

Abstract

Membrane lipids interact with proteins in a variety of ways, ranging from providing a stable membrane environment for proteins to being embedded in to detailed roles in complicated and well-regulated protein functions. Experimental and computational advances are converging in a rapidly expanding research area of lipid–protein interactions. Experimentally, the database of high-resolution membrane protein structures is growing, as are capabilities to identify the complex lipid composition of different membranes, to probe the challenging time and length scales of lipid–protein interactions, and to link lipid–protein interactions to protein function in a variety of proteins. Computationally, more accurate membrane models and more powerful computers now enable a detailed look at lipid–protein interactions and increasing overlap with experimental observations for validation and joint interpretation of simulation and experiment. Here we review papers that use computational approaches to study detailed lipid–protein interactions, together with brief experimental and physiological contexts, aiming at comprehensive coverage of simulation papers in the last five years. Overall, a complex picture of lipid–protein interactions emerges, through a range of mechanisms including modulation of the physical properties of the lipid environment, detailed chemical interactions between lipids and proteins, and key functional roles of very specific lipids binding to well-defined binding sites on proteins. Computationally, despite important limitations, molecular dynamics simulations with current computer power and theoretical models are now in an excellent position to answer detailed questions about lipid–protein interactions.
1. Introduction
Cell membranes, both enveloping internal organelles and the entire cell, are essential structural elements in all kingdoms of life. They are composed of a complex mixture of lipids and proteins with a lateral structure that has yet to be resolved in detail and depends on the cell type and the location of the membrane. Cell membranes enable tight regulation of the flow of energy, information, nutrients, and metabolites. Although historically these roles were largely attributed to membrane proteins, it is becoming increasingly clear that lipid–protein interactions are essential determinants of membrane-bound processes.1,2 As a direct consequence, membrane proteins are important drug targets,3 and a growing body of evidence shows that the lipid component of membranes is an essential player in understanding the mechanism of action and targeting of many drugs.4,5
Cell membranes consist of two leaflets of lipids, outer and inner, arranged in a tail-to-tail manner. Lipids are often grouped into three main classes: glycerophospholipids, sphingolipids, and sterols. Several modifications of the polar head groups and hydrophobic tails exist, thus increasing the complexity in lipid diversity to more than a thousand types identified in living cells.6−8 The lipid repertoire is different across the three domains of life, with many lipid types that are unique for archaea, bacteria, or eukaryotes. This variety in principle allows an almost infinite combination of lipid–protein interactions and roles varying from basic structural roles to specifically switching on and off proteins in response to highly controlled signaling events involving lipid modifications.
Although initially the lipid matrix was largely considered as the solvent media for membrane proteins and a simple barrier separating two compartments electrically and chemically, for instance in the fluid mosaic model,9,10 the potential importance of lipid–protein interactions was recognized several decades ago.11,12 Some examples are the early studies on the modulatory effect of cholesterol on rhodopsin,13−16 impacts of bilayer composition and fluidity on the kinetics of the gramicidin assembly and ion transport17,18 or the effect of lipid thickness in regulating the conformational transitions of the Ca2+-ATPase19 and many others.12
Detailed atomistic computer simulations of membrane proteins became feasible in the 1990s, although an extensive body of important computational work using less-detailed models by, among others, Mouritsen et al. led to the development of influential models for lipid–protein interactions, including the mattress model based on hydrophobic mismatch.20 An early example of atomistic simulations addressing similar questions used a set of proteins of different size to investigate the range of membrane perturbations due to the proteins.21 Significant emphasis was also placed on the connections between experimental data and results of atomistic simulations probing protein impact on the lipid structuring and dynamics in the first and second coordination shell.22 Over the past 20 years computer power has increased by at least 4 orders of magnitude, and simulation has become a standard technique to study aspects of membrane protein biophysics.23
In this review, we attempt to provide a comprehensive overview of molecular dynamics simulation studies aimed primarily at some aspect of lipid–protein interactions, published in the past 5 years. We specifically exclude studies of membrane proteins that include lipids but do not investigate lipid–protein interactions, for instance studies of selectivity mechanisms in ion channels,24 conformational changes upon ligand-binding of G-protein coupled receptors,25 or studies dealing with membrane proteins modulated by (membrane soluble) compounds such as cofactors, drugs, or phytochemicals.26 Coarse-grained (much less detailed than atomistic models or coarse-grained models that retain chemical specificity like Martini; see below) and mean-field models have been used to model protein–protein interactions mediated through the membrane and large-scale phenomena such as the remodeling of the membrane by caveolins27 or BAR domains.28 While these are essential and highly interesting processes, and the insights from more simplified models on fundamental principles of protein–protein interactions and membrane remodeling are fascinating, they are outside the scope of this review. We also largely exclude studies of membrane-active peptides,29,30 peripheral membrane proteins,31 and small model systems like WALP32 and other synthetic peptides as well as gramicidin A.33 The universe of single-span membrane proteins is extremely large,34 but only a fraction has been modeled and experimental structural biology on these proteins often excludes the transmembrane part. We have attempted to include most recent simulation studies in this area based on biological interest, computational approach, and available structural information. We include a selection of experimental studies to provide context for the simulation studies without any attempt to be comprehensive, as well as a selection of earlier simulation papers (reviewed for instance in refs (35 and 36)) for illustrative purposes.
First, we discuss some general aspects of lipid–protein interactions, including experimental and computational approaches to study these interactions. We then proceed with individual sections on specific protein families, in part organized based on the detail of available studies and in part on biological classification. We conclude with a broader outlook.
1.1. Lipid–Protein Interactions
Lipid–protein interactions fall on a continuum ranging from very specific interactions between specific lipids directly affecting a selective binding site on a protein to much more generalized interactions involving changes in the physical properties of the membrane, which may affect membrane structure on mesoscopic length scales or properties such as dimerization of two membrane proteins.
At a more detailed level, interactions between lipids and proteins can, rather artificially, be classified based on specificity, on functional effects on proteins, or on effects on local membrane properties. Examples of these cases are given below in the detailed review of actual cases of lipid–protein interactions and summarized in the Discussion. These different cases also relate to strengths and weaknesses of computational methods in ways that should perhaps be more clearly recognized in the literature. We highlight two issues that are important to keep in mind in the interpretation of the detailed papers below.
First, binding sites for lipids present in various membrane proteins range from explicit, in the sense that they are visible in crystal structure or mass-spectrometry (with its own issues), to much less well-defined like the CRAC, CARC, and CCM motifs that are thought to be involved in cholesterol binding, despite large numbers of such motifs that do not bind cholesterol and large numbers of cholesterol that bind but not to such motifs.37−39 In principle, these binding sites have a well-defined binding affinity, but this is rarely reported in experimental studies or calculated. In MD simulations, a particular binding event is often characterized in terms of occupancy of a specific lipid in a specific protein site observed in the trajectory. However, given the length of what is considered “long” simulations in standards of 2018, occupancy is a poor statistical metric to discuss binding as it does not allow quantitative assessment of binding affinity due to exhaustive sampling challenges. More advanced free energy calculations could be used to test for a specific site/lipid combination what the binding free energy is, the kon rate, and from those the koff rate. This is rarely done as of yet but eminently possible and as simpler simulations identify more interesting candidates we expect this to be a growing use of simulations. This would also allow testing the effect of mutations in proteins, analogous to what has been common in the drug binding field for a long time.
Second, many experimental studies characterize lipid–protein interactions in terms of functional relevance of a particular lipid, following some kind of activity assay or in ion channels electrophysiological measurements. Although such associations have drawn considerable attention in the literature, this type of data does not cleanly translate to lipid–protein interactions, generally speaking. Below, we discuss a number of cases, including a lipid-dependent ABC transporter, cholesterol effects on GPCRs, and binding of PIP lipids to potassium channels. Although simulations can identify interactions between these lipids and proteins, they do not directly access the functional data from the experiments, which remains an area for improvements both experimentally and computationally.
1.2. Characterization of Lipid–Protein Interactions
1.2.1. Experimental Techniques
In the last few decades, several experimental techniques have been used to answer questions related to the identification of lipid binding sites on the protein surface, the type of lipids found associated with proteins, and how such lipids influence protein function.40,41
Structural characterization of lipid binding sites can be achieved with X-ray crystallography, where high-resolution structures are obtained by extracting membrane proteins from lipid bilayers using detergents to allow solubilization.42,43 Because of the use of detergents for protein purification and crystallization, lipids that would be naturally associated with the protein are often lost, or if they are copurified and crystallized, they might not be identified unequivocally in the final structure. However, despite the many challenges encountered in working with membrane proteins, tightly bound lipids that survived the solubilization and purification steps have been identified via X-ray crystallography for several proteins.44−48 Structures of membrane proteins in the presence of lipids have also been solved via electron crystallization. Here, a purified membrane protein is reconstituted within a lipid bilayer to form a periodic array (known as 2D crystal).49−51 Lipids surround each copy of the membrane protein, thus mimicking the native lipid environment. This technique has allowed for the visualization of layers of lipids in contact with the protein surface (annular lipids) and the study of how such lipids interact with the protein.52−57
Recent developments in single particle cryo-electron microscopy (cryo-EM) overcome many of the challenges of membrane proteins crystallography, by generating high-resolution 3-dimensional structures from images of proteins in solution.58 Applications of cryo-EM span from investigating the structure and function of large complexes to the elucidation of side chains packing in proteins, with possible implications for drug discovery.59 The near atomic resolution that is now achieved with this technique has dramatically improved the structural landscape of membrane proteins and can provide molecular details on lipid binding, as seen, for instance, for the TRPV1 ion channel,60 the TPC1 channel,61 or the ABC transporter ABCG2.62
Lipid binding to membrane proteins and the local lipid composition in proximity of the protein can also be studied using fluorescent methods. Förster resonance energy transfer (FRET),63 for example, allowed the characterization of a specific interaction between a sphingolipid and the COPI machinery protein p24,64 while fluorescence cross-correlation spectroscopy (FCCS) was employed to probe protein oligomerization as a function of the lipid composition for the mitochondrial voltage-dependent anion channel (VDAC).65
Additionally, biophysical studies of lipid–protein interactions can be obtained using nanodiscs, discoidal membranes with a diameter of 8–17 nm and enclosed by helical scaffolding proteins.66,67 Using membrane proteins in nanodiscs protein function is assessed in controlled lipid environments.68−70 Recently, the application of detergent-free approaches that use specific copolymers to extract proteins and native lipids into nanodiscs has provided a new tool to characterize the lipid environment of a given membrane protein, as shown for the potassium channel KcsA.71
Quantitative analyses and identification of native lipid species tightly associated with membrane proteins can be achieved via mass-spectroscopy (MS) studies, where membrane–protein complexes can be solubilized in nonionic detergents to provide resistance to the electronspray ionization step, thus allowing for stable complexes in gas phase.72 This technique has been applied to several transporters,48,73 and other membrane proteins,74−76 providing information on lipid selectivity, stoichiometry of binding and possible roles played by lipids in protein mechanism. Native MS techniques allow the thermodynamics of lipid binding to membrane proteins to be studied, shedding light on the mechanism of molecular recognition between lipids and proteins. For the E. coli ammonia channel AmtB, for example, individual lipid binding events have been characterized in detail, highlighting the different thermodynamics signature of different lipid types, addressing the contribution of the acyl tail length to binding thermodynamics,77 and providing insights on how lipid binding can modulate protein–protein interactions.78 Mass-spectrometry techniques can also be combined with click-chemistry methods to identify lipid-binding proteins in native environments. Such methods allow proteins to be cross-linked with selected phospholipids carrying photoactivable groups, upon activation via UV light, and the product can be further characterized by mass-spectometry.79,80
Infrared (IR) spectroscopy has also been applied to the study of protein or peptide interactions with membranes.81,82 For instance, using attenuated total reflection Fourier transform (ATR FT-IR) spectroscopy Güler et al. investigated the effect of K+ binding on the interactions between anionic lipids and the Na+-coupled betaine symporter BetP, revealing weakening of such interactions due to increase K+ concentrations following osmotic stress.83 Other spectroscopy methods that can be applied to the study of both specific and nonspecific lipid–protein interactions include nuclear magnetic resonance (NMR) methods, such as solid-state NMR84 and solution NMR, as in the case of outer membrane proteins of Gram-negative bacteria,85,86 as well as a powerful technique specific to lipid–protein interactions using saturating transfer between protein and lipids.87
1.2.2. Computer Simulations: Atomistic, Coarse-Grained, and Continuum Models
Computer simulations of biomolecular systems span a wide range of time and length scales, with essential approximations at each level. For the purpose of studying lipid–protein interactions the most useful levels are atomistic simulations, in which every atom is described explicitly, and coarse-grained simulations, which average in some form over multiple atoms. At the more detailed side of the spectrum simulations incorporating electronic structure at some level, either through polarizable force fields88 or explicit quantum treatment of some or all degrees of freedom,89 are widely used in biomolecular simulation but have not yet been used in detailed studies of lipid–protein interactions, in part because only very basic lipid parameters are available. At the coarse-grained end of the spectrum, there is a rich literature on qualitative lipid models with very simple bead types describing generic head groups and generic tails, as well as on continuum mechanics models, usually based on elasticity theory. Continuum approaches can be used to study generic lipid–protein interactions as well as the detailed response of a membrane to an atomistic protein structure.90 We include some studies using the latter approach in this review but omit the large literature on more generic models.91
At the atomistic level, simulations numerically solve classical equations of motion with a time step of 1 or 2 fs, typically over billions of steps, resulting in a trajectory describing the position of all atoms as a function of time. The main ingredients of a successful simulation are an accurate description of the interactions between the atoms and sufficient sampling. These interactions are described by an interaction function and a set of parameters called a force field. For lipid–protein simulations, these parameters have to be consistent between the chemical groups in the lipids and in the proteins, for instance ideally the same parameters describe the alkane side chain of a leucine residue and the alkane chain of lipids; well-tested against experimental data; and cover the universe of relevant lipids and, typically less limiting, proteins. In practice, the most commonly used force fields for lipid–protein interaction studies are based on the CHARMM92−94 and AMBER95−99 families of force fields, as these continue to be developed for both lipids and proteins. Other force fields can be quite appropriate for specific purposes but tend to have more limited options in terms of the availability of lipid types (Slipids,100−103 GROMOS,104−109 and OPLS-AA110). Developing parameters for new lipid types is not trivial, as even if the chemical groups are the same or similar to existing lipid models these parameters might not reliably transfer to new lipid types and testing against experiments remains essential.
Although a detailed atomistic model matches the chemistry and physics of interest in lipid–protein interactions, the maximum time and length scales that can be simulated are limited with current computer power. In many cases, lipid–protein interactions are studied in relatively simple models with a single protein in a lipid environment, increasingly commonly a lipid mixture. At this level, atomistic simulations are a reasonable approach, but when protein–protein interactions or larger systems are of interest less detailed models are currently essential. An accompanying paper in this issue reviews progress and issues in simulations of larger models.111 Many papers in this review use, sometimes combined with atomistic simulations in a serial multiscale approach, coarse-grained (CG) models.112 The most widely used CG model is the Martini model,113−115 in which on average four non-hydrogen atoms are combined into a single interaction site. This, clearly, reduces the chemical specificity and level of detail, but results in a 2–3 orders of magnitude speed-up compared to atomistic simulations. Martini has been parametrized primarily on the accurate solubility of molecular fragments in different environments, combined with extensive testing and fine-tuning on actual lipids and other biomolecules. The transferable nature and simplification of the molecular topologies compared to atomistic simulations makes it relatively easy to model new lipids, and consistency between lipids and proteins is nearly guaranteed by the construction of the interaction parameters of Martini. The coarse-grained nature also leads to significant limitations. Two major limitations are the lack of accurate internal dynamics of proteins, which are usually treated with an elastic network to maintain their secondary and tertiary structure with only limited fluctuations, and the lack of detailed chemical features, including hydrogen bonding and water entropy, that play a role in specific interactions between lipids and proteins.
There are several other approaches. Marrink et al. review all currently available coarse-grained force fields for biomembrane simulation, but in practice these have not been used to our knowledge for studying detailed lipid–protein interactions.111 We do review papers116,117 here that use models derived from the multiscale CG (MS-CG) method.118 This method in general starts from atomistic reference simulations of the same or a closely related system and systematically derives interaction potentials at coarser levels. At a more detailed level, Feig and co-workers have developed a coarse-grained model that can be combined with atomistic detail in part of the system, including recent tests on membrane proteins, using an implicit membrane environment.119 Such hybrid approaches may become more widely applicable in the near future.
In addition to the accuracy of the parameters that describe the interactions between atoms, obtaining enough sampling to reliably draw conclusions is a major challenge in lipid–protein simulations. As the complexity of systems increases, the simulation time required to obtain converged averages also increases. As an example, in a recent study we estimated that it takes 30–50 μs to equilibrate a complex lipid mixture120 around a protein and probably more given the nature of the force field used in this study.121 In contrast, interactions between liquid crystalline POPC and a small membrane protein probably require simulation lengths of the order of hundreds of nanoseconds to obtain correct averages for many properties. In practice, a handful of common molecular dynamics software packages is used for the vast majority of published studies due to their flexibility, efficiency, and ability to work on a variety of modern computational architectures. These include AMBER,122,123 NAMD,124 GROMACS,125,126 as well the special purpose machine with its own software, ANTON.127 In addition to advanced software, advanced algorithms can also help in increasing sampling. There is a vast literature on this topic,128 but applications in lipid–protein interactions reviewed here have been limited so far. However, there is major potential in this area. For instance, algorithms that can swap lipids rather than wait for lipids to switch place by diffusion could speed up sampling mixtures;129 Hamiltonian replica exchange and similar methods could improve the sampling of slow interactions at interfaces between lipids and proteins;130 and path-based free energy methods such as umbrella sampling can be used to obtain quantitative data on binding affinities from simulations as well as on the thermodynamics of lipid mixing.131
While accuracy of the force field and degree of sampling are fundamental issues, simulations of lipid–protein interactions are sufficiently complicated that there are practical issues to consider too. In principle, how a starting model is created in an MD simulation, whether atomistic or coarse-grained, is irrelevant and only affects the time it takes to equilibrate the system, which is not of scientific interest. However, in practice these equilibration times can become so long that it is beneficial to create systems that are as close as possible to the equilibrium structure (which is generally not known). In the early days of membrane simulations, ca. 20 years ago, this was a very active field with a large number of technical approaches to create starting structures. As computers become faster, it became more feasible to just equilibrate a system, but in lipid mixtures with equilibration times of microseconds attention to starting structures is warranted again. In addition, in complex lipid mixtures with proteins and hundreds of thousands of atoms a procedure that generates starting structures without errors that may be very difficult to find is very valuable. We do not attempt to review all possible approaches, but point out that the two most common methods at the moment are the online tool CHARMM-GUI,132,133 which can create a variety of membrane and other systems for different MD packages, and the Martini tool Insane.134 In general, both setup and equilibration are easier with Martini due to the nature of the interaction function, and in fact Martini simulations could be used as an intermediate to generate complex atomistic simulations.135
2. G Protein-Coupled Receptors (GPCRs)
G protein–coupled receptors (GPCRs) are the largest superfamily of membrane proteins and the number one drug target.136 They are characterized by a highly conserved seven transmembrane helix (TM) topology with a ligand-binding extracellular site and G-protein coupling intracellular site (Figure 1). Ligand binding on the extracellular site triggers a sequence of conformational changes in the TM domain that results in the activation of a membrane-anchored G protein. Due to their localization on the plasma membrane, GPCR function and activity are exposed to all of the biophysical processes that characterize cell membranes. In the past few years, significant advances have been made in understanding this relationship between GPCRs and their lipid environment.
Figure 1.

Representative structures of GPCRs. From left to right, the structures of rhodopsin,137 the β2 adrenergic receptor (β2AR),138 and the smoothened receptor139 are shown as cartoons, with the 7 transmembrane (TM) helices highlighted in different colors. For β2AR, the three subunits of the G protein (Gα, Gβ, and Gγ) are shown in light gray and blue cartoons. For the smoothened receptor, the extracellular cysteine-rich domain (CRD) and the long extracellular loop 3 are shown in light blue and gray cartoons, respectively. The top panels show a side view of the receptors, while the bottom panels provide a view from the extracellular side. Substrates or agonists are labeled and shown in white spheres, and the hydrophobic region of the membrane is highlighted in gray. In, intracellular side; Out, extracellular side; ICL, intracellular loop; ECL, extracellular loop.
Early evidence for the importance of lipid–protein interactions in GPCR function and activity dates back to the early to mid-1990s, when a series of discoveries was made regarding cholesterol modulation of rhodopsin and oxytocin function. The equilibrium between meta-rhodopsin I and meta-rhodopsin II depends on cholesterol concentration13 and was assumed to be a result of cholesterol’s ability to alter the physical properties of the bilayer. Albert et al. a few years later, however, showed that another mechanism is possible, namely a direct, structurally specific, interaction between rhodopsin and cholesterol.14 Gimpl et al. studied this cholesterol-modulatory effect on the ligand–binding activity of two different GPCRs with peptide–hormone ligands: the oxytocin and the cholecystokinin receptor.15,16 They found that this effect is caused by altering the membrane properties in the case of cholecystokinin, but by a “putatively specific cholesterol–receptor interaction” for the oxytocin receptor. More recently, the cholecystokinin type 1 receptor was shown to be sensitive to membrane cholesterol levels, as opposed to its type 2 relative cholecystokinin receptor which does not share this cholesterol sensitivity.140−142
In the last two decades a plethora of experimental and theoretical techniques have been used to investigate the existence and significance of lipid GPCR interactions. Two illustrative, although not exclusive, examples of the progress made are the several solved crystallographic structures of GPCRs with bound lipids143−152 and a series of papers published recently clearly and unequivocally demonstrating the importance of cholesterol in activating the class F GPCR Smoothened in hedgehog signaling.139,153−156
2.1. GPCR–Lipid Interactions
The most commonly used classification of GPCRs uses their sequence homology to categorize them into classes A–F or, alternatively, into the GRAFS system (with each letter of the acronym standing for the most representative member of the family, e.g., R = rhodopsin).136,157 Class A (or rhodopsin-like) GPCRs are the largest, most studied, and hence best-understood GPCR family by practically any metric. MD literature follows published GPCR structures. Therefore, GPCR–lipid interactions have mainly been studied in the context of class A GPCRs. We begin with a discussion of the prototypical rhodopsin and continue with aminergic receptors, in no particular order, concluding with a brief section on other miscellaneous GPCR–lipid interaction papers. Considering the vast GPCR literature, no single review, no matter how comprehensive, can do justice at detailing every aspect of their biology. We limit the discussion to the aspects outlined in the introduction of this review and refer the reader to several other excellent reviews.39,158−163 When necessary, the Ballesteros–Weinstein numbering scheme will be used to identify residues.164
2.1.1. Rhodopsin
Rhodopsin is found in rod cells embedded in a membrane that is rich in ω-3 polyunsaturated lipids and cholesterol (although the content of the latter varies with cell age). Polyunsaturated lipids, e.g., docosahexaenoic acid (DHA), have a stabilizing effect on rhodopsin structure and increase its activity; cholesterol has the opposite effect. Multiple short-time scale MD simulations of rhodopsin165 embedded in a bilayer with PE and PC lipids with mixed saturation lipid tails and cholesterol showed that rhodopsin forms a few, potentially specific, interactions with DHA, the unsaturated tail, but no specific interaction with stearic acid, the saturated tail, and cholesterol. A later study, employing 1.6 μs simulation of the same system, however, provided more insight into the cholesterol–rhodopsin interactions.166 Rhodopsin contains three structural regions that exhibit increased affinity for cholesterol molecules: the extracellular sides of the TM2–TM3 bundle and TM7 helix and the intracellular side of the TM1–TM2–TM4 helices. Extending these simulations even further in time scale using the Martini model confirmed the preferential interaction of cholesterol and DHA with rhodopsin and suggested a preference of rhodopsin for PE headgroup over PC headgroups.167 The effect of polyunsaturated tails and PE headgroups on rhodopsin activity has also been observed in experiments on reconstituted rhodopsin receptors in model membranes.168 These experiments show that PE lipids, by creating a negative curvature, affect the MI/MII equilibrium of rhodopsin. Increased levels of PE lipids shift the equilibrium toward the MII state. The implication here is the possibility of a lipid-regulated inactive–active state transitioning of the receptor. More recent MD simulation studies on lipid–rhodopsin interactions, however, highlight a more dualistic nature of lipid–protein interactions whereby membrane lipids express their modulatory effects on rhodopsin by acting, simultaneously, as allosteric modulators and by altering membrane physical properties (e.g., membrane fluidity).169
2.1.2. Adrenergic Receptors
Adrenergic receptors are among the best-studied GPCRs, and their lipid–interaction profile has consequently been studied and characterized extensively. In 2008, Hanson et al. solved the structure of the human β2AR receptor bound to two cholesterol molecules, strongly indicating structurally specific cholesterol-binding sites on the receptor surface.144 Helices I–IV of the β2AR form a shallow groove that is sufficient to accommodate the binding of two cholesterol molecules, with different binding affinities. These observations enabled the definition of a cholesterol-binding motif, which is found in 21% of human class A GPCRs.144 This domain, coined cholesterol consensus motif (CCM), is formed by residues in helices II and IV of the receptor and differs from the CRAC/CARC domains in that it is formed by a spatial arrangement of residues rather than a linear one. Cholesterol interactions with adrenergic receptors have been observed in several other crystal structures.145,147 MD simulations at both the atomic and coarse-grained level of the receptor embedded in a POPC bilayer in the presence or absence of cholesterol have shown that β2AR interacts with cholesterol at several potential interaction sites or hot-spots (some of which match those observed in several crystal structures), quantified by cholesterol occupancy time from the simulation trajectories.170,171 A possible functional role of these additional putative interaction sites remains to be established, but it is reassuring that the simulations agree with each other and can reproduce with good agreement the cholesterol interaction sites observed in solved crystal structures. Interestingly, the interaction of cholesterol with these binding sites is dynamic and ranges from nanosecond to microsecond time scales and might serve as a basis for dividing cholesterol−β2AR binding events into short- and long-lived. This is an interesting prospect considering recent experimental evidence supporting it.172 Experiments with unfolding temperature assays and saturation transfer difference NMR showed that cholesterol binds to the β2AR with high affinity–slow exchange rate and low affinity–fast exchange rate, respectively. Control experiments reveal both these types of binding events to be specific to cholesterol.172
When the β2AR and β1AR are each simulated separately in cholesterol containing POPC bilayers in microsecond-long atomistic MD simulations, the resulting cholesterol-interaction profiles of the receptors differ significantly from each other.173 Most notably, the H1–H8 interface of β2AR seems to interact preferentially with two cholesterol molecules, while the same interaction is missing from the β1AR, which can be attributed to the slightly different resides lining the interface. More generally, this means that lipid interaction data from one GPCR may not be easily extended to other members of even the same GPCR family despite sharing a high structural similarity.
Important advances in the understanding of β2AR–lipid interactions were made by Kobilka and colleagues who showed that cholesterol and phospholipids affect the kinetics and stability of the receptor.174,175 Using force spectroscopy methods and cholesteryl hemisuccinate as a cholesterol analog they showed that cholesterol increases the stability of almost all structural elements of the β2AR, presumably by making it difficult for the protein to sample its conformational landscape.174 This finding is also supported by recent experimental176 and computational177 studies. In the latter study, Manna et al. carried out atomistic MD simulations of the human β2AR in, among other setups, DOPC bilayers of varying cholesterol concentration and showed that the conformational landscape sampled by β2AR is reduced significantly at or above 10 mol % concentrations of cholesterol (Figure 2).177 Further simulations suggest that this reduced conformational flexibility of β2AR is a direct result of cholesterol–receptor interactions not of indirect modulation through altered bilayer bulk properties. In addition, the binding of cholesterol agreed well with previous studies,170−172 as several known interaction sites were retrieved. Time-correlation data also reveal some binding sites to be dependent on cholesterol concentration and others independent of it, further supporting the division of cholesterol binding sites according to their binding affinity or exchange rate.177
Figure 2.

Effect of cholesterol on β2AR activation. A. Definition of two distance parameters used to measure the conformational changes as a function of time.177 The D3.32–S5.46 Cα atom distance (denoted LL) measures fluctuations in the ligand-binding site of the receptor, and the R3.50–E6.30 Cα atom distance (denoted LG) captures fluctuations in the G protein binding interface. B and C. The conformational space probed by the simulations in pure DOPC and DOPC–10% Chol concentration bilayer, respectively, plotted as a function of these two distance parameters.177 Cholesterol significantly decreases the conformational space sampled by the receptor. Adapted with permission from ref (177). Copyright 2016 Manna et al. Licensed under Creative Commons Attribution 4.0.
While cholesterol is clearly the major lipid type to consider when looking at lipid–protein interactions in GPCRs, Dawaliby et al.175 showed recently, experimentally, that phospholipids also regulate the activity of the β2AR by acting as allosteric modulators. By testing phospholipids with different headgroup types they found that phospholipids, depending on the nature of the headgroup, can shift the equilibrium toward either the active or inactive state of the receptor, with DOPG, DOPS, and DOPI favoring the former and DOPE favoring the latter. Remarkably, this effect, in the case of negatively charged phospholipids is dose-dependent and present even in the absence of a bilayer, probably by the headgroup interacting with the cytoplasmic side of the receptor. These studies show the effect of cholesterol and phospholipids to be independent of ligand binding. The importance of phospholipids has been underscored in several other experimental studies for other GPCRs.178,179
MD simulations have been quite successful in reaffirming and even predicting these phospholipid−β2AR interactions. Neale et al. showed that POPG stabilizes the active state of the embedded β2AR through specific interactions with Arg3.50 on the intracellular side of the receptor.180 This interaction of POPG is stronger and more frequent when compared to equivalent interactions of the zwitterionic POPC. While it is not possible to attribute this stabilizing activity of POPG solely on its interaction with Arg3.50, it is clear that it opposes the closure of H6, a critical step in β2AR activation. More recently, microsecond-length atomistic simulations of β2AR embedded in DOPG, DOPE, or DOPC phospholipids reaffirm the differential effect of negatively charged versus zwitterionic lipids on the activation of β2AR.181 DOPC, DOPE, and DOPG partially inactivate, fully deactivate, and stabilize the active-state of the β2AR, respectively. While the exact details of these interactions remain to be deciphered, MD simulations in combination with many experimental findings have already provided a wealth of information with regards to cholesterol– and phospholipid–β2AR interactions.
MD simulations have also hinted to a possible role of PIP lipids in mediating lipid–protein interactions by demonstrating preferential localization of these lipids in microsecond-long simulations.121,182 Native mass spectrometry studies complemented with CG MD simulations have highlighted the importance of PIP2 lipids. In particular, experiments by Yen et al. demonstrate that PIP2 lipids do not only affect the stability of the active state of these receptors but also exert influence on its coupling to G proteins.183 The GPCRs studied were β1AR, adenosine A2A receptor (A2AR), and neurotensin receptor 1 (NTSR1), although the CG MD simulations points to this effect likely being conserved in other class A GPCRs as well.183
2.1.3. Adenosine Receptors
Evidence for the importance of cholesterol in the adenosine receptor function and activity dates to at least 2008.184 The same year a 2.6 Å crystal structure of the A2A adenosine receptor (A2AAR) was published185 Lyman et al.186 took this opportunity to investigate, using MD simulations, the behavior of the receptor in the presence and absence of cholesterol. In bilayers where cholesterol is absent, helix II of the A2AAR is remarkably unstable if the ligand is removed from the simulations.186 If, however, cholesterol is introduced in these simulations, the stability of helix II is restored, thanks to cholesterol–protein interactions. Later MD simulations provided a clearer picture of the cholesterol–A2AAR interaction profile,187 where three cholesterol binding sites are identified. Two of these cholesterol hotspots are on the extracellular side and one on the intracellular side of the receptor. One of the cholesterol interaction sites observed on the extracellular leaflet of the receptor interacting on the interface between helices II and III, is confirmed by a previously solved X-ray crystallographic structure of the receptor;148 however, the other cholesterol hotspots observed lack cross-validation, possibly due to limited sampling, especially considering more recent results below. MD simulations of the A2AAR embedded in POPC and POPE bilayers suggest that the receptor samples a larger part of the conformational landscape if it is embedded in the former, although this may be due to generally slower dynamics in the more ordered POPE bilayer.188 It is tempting to look at similar findings obtained for the human β2AR and extend those, but it remains to be established if such extrapolation of data is sensible. More recent MD simulations of the A2AAR, by combining data at both the atomistic and coarse-grained level, have identified two new cholesterol–interaction sites.189 One of these is located on the intracellular side of the interface formed by helices V and VI (not validated experimentally) and the other on the extracellular side of helix VI, matching experimental evidence.148
2.1.4. Serotonin Receptors
Experimental findings revealed that cholesterol depletion alters ligand binding and G-protein coupling to the serotonin 1A receptor.190 Cholesterol also increases the stability of the human serotonin 1A receptor191 and in giant unilamellar protein-vesicles has been observed to increase oligonucleotide exchange.192 Due to the lack of a crystal structure of serotonin receptors, initial MD simulations used homology models. One such study, using Martini coarse–grained MD simulations, showed that in cholesterol containing POPC bilayers the embedded homology model of the serotonin 1A receptor displays several preferential cholesterol interaction sites.193 One of these interactions is with helix V which represents one (out of three) CRAC motifs found on the receptor. Atomistic MD simulations of the activity of serotonin 1A and serotonin 2A receptors as a function of bilayer cholesterol content, currently portray a conflicting picture as to if cholesterol decreases the conformational flexibility of the receptor194 or increases it,195 respectively. Ganglioside GM1 and sphingolipids also interact with the serotonin 1A receptor.196,197 Shan et al. simulated the conformational changes of the serotonin 2A receptor induced by its binding to three different ligands (full agonist, partial agonist, and inverse agonist).198 They found a noticeably different response of the receptor depending on the ligand bound and that these conformational changes are relayed into the surrounding membrane environment of the embedded receptor, confirmed by specific interactions of the receptor with cholesterol and distinct membrane perturbations around the TM core of the receptor.
2.1.5. Other GPCRs
MD simulations either alone or in tandem with experimental studies have been used to explore the lipid–binding properties of other GPCRs as well. Marino et al. carried out extensive Martini coarse–grained MD simulations of the mu-opioid receptor (μOR) in highly realistic membrane compositions,182 showing specific interactions of μOR with cholesterol and with the negatively charged PIP lipids, which surprisingly, differ to some extent depending on the conformational state of the receptor. The implication of these differences is, however, unclear. Similar simulations of the delta-opioid receptor (δOR) confirm the relative enrichment of cholesterol adjacent to the receptor compared to bulk concentrations.121 This type of simulations in complex bilayers reveal a unique interaction profile of opioid receptors with individual membrane lipids (cholesterol, PIP lipids) and groups of lipids that share a chemical feature (polyunsaturated, fully saturated, headgroup type, etc.), hinting toward a functionally relevant involvement in GPCR activity and oligomerization (see below).121,182
Cholesterol is an essential component in Smoothened receptor activation.139,153−156 Cholesterol binds to the cysteine-rich domain (CRD) of Smoothened. MD simulations showed that cholesterol confers stability to the CRD domain but did not affect the stability of the TM domain.139 In a series of experiments, Huang et al.153 and Luchetti et al.154 concurrently showed that cholesterol is the endogenous ligand that activates Smoothened. Indeed, cholesterol is not only necessary but also sufficient for Smoothened activation.154 Additionally, MD simulations coupled with PMF calculations point to the existence of an interaction site for cholesterol formed by TM2 and TM3 helices of Smoothened on the extracellular site of the receptor.199
Cholesterol also affects the activity and stability of the neurotensin receptor 1 (NTS1).200 NTS1 also displays a potential preference for PS lipids201 and its G protein coupling affinity is significantly increased in the presence of PE lipids.202 This dependency of G protein coupling by GPCRs on the lipid environment is also evident for the cannabinoid type 2 receptor (CB2R), which increases G protein activation in the presence of anionic lipids.179 Interestingly, CB2R and the structurally similar CB1R seem to differ in their cholesterol-interaction profiles.203 Sphingosine-1-phosphate receptor 1, in extensive Martini coarse-grained simulations, consistently interacts with cholesterol and PIP2 lipids.204 Membrane localization of the dopamine D1 receptor is dependent on membrane cholesterol and sphingolipid levels.205 GPCR–cholesterol interactions, mainly by way of CRAC motifs, have also been demonstrated for class A chemokine receptors,206 for class C metabotropic glutamate receptors,207 and T2R4 bitter taste receptor208 perhaps highlighting the presence and importance of this motif across very different GPCRs.
2.2. GPCR Scramblase Activity and Lipid Entry Events
An unexpected finding from MD simulations of GPCRs is the occasionally observed complete entrance of lipid molecules from the bilayer into the receptor. Considering the lack of structural data pointing toward such a possibility, however, not much attention has been paid to this phenomenon. So far, MD simulations have demonstrated lipid entry for several GPCRs, including opsin,209 cannabinoid CB2 receptor,210 sphingosine-1-phosphate receptor,211 β2AR,180 and A2AAR.212 11-cis-retinal uptake by the opsin receptor is likely achieved via the TMH5/6 interface as an entrance port and either TMH1/7213 or TMH5/6209 serving as an all-trans-retinal exit site. In MD simulations of the cannabinoid type 2 receptor, 2-arachidonoylglycerol (2-AG) partitions out of the POPC bilayer and interacts specifically with the TMH6/7 interface, where it also enters the receptor.210 In contrast, simulations of the sphingosine-1-phosphate receptor showed a POPC lipid to interact specifically with and enter the receptor through the TMH1/7 interface.211 The TMH1–TMH7 distance increases significantly during the course of the simulation to accommodate this lipid entry. Endogenous ligand binding to GPCRs is expected. More puzzling is the observed complete entry of bilayer lipids inside receptors that do not have lipids as natural ligands. In simulations of the β2AR, a POPC lipid sometimes (estimated at 6% of the time) accesses the inside of the receptor via the TMH6/7.180 This complete lipid entry is, however, only observed with one of the two force-fields the authors used. More recently, MD simulations revealed that cholesterol completely enters the A2A adenosine receptor through TM5/6, in a process that appears to be dependent on the bulk properties of the surrounding bilayer.212 While in the interior of the receptor, cholesterol preferentially samples an area of the receptor that binds the ZM241385 ligand in the A2A adenosine receptor crystal structure,185 hinting that cholesterol could affect ligand binding properties. The latter was confirmed using biotinylation assay experiments.212 It is unclear what the implications of these findings are, but they may add an additional layer of complexity to GCPR–lipid interactions.
Experiments by Menon et al. showed that opsin acts as a phospholipid scramblase.214 In a later study, it was discovered that rhodopsin as well is a phospholipid scramblase with an activity of >10 000 phospholipids per protein per second.215 The authors demonstrated that this activity of rhodopsin is independent of the conformational state of the receptor, which means that phototransduction and scramblase activity are not coupled to each other. Furthermore, β2AR and A2AAR, as well, scramble phospholipids, hinting that this activity could be shared by all class A GPCRs.215 MD simulations and Markov state model analysis reveal that the mechanism for phospholipid translocation involves a hydrophilic pathway that is created between TMH6 and TMH7 of opsin, through which the phospholipid headgroup crosses from the intracellular to the extracellular leaflet, while the lipid tail remains in the bilayer.216 The simulations also characterize the conformational changes necessary for this translocation event to occur. Considering this observed scramblase activity is dependent on a thin low-cholesterol membrane, it remains to be seen how this is affected by cholesterol- and sphingolipid-containing membranes.215 While sampling these events is challenging and may require specialized methods, MD simulations have already demonstrated that they can be a useful tool to shed light on this novel aspect of GPCR activity.
2.3. GPCR Oligomerization
The physiological role of oligomerization of several GPCRs is a topic of ongoing discussion (Figure 3). It has been observed and characterized as functionally important in many experiments,217−220 but at the same time doubts have been raised about several aspects of its occurrence and importance.221,222 Computer simulations have been used to study membrane-mediated aspects of oligomerization, which we discuss below. A more detailed discussion of oligomerization and other aspects of their relevance and function is available in several other reviews.219,222−225
Figure 3.
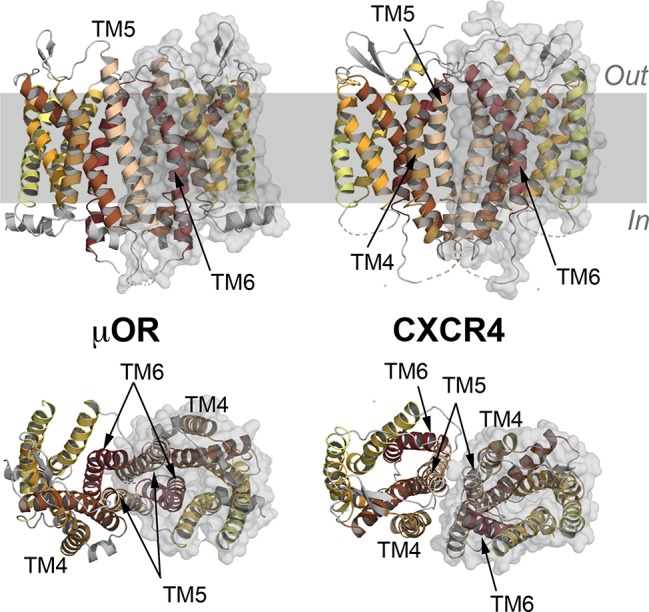
Experimental structures of GPCR dimers. Side view (upper panels) and the view from the extracellular side (bottom panels) of the (left) μ opioid receptor (μOR) dimer149 and (right) the chemokine receptor type 4 (CXCR4) dimer.226 The 7 transmembrane (TM) helices of the receptors are highlighted in different colors and shown as cartoons, while one of the monomers is also represented with a transparent gray surface. Substrates bound to these dimers are not shown for clarity.
Initial MD simulations of GPCR dimerization were focused on rhodopsin. Periole et al. carried out coarse-grained MD simulations of several rhodopsin receptors embedded in PC bilayers of different lipid tail lengths and found that the increase in hydrophobic mismatch with shorter lipid tail length resulted in local thickening of the bilayer, particularly noticeable near TM2, TM4, and TM7 helices and thinning near TM1, TM5, TM6, and TM8.227 These results suggest that hydrophobic mismatch acts as a driving force for receptor oligomerization. Additionally, the most prominent interaction surface involved a symmetric arrangement of TM1, TM2, and H8, although other dimer interfaces were also observed. Later studies, however, affirmed that the primary mode of interaction in rhodopsin dimers involves the TM1/TM2 on the extracellular side and H8 on the intracellular side.228,229 NMR experiments show that hydrophobic mismatch modulates the MI/MII rhodopsin intermediate equilibrium, with oligomerization shifting the equilibrium toward MI, and local thickening of the membrane to compensate for the hydrophobic mismatch favors the MII intermediate.230
To characterize the role of hydrophobic mismatch in GPCR oligomerization, Mondal et al. used Martini coarse-grained simulations of the prototypical β1AR and β2AR and studied the energy penalty associated with the residual hydrophobic mismatch, RHM (the energy penalty resulting from the inability of the membrane to completely counter hydrophobic mismatch).231 They found that the highest energy cost of RHM for the β2AR monomer was at TM1, TM4, and TM5. Oligomerization of β2AR occurs via the TM1 and TM4/TM5 interface and significantly decreases this energy penalty, thus highlighting hydrophobic mismatch as a possible driving force behind oligomerization. The highly homologous β1AR in contrast showed a high RHM only on its TMH1, indicating a preference of the receptor to form dimers, not oligomers, through its TMH1/TMH1 interface. In their simulations, Mondal et al. did not see a significant effect of cholesterol on receptor oligomerization.231 A different study, however, proposes a modulatory role of cholesterol in β2AR dimer interfaces.171 Without cholesterol present, the preferred β2AR dimer interface involves TMH4 and TMH5. Increasing cholesterol concentrations, however, changed the relative involvement of TM helices at the dimer interface, with 50% cholesterol favoring a predominantly TMH1 and TMH2 interface. This modulatory role of cholesterol was attributed to its preferred localization at TMH4.171
Opioid receptors are another GPCR family with extensively studied oligomerization properties, mainly by the Filizola lab and collaborators. Umbrella sampling free energy calculations and metadynamics simulations revealed a short-lived interaction of delta-opioid receptors (δOR), mainly through TMH4, but with a relative involvement of TMH5, as well.232,233 In a later, more extensive study, Provasi et al. used the Martini model to simulate the preferred di/oligomerization pattern of the main opioid receptor (OR) subtypes: μOR, δOR, and κOR, simulated in their homomeric and main heteromeric form.234 OR utilize a limited number of interfaces which are consistent among the subtypes simulated but differ in their relative fraction of occurrence. Also, consistent seems to be the shared lack of either TM3 or TM7 involvement in dimer interfaces. Analysis of different kinetic parameters pointed toward different propensities and association rates of dimers to form, depending on the interface involved, and an active role of membrane lipids, including cholesterol, in guiding these associations. Local lipid exchange and persistence time might affect and serve a modulatory effect, on the kinetic favorability of different interfaces to form.234 Experimental and computational tools have shown that μOR homodimerization is facilitated by cholesterol through a Cys3.55-palmitoyl–cholesterol interaction.235 Simulations of the active- and inactive-state μOR in more realistic plasma model provided additional insights into how the membrane environment guides receptor dimerization.182 Notably, TMH1, TMH5, and TMH6 induced an ordering of lipids, in contrast to TMH4, which induced a more disordered region in the membrane. These local membrane adaptations facilitate receptor dimerization and affect the preferred dimer interfaces formed. Interestingly, the latter is also dependent on the conformational state of the receptor.
A recent computational study of A2AAR and dopamine D2 receptor oligomerization suggests that it is highly DHA concentration dependent.236 DHA displays a preferential interaction with each receptor and high-levels of it increase receptor heteromerization. Bioluminescence resonance energy transfer (BRET) experiments showed the number of oligomers formed, however, to be independent of DHA levels, underscoring a kinetic modulation by DHA. Prasanna et al., using coarse-grained MD simulations, demonstrated the lipid-dependent oligomerization of serotonin 1A receptor.237 Specifically, they observed two main dimer interfaces involving TMH1/TMH2 and TMH4/TM5/TMH6, and they highlight the importance of cholesterol in modulating the stability and flexibility of these dimers. Pluhackova et al. simulated the chemokine receptor type 4 (CXCR4) in pure phospholipid bilayers and noticed that the most prominent dimerization interface involved TM1/TM5–7.238 In bilayers containing cholesterol, however, this interface was unavailable due to a cholesterol molecule binding between TM1 and TM7. Instead, a new symmetric TM3/TM4 interface was observed that seems to be supported by experiment, highlighting not only its regulatory role but also the importance of considering its effect in studies like this. A more recent study further characterized chemokine receptor homo- and heterodimerization patterns and their dependence on membrane cholesterol.239 Coarse-grained MD simulations were also used to study the oligomerization of S1P1 receptors.204
A common theme that emerges from both experimental and theoretical results is the dual modulatory effect of lipids on GPCRs, by way of either altering membrane bulk properties or specific interactions with the receptor. While quantifying this effect and the contributions from each component is challenging for GPRCs, current evidence suggests the modulatory role of membrane lipids on receptor function and activity is achieved primarily by specific lipid–protein interactions. However, to make matters more complicated, that does not seem to be the case for rhodopsin, where the modulatory role is currently mainly attributed to local membrane curvature instead.240
It is challenging to compare the lipid–protein interaction results across GPCR members given the differences in experimental and simulation setups employed. The current literature suggests that extending results from one GPCR to others, simply based on structural similarity and sequence identity might not be enough. MD simulation data show that many GPCRs interact preferentially with cholesterol through several interaction sites. Some of these are likely of functional importance, others seem not to be. Recent simulations show that microseconds are required to obtain statistically significant results on cholesterol–GPCR interactions, a bar that most of the older papers do not reach.
GPCR oligomerization is a topic of debate and caution is required when interpreting results from MD simulations. Regardless of that, however, it seems that insofar as GPCR association is a true phenomenon, it is regulated and dependent on specific membrane lipids (cholesterol, DHA) and membrane physical properties (hydrophobic mismatch, lipid order/disorder) that act as molecular driving forces.
3. Ion Channels
Ion channels are integral membrane proteins enabling water-filled pathways for ion permeation. Ion channels change conformations from open (or activated) and nonconducting (inactivated) states in response to a variety of stimuli, ranging from perturbation of the membrane potential, binding of modulating ligands, changes in the intracellular pH, and osmotic stress.241 Substantial evidence shows that membrane composition plays a role in ion channel structure and function. As for other membrane proteins, in general, two mechanisms have been proposed for lipid–protein interactions: either through specific interactions between lipids and proteins or nonspecific through changes in membrane physicochemical properties that affect ion channel dynamics. A plethora of recent studies showed that ion channel gating dynamics or conductance properties are sensitive to the presence of specific lipids such as phosphatidylcholine (PC), phosphatidylethanolamine (PE), cholesterol, phosphatidylinositol 4,5 biphosphate (PIP2), ceramides, or polyunsaturated fatty acids (PUFA).242−248
Atomistic and coarse grained molecular dynamic (MD) simulations have been useful tools to understand how lipid–protein interactions can regulate the function of a variety of ion channels.36,249,250 In the following sections, we will focus on the insight given by MD simulations in relation to the role of lipid–protein interactions in regulating the activity and function of different members of the ion channel superfamily. Simulations have primarily addressed more specific interactions between lipids and proteins. Two different classes of bound lipids have been described to explain such interactions: (i) A first layer of lipids surrounding the transmembrane portion of the membrane protein and (ii) lipids that bind to certain grooves, especially between protein subunits.
3.1. Channels with General Voltage-Gated Potassium Channel Pore Architecture
Voltage-gated ion channels (VGICs) are responsible for the rapid and selective conduction of Na+, K+, Cl–, H+, or Ca2+. The group includes very diverse structural topologies ranging from the homotetrameric channels such as the superfamily of K+-selective channels and Na+-selective bacterial channels (NaVab, NachBac, etc.) to the membrane-inserted structures formed by large monomeric α-subunits in eukaryotic Na+ and Ca2+-selective channels (Figure 4). Regardless of the topology (e.g., monomer vs homotetramer), two major structural elements can be easily identified.241
Figure 4.

Different structural topologies of selected ion channels. From left to right the structures of the Shaker Kv1.2,251 the two-pore domains K2P1,252 the inwardly rectifying Kir2.2253 potassium channels, and the structure of the NavAb sodium channel254 are shown in cartoons. All structures, with one notable exception of K2P1 channels, are functional tetramers comprised of four identical monomers. Monomers are colored from yellow to orange to dark brown. The trans-membrane helices forming voltage-sensing domains (VSD) are labeled S1 to S4, while two transmembrane helices forming the pore domain (PD) in voltage-gated channels are labeled as S5 to S6; Kir2.2 pore is comprised of two transmembrane helices, outer and inner corresponding to S5 and S6 helices in Kv1.2 or NaVab channels. The location of the pore helix present in many structures of ion channels is shown on the top-view of Kir 2.2. The functional dimer of the K2P1 channel is comprised of two pore domains (P1 and P2) with each domain formed by TM helices M1 to M4, where helices M1 and M3 are outer helices of P1 and P2 protomers. For the K2P1 and the Kir2.2 channels the positions of potassium ions in the filter region are shown by purple spheres. In, intracellular side; out, extracellular side. The membrane region is highlighted in gray.
The alpha-subunit of voltage-gated Na+ and Ca2+-selective channels contains four repeat domains with structural features resembling those found in homotetrameric channels. The first highly conserved structural element present in most of the VGICs is the voltage-sensing domain (VSD) formed by four trans-membrane helices labeled S1 to S4. The main functional role for VSDs is to change conformation in response to changes in the membrane potential, opening or closing the ion permeation pathway, known as activation/inactivation. The rapid response to changes in the membrane surface electrostatics is achieved by the presence of an unusually (for membrane-inserted proteins) large number of positive charges (labeled as R1, R2, ..., R7–8) in the trans-membrane S4 helix (the voltage-sensor). The second essential structural element is the pore domain (PD) formed by the S5–S6 helices, which contains the selective permeation pathway. Additional cytoplasmic N- and C-terminal subunits are common in all voltage-gated channels and may contain a variety of regulatory domains.241 The activation process depends on the electro-mechanical coupling between the VSD and pore domain. The movement of the VSD in response to the membrane potential results in opening or closing the intracellular gate in the pore domain, thus opening or closing ion flow through the pore. Other closely related families include the family of the bacterial K+ selective channels46 and eukaryotic inwardly rectifying K+ channels, both containing only 2 transmembrane (TM) helices per monomer (S5–S6 equivalent) that contribute to the tetrameric pore domain,255−257 voltage-sensitive phosphatases, and voltage-gated H+ channels comprised of S1–S4 forming both VSD and permeation pathway.258,259
While assembly (monomeric vs oligomeric) of functional units may vary depending on the family, these proteins are known to be modulated by a variety of membrane lipids. Here we review primarily the role of lipids in the regulation of voltage-gated ion channel function.260,261 Specific lipid binding in a state-dependent manner to TM domain (TMD) of voltage-gated ion channels may change the kinetics and/or energetics of activation and/or impact maximal conductance. The taxonomy and classification of voltage-gated ion channels is complex.260,261 Below we take a pragmatic approach rather than a rigorous systematic approach in the organization of the sections. Transient receptor potential (TRP) channels also adopt a structural architecture similar to that of voltage-gated potassium channels, but we discuss them below in section 3.2 together with other ligand-gated channels.
3.1.1. Lipid Interactions with K+-Selective Channels
The determination of the X-ray crystal structure of bacterial channel KcsA containing only S5–S6 TM helices in 1998 and the S1–S5 voltage-gated K+-selective (Kv) mammalian channels47,262 resulted in a large number of modeling studies aimed at understanding of gating and permeation mechanisms present in this superfamily of voltage-gated channels.46,263 Several lipids including cholesterol, ceramides, polyunsaturated fatty acids (PUFAs) and PIP2 have been suggested to play modulatory roles in the activation process and permeation across various K+ selective channels by binding to lipid-specific pockets.
One of the first simulation studies of K+-channel interactions with lipids was performed on the bacterial 2TM channel KcsA.46 KcsA is a homotetrameric channel from the Gram-positive soil bacterium Streptomyces lividans with 2 TMs per monomer (S5–S6, sometimes labeled TM1-TM2). It contains the highly conserved amino-acid sequence in the pore helix and selectivity filter characteristic for potassium channels.47,253,263 The overall pore organization is also well-conserved in K+-selective channels from higher organisms.264 Selective ion conductance in KcsA is controlled by the dynamics of (i) the intracellular or inner gate, which opens at an acidic pH through a movement of the TM2 helices; and (ii) the extracellular or outer gate, that has been proposed to involve dynamics around the selectivity filter, and generally in a nonconductive state when the inner gate is open.265−267 KcsA is arguably the simplest K+ selective channel that contains the pore domain conserved across the superfamily. Therefore, it has been used as a template to investigate lipid interactions with PDs in various families of K+-channels. The crystallographic structure of the channel suggests the presence of binding sites for anionic phospholipids at the extracellular side of the channel. The binding pockets are located in the intersubunit crevices between S6 (TM2), where binding of phospholipid head-groups is enabled by interactions with highly conserved Arg and Thr residues.46,263,268−270 The lipid-facing surface of the S6 helix (TM2) contains a large number of hydrophobic and amphipathic residues, further stabilizing the fatty acid acyl chains of lipids in the proposed binding site.268,269,271 The lipid binding to the intersubunit pockets was proposed to be essential for stabilization of the tetrameric structure and protection from denaturation.271
Atomistic and coarse-grained MD simulations combined with NMR data showed that KcsA preferentially interacts with anionic phospholipids.269 MD simulations performed in DPPC:DPPG bilayers showed marked differences in the protein–lipid binding patterns between KcsA and the chimeric KcsA-Kv1.3 channel. DPPG lipid bound to both channels, but the occupancy in the binding site was higher in KcsA. However, neither lipid is biological for KcsA. Using solid state NMR, electrophysiology, and MD simulations, Van der Cruijsen et al. reported that lipid composition affects the open probability of the prokaryotic potassium channel KcsA.272 Atomistic MD simulations of 25 ns time scale suggested the possible relevance of protein–lipid interactions between the negatively charged amino acids and the positively charged phosphatidylcholine head groups in the pore loop region of KcsA and in the chimera KcsA-Kv1.3, which is directly involved in channel activation. Molina et al. combined mutagenesis, electrophysiology, and MD simulation studies in PC membranes to characterize the role of phospholipids in KcsA clustering, which might be involved in the gating properties of the channel.273 Weingarth et al. proposed an alternative mechanism to clustering to explain lipid regulation in the KcsA channel. They pointed out that Arg residues are a dominant factor in channel-anionic phospholipids interactions.269 In spite of active modeling and experimental work over last two decades, the details of the role of lipid–protein interactions in KcsA function remain unclear.140,263,269,271,273−275
3.1.2. Tentative Lipid Binding Sites in K2P Channels
The K2P channels are structural unique potassium channels containing two pore-domain sequences per subunit (Figure 4).252,276 K2P channels are probably dimers, and the overall pore architecture resembles the inwardly rectifying bacterial K+ channels (like KirBac1.1), whose three-dimensional structures have been determined by crystallography.252,276,277 The structure of K2P1 channel showed tubular regions near fenestration windows between the intracellular cavity and lipid bilayer, which were attributed to alkyl chains from copurified lipids.278 Miller et al.278 proposed that these lipid tails could regulate channel function through steric obstruction of the ion conduction pathway.278 The influence of lipids on the fenestrations within the pore of the K2P1 channel has been investigated with a multiscale approach combining atomistic and coarse-grained MD simulations.279 For several other channels including Navs and Kv channels, the presence of a fenestration between the transmembrane helices forming the pore structure has been also reported and implicated in various lipophilic access mechanisms.252,254,276,280 Aryal et al. reported that in K2P1 the lipid alkyl chains can indeed access the fenestration zone in the open state, albeit without occluding the permeation pathway in the central pore.279 It has been also suggested that the fenestration windows might be contributing to the overall hydrophobicity of the intracavitary space and therefore have an indirect impact on the pore dewetting.
3.1.3. Specific Binding of PIP2 As a Modulator for Channel Function
One of the most detailed mechanistic models involving specific K+ channel-lipid interactions was developed for the charged PIP2 lipid. Its specific modulatory roles have been firmly established for the inwardly 2TM rectifying K+ (Kir) channels (Figure 4).281,282 These channels play an essential role in molecular processes ranging from control of the resting membrane potential to regulation of insulin secretion in pancreatic β cells.283 Selective recruitment of PIP2 lipid is required for the activation process present in the closely related G protein-gated inwardly rectifying potassium (GIRK or Kir3).255,256 Atomic resolution structures are available for Kir and GIRK channels in the presence or absence of PIP2.256,257
One of the first modeling studies combining CG and atomistic simulations to explore lipid–protein interaction in the Kir channels was performed by Stansfeld et al.284 In this study, three Kir channels—the KirBac1.1 channel, a Kir3.1-KirBac1.3 chimera channel, and a homology model of the Kir6.2 channel—were embedded in a POPC bilayer with 4 PIP2 lipid molecules localized in the cytoplasmic leaflet. Analysis of time-averaged density maps revealed PIP2 preferential localization at four sites present in all three Kir channels structures, located at a conserved binding site.285 At physiological pH, PIP2 is expected to have a charge of −4, with one of two phosphates protonated, enabling interactions between positive charges located in the cytoplasmic N-terminus and the pore domains, allowing complex control over the gating process. The authors proposed that relatively minor differences in the sequence of cytoplasmic gating may result in marked differences between eukaryotic Kir channel and bacterial Kir channel activation or inhibition by PIP2, respectively. The modeling study showed that, once recruited, PIP2 remains bound over 5 μs-long CG simulations. Several of the residues identified in this study involved in lipid–protein interactions were later confirmed by the high-resolution crystal structure of the Kir2.2 channel in complex with four short chain (dioctanoyl) PIP2 lipids.286 Moreover, additional analysis of the interactions between the channel and the PIP2 lipids found in crystal structures253,286 and a phosphatidic acid bound state286,287 provided further support for a specific role of PIP2 as a gating modifier with conserved binding sites.286 Recently, Lacin et al. combined atomistic MD simulations with electrophysiology to investigate the effect of PIP2 with the GIRK channel which revealed substantial differences between GIRKs and channels from Kir family.288 The authors proposed that, in contrast to Kir channels, PIP2 binds and specifically interacts with the highly conserved arginine located in the C-linker, which is essential for coupling between the cytoplasmic and trans-membrane domains.
The PIP2 lipids play an important role in the gating dynamics of canonical voltage-gated K+ channels (Kv). The binding of PIP2 at the interface between VSD and PD allows the state-dependent lipid-mediated coupling of the VSD domain and the proximal residues from the C-terminal/intracellular gate located in the distal S6 helix. The intracellular surface in many Kv channels contains a large number of basic residues and therefore the presence of PIP2 plays a significant role in reducing electrostatic repulsion between basic residues in the VSD and pore domain, stabilizing the open or closed states of the channel.244 Electrophysiological studies combined with mutagenesis unambiguously showed that the presence of PIP2 lipids is an essential element in the gating process of the Kv7.1 channel.289,290 The decreased affinity of Kv7.1 for PIP2 due to congenital mutations is associated with long QT syndrome.291 All-atom MD simulations were performed on the Kv1.2-based homology models for the open and closed states of Kv7.1.292 This work showed that PIP2 preferentially interacts with the S4–S5 linker in the open state of the Kv7.2 channel, whereas it contacts the S2–S3 loop in the closed state (Figure 5). The modeling was supported by electrophysiology and mutagenesis studies. The direct interactions of PIP2 lipids with the S4–S5 linker seems to lower both the voltage sensitivity and the current amplitude. The secondary binding site in the S2–S3 loop linker affects the current amplitude only. The proposed VSD-localized binding pockets for PIP2 are in agreement with previously reported sites from electrophysiology experiments and atomistic MD simulations.293−296
Figure 5.
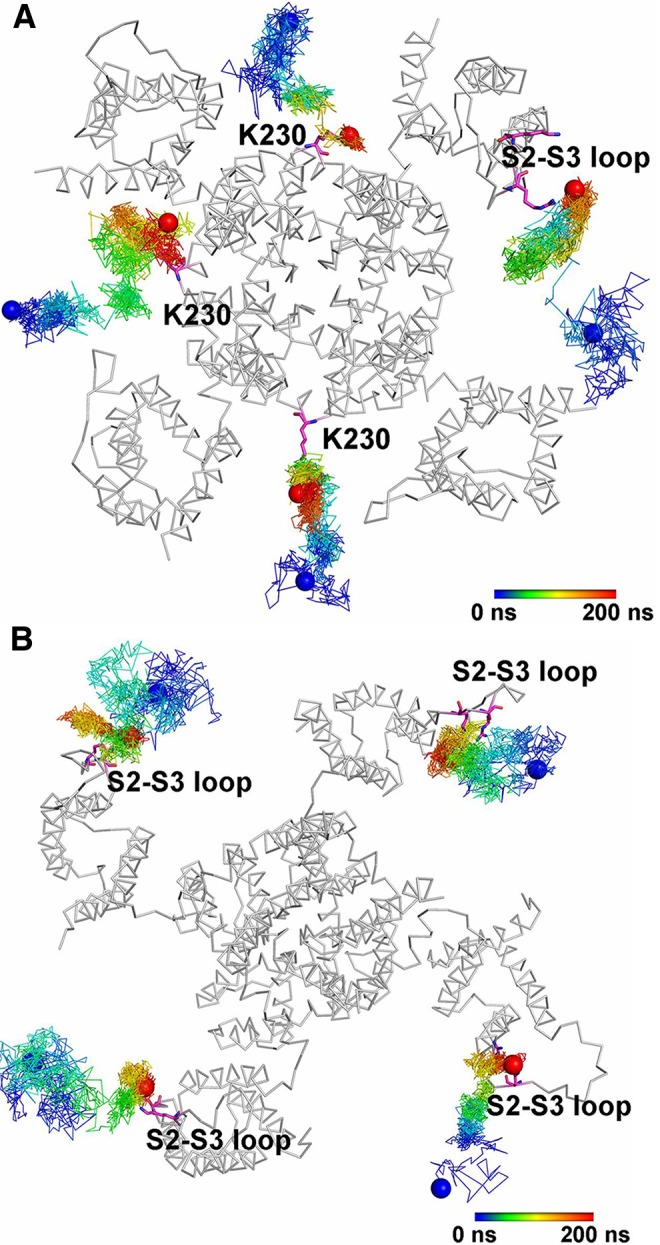
State-dependent PIP2 lipid interactions in the Kv7.2 channel. Zhang et al. used atomistic MD simulations to model the interactions of PIP2 lipids in the open (A) and closed (B) state of the channel.292 Shown are the trajectories of PIP2 lipid molecules as a function of time near the channel (viewed from the intracellular side). PIP2 migrates toward the S4–S5 linker in the open state (A) and toward the S2–S3 linker in the closed state (B). Adapted with permission from ref (292). Copyright 2013 Zhang et al.
3.1.4. Cholesterol and PUFAs-Mediated Regulation of K+ Channels
The debate on K+ channel regulation by cholesterol or dietary PUFAs abundantly present in neuronal or myocyte membranes revolves around three major mechanisms. The first mechanism postulates that lipid impact on the channel function is achieved via direct interactions of fatty acids with amino acids on the lipid facing side of the channel, reminiscent of interactions around the fenestration zone. In the second mechanism, the channels’ function is sensitive to modifications in chemical composition of the lipid bilayer where bilayer fluidity may play a role in the activation process. The third mechanism relies on specific interactions between cholesterol or PUFAs disrupting or modifying interactions within the protein, thereby shifting state equilibria.297 Experimental data show that K+ channels from the Kir family are regulated by cholesterol281 in a stereospecific orientation, suggesting the existence of specific cholesterol binding sites.298 MD simulations combined with molecular docking approaches were used to identify at least two cholesterol binding sites in Kir2.1 channels, which were confirmed by mutagenesis and electrophysiology studies.299,300 MD simulations also enabled a comparison of predicted cholesterol interactions of bacterial (KirBac) and mammalian Kir channels.281 MD studies performed independently suggested a consensus cholesterol binding region in the cytoplasmic half of the TM domains of Kir channels.281,298,299 Recently, Bukiya et al. used a combination of homology modeling and molecular docking to identify two putative cholesterol binding sites in the G protein-gated inwardly rectifying potassium (GIRK) channel, which are located primarily at the center of the transmembrane domain and at its interface with the cytosolic domains of the channel.301 The results suggest that cholesterol interaction sites in the Kir3.1 channel overlaps with the previously suggested sites for Kir2.1 (Figure 6).299 For a more detailed review of cholesterol interactions, see the review by Grouleff et al.36
Figure 6.
Rosenhouse-Dantsker et al. identified specific cholesterol-protein interactions in the Kir2.1 channel by combining molecular docking, MD simulations, mutagenesis, and electrophysiology experiments.299 A. Cholesterol binding sites identified via molecular docking and MD simulations. In blue are residues lining the cholesterol binding sites, while in red are residues whose mutation alters cholesterol sensitivity. B. Schematic representation of the identified cholesterol interaction sites with respect to the architecture of the channel. Reproduced with permission from ref (299). Copyright 2013 The American Society for Biochemistry and Molecular Biology, Inc.
μs-long CG simulations were recently used to characterize cholesterol interaction sites on the Kir2.2 channel, which share ca. 70% of sequence identity with Kir2.1.302 The open and closed states of Kir2.2 were simulated in a POPC membrane with increasing concentrations of cholesterol (15 and 30 mol %). The analysis of the simulation data reveals the diversity of cholesterol interactions with the channel: persistent contacts are detected for cholesterol molecules bound into deeper pockets between TM helices, while more frequent and transient interactions are observed for a number of residues located at the protein–lipid interface. The simulations allow the characterization of cholesterol interaction sites that not only differ between the open (sites I and II) and the closed (sites III and IV) state but also show a different sensitivity to cholesterol depending on the cholesterol concentration (Figure 7).
Figure 7.
Kir2.2 cholesterol binding sites identified with CG MD simulations. Simulation snapshots of Kir2.2 with cholesterol molecules (in yellow) bound to sites I and II in the open state and to sites III, IV and Ib in the closed state.302 Reproduced with permission from ref (302). Copyright 2018 Biophysical Society.
Another group of K+-selective channels sensitive to cholesterol levels in plasma membranes are the large conductance voltage- and Ca2+-gated K+ channels, also known as “big potassium” (BK) channels. BK channels are found in most human cell membranes and control a variety of biological processes, such as circadian behavioral rhythms and neuronal excitability.303 Experimental studies initially related the reduction of activity in the BK channels with changes in the physical properties of the bulk lipid bilayer upon cholesterol insertion in the bilayer.304
Experimental results showed that sterols and PUFAs embedded in the lipid bilayer also directly modulate gating of Kv1.5 and Kv2.1 channels.297,305 PUFA binding to Kv1.2 in both closed and open states was reported recently from μs-long atomistic MD simulations.306 The authors found a potential PUFA-Shaker Kv interaction site located on the lipid–facing side of S3–S4 linker and present only in the open state, confirmed by electrophysiology. It was also shown that headgroup charge and PUFA tail structure are both important determinants of specific binding to Kv channels.297,307 Thus, there is mounting evidence for the specific Kv channel regulation by cholesterol and PUFAs, a mechanism that can be potentially explored for therapeutic applications.307 In addition to PUFAs, polyunsaturated and fully saturated lipids also adopt a unique distribution around Kv channels, as recently shown by a CG study where Kv1.2 channels were simulated for 30 μs in a complex membrane model (Figure 8),121 further highlighting the complexity of the lipid–protein interplay and the need for a better understanding of lipid-mediated channel modulation.
Figure 8.
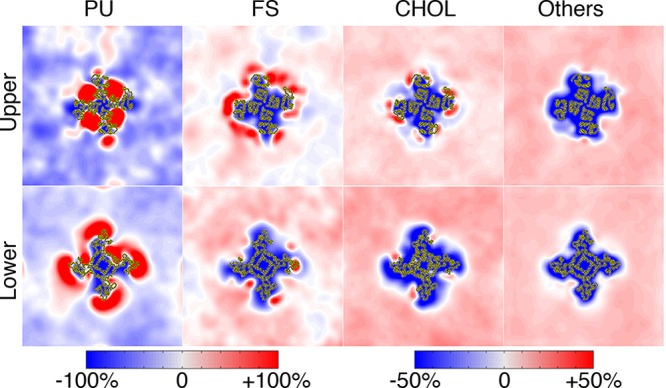
Lipid distribution around Kv1.2 described by CG MD simulations. MD simulations of Kv1.2 in a plasma membrane mixture revealed an asymmetric distribution of polyunsaturated (PU), fully saturated (FS) lipids and cholesterol (CHOL) between upper and lower.121 The simulation system consisted of 4 copies of Kv1.2 and ca. 6000 lipids. Shown are 2D distribution maps for PU, FS lipids, CHOL, and the class Others (which groups all the remaining lipid classes of the plasma membrane mixtures), highlighting enrichment (red) and depletion (blue) with respect to the average value of the corresponding class in each leaflet. Figure generated as described in Corradi et al. for Aquaporin 1.121
The exact mechanisms of Kv function regulation by membrane lipids are complex and yet to be established. Recently, Zakany et al. used a combination of two-electrode voltage-clamp fluorometry with a nonstationary noise analysis to show that cholesterol impacts predominantly the single-channel current rather than modifying the gating process in the Kv1.3 and Kv10.1 channels. Sterols may, therefore, have binding sites in the pore domain of Kv channels.308
3.1.5. Lipid Regulation of Nav Channel Function
Progress in understanding the roles of lipids in the family of voltage-gated Na+ channels was facilitated by the X-ray structure of the bacterial channel NavAb by Payandeh et al. (Figure 4).254 This structure was followed by several other bacterial channels.309−312 Unlike their eukaryotic counterparts, which consist of a single polypeptide chain, the functional unit of the bacterial Na+ channels is a homotetramer,313,314 but it has been shown to possess many of the key elements of mammalian channels.315 The NavAb pore domain features lateral openings, already mentioned above, directly connecting the central pore to the surrounding lipid phase. The crystal structure shows that electron densities assigned to phospholipids protrude through the fenestration zone and may physically impact ion transport through the pore.254 MD simulations, focused on the mechanism of Na+ permeation, have also suggested possible roles of lipids bound to the fenestrations.316−318 The presence of lipids in these lateral fenestrations is linked to their size and shape as shown by MD simulation studies on bacterial Nav channels (NavAb closed, NavAb inactivated, NavRh and NavMs) embedded in POPC membranes.319 In this work the authors reported a clear connection between the presence of lipid in fenestration and the fenestration size. The fenestration reduced in size when the lipids retracted from the cavities. The widening of the fenestrations was influenced by protein conformational changes that potentiate the entrance of lipids to these cavities. Lipid tails also force side-chains toward the sides of the fenestrations, resulting in a larger pore. Ulmschneider et al. observed that lipids can access the fenestrations multiple times over μs-long simulations, showing that channel-lipid interactions are transient.320 Experimental results suggested that cholesterol regulation of membrane properties indirectly regulates the function of Nav channels.243 Atomistic MD simulation of the NavAb channel in POPC:chol membranes have also shown that cholesterol does not directly interact with the channel.321 The results suggested that cholesterol may influence Nav channel function by affecting the physical properties of lipid bilayers such as lipid packing, bilayer thickness, and lipid movement.
3.2. Lipid Regulation of Ligand-Gated Channels
Ligand-gated channels change state in response to chemical stimuli. The functions played by members of this family are as diverse as the family itself (Figure 9). For example, glutamate-receptor ion channels (iGluRs) mediate the fast component of excitatory glutamatergic neurotransmission in the central nervous system. Cys-loop receptors activated by a variety of neurotransmitters are essential components of the excitatory and inhibitory transmission in the central and peripheral nervous systems.322 Excitable membranes show a distinct lipid composition, which may be related at least in part to lipid–protein interactions.
Figure 9.

Ligand-gated channels. From left to right are the structures of the nicotinic acetylcholine receptor (nAchR),323 the P2X3 channel324 and the TRPV1 channel.60 For each structure, the individual monomers are colored from light yellow to dark brown. ATP molecules bound to P2X3 and resiniferatoxin (RTX) bound to TRPV1 are shown in white spheres. ECD, extracellular domain; ICD, intracellular domain. The top panels show a side view of the proteins, with the membrane region highlighted in gray. The lower panels are the view from the extracellular side. The functional assembly of nAchR includes five protomers labeled α to γ, with each monomer comprised of trans-membrane sections (helices M1 to M4) and the extra-cellular domain (ECD). P2X3 channels are assembled as functional trimers with each monomer containing a trans-membrane section comprised of 2 TM helices, TM1 and TM2. The nomenclature of the TRPV1 channel is similar to that of voltage-gated ion channels shown in Figure 4.
3.2.1. Lipid Modulation of Cys-Loop Receptors
The Cys-loop receptors constitute an important superfamily of mainly pentameric ligand-gated ion channels (LGICs) named after a cysteine bridge present in the N-terminal extracellular domain for all members of the family. Depending on the preferred charge carrier, this family of the receptors is subdivided into cation and anion selective cys-loop receptors.322 A unique binding site located in the N-terminal ligand-recognition domain defines the type of the receptor, e.g., acetylcholine (ACh), serotonin, glycine, glutamate or γ-aminobuturic acid receptors.322,325−327 The preferred substrate binding to one or more binding sites triggers pore opening (or closure) enabling selective permeation, which is controlled by the selectivity filter located in the trans-membrane domain of the receptor. Each subunit is usually formed by three domains: (i) the large ECD containing the ligand-binding site; (ii) the transmembrane domains that consist of four transmembrane- α-helices (M1-M4); and (iii) the intracellular domain (ICD) that is primarily formed by the large M3-M4 intracellular loop which permits the access of ions in and out of the pore and is involved directly in ion permeation (Figure 9). The interface between the EC and TM domains seems to play a key role in the gating process and is thought to be additionally regulated by lipid molecules closely associated with the TMD.322,325,326
One of the best characterized members of the family is the nicotinic acetylcholine receptor (nAChR), a cation-selective channel expressed in many kinds of both excitable and nonexcitable cells (Figure 9). Experimental studies have demonstrated that the incorporation of cholesterol in lipid vesicles is essential for nAChr to retain its functional activity.328 Thus, when the nAChR was reconstituted into PC membranes without cholesterol, agonists no longer simulated cation flux. Photolabeling studies have shown that sterols interact directly with the nAChR’s lipid-exposed TM helices.329 Brannigan et al. demonstrated the presence of internal cholesterol binding sites in the cryo-EM structures of the nAChr channel.330 Also, the authors compared the stability of these structures with and without cholesterol by atomistic MD simulations. Overall, the authors suggested that the role of cholesterol in the membrane is to stabilize the trans-membrane domain of the channel. Cholesterol and 1-palmitoyl-2-oleyl phosphatidic acid (POPA) interactions with the channel have been also proposed to play a more direct role in the gating process of Torpedo nAChr channels. From 10 ns long MD simulations performed in POPC:POPA:cholesterol membranes the authors suggested that cholesterol and POPA bind specifically to one of the key lysines directly implicated in activation of nAChR.331 Give the simulation length and the transient nature of cholesterol-channel interactions discussed in this review, the exact mechanism of cholesterol-dependent gating in nAChR requires additional studies.
The lipid-dependent gating was also proposed to exist in the glycine receptors (GlyRs) and the γ-aminobuturic receptors (GABAAR). Structural models for GABAAR and GlyR were built using the eukaryotic chloride channel (GluCl, from Ceanorhabditis elegans) as a template and then used for MD simulations.332 This GluCl was cocrystallized with invermectin (an anthelmintic drug), which behaves as an allosteric agonist on this receptor and related Cys-loop receptors.327 MD simulation of GABAAR identified that the intersubunit (invermectin) sites in the transmembrane domain are potential cholesterol-binding sites that were present in other GABAAR models. In each interface between subunits, residues belonging to helices M1 and M3 form a belt of nonpolar contacts around the steroidal nucleus of cholesterol. Also, the conserved Ser residue from helix M2 was proposed as a key interacting residue with cholesterol. Combining MD simulations with homology modeling, Henin et al. proposed that the depletion and enrichment of cholesterol have a negative impact on the interaction between the GABAAR and the endogenous ligand GABA.333 They proposed that the decreased potency at low cholesterol concentrations may reflect the direct interactions established between GABAAR and this lipid. In contrast, the decreased potency of the receptor at high cholesterol concentration may be due to the changes in the physical properties of the membrane, such as fluidity and thickness.
3.2.2. Lipid Modulation of ATP-Gated Channels
Ionotropic P2X receptors are canonical ligand (ATP) gated channels permeable to mono- and divalent cations.334 The P2X subunit is comprised of two TM domains (TM1 and TM2), often separated by Cys-rich ectodomains,335 intracellular C- and N-terminal domains, and a large extracellular ligand-binding loop with a highly variable sequence in different members of the family.336 The functional assembly of the P2X is a trimer, where the trans-membrane pore is formed by six α-helical TMs (TM1 and TM2 from each of the protomers) (Figure 9).335,337,338 The available high-resolution structure of the open state of the P2X3 receptor324 displays a continuous narrow pore with a narrowest constriction radius of only ∼3.2 Å, e.g. consistent with the radius of a dehydrated Na+ ion. Residues from the TM2 helix line the permeation pathway. Structural modeling based on the available structure of the P2X4 receptor from zebrafish also supports an essential role of TM2 in water and ion permeation.337,338 However, the functional mechanism of ATP activation of P2X receptors enabling cation permeation remains highly controversial. For example, prolonged exposure to ATP was proposed to result in a “dilated” state of the channel, highly permeable to large cations such as spermidine or NMDG+ with a cross-sectional radius considerably greater than that found in the published crystal structure.339
The functional assembly of the human P2X3 receptor in its open state shows a separation at the outer ends of the TM helices with well-defined lipid-exposed surfaces. Rothwell et al. proposed that membrane lipid must enter in the large cavities that appear between the outer ends of the TM helices by comparing the open and closed structure of the receptor.340 The authors combined electrophysiology, homology modeling and MD simulations to characterize the dynamics of this region during channel gating.340 They suggested that in the open state the lipids enter in the interstices between the outer end of the transmembrane domain, while in the closed state these residues packed together, diminishing the possibility of interaction with lipids. Mansoor et al. suggested the fenestrations in the open state of the channel as plausible route for ion transport with polar lipids head groups lining the fenestration zone.324 In a follow-up study, Scheurer et al. performed extensive analysis of lipid-ion interactions during permeation events observed in 0.5 μs of all-atom MD simulations of P2X3 receptor in POPC membrane.341 The authors concluded that Na+ interactions with the phospholipid head-groups may be an essential mechanism of permeant cation recruitment to the channel’s lumen.
Lipid-specific effects have also been proposed to play essential roles in the regulation of P2X function and Na+ or NMDG+ transport via yet unknown mechanisms. Robinson et al. showed that the cholesterol content of the plasma membrane is an essential factor in regulation of P2X7 receptors.342 Cholesterol depletion of cell membranes was associated with a high rate of agonist-induced pore formation. Subsequent electrophysiology studies of P2X7 receptor currents also indicated an enhanced activation and current facilitation following cholesterol depletion. However, the sensitivity of the P2X receptors to cholesterol content varies greatly among various family members.343,344 The experimental data collected to date suggest that P2X receptor subtypes may be differentially targeted to cholesterol-rich regions of the membrane.
3.2.3. TRP Channels
Transient receptor potential (TRP) channels form an important family of potassium channels involved in sensing a variety of stimuli in living organisms.345,346 While TRP channels can be regulated by heat and membrane voltage, they are most commonly associated with activation by ligand binding (vanilloid-binding, melastin-related, etc.). The TRPV1 structures in open and closed configurations60,347 show striking organizational similarity to the Kv channels discussed above. Structurally, TRPV1 channels are homotetramers of subunits organized around a central aqueous pore (Figure 9), with several additional domains besides the canonical Kv voltage sensing domain (S1–S4) and pore domain, including the TRP domain and an ankyrin repeat domain (ARD) in the intracellular side.
MD simulations combined with molecular docking and mutagenesis indicated that PIP2 lipids are able to induce conformational changes in the TRPV1 channel, suggesting this lipid as agonist.348 The authors identified a PIP2 lipid binding site located between the TRP domain and the S4–S5 linker, where PIP2 interacts with two conserved arginines in the S4–S5 linker and a lysine in the TPR domain. The binding of PIP2 lipid couples the VSD to the TRP domain and induces structural changes in the S6 helix. Takahashi et al. showed that PIP2 binding to the ARD domain of TRPV4 regulates the activity of the channel.349 This study showed that the ARD domain interacts with the inositol headgroup of PIP2, which negatively regulates the channel activity. Additionally, the most recent cryo-EM study of TRPV1 in a lipid nanodisk showed that PIP2 lipids are embedded in the vanilloid binding pockets (VBP) located at the S4–S5 linker of the channel.60 Atomistic MD simulations of TRPV1 combined with mutagenesis experiments, showed that at high temperatures the lipids present in the VBP are displaced and lipid unbinding may act as a heat sensing mechanism.350 Zimova et al. combined atomistic MD simulations and homology modeling to identify residues putatively involved in PIP2 binding in TRPA1 at the S1–S4 sensor.351 The PIP2 binding sites are very similar to previously reported in the TRPV1 and TRPV2 structures.60,352,353 These results reaffirm the role of PIP2 as coupling elements regulating the voltage sensing properties of the TRP channels by occupying channel hydrophobic pockets with PIP2’s aliphatic chain. TRP channels have been also reported to be regulated by cholesterol. Experimental evidence suggests that cholesterol may influence TRP channel properties such as threshold activation temperatures and channel currents.344,354 Saha et al. predicted possible interactions of cholesterol with CRAC-like domains present in the hTRPV1 channel sequence.355 Docking studies suggested high affinity cholesterol binding to the conserved CRAC-motif in TRP channel. However, future atomistic simulations may be needed to provide detailed information on apparent affinities and residence times, as well as specific mechanism of TRPs function regulation by cholesterol.
3.3. Bacterial Mechanosensitive Channels
Bacterial mechanosensitive channels (MS) regulate and maintain osmotic homeostasis. During hypo-osmotic shock, MS channels transition from a closed to an open conformational state, allowing for cytoplasmic osmolytes to escape and prevent cell lysis.356 MS channels, rather than directly sensing the increase in cell volume, are activated by membrane tension which is a result of increased internal cell pressure.357 In prokaryotic cells, MS are classified into large (MscL), medium (MscM), and small (MscS) conductance, an example of which is shown in Figure 10. The classification is mainly functional, but they also differ in their primary sequence, spatial topology, and, in what is becoming increasingly more evident, the driving forces governing their opening mechanism.356,358
Figure 10.
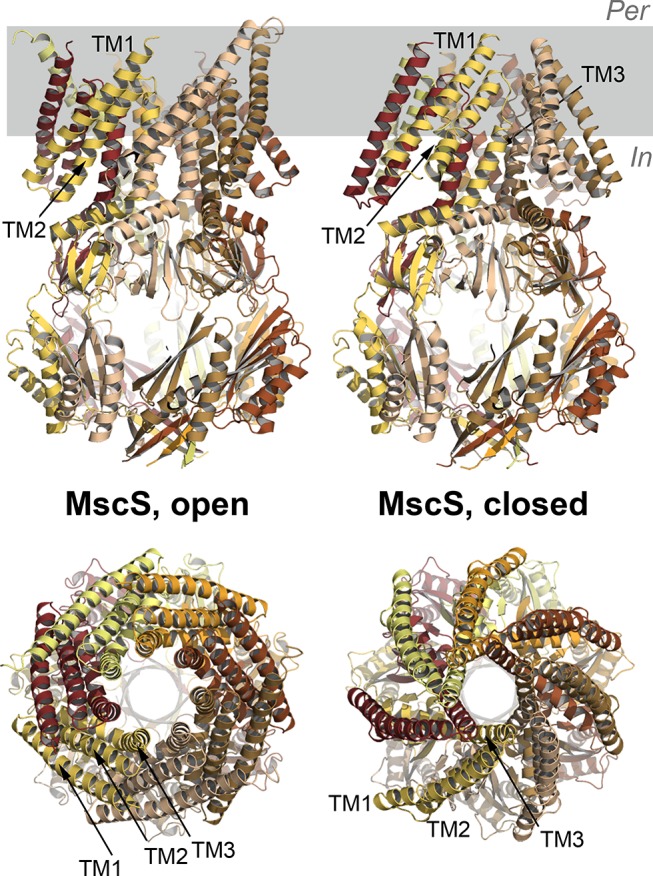
Open and closed structures of the MscS channel. Side view (upper panels) and view from the periplasmic side (bottom panels) of an open and closed MscS channel.359,360 Each monomer is shown in cartoons and colored on a scale from light yellow to dark brown. Per, periplasm; In, intracellular side. The hydrophobic region of the membrane is highlighted in gray.
MscS induces a local bilayer curvature361 that could, at least in part, account for its sensitivity to lateral membrane tension,362,363 whereas global curvature was found to have a negligible effect.363 The discovery of lipid acyl chains filling crevices on the TM domain of MscS suggested the involvement of lipid–protein interactions in its gating mechanism.359 Biophysical experiments and MD simulations showed that this acyl chain binding is in a dynamic equilibrium with lipids in the membrane and more prevalent in the closed state of the channel. Membrane tension shifts this equilibrium toward the bilayer, inducing the transitioning of the receptor to the open state.359 MscL activation, in comparison, involves an increase in channel cross-section area leading to hydrophobic mismatch.357 Essentially, an increase in lateral membrane tension produces the acute tilting of MscL TM helices, necessary to trigger channel opening.358 This tilting-mediated channel opening was also seen in previous CG MD simulations.364365 Bavi et al., using an array of experimental techniques combined with MD simulations, found the N-terminus of MscL is located at the lipid–solvent interface and responsible for translating the membrane tension-generated torque into a conformational change mechanical force.366 Interestingly, lipid tail binding and expulsion from MscL pockets resembles that of MscS; however, their continued interaction with the N-terminus points suggests removal of lipid tails does not trigger MscL activation but, rather, is a result of it.,366367 MS channel sensing of membrane tension is not contingent on the availability of other proteins or cofactors, but can be modulated by the presence of membrane additives such as alcohols or short chain lipids as demonstrated in combined experimental and simulation studies.368,369 Current evidence supports a complex mechanism of MscL and MscS channel gating involving membrane curvature and hydrophobic mismatch, respectively, and an intimate relationship with individual lipid chains.357,367,370
4. Respiratory Proteins
Mitochondria are essential intracellular compartments found in most eukaryotic cells. These membrane-bound organelles synthesize most of the energy used by our cells. A complex mechanism involving a series of oxidoreduction reactions carried out by several enzymes embedded in the inner membrane of mitochondria, known as the respiratory chain, is responsible for this synthesis. The enzymes forming the respiratory chain are complex I (NADH dehydrogenase), complex III (cytochrome bc1), and complex IV (cytochrome c oxidase); these proteins act in unison to create the proton gradient required by the ATP synthase, often called the complex V of the respiratory chain, to convert ADP into ATP. Complex II, succinate dehydrogenase, is not directly involved in the building of the proton gradient. Another important player is the ADP/ATP carrier (AAC), also known as adenine nucleotide translocase, which exchanges ADP and ATP across the inner mitochondrial membrane. Figure 11 illustrates key aspects of complexes III and IV.
Figure 11.

CLs stabilize supercomplexes in large scale CG MD simulations. Top: Snapshot of final configuration of the system containing CIII and CIV, indicating the presence of many supercomplexes. Bottom: Close-up view on a particular supercomplex with CLs present at the protein–protein interface. Figure courtesy of Clement Arnarez.
The proton gradient used by the ATP synthase to construct ATP is the result of a series of electron transfers. Most of these transfers are carried out between specific redox centers included in, or diffusing between, the three respiratory chain protein complexes I (CI), III (CIII), and IV (CIV). One cycle of the respiratory chain involves at least three protein complexes (CI, CIII, and CIV) and two electron carriers (QH2/Q and the cytochrome c). The buildup of the proton gradient, and, by extension, the rate of ATP synthesis are thus limited by the encounter rate of these molecules and depends critically on the large scale organization of the respiratory chain complexes. Over the last decades, the existence of a higher-order organization of the respiratory chain has been demonstrated through various techniques, notably blue-native gel electrophoresis (BN-PAGE) and electron microscopy. The three protein complexes seem to assemble into larger structures named supercomplexes (together defining the respirasome),371−373 which potentially would facilitate the electron transport between them.
The composition of the inner mitochondrial membrane plays a critical role in the respiratory chain process. The lipid environment includes a specific variety of head groups, with three major components being phosphatidylcholine (PC), phosphatidylethanolamine (PE), and cardiolipins (CL). In particular CL has been shown to have an effect on both stability and functionality of the whole respiratory chain, with CL alterations causing malfunction and disease.374,375 In Barth syndrome (caused by a mutation leading to cardiomyopathy, skeletal myopathy, and growth retardation), for instance, the mitochondrial inner membrane shows a lower content of CL and/or a spread in the composition of their alkyl tails.376 BN-PAGE analyses performed on such cells report a decrease in supercomplex formation and an increase in monomeric free complexes. CL seems to be involved at every level of the respiratory chain process: maintaining the functionality of independent complexes, allowing the formation of supercomplexes and stabilizing them, and preserving the functionality of these assemblies.377−379 Zhang et al. labeled CL as the “glue holding supercomplexes together”.380 CLs are present in the crystal structures of the respiratory chain complexes,381 however, deeply embedded inside the core of the proteins. It is therefore not clear how these CLs could regulate supercomplex formation.
In addition, CL has been implicated as an important signaling lipid,382 involved in shaping or stabilizing cristae,383 and as proton carrier. To shed light on this multifaceted role of CL, simulation studies have recently entered this field.
4.1. CL Binding Sites
To explain the putative role of CLs in gluing respiratory chains together, Arnarez et al. hypothesized that CL binding sites exist at the membrane-exposed surface of the proteins that constitute the respiratory chain.384 The use of protocols to purify proteins in which detergents often wash away lipids that are exposed to the bulk membrane can easily rationalize the absence of such bound CLs in the crystal structures. To test the hypothesis, Arnarez et al. performed extensive (100 μs) CG MD simulations of the cytochrome bc1 (CIII) dimer from bovine heart, embedded in a model mitochondrial membrane composed of POPC lipids together with CL at a 20:1 molar ratio.384 The length of the simulation allowed observation of reversible binding and unbinding events of CLs around the protein. Subsequent analysis of the average CL density revealed the existence of six binding sites, all on the matrix side of the protein and including sites known from earlier structural studies and buried into protein cavities. The sites proved stable upon backmapping the configuration to all-atom resolution and performing an additional 100 ns atomistic simulation. Interestingly, identical CL binding sites were found in simulations of the same complex extracted from yeast. Using a similar protocol, the same authors analyzed the CL density around bovine heart CIV.385 Here, seven binding sites were found on either leaflet of the membrane. In a related study, also based on the Martini CG force field, Duncan et al. revealed transient CL binding sites on the rotor part of ATP synthase (CV), discussed in more detail below.386
Poyry et al. also studied the role of CL, through atomistic MD of the CIII dimer of the purple photosynthetic bacterium Rhodobacter capsulatus, embedded in mixed PC/PE/CL membranes.387 Although the time scales explored in this study (200 ns) are much smaller than accessible with a CG model, evidence for CL binding sites was obtained. In particular, the authors observe CLs to spontaneously diffuse to the dimer interface, in the immediate vicinity of the complex’s catalytic quinone reduction sites and in agreement with crystallographic studies of the complex. Sharma et al. performed atomistic simulations of CIV, embedded in mixed PC/PE/CL membranes, focusing on the stability of lipids near one of the proton uptake pathways.388 The results show that different lipid types can be bound, in line with crystallographic data from different organisms. The authors further predict that CL also forms stable interactions.
CL binding sites were also observed in simulations of the ATP/ADP carrier (AAC) protein. Using simulations at both coarse-grained and atomistic resolutions, Hedger et al. identified three CL binding sites on this translocase, in agreement with those seen in crystal structures and inferred from nuclear magnetic resonance measurements.389 An overlapping study of Duncan et al. confirmed these findings.390
The studies described above confirm previously found CL binding sites and reveal hitherto unknown sites on the membrane–exposed surfaces of the respiratory chain complexes. These surface bound CLs could play an important role in, e.g., proton uptake,385−388 or by providing structural integrity of the complexes and supercomplexes.384 Analysis of the distribution of residues in contact with the CLs show that, in general, the binding sites observed in MD simulations are enriched in positively charged residues. This behavior is consistent with the negative charge of cardiolipins and in agreement with a generic analysis on CL binding motifs across the protein databank.381 Nevertheless, the bulky tails of CL are also important. In the study of Arnarez et al. on CIV, the potential of mean forces (PMFs) of CL binding between different CL variants were compared, revealing an almost equally strong contribution of the tails and the charged headgroup to the overall binding strength.385 PMFs computed in the study of Hedger et al. furthermore show that other anionic lipids, such as PS, have an affinity intermediate between CL and PC.389
4.2. CL Mediated Supercomplex Formation
Respiratory chain complexes self-organize into supramolecular structures called respiratory supercomplexes or respirasomes.371−373 Numerous experimental studies have shown the existence of supercomplexes consisting of CI, CIII, and CIV. The formation and stability of these supercomplexes strongly depends on the presence of CLs in the membrane environment.377−380 Arnarez et al. hypothesized that the bound CLs identified on the membrane-exposed surface of the complexes actually play a key role in gluing the respiratory chain together.384 The presence of CLs at specific locations of the protein surface might either prevent the binding of potential partners at that same location (“protective mode”) or the CL binding sites might define the actual location at which the proteins interact and CLs glue the proteins together (“bridging mode”).
Initial small scale self-assembly CG MD simulations of respiratory chain complexes CIII and CIV indeed show formation of a CIII–CIV supercomplex, with CL present at the interface between the complexes.384 In a follow up study, the formation of supercomplexes was simulated at a much larger scale.391 Here, the system included 9 CIII dimers, 27 CIV monomers, 18 679 POPC lipids, and 1165 CLs, being one of the first example simulations of a realistically complex and crowded membrane system. This system was simulated for 20 μs, starting from an initial randomized distribution of the protein complexes. Although this time scale is too short to obtain an equilibrium lateral distribution of the proteins in the membrane (lateral diffusion and binding/unbinding of membrane proteins are notoriously slow processes), the formation of supercomplexes was clearly observed as shown in Figure 11 (top). Analysis of the protein–protein interfaces of the supercomplexes shows that CLs are becoming enriched during the simulation. At the end of the simulations, the ratio of PC/CL lipids is increased from 1:15 in the bulk to 1:5 in the annular shell of the isolated proteins and 1:2–3 considering the protein–protein interfaces. A typical example of a CIII–CIV supercomplex is shown in Figure 11, with the CLs present at the interface highlighted. Further analysis reveals that the CL binding sites are conserved on each of the complexes and are shared in the supercomplex formed, suggesting that they operate according to a bridging mode. CG MD simulation of large patches of a model mitochondrial membrane containing multiple copies of the ATP/ADP carrier also show that CL interactions persist in the presence of protein–protein interactions and suggests CL may mediate interactions between translocases.389 CG simulations in combinations with experimental biophysical methods also propose that CLs mediate the interactions between components of the translocase of the inner mitochondrial membrane 23 (TIM23) complex.392
Taken together, MD data support a role for CL in steering the formation of supercomplexes in mitochondrial membranes and reveal a mechanism that implies supercomplex organization is regulated by CL binding sites.
5. ATP-Binding Cassette Transporters
ATP-binding cassette (ABC) transporters constitute a large superfamily of membrane proteins and include both exporters (in prokaryotes and eukaryotes) and importers (in prokaryotes).393 Their substrates cover a broad range of chemical diversity, ranging from ions to sugars, amino acids, peptides, proteins, toxins, metabolites, drugs, sterols, and lipids. With the exception of a few subfamilies, all ABC transporters feature two cytosolic nucleotide-binding domains (NBDs), and two transmembrane domains (TMDs), whose structural architecture and hallmarks differ between importers and exporters, as well as across subfamilies.394 This structural diversity is reflected in different mechanistic details of the transport cycle, although general features exist.394 Transport is powered by ATP binding and hydrolysis at the NBDs, triggering their dimerization and dissociation, respectively. These motions are in turn coupled with changes in the TMDs, which provide a translocation pathway first accessible from one side of the membrane for substrate uptake and then open to the opposite side for substrate release (Figure 12).
Figure 12.
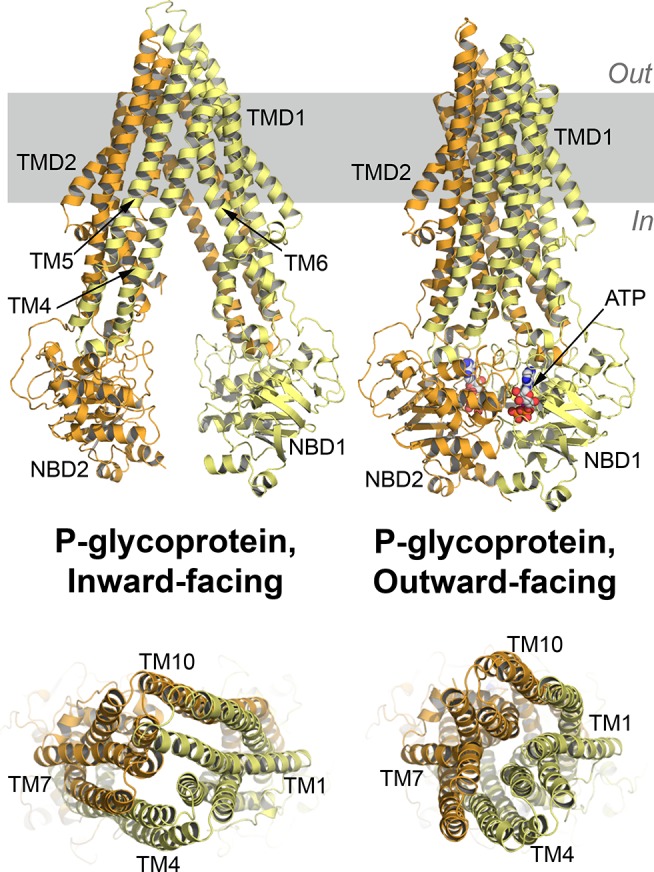
Structural features of an ABC exporter. The two halves of P-glycoprotein are shown in orange and light yellow cartoons for the (left) inward-facing395 and (right) outward-facing396 state. ATP molecules bound to the nucleotide binding domains (NBD1 and NBD2) of the outward-facing state are shown in white spheres. TMD1 and TMD2 indicate the two transmembrane domains, embedded in the hydrophobic region of the membrane here highlighted in gray.
Purification and reconstitution studies for the characterization of ABC transporters have highlighted the effects of membrane composition on protein structure and function,397−412 while specific lipid species tightly bound to the protein have been identified by mass spectrometry,413−415 crystallography, and cryo-EM studies.415−419 In this context, molecular dynamics simulations have provided molecular insights into the lipids organization and dynamics around ABC transporters.
The transporter associated with antigen processing (TAP) translocates antigenic peptides, derived from proteosomal degradation, into the endoplasmic reticulum, for loading of major histocompatibility class I molecules.420 Experimental estimates found 22 lipids are required around TAP in a nanodisc to bind an inhibiting protein.421 Atomistic simulations showed that 22 lipids are indeed sufficient to surround TAP, although their detailed structure differed significantly from a control simulation in a bilayer.421
Other simulations studies employed MD simulations for a more detailed characterization of lipid–protein interactions. The E. coli ABC transporter McjD translocates microcin J25 (MccJ25), a 21 amino acids long peptide with antibacterial activity.422,423 Mass spectrometry studies revealed the presence of PG and PE lipids together with lipopolysaccharide tightly bound to the transporter.415 Retention of PG lipids after protein delipidation together with a higher ligand-induced ATPase activity stimulated by PG lipids in comparison to other lipids suggested a functional role for this negatively charged lipid species.415 To investigate the molecular interactions between McjD and lipids, 40 μs-long CG molecular dynamics simulations using the Martini model were carried out in lipid bilayers of POPE, POPG, and CL in different ratios.415 The negatively charged head groups of PG and/or CL were shown to preferentially interact with arginine and lysine residues located at the interface with the cytosolic side of the membrane. In binary and ternary mixtures, these interactions reduced the contacts between the protein and PE lipids, although these zwitterionic lipids remained colocalized within the same lipid shell (Figure 13).415 These findings support the crystallization of McjD with a PG lipid molecule.415 Combining equilibrium and nonequilibrium simulations to pull the MccJ25 substrate out of the occluded cavity of McjD424 could also provide a tool to look at coupling of conformational changes to lipid–protein interactions.
Figure 13.

Lipid organization around the ABC transporter McjD. CG simulations of the McjD transporter were carried out in different mixtures (A–D) of POPE, POPG, and cardiolipin.415 Preferential interactions in the binary and tertiary mixtures with the headgroup of anionic lipids were driven by electrostatic interactions with several positively charged residues. Shown are the headgroup number density maps for the four simulated systems, with the densities of POPE, POPG, and cardiolipin in cyan, magenta, and yellow, respectively, for upper and lower leaflet. Adapted with permission from ref (415). Copyright 2016 The American Society for Biochemistry and Molecular Biology, Inc.
In a recent study,425 the results of Martini CG simulations were converted to the atomistic level of detail to investigate the protein–lipid interplay for P-glycoprotein, an ABC multidrug transporter involved in cancer drug resistance.426 A homology model of human P-glycoprotein with the TMD cavity open to the cytosol and the NBDs in contact with each other was embedded in a more complex membrane, consisting of POPE, POPC, POPS, SM lipids, and cholesterol, to mimic the composition of brain epithelial cell membranes.425 Multiple replicas of the same system were simulated for 10 μs, followed by an additional 100 ns of simulation time after converting the system to the atomistic level of detail using the backward Martini tool.135 This computational approach revealed ordered rings of lipids around P-glycoprotein, similarly to what has been proposed for McjD415 and smaller transmembrane domains.427 PS lipids were identified, in particular, in the first lipid shell around the protein, due to positively charged residues located at the water interface of the inner leaflet, as observed for McjD,415 and in line with experimental findings from mass spectrometry suggesting that P-glycoprotein preferentially binds negatively charged lipids.413 The simulations also provided insights on the interactions between P-glycoprotein and cholesterol: multiple cholesterol binding sites, defined based on a distance cutoff and contact time duration, were retrieved on the protein surface, with a prominent binding site located in a cleft formed by transmembrane helices 10 and 12, which line one of the entries to the main cavity of the transporter.425 The distribution of cholesterol around the TMDs of ABCC4, also known as MRP4, was also studied via atomistic MD simulations in POPC:cholesterol mixtures. Increasing cholesterol concentrations, reflected in higher thickness of the membrane, reduced the degree of opening between the NBDs, showing how the modulatory effect of lipids might extend to domains not embedded in the membrane.428
The presence of positively charged residues promoting interactions with negatively charged lipids has also been reported for the bacterial transporter TmrAB, an ABC exporter from T. thermophilus with a broad spectrum of substrate specificity.412 As for many other ABC transporters, TmrAB function is modulated by the lipid composition, with negatively charged lipids such as PG lipids essential for transport,412 and found tightly bound to the transporter.414 To characterize lipid binding sites, Noll et al. used atomistic molecular dynamics simulations of TmrAB in POPE or POPG bilayers, on time scales ranging from 0.5 to 2 μs. As previously discussed for McjD and P-glycoprotein,415,425 the binding of either POPE or POPG molecules to TmrAB was driven by clusters of positively charged residues located at the interface region of the inner or outer leaflet of the membrane.412
The interplay between ABC transporters and lipids is not limited to protein function or structure modulation. Many ABC transporters carry out transport of sterols, lipids, or lipid-like molecules, and mutations in these transporters are linked to several diseases, including, for instance, Tangier and Stargardt disease.429 MD simulations can be used to characterize the details and pathways of lipids uptake into the main cavity of the transporters. An early study where such events were detected is the work of Ward et al.,116 who applied the multiscale CG (MS-CG) method118 to develop CG potentials suitable for the investigations of lipid–protein interactions starting from atomistic simulations of the reference systems. To test these potentials, the bacterial ABC exporter MsbA, embedded in a DOPE:DOPC membrane, was used. During the simulations of the conformational state with a large intracellular opening, lipids were observed to diffuse into the main cavity of the transporter, preventing further conformational changes toward a more closed state.116 MsbA is a lipid flippase that translocates lipopolysaccharide across the inner membrane of Gram negative bacteria,430 and lipids entry into the main cavity as detected in the simulations might have mechanistic implications or even provide an example of entry pathway possibly shared with lipopolysaccharide molecules. This study also showed how simulations at a coarser resolution could sample events (such as lipids uptakes into the transporter cavity) not easily accessible at an atomistic scale, while preserving the molecular and physical representation of the system.116 Other CG models have shown a similar distribution of lipids in the central cavity of ABC transporters. Using the Martini model, for instance, Stansfeld et al. developed an automated protocol to insert, in a POPC lipid bilayer, crystal structures of membrane proteins, including ABCB10, a mitochondrial ABC transporter.431 During the 100 ns-long simulation, lipids were shown to partition inside the cavity of the transporter.431 The CgProt force-field, also derived from the MS-CG model,118 has been applied to study lipid–protein interactions for P-glycoprotein in a POPE:POPC bilayer containing 20% cholesterol.117 P-glycoprotein structure and function are strongly modulated by sterols and lipids, and lipids such as fluorescently labeled PE, PC, and SM lipids have been shown to be substrates of P-glycoprotein.426 The CgProt simulations allowed the study of the transition from a conformational state with a large intracellular opening (suitable for substrates like lipids uptake) to a conformational state with the cavity open to the opposite side of the membrane, while lipids and cholesterol redistributed around the protein.117 Within the first lipid shells near the protein, enrichment was observed for PC lipids and cholesterol, while lipid diffusion inside the central cavity was retrieved only for PC and PE lipids. Although these studies captured lipids uptake events, details on the time scale or the protein residues involved were not described. Barreto-Ojeda et al. used the Martini model to characterize such interactions.432 Here, the conformational state of P-glycoprotein with a large intracellular opening (also used in the CgProt simulations discussed above)117 was embedded in lipid bilayers of different ratios of POPE and POPC lipids. In this state, access to the cavity is granted through two different portals, lined by transmembrane helices 4 and 6 on one side and 10 and 12 on the other.395 The 20 μs-long simulations sampled several lipid uptake events for both POPE and POPC species (Figure 14), evaluated based on the presence of a lipid headgroup within a 8 Å cutoff from the residues inside the cavity, for at least 200 consecutive ns. A different probability of lipid uptake events was detected between the two portals, reflecting the asymmetric conformation of the helices lining the portals. During the simulations, as also found by Domicevica et al.,425 positively charged residues located at the water–lipid interface in the inner leaflet favor interactions with the polar headgroup of the lipids, recruiting them near the transmembrane helices at the portals, whereas no significant difference was observed in the pattern of interactions of POPE and POPC lipids with the protein. Inside the cavity, the lipids orient their tails toward the upper and more hydrophobic region, while their headgroup remains in contact with the more polar environment of the cavity near the cytosolic interface, reminiscent of the cryo-EM structure of MsbA in nanodisc in complex with a lipopolysaccharide molecule trapped in the cavity.417 The residues identified as key players in P-glycoprotein for lipid–protein interactions are conserved in related transporters such as ABCB4, known to translocate PC lipids in hepatocytes.429,432 This highlights how the simulations results can be of relevance for transporters for which high resolution structures are not yet available. A recent simulation study from Pan et al. targeted the effects of the presence of drug molecules inside the cavity on the dimerization motion of the NBDs.433 100 and 300 ns-long atomistic simulations of P-glycoprotein were carried out in the presence (holo) and in the absence (apo) of drugs. The two sets of systems, holo and apo, revealed a different closing motion, with the apo simulations characterized by NBDs misaligned with respect to the orientation required to form a proper dimer. This misalignment appears correlated with the degree of lipid protrusion through the portals. While this study emphasizes the coupling between drug occupancy in the cavity and the motion of TMDs and NBDs, it also highlights how lipids can have an impact on the conformational transitions of the transporter.433
Figure 14.
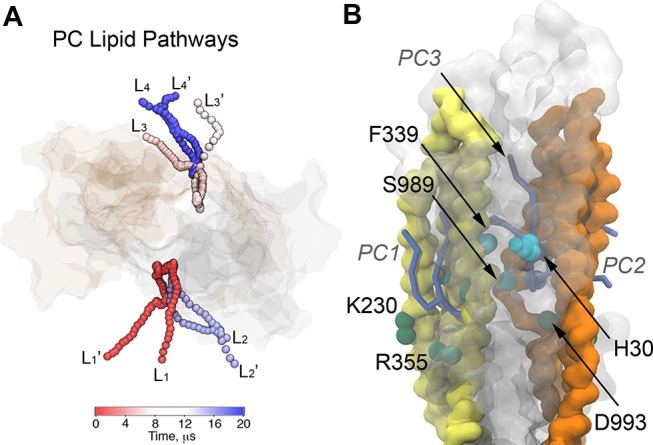
Lipid access to the central cavity of P-glycoprotein. CG simulations of P-glycoprotein in different mixtures of POPE and POPC lipids were used to identify the residues involved in interactions with lipids at the portals and inside the cavity.432 A. Displayed are examples of pathways sampled by different lipids to gain access to (L1 to L4) and to leave the central cavity ((L1′ to L4′) during the simulation time. P-glycoprotein is viewed from the extracellular side. Reprinted with permission from ref (432). Copyright 2018 Barreto-Ojeda et al. B. Snapshot of P-glycoprotein with a POPC lipid (PC1) located at portal 1 (yellow helices), a second lipid (PC2) located at portal 2 (orange helices), and a third lipid (PC3) inside the cavity. Highlighted are residues identified in the simulations as hot-spots for lipid interactions at the portals (green spheres) and inside the cavity (cyan spheres).
The studies described above provide details of lipid binding or access to the main cavity of ABC transporters but do not show full translocation events from the inner to the outer leaflet, due to the large conformational changes required during the transport cycle. Another limitation is the relatively simple lipid mixture used in the simulations. Recently, the use of the Martini model showed P-glycoprotein molecules simulated in a plasma membrane mixture, highlighting, for instance, the different distribution of fully saturated and polyunsaturated lipids around the two TMDs, with changes in the membrane thickness profile that shows region of increased and decreased thickness, respectively.121 Moreover, the simulation shows an enrichment in negatively charged lipids (such as PIP, PI, and PA lipids) around the protein–lipid interface in the inner leaflet, in line with the previously mentioned studies that highlight a ring of positively charged residues engaged in electrostatic interactions with lipid headgroups. The reorganization of the lipids near the protein also creates a clear pattern of negative curvature, possibly linked to specific structural features of P-glycoprotein, for instance the shorter extracellular end of TM3 and TM4 together with the presence of positively charged residues in this region. The interplay between lipids and ABC transporters is not limited to the TMDs, as lipids can affect the dynamics of the transporters and mutations at the NBDs can, in turn, affect substrate transport.434 Simulations have also suggested that membrane defects near specific transmembrane helices could be associated with a given conformational state of the cystic fibrosis transmembrane conductance regulator (CFTR), an ABC transporter that functions as a chloride channel, which would be a novel and uncommon role for lipid–protein interactions.435
6. Outer Membrane Proteins
The cell envelope of Gram-negative bacteria consists of two membranes, an outer membrane (OM) and an inner membrane (IM), separated by the periplasm.436 The lipid composition of the two leaflets of the OM is highly asymmetric: the inner leaflet is composed of phospholipids, mainly PE, PG, and CL, while the outer leaflet is composed of lipopolysaccharide (LPS) molecules. LPS consists of Lipid A, a glycolipid with six or seven saturated acyl tails connected to a glucosamine disaccharide unit and a polysaccharide. The polysaccharide is divided into a core sugar region, with an inner and outer core layer, directly attached to Lipid A, and the O antigen, consisting of repeating units of monosaccharides, attached to the core region and extending into the extracellular medium.437 LPS molecules are linked together by divalent cations, which strongly bind to the several negatively charged groups of the LPS molecules.437
Due to its composition, the OM is impermeable to hydrophobic and large polar molecules. Beta-barrel transmembrane proteins, called outer membrane proteins (OMPs), regulate the movement of a large range of molecules across the bilayer, and can constitute up to half of the OM mass.437,438 OMPs differ in diameter and can function as selective or nonselective porins, as well as transporters, enzymes, or receptors.437Figure 15 illustrates a few examples of OMPs discussed below.
Figure 15.

Examples of OMPs discussed in this review. From left to right: OmpA,439 the OmpF trimer,440 Hia,441 LptD and LptE,442 and BamA.443 The five POTRA domains of BamA are labeled P1–P5. Individual subunits are shown as cartoons and colored in light yellow, orange, and brown. Upper and lower panels show a side view and a view from the extracellular side of the proteins, respectively. Out, extracellular side; Per, periplasm. The membrane region is highlighted in gray.
6.1. OMPs in Simple Mixtures
The interplay between OMPs and the OM has been extensively studied with MD simulations. Simulations in simple phospholipids mixtures have been used to characterize the hydrophobic mismatch between OMPs and the membrane together with related lipid sorting effects. Membrane thinning over several nm away from the proteins coupled with enrichment in shorter tails lipids has been shown, for instance, for OmpA in phospholipid bilayers.444 The size and concentration of OMPs affect both protein and lipid diffusion. A panel of OMPs including OmpA, NanC, FhuA, OmpF, and LamB, which differ in size and amino acid composition, were also studied in lipid bilayers consisting of only POPE or a mixture of POPE and POPG lipids, to approximate the composition of the OM inner leaflet.445 CG simulations carried out at varying protein densities, from 2 to 50% of the membrane area covered by proteins, showed slower lipid diffusion over 2 to 3 nm from the protein surface, with the larger trimeric assemblies of OmpF having the strongest effect compared to the smaller, monomeric NanC.445 The slower lipid diffusion near the protein was also shown to be different between the leaflets, with the lipids in the outer leaflet diffusing slower than those in the inner leaflet. This effect was correlated with the higher content of charged residues on the outer surface of the proteins, providing hot spots for electrostatic interactions with lipids.445 Crowding was also responsible for slower lipid and protein diffusion, with the largest proteins (FhuA, for instance) forming extended clusters that led to the compartmentalization of lipids.445 The analysis of the effect of crowding in larger bilayers of similar composition but with 256 OmpA molecules prompted the development of new computational tools for the visualization of lipid motion linked to protein oligomerization.446
Lipid–protein interactions are important for protein oligomerization as well. For instance, the free energy of dimerization between NanC monomers has been calculated by means of umbrella sampling and CG simulations.447 Dunton et al. proposed that lipid–protein interactions can induce the formation of metastable states, with the interface between the two proteins separated by one, two, or three lipid molecules that bridge the proteins together, stabilizing the dimers.447
The use of CG models for the study of the lipid–protein interplay allows the simulations of large systems with high protein concentrations, useful to address questions concerning protein–protein interactions in the membrane as well as to investigate how proteins modify physical properties of the membrane. For OMPs, such studies have been applied to the interactions of BtuB and OmpF with a POPE and POPG lipid bilayer,448−450 (see also below for more details) as well as to OmpF in vesicles of diameter ranging from 16 to 65 nm, with a POPE and POPG mixture or DLPC and DSPC lipids, where protein–protein interactions were linked to membrane thinning and lipid sorting.451,452 BtuB and OmpF seem to modulate membrane bending rigidity in different ways. With 10 μs-long simulations where 28%, 37% and 40% of the membrane area was covered by BtuB, OmpF or an equal distribution of the two proteins, respectively, Fowler and colleagues show how BtuB, a monomeric OMP, significantly reduces membrane stiffness.449 The OmpF trimer, on the other hand, shows no comparable effects, although it remains unclear if this different behavior between the two OMPs is due to their oligomerization state or to the limited time scale (5 μs) of the simulations.449
CG models have also been applied to lipid binding to OMPs and inner membrane proteins (IMPs) as a function of charge.453 Binding of POPG or POPC lipids to OmpF, the ferripyoverdine receptor (FpvA) and VDAC (a mitochondrial outer membrane protein covered in more detail below) as examples of OMPs and to AmtB, MscL, and NavMS as examples of IMPs was addressed at different pHs using native MS.453 Lipid binding to OmpF, the protein with the highest local densities of charged residues among those tested in this study, showed the greatest sensitivity to electronspray polarity in the MS approach, while lipid binding to AmtB showed minimal changes (see section 13.4 for more details on AmtB). 1 μs-long CG simulations of AmtB and OmpF in a POPC:POPG 1:1 mixture were performed for different protonation states of both lipids and proteins and showed increased binding of anionic POPG for the charged state of the proteins, especially for OmpF. The analysis of lipid distribution around the proteins between the different protonation states support a model where the location of basic residues along the protein surface and their charge state affect lipid binding selectivity, in line with the native MS experiments.453
6.2. OMPs in Complex Mixtures
Given the asymmetric composition of the OM, more realistic OM models for MD simulations required the development of atomistic and CG force field parameters for LPS, which allowed the investigation of different structural properties of the OM.454−467 For instance, the coupling between the two asymmetric leaflets, as well as the network of electrostatic interactions between LPS molecules and the details of the electroporation mechanism after applying varying external electric fields have been studied using atomistic simulations.455 The role of the different components of the LPS molecules, crucial for the structural integrity of the membrane, has also been investigated by means of atomistic simulations in combination with NMR studies457 and using the Martini model as well.459 Experimental evidence of specific interactions between LPS and OMPs,468−473 and the role of LPS in regulating the assembly of several OMPs into trimers,473−477 highlights the importance of using more complex OM models for proper characterization of OMPs dynamics and lipid–protein interactions in computer simulations.
Atomistic simulations of E. coli FecA, an OM 22-stranded beta-barrel, in a membrane composed of LPS in the outer leaflet and PE, PG lipids, and CL in the inner leaflet characterized conformational changes of extracellular loops upon ligand binding and FecA-LPS interactions.478 The slower diffusion rate of LPS in the outer leaflet allows for frequent but short-lived hydrogen bonds between the core sugar region of LPS molecules and specific FecA residues in the extracellular loops, experimentally shown to be essential for FecA function.478 Such interactions modify the loop dynamics compared to control simulations of FecA in POPC membranes, where no hydrogen bonds were retrieved between the polar POPC headgroup and the selected protein residues.478 Similarly, restrained loop dynamics has been reported for other OMPs, including the outer membrane protein phospholipase A OmpLA, and OmpA in atomistic, asymmetric membranes with LPS in the outer leaflet and phospholipids mixtures in the inner leaflet.458,479,480 Differences in the interactions between OMPs and LPS or phospholipids have also been described for the H. influenza autotransporter Hia,479 which consists of three monomers, each carrying four beta-strands and an α helix, that combine together in a 12-stranded beta-barrel and three helices located in the central pore.441,481 In atomistic simulations of Hia embedded in an OM model with LPS and PE lipids in the outer and inner leaflet, respectively, positively charged residues of the helices interact with LPS molecules, possibly acting as anchor points for the protein in the membrane.479 However, a detailed characterization of the LPS–protein interactions is limited by the short time scales of the simulations and the slower diffusion of LPS.479 Balusek et al. used 300 ns-long atomistic simulations to analyze conformational changes of BtuB, a monomeric beta-barrel that translocates vitamin B12 through the OM, as a function of the membrane environment and the presence of Ca2+ ions, required for transport.482 As discussed above for other OMPs, the flexibility of the extracellular loops and several substrate-binding residues is restrained in the presence of LPS molecules, while significant conformational changes are detected when LPS is missing from the membrane environment. Comparisons between the simulations carried out with and without Ca2+ ions also show how Ca2+ binding not only stabilizes further an extracellular loop important for substrate binding but also drives specific structural rearrangements that are typical of BtuB structures in complex with vitamin B12.482
MD simulations carried out in these more complex models often highlight how the thickness profile of the OM changes in the proximity of the proteins, as previously shown for the simpler mixtures. In particular, the simulations on OmpLA show an overall thinner membrane for the complex model compared to a phospholipids-only bilayer, due to the shorter hydrophobic length of the LPS molecules.458 The more pronounced smaller thickness near OmpLA is also not uniformly distributed, reflecting lipid adaptation to the asymmetric structure of the protein.458 Atomistic simulations on OmpA reveal how the interactions between LPS and the protein as well as protein–protein interactions can be modulated by different ionic strengths.480 CG simulations of OmpA and OmpF show that the proteins perturb lipid ordering and thickness over a radius of ca. 3 nm in a PE, PG, and cardiolipin mixture, while in an asymmetric OM model including LPS the proteins’ effect on the bilayer properties is not as pronounced.483 The difference is attributed to the slower diffusion of the large LPS molecules, which in turn reduces the diffusion of phospholipids in the opposite leaflet.483
CG simulations of OmpF in asymmetric membranes with lipid A or lipid A and the core sugar region moieties of LPS further confirm the interactions between lipid A and the core region with the protein in the outer leaflet.464 Lipid A is found distributed along the protein surface of monomers, with the tails interacting with the hydrophobic residues of the beta-barrel, while the head groups are engaged in electrostatic interactions with the protein positively charged residues. The so-called “corona” of lipid A around the protein does not prevent the formation of dimer or trimers, and as proteins oligomerize, protein–protein interactions replace lipid A-protein interactions, with lipid A reorganizing around the newly formed dimer or trimer.464
LPS and OMPs are essential to regulate proper homeostasis of the OM and provide a barrier against antibiotics and antibodies that hamper bacterial invasion. Depending on the environmental conditions, Gram negative bacteria can express different forms of LPS, thus promoting different interactions with OMPs and possibly the accessibility of the proteins extracellular loops, which are targeted by antibodies. To address the question of how the LPS leaflet hampers antibodies access to the OMPs, Patel et al. carried out atomistic simulations of OmpF in OM models characterized by a different degree of complexity, depending on the presence and type of the LPS core region and the O antigen, while the inner leaflet is composed of a mixture of PE, PG lipids, and CL (Figure 16).484 These simulations show at the molecular level how the LPS sugars are effective in blocking access to the extracellular loops of OmpF: their exposure is mainly regulated by the interactions between the core sugar region and specific residues of the protein outer surface, without affecting its channel function.484 The degree of such exposure, however, depends on the complexity of the OM model, which controls the flexibility of the core region as well as the O antigen, and in turn the dynamics of the protein extracellular loops.484 A similar approach has been applied to study the interactions between the different domains of LPS and the outer membrane protein H (OmpH) of P. aeruginosa.485 Atomistic simulations of OmpH in asymmetric membranes modeling P. aeruginosa and E. coli OMs show how the complexity of the LPS domains modulate the dynamics of the extracellular loops, as well as the interactions between LPS and positively charged residues on the outer leaflet surface of the protein. Although the E. coli OM is thicker than the P. aeruginosa OM, no significant difference in thickness was retrieved near the protein in the simulations, as a consequence of a reciprocal hydrophobic matching between the membrane components and the protein.485
Figure 16.

Simulation snapshots of the OmpF trimer embedded in atomistic OM models of increasing complexity.484 PE, PG and cardiolipin types of lipids of the inner leaflet are shown in blue, orange and magenta spheres; Lipid A is shown as pink spheres; a model of the LPS core is shown in gray stick, and polysaccharides of the O-antigen are shown in orange sticks. Ca2+, K+ and Cl– ions are shown as cyan, green and magenta spheres. Reprinted with permission from ref (484). Copyright 2016 Biophysical Society, by Elsevier Inc.
LPS is transported from the IM to the OM by a complex of seven lipopolysaccharide transport (Lpt) proteins, namely LptA through LptG.486 LptD, a beta-barrel protein, and LptE, required for LptD folding and acting as a plug inside the barrel (Figure 15), are responsible for the translocation of LPS across the OM and its insertion in the outer leaflet.486 Structural and functional assays combined with molecular simulations show that the polar domains of LPS may be translocated while inside the barrel, whereas the lipid A moiety exploits a lateral opening in the barrel structure to maintain the acyl tails in contact with the hydrophobic region of the OM.442,487,488 Dong and colleagues applied a multiscale approach combining CG and atomistic MD simulations of the LptD-LptE complex in a DMPE-DMPG bilayer and at different lateral pressures.442 The barrel opening was observed at −65 bar, a lateral pressure for which other beta-barrels, BtuB and FhaC, simulated as controls, did not show any instability.442 This study guided further experimental characterization and simulation studies focused on specific residues required for LPS transport as well as a series of hydrophobic residues predicted to facilitate the projection of Lipid A acyl tails into the membrane.487 Using a OM model with LPS molecules in the outer leaflet and PE lipids in the inner leaflet, Botos et al. applied atomistic simulations, in combination with structural and functional studies, to address the role of conserved proline residues in the beta-sheets responsible for the lateral opening.488 Simulations of the WT LptD-LptE complex and the complex with alanine mutations of the conserved proline residues showed a distinct pattern of hydrogen bond interactions between the beta-sheets, with less interactions for the WT complex, supporting experimental findings and confirming that the presence of the proline residues is essential to facilitate the lateral opening for LPS insertion.488
6.3. Beta-Barrel Assembly Machinery
Another macromolecular complex is required for proper homeostasis of the OM, the beta-barrel assembly machinery (BAM), which ensures proper folding and insertion of OMPs in the OM.489 BAM consists of five proteins, the lipoproteins BamB–E, and the beta-barrel BamA (Figure 15), whose function is essential for final insertion of OMPs in the OM. For this, BamA, as previously described for LptD, requires a lateral opening in its barrel architecture linked to an exit pore for OMPs, as suggested by structural, functional and molecular dynamics studies.443,490−492 The hydrophobic belt of BamA is asymmetric, with the thinner portion located near the C-terminal strand. In this region, μs-long atomistic simulations of H. ducreyi and N. gonorrheae BamA in a DMPE bilayer have shown a significant decrease in lipid order and membrane thickness as well as a lateral opening between the first and the last strands, providing access to the membrane from the barrel interior.443 Additional simulations of BamA have further characterized the weak points in the structure of the barrel that allow separation between the strands and the formation of an exit pore above the lateral opening, required for the extracellular loops of the nascent OMP.491 Recently, atomistic simulations of different BamA proteins in lipid bilayers and in a complex OM model, together with PMF calculations and mutagenesis studies, have also linked a kinked conformation in the C-terminal strand with the lateral opening of the barrel.492 While these studies helped develop a possible model for BamA-mediated OMP insertion, alternative models also exist (reviewed, for instance, by Kim et al. and Pavlova et al.).493,494
The N-terminus of BamA includes a large periplasmic domain formed by five polypeptide transport-associated (POTRA) motifs (Figure 15), whose degree of flexibility depends on the interactions with the other components of the BAM complex.495 Using atomistic MD simulations, Fleming and co-workers showed that the flexibility of the POTRA domains is also regulated by the interaction with the inner leaflet of the OM.496 A complex model of the OM was used, containing LPS molecules in the outer leaflet and a mixture of PE, PG lipids and CL in the inner leaflet. The POTRA domains partitioned into the membrane via hydrophobic as well as hydrogen bond interactions, suggesting that the BAM complex operates through a mechanism that requires the coordination between multiple domains, as well as the lipid matrix.496
CG simulations have been applied to understand the role of BamA and protein–protein interactions in the turnover of OMPs in the OM. Using fluorescence microscopy, Rassam et al. showed the existence of OMPs islands containing the BAM complex, which are linked to an OMPs biogenesis gradient through the cell, highest at the cell center.448 Within these islands, of ca. 0.5 μm diameter, different OMPs were identified. Protein diffusion rates decreased as a function of protein concentration and size and appear limited by protein–protein interactions in protein cluster formation.448 To study protein–protein interactions within the OMPs islands generated by the BAM complex, CG MD simulations were applied to systems containing PE and PG lipids and different ratios of BtuB, a monomer, and OmpF, a trimer, both identified within the OMPs islands used in the experiments. The simulations showed that the barrels engage in homologous and heterologous protein–protein interactions, with aromatic residues mainly involved in the latter.448 In a follow up study, the same group considerably extended the length scale of the simulation systems and used the information derived from OMPs clustering formation to derive a mesoscale model of how OMPs interact in a crowded environment:450 Larger clusters formed by different OMPs fence smaller clusters of proteins or single proteins, limiting their diffusion and creating a coralling effect that is proposed to be related to the insertion of OMPs in the membrane via the BAM machinery.448,450
The simulation studies described above have targeted different aspects of the protein–lipid interplay for OMPs, including membrane remodeling, protein stability, and protein–protein interactions, and highlighted the progress in modeling and simulating the complexity of the OM. However, the interactions between OMPs and LPS are hampered by the limited diffusion of LPS molecules on the time scale of even CG simulations, which remains one of the main challenges in this area.
Computer simulations can also address larger-scale questions about the structure of the bacterial envelope, which requires models beyond atomistic or coarse-grained simulations at the level of Martini. We highlight one example here, based on elastic models derived from more detailed simulations. The bacterial envelope (OM, cell wall and IM) is a complex structure that allows the cell to respond to changes in the osmotic pressure. Atomistic simulations of different OMPs embedded in a DMPC bilayer and subject to external forces, have been applied to derive an elastic energy model for OMPs, in which specific features of the barrel structure allow OMPs to withstand stress, possibly contributing the structural stability of the OM.497 The calculation of area compressibility from atomistic simulations of different models of the OM and the IM as well as of a model of the cell wall have linked elastic properties of lipid and protein components to the mechanical properties of the envelope as a whole.498 This type of studies provides a framework where structural properties of lipids and proteins are related to macroscopic features of the cell envelope.
7. TMEM16 Protein Family and Other Lipid Transport Proteins
Lipid asymmetry across membrane leaflets is regulated by several proteins that recognize different lipid types and move them between leaflets.499 We have previously described GPCRs and ABC transporters for which experimental studies and MD simulations have detailed lipid binding and/or translocation events. This section discusses the TMEM16 protein family and a few other examples for which MD simulations have contributed to a better understanding of their role in shuttling lipids across membrane leaflets.
Bidirectional lipid transport is achieved by scramblases, including the transmembrane protein 16 (TMEM16) family.500 TMEM16 proteins include members that carry out diverse Ca2+-activated functions, while sharing a common structural architecture, with limited conformational changes.500 Some members function as Ca2+-activated anion channels, others as Ca2+-activated lipid scramblases.500 Structural and functional studies of N. haematococca TMEM16 showed the presence of a hydrophilic groove exposed to the membrane that could provide the lipid translocation pathway501 while maintaining channel activity,502 thus proving that the dual channel-scramblase function can be carried out by the same protein.502 Similarly, the presence of the hydrophilic groove has been characterized for other TMEM16 proteins, whose details are reviewed by Whitlock et al. and more recently by Falzone et al.503,504 Many of the mutations that affect channel function are part of or near the hydrophilic groove, supporting the hypothesis that the TMEM16 pore is partially formed by lipids.503 MD simulations on TMEM16 proteins have so far addressed two main questions: how lipid scrambling occurs via the hydrophilic groove and if lipids and ions can move along the same pathway. A 1-μs long CG MD simulation of N. haematococca TMEM16 in a DPPC lipid bilayer detected not only lipid binding but also ca. 15 lipid translocation events between the leaflets along the groove.431 This simulation, however, did not characterize the details of lipid binding in the groove. Using a hybrid continuum-atomistic model, Argudo et al. and Bethel et al. described large membrane deflections induced by hydrophobic and charged residues located on the surface of N. haematococca TMEM16, with significant bending and membrane thinning located near the hydrophilic groove, which persisted after neutralizing all of the charged residues in the groove.90,505 Atomistic simulations of 120 and 400 ns, in a POPC lipid bilayer, showed the same membrane deformation profile, further supporting the hypothesis that the thinning of the membrane might provide a shorter pathway for lipid translocation through the groove.505 The simulations also identified, via density analyses, two interaction sites for the phospholipids, located at the extracellular and cytoplasmic ends of the groove.505 For these sites, the sequence alignment of different members of the TMEM16 family showed a smaller degree of conservation for those TMEM16 proteins with no or decreased scramblase activity, while they all share the same membrane deformation profile.505 A total of four lipid translocation events were detected in independent simulations and suggested a dipole-stacking mechanism for lipid access to the groove and translocation.505 According to this mechanism, lipids arrange in a dipole-like fashion, with the positively charged choline group of one lipid interacting with the negatively charged phosphate group of a second lipid, eventually forming a single chain of lipid head groups inside the groove, initiating at the cytosolic site.505 The energetic barrier for lipid translocation was estimated to be small, ca. 1 kcal/mol; thus, lipids are stable in the groove, as a result of the dipole-stacking and water molecules also contributing to the interactions. Jiang et al. also used atomistic simulations, 1 μs-long, to characterize lipid translocation in N. haematococca TMEM16 with Ca2+ bound, embedded in an asymmetric bilayer consisting of POPC lipids in the outer leaflet and a POPC:POPS mixture in the inner leaflet.506 These simulations showed the same membrane deformation profile observed before90,505 and identified a single chain of lipids in the groove as described by Bethel et al.;505 however, the major interaction sites between protein and lipids differ from those previously reported,505 with the exception of the extracellular site. The new sites were further investigated with mutagenesis studies and lipid scrambling assays confirming their role as hot spots for interactions with the lipids.506 The lipid scrambling pathway of N. haematococca TMEM16 has also been studied via mutations to tryptophan of 18 residues along the groove and by assessing their effects on scrambling rate constants.507 This study focuses mainly on the extracellular side of the groove and highlights three regions with the strongest effects on lipid scrambling and ion permeation: (i) an extracellular gate formed by charged residues, (ii) a constriction in the central region of the pathway, and (iii) a group of four residues located between the gate and the constriction (Figure 17). From 2 to 4 μs long atomistic simulations of wild type and selected tryptophan mutants of N. haematococca TMEM16 the authors derived the structural rearrangements of these three regions that enable lipid scrambling, resulting in a network of interactions between residues and lipids that starts with the opening of the extracellular gate.507 To further investigate the sequence of events that enables a complete translocation of lipids, Lee et al. applied the same iterative protocol previously used for opsin216 and detected full lipid scrambling events from the intracellular to the extracellular side, with lipid release in the extracellular leaflet only after the opening of the gate (Figure 17).
Figure 17.

Lipid scrambling and structural rearrangements in N. haematococca TMEM16 as described by MD simulations. A. Snapshots showing, from left to right, the structural rearrangements needed for lipid molecules to access the groove from the extracellular side. R432 initially interacts with E313 but as lipids approach it switches to the interactions with E318.507 B. Snapshots showing, from left to right, lipid translocation from the intracellular side to the extracellular side.507 The transmembrane region of TMEM16 is shown as white cartoons, while relevant residues involved in key interactions and lipids molecules are shown as spheres and sticks, respectively. Adapted with permission from ref (507). Copyright 2018 Lee et al. Licensed under Creative Commons Attribution 4.0. Panels in B are only a subset of those in the original figure. We thank G. Khelashvili and H. Weinstein for providing the original figures.
Electrophysiology experiments showed that high intracellular Ca2+ concentrations stimulate both the scramblase and channel activity of N. haematococca TMEM16.506 Additional MD simulations carried out at different voltages revealed that in the presence of Ca2+, lipid translocation events as well as Na+ ion permeation followed the same pathway along the hydrophilic groove, with longer residence time for Na+ ions retrieved at locations near the newly identified lipid interaction sites.506 POPC and POPS translocation events showed distinct features, with the POPS headgroup being more coordinated and for a longer time than POPC head groups. In the absence of Ca2+ ions, however, a smaller number of lipids accessed the groove. The simulations also captured the effects of Ca2+ ions on the conformational state of the protein: in the absence of Ca2+ ions, as a consequence of changes in electrostatic interactions, the intracellular end of the hydrophilic groove (lined by transmembrane helices TM4 and 6), widens considerably, while at the extracellular end TM4 can be in close contact with TM6, closing the groove, although for only less than 50% of the time.506
The computational studies described above have provided many insights in the molecular mechanism of TMEM16 proteins, but several aspects of their molecular mechanism remain to be elucidated. For instance, TMEM16 proteins have been shown to be involved in the scrambling of different lipid types,501,508−511 possibly with different lipid specificity.509 Malvezzi et al. recently showed that TMEM16 proteins can also scramble lipids conjugated with polyethylene glycol molecules, thus bearing head groups significantly larger than the width of the groove.512 These findings indicate that TMEM16 proteins enable lipid scrambling outside the hydrophilic groove as well, as part of a mechanism where the groove would trigger those membrane deformations that lower the energy barrier for lipid scrambling inside505,506,513 and outside the groove as well.512 This dual mechanism for lipid scrambling has also been verified for GPCR scramblases.512 In the near future, we expect MD simulations to be able to target the molecular details of this new mechanism for lipid scrambling. Further studies are also required to elucidate the complex interplay between TMEM16 and lipids, as cholesterol, fatty acids and PIP lipids have been shown to regulate protein activity.500
MD simulations have also provided some insights into the mechanism of lipid transport and scramblase activity of MlaA514 and PLSCR1,515 respectively. Additional structural data for PLSCR1 and information on possible interactions with other proteins and structural changes in MlaA are however needed to fully elucidate their mechanism of action.
8. P-Type ATPases
P-type ATPases constitute a family of membrane pumps that use the energy derived from ATP hydrolysis to flip lipids, transport ions and generate electrochemical gradients across biological membranes.516 Different from previously discussed ABC transporters, their mechanism requires phosphorylation and dephosphorylation of a conserved aspartate residue during the transport cycle.516 P-type ATPases are divided into five classes (P1 to P5), further organized into subfamilies, and share a common architectural core with three cytoplasmic domains (namely the Actuator, A, domain; the Phosphorylation, P, domain; the Nucleotide, N, binding domain) and a six helixes transmembrane domain, the so-called Transport domain, followed by additional transmembrane helices forming the Support domain.516 During the catalytic cycle, P-type ATPases switch between two main conformational states, namely the E1 state, with the binding sites for the ions to be expelled exposed to the cytoplasmic side, and the E2 state, where the ion binding sites are open to the opposite side of the membrane.516,517 Current models based on structural data, mutagenesis and transport studies propose that the pumps are phosphorylated in the E1 state (E1-P), to then switch to a phosphorylated E2 state (E2-P), in turn dephosphorylated to return to the E1 state. These conformational changes require large motions of the cytoplasmic domains, coupled with changes of specific transmembrane helices in the transport domain to allow ion transport.516
Ca2+-ATPases (members of the P2A class) transport Ca2+ ions from the cytosol into the lumen of organelles or outside the cell. The Sarco(endo)plasmic Reticulum Ca2+-ATPase (SERCA) located in the endoplasmic reticulum drives the reuptake of Ca2+ from the cytosol, required for muscle contraction, in exchange for H+.516 Several MD simulations studies have focused on Ca2+ and H+ ion exchange and possible pathways.518−520 SERCA activity is, however, heavily modulated by lipids.521 A membrane thickness of ca. 30 Å is required for maximum ATPase activity, although the hydrophobic belt of the protein is considerably shorter.521−525 In MD simulation studies of SERCA in PC lipid bilayers with acyl tails of different length, lipids adapt to the protein surface in a dynamic manner, depending on the conformational state in the starting structure.522,526 The thinning of the bilayer is not uniformly distributed around the protein but highly localized to match the hydrophobic and hydrophilic residues on the surface522 and linked to changes in the lipids tilt angle and structure of the acyl chains.526 Autzen et al. applied CG and atomistic MD simulations to address the interactions between SERCA and cholesterol, for which both specific and nonspecific interactions with the protein have been postulated.527 From 30 independent CG simulations in a POPC:cholesterol bilayer, Autzen et al. detected cholesterol enrichment near the protein, identifying putative cholesterol interaction sites. However, phospholipids were shown to compete with cholesterol for binding to the same pockets, which makes it difficult to explain how cholesterol might regulate SERCA via specific interactions. Moreover, the binding pocket of thapsigargin, a SERCA inhibitor, was postulated to be a potential cholesterol site due to shared structural features, but neither CG nor atomistic simulations found a stable binding mode, supporting the hypothesis of nonspecific interactions as the main mechanism for cholesterol regulation of the pump.
The studies above provide snapshots of local lipid adaptation around SERCA but do not describe the full complexity of the protein–lipid interplay. Many structural studies show several lipid binding sites, some of which are specific for a given conformational state while others are state independent, with distinct regulatory and structural roles (compared and analyzed by Drachmann et al.).528 More recently, Norimatsu et al. using X-ray solvent contrast modulation have revealed new details on how lipids actively contribute to the activity of the pump.529 Stable interactions with lipid headgroups were identified for specific arginine and lysine residues that act as anchor points for lipids through the entire transport cycle. To account for the perpendicular movement of selected transmembrane helices during transport, changes in the tilt of the entire protein, up to 18°, are required to preserve the interactions with the anchor points in the different conformational states, together with local variations in lipid thickness. At the same time, more dynamic interactions with lipids, different for different conformational states, were suggested with other positively charged residues and with tryptophan residues, important to drive the proper orientation of the protein with respect to the membrane through the catalytic cycle. Combined, protein tilt changes, stable interactions with lipids via the anchor points and more dynamic interactions with the supporting residues result in an effective mechanism that couples ion translocation with the large movements of the transport domain and the cytosolic domains.529,530
The Na+,K+-ATPase (a P2C ATPase) resides in the plasma membrane and extrudes Na+ ions while counter-transporting K+ ions.531 As for SERCA, ATP hydrolysis and movement of ions across the membrane are coupled with conformational transitions between phosphorylated or dephosphorylated states, E1 and E2, which contain ion binding sites of different affinity and exposed to opposite sides of the membrane.532 The Na+,K+-ATPase consists of three subunits, namely the alpha subunit, carrying the three cytoplasmic domains and the transport domain with ten transmembrane helices, the one transmembrane helix beta subunit, and the FXYD subunit.533 Nonspecific lipid–protein interactions, including membrane thickness and fluidity linked to cholesterol concentration, modulate protein activity (reviewed by Cornelius et al.).531 The specificity of the interactions between the pump and lipid headgroups, tails and cholesterol has been widely investigated via biochemical, kinetic and structural studies, which revealed how lipids can stabilize, stimulate, or inhibit the pump.531 In particular, expression of recombinant Na+,K+-ATPase in Pichia pastoris and purification studies in controlled lipid environments revealed a stabilizing effect of (18:0/18:1) PS lipids and cholesterol via specific interactions,534 while neutral polyunsaturated phospholipids (18:0/20:4 or 18:0/22:6 PC or PE) stimulate the ATPase activity promoting the transition between the E1-P and E2-P states.535,536 Anionic and polyunsaturated neutral lipids bind at distinct sites, namely site A for stabilizing lipids and site B for stimulating lipids.535−538 Cholesterol contributes to the thermal stability of the pump via direct interactions with phosphatidylserine at site A,536 while saturated PC and SM lipids inhibit the pump in a cholesterol dependent manner, leading to the hypothesis of a third lipid binding site (site C).536 The stoichiometry of lipid binding to site A and B has been further characterized by native mass spectroscopy combined with mutagenesis studies.539 Additional details and analysis of structural data are described in Cornelius et al.531 MD simulations have yet to address the specificity of the interactions described above and are so far limited to local perturbations of the lipids. While investigating the mechanism of response of a dye to the conformational transitions of the Na+,K+-ATPase, Garcia et al. showed, by means of atomistic simulations, the different thickness profiles induced by different conformational states of the pump on a DOPC membrane (Figure 18).540 However, in a more realistic membrane environment, the thickness profile also correlates with the local lipid distribution, as shown by CG simulations of the pump in a generic plasma membrane model.121 It has also been hypothesized that the N-terminal domain of the protein could exert an autoinhibitory effect by controlling the transition between states via interactions with anionic lipids.540−542 MD simulations of the E2 state with a modeled N-terminal domain and embedded in a POPC:POPS bilayer showed how the interaction with the membrane might be driven by electrostatic interactions between positively charged residues of the N-terminus and POPS lipids.541 With a similar simulation approach applied to both the phosphorylated E1 and E2 states, Nguyen et al. found that the interactions with the membrane are different between the phosphorylated states and stronger for the phosphorylated E1.542
Figure 18.
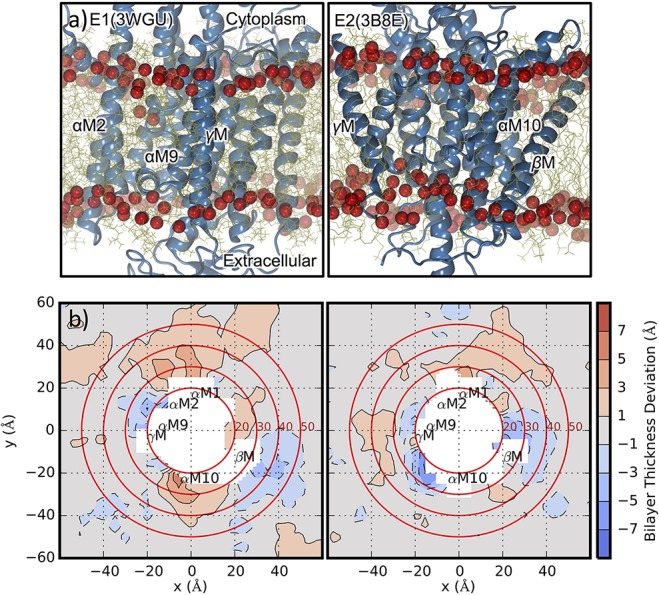
Membrane deformation around the Na+,K+-ATPase pump. MD simulations revealed distinct local perturbations of membrane thickness for two conformational states of the pump, linked to specific transmembrane helices.540 A. Snapshots of the pump in the E1 (left) and E2 (right) state. B. Thickness deviations profiles obtained from the last 50 ns of the simulations for the E1 (left) and E2 (right) state of the pump. Transmembrane helices linked to thickness changes are labeled. Reprinted with permission from ref (540). Copyright 2017 Elsevier B.V.
The plasma membrane Ca2+ pump (PMCA) is a P2B-ATPase whose activity is also modulated by lipids.543−545 Atomistic simulations of a PMCA model based on SERCA in bilayers consisting of DOPC or DMPC lipids showed how lipids adapt to the protein hydrophobic belt.545 In this case, the shorter DMPC showed a better hydrophobic matching than the longer DOPC,545 contrary to what Sonntag et al. described for SERCA, where the shorter 14 carbons lipids used in their study provided less favorable coverage,522 probably reflecting different structural features of the two ATPases.
P4-ATPases share the same catalytic mechanism of P2-ATPases like SERCA but use ATP as a source of energy to flip lipids from the extracellular or luminal leaflet to the cytoplasmic leaflet of membranes.546 Among different models proposed for the mechanism of action of P4-ATPases is the presence of a hydrophilic groove along the protein surface. This groove would allow the interaction with the lipid headgroup during flipping, while the acyl tails remain in contact with the hydrophobic region of the membrane, similarly to what has been previously discussed for TMEM16 proteins.546 Atomistic MD simulations on homology models of the ATP8A2 P4-ATPase supported this model by showing how water molecules occupy different locations of the groove depending on the conformational state of the pump.547 Full lipid translocation events, however, yet remain to be described in simulations, limited by the time scale of protein conformational changes that are required for transport or experimental conformations that can be use to construct a plausible reaction coordinate to describe the transport process.
The Cu(I)-transporting P-type (CopA) ATPase couples ATP hydrolysis with the transport of Cu(I). It shares the overall architecture of other P-type ATPases, with an additional cytosolic domain required for Cu(I) transport, and two additional transmembrane helices, MA and MB.548 In in vitro studies, the lipid composition greatly affects the activity of the enzyme, with increased ATPase activity induced by more disordered membranes as well as by the anionic lipids PG and especially CL.549 A combination of CG and atomistic simulations of CopA from Legionella pneumophila (LpCopA) in a PE, PG, and CL mixture revealed a preference for anionic lipids in close proximity of the protein.549 Anionic lipids, however, are not uniformly distributed and preferential hot-spots for PG and CL are identified from the simulations, with prominent cardiolipin interactions located at the C-terminal side of MB. Further analyses of the protein–lipid contacts highlight how for CL and PG lipids the interactions mainly involve their head groups, with contributions from the acyl tails in the case of PG lipids, while for the more abundant PE lipids headgroup and tails equally contribute. Combined, the in vitro and in silico approach applied by Autzen et al. shows a clear modulatory effect of the lipid composition on the activity of LpCopA, exerted through direct interactions with specific lipid types.549
9. ATP Synthases
The synthesis of ATP from ADP and inorganic phosphate is achieved by ATP synthases, rotary motors which utilize electrochemical gradients across membranes as their source of energy.550−552 One of the most studied ATP synthases is the F0F1-ATP synthase, found in chloroplasts, yeasts, bacteria, and the inner membrane of mitochondria. F0 is the transmembrane and F1 the catalytic domain, connected by a central and a peripheral stalk.551,553,554 MD simulations have been used extensively to study the free energy landscape and conformational coupling of the different subunits of these motors551,555,556 and, recently, have targeted the interactions with the surrounding membrane, discussed below.
In mitochondria, the F0F1-ATP synthase forms dimers arranged in rows along the tips of the cristae, contributing to the morphology of the membrane and optimization of ATP synthesis.557−559 To explain the experimental observation that the formation of rows of dimers is not due to protein–protein interactions, Davies et al. applied CG simulations of one, two, and four F0F1 dimers in a POPC bilayer.557 A single dimer induced a strong curvature in the surrounding membrane, which in turn promoted the association of dimers into rows and the formation of a sharp ridge in the membrane. A decrease in the membrane elastic energy rather than direct dimer–dimer contacts would then be the driving force for the formation of rows of F0F1 dimers along the curved edges of cristae (Figure 19).557 In a follow up study, the authors used free energy calculations to quantify the energetics of dimer association and further prove that direct dimer–dimer interactions are not required.560 Instead, dimers sense each other over a large distance, ca. 40 nm, via membrane deformations that are enough to promote spontaneous association of multiple dimers. This proposed mechanism has significant implications, as it has been suggested to be linked to the structural organization of the cristae.560
Figure 19.
Mitochondrial membrane shaping induced by ATP synthase dimers. CG MD simulations showed how ATP synthase dimers (yellow surface) contribute to the formation of mitochondrial cristae via curvature effects.560 A, one dimer; B, two dimers; C, four dimers. Adapted with permission from ref (557). Copyright 2012 Davies et al.
The local lipid composition and, in particular, the role and effects of CL should also be considered for the mechanism of the ATP synthase, as CL contributes to the morphology of mitochondrial membranes383 and directly interacts with ATP synthases.76,561−564 Duncan et al. used CG simulations to investigate the molecular details of such interactions, targeting the c-ring in the F0 domain in a POPE, POPC, and cardiolipin bilayer.386 The c-ring consists of multiple replicas of a two-helix hairpin subunit and functions as a rotor coupled with ion transfer. The simulation results on the bovine c8-ring, the S. cerevisiae c10-ring, and the I. tartaricus c11-ring showed CL molecules within the first two lipid shells at 0.6 and 0.9 nm, with interaction patterns remarkably similar across the different c-rings (Figure 20). Multiple CL molecules can interact and cluster around specific residues, especially in the inner leaflet, where the residence time was longer than that of phospholipids and longer than their residence time in the outer leaflet. The longest residence time of ca. 600 ns was detected for the c11-ring, indicating that during the 4 μs-long simulations CLs frequently exchange at the interaction sites. As mentioned above, CLs are required for proper function of the ATP-synthase, but tightly bound lipids could hamper the rotation mechanism of the c-ring. Based on their simulations, Duncan et al. suggest that the transient yet selective binding of CL would benefit the mechanism of the rotor by acting as a lubricant.386
Figure 20.
Selective binding of cardiolipin around c-rings of different size, as detected from MD simulation studies.386 A. Location of cardiolipin headgroup densities, determined based on radial distribution function analyses, around the c-rings are shown as spheres, color-coded accordingly to specific residues involved in the interactions. The size of the spheres reflects the intensity of the density signal. B. Time-averaged lipid density. Density for POPC and POPE headgroups is shown in gray, and density for cardiolipin headgroup is shown in pink. Reprinted with permission from ref (386). Copyright 2016 Duncan et al.
10. Aquaporins
Aquaporins (AQPs) represent a family of ubiquitous channels crucial for the regulation of flow of water and other solutes across membranes.565 As the time scale for water permeation across these tetrameric channels is on the order of ns and thus within comfortable reach of equilibrium MD simulations, computational studies have been extensively applied to characterize the mechanism of the channels and their selectivity for different solutes.566 The availability of several high-resolution AQP structures enabled a systematic analysis of lipid-interaction sites via a multiscale approach, combining CG and atomistic simulations.567 A similar pattern of interactions was found from the simulations of a total of 40 structures of different AQPs used as input structures for independent simulations: Preferential electrostatic interactions, although transient in nature, were identified between the lipid phosphate groups and positively charged residues. As AQPs are found in membranes of different lipid composition, the conservation of these residues would endow the channels with a similar lipid–protein interface evolved to ensure nonspecific yet recurring interactions.567
The structures of AQP0 solved by electron crystallography revealed the first shell of lipids interacting with AQP0 tetramers.56,57,568 Comparisons of the 2D crystals obtained in DMPC568 and E. coli polar lipids56 revealed minimal changes in the organization of the lipid acyl tails around the AQP0 tetramers, while differences were detected for the lipid head groups, suggesting that the interactions with the protein for the first layer of lipids is mainly driven by hydrophobic contacts. Aponte-Santamaria et al. embedded a single tetramer and the 2D crystal organization of four tetramers in DMPC bilayers and performed 100 ns-long simulations.569 Time-average lipid density analyses described at the molecular level forces driving the lipid tail localization on the surface of AQP0. This appears to be guided primarily by surface complementarity and impacted by the degree of flexibility of the protein. Although polar and positively charged residues allow for electrostatic interactions with the lipid headgroups, in silico mutagenesis of such residues and additional simulations retrieved the same lipid distribution around the protein surface as for the WT systems, ruling out their role as specific lipid-interaction sites.569 In a follow up study from the same group, the effects of temperature, phase transition, and protein mobility on lipid localization were further investigated, with protein mobility shown to have the strongest effect (Figure 21).570 Lipids adapted to the protein hydrophobic belt, resulting in an increased thickness as well as in localized, gel-like regions with higher order, linked to different features of the protein surface. Lipid headgroups, however, although not the major players in controlling lipid organization around AQP0 tetramers,56,568,569 do affect crystal packing, as shown by the analysis of AQP0 2D crystals obtained in the presence of anionic lipids (DMPG, DMPS, or DMPA). The headgroup of these lipids interact with different protein residues, thus altering the interactions between tetramers.57
Figure 21.

Organization of lipids around AQP0. To study the effect of temperature, lipid phase, and protein mobility on the reorganization of lipids around AQP0, Briones et al. performed MD simulations using different simulation setups and found that protein mobility has the strongest effect on lipid packing in close proximity of the protein.570 Displayed are lipid densities around AQP0 monomers as a function of simulation conditions. Reprinted with permission from ref (570). Copyright 2017 Briones et al. Licensed under Creative Commons Attribution 4.0.
Other simulation studies have targeted different aspects of the interplay between AQPs and lipids. MD simulations of AQP5 with a PS lipid bound to the central pore revealed that the bound lipid does not modify the water flow through the channel but alters, instead, gas permeation.571 Simulations of an AqpZ monomer were also used as part of a study to describe the effects of surface tension on phase transition of DPPC lipids.572 Compared to the pure lipid bilayer, AqpZ has a stabilizing effect on the lipids, with a thinning of the membrane that persists at different surface tensions near the protein. In the gel phase reduced lipid order was observed, while in the liquid phase the protein hampered lipid diffusion.572 A previous simulation study targeted, at the atomistic level, the interactions between AQP0 and membranes consisting of DMPC lipids or DMPC lipids and cholesterol.573 Over 25 to 100 ns of simulation time, cholesterol engaged in both stacking interactions with aromatic residues and hydrogen bonds with polar residues on the surface of AQP and strengthened the hydrogen bonds between DMPC headgroups and the protein.573 The use of CG simulations has enabled the study of the interactions with more complex lipids and membranes. Gu et al. investigated the behavior of gangliosides in μs-long atomistic and CG simulations, highlighting possible interactions with AQP1.574 Gangliosides form small clusters and interact directly or via other lipids with the protein, establishing extended hydrogen bond interaction networks. The lipid organization around AQP1 derived from CG simulations using a generic plasma membrane revealed a more complex lipid redistribution: small, highly localized islands of cholesterol enrichment are symmetrically distributed near the monomers interface, while broader distributions are found for polyunsaturated and saturated lipids, with significant differences between upper and lower leaflets.121 Despite local differences in lipid distribution between four simulated tetramers, this simulation study showed a highly symmetric curvature profile induced by the proteins, outlining a complex landscape of possible interactions with lipids and the need for further characterization of the lipid–protein interplay and its effect on protein function.
11. Receptor Tyrosine Kinases and Cytokine Receptors
Bitopic membrane proteins are characterized by one transmembrane helix that connects the extracellular domain to the intracellular domain, including a juxtamembrane region, and are usually active as homodimers or heterodimers. Different types of cell receptors are bitopic proteins, including receptor tyrosine kinases (RTKs) and cytokine receptors, large families of membrane proteins essential for signal transduction (Figure 22A).575,576 To date, there is no high-resolution experimental structure of a full length RTK or cytokine receptor, but structures are available for the separate domains as well as for dimeric transmembrane domains.577 Experimental and computational efforts have extensively targeted the dynamics and energetics of the transmembrane dimers, and experimental findings on the membrane-mediated modulation of these proteins have prompted a number of studies targeting interactions with the lipids as well.577,578 In atomistic and CG studies addressing helix–helix association of RTKs (epidermal growth factor ErbB and ephrin, Eph, receptors), membrane thickness has been shown to affect dimer stability and preferentially select specific dimer conformations, which also depend on lipid composition.579,580 Computational approaches have also been used to assemble full-length models of the epidermal growth factor receptor (EGFR or ErbB-1) starting from experimental structures of the individual domains and to characterize their interactions with the membrane.581−583 Atomistic μs-long simulations of the transmembrane dimers extracted from these models revealed a different degree of opening of the dimer depending on the lipid composition.581 Larger openings were detected in a DMPC bilayer compared to a POPC:POPS mixture, as a consequence of different helix tilting in membranes of different thickness. The PS lipids not only selectively stabilized the transmembrane dimer in the inactive state but also enhanced the conformational coupling between all of the protein domains via strong electrostatic interactions with the juxtamembrane region of EGFR.581 As anionic lipids are involved in the signaling cascade mediated by EGFR, MD simulations studies have focused on the interactions between PIP lipids and the juxtamembrane region.584 In a multiscale approach combining atomistic and CG simulations, Abd Halim et al. showed PIP2 lipids clustering near a group of positively charged residues of the juxtamembrane region. For a construct where mutations were introduced in place of the basic residues, a significant decrease in PIP2-protein contacts was observed, together with changes in the structure of the dimer.584 Computer simulations also showed how this pattern of interactions between anionic lipids and the juxtamembrane domain is a feature not only of EGFR but of many other RTKs as well. Hedger et al. applied a multiscale approach similar to that of Abd Halim and colleagues to a panel of 58 RTKs, retrieving clustering of PS or PIP lipids around specific and conserved juxtamembrane residues, driven by electrostatic interactions (Figure 22A,B).585
Figure 22.

Lipid–protein interactions in RTKs and cytokine receptors as detected in MD simulations. A. General structure of an RTK. B. The transmembrane helix and juxtamembrane region of EphA2 receptor, colored according to the extent of the interactions with PIP lipids.585 A and B are reprinted with permission from ref (585). Licensed under Creative Commons Attribution 4.0. C. Snapshots after 5 μs of simulation time (left, upper leaflet; right, lower leaflet) of a plasma membrane model with multiple transmembrane segments of a cytokine receptor.591 GM3, PIP lipids and sodium ions are shown in magenta, yellow and blue spheres, respectively. D. Extent of the interactions from blue to red for PIP lipids and cholesterol with the transmembrane helix of the receptor. C and D are reprinted with permission from ref (591). Copyright 2014 Koldso et al. Licensed under Creative Commons Attribution 4.0.
Anionic lipids are not the only lipids known to modulate RTK activity. For EGFR, for instance, the interactions between GM3 gangliosides and the extracellular domain affect the dimerization mechanism and prevent the activation of the kinase domain.586 CG simulations provided more details on the interactions between EGFR, PIP and GM3 lipids.587 While the contacts between PIP lipids and the juxtamembrane region persist over several μs of simulation time, the interactions of the GM3 lipids in the extracellular side are dynamic, although always localized around positively charged residues. The free energy profiles of lipid association to WT and mutant transmembrane dimers revealed weaker interactions with GM3 lipids than with PIP lipids. This may be linked to receptor localization in raft-like domains enriched in gangliosides in the upper leaflet and to a specific role of PIP lipids in the cytoplasmic leaflet in modulating receptor dimerization.587 A simulation-based multiscale approach also showed how anionic lipids have an effect on the conformation of the extracellular domain of RTKs.588 The interactions with PG lipids anchored the large and flexible membrane-proximal fibronectin domain 2 (FN2) of the extracellular domain of ephrin (Eph) receptors to the membrane surface, which may be involved in receptor clustering for signal transduction.588 The extracellular domain of RTKs plays a significant role in the conformational coupling of the different receptor domains not only via substrate binding, but also through N-glycosylation. Simulation studies have suggested how N-glycosylation triggers a different pattern of interactions with the membrane, which may be relevant for kinase activation.589 A recent computational study on the EphA2 receptor targeted in particular the interactions between the kinase domain, the juxtamembrane region and the membrane.590 In combination with liposome pull-down experiments, the simulations showed the ability of the kinase domain to interact with the membrane mediated by PIP lipids, resulting in two main orientations of the kinase domain.590
The studies described above highlighted the interactions of the juxtamembrane region with anionic lipids. Here, this pattern of interactions is influenced by the presence of the kinase domain, with decreased PIP lipid clustering when the kinase domain contacts the juxtamembrane region. This study combined with previous modeling of the full EphA2 receptor588 provides new insights on the possible conformational coupling of the extracellular, transmembrane, and cytosolic domains of the receptor, relevant for receptor activation and inactivation.590 Other membrane properties might be relevant as well. CG simulations of EGFR in a plasma membrane model, for instance, described large membrane perturbations in terms of thickness and curvature induced by the 2 transmembrane helices and possibly by interactions with the extracellular and juxtamembrane domains.121 While at the CG level changes in the conformational state of the receptor are not addressed, this study shows that the effect of the protein on lipids goes beyond lipid sorting near the protein and specific interactions with selected protein residues, and extends to significant changes on membrane properties.
Cytokine receptors represent another family of membrane receptors involved in signal transduction upon homo- or heterodimerization.576 Simulation studies revealed a pattern of interactions with anionic lipids similar to RTKs: CG and atomistic simulations identified both zwitterionic and negatively charged lipids in the first few shells near the protein, but with enhanced clustering of anionic lipids close to charged residues of the juxtamembrane region.427 The interactions between lipids and cytokine receptors were studied in a complex mixture of lipids asymmetrically distributed between leaflets (Figure 22C).591 This membrane model included, among others, ganglioside GM3 and anionic lipids, which clustered and colocalized with the cytokine receptors as discussed above for EGFR, as well as cholesterol, for which preferred interaction sites were identified (Figure 22D).591
The increasing complexity of the membrane composition used in simulations and the use of the CG representation of the system combined with atomistic studies have helped identifying conserved lipid interaction sites and confirmed the role of the membrane in modulating proteins’ conformational state. Currently, one major limitation is related to the large size of these dimers and their flexibility, which hinder sampling and limit the time scale on full-length models.
12. Integrins and L-Selectin
Integrins and selectins are different families of cell adhesion molecules. Integrins are heterodimeric receptors involved in cell adhesion and migration and regulate signaling cascades in many cellular processes.592 The intracellular domain of integrins associate with several cytosolic proteins that, in turn, mediate the interactions with the actin cytoskeleton.593 Active and inactive conformations of these heterodimers (consisting of an α and a β subunit) are associated with different orientations of the transmembrane helices, in turn controlled by the interactions with talin on the intracellular side or with ligands on the extracellular domain.592 MD simulations have targeted the packing of the transmembrane helices, highlighting the role of a specific motif in heterodimer stability594−596 and showing how the free energy of association of integrin transmembrane domains depends on the specific subunits involved in the heterodimer, relevant for integrin activation.597 Below we describe in more detail simulations studies that addressed interactions with the membrane.
Talin connects integrin receptors with the cytoskeleton. Integrin activation involves the interaction between talin, the membrane, and the β subunit of the receptor, which results in conformational changes in the extracellular domains of integrins.598 Anionic lipids are important players in this mechanism.598−601 MD simulations have been extensively used to describe the interactions between talin and the membrane, highlighting the role of anionic lipids in triggering the interactions with several basic residues of talin.602−604 Multiscale approaches combining CG and atomistic simulations or atomistic simulations alone of integrin transmembrane helices, with or without talin, revealed how talin modulates the interactions between the helices, controlling the switch between integrin conformational states.603,605,606 The results of atomistic MD simulations suggested that the interplay between PIP2 lipids and talin contributes to the activation of the integrin heterodimer by breaking an interhelix salt-bridge and promoting transmembrane helix separation.607 The residues involved in the salt-bridge are highly conserved, and the interaction is required to form a stable helix–helix complex that prevents integrin activation.608,609 Using a combination of biophysical and simulation methods, Schmidt et al. evaluated the effects of zwitterionic and anionic lipids on the salt-bridge: anionic lipids do interfere with the salt-bridge interaction but at the same time are able to stabilize the transmembrane complex by bridging the helices together, resulting in more stable contacts compared to zwitterionic lipids.599 Microsecond-long atomistic simulations also suggested an alternative mechanism of tail-mediated integrin activation that does not require the transmembrane helices to separate.610 While these studies used the transmembrane domain of the receptor with or without talin or talin alone, Kalli et al. addressed lipid–protein interactions for a full integrin receptor in complex with talin, in three lipid bilayers with different ratios of PC, PE, PS, SM lipids, and cholesterol between leaflets.611 The simulations identified residues that specifically interact with PS lipids, which also promote stable interactions of talin with the membrane, in line with previous studies postulating the critical role of electrostatic interactions in the formation of the complex with talin. The interactions with PS lipids, together with higher cholesterol density near the transmembrane domains, resulted in changes in lipid organization near the receptor, with slower lipid diffusion extending for up to 2.8 nm from the receptor.611
Integrin clustering in the membrane has been hypothesized to be part of the signaling transduction mechanism, although some controversy remains.612 MD simulations proposed that integrin clustering in the membrane might be driven mainly by the β subunit, which induces a less tight lipid packing in its immediate surrounding, compared to the α subunit.613
Selectins are a group of three glycoproteins, namely P-, E-, and L-selectin, expressed in different types of cells, characterized by an extracellular region containing a lectin type domain, an EGF type domain and a series of short repeats; a single transmembrane spanning helix, and a short intracellular domain.614 L-Selectin, expressed on leukocytes, acts as the membrane receptor for ezrin/radixin/moesin (ERM) proteins, which connect L-selectin to the cytoskeleton.614 Sun and colleagues have used MD simulations to study the role of anionic lipids in triggering the formation of the complex between the cytosolic domain of L-selectin and the N-terminal subdomain of ERM (FERM).615 Here either PS or PIP2 lipids were found to enhance the binding strength of FERM to the membrane, with PIP2 providing the largest driving force. Moreover, PIP2 induced a conformational change of L-selectin, which allowed the formation of the heterocomplex by merging of PIP2 clusters around FERM and L-selectin, highlighting once again the role of PIP lipids in signal transduction.615
The studies discussed above describe the molecular details of membrane reorganization associated with lipid–lipid and lipid–protein interactions and provide further evidence on the role of lipids as active players in signal transduction.
13. SLC Transporters
The solute carrier transporters (SLCs) superfamily is comprised of a large number of membrane proteins involved in the transport of a wide range of substrates, including ions, amino acids, neurotransmitters, drugs, sugars, and lipids.616 As defects in SLCs underlie many diseases, SLCs represent a class of attractive drug targets.617 Lipids can affect the folding of SLCs, modulate their activity, and contribute to the stability of protein oligomers.44,618 Below we review the application of MD simulations to study the role of lipids in SLCs. Following other literature, we classify SLCs into four main categories based on their structural fold, namely (i) the major facilitator superfamily (MFS) fold, (ii) the LeuT fold, (iii) other inverted repeat folds, and (iv) other folds, including all of the remaining SLCs.616 Importantly, in SLC families many proteins with low sequence identity adopt the same fold (often LeuT-like or MFS). To connect fold-based classification to SLC-family based classification we show examples of SLC families grouped by fold.619,620
13.1. MFS Fold (SLC2, SLC15–19, and SLC37 Family of Transporters)
The MFS is the largest superfamily of SLCs. MFS members transport nutrients, metabolites, toxins, drugs, and other substrates across membranes623 and are characterized by a structural architecture of two 6 transmembrane helices bundles, each containing two symmetric inverted repeats.616 One of the first structures was the lactose permease, LacY (Figure 23). In an early simulation study, Lensink et al. simulated LacY in three different bilayers (POPE, POPC, and POPG).624 The authors proposed that the PE-dependence of LacY for lactose transport is related to specific interactions between highly conserved aspartic acid and (non-, mono-, and dimethylated) POPE amine groups. The phospholipid head groups are stabilized by interactions with a conserved lysine. Lensink et al. concluded that lipid-mediated salt-bridges are important determinants of LacY state stabilization. This mechanism of lipid-specific transporter function modulation was tested in a FRET study by Suárez-Germà et al.625 The D/C mutation of the conserved aspartate identified by MD studies was experimentally proven to considerably change LacY selectivity for lipids. Recently, Martens et al. combined hydrogen–deuterium exchange mass spectrometry (HDX-MS) with simulations and showed how the specific interactions between PE lipid headgroups and conserved charged residues (including the aspartate describe above) regulate the conformational equilibrium of the transporter.626 The HDX-MS experiments show not only how mutations that abolish the interactions at these charged positions promote the inward-facing state of LacY but also how PE lipids support the opening on the intracellular side. The same experimental approach applied to the xylose transporter XylE resulted in similar findings. Atomistic simulations showed specific interactions between the conserved charged residues of both transporters, LacY and XylE, in the inward-facing state and PE lipids, but not for PC headgroups, in line with the HDX-MS experiments. Moreover, the XylE simulations in the presence of PC lipids revealed a closing motion at the intracellular side of the transporter, further validated by additional HDX-MS studies. Combined, these results depict a complex mechanism for the transport cycle, possibly valid for other homologous SLCs, where the switching between outward- and inward-facing states should be described as a function of the interactions with lipids.626 μs-long atomistic simulations carried out on the ANTON supercomputer also show how PE lipids might be involved in substrate uptake on the periplasmic side of LacY, highlighting further the complex interplay between the transporter and lipids.627
Figure 23.
MFS fold and the LeuT fold in the SLC superfamily. Left. LacY structure621 in the inward-facing state with the two repeats (Rep1 and Rep2) shown in light yellow and orange cartoons. The side chains of residues known to interact with the sugar substrate are represented in white spheres. Right. Structure of a LeuT dimer,622 with the Na+ ions represented as blue spheres and the Leu substrate as white spheres. For one monomer, the two repeats are shown in light yellow and orange cartoons, while the second monomer is shown as a white surface. The top panels represent a side view of the proteins, with the membrane region highlighted in gray. The bottom panels represent the view from the periplasmic side. Per, periplasm; In, intracellular side.
Some of the best characterized MFS transporters are the glucose transporters (GLUTs), which transport glucose and other sugars across membranes via an alternating access mechanism, possibly combined with the accessibility of multiple substrate-binding sites.628,629 Iglesias-Fernandez et al. used atomistic MD simulations to study how the structure of GLUT1 is affected by the bilayer structure, following experimental findings of decreased transport in the gel phase.630 The simulations of two opposite protein conformations showed how the protein responds to different lipid environments, as in the gel phase the access to the central glucose binding site becomes narrower, with no continuous pathway from the extracellular to the intracellular side.630 CG simulations have provided a first characterization of the lipid distribution around the transporter embedded in a complex mixture, showing an enrichment of polyunsaturated and fully saturated lipids in the lower and upper leaflet, respectively.121 As the activity of GLUTs is regulated by the composition of the membrane, molecular simulations in the future are expected to yield further insight into the specificity of such interactions, especially with anionic lipids and cone-shaped lipids.631
13.2. LeuT Fold (SLC5, SLC6, SLC7, and SLC11–13 Families)
The leucine transporter (LeuT) fold consists of two inverted repeats of 5 transmembrane helices (Figure 23).632 LeuT has been extensively characterized from a functional and structural point of view and has served as a model to guide studies on other SLCs that share the same fold, including the neurotransmitter:sodium symporters (NSSs).633 The role of the membrane and detergent molecules in modulating its structure and function has been addressed with both experimental and computational approaches. Following up on experimental studies showing how detergent concentration alters the stoichiometry of substrate binding, MD simulations of LeuT in micelles revealed that this is due to detergent molecules occupying the secondary substrate binding site (S2).634 The substrate or agonist binding to the site S2 was proposed to be an essential part of the allosteric regulation of the transport cycle in NSS family. Simulations of LeuT in a POPE:POPG lipid environment mixed with increasing ratios of detergent were also carried out to mimic protein solubilization in detergent and investigate the behavior of lipids that remain bound to the protein.635 Penetration of detergent into the secondary substrate binding site occurs at high detergent concentrations and depends on the type of detergent, which has implications for the use of detergents in studies of NSSs. Moreover, at higher concentration, lipids within the first shells predominantly occupying site S2 may obstruct the substrate access to primary binding site S1 and hence impair transport function.635 It has been shown previously that detergent binding to site S2 (octylglucosides) results in the functionally blocked (transport-incompetent) form of the transporter.636
Atomistic simulations of LeuT in a POPC bilayer and in a POPE:POPG mixture detailed membrane thinning around specific helices important for transport as a result of hydrophobic mismatch, depending on the protein conformational state.637 A lysine residue in TM7 was mainly responsible for the membrane defect and water penetration deeper in the membrane, both of which were abolished in simulations with the corresponding alanine mutant.637 LeuT is highly dynamic during its transport cycle, and some controversy remains regarding the biological relevance of its crystallographic inward-open state in the membrane environment.633 MD simulations of LeuT in a POPC bilayer and in detergent micelles showed how this state is preferred in the micelle environment, due to a better compensation of the hydrophobic mismatch and higher mobility of the TM1A segment.638 In the bilayer, on the other hand, the dynamics of this segments is limited, thus restricting the opening of the inner vestibule compared to the crystallographic state.638
LeuT is the bacterial homologue of NSSs, such as the transporter for dopamine (DAT), serotonine (SERT), and others, which couple the sodium gradient with the reuptake of neurotransmitter from the synaptic cleft in an alternating access mechanism.639 The mechanism of transport of DAT and SERT is regulated by interactions with lipids, including PIP2 lipids and cholesterol.640−644 Direct interactions between PIP2 lipids and DAT involve electrostatic interactions with positively charged residues located at the N-terminus,640 as supported by atomistic simulations and self-consistent mean field model calculations to quantify the interaction energy of the DAT N-terminus with PIP2 lipids.640,645 The above-mentioned simulations were carried out on a model of the DAT N-terminus. Khelashvilli et al. also carried out equilibrium atomistic simulations of a model of human DAT embedded in a membrane enriched in PIP2 lipids, showing spontaneous conformational changes between different states.646 Electrostatic interactions between PIP2 lipids, the DAT N-terminus and the intracellular loop 4 were identified as the triggering event to induce the conformational transition from an outward-facing to an inward-facing state and release of sodium from the second binding site, highlighting the role of lipids as active players in the mechanism of transport.646 The comparison of extensive molecular dynamics simulations of DAT in membranes with and without PIP2 lipids highlighted how the different patterns of interactions between the N-terminus and the protein surface are directly linked to sodium release as they regulate the opening and closing of intracellular gates and water access to the binding site.647,648 As the N- and C-termini are segments typical of eukaryotic NSSs, these findings support the hypothesis that lipid-mediated regulation is different for NSSs and their bacterial homologues.649
The conformational transitions and function of NSSs are also regulated by cholesterol. Different populations of DATs are associated with cholesterol-depleted or cholesterol-rich domains.642,650−654 In cholesterol-rich domains, outward-facing states of the transporter are preferred and stabilized over inward-facing states, promoting binding of DAT blockers such as cocaine;642 cholesterol depletion shifts the equilibrium toward inward-facing states.653 Furthermore, cholesterol modulation encompasses interactions between cholesterol and specific sites on DAT,652,653 as also recently suggested by experimental structures, reporting different cholesterol binding sites.655−657 Similar considerations apply to other NSSs, including SERT.643,644,658−661 MD simulations at the atomistic and CG level of detail have explored the specific interactions between cholesterol and the suggested binding sites in NSSs. The crystal structure of DAT showed cholesterol bound to a groove lined by TM1a, TM5, and TM7.655 CG simulations on a SERT homology model in mixtures with increasing cholesterol concentrations identified six binding sites,662 including CRAC/CARC motifs and the crystallographic cholesterol site in DAT. In particular, Ferraro et al. suggested that cholesterol modulates the interactions at the intracellular gate of the transporter, stabilizing the outward-facing conformation.662 The cholesterol crystallographic site of DAT was also reproduced by CG simulations of DAT embedded in a plasma membrane model.121 The stabilizing effect of cholesterol on the conformational state of the transporter was further addressed by Laursen et al., who combined biochemical studies and CG simulations of the human SERT structure to show that cholesterol binding to the site identified in DAT is crucial in regulating the conformational transitions between states, favoring an outward-facing conformation.658 Extensive CG and atomistic simulations recently added further support to this, showing a common behavior across DAT, SERT and the norepinephrine transporter (NET).663 The CG simulations showed that two cholesterol sites described in DAT crystal structures655,657 are common to all of the NSSs used in this study (Figure 24). A comparison of these sites with the sequences of CRAC/CARC motifs revealed that such motifs are not essential for cholesterol binding.663 The μs-long atomistic simulations showed that the site lined by TM1a, TM5, and TM7655 is the most stable and hampers the transition to an inward-facing state. Considering the degree of conservation of this site, this simulation study suggests the existence of a common cholesterol-based regulation mechanism, possibly extending to other species.663
Figure 24.

Cholesterol interaction sites in NSSs obtained from CG simulations.663 Shown are different views of the selected NSSs with the cholesterol sites identified based on occupancy maps obtained in the presence of 20% cholesterol. The maps are drawn at occupancy values at least three times higher than bulk values. Adapted with permission from ref (663). Copyright 2018 Zeppelin et al. Licensed under Creative Commons Attribution 4.0.
The interplay between LeuT or NSSs and lipids extends further, as lipids also contribute to protein oligomerization, as shown by cardiolipin and phospholipid binding to the dimer interface of LeuT.618 A combination of different simulation techniques showed that in a pure POPC bilayer SERT forms symmetric dimers preferably via its transmembrane helix 12.664 This dimerization, however, is highly dependent on membrane composition as simulations performed in mixtures of POPC with cholesterol or with PIP2 lipids show marked differences and a higher energy barrier for SERT dimerization.664 PIP2 lipids were not found at the TM12 dimer interface, suggesting that they do not act as a bridge between SERT monomers. As experimental data show that PIP2 lipid concentration regulates the equilibrium of protein oligomerization,665 further studies are needed to clarify the direct interactions of PIP2 lipids and their role in protein oligomerization.
13.3. Other Inverted Repeat Folds (SLC9, SLC10, and SLC4 Families)
Na+/H+ antiporters regulate sodium level and pH via an alternating access mechanism as in other SLCs. The structures of E. coli NhaA and T. thermophilus NapA (Figure 25) represent different states of the transport cycle, and reveal a core region for ion translocation that adopt different conformations while the dimerization interface remains unchanged.666,669 Landreh et al. applied mass spectrometry and MD simulations to investigate the role of lipids in protein dimerization for NhA, NapA, and the human homologue NHA2.670 The mass spectrometry (MS) study highlighted the high dimer stability of NapA (Figure 25) and NHA2 together with a limited amount of bound lipids, while NhaA was found primarily as a monomer and with bound cardiolipin.670 Atomistic simulations of a NapA dimer in POPE lipids attributed the higher dimer stability of NapA to the tight packing of lipids at the interface between the core and the helices of the dimer interface, where membrane thinning was observed as a consequence of hydrophobic mismatch.670 Additional simulations of NapA, with and without tightly bound PE lipids, were carried out in vacuum to mimic the MS conditions. Lipids in these regions prevented protein helices from unfolding, and established interactions with the protein that persisted at higher temperatures, overall increasing dimer stability.670 The lower dimerization strength of NhaA was also reported by Gupta et al., who found evidence of cardiolipin binding in NhaA dimers at the interface between monomers, and an increase in the amount of monomers following loss of bound cardiolipin.618 These findings support the hypothesis that cardiolipin stabilizes the dimeric form by bridging the two monomers together. In contrast, NapA dimers are stable without cardiolipin, as the glue-like mechanism is provided by an N-terminal segment not present in NhaA.618
Figure 25.

Representative structures of other inverted repeat folds in the SLC superfamily. Left. NapA dimer,666 with the ion translocation domain in light yellow cartoons and the dimerization domain in orange cartoons. The second monomer is shown as a white surface. Aspartate 157 required for ion binding is highlighted in white spheres. Center. The UraA structure,667 with the gate and the core domains in orange and light yellow cartoons, respectively, and the Ura substrate highlighted in white spheres. Right. The structure of a Band 3 dimer,668 with the gate and core domain of one monomer in orange and yellow cartoons, respectively. The second monomer is shown as a white surface and the bound inhibitor is shown as white spheres.
The E. coli UraA/H+ symporter couples the uptake of uracil with H+ across membranes (Figure 25).671 By means of CG and atomistic simulations, Kalli et al. investigated the symporter interaction with lipids using a membrane model mimicking the composition of E. coli membranes, consisting primarily of PE, PG and cardiolipin.672 The anionic lipids associated more readily with the protein, driven by electrostatic interactions. While PG lipids distributed more evenly on the protein surface, three main interaction sites were identified for cardiolipin headgroups, further confirmed with additional simulations after in-silico mutagenesis of the residues involved in the interactions.
UapA is a uric acid/xanthine H+ symporter. Structural studies on UapA from Aspergillus nidulans showed the preference of the symporter to form homodimers,673 with protein oligomerization important for function and protein localization on the fungal plasma membrane.673,674 Using mass spectrometry, Pyle et al. identified PC, PE and PI lipids as lipid classes that copurify with the protein, although only PI and PE were able to promote the formation of a functional dimer after protein delipidation, suggesting a specific effect of PI and PE lipids in mediating the interactions at the dimer interface.675 5 μs-long CG MD simulations of UpaA in different mixtures of PC, PE, and PI lipids revealed different patterns of interactions between PI lipids and PE and PC lipids, with frequent contacts between PI lipids and positively charged residues at the cytosolic side of the dimer interface. A combination of mutagenesis studies and in vivo assays showed that mutations at these position do not affect protein trafficking nor substrate binding but do alter transport.675 While this study highlights the role of lipids in maintaining a functional dimer, further studies are required to characterize possible interaction sites and the regulatory role of lipids on the function of the symporter.
Band 3 (AE1, SLC4A1) catalyzes the electroneutral exchange of bicarbonate ions with chloride ions in red blood cell membranes.676,677 Band 3 is found primarily as a dimer. The transmembrane domain of each monomer consists of 14 transmembrane helices, organized into two inverted repeats of 7 helices forming a core and a gate domain (Figure 25).676 Several experimental studies provide evidence that phospholipids and cholesterol affect structure, function, and localization of the transporter,678−682 and other members of the SLC4 family are directly regulated by phosphoinositides, e.g., the PIP2-induced stimulation of NBCe1-A (SLC4A4) activity.683 The interactions between Band 3 and cholesterol and phospholipids were recently addressed by Kalli et al. in a computational study that combines CG and atomistic simulations.684 At the coarse grain level of detail, Band 3 was simulated in 8 different membrane environments, with varying ratios of cholesterol, PC, PE, SM, PS, and PIP2 lipids. Specific interaction sites were identified for anionic lipids and cholesterol: PS and PIP2 lipids coordinate with several positively charged residues, with lipid density from PIP2 molecules being more localized than PS lipid density. Cholesterol also interacted consistently with the same protein residues, independently from its concentration. The simulation results suggest that lipids and cholesterol might modulate the interaction between the two monomers, as cholesterol molecules, together with acyl tails of PS lipids, fill the cavity between the two monomers preventing changes at their interface. The molecular details of Band 3–lipid interactions described by the work of Kalli et al. provide a range of hypotheses that could be tested further experimentally to address the role of specific residues and lipids in modulating Band 3 activity.
13.4. Other Folds
Additional folds found in SLCs include, but are not limited to, those shown by mitochondrial ADP/ATP carriers, whose interactions with cardiolipins have been described above together with the respiratory proteins and proton carriers.616 The uncoupling protein 1 (UCP1) dissipates the proton gradient across the inner mitochondrial membrane by moving protons into the mitochondrial matrix, thus uncoupling the proton electrochemical gradient with the synthesis of ATP.685 With a combination of NMR, functional, and MD simulation studies, Zhao et al. characterized the binding of fatty acid molecules to UCP1, required for protein activation, and identified specific residues engaged in electrostatic interactions with the fatty acid.686
The ammonium channel AmtB from E. coli is the most characterized member of the ammonium transporter/methylammonium permease/rhesus (Amt/Mep/Rh) protein family.688 AmtB is found as a trimer, with each monomer consisting of a transmembrane domain of 11 membrane-spanning helices (Figure 26).688 Ion-mobility mass spectrometry (IM-MS) has shown how the trimer stability is linked to PG lipids tightly associated with the protein, while X-ray crystallography has identified at least 8 sites specific for the interactions with PG headgroups.75 The effects of PG lipids binding on AmtB function have recently been investigated with solid supported membrane electrophysiology (SSME) and MD simulations.689 The SSME measurements carried out on AmtB reconstituted in POPA/POPC and POPA/POPC/POPG liposomes show that PG lipids are required for proper transport activity. 700 ns-long atomistic simulations of the AmtB trimer in POPA/POPC and POPA/POPC/POPG mixtures retrieved the crystallographic PG binding sites and identified new ones, highlighting how PG lipids preferentially bind at the interface between monomers, with no significant structural changes except in a short periplasmic loop. This study supports the hypothesis that PG lipids might act as a bridge in controlling the interactions between monomers.689 However, there is more to the complex interplay between AmtB and lipids, as specific lipid binding can allosterically regulate not only the interactions of AmtB with other proteins78 but also the interactions with other lipid types690
Figure 26.
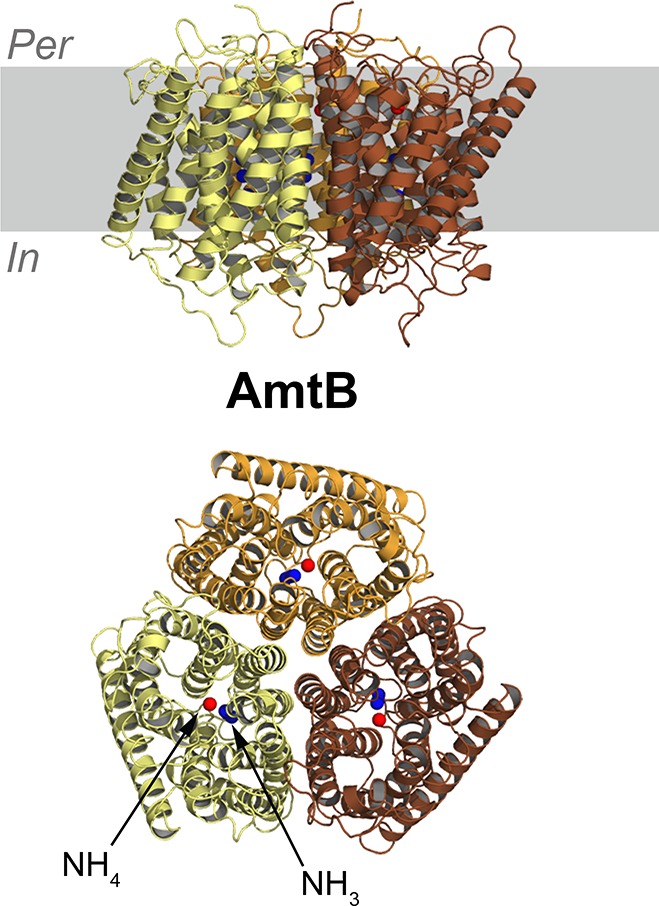
Ammonium channel AmtB. The three monomers of AmtB687 are shown in light yellow, orange, and brown cartoons, with the sites for NH3 and NH4 binding highlighted by blue and red spheres, respectively. The upper panel is the side view of the protein, with the membrane region highlighted in gray. The bottom panel is the top view from the periplasmic side. Per, periplasm; In, intracellular side.
14. Viral Proteins
The general virus infection cycle is divided into the early stage (entry process) and the late stage (assembly, budding, and release processes of virus particles). The genome of many circulating human pathogens such as human immunodeficiency virus (HIV) and Ebola virus are enveloped, i.e., contain a lipid bilayer covering the protein capsid. For the infection to occur, the genome is delivered to the host cytoplasm by inducing the fusion of the viral envelope with the cell membrane. In all known cases, the membrane fusion reaction that allows viral entry is accomplished by viral surface glycoproteins, including one key factor that is generally denoted as the fusion protein.691 Initial steps involve interactions of a viral fusion peptide with the membrane of the target cell, a biophysical process that has received considerable attention using model peptides but that is outside the scope of this review692−697 The final step of fusion involves the merging of the viral and host membrane and depends critically on their lipid composition. Lipids contribute to fusion through their physical, mechanical, and/or chemical properties.698 Specific interactions between viral envelope glycoproteins, including fusion peptides, and lipids, including cholesterol, ceramides, and glycophospholipids, have been the subject of several excellent reviews and will not be discussed in more detail here.699−702
Enveloped viruses generally acquire their envelope by budding at the plasma membrane or internal compartments depending on the virus type. Therefore, all of the viral membranes originate from cellular membranes, but they diverge in protein and lipid composition. Overall, the release of an infectious mature virion requires trafficking and assembly of its vital components to the budding site, induction of membrane curvature and scission of the virion from the cellular membrane. This is accomplished with the help of glycoproteins on the viral membrane envelope as in the case of influenza, or by inner structural proteins such as Gag proteins in HIV as well as complexes from the host, for example the endosomal sorting complex required for transport machinery.703,704 The lipid compositions of infected cell membranes and budded viral membranes are different, indicating lipid sorting in virus release699,705,706 Viruses modify lipid synthesis and reorganization of cellular membranes for virus assembly and exit. They can also promote the active synthesis of lipids to provide membranes for the viral envelope. Quantitative analyses of viral lipidomes at the individual molecular species level have become possible by means of mass spectrometry, but difficulties involved in cellular membrane purification complicates the understanding of lipid sorting and membrane remodeling during viral budding. Lipids such as cholesterol, sphingolipids, phospholipids, and PIP2 participate in both early and late steps in the replication of a large number of enveloped viruses.707−712
A combination of X-ray crystallography with cryo-electron microscopy (cryo-EM) and cryo-electron tomography has provided many high-resolution static structures of capsids and envelopes.713 Meanwhile, MD simulations have been widely used to explore the conformational dynamics and stability of the viral capsids and organization of viral envelopes. In the following two sections, we will consider large-scale MD simulations that focus on structural stability and integrity of enveloped virions and viral membrane fusion and budding with and from host cells, respectively.
14.1. Viral Membrane Structural Stability and Organization
There are considerable challenges in simulating a model of a complete virion envelope. Compared to MD simulation of proteins and lipid bilayers, enveloped viruses impose greater structural complexity due to asymmetric bilayers and large assemblies of proteins.714,715 Knowledge of a viral lipidome is a critical step in building a computational model of an enveloped virion. Although it is possible for viruses to select their own unique lipid species, the lipid composition of the envelopes is usually approximated based on the lipid composition of the host cell membrane. Other crucial pieces of information are the structures and copy numbers of proteins embedded within the lipid bilayer. To date, several structures of the extra-membrane domains of such proteins have been determined but, in most cases, complete structures of the proteins including their transmembrane (TM) domains are lacking.715 Thus, a more speculative modeling approach based on domain prediction and/or structural data is required.716
The interplay between lipid environments and influenza virus has been an interesting subject for several computational studies.692,717−719 The influenza virus triggers annual epidemics, occasional pandemics, and is considered a significant worldwide health problem. The viral proteins hemagglutinin (HA) and neuraminidase (NA) are targeted to lipid rafts, causing the merger and extension of the raft domains. This clustering of HA and NA may cause a deformation of the membrane and the initiation of the virus membrane fusion and budding events.720,721 Many simulation studies have addressed the effect of HA on membrane deformation, as recently reviewed by Boonstra et al.722 To understand the structural dynamics of the entire influenza A membrane envelope, Reddy et al. used CG MD simulations on the microsecond time scale.718 The Martini CG force field was used to simulate the viral membrane model with POPS:DOPE:ether-linked DOPE:cholesterol:hydroxylated sphingomyelin:Forssman glycolipids as the experimentally determined lipidome, an unusual complexity compared to most simulations currently in the literature. A high concentration of influenza A integral membrane proteins, the HA trimers, NA tetramers, and the TM domain of M2 tetramers were incorporated in the vesicle, together forming the complete virion model. The final model contained >5 M particles and is relatively crowded where the fractional volume of the proteins occupies ca. 15% of the viral membrane area. In the absence of glycolipids, a stable lipid population is maintained in each leaflet while the presence of glycolipids leads to a broader lipid distribution pointing to possible lipid mixing between the leaflets. The biologically realistic and complex membrane model in this study points to subdiffusive anomalous diffusion of influenza proteins where M2 protein diffuses slower than the other two larger proteins. This is mainly due to the larger cross-sectional area of TM domains and the strong interaction of M2 proteins with surrounding lipids. In another CG MD study, the interactions between HA in domain forming membranes with various local concentrations of HA was investigated.719 The system incorporated Martini representations of the crystal structure of the HA ectodomain and models of TM and cytoplasmic domains, DPPC, DLiPC, and cholesterol and explicit solvent. After 12 μs of simulation, the proteins aggregated through their bulky ectodomains without forming contacts among TM domains. HA preferentially partitioned to the liquid ordered (Lo) and liquid disordered (Ld) domain borders, initiating the formation of lipid nanoassemblies of DPPC and cholesterol within the protein clusters. In another study, lipid bilayers containing POPC, DOPE, cholesterol, and the model receptor (GD1a) with varying concentrations of GD1a and cholesterol were used to show that influenza virus binding affinity for membranes is sensitive to membrane spatial organization and sterol composition in the membrane.723 GD1a clusters form and dissolve repeatedly within 18 μs of simulation. Dissociation rates of multimers based on a Bayesian formulation suggest that increasing cholesterol concentration decreases GD1a-GD1a off rates and stabilizes GD1a clusters, thus increasing viral binding affinity.
Stability of the Dengue virions from the flavivirus family has also been studied by MD simulations.724−726 Dengue virions are characterized by a relatively smooth surface, with a diameter of approximately 500 Å. In addition to the RNA genome, there are three structural proteins that occur in stoichiometric amounts in the particle: the core, membrane (M), and envelope (E).727 Reddy et al. used CG MD simulations to build a model of the complete envelope of Dengue virus by combining the cryo-EM structures of the membrane proteins with the virus-infected insect cell membrane lipid composition,728 resulting in a model containing 7 different lipid types, three M proteins and three E proteins. In total, the model has 750 TM helices that occupy 18% of the membrane surface area, resulting in a degree of crowding close to that of a mammalian cell. The size and shape of the model was stable during 5 μs simulation and comparable to the cryo-EM image of the Dengue virion. Due to the crowded nature and the extensive interactions of the proteins with the lipid headgroups, the lipids diffuse anomalously, similar to the influenza A simulations. In a separate effort, Marzinek et al. developed a model of the Dengue virion containing various phospholipids, obtaining a system that is consistent morphologically with experimental observations.724 The arrangements of envelope protein complexes were relatively unstable in pure zwitterionic phospholipid vesicles compared to a more realistic mixed-membrane model, suggesting that anionic lipids may be required for both stability and fusion of viral particles. The simulations also suggest that the ectodomain and stem regions of the E and M proteins are involved in inducing lipid curvature. Moreover, several His residues interacted with lipid head groups at the interface, which may change their ionization state upon exposure to the acidic environment of the endosome and initiate envelope fusion (Figure 27).
Figure 27.

Interactions between protein and lipids. (A) Initial (top) and final (bottom) back-mapped coordinates of a representative E/M dimer (DENVimpl) complexed with lipids. The E protein ectodomains (red, yellow, and blue corresponding to domains I, II, and III, respectively) and E protein TM/stem regions (purple) as well as entire M protein (green) are shown in cartoon representation, with lipids shown in CPK format. (B) The percentage contacts, averaged over 200 ns, between lipid phosphate groups and residues mapped onto the E/M protein heterodimer for the three complete dengue envelope systems, together with a list of the residues in the E/M proteins that contacted lipid phosphates in >80% of frames analyzed across all systems. A contact was recorded if a given amino acid was within 0.45 nm of a phosphate particle. Arginine/lysine residues are excluded from this analysis and are instead shown in C. (C) The basic residues that made a contact with a lipid phosphate in >80% of the simulation frames across all systems. Basic residues in the E and M proteins are shown in orange and pink, respectively, while the protein is represented in white molecular surface format. Reproduced with permission from ref (724). Copyright 2018 Elsevier.
In another study, Wewer et al. used the Martini force field to study glycosylated E3M3 hexamer proteins of Zika and Dengue viruses embedded in a PC:PL:PE:PS:SM membrane.729 It has been proposed that PS and PE lipids are the key lipids for viral recognition by host proteins. Therefore, understanding the lateral distribution of lipids and lipid–protein interactions in Zika and Dengue membranes might be useful for understanding the requirements for host entry. The authors found that Zika and Dengue virus leave different footprints on the lipid distribution of the membrane. As for the distribution of lipids around the proteins, PE and PS were densely populated around both viral proteins, relative to SM and PC, while PE lipids were more enriched around the Dengue proteins and PS lipids were closer to the Zika proteins. Additional examination of the protein sequences suggests specific lipid binding sites on M proteins of both viruses. The viral membrane became thinner around the Zika hexamer compared to the Dengue hexamer, while M proteins from both viruses induced positive curvature in the lower monolayer of their associated membranes.
MD simulations have also been used to investigate the interaction of lipids with nonstructural proteins from Dengue virus.730,731 Nonstructural proteins are key viral factors that induce membrane curvature and thereby contribute to the formation of the membranous replication complex. Lin et al. modeled the nonstructural protein 4A (NS4A) in a POPC lipid bilayer by combining a docking approach and MD simulations to develop a reasonable model for the structure of the membrane-embedded and attached parts of the NS4A protein.732 The model induces membrane curvature after 200 ns of all atom simulations in a structure-dependent manner with a radius similar to the estimated radius of a replication vesicle. In another study by Fajardo-Sanchez et al., all-atom MD simulations were employed to understand interactions of the PepC segment of Dengue virus with artificial short-chain phospholipid membrane mimicks.733
HIV is an enveloped retrovirus, which buds primarily from the plasma membrane of infected T cells.734 Different simulation approaches have evaluated the protein packing and assembly in HIV virions.735,736 Of particular interest is the interaction between lipids and Gag polyproteins. The HIV Gag polypeptide contains all of the structural proteins required to form the mature, infectious virion. Charlier et al. studied PIP2 membrane anchoring of HIV myristoylated MA protein that lies at the N-terminal of the Gag protein precursor using CG MD simulations.737 Before the MA protein reaches the membrane, the myristoyl group spontaneously releases from its initial hydrophobic pocket which costs ∼5 kcal/mol energetically as estimated by umbrella sampling. After adopting a stable orientation at the membrane surface, MA confines PIP2 lipids around its surface where the PIP2 head groups bind to its highly basic region in agreement with NMR data, but without flipping of their acyl chain into the MA protein. Such a confinement induces a lateral segregation of PIP2 in domains and is consistent with a PIP2 enrichment of the viral envelope relative to the host cell membrane.
The structural characteristics, molecular assembly and ion conductivity of Hepatitis virus proteins in various lipid compositions have also been studied by means of MD simulations.738−740 The Hepatitis C virus p7 viroporin is a potential drug target that controls the permeability of the membrane to ions and facilitates virus production, analogous to M2 from influenza A virus. Chandler et al. used monomeric p7 to build oligomeric models with 4 to 7 subunits in hydrated POPC lipid bilayers.741 Hexamers and heptamers with optimized intersubunit contacts are the most structurally stable and retained their symmetry. In the simulations, the heptameric pore is always accessible to solvent, whereas the hexameric pore is blocked occasionally. This may suggest that cell membranes contain transiently open p7 hexamers combined with always open p7 heptamers. In response to varying lipid environments, the flexible p7 hexamer in thinner DHPC bilayers retains a highly tilted conformation in agreement with cryo-EM observations while they adopt the more upright conformation in thicker POPC bilayers.
14.2. Viral Membrane Fusion, Assembly, and Budding
MD simulations have proven to be beneficial in studying specific protein–lipid dynamics that control membrane fusion, membrane bending followed by virus budding, and final membrane scission, in which the virion is detached from the cellular membrane.693,694,742−758 Soares et al. used all atom and CG MD simulations to investigate the effects of protein–protein and protein–membrane interactions in the generation of membrane-bending forces in the Dengue virus envelope proteins.759 Their bilayer model incorporates POPC:POPE:POPS:cholesterol with the lipid ratios from Dengue virus, derived from the mosquito lipidome. The structural organization of three heterotetrameric EM proteins (EM3 unit) serves as an anisotropic bending unit for the Dengue virus envelope, because it is able to locally decrease the thickness of the membrane with its short transmembrane helices and impose a specific radius of curvature to the membrane. This suggests that the EM3 unit may be the main determinant of the viral particle size. The simulations show that the EM15 system which includes five EM3 assemblies and corresponds to one-sixth of the viral envelope induces complete vesiculation of an open membrane patch. The pentagonal arrangement of five EM3 units inflicts a specific curvature on the membrane, breaking its initial symmetry and creating stress in the membrane. The resulting elastic energy is minimized by the systematic migration of lipids from the lower into the upper layer. Overall, the vesiculation process studied in this work suggests an explanation for the role of the envelope proteins of Dengue virus in the mechanism of membrane shaping.
Rogers et al. used atomistic and continuum-level simulations to study the energetics involved in Dengue virus assisted membrane fusion.760 The system consisted of the E protein trimers and a membrane of uniformly mixed POPG:POPC lipids. The potential of mean force (PMF) of association of the viral protein and the membrane displays a broad minimum. POPG lipids exert a strong attractive force on E proteins to produce an irreversible surface-associated protein–membrane contact. Continuum calculations in combination with the all-atom PMF resulted in an estimated binding free energy of −15 kcal/mol per trimer, which represents an upper bound to membrane bending energy necessary for membrane–membrane fusion.
15. C99 and γ-Secretase
The product of β-secretase cleavage of the full-length amyloid precursor protein (APP) is a 99 residues long transmembrane helix (C99) with an intracellular domain, which is further cleaved in fragments of different length by γ-secretase to form the final Aβ peptides.761 NMR and EPR spectroscopy studies combined with mutagenesis assays identified the tandem glycine motif of the helix as the interaction site for cholesterol, in a 1:1 ratio, with an asparagine and a glutamate residue on the extracellular end of the helix coordinating the polar group of cholesterol.762 The tandem glycine motif induces a kink in the helix that together with cholesterol binding has been suggested to facilitate the interaction between C99 and the γ-secretase.762 MD simulations also suggested that cholesterol binding is sensitive to the protonation state of this glutamate and an aspartate at this end of the helix, with the neutral state of their side chains stabilizing the C99-cholesterol complex.763 Furthermore, MD simulations studies have provided additional molecular details on the interactions of cholesterol with C99, suggesting alternative binding modes to interact with the glycine motif,412,764 as well as the existence of multiple binding sites.412 The sensitivity to cholesterol of different Aβ peptides has also been addressed by simulations describing different cholesterol hot-spots depending on their length.765−767 Cholesterol bound at the tandem glycine motif might compete with helix dimerization,768 but other sites have also been proposed to promote helix–helix interactions.769 More in general, the lipid environment and composition greatly affect helix orientation and dimerization of precursors of different lengths,770,771 an aspect that has been addressed with MD simulations as well. Simulations in detergent micelles and lipid bilayers captured different dimeric states and helix orientations, linked to the different curvature in bilayers and micelles;772−774 simulations of C99 (residues 23–55) monomers and homodimers in bilayers of different composition highlighted the different kink and tilt angle of the helix as a function of membrane thickness, favoring different dimeric structures.775 The length of the precursor also impacts its structure in the membrane, as shown by simulations of a full-length C99 (residues 1–99) monomer in implicit membranes of different thickness, where different secondary structure elements for the N-terminus were found depending on membrane thickness.776 Using CG simulations and comparing raft-like with nonraft environments, Sun et al. showed that cholesterol not only affects C99 dimerization, but also alters dimer stability and C99 partitioning in lipid domains.777 In raft-like membranes, C99 preferentially partitioned at the boundary of liquid ordered and disordered phases. Dimerization was significantly impacted, resulting in structural deformations of the dimer compared to a nonraft membrane. As γ-secretase only cleaves monomeric C99 and considering the greater risk of developing Alzheimer diseases with increasing levels of cholesterol, the dimer destabilization observed in the presence of a raft-like environment might be linked to the molecular details leading to the formation of Aβ peptides.777 Audagnotto et al. used CG and atomistic simulations to investigate C99 dimerization in a simple POPC bilayer and in a complex, synaptic plasma membrane (SPM) model with 32 lipid types, showing that the motifs involved in helix dimerization are different in the two membrane environments, resulting in different dimer stability.778
Recently, MD simulations have also been used to study how the membrane affects APP recruitment by the γ-secretase,779 a multiprotein complex formed by presenilin (PSEN1), presenilin enhancer 2, nicastrin, and anterior pharynx-defective 1 (APH-1).780 Audagnotto et al. used a CG approach to simulate a shorter fragment of C99 that includes the transmembrane helix as well as a full model of the γ-secretase embedded in the synaptic plasma membrane (SPM) model described above.779 The formation of a liquid ordered and disordered phase was observed in the simulations, with the ordered phase preferred in proximity of the convex face of the γ-secretase.779 In the complex membrane, APP seems to preferentially partition in raft-like, cholesterol-rich domains when not interacting with the secretase, and in nonraft domains when in proximity of the secretase. In line with what was discussed above, APP dimerization was detected only for low cholesterol concentrations, due to the inhibiting effect of cholesterol in helix–helix association. In a different study, MD simulations of the PSEN1 component of the γ-secretase suggested an additional role for lipids in regulating the cleavage, as a PE molecule was observed to enter the catalytic site and form stable interactions.781
16. Miscellanea
For members of many other protein families, computer simulations, often in combination with experiments, have provided details on how lipids interact with proteins and possibly modulate protein function. Below we describe studies of a variety of membrane proteins that do not fit neatly in the larger groups previously described, recognizing this distinction is somewhat arbitrary and based both on biological grounds and the volume of simulation studies devoted to particular proteins.
16.1. Glycophorin A
Glycophorin A (GpA) is a major sialoglycoprotein from erythrocytes. Its main biological interest is derived from its glycosylation, but the transmembrane part of the protein is widely used as a model system for membrane protein thermodynamics. Several experimental structures are available from solution NMR in micelles,782 X-ray diffraction,783 and solid state NMR,784 showing a consistent well-defined helical dimer structure.577 Combined with an abundance of thermodynamic experimental data, see for instance Fleming et al.,785 including on sets of mutants, this has made GpA a popular model system for simulation methods development and testing.786 With currently available computer resources potentials of mean force calculations of helices in fully detailed atomistic models have become readily accessible, which enables their use in testing and developing computational models. A recent example is the free energy of association of two GpA transmembrane helices in explicit lipids.787 Although the structure of the dimer remained stable and close to the experimental structures, somewhat surprisingly the force field used predicted that the dimer was metastable. Minor changes to interaction parameters that govern all lipid–protein interactions, not specific to GpA, can modulate the stability. This highlighted a second problem, in that the experimental values vary over a broad range.787 Simulations using the coarse-grained Martini model correctly predicted that the dimer is the most stable thermodynamic state, but the detailed structure cannot be reproduced with a coarse-grained model. Overall, this important paper highlights both strengths and weaknesses in simulation approaches and in experimental data, and emphasizes the need for careful studies on model systems, and financial support for such work from granting agencies, to improve computational models and the interpretation of experimental results for more complex systems of biological interest.
16.2. Membrane Fluidity Sensors
There are several proteins able to sense membrane fluidity for which simulations have provided more details on their mechanism of action. Two examples are the thermosensor DesK, which responds to changes in thickness as a consequence of changes in temperature,788 and the transcription factor Mga2, for which the degree of lipid saturation preferentially selects dimers with different helix orientations.789
16.3. Lysosome-Associated Proteins
Atomistic simulations supported the identification of a ceramide interaction motif in the lysosome-associated protein transmembrane 4B (LAPTM4B),790 required for recruiting amino acid transporters to the lysosomal membrane in a ceramide-dependent fashion.791 In particular, Zhou et al., based on a comparison with previously identified sphingolipid-binding motifs, identified residues in transmembrane helix (TM) 3 of LAPTM4B involved in ceramide interactions, which were confirmed by in vitro and in vivo assays.790 Atomistic MD simulations of POPC bilayers containing several copies of TM3 showed that TM3 was able to trigger insertion of ceramide micelles, a phenomenon that was not observed when TM3 was replaced in a control simulations by the WALP23 peptide. A flexible region in the helix together with the presence of an aspartate residue in the middle of TM3 is responsible for changes in helix kink and preferential interactions with ceramide, while nearby acidic residues control helix tilting, to facilitate ceramide intake. These conformational changes may be important for interactions with the amino acid transporters.790
16.4. AcrAB-TolC Efflux Pump
Gram-negative bacteria use multidrug efflux pumps that span both the inner membrane (IM) and the outer membrane (OM) to extrude antibiotics out of the cell.792,793 We review lipid–protein interactions of such pumps, which are large complexes that consist of multiple proteins including an OMP. AcrABZ-TolC is the best-studied example. It is a tripartite complex formed by AcrB in the IM, controlling substrates specificity; TolC, embedded in the OM; AcrA, a periplasmic membrane fusion protein (MFP) connecting AcrB with TolC; and AcrZ, a small protein with modulating effects on AcrB.794 MD simulations have also been applied to characterize the dynamics of individual components (mainly AcrB and TolC) of the pump, as well as of the entire pump and their interfaces and substrate interactions.793,795 Hsu et al. used CG MD simulations to investigate the effect of the full AcrABZ-TolC complex on lipid organization in the presence of nearby proteins, namely AqpZ and LacY in the IM and OmpA in the OM.565 Membrane reshaping is observed in both the IM and the OM in terms of curvature effects, which differ depending on the type of protein present and their concentration. Similarly, lipid sorting effects, and in particular cardiolipin enrichment over PE and PG lipids, were also observed to be protein-dependent, highlighting the complexity of the interplay between lipids and bacterial proteins.565
16.5. EmrE
EmrE is an ion-coupled, small multidrug transporter from E. coli that confers resistance to many cationic drugs.796 EmrE oligomerization is regulated by the lipid environment, as shown by Brewster angle microscopy797 and FRET measurements,798 but the interplay with the membrane extends further as physical properties of the lipids are modified upon ligand-binding to EmrE, as shown by differential scanning calorimetry and solid state NMR studies.799 MD simulations showed that ligand-binding alters the interaction of lipids with selected Trp residues and highlighted a preference for PE lipids in a PE/PG mixture, based on hydrogen-bond patterns.800
16.6. VDAC
In addition to the OMPs described in a previous section above, lipid–protein interactions of other beta-barrel pores have been addressed with computer simulations. This is the case for Voltage-Dependent Anion-Channels (VDACs) and several toxins (discussed below). VDACs are located in the mitochondrial outer membrane and are related to ancient porins present in bacteria.801 VDACs are the major pathway for metabolite and ion transport across the mitochondrial outer membrane.802 The channel is formed by a 19-stranded β-barrel connected by loops and an N-terminal helix inside.803−805 Lipids contribute to the regulation of VDAC gating.806 Villinger and colleagues, using NMR spectroscopy, Gaussian network model analysis, and molecular dynamics simulations, showed how the dynamics of the channel is tightly regulated by mutations or changes in the protonation state of glutamate 73, whose side chain projects toward the lipids.807 MD simulations revealed significant changes in membrane thickness near the protein, creating, when the glutamate is negatively charged, a membrane defect that extends up to the bilayer middle.807 VDAC is also one of the few examples of cholesterol interactions with β-barrel proteins. Molecular docking combined with MD simulations suggested that sterols are essential for VDACs’ structure and dynamic.808 This work suggested a model in which 1 to 5 cholesterol molecules bind to the channel, consistent with previous binding studies of VDAC in detergent.803 The authors performed 15 atomistic molecular dynamic simulations, averaging 37 ns, in which the criterion to select the most favorable poses was based on the sustained residency of cholesterol in the sites808 Additionally, the channels were simulated in a POPC bilayer with cholesterol bound in five hypothetical sites identified previously by Hiller et al.803 and compared with equivalent systems without cholesterol in simulations of 1.4 μs in total. NMR studies on the mammalian VDAC1 suggested the existence of a shell of tightly bound phospholipids around the protein.809 Electrophysiology studies have also shown that protein-sterol interactions may be involved in both voltage dependence and selectivity of the plant VDAC (PcVDAC from Phaseolus coccineus).810 Mlayeh et al. combined electrophysiological studies in planar bilayers with MD simulations to characterize the effect of phosphatidylethanolamine (PE) and ion concentration on PcVDAC.811 This study suggested that ionic interactions formed by acidic residues with PE head groups have an impact on VDAC ion selectivity.
16.7. Beta-Barrel Toxin Channels
Examples of beta-pore-forming toxins are anthrax toxin, α-hemolysin and actinoporins. The anthrax toxin from B. anthracis consists of the so-called lethal factor, edema factor and protective antigen (PA).812 PA, after binding to specific receptors and cleavage-based activation, assembles into a heptameric prepore that binds lethal and edema factors for endocytosis and delivery into the endosomal compartment.812 The acidic pH of the late endosome triggers conformational changes in the prepore that lead to the insertion of the channel in the endosomal membrane to release lethal and edema factors in the cytoplasm.812 The membrane environment affects different stages of this mechanism.812,813 Using model membranes measurements and atomistic MD simulations, Kalu and colleagues recently showed how lipids have a strong impact on channel properties including its insertion in the membrane, gating, and selectivity, possibly linked to lipid-induced structural changes in the final PA pore.813
α-Hemolysin (αHL) is a pore-forming toxin from S. aureus that in its monomeric form interacts with host membranes to assemble into an heptameric structure whose stem domain forms a beta-barrel pore across the lipid bilayer.814 Although there is evidence that lipids affect the mechanism of αHL, a systematic analysis of the interplay with the membrane is not yet available.815 Guros et al. applied MD simulations to study the structure and conductance of αHL in a pure POPC bilayer and a pure 1,2-diphytanoyl-sn-glycero-3-phospho-choline, DPhPC, membrane, for direct comparisons with previous experimental data.815 Lipids and protein adapt to each other, with lipids creating a negative curvature to match the protein hydrophobic profile while the protein changes its tilt in the membrane. There is, however, a difference in this mutual adaptation between the POPC and the DPhPC bilayers. More prominent effects are detected in the thicker POPC bilayer, where decreased thickness and negative curvature near the protein force some loops to bend inward, thus occluding the channel, or outward, thus destabilizing nearby lipids organization. In the methyl-branched DPhPC bilayer, with a thickness that better matched the hydrophobic profile of the protein, protein tilt and distortions as well as the negative curvature of the membrane are less evident. Due to the changes in the structure of the pore, in POPC lipids αHL shows a reduced ionic current, highlighting, in line with experimental reports, coupling between protein function, structure and the lipid environment.815
Actinoporins are toxins from sea anemones that assemble into beta-barrels, similar to αHL. Family members include fragaceatoxin, equinatoxin, and sticholysin. Recent structures816 have enabled simulations to study details of pore formation.817,818 As most actinoporins appear to require specific lipids, these toxins may be of further methodological interest, in addition to their intrinsic biological and biotechnological importance.
16.8. Tetraspanin
The structure of tetraspanin CD81, a cell surface protein with four transmembrane (TM) helices, is characterized by a conic shape, with a large separation between TM1–2 and TM3–4, and capped by the extracellular loop (closed state).819 A bound cholesterol molecule is located in the gap between the two sets of helices. Binding assays together with mutagenesis studies showed that the pocket between TM1–2 and TM3–4 is specific for cholesterol. Several μs-long MD simulations of CD81 showed how the dynamics of the protein may be regulated by cholesterol: in the apo state, the protein switched from a closed to a fully open conformation with dissociation of the extracellular loop. In the presence of cholesterol, the closed conformation was preserved, and only one cholesterol unbinding event was captured, followed by a transition to the open state. Combined, these findings have important mechanistic implications as they describe a binding site specific for cholesterol that could be possibly targeted by drugs and supports a cholesterol-regulated mechanism for the interactions between the CD81 extracellular loop with other proteins (Figure 28).819
Figure 28.
Cholesterol-regulated mechanism of CD81. Cholesterol regulates CD81 equilibrium between the capped state (left) and the open state (right). The conformational transition between these states was studied with MD simulations, and it is believed to be an important regulatory step for the interactions between CD81 and other protein, such as CD19 (modeled in the picture).819 Reprinted with permission from ref (819). Copyright 2016 Elsevier Inc.
16.9. Undecaprenyl Pyrophosphate Phosphatase
Undecaprenyl pyrophosphate phosphatase (UppP/BacA) is an integral membrane protein essential in the synthesis of the cell wall. UppP dephosphorylates undecaprenyl pyrophosphate to its monophosphate form, which is then used as a carrier for sugars and glycan chains across membranes.820 A combination of molecular modeling and MD simulations helped the characterization of the binding site for undecaprenyl pyrophosphate in the UppP of E. coli.821
16.10. Photosystem II
The protein complex photosystem II (PSII), embedded in the thylakoid membrane, is a key component of the photosynthetic machinery. PSII functions as a homodimer, where each monomer consists of 27 subunits in plants and 20 in cyanobacteria. A large number of cofactors endow PSII with its light-harvesting and water-splitting capabilities. To provide a dynamical view on this large and important protein complex, van Eerden et al. developed a CG model of PSII from cyanobacterium Thermosynechococcus vulcanus based on the Martini force field.822 The complex was embedded in a realistic thylakoid membrane composed of a mixture of phosphatidylglycerol (PG) and the glycolipids digalactosyl-diacylglycerol (DGDG), monogalactosyl-diacylglycerol (MGDG), and sulfoquinovosyl-diacylglycerol (SQDG). Based on cumulative simulations of more than 500 μs of the full PSII dimer in the thylakoid membrane, the authors identified the exchange pathways of the electron carriers plastoquinone (PLQ) and plastoquinol (PLQol).823 The long time scale allowed the observation of many spontaneous entries of PLQ into PSII, as well as the unbinding of the reduced carrier PLQol from the exchange cavity inside the complex. The simulations point to a promiscuous diffusion mechanism in which three channels function as both entry and exit channels. The exchange cavity itself serves as a PLQ reservoir. The thylakoid lipids also play a dynamic role in this process, as they can leave and enter the exchange cavity, allowing exchange of cocrystallized lipids with the bulk membrane (Figure 29A). Interestingly, the simulations predict an accumulation of MGDG and SQDG lipids in the annular shell around the protein, at distinct binding sites (Figure 29B–D). The biological relevance of this finding, however, remains unclear.822
Figure 29.

Lipid–protein interactions in the photosystem II. A. Examples of two lipid diffusion events from the stromal (left) and luminal (right) leaflet through the plastoquinone exchange cavity. The two lipids are colored relatively to the time the diffusion events took place. B–D. Binding sites for (B–C) MGDG (headgroup in red) and (D) SQDG (headgroup in yellow) lipids identified in the simulations. Chlorophyll molecules are shown in green. Adapted with permission from ref (822). Copyright 2017 Biophysical Society. Licensed under Creative Commons Attribution 4.0.
16.11. Sec Translocon
Post-translational or cotranslational translocation of most proteins across membranes is achieved by the Sec translocon, a complex of proteins found in eukaryotes, bacteria, and archaea.824 In bacteria, the Sec translocon consists of SecY, SecE, and SecG, which form a transmembrane complex, also associated with the SecA ATPase motor for post-translational protein translocation. SecY forms a central pore for proteins to be translocated fueled by ATP hydrolysis via SecA and by the proton motive force.824,825 Using CG MD simulations of the SecYEG-SecA complex, Corey and colleagues, in addition to a known cardiolipin binding site involving SecA, identified two new sites as possible cardiolipin interaction sites (Figure 30), and further confirmed their findings with native MS and FRET.826 The specific binding of cardiolipin to these sites is shown to regulate the energetics of the translocon by stimulating SecA ATPase activity as well as translocation induced by the electrochemical gradient of protons, while nonspecific cardiolipin interactions with the complex could drive SecYEG dimerization.826
Figure 30.

Cardiolipin interaction sites on the SecYEG complex. CG simulations of the SecYEG-SecA complex embedded in lipid bilayers with different concentrations of cardiolipins identified specific protein residues responsible for stable interactions with cardiolipins.826 A. CG representation of the complex, with SecY in pink, SecE in tan, SecG in green, and SecA in light blue. Residues involved in the interactions with cardiolipins are highlighted as spheres and colored from yellow to red according to occupancy values determined from the simulations. The newly identified sites 1 and 2 are marked. B. Selected frames from the CG simulations were back-mapped to an atomistic representation to highlight site 1 and 2 with cardiolipin molecules bound. C. Cytoplasmic view of the SecYEG complex in B. SecA is not shown for clarity. Adapted with permission from ref (826). Copyright 2018 Corey et al.
In bacteria, the YidC insertase promotes protein insertion into the inner membrane via a mechanism that does not require ATP, and can work independently from the Sec translocon or in conjunction with it.827 Atomistic simulations of the YidC instertase of E. coli revealed the changes in membrane thickness induced by the shorter transmembrane helices of the enzyme.828
The time scale associated with protein ribosomal translation and the complexity of the mechanism of the systems here described have prompted the development of a 2D coarse model of the Sec translocon,829 which has been successfully used to predict the topology of nascent proteins and to provide a link between protein expression levels and protein integration in the membrane mediated by the translocon.829−831 Recently, a 3D representation of the 2D model has been developed, at a level of detail much coarser than the Martini force field.832 This model still describes critical features of the system, including an explicit representation of the nascent protein and the lateral gate of the translocon, modeled in different conformations, in an implicit membrane. This type of biological events are, at the present time, still inaccessible to the current atomistic and Martini CG simulations techniques. The continuous development of such coarser models will allow simulations to address other aspects of protein insertion via the Sec translocon, including the role of the membrane.
17. Discussion
As the hundreds of papers referenced in the main body above demonstrate, computer simulations have been applied to a large number of proteins and lipids, addressing a wide range of questions. Although the volume of papers did not allow a critical detailed discussion of each paper, we would like to conclude with a number of more general points. Technically, it has become fairly straightforward to set up and run atomistic simulations of a single protein with a high-quality known structure in a simple lipid mixture and obtain reasonable simulation lengths (microseconds, as a general guideline) on modest computational resources, starting from a few desktop computers with high-end gaming graphics hardware, although the state of the art requires custom hardware like ANTON or large-scale super computers. This presents a huge opportunity, as such simulations can become routine ingredients of broader functional studies, which is already happening in the literature. Below we outline the current state of the field as well as challenges and future directions.
17.1. Diversity in Lipid–Protein Interactions
Broadly speaking, the literature reviewed above supports a wide range of roles for lipid–protein interactions, studied by an equally wide range of experimental and computational approaches that in some of the most interesting cases are combined to span the whole spectrum from detailed local interactions to functional biological relevance. Lipid composition and lipid–protein interactions modulate the physical properties of membranes. An obvious example are the mechanosensitive channels, where lipid–protein interactions directly transfer a tension from the membrane to the protein, which then undergoes a structural change. Lipid compositions may also change the equilibrium between conformational states, as shown for instance in several GPCRs and ion channels.
Moving toward more specific interactions, simulations of many proteins have noticed an enrichment or depletion of specific lipids near a protein. A striking example is a recent set of simulations of 10 different proteins in a very complex membrane environment, using the Martini model on a time scale of 30–50 μs, that showed a lipid environment different from bulk and different between all proteins (Figure 31)121 but the general feature has been observed in a variety of simpler systems. Part of this phenomena may just be a local adjustment of the membrane to the specific thickness and more detailed chemical features of a specific protein, but these enrichment or depletion effects may also play an important role in organizing the membrane at larger scales.
Figure 31.

Corradi et al. used CG MD simulations to investigate lipid reorganization around different membrane proteins embedded in a plasma membrane model.121 Each system consisted of 4 copies of a given membrane protein, and ca. 6000 lipid molecules of more than 60 different types. A. Snapshots at 30 μs of four selected systems, namely AQP1, COX1, Kv1.2, and P-gp, viewed from the extracellular side. Fully saturated and polyunsaturated lipids are shown in white and black licorice, respectively, while all other lipid types are shown as a transparent surface. Different lipid distribution around the proteins is reflected in different thickness profiles, averaged between 25 and 30 μs of simulation time and over the 4 protein copies of a given system. B. Unique lipid composition is found around ten different proteins in the plasma mixture. The total number of lipids found within 0.7 nm cutoff is reported in parentheses as average number of lipids obtained from the four protein copies of each system, between 25 and 30 μs of simulation time.
Simulations have also observed strongly localized specific lipids near a variety of proteins. A noteworthy example is the binding of cholesterol to GPCRs, but examples are found throughout this review. In many cases, the biological significance of this remains to be investigated, but in many examples protein function depends on the specific lipids identified, and in some cases the same lipids are observed in high-resolution structures that include lipid density. We have also highlighted some areas where there is detailed experimental data on the importance of specific lipid–protein interactions, notably in P-type ATPases and potassium channels, where further simulations might be useful to provide further details at the molecular level.
In the most specific case, certain lipids play a key role that has been well-established experimentally, and simulations have made it possible to identify the specific features of lipid–protein interactions and resulting protein structures involved in the mechanism of the protein. The most obvious examples of this are inwardly rectifying potassium channels that depend on interactions of PIP lipids with conserved residues in the channel for their activation. In another class of proteins, lipids are actually the substrates, such as in lipid flippases and scramblases or are intimately involved in the transport of other substrates.
Finally, at a much larger scale, lipids mediate interactions between proteins that lead to large scale organization of protein complexes and membranes. The most obvious examples in this review are the respiratory complexes bridged by cardiolipin and the organization of ATPases in rows by indirect interactions through lipids. In the accompanying review on large-scale membrane models,111 we further expand on this topic.
17.2. Challenges
The main critical elements in molecular dynamics simulations are an appropriate choice of force field and the ability to obtain enough sampling to draw meaningful conclusions. After reviewing the literature, we argue that the most common problems are currently indeed a lack of sampling, leading to poorly justified conclusions, limitations in the accuracy of force fields, and stretching the interpretation of simulation results to argue they support a functional mechanism that cannot be directly inferred from the simulations. The latter is not a simulation challenge per se but is related to the more general difficulty of interpreting functional assays in terms of molecular interactions at the lipid–protein level.
Sampling involves both sampling over lipid and protein diffusion, lipid–protein interactions as well as lipid-mediated protein–protein interactions, and internal dynamics of proteins that may change conformation depending on their lipid environment and interactions. As computer speed increases sampling almost automatically improves, as shown by the roughly 5 orders of magnitude increase in scope of computer simulations in the past 25 years. We also anticipate algorithmic advances that speed up simulations of lipid–protein interactions. Simulations are not fundamentally limited by the maximum time scale they can reach in equilibrium simulations. Techniques such as Hamiltonian replica exchange might be used to increase sampling in complex membrane systems, as they have elsewhere,130 as might practical methods that combine Monte Carlo moves with molecular dynamics to enable entire lipids to swap place rather than wait for lipids to swap places by diffusion. A recent example of this is the work of Fathizadeh and Elber,833 building on several previous advances that have not been widely used for practical reasons. A tighter integration of coarse-grained and atomistic simulations might also increase sampling, although there is currently no rigorous way to do this (e.g., PACE force field)834 and balancing interactions between different levels of resolution within one calculation remains challenging.835 Another approach that has proven very powerful in processes such as protein folding is the use of Markov state models and other collective variable methods that can combine large numbers of short simulations of the order of hundreds of nanoseconds or microseconds to describe dynamics on time scales of seconds. A recent application of peptide–protein complex formation showed that a particular class of Markov models combined with machine learning correctly identified complex structures as well as the kinetics associated with complex formation, a problem somewhat similar to lipid–protein interactions.836
In addition to sampling, the quality of force fields is a key technical requirement for the accuracy and usefulness of simulations. Current force fields for biomolecular simulation are quite sophisticated and generally well-tested, but every time simulations cover new ground in simulation time or system complexity areas where improvements are required become clear. A recent example in simulations of soluble proteins is the overstabilization of unfolded states, relevant for protein folding and the nature of intrinsically disordered proteins.837 Although the disagreement between experiment and simulation in this area was quite dramatic, once both experiment and simulation were able to probe detailed structural properties of these difficult-to-study states of proteins, relatively minor adjustments to water–protein interaction parameters equally dramatically improved the agreement. In lipid–protein interactions a similar phenomenon was observed by Domanski et al., who showed that the widely used CHARMM force field actually described the stable glycophorin A dimer as a metastable state.787 A small adjustment to lipid–protein interaction parameters, without affecting either the lipid properties or the protein internal dynamics, is in principle sufficient to improve the thermodynamic description.787 This study also highlighted a perhaps more serious problem, as a review of experimental data gave a range of between −16 and −51 kJ/mol for the stability of the glycophorin A dimer with the same reference state.787,838 This is an unacceptably large range of uncertainty for many purposes, both for biophysical understanding of protein stability and as parametrization or validation goal for simulations. Sandoval-Perez et al. tested several force fields to describe membrane proteins, focusing on the membrane–water interface.839 They compare the conservation of the secondary structure of transmembrane proteins, the positioning of transmembrane peptides relative to the lipid bilayer, the insertion depth of side chains of unfolded peptides absorbed at the membrane interface, and the ability to reproduce experimental insertion energies of Wimley-White peptides at the membrane interface.839 Only the Wimley-White experiments give unambiguous quantitative data on the thermodynamics of lipid–protein interactions but are limited to side-chain partitioning at the interface and to a single lipid type. This type of force field comparison is useful as it highlights areas of strength and weakness but also highlights the lack of quantitative experimental data on lipid–protein interactions in more complex systems. One example is the relative balance between lipid–lipid, lipid–protein and protein–protein interactions. Both atomistic840 and Martini simulations841 overestimate the strength of protein–protein interactions, leading to artificial aggregation of proteins. Martini 3.0 improves this balance111 but quantitative experimental data to calibrate these interactions remains a challenge. Similarly, hydrophobicity scales based on residue substitutions in small beta-barrel proteins from Fleming provide thermodynamic data for side chain partitioning in bilayers but in the context of membrane protein folding using an experimental approach that is only available in one lipid type, for now.842 Although aspects of these systems are readily accessible for simulations, a direct comparison with relative protein stabilities for different mutants requires assumptions about the unfolded state, for which there is no structural data available. We have highlighted some model systems for lipid–protein interactions in this section, as these are particularly relevant for the topic of this review. In addition, lipid and protein parameters themselves are under continuous development. While simulations of soluble proteins are quite mature, aspects of membrane proteins might require further development as more experimental data becomes available, for instance from multiple conformations obtained from electron microscopy data. It is also quite possible that further testing of the description by current force fields of membrane protein conformations and dynamics will show that they already adequately describe these propertie. Diffusion times in lipid mixtures almost guarantee that microsecond time scale simulations are essential to explore lipid mixtures, a time scale that has only been readily available for a few years. We expect significant effort in studying lipid mixtures in the next few years, perhaps highlighting areas where lipid parameters need to be adjusted to accurately represent lipid mixtures.
In many of the papers reviewed above, and in the field of lipid–protein interactions in general, simulations and experiments are used hand in hand. Simulations give exquisite detail but are subject to the limitations described above. Experiments, broadly speaking, require significant interpretation to translate from experimental observables to a molecular-level model of the system of interest. The range of experiments that has been applied or can be applied to lipid–protein interactions is too extensive to discuss here, but it may be useful to consider how to optimize the combined use of simulations and experiments. Experiments that yield quantitative measurements of molecular properties that can be directly calculated from simulations are essential to develop and validate simulation methods. Such experiments include detailed structural and dynamic measurements on lipids and lipid–protein systems by NMR and diffraction, thermodynamic measurements, and potentially a host of other data. They might also include in the future data on the organization of complex membrane mixtures, from, e.g., NanoSIMS (secondary ion mass spectrometry)843 and super-resolution imaging,844 as simulations increase to larger length scales and experiments increase their resolution to directly overlap with the size of simulation systems. Experiments that yield data on direct interactions between lipids and proteins are also essential. Some of these include cryo electron microscopy, mass spectrometry, and quantitative experiments using SMALP and related polymers.845 Challenges in this area may lie in defining the quantitative nature of these experiments, as they currently appear to be in a gray zone between qualitative and quantitative. Lastly, experiments that modify lipid compositions to perturb properties of membrane proteins that can be functionally assayed are of direct biological relevance, the real goal of studying lipid–protein interactions but are difficult to link directly to simulations or details of interactions, in most cases.
18. Conclusions
Simulations using a combination of approaches have given insight in a wide range of lipid–protein interactions. Despite a range of challenges, including technical computational challenges, the nature of experimental data, and challenges linking functional experimental data to local molecular interactions, the vast majority of papers does give interesting new insights in lipid–protein interactions and provides a molecular picture with a variety of scenarios involving these interactions, together with hypotheses that can be tested experimentally, such as, for instance, protein residues involved in lipid binding with consequences for protein function. Given the accessibility of current computational techniques and the strong overlap of achievable time scales in simulations and biologically relevant time scales, we expect this field to continue to grow very rapidly.
Acknowledgments
Work in S.J.M.’s group was supported by an ERC Advanced Grant “COMP-MICR-CROW-MEM”. Work in D.P.T.’s group was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC; RGPIN/05305-2015) and the Canadian Institutes of Health Research (CIHR; Project Program FRN-CIHR: 156236). Work in SYN’s group was supported by NSERC (RGPIN-315019), CIHR (Project Program FRN-CIHR: 156236), and in part by the National Institutes of Health (U.S.A.) Grant R01HL128537-01. H.M.G. is supported by a Vanier Canada Graduate scholarship and an Alberta Innovates Health Solutions studentship. D.P.T. holds the Alberta Innovates Technology Futures Strategic Chair in (Bio)Molecular Simulation. This work was undertaken, in part, thanks to funding from the Canada Research Chairs program.
Biographies
Valentina Corradi holds a Master of Science in Pharmaceutical Chemistry and Technology from the University of Parma, Italy, and obtained a Ph.D. in Pharmaceutical Sciences from the University of Siena, Italy, under the supervision of Dr. Maurizio Botta. In 2009, she joined Dr. D. P. Tieleman’s lab at the University of Calgary as a postdoctoral associate, to work in the area of biomolecular simulation and computational biology. Since 2013 she works as a Research Associate in the same group. Her research interests focus on understanding the correlation between structure, function, and dynamics of membrane proteins and their interactions with lipids.
Besian I. Sejdiu received his Magister of Pharmacy degree from the University of Prishtina (Kosovo) in 2012. Currently, he is a Ph.D. student in the Department of Biological Sciences at the University of Calgary (Canada), under the supervision of Professor D.P. Tieleman. His research interests include using different scale computational methods to understand how G protein–coupled receptors interact with their native membrane environment and how the latter shapes the structure and function of membrane proteins.
Haydee Mesa-Galloso received her B.Sc. and M.Sc. degrees in Biochemistry from the University of Havana, Cuba. Currently she is a Ph.D. student in Biological Sciences under the supervision of Professor D. Peter Tieleman, Department of Biological Sciences and Centre for Molecular Simulation, University of Calgary, Canada. She is a 2018 Vanier Canada Graduate Scholar and a 2017 Alberta Innovates Health Solution Graduate Studentship awardee. Her current research area focuses on the characterization of lipid–protein interactions in ion-channels.
Haleh Abdizadeh is a postdoctoral researcher at Groningen Biomolecular Science and Biotechnology Institute, University of Groningen. She did her graduate studies in Materials Science and Engineering at Sabanci University and received her Ph.D. degree in 2017. During her graduate studies, she was interested in iron transport proteins and studied the structural and allosteric features of human serum transferrin and its complexes under physiologically relevant environmental conditions using molecular dynamics simulations and coarse-grained methodologies. At the University of Groningen she is now focusing on coarse-grained molecular dynamics simulations of prototypical crowded plasma.
Sergei Yu. Noskov was born in the city of Vladimir, Russia. In 1995, he received a M.Sc. in physical chemistry from the Ivanovo State University. In 1999, he obtained a Ph.D. in physical chemistry at the Russian Academy of Sciences under the direction of Profs. Arkadiy Kolker and Michael Kiselev working on molecular modelling and statistical mechanics of ion transport in complex liquids. During his doctoral training he was a OeAD graduate fellow at the University of Innsbruck, Austria in 1997–98 working with Prof. Bernd Rode. Subsequent training involved postdoctoral fellowships in the Institute for BioMedical Sciences, Academia Sinica with Prof. Carmay Lim and in the Weill Medical College of Cornell University with Prof. Benoît Roux. Following a one-year appointment as a staff research professional at the Institute for Molecular Pediatric Sciences, University of Chicago, he assumed his faculty position in the Biochemistry Research Cluster, Department of Biological Sciences, University of Calgary, Canada in 2006. He is currently Full Professor of Biophysical Chemistry. He spent 1 year as visiting research professor with the Program in Physical Biology, NICHD, National Institutes of Health, U.S.A. The Noskov lab is focussed on the development and applications of computational methods to study membrane transport with an emphasis on membrane protein structure/function relations and specificity of membrane transport.
Siewert J. Marrink received his Ph.D. in Chemistry in 1994 from the University of Groningen, The Netherlands. His academic career continued with postdoc positions at the Max Planck Institute of Tuebingen (Germany) and the Australian National University (Australia). Since 2005 he is a full professor, heading a research group in Molecular Dynamics at the University of Groningen. He also acts as director of the Berendsen Center for Multiscale Modeling at the same university and holds an ERC Advanced grant. His main research interest is on multiscale modeling of (bio)molecular processes, with a focus on unraveling the lateral organization principles of cell membranes. To this end, the group of Marrink established the popular Martini model, a generic coarse-grain force field.
D. Peter Tieleman studied Chemistry and Philosophy at the University of Groningen, where he obtained his Ph.D. under the supervision of Herman Berendsen. After a postdoctoral position with Mark Sansom at Oxford University he moved to the University of Calgary in 2000. He holds the Canada Research Chair in Molecular Simulation and directs the Centre for Molecular Simulation. His research interests include methods for biomolecular simulation and the biophysics of membranes.
The authors declare no competing financial interest.
References
- Pereto J.; Lopez-Garcia P.; Moreira D. Ancestral Lipid Biosynthesis and Early Membrane Evolution. Trends Biochem. Sci. 2004, 29, 469–477. 10.1016/j.tibs.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Lombard J.; Lopez-Garcia P.; Moreira D. The Early Evolution of Lipid Membranes and the Three Domains of Life. Nat. Rev. Microbiol. 2012, 10, 507–515. 10.1038/nrmicro2815. [DOI] [PubMed] [Google Scholar]
- Rask-Andersen M.; Masuram S.; Schioth H. B. The Druggable Genome: Evaluation of Drug Targets in Clinical Trials Suggests Major Shifts in Molecular Class and Indication. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 9–26. 10.1146/annurev-pharmtox-011613-135943. [DOI] [PubMed] [Google Scholar]
- Escriba P. V.; Gonzalez-Ros J. M.; Goni F. M.; Kinnunen P. K. J.; Vigh L.; Sanchez-Magraner L.; Fernandez A. M.; Busquets X.; Horvath I.; Barcelo-Coblijn G. Membranes: A Meeting Point for Lipids, Proteins and Therapies. J. Cell. Mol. Med. 2008, 12, 829–875. 10.1111/j.1582-4934.2008.00281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escriba P. V. Membrane-Lipid Therapy: A New Approach in Molecular Medicine. Trends Mol. Med. 2006, 12, 34–43. 10.1016/j.molmed.2005.11.004. [DOI] [PubMed] [Google Scholar]
- Fahy E.; Subramaniam S.; Brown H. A.; Glass C. K.; Merrill A. H. Jr.; Murphy R. C.; Raetz C. R.; Russell D. W.; Seyama Y.; Shaw W.; et al. A Comprehensive Classification System for Lipids. J. Lipid Res. 2005, 46, 839–861. 10.1194/jlr.E400004-JLR200. [DOI] [PubMed] [Google Scholar]
- Fahy E.; Subramaniam S.; Murphy R. C.; Nishijima M.; Raetz C. R.; Shimizu T.; Spener F.; van Meer G.; Wakelam M. J.; Dennis E. A. Update of the Lipid Maps Comprehensive Classification System for Lipids. J. Lipid Res. 2009, 50 (Suppl), S9–14. 10.1194/jlr.R800095-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo T. C.; Tseng Y. J. Lipidpedia: A Comprehensive Lipid Knowledgebase. Bioinformatics 2018, 34, 2982–2987. 10.1093/bioinformatics/bty213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer S. J.; Nicolson G. L. The Fluid Mosaic Model of the Structure of Cell Membranes. Science 1972, 175, 720–731. 10.1126/science.175.4023.720. [DOI] [PubMed] [Google Scholar]
- Nicolson G. L. The Fluid-Mosaic Model of Membrane Structure: Still Relevant to Understanding the Structure, Function and Dynamics of Biological Membranes after More Than 40 Years. Biochim. Biophys. Acta, Biomembr. 2014, 1838, 1451–1466. 10.1016/j.bbamem.2013.10.019. [DOI] [PubMed] [Google Scholar]
- Lee A. G. Biological Membranes: The Importance of Molecular Detail. Trends Biochem. Sci. 2011, 36, 493–500. 10.1016/j.tibs.2011.06.007. [DOI] [PubMed] [Google Scholar]
- Brown M. F. In Annu. Rev. Biophys.; Dill K. A., Ed.; Annual Reviews: Palo Alto, 2017; Vol. 46, pp 379–410. [DOI] [PubMed] [Google Scholar]
- Mitchell D. C.; Straume M.; Miller J. L.; Litman B. J. Modulation of Metarhodopsin Formation by Cholesterol-Induced Ordering of Bilayer Lipids. Biochemistry 1990, 29, 9143–9149. 10.1021/bi00491a007. [DOI] [PubMed] [Google Scholar]
- Albert A. D.; Young J. E.; Yeagle P. L. Rhodopsin-Cholesterol Interactions in Bovine Rod Outer Segment Disk Membranes. Biochim. Biophys. Acta, Biomembr. 1996, 1285, 47–55. 10.1016/S0005-2736(96)00145-9. [DOI] [PubMed] [Google Scholar]
- Gimpl G.; Klein U.; Reilander H.; Fahrenholz F. Expression of the Human Oxytocin Receptor in Baculovirus-Infected Insect Cells - High-Affinity Binding Is Induced by a Cholesterol Cyclodextrin Complex. Biochemistry 1995, 34, 13794–13801. 10.1021/bi00042a010. [DOI] [PubMed] [Google Scholar]
- Gimpl G.; Burger K.; Fahrenholz F. Cholesterol as Modulator of Receptor Function. Biochemistry 1997, 36, 10959–10974. 10.1021/bi963138w. [DOI] [PubMed] [Google Scholar]
- Andersen O. S. Gramicidin Channels. Annu. Rev. Physiol. 1984, 46, 531–548. 10.1146/annurev.ph.46.030184.002531. [DOI] [PubMed] [Google Scholar]
- O’Connell A. M.; Koeppe R. E. 2nd; Andersen O. S. Kinetics of Gramicidin Channel Formation in Lipid Bilayers: Transmembrane Monomer Association. Science 1990, 250, 1256–1259. 10.1126/science.1700867. [DOI] [PubMed] [Google Scholar]
- Lee A. G. How Lipids Interact with an Intrinsic Membrane Protein: The Case of the Calcium Pump. Biochim. Biophys. Acta, Rev. Biomembr. 1998, 1376, 381–390. 10.1016/S0304-4157(98)00010-0. [DOI] [PubMed] [Google Scholar]
- Mouritsen O. G.; Bloom M. Mattress Model of Lipid-Protein Interactions in Membranes. Biophys. J. 1984, 46, 141–153. 10.1016/S0006-3495(84)84007-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tieleman D. P.; Forrest L. R.; Sansom M. S.; Berendsen H. J. Lipid Properties and the Orientation of Aromatic Residues in Ompf, Influenza M2, and Alamethicin Systems: Molecular Dynamics Simulations. Biochemistry 1998, 37, 17554–17561. 10.1021/bi981802y. [DOI] [PubMed] [Google Scholar]
- Woolf T. B.; Roux B. Structure, Energetics, and Dynamics of Lipid-Protein Interactions: A Molecular Dynamics Study of the Gramicidin a Channel in a Dmpc Bilayer. Proteins: Struct., Funct., Genet. 1996, 24, 92–114. 10.1002/(SICI)1097-0134(199601)24:1<92::AID-PROT7>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Ingolfsson H. I.; Arnarez C.; Periole X.; Marrink S. J. Computational ’Microscopy’ of Cellular Membranes. J. Cell Sci. 2016, 129, 257–268. 10.1242/jcs.176040. [DOI] [PubMed] [Google Scholar]
- Maffeo C.; Bhattacharya S.; Yoo J.; Wells D.; Aksimentiev A. Modeling and Simulation of Ion Channels. Chem. Rev. 2012, 112, 6250–6284. 10.1021/cr3002609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latorraca N. R.; Venkatakrishnan A. J.; Dror R. O. Gpcr Dynamics: Structures in Motion. Chem. Rev. 2017, 117, 139–155. 10.1021/acs.chemrev.6b00177. [DOI] [PubMed] [Google Scholar]
- Ingolfsson H. I.; Thakur P.; Herold K. F.; Hobart E. A.; Ramsey N. B.; Periole X.; de Jong D. H.; Zwama M.; Yilmaz D.; Hall K.; et al. Phytochemicals Perturb Membranes and Promiscuously Alter Protein Function. ACS Chem. Biol. 2014, 9, 1788–1798. 10.1021/cb500086e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynwar B. J.; Illya G.; Harmandaris V. A.; Muller M. M.; Kremer K.; Deserno M. Aggregation and Vesiculation of Membrane Proteins by Curvature-Mediated Interactions. Nature 2007, 447, 461–464. 10.1038/nature05840. [DOI] [PubMed] [Google Scholar]
- Simunovic M.; Srivastava A.; Voth G. A. Linear Aggregation of Proteins on the Membrane as a Prelude to Membrane Remodeling. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 20396–20401. 10.1073/pnas.1309819110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tieleman D. P. Antimicrobial Peptides in the Cross Hairs of Computer Simulations. Biophys. J. 2017, 113, 1–3. 10.1016/j.bpj.2017.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulmschneider J. P.; Ulmschneider M. B. Molecular Dynamics Simulations Are Redefining Our View of Peptides Interacting with Biological Membranes. Acc. Chem. Res. 2018, 51, 1106–1116. 10.1021/acs.accounts.7b00613. [DOI] [PubMed] [Google Scholar]
- Monje-Galvan V.; Klauda J. B. Peripheral Membrane Proteins: Tying the Knot between Experiment and Computation. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 1584–1593. 10.1016/j.bbamem.2016.02.018. [DOI] [PubMed] [Google Scholar]
- Killian J. A.; Nyholm T. K. Peptides in Lipid Bilayers: The Power of Simple Models. Curr. Opin. Struct. Biol. 2006, 16, 473–479. 10.1016/j.sbi.2006.06.007. [DOI] [PubMed] [Google Scholar]
- Andersen O. S.; Koeppe R. E. 2nd. Bilayer Thickness and Membrane Protein Function: An Energetic Perspective. Annu. Rev. Biophys. Biomol. Struct. 2007, 36, 107–130. 10.1146/annurev.biophys.36.040306.132643. [DOI] [PubMed] [Google Scholar]
- Hubert P.; Sawma P.; Duneau J. P.; Khao J.; Henin J.; Bagnard D.; Sturgis J. Single-Spanning Transmembrane Domains in Cell Growth and Cell-Cell Interactions: More Than Meets the Eye?. Cell Adh. Migr. 2010, 4, 313–324. 10.4161/cam.4.2.12430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansom M. S. P.; Bond P. J.; Deol S. S.; Grottesi A.; Haider S.; Sands Z. A. Molecular Simulations and Lipid-Protein Interactions: Potassium Channels and Other Membrane Proteins. Biochem. Soc. Trans. 2005, 33, 916–920. 10.1042/BST20050916. [DOI] [PubMed] [Google Scholar]
- Grouleff J.; Irudayam S. J.; Skeby K. K.; Schiott B. The Influence of Cholesterol on Membrane Protein Structure, Function, and Dynamics Studied by Molecular Dynamics Simulations. Biochim. Biophys. Acta, Biomembr. 2015, 1848, 1783–1795. 10.1016/j.bbamem.2015.03.029. [DOI] [PubMed] [Google Scholar]
- Epand R. M. Proteins and Cholesterol-Rich Domains. Biochim. Biophys. Acta, Biomembr. 2008, 1778, 1576–1582. 10.1016/j.bbamem.2008.03.016. [DOI] [PubMed] [Google Scholar]
- Levitan I.; Singh D. K.; Rosenhouse-Dantsker A. Cholesterol Binding to Ion Channels. Front. Physiol. 2014, 5, 65. 10.3389/fphys.2014.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimpl G. Interaction of G Protein Coupled Receptors and Cholesterol. Chem. Phys. Lipids 2016, 199, 61–73. 10.1016/j.chemphyslip.2016.04.006. [DOI] [PubMed] [Google Scholar]
- Deleu M.; Crowet J. M.; Nasir M. N.; Lins L. Complementary Biophysical Tools to Investigate Lipid Specificity in the Interaction between Bioactive Molecules and the Plasma Membrane: A Review. Biochim. Biophys. Acta, Biomembr. 2014, 1838, 3171–3190. 10.1016/j.bbamem.2014.08.023. [DOI] [PubMed] [Google Scholar]
- Montenegro F. A.; Cantero J. R.; Barrera N. P. Combining Mass Spectrometry and X-Ray Crystallography for Analyzing Native-Like Membrane Protein Lipid Complexes. Front. Physiol. 2017, 8, 892. 10.3389/fphys.2017.00892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loll P. J. Membrane Proteins, Detergents and Crystals: What Is the State of the Art?. Acta Crystallogr., Sect. F: Struct. Biol. Commun. 2014, 70, 1576–1583. 10.1107/S2053230X14025035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moraes I.; Evans G.; Sanchez-Weatherby J.; Newstead S.; Stewart P. D. S. Membrane Protein Structure Determination the Next Generation. Biochim. Biophys. Acta, Biomembr. 2014, 1838, 78–87. 10.1016/j.bbamem.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshy C.; Ziegler C. Structural Insights into Functional Lipid-Protein Interactions in Secondary Transporters. Biochim. Biophys. Acta, Gen. Subj. 2015, 1850, 476–487. 10.1016/j.bbagen.2014.05.010. [DOI] [PubMed] [Google Scholar]
- Luecke H.; Schobert B.; Richter H. T.; Cartailler J. P.; Lanyi J. K. Structure of Bacteriorhodopsin at 1.55 Angstrom Resolution. J. Mol. Biol. 1999, 291, 899–911. 10.1006/jmbi.1999.3027. [DOI] [PubMed] [Google Scholar]
- Zhou Y. F.; Morais-Cabral J. H.; Kaufman A.; MacKinnon R. Chemistry of Ion Coordination and Hydration Revealed by a K+ Channel-Fab Complex at 2.0 Angstrom Resolution. Nature 2001, 414, 43–48. 10.1038/35102009. [DOI] [PubMed] [Google Scholar]
- Long S. B.; Tao X.; Campbell E. B.; MacKinnon R. Atomic Structure of a Voltage-Dependent K+ Channel in a Lipid Membrane-Like Environment. Nature 2007, 450, 376–382. 10.1038/nature06265. [DOI] [PubMed] [Google Scholar]
- Barrera N. P.; Zhou M.; Robinson C. V. The Role of Lipids in Defining Membrane Protein Interactions: Insights from Mass Spectrometry. Trends Cell Biol. 2013, 23, 1–8. 10.1016/j.tcb.2012.08.007. [DOI] [PubMed] [Google Scholar]
- Raunser S.; Walz T. Electron Crystallography as a Technique to Study the Structure on Membrane Proteins in a Lipidic Environment. Annu. Rev. Biophys. 2009, 38, 89–105. 10.1146/annurev.biophys.050708.133649. [DOI] [PubMed] [Google Scholar]
- Wisedchaisri G.; Reichow S. L.; Gonen T. Advances in Structural and Functional Analysis of Membrane Proteins by Electron Crystallography. Structure 2011, 19, 1381–1393. 10.1016/j.str.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahlberg H.; Biyani N.; Engel A. 3d Reconstruction of Two-Dimensional Crystals. Arch. Biochem. Biophys. 2015, 581, 68–77. 10.1016/j.abb.2015.06.006. [DOI] [PubMed] [Google Scholar]
- Mitsuoka K.; Hirai T.; Murata K.; Miyazawa A.; Kidera A.; Kimura Y.; Fujiyoshi Y. The Structure of Bacteriorhodopsin at 3.0 a Resolution Based on Electron Crystallography: Implication of the Charge Distribution. J. Mol. Biol. 1999, 286, 861–882. 10.1006/jmbi.1998.2529. [DOI] [PubMed] [Google Scholar]
- Murata T.; Yamato I.; Kakinuma Y.; Leslie A. G. W.; Walker J. E. Structure of the Rotor of the V-Type Na+-Atpase from Enterococcus Hirae. Science 2005, 308, 654–659. 10.1126/science.1110064. [DOI] [PubMed] [Google Scholar]
- Gonen T.; Cheng Y. F.; Sliz P.; Hiroaki Y.; Fujiyoshi Y.; Harrison S. C.; Walz T. Lipid-Protein Interactions in Double-Layered Two-Dimensional Aqp0 Crystals (Vol 438, Pg 633, 2005). Nature 2006, 441, 248–248. 10.1038/nature04775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hite R. K.; Gonen T.; Harrison S. C.; Walz T. Interactions of Lipids with Aquaporin-0 and Other Membrane Proteins. Pfluegers Arch. 2008, 456, 651–661. 10.1007/s00424-007-0353-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hite R. K.; Li Z.; Walz T. Principles of Membrane Protein Interactions with Annular Lipids Deduced from Aquaporin-0 2d Crystals. EMBO J. 2010, 29, 1652–1658. 10.1038/emboj.2010.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hite R. K.; Chiu P. L.; Schuller J. M.; Walz T. Effect of Lipid Head Groups on Double-Layered Two-Dimensional Crystals Formed by Aquaporin-0. PLoS One 2015, 10, e0117371 10.1371/journal.pone.0117371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai X. C.; McMullan G.; Scheres S. H. W. How Cryo-Em Is Revolutionizing Structural Biology. Trends Biochem. Sci. 2015, 40, 49–57. 10.1016/j.tibs.2014.10.005. [DOI] [PubMed] [Google Scholar]
- Renaud J. P.; Chari A.; Ciferri C.; Liu W. T.; Remigy H. W.; Stark H.; Wiesmann C. Cryo-Em in Drug Discovery: Achievements, Limitations and Prospects. Nat. Rev. Drug Discovery 2018, 17, 471–492. 10.1038/nrd.2018.77. [DOI] [PubMed] [Google Scholar]
- Gao Y.; Cao E.; Julius D.; Cheng Y. Trpv1 Structures in Nanodiscs Reveal Mechanisms of Ligand and Lipid Action. Nature 2016, 534, 347–351. 10.1038/nature17964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- She J.; Guo J.; Chen Q.; Zeng W.; Jiang Y.; Bai X. C. Structural Insights into the Voltage and Phospholipid Activation of the Mammalian Tpc1 Channel. Nature 2018, 556, 130–134. 10.1038/nature26139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson S. M.; Manolaridis I.; Kowal J.; Zechner M.; Taylor N. M. I.; Bause M.; Bauer S.; Bartholomaeus R.; Bernhardt G.; Koenig B.; et al. Structural Basis of Small-Molecule Inhibition of Human Multidrug Transporter Abcg2. Nat. Struct. Mol. Biol. 2018, 25, 333–340. 10.1038/s41594-018-0049-1. [DOI] [PubMed] [Google Scholar]
- Loura L. M. S.; Prieto M.; Fernandes F. Quantification of Protein-Lipid Selectivity Using Fret. Eur. Biophys. J. 2010, 39, 565–578. 10.1007/s00249-009-0532-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras F. X.; Ernst A. M.; Haberkant P.; Bjorkholm P.; Lindahl E.; Gonen B.; Tischer C.; Elofsson A.; von Heijne G.; Thiele C.; et al. Molecular Recognition of a Single Sphingolipid Species by a Protein’s Transmembrane Domain. Nature 2012, 481, 525–529. 10.1038/nature10742. [DOI] [PubMed] [Google Scholar]
- Betaneli V.; Petrov E. P.; Schwille P. The Role of Lipids in Vdac Oligomerization. Biophys. J. 2012, 102, 523–531. 10.1016/j.bpj.2011.12.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinkova Y. V.; Denisov I. G.; Sligar S. G. Engineering Extended Membrane Scaffold Proteins for Self-Assembly of Soluble Nanoscale Lipid Bilayers. Protein Eng., Des. Sel. 2010, 23, 843–848. 10.1093/protein/gzq060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuler M. A.; Denisov I. G.; Sligar S. G. In Lipid-Protein Interactions: Methods and Protocols; Kleinschmidt J. H., Ed.; Humana Press: Totowa, NJ, 2013; pp 415–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranaghan M. J.; Schwall C. T.; Alder N. N.; Birge R. R. Green Proteorhodopsin Reconstituted into Nanoscale Phospholipid Bilayers (Nanodiscs) as Photoactive Monomers. J. Am. Chem. Soc. 2011, 133, 18318–18327. 10.1021/ja2070957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayburt T. H.; Vishnivetskiy S. A.; McLean M. A.; Morizumi T.; Huang C. C.; Tesmer J. J. G.; Ernst O. P.; Sligar S. G.; Gurevich V. V. Monomeric Rhodopsin Is Sufficient for Normal Rhodopsin Kinase (Grk1) Phosphorylation and Arrestin-1 Binding. J. Biol. Chem. 2011, 286, 1420–1428. 10.1074/jbc.M110.151043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T.; Caaveiro J. M. M.; Abe R.; Katagiri T.; Tsumoto K. Catalytic Activity of Msba Reconstituted in Nanodisc Particles Is Modulated by Remote Interactions with the Bilayer. FEBS Lett. 2011, 585, 3533–3537. 10.1016/j.febslet.2011.10.015. [DOI] [PubMed] [Google Scholar]
- Dorr J. M.; Koorengevel M. C.; Schafer M.; Prokofyev A. V.; Scheidelaar S.; van der Cruijsen E. A. W.; Dafforn T. R.; Baldus M.; Killian J. A. Detergent-Free Isolation, Characterization, and Functional Reconstitution of a Tetrameric K+ Channel: The Power of Native Nanodiscs. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 18607–18612. 10.1073/pnas.1416205112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrera N. P.; Di Bartolo N.; Booth P. J.; Robinson C. V. Micelles Protect Membrane Complexes from Solution to Vacuum. Science 2008, 321, 243–246. 10.1126/science.1159292. [DOI] [PubMed] [Google Scholar]
- Barrera N. P.; Isaacson S. C.; Zhou M.; Bavro V. N.; Welch A.; Schaedler T. A.; Seeger M. A.; Miguel R. N.; Korkhov V. M.; van Veen H. W.; et al. Mass Spectrometry of Membrane Transporters Reveals Subunit Stoichiometry and Interactions. Nat. Methods 2009, 6, 585–587. 10.1038/nmeth.1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landreh M.; Marty M. T.; Gault J.; Robinson C. V. A Sliding Selectivity Scale for Lipid Binding to Membrane Proteins. Curr. Opin. Struct. Biol. 2016, 39, 54–60. 10.1016/j.sbi.2016.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laganowsky A.; Reading E.; Allison T. M.; Ulmschneider M. B.; Degiacomi M. T.; Baldwin A. J.; Robinson C. V. Membrane Proteins Bind Lipids Selectively to Modulate Their Structure and Function. Nature 2014, 510, 172–175. 10.1038/nature13419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M.; Morgner N.; Barrera N. P.; Politis A.; Isaacson S. C.; Matak-Vinkovic D.; Murata T.; Bernal R. A.; Stock D.; Robinson C. V. Mass Spectrometry of Intact V-Type Atpases Reveals Bound Lipids and the Effects of Nucleotide Binding. Science 2011, 334, 380–385. 10.1126/science.1210148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong X.; Liu Y.; Liu W.; Liang X.; Russell D. H.; Laganowsky A. Determining Membrane Protein-Lipid Binding Thermodynamics Using Native Mass Spectrometry. J. Am. Chem. Soc. 2016, 138, 4346–4349. 10.1021/jacs.6b01771. [DOI] [PubMed] [Google Scholar]
- Cong X.; Liu Y.; Liu W.; Liang X.; Laganowsky A. Allosteric Modulation of Protein-Protein Interactions by Individual Lipid Binding Events. Nat. Commun. 2017, 8, 2203. 10.1038/s41467-017-02397-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubbens J.; de Kroon A. I. P. M. Proteome-Wide Detection of Phospholipid-Protein Interactions in Mitochondria by Photocrosslinking and Click Chemistry. Mol. BioSyst. 2010, 6, 1751–1759. 10.1039/c003064n. [DOI] [PubMed] [Google Scholar]
- Peng T.; Yuan X. Q.; Hang H. C. Turning the Spotlight on Protein-Lipid Interactions in Cells. Curr. Opin. Chem. Biol. 2014, 21, 144–153. 10.1016/j.cbpa.2014.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L.; Jiang X. In Membrane Biophysics: New Insights and Methods; Wang H., Li G., Eds.; Springer: Singapore, 2018; pp 319–354. [Google Scholar]
- Smith A. W. Lipid-Protein Interactions in Biological Membranes: A Dynamic Perspective. Biochim. Biophys. Acta, Biomembr. 2012, 1818, 172–177. 10.1016/j.bbamem.2011.06.015. [DOI] [PubMed] [Google Scholar]
- Guler G.; Gartner R. M.; Ziegler C.; Mantele W. Lipid-Protein Interactions in the Regulated Betaine Symporter Betp Probed by Infrared Spectroscopy. J. Biol. Chem. 2016, 291, 4295–4307. 10.1074/jbc.M114.621979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huster D. Solid-State Nmr Spectroscopy to Study Protein Lipid Interactions. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2014, 1841, 1146–1160. 10.1016/j.bbalip.2013.12.002. [DOI] [PubMed] [Google Scholar]
- Edrington T. C.; Kintz E.; Goldberg J. B.; Tamm L. K. Structural Basis for the Interaction of Lipopolysaccharide with Outer Membrane Protein H (Oprh) from Pseudomonas Aeruginosa. J. Biol. Chem. 2011, 286, 39211–39223. 10.1074/jbc.M111.280933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucharska I.; Liang B.; Ursini N.; Tamm L. K. Molecular Interactions of Lipopolysaccharide with an Outer Membrane Protein from Pseudomonas Aeruginosa Probed by Solution Nmr. Biochemistry 2016, 55, 5061–5072. 10.1021/acs.biochem.6b00630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soubias O.; Gawrisch K. Probing Specific Lipid-Protein Interaction by Saturation Transfer Difference Nmr Spectroscopy. J. Am. Chem. Soc. 2005, 127, 13110–13111. 10.1021/ja0538942. [DOI] [PubMed] [Google Scholar]
- Baker C. M. Polarizable Force Fields for Molecular Dynamics Simulations of Biomolecules. Wires Comput. Mol. Sci. 2015, 5, 241–254. 10.1002/wcms.1215. [DOI] [Google Scholar]
- Cui Q. Perspective: Quantum Mechanical Methods in Biochemistry and Biophysics. J. Chem. Phys. 2016, 145, 140901. 10.1063/1.4964410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argudo D.; Bethel N. P.; Marcoline F. V.; Grabe M. Continuum Descriptions of Membranes and Their Interaction with Proteins: Towards Chemically Accurate Models. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 1619–1634. 10.1016/j.bbamem.2016.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown F. L. H. Elastic Modeling of Biomembranes and Lipid Bilayers. Annu. Rev. Phys. Chem. 2008, 59, 685–712. 10.1146/annurev.physchem.59.032607.093550. [DOI] [PubMed] [Google Scholar]
- Klauda J. B.; Venable R. M.; Freites J. A.; O’Connor J. W.; Tobias D. J.; Mondragon-Ramirez C.; Vorobyov I.; MacKerell A. D.; Pastor R. W. Update of the Charmm All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J. Phys. Chem. B 2010, 114, 7830–7843. 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanommeslaeghe K.; MacKerell A. D. Charmm Additive and Polarizable Force Fields for Biophysics and Computer-Aided Drug Design. Biochim. Biophys. Acta, Gen. Subj. 2015, 1850, 861–871. 10.1016/j.bbagen.2014.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venable R. M.; Brown F. L. H.; Pastor R. W. Mechanical Properties of Lipid Bilayers from Molecular Dynamics Simulation. Chem. Phys. Lipids 2015, 192, 60–74. 10.1016/j.chemphyslip.2015.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson C. J.; Rosso L.; Betz R. M.; Walker R. C.; Gould I. R. Gafflipid: A General Amber Force Field for the Accurate Molecular Dynamics Simulation of Phospholipid. Soft Matter 2012, 8, 9617–9627. 10.1039/c2sm26007g. [DOI] [Google Scholar]
- Wang J. M.; Wolf R. M.; Caldwell J. W.; Kollman P. A.; Case D. A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- Coimbra J. T. S.; Sousa S. F.; Fernandes P. A.; Rangel M.; Ramos M. J. Biomembrane Simulations of 12 Lipid Types Using the General Amber Force Field in a Tensionless Ensemble. J. Biomol. Struct. Dyn. 2014, 32, 88–103. 10.1080/07391102.2012.750250. [DOI] [PubMed] [Google Scholar]
- Dickson C. J.; Madej B. D.; Skjevik A. A.; Betz R. M.; Teigen K.; Gould I. R.; Walker R. C. Lipid14: The Amber Lipid Force Field. J. Chem. Theory Comput. 2014, 10, 865–879. 10.1021/ct4010307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madej B. D.; Gould I. R.; Walker R. C. A Parameterization of Cholesterol for Mixed Lipid Bilayer Simulation within the Amber Lipid14 Force Field. J. Phys. Chem. B 2015, 119, 12424–12435. 10.1021/acs.jpcb.5b04924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jambeck J. P. M.; Lyubartsev A. P. Derivation and Systematic Validation of a Refined All-Atom Force Field for Phosphatidylcholine Lipids. J. Phys. Chem. B 2012, 116, 3164–3179. 10.1021/jp212503e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jambeck J. P. M.; Lyubartsev A. P. An Extension and Further Validation of an All-Atomistic Force Field for Biological Membranes. J. Chem. Theory Comput. 2012, 8, 2938–2948. 10.1021/ct300342n. [DOI] [PubMed] [Google Scholar]
- Jambeck J. P. M.; Lyubartsev A. P. Another Piece of the Membrane Puzzle: Extending Slipids Further. J. Chem. Theory Comput. 2013, 9, 774–784. 10.1021/ct300777p. [DOI] [PubMed] [Google Scholar]
- Ermilova I.; Lyubartsev A. P. Extension of the Slipids Force Field to Polyunsaturated Lipids. J. Phys. Chem. B 2016, 120, 12826–12842. 10.1021/acs.jpcb.6b05422. [DOI] [PubMed] [Google Scholar]
- Schmid N.; Eichenberger A. P.; Choutko A.; Riniker S.; Winger M.; Mark A. E.; van Gunsteren W. F. Definition and Testing of the Gromos Force-Field Versions 54a7 and 54b7. Eur. Biophys. J. 2011, 40, 843–856. 10.1007/s00249-011-0700-9. [DOI] [PubMed] [Google Scholar]
- Poger D.; Van Gunsteren W. F.; Mark A. E. A New Force Field for Simulating Phosphatidylcholine Bilayers. J. Comput. Chem. 2010, 31, 1117–1125. 10.1002/jcc.21396. [DOI] [PubMed] [Google Scholar]
- Poger D.; Mark A. E. On the Validation of Molecular Dynamics Simulations of Saturated and Cis-Monounsaturated Phosphatidylcholine Lipid Bilayers: A Comparison with Experiment. J. Chem. Theory Comput. 2010, 6, 325–336. 10.1021/ct900487a. [DOI] [PubMed] [Google Scholar]
- Poger D.; Caron B.; Mark A. E. Effect of Methyl-Branched Fatty Acids on the Structure of Lipid Bilayers. J. Phys. Chem. B 2014, 118, 13838–13848. 10.1021/jp503910r. [DOI] [PubMed] [Google Scholar]
- Poger D.; Mark A. E. A Ring to Rule Them All: The Effect of Cyclopropane Fatty Acids on the Fluidity of Lipid Bilayers. J. Phys. Chem. B 2015, 119, 5487–5495. 10.1021/acs.jpcb.5b00958. [DOI] [PubMed] [Google Scholar]
- Caron B.; Mark A. E.; Poger D. Some Like It Hot: The Effect of Sterols and Hopanoids on Lipid Ordering at High Temperature. J. Phys. Chem. Lett. 2014, 5, 3953–3957. 10.1021/jz5020778. [DOI] [PubMed] [Google Scholar]
- Maciejewski A.; Pasenkiewicz-Gierula M.; Cramariuc O.; Vattulainen I.; Rog T. Refined Opls All-Atom Force Field for Saturated Phosphatidylcholine Bilayers at Full Hydration. J. Phys. Chem. B 2014, 118, 4571–4581. 10.1021/jp5016627. [DOI] [PubMed] [Google Scholar]
- Marrink S. J.; Corradi V.; Souza P.; Ingolfsson H.; Tieleman D. P.; Sansom M. S. P. Computational Modeling of Realistic Cell Membranes. Chem. Rev. 2019, 10.1021/acs.chemrev.8b00460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolfsson H. I.; Lopez C. A.; Uusitalo J. J.; de Jong D. H.; Gopal S. M.; Periole X.; Marrink S. J. The Power of Coarse Graining in Biomolecular Simulations. Wires Comput. Mol. Sci. 2014, 4, 225–248. 10.1002/wcms.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrink S. J.; Risselada H. J.; Yefimov S.; Tieleman D. P.; de Vries A. H. The Martini Force Field: Coarse Grained Model for Biomolecular Simulations. J. Phys. Chem. B 2007, 111, 7812–7824. 10.1021/jp071097f. [DOI] [PubMed] [Google Scholar]
- de Jong D. H.; Singh G.; Bennett W. F.; Arnarez C.; Wassenaar T. A.; Schafer L. V.; Periole X.; Tieleman D. P.; Marrink S. J. Improved Parameters for the Martini Coarse-Grained Protein Force Field. J. Chem. Theory Comput. 2013, 9, 687–697. 10.1021/ct300646g. [DOI] [PubMed] [Google Scholar]
- Marrink S. J.; Tieleman D. P. Perspective on the Martini Model. Chem. Soc. Rev. 2013, 42, 6801–6822. 10.1039/c3cs60093a. [DOI] [PubMed] [Google Scholar]
- Ward A. B.; Guvench O.; Hills R. D. Jr. Coarse Grain Lipid-Protein Molecular Interactions and Diffusion with Msba Flippase. Proteins: Struct., Funct., Genet. 2012, 80, 2178–2190. 10.1002/prot.24108. [DOI] [PubMed] [Google Scholar]
- Fosso-Tande J.; Black C.; Aller S. G.; Lu L.; Hills R. D. Simulation of Lipid-Protein Interactions with the Cgprot Force Field. Aims Molecular Science 2017, 4, 352–369. 10.3934/molsci.2017.3.352. [DOI] [Google Scholar]
- Noid W. G.; Chu J. W.; Ayton G. S.; Krishna V.; Izvekov S.; Voth G. A.; Das A.; Andersen H. C. The Multiscale Coarse-Graining Method. I. A Rigorous Bridge between Atomistic and Coarse-Grained Models. J. Chem. Phys. 2008, 128, 244114. 10.1063/1.2938860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar P.; Feig M. Hybrid All-Atom/Coarse-Grained Simulations of Proteins by Direct Coupling of Charmm and Primo Force Fields. J. Chem. Theory Comput. 2017, 13, 5753–5765. 10.1021/acs.jctc.7b00840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolfsson H. I.; Melo M. N.; van Eerden F. J.; Arnarez C.; Lopez C. A.; Wassenaar T. A.; Periole X.; de Vries A. H.; Tieleman D. P.; Marrink S. J. Lipid Organization of the Plasma Membrane. J. Am. Chem. Soc. 2014, 136, 14554–14559. 10.1021/ja507832e. [DOI] [PubMed] [Google Scholar]
- Corradi V.; Mendez-Villuendas E.; Ingólfsson H. I.; Gu R.-X.; Siuda I.; Melo M. N.; Moussatova A.; DeGagné L. J.; Sejdiu B. I.; Singh G.; et al. Lipid–Protein Interactions Are Unique Fingerprints for Membrane Proteins. ACS Cent. Sci. 2018, 4, 709–717. 10.1021/acscentsci.8b00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomon-Ferrer R.; Case D. A.; Walker R. C. An Overview of the Amber Biomolecular Simulation Package. Wires Comput. Mol. Sci. 2013, 3, 198–210. 10.1002/wcms.1121. [DOI] [Google Scholar]
- Case D. A.; Cheatham T. E.; Darden T.; Gohlke H.; Luo R.; Merz K. M.; Onufriev A.; Simmerling C.; Wang B.; Woods R. J. The Amber Biomolecular Simulation Programs. J. Comput. Chem. 2005, 26, 1668–1688. 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips J. C.; Braun R.; Wang W.; Gumbart J.; Tajkhorshid E.; Villa E.; Chipot C.; Skeel R. D.; Kale L.; Schulten K. Scalable Molecular Dynamics with Namd. J. Comput. Chem. 2005, 26, 1781–1802. 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendsen H. J. C.; Vanderspoel D.; Vandrunen R. Gromacs - a Message-Passing Parallel Molecular-Dynamics Implementation. Comput. Phys. Commun. 1995, 91, 43–56. 10.1016/0010-4655(95)00042-E. [DOI] [Google Scholar]
- Abraham M. J.; Murtola T.; Schulz R.; Páll S.; Smith J. C.; Hess B.; Lindahl E. Gromacs: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- Shaw D. E.; Deneroff M. M.; Dror R. O.; Kuskin J. S.; Larson R. H.; Salmon J. K.; Young C.; Batson B.; Bowers K. J.; Chao J. C.; et al. Anton, a Special-Purpose Machine for Molecular Dynamics Simulation. Commun. ACM 2008, 51, 91–97. 10.1145/1364782.1364802. [DOI] [Google Scholar]
- Wales D. J. Exploring Energy Landscapes. Annu. Rev. Phys. Chem. 2018, 69, 401–425. 10.1146/annurev-physchem-050317-021219. [DOI] [PubMed] [Google Scholar]
- Coppock P. S.; Kindt J. T. Atomistic Simulations of Mixed-Lipid Bilayers in Gel and Fluid Phases. Langmuir 2009, 25, 352–359. 10.1021/la802712q. [DOI] [PubMed] [Google Scholar]
- Mori T.; Miyashita N.; Im W.; Feig M.; Sugita Y. Molecular Dynamics Simulations of Biological Membranes and Membrane Proteins Using Enhanced Conformational Sampling Algorithms. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 1635–1651. 10.1016/j.bbamem.2015.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett W. F. D.; Shea J. E.; Tieleman D. P. Phospholipid Chain Interactions with Cholesterol Drive Domain Formation in Lipid Membranes. Biophys. J. 2018, 114, 2595–2605. 10.1016/j.bpj.2018.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S.; Kim T.; Iyer V. G.; Im W. Charmm-Gui: A Web-Based Graphical User Interface for Charmm. J. Comput. Chem. 2008, 29, 1859–1865. 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- Jo S.; Cheng X.; Lee J.; Kim S.; Park S. J.; Patel D. S.; Beaven A. H.; Lee K. I.; Rui H.; Park S.; et al. Charmm-Gui 10 Years for Biomolecular Modeling and Simulation. J. Comput. Chem. 2017, 38, 1114–1124. 10.1002/jcc.24660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassenaar T. A.; Ingolfsson H. I.; Bockmann R. A.; Tieleman D. P.; Marrink S. J. Computational Lipidomics with Insane: A Versatile Tool for Generating Custom Membranes for Molecular Simulations. J. Chem. Theory Comput. 2015, 11, 2144–2155. 10.1021/acs.jctc.5b00209. [DOI] [PubMed] [Google Scholar]
- Wassenaar T. A.; Pluhackova K.; Bockmann R. A.; Marrink S. J.; Tieleman D. P. Going Backward: A Flexible Geometric Approach to Reverse Transformation from Coarse Grained to Atomistic Models. J. Chem. Theory Comput. 2014, 10, 676–690. 10.1021/ct400617g. [DOI] [PubMed] [Google Scholar]
- Isberg V.; Mordalski S.; Munk C.; Rataj K.; Harpsoe K.; Hauser A. S.; Vroling B.; Bojarski A. J.; Vriend G.; Gloriam D. E. Gpcrdb: An Information System for G Protein-Coupled Receptors. Nucleic Acids Res. 2016, 44, D356–D364. 10.1093/nar/gkv1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada T.; Sugihara M.; Bondar A. N.; Elstner M.; Entel P.; Buss V. The Retinal Conformation and Its Environment in Rhodopsin in Light of a New 2.2 a Crystal Structure. J. Mol. Biol. 2004, 342, 571–583. 10.1016/j.jmb.2004.07.044. [DOI] [PubMed] [Google Scholar]
- Rasmussen S. G.; DeVree B. T.; Zou Y.; Kruse A. C.; Chung K. Y.; Kobilka T. S.; Thian F. S.; Chae P. S.; Pardon E.; Calinski D.; et al. Crystal Structure of the Beta2 Adrenergic Receptor-Gs Protein Complex. Nature 2011, 477, 549–555. 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne E. F. X.; Sircar R.; Miller P. S.; Hedger G.; Luchetti G.; Nachtergaele S.; Tully M. D.; Mydock-McGrane L.; Covey D. F.; Rambo R. P.; et al. Structural Basis of Smoothened Regulation by Its Extracellular Domains. Nature 2016, 535, 517–522. 10.1038/nature18934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harikumar K. G.; Puri V.; Singh R. D.; Hanada K.; Pagano R. E.; Miller L. J. Differential Effects of Modification of Membrane Cholesterol and Sphingolipids on the Conformation, Function, and Trafficking of the G Protein-Coupled Cholecystokinin Receptor. J. Biol. Chem. 2005, 280, 2176–2185. 10.1074/jbc.M410385200. [DOI] [PubMed] [Google Scholar]
- Potter R. M.; Harikumar K. G.; Wu S. V.; Miller L. J. Differential Sensitivity of Types 1 and 2 Cholecystokinin Receptors to Membrane Cholesterol. J. Lipid Res. 2012, 53, 137–148. 10.1194/jlr.M020065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harikumar K. G.; Potter R. M.; Patil A.; Echeveste V.; Miller L. J. Membrane Cholesterol Affects Stimulus-Activity Coupling in Type 1, but Not Type 2, Cck Receptors: Use of Cell Lines with Elevated Cholesterol. Lipids 2013, 48, 231–244. 10.1007/s11745-012-3744-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherezov V.; Rosenbaum D. M.; Hanson M. A.; Rasmussen S. G. F.; Thian F. S.; Kobilka T. S.; Choi H. J.; Kuhn P.; Weis W. I.; Kobilka B. K.; et al. High-Resolution Crystal Structure of an Engineered Human Beta(2)-Adrenergic G Protein-Coupled Receptor. Science 2007, 318, 1258–1265. 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson M. A.; Cherezov V.; Griffith M. T.; Roth C. B.; Jaakola V. P.; Chien E. Y. T.; Velasquez J.; Kuhn P.; Stevens R. C. A Specific Cholesterol Binding Site Is Established by the 2.8 Angstrom Structure of the Human Beta(2)-Adrenergic Receptor. Structure 2008, 16, 897–905. 10.1016/j.str.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker D.; Fenalti G.; Brown M. A.; Katritch V.; Abagyan R.; Cherezov V.; Stevens R. C. Conserved Binding Mode of Human Beta(2) Adrenergic Receptor Inverse Agonists and Antagonist Revealed by X-Ray Crystallography. J. Am. Chem. Soc. 2010, 132, 11443–11445. 10.1021/ja105108q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warne T.; Moukhametzianov R.; Baker J. G.; Nehme R.; Edwards P. C.; Leslie A. G. W.; Schertler G. F. X.; Tate C. G. The Structural Basis for Agonist and Partial Agonist Action on a Beta(1)-Adrenergic Receptor. Nature 2011, 469, 241–244. 10.1038/nature09746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum D. M.; Zhang C.; Lyons J. A.; Holl R.; Aragao D.; Arlow D. H.; Rasmussen S. G.; Choi H. J.; Devree B. T.; Sunahara R. K.; et al. Structure and Function of an Irreversible Agonist-Beta(2) Adrenoceptor Complex. Nature 2011, 469, 236–240. 10.1038/nature09665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W.; Chun E.; Thompson A. A.; Chubukov P.; Xu F.; Katritch V.; Han G. W.; Roth C. B.; Heitman L. H.; Ijzerman A. P.; et al. Structural Basis for Allosteric Regulation of Gpcrs by Sodium Ions. Science 2012, 337, 232–236. 10.1126/science.1219218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A.; Kruse A. C.; Kobilka T. S.; Thian F. S.; Mathiesen J. M.; Sunahara R. K.; Pardo L.; Weis W. I.; Kobilka B. K.; Granier S. Crystal Structure of the Micro-Opioid Receptor Bound to a Morphinan Antagonist. Nature 2012, 485, 321–326. 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker D.; Wang C.; Katritch V.; Han G. W.; Huang X. P.; Vardy E.; McCorvy J. D.; Jiang Y.; Chu M. H.; Siu F. Y.; et al. Structural Features for Functional Selectivity at Serotonin Receptors. Science 2013, 340, 615–619. 10.1126/science.1232808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q. Y.; Liang Q. Interplay between Curvature and Lateral Organization of Lipids and Peptides/Proteins in Model Membranes. Langmuir 2014, 30, 1116–1122. 10.1021/la4039123. [DOI] [PubMed] [Google Scholar]
- Zhang K.; Zhang J.; Gao Z. G.; Zhang D.; Zhu L.; Han G. W.; Moss S. M.; Paoletta S.; Kiselev E.; Lu W.; et al. Structure of the Human P2y12 Receptor in Complex with an Antithrombotic Drug. Nature 2014, 509, 115–118. 10.1038/nature13083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P.; Nedelcu D.; Watanabe M.; Jao C.; Kim Y.; Liu J.; Salic A. Cellular Cholesterol Directly Activates Smoothened in Hedgehog Signaling. Cell 2016, 166, 1176. 10.1016/j.cell.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchetti G.; Sircar R.; Kong J. H.; Nachtergaele S.; Sagner A.; Byrne E. F.; Covey D. F.; Siebold C.; Rohatgi R. Cholesterol Activates the G-Protein Coupled Receptor Smoothened to Promote Hedgehog Signaling. eLife 2016, 5, e20304 10.7554/eLife.20304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X.; Tang J. J.; Peng C.; Wang Y.; Fu L.; Qiu Z. P.; Xiong Y.; Yang L. F.; Cui H. W.; He X. L.; et al. Cholesterol Modification of Smoothened Is Required for Hedgehog Signaling. Mol. Cell 2017, 66, 154–162. 10.1016/j.molcel.2017.02.015. [DOI] [PubMed] [Google Scholar]
- Myers B. R.; Neahring L.; Zhang Y. X.; Roberts K. J.; Beachy P. A. Rapid, Direct Activity Assays for Smoothened Reveal Hedgehog Pathway Regulation by Membrane Cholesterol and Extracellular Sodium. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, E11141–E11150. 10.1073/pnas.1717891115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredriksson R.; Lagerstrom M. C.; Lundin L. G.; Schioth H. B. The G-Protein-Coupled Receptors in the Human Genome Form Five Main Families. Phylogenetic Analysis, Paralogon Groups, and Fingerprints. Mol. Pharmacol. 2003, 63, 1256–1272. 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- Periole X. Interplay of G Protein-Coupled Receptors with the Membrane: Insights from Supra-Atomic Coarse Grain Molecular Dynamics Simulations. Chem. Rev. 2017, 117, 156–185. 10.1021/acs.chemrev.6b00344. [DOI] [PubMed] [Google Scholar]
- Genheden S.; Essex J. W.; Lee A. G. G. Protein Coupled Receptor Interactions with Cholesterol Deep in the Membrane. Biochim. Biophys. Acta, Biomembr. 2017, 1859, 268–281. 10.1016/j.bbamem.2016.12.001. [DOI] [PubMed] [Google Scholar]
- Sengupta D.; Chattopadhyay A. Molecular Dynamics Simulations of Gpcr-Cholesterol Interaction: An Emerging Paradigm. Biochim. Biophys. Acta, Biomembr. 2015, 1848, 1775–1782. 10.1016/j.bbamem.2015.03.018. [DOI] [PubMed] [Google Scholar]
- Parrill A. L.; Tigyi G. Integrating the Puzzle Pieces: The Current Atomistic Picture of Phospholipid-G Protein Coupled Receptor Interactions. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2013, 1831, 2–12. 10.1016/j.bbalip.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal S.; Khelashvili G.; Weinstein H. Not Just an Oil Slick: How the Energetics of Protein-Membrane Interactions Impacts the Function and Organization of Transmembrane Proteins. Biophys. J. 2014, 106, 2305–2316. 10.1016/j.bpj.2014.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vickery O. N.; Machtens J. P.; Zachariae U. Membrane Potentials Regulating Gpcrs: Insights from Experiments and Molecular Dynamics Simulations. Curr. Opin. Pharmacol. 2016, 30, 44–50. 10.1016/j.coph.2016.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesteros J. A.; Weinstein H. In Methods in Neurosciences; Sealfon S. C., Ed.; Academic Press: New York, 1995; Vol. 25, pp 366–428. [Google Scholar]
- Grossfield A.; Feller S. E.; Pitman M. C. A Role for Direct Interactions in the Modulation of Rhodopsin by Omega-3 Polyunsaturated Lipids. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 4888–4893. 10.1073/pnas.0508352103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khelashvili G.; Grossfield A.; Feller S. E.; Pitman M. C.; Weinstein H. Structural and Dynamic Effects of Cholesterol at Preferred Sites of Interaction with Rhodopsin Identified from Microsecond Length Molecular Dynamics Simulations. Proteins: Struct., Funct., Genet. 2009, 76, 403–417. 10.1002/prot.22355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn J. N.; Kao T. C.; Grossfield A. In G Protein-Coupled Receptors - Modeling and Simulation; Filizola M., Ed.; Springer-Verlag: Berlin, 2014; Vol. 796, pp 75–94. [Google Scholar]
- Teague W. E.; Soubias O.; Petrache H.; Fuller N.; Hines K. G.; Rand R. P.; Gawrisch K. Elastic Properties of Polyunsaturated Phosphatidylethanolamines Influence Rhodopsin Function. Faraday Discuss. 2013, 161, 383–395. 10.1039/C2FD20095C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salas-Estrada L. A.; Leioatts N.; Romo T. D.; Grossfield A. Lipids Alter Rhodopsin Function Via Ligand-Like and Solvent-Like Interactions. Biophys. J. 2018, 114, 355–367. 10.1016/j.bpj.2017.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cang X. H.; Du Y.; Mao Y. Y.; Wang Y. Y.; Yang H. Y.; Jiang H. L. Mapping the Functional Binding Sites of Cholesterol in Beta(2)-Adrenergic Receptor by Long-Time Molecular Dynamics Simulations. J. Phys. Chem. B 2013, 117, 1085–1094. 10.1021/jp3118192. [DOI] [PubMed] [Google Scholar]
- Prasanna X.; Chattopadhyay A.; Sengupta D. Cholesterol Modulates the Dimer Interface of the Beta(2)-Adrenergic Receptor Via Cholesterol Occupancy Sites. Biophys. J. 2014, 106, 1290–1300. 10.1016/j.bpj.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gater D. L.; Saurel O.; Iordanov I.; Liu W.; Cherezov V.; Milon A. Two Classes of Cholesterol Binding Sites for the Beta(2)Ar Revealed by Thermostability and Nmr. Biophys. J. 2014, 107, 2305–2312. 10.1016/j.bpj.2014.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cang X. H.; Yang L. L.; Yang J.; Luo C.; Zheng M. Y.; Yu K. Q.; Yang H. Y.; Jiang H. L. Cholesterol-Beta(1)Ar Interaction Versus Cholesterol-Beta(2)Ar Interaction. Proteins: Struct., Funct., Genet. 2014, 82, 760–770. 10.1002/prot.24456. [DOI] [PubMed] [Google Scholar]
- Zocher M.; Zhang C.; Rasmussen S. G. F.; Kobilka B. K.; Muller D. J. Cholesterol Increases Kinetic, Energetic, and Mechanical Stability of the Human Beta(2)-Adrenergic Receptor. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, E3463–E3472. 10.1073/pnas.1210373109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawaliby R.; Trubbia C.; Delporte C.; Masureel M.; Van Antwerpen P.; Kobilka B. K.; Govaerts C. Allosteric Regulation of G Protein-Coupled Receptor Activity by Phospholipids. Nat. Chem. Biol. 2016, 12, 35–39. 10.1038/nchembio.1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casiraghi M.; Damian M.; Lescop E.; Point E.; Moncoq K.; Morellet N.; Leyy D.; Marie J.; Guittet E.; Baneres J. L.; et al. Functional Modulation of a G Protein-Coupled Receptor Conformational Landscape in a Lipid Bilayer. J. Am. Chem. Soc. 2016, 138, 11170–11175. 10.1021/jacs.6b04432. [DOI] [PubMed] [Google Scholar]
- Manna M.; Niemela M.; Tynkkynen J.; Javanainen M.; Kulig W.; Muller D. J.; Rog T.; Vattulainen I. Mechanism of Allosteric Regulation of Beta2-Adrenergic Receptor by Cholesterol. eLife 2016, 5, e18432 10.7554/eLife.18432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki S.; Ghirlando R.; White J. F.; Gvozdenovic-Jeremic J.; Northup J. K.; Grisshammer R. Modulation of the Interaction between Neurotensin Receptor Nts1 and Gq Protein by Lipid. J. Mol. Biol. 2012, 417, 95–111. 10.1016/j.jmb.2012.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura T.; Yeliseev A. A.; Vukoti K.; Rhodes S. D.; Cheng K.; Rice K. C.; Gawrisch K. Recombinant Cannabinoid Type 2 Receptor in Liposome Model Activates G Protein in Response to Anionic Lipid Constituents. J. Biol. Chem. 2012, 287, 4076–4087. 10.1074/jbc.M111.268425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale C.; Herce H. D.; Pomes R.; Garcia A. E. Can Specific Protein-Lipid Interactions Stabilize an Active State of the Beta 2 Adrenergic Receptor?. Biophys. J. 2015, 109, 1652–1662. 10.1016/j.bpj.2015.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruzzese A.; Gil C.; Dalton J. A. R.; Giraldo J. Structural Insights into Positive and Negative Allosteric Regulation of a G Protein-Coupled Receptor through Protein-Lipid Interactions. Sci. Rep. 2018, 8, 4456. 10.1038/s41598-018-22735-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino K. A.; Prada-Gracia D.; Provasi D.; Filizola M. Impact of Lipid Composition and Receptor Conformation on the Spatio-Temporal Organization of Mu-Opioid Receptors in a Multi-Component Plasma Membrane Model. PLoS Comput. Biol. 2016, 12, e1005240 10.1371/journal.pcbi.1005240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen H. Y.; Hoi K. K.; Liko I.; Hedger G.; Horrell M. R.; Song W.; Wu D.; Heine P.; Warne T.; Lee Y.; et al. Ptdins(4,5)P2 Stabilizes Active States of Gpcrs and Enhances Selectivity of G-Protein Coupling. Nature 2018, 559, 423–427. 10.1038/s41586-018-0325-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zezula J.; Freissmuth M. The a(2a)-Adenosine Receptor: A Gpcr with Unique Features?. Br. J. Pharmacol. 2008, 153, S184–S190. 10.1038/sj.bjp.0707674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaakola V. P.; Griffith M. T.; Hanson M. A.; Cherezov V.; Chien E. Y. T.; Lane J. R.; Ijzerman A. P.; Stevens R. C. The 2.6 Angstrom Crystal Structure of a Human a(2a) Adenosine Receptor Bound to an Antagonist. Science 2008, 322, 1211–1217. 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyman E.; Higgs C.; Kim B.; Lupyan D.; Shelleys J. C.; Farid R.; Voth G. A. A Role for a Specific Cholesterol Interaction in Stabilizing the Apo Configuration of the Human a(2a) Adenosine Receptor. Structure 2009, 17, 1660–1668. 10.1016/j.str.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. Y.; Lyman E. Predictions for Cholesterol Interaction Sites on the a(2a) Adenosine Receptor. J. Am. Chem. Soc. 2012, 134, 16512–16515. 10.1021/ja307532d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng H. W.; Laughton C. A.; Doughty S. W. Molecular Dynamics Simulations of the Adenosine A2a Receptor in Popc and Pope Lipid Bilayers: Effects of Membrane on Protein Behavior. J. Chem. Inf. Model. 2014, 54, 573–581. 10.1021/ci400463z. [DOI] [PubMed] [Google Scholar]
- Rouviere E.; Arnarez C.; Yang L. W.; Lyman E. Identification of Two New Cholesterol Interaction Sites on the a(2a) Adenosine Receptor. Biophys. J. 2017, 113, 2415–2424. 10.1016/j.bpj.2017.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrivastava S.; Pucadyil T. J.; Paila Y. D.; Ganguly S.; Chattopadhyay A. Chronic Cholesterol Depletion Using Statin Impairs the Function and Dynamics of Human Serotonin(1a) Receptors. Biochemistry 2010, 49, 5426–5435. 10.1021/bi100276b. [DOI] [PubMed] [Google Scholar]
- Saxena R.; Chattopadhyay A. Membrane Cholesterol Stabilizes the Human Serotonin(1a) Receptor. Biochim. Biophys. Acta, Biomembr. 2012, 1818, 2936–2942. 10.1016/j.bbamem.2012.07.032. [DOI] [PubMed] [Google Scholar]
- Gutierrez M. G.; Mansfield K. S.; Malmstadt N. The Functional Activity of the Human Serotonin 5-Ht1a Receptor Is Controlled by Lipid Bilayer Composition. Biophys. J. 2016, 110, 2486–2495. 10.1016/j.bpj.2016.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta D.; Chattopadhyay A. Identification of Cholesterol Binding Sites in the Serotonin1a Receptor. J. Phys. Chem. B 2012, 116, 12991–12996. 10.1021/jp309888u. [DOI] [PubMed] [Google Scholar]
- Patra S. M.; Chakraborty S.; Shahane G.; Prasanna X.; Sengupta D.; Maiti P. K.; Chattopadhyay A. Differential Dynamics of the Serotonin1a Receptor in Membrane Bilayers of Varying Cholesterol Content Revealed by All Atom Molecular Dynamics Simulation. Mol. Membr. Biol. 2015, 32, 127–137. 10.3109/09687688.2015.1096971. [DOI] [PubMed] [Google Scholar]
- Ramirez-Anguita J. M.; Rodriguez-Espigares I.; Guixa-Gonzalez R.; Bruno A.; Torrens-Fontanals M.; Varela-Rial A.; Selent J. Membrane Cholesterol Effect on the 5-Ht2a Receptor: Insights into the Lipid-Induced Modulation of an Antipsychotic Drug Target. Biotechnol. Appl. Biochem. 2018, 65, 29–37. 10.1002/bab.1608. [DOI] [PubMed] [Google Scholar]
- Prasanna X.; Jafurulla M.; Sengupta D.; Chattopadhyay A. The Ganglioside Gm1 Interacts with the Serotonin1a Receptor Via the Sphingolipid Binding Domain. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 2818–2826. 10.1016/j.bbamem.2016.08.009. [DOI] [PubMed] [Google Scholar]
- Jafurulla M.; Bandari S.; Pucadyil T. J.; Chattopadhyay A. Sphingolipids Modulate the Function of Human Serotonin(1a) Receptors: Insights from Sphingolipid-Deficient Cells. Biochim. Biophys. Acta, Biomembr. 2017, 1859, 598–604. 10.1016/j.bbamem.2016.10.016. [DOI] [PubMed] [Google Scholar]
- Shan J.; Khelashvili G.; Mondal S.; Mehler E. L.; Weinstein H. Ligand-Dependent Conformations and Dynamics of the Serotonin 5-Ht(2a) Receptor Determine Its Activation and Membrane-Driven Oligomerization Properties. PLoS Comput. Biol. 2012, 8, e1002473 10.1371/journal.pcbi.1002473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedger G.; Koldso H.; Chavent M.; Siebold C.; Rohatgi R.; Sansom M. S. P. Cholesterol Interaction Sites on the Transmembrane Domain of the Hedgehog Signal Transducer and Class F G Protein-Coupled Receptor Smoothened. Structure 2018, 10.1016/j.str.2018.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oates J.; Faust B.; Attrill H.; Harding P.; Orwick M.; Watts A. The Role of Cholesterol on the Activity and Stability of Neurotensin Receptor 1. Biochim. Biophys. Acta, Biomembr. 2012, 1818, 2228–2233. 10.1016/j.bbamem.2012.04.010. [DOI] [PubMed] [Google Scholar]
- Bolivar J. H.; Munoz-Garcia J. C.; Castro-Dopico T.; Dijkman P. M.; Stansfeld P. J.; Watts A. Interaction of Lipids with the Neurotensin Receptor 1. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 1278–1287. 10.1016/j.bbamem.2016.02.032. [DOI] [PubMed] [Google Scholar]
- Dijkman P. M.; Watts A. Lipid Modulation of Early G Protein-Coupled Receptor Signalling Events. Biochim. Biophys. Acta, Biomembr. 2015, 1848, 2889–2897. 10.1016/j.bbamem.2015.08.004. [DOI] [PubMed] [Google Scholar]
- Oddi S.; Dainese E.; Fezza F.; Lanuti M.; Barcaroli D.; De Laurenzi V.; Centonze D.; Maccarrone M. Functional Characterization of Putative Cholesterol Binding Sequence (Crac) in Human Type-1 Cannabinoid Receptor. J. Neurochem. 2011, 116, 858–865. 10.1111/j.1471-4159.2010.07041.x. [DOI] [PubMed] [Google Scholar]
- Koldso H.; Sansom M. S. P. Organization and Dynamics of Receptor Proteins in a Plasma Membrane. J. Am. Chem. Soc. 2015, 137, 14694–14704. 10.1021/jacs.5b08048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mystek P.; Dutka P.; Tworzydlo M.; Dziedzicka-Wasylewska M.; Polit A. The Role of Cholesterol and Sphingolipids in the Dopamine D-1 Receptor and G Protein Distribution in the Plasma Membrane. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2016, 1861, 1775–1786. 10.1016/j.bbalip.2016.08.015. [DOI] [PubMed] [Google Scholar]
- Zhukovsky M. A.; Lee P. H.; Ott A.; Helms V. Putative Cholesterol-Binding Sites in Human Immunodeficiency Virus (Hiv) Coreceptors Cxcr4 and Ccr5. Proteins: Struct., Funct., Genet. 2013, 81, 555–567. 10.1002/prot.24211. [DOI] [PubMed] [Google Scholar]
- Kumari R.; Castillo C.; Francesconi A. Agonist-Dependent Signaling by Group I Metabotropic Glutamate Receptors Is Regulated by Association with Lipid Domains. J. Biol. Chem. 2013, 288, 32004–32019. 10.1074/jbc.M113.475863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pydi S. P.; Jafurulla M.; Wai L. S.; Bhullar R. P.; Chelikani P.; Chattopadhyay A. Cholesterol Modulates Bitter Taste Receptor Function. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 2081–2087. 10.1016/j.bbamem.2016.06.005. [DOI] [PubMed] [Google Scholar]
- Hildebrand P. W.; Scheerer P.; Park J. H.; Choe H. W.; Piechnick R.; Ernst O. P.; Hofmann K. P.; Heck M. A Ligand Channel through the G Protein Coupled Receptor Opsin. PLoS One 2009, 4, e4382 10.1371/journal.pone.0004382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst D. P.; Grossfield A.; Lynch D. L.; Feller S.; Romo T. D.; Gawrisch K.; Pitman M. C.; Reggio P. H. A Lipid Pathway for Ligand Binding Is Necessary for a Cannabinoid G Protein-Coupled Receptor. J. Biol. Chem. 2010, 285, 17954–17964. 10.1074/jbc.M109.041590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caliman A. D.; Miao Y. L.; McCammon J. A. Activation Mechanisms of the First Sphingosine-1-Phosphate Receptor. Protein Sci. 2017, 26, 1150–1160. 10.1002/pro.3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guixa-Gonzalez R.; Albasanz J. L.; Rodriguez-Espigares I.; Pastor M.; Sanz F.; Marti-Solano M.; Manna M.; Martinez-Seara H.; Hildebrand P. W.; Martin M.; et al. Membrane Cholesterol Access into a G-Protein-Coupled Receptor. Nat. Commun. 2017, 8, 14505. 10.1038/ncomms14505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J. H.; Scheerer P.; Hofmann K. P.; Choe H. W.; Ernst O. P. Crystal Structure of the Ligand-Free G-Protein-Coupled Receptor Opsin. Nature 2008, 454, 183–187. 10.1038/nature07063. [DOI] [PubMed] [Google Scholar]
- Menon I.; Huber T.; Sanyal S.; Banerjee S.; Barre P.; Canis S.; Warren J. D.; Hwa J.; Sakmar T. P.; Menon A. K. Opsin Is a Phospholipid Flippase. Curr. Biol. 2011, 21, 149–153. 10.1016/j.cub.2010.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goren M. A.; Morizumi T.; Menon I.; Joseph J. S.; Dittman J. S.; Cherezov V.; Stevens R. C.; Ernst O. P.; Menon A. K. Constitutive Phospholipid Scramblase Activity of a G Protein-Coupled Receptor. Nat. Commun. 2014, 5, 5115. 10.1038/ncomms6115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morra G.; Razavi A. M.; Pandey K.; Weinstein H.; Menon A. K.; Khelashvili G. Mechanisms of Lipid Scrambling by the G Protein-Coupled Receptor Opsin. Structure 2018, 26, 356–367. 10.1016/j.str.2017.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice P.; Kamal M.; Jockers R. Asymmetry of Gpcr Oligomers Supports Their Functional Relevance. Trends Pharmacol. Sci. 2011, 32, 514–520. 10.1016/j.tips.2011.05.006. [DOI] [PubMed] [Google Scholar]
- Gaitonde S. A.; Gonzalez-Maeso J. Contribution of Heteromerization to G Protein-Coupled Receptor Function. Curr. Opin. Pharmacol. 2017, 32, 23–31. 10.1016/j.coph.2016.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai R. S.; Kusumi A. Single-Molecule Imaging Revealed Dynamic Gpcr Dimerization. Curr. Opin. Cell Biol. 2014, 27, 78–86. 10.1016/j.ceb.2013.11.008. [DOI] [PubMed] [Google Scholar]
- Kniazeff J.; Prezeau L.; Rondard P.; Pin J. P.; Goudet C. Dimers and Beyond: The Functional Puzzles of Class C Gpcrs. Pharmacol. Ther. 2011, 130, 9–25. 10.1016/j.pharmthera.2011.01.006. [DOI] [PubMed] [Google Scholar]
- Chabre M.; Deterre P.; Antonny B. The Apparent Cooperativity of Some Gpcrs Does Not Necessarily Imply Dimerization. Trends Pharmacol. Sci. 2009, 30, 182–187. 10.1016/j.tips.2009.01.003. [DOI] [PubMed] [Google Scholar]
- Gurevich V. V.; Gurevich E. V. Gpcrs and Signal Transducers: Interaction Stoichiometry. Trends Pharmacol. Sci. 2018, 39, 672–684. 10.1016/j.tips.2018.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farran B. An Update on the Physiological and Therapeutic Relevance of Gpcr Oligomers. Pharmacol. Res. 2017, 117, 303–327. 10.1016/j.phrs.2017.01.008. [DOI] [PubMed] [Google Scholar]
- Gahbauer S.; Bockmann R. A. Membrane-Mediated Oligomerization of G Protein Coupled Receptors and Its Implications for Gpcr Function. Front. Physiol. 2016, 7, 494. 10.3389/fphys.2016.00494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco R.; Martinez-Pinilla E.; Lanciego J. L.; Navarro G. Basic Pharmacological and Structural Evidence for Class a G-Protein-Coupled Receptor Heteromerization. Front. Pharmacol. 2016, 7, 76. 10.3389/fphar.2016.00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B.; Chien E. Y.; Mol C. D.; Fenalti G.; Liu W.; Katritch V.; Abagyan R.; Brooun A.; Wells P.; Bi F. C.; et al. Structures of the Cxcr4 Chemokine Gpcr with Small-Molecule and Cyclic Peptide Antagonists. Science 2010, 330, 1066–1071. 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Periole X.; Huber T.; Marrink S. J.; Sakmar T. P. G. Protein-Coupled Receptors Self-Assemble in Dynamics Simulations of Model Bilayers. J. Am. Chem. Soc. 2007, 129, 10126–10132. 10.1021/ja0706246. [DOI] [PubMed] [Google Scholar]
- Knepp A. M.; Periole X.; Marrink S. J.; Sakmar T. P.; Huber T. Rhodopsin Forms a Dimer with Cytoplasmic Helix 8 Contacts in Native Membranes. Biochemistry 2012, 51, 1819–1821. 10.1021/bi3001598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Periole X.; Knepp A. M.; Sakmar T. P.; Marrink S. J.; Huber T. Structural Determinants of the Supramolecular Organization of G Protein-Coupled Receptors in Bilayers. J. Am. Chem. Soc. 2012, 134, 10959–10965. 10.1021/ja303286e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soubias O.; Teague W. E.; Hines K. G.; Gawrisch K. Rhodopsin/Lipid Hydrophobic Matching-Rhodopsin Oligomerization and Function. Biophys. J. 2015, 108, 1125–1132. 10.1016/j.bpj.2015.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal S.; Johnston J. M.; Wang H.; Khelashvili G.; Filizola M.; Weinstein H. Membrane Driven Spatial Organization of Gpcrs. Sci. Rep. 2013, 3, 2909. 10.1038/srep02909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provasi D.; Johnston J. M.; Filizola M. Lessons from Free Energy Simulations of Delta-Opioid Receptor Homodimers Involving the Fourth Transmembrane Helix. Biochemistry 2010, 49, 6771–6776. 10.1021/bi100686t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston J. M.; Aburi M.; Provasi D.; Bortolato A.; Urizar E.; Lambert N. A.; Javitch J. A.; Filizola M. Making Structural Sense of Dimerization Interfaces of Delta Opioid Receptor Homodimers. Biochemistry 2011, 50, 1682–1690. 10.1021/bi101474v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provasi D.; Boz M. B.; Johnston J. M.; Filizola M. Preferred Supramolecular Organization and Dimer Interfaces of Opioid Receptors from Simulated Self-Association. PLoS Comput. Biol. 2015, 11, e1004148 10.1371/journal.pcbi.1004148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H.; Pearsall E. A.; Hurst D. P.; Zhang Y.; Chu J.; Zhou Y.; Reggio P. H.; Loh H. H.; Law P. Y. Palmitoylation and Membrane Cholesterol Stabilize Mu-Opioid Receptor Homodimerization and G Protein Coupling. BMC Cell Biol. 2012, 13, 6. 10.1186/1471-2121-13-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guixa-Gonzalez R.; Javanainen M.; Gomez-Soler M.; Cordobilla B.; Domingo J. C.; Sanz F.; Pastor M.; Ciruela F.; Martinez-Seara H.; Selent J. Membrane Omega-3 Fatty Acids Modulate the Oligomerisation Kinetics of Adenosine A2a and Dopamine D2 Receptors. Sci. Rep. 2016, 6, 19839. 10.1038/srep19839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasanna X.; Sengupta D.; Chattopadhyay A. Cholesterol-Dependent Conformational Plasticity in Gpcr Dimers. Sci. Rep. 2016, 6, 31858. 10.1038/srep31858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluhackova K.; Gahbauer S.; Kranz F.; Wassenaar T. A.; Bockmann R. A. Dynamic Cholesterol-Conditioned Dimerization of the G Protein Coupled Chemokine Receptor Type 4. PLoS Comput. Biol. 2016, 12, e1005169 10.1371/journal.pcbi.1005169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gahbauer S.; Pluhackova K.; Btockmann R. A. Closely Related, yet Unique: Distinct Homo- and Heterodimerization Patterns of G Protein Coupled Chemokine Receptors and Their Fine-Tuning by Cholesterol. PLoS Comput. Biol. 2018, 14, e1006062. 10.1371/journal.pcbi.1006062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soubias O.; Gawrisch K. The Role of the Lipid Matrix for Structure and Function of the Gpcr Rhodopsin. Biochim. Biophys. Acta, Biomembr. 2012, 1818, 234–240. 10.1016/j.bbamem.2011.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B.Ion Channels of Excitable Membranes; Sinauer Associates, Inc.: Sunderland, MA, 2001; p 814. [Google Scholar]
- Hansen S. B. Lipid Agonism: The Pip2 Paradigm of Ligand-Gated Ion Channels. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2015, 1851, 620–628. 10.1016/j.bbalip.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitan I.; Fang Y.; Rosenhouse-Dantsker A.; Romanenko V. In Cholesterol Binding and Cholesterol Transport Proteins: Structure and Function in Health and Disease; Harris J. R., Ed.; Springer: New York, 2010; Vol. 51, pp 509–549. [Google Scholar]
- Poveda J. A.; Giudici A. M.; Renart M. L.; Morales A.; Gonzalez-Ros J. M. Towards Understanding the Molecular Basis of Ion Channel Modulation by Lipids: Mechanistic Models and Current Paradigms. Biochim. Biophys. Acta, Biomembr. 2017, 1859, 1507–1516. 10.1016/j.bbamem.2017.04.003. [DOI] [PubMed] [Google Scholar]
- Taberner F. J.; Fernandez-Ballester G.; Fernandez-Carvajal A.; Ferrer-Montiel A. Trp Channels Interaction with Lipids and Its Implications in Disease. Biochim. Biophys. Acta, Biomembr. 2015, 1848, 1818–1827. 10.1016/j.bbamem.2015.03.022. [DOI] [PubMed] [Google Scholar]
- Poveda J. A.; Giudici A. M.; Renart M. L.; Molina M. L.; Montoya E.; Fernandez-Carvajal A.; Fernandez-Ballester G.; Encinar J. A.; Gonzalez-Ros J. M. Lipid Modulation of Ion Channels through Specific Binding Sites. Biochim. Biophys. Acta, Biomembr. 2014, 1838, 1560–1567. 10.1016/j.bbamem.2013.10.023. [DOI] [PubMed] [Google Scholar]
- Kudaibergenova M.; Perissinotti L. L.; Noskov S. Y. Lipid Roles in Herg Function and Interactions with Drugs. Neurosci. Lett. 2018, 10.1016/j.neulet.2018.05.019. [DOI] [PubMed] [Google Scholar]
- Chapman H.; Ramstrom C.; Korhonen L.; Laine M.; Wann K. T.; Lindholm D.; Pasternack M.; Tornquist K. Downregulation of the Herg (Kcnh2) K(+) Channel by Ceramide: Evidence for Ubiquitin-Mediated Lysosomal Degradation. J. Cell Sci. 2005, 118, 5325–5334. 10.1242/jcs.02635. [DOI] [PubMed] [Google Scholar]
- Poyry S.; Vattulainen I. Role of Charged Lipids in Membrane Structures - Insight Given by Simulations. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 2322–2333. 10.1016/j.bbamem.2016.03.016. [DOI] [PubMed] [Google Scholar]
- Hedger G.; Sansom M. S. P. Lipid Interaction Sites on Channels, Transporters and Receptors: Recent Insights from Molecular Dynamics Simulations. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 2390–2400. 10.1016/j.bbamem.2016.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Wang Q.; Ni F.; Ma J. Structure of the Full-Length Shaker Potassium Channel Kv1.2 by Normal-Mode-Based X-Ray Crystallographic Refinement. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 11352–11357. 10.1073/pnas.1000142107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller A. N.; Long S. B. Crystal Structure of the Human Two-Pore Domain Potassium Channel K2p1. Science 2012, 335, 432–436. 10.1126/science.1213274. [DOI] [PubMed] [Google Scholar]
- Tao X.; Avalos J. L.; Chen J. Y.; MacKinnon R. Crystal Structure of the Eukaryotic Strong Inward-Rectifier K+ Channel Kir2.2 at 3.1 Angstrom Resolution. Science 2009, 326, 1668–1674. 10.1126/science.1180310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payandeh J.; Scheuer T.; Zheng N.; Catterall W. A. The Crystal Structure of a Voltage-Gated Sodium Channel. Nature 2011, 475, 353–358. 10.1038/nature10238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscher C.; Slesinger P. A. Emerging Roles for G Protein-Gated Inwardly Rectifying Potassium (Girk) Channels in Health and Disease. Nat. Rev. Neurosci. 2010, 11, 301–315. 10.1038/nrn2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whorton M. R.; MacKinnon R. Crystal Structure of the Mammalian Girk2 K+ Channel and Gating Regulation by G Proteins, Pip2, and Sodium. Cell 2011, 147, 199–208. 10.1016/j.cell.2011.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whorton M. R.; MacKinnon R. X-Ray Structure of the Mammalian Girk2-Betagamma G-Protein Complex. Nature 2013, 498, 190–119. 10.1038/nature12241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decoursey T. E. Voltage-Gated Proton Channels and Other Proton Transfer Pathways. Physiol. Rev. 2003, 83, 475–579. 10.1152/physrev.00028.2002. [DOI] [PubMed] [Google Scholar]
- Murata Y.; Iwasaki H.; Sasaki M.; Inaba K.; Okamura Y. Phosphoinositide Phosphatase Activity Coupled to an Intrinsic Voltage Sensor. Nature 2005, 435, 1239–1243. 10.1038/nature03650. [DOI] [PubMed] [Google Scholar]
- Clapham D. E.; Montell C.; Schultz G.; Julius D. International Union of, P. International Union of Pharmacology. Xliii. Compendium of Voltage-Gated Ion Channels: Transient Receptor Potential Channels. Pharmacol. Rev. 2003, 55, 591–596. 10.1124/pr.55.4.6. [DOI] [PubMed] [Google Scholar]
- Gutman G. A.; Chandy K. G.; Adelman J. P.; Aiyar J.; Bayliss D. A.; Clapham D. E.; Covarriubias M.; Desir G. V.; Furuichi K.; Ganetzky B.; et al. International Union of Pharmacology. Xli. Compendium of Voltage-Gated Ion Channels: Potassium Channels. Pharmacol. Rev. 2003, 55, 583–586. 10.1124/pr.55.4.9. [DOI] [PubMed] [Google Scholar]
- Long S. B.; Campbell E. B.; MacKinnon R. Crystal Structure of a Mammalian Voltage-Dependent Shaker Family K+ Channel. Science 2005, 309, 897–903. 10.1126/science.1116269. [DOI] [PubMed] [Google Scholar]
- Valiyaveetil F. I.; Zhou Y. F.; Mackinnon R. Lipids in the Structure, Folding, and Function of the Kcsa K+ Channel. Biochemistry 2002, 41, 10771–10777. 10.1021/bi026215y. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Guo J.; Perissinotti L. L.; Lees-Miller J.; Teng G.; Durdagi S.; Duff H. J.; Noskov S. Y. Role of the Ph in State-Dependent Blockade of Herg Currents. Sci. Rep. 2016, 6, 32536. 10.1038/srep32536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordero-Morales J. F.; Cuello L. G.; Zhao Y. X.; Jogini V.; Cortes D. M.; Roux B.; Perozo E. Molecular Determinants of Gating at the Potassium-Channel Selectivity Filter. Nat. Struct. Mol. Biol. 2006, 13, 311–318. 10.1038/nsmb1069. [DOI] [PubMed] [Google Scholar]
- Cordero-Morales J. F.; Jogini V.; Chakrapani S.; Perozo E. A Multipoint Hydrogen-Bond Network Underlying Kcsa C-Type Inactivation. Biophys. J. 2011, 100, 2387–2393. 10.1016/j.bpj.2011.01.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordero-Morales J. F.; Jogini V.; Lewis A.; Vasquez V.; Cortes D. M.; Roux B.; Perozo E. Molecular Driving Forces Determining Potassium Channel Slow Inactivation. Nat. Struct. Mol. Biol. 2007, 14, 1062–1069. 10.1038/nsmb1309. [DOI] [PubMed] [Google Scholar]
- Deol S. S.; Domene C.; Bond P. J.; Sansom M. S. P. Anionic Phospholipid Interactions with the Potassium Channel Kcsa: Simulation Studies. Biophys. J. 2006, 90, 822–830. 10.1529/biophysj.105.071407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weingarth M.; Prokofyev A.; van der Cruijsen E. A. W.; Nand D.; Bonvin A.; Pongs O.; Baldus M. Structural Determinants of Specific Lipid Binding to Potassium Channels. J. Am. Chem. Soc. 2013, 135, 3983–3988. 10.1021/ja3119114. [DOI] [PubMed] [Google Scholar]
- Marius P.; de Planque M. R. R.; Williamson P. T. F. Probing the Interaction of Lipids with the Non-Annular Binding Sites of the Potassium Channel Kcsa by Magic-Angle Spinning Nmr. Biochim. Biophys. Acta, Biomembr. 2012, 1818, 90–96. 10.1016/j.bbamem.2011.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triano I.; Barrera F. N.; Renart M. L.; Molina M. L.; Fernandez-Ballester G.; Poveda J. A.; Fernandez A. M.; Encinar J. A.; Ferrer-Montiel A. V.; Otzen D.; et al. Occupancy of Nonannular Lipid Binding Sites on Kcsa Greatly Increases the Stability of the Tetrameric Protein. Biochemistry 2010, 49, 5397–5404. 10.1021/bi1003712. [DOI] [PubMed] [Google Scholar]
- van der Cruijsen E. A. W.; Nand D.; Weingarth M.; Prokofyev A.; Hornig S.; Cukkemane A. A.; Bonvin A.; Becker S.; Hulse R. E.; Perozo E.; et al. Importance of Lipid-Pore Loop Interface for Potassium Channel Structure and Function. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 13008–13013. 10.1073/pnas.1305563110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina M. L.; Giudici A. M.; Poveda J. A.; Fernandez-Ballester G.; Montoya E.; Renart M. L.; Fernandez A. M.; Encinar J. A.; Riquelme G.; Morales A.; et al. Competing Lipid-Protein and Protein-Protein Interactions Determine Clustering and Gating Patterns in the Potassium Channel from Streptomyces Lividans (Kcsa). J. Biol. Chem. 2015, 290, 25745–25755. 10.1074/jbc.M115.669598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Encinar J. A.; Molina M. L.; Poveda J. A.; Barrera F. N.; Renart M. L.; Fernandez A. M.; Gonzalez-Ros J. M. The Influence of a Membrane Environment on the Structure and Stability of a Prokaryotic Potassium Channel, Kcsa. FEBS Lett. 2005, 579, 5199–5204. 10.1016/j.febslet.2005.08.038. [DOI] [PubMed] [Google Scholar]
- van Dalen A.; Hegger S.; Killian J. A.; de Kruijff B. Influence of Lipids on Membrane Assembly and Stability of the Potassium Channel Kcsa. FEBS Lett. 2002, 525, 33–38. 10.1016/S0014-5793(02)03061-2. [DOI] [PubMed] [Google Scholar]
- Brohawn S. G.; del Marmol J.; MacKinnon R. Crystal Structure of the Human K2p Traak, a Lipid- and Mechano-Sensitive K+ Ion Channel. Science 2012, 335, 436–441. 10.1126/science.1213808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo A. L.; Gulbis J. M.; Antcliff J. F.; Rahman T.; Lowe E. D.; Zimmer J.; Cuthbertson J.; Ashcroft F. M.; Ezaki T.; Doyle D. A. Crystal Structure of the Potassium Channel Kirbac1.1 in the Closed State. Science 2003, 300, 1922–1926. 10.1126/science.1085028. [DOI] [PubMed] [Google Scholar]
- Nilius B.; Honore E. Sensing Pressure with Ion Channels. Trends Neurosci. 2012, 35, 477–486. 10.1016/j.tins.2012.04.002. [DOI] [PubMed] [Google Scholar]
- Aryal P.; Abd-Wahab F.; Bucci G.; Sansom M. S. P.; Tucker S. J. Influence of Lipids on the Hydrophobic Barrier within the Pore of the Twik-1 K2p Channel. Channels 2015, 9, 44–49. 10.4161/19336950.2014.981987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen C.; Darre L.; Oakes V.; Torella R.; Pryde D.; Domene C. Lateral Fenestrations in K+-Channels Explored Using Molecular Dynamics Simulations. Mol. Pharmaceutics 2016, 13, 2263–2273. 10.1021/acs.molpharmaceut.5b00942. [DOI] [PubMed] [Google Scholar]
- Furst O.; Mondou B.; D’Avanzo N. Phosphoinositide Regulation of Inward Rectifier Potassium (Kir) Channels. Front. Physiol. 2014, 4, 404. 10.3389/fphys.2013.00404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh B. C.; Hille B. Pip(2) Is a Necessary Cofactor for Ion Channel Function: How and Why?. Annu. Rev. Biophys. 2008, 37, 175–195. 10.1146/annurev.biophys.37.032807.125859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agwa A. J.; Henriques S. T.; Schroeder C. I. Gating Modifier Toxin Interactions with Ion Channels and Lipid Bilayers: Is the Trimolecular Complex Real?. Neuropharmacology 2017, 127, 32–45. 10.1016/j.neuropharm.2017.04.004. [DOI] [PubMed] [Google Scholar]
- Stansfeld P. J.; Hopkinson R.; Ashcroft F. M.; Sansom M. S. P. Pip2-Binding Site in Kir Channels: Definition by Multiscale Biomolecular Simulations. Biochemistry 2009, 48, 10926–10933. 10.1021/bi9013193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes C. M. B.; Zhang H. L.; Rohacs T.; Jin T. H.; Yang J.; Logothetis D. E. Alterations in Conserved Kir Channel-Pip2 Interactions Underlie Channelopathies. Neuron 2002, 34, 933–944. 10.1016/S0896-6273(02)00725-0. [DOI] [PubMed] [Google Scholar]
- Hansen S. B.; Tao X.; MacKinnon R. Structural Basis of Pip2 Activation of the Classical Inward Rectifier K+ Channel Kir2.2. Nature 2011, 477, 495–498. 10.1038/nature10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M. R.; Stansfeld P. J.; Tucker S. J.; Sansom M. S. P. Simulation-Based Prediction of Phosphatidylinositol 4,5-Bisphosphate Binding to an Ion Channel. Biochemistry 2013, 52, 279–281. 10.1021/bi301350s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacin E.; Aryal P.; Glaaser I. W.; Bodhinathan K.; Tsai E.; Marsh N.; Tucker S. J.; Sansom M. S. P.; Slesinger P. A. Dynamic Role of the Tether Helix in Pip2-Dependent Gating of a G Protein-Gated Potassium Channel. J. Gen. Physiol. 2017, 149, 799–811. 10.1085/jgp.201711801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaydman M. A.; Cui J. Pip2 Regulation of Kcnq Channels: Biophysical and Molecular Mechanisms for Lipid Modulation of Voltage-Dependent Gating. Front. Physiol. 2014, 5, 195. 10.3389/fphys.2014.00195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaydman M. A.; Silva J. R.; Delaloye K.; Li Y.; Liang H.; Larsson H. P.; Shi J.; Cui J. Kv7.1 Ion Channels Require a Lipid to Couple Voltage Sensing to Pore Opening. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 13180–13185. 10.1073/pnas.1305167110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logothetis D. E.; Petrou V. I.; Adney S. K.; Mahajan R. Channelopathies Linked to Plasma Membrane Phosphoinositides. Pfluegers Arch. 2010, 460, 321–341. 10.1007/s00424-010-0828-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q.; Zhou P.; Chen Z.; Li M.; Jiang H.; Gao Z.; Yang H. Dynamic Pip2 Interactions with Voltage Sensor Elements Contribute to Kcnq2 Channel Gating. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 20093–20098. 10.1073/pnas.1312483110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Menchaca A. A.; Adney S. K.; Tang Q. Y.; Meng X. Y.; Rosenhouse-Dantsker A.; Cui M.; Logothetis D. E. Pip2 Controls Voltage-Sensor Movement and Pore Opening of Kv Channels through the S4-S5 Linker. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, E2399–E2408. 10.1073/pnas.1207901109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abderemane-Ali F.; Es-Salah-Lamoureux Z.; Delemotte L.; Kasimova M. A.; Labro A. J.; Snyders D. J.; Fedida D.; Tarek M.; Baro I.; Loussouarn G. Dual Effect of Phosphatidyl (4,5)-Bisphosphate Pip2 on Shaker K+ Channels. J. Biol. Chem. 2012, 287, 36158–36167. 10.1074/jbc.M112.382085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasimova M. A.; Zaydman M. A.; Cui J.; Tarek M. Pip(2)-Dependent Coupling Is Prominent in Kv7.1 Due to Weakened Interactions between S4-S5 and S6. Sci. Rep. 2015, 5, 7474. 10.1038/srep07474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L.; Zhang Q.; Qiu Y.; Li Z.; Chen Z.; Jiang H.; Li Y.; Yang H. Migration of Pip2 Lipids on Voltage-Gated Potassium Channel Surface Influences Channel Deactivation. Sci. Rep. 2015, 5, 15079. 10.1038/srep15079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elinder F.; Liin S. I. Actions and Mechanisms of Polyunsaturated Fatty Acids on Voltage-Gated Ion Channels. Front. Physiol. 2017, 8, 43. 10.3389/fphys.2017.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Avanzo N.; Hyrc K.; Enkvetchakul D.; Covey D. F.; Nichols C. G. Enantioselective Protein-Sterol Interactions Mediate Regulation of Both Prokaryotic and Eukaryotic Inward Rectifier K+ Channels by Cholesterol. PLoS One 2011, 6, 7. 10.1371/journal.pone.0019393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenhouse-Dantsker A.; Noskov S.; Durdagi S.; Logothetis D. E.; Levitan I. Identification of Novel Cholesterol-Binding Regions in Kir2 Channels. J. Biol. Chem. 2013, 288, 31154–31164. 10.1074/jbc.M113.496117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furst O.; Nichols C. G.; Lamoureux G.; D’Avanzo N. Identification of a Cholesterol-Binding Pocket in Inward Rectifier K(+) (Kir) Channels. Biophys. J. 2014, 107, 2786–2796. 10.1016/j.bpj.2014.10.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukiya A. N.; Durdagi S.; Noskov S.; Rosenhouse-Dantsker A. Cholesterol up-Regulates Neuronal G Protein-Gated Inwardly Rectifying Potassium (Girk) Channel Activity in the Hippocampus. J. Biol. Chem. 2017, 292, 6135–6147. 10.1074/jbc.M116.753350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbera N.; Ayee M. A. A.; Akpa B. S.; Levitan I. Molecular Dynamics Simulations of Kir2.2 Interactions with an Ensemble of Cholesterol Molecules. Biophys. J. 2018, 115, 1264–1280. 10.1016/j.bpj.2018.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribkoff V. K.; Starrett J. E.; Dworetzky S. I. Maxi-K Potassium Channels: Form, Function, and Modulation of a Class of Endogenous Regulators of Intracellular Calcium. Neuroscientist 2001, 7, 166–177. 10.1177/107385840100700211. [DOI] [PubMed] [Google Scholar]
- Morris C. E.; Juranka P. F. In Mechanosensitive Ion Channels, Pt B; Hamill O. P., Ed.; Elsevier Academic Press Inc: San Diego, 2007; Vol. 59, pp 297–338. [Google Scholar]
- McKay M. C.; Worley J. F. Linoleic Acid Both Enhances Activation and Blocks Kv1.5 and Kv2.1 Channels by Two Separate Mechanisms. Am. J. Physiol. Cell Physiol. 2001, 281, C1277–C1284. 10.1152/ajpcell.2001.281.4.C1277. [DOI] [PubMed] [Google Scholar]
- Yazdi S.; Stein M.; Elinder F.; Andersson M.; Lindahl E. The Molecular Basis of Polyunsaturated Fatty Acid Interactions with the Shaker Voltage-Gated Potassium Channel. PLoS Comput. Biol. 2016, 12, e1004704 10.1371/journal.pcbi.1004704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liin S. I.; Silvera Ejneby M.; Barro-Soria R.; Skarsfeldt M. A.; Larsson J. E.; Starck Harlin F.; Parkkari T.; Bentzen B. H.; Schmitt N.; Larsson H. P.; et al. Polyunsaturated Fatty Acid Analogs Act Antiarrhythmically on the Cardiac Iks Channel. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 5714–5719. 10.1073/pnas.1503488112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakany F.; Pap P.; Papp F.; Kovacs T.; Nagy P.; Peter M.; Szente L.; Panyi G.; Varga Z. Determining the Target of Membrane Sterols on Voltage-Gated Potassium Channels. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2019, 1864, 312–325. 10.1016/j.bbalip.2018.12.006. [DOI] [PubMed] [Google Scholar]
- Bagneris C.; Decaen P. G.; Hall B. A.; Naylor C. E.; Clapham D. E.; Kay C. W.; Wallace B. A. Role of the C-Terminal Domain in the Structure and Function of Tetrameric Sodium Channels. Nat. Commun. 2013, 4, 2465. 10.1038/ncomms3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCusker E. C.; Bagneris C.; Naylor C. E.; Cole A. R.; D’Avanzo N.; Nichols C. G.; Wallace B. A. Structure of a Bacterial Voltage-Gated Sodium Channel Pore Reveals Mechanisms of Opening and Closing. Nat. Commun. 2012, 3, 1102. 10.1038/ncomms2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payandeh J.; Gamal El-Din T. M.; Scheuer T.; Zheng N.; Catterall W. A. Crystal Structure of a Voltage-Gated Sodium Channel in Two Potentially Inactivated States. Nature 2012, 486, 135–139. 10.1038/nature11077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaya D.; Findeisen F.; Abderemane-Ali F.; Arrigoni C.; Wong S.; Nurva S. R.; Loussouarn G.; Minor D. L. Structure of a Prokaryotic Sodium Channel Pore Reveals Essential Gating Elements and an Outer Ion Binding Site Common to Eukaryotic Channels. J. Mol. Biol. 2014, 426, 467–483. 10.1016/j.jmb.2013.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti N.; Ing C.; Payandeh J.; Zheng N.; Catterall W. A.; Pomes R. Catalysis of Na+ Permeation in the Bacterial Sodium Channel Na(V)Ab. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 11331–11336. 10.1073/pnas.1309452110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenaeus M. J.; Gamal El-Din T. M.; Ing C.; Ramanadane K.; Pomes R.; Zheng N.; Catterall W. A. Structures of Closed and Open States of a Voltage-Gated Sodium Channel. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, E3051–E3060. 10.1073/pnas.1700761114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D.; Gamal El-Din T. M.; Ing C.; Lu P.; Pomes R.; Zheng N.; Catterall W. A. Structural Basis for Gating Pore Current in Periodic Paralysis. Nature 2018, 557, 590–594. 10.1038/s41586-018-0120-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raju S. G.; Barber A. F.; LeBard D. N.; Klein M. L.; Carnevale V. Exploring Volatile General Anesthetic Binding to a Closed Membrane-Bound Bacterial Voltage-Gated Sodium Channel Via Computation. PLoS Comput. Biol. 2013, 9, e1003090 10.1371/journal.pcbi.1003090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin L. J.; Corry B. Locating the Route of Entry and Binding Sites of Benzocaine and Phenytoin in a Bacterial Voltage Gated Sodium Channel. PLoS Comput. Biol. 2014, 10, e1003688 10.1371/journal.pcbi.1003688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boiteux C.; Vorobyov I.; French R. J.; French C.; Yarov-Yarovoy V.; Allen T. W. Local Anesthetic and Antiepileptic Drug Access and Binding to a Bacterial Voltage-Gated Sodium Channel. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 13057–13062. 10.1073/pnas.1408710111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczmarski J. A.; Corry B. Investigating the Size and Dynamics of Voltage-Gated Sodium Channel Fenestrations a Molecular Dynamics Study. Channels 2014, 8, 264–277. 10.4161/chan.28136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulmschneider M. B.; Bagneris C.; McCusker E. C.; Decaen P. G.; Delling M.; Clapham D. E.; Ulmschneider J. P.; Wallace B. A. Molecular Dynamics of Ion Transport through the Open Conformation of a Bacterial Voltage-Gated Sodium Channel. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 6364–6369. 10.1073/pnas.1214667110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suwattanasophon C.; Wolschann P.; Faller R. Molecular Dynamics Simulations on the Interaction of the Transmembrane Na(V)Ab Channel with Cholesterol and Lipids in the Membrane. J. Biomol. Struct. Dyn. 2016, 34, 318–326. 10.1080/07391102.2015.1030691. [DOI] [PubMed] [Google Scholar]
- Thompson A. J.; Lester H. A.; Lummis S. C. R. The Structural Basis of Function in Cys-Loop Receptors. Q. Rev. Biophys. 2010, 43, 449–499. 10.1017/S0033583510000168. [DOI] [PubMed] [Google Scholar]
- Unwin N. Refined Structure of the Nicotinic Acetylcholine Receptor at 4 Angstrom Resolution. J. Mol. Biol. 2005, 346, 967–989. 10.1016/j.jmb.2004.12.031. [DOI] [PubMed] [Google Scholar]
- Mansoor S. E.; Lu W.; Oosterheert W.; Shekhar M.; Tajkhorshid E.; Gouaux E. X-Ray Structures Define Human P2x(3) Receptor Gating Cycle and Antagonist Action. Nature 2016, 538, 66–71. 10.1038/nature19367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertozzi C.; Zimmermann I.; Engeler S.; Hilf R. J.; Dutzler R. Signal Transduction at the Domain Interface of Prokaryotic Pentameric Ligand-Gated Ion Channels. PLoS Biol. 2016, 14, e1002393 10.1371/journal.pbio.1002393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velisetty P.; Chalamalasetti S. V.; Chakrapani S. Structural Basis for Allosteric Coupling at the Membrane-Protein Interface in Gloeobacter Violaceus Ligand-Gated Ion Channel (Glic). J. Biol. Chem. 2014, 289, 3013–3025. 10.1074/jbc.M113.523050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibbs R. E.; Gouaux E. Principles of Activation and Permeation in an Anion-Selective Cys-Loop Receptor. Nature 2011, 474, 54–U80. 10.1038/nature10139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrantes F. J. Cholesterol Effects on Nicotinic Acetylcholine Receptor. J. Neurochem. 2007, 103, 72–80. 10.1111/j.1471-4159.2007.04719.x. [DOI] [PubMed] [Google Scholar]
- Hamouda A. K.; Chiara D. C.; Sauls D.; Cohen J. B.; Blanton M. P. Cholesterol Interacts with Transmembrane Alpha-Helices M1, M3, and M4 of the Torpedo Nicotinic Acetylcholine Receptor: Photolabeling Studies Using H-3- Azicholesterol. Biochemistry 2006, 45, 976–986. 10.1021/bi051978h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brannigan G.; Henin J.; Law R.; Eckenhoff R.; Klein M. L. Embedded Cholesterol in the Nicotinic Acetylcholine Receptor. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 14418–14423. 10.1073/pnas.0803029105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng M. H.; Xu Y.; Tang P. Anionic Lipid and Cholesterol Interactions with Alpha 4 Beta 2 Nachr: Insights from Md Simulations. J. Phys. Chem. B 2009, 113, 6964–6970. 10.1021/jp900714b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoluk O.; Bromstrup T.; Bertaccini E. J.; Trudell J. R.; Lindahl E. Stabilization of the Glucl Ligand-Gated Ion Channel in the Presence and Absence of Ivermectin. Biophys. J. 2013, 105, 640–647. 10.1016/j.bpj.2013.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henin J.; Salari R.; Murlidaran S.; Brannigan G. A Predicted Binding Site for Cholesterol on the Gaba(a) Receptor. Biophys. J. 2014, 106, 1938–1949. 10.1016/j.bpj.2014.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surprenant A.; North R. A. In Annu. Rev. Physiol.; Annual Reviews: Palo Alto, 2009; Vol. 71, pp 333–359. [DOI] [PubMed] [Google Scholar]
- North R. A. Molecular Physiology of P2x Receptors. Physiol. Rev. 2002, 82, 1013–1067. 10.1152/physrev.00015.2002. [DOI] [PubMed] [Google Scholar]
- Roberts J. A.; Vial C.; Digby H. R.; Agboh K. C.; Wen H.; Atterbury-Thomas A.; Evans R. J. Molecular Properties of P2x Receptors. Pfluegers Arch. 2006, 452, 486–500. 10.1007/s00424-006-0073-6. [DOI] [PubMed] [Google Scholar]
- Kawate T.; Michel J. C.; Birdsong W. T.; Gouaux E. Crystal Structure of the Atp-Gated P2x(4) Ion Channel in the Closed State. Nature 2009, 460, 592–598. 10.1038/nature08198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori M.; Gouaux E. Molecular Mechanism of Atp Binding and Ion Channel Activation in P2x Receptors. Nature 2012, 485, 207–U291. 10.1038/nature11010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harkat M.; Peverini L.; Cerdan A. H.; Dunning K.; Beudez J.; Martz A.; Calimet N.; Specht A.; Cecchini M.; Chataigneau T.; et al. On the Permeation of Large Organic Cations through the Pore of Atp-Gated P2x Receptors. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, E3786–E3795. 10.1073/pnas.1701379114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothwell S. W.; Stansfeld P. J.; Bragg L.; Verkhratsky A.; North R. A. Direct Gating of Atp-Activated Ion Channels (P2 × 2 Receptors) by Lipophilic Attachment at the Outer End of the Second Transmembrane Domain. J. Biol. Chem. 2014, 289, 618–626. 10.1074/jbc.M113.529099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheurer M.; Rodenkirch P.; Siggel M.; Bernardi R. C.; Schulten K.; Tajkhorshid E.; Rudack T. Pycontact: Rapid, Customizable, and Visual Analysis of Noncovalent Interactions in Md Simulations. Biophys. J. 2018, 114, 577–583. 10.1016/j.bpj.2017.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson L. E.; Shridar M.; Smith P.; Murrell-Lagnado R. D. Plasma Membrane Cholesterol as a Regulator of Human and Rodent P2X7 Receptor Activation and Sensitization. J. Biol. Chem. 2014, 289, 31983–31994. 10.1074/jbc.M114.574699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vial C.; Fung C. Y.; Goodall A. H.; Mahaut-Smith M. P.; Evans R. J. Differential Sensitivity of Human Platelet P2 × 1 and P2y1 Receptors to Disruption of Lipid Rafts. Biochem. Biophys. Res. Commun. 2006, 343, 415–419. 10.1016/j.bbrc.2006.02.174. [DOI] [PubMed] [Google Scholar]
- Liu M.; Huang W. L.; Wu D. S.; Priestley J. V. Trpv1, but Not P2x(3), Requires Cholesterol for Its Function and Membrane Expression in Rat Nociceptors. Eur. J. Neurosci. 2006, 24, 1–6. 10.1111/j.1460-9568.2006.04889.x. [DOI] [PubMed] [Google Scholar]
- Basbaum A. I.; Bautista D. M.; Scherrer G.; Julius D. Cellular and Molecular Mechanisms of Pain. Cell 2009, 139, 267–284. 10.1016/j.cell.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julius D. Trp Channels and Pain. Annu. Rev. Cell Dev. Biol. 2013, 29, 355–384. 10.1146/annurev-cellbio-101011-155833. [DOI] [PubMed] [Google Scholar]
- Liao M. F.; Cao E. H.; Julius D.; Cheng Y. F. Structure of the Trpv1 Ion Channel Determined by Electron Cryo-Microscopy. Nature 2013, 504, 107–112. 10.1038/nature12822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poblete H.; Oyarzun I.; Olivero P.; Comer J.; Zuniga M.; Sepulveda R. V.; Baez-Nieto D.; Leon C. G.; Gonzalez-Nilo F.; Latorre R. Molecular Determinants of Phosphatidylinositol 4,5-Bisphosphate (Pi(4,5)P-2) Binding to Transient Receptor Potential V1 (Trpv1) Channels. J. Biol. Chem. 2015, 290, 2086–2098. 10.1074/jbc.M114.613620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi N.; Hamada-Nakahara S.; Itoh Y.; Takemura K.; Shimada A.; Ueda Y.; Kitamata M.; Matsuoka R.; Hanawa-Suetsugu K.; Senju Y.; et al. Trpv4 Channel Activity Is Modulated by Direct Interaction of the Ankyrin Domain to Pi(4,5)P(2). Nat. Commun. 2014, 5, 4994. 10.1038/ncomms5994. [DOI] [PubMed] [Google Scholar]
- Melnick C.; Kaviany M. Thermal Actuation in Trpv1: Role of Embedded Lipids and Intracellular Domains. J. Theor. Biol. 2018, 444, 38–49. 10.1016/j.jtbi.2018.02.004. [DOI] [PubMed] [Google Scholar]
- Zimova L.; Sinica V.; Kadkova A.; Vyklicka L.; Zima V.; Barvik I.; Vlachova V. Intracellular Cavity of Sensor Domain Controls Allosteric Gating of Trpa1 Channel. Sci. Signaling 2018, 11, eaan8621 10.1126/scisignal.aan8621. [DOI] [PubMed] [Google Scholar]
- Huynh K. W.; Cohen M. R.; Jiang J.; Samanta A.; Lodowski D. T.; Zhou Z. H.; Moiseenkova-Bell V. Y. Structure of the Full-Length Trpv2 Channel by Cryo-Em. Nat. Commun. 2016, 7, 11130. 10.1038/ncomms11130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubcevic L.; Herzik M. A. Jr.; Chung B. C.; Liu Z.; Lander G. C.; Lee S. Y. Cryo-Electron Microscopy Structure of the Trpv2 Ion Channel. Nat. Struct. Mol. Biol. 2016, 23, 180–186. 10.1038/nsmb.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picazo-Juarez G.; Romero-Suarez S.; Nieto-Posadas A.; Llorente I.; Jara-Oseguera A.; Briggs M.; McIntosh T. J.; Simon S. A.; Ladron-de-Guevara E.; Islas L. D.; et al. Identification of a Binding Motif in the S5 Helix That Confers Cholesterol Sensitivity to the Trpv1 Ion Channel. J. Biol. Chem. 2011, 286, 24966–24976. 10.1074/jbc.M111.237537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha S.; Ghosh A.; Tiwari N.; Kumar A.; Kumar A.; Goswami C. Preferential Selection of Arginine at the Lipid-Water-Interface of Trpv1 During Vertebrate Evolution Correlates with Its Snorkeling Behaviour and Cholesterol Interaction. Sci. Rep. 2017, 7, 16808. 10.1038/s41598-017-16780-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox C. D.; Bavi N.; Martinac B. Bacterial Mechanosensors. Annu. Rev. Physiol. 2018, 80, 71–93. 10.1146/annurev-physiol-021317-121351. [DOI] [PubMed] [Google Scholar]
- Rasmussen T. How Do Mechanosensitive Channels Sense Membrane Tension?. Biochem. Soc. Trans. 2016, 44, 1019–1025. 10.1042/BST20160018. [DOI] [PubMed] [Google Scholar]
- Zhang X. J. C.; Liu Z. F.; Li J. From Membrane Tension to Channel Gating: A Principal Energy Transfer Mechanism for Mechanosensitive Channels. Protein Sci. 2016, 25, 1954–1964. 10.1002/pro.3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pliotas C.; Dahl A. C.; Rasmussen T.; Mahendran K. R.; Smith T. K.; Marius P.; Gault J.; Banda T.; Rasmussen A.; Miller S.; et al. The Role of Lipids in Mechanosensation. Nat. Struct. Mol. Biol. 2015, 22, 991–998. 10.1038/nsmb.3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbacher S.; Bass R.; Strop P.; Rees D. C. Structures of the Prokaryotic Mechanosensitive Channels Mscl and Mscs. Curr. Top. Membr. 2007, 58, 1–24. 10.1016/S1063-5823(06)58001-9. [DOI] [Google Scholar]
- Sotomayor M.; Schulten K. Molecular Dynamics Study of Gating in the Mechanosensitive Channel of Small Conductance Mscs. Biophys. J. 2004, 87, 3050–3065. 10.1529/biophysj.104.046045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips R.; Ursell T.; Wiggins P.; Sens P. Emerging Roles for Lipids in Shaping Membrane-Protein Function. Nature 2009, 459, 379–385. 10.1038/nature08147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bavi O.; Cox C. D.; Vossoughi M.; Naghdabadi R.; Jamali Y.; Martinac B. Influence of Global and Local Membrane Curvature on Mechanosensitive Ion Channels: A Finite Element Approach. Membranes 2016, 6, 14. 10.3390/membranes6010014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deplazes E.; Louhivuori M.; Jayatilaka D.; Marrink S. J.; Corry B. Structural Investigation of Mscl Gating Using Experimental Data and Coarse Grained Md Simulations. PLoS Comput. Biol. 2012, 8, e1002683 10.1371/journal.pcbi.1002683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louhivuori M.; Risselada H. J.; van der Giessen E.; Marrink S. J. Release of Content through Mechano-Sensitive Gates in Pressurized Liposomes. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 19856–19860. 10.1073/pnas.1001316107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bavi N.; Cortes D. M.; Cox C. D.; Rohde P. R.; Liu W.; Deitmer J. W.; Bavi O.; Strop P.; Hill A. P.; Rees D.; et al. The Role of Mscl Amphipathic N Terminus Indicates a Blueprint for Bilayer-Mediated Gating of Mechanosensitive Channels. Nat. Commun. 2016, 7, 11984. 10.1038/ncomms11984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bavi N.; Cox C. D.; Perozo E.; Martinac B. Toward a Structural Blueprint for Bilayer-Mediated Channel Mechanosensitivity. Channels 2017, 11, 91–93. 10.1080/19336950.2016.1224624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo M. N.; Arnarez C.; Sikkema H.; Kumar N.; Walko M.; Berendsen H. J.; Kocer A.; Marrink S. J.; Ingolfsson H. I. High-Throughput Simulations Reveal Membrane-Mediated Effects of Alcohols on Mscl Gating. J. Am. Chem. Soc. 2017, 139, 2664–2671. 10.1021/jacs.6b11091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee N.; Jose M. D.; Birkner J. P.; Walko M.; Ingolfsson H. I.; Dimitrova A.; Arnarez C.; Marrink S. J.; Kocer A. The Activation Mode of the Mechanosensitive Ion Channel, Mscl, by Lysophosphatidylcholine Differs from Tension-Induced Gating. FASEB J. 2014, 28, 4292–4302. 10.1096/fj.14-251579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ollila O. H. S.; Louhivuori M.; Marrink S. J.; Vattulainen I. Protein Shape Change Has a Major Effect on the Gating Energy of a Mechanosensitive Channel. Biophys. J. 2011, 100, 1651–1659. 10.1016/j.bpj.2011.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letts J. A.; Sazanov L. A. Clarifying the Supercomplex: The Higher-Order Organization of the Mitochondrial Electron Transport Chain. Nat. Struct. Mol. Biol. 2017, 24, 800–808. 10.1038/nsmb.3460. [DOI] [PubMed] [Google Scholar]
- Enriquez J. A. Supramolecular Organization of Respiratory Complexes. Annu. Rev. Physiol. 2016, 78, 533–561. 10.1146/annurev-physiol-021115-105031. [DOI] [PubMed] [Google Scholar]
- Chaban Y.; Boekema E. J.; Dudkina N. V. Structures of Mitochondrial Oxidative Phosphorylation Supercomplexes and Mechanisms for Their Stabilisation. Biochim. Biophys. Acta, Bioenerg. 2014, 1837, 418–426. 10.1016/j.bbabio.2013.10.004. [DOI] [PubMed] [Google Scholar]
- Chicco A. J.; Sparagna G. C. Role of Cardiolipin Alterations in Mitochondrial Dysfunction and Disease. Am. J. Physiol. Cell Physiol. 2007, 292, C33–44. 10.1152/ajpcell.00243.2006. [DOI] [PubMed] [Google Scholar]
- Sullivan E. M.; Pennington E. R.; Green W. D.; Beck M. A.; Brown D. A.; Shaikh S. R. Mechanisms by Which Dietary Fatty Acids Regulate Mitochondrial Structure-Function in Health and Disease. Adv. Nutr. 2018, 9, 247–262. 10.1093/advances/nmy007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlame M.; Ren M. Barth Syndrome, a Human Disorder of Cardiolipin Metabolism. FEBS Lett. 2006, 580, 5450–5455. 10.1016/j.febslet.2006.07.022. [DOI] [PubMed] [Google Scholar]
- Mileykovskaya E.; Dowhan W. Cardiolipin-Dependent Formation of Mitochondrial Respiratory Supercomplexes. Chem. Phys. Lipids 2014, 179, 42–48. 10.1016/j.chemphyslip.2013.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer K.; Gohil V.; Stuart R. A.; Hunte C.; Brandt U.; Greenberg M. L.; Schagger H. Cardiolipin Stabilizes Respiratory Chain Supercomplexes. J. Biol. Chem. 2003, 278, 52873–52880. 10.1074/jbc.M308366200. [DOI] [PubMed] [Google Scholar]
- Arias-Cartin R.; Grimaldi S.; Arnoux P.; Guigliarelli B.; Magalon A. Cardiolipin Binding in Bacterial Respiratory Complexes: Structural and Functional Implications. Biochim. Biophys. Acta, Bioenerg. 2012, 1817, 1937–1949. 10.1016/j.bbabio.2012.04.005. [DOI] [PubMed] [Google Scholar]
- Zhang M.; Mileykovskaya E.; Dowhan W. Gluing the Respiratory Chain Together. Cardiolipin Is Required for Supercomplex Formation in the Inner Mitochondrial Membrane. J. Biol. Chem. 2002, 277, 43553–43556. 10.1074/jbc.C200551200. [DOI] [PubMed] [Google Scholar]
- Planas-Iglesias J.; Dwarakanath H.; Mohammadyani D.; Yanamala N.; Kagan V. E.; Klein-Seetharaman J. Cardiolipin Interactions with Proteins. Biophys. J. 2015, 109, 1282–1294. 10.1016/j.bpj.2015.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire J. J.; Tyurina Y. Y.; Mohammadyani D.; Kapralov A. A.; Anthonymuthu T. S.; Qu F.; Amoscato A. A.; Sparvero L. J.; Tyurin V. A.; Planas-Iglesias J.; et al. Known Unknowns of Cardiolipin Signaling: The Best Is yet to Come. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2017, 1862, 8–24. 10.1016/j.bbalip.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikon N.; Ryan R. O. Cardiolipin and Mitochondrial Cristae Organization. Biochim. Biophys. Acta, Biomembr. 2017, 1859, 1156–1163. 10.1016/j.bbamem.2017.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnarez C.; Mazat J. P.; Elezgaray J.; Marrink S. J.; Periole X. Evidence for Cardiolipin Binding Sites on the Membrane-Exposed Surface of the Cytochrome Bc1. J. Am. Chem. Soc. 2013, 135, 3112–3120. 10.1021/ja310577u. [DOI] [PubMed] [Google Scholar]
- Arnarez C.; Marrink S. J.; Periole X. Identification of Cardiolipin Binding Sites on Cytochrome C Oxidase at the Entrance of Proton Channels. Sci. Rep. 2013, 3, 1263. 10.1038/srep01263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan A. L.; Robinson A. J.; Walker J. E. Cardiolipin Binds Selectively but Transiently to Conserved Lysine Residues in the Rotor of Metazoan Atp Synthases. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 8687–8692. 10.1073/pnas.1608396113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poyry S.; Cramariuc O.; Postila P. A.; Kaszuba K.; Sarewicz M.; Osyczka A.; Vattulainen I.; Rog T. Atomistic Simulations Indicate Cardiolipin to Have an Integral Role in the Structure of the Cytochrome Bc(1) Complex. Biochim. Biophys. Acta, Bioenerg. 2013, 1827, 769–778. 10.1016/j.bbabio.2013.03.005. [DOI] [PubMed] [Google Scholar]
- Sharma V.; Ala-Vannesluoma P.; Vattulainen I.; Wikstrom M.; Rog T. Role of Subunit Iii and Its Lipids in the Molecular Mechanism of Cytochrome C Oxidase. Biochim. Biophys. Acta, Bioenerg. 2015, 1847, 690–697. 10.1016/j.bbabio.2015.04.007. [DOI] [PubMed] [Google Scholar]
- Hedger G.; Rouse S. L.; Domanski J.; Chavent M.; Koldso H.; Sansom M. S. Lipid-Loving Ants: Molecular Simulations of Cardiolipin Interactions and the Organization of the Adenine Nucleotide Translocase in Model Mitochondrial Membranes. Biochemistry 2016, 55, 6238–6249. 10.1021/acs.biochem.6b00751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan A. L.; Ruprecht J. J.; Kunji E. R. S.; Robinson A. J. Cardiolipin Dynamics and Binding to Conserved Residues in the Mitochondrial Adp/Atp Carrier. Biochim. Biophys. Acta, Biomembr. 2018, 1860, 1035–1045. 10.1016/j.bbamem.2018.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnarez C.; Marrink S. J.; Periole X. Molecular Mechanism of Cardiolipin-Mediated Assembly of Respiratory Chain Supercomplexes. Chem. Sci. 2016, 7, 4435–4443. 10.1039/C5SC04664E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra K.; Modak A.; Nangia S.; Daman T. H.; Gunsel U.; Robinson V. L.; Mokranjac D.; May E. R.; Alder N. N. Cardiolipin Mediates Membrane and Channel Interactions of the Mitochondrial Tim23 Protein Import Complex Receptor Tim50. Science Advances 2017, 3, e1700532 10.1126/sciadv.1700532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees D. C.; Johnson E.; Lewinson O. Abc Transporters: The Power to Change. Nat. Rev. Mol. Cell Biol. 2009, 10, 218–227. 10.1038/nrm2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locher K. P. Mechanistic Diversity in Atp-Binding Cassette (Abc) Transporters. Nat. Struct. Mol. Biol. 2016, 23, 487–493. 10.1038/nsmb.3216. [DOI] [PubMed] [Google Scholar]
- Li J.; Jaimes K. F.; Aller S. G. Refined Structures of Mouse P-Glycoprotein. Protein Sci. 2014, 23, 34–46. 10.1002/pro.2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y.; Chen J. Molecular Structure of Human P-Glycoprotein in the Atp-Bound, Outward-Facing Conformation. Science 2018, 359, 915–919. 10.1126/science.aar7389. [DOI] [PubMed] [Google Scholar]
- Doige C. A.; Yu X.; Sharom F. J. The Effects of Lipids and Detergents on Atpase-Active P-Glycoprotein. Biochim. Biophys. Acta, Biomembr. 1993, 1146, 65–72. 10.1016/0005-2736(93)90339-2. [DOI] [PubMed] [Google Scholar]
- Urbatsch I. L.; Senior A. E. Effects of Lipids on Atpase Activity of Purified Chinese Hamster P-Glycoprotein. Arch. Biochem. Biophys. 1995, 316, 135–140. 10.1006/abbi.1995.1020. [DOI] [PubMed] [Google Scholar]
- Ahn J.; Wong J. T.; Molday R. S. The Effect of Lipid Environment and Retinoids on the Atpase Activity of Abcr, the Photoreceptor Abc Transporter Responsible for Stargardt Macular Dystrophy. J. Biol. Chem. 2000, 275, 20399–20405. 10.1074/jbc.M000555200. [DOI] [PubMed] [Google Scholar]
- dos Santos S. M.; Weber C. C.; Franke C.; Muller W. E.; Eckert G. P. Cholesterol: Coupling between Membrane Microenvironment and Abc Transporter Activity. Biochem. Biophys. Res. Commun. 2007, 354, 216–221. 10.1016/j.bbrc.2006.12.202. [DOI] [PubMed] [Google Scholar]
- Geertsma E. R.; Nik Mahmood N. A.; Schuurman-Wolters G. K.; Poolman B. Membrane Reconstitution of Abc Transporters and Assays of Translocator Function. Nat. Protoc. 2008, 3, 256–266. 10.1038/nprot.2007.519. [DOI] [PubMed] [Google Scholar]
- Zhao C.; Haase W.; Tampe R.; Abele R. Peptide Specificity and Lipid Activation of the Lysosomal Transport Complex Abcb9 (Tapl). J. Biol. Chem. 2008, 283, 17083–17091. 10.1074/jbc.M801794200. [DOI] [PubMed] [Google Scholar]
- Gustot A.; Smriti; Ruysschaert J. M.; McHaourab H.; Govaerts C. Lipid Composition Regulates the Orientation of Transmembrane Helices in Hora, an Abc Multidrug Transporter. J. Biol. Chem. 2010, 285, 14144–14151. 10.1074/jbc.M109.079673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galian C.; Manon F.; Dezi M.; Torres C.; Ebel C.; Levy D.; Jault J. M. Optimized Purification of a Heterodimeric Abc Transporter in a Highly Stable Form Amenable to 2-D Crystallization. PLoS One 2011, 6, e19677 10.1371/journal.pone.0019677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz C.; Parcej D.; Ejsing C. S.; Robenek H.; Urbatsch I. L.; Tampe R. Specific Lipids Modulate the Transporter Associated with Antigen Processing (Tap). J. Biol. Chem. 2011, 286, 13346–13356. 10.1074/jbc.M110.216416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirayama H.; Kimura Y.; Kioka N.; Matsuo M.; Ueda K. Atpase Activity of Human Abcg1 Is Stimulated by Cholesterol and Sphingomyelin. J. Lipid Res. 2013, 54, 496–502. 10.1194/jlr.M033209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telbisz A.; Ozvegy-Laczka C.; Hegedus T.; Varadi A.; Sarkadi B. Effects of the Lipid Environment, Cholesterol and Bile Acids on the Function of the Purified and Reconstituted Human Abcg2 Protein. Biochem. J. 2013, 450, 387–395. 10.1042/BJ20121485. [DOI] [PubMed] [Google Scholar]
- Bao H.; Dalal K.; Wang V.; Rouiller I.; Duong F. The Maltose Abc Transporter: Action of Membrane Lipids on the Transporter Stability, Coupling and Atpase Activity. Biochim. Biophys. Acta, Biomembr. 2013, 1828, 1723–1730. 10.1016/j.bbamem.2013.03.024. [DOI] [PubMed] [Google Scholar]
- Rice A. J.; Alvarez F. J. D.; Davidson A. L.; Pinkett H. W. Effects of Lipid Environment on the Conformational Changes of an Abc Importer. Channels 2014, 8, 327–333. 10.4161/chan.29294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollock N. L.; McDevitt C. A.; Collins R.; Niesten P. H.; Prince S.; Kerr I. D.; Ford R. C.; Callaghan R. Improving the Stability and Function of Purified Abcb1 and Abca4: The Influence of Membrane Lipids. Biochim. Biophys. Acta, Biomembr. 2014, 1838, 134–147. 10.1016/j.bbamem.2013.09.001. [DOI] [PubMed] [Google Scholar]
- Clay A. T.; Lu P.; Sharom F. J. Interaction of the P-Glycoprotein Multidrug Transporter with Sterols. Biochemistry 2015, 54, 6586–6597. 10.1021/acs.biochem.5b00904. [DOI] [PubMed] [Google Scholar]
- Noll A.; Thomas C.; Herbring V.; Zollmann T.; Bartha K.; Mehdipour A. R.; Tomasiak T. M.; Bruchert S.; Joseph B.; Abele R.; et al. Crystal Structure and Mechanistic Basis of a Functional Homolog of the Antigen Transporter Tap. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, E438–E447. 10.1073/pnas.1620009114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcoux J.; Wang S. C.; Politis A.; Reading E.; Ma J.; Biggin P. C.; Zhou M.; Tao H. C.; Zhang Q. H.; Chang G.; et al. Mass Spectrometry Reveals Synergistic Effects of Nucleotides, Lipids, and Drugs Binding to a Multidrug Resistance Efflux Pump. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 9704–9709. 10.1073/pnas.1303888110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechara C.; Noell A.; Morgner N.; Degiacomi M. T.; Tampe R.; Robinson C. V. A Subset of Annular Lipids Is Linked to the Flippase Activity of an Abc Transporter. Nat. Chem. 2015, 7, 255–262. 10.1038/nchem.2172. [DOI] [PubMed] [Google Scholar]
- Mehmood S.; Corradi V.; Choudhury H. G.; Hussain R.; Becker P.; Axford D.; Zirah S.; Rebuffat S.; Tieleman D. P.; Robinson C. V.; et al. Structural and Functional Basis for Lipid Synergy on the Activity of the Antibacterial Peptide Abc Transporter Mcjd. J. Biol. Chem. 2016, 291, 21656–21668. 10.1074/jbc.M116.732107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintre C. A.; Pike A. C. W.; Li Q.; Kim J. I.; Barr A. J.; Goubin S.; Shrestha L.; Yang J.; Berridge G.; Ross J.; et al. Structures of Abcb10, a Human Atp-Binding Cassette Transporter in Apo- and Nucleotide-Bound States. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 9710–9715. 10.1073/pnas.1217042110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi W.; Li Y.; Yoon S. H.; Ernst R. K.; Walz T.; Liao M. Structural Basis of Msba-Mediated Lipopolysaccharide Transport. Nature 2017, 549, 233–237. 10.1038/nature23649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam A.; Kung R.; Kowal J.; McLeod R. A.; Tremp N.; Broude E. V.; Roninson I. B.; Stahlberg H.; Locher K. P. Structure of a Zosuquidar and Uic2-Bound Human-Mouse Chimeric Abcb1. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E1973–E1982. 10.1073/pnas.1717044115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson Z. L.; Chen J. Atp Binding Enables Substrate Release from Multidrug Resistance Protein 1. Cell 2018, 172, 81–89. 10.1016/j.cell.2017.12.005. [DOI] [PubMed] [Google Scholar]
- Parcej D.; Tampe R. Abc Proteins in Antigen Translocation and Viral Inhibition. Nat. Chem. Biol. 2010, 6, 572–580. 10.1038/nchembio.410. [DOI] [PubMed] [Google Scholar]
- Eggensperger S.; Fisette O.; Parcej D.; Schafer L. V.; Tampe R. An Annular Lipid Belt Is Essential for Allosteric Coupling and Viral Inhibition of the Antigen Translocation Complex Tap (Transporter Associated with Antigen Processing). J. Biol. Chem. 2014, 289, 33098–33108. 10.1074/jbc.M114.592832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson K. A.; Kalkum M.; Ottesen J.; Yuzenkova J.; Chait B. T.; Landick R.; Muir T.; Severinov K.; Darst S. A. Structure of Microcin J25, a Peptide Inhibitor of Bacterial Rna Polymerase, Is a Lassoed Tail. J. Am. Chem. Soc. 2003, 125, 12475–12483. 10.1021/ja036756q. [DOI] [PubMed] [Google Scholar]
- Rosengren K. J.; Clark R. J.; Daly N. L.; Goransson U.; Jones A.; Craik D. J. Microcin J25 Has a Threaded Sidechain-to-Backbone Ring Structure and Not a Head-to-Tail Cyclized Backbone. J. Am. Chem. Soc. 2003, 125, 12464–12474. 10.1021/ja0367703. [DOI] [PubMed] [Google Scholar]
- Gu R.-X.; Corradi V.; Singh G.; Choudhury H. G.; Beis K.; Tieleman D. P. Conformational Changes of the Antibacterial Peptide Atp Binding Cassette Transporter Mcjd Revealed by Molecular Dynamics Simulations. Biochemistry 2015, 54, 5989–5998. 10.1021/acs.biochem.5b00753. [DOI] [PubMed] [Google Scholar]
- Domicevica L.; Koldso H.; Biggin P. C. Multiscale Molecular Dynamics Simulations of Lipid Interactions with P-Glycoprotein in a Complex Membrane. J. Mol. Graphics Modell. 2018, 80, 147–156. 10.1016/j.jmgm.2017.12.022. [DOI] [PubMed] [Google Scholar]
- Sharom F. J. Complex Interplay between the P-Glycoprotein Multidrug Efflux Pump and the Membrane: Its Role in Modulating Protein Function. Front. Oncol. 2014, 4, 41. 10.3389/fonc.2014.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koldso H.; Sansom M. S. Local Lipid Reorganization by a Transmembrane Protein Domain. J. Phys. Chem. Lett. 2012, 3, 3498–3502. 10.1021/jz301570w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chantemargue B.; Di Meo F.; Berka K.; Picard N.; Arnion H.; Essig M.; Marquet P.; Otyepka M.; Trouillas P. Structural Patterns of the Human Abcc4/Mrp4 Exporter in Lipid Bilayers Rationalize Clinically Observed Polymorphisms. Pharmacol. Res. 2018, 133, 318–327. 10.1016/j.phrs.2018.02.029. [DOI] [PubMed] [Google Scholar]
- Tarling E. J.; Vallim T. Q. D.; Edwards P. A. Role of Abc Transporters in Lipid Transport and Human Disease. Trends Endocrinol. Metab. 2013, 24, 342–350. 10.1016/j.tem.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz N.; Kahne D.; Silhavy T. J. Transport of Lipopolysaccharide across the Cell Envelope: The Long Road of Discovery. Nat. Rev. Microbiol. 2009, 7, 677–683. 10.1038/nrmicro2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stansfeld P. J.; Goose J. E.; Caffrey M.; Carpenter E. P.; Parker J. L.; Newstead S.; Sansom M. S. P. Memprotmd: Automated Insertion of Membrane Protein Structures into Explicit Lipid Membranes. Structure 2015, 23, 1350–1361. 10.1016/j.str.2015.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreto-Ojeda E.; Corradi V.; Gu R. X.; Tieleman D. P. Coarse-Grained Molecular Dynamics Simulations Reveal Lipid Access Pathways in P-Glycoprotein. J. Gen. Physiol. 2018, 150, 417–429. 10.1085/jgp.201711907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan L.; Aller S. G. Allosteric Role of Substrate Occupancy toward the Alignment of P-Glycoprotein Nucleotide Binding Domains. Sci. Rep. 2018, 8, 14643. 10.1038/s41598-018-32815-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann J.; Rose-Sperling D.; Hellmich U. A. Diverse Relations between Abc Transporters and Lipids: An Overview. Biochim. Biophys. Acta, Biomembr. 2017, 1859, 605–618. 10.1016/j.bbamem.2016.09.023. [DOI] [PubMed] [Google Scholar]
- Corradi V.; Gu R. X.; Vergani P.; Tieleman D. P. Structure of Transmembrane Helix 8 and Possible Membrane Defects in Cftr. Biophys. J. 2018, 114, 1751–1754. 10.1016/j.bpj.2018.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silhavy T. J.; Kahne D.; Walker S. The Bacterial Cell Envelope. Cold Spring Harbor Perspect. Biol. 2010, 2, a000414. 10.1101/cshperspect.a000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikaido H. Molecular Basis of Bacterial Outer Membrane Permeability Revisited. Microbiol. Mol. Biol. Rev. 2003, 67, 593–656. 10.1128/MMBR.67.4.593-656.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koebnik R.; Locher K. P.; Van Gelder P. Structure and Function of Bacterial Outer Membrane Proteins: Barrels in a Nutshell. Mol. Microbiol. 2000, 37, 239–253. 10.1046/j.1365-2958.2000.01983.x. [DOI] [PubMed] [Google Scholar]
- Pautsch A.; Schulz G. E. Structure of the Outer Membrane Protein a Transmembrane Domain. Nat. Struct. Biol. 1998, 5, 1013–1017. 10.1038/2983. [DOI] [PubMed] [Google Scholar]
- Efremov R. G.; Sazanov L. A. Structure of Escherichia Coli Ompf Porin from Lipidic Mesophase. J. Struct. Biol. 2012, 178, 311–318. 10.1016/j.jsb.2012.03.005. [DOI] [PubMed] [Google Scholar]
- Meng G.; Surana N. K.; St Geme J. W. 3rd; Waksman G. Structure of the Outer Membrane Translocator Domain of the Haemophilus Influenzae Hia Trimeric Autotransporter. EMBO J. 2006, 25, 2297–2304. 10.1038/sj.emboj.7601132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H.; Xiang Q.; Gu Y.; Wang Z.; Paterson N. G.; Stansfeld P. J.; He C.; Zhang Y.; Wang W.; Dong C. Structural Basis for Outer Membrane Lipopolysaccharide Insertion. Nature 2014, 511, 52–56. 10.1038/nature13464. [DOI] [PubMed] [Google Scholar]
- Noinaj N.; Kuszak A. J.; Gumbart J. C.; Lukacik P.; Chang H. S.; Easley N. C.; Lithgow T.; Buchanan S. K. Structural Insight into the Biogenesis of Beta-Barrel Membrane Proteins. Nature 2013, 501, 385–390. 10.1038/nature12521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin F.; Kindt J. T. Hydrophobic Mismatch and Lipid Sorting near Ompa in Mixed Bilayers: Atomistic and Coarse-Grained Simulations. Biophys. J. 2012, 102, 2279–2287. 10.1016/j.bpj.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goose J. E.; Sansom M. S. Reduced Lateral Mobility of Lipids and Proteins in Crowded Membranes. PLoS Comput. Biol. 2013, 9, e1003033 10.1371/journal.pcbi.1003033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavent M.; Reddy T.; Goose J.; Dahl A. C.; Stone J. E.; Jobard B.; Sansom M. S. Methodologies for the Analysis of Instantaneous Lipid Diffusion in Md Simulations of Large Membrane Systems. Faraday Discuss. 2014, 169, 455–475. 10.1039/C3FD00145H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunton T. A.; Goose J. E.; Gavaghan D. J.; Sansom M. S.; Osborne J. M. The Free Energy Landscape of Dimerization of a Membrane Protein, Nanc. PLoS Comput. Biol. 2014, 10, e1003417 10.1371/journal.pcbi.1003417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rassam P.; Copeland N. A.; Birkholz O.; Toth C.; Chavent M.; Duncan A. L.; Cross S. J.; Housden N. G.; Kaminska R.; Seger U.; et al. Supramolecular Assemblies Underpin Turnover of Outer Membrane Proteins in Bacteria. Nature 2015, 523, 333–336. 10.1038/nature14461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler P. W.; Helie J.; Duncan A.; Chavent M.; Koldso H.; Sansom M. S. P. Membrane Stiffness Is Modified by Integral Membrane Proteins. Soft Matter 2016, 12, 7792–7803. 10.1039/C6SM01186A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavent M.; Duncan A. L.; Rassam P.; Birkholz O.; Helie J.; Reddy T.; Beliaev D.; Hambly B.; Piehler J.; Kleanthous C.. et al. How Nanoscale Protein Interactions Determine the Mesoscale Dynamic Organisation of Bacterial Outer Membrane Proteins. Nat. Commun. 2018, 9. 10.1038/s41467-018-05255-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holdbrook D. A.; Huber R. G.; Piggot T. J.; Bond P. J.; Khalid S. Dynamics of Crowded Vesicles: Local and Global Responses to Membrane Composition. PLoS One 2016, 11, e0156963 10.1371/journal.pone.0156963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalid S.; Berglund N. A.; Holdbrook D. A.; Leung Y. M.; Parkin J. The Membranes of Gram-Negative Bacteria: Progress in Molecular Modelling and Simulation. Biochem. Soc. Trans. 2015, 43, 162–167. 10.1042/BST20140262. [DOI] [PubMed] [Google Scholar]
- Liko I.; Degiacomi M. T.; Lee S.; Newport T. D.; Gault J.; Reading E.; Hopper J. T. S.; Housden N. G.; White P.; Colledge M.; et al. Lipid Binding Attenuates Channel Closure of the Outer Membrane Protein Ompf. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 6691–6696. 10.1073/pnas.1721152115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lins R. D.; Straatsma T. P. Computer Simulation of the Rough Lipopolysaccharide Membrane of Pseudomonas Aeruginosa. Biophys. J. 2001, 81, 1037–1046. 10.1016/S0006-3495(01)75761-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piggot T. J.; Holdbrook D. A.; Khalid S. Electroporation of the E. Coli and S. Aureus Membranes: Molecular Dynamics Simulations of Complex Bacterial Membranes. J. Phys. Chem. B 2011, 115, 13381–13388. 10.1021/jp207013v. [DOI] [PubMed] [Google Scholar]
- Kirschner K. N.; Lins R. D.; Maass A.; Soares T. A. A Glycam-Based Force Field for Simulations of Lipopolysaccharide Membranes: Parametrization and Validation. J. Chem. Theory Comput. 2012, 8, 4719–4731. 10.1021/ct300534j. [DOI] [PubMed] [Google Scholar]
- Wu E. L.; Engstrom O.; Jo S.; Stuhlsatz D.; Yeom M. S.; Klauda J. B.; Widmalm G.; Im W. Molecular Dynamics and Nmr Spectroscopy Studies of E. Coli Lipopolysaccharide Structure and Dynamics. Biophys. J. 2013, 105, 1444–1455. 10.1016/j.bpj.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu E. L.; Fleming P. J.; Yeom M. S.; Widmalm G.; Klauda J. B.; Fleming K. G.; Im W. E. Coli Outer Membrane and Interactions with Ompla. Biophys. J. 2014, 106, 2493–2502. 10.1016/j.bpj.2014.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H.; Irudayanathan F. J.; Jiang W.; Nangia S. Simulating Gram-Negative Bacterial Outer Membrane: A Coarse Grain Model. J. Phys. Chem. B 2015, 119, 14668–14682. 10.1021/acs.jpcb.5b07122. [DOI] [PubMed] [Google Scholar]
- Murzyn K.; Pasenkiewicz-Gierula M. Structural Properties of the Water/Membrane Interface of a Bilayer Built of the E. Coli Lipid A. J. Phys. Chem. B 2015, 119, 5846–5856. 10.1021/jp5119629. [DOI] [PubMed] [Google Scholar]
- Kim S.; Patel D. S.; Park S.; Slusky J.; Klauda J. B.; Widmalm G.; Im W. Bilayer Properties of Lipid a from Various Gram-Negative Bacteria. Biophys. J. 2016, 111, 1750–1760. 10.1016/j.bpj.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu P. C.; Jefferies D.; Khalid S. Molecular Dynamics Simulations Predict the Pathways Via Which Pristine Fullerenes Penetrate Bacterial Membranes. J. Phys. Chem. B 2016, 120, 11170–11179. 10.1021/acs.jpcb.6b06615. [DOI] [PubMed] [Google Scholar]
- Van Oosten B.; Harroun T. A. A Martini Extension for Pseudomonas Aeruginosa Pao1 Lipopolysaccharide. J. Mol. Graphics Modell. 2016, 63, 125–133. 10.1016/j.jmgm.2015.12.002. [DOI] [PubMed] [Google Scholar]
- Ma H.; Khan A.; Nangia S. Dynamics of Ompf Trimer Formation in the Bacterial Outer Membrane of Escherichia Coli. Langmuir 2018, 34, 5623–5634. 10.1021/acs.langmuir.7b02653. [DOI] [PubMed] [Google Scholar]
- Ma H.; Cummins D. D.; Edelstein N. B.; Gomez J.; Khan A.; Llewellyn M. D.; Picudella T.; Willsey S. R.; Nangia S. Modeling Diversity in Structures of Bacterial Outer Membrane Lipids. J. Chem. Theory Comput. 2017, 13, 811–824. 10.1021/acs.jctc.6b00856. [DOI] [PubMed] [Google Scholar]
- Hsu P. C.; Bruininks B. M. H.; Jefferies D.; Cesar Telles de Souza P.; Lee J.; Patel D. S.; Marrink S. J.; Qi Y.; Khalid S.; Im W. Charmm-Gui Martini Maker for Modeling and Simulation of Complex Bacterial Membranes with Lipopolysaccharides. J. Comput. Chem. 2017, 38, 2354–2363. 10.1002/jcc.24895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalid S.; Piggot T. J.; Samsudin F. Atomistic and Coarse Grain Simulations of the Cell Envelope of Gram-Negative Bacteria: What Have We Learned?. Acc. Chem. Res. 2019, 52, 180–188. 10.1021/acs.accounts.8b00377. [DOI] [PubMed] [Google Scholar]
- Ferguson A. D.; Hofmann E.; Coulton J. W.; Diederichs K.; Welte W. Siderophore-Mediated Iron Transport: Crystal Structure of Fhua with Bound Lipopolysaccharide. Science 1998, 282, 2215–2220. 10.1126/science.282.5397.2215. [DOI] [PubMed] [Google Scholar]
- Ferguson A. D.; Welte W.; Hofmann E.; Lindner B.; Holst O.; Coulton J. W.; Diederichs K. A Conserved Structural Motif for Lipopolysaccharide Recognition by Procaryotic and Eucaryotic Proteins. Structure 2000, 8, 585–592. 10.1016/S0969-2126(00)00143-X. [DOI] [PubMed] [Google Scholar]
- Eren E.; Murphy M.; Goguen J.; van den Berg B. An Active Site Water Network in the Plasminogen Activator Pla from Yersinia Pestis. Structure 2010, 18, 809–818. 10.1016/j.str.2010.03.013. [DOI] [PubMed] [Google Scholar]
- Eren E.; van den Berg B. Structural Basis for Activation of an Integral Membrane Protease by Lipopolysaccharide. J. Biol. Chem. 2012, 287, 23971–23976. 10.1074/jbc.M112.376418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baboolal T. G.; Conroy M. J.; Gill K.; Ridley H.; Visudtiphole V.; Bullough P. A.; Lakey J. H. Colicin N Binds to the Periphery of Its Receptor and Translocator, Outer Membrane Protein F. Structure 2008, 16, 371–379. 10.1016/j.str.2007.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arunmanee W.; Pathania M.; Solovyova A. S.; Le Brun A. P.; Ridley H.; Basle A.; van den Berg B.; Lakey J. H. Gram-Negative Trimeric Porins Have Specific Lps Binding Sites That Are Essential for Porin Biogenesis. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, E5034–E5043. 10.1073/pnas.1602382113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolla J. M.; Lazdunski C.; Pages J. M. The Assembly of the Major Outer-Membrane Protein Ompf of Escherichia-Coli Depends on Lipid-Synthesis. EMBO J. 1988, 7, 3595–3599. 10.1002/j.1460-2075.1988.tb03237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rachel R.; Engel A. M.; Huber R.; Stetter K. O.; Baumeister W. A Porin-Type Protein Is the Main Constituent of the Cell-Envelope of the Ancestral Eubacterium Thermotoga-Maritima. FEBS Lett. 1990, 262, 64–68. 10.1016/0014-5793(90)80155-C. [DOI] [Google Scholar]
- de Cock H.; Tommassen J. Lipopolysaccharides and Divalent Cations Are Involved in the Formation of an Assembly-Competent Intermediate of Outer-Membrane Protein Phoe of E-Coli. EMBO J. 1996, 15, 5567–5573. 10.1002/j.1460-2075.1996.tb00941.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird M. W.; Kloser A. W.; Misra R. Assembly of Lamb and Ompf in Deep Rough Lipopolysaccharide Mutants of Escherichia-Coli K-12. J. Bacteriol. 1994, 176, 2259–2264. 10.1128/jb.176.8.2259-2264.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piggot T. J.; Holdbrook D. A.; Khalid S. Conformational Dynamics and Membrane Interactions of the E. Coli Outer Membrane Protein Feca: A Molecular Dynamics Simulation Study. Biochim. Biophys. Acta, Biomembr. 2013, 1828, 284–293. 10.1016/j.bbamem.2012.08.021. [DOI] [PubMed] [Google Scholar]
- Holdbrook D. A.; Piggot T. J.; Sansom M. S.; Khalid S. Stability and Membrane Interactions of an Autotransport Protein: Md Simulations of the Hia Translocator Domain in a Complex Membrane Environment. Biochim. Biophys. Acