommons.org/licenses/by/4.0/).
In addition to self-assembly approaches, biased simulations can be used to predict binding interfaces and obtain insight into the thermodynamic driving forces for protein–protein aggregation by computation of the PMF. Although in certain cases protein–protein PMFs can be extracted from self-assembly simulations (when multiple binding/unbinding events are spontaneously observed),502 in most cases calculation of the PMF requires forced unbinding using, for example, umbrella sampling or metadynamics type approaches. Like soluble proteins, the large number of possible interfaces still poses a major sampling problem. On the one hand, due to the two-dimensional nature of the membrane, prediction of membrane protein interfaces is easier. On the other hand, sampling of the desolvation of the protein–protein interface is problematic due to trapping of lipids. Even between simple TM helices convergence of the PMF requires sampling on the microsecond time scale, often necessitating the use of CG models.503−507 These studies provide important insights on the driving forces for helix–helix association and, in particular, the contribution of lipid-mediated effects versus direct protein contacts. In a systematic study on the dimerization of WALP peptides under different mismatch conditions, Castillo et al.504 concluded helix–helix association to be enthalpically favorable in all cases, while the entropic contribution appears favorable only in the presence of significant positive hydrophobic mismatch. The interpretation of this requires care given the coarse-grained nature of these simulations, but the sign of the thermodynamic quantities agrees with experimental measurements on dimerization of (AALALAA)3 peptides, and the observed association free energies are within the experimental range. The work of Benjamini and Smit is also noteworthy.508 The authors use a CG DPD model to challenge the notion that packing of TM helices is determined by specific interactions. Instead, the authors show that hydrophobic mismatch, through its effect on helix tilt, can explain many experimental cross-angle distribution features. This notion is supported by all-atom simulations of glycophorin A dimers embedded in different membrane environments, showing robust packing motifs despite the poor hydrophobic match, using mechanisms based on dimer tilting or local membrane thickness perturbations.509 In a combined experimental/computational study, Cybulski et al. show that mismatch-induced tilting of TM helices forms the basis for the thermosensing mechanism of DesK in bacterial membranes.510
Due to the sampling issues mentioned above, calculation of PMFs between polytopic membrane proteins has thusfar been limited to specific interfaces only. The first PMFs between fully solvated polytopic membrane proteins, GPCRs, was reported by Periole et al.488 The authors show that sampling times exceeding 1 ms are required to obtain converged profiles for specific binding interfaces. Remarkably, it was found that the amount of protein burial (i.e., number of protein–protein contacts not exposed to lipids) does not correlate with the binding strength of the interface. This finding challenges the potential utility of buried accessible surface area as a predictor of the strength of membrane-embedded protein–protein interfaces, a strategy that works well for soluble proteins. This view was not confirmed, however, in the case of another membrane protein, NanC.511 Here, the strength of binding was found to be proportional to the number of protein–protein contacts. Clearly, more work is needed in this area. In general, the above, and other recent studies on GPCRs by Filizola and co-workers,512,513 on the dopamine transporters by the group of Stockner514 and on the human serotonin transporter496 reveal specific, favorable, association interfaces stabilized by energies of the order of 30–60 kJ mol–1. Considering the quantitative predictive capability of CG models, a warning is in place, however. Overstabilization of TM helix dimer formation has been reported for the widely used Martini model with respect to the all-atom OPLS FF,515 as well as compared to experimental data.211 On the contrary, other studies show a much better match, either with atomistic data516 or in comparison to available experimental data.504 Given the high sensitivity of dimerization free energies to the exact mismatch conditions (see for instance the work of Benjamini and Smit508), and the importance of a proper choice of reaction coordinates,85 more systematic studies are needed to solve this controversy. Further progress in the use of enhanced sampling techniques will be very valuable in this respect. For instance, Lelimousin et al. recently showed that metadynamics can be used to induce reversible binding/unbinding of the TM domain of EGFR to obtain free energy landscapes directly.84 Domanski et al.85 used replica exchange umbrella sampling to speed up convergence of the PMF between glycophorin TM domains. Application of these methods to polytopic membrane proteins should, in principle, be possible. Another example is the combination of MD with Markov state models, as used in the study of Filizola and co-workers to elucidate the association kinetics of μ-opioid receptors.513 Advanced protein–protein docking tools such as HADDOCK are currently being extended into the realm of membrane proteins and could provide an alternative route toward prediction of protein–protein complexes.517
For more detailed information on the topic of membrane protein oligomerization, see for instance the general review on protein–protein interactions by Baaden and Marrink,518 reviews on GPCRs by Periole,519 Gabhauer and Böckmann,520 and Meng et al.,521 and a review focusing on driving forces by Johannes et al.522
3.1.4. Membrane Curvature Generation and Sensing
Curvature generation and sensing is important for many cellular processes that involve membrane remodeling, such as fusion and fission, and shaping of internal cellular compartments.523−525 In general, membrane curvature may result from the presence of nonlamellar forming lipids (e.g., DOPE) or arise from any asymmetry between the membrane monolayers. In vivo, generation of large curvatures typically requires the action of specialized proteins. Protein-induced membrane curvature could be an activated process, for example, making use of molecular motors or polymerizing actin filaments but also arise from direct protein–lipid interplay. In the latter case, three mechanisms can be distinguished: scaffolding, crowding, and insertion.523 In scaffolding, the proteins adhere to the bilayer and induce curvature through their curved interaction interface. In crowding, proteins located at the membrane surface generate a pressure that produces a bending moment acting on the membrane. In the insertion mechanism, proteins generate a curvature stress by asymmetric insertion of hydrophobic or amphipatic domains in the membrane. The three mechanisms are not mutually exclusive, however, and may act together. The number of simulation studies that address these mechanisms is steadily growing, following the pioneering simulations of Reynwar et al.287 on large-scale membrane remodeling using a generic CG model, the all-atom simulation by Blood and Voth,526 showing membrane curvature generation by a BAR domain, and the four-scale description of membrane sculpting of BAR domains by Arkhipov et al.527
A recent example in this area is the work of Davies and co-workers,528,529 probing the role of F1F0-ATP synthase dimers in shaping the mitochondrial cristae. On the basis of large-scale CG MD simulations, the authors propose that the assembly of ATP synthase dimer rows is driven by the reduction in the membrane elastic energy, rather than by direct protein contacts, and that the dimer rows enable the formation of highly curved ridges in mitochondrial cristae. De Oliveira Dos Santos Soares et al.530 used all-atom and CG MD simulations to investigate membrane-bending forces in the Dengue virus envelope. The structural organization of three heterotetramers EM proteins (EM3 unit) serves as an anisotropic bending unit for the Dengue virus envelope because it is able to locally decrease the thickness of the membrane with its short transmembrane helices. The simulations show that the specific arrangement of the EM membrane proteins inflict a curvature stress on the membrane. The resulting elastic energy is minimized by the systematic migration of lipids from the lower into the upper layer. Membrane undulations induced by the NS4A domain of Dengue virus have also been reported531 and were linked to the U-shape of this membrane spanning protein. Simulations of the curvature field induced by α-synuclein, by Sachs and co-workers,532,533 are also good examples of the power of near-atomistic membrane modeling in this field. On the basis of simulations involving 48 copies of the N-terminal membrane-binding domain of α-synuclein, together with more than 85000 lipids, the onset of membrane tubulation could be observed due to the collective action of the proteins (Figure 8). Another recent example of curvature generation due to surface-bound proteins is a simulation study of Li and Gorfe on asymmetrically bound H-ras proteins.534 Imposing curvature stress on membranes is not limited to proteins; small amphipatic peptides that adsorb at the membrane/water interface potentially have the same effect. Buckling of model bilayers can, for instance, be induced by antimicrobial peptides (AMPs) as is shown in simulations of Woo and Wallqvist.535 Sodt and Pastor have quantified the curvature stress generated by a model amphipatic peptide,536 showing that the peptide induces positive curvature in line with the conclusions from a simulation study on fusion peptides.537 The extent of curvature induction was found to depend sensitively on the molecular interactions and cannot be explained using simple shape-based concepts. Pannuzzo et al.538 proposed an efficient approach to simulate the bending power of peptides based on the use of lipid bicelles that are stabilized by short-chain lipids.
Figure 8.

Membrane curvature generation by proteins. Onset of membrane tubulation induced by 48 copies of α-Syn100 (yellow) interacting with a membrane composed of 85296 POPG lipids (blue tails, red headgroups). Water is not shown for clarity. Snapshot is obtained at 300 ns simulation time with the Martini model. The budding tubule extends ∼25 nm above the bulk lipid bilayer. Adapted from ref (533). Copyright 2014 American Chemical Society.
As mentioned before, membrane curvature may also arise from an asymmetric distribution of lipids between the leaflets. MD simulations of multicomponent membranes show, for example, that the ganglioside GM1 induces curvature.539,540 Conversely, curvature leads to lipid sorting as is demonstrated in a number of simulation studies.369,350,352,541 Again, simple shape-based concepts do not suffice to explain lipid-induced curvatures and sorting effects, in particular in multicomponent systems where effects are nonadditive.542−544 Noteworthy are also simulations that show how electrostatic fields can induce membrane curvature (flexoelectric effect).545
In addition to curvature generation, an important question is how proteins may sense different curvatures. Cui et al.546 used the concept of membrane-packing defects547,548 as measure for curvature sensing. The idea is that curved membranes expose a larger fraction of hydrophobic defects to which the hydrophobic domains of proteins can bind. Indeed, the authors demonstrate, based on all-atom MD simulations, an increasing number of defects with increasing curvature. Another study showed that the ability of lipid tails to backfold to the membrane/water interface also increases with curvature.549 Vamparys et al.550 furthermore show that the size and number of such defects increase with the number of monounsaturated acyl chains and with the introduction of conical lipids. Moreover, the size and probability of the defects promoted by conical lipids resembled those induced by positive curvature, thus explaining why conical lipids and positive curvature can both drive the adsorption of surface active peptides and proteins. This hypothesis was confirmed by subsequent studies in which experimental data and simulation data were combined to explain the binding affinity of peripheral proteins as a function of lipid composition and curvature551 as well as membrane tension as another determining factor.552 A simulation study of a buckled membrane demonstrated differences in sensing characteristics between different AMPs.553 Thus, proteins can sense curvature, and induce curvature, but can also undergo conformational changes in response to curvature. A number of MD studies have demonstrated that membrane curvature can indeed shift the conformational equilibrium in peptides,554−556 as well as affect peptide folding kinetics.546
Fusion and fission are key cellular processes that involve extensive membrane curvatures. The main question remains to what extent fusion and fission are lipid-driven or protein-mediated. Simulation studies have contributed significantly in this area, and protein-free fusion pathways between lamellar membranes and between vesicles are now quite well-established (reviewed in refs (557 and 558)). Current efforts are directed to calculate the energetics and kinetics of the various intermediates,559−567 the importance of hydration forces in the initial approach,568−572 stalk formation between multicomponent phase separated membranes,573 the role of calcium and PEG in mediating fusion,574,575 and carbon nanotube-mediated fusion.576 Computational modeling of peptide and protein-induced fusion or fission is still in its pioneering phase. A number of researchers537,577−579 investigated the ability of small amphipatic peptides, including HA fusion peptides, to stabilize cubic phases and stalk/pore complexes that are relevant as fusion intermediates. Moiset et al.580 found that certain AMPs, that are traditionally associated with forming transmembrane pores, can also induce stalk formation between juxtaposed membranes. Stalk formation in this case is initiated by the ability of multiple lysine residues to form a bridge between the apposing bilayers and trigger the flipping of lipid tails between the proximal leaflets. A similar mechanism was recently observed in all-atom simulations of stalk formation in the presence of arginin-rich cell penetrating peptides.581 Baoukina and Tieleman582 simulated the fusion of small unilamellar vesicles mediated by lung surfactant protein B (SP-B). They found SP-B monomers capable of triggering fusion events by anchoring two vesicles, facilitating the formation of a lipid bridge between the proximal leaflets. In a series of breakthrough papers, Risselada et al.583−586 simulated neuronal SNARE-mediated membrane fusion. The simulations reveal that SNARE complexes operate in a cooperative and synchronized way. In the postfusion state, zipping of the SNAREs extends into the membrane region, in agreement with the recently resolved X-ray structure of the fully assembled state. Additional details of the fusion pathway were resolved in simulations of SNARE mediated fusion between bilayers and nanodisks.587 In the work of Pinot et al.,588 a combination of in vivo, in vitro, and in silico experiments were used to show the combined effect of lipids and proteins in shaping membranes during fission. In particular, the role of polyunsaturated lipids in membrane vesiculation by dynamin and endophilin was revealed. The simulations provided the molecular mechanism: polyunsaturated lipids can backfold in the membrane and thereby adapt their conformation to the change in membrane curvature during vesiculation. This plasticity of polyunsaturated lipids was already noted in earlier simulations of small liposomes.589
3.2. Realistic Cell Membranes
Recent advances in computation and molecular FFs have allowed for more faithful modeling of realistic biological cell membranes. Focusing on the lipid component only, recent models are approaching realistic complexity of biological bilayers with respect to the number of different lipid types, bilayer asymmetry, and geometry. Marked differences are found in the lipid composition and dynamics of bilayers from different organisms, cell and tissue types, organelle, as well as dependent on environmental factors and cell cycle,3,590,591 therefore, a diverse set of bilayer models is needed. Here we list some of the different types of membrane models that have been developed.
3.2.1. Plasma Membranes
In cells, the plasma membrane (PM) defines the boundary, separating the cell interior from the outside environment. A typical PM contains hundreds, if not thousands, of different lipid species that are actively regulated by the cell and nonuniformly distributed in the membrane plane.3,4 Eukaryotic cell PMs typically contain a significant fraction of cholesterol and have an asymmetric leaflet composition. The outer leaflet is composed of more saturated lipids and enriched in SM and glycolipids (GM). The inner leaflet has more unsaturated lipids and most of the charged lipids (PS, PA, PI, PIP’s). The extent to which cholesterol is enriched in the outer or inner leaflet is still debated, for example, see a recent review by Steck and Lange.592
In earlier simulation studies, eukaryotic PMs were often approximated as a pure POPC bilayer (POPC, being the most common PM phospholipid) or a pure DOPC bilayer (were DOPC average bilayer properties can be closer to those of a PM, although DOPC itself is not prevalent in eukaryotic cells). When a specific lipid type is known to be of importance, it is then simply added to the pure mixture and for cases when phase separation is believed to be important, a three-component mixture is often used (with a high and low melting temperature phospholipid and cholesterol, see above). Recently, however, models attempting to approximate realistic PMs have emerged,593−600 some of which we will discuss here.
In terms of lipid composition, Ingólfsson et al. average mammalian PM model593 is the most complex simulation to date. The model is Martini-based and contains 63 different lipid types, with 14 different types of lipid headgroups and 11 different tails that are asymmetrically distributed across the leaflets. A large-scale simulation of the model membrane, ∼20000 lipids and simulated for 80 μs,593,596 gives a high-resolution view of the dynamic interplay of all lipid species and overall organization. PM nonideal lipid mixing, membrane properties, lipid flip-flop dynamics, leaflet coupling, and domain formation were explored. At the microsecond time scale cholesterol, ceramide and diacylglycerol lipids flip-flop between the leaflets. Due to cholesterol’s preferred interactions with the more saturated outer-leaflet, the cholesterol distribution equilibrates to a slight enrichment in the outer leaflet (∼54%). Globally, neither leaflet phase separates, but the lipids are heterogeneously mixed and show nonideal mixing of different lipid species at different spatiotemporal scales. Both leaflets contain domains that are continuously changing in size and composition. The domain sizes range from small, only a few lipids big, to larger, spanning the simulation box. The smaller domains are transient, while the larger are more persistent and are coupled between the leaflets. GM lipids form small clusters in the outer leaflet, whereas in the inner leaflet, dimers and trimers of PIP lipids form more frequently than would be expected based on their concentration. It should be noted that the extent to which GMs are observed to cluster in these simulations might be overestimated. Recently retuned parameters for gangliosides in the Martini model show a reduced clustering propensity, in line with atomistic simulations.185 Additionally, MD simulations provide evidence that GM clusters depend on sterol concentration.601
The PM model was used to explore curvature-based lipids sorting by pulling tethers, pulling from the outer or the inner leaflet.602 Spatially varying lipid redistribution was observed, dependent on pulling direction, as well as a softening of the tethers due to the sorting of the lipids. Vögele et al.152 used a number of systems to show how hydrodynamics can explain the finite-size effects of lipid diffusion, including patches of the complex PM model with a size up to 286 × 286 nm. Corradi et al.603 simulated ten different membrane proteins in the complex PM mixture. The simulations showed how each protein uniquely modulated its local lipid environment. At different spatial locations around the proteins local enrichment or depletion of specific lipids resulted in bilayer thickness and curvature gradients, together forming unique lipid fingerprints (Figure 9).
Figure 9.

An example of a complex plasma membrane model. Corradi et al.603 simulated ten different membrane proteins in a 63 lipid PM mixture. Each protein’s different TM shape and lipid–protein interactions resulted in a unique lipid fingerprint (a). Here AQP1 is depicted showing the simulation setup, snapshot of the (b) outer membrane as well as lipid enrichment/depletion and bilayer properties around the (c) protein. Adapted from ref (603). Copyright 2018 American Chemical Society.
In a seven component PM Martini mixture, Koldsø et al.594 capture many of the same properties as the complex mixture above, including nanodomains of GM lipids on the outer leaflet and PIPs on the inner leaflet. Additionally, they explore the effects of curvature and the addition of membrane proteins. Curvature was found to affect lipid organization and sorting with GM and PE lipids enriched in concave deflections of the outer leaflet, while PIPs and cholesterol are enriched in concave deflections of the inner leaflet. Model α-helical transmembrane domains were inserted in the PM mixture and found to cocluster with a number of lipid species, including anionic lipids, as well as slow down lipid diffusion. The model, or variants of, has been used in a number of studies, including large-scale simulations to explore protein crowding and clustering,604 the effect of cytoskeletal immobilization on protein and lipid mobility,605 how loading with model transmembrane helixes or GPCRs effect membrane dynamics (such as bilayer undulation and lipid diffusion),606 and lipid binding to receptor tyrosine kinases (RTKs)607 and the epidermal growth factor receptor (EGFR).608
A number of other average or specific tissue type PM models have been developed. Jeevan et al. made an average PM model using Martini that is asymmetric and has six lipid components.609 The model has been used to explore Ebola virus protein VP40 PM binding.609 Hedger et al. explored the cholesterol interaction of the Class F G protein-coupled receptor Smoothened in a number of bilayers, including an asymmetric five-component Martini lipid bilayer containing PC, PE, PS, PIP2, and cholesterol.380 Kalli et al. constructed an asymmetrical five lipid type PM model using Martini to explore integrin receptor dynamics, showing how the receptor altered lipid organization especially that of cholesterol and PS.610 Domicevica, Koldsø, and Biggin constructed a five-component asymmetrical epithelial brain PM to explore the lipid interaction of P-glycoprotein.597 They used both CG Martini and atomistic Slipid models and found enrichment of charged PS lipids next to the protein and specific cholesterol interaction sites. A five-component Slipid model was also used to explore the effect of curvature on PM properties by Yesylevskyy et al.611 Yesylevskyy and co-workers recently made a variant of the model to mimic a cancerogenic PM.612 Klähn and Zacharias build asymmetric five component PM models representing a cancerogenic and normal eukaryotic PM and simulated them using the atomistic CHARMM FF.613
Ueoka and co-workers made compositionally complex asymmetrical PM models of normal and cancerogenic thymocyte membranes using the CHARMM FF and containing 23 and 25 different lipid types, respectively.600 Flinner and Schleiff constructed an asymmetrical ten component Martini bilayer model of the red blood cell (RBC) PM to explore the dynamics of glycophorin A dimers.595 Characteristic of RBC PMs, the model mixture is high in cholesterol and contains PE plasmalogen lipids. Kalli and Reithmeier constructed asymmetric six component RBC PMs both using Martini and GROMOS to study the interactions between the Band 3 and glycophorin A proteins and the lipids.614 Kadri et al. constructed symmetric epithelial cell membrane models with 10 different Martini PC lipids to study how the increased tail saturation associated with lipo-intoxication effects bilayer properties.615 Ingólfsson, Carpenter, and co-workers assembled a human neuronal PM model, based on Martini, with an asymmetric lipid distribution and 58 different lipid types.596 Compared to the 63-lipid type average PM model the bilayer properties of the neuronal PM are overall strikingly similar, despite significant difference in lipid composition. The effects of the higher cholesterol content of the neuronal bilayer are somewhat compensated by the higher tail unsaturation. Interestingly, the domain sizes fluctuations in both the neuronal brain and average PM mixtures were sensitive to the level of bilayer undulation. Guixà-González et al. constructed two six component symmetrical brain models with high and low docosahexaenoic acid (DHA) concentration to explore DHA role in GPCR oligomerization.501
Klauda and co-workers created PM models of the soybean hypocotyl (the stem of the germinating seedling) and root using the atomistic CHARMM FF; the models are symmetrical and contain 9 and 10 lipid types, respectively.598 Soybeans lipid composition differs significantly from eukaryotic membranes, containing different sterols (sitosterol and stigmasterol instead of cholesterol) as well as a large fraction of di- and tripolyunsaturated fatty acid tails. Jo et al. made a six-lipid type symmetric average yeast membrane using CHARMM and characterized its properties with more tail saturation, less sterol content, and imposed surface tension.599
3.2.2. Organelle Membranes
The lipid composition varies widely between organelles,1−3 requiring a large set of membrane models, which to date is significantly underrepresented. One of the more studied organelles are mitochondria, the “powerhouse” of the cell, generating most of the ATP that the cell uses. Cardiolipin (CL) is the signature lipid of mitochondria. It is anionic, with two phosphate groups and four acyl tails. Cardiolipin is present at a high concentration in the inner membrane of mitochondria (up to 20%)616 and is required to stabilize the respiratory chain supercomplexes.617 Mitochondrial membranes have mostly been modeled as symmetric binary or ternary mixtures consisting of PC or PC/PE with cardiolipin. At the CG Martini level, inner mitochondrial mixtures have been used to explore cardiolipin protein binding for the respiratory chain supercomplexes,382,401,618 the rotor of the metazoan ATP synthases,619 and the adenine nucleotide translocase (ANT).402 Vähäheikkilä et al. explored the inner mitochondrial bilayer properties using the atomistic OPLS FF with different cardiolipin variants and levels of cardiolipin tail peroxidation.620
Thylakoid compartments are the sites of lipid-dependent photosynthetic reactions in chloroplasts and cyanobacteria. The thylakoid membranes are rich in galactolipids, their lipids have a high fraction of polyunsaturated tails, and many of their lipids are nonlamellar phase lipids. Van Eerden et al. created two thylakoid membrane models: a five-lipid type cyanobacterial model and a seven-lipid type plant model, both at the CG Martini level and atomistic GROMOS level.621 The bilayer properties of both membrane models were evaluated as well as the dynamics of two photosynthesis cofactors (plastoquinone and plastoquionol) inserted in the membranes. Later studies have used thylakoid membranes to explore the dynamics of the Photosystem II (PSII) complex, focusing on the protein lipid interaction and the entry and exit of membrane embedded cofactors to the protein,622−624 as well the membrane interaction of the cold-regulated (COR) protein COR15A.625
Ray et al. constructed symmetric four to six component membrane models for the endoplasmic reticulum (ER), Golgi apparatus, and mitochondria using the CHARMM FF and analyzed the distribution of forces within the membranes.626 Su et al. modeled a peroxisomal membrane from the yeast Pichia pastoris, as a symmetrical five component mixture using the Martini model, which they used to explore the lipid association and aggregation of the N-terminal helix of the peroxisome elongation protein.627 Monje-Galvan and Klauda modeled the PM, ER, and trans-Golgi Network (TGN) bilayers of yeast628 and compared their properties with the previously constructed average yeast membrane model.599 The models were built using the atomistic CHARMM FF. The membrane is kept symmetric, with 6–11 different lipid types, and for each organelle, two models were made with different levels of tail unsaturation. Simulations of the model membranes highlight differences in bilayer properties (e.g., thickness, area per lipid, compressibility) between the different organellar membranes.628
3.2.3. Bacterial Membranes
The lipid composition of different bacteria is quite diverse. Gram-negative bacteria, such as E. coli and S. aureus, have an inner and outer cell membrane, separated by a viscous periplasm.629 The outer leaflet of the outer membrane is mainly composed of lipopolysaccharide (LPS) lipids. LPS consist of Lipid A, with 4–7 fatty acid tails attached to a sugar backbone and a polysaccharide forming an inner and outer core and a variable length O-antigen.630,631 The different constituents of LPS can vary significantly both within and between bacterial species. A range of different LPS variants and fragments have been parametrized. For CHARMM, the CHARMM-GUI web portal now has an LPS Modeler that as of May 2018 has “15 bacteria species, 37 lipid A types, 52 core oligosaccharide types, and 304 O-antigen polysaccharide types”.632
A number of models of the outer bacterial membrane has been constructed, both at the CG and atomistic level of resolution.115,132,121,192−194,633−638,631,639−641 Typically, these models contain 2–5 lipid species and are asymmetric, with the outer leaflet consisting mostly of different variants of LPS, and the inner leaflet either DPPE or a mixture of PE, PG, and sometimes cardiolipin. The models have been used to explore and characterize different basic properties of the outer bacterial membrane such as density, packing, average area per lipid, diffusion, and divalent cation binding. In addition, partitioning and permeation of molecules into and through the membrane,194,638,642 the influence and packing of membrane proteins,635,636,639−641,643−646 and the effects of Lipid A structural variations from different pathogenic bacterial species631,647 and within species (S. enterica)648,647 have been explored.
Other bacterial membranes have also been modeled. Models for chlamydia’s (C. trachomatis) two main life cycles, the elementary body and reticular body, have been developed within the CHARMM FF.649 Both simulated membranes are symmetrical and contain nine different lipid types, corresponding to the most prevalent lipids of the different life cycles, including three lipid types with methyl branched tails. Klauda and co-workers constructed a cytoplasmic E. coli membrane in the CHARMM atomistic FF. The membrane model is symmetrical, containing the six most prominent lipid types in the inner membrane, including a lipid containing a cyclopropane ring within the acyl chain tail that they parametrized.650 In a later study, they modeled the E. coli inner membrane at different stages along the growth cycle, showing significant differences in average area per lipid and rigidity.651 Hwang et al. also used the CHARMM FF to study the effect of stress on the E. coli cell envelope, modeling both the outer and inner membrane as well as the cell wall.652 Berglund et al. explored the interaction of the antimicrobial peptide polymyxin B1 with both the outer and inner E. coli membranes, using the GROMOS atomistic FF; their inner membrane model was a symmetric three component mixture of mostly PE with some PG and cardiolipin.637 Hsu et al. also modeled both membranes using the Martini model and simulated them with various native membrane proteins embedded, including the outer/inner membrane spanning AcrABZ-TolC complex653 (Figure 10). For a review focusing on simulations of bacterial membrane channels, see ref (654).
Figure 10.

Example of a complex bacterial membrane model. Showing the outer and inner E. coli cell membrane with embedded membrane proteins including the membrane spanning multidrug efflux pump AcrABZ-TolC. Adapted from ref (653). Copyright 2017 American Chemical Society.
3.2.4. Skin Models
The outer layer of the skin (stratum corneum, SC) consists of dead cells (corneocytes). The lipid “structure” of the stratum corneum is a mixture of long-chain saturated ceramides, free fatty acids, and cholesterol, in a 1:1:1 ratio.655 Due to their relevance for skin barrier properties, numerous simulation efforts have studied the properties of these lipid mixtures. Here, we mention a few of the more recent studies.
McCabe and co-workers have modeled the SC using mixtures of ceramides and fatty acids as well as ceramides, fatty acids, and cholesterol, using both the CHARMM FF with modified ceramide parameters and the Berger lipid FF as well as a customized CG FF for lipid self-assembly.656−659 Wang and Klauda characterized bilayer properties of pure ceramide bilayers and SC ternary models using the CHARMM FF at different temperatures and ceramide tail length.660 Höltje et al. modeled the SC with a combination of fatty acids and cholesterol using the GROMOS FF.661 Das et al. constructed a SC model containing 15 ceramide variants as well as a fatty acid and cholesterol using the atomistic Berger lipid FF and found a preference for the inverse micellar phase.662 Del Regno and Notman modeled SC at two different lipid concentrations and two levels of hydrations using the Berger FF.663 They suggest a permeation path for small polar molecules through the SC lamellae that avoids pockets of water between the bilayers. Wennberg et al. simulated glycosylceramides and ceramides at varying levels of hydration using the Martini FF.664 They showed that glycosylceramides can maintain a cubiclike bilayer structure while the ceramides collapse into a stacked lamellar structure, which might be an important step for SC. In two studies, Gupta and Rai explored fullerene C60 permeation through SC bilayers using the Martini FF,665 and using an atomistic SC model they studied electroporation by imposing a varying external electric field.666
3.2.5. Complications of Complexity
Membrane models need to be complex enough for the question at hand, but additional complexity comes with a prize. Before adopting a more realistic, more complex model, the price of doing so should be carefully evaluated. Here we discuss some of the caveats that need to be considered.
Bilayer models with more lipid species require longer sampling times, especially if rare lipid species are included. Proteins can affect their local lipid environment, promoting lipid sorting and/or bilayer perturbation, see for example, refs (603, 610, and 667), also extending the required sampling time. The sampling challenge is even bigger when considering more realistic conditions characterized by a high protein density. Domanski et al.,498 Goose and Sansom,668 and Javanainen et al.669 simulated membranes under such crowded conditions, with formation of extended clusters and networks of proteins dramatically slowing down the lateral diffusion rates of the components. Interested readers are pointed to a recent review on protein crowding.670 In fact, under crowded conditions, diffusion becomes anomalous498,671 and may lead to deviations from the Saffman-Delbruck model at physiological levels.672 Although, in the latter case, these claims are not substantiated as a proper correction of periodicity artifacts on hydrodynamics has not been taken into account.149−152
Many biological membranes are asymmetric; therefore, more physiologically relevant models of those membranes may need to include asymmetry. To model an asymmetrical membrane, first, it is necessary to determine the lipid concentrations in each leaflet. This is not trival as our current knowledge about lipid asymmetry is incomplete. For specific lipid classes and membranes, the asymmetry has been determined, see for example, ref (3), but for most membranes, many lipid classes, and most individual lipid types, the asymmetry is not well-determined or not known at all. Second, including asymmetry in a periodically constrained system, a primary concern is to determine the relative number of lipids in each leaflet. Several different criteria have been proposed to determine what a “correct” balance of outer/inner leaflet lipids should be, such as matching the average area per lipid (APL) in both leaflets, the leaflets surface tension, the lateral pressure profile across the two leaflets, and the lipid’s chemical potential. Generating a “well-balanced” asymmetric membrane using one of these criteria can be quite involved. Recent simulation setup protocols for asymmetrical bilayers include using prior estimates of idealized APL110−112 biased self-assembly,673 an iterative building procedure,593,596 or zeroing bilayer leaflet tension.674 Additional complexity also arises when including lipids that can flip-flop between the leaflets at time scales relevant for the simulation at hand. Cholesterol is a good example of a fast flip-flopping lipid; it has been shown to flip-flop on the microsecond time scale.675
Realistic bilayers, depending on lipid mixture, protein content, and cell attachment, can undulate significantly. Allowing for larger bilayer undulations is computationally very expensive. The simulation box has to be large both in the plane of the bilayer (to reduce undulation dampening due to periodic image constraints) as well as perpendicular to the plane, increasing simulation cost. Longer simulations are also needed to capture the longer length scale bilayer undulation modes and lipids redistribution, as lipids have been shown to organize in the plane of the bilayer based on curvature.594,602 All analysis of undulating bilayers also becomes more complex as the curved bilayer surface needs to be fitted and accounted for.
With increased model complexity, the sampling required goes up exponentially. Analysis therefore also becomes a bottleneck, as the amount of generated data mirrors the sampling and the number of interactions between components to analyze also goes up with the square of the number of species in the model. For the more complex models, they fast become intractable for manual analysis, requiring reduction in complexity (e.g., combining lipid species into classes), automated analysis methods, and/or use of unsupervised machine learning methods for identifying possible hidden correlations.
3.3. Toward Full Cell Models
With the use of the CG and multiscale approaches described above, it is possible to perform very large-scale simulations of cell membranes, at the level of, for example, the envelope of a complete virus particle.676,677 While appealing as a tour de force, one should ask what might be learned from such simulations. A major motivation is to overcome barriers between simulations and experiments. Thus, vary large scale simulations may allow us to approach the length and/or time scales of experimental studies of biological membranes, which in turn will enable direct comparison between experiments and simulation, permitting rigorous molecular interpretations of mesoscopic observations. This is especially important in linking molecular structures of membranes and their components through to cell biological investigations using a variety of imaging modalities, for example, cryoelectron tomography and super-resolution optical microscopies.
There are spatial and temporal challenges in matching mesoscale experimental data while not losing molecular specificity in the underlying models. How we can address such challenges is illustrated via a number of examples of increasing scale: (i) viral envelope membranes via very large-scale CG simulations; (ii) bacterial outer membrane protein (OMP) clustering by large scale CG simulations enabling parametrization of simple mesoscale models; and (iii) CG and meso scale simulations to study processes of remodeling of eukaryotic cell membranes.
3.3.1. Viral Envelopes
CG-MD has been used to explore the membranes of a number of enveloped viruses, providing examples of very large-scale (ca. 5 million particles) simulations of biological membrane assemblies. A ground-breaking study of the membrane envelope of the immature HIV-1 virion678 combined electron cryotomography data and multiscale simulations to provide insights into the Gag lattice assembly process in the immature HIV-1 virion. These simulations employed a multiscale approach in which multiple CG parameters were explored in critical regions, with the aim of identifying those interactions that are critical to maintaining the structure of the virion. Subsequently, the CG simulation results were used to guide all-atom MD simulations of selected regions in order to refine the model.
Simulations of a complete virion envelope model for influenza A combined X-ray structures and TM domain models for the hemagglutinin (HA) and neuraminidase (NA) proteins, an NMR structure for the TM domain of the M2 protein, and a lipid bilayer composition based on the experimentally determined lipidome of the viral membrane.679 The prevalence of glycolipid headgroups on the outer surface of the influenza A viral membrane suggested that access of therapeutic compounds to the M2 proton channel may have to overcome substantial steric barriers. The influenza A envelope proteins moved slowly within the cholesterol-rich membrane, with diffusion constants matching previous NMR measurements. Lipid molecules had reduced diffusion coefficients (D) and exponents (α) less than 1, the latter indicative of anomalous diffusion. The spacing between membrane glycoprotein molecules on the influenza A surface suggested that polyvalent interactions between HA and/or NA on the viral surface and sialic acid residues on the host cell membrane are likely to occur. This would enable strong virus-host association despite relatively weak (∼2–3 mM affinity) viral HA-single host receptor interactions in vitro.
The membrane envelope of the dengue virus has been simulated in two recent studies680,681 using the Martini FF. Reddy and Sansom681 used a combination of CG modeling and simulation to “add back” the lipid bilayer to the cryo-EM structure of the Dengue virus envelope proteins. These simulations revealed that the crowding of protein TM domains and the enclosure of the outer leaflet of the lipid bilayer within a protein shell resulted in lipid diffusive properties similar to those in the “raftlike” influenza A membrane, despite the absence of cholesterol from the dengue membrane model. Bond and colleagues680 used a novel protocol to embed the cryo-EM structure of the envelope protein complexes of the DENV-2 icosahedral shell within a spherical lipid vesicle, the composition of which was guided by lipidomics data. Microsecond-time scale simulations of the virion envelope enabled refinement of the lipid/protein complex, assessed by comparing density maps calculated from simulations with those determined by cryo-EM. The refined structures revealed locally induced curvature resulting from specific interactions with phosphatidylserine molecules. These lipids may facilitate subsequent fusion of the viral envelope with the host membrane inside the endosome during infection. A subsequent study,682 based on targeted MD simulations, provided evidence that the low pH structures obtained with cryo-EM are biologically meaningful intermediates of the fusion process with the endosomal membrane.
A hybrid multiscale approach using the SIRAH FF has been used to study the envelope of zika virus (ZIKV).683 Those parts of the system of particular interest were modeled using atomistic and/or CG resolution, while those of less direct interest used a supra-CG resolution. This hybrid multiscale approach allows for efficient simulations of large-scale biological membrane systems to be run on modest computational resources, thereby making computational virology accessible to a wider range of researchers. These and other studies demonstrate the potential of very large-scale simulation of viral envelopes. Future challenges for such studies include development of a full CG model of glycosylation of viral surface proteins, which will enable more realistic and hence predictive modeling of virions binding to models of target cell membranes.
3.3.2. Large-Scale Membrane Organization
Large-scale simulations can be used to probe the structural and dynamic consequences of protein–lipid and protein–protein interactions in complex and crowded cellular membranes. For example, simulations of a mitochondrial inner membrane model indicate how cardiolipin may “glue” together respiratory proteins into supercomplexes.618 Analysis of the free energy landscape of interactions of the bacterial outer membrane protein (OMP) NanC have also revealed how intervening lipids may stabilize a membrane protein dimer.511 Such protein–lipid–protein interaction may underlie functionally important larger scale membrane organization. Earlier work in this area684,685 provided a theoretical framework for our understanding of the role of lipids in mediating membrane protein interactions and for extracting appropriate parameters from large scale MD simulations. Recently, highly coarse-grained simulations have been used alongside experiments to explore the interplay of lipids and proteins which underlie clustering of the influenza M2 protein and its possible role in mediating viral budding from infected host cells.686
Large-scale CG-MD simulations can in turn enable more highly coarse-grained (or mesoscopic) simulation approaches to be developed for modeling of emergent behaviors in these complex protein–membrane systems (Figure 11). For example, the spatiotemporal organization of membrane proteins is often characterized by the formation of large protein clusters. In the outer membrane of E. coli, protein clustering leads to OMP islands, the formation of which underpins membrane protein turnover and drives organization across the cell envelope. By combining CG simulations with in vitro and in vivo experimental studies, it has been possible to suggest how protein–protein interactions enable formation of large clusters of bacterial OMPs which may play a key role in the formation of these membrane protein “islands”.687 However, a detailed mechanistic understanding of how OMP islands form has been confounded by the difficulties of simulating very large number of OMPs on experimentally addressable time scales. To address this limitation, Chavent, Duncan, and colleagues recently developed a mesoscale model which they trained on large scale CG-MD simulations.688 In the meso model, each OMP molecule was represented by a single particle within a 2D membrane model. The meso model was used to run simulations of ca. 5000 copies of an OMP on multimillisecond time scales, thus allowing direct comparison of simulated and in vitro experimental single tracking measurements of OMPs. These studies revealed that specific interaction surfaces between OMPs were the key to formation of OMP clusters and that mesoscale simulations captured the restricted diffusion characteristics of OMPs. This agrees well with recent measurement of the glasslike behavior of crowded membranes.689 The OMP clusters in turn presented a mesh of moving barriers that confine newly inserted proteins within membrane islands. Such “corralling” of newly inserted proteins is likely to be of importance for OMPs newly inserted by the BAM machinery. Thus, this type of model enables us to provide a nanoscale molecular mechanism for mesoscale experimental observations. Future refinements of this approach could include using large-scale CG-MD simulations of realistic models of the lipid composition of E. coli outer membranes258,653 in order to allow meso models to explore the behavior of OMP islands in vivo.
Figure 11.
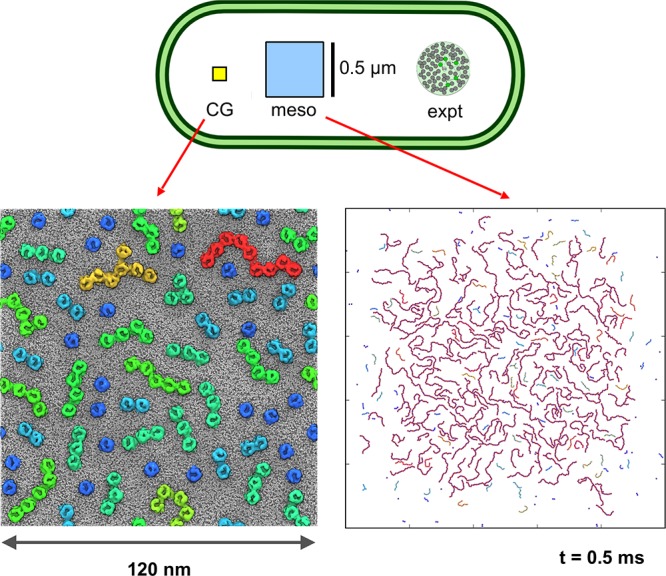
Developing a mesoscale model for simulation of bacterial outer membrane protein islands. The top panel shows a schematic diagram of an E. coli cell, with the areas of outer membrane studied via CG simulation (yellow square), by mesoscale simulation (blue square), and by experimental single molecule tracking (green circle) shown to scale. The lower two panels are snapshots from CG (left) and meso (right) simulations of OMP clustering (see main text and ref (688) for details).
In addition to providing data for parametrization of mesoscale models, very large-scale simulations can provide insights into the emergent behavior of complex and crowded biological membranes which may in the future be included in more biorealistic mesoscale models of complex in vivo membranes. This approach builds upon pioneering work in large scale simulations of crowding of proteins in models of the cytoplasm.690,691 These emergent properties include large scale dynamic fluctuations of membranes which may be used to derived mesoscale mechanical parameters of membranes such as the bending rigidity.41,109 Application of such analysis to large-scale CG simulations has revealed a complex dependence of the membrane-bending rigidity on both protein contents and lipid composition.692 Inclusion of simple models of cytoskeletal tethering of integral membrane proteins also modulates membrane bending rigidity.605 Large-scale CG simulations may also be used to explore, for example, the influence of lipid bilayer composition and of specific protein–lipid interactions on patterns and dynamics of membrane protein clustering.604 These and other emergent properties from CG simulations will need to be included in a next generation of mesoscale models in order to address the picture emerging from current dynamic experimental measurements which are suggesting cell membranes to be heterogeneous and “scale rich”.8
3.3.3. Membrane Remodeling
In addition to the large-scale dynamic organization of cell membranes “at rest”, large-scale molecular simulations have been used to explore dynamic events including, for example, membrane fusion and remodeling of membranes.693,694 In particular, Voth and colleagues have taken a multiscale approach288 to explore the biologically important question of how BAR domain proteins interact with lipid bilayers to bring about membrane remodeling.695 Using a supra-CG model, they demonstrated how multiple copies of N-BAR domain proteins on a membrane surface form linear aggregates at high protein densities can lead to formation of budlike deformations of the membrane.696 Combining CG simulations with microscopy data was used to develop a model of a BAR-domain scaffold, emphasizing the key role of amphipathic helices in the formation of these scaffolds.697 These CG studies have in turn fed into mesoscale approaches,288,295 allowing membrane remodeling to be explored on submicron length and microsecond time scales. Some other reviews covering mesoscale modeling of curvature generation can be found elsewhere.267,698
3.3.4. In Silico in Vivo
It is clear that large multiscale simulations of cell membranes can now be used to simulate complex dynamic events in cell and organelle membranes. By integrating such computational approaches with a growing wealth of cryo-EM and optical microscopy data, there is the prospect for future “in silico in vivo” studies of the cell biology of membranes, relating underlying structural and biophysical properties to cellular level events. A number of computational tools will facilitate simulation studies of increasingly complex membrane systems, including tools for semiautomated setup of complex mixed lipid bilayers.258 On a larger scale, for example, cellPACK699,700 provides mesoscale packing algorithms to generate and visualize three-dimensional models of complex biological environments. This has been evaluated on, for example, models of synaptic vesicles and of an HIV virion. Future developments are likely to further integrate a range of tools for setup, running, visualization, and analysis of larger and more complex membrane and cellular systems,701−704 in addition to development of databases for storage and dissemination of the results of membrane simulations (e.g., MemProtMD256 and Limonada705).
4. Outlook
Thirty years of computer modeling of cell membranes have provided a wealth of information on the lateral organization principles underlying these fascinating quasi two-dimensional systems. From the detailed dynamics of individual lipid tails, via collective processes such as pore formation, protein–lipid sorting, and membrane remodeling, we have now reached a stage where the full complexity of real cell membranes is being captured. Referring back to Figure 1, the obvious question is, what stage comes next?
On the one hand, the quest for more realism has certainly not ended (Figure 12). Detailed models for cell envelopes of most cell types, as well as the many internal organelles, are still very sparse. A key bottleneck is the availability of experimental data concerning their exact lipid composition. Although advanced lipidomics can provide a wealth of data in this respect, it is often not trivial to isolate specific cell fractions. Moreover, any information on membrane asymmetry is lost, hampering realistic modeling efforts. A level up in realism can be achieved by putting cell membrane models into a more realistic environment. Addition of the cytoskeleton would be an obvious example but also the interaction of cell membranes with the crowded environment of the cytoplasm. In fact, not only the membrane leaflets are asymmetric but also the solvent facing both sides, including differences in pH, ionic strength, and electric potential. This has hardly been considered at all in current simulations studies. A major challenge of increased complexity is the increase in required sampling time, as discussed above. Here, there is a need for enhanced sampling algorithms that can deal with crowded and very heterogeneous environments. Data analysis becomes another bottleneck. Whereas waiting for a simulation to complete used to be the bottleneck until some ten years ago, nowadays producing terabytes of data occurs overnight. To make sense of this source of big data, the use of machine learning techniques is promising, but still at its infancy.
Figure 12.
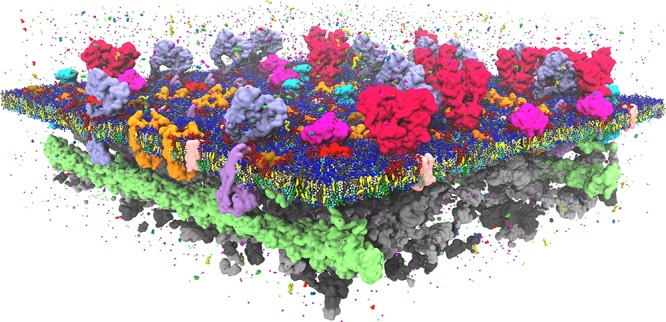
Glimpse at the near future: model of the plasma membrane in full complexity. Featuring: a lipid bilayer composed of hundreds of different lipids, crowded with a large variety of embedded as well as peripherally bound proteins, a supporting actin skeleton, a cytoplasmic site full of proteins, and realistic gradients of metabolites, ions, and pH.
On the other hand, there will be a continuing demand for simulations of model membranes containing few components only. Even simple systems can give rise to rich and complex behavior; many of the simulation studies discussed in this review are proof of this. In principle, simplified model bilayers are ideally suited to connect computational and experimental data. From the experimental side, it would be helpful to have more systematic data on some of the basic properties, such as lipid mixing, the effect of membrane curvature, the effect of leaflet asymmetry, effect of ions and pH, as well as the behavior of dyes. Related challenges on the computational side are to provide high throughput data on, for example, multicomponent lipid phase behavior and protein sorting and clustering, and to more systematically explore the effect of curvature gradients. Furthermore, efficient constant-pH algorithms need to be developed. Enforcing the connection between experiment and simulation will benefit the ongoing validation of both existing and novel lipid types, and the careful calibration of protein–lipid interactions. Machine learning techniques could also be used to improve the parametrization of FFs; pioneering efforts are already taking place in this direction.706−710
Considering the progress not only in complexity but also in system sizes that can be simulated with particle-based models, it is not too bold to predict that a full cell simulation at near-atomic resolution is feasible within the next ten years. Although such a simulation, featuring many billion atoms, would certainly be very impressive and aid our understanding of how cells are structured at the molecular level, this is by no means the final aim. Real cells, in contrast to equilibrated pieces of cells in a simulation box, are inherently out-of-equilibrium. Incorporating the constant energy flow into nonequilibrium simulations is one of the major challenges for the future.
Acknowledgments
Part of this work was performed under the auspices of the U.S. Department of Energy under contract DE-AC52-07NA27344 (LLNL-JRNL-755168). Work in the M.S.P.S. group is supported by the Wellcome Trust and by BBSRC. Work in the S.J.M. group was supported by an ERC Advanced Grant “COMP-MICR-CROW-MEM”. Work in the D.P.T. group was supported by the Natural Sciences and Engineering Research Council of Canada and the Canadian Institutes of Health Research. D.P.T. holds the Alberta Innovates Technology Futures Strategic Chair in (Bio)Molecular Simulation. This work was undertaken, in part, thanks to funding from the Canada Research Chairs program. For help with the figures, we thank Timothy Carpenter (Figure 3), Durba Sengupta (Figure 4), George Khelashvili (Figure 5A), César A. López (Figure 5B), Tao Jiang and Emad Tajkhorshid (Figure 5C), Thorsten Lang (Figure 6), Hector Martinez-Seara (Figure 7), Jonathan Sachs (Figure 8), and Firdaus Samsudin and Syma Khalid (Figure 9).
Biographies
Siewert J. Marrink received his Ph.D. in Chemistry in 1994 from the University of Groningen. His academic career continued with postdoc positions at the Max Planck Institute of Tuebingen and the Australian National University. Since 2005, he has been full professor, heading a research group in Molecular Dynamics at the University of Groningen. He also acts as director of the Berendsen Center for Multiscale Modeling at the same university. His main research interest is on multiscale modeling of (bio)molecular processes, with a focus on unraveling the lateral organization principles of cell membranes.
Valentina Corradi holds a Master of Science in Pharmaceutical Chemistry and Technology from the University of Parma and obtained a Ph.D. in Pharmaceutical Sciences from the University of Siena under the supervision of Dr. Maurizio Botta. In 2009, she joined Dr. D. P. Tieleman’s lab at the University of Calgary as a postdoctoral associate, to work in the area of biomolecular simulation. Since 2013, she has worked as a Research Associate in the same group. Her research interests focus on understanding the correlation between structure, function, and dynamics of membrane proteins and their interactions with lipids.
Paulo C.T. Souza is a senior postdoc at the University of Groningen. He received his BSc (2007) and MSc in Chemistry (2009) from the University of Campinas. At the same university, he completed his Ph.D. in Physical-Chemistry (2013) with the guidance of Munir S. Skaf. Since his undergraduate studies, Paulo has dedicated his efforts on the use of molecular dynamic simulations to study proteins as nuclear receptors, cyclin-dependent kinases, and toll-like receptors. Currently, he is in the group of Siewert-Jan Marrink at the University of Groningen, where he works on the development of the MARTINI force field.
Helgi I. Ingólfsson obtained his B.S. degree in Computer Science from the University of Iceland in 2002 and his Ph.D. degree in Computational Biology and Medicine from Weill Cornell Medical College in 2010. He worked as a postdoctoral fellow at the University of Groningen from 2011 to 2016, after which he joined Lawrence Livermore National Laboratory as a Staff Scientist. His current research is primarily focused on the lipid organization of cellular membranes and their perturbation by small molecules, as well as lipid protein interactions and how membrane proteins can be functionally regulated by their lipid environment.
D. Peter Tieleman studied Chemistry and Philosophy at the University of Groningen, where he obtained his Ph.D. under the supervision of Herman Berendsen. After a postdoctoral position with Mark Sansom at Oxford University he moved to the University of Calgary in 2000. He holds the Canada Research Chair in Molecular Simulation and directs the Centre for Molecular Simulation. His research interests include methods for biomolecular simulation and the biophysics of membranes.
Mark S.P. Sansom is the David Philips professor of molecular biophysics in the Biochemistry Department, University of Oxford. He obtained his D.Phil. in Molecular Biophysics from Oxford in 1983. After a postdoctoral fellowship at the University of Nottingham, he was a Lecturer at Nottingham from 1984 to 1991. In 1991, he joined the Biochemistry Department at Oxford. His research interests are in the use of molecular simulations to understand structure, dynamics, and function of membrane proteins. His current interests are focused on protein–lipid interactions and on water in ion channels and nanopores.
The authors declare no competing financial interest.
References
- Yang Y.; Lee M.; Fairn G. D. Phospholipid Subcellular Localization And Dynamics. J. Biol. Chem. 2018, 293, 6230–6240. 10.1074/jbc.R117.000582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harayama T.; Riezman H. Understanding The Diversity Of Membrane Lipid Composition. Nat. Rev. Mol. Cell Biol. 2018, 19, 281–296. 10.1038/nrm.2017.138. [DOI] [PubMed] [Google Scholar]
- Van Meer G.; Voelker D. R.; Feigenson G. V. Membrane Lipids: Where They Are And How They Behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–24. 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingwood D.; Simons K. Lipid Rafts As A Membrane-Organizing Principle. Science 2010, 327, 46–50. 10.1126/science.1174621. [DOI] [PubMed] [Google Scholar]
- Jacobson K.; Mouritsen O. G.; Anderson R. G. Lipid Rafts: At A Crossroad Between Cell Biology And Physics. Nat. Cell Biol. 2007, 9, 7–14. 10.1038/ncb0107-7. [DOI] [PubMed] [Google Scholar]
- Saka S. K.; Honigmann A.; Eggeling C.; Hell S. W.; Lang T.; Rizzoli S. O. Multi-Protein Assemblies Underlie The Mesoscale Organization Of The Plasma Membrane. Nat. Commun. 2014, 5, 4509. 10.1038/ncomms5509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips R.; Ursell T.; Wiggins P.; Sens P. Emerging Roles For Lipids In Shaping Membrane-Protein Function. Nature 2009, 459, 379–385. 10.1038/nature08147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyman E.; Hsieh C. L.; Eggeling C. From Dynamics to Membrane Organization: Experimental Breakthroughs Occasion a ″Modeling Manifesto. Biophys. J. 2018, 115, 595–604. 10.1016/j.bpj.2018.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickels J. D.; Smith J. C.; Cheng X. Lateral Organization, Bilayer Asymmetry, And Inter-Leaflet Coupling Of Biological Membranes. Chem. Phys. Lipids 2015, 192, 87–99. 10.1016/j.chemphyslip.2015.07.012. [DOI] [PubMed] [Google Scholar]
- Heberle F. A.; Myles D. A. A.; Katsaras J. Biomembranes Research Using Thermal And Cold Neutrons. Chem. Phys. Lipids 2015, 192, 41–50. 10.1016/j.chemphyslip.2015.07.020. [DOI] [PubMed] [Google Scholar]
- Sezgin E.; Schwille P. Model Membrane Platforms To Study Protein-Membrane Interactions. Mol. Membr. Biol. 2012, 29, 144–154. 10.3109/09687688.2012.700490. [DOI] [PubMed] [Google Scholar]
- Marty M. T.; Hoi K. K.; Robinson C. V. Interfacing Membrane Mimetics With Mass Spectrometry. Acc. Chem. Res. 2016, 49, 2459–2467. 10.1021/acs.accounts.6b00379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deleu M.; Crowet J. M.; Nasir M. N.; Lins L. Complementary Biophysical Tools To Investigate Lipid Specificity In The Interaction Between Bioactive Molecules And The Plasma Membrane: A Review. Biochim. Biophys. Acta, Biomembr. 2014, 1838, 3171–3190. 10.1016/j.bbamem.2014.08.023. [DOI] [PubMed] [Google Scholar]
- Van Der Wel P. C. A. Lipid Dynamics And Protein–Lipid Interactions In Integral Membrane Proteins: Insights From Solid-State NMR. Emagres 2014, 3, 111–118. 10.1002/9780470034590.emrstm1356. [DOI] [Google Scholar]
- Maity P. C.; Yang J.; Klaesener K.; Reth M. The Nanoscale Organization Of The B Lymphocyte Membrane. Biochim. Biophys. Acta, Mol. Cell Res. 2015, 1853, 830–840. 10.1016/j.bbamcr.2014.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E. H.; Hsin J.; Sotomayor M.; Comellas G.; Schulten K. Discovery Through The Computational Microscope. Structure 2009, 17, 1295–1306. 10.1016/j.str.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingólfsson H. I.; Arnarez C.; Periole X.; Marrink S. J. Computational ’Microscopy’ Of Cellular Membranes. J. Cell Sci. 2016, 129, 257–268. 10.1242/jcs.176040. [DOI] [PubMed] [Google Scholar]
- Mccammon J. A.; Gelin B. R.; Karplus M. Dynamics Of Folded Proteins. Nature 1977, 267, 585–590. 10.1038/267585a0. [DOI] [PubMed] [Google Scholar]
- Van Der Ploeg P.; Berendsen H. J. C. Molecular-Dynamics Simulation Of A Bilayer-Membrane. J. Chem. Phys. 1982, 76, 3271–3276. 10.1063/1.443321. [DOI] [Google Scholar]
- Egberts E.; Berendsen H. J. C. Molecular Dynamics Simulation Of A Smectic Liquid Crystal With Atomic Detail. J. Chem. Phys. 1988, 89, 3718–3732. 10.1063/1.454893. [DOI] [Google Scholar]
- Jonsson B.; Edholm O.; Teleman O. Molecular Dynamics Simulations Of A Sodium Octanoate Micelle In Aqueous Solution. J. Chem. Phys. 1986, 85, 2259–2271. 10.1063/1.451122. [DOI] [Google Scholar]
- Egberts E.Molecular Dynamics Simulation Of A Smectic Liquid Crystal With Atomic Detail. Ph.D. Thesis, University Of Groningen, 1988. [Google Scholar]
- Berger O.; Edholm O.; Jähnig F. Molecular Dynamics Simulations Of A Fluid Bilayer Of Dipalmitoylphosphatidylcholine At Full Hydration, Constant Pressure And Constant Temperature. Biophys. J. 1997, 72, 2002–2013. 10.1016/S0006-3495(97)78845-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venable R. M.; Zhang Y.; Hardy B. J.; Pastor R. W. Molecular Dynamics Simulations Of A Lipid Bilayer And Of Hexadecane: An Investigation Of Membrane Fluidity. Science 1993, 262, 223–226. 10.1126/science.8211140. [DOI] [PubMed] [Google Scholar]
- Egberts E.; Marrink S. J.; Berendsen H. J. C. Molecular Dynamics Simulation Of A Phospholipid Membrane. Eur. Biophys. J. 1994, 22, 423–426. 10.1007/BF00180163. [DOI] [PubMed] [Google Scholar]
- Heller H.; Schaefer M.; Schulten K. Molecular Dynamics Simulations Of A Bilayer Of 200 Lipids In The Gel And In The Liquid-Crystal Phases. J. Phys. Chem. 1993, 97, 8343–8360. 10.1021/j100133a034. [DOI] [Google Scholar]
- Marrink S. J.; Berendsen H. J. C. Permeation Process Of Small Molecules Across Lipid Membranes Studied By Molecular Dynamics Simulations. J. Phys. Chem. 1996, 100, 16729–16738. 10.1021/jp952956f. [DOI] [Google Scholar]
- Edholm O.; Nyberg A. M. Cholesterol In Model Membranes: A Molecular Dynamics Study. Biophys. J. 1992, 63, 1081–1089. 10.1016/S0006-3495(92)81678-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrink S. J.; Berkowitz M.; Berendsen H. J. C. Molecular Dynamics Simulation Of A Membrane/Water Interface: The Ordering Of Water And Its Relation To The Hydration Force. Langmuir 1993, 9, 3122–3131. 10.1021/la00035a062. [DOI] [Google Scholar]
- Damodaran K. V.; Merz K. M. Jr.; Gaber B. P. Interaction Of Small Peptides With Lipid Bilayers. Biophys. J. 1995, 69, 1299–1308. 10.1016/S0006-3495(95)79997-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf T. B.; Roux B. Molecular Dynamics Simulation Of The Gramicidin Channel In A Phospholipid Bilayer. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 11631–11635. 10.1073/pnas.91.24.11631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edholm O.; Berger O.; Jahnig F. Structure And Fluctuations Of Bacteriorhodopsin In The Purple Membrane: A Molecular Dynamics Study. J. Mol. Biol. 1995, 250, 94–111. 10.1006/jmbi.1995.0361. [DOI] [PubMed] [Google Scholar]
- Tieleman D. P.; Berendsen H. J. C. Molecular Dynamics Study Of The Pores Formed By E. Coli Ompf Porin In A Fully Hydrated Palmitoyloleoylphosphatidylcholine Bilayer. Biophys. J. 1998, 74, 2786–2801. 10.1016/S0006-3495(98)77986-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou F.; Schulten K. Molecular Dynamics Study Of Phospholipase A2 On A Membrane Surface. Proteins: Struct., Funct., Genet. 1996, 25, 12–27. 10.1002/(SICI)1097-0134(199605)25:1<12::AID-PROT2>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Tieleman D. P.; Forrest L.; Sansom M. S. P.; Berendsen H. J. C. Lipid Properties And The Orientation Of Aromatic Residues In Ompf, Influenza M2 And Alamethicin Systems: Molecular Dynamics Simulations. Biochemistry 1998, 37, 17554–17561. 10.1021/bi981802y. [DOI] [PubMed] [Google Scholar]
- Tieleman D. P.; Marrink S. J.; Berendsen H. J. C. A Computer Perspective Of Membranes: Molecular Dynamics Studies Of Lipid Bilayer Systems. Biochim. Biophys. Acta, Rev. Biomembr. 1997, 1331, 235–270. 10.1016/S0304-4157(97)00008-7. [DOI] [PubMed] [Google Scholar]
- Pastor R. W. Molecular Dynamics And Monte Carlo Simulations Of Lipid Bilayers. Curr. Opin. Struct. Biol. 1994, 4, 486–492. 10.1016/S0959-440X(94)90209-7. [DOI] [Google Scholar]
- Oloo E. O.; Tieleman D. P. Conformational Transitions Induced by the Binding of MgATP to the Vitamin B12 ATP-binding Cassette (ABC) Transporter BtuCD. J. Biol. Chem. 2004, 279, 45013–45019. 10.1074/jbc.M405084200. [DOI] [PubMed] [Google Scholar]
- Patra M.; Karttunen M.; Hyvönen M. T.; Falck E.; Lindqvist P.; Vattulainen I. Molecular Dynamics Simulations Of Lipid Bilayers: Major Artifacts Due To Truncating Electrostatic Interactions. Biophys. J. 2003, 84, 3636–3645. 10.1016/S0006-3495(03)75094-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feller S. E.; Pastor R. W. On Simulating Lipid Bilayers With An Applied Surface Tension: Periodic Boundary Conditions And Undulations. Biophys. J. 1996, 71, 1350–1355. 10.1016/S0006-3495(96)79337-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl E.; Edholm O. Mesoscopic Undulations And Thickness Fluctuations In Lipid Bilayers From Molecular Dynamics Simulations. Biophys. J. 2000, 79, 426–433. 10.1016/S0006-3495(00)76304-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrink S. J.; Lindahl E.; Edholm O.; Mark A. E. Simulation Of The Spontaneous Aggregation Of Phospholipids Into Bilayers. J. Am. Chem. Soc. 2001, 123, 8638–8639. 10.1021/ja0159618. [DOI] [PubMed] [Google Scholar]
- Leontiadou H.; Mark A. E.; Marrink S. J. Antimicrobial Peptides In Action. J. Am. Chem. Soc. 2006, 128, 12156–12161. 10.1021/ja062927q. [DOI] [PubMed] [Google Scholar]
- Gurtovenko A. A.; Vattulainen I. Pore Formation Coupled To Ion Transport Through Lipid Membranes As Induced By Transmembrane Ionic Charge Imbalance: Atomistic Molecular Dynamics Study. J. Am. Chem. Soc. 2005, 127, 17570–17571. 10.1021/ja053129n. [DOI] [PubMed] [Google Scholar]
- Tieleman D. P. The Molecular Basis Of Electroporation. BMC Biochem 2004, 5, 10. 10.1186/1471-2091-5-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tieleman D. P.; Marrink S. J. Lipids Out Of Equilibrium: Energetics Of Desorption And Pore Mediated Flip-Flop. J. Am. Chem. Soc. 2006, 128, 12462–12467. 10.1021/ja0624321. [DOI] [PubMed] [Google Scholar]
- Falck E.; Rog T.; Karttunen M.; Vattulainen I. Lateral Diffusion In Lipid Membranes Through Collective Flows. J. Am. Chem. Soc. 2008, 130, 44–45. 10.1021/ja7103558. [DOI] [PubMed] [Google Scholar]
- Risselada H. J.; Marrink S. J. The Molecular Face Of Lipid Rafts In Model Membranes. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 17367–17372. 10.1073/pnas.0807527105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrink S. J.; Mark A. E. The Mechanism Of Vesicle Fusion As Revealed By Molecular Dynamics Simulations. J. Am. Chem. Soc. 2003, 125, 11144–11145. 10.1021/ja036138+. [DOI] [PubMed] [Google Scholar]
- Marrink S. J.; De Vries A. H.; Tieleman D. P. Lipids On The Move: Simulations Of Membrane Pores, Domains, Stalks And Curves. Biochim. Biophys. Acta, Biomembr. 2009, 1788, 149–168. 10.1016/j.bbamem.2008.10.006. [DOI] [PubMed] [Google Scholar]
- Gumbart J.; Wang Y.; Aksimentiev A.; Tajkhorshid E.; Schulten K. Molecular Dynamics Simulations Of Proteins In Lipid Bilayers. Curr. Opin. Struct. Biol. 2005, 15, 423–431. 10.1016/j.sbi.2005.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl E.; Sansom M. S. P. Membrane Proteins: Molecular Dynamics Simulations. Curr. Opin. Struct. Biol. 2008, 18, 425–431. 10.1016/j.sbi.2008.02.003. [DOI] [PubMed] [Google Scholar]
- Schlick T.; Collepardo-Guevara R.; Halvorsen L. A.; Jung S.; Xiao X. Biomolecular Modeling And Simulation: A Field Coming Of Age. Q. Rev. Biophys. 2011, 44, 191–228. 10.1017/S0033583510000284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perilla J. R.; Goh B. C.; Cassidy K. C.; Liu B.; Bernardi R. C.; Rudack T.; Yu H.; Wu Z.; Schulten K. Molecular Dynamics Simulations Of Large Macromolecular Complexes. Curr. Opin. Struct. Biol. 2015, 31, 64–74. 10.1016/j.sbi.2015.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huggins D. J.; Biggin P. C.; Dämgen M. A.; et al. Biomolecular Simulations: From Dynamics And Mechanisms To Computational Assays Of Biological Activity. WIREs Comput. Mol. Sci. 2018, e1393. 10.1002/wcms.1393. [DOI] [Google Scholar]
- Bottaro S.; Lindorff-Larsen K. Biophysical Experiments And Biomolecular Simulations: A Perfect Match?. Science 2018, 361, 355–360. 10.1126/science.aat4010. [DOI] [PubMed] [Google Scholar]
- Chavent M.; Duncan A. L.; Sansom M. S. P. Molecular Dynamics Simulations Of Membrane Proteins And Their Interactions: From Nanoscale To Mesoscale. Curr. Opin. Struct. Biol. 2016, 40, 8–16. 10.1016/j.sbi.2016.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman R.; Khalid S.; Aponte-Santamaría C.; et al. Understanding Conformational Dynamics of Complex Lipid Mixtures Relevant to Biology. J. Membr. Biol. 2018, 251, 609–631. 10.1007/s00232-018-0050-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffeo C.; Bhattacharya S.; Yoo J.; Wells D.; Aksimentiev A. Modeling And Simulation Of Ion Channels. Chem. Rev. 2012, 112, 6250–6284. 10.1021/cr3002609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida J. G.; Preto A. J.; Koukos P. I.; Bonvin A. M. J. J.; Moreira I. S. Membrane Proteins Structures: A Review On Computational Modeling Tools. Biochim. Biophys. Acta, Biomembr. 2017, 1859, 2021–2039. 10.1016/j.bbamem.2017.07.008. [DOI] [PubMed] [Google Scholar]
- Ladefoged L. K.; Zeppelin T.; Schiøtt B.. Molecular Modeling Of Neurological Membrane Proteins: From Binding Sites To Synapses, Neurosci. Lett. 2018, in press, 10.1016/j.neulet.2018.05.034. [DOI] [PubMed] [Google Scholar]
- Bondar A. N.; Keller S. Lipid Membranes and Reactions at Lipid Interfaces: Theory, Experiments, and Applications. J. Membr. Biol. 2018, 251, 295. 10.1007/s00232-018-0039-6. [DOI] [PubMed] [Google Scholar]
- Ulmschneider J. P.; Ulmschneider M. B. Molecular Dynamics Simulations Are Redefining Our View Of Peptides Interacting With Biological Membranes. Acc. Chem. Res. 2018, 51, 1106–1116. 10.1021/acs.accounts.7b00613. [DOI] [PubMed] [Google Scholar]
- Li Z. L.; Ding H. M.; Ma Y. Q. Interaction Of Peptides With Cell Membranes: Insights From Molecular Modeling. J. Phys.: Condens. Matter 2016, 28, 083001. 10.1088/0953-8984/28/8/083001. [DOI] [PubMed] [Google Scholar]
- Ding H. M.; Ma Y. Q. Theoretical And Computational Investigations Of Nanoparticle-Biomembrane Interactions In Cellular Delivery. Small 2015, 11, 1055–1071. 10.1002/smll.201401943. [DOI] [PubMed] [Google Scholar]
- Nakamura H.; Watano S. Direct Permeation Of Nanoparticles Across Cell Membrane: A Review. Kona Powd. Part. J. 2018, 35, 49–65. 10.14356/kona.2018011. [DOI] [Google Scholar]
- Rossi G.; Monticelli L. Simulating The Interaction Of Lipid Membranes With Polymer And Ligand-Coated Nanoparticles. Adv. Phys. X. 2016, 1, 276–296. 10.1080/23746149.2016.1177468. [DOI] [Google Scholar]
- Lopes D.; Jakobtorweihen S.; Nunes C.; Sarmento B.; Reis S. Shedding Light On The Puzzle Of Drug-Membrane Interactions: Experimental Techniques And Molecular Dynamics Simulations. Prog. Lipid Res. 2017, 65, 24–44. 10.1016/j.plipres.2016.12.001. [DOI] [PubMed] [Google Scholar]
- Di Meo F.; Fabre G.; Berka K.; Ossman T.; Chantemargue B.; Paloncýová M.; Marquet P.; Otyepka M.; Trouillas P. In Silico Pharmacology: Drug Membrane Partitioning And Crossing. Pharmacol. Res. 2016, 111, 471–486. 10.1016/j.phrs.2016.06.030. [DOI] [PubMed] [Google Scholar]
- Benedetto A.; Ballone P. An Overview Of Neutron Scattering And Molecular Dynamics Simulation Studies Of Phospholipid Bilayers In Room-Temperature Ionic Liquid/Water Solutions. Phys. B 2018, 551, 227. 10.1016/j.physb.2018.02.043. [DOI] [Google Scholar]
- Kirsch S. A.; Böckmann R. A. Membrane Pore Formation In Atomistic And Coarse-Grained Simulations. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 2266–2277. 10.1016/j.bbamem.2015.12.031. [DOI] [PubMed] [Google Scholar]
- Parisio G.; Ferrarini A.; Sperotto M. M. Model Studies Of Lipid Flip-Flop In Membranes. Int. J. Adv. Eng. Sci. Appl. Math. 2016, 8, 134–146. 10.1007/s12572-015-0155-9. [DOI] [Google Scholar]
- Lee S. C.; Khalid S.; Pollock N. L.; Knowles T. J.; Edler K.; Rothnie A. J.; Thomas O. R. T.; Dafforn T. R. Encapsulated Membrane Proteins: A Simplified System For Molecular Simulation. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 2549–2557. 10.1016/j.bbamem.2016.02.039. [DOI] [PubMed] [Google Scholar]
- Salomon-Ferrer R.; Case D. A.; Walker R. C. An Overview of The Amber Biomolecular Simulation Package. Wires Comput. Mol. Sci. 2013, 3, 198–210. 10.1002/wcms.1121. [DOI] [Google Scholar]
- Case D. A.; Cheatham T. E. 3rd; Darden T.; Gohlke H.; Luo R.; Merz K. M. Jr.; Onufriev A.; Simmerling C.; Wang B.; Woods R. The Amber Biomolecular Simulation Programs. J. Comput. Chem. 2005, 26, 1668–1688. 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks B. R.; Brooks C. L. III; Mackerell A. D.; Nilsson L.; et al. CHARMM: The Biomolecular Simulation Program. J. Comput. Chem. 2009, 30, 1545–1615. 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips J. C.; Braun R.; Wang W.; et al. Scalable Molecular Dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eastman P.; Swails J.; Chodera J. D.; Mcgibbon R. T.; Zhao Y.; Beauchamp K. A.; Wang L.-P.; Simmonett A. C.; Harrigan M. P.; Stern C. D.; et al. Openmm 7: Rapid Development of High Performance Algorithms For Molecular Dynamics. PLoS Comput. Biol. 2017, 13, E1005659. 10.1371/journal.pcbi.1005659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plimpton S. J. Fast Parallel Algorithms For Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. 10.1006/jcph.1995.1039. [DOI] [Google Scholar]
- Guzman H. V.; Kobayashi H.; Tretyakov N.; Fogarty A. C.; Kreis K.; Krajniak J.; Junghans C.; Kremer K.; Stuehn T. Espresso++ 2.0: Advanced Methods For Multiscale Molecular Simulation. arXiv 2018, arXiv:1806.10841. [Google Scholar]
- Berendsen H. J. C.; Van Der Spoel D.; Van Drunen R. GROMACS: a Message-Passing Parallel Molecular Dynamics Implementation. Comput. Phys. Commun. 1995, 91, 43–56. 10.1016/0010-4655(95)00042-E. [DOI] [Google Scholar]
- Abraham M. J.; Murtola T.; Schulz R.; Páll S.; et al. GROMACS: High Performance Molecular Simulations Through Multi-Level Parallelism From Laptops To Supercomputers. Softwarex 2015, 1–2, 19–25. 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- Shaw D. E.; Deneroff M. M.; Dror R. O.; Kuskin J. S.; et al. Anton, a Special-Purpose Machine For Molecular Dynamics Simulation. Commun. ACM 2008, 51, 91–97. 10.1145/1364782.1364802. [DOI] [Google Scholar]
- Lelimousin M.; Limongelli V.; Sansom M. S. P. Conformational Changes in The Epidermal Growth Factor Receptor: Role of The Transmembrane Domain Investigated By Coarse-Grained Metadynamics Free Energy Calculations. J. Am. Chem. Soc. 2016, 138, 10611–10622. 10.1021/jacs.6b05602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domanski J.; Hedger G.; Best R. B.; Stansfeld P. J.; Sansom M. S. P. Convergence and Sampling in Determining Free Energy Landscapes For Membrane Protein Association. J. Phys. Chem. B 2017, 121, 3364–3375. 10.1021/acs.jpcb.6b08445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori T.; Miyashita N.; Im W.; Feig M.; Sugita Y. Molecular Dynamics Simulations of Biological Membranes and Membrane Proteins Using Enhanced Conformational Sampling Algorithms. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 1635–1651. 10.1016/j.bbamem.2015.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fathizadeh A.; Elber R. A Mixed Alchemical and Equilibrium Dynamics To Simulate Heterogeneous Dense Fluids: Illustrations For Lennard-Jones Mixtures and Phospholipid Membranes. J. Chem. Phys. 2018, 149, 072325. 10.1063/1.5027078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardelli C.; Barducci A.; Procacci P. Lipid Tempering Simulation of Model Biological Membranes on Parallel Platforms. Biochim. Biophys. Acta, Biomembr. 2018, 1860, 1480–1488. 10.1016/j.bbamem.2018.04.013. [DOI] [PubMed] [Google Scholar]
- Nitschke N.; Atkovska K.; Hub J. S. Accelerating Potential of Mean Force Calculations For Lipid Membrane Permeation: System Size, Reaction Coordinate, Solute-Solute Distance, and Cutoffs. J. Chem. Phys. 2016, 145, 125101. 10.1063/1.4963192. [DOI] [PubMed] [Google Scholar]
- Elber R. A New Paradigm For Atomically Detailed Simulations of Kinetics in Biophysical Systems. Q. Rev. Biophys. 2017, 50, E8. 10.1017/S0033583517000063. [DOI] [PubMed] [Google Scholar]
- Salari R.; Joseph T.; Lohia R.; Hénin J.; Brannigan G. A. Streamlined, General Approach For Computing Ligand Binding Free Energies And Its Application To GPCR-Bound Cholesterol. J. Chem. Theory Comput. 2018, 14, 6560. 10.1021/acs.jctc.8b00447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackerell A. D. Empirical Force Fields For Biological Macromolecules: Overview and Issues. J. Comput. Chem. 2004, 25, 1584–1604. 10.1002/jcc.20082. [DOI] [PubMed] [Google Scholar]
- Riniker S. Fixed-Charge Atomistic Force Fields For Molecular Dynamics Simulations in The Condensed Phase: An Overview. J. Chem. Inf. Model. 2018, 58, 565–578. 10.1021/acs.jcim.8b00042. [DOI] [PubMed] [Google Scholar]
- Pluhackova K.; Kirsch S. A.; Han J.; Sun L. P.; Jiang Z. Y.; Unruh T.; Bockmann R. A. a Critical Comparison of Biomembrane Force Fields: Structure and Dynamics of Model DMPC, POPC, and POPE Bilayers. J. Phys. Chem. B 2016, 120, 3888–3903. 10.1021/acs.jpcb.6b01870. [DOI] [PubMed] [Google Scholar]
- Sandoval-Perez A.; Pluhackova K.; Boeckmann R. A. Critical Comparison of Biomembrane Force Fields: Protein-Lipid Interactions At The Membrane Interface. J. Chem. Theory Comput. 2017, 13, 2310–2321. 10.1021/acs.jctc.7b00001. [DOI] [PubMed] [Google Scholar]
- Pezeshkian W.; Khandelia H.; Marsh D. Lipid Configurations From Molecular Dynamics Simulations. Biophys. J. 2018, 114, 1895–1907. 10.1016/j.bpj.2018.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard A. N.; Wang E.; Monje-Galvan V.; Klauda J. B.. Developing and Testing of Lipid Force Fields with Applications to Modeling Cellular Membranes. Chem. Rev. 2018, in press. [DOI] [PubMed] [Google Scholar]
- Ollila O. H. S.; Pabst G. Atomistic Resolution Structure And Dynamics Of Lipid Bilayers In Simulations And Experiments. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 2512–2528. 10.1016/j.bbamem.2016.01.019. [DOI] [PubMed] [Google Scholar]
- Poger D.; Caron B.; Mark A. E. Validating Lipid Force Fields Against Experimental Data: Progress, Challenges And Perspectives. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 1556–1565. 10.1016/j.bbamem.2016.01.029. [DOI] [PubMed] [Google Scholar]
- Heberle F. A.; Pan J.; Standaert R. F.; Drazba P.; Kučerka N.; Katsaras J. Model Based Approaches For The Determination Of Lipid Bilayer Structure From Small-Angle Neutron And X-Ray Scattering Data. Eur. Biophys. J. 2012, 41, 875–890. 10.1007/s00249-012-0817-5. [DOI] [PubMed] [Google Scholar]
- Kučerka N.; Heberle F. A.; Pan J.; Katsaras J. Structural Significance Of Lipid Diversity As Studied By Small Angle Neutron And X-Ray Scattering. Membranes 2015, 5, 454–472. 10.3390/membranes5030454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassall S. R.; Leng X.; Canner S. W.; Pennington E. R.; Kinnun J. J.; Cavazos A. T.; Dadoo S.; Johnson D.; Heberle F. A.; Katsaras J.; Shaikh S. R. Docosahexaenoic Acid Regulates The Formation Of Lipid Rafts: A Unified View From Experiment And Simulation. Biochim. Biophys. Acta, Biomembr. 2018, 1860, 1985. 10.1016/j.bbamem.2018.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toppozini L.; Meinhardt S.; Armstrong C. L.; Yamani Z.; Kucerka N.; Schmid F.; Rheinstadter M. C. Structure Of Cholesterol In Lipid Rafts. Phys. Rev. Lett. 2014, 113, 228101. 10.1103/PhysRevLett.113.228101. [DOI] [PubMed] [Google Scholar]
- Kinnun J. J.; Mallikarjunaiah K. J.; Petrache H. I.; Brown M. F. Elastic Deformation And Area Per Lipid Of Membranes: Atomistic View From Solid-State Deuteriumnmr Spectroscopy. Biochim. Biophys. Acta, Biomembr. 2015, 1848, 246–59. 10.1016/j.bbamem.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leftin A.; Brown M. F. An NMR Database For Simulations Of Membrane Dynamics. Biochim. Biophys. Acta, Biomembr. 2011, 1808, 818–839. 10.1016/j.bbamem.2010.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keyvanloo A.; Shaghaghi M.; Zuckermann M. J.; Thewalt J. L. The Phase Behavior And Organization Of Sphingomyelin/Cholesterol Membranes: A Deuterium NMR Study. Biophys. J. 2018, 114, 1344–1356. 10.1016/j.bpj.2018.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klauda J. B.; Venable R. M.; Freites J. A.; O’Connor J. W.; Tobias D. J.; Mondragon-Ramirez C.; Vorobyov I.; Mackerell A. D. Jr.; Pastor R. W. Update Of The CHARMM All-Atom Additive Force Field For Lipids: Validation On Six Lipid Types. J. Phys. Chem. B 2010, 114, 7830–7843. 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanommeslaeghe K.; Mackerell A. D. Jr. CHARMM Additive And Polarizable Force Fields For Biophysics And Computer-Aided Drug Design. Biochim. Biophys. Acta, Gen. Subj. 2015, 1850, 861–871. 10.1016/j.bbagen.2014.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venable R. M.; Brown F. L. H.; Pastor R. W. Mechanical Properties Of Lipid Bilayers From Molecular Dynamics Simulation. Chem. Phys. Lipids 2015, 192, 60–74. 10.1016/j.chemphyslip.2015.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S.; Kim T.; Iyer V. G.; Im W. CHARMM-GUI: A Web-Based Graphical User Interface For CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- Jo S.; Cheng X.; Lee J.; Kim S.; Park S. J.; Patel D. S.; Beaven A. H.; Lee K. I.; Rui H.; Park S.; Lee H. S.; Roux B.; Mackerell A. D. Jr.; Klauda J. B.; Qi Y.; Im W. CHARMM-GUI 10 Years For Biomolecular Modeling And Simulation. J. Comput. Chem. 2017, 38, 1114–1124. 10.1002/jcc.24660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu E. L.; Cheng X.; Jo S.; Rui H.; Song K. C.; Dávila-Contreras E. M.; Qi Y.; Lee J.; Monje-Galvan V.; Venable R. M.; Klauda J. B.; Im W. CHARMM-GUI Membrane Builder Toward Realistic Biological Membrane Simulations. J. Comput. Chem. 2014, 35, 1997–2004. 10.1002/jcc.23702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguayo D.; Gonzalez-Nilo F. D.; Chipot C. Insight Into The Properties Of Cardiolipin Containing Bilayers From Molecular Dynamics Simulations, Using A Hybrid All-Atom/United-Atom Force Field. J. Chem. Theory Comput. 2012, 8, 1765–73. 10.1021/ct200849k. [DOI] [PubMed] [Google Scholar]
- Boyd K. J.; Alder N. N.; May E. R. Molecular Dynamics Analysis Of Cardiolipin And Monolysocardiolipin On Bilayer Properties. Biophys. J. 2018, 114, 2116–27. 10.1016/j.bpj.2018.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu E. L.; Engström O.; Jo S.; Stuhlsatz D.; Yeom M. S.; Klauda J. B.; Widmalm G.; Im W. Molecular Dynamics And NMR Spectroscopy Studies Of E. Coli Lipopolysaccharide Structure And Dynamics. Biophys. J. 2013, 105, 1444–55. 10.1016/j.bpj.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson C. J.; Rosso L.; Betz R. M.; Walker R. C.; Gould I. R. Gafflipid: A General Amber Force Field For The Accurate Molecular Dynamics Simulation Of Phospholipid. Soft Matter 2012, 8, 9617–9627. 10.1039/c2sm26007g. [DOI] [Google Scholar]
- Wang J.; Wolf R. M.; Caldwell J. W.; Kollman P. A.; Case D. A. Development And Testing Of A General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–74. 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- Coimbra J. T. S.; Sousa S. F.; Fernandes P. A.; Rangel M.; Ramos M. J. Biomembrane Simulations Of 12 Lipid Types Using The General Amber Force Field In A Tensionless Ensemble. J. Biomol. Struct. Dyn. 2014, 32, 88–103. 10.1080/07391102.2012.750250. [DOI] [PubMed] [Google Scholar]
- Dickson C. J.; Madej B. D.; Skjevik A. A.; Betz R. M.; Teigen K.; Gould I. R.; Walker R. C. J. Lipid14: The Amber Lipid Force Field. J. Chem. Theory Comput. 2014, 10, 865–879. 10.1021/ct4010307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madej B. D.; Gould I. R.; Walker R. C. A Parameterization Of Cholesterol For Mixed Lipid Bilayer Simulation Within The Amber Lipid14 Force Field. J. Phys. Chem. B 2015, 119, 12424–12435. 10.1021/acs.jpcb.5b04924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias R. P.; Da Hora G. C. A.; Ramstedt M.; Soares T. A. Outer Membrane Remodeling: The Structural Dynamics And Electrostatics Of Rough Lipopolysaccharide Chemotypes. J. Chem. Theory Comput. 2014, 10 (6), 2488–2497. 10.1021/ct500075h. [DOI] [PubMed] [Google Scholar]
- Jämbeck J. P. M.; Lyubartsev A. P. Derivation And Systematic Validation Of A Refined All-Atom Force Field For Phosphatidylcholine Lipids. J. Phys. Chem. B 2012, 116, 3164–3179. 10.1021/jp212503e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jämbeck J. P. M.; Lyubartsev A. P. An Extension And Further Validation Of An All-Atomistic Force Field For Biological Membranes. J. Chem. Theory Comput. 2012, 8, 2938–2948. 10.1021/ct300342n. [DOI] [PubMed] [Google Scholar]
- Jämbeck J. P. M.; Lyubartsev A. P. Another Piece Of The Membrane Puzzle: Extending Slipids Further. J. Chem. Theory Comput. 2013, 9, 774–784. 10.1021/ct300777p. [DOI] [PubMed] [Google Scholar]
- Ermilova I.; Lyubartsev A. P. Extension Of The Slipids Force Field To Polyunsaturated Lipids. J. Phys. Chem. B 2016, 120, 12826–12842. 10.1021/acs.jpcb.6b05422. [DOI] [PubMed] [Google Scholar]
- Atkovska K.; Klingler J.; Oberwinkler J.; Keller S.; Hub J. S. Rationalizing Steroid Interactions with Lipid Membranes: Conformations, Partitioning, and Kinetics. ACS Cent. Sci. 2018, 4, 1155–1165. 10.1021/acscentsci.8b00332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid N.; Eichenberger A. P.; Choutko A.; Riniker S.; et al. Definition And Testing Of The GROMOS Force-Field Versions 54A7 And 54B7. Eur. Biophys. J. 2011, 40, 843–856. 10.1007/s00249-011-0700-9. [DOI] [PubMed] [Google Scholar]
- Poger D.; Van Gunsteren W. F.; Mark A. E. A New Force Field For Simulating Phosphatidylcholine Bilayers. J. Comput. Chem. 2010, 31, 1117–1125. 10.1002/jcc.21396. [DOI] [PubMed] [Google Scholar]
- Poger D.; Mark A. E. On The Validation Of Molecular Dynamics Simulations Of Saturated And Cis-Monounsaturated Phosphatidylcholine Lipid Bilayers: A Comparison With Experiment. J. Chem. Theory Comput. 2010, 6, 325–336. 10.1021/ct900487a. [DOI] [PubMed] [Google Scholar]
- Poger D.; Caron B.; Mark A. E. Effect Of Methyl-Branched Fatty Acids On The Structure Of Lipid Bilayers. J. Phys. Chem. B 2014, 118, 13838–13848. 10.1021/jp503910r. [DOI] [PubMed] [Google Scholar]
- Poger D.; Mark A. E. A Ring To Rule Them All: The Effect Of Cyclopropane Fatty Acids On The Fluidity Of Lipid Bilayers. J. Phys. Chem. B 2015, 119, 5487–5495. 10.1021/acs.jpcb.5b00958. [DOI] [PubMed] [Google Scholar]
- Piggot T. J.; Holdbrook D. A.; Khalid S. Electroporation Of The E. Coli And S. Aureus Membranes: Molecular Dynamics Simulations Of Complex Bacterial Membranes. J. Phys. Chem. B 2011, 115, 13381–88. 10.1021/jp207013v. [DOI] [PubMed] [Google Scholar]
- Caron B.; Mark A. E.; Poger D. Some Like It Hot: The Effect Of Sterols And Hopanoids On Lipid Ordering At High Temperature. J. Phys. Chem. Lett. 2014, 5, 3953–3957. 10.1021/jz5020778. [DOI] [PubMed] [Google Scholar]
- Baker C. M. Polarizable Force Fields For Molecular Dynamics Simulations Of Biomolecules. Wires Comput. Mol. Sci. 2015, 5, 241–254. 10.1002/wcms.1215. [DOI] [Google Scholar]
- Drude P.The Theory Of Optics; Longmans, Green: New York, 1902. [Google Scholar]
- Mortier W. J.; Ghosh S. K.; Shankar S. Electronegativity-Equalization Method For The Calculation Of Atomic Charges In Molecules. J. Am. Chem. Soc. 1986, 108, 4315–4320. 10.1021/ja00275a013. [DOI] [Google Scholar]
- Chowdhary J.; Harder E.; Lopes P. E. M.; Huang L.; Mackerell A. D. Jr.; Roux B. A Polarizable Force Field Of Dipalmitoylphosphatidylcholine Based On The Classical Drude Model For Molecular Dynamics Simulations Of Lipids. J. Phys. Chem. B 2013, 117, 9142–9160. 10.1021/jp402860e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Chowdhary J.; Huang L.; He X.; Mackerell A. D. Jr.; Roux B. Drude Polarizable Force Field For Molecular Dynamics Simulations Of Saturated And Unsaturated Zwitterionic Lipids. J. Chem. Theory Comput. 2017, 13, 4535–4552. 10.1021/acs.jctc.7b00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes P. E. M.; Huang J.; Shim J.; Luo Y.; Li H.; Roux B.; Mackerell A. D. Jr. Polarizable Force Field For Peptides And Proteins Based On The Classical Drude Oscillator. J. Chem. Theory Comput. 2013, 9, 5430–5449. 10.1021/ct400781b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savelyev A.; Mackerell A. D. Jr. All-Atom Polarizable Force Field For DNA Based On The Classical Drude Oscillator Model. J. Comput. Chem. 2014, 35, 1219–1239. 10.1002/jcc.23611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas T. R.; Bauer B. A.; Patel S. Charge Equilibration Force Fields For Molecular Dynamics Simulations Of Lipids, Bilayers, And Integral Membrane Protein Systems. Biochim. Biophys. Acta, Biomembr. 2012, 1818, 318–329. 10.1016/j.bbamem.2011.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorobyov I.; Allen T. W. The Electrostatics Of Solvent And Membrane Interfaces And The Role Of Electronic Polarizability. J. Chem. Phys. 2010, 132, 185101. 10.1063/1.3402125. [DOI] [Google Scholar]
- Vorobyov I.; Bennett W. F. D.; Tieleman D. P.; Allen T. W.; Noskov S. Y. The Role Of Atomic Polarization In The Thermodynamics Of Chloroform Partitioning To Lipid Bilayers. J. Chem. Theory Comput. 2012, 8, 618–628. 10.1021/ct200417p. [DOI] [PubMed] [Google Scholar]
- Ponder J. W.; Wu C.; Ren P.; Pande V. S.; Chodera J. D.; et al. Current Status Of The AMOEBA Polarizable Force Field. J. Phys. Chem. B 2010, 114, 2549–2564. 10.1021/jp910674d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y.; Xia Z.; Zhang J.; Best R.; Wu C.; Ponder J. W.; Ren P. The Polarizable Atomic Multipole-Based AMOEBA Force Field For Proteins. J. Chem. Theory Comput. 2013, 9, 4046–4063. 10.1021/ct4003702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. 10.1021/acs.jctc.5b00935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botan A.; Favela-Rosales F.; Fuchs P.; Javanainen M.; Kanduč M.; Kulig W.; Lamberg A.; Loison C.; Lyubartsev A.; Miettinen M. S.; et al. Towards Atomistic Resolution Structure Of Phosphatidylcholine Headgroup And Glycerol Backbone At Different Ambient Conditions. J. Phys. Chem. B 2015, 119, 15075–15088. 10.1021/acs.jpcb.5b04878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reißer S.; Poger D.; Stroet M.; Mark A. E. Real cost of speed: the effect of a time-saving multiple-time-stepping algorithm on the accuracy of molecular dynamics simulations. J. Chem. Theory Comput. 2017, 13, 2367–2372. 10.1021/acs.jctc.7b00178. [DOI] [PubMed] [Google Scholar]
- Camley B. A.; Lerner M. G.; Pastor R. W.; Brown F. L. H. Strong Influence Of Periodic Boundary Conditions On Lateral Diffusion In Lipid Bilayer Membranes. J. Chem. Phys. 2015, 143, 243113. 10.1063/1.4932980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vögele M.; Hummer G. Divergent Diffusion Coefficients In Simulations Of Fluids And Lipid Membranes. J. Phys. Chem. B 2016, 120, 8722–8732. 10.1021/acs.jpcb.6b05102. [DOI] [PubMed] [Google Scholar]
- Venable R. M.; Ingólfsson H. I.; Lerner M. G.; Perrin B. S. Jr.; Camley B. A.; Marrink S. J.; Brown F. L. H.; Pastor R. W. Lipid And Peptide Diffusion In Bilayers: The Saffman-Delbrück Model And Periodic Boundary Conditions. J. Phys. Chem. B 2017, 121, 3443–3457. 10.1021/acs.jpcb.6b09111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vögele M.; Köfinger J.; Hummer G. Hydrodynamics Of Diffusion In Lipid Membrane Simulations. Phys. Rev. Lett. 2018, 120, 268104. 10.1103/PhysRevLett.120.268104. [DOI] [PubMed] [Google Scholar]
- Melcr J.; Martinez-Seara H.; Nencini R.; Kolafa J.; Jungwirth P.; Ollila O. H. S. Accurate Binding Of Sodium And Calcium To A POPC Bilayer By Effective Inclusion Of Electronic Polarization. J. Phys. Chem. B 2018, 122, 4546–4557. 10.1021/acs.jpcb.7b12510. [DOI] [PubMed] [Google Scholar]
- Javanainen M.; Melcrová A.; Magarkar A.; Jurkiewicz P.; Hof M.; Jungwirth P.; Martinez-Seara H. Two Cations, Two Mechanisms: Interactions Of Sodium And Calcium With Zwitterionic Lipid Membranes. Chem. Commun. 2017, 53, 5380–5383. 10.1039/C7CC02208E. [DOI] [PubMed] [Google Scholar]
- Bilkova E.; Pleskot R.; Rissanen S.; Sun S.; Czogalla A.; Cwiklik L.; ŔOg T.; Vattulainen I.; Cremer P. S.; Jungwirth P.; et al. Calcium Directly Regulates Phosphatidylinositol4,5-Bisphosphate Headgroup Conformation And Recognition. J. Am. Chem. Soc. 2017, 139, 4019–4024. 10.1021/jacs.6b11760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catte A.; Girych M.; Javanainen M.; Loison C.; Melcr J.; Miettinen M. S.; Monticelli L.; Maatta J.; Oganesyan V. S.; Ollila O. H. S.; et al. Molecular Electrometer And Binding Of Cations To Phospholipid Bilayers. Phys. Chem. Chem. Phys. 2016, 18, 32560–32569. 10.1039/C6CP04883H. [DOI] [PubMed] [Google Scholar]
- Kandt C.; Ash W. L.; Tieleman D. P. Setting Up And Running Molecular Dynamics Simulations Of Membrane Proteins. Methods (Amsterdam, Neth.) 2007, 41, 475–488. 10.1016/j.ymeth.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Wolf M. G.; Hoefling M.; Aponte-Santamaria C.; Grubmueller H.; Groenhof G. G_Membed: Efficient Insertion Of A Membrane Protein Into An Equilibrated Lipid Bilayer With Minimal Perturbation. J. Comput. Chem. 2010, 31, 2169–2174. 10.1002/jcc.21507. [DOI] [PubMed] [Google Scholar]
- Staritzbichler R.; Anselmi C.; Forrest L. R.; Faraldo-Gomez J. D. GRIFFIN: A Versatile Methodology For Optimization Of Protein-Lipid Interfaces For Membrane Protein Simulations. J. Chem. Theory Comput. 2011, 7, 1167–1176. 10.1021/ct100576m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt T. H.; Kandt C. LAMBADA And Inflategro2: Efficient Membrane Alignment And Insertion Of Membrane Proteins For Molecular Dynamics Simulations. J. Chem. Inf. Model. 2012, 52, 2657–2669. 10.1021/ci3000453. [DOI] [PubMed] [Google Scholar]
- Javanainen M. Universal Method For Embedding Proteins Into Complex Lipid Bilayers For Molecular Dynamics Simulations. J. Chem. Theory Comput. 2014, 10, 2577–2582. 10.1021/ct500046e. [DOI] [PubMed] [Google Scholar]
- Skjevik Å.A.; Madej B. D.; Dickson C. J.; Lin C.; Teigen K.; Walker R. C.; Gould I. R. Simulation Of Lipid Bilayer Self-Assembly Using All-Atom Lipid Force Fields. Phys. Chem. Chem. Phys. 2016, 18, 10573–84. 10.1039/C5CP07379K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.; Cheng X.; Swails J. M.; Yeom M. S.; et al. CHARMM-GUI Input Generator For NAMD, GROMACS, Amber, Openmm, And CHARMM/Openmm Simulations Using The CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. 10.1021/acs.jctc.5b00935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doerr S.; Giorgino T.; Martínez-Rosell G.; Damas J. M.; De Fabritiis G. High-Throughput Automated Preparation and Simulation of Membrane Proteins with HTMD. J. Chem. Theory Comput. 2017, 13, 4003–4011. 10.1021/acs.jctc.7b00480. [DOI] [PubMed] [Google Scholar]
- Wassenaar T. A.; Ingólfsson H. I.; Böckmann R. A.; Tieleman D. P.; Marrink S. J. Computational Lipidomics With Insane: A Versatile Tool For Generating Custom Membranes For Molecular Simulations. J. Chem. Theory Comput. 2015, 11, 2144–2155. 10.1021/acs.jctc.5b00209. [DOI] [PubMed] [Google Scholar]
- Wassenaar T. A.; Pluhackova K.; Böckmann R.; Marrink S. J.; Tieleman D. P. Going Backward: A Flexible Geometric Approach To Reverse Transformation From Coarse Grained To Atomistic Models. J. Chem. Theory Comput. 2014, 10, 676–690. 10.1021/ct400617g. [DOI] [PubMed] [Google Scholar]
- Pronk S.; Pall S.; Schulz R.; Larsson P.; Bjelkmar P.; Apostolov R.; Shirts M. R.; Smith J. C.; Kasson P. M.; Van Der Spoel D.; Hess B.; Lindahl E. GROMACS 4.5: A High-Throughput And Highly Parallel Open Source Molecular Simulation Toolkit. Bioinformatics 2013, 29, 845–854. 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkubo Y. Z.; Pogorelov T. V.; Arcario M. J.; Christensen G. A.; Tajkhorshid E. Accelerating Membrane Insertion Of Peripheral Proteins With A Novel Membrane Mimetic Model. Biophys. J. 2012, 102, 2130–2139. 10.1016/j.bpj.2012.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogorelov T. V.; Vermaas J. V.; Arcario M. J.; Tajkhorshid E. Partitioning Of Amino Acids Into A Model Membrane: Capturing The Interface. J. Phys. Chem. B 2014, 118, 1481–1492. 10.1021/jp4089113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baylon J. L.; Lenov I. L.; Sligar S. G.; Tajkhorshid E. Characterizing The Membrane-Bound State Of Cytochrome P450 3A4: Structure, Depth Of Insertion, And Orientation. J. Am. Chem. Soc. 2013, 135, 8542–8551. 10.1021/ja4003525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit B.; Hilbers P. A. J.; Esselink K.; Rupert L. A. M.; Van Os N. M.; Schlijper A. G. Computer Simulations of a Water/Oil Interface in The Presence of Micelles. Nature 1990, 348, 624–625. 10.1038/348624a0. [DOI] [Google Scholar]
- Goetz R.; Lipowsky R. Computer Simulations Of Bilayer Membranes: Self-Assembly And Interfacial Tension. J. Chem. Phys. 1998, 108, 7397. 10.1063/1.476160. [DOI] [Google Scholar]
- Bennun S. V.; Hoopes M. I.; Xing C.; Faller R. Coarse-Grained Modeling of Lipids. Chem. Phys. Lipids 2009, 159, 59–66. 10.1016/j.chemphyslip.2009.03.003. [DOI] [PubMed] [Google Scholar]
- Lyubartsev A. P.; Rabinovich A. L. Recent Development in Computer Simulations of Lipid Bilayers. Soft Matter 2011, 7, 25–39. 10.1039/C0SM00457J. [DOI] [Google Scholar]
- Cascella M.; Vanni S. Toward Accurate Coarse-Graining Approaches For Protein and Membrane Simulations. Chem. Modell. 2015, 12, 1–52. 10.1039/9781782622703-00001. [DOI] [Google Scholar]
- Bradley R.; Radhakrishnan R. Coarse-Grained Models For Protein-Cell Membrane Interactions. Polymers 2013, 5, 890–936. 10.3390/polym5030890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson M. E.; Head-Gordon T.; Louis A. A. Representability Problems For Coarse-Grained Water Potentials. J. Chem. Phys. 2007, 126, 144509–144519. 10.1063/1.2715953. [DOI] [PubMed] [Google Scholar]
- Wagner W.; Dama J. F.; Durumeric A. E. P.; Voth G. A. on The Representability Problem and The Physical Meaning of Coarse-Grained Models. J. Chem. Phys. 2016, 145, 044108. 10.1063/1.4959168. [DOI] [PubMed] [Google Scholar]
- Brini E.; Algaer E. A.; Ganguly P.; Li C. L.; Rodriguez-Ropero F.; Van Der Vegt N. F. A. Systematic Coarse-Graining Methods For Soft Matter Simulations - a Review. Soft Matter 2013, 9, 2108–2119. 10.1039/C2SM27201F. [DOI] [Google Scholar]
- Ingólfsson H. I.; López C. A.; Uusitalo J. J.; De Jong D. H.; Gopal S. M.; Periole X.; Marrink S. J. The Power of Coarse Graining in Biomolecular Simulations. Wires Comput. Mol. Sci. 2014, 4, 225–248. 10.1002/wcms.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noid W. G. Perspective: Coarse-Grained Models For Biomolecular Systems. J. Chem. Phys. 2013, 139, 090901. 10.1063/1.4818908. [DOI] [PubMed] [Google Scholar]
- Marrink S. J.; De Vries A. H.; Mark A. E. Coarse Grained Model For Semi-Quantitative Lipid Simulations. J. Phys. Chem. B 2004, 108, 750–760. 10.1021/jp036508g. [DOI] [Google Scholar]
- Marrink S. J.; Risselada H. J.; Yefimov S.; Tieleman D. P.; De Vries A. H. The MARTINI Forcefield: Coarse Grained Model For Biomolecular Simulations. J. Phys. Chem. B 2007, 111, 7812–7824. 10.1021/jp071097f. [DOI] [PubMed] [Google Scholar]
- Lopez C. A.; Sovova Z.; Van Eerden F. J.; De Vries A. H.; Marrink S. J. Martini Force Field Parameters For Glycolipids. J. Chem. Theory Comput. 2013, 9, 1694–1708. 10.1021/ct3009655. [DOI] [PubMed] [Google Scholar]
- Gu R. X.; Ingólfsson H. I.; De Vries A. H.; Marrink S. J.; Tieleman D. P. Ganglioside-Lipid and Ganglioside-Protein Interactions Revealed By Coarse-Grained and Atomistic Molecular Dynamics Simulations. J. Phys. Chem. B 2017, 121, 3262–3275. 10.1021/acs.jpcb.6b07142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H.; Pastor R. W. Coarse-Grained Model For Pegylated Lipids: Effect of Pegylation on The Size and Shape of Self-Assembled Structures. J. Phys. Chem. B 2011, 115, 7830–7837. 10.1021/jp2020148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunewald F.; Rossi G.; De Vries A. H.; Marrink S. J.; Monticelli L. a Transferable MARTINI Model of Polyethylene Oxide. J. Phys. Chem. B 2018, 122, 7436–7449. 10.1021/acs.jpcb.8b04760. [DOI] [PubMed] [Google Scholar]
- Dahlberg M. Polymorphic Phase Behavior of Cardiolipin Derivatives Studied By Coarse-Grained Molecular Dynamics. J. Phys. Chem. B 2007, 111, 7194–7200. 10.1021/jp071954f. [DOI] [PubMed] [Google Scholar]
- Dahlberg M.; Maliniak A. Mechanical Properties of Coarse-Grained Bilayers Formed By Cardiolipin and Zwitterionic Lipids. J. Chem. Theory Comput. 2010, 6, 1638–49. 10.1021/ct900654e. [DOI] [PubMed] [Google Scholar]
- Bulacu M.; Periole X.; Marrink S. J. In-Silico Design of Robust Bolalipid Membranes. Biomacromolecules 2012, 13, 196–205. 10.1021/bm201454j. [DOI] [PubMed] [Google Scholar]
- Ma H.; Cummins D. D.; Edelstein N. B.; Gomez J.; Khan A.; Llewellyn M. D.; Picudella T.; Willsey S. R.; Nangia S. Modeling Diversity in Structures of Bacterial Outer Membrane Lipids. J. Chem. Theory Comput. 2017, 13, 811–824. 10.1021/acs.jctc.6b00856. [DOI] [PubMed] [Google Scholar]
- Ma H.; Irudayanathan F. J.; Jiang W.; Nangia S. Simulating Gram-Negative Bacterial Outer Membrane: a Coarse Grain Model. J. Phys. Chem. B 2015, 119, 14668–14682. 10.1021/acs.jpcb.5b07122. [DOI] [PubMed] [Google Scholar]
- Van Oosten B.; Harroun T. a. MARTINI Extension For Pseudomonas Aeruginosa PAO1 Lipopolysaccharide. J. Mol. Graphics Modell. 2016, 63, 125–133. 10.1016/j.jmgm.2015.12.002. [DOI] [PubMed] [Google Scholar]
- Hsu P. C.; Jefferies D.; Khalid S. Molecular Dynamics Simulations Predict The Pathways Via Which Pristine Fullerenes Penetrate Bacterial Membranes. J. Phys. Chem. B 2016, 120, 11170–79. 10.1021/acs.jpcb.6b06615. [DOI] [PubMed] [Google Scholar]
- Melo M. N.; Ingólfsson H. I.; Marrink S. J. Parameters For Martini Sterols and Hopanoids Based On a Virtual-Site Description. J. Chem. Phys. 2015, 143, 243152. 10.1063/1.4937783. [DOI] [PubMed] [Google Scholar]
- Materials Science Suite 2018–2; Schrödinger, LLC: New York, NY, 2018. [Google Scholar]
- Monticelli L.; Kandasamy S. K.; Periole X.; Larson R. G.; Tieleman D. P.; Marrink S. J. The MARTINI Coarse-Grained Force Field: Extension To Proteins. J. Chem. Theory Comput. 2008, 4, 819–834. 10.1021/ct700324x. [DOI] [PubMed] [Google Scholar]
- De Jong D. H.; Singh G.; Bennett W. F. D.; Arnarez C.; Wassenaar T. A.; Schäfer L. V.; Periole X.; Tieleman D. P.; Marrink S. J. Improved Parameters For The Martini Coarse-Grained Protein Force Field. J. Chem. Theory Comput. 2013, 9, 687–697. 10.1021/ct300646g. [DOI] [PubMed] [Google Scholar]
- Loṕez C. A.; Rzepiela A. J.; De Vries A. H.; Dijkhuizen L.; Hünenberger P. H.; Marrink S. J. Martini Coarse-Grained Force Field: Extension To Carbohydrates. J. Chem. Theory Comput. 2009, 5, 3195–3210. 10.1021/ct900313w. [DOI] [PubMed] [Google Scholar]
- Uusitalo J. J.; Ingólfsson H. I.; Akhshi P.; Tieleman D. P.; Marrink S. J. Martini Coarse-Grained Force Field: Extension To DNA. J. Chem. Theory Comput. 2015, 11, 3932–3945. 10.1021/acs.jctc.5b00286. [DOI] [PubMed] [Google Scholar]
- Uusitalo J. J.; Ingólfsson H. I.; Marrink S. J.; Faustino I. Martini Coarse-Grained Force Field: Extension To RNA. Biophys. J. 2017, 113, 246–256. 10.1016/j.bpj.2017.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H.; De Vries A. H.; Marrink S. J.; Pastor R. W. a Coarse- Grained Model For Polyethylene Oxide and Polyethylene Glycol: Conformation and Hydrodynamics. J. Phys. Chem. B 2009, 113, 13186–13194. 10.1021/jp9058966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong-Ekkabut J.; Baoukina S.; Triampo W.; Tang I. M.; Tieleman D. P.; Monticelli L. Computer Simulation Study of Fullerene Translocation Through Lipid Membranes. Nat. Nanotechnol. 2008, 3, 363. 10.1038/nnano.2008.130. [DOI] [PubMed] [Google Scholar]
- Xue M.; Cheng L.; Faustino I.; Guo W.; Marrink S. J. Molecular Mechanism of Lipid Nanodisk Formation by Styrene-Maleic Acid Copolymers. Biophys. J. 2018, 115, 494–502. 10.1016/j.bpj.2018.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salassi S.; Simonelli F.; Bochicchio D.; Ferrando R.; Rossi G. Au Nanoparticles in Lipid Bilayers: a Comparison between Atomistic and Coarse-Grained Models. J. Phys. Chem. C 2017, 121, 10927–10935. 10.1021/acs.jpcc.6b12148. [DOI] [Google Scholar]
- Yesylevskyy S. O.; Schäfer L. V.; Sengupta D.; Marrink S. J. Polarizable Water Model For The Coarse-Grained Martini Force Field. PLoS Comput. Biol. 2010, 6, E1000810. 10.1371/journal.pcbi.1000810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalowsky J.; Schaefer L. V.; Holm C.; Smiatek J. a Refined Polarizable Water Model For The Coarse-Grained MARTINI Force Field with Long-Range Electrostatic Interactions. J. Chem. Phys. 2017, 146, 054501. 10.1063/1.4974833. [DOI] [PubMed] [Google Scholar]
- Michalowsky J.; Zeman J.; Holm C.; Smiatek J. a Polarizable MARTINI Model For Monovalent Ions in Aqueous Solution. J. Chem. Phys. 2018, 149, 163319. 10.1063/1.5028354. [DOI] [PubMed] [Google Scholar]
- Periole X.; Cavalli M.; Marrink S. J.; Ceruso M. Combining An Elastic Network with a Coarse-Grained Molecular Force Field: Structure, Dynamics and Intermolecular Recognition. J. Chem. Theory Comput. 2009, 5, 2531–2543. 10.1021/ct9002114. [DOI] [PubMed] [Google Scholar]
- Poma A. B.; Cieplak M.; Theodorakis P. E. Combining The MARTINI and Structure-Based Coarse-Grained Approaches For The Molecular Dynamics Studies of Conformational Transitions in Proteins. J. Chem. Theory Comput. 2017, 13, 1366–1374. 10.1021/acs.jctc.6b00986. [DOI] [PubMed] [Google Scholar]
- Javanainen M.; Martinez-Seara H.; Vattulainen I. Excessive Aggregation of Membrane Proteins in The Martini Model. PLoS One 2017, 12, e0187936. 10.1371/journal.pone.0187936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmalhorst P. S.; Deluweit F.; Scherrers R.; Heisenberg C. P.; Sikora M. Overcoming The Limitations of The MARTINI Force Field in Simulations of Polysaccharides. J. Chem. Theory Comput. 2017, 13, 5039–5053. 10.1021/acs.jctc.7b00374. [DOI] [PubMed] [Google Scholar]
- Stark A. C.; Andrews C. T.; Elcock A. H. Toward Optimized Potential Functions For Protein–Protein Interactions in Aqueous Solutions: Osmotic Second Virial Coefficient Calculations Using The Martini Coarse-Grained Force Field. J. Chem. Theory Comput. 2013, 9, 4176–4185. 10.1021/ct400008p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrink S. J.; Tieleman D. P. Perspective on The Martini Model. Chem. Soc. Rev. 2013, 42, 6801–6822. 10.1039/c3cs60093a. [DOI] [PubMed] [Google Scholar]
- Shinoda W.; Devane R.; Klein M. L. Zwitterionic Lipid Assemblies: Molecular Dynamics Studies of Monolayers, Bilayers, and Vesicles Using a New Coarse Grain Force Field. J. Phys. Chem. B 2010, 114, 6836–6849. 10.1021/jp9107206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinoda W.; Devane R.; Klein M. L. Computer Simulation Studies of Self-Assembling Macromolecules. Curr. Opin. Struct. Biol. 2012, 22, 175–186. 10.1016/j.sbi.2012.01.011. [DOI] [PubMed] [Google Scholar]
- Bacle A.; Gautier R.; Jackson C. L.; Fuchs P. F. J.; Vanni S. Interdigitation Between Triglycerides and Lipids Modulates Surface Properties of Lipid Droplets. Biophys. J. 2017, 112, 1417–1430. 10.1016/j.bpj.2017.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo S.; Shinoda W. SPICA Force Field for Lipid Membranes: Domain Formation Induced by Cholesterol. J. Chem. Theory Comput. 2018, 10.1021/acs.jctc.8b00987. [DOI] [PubMed] [Google Scholar]
- Orsi M.; Essex J. W. The ELBA Force Field For Coarse-Grain Modeling of Lipid Membranes. PLoS One 2011, 6, e28637. 10.1371/journal.pone.0028637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siani P.; Khandelia H.; Orsi M.; Dias L. G. Parameterization of a Coarse-Grained Model of Cholesterol with Point-Dipole Electrostatics. J. Comput.-Aided Mol. Des. 2018, 32, 1259. 10.1007/s10822-018-0164-4. [DOI] [PubMed] [Google Scholar]
- Darre L.; Machado M. R.; Brandner A. F.; Gonzalez H. C.; Ferreira S.; Pantano S. SIRAH: a Structurally Unbiased Coarse-Grained Force Field For Proteins with Aqueous Solvation and Long-Range Electrostatics. J. Chem. Theory Comput. 2015, 11, 723–739. 10.1021/ct5007746. [DOI] [PubMed] [Google Scholar]
- Brandner A.; Schüller A.; Melo F.; Pantano S. Exploring DNA Dynamics Within Oligonucleosomes with Coarse-Grained Simulations: SIRAH Force Field Extension For Protein-DNA Complexes. Biochem. Biophys. Res. Commun. 2018, 498, 319–326. 10.1016/j.bbrc.2017.09.086. [DOI] [PubMed] [Google Scholar]
- Barrera E. E.; Frigini E. N.; Porasso R. D.; Pantano S. Modeling DMPC Lipid Membranes with SIRAH Force-Field. J. Mol. Model. 2017, 23, 259. 10.1007/s00894-017-3426-5. [DOI] [PubMed] [Google Scholar]
- Izvekov S.; Voth G. A. Solvent-Free Lipid Bilayer Model Using Multiscale Coarse- Graining. J. Phys. Chem. B 2009, 113, 4443–4455. 10.1021/jp810440c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L.; Voth G. A. Systematic Coarse-Graining of a Multicomponent Lipid Bilayer. J. Phys. Chem. B 2009, 113, 1501–1510. 10.1021/jp809604k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hills R. D. Jr.; Lu L.; Voth G. A. Multiscale Coarse-Graining of The Protein Energy Landscape. PLoS Comput. Biol. 2010, 6, e1000827. 10.1371/journal.pcbi.1000827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hills R. D. Jr.; Mcglinchey N. Model Parameters For Simulation of Physiological Lipids. J. Comput. Chem. 2016, 37, 1112–1118. 10.1002/jcc.24324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyubartsev A. P.; Mirzoev A.; Chen L.; Laaksonen A. Systematic Coarse- Graining of Molecular Models By The Newton Inversion Method. Faraday Discuss. 2010, 144, 43–56. 10.1039/B901511F. [DOI] [PubMed] [Google Scholar]
- Wang Z. J.; Deserno M. a Systematically Coarse-Grained Solvent-Free Model For Quantitative Phospholipid Bilayer Simulations. J. Phys. Chem. B 2010, 114, 11207–11220. 10.1021/jp102543j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodt A. J.; Head-Gordon T. An Implicit Solvent Coarse-Grained Lipid Model with Correct Stress Profile. J. Chem. Phys. 2010, 132, 205103. 10.1063/1.3408285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bereau T.; Wang Z. J.; Deserno M. More Than The Sum of Its Parts: Coarse-Grained Peptide-Lipid Interactions From a Simple Cross-Parametrization. J. Chem. Phys. 2014, 140, 115101. 10.1063/1.4867465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bereau T.; Bennett W. F. D.; Pfaendtner J.; Deserno M.; Karttunen M. Folding and Insertion Thermodynamics of The Transmembrane WALP Peptide. J. Chem. Phys. 2015, 143, 243127. 10.1063/1.4935487. [DOI] [PubMed] [Google Scholar]
- Curtis E. M.; Hall C. K. Molecular Dynamics Simulations of DPPC Bilayers Using ″LIME″, a New Coarse-Grained Model. J. Phys. Chem. B 2013, 117, 5019–5030. 10.1021/jp309712b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnarez C.; Uusitalo J. J.; Masman M. F.; Ingólfsson H. I.; De Jong D. H.; Melo M. N.; Periole X.; De Vries A. H.; Marrink S. J. Dry Martini, a Coarse-Grained Force Field For Lipid Membrane Simulations with Implicit Solvent. J. Chem. Theory Comput. 2015, 11, 260–275. 10.1021/ct500477k. [DOI] [PubMed] [Google Scholar]
- Zgorski A.; Lyman E. Toward Hydrodynamics with Solvent Free Lipid Models: STRD Martini. Biophys. J. 2016, 111, 2689–2697. 10.1016/j.bpj.2016.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan M.; Gao L.; Fang W. Implicit-Solvent Dissipative Particle Dynamics Force Field Based On a Four-To-One Coarse-Grained Mapping Scheme. PLoS One 2018, 13, e0198049. 10.1371/journal.pone.0198049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar P.; Gopal S. M.; Cheng Y.-M.; Panahi A.; Feig M. Transferring The PRIMO Coarse-Grained Force Field To The Membrane Environment: Simulation of Proteins and Helix-Helix Association. J. Chem. Theory Comput. 2014, 10, 3459–3472. 10.1021/ct500443v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassenaar T. A.; Ingólfsson H. I.; Prieß M.; Marrink S. J.; Schäfer L. V. Mixing Martini: Electrostatic Coupling in Hybrid Atomistic – Coarse-Grained Biomolecular Simulations. J. Phys. Chem. B 2013, 117, 3516. 10.1021/jp311533p. [DOI] [PubMed] [Google Scholar]
- Han W.; Schulten K. Further Optimization of a Hybrid United-Atom and Coarse-Grained Force Field For Folding Simulations: Improved Backbone Hydration and Interactions Between Charged Side Chains. J. Chem. Theory Comput. 2012, 8, 4413. 10.1021/ct300696c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsi M.; Sanderson W. E.; Essex J. W. Permeability of Small Molecules Through a Lipid Bilayer: a Multiscale Simulation Study. J. Phys. Chem. B 2009, 113, 12019–12029. 10.1021/jp903248s. [DOI] [PubMed] [Google Scholar]
- Machado M. R.; Pantano S. Exploring Laci–DNA Dynamics By Multiscale Simulations Using The SIRAH Force Field. J. Chem. Theory Comput. 2015, 11, 5012–5023. 10.1021/acs.jctc.5b00575. [DOI] [PubMed] [Google Scholar]
- Kar P.; Feig M. Hybrid All-Atom/Coarse-Grained Simulations of Proteins By Direct Coupling of CHARMM and PRIMO Force Fields. J. Chem. Theory Comput. 2017, 13, 5753–5765. 10.1021/acs.jctc.7b00840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih A. Y.; Freddolino P. L.; Sligar S. G.; Schulten K. Disassembly of Nanodiscs with Cholate. Nano Lett. 2007, 7, 1692–1696. 10.1021/nl0706906. [DOI] [PubMed] [Google Scholar]
- Stansfeld P. J.; Sansom M. S. P. Insertion and Assembly of Membrane Proteins Via Simulation. J. Chem. Theory Comput. 2011, 7, 1157–1166. 10.1021/ct100569y. [DOI] [PubMed] [Google Scholar]
- Rzepiela A. J.; Schafer L. V.; Goga N.; Risselada H. J.; De Vries A. H.; Marrink S. J. Reconstruction of Atomistic Details From Coarse-Grained Structures. J. Comput. Chem. 2010, 31, 1333–1343. 10.1002/jcc.21415. [DOI] [PubMed] [Google Scholar]
- Brocos P.; Mendoza-Espinosa P.; Castillo R.; Mas-Oliva J.; PiñEiro A. Multiscale Molecular Dynamics Simulations of Micelles: Coarse-Grain For Self-Assembly and Atomic Resolution For Finer Details. Soft Matter 2012, 8, 9005–9014. 10.1039/c2sm25877c. [DOI] [Google Scholar]
- Machado M. R.; Pantano S. SIRAH Tools: Mapping, Backmapping and Visualization of Coarse-Grained Models. Bioinformatics 2016, 32, 1568–1570. 10.1093/bioinformatics/btw020. [DOI] [PubMed] [Google Scholar]
- Otero-Mato J. M.; Montes-Campos H.; Calvelo M.; García-Fandiño R.; Gallego L. J.; Piñeiro A.; Varela L. M. GADDLE Maps: General Algorithm For Discrete Object Deformations Based on Local Exchange Maps. J. Chem. Theory Comput. 2018, 14, 466–478. 10.1021/acs.jctc.7b00861. [DOI] [PubMed] [Google Scholar]
- Milano G.; Kawakatsu T. Hybrid Particle-Field Molecular Dynamics Simulations for Dense Polymer Systems. J. Chem. Phys. 2009, 130, 214106. 10.1063/1.3142103. [DOI] [PubMed] [Google Scholar]
- De Nicola A.; Zhao Y.; Kawakatsu T.; Roccatano D.; Milano G. Hybrid Particle-Field Coarse-Grained Models for Biological Phospholipids. J. Chem. Theory Comput. 2011, 7, 2947–2962. 10.1021/ct200132n. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; De Nicola A.; Kawakatsu T.; Milano G. Hybrid Particle-Field Molecular Dynamics Simulations: Parallelization and Benchmarks. J. Comput. Chem. 2012, 33, 868–880. 10.1002/jcc.22883. [DOI] [PubMed] [Google Scholar]
- Milano G.; Kawakatsu T.; De Nicola A. A Hybrid Particle–Field Molecular Dynamics Approach: A Route Toward Efficient Coarse-Grained Models for Biomembranes. Phys. Biol. 2013, 10, 045007. 10.1088/1478-3975/10/4/045007. [DOI] [PubMed] [Google Scholar]
- De Nicola A.; Zhao Y.; Kawakatsu T.; Roccatano D.; Milano G. Validation of a Hybrid MD-SCF Coarse-Grained Model for DPPC in Non-Lamellar Phases. Theor. Chem. Acc. 2012, 131, 11–16. 10.1007/s00214-012-1167-1. [DOI] [Google Scholar]
- Bore S. L.; Milano G.; Cascella M. Hybrid Particle-Field Model for Conformational Dynamics of Peptide Chains. J. Chem. Theory Comput. 2018, 14, 1120–1130. 10.1021/acs.jctc.7b01160. [DOI] [PubMed] [Google Scholar]
- Scott K. A.; Bond P. J.; Ivetac A.; Chetwynd A. P.; Khalid S.; Sansom M. S. P. Coarse-Grained MD Simulations of Membrane Protein-Bilayer Self-Assembly. Structure 2008, 16, 621–630. 10.1016/j.str.2008.01.014. [DOI] [PubMed] [Google Scholar]
- Stansfeld P. J.; Goose J. E.; Caffrey M.; Carpenter E. P.; Parker J. L.; Newstead N.; Sansom M. S. P. Memprotmd: Automated Insertion of Membrane Protein Structures Into Explicit Lipid Membranes. Structure 2015, 23, 1350–1361. 10.1016/j.str.2015.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Y.; Ingólfsson H. I.; Cheng X.; Lee J.; Marrink S. J.; Im W. CHARMM-GUI Martini Maker For Coarse-Grained Simulations with The Martini Force Field. J. Chem. Theory Comput. 2015, 11, 4486–4494. 10.1021/acs.jctc.5b00513. [DOI] [PubMed] [Google Scholar]
- Hsu P. C.; Bruininks B. M. H.; Jefferies D.; Telles De Souza P. C.; Lee J.; Patel D. S.; Marrink S. J.; Qi Y.; Khalid S.; Im W. CHARMM-GUI Martini Maker For Modeling and Simulation of Complex Bacterial Membranes with Lipopolysaccharides. J. Comput. Chem. 2017, 38, 2354–2363. 10.1002/jcc.24895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall B. A.; Halim K. B. A.; Buyan A.; Emmanouil B.; Sansom M. S. P. Sidekick For Membrane Simulations: Automated Ensemble Molecular Dynamics Simulations of Transmembrane Helices. J. Chem. Theory Comput. 2014, 10, 2165–2175. 10.1021/ct500003g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassenaar T. A.; Pluhackova K.; Moussatova A.; Sengupta D.; Marrink S. J.; Tieleman D. P.; Böckmann R. A. High-Throughput Simulations of Dimer and Trimer Assembly of Membrane Proteins. The DAFT Approach. J. Chem. Theory Comput. 2015, 11, 2278–2291. 10.1021/ct5010092. [DOI] [PubMed] [Google Scholar]
- Sinitskiy A. V.; Saunders M. G.; Voth G. A. Optimal Number of Coarse-Grained Sites in Different Components of Large Biomolecular Complexes. J. Phys. Chem. B 2012, 116, 8363–8374. 10.1021/jp2108895. [DOI] [PubMed] [Google Scholar]
- Bereau T.; Kremer K. Automated Parametrization of The Coarse-Grained Martini Force Field For Small Organic Molecules. J. Chem. Theory Comput. 2015, 11, 2783–2791. 10.1021/acs.jctc.5b00056. [DOI] [PubMed] [Google Scholar]
- Graham J. A.; Essex J. W.; Khalid S. Pycgtool: Automated Generation of Coarse-Grained Molecular Dynamics Models From Atomistic Trajectories. J. Chem. Inf. Model. 2017, 57, 650–656. 10.1021/acs.jcim.7b00096. [DOI] [PubMed] [Google Scholar]
- Mirzoev A.; Nordenskiöld L.; Lyubartsev A. Magic V.3: An Integrated Software Package For Systematic Structure-Based Coarse-Graining. Comput. Phys. Commun. 2018, 10.1016/j.cpc.2018.11.018. [DOI] [Google Scholar]
- Menichetti R.; Kanekal K. H.; Kremer K.; Bereau T. in Silico Screening of Drug-Membrane Thermodynamics Reveals Linear Relations Between Bulk Partitioning and The Potential of Mean Force. J. Chem. Phys. 2017, 147, 125101. 10.1063/1.4987012. [DOI] [PubMed] [Google Scholar]
- Miller S. E.; Mathiasen S.; Bright N. A.; Pierre F.; Kelly B. T.; Kladt N.; Schauss A.; Merrifield C. J.; Stamou D.; Honing S.; Owen D. J. CALM Regulates Clathrin-Coated Vesicle Size and Maturation By Directly Sensing and Driving Membrane Curvature. Dev. Cell 2015, 33, 163–175. 10.1016/j.devcel.2015.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishnan N.; Kumar P. B. S.; Radhakrishnan R. Mesoscale Computational Studies of Membrane Bilayer Remodeling By Curvature-Inducing Proteins. Phys. Rep. 2014, 543, 1–60. 10.1016/j.physrep.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishnan N.; Bradley R. P.; Tourdot R. W.; Radhakrishnan R. Biophysics of Membrane Curvature Remodeling At Molecular and Mesoscopic Lengthscales. J. Phys.: Condens. Matter 2018, 30, 273001. 10.1088/1361-648X/aac702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayton G. S.; Voth G. A. Hybrid Coarse-Graining Approach For Lipid Bilayers At Large Length and Time Scales. J. Phys. Chem. B 2009, 113, 4413–4424. 10.1021/jp8087868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z. Y.; Lu L. Y.; Noid W. G.; Krishna V.; Pfaendtner J.; Voth G. A. a Systematic Methodology For Defining Coarse-Grained Sites in Large Biomolecules. Biophys. J. 2008, 95, 5073–5083. 10.1529/biophysj.108.139626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z. Y.; Pfaendtner J.; Grafmuller A.; Voth G. A. Defining Coarse-Grained Representations of Large Biomolecules and Biomolecular Complexes From Elastic Network Models. Biophys. J. 2009, 97, 2327–2337. 10.1016/j.bpj.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venturoli M.; Sperotto M. M.; Kranenburg M.; Smit B. Mesoscopic Models of Biological Membranes. Phys. Rep. 2006, 437, 1–54. 10.1016/j.physrep.2006.07.006. [DOI] [Google Scholar]
- Dama J. F.; Sinitskiy A. V.; Mccullagh M.; Weare J.; Roux B.; Dinner A. R.; Voth G. A. The Theory of Ultra-Coarse-Graining. 1. General Principles. J. Chem. Theory Comput. 2013, 9, 2466–2480. 10.1021/ct4000444. [DOI] [PubMed] [Google Scholar]
- Davtyan A.; Dama J. F.; Sinitskiy A. V.; Voth G. A. The Theory of Ultra-Coarse-Graining. 2. Numerical Implementation. J. Chem. Theory Comput. 2014, 10, 5265–5275. 10.1021/ct500834t. [DOI] [PubMed] [Google Scholar]
- Brannigan G.; Brown F. L. H. Solvent-Free Simulations Of Fluid Membrane Bilayers. J. Chem. Phys. 2004, 120, 1059–1071. 10.1063/1.1625913. [DOI] [PubMed] [Google Scholar]
- Cooke I. R.; Kremer K.; Deserno M. Tunable Generic Model For Fluid Bilayer Membranes. Phys. Rev. E 2005, 72, 011506. 10.1103/PhysRevE.72.011506. [DOI] [PubMed] [Google Scholar]
- Srivastava A.; Voth G. A. Solvent-Free, Highly Coarse-Grained Models For Charged Lipid Systems. J. Chem. Theory Comput. 2014, 10, 4730–4744. 10.1021/ct500474a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Meyer F.; Smit B. Effect Of Cholesterol On The Structure Of A Phospholipid Bilayer. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 3654–3658. 10.1073/pnas.0809959106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grafmuller A.; Shillcock J.; Lipowsky R. The Fusion Of Membranes And Vesicles: Pathway And Energy Barriers From Dissipative Particle Dynamics. Biophys. J. 2009, 96, 2658–2675. 10.1016/j.bpj.2008.11.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laradji M.; Kumar P. B. S.; Spangler E. J. Exploring Large-Scale Phenomena In Composite Membranes Through An Efficient Implicit-Solvent Model. J. Phys. D: Appl. Phys. 2016, 49, 293001. 10.1088/0022-3727/49/29/293001. [DOI] [Google Scholar]
- De Meyer F. J. M.; Venturoli M.; Smit B. Molecular Simulations Of Lipid-Mediated Protein-Protein Interactions. Biophys. J. 2008, 95, 1851–1865. 10.1529/biophysj.107.124164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Meyer F. J. M.; Rodgers J. M.; Willems T. F.; Smit B. Molecular Simulation Of The Effect Of Cholesterol On Lipid-Mediated Protein-Protein Interactions. Biophys. J. 2010, 99, 3629–3638. 10.1016/j.bpj.2010.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yiannourakou M.; Marsella L.; De Meyer F.; Smit B. Towards An Understanding Of Membrane-Mediated Protein-Protein Interactions. Faraday Discuss. 2010, 144, 359–367. 10.1039/B902190F. [DOI] [PubMed] [Google Scholar]
- Guigas G.; Weiss M. Membrane Protein Mobility Depends On The Length Of Extra-Membrane Domains And On The Protein Concentration. Soft Matter 2015, 11, 33–37. 10.1039/C4SM01846J. [DOI] [PubMed] [Google Scholar]
- Schmidt U.; Guigas G.; Weiss M.. Cluster Formation Of Transmembrane Proteins Due To Hydrophobic Mismatching. Phys. Rev. Lett. 2008, 101, 10.1103/PhysRevLett.101.128104. [DOI] [PubMed] [Google Scholar]
- Schmidt U.; Weiss M. Anomalous Diffusion Of Oligomerized Transmembrane Proteins. J. Chem. Phys. 2011, 134, 165101. 10.1063/1.3582336. [DOI] [PubMed] [Google Scholar]
- Reynwar B. J.; Illya G.; Harmandaris V. A.; Müller M. M.; Kremer K.; Deserno M. Aggregation And Vesiculation Of Membrane Proteins By Curvature-Mediated Interactions. Nature 2007, 447, 461–464. 10.1038/nature05840. [DOI] [PubMed] [Google Scholar]
- Davtyan A.; Simunovic M.; Voth G. A. Multiscale Simulations Of Protein-Facilitated Membrane Remodeling. J. Struct. Biol. 2016, 196, 57–63. 10.1016/j.jsb.2016.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C. K.; Pao C. W.; Smit B. PSII-LHCII Supercomplex Organizations In Photosynthetic Membrane By Coarse-Grained Simulation. J. Phys. Chem. B 2015, 119, 3999–4008. 10.1021/jp511277c. [DOI] [PubMed] [Google Scholar]
- Yiannourakou M.; Marsella L.; De Meyer F.; Smit B. Clustering Of Proteins Embedded In Lipid Bilayers: A Monte Carlo Study. Computation In Modern Science And Engineering 2007, 2, 448–451. [Google Scholar]
- Chavent M.; Duncan A. L.; Rassam P.; Birkholz O.; Hélie J.; Reddy T.; Beliaev D.; Hambly B.; Piehler J.; Kleanthous C.; Sansom M. S. P. How Nanoscale Protein Interactions Determine The Mesoscale Dynamic Organisation Of Bacterial Outer Membrane Proteins. Nat. Commun. 2018, 9, 2846. 10.1038/s41467-018-05255-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Jumper J. M.; Wang S.; Freed K. F.; Sosnick T. R. A Membrane Burial Potential With H-Bonds And Applications To Curved Membranes And Fast Simulations. Biophys. J. 2018, 115, 1872–1884. 10.1016/j.bpj.2018.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J. L.; Weikl T. R.; Lipowsky R. Vesicles With Multiple Membrane Domains. Soft Matter 2011, 7, 6092–6102. 10.1039/c0sm01500h. [DOI] [Google Scholar]
- Nepal B.; Leveritt J. III; Lazaridis T. Membrane Curvature Sensing By Amphipathic Helices: Insights From Implicit Membrane Modeling. Biophys. J. 2018, 114, 2128–2141. 10.1016/j.bpj.2018.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davtyan A.; Simunovic M.; Voth G. A. The Mesoscopic Membrane With Proteins (Mesm-P) Model. J. Chem. Phys. 2017, 147, 044101. 10.1063/1.4993514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Z. L.; Li X. J.; Pivkin I. V.; Dao M.; Karniadakis G. E.; Suresh S. Lipid Bilayer And Cytoskeletal Interactions In A Red Blood Cell. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 13356–13361. 10.1073/pnas.1311827110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Huang C. J.; Kim S.; Golkaram M.; Dixon M. W. A.; Tilley L.; Li J.; Zhang S. L.; Suresh S. Multiple Stiffening Effects Of Nanoscale Knobs On Human Red Blood Cells Infected With Plasmodium Falciparum Malaria Parasite. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 6068–6073. 10.1073/pnas.1505584112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezeshkian W.; Hansen A. G.; Johannes L.; Khandelia H.; Shillcock J. C.; Kumar P. B. S.; Ipsen J. H. Membrane Invagination Induced By Shiga Toxin B-Subunit: From Molecular Structure To Tube Formation. Soft Matter 2016, 12, 5164–5171. 10.1039/C6SM00464D. [DOI] [PubMed] [Google Scholar]
- Mauer J.; Mendez S.; Lanotte L.; Nicoud F.; Abkarian M.; Gompper G.; Fedosov D. A. Flow-Induced Transitions of Red Blood Cell Shapes under Shear. Phys. Rev. Lett. 2018, 121, 118103. 10.1103/PhysRevLett.121.118103. [DOI] [PubMed] [Google Scholar]
- Oliver R. C.; Read D. J.; Harlen O. G.; Harris S. A. A Stochastic Finite Element Model For The Dynamics Of Globular Macromolecules. J. Comput. Phys. 2013, 239, 147–165. 10.1016/j.jcp.2012.12.027. [DOI] [Google Scholar]
- Sadeghi M.; Weikl T. R.; Noé F. Particle-Based Membrane Model For Mesoscopic Simulation Of Cellular Dynamics. J. Chem. Phys. 2018, 148, 044901. 10.1063/1.5009107. [DOI] [PubMed] [Google Scholar]
- Kovalenko I. B.; Knyazeva O. S.; Antal T. K.; Ponomarev V. Y.; Riznichenko G. Y.; Rubin A. B. Multiparticle Brownian Dynamics Simulation Of Experimental Kinetics Of Cytochrome Bf Oxidation And Photosystem I Reduction By Plastocyanin. Physiol. Plant. 2017, 161, 88–96. 10.1111/ppl.12570. [DOI] [PubMed] [Google Scholar]
- Sezgin E.; Levental I.; Mayor S.; Eggeling C. The Mystery of Membrane Organization: Composition, Regulation and Roles of Lipid Rafts. Nat. Rev. Mol. Cell Biol. 2017, 18, 361. 10.1038/nrm.2017.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K.; Sampaio J. L. Membrane Organization and Lipid Rafts. Cold Spring Harbor Perspect. Biol. 2011, 3, a004697. 10.1101/cshperspect.a004697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veatch S. L.; Cicuta P.; Sengupta P.; Honerkamp-Smith A.; Holowka D.; Baird B. Critical Fluctuations in Plasma Membrane Vesicles. ACS Chem. Biol. 2008, 3, 287–293. 10.1021/cb800012x. [DOI] [PubMed] [Google Scholar]
- Levental I.; Grzybek M.; Simons K. Raft Domains of Variable Properties and Compositions in Plasma Membrane Vesicles. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 11411–11416. 10.1073/pnas.1105996108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlomovitz R.; Maibaum L.; Schick M. Macroscopic Phase Separation, Modulated Phases, and Microemulsions: a Unified Picture of Rafts. Biophys. J. 2014, 106, 1979–1985. 10.1016/j.bpj.2014.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honerkamp-Smith A. R.; Cicuta P.; Collins M. D.; et al. Line Tensions, Correlation Lengths, and Critical Exponents in Lipid Membranes Near Critical Points. Biophys. J. 2008, 95, 236–246. 10.1529/biophysj.107.128421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Destainville N.; Manghi M.; Cornet J. A Rationale For Mesoscopic Domain Formation in Biomembranes. Biomolecules 2018, 8, 104. 10.3390/biom8040104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javanainen M.; Martinez-Seara H.; Vattulainen I. Nanoscale Membrane Domain Formation Driven By Cholesterol. Sci. Rep. 2017, 7, 1143. 10.1038/s41598-017-01247-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodt A. J.; Sandar M. L.; Gawrisch K.; Pastor R. W.; Lyman E. The Molecular Structure of The Liquid-Ordered Phase of Lipid Bilayers. J. Am. Chem. Soc. 2014, 136, 725–732. 10.1021/ja4105667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodt A. J.; Pastor R. W.; Lyman E. Hexagonal Substructure and Hydrogen Bonding in Liquid-Ordered Phases Containing Palmitoyl Sphingomyelin. Biophys. J. 2015, 109, 948–955. 10.1016/j.bpj.2015.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakobyan D.; Heuer A. Phase Separation in a Lipid/Cholesterol System: Comparison of Coarse-Grained and United-Atom Simulations. J. Phys. Chem. B 2013, 117, 3841. 10.1021/jp312245y. [DOI] [PubMed] [Google Scholar]
- Bennett W. F. D.; Shea J. E.; Tieleman D. P. Phospholipid Chain Interactions with Cholesterol Drive Domain Formation in Lipid Membranes. Biophys. J. 2018, 114, 2595–2605. 10.1016/j.bpj.2018.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlmutter J. D.; Sachs J. N. Inhibiting Lateral Domain Formation in Lipid Bilayers: Simulations of Alternative Steroid Headgroup Chemistries. J. Am. Chem. Soc. 2009, 131, 16362. 10.1021/ja9079258. [DOI] [PubMed] [Google Scholar]
- Marrink S. J.; de Vries A. H.; Harroun T. A.; Katsaras J.; Wassall S. R. Cholesterol Shows Preference For The Interior of Polyunsaturated Lipid Membranes. J. Am. Chem. Soc. 2008, 130, 10. 10.1021/ja076641c. [DOI] [PubMed] [Google Scholar]
- Rosetti C.; Pastorino C. Comparison of Ternary Bilayer Mixtures with Asymmetric Or Symmetric Unsaturated Phosphatidylcholine Lipids By Coarse Grained Molecular Dynamics Simulations. J. Phys. Chem. B 2012, 116, 3525. 10.1021/jp212406u. [DOI] [PubMed] [Google Scholar]
- Kucerka N.; Marquardt D.; Harroun T. A.; Nieh M. P.; Wassall S. R.; De Jong D. H.; Schäfer L. V.; Marrink S. J.; Katsaras J. Cholesterol in Bilayers with PUFA Chains: Doping with DMPC Or POPC Results in Sterol Reorientation and Membrane-Domain Formation. Biochemistry 2010, 49, 7485. 10.1021/bi100891z. [DOI] [PubMed] [Google Scholar]
- Rosetti C.; Pastorino C. Polyunsaturated and Saturated Phospholipids in Mixed Bilayers: a Study From The Molecular Scale To The Lateral Lipid Organization. J. Phys. Chem. B 2011, 115, 1002. 10.1021/jp1082888. [DOI] [PubMed] [Google Scholar]
- Davis R. S.; Kumar P. B. S.; Sperotto M. M.; Laradji M. Predictions of Phase Separation in Three-Component Lipid Membranes By The MARTINI Force Field. J. Phys. Chem. B 2013, 117, 4072. 10.1021/jp4000686. [DOI] [PubMed] [Google Scholar]
- Baoukina S.; Mendez-Villuendas E.; Bennett W. F. D.; Tieleman D. P. Computer Simulations of The Phase Separation in Model Membranes. Faraday Discuss. 2013, 161, 63. 10.1039/C2FD20117H. [DOI] [PubMed] [Google Scholar]
- Lin X.; Lorent J. H.; Skinkle A. D.; Levental K. R.; Waxham M. N.; Gorfe A. A.; Levental A. Domain Stability in Biomimetic Membranes Driven By Lipid Polyunsaturation. J. Phys. Chem. B 2016, 120, 11930–11941. 10.1021/acs.jpcb.6b06815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäfer L. V.; Marrink S. J. Partitioning of Lipids At Domain Boundaries in Model Membranes. Biophys. J. 2010, 99, L91. 10.1016/j.bpj.2010.08.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baoukina S.; Rozmanov D.; Tieleman D. P. Composition Fluctuations in Lipid Bilayers. Biophys. J. 2017, 113, 2750–2761. 10.1016/j.bpj.2017.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López C. A.; Unkefer C. J.; Swanson B. I.; Swanson J. M. J.; Gnanakaran S. Membrane Perturbing Properties of Toxin Mycolactone From Mycobacterium Ulcerans. PLoS Comput. Biol. 2018, 14, e1005972. 10.1371/journal.pcbi.1005972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosetti C. M.; Montich G. G.; Pastorino C. Molecular Insight Into The Line Tension of Bilayer Membranes Containing Hybrid Polyunsaturated Lipids. J. Phys. Chem. B 2017, 121, 1587–1600. 10.1021/acs.jpcb.6b10836. [DOI] [PubMed] [Google Scholar]
- Brummel B. E.; Braun A. R.; Sachs J. N. Polyunsaturated Chains in Asymmetric Lipids Disorder Raft Mixtures and Preferentially Associate with Α-Synuclein. Biochim. Biophys. Acta, Biomembr. 2017, 1859, 529–536. 10.1016/j.bbamem.2016.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnarez C.; Webb A.; Rouvière A.; Lyman E. Hysteresis and The Cholesterol Dependent Phase Transition in Binary Lipid Mixtures with The Martini Model. J. Phys. Chem. B 2016, 120, 13086–13093. 10.1021/acs.jpcb.6b09728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Gkeka P.; Fuchs J. E.; Liedl K. R.; Cournia Z. DPPC-Cholesterol Phase Diagram Using Coarse-Grained Molecular Dynamics Simulations. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 2846–2857. 10.1016/j.bbamem.2016.08.005. [DOI] [PubMed] [Google Scholar]
- Ackerman D. G.; Feigenson G. W. Multiscale Modeling of Four-Component Lipid Mixtures: Domain Composition, Size, Alignment, and Properties of The Phase Interface. J. Phys. Chem. B 2015, 119, 4240–4250. 10.1021/jp511083z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S.; Maibaum L. Identifying The Onset of Phase Separation in Quaternary Lipid Bilayer Systems From Coarse-Grained Simulations. J. Phys. Chem. B 2018, 122, 3961. 10.1021/acs.jpcb.8b00364. [DOI] [PubMed] [Google Scholar]
- Carpenter T. S.; López C. A.; Neale C.; Montour C.; Ingólfsson H. I.; Di Natale F.; Lightstone F. C.; Gnanakaran S. Capturing Phase Behavior of Ternary Lipid Mixtures with a Refined Martini Coarse-Grained Force Field. J. Chem. Theory Comput. 2018, 14, 6050–6062. 10.1021/acs.jctc.8b00496. [DOI] [PubMed] [Google Scholar]
- Podewitz M.; Wang Y.; Gkeka P.; von Grafenstein S.; Liedl K. R.; Cournia Z. The Phase Diagram of a Stratum Corneum Lipid Mixture. J. Phys. Chem. B 2018, 122, 10505–10521. 10.1021/acs.jpcb.8b07200. [DOI] [PubMed] [Google Scholar]
- Pantelopulos G. A.; Straub J. E. Regimes of Complex Lipid Bilayer Phases Induced by Cholesterol Concentration in MD Simulation. Biophys. J. 2018, 115, 2167. 10.1016/j.bpj.2018.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter T. S.; López C. A.; Neale C.; Montour C.; Ingólfsson H. I.; Di Natale F.; Lightstone F. C.; Gnanakaran S. Capturing Phase Behavior of Ternary Lipid Mixtures with a Refined Martini Coarse-Grained Force Field. J. Chem. Theory Comput. 2018, 14, 6050–6062. 10.1021/acs.jctc.8b00496. [DOI] [PubMed] [Google Scholar]
- Uppamoochikkal P.; Tristram-Nagle S.; Nagle J. F. Orientation of Tie-Lines in the Phase Diagram of DOPC/DPPC/Cholesterol Model Biomembranes. Langmuir 2010, 26, 17363–17368. 10.1021/la103024f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S.; Im W.. Analysis of Lipid Order States and Domains in Lipid Bilayer Simulations. J. Chem. Theory Comput. 2018, in press, 10.1021/acs.jctc.8b00828. [DOI] [PubMed] [Google Scholar]
- Chiantia S.; London E. Acyl Chain Length and Saturation Modulate Interleaflet Coupling in Asymmetric Bilayers: Effects on Dynamics and Structural Order. Biophys. J. 2012, 103, 2311–2319. 10.1016/j.bpj.2012.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galimzyanov T. R.; Kuzmin P. I.; Pohl P.; Akimov S. A. Undulations Drive Domain Registration From The Two Membrane Leaflets. Biophys. J. 2017, 112, 339–345. 10.1016/j.bpj.2016.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han T.; Haataja M. Compositional Interface Dynamics Within Symmetric and Asymmetric Planar Lipid Bilayer Membranes. Soft Matter 2013, 9, 2120–2124. 10.1039/C2SM27269E. [DOI] [Google Scholar]
- Perlmutter J. D.; Sachs J. N. Interleaflet Interaction and Asymmetry in Phase Separated Lipid Bilayers: Molecular Dynamics Simulations. J. Am. Chem. Soc. 2011, 133, 6563. 10.1021/ja106626r. [DOI] [PubMed] [Google Scholar]
- Lin X.; Zhang S.; Ding H.; Levental I.; Gorfe A. A. The Aliphatic Chain of Cholesterol Modulates Bilayer Interleaflet Coupling and Domain Registration. FEBS Lett. 2016, 590, 3368–3374. 10.1002/1873-3468.12383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler P. W.; Williamson J. J.; Sansom M. S. P.; Olmsted P. D. Roles of Interleaflet Coupling and Hydrophobic Mismatch in Lipid Membrane Phase-Separation Kinetics. J. Am. Chem. Soc. 2016, 138, 11633–11642. 10.1021/jacs.6b04880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manna M.; Javanainen M.; Martinez-Seara Monne H.; Gabius H. J.; Rog T.; Vattulainen I. Long-Chain GM1 Gangliosides Alter Transmembrane Domain Registration Through Interdigitation. Biochim. Biophys. Acta, Biomembr. 2017, 1859, 870–878. 10.1016/j.bbamem.2017.01.033. [DOI] [PubMed] [Google Scholar]
- Reigada R. Alteration of Interleaflet Coupling Due To Compounds Displaying Rapid Translocation in Lipid Membranes. Sci. Rep. 2016, 6, 32934. 10.1038/srep32934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantano D. A.; Moore P. B.; Klein M. L.; Discher D. E. Soft Matter 2011, 7, 8182–8191. 10.1039/c1sm05490b. [DOI] [Google Scholar]
- Sornbundit K.; Modchang C.; Triampo W.; Triampo D.; Nuttavut N.; Kumar P. B. S.; Laradji M. Kinetics of Domains Registration in Multicomponent Lipid Bilayer Membranes. Soft Matter 2014, 10, 7306–7315. 10.1039/C4SM01059K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thallmair S.; Ingólfsson H. I.; Marrink S. J. Cholesterol Flip-Flop Impacts Domain Registration in Plasma Membrane Models. J. Phys. Chem. Lett. 2018, 9, 5527–5533. 10.1021/acs.jpclett.8b01877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner M. D.; Feigenson G. W. The Presence and Role of Midplane Cholesterol in Lipid Bilayers Containing Registered or Antiregistered Phase Domains. J. Phys. Chem. B 2018, 122, 8193–8200. 10.1021/acs.jpcb.8b03949. [DOI] [PubMed] [Google Scholar]
- Risselada H. J.; Marrink S. J.; Muller M. Curvature-Dependent Elastic Properties of Liquid-Ordered Domains Result in Inverted Domain Sorting on Uniaxially Compressed Vesicles. Phys. Rev. Lett. 2011, 106, 148102. 10.1103/PhysRevLett.106.148102. [DOI] [PubMed] [Google Scholar]
- Fischer T.; Jelger Risselada H.; Vink R. L. C. Membrane Lateral Structure: The Influence of Immobilized Particles on Domain Size. Phys. Chem. Chem. Phys. 2012, 14, 14500. 10.1039/c2cp41417a. [DOI] [PubMed] [Google Scholar]
- De Jong D. H.; Heuer A. The Influence of Solid Scaffolds on Flat and Curved Lipid Membranes. AIP Adv. 2017, 7, 075007. 10.1063/1.4989439. [DOI] [Google Scholar]
- Barnoud J.; Rossi G.; Marrink S. J.; Monticelli L. Hydrophobic Compounds Reshape Membrane Domains. PLoS Comput. Biol. 2014, 10, e1003873. 10.1371/journal.pcbi.1003873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muddana H. S.; Chiang H. H.; Butler P. J. Biophys. J. 2012, 102, 489. 10.1016/j.bpj.2011.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moiset G.; López C. A.; Bartelds R.; Syga L.; Rijpkema E.; Cukkemane A.; Baldus M.; Poolman B.; Marrink S. J. Disaccharides Impact The Lateral Organization of Lipid Membranes. J. Am. Chem. Soc. 2014, 136, 16167–16175. 10.1021/ja505476c. [DOI] [PubMed] [Google Scholar]
- López C. A.; de Vries A. H.; Marrink S. J. Computational Microscopy of Cyclodextrin Mediated Cholesterol Extraction From Lipid Model Membranes. Sci. Rep. 2013, 3, 2071. 10.1038/srep02071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganesan S. J.; Xu H.; Matysiak S. Influence of Monovalent Cation Size on Nanodomain Formation in Anionic–Zwitterionic Mixed Bilayers. J. Phys. Chem. B 2017, 121, 787–799. 10.1021/acs.jpcb.6b10099. [DOI] [PubMed] [Google Scholar]
- Bartelds R.; Barnoud J.; Boersma A. J.; Marrink S. J.; Poolman B. Lipid Phase Separation in The Presence of Hydrocarbons in Giant Unilamellar Vesicles. AIMS Biophysics 2017, 4, 528–542. 10.3934/biophy.2017.4.528. [DOI] [Google Scholar]
- Lin X.; Gorfe A. A.; Levental I. Protein Partitioning Into Ordered Membrane Domains: Insights From Simulations. Biophys. J. 2018, 114, 1936–1944. 10.1016/j.bpj.2018.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorent J. H.; Levental I. Structural Determinants of Protein Partitioning Into Ordered Membrane Domains and Lipid Rafts. Chem. Phys. Lipids 2015, 192, 23–32. 10.1016/j.chemphyslip.2015.07.022. [DOI] [PubMed] [Google Scholar]
- Schäfer L. V.; De Jong D. H.; Holt A.; Rzepiela A. J.; De Vries A. H.; Poolman B.; Killian J. A.; Marrink S. J. Lipid Packing Drives The Segregation of Transmembrane Helices Into Disordered Lipid Domains in Model Biomembranes. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 1343. 10.1073/pnas.1009362108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Jong D. H.; Lopez C. A.; Marrink S. J. Molecular View on Protein Sorting Into Liquid-Ordered Membrane Domains Mediated By Gangliosides and Lipid Anchors. Faraday Discuss. 2013, 161, 347. 10.1039/C2FD20086D. [DOI] [PubMed] [Google Scholar]
- Janosi L.; Li Z.; Hancock J. F.; Gorfe A. A. Organization, Dynamics and Segregation of Ras Nanoclusters in Membrane Domains. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 8097. 10.1073/pnas.1200773109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z.; Janosi L.; Gorfe A. A. Formation and Domain Partitioning of H-Ras Peptide Nanoclusters: Effects of Peptide Concentration and Lipid Composition. J. Am. Chem. Soc. 2012, 134, 17278–17285. 10.1021/ja307716z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Gorfe A. A. Aggregation of Lipid-Anchored Full-Length H-Ras in Lipid Bilayers: Simulations with The MARTINI Force Field. PLoS One 2013, 8, e71018. 10.1371/journal.pone.0071018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jefferys E.; Sansom M. S. P.; Fowler P. W. Nras Slows The Rate At Which a Model Lipid Bilayer Phase Separates. Faraday Discuss. 2014, 169, 209. 10.1039/C3FD00131H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parton D. L.; Tek A.; Baaden M.; Sansom M. S. P. Formation of Raft-Like Assemblies Within Clusters of Influenza Hemagglutinin Observed By MD Simulations. PLoS Comput. Biol. 2013, 9, e1003034. 10.1371/journal.pcbi.1003034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Q.; Wu Q. Y.; Wang Z. Y. Effect of Hydrophobic Mismatch on Domain Formation and Peptide Sorting in The Multicomponent Lipid Bilayers in The Presence of Immobilized Peptides. J. Chem. Phys. 2014, 141, 074702. 10.1063/1.4891931. [DOI] [PubMed] [Google Scholar]
- Wu Q. Y.; Liang Q. Interplay Between Curvature and Lateral Organization of Lipids and Peptides/Proteins in Model Membranes. Langmuir 2014, 30, 1116–1122. 10.1021/la4039123. [DOI] [PubMed] [Google Scholar]
- Lopez C. A.; Sethi A.; Goldstein B.; Wilson B. S.; Gnanakaran S. Membrane-Mediated Regulation of The Intrinsically Disordered CD3e Cytoplasmic Tail of The TCR. Biophys. J. 2015, 108, 2481–2491. 10.1016/j.bpj.2015.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser H. J.; Orłowski A.; Róg T.; Nyholm T. K. M.; Chai W.; Feizi T.; Lingwood D.; Vattulainen I.; Simons K. Lateral Sorting in Model Membranes By Cholesterol- Mediated Hydrophobic Matching. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 16628–16633. 10.1073/pnas.1103742108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett W. F. D.; Tieleman D. P. Computer Simulations of Lipid Membrane Domains. Biochim. Biophys. Acta, Biomembr. 2013, 1828, 1765–76. 10.1016/j.bbamem.2013.03.004. [DOI] [PubMed] [Google Scholar]
- Róg T.; Vattulainen I. Cholesterol, Sphingolipids, and Glycolipids: What Do We Know About Their Role in Raft-Like Membranes?. Chem. Phys. Lipids 2014, 184, 82–104. 10.1016/j.chemphyslip.2014.10.004. [DOI] [PubMed] [Google Scholar]
- Cebecauer M.; Amaro M.; Jurkiewicz P.; Sarmento M. J.; Šachl R.; Cwiklik L.; Hof M. Membrane Lipid Nanodomains. Chem. Rev. 2018, 118, 11259. 10.1021/acs.chemrev.8b00322. [DOI] [PubMed] [Google Scholar]
- Yeagle P. L. Non-Covalent Binding of Membrane Lipids To Membrane Proteins. Biochim. Biophys. Acta, Biomembr. 2014, 1838, 1548–59. 10.1016/j.bbamem.2013.11.009. [DOI] [PubMed] [Google Scholar]
- Swainsbury D. J. K.; Scheidelaar S.; Van Grondelle R.; Killian J. A.; Jones M. R. Bacterial Reaction Centers Purified with Styrene Maleic Acid Copolymer Retain Native Membrane Functional Properties and Display Enhanced Stability. Angew. Chem., Int. Ed. 2014, 53, 11803–11807. 10.1002/anie.201406412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechara C.; Nöll A.; Morgner N.; Degiacomi M. T.; Tampé R.; Robinson C. V. a Subset of Annular Lipids Is Linked To The Flippase Activity of An ABC Transporter. Nat. Chem. 2015, 7, 255–262. 10.1038/nchem.2172. [DOI] [PubMed] [Google Scholar]
- Mondal S.; Khelashvili G.; Weinstein H. Not Just An Oil Slick: How The Energetics of Protein-Membrane Interactions Impacts The Function and Organization of Transmembrane Proteins. Biophys. J. 2014, 106, 2305–2316. 10.1016/j.bpj.2014.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanegas J. M.; Arroyo M. Force Transduction and Lipid Binding in Mscl: a Continuum-Molecular Approach. PLoS One 2014, 9, e113947. 10.1371/journal.pone.0113947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedger G.; Koldsoe H.; Chavent M.; Siebold C.; Rohatgi R.; Sansom M. S. P.. Cholesterol Interaction Sites on The Transmembrane Domain of The Hedgehog Signal Transducer and Class F G Protein-Coupled Receptor Smoothened. Structure, 2018, 10.1016/j.str.2018.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogaski B.; Klauda J. B. Membrane-Binding Mechanism of a Peripheral Membrane Protein Through Microsecond Molecular Dynamics Simulations. J. Mol. Biol. 2012, 423, 847–861. 10.1016/j.jmb.2012.08.015. [DOI] [PubMed] [Google Scholar]
- Arnarez C.; Mazat J. P.; Elezgaray J.; Marrink S. J.; Periole X. Evidence For Cardiolipin Binding Sites on The Membrane-Exposed Surface of The Cytochrome Bc1. J. Am. Chem. Soc. 2013, 135, 3112. 10.1021/ja310577u. [DOI] [PubMed] [Google Scholar]
- Schmidt M. R.; Stansfeld P. J.; Tucker S. J.; Sansom M. S. P. Simulation-Based Prediction of Phosphatidylinositol 4,5-Bisphosphate Binding To An Ion Channel. Biochemistry 2013, 52, 279. 10.1021/bi301350s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stansfeld P. J.; Jefferys E. E.; Sansom M. S. P. Multiscale Simulations Reveal Conserved Patterns of Lipid Interactions with Aquaporins. Structure 2013, 21, 810–819. 10.1016/j.str.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aponte-Santamaria C.; Briones R.; Schenk A. D.; Walz T.; De Groot B. L. Molecular Driving Forces Defining Lipid Positions Around Aquaporin-0. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 9887–9892. 10.1073/pnas.1121054109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briones R.; Aponte-Santamaria C.; De Groot B. L. Localization and Ordering of Lipids Around Aquaporin-0: Protein and Lipid Mobility Effects. Front. Physiol. 2017, 8, 124. 10.3389/fphys.2017.00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras F. X.; Ernst A. M.; Haberkant P.; Bjorkholm P.; et al. Molecular Recognition of a Single Sphingolipid Species By a Protein’s Transmembrane Domain. Nature 2012, 481, 525–529. 10.1038/nature10742. [DOI] [PubMed] [Google Scholar]
- Lee J. Y.; Patel R.; Lyman E. Ligand-Dependent Cholesterol Interactions with The Human A(2A) Adenosine Receptor. Chem. Phys. Lipids 2013, 169, 39–45. 10.1016/j.chemphyslip.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. Y.; Lyman E. Predictions For Cholesterol Interaction Sites on The A(2A) Adenosine Receptor. J. Am. Chem. Soc. 2012, 134, 16512–16515. 10.1021/ja307532d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyman E.; Higgs C.; Kim B.; et al. a Role For a Specific Cholesterol Interaction in Stabilizing The Apo Configuration of The Human A(2A) Adenosine Receptor. Structure 2009, 17, 1660–1668. 10.1016/j.str.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cang X. H.; Du Y.; Mao Y. Y.; Wang Y. Y.; Yang H. Y.; Jiang H. L. Mapping The Functional Binding Sites of Cholesterol in Beta(2)-Adrenergic Receptor By Long-Time Molecular Dynamics Simulations. J. Phys. Chem. B 2013, 117, 1085–1094. 10.1021/jp3118192. [DOI] [PubMed] [Google Scholar]
- Manna M.; Niemelä M.; Tynkkynen J.; Javanainen M.; Kulig W.; Müller D. J.; Rog T.; Vattulainen I. Mechanism of Allosteric Regulation of Β2-Adrenergic Receptor By Cholesterol. eLife 2016, 5, e18432. 10.7554/eLife.18432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khelashvili G.; Grossfield A.; Feller S. E.; et al. Structural and Dynamic Effects of Cholesterol At Preferred Sites of Interaction with Rhodopsin Identified From Microsecond Length Molecular Dynamics Simulations. Proteins: Struct., Funct., Genet. 2009, 76, 403–417. 10.1002/prot.22355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta D.; Prasanna X.; Mohole M.; Chattopadhyay A. Exploring GPCR-Lipid Interactions By Molecular Dynamics Simulations: Excitements, Challenges and The Way Forward. J. Phys. Chem. B 2018, 122, 5727–5737. 10.1021/acs.jpcb.8b01657. [DOI] [PubMed] [Google Scholar]
- Rogaski B.; Lim J. B.; Klauda J. B. Sterol Binding and Membrane Lipid Attachment To The Osh4 Protein of Yeast. J. Phys. Chem. B 2010, 114, 13562–13573. 10.1021/jp106890e. [DOI] [PubMed] [Google Scholar]
- Weiser B. P.; Salari R.; Eckenhoff R. G.; Brannigan G. Computational Investigation of Cholesterol Binding Sites on Mitochondrial VDAC. J. Phys. Chem. B 2014, 118, 9852–9860. 10.1021/jp504516a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeppelin T.; Ladefoged L. K.; Sinning S.; Periole X.; Schiøtt B. A Direct Interaction of Cholesterol with The Dopamine Transporter Prevents Its Out-To-Inward Transition. PLoS Comput. Biol. 2018, 14, e1005907. 10.1371/journal.pcbi.1005907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koivuniemi A.; Vuorela T.; Kovanen P. T.; Vattulainen I.; Hyvönen M. T. Lipid Exchange Mechanism of The Cholesteryl Ester Transfer Protein Clarified By Atomistic and Coarse-Grained Simulations. PLoS Comput. Biol. 2012, 8, e1002299. 10.1371/journal.pcbi.1002299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumb C. N.; Sansom M. S. P. Finding a Needle in a Haystack: The Role of Electrostatics in Target Lipid Recognition By PH Domains. PLoS Comput. Biol. 2012, 8, e1002617. 10.1371/journal.pcbi.1002617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stansfeld P. J.; Hopkinson R.; Ashcroft F. M.; Sansom M. S. P. PIP2-Binding Site in Kir Channels: Definition By Multiscale Biomolecular Simulations. Biochemistry 2009, 48, 10926. 10.1021/bi9013193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnarez C.; Marrink S. J.; Periole X. Identification of Cardiolipin Binding Sites on Cytochrome C Oxidase At The Entrance of Proton Channels. Sci. Rep. 2013, 3, 1263. 10.1038/srep01263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedger G.; Rouse S. L.; Domański J.; Chavent M.; Koldsø H.; Sansom M. S. P. Lipid-Loving Ants: Molecular Simulations of Cardiolipin Interactions and The Organization of The Adenine Nucleotide Translocase in Model Mitochondrial Membranes. Biochemistry 2016, 55, 6238–6249. 10.1021/acs.biochem.6b00751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan A. L.; Ruprecht J. J.; Kunji E. R. S.; Robinson A. J. Cardiolipin Dynamics and Binding To Conserved Residues in The Mitochondrial ADP/ATP Carrier. Biochim. Biophys. Acta, Biomembr. 2018, 1860, 1035–1045. 10.1016/j.bbamem.2018.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weingarth M.; Prokofyev A.; Van Der Cruijsen E.; Nand D.; Bonvin A. M. J. J.; Pongs O.; Baldus M. Structural Determinants of Specific Lipid Binding To Potassium Channels. J. Am. Chem. Soc. 2013, 135, 3983. 10.1021/ja3119114. [DOI] [PubMed] [Google Scholar]
- Poyry S.; Cramariuc O.; Postila P. A.; Kaszuba K.; Sarewicz M.; Osyczka A.; Vattulainen I.; Rog T. Atomistic Simulations Indicate Cardiolipin To Have An Integral Role In The Structure Of The Cytochrome Bc(1) Complex. Biochim. Biophys. Acta, Bioenerg. 2013, 1827, 769–778. 10.1016/j.bbabio.2013.03.005. [DOI] [PubMed] [Google Scholar]
- Duncan A. L.; Robinson A. J.; Walker J. E. Cardiolipin Binds Selectively To ATP Synthase. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 8687–8692. 10.1073/pnas.1608396113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma V.; Ala-Vannesluoma P.; Vattulainen I.; Wikström M.; Róg T. Role Of Subunit III And Its Lipids In The Molecular Mechanism Of Cytochrome C Oxidase. Biochim. Biophys. Acta, Bioenerg. 2015, 1847, 690–697. 10.1016/j.bbabio.2015.04.007. [DOI] [PubMed] [Google Scholar]
- Malkamäki A.; Sharma V. Atomistic Insights Into Cardiolipin Binding Sites Of Cytochrome C Oxidase. Biochim. Biophys. Acta, Bioenerg. 2018, 10.1016/j.bbabio.2018.11.004. [DOI] [PubMed] [Google Scholar]
- Kalli A. C.; Sansom M. S. P.; Reithmeier R. A. F. Molecular Dynamics Simulations Of The Bacterial Uraa H+-Uracil Symporter In Lipid Bilayers Reveal A Closed State And A Selective Interaction With Cardiolipin. PLoS Comput. Biol. 2015, 11, e1004123. 10.1371/journal.pcbi.1004123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autzen H.; Koldsø H.; Stansfeld P. J.; Gourdon P.; Sansom M. S. P.; Nissen P. Interactions Of A Bacterial Cu(I)-Atpase With A Complex Lipid Environment. Biochemistry 2018, 57, 4063–4073. 10.1021/acs.biochem.8b00326. [DOI] [PubMed] [Google Scholar]
- Corey R. A.; Pyle E.; Allen W. J.; Watkins D. W.; Casiraghi M.; Miroux B.; Arechaga I.; Politis A.; Collinson I. Specific Cardiolipin–Secy Interactions Are Required For Proton-Motive Force Stimulation Of Protein Secretion. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 7967–7972. 10.1073/pnas.1721536115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt V.; Sidore M.; Bechara C.; Duneau J. P.; Sturgis J. N. The Lipid Environment Of Escherichia Coli Aquaporin Z. Biochim. Biophys. Acta, Biomembr. 2019, 1861, 431. 10.1016/j.bbamem.2018.10.017. [DOI] [PubMed] [Google Scholar]
- Sengupta D.; Chattopadhyay A. Identification Of Cholesterol Binding Sites In The Serotonin1A Receptor. J. Phys. Chem. B 2012, 116, 12991. 10.1021/jp309888u. [DOI] [PubMed] [Google Scholar]
- Rouviere E.; Arnarez C.; Yang L.; Lyman E. Identification of Two New Cholesterol Interaction Sites on the A2A Adenosine Receptor. Biophys. J. 2017, 113, 2415–2424. 10.1016/j.bpj.2017.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen H. Y.; Hoi K. K.; Liko I.; Hedger G.; et al. Ptdins(4,5)P2 Stabilizes Active States Of Gpcrs And Enhances Selectivity Of G-Protein Coupling. Nature 2018, 559, 423–427. 10.1038/s41586-018-0325-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbera N.; Ayee M. M. A.; Akpa B. S.; Levitan I. Molecular Dynamics Simulations of Kir2.2 Interactions with an Ensemble of Cholesterol Molecules. Biophys. J. 2018, 115, 1264–1280. 10.1016/j.bpj.2018.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpa L.; Salaria R.; Brannigan G.. Boundary Lipids Of The Nicotinic Acetylcholine Receptor: Spontaneous Partitioning Via Coarse-Grained Molecular Dynamics Simulation. arXiv 2018,arXiv:1807.11090v2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lensink M. F.; Govaerts C.; Ruysschaert J. M. Identification Of Specific Lipid-Binding Sites In Integral Membrane Proteins. J. Biol. Chem. 2010, 285, 10519–10526. 10.1074/jbc.M109.068890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirandela G. D.; Tamburrino G.; Hoskisson P. A.; Zachariae U.; Javelle A. The Lipid Environment Determines The Activity Of The E. Coli Ammonium Transporter, AmtB. FASEB J. 2018, 33, 1–11. 10.1096/fj.201800782R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyle E.; Kalli A. C.; Amillis S.; Hall Z.; Lau A. M.; Hanyaloglu A. C.; Diallinas G.; Byrne B.; Politis A. Structural Lipids Enable The Formation Of Functional Oligomers Of The Eukaryotic Purine Symporter Uapa. Cell Chem. Biol. 2018, 25, 840–848. 10.1016/j.chembiol.2018.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsch S. A.; Kugemann A.; Carpaneto A.; Böckmann R. A.; Dietrich P. Phosphatidylinositol-3,5-Bisphosphate Lipid-Binding-Induced Activation Of The Human Two-Pore Channel 2. Cell. Mol. Life Sci. 2018, 75, 3803–3815. 10.1007/s00018-018-2829-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou K.; Dichlberger A.; Martinez-Seara H.; et al. A Ceramide-Regulated Element In The Late Endosomal Protein LAPTM4B Controls Amino Acid Transporter Interaction. ACS Cent. Sci. 2018, 4, 548–558. 10.1021/acscentsci.7b00582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morra G.; Razavi A. M.; Pandey K.; Weinstein H.; Menon A. K.; Khelashvili G. Mechanisms Of Lipid Scrambling By The G Protein- Coupled Receptor Opsin. Structure 2018, 26, 356–367. 10.1016/j.str.2017.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang T.; Yu K.; Hartzell H. C.; Tajkhorshid E. Lipids And Ions Traverse The Membrane By The Same Physical Pathway In The Nhtmem16 Scramblase. eLife 2017, 6, e28671. 10.7554/eLife.28671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B. C.; Khelashvili G.; Falzone M.; Menon A. K.; Weinstein H.; Accardi A. Gating Mechanism Of The Extracellular Entry To The Lipid Pathway In A TMEM16 Scramblase. Nat. Commun. 2018, 9, 3251. 10.1038/s41467-018-05724-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushell S. R.; Pike A. C. W.; Falzone M. E.; Rorsman N. J. G.; Ta C. M.; Corey R. A.; Newport T. D.; Shintre C. A.; Tessitore A.; et al. The Structural Basis Of Lipid Scrambling And Inactivation In The Endoplasmic Reticulum Scramblase TMEM16K. bioRxiv 2018, 10.1101/447417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch S.; Exterkate M.; López C. A.; Patro M.; Marrink S. J.; Driessen A. J. M.. Two Distinct Anionic Phospholipid-Dependent Steps During SecA-Mediated Protein Translocation. 2018, submitted. [DOI] [PubMed] [Google Scholar]
- Huber R. G.; Berglund N. A.; Kargas V.; Marzinek J. K.; Holdbrook D. A.; Khalid S.; Piggot T. J.; Schmidtchen A.; Bond P. J. A Thermodynamic Funnel Drives Bacterial Lipopolysaccharide Transfer In The TLR4 Pathway. Structure 2018, 26, 1151–1161. 10.1016/j.str.2018.04.007. [DOI] [PubMed] [Google Scholar]
- Ohmann A.; Li C. Y.; Maffeo C.; Al Nahas K.; Baumann K. N.; Göpfrich K.; Yoo J.; Keyser U. F.; Aksimentiev A. A Synthetic Enzyme Built From DNA Flips 107 Lipids Per Second In Biological Membranes. Nat. Commun. 2018, 9, 2426. 10.1038/s41467-018-04821-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemelä P. S.; Miettinen M. S.; Monticelli L.; Hammaren H.; Bjelkmar P.; Murtola T.; Lindahl E.; Vattulainen I. Membrane Proteins Diffuse As Dynamic Complexes with Lipids. J. Am. Chem. Soc. 2010, 132, 7574–7575. 10.1021/ja101481b. [DOI] [PubMed] [Google Scholar]
- Apajalahti T.; Niemela P.; Govindan P. N.; Miettinen M.; Salonen E.; Marrink S. J.; Vattulainen I. Concerted Diffusion of Lipids in Raft-Like Membranes. Faraday Discuss. 2010, 144, 411–430. 10.1039/B901487J. [DOI] [PubMed] [Google Scholar]
- Duneau J. P.; Khao J.; Sturgis J. N. Lipid Perturbation By Membrane Proteins and The Lipophobic Effect. Biochim. Biophys. Acta, Biomembr. 2017, 1859, 126–134. 10.1016/j.bbamem.2016.10.014. [DOI] [PubMed] [Google Scholar]
- Grossfield A.; Feller S. E.; Pitman M. C. a Role For Direct Interactions in The Modulation of Rhodopsin By Omega-3 Polyunsaturated Lipids. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 4888–4893. 10.1073/pnas.0508352103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salas-Estrada L. A.; Leioatts N.; Romo T. D.; Grossfield A. Lipids Alter Rhodopsin Function via Ligand-like and Solvent-like Interactions. Biophys. J. 2018, 114, 355–367. 10.1016/j.bpj.2017.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin F.; Kindt J. T. Hydrophobic Mismatch and Lipid Sorting Near Ompa in Mixed Bilayers: Atomistic and Coarse-Grained Simulations. Biophys. J. 2012, 102, 2279. 10.1016/j.bpj.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung A.; Yarovsky I. Gap Junction Hemichannel Interactions with Zwitterionic Lipid, Anionic Lipid, and Cholesterol: Molecular Simulation Studies. Biochemistry 2011, 50, 1492. 10.1021/bi1004156. [DOI] [PubMed] [Google Scholar]
- Koldsø H.; Sansom M. S. P. Local Lipid Reorganization By a Transmembrane Protein Domain. J. Phys. Chem. Lett. 2012, 3, 3498. 10.1021/jz301570w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J.; Zhang Y.; Li H.; Chu H.; Wang Q.; Jiang S.; et al. Intramembrane Ionic Protein–Lipid Interaction Regulates Integrin Structure and Function. PLoS Biol. 2018, 16, e2006525. 10.1371/journal.pbio.2006525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward A. B.; Guvench O.; Hills Jr R. D. Coarse Grain Lipid-Protein Molecular Interactions and Diffusion with Msba Flippase. Proteins: Struct., Funct., Genet. 2012, 80, 2178–2190. 10.1002/prot.24108. [DOI] [PubMed] [Google Scholar]
- Venken T.; Schillinger A. S.; Fuglebakk E.; Reuter N. Interactions Stabilizing The C-Terminal Helix of Human Phospholipid Scramblase 1 in Lipid Bilayers: a Computational Study. Biochim. Biophys. Acta, Biomembr. 2017, 1859, 1200–1210. 10.1016/j.bbamem.2017.03.019. [DOI] [PubMed] [Google Scholar]
- Martens C.; Shekhar M.; Borysik A. J.; Lau A. M.; Reading E.; Tajkhorshid E.; Booth P. J.; Politis A. Direct Protein-Lipid Interactions Shape The Conformational Landscape Of Secondary Transporters. Nat. Commun. 2018, 9, 4151. 10.1038/s41467-018-06704-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Liefferinge F.; Krammer E. M.; Sengupta D.; Prévost M.. Lipid Composition and Salt Concentration as Regulatory Factors of the Anion Selectivity of VDAC Studied by Coarse-Grained Molecular Dynamics Simulations. Chem. Phys. Lipids 2018, in press, 10.1016/j.chemphyslip.2018.11.002. [DOI] [PubMed] [Google Scholar]
- Beaven A. H.; Maer A. M.; Sodt A. J.; Rui H.; Pastor R. W.; Andersen O. S.; Im W. Gramicidin a Channel Formation Induces Local Lipid Redistribution I: Experiment and Simulation. Biophys. J. 2017, 112, 1185–1197. 10.1016/j.bpj.2017.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karo J.; Peterson P.; Vendelin M. Molecular Dynamics Simulations Of Creatine Kinase And Adenine Nucleotide Translocase In Mitochondrial Membrane Patch. J. Biol. Chem. 2012, 287, 7467. 10.1074/jbc.M111.332320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleskot R.; Pejchar P.; Žárský V.; Staiger C. J.; Potocký M. Structural Insights Into The Inhibition Of Actin-Capping Protein By Interactions With Phosphatidic Acid And Phosphatidylinositol (4,5)-Bisphosphate. PLoS Comput. Biol. 2012, 8, e1002765. 10.1371/journal.pcbi.1002765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge C.; Gómez-Llobregat J.; Skwark M. J.; Ruysschaert J. M.; Wieslander A.; Lindén M. Membrane Remodeling Capacity Of A Vesicle-Inducing Glycosyltransferase. FEBS J. 2014, 281, 3667–368. 10.1111/febs.12889. [DOI] [PubMed] [Google Scholar]
- Darvill N.; Dubois D. J.; Rouse S. L.; Hammoudi P. M.; Blake T.; Benjamin S.; Liu B.; Soldati-Favre D.; Matthews S. Structural Basis Of Phosphatidic Acid Sensing By APH In Apicomplexan Parasites. Structure 2018, 26, 1059–1071. 10.1016/j.str.2018.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Z.; Ghai R.; Collins B. M.; Mark A. E. The Recognition Of Membrane-Bound Ptdins3p By PX Domains. Proteins: Struct., Funct., Genet. 2014, 82, 2332–2342. 10.1002/prot.24593. [DOI] [PubMed] [Google Scholar]
- Kalli A. C.; Morgan G.; Sansom M. S. P. Interactions Of The Auxilin-1 PTEN-Like Domain With Model Membranes Result In Nanoclustering Of Phosphatidyl Inositol Phosphates. Biophys. J. 2013, 105, 137–145. 10.1016/j.bpj.2013.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picas L.; Viaud J.; Schauer K.; Vanni S.; Hnia K.; Fraisier V.; Roux A.; Bassereau P.; Gaits-Iacovoni F.; Payrastre B.; Laporte J.; Manneville J. B.; Goud B. BIN1/M-Amphiphysin2 Induces Clustering Of Phosphoinositides To Recruit Its Downstream Partner Dynamin. Nat. Commun. 2014, 5, 5647. 10.1038/ncomms6647. [DOI] [PubMed] [Google Scholar]
- Lin X.; Wang H.; Lou Z.; Cao M.; Zhang Z.; Gu N. Roles Of PIP2 In The Membrane Binding Of MIM I-BAR: Insights From Molecular Dynamics Simulations. FEBS Lett. 2018, 592, 2533–2542. 10.1002/1873-3468.13186. [DOI] [PubMed] [Google Scholar]
- Karandur D.; Nawrotek A.; Kuriyan J.; Cherfils J. Multiple Interactions Between An Arf/GEF Complex And Charged Lipids Determine Activation Kinetics On The Membrane. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 11416–11421. 10.1073/pnas.1707970114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posner M. G.; Upadhyay A.; Ishima R.; Kalli A. C.; et al. KIBRA C2: Distinctive Phosphoinositide And Ca2+ Binding Distinctive Phosphoinositide And Ca2+ Binding Properties Of Normal And Cognitive Performance-Linked Variant Forms Of KIBRA C2 Domain. J. Biol. Chem. 2018, 293, 9335–9344. 10.1074/jbc.RA118.002279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlier L.; Louet M.; Chaloin L.; Fuchs P. F. J.; Martinez J.; Muriaux D.; Favard C.; Floquet N. Coarse Grained Simulations Of The HIV-1 Matrix Protein Anchoring: Revisiting Its Assembly On Membrane Domains. Biophys. J. 2014, 106, 577–585. 10.1016/j.bpj.2013.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakala M.; Kalimeri M.; Enkavi G.; Vattulainen I.; Lappalainen P. Molecular Mechanism For Inhibition Of Twinfilin By Phosphoinositides. J. Biol. Chem. 2018, 293, 4818. 10.1074/jbc.RA117.000484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senju Y.; Kalimeri M.; Koskela E. V.; Somerharju P.; Zhao H.; Vattulainen I.; Lappalainen P. Regulation Of The Actin Cytoskeleton By PI(4,5)P2. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, E8977–E8986. 10.1073/pnas.1705032114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun F.; Schroer C. F. E.; Xu L.; Yin H.; Marrink S. J.; Luo S. Z. Molecular Dynamics Of The Association Of L-Selectin And FERM Regulated By PIP2. Biophys. J. 2018, 114, 1858–1868. 10.1016/j.bpj.2018.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sridhar A.; Kumar A.; Dasmahapatra A. K. Multi-Scale Molecular Dynamics Study Of Cholera Pentamer Binding To A GM1-Phospholipid Membrane. J. Mol. Graphics Modell. 2016, 68, 236–251. 10.1016/j.jmgm.2016.07.007. [DOI] [PubMed] [Google Scholar]
- Enkavi G.; Mikkolainen H.; Güngör B.; Ikonen E.; Vattulainen I. Concerted Regulation Of Npc2 Binding To Endosomal/Lysosomal Membranes By Bis(Monoacylglycero)Phosphate And Sphingomyelin. PLoS Comput. Biol. 2017, 13, e1005831. 10.1371/journal.pcbi.1005831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan H. M.; He T.; Fuglebakk E.; Grauffel C.; Yang B.; Roberts M. F.; Gershenson A.; Reuter N. A Role for Weak Electrostatic Interactions in Peripheral Membrane Protein Binding. Biophys. J. 2016, 110, 1367–1378. 10.1016/j.bpj.2016.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta D. Cholesterol Modulates The Structure, Binding Modes, And Energetics Of Caveolin-Membrane Interactions. J. Phys. Chem. B 2012, 116, 14556–14564. 10.1021/jp3077886. [DOI] [PubMed] [Google Scholar]
- Polyansky A. A.; Ramaswamy R.; Volynsky P. E.; Sbalzarini I. F.; Marrink S. J.; Efremov R. G. Antimicrobial Peptides Induce Growth Of Phosphatidylglycerol Domains In A Model Bacterial Membrane. J. Phys. Chem. Lett. 2010, 1, 3108–3111. 10.1021/jz101163e. [DOI] [Google Scholar]
- Horn J. N.; Cravens A.; Grossfield A. Interactions Between Fengycin And Model Bilayers Quantified By Coarse-Grained Molecular Dynamics. Biophys. J. 2013, 105, 1612–1623. 10.1016/j.bpj.2013.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofbauer H. F.; Gecht M.; Fischer S. C.; Seybert A.; Frangakis A. S.; Stelzer E. H. K.; Covino R.; Hummer G.; Ernst R. The Molecular Recognition Of Phosphatidic Acid By An Amphipathic Helix In Opi1. J. Cell Biol. 2018, 217, 3109–3126. 10.1083/jcb.201802027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Z.; Zou Y.; Zhang Q.; Chen P.; Ma B.; Wei G.; Nussinov R. Atomistic-Level Study Of The Interactions Between Hiapp Protofibrils And Membranes: Influence Of Ph And Lipid Composition. Biochim. Biophys. Acta, Biomembr. 2018, 1860, 1818–1825. 10.1016/j.bbamem.2018.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino T.; Mahmood M. I.; Mori K.; Matsuzaki K. Binding and Aggregation Mechanism of Amyloid β-Peptides onto the GM1 Ganglioside-Containing Lipid Membrane. J. Phys. Chem. B 2013, 117, 8085–8094. 10.1021/jp4029062. [DOI] [PubMed] [Google Scholar]
- Christensen M.; Skeby K. K.; Schiøtt B. Identification of Key Interactions in the Initial Self-Assembly of Amylin in a Membrane Environment. Biochemistry 2017, 56, 4884–4894. 10.1021/acs.biochem.7b00344. [DOI] [PubMed] [Google Scholar]
- Owen M. C.; Kulig W.; Poojari C.; Rog T.; Strodel B. Physiologically-Relevant Levels Of Sphingomyelin, But Not GM1, Induces A Β-Sheet-Rich Structure In The Amyloid-Β(1–42) Monomer. Biochim. Biophys. Acta, Biomembr. 2018, 1860, 1709–1720. 10.1016/j.bbamem.2018.03.026. [DOI] [PubMed] [Google Scholar]
- Hedger G.; Sansom M. S. P. Lipid Interaction Sites On Channels, Transporters And Receptors: Recent Insights From Molecular Dynamics Simulations. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 2390–2400. 10.1016/j.bbamem.2016.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grouleff J.; Irudayam S. J.; Skeby K. K.; Schiøtt B. The Influence Of Cholesterol On Membrane Protein Structure, Function, And Dynamics Studied By Molecular Dynamics Simulations. Biochim. Biophys. Acta, Biomembr. 2015, 1848, 1783–1795. 10.1016/j.bbamem.2015.03.029. [DOI] [PubMed] [Google Scholar]
- Wen P. C.; Mahinthichaichan P.; Trebesch N.; Jiang T.; Zhao Z.; Shinn E.; Wang Y.; Shekhar M.; Kapoor K.; Chan C. K.; Tajkhorshid E. Microscopic View Of Lipids And Their Diverse Biological Functions. Curr. Opin. Struct. Biol. 2018, 51, 177–186. 10.1016/j.sbi.2018.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corradi V.; Sejdiu B. I.; Mesa-Galloso H.; Abdizadeh H.; Noskov S. Y.; Marrink S. J.; Tieleman D. P.. Emerging Diversity In Lipid-Protein Interactions. Chem. Rev. 2018, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenz S.; Bihr T.; Schmidt D.; Merkel R.; Seifert U.; Sengupta K.; Smith A. S. Membrane Fluctuations Mediate Lateral Interactions Between Cadherin Bonds. Nat. Phys. 2017, 13, 906–913. 10.1038/nphys4138. [DOI] [Google Scholar]
- Katira S.; Mandadapu K. K.; Vaikuntanathan S.; Smit B.; Chandler D. Pre-Transition Effects Mediate Forces of Assembly Between Transmembrane Proteins. eLife 2016, 5, e13150. 10.7554/eLife.13150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haussman R. C.; Deserno M. Effective Field Theory of Thermal Casimir Interactions Between Anisotropic Particles. Phys. Rev. E 2014, 89, 062102. 10.1103/PhysRevE.89.062102. [DOI] [PubMed] [Google Scholar]
- Bocharov E. V.; Mineev K. S.; Pavlov K. V.; Akimov S. A.; Kuznetsov A. S.; Efremov R. G.; Arseniev A. S. Helix-Helix Interactions in Membrane Domains of Bitopic Proteins: Specificity and Role of Lipid Environment. Biochim. Biophys. Acta, Biomembr. 2017, 1859, 561–576. 10.1016/j.bbamem.2016.10.024. [DOI] [PubMed] [Google Scholar]
- Psachoulia E.; Marshall D. P.; Sansom M. S. P. Molecular Dynamics Simulations of The Dimerization of Transmembrane Alpha-Helices. Acc. Chem. Res. 2010, 43, 388–396. 10.1021/ar900211k. [DOI] [PubMed] [Google Scholar]
- Han J.; Pluhackova K.; Wassenaar T. A.; Böckmann R. A. Synaptobrevin Transmembrane Domain Dimerization Studied By Multiscale Molecular Dynamics Simulations. Biophys. J. 2015, 109, 760–771. 10.1016/j.bpj.2015.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altwaijry N. A.; Baron M.; Wright D. W.; Coveney P. V.; Townsend-Nicholson A. An Ensemble-Based Protocol For The Computational Prediction of Helix–Helix Interactions in G Protein-Coupled Receptors Using Coarse-Grained Molecular Dynamics. J. Chem. Theory Comput. 2017, 13, 2254–2270. 10.1021/acs.jctc.6b01246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash A.; Janosi L.; Doxastakis M. Gxxxg Motifs, Phenylalanine, and Cholesterol Guide The Self-Association of Transmembrane Domains of Erbb2 Receptors. Biophys. J. 2011, 101, 1949–1958. 10.1016/j.bpj.2011.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chng C. P.; Tan S. M. Leukocyte Integrin Αlβ2 Transmembrane Association Dynamics Revealed By Coarse-Grained Molecular Dynamics Simulations. Proteins: Struct., Funct., Genet. 2011, 79, 2203–2213. 10.1002/prot.23044. [DOI] [PubMed] [Google Scholar]
- Hall B. A.; Armitage J. P.; Sansom M. S. P. Mechanism of Bacterial Signal Transduction Revealed By Molecular Dynamics of Tsr Dimers and Trimers of Dimers in Lipid Vesicles. PLoS Comput. Biol. 2012, 8, e1002685. 10.1371/journal.pcbi.1002685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Den Bogaart G.; Meyenberg K.; Risselada H. J.; Amin H.; Willig K. I.; Hubrich B. E.; Dier M.; Hell S. W.; Grubmuller H.; Diederichsen U.; Jahn R. Membrane Protein Sequestering By Ionic Protein-Lipid Interactions. Nature 2011, 479, 552. 10.1038/nature10545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orłowski A.; Kukkurainen S.; Pöyry A.; Rissanen S.; Vattulainen I.; Hytönen V. P.; Róg T. PIP2 and Talin Join Forces To Activate Integrin. J. Phys. Chem. B 2015, 119, 12381–12389. 10.1021/acs.jpcb.5b06457. [DOI] [PubMed] [Google Scholar]
- Chavent M.; Karia D.; Kalli A. C.; Domanski J.; Duncan A. L.; Hedger G.; Stansfeld P. J.; Seiradake E.; Jones E. Y.; Sansom M. S. P. Interactions of The Epha2 Kinase Domain with Pips in Membranes: Implications For Receptor Function. Structure 2018, 26, 1025–1034. 10.1016/j.str.2018.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travers T.; López C. A.; Van Q. N.; Neale C.; Tonelli M.; Stephen A. G.; Gnanakaran S. Molecular Recognition of RAS/RAF Complex At The Membrane: Role of RAF Cysteine-Rich Domain. Sci. Rep. 2018, 8, 8461. 10.1038/s41598-018-26832-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Periole X.; Huber T.; Marrink S. J.; Sakmar T. P. G. Protein-Coupled Receptors Self-Assemble in Dynamics Simulations of Model Bilayers. J. Am. Chem. Soc. 2007, 129, 10126–10132. 10.1021/ja0706246. [DOI] [PubMed] [Google Scholar]
- Periole X.; Knepp A. M.; Sakmar T. P.; Marrink S. J.; Huber T. Structural Determinants of The Supramolecular Organization of G Protein-Coupled Receptors in Bilayers. J. Am. Chem. Soc. 2012, 134, 10959. 10.1021/ja303286e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A.; Sonavane U.; Joshi R. Multiscale Modelling To Understand The Self-Assembly Mechanism of Human β2-Adrenergic Receptor in Lipid Bilayer. Comput. Biol. Chem. 2014, 48, 29–39. 10.1016/j.compbiolchem.2013.11.002. [DOI] [PubMed] [Google Scholar]
- Mondal S.; Johnston J. M.; Wang H.; Khelashvili G.; Filizola M.; Weinstein H. Membrane Driven Spatial Organization of Gpcrs. Sci. Rep. 2013, 3, 2909. 10.1038/srep02909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rassam P.; Copeland N. A.; Birkholz O.; Tóth C.; Chavent M.; Duncan A. L.; et al. Supramolecular Assemblies Underpin Turnover of Outer Membrane Proteins in Bacteria. Nature 2015, 523, 333–336. 10.1038/nature14461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irudayanathan F. J.; Wang X.; Wang N.; Willsey S. R.; Seddon I. A.; Nangia S. Self-Assembly Simulations of Classic Claudins–Insights Into The Pore Structure, Selectivity and Higher Order Complexes. J. Phys. Chem. B 2018, 122, 7463–7474. 10.1021/acs.jpcb.8b03842. [DOI] [PubMed] [Google Scholar]
- Prasanna X.; Chattopadhyay A.; Sengupta D. Cholesterol Modulates The Dimer Interface of The B2-Adrenergic Receptor Via Cholesterol Occupancy Sites. Biophys. J. 2014, 106, 1290–1300. 10.1016/j.bpj.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluhackova K.; Gahbauer S.; Kranz F.; Wassenaar T. A.; Böckmann R. A. Dynamic Cholesterol-Conditioned Dimerization of The G Protein Coupled Chemokine Receptor Type 4. PLoS Comput. Biol. 2016, 12, e1005169. 10.1371/journal.pcbi.1005169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gahbauer S.; Pluhackova K.; Böckmann R. A. Closely Related, Yet Unique: Distinct Homo- and Heterodimerization Patterns of G Protein Coupled Chemokine Receptors and Their Fine-Tuning By Cholesterol. PLoS Comput. Biol. 2018, 14, e1006062. 10.1371/journal.pcbi.1006062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Periole X.; Zeppelin T.; Schiøtt B. Dimer Interface of The Human Serotonin Transporter and Effect of The Membrane Composition. Sci. Rep. 2018, 8, 5080. 10.1038/s41598-018-22912-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt T. H.; Homsi Y.; Lang T. Oligomerization of The Tetraspanin CD81 Via The Flexibility of Its D-Loop. Biophys. J. 2016, 110, 2463–2474. 10.1016/j.bpj.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domanski J.; Marrink S. J.; Schäfer L. V. Transmembrane Helices Can Induce Domain Formation in Crowded Model Membranes. Biochim. Biophys. Acta, Biomembr. 2012, 1818, 984. 10.1016/j.bbamem.2011.08.021. [DOI] [PubMed] [Google Scholar]
- Gil T.; Sabra M. C.; Ipsen J. H.; Mouritsen O. G. Wetting and Capillary Condensation As Means of Protein Organization in Membranes. Biophys. J. 1997, 73, 1728–1741. 10.1016/S0006-3495(97)78204-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackerman D. G.; Feigenson G. W. Effects of Transmembrane Α-Helix Length and Concentration on Phase Behavior in Four-Component Lipid Mixtures: a Molecular Dynamics Study. J. Phys. Chem. B 2016, 120, 4064–4077. 10.1021/acs.jpcb.6b00611. [DOI] [PubMed] [Google Scholar]
- Guixà-González R.; Javanainen M.; GóMez-Soler M.; Cordobilla B.; Domingo J. C.; Sanz F.; Pastor M.; Ciruela F.; Martinez- Seara H.; Selent J. Membrane Omega-3 Fatty Acids Modulate The Oligomerisation Kinetics of Adenosine A2A and Dopamine D2 Receptors. Sci. Rep. 2016, 6, 19839. 10.1038/srep19839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawar A. B.; Deshpande S. A.; Gopal S. M.; Wassenaar T. A.; Athale C. A.; Sengupta D. Thermodynamic and Kinetic Characterization of Transmembrane Helix Association. Phys. Chem. Chem. Phys. 2015, 17, 1390. 10.1039/C4CP03732D. [DOI] [PubMed] [Google Scholar]
- Sengupta D.; Marrink S. J. Lipid-Mediated Interactions Tune The Association of Glycophorin a Helix and Its Disruptive Mutants in Membranes. Phys. Chem. Chem. Phys. 2010, 12, 12987. 10.1039/c0cp00101e. [DOI] [PubMed] [Google Scholar]
- Castillo N.; Monticelli L.; Barnoud J.; Tieleman D. P. Free Energy of WALP23 Dimer Association in DMPC, DPPC, and DOPC Bilayers. Chem. Phys. Lipids 2013, 169, 95–105. 10.1016/j.chemphyslip.2013.02.001. [DOI] [PubMed] [Google Scholar]
- Yoo J.; Cui Q. Membrane-Mediated Protein-Protein Interactions and Connection To Elastic Models: a Coarse-Grained Simulation Analysis of Gramicidin a Association. Biophys. J. 2013, 104, 128–138. 10.1016/j.bpj.2012.11.3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aci-Sèche S.; Sawma P.; Hubert P.; Sturgis J. N.; Bagnard D.; Jacob L.; Genest M.; Garnier N. Transmembrane Recognition of The Semaphorin Co-Receptors Neuropilin 1 and Plexin A1: Coarse-Grained Simulations. PLoS One 2014, 9, e97779. 10.1371/journal.pone.0097779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasanna X.; Praveen P. J.; Sengupta D. Sequence Dependent Lipid-Mediated Effects Modulate The Dimerization of Erbb2 and Its Associative Mutants. Phys. Chem. Chem. Phys. 2013, 15, 19031. 10.1039/c3cp52447g. [DOI] [PubMed] [Google Scholar]
- Benjamini A.; Smit B. Lipid Mediated Packing of Transmembrane Helices - a Dissipative Particle Dynamics Study. Soft Matter 2013, 9, 2673–2683. 10.1039/c2sm27165f. [DOI] [Google Scholar]
- Grau B.; Javanainen M.; García-Murria M. J.; Kulig W.; Vattulainen I.; Mingarro I.; Martínez-Gil L. The Role of Hydrophobic Matching on Transmembrane Helix Packing in Cells. Cell Stress 2017, 1, 90–106. 10.15698/cst2017.11.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cybulski L. E.; Ballering J.; Moussatova A.; Inda M. E.; Vazquez D. B.; Wassenaar T. A.; De Mendoza D.; Tieleman D. P.; Killian J. A. Activation of The Bacterial Thermosensor Desk Involves a Serine Zipper Dimerization Motif That Is Modulated By Bilayer Thickness. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 6353–6358. 10.1073/pnas.1422446112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunton T. A.; Goose J. E.; Gavaghan D. J.; Sansom M. S. P.; Osborne J. M. The Free Energy Landscape of Dimerization of a Membrane Protein, NanC. PLoS Comput. Biol. 2014, 10, e1003417. 10.1371/journal.pcbi.1003417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston J. M.; Wang H.; Provasi D.; Filizola M. Assessing The Relative Stability of Dimer Interfaces in G Protein-Coupled Receptors. PLoS Comput. Biol. 2012, 8, e1002649. 10.1371/journal.pcbi.1002649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meral D.; Provasi D.; Prada-Gracia D.; Möller J.; Marino K.; Lohse M. J.; Filizola M. Molecular Details of Dimerization Kinetics Reveal Negligible Populations of Transient μ-Opioid Receptor Homodimers At Physiological Concentrations. Sci. Rep. 2018, 8, 7705. 10.1038/s41598-018-26070-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaraman K.; Morley A. N.; Szöllősi D.; Wassenaar T. A.; Sitte H. H.; Stockner T. Dopamine Transporter Oligomerization Involves The Scaffold Domain, But Spares The Bundle Domain. PLoS Comput. Biol. 2018, 14, e1006229. 10.1371/journal.pcbi.1006229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishizawa M.; Nishizawa K. Potential of Mean Force Analysis of The Self-Association of Leucine-Rich Transmembrane Α-Helices: Difference Between Atomistic and Coarse-Grained Simulations. J. Chem. Phys. 2014, 141, 075101. 10.1063/1.4891932. [DOI] [PubMed] [Google Scholar]
- May A.; Pool R.; Van Dijk E.; Bijlard J.; Abeln S.; Heringa J.; Feenstra K. A. Coarse-Grained Versus Atomistic Simulations: Realistic Interaction Free Energies For Real Proteins. Bioinformatics 2014, 30, 326–334. 10.1093/bioinformatics/btt675. [DOI] [PubMed] [Google Scholar]
- Koukos P. I.; Faro I.; van Noort C. W.; Bonvin A. M. J. J. A Membrane Protein Complex Docking Benchmark. J. Mol. Biol. 2018, 430, 5246. 10.1016/j.jmb.2018.11.005. [DOI] [PubMed] [Google Scholar]
- Baaden M.; Marrink S. J. Coarse-Grain Modelling of Protein–Protein Interactions. Curr. Opin. Struct. Biol. 2013, 23, 878–886. 10.1016/j.sbi.2013.09.004. [DOI] [PubMed] [Google Scholar]
- Periole X. Interplay of G Protein-Coupled Receptors with The Membrane: Insights From Supra-Atomic Coarse Grain Molecular Dynamics Simulations. Chem. Rev. 2017, 117, 156–185. 10.1021/acs.chemrev.6b00344. [DOI] [PubMed] [Google Scholar]
- Gahbauer S.; Böckmann R. A. Membrane-Mediated Oligomerization of G Protein Coupled Receptors and Its Implications For GPCR Function. Front. Physiol. 2016, 7, 494. 10.3389/fphys.2016.00494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X. Y.; Mezei M.; Cui M. Computational Approaches For Modeling GPCR Dimerization. Curr. Pharm. Biotechnol. 2014, 15, 996–1006. 10.2174/1389201015666141013102515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannes L.; Pezeshkian W.; Ipsen J. H.; Shillcock J. C. Clustering on Membranes: Fluctuations and More. Trends Cell Biol. 2018, 28, 405–415. 10.1016/j.tcb.2018.01.009. [DOI] [PubMed] [Google Scholar]
- Kozlov M. M.; Campelo F.; Liska N.; Chernomordik L. V.; Marrink S. J.; Mcmahon H. T. Mechanisms Shaping Cell Membranes. Curr. Opin. Cell Biol. 2014, 29, 53–60. 10.1016/j.ceb.2014.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgart T.; Capraro B. R.; Zhu C.; Das S. L. Thermodynamics and Mechanics of Membrane Curvature Generation and Sensing By Proteins and Lipids. Annu. Rev. Phys. Chem. 2011, 62, 483–506. 10.1146/annurev.physchem.012809.103450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simunovic M.; Bassereau P. Reshaping Biological Membranes in Endocytosis: Crossing The Configurational Space of Membrane- Protein Interactions. Biol. Chem. 2014, 395, 275–283. 10.1515/hsz-2013-0242. [DOI] [PubMed] [Google Scholar]
- Blood P. D.; Voth G. A. Direct Observation of Bin/Amphiphysin/Rvs (BAR) Domain-Induced Membrane Curvature By Means of Molecular Dynamics Simulations. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 15068–15072. 10.1073/pnas.0603917103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkhipov A.; Yin Y.; Schulten K. Four-Scale Description of Membrane Sculpting By BAR Domains. Biophys. J. 2008, 95, 2806. 10.1529/biophysj.108.132563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies K. M.; Anselmi C.; Wittig I.; Faraldo-Gomez J. D.; Kuhlbrandt W. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 13602. 10.1073/pnas.1204593109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anselmi C.; Davies K. M.; Faraldo-Gómez J. D. Mitochondrial ATP Synthase Dimers Spontaneously Associate Due To a Long-Range Membrane Induced Force. J. Gen. Physiol. 2018, 150, 763–770. 10.1085/jgp.201812033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Oliveira Dos Santos Soares R.; Bortot L. O.; Van Der Spoel D.; Caliri A. Membrane Vesiculation Induced By Proteins of The Dengue Virus Envelope Studied By Molecular Dynamics Simulations. J. Phys.: Condens. Matter 2017, 29, 504002. 10.1088/1361-648X/aa99c6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin M. H.; Hsu H. J.; Bartenschlager R.; Fischer W. B. Membrane Undulation Induced By NS4A of Dengue Virus: a Molecular Dynamics Simulation Study. J. Biomol. Struct. Dyn. 2014, 32, 1552–1562. 10.1080/07391102.2013.826599. [DOI] [PubMed] [Google Scholar]
- Braun A. R.; Sevcsik E.; Chin P.; Rhoades E.; Tristram-Nagle S.; Sachs J. N. Α-Synuclein Induces Both Positive Mean Curvature and Negative Gaussian Curvature in Membranes. J. Am. Chem. Soc. 2012, 134, 2613. 10.1021/ja208316h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun A. R.; Lacy M. M.; Ducas V. C.; Rhoades E.; Sachs J. N. Α-Synuclein-Induced Membrane Remodeling Is Driven By Binding Affinity, Partition Depth, and Interleaflet Order Asymmetry. J. Am. Chem. Soc. 2014, 136, 9962–9972. 10.1021/ja5016958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Gorfe A. A. Membrane Remodeling By Surface-Bound Protein Aggregates: Insights From Coarse-Grained Molecular Dynamics Simulation. J. Phys. Chem. Lett. 2014, 5, 1457–1462. 10.1021/jz500451a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo H. J.; Wallqvist A. Spontaneous Buckling of Lipid Bilayer and Vesicle Budding Induced By Antimicrobial Peptide Magainin 2: a Coarse-Grained Simulation Study. J. Phys. Chem. B 2011, 115, 8122–8129. 10.1021/jp2023023. [DOI] [PubMed] [Google Scholar]
- Sodt A. J.; Pastor R. W. Molecular Modeling of Lipid Membrane Curvature Induction By a Peptide: More Than Simply Shape. Biophys. J. 2014, 106, 1958–1969. 10.1016/j.bpj.2014.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrmans M.; Marrink S. J. Molecular View of the Role of Fusion Peptides in Promoting Positive Membrane Curvature. J. Am. Chem. Soc. 2012, 134, 1543. 10.1021/ja207290b. [DOI] [PubMed] [Google Scholar]
- Pannuzzo M.; Raudino A.; Böckmann R. A. Peptide-Induced Membrane Curvature In Edge-Stabilized Open Bilayers: A Theoretical And Molecular Dynamics Study. J. Chem. Phys. 2014, 141, 024901. 10.1063/1.4885340. [DOI] [PubMed] [Google Scholar]
- Patel D. S.; Park S.; Wu E. L.; Yeom M. S.; Widmalm G.; Klauda J. B.; Im W. Influence of Ganglioside GM1 Concentration on Lipid Clustering and Membrane Properties and Curvature. Biophys. J. 2016, 111, 1987–99. 10.1016/j.bpj.2016.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta R.; Miettinen M. S.; Fricke N.; Lipowsky R.; Dimova R. The Glycolipid GM1 Reshapes Asymmetric Biomembranes and Giant Vesicles By Curvature Generation. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 5756–5761. 10.1073/pnas.1722320115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baoukina S.; Ingólfsson H. I.; Marrink S. J.; Tieleman D. P. Curvature-Induced Sorting of Lipids in Plasma Membrane Tethers. Adv. Theory Sim. 2018, 1, 1800034. 10.1002/adts.201800034. [DOI] [Google Scholar]
- Sodt A. J.; Venable R. M.; Lyman E.; Pastor R. W. Nonadditive Compositional Curvature Energetics of Lipid Bilayers. Phys. Rev. Lett. 2016, 117, 138104. 10.1103/PhysRevLett.117.138104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campelo F.; Arnarez C.; Marrink S. J.; Kozlov M. M. Helfrich Model Of Membrane Bending: From Gibbs Theory Of Liquid Interfaces To Membranes As Thick Anisotropic Elastic Layers. Adv. Colloid Interface Sci. 2014, 208, 25–33. 10.1016/j.cis.2014.01.018. [DOI] [PubMed] [Google Scholar]
- Krishna A.; Sengupta D. Interplay Between Membrane Curvature And Cholesterol: Role Of Palmitoylated Caveolin-1. Biophys. J. 2018, 10.1016/j.bpj.2018.11.3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruhn D. S.; Lomholt M. A.; Khandelia H. Quantifying The Relationship Between Curvature and Electric Potential in Lipid Bilayers. J. Phys. Chem. B 2016, 120, 4812–4817. 10.1021/acs.jpcb.6b03439. [DOI] [PubMed] [Google Scholar]
- Cui H.; Lyman E.; Voth G. A. Mechanism of Membrane Curvature Sensing By Amphipathic Helix Containing Proteins. Biophys. J. 2011, 100, 1271–1279. 10.1016/j.bpj.2011.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng K.; Somerharju P.; Sugar I. Detection and Characterization of The Onset of Bilayer Packing Defects By Nanosecond-Resolved Intramolecular Excimer Fluorescence Spectroscopy. Chem. Phys. Lipids 1994, 74, 49–64. 10.1016/0009-3084(94)90111-2. [DOI] [PubMed] [Google Scholar]
- Nuscher B.; Kamp F.; Mehnert T.; Odoy S.; Haass C.; Kahle P. J.; Beyer K. Alpha-Synuclein Has a High Affinity For Packing Defects in a Bilayer Membrane - a Thermodynamics Study. J. Biol. Chem. 2004, 279, 21966–21975. 10.1074/jbc.M401076200. [DOI] [PubMed] [Google Scholar]
- Tahir A.; Van Lehn R. C.; Choi S. H.; Alexander-Katz A. M. Solvent-Exposed Lipid Tail Protrusions Depend on Lipid Membrane Composition and Curvature. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 1207–1215. 10.1016/j.bbamem.2016.01.026. [DOI] [PubMed] [Google Scholar]
- Vamparys L.; Gautier R.; Vanni S.; Bennett W. F. D.; Tieleman D. P.; Antonny B.; Etchebest C.; Fuchs P. F. J. Conical Lipids in Flat Bilayers Induce Packing Defects Similar To That Induced By Positive Curvature. Biophys. J. 2013, 104, 585–593. 10.1016/j.bpj.2012.11.3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanni S.; Hirose H.; Barelli H.; Antonny B.; Gautier R. a Sub-Nanometre View of How Membrane Curvature and Composition Modulate Lipid Packing and Protein Recruitment. Nat. Commun. 2014, 5, 4916. 10.1038/ncomms5916. [DOI] [PubMed] [Google Scholar]
- Pinot M.; Vanni S.; Ambroggio E.; Guet D.; Goud B.; Manneville J. B.. Feedback Between Membrane Tension, Lipid Shape and Curvature in The Formation of Packing Defects. bioRxiv 2018, 10.1101/389627. [DOI] [Google Scholar]
- Gómez-Llobregat J.; Elías-Wolff F.; Lindén M. Anisotropic Membrane Curvature Sensing By Amphipathic Peptides. Biophys. J. 2016, 110, 197–204. 10.1016/j.bpj.2015.11.3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlmutter J. D.; Braun A. R.; Sachs J. N. Curvature Dynamics of Α-Synuclein Familial Parkinson Disease Mutants: Molecular Simulations of The Micelle- and Bilayer-Bound Forms. J. Biol. Chem. 2009, 284, 7177–7189. 10.1074/jbc.M808895200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R.; Mark A. E. The Effect of Membrane Curvature on The Conformation of Antimicrobial Peptides: Implications For Binding and The Mechanism of Action. Eur. Biophys. J. 2011, 40, 545–553. 10.1007/s00249-011-0677-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez L.; Meredith S. C.; Straub J. E.; Thirumalai D. Transmembrane Fragment Structures of Amyloid Precursor Protein Depend on Membrane Surface Curvature. J. Am. Chem. Soc. 2014, 136, 854–857. 10.1021/ja410958j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markvoort A. J.; Marrink S. J. Lipid Acrobatics in The Membrane Fusion Arena. Curr. Top. Membr. 2011, 68, 259–294. 10.1016/B978-0-12-385891-7.00011-8. [DOI] [PubMed] [Google Scholar]
- Fuhrmans M.; Marelli G.; Smirnova Y. G.; Müller M. Mechanics of Membrane Fusion/Pore Formation. Chem. Phys. Lipids 2015, 185, 109–28. 10.1016/j.chemphyslip.2014.07.010. [DOI] [PubMed] [Google Scholar]
- Smirnova Y. G.; Marrink S. J.; Lipowsky R.; Knecht V. Solvent-Exposed Tails As Prestalk Transition States For Membrane Fusion At Low Hydration. J. Am. Chem. Soc. 2010, 132, 6710. 10.1021/ja910050x. [DOI] [PubMed] [Google Scholar]
- Mirjanian D.; Dickey A. N.; Hoh J. H.; Woolf T. B.; Stevens M. J. Splaying of Aliphatic Tails Plays a Central Role in Barrier Crossing During Liposome Fusion. J. Phys. Chem. B 2010, 114, 11061. 10.1021/jp1055182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo J.; Jackson M. B.; Cui Q. a Comparison of Coarse-Grained and Continuum Models For Membrane Bending in Lipid Bilayer Fusion Pores. Biophys. J. 2013, 104, 841. 10.1016/j.bpj.2012.12.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishizawa M.; Nishizawa K. Molecular Dynamics Simulation Analysis of Membrane Defects and Pore Propensity of Hemifusion Diaphragms. Biophys. J. 2013, 104, 1038. 10.1016/j.bpj.2013.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risselada H. J.; Smirnova Y.; Grubmüller H. Free Energy Landscape of Rim-Pore Expansion in Membrane Fusion. Biophys. J. 2014, 107, 2287–2295. 10.1016/j.bpj.2014.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risselada H. J.; Bubnis G.; Grubmüller H. Expansion of The Fusion Stalk and Its Implication For Biological Membrane Fusion. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 11043–11048. 10.1073/pnas.1323221111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamoto S.; Shinoda W. Free Energy Analysis Along The Stalk Mechanism of Membrane Fusion. Soft Matter 2014, 10, 3048–3054. 10.1039/c3sm52344f. [DOI] [PubMed] [Google Scholar]
- Gardner J. M.; Abrams C. F. Lipid Flip-Flop Vs. Lateral Diffusion in The Relaxation of Hemifusion Diaphragms. Biochim. Biophys. Acta, Biomembr. 2018, 1860, 1452–1459. 10.1016/j.bbamem.2018.04.007. [DOI] [PubMed] [Google Scholar]
- Urakami N.; Jimbo T.; Sakuma Y.; Imai M. Molecular Mechanism of Vesicle Division Induced By Coupling Between Lipid Geometry and Membrane Curvatures. Soft Matter 2018, 14, 3018–3027. 10.1039/C7SM02188G. [DOI] [PubMed] [Google Scholar]
- Eun C.; Berkowitz M. L. Thermodynamic and Hydrogen-Bonding Analyses of The Interaction Between Model Lipid Bilayers. J. Phys. Chem. B 2010, 114, 3013. 10.1021/jp910347n. [DOI] [PubMed] [Google Scholar]
- Gentilcore A. N.; Michaud-Agrawal N.; Crozier P. S.; Stevens M. J.; Woolf T. B. Examining The Origins of The Hydration Force Between Lipid Bilayers Using All-Atom Simulations. J. Membr. Biol. 2010, 235, 1. 10.1007/s00232-010-9249-2. [DOI] [PubMed] [Google Scholar]
- Smirnova Y. G.; Aeffner S.; Risselada H. J.; Salditt T.; Marrink S. J.; Lipowsky R.; Mueller M.; Knecht V. Interbilayer Repulsion Forces Between Tension-Free Lipid Bilayers From Simulation. Soft Matter 2013, 9, 10705–10718. 10.1039/c3sm51771c. [DOI] [Google Scholar]
- Kasson P. M.; Lindahl E.; Pande V. S. Water Ordering At Membrane Interfaces Controls Fusion Dynamics. J. Am. Chem. Soc. 2011, 133, 3812–3815. 10.1021/ja200310d. [DOI] [PubMed] [Google Scholar]
- Schneck E.; Sedlmeier F.; Netz R. R. Hydration Repulsion Between Biomembranes Results From An Interplay of Dehydration and Depolarization. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 14405. 10.1073/pnas.1205811109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risselada H. J. Membrane Fusion Stalks and Lipid Rafts: a Love-Hate Relationship. Biophys. J. 2017, 112, 2475–2478. 10.1016/j.bpj.2017.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannuzzo M.; De Jong D. H.; Raudino A.; Marrink S. J. Simulation of Polyethylene Glycol and Calcium-Mediated Membrane Fusion. J. Chem. Phys. 2014, 140, 124905. 10.1063/1.4869176. [DOI] [PubMed] [Google Scholar]
- Tsai H. H. G.; Chang C. M.; Lee J. B. Multi-Step Formation of a Hemifusion Diaphragm For Vesicle Fusion Revealed By All-Atom Molecular Dynamics Simulations. Biochim. Biophys. Acta, Biomembr. 2014, 1838, 1529–1535. 10.1016/j.bbamem.2014.01.018. [DOI] [PubMed] [Google Scholar]
- Bhaskara R. M.; Linker S. M.; Vögele M.; Köfinger J.; Hummer G. Carbon Nanotubes Mediate Fusion of Lipid Vesicles. ACS Nano 2017, 11, 1273–1280. 10.1021/acsnano.6b05434. [DOI] [PubMed] [Google Scholar]
- Wu Z.; Cui Q.; Yethiraj A. Why Do Arginine and Lysine Organize Lipids Differently? Insights From Coarse-Grained and Atomistic Simulations. J. Phys. Chem. B 2013, 117, 12145–12156. 10.1021/jp4068729. [DOI] [PubMed] [Google Scholar]
- Risselada H. J.; Marelli G.; Fuhrmans M.; Smirnova Y. G.; Grubmüller H.; Marrink S. J.; Muller M. Line-Tension Controlled Mechanism For Influenza Fusion. PLoS One 2012, 7, e38302. 10.1371/journal.pone.0038302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson P.; Kasson P. M. Lipid Tail Protrusion in Simulations Predicts Fusogenic Activity of Influenza Fusion Peptide Mutants and Conformational Models. PLoS Comput. Biol. 2013, 9, e1002950. 10.1371/journal.pcbi.1002950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moiset G.; Cirac A. D.; Stuart M. C. A.; Marrink S. J.; Sengupta D.; Poolman B. Dual Action of BPC194: a Membrane Active Peptide Killing Bacterial Cells. PLoS One 2013, 8, e61541. 10.1371/journal.pone.0061541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allolio C.; Magarkar A.; Jurkiewicz P.; Baxová K.; Javanainen M.; Mason P. E.; Šachl R.; Cebecauer M.; Hof M.; Horinek D.; Heinz V.; et al. Arginine-Rich Cell-Penetrating Peptides Induce Membrane Multilamellarity And Subsequently Enter Via Formation Of A Fusion Pore. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 11923–11928. 10.1073/pnas.1811520115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baoukina S.; Tieleman D. P. Direct Simulation of Protein-Mediated Vesicle Fusion: Lung Surfactant Protein B. Biophys. J. 2010, 99, 2134. 10.1016/j.bpj.2010.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risselada H. J.; Kutzner C.; Grubmüller H. Caught in The Act: Visualization of SNARE-Mediated Fusion Events in Molecular Detail. ChemBioChem 2011, 12, 1049. 10.1002/cbic.201100020. [DOI] [PubMed] [Google Scholar]
- Risselada H. J.; Grubmüller H. How SNARE Molecules Mediate Membrane Fusion: Recent Insights From Molecular Simulations. Curr. Opin. Struct. Biol. 2012, 22, 187–196. 10.1016/j.sbi.2012.01.007. [DOI] [PubMed] [Google Scholar]
- D’Agostino M.; Risselada H. J.; Lürick A.; Ungermann C.; Mayer A. A Tethering Complex Drives The Terminal Stage of SNARE-Dependent Membrane Fusion. Nature 2017, 551, 634. 10.1038/nature24469. [DOI] [PubMed] [Google Scholar]
- D’Agostino M.; Risselada H. J.; Endter L. J.; Comte-Miserez V.; Mayer A. SNARE-Mediated Membrane Fusion Arrests At Pore Expansion To Regulate The Volume of An Organelle. EMBO J. 2018, 37, e99193. 10.15252/embj.201899193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S.; Lindau M. Molecular Mechanism Of Fusion Pore Formation Driven By The Neuronal SNARE Complex. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 12751. 10.1073/pnas.1816495115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinot M.; Vanni S.; Pagnotta S.; Lacas-Gervais S.; Payet L. A.; Ferreira T.; Gautier R.; Goud B.; Antonny B.; Barelli H. Polyunsaturated Phospholipids Facilitate Membrane Deformation and Fission By Endocytic Proteins. Science 2014, 345, 693–697. 10.1126/science.1255288. [DOI] [PubMed] [Google Scholar]
- Risselada H. J.; Marrink S. J. Curvature Effects on Lipid Packing and Dynamics in Liposomes Revealed By Coarse Grained Molecular Dynamics Simulations. Phys. Chem. Chem. Phys. 2009, 11, 2056–2067. 10.1039/b818782g. [DOI] [PubMed] [Google Scholar]
- Shevchenko A.; Simons K. Lipidomics: Coming to Grips with Lipid Diversity. Nat. Rev. Mol. Cell Biol. 2010, 11, 593–598. 10.1038/nrm2934. [DOI] [PubMed] [Google Scholar]
- Harayama T.; Riezman H. Understanding the Diversity of Membrane Lipid Composition. Nat. Rev. Mol. Cell Biol. 2018, 19, 281–296. 10.1038/nrm.2017.138. [DOI] [PubMed] [Google Scholar]
- Steck T. L.; Lange Y. Transverse Distribution of Plasma Membrane Bilayer Cholesterol: Picking Sides. Traffic 2018, 19, 750–760. 10.1111/tra.12586. [DOI] [PubMed] [Google Scholar]
- Ingólfsson H. I.; Melo M. N.; Van Eerden F. J.; Arnarez C.; Lopez C. A.; Wassenaar T. A.; et al. Lipid Organization of The Plasma Membrane. J. Am. Chem. Soc. 2014, 136, 14554–14559. 10.1021/ja507832e. [DOI] [PubMed] [Google Scholar]
- Koldsø H.; Shorthouse D.; Hélie J.; Sansom M. S. P. Lipid Clustering Correlates with Membrane Curvature as Revealed by Molecular Simulations of Complex Lipid Bilayers. PLoS Comput. Biol. 2014, 10, e1003911. 10.1371/journal.pcbi.1003911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flinner N.; Schleiff E. Dynamics of the Glycophorin a Dimer in Membranes of Native-Like Composition Uncovered by Coarse-Grained Molecular Dynamics Simulations. PLoS One 2015, 10, e0133999. 10.1371/journal.pone.0133999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingólfsson H. I.; Carpenter T. S.; Bhatia H.; Bremer P. T.; Marrink S. J.; Lightstone F. C. Computational Lipidomics of the Neuronal Plasma Membrane. Biophys. J. 2017, 113, 2271–2280. 10.1016/j.bpj.2017.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domicevica L.; Koldsø H.; Biggin P. C. Multiscale Molecular Dynamics Simulations of Lipid Interactions with P-Glycoprotein in a Complex Membrane. J. Mol. Graphics Modell. 2018, 80, 147. 10.1016/j.jmgm.2017.12.022. [DOI] [PubMed] [Google Scholar]
- Zhuang X.; Ou A.; Klauda J. B. Simulations of Simple Linoleic Acid-Containing Lipid Membranes and Models for the Soybean Plasma Membranes. J. Chem. Phys. 2017, 146, 215103–215111. 10.1063/1.4983655. [DOI] [PubMed] [Google Scholar]
- Jo S.; Lim J. B.; Klauda J. B.; Im W. CHARMM-GUI Membrane Builder for Mixed Bilayers and its Application to Yeast Membranes. Biophys. J. 2009, 97, 50–58. 10.1016/j.bpj.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andoh Y.; Okazaki S.; Ueoka R. Molecular Dynamics Study of Lipid Bilayers Modeling the Plasma Membranes of Normal Murine Thymocytes and Leukemic GRSL Cells. Biochim. Biophys. Acta, Biomembr. 2013, 1828, 1259–1270. 10.1016/j.bbamem.2013.01.005. [DOI] [PubMed] [Google Scholar]
- Goronzy I. N.; Rawle R. J.; Boxer S. G.; Kasson P. M. Cholesterol Enhances Influenza Binding Avidity by Controlling Nanoscale Receptor Clustering. Chem. Sci. 2018, 9, 2340–2347. 10.1039/C7SC03236F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baoukina S.; Ingólfsson H. I.; Marrink S. J.; Tieleman D. P. Curvature-Induced Sorting of Lipids in Plasma Membrane Tethers. Adv. Theory Sim. 2018, 1, 1800034. 10.1002/adts.201800034. [DOI] [Google Scholar]
- Corradi V.; Mendez-Villuendas E.; Ingólfsson H. I.; Gu R. X.; Siuda I.; Melo M. N.; Moussatova A.; et al. Lipid–Protein Interactions are Unique Fingerprints for Membrane Proteins. ACS Cent. Sci. 2018, 4, 709–717. 10.1021/acscentsci.8b00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan A. L.; Reddy T.; Koldsø H.; Hélie J.; Fowler P. W.; Chavent M.; Sansom M. S. P. Protein Crowding and Lipid Complexity Influence the Nanoscale Dynamic Organization of Ion Channels in Cell Membranes. Sci. Rep. 2017, 7, 16647. 10.1038/s41598-017-16865-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koldsø H.; Reddy T.; Fowler P. W.; Duncan A. L.; Sansom M. S. P. Membrane Compartmentalization Reducing the Mobility of Lipids and Proteins Within a Model Plasma Membrane. J. Phys. Chem. B 2016, 120, 8873–8881. 10.1021/acs.jpcb.6b05846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koldsø H.; Sansom M. S. P. Organization and Dynamics of Receptor Proteins in a Plasma Membrane. J. Am. Chem. Soc. 2015, 137, 14694–704. 10.1021/jacs.5b08048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedger G.; Sansom M. S. P.; Koldsø H. The Juxtamembrane Regions of Human Receptor Tyrosine Kinases Exhibit Conserved Interaction Sites with Anionic Lipids. Sci. Rep. 2015, 5, 9198. 10.1038/srep09198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorthouse D.; Hedger G.; Koldsø H.; Sansom M. S. P. Molecular Simulations of Glycolipids: Towards Mammalian Cell Membrane Models. Biochimie 2016, 120, 105–109. 10.1016/j.biochi.2015.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeevan B. G. C.; Gerstman B. S.; Stahelin R. V.; Chapagain P. The Ebola Virus Protein VP40 Hexamer Enhances the Clustering of PI(4,5)P2 Lipids in the Plasma Membrane. Phys. Chem. Chem. Phys. 2016, 18, 28409. 10.1039/C6CP03776C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalli A. C.; Róg T.; Vattulainen I.; Campbell I. D.; Sansom M. S. P. The Integrin Receptor in Biologically Relevant Bilayers: Insights From Molecular Dynamics Simulations. J. Membr. Biol. 2017, 250, 337. 10.1007/s00232-016-9908-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yesylevskyy S. O.; Rivel T.; Ramseyer C. The Influence of Curvature on the Properties of the Plasma Membrane. Insights from Atomistic Molecular Dynamics Simulations. Sci. Rep. 2017, 7, 16078. 10.1038/s41598-017-16450-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivel T.; Ramseyer C.; Yesylevskyy S. O.. Permeation of Cisplatin Through the Membranes of Normal and Cancer Cells: a Molecular Dynamics Study. 2018, bioRxiv 10.1101/375980. [DOI] [Google Scholar]
- Klähn M.; Zacharias M. Transformations in Plasma Membranes of Cancerous Cells and Resulting Consequences for Cation Insertion Studied with Molecular Dynamics. Phys. Chem. Chem. Phys. 2013, 15, 14427–14415. 10.1039/c3cp52085d. [DOI] [PubMed] [Google Scholar]
- Kalli A. C.; Reithmeier R. A. F. Interaction of the Human Erythrocyte Band 3 Anion Exchanger 1 (AE1, SLC4A1) With Lipids and Glycophorin A: Molecular Organization of the Wright (Wr) Blood Group Antigen. PLoS Comput. Biol. 2018, 14, e1006284–24. 10.1371/journal.pcbi.1006284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadri L.; Ferru-Clément R.; Bacle A.; Payet L. A.; Cantereau A.; Hélye R.; Becq F.; Jayle C.; Vandebrouck C.; Ferreira T. Modulation of Cellular Membrane Properties as a Potential Therapeutic Strategy to Counter Lipointoxication in Obstructive Pulmonary Diseases. Biochim. Biophys. Acta, Mol. Basis Dis. 2018, 1864, 3069. 10.1016/j.bbadis.2018.06.021. [DOI] [PubMed] [Google Scholar]
- Daum G. Lipids of Mitochondria. Biochim. Biophys. Acta, Rev. Biomembr. 1985, 822, 1–42. 10.1016/0304-4157(85)90002-4. [DOI] [PubMed] [Google Scholar]
- Pfeiffer K.; Gohil V.; Stuart R. A.; Hunte C.; Brandt U.; Greenberg M. L.; Schagger H. Cardiolipin Stabilizes Respiratory Chain Supercomplexes. J. Biol. Chem. 2003, 278, 52873–52880. 10.1074/jbc.M308366200. [DOI] [PubMed] [Google Scholar]
- Arnarez C.; Marrink S. J.; Periole X. Molecular Mechanism of Cardiolipin-Mediated Assembly of Respiratory Chain Supercomplexes. Chem. Science 2016, 7, 4435–4443. 10.1039/C5SC04664E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan A. L.; Robinson A. J.; Walker J. E. Cardiolipin Binds Selectively but Transiently to Conserved Lysine Residues in the Rotor of Metazoan ATP Synthases. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 8687–8692. 10.1073/pnas.1608396113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vähäheikkilä M.; Peltomaa T.; Róg T.; Vazdar M.; Pöyry S.; Vattulainen I. How Cardiolipin Peroxidation Alters The Properties of the Inner Mitochondrial Membrane?. Chem. Phys. Lipids 2018, 214, 15. 10.1016/j.chemphyslip.2018.04.005. [DOI] [PubMed] [Google Scholar]
- Van Eerden F. J.; De Jong D. H.; De Vries A. H.; Wassenaar T. A.; Marrink S. J. Characterization of Thylakoid Lipid Membranes From Cyanobacteria and Higher Plants by Molecular Dynamics Simulations. Biochim. Biophys. Acta, Biomembr. 2015, 1848, 1319–1330. 10.1016/j.bbamem.2015.02.025. [DOI] [PubMed] [Google Scholar]
- Van Eerden F. J.; Melo M. N.; Frederix P. W. J. M.; Periole X.; Marrink S. J. Exchange Pathways of Plastoquinone and Plastoquinol in The Photosystem II Complex. Nat. Commun. 2017, 8, 1–8. 10.1038/ncomms15214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Eerden F. J.; Van Den Berg T.; Frederix P W.J.M.; De Jong D. H.; Periole X.; Marrink S. J. Molecular Dynamics of Photosystem II Embedded in the Thylakoid Membrane. J. Phys. Chem. B 2017, 121, 3237–3249. 10.1021/acs.jpcb.6b06865. [DOI] [PubMed] [Google Scholar]
- Van Eerden F. J.; Melo M. N.; Frederix P. W. J. M.; Marrink S. J. Prediction of Thylakoid Lipid Binding Sites on Photosystem II. Biophys. J. 2017, 113, 2669–2681. 10.1016/j.bpj.2017.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro-Retamal C.; Bremer A.; Ingólfsson H. I.; Alzate-Morales J.; Caballero J.; Thalhammer A.; Gonzalez W.; Hincha D. K. Folding and Lipid Composition Determine Membrane Interaction of the Disordered Protein COR15A. Biophys. J. 2018, 115, 968–980. 10.1016/j.bpj.2018.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray A.; Gräter F.; Thukral L. Probing Molecular Forces in Multi-Component Physiological Membranes. Phys. Chem. Chem. Phys. 2018, 20, 2155–2161. 10.1039/C7CP05981G. [DOI] [PubMed] [Google Scholar]
- Su J. J.; Thomas A. S.; Grabietz T.; Landgraf C.; Volkmer R.; Marrink S. J.; Williams C.; Melo M. N. The N-Terminal Amphipathic Helix of Pex11p Self-Interacts To Induce Membrane Remodelling During Peroxisome Fission. Biochim. Biophys. Acta, Biomembr. 2018, 1860, 1292–1300. 10.1016/j.bbamem.2018.02.029. [DOI] [PubMed] [Google Scholar]
- Monje-Galvan V.; Klauda J. B. Modeling Yeast Organelle Membranes and How Lipid Diversity Influences Bilayer Properties. Biochemistry 2015, 54, 6852–6861. 10.1021/acs.biochem.5b00718. [DOI] [PubMed] [Google Scholar]
- Silhavy T. J.; Kahne D.; Walker S. The Bacterial Cell Envelope. Cold Spring Harbor Perspect. Biol. 2010, 2, a000414. 10.1101/cshperspect.a000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikaido H. Molecular Basis of Bacterial Outer Membrane Permeability Revisited. Microbiol Mol. Biol. Rev. 2003, 67, 593. 10.1128/MMBR.67.4.593-656.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H.; Cummins D. D.; Edelstein N. B.; Gomez J.; Khan A.; Llewellyn M. D.; Picudella T.; Willsey S. R.; Nangia S. Modeling Diversity in Structures of Bacterial Outer Membrane Lipids. J. Chem. Theory Comput. 2017, 13, 811–24. 10.1021/acs.jctc.6b00856. [DOI] [PubMed] [Google Scholar]
- Lee J.; Patel D. S.; Ståhle J.; Park S.-J.; Kern N. R.; Kim S.; Lee J.; Cheng X.; Valvano M. A.; Holst O.; et al. CHARMM-GUI Membrane Builder with Glycolipids and Lipopolysaccharides for Complex Biological Membrane Simulations. J. Chem. Theory Comput. 2018, in press, 10.1021/acs.jctc.8b01066. [DOI] [PubMed] [Google Scholar]
- Lins R. D.; Straatsma T. P. Computer Simulation of the Rough Lipopolysaccharide Membrane of Pseudomonas Aeruginosa. Biophys. J. 2001, 81, 1037–1046. 10.1016/S0006-3495(01)75761-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschner K. N.; Lins R. D.; Maass A.; Soares T. A. A Glycam-Based Force Field For Simulations of Lipopolysaccharide Membranes: Parametrization and Validation. J. Chem. Theory Comput. 2012, 8, 4719–4731. 10.1021/ct300534j. [DOI] [PubMed] [Google Scholar]
- Piggot T. J.; Holdbrook D. A.; Khalid S. Conformational Dynamics and Membrane Interactions of The E. Coli Outer Membrane Protein Feca: a Molecular Dynamics Simulation Study. Biochim. Biophys. Acta, Biomembr. 2013, 1828, 284–293. 10.1016/j.bbamem.2012.08.021. [DOI] [PubMed] [Google Scholar]
- Wu E. L.; Fleming P. J.; Yeom M. S.; Widmalm G.; Klauda J. B.; Fleming K. G.; Im W. E. Coli Outer Membrane and Interactions with Ompla. Biophys. J. 2014, 106, 2493–2502. 10.1016/j.bpj.2014.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berglund N. A.; Piggot T. J.; Jefferies D.; Sessions R. B.; Bond P. J.; Khalid S. Interaction of the Antimicrobial Peptide Polymyxin B1 with Both Membranes of E. Coli: a Molecular Dynamics Study. PLoS Comput. Biol. 2015, 11, e1004180. 10.1371/journal.pcbi.1004180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter T. S.; Parkin J.; Khalid S. The Free Energy of Small Solute Permeation Through the Escherichia Coliouter Membrane Has a Distinctly Asymmetric Profile. J. Phys. Chem. Lett. 2016, 7, 3446–3451. 10.1021/acs.jpclett.6b01399. [DOI] [PubMed] [Google Scholar]
- Ma H.; Khan A.; Nangia S. Dynamics of Ompf Trimer Formation in The Bacterial Outer Membrane of Escherichia Coli. Langmuir 2018, 34, 5623–5634. 10.1021/acs.langmuir.7b02653. [DOI] [PubMed] [Google Scholar]
- Balusek C.; Gumbart J. C. Role of the Native Outer-Membrane Environment on the Transporter BtuB. Biophys. J. 2016, 111, 1409–1417. 10.1016/j.bpj.2016.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming P. J.; Patel D. S.; Wu E. L.; Qi Y.; Yeom M. S.; Sousa M. C.; Fleming K. G.; Im W. BamA POTRA Domain Interacts with a Native Lipid Membrane Surface. Biophys. J. 2016, 110, 2698–2709. 10.1016/j.bpj.2016.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orekhov P. S.; Kholina E. G.; Bozdaganyan M. E.; Nesterenko A. M.; Kovalenko I. B.; Strakhovskaya M. G. Molecular Mechanism of Uptake of Cationic Photoantimicrobial Phthalocyanine Across Bacterial Membranes Revealed by Molecular Dynamics Simulations. J. Phys. Chem. B 2018, 122, 3711–3722. 10.1021/acs.jpcb.7b11707. [DOI] [PubMed] [Google Scholar]
- Patel D. S.; Re S.; Wu E. L.; Qi Y.; Klebba P. E.; Widmalm G.; Yeom M. S.; Sugita Y.; Im W. Dynamics and Interactions of Ompf and LPS: Influence on Pore Accessibility and Ion Permeability. Biophys. J. 2016, 110, 930–938. 10.1016/j.bpj.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehmood S.; Corradi V.; Choudhury H. G.; Hussain R.; Becker P.; Axford D.; Zirah S.; et al. Structural and Functional Basis For Lipid Synergy on The Activity of The Antibacterial Peptide ABC Transporter Mcjd. J. Biol. Chem. 2016, 291, 21656–21668. 10.1074/jbc.M116.732107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abellon-Ruiz J.; Kaptan S. S.; Basle A.; Claudi B.; Bumann D.; Kleinekathofer U.; Van Den Berg B. Structural Basis for Maintenance of Bacterial Outer Membrane Lipid Asymmetry. Nat. Microbiol 2017, 2, 1616. 10.1038/s41564-017-0046-x. [DOI] [PubMed] [Google Scholar]
- Shearer J.; Khalid S. Communication Between the Leaflets of Asymmetric Membranes Revealed From Coarse-Grain Molecular Dynamics Simulations. Sci. Rep. 2018, 8, 1805. 10.1038/s41598-018-20227-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.; Patel D. S.; Park S.; Slusky J.; Klauda J. B.; Widmalm G.; Im W. Bilayer Properties of Lipid a From Various Gram-Negative Bacteria. Biophys. J. 2016, 111, 1750–1760. 10.1016/j.bpj.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice A.; Wereszczynski J. Atomistic Scale Effects of Lipopolysaccharide Modifications on Bacterial Outer Membrane Defenses. Biophys. J. 2018, 114, 1389–1399. 10.1016/j.bpj.2018.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J. B.; Klauda J. B. Lipid Chain Branching at the Iso- and Anteiso-Positions in Complex Chlamydia Membranes: a Molecular Dynamics Study. Biochim. Biophys. Acta, Biomembr. 2011, 1808, 323–331. 10.1016/j.bbamem.2010.07.036. [DOI] [PubMed] [Google Scholar]
- Pandit K. R.; Klauda J. B. Membrane Models of E. Coli Containing Cyclic Moieties in the Aliphatic Lipid Chain. Biochim. Biophys. Acta, Biomembr. 2012, 1818, 1205–1210. 10.1016/j.bbamem.2012.01.009. [DOI] [PubMed] [Google Scholar]
- Khakbaz P.; Klauda J. B. Probing The Importance of Lipid Diversity in Cell Membranes Via Molecular Simulation. Chem. Phys. Lipids 2015, 192, 12–22. 10.1016/j.chemphyslip.2015.08.003. [DOI] [PubMed] [Google Scholar]
- Hwang H.; Paracini N.; Parks J. M.; Lakey J. H.; Gumbart J. C. Distribution Of Mechanical Stress In The Escherichia Coli Cell Envelope. Biochim. Biophys. Acta, Biomembr. 2018, 1860, 2566–2575. 10.1016/j.bbamem.2018.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu P. C.; Samsudin F.; Shearer J.; Khalid S. It Is Complicated: Curvature, Diffusion, and Lipid Sorting Within the Two Membranes of Escherichia Coli. J. Phys. Chem. Lett. 2017, 8, 5513–5518. 10.1021/acs.jpclett.7b02432. [DOI] [PubMed] [Google Scholar]
- Karunakar R. P.; Solano C. J. F.; Kleinekathöfer U. Simulations of Outer Membrane Channels and Their Permeability. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 1760–1771. 10.1016/j.bbamem.2015.12.020. [DOI] [PubMed] [Google Scholar]
- Weerheim A.; Ponec M. Determinationofstratumcorneum Lipid Profile by Tape Stripping in Combination with High-Performance Thin-Layer Chromatography. Arch. Dermatol. Res. 2001, 293, 191–199. 10.1007/s004030100212. [DOI] [PubMed] [Google Scholar]
- Guo S.; Moore T. C.; Iacovella C. R.; Strickland L. A.; Mccabe C. Simulation Study of The Structure and Phase Behavior of Ceramide Bilayers and The Role of Lipid Headgroup Chemistry. J. Chem. Theory Comput. 2013, 9, 5116–5126. 10.1021/ct400431e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartkamp R.; Moore T. C.; Iacovella C. R.; Thompson M. A.; Bulsara P. A.; Moore D. J.; Mccabe C. Investigating The Structure of Multicomponent Gel-Phase Lipid Bilayers. Biophys. J. 2016, 111, 813–823. 10.1016/j.bpj.2016.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore T. C.; Hartkamp R.; Iacovella C. R.; Bunge A. L.; Mccabe C. Effect of Ceramide Tail Length on The Structure of Model Stratum Corneum Lipid Bilayers. Biophys. J. 2018, 114, 113–125. 10.1016/j.bpj.2017.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore T. C.; Iacovella C. R.; Leonhard A. C.; Bunge A. L.; Mccabe C. Molecular Dynamics Simulations of Stratum Corneum Lipid Mixtures: a Multiscale Perspective. Biochem. Biophys. Res. Commun. 2018, 498, 313–318. 10.1016/j.bbrc.2017.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang E.; Klauda J. B. Simulations of Pure Ceramide and Ternary Lipid Mixtures As Simple Interior Stratum Corneummodels. J. Phys. Chem. B 2018, 122, 2757–2768. 10.1021/acs.jpcb.8b00348. [DOI] [PubMed] [Google Scholar]
- Höltje M.; Förster T.; Brandt B.; Engels T.; Von Rybinski W.; Höltje H. D. Molecular Dynamics Simulations of Stratum Corneum Lipid Models: Fatty Acids and Cholesterol. Biochim. Biophys. Acta, Biomembr. 2001, 1511, 156–167. 10.1016/S0005-2736(01)00270-X. [DOI] [PubMed] [Google Scholar]
- Das C.; Noro M. G.; Olmsted P. D. Lamellar and Inverse Micellar Structures of Skin Lipids: Effect of Templating. Phys. Rev. Lett. 2013, 111, 183–185. 10.1103/PhysRevLett.111.148101. [DOI] [PubMed] [Google Scholar]
- Del Regno A.; Notman R. Permeation Pathways Through Lateral Domains in Model Membranes of Skin Lipids. Phys. Chem. Chem. Phys. 2018, 20, 2162–2174. 10.1039/C7CP03258G. [DOI] [PubMed] [Google Scholar]
- Wennberg C. L.; Narangifard A.; Lundborg M.; Norlén L.; Lindahl E. Structural Transitions in Ceramide Cubic Phases During Formation of The Human Skin Barrier. Biophys. J. 2018, 114, 1116–1127. 10.1016/j.bpj.2017.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R.; Rai B. Molecular Dynamics Simulation Study of Translocation of Fullerene C 60 Through Skin Bilayer: Effect of Concentration on Barrier Properties. Nanoscale 2017, 9, 4114–4127. 10.1039/C6NR09186E. [DOI] [PubMed] [Google Scholar]
- Gupta R.; Rai B. Electroporation of Skin Stratum Corneum Lipid Bilayer and Molecular Mechanism of Drug Transport: a Molecular Dynamics Study. Langmuir 2018, 34, 5860–5870. 10.1021/acs.langmuir.8b00423. [DOI] [PubMed] [Google Scholar]
- Holdbrook D. A.; Huber R. G.; Piggot T. J.; Bond P. J.; Khalid S. Dynamics of Crowded Vesicles: Local and Global Responses To Membrane Composition. PLoS One 2016, 11, e0156963. 10.1371/journal.pone.0156963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goose J. E.; Sansom M. S. P. Reduced Lateral Mobility of Lipids and Proteins in Crowded Membranes. PLoS Comput. Biol. 2013, 9, e1003033. 10.1371/journal.pcbi.1003033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javanainen M.; Hammaren H.; Monticelli L.; Jeon J. H.; Miettinen M. S.; Martinez-Seara H.; Metzler R.; Vattulainen I. Anomalous and Normal Diffusion of Proteins and Lipids in Crowded Lipid Membranes. Faraday Discuss. 2013, 161, 397–417. 10.1039/C2FD20085F. [DOI] [PubMed] [Google Scholar]
- Guigas G.; Weiss M. Effects of Protein Crowding on Membrane Systems. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 2441–2450. 10.1016/j.bbamem.2015.12.021. [DOI] [PubMed] [Google Scholar]
- Jeon J. H.; Javanainen M.; Martinez-Seara H.; Metzler R.; Vattulainen I. Protein Crowding in Lipid Bilayers Gives Rise To Non-Gaussian Anomalous Lateral Diffusion of Phospholipids and Proteins. Phys. Rev. X 2016, 6, 021006. 10.1103/PhysRevX.6.021006. [DOI] [Google Scholar]
- Javanainen M.; Martinez-Seara H.; Metzler R.; Vattulainen I. Diffusion of Integral Membrane Proteins in Protein-Rich Membranes. J. Phys. Chem. Lett. 2017, 8, 4308–4313. 10.1021/acs.jpclett.7b01758. [DOI] [PubMed] [Google Scholar]
- Sharma S.; Kim B. N.; Stansfeld P. J.; Sansom M. S. P.; Lindau M. A Coarse Grained Model For a Lipid Membrane with Physiological Composition and Leaflet Asymmetry. PLoS One 2015, 10, e0144814. 10.1371/journal.pone.0144814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doktorova M.; Weinstein H. Accurate in silico modeling of asymmetric bilayers based on biophysical principles. Biophys. J. 2018, 115, 1638–1643. 10.1016/j.bpj.2018.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett W. F. D.; Maccallum J. L.; Hinner M. J.; Marrink S. J.; Tieleman D. P. Molecular View of Cholesterol Flip-Flop and Chemical Potential in Different Membrane Environments. J. Am. Chem. Soc. 2009, 131, 12714–12720. 10.1021/ja903529f. [DOI] [PubMed] [Google Scholar]
- Huber R. G.; Marzinek J. K.; Holdbrook D. A.; Bond P. J. Multiscale Molecular Dynamics Simulation Approaches to The Structure and Dynamics of Viruses. Prog. Biophys. Mol. Biol. 2017, 128, 121–132. 10.1016/j.pbiomolbio.2016.09.010. [DOI] [PubMed] [Google Scholar]
- Reddy T.; Sansom M. S. P. Computational Virology: From the Inside Out. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 1610–1618. 10.1016/j.bbamem.2016.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayton G. S.; Voth G. A. Multiscale Computer Simulation of The Immature HIV-1 Virion. Biophys. J. 2010, 99, 2757–2765. 10.1016/j.bpj.2010.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy T.; Shorthouse D.; Parton D. L.; Jefferys E.; Fowler P. W.; Chavent M.; Baaden M.; Sansom M. S. P. Nothing to Sneeze at: a Dynamic and Integrative Computational Model of an Influenza a Virion. Structure 2015, 23, 584–597. 10.1016/j.str.2014.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzinek J. K.; Holdbrook D. A.; Huber R. G.; Verma C.; Bond P. J. Pushing the Envelope: Dengue Viral Membrane Coaxed Into Shape by Molecular Simulations. Structure 2016, 24, 1410–1420. 10.1016/j.str.2016.05.014. [DOI] [PubMed] [Google Scholar]
- Reddy T.; Sansom M. S. P. The Role of the Membrane in the Structure and Biophysical Robustness of The Dengue Virion Envelope. Structure 2016, 24, 375–382. 10.1016/j.str.2015.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirawan M.; Fibriansah G.; Marzinek J. K.; Lim X. X.; Ng T. S.; Sim A. Y. L.; Zhang Q.; Kostyuchenko V. A.; Shi J.; Smit S. A.; et al. Mechanism of Enhanced Immature Dengue Virus Attachment to Endosomal Membrane Induced by prM Antibody. Structure 2018, 27, 1–15. 10.1016/j.str.2018.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado M. R.; Gonzalez H. C.; Pantano S. MD Simulations of Viruslike Particles with Supra CG Solvation Affordable to Desktop Computers. J. Chem. Theory Comput. 2017, 13, 5106–5116. 10.1021/acs.jctc.7b00659. [DOI] [PubMed] [Google Scholar]
- Lagüe P.; Zuckermann M. J.; Roux B. Lipid-mediated Interactions Between Intrinsic Membrane Proteins: A Theoretical Study Based On Integral Equations. Biophys. J. 2000, 79, 2867–2879. 10.1016/S0006-3495(00)76524-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagüe P.; Zuckermann M. J.; Roux B. Lipid-mediated Interactions Between Intrinsic Membrane Proteins: Dependence On Protein Size And Lipid Composition. Biophys. J. 2001, 81, 276–284. 10.1016/S0006-3495(01)75698-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen J. J.; Grime J. M. A.; Rossman J. S.; Voth G. A. Entropic Forces Drive Clustering And Spatial Localization Of Influenza A M2 During Viral Budding. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E8595–E8603. 10.1073/pnas.1805443115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rassam P.; Copeland N. A.; Birkholz O.; Toth C.; Chavent M.; Duncan A. L.; Cross S. J.; Housden N. G.; Kaminska R.; Seger U.; et al. Supramolecular Assemblies Underpin Turnover Of Outer Membrane Proteins In Bacteria. Nature 2015, 523, 333–336. 10.1038/nature14461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavent M.; Duncan A. L.; Rassam P.; Birkholz O.; Hélie J.; Reddy T.; Beliaev D.; Hambly B.; Piehler J.; Kleanthous C.; et al. How Nanoscale Protein Interactions Determine The Mesoscale Dynamic Organisation Of Bacterial Outer Membrane Proteins. Nat. Commun. 2018, 9, 2846. 10.1038/s41467-018-05255-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munguira I.; Casuso I.; Takahashi H.; Rico F.; Miyagi A.; Chami M.; Scheuring S. Glasslike Membrane Protein Diffusion In A Crowded Membrane. ACS Nano 2016, 10, 2584–2590. 10.1021/acsnano.5b07595. [DOI] [PubMed] [Google Scholar]
- McGuffee S. R.; Elcock A. H. Diffusion, Crowding and Protein Stability in a Dynamic Molecular Model of the Bacterial Cytoplasm. PLoS Comput. Biol. 2010, 6, e1000694. 10.1371/journal.pcbi.1000694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu I.; Mori T.; Ando T.; Harada R.; Jung J.; Sugita Y.; Feig M. Biomolecular Interactions Modulate Macromolecular Structure And Dynamics In Atomistic Model Of A Bacterial Cytoplasm. eLife 2016, 5, e19274. 10.7554/eLife.19274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler P. F.; Chavent M.; Duncan A.; Helie J.; Koldsø H.; Sansom M. S. P. Membrane Stiffness Is Modified By Integral Membrane Proteins. Soft Matter 2016, 12, 7792–7803. 10.1039/C6SM01186A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y.; Arkhipov A.; Schulten K. Simulations of Membrane Tubulation by Lattices of Amphiphysin N-BAR Domains. Structure 2009, 17, 882–892. 10.1016/j.str.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H.; Schulten K. Membrane Sculpting By F-BAR Domains Studied by Molecular Dynamics Simulations. PLoS Comput. Biol. 2013, 9, e1002892. 10.1371/journal.pcbi.1002892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simunovic M.; Voth G. A.; Callan-Jones A.; Bassereau P. When Physics Takes Over: BAR Proteins and Membrane Curvature. Trends Cell Biol. 2015, 25, 780–792. 10.1016/j.tcb.2015.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simunovic M.; Srivastava A.; Voth G. A. Linear Aggregation of Proteins on the Membrane as a Prelude to Membrane Remodeling. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 20396–20401. 10.1073/pnas.1309819110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simunovic M.; Evergren E.; Golushko I.; Prevost C.; Renard H. F.; Johannes L.; Mcmahon H. T.; Lorman V.; Voth G. A.; Bassereau P. How Curvature-Generating Proteins Build Scaffolds on Membrane Nanotubes. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 11226–11231. 10.1073/pnas.1606943113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Q.; Zhang L.; Wu Z.; Yethiraj A. Generation and Sensing of Membrane Curvature: Where Materials Science and Biophysics Meet. Curr. Opin. Solid State Mater. Sci. 2013, 17, 164–174. 10.1016/j.cossms.2013.06.002. [DOI] [Google Scholar]
- Johnson G. T.; Autin L.; Al-Alusi M.; Goodsell D. S.; Sanner M. F.; Olson A. J. Cellpack: a Virtual Mesoscope to Model and Visualize Structural Systems Biology. Nat. Methods 2015, 12, 85–91. 10.1038/nmeth.3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodsell D. S.; Franzen M. A.; Herman T. From Atoms to Cells: Using Mesoscale Landscapes To Construct Visual Narratives. J. Mol. Biol. 2018, 430, 3954–3968. 10.1016/j.jmb.2018.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astrom J. A.; Karttunen M. Cell Aggregation: Packing Soft Grains. Phys. Rev. E 2006, 73, 062301. 10.1103/PhysRevE.73.062301. [DOI] [PubMed] [Google Scholar]
- Madhikar P.; Astrom J.; Westerholm J.; Karttunen M. CellSim3D: GPU Accelerated Software For Simulations Of Cellular Growth And Division In Three Dimensions. Comput. Phys. Commun. 2018, 232, 206–213. 10.1016/j.cpc.2018.05.024. [DOI] [Google Scholar]
- Boyd K. J.; May E. R. BUMPy: A Model-Independent Tool For Constructing Lipid Bilayers Of Varying Curvature And Composition. J. Chem. Theory Comput. 2018, 14, 6642. 10.1021/acs.jctc.8b00765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrant J. D.; Amaro R. E. LipidWrapper: An Algorithm for Generating Large-Scale Membrane Models of Arbitrary Geometry. PLoS Comput. Biol. 2014, 10, e1003720. 10.1371/journal.pcbi.1003720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowet J. M.; Buchoux S.. Limondada: Lipid Membranes Open Network And Database; https://www.limonadamd.eu. [Google Scholar]
- Zhang L.; Han J.; Wang H.; Car R.; Weinan E. Deepcg: Constructing Coarse-Grained Models Via Deep Neural Networks. J. Chem. Phys. 2018, 149, 034101. 10.1063/1.5027645. [DOI] [PubMed] [Google Scholar]
- Bereau T.; Distasio R. A. Jr.; Tkatchenko A.; Von Lilienfeld O. A. Non-Covalent Interactions Across Organic and Biological Subsets of Chemical Space: Physics-Based Potentials Parametrized From Machine Learning. J. Chem. Phys. 2018, 148, 241706. 10.1063/1.5009502. [DOI] [PubMed] [Google Scholar]
- Bejagam K. K.; Singh S. K.; An Y.; Deshmukh S. A. Machine Learnt Coarse-Grained Models. J. Phys. Chem. Lett. 2018, 9, 4667–4672. 10.1021/acs.jpclett.8b01416. [DOI] [PubMed] [Google Scholar]
- Lemke T.; Peter C. Neural Network Based Prediction of Conformational Free Energies-A New Route toward Coarse-Grained Simulation Models. J. Chem. Theory Comput. 2017, 13, 6213–6221. 10.1021/acs.jctc.7b00864. [DOI] [PubMed] [Google Scholar]
- Wang J.; Wehmeyer C.; Noé F.; Clementi C.. Machine Learning of Coarse-Grained Molecular Dynamics Force Fields. arXiv 2018, arXiv:1812.01736v1. [DOI] [PMC free article] [PubMed] [Google Scholar]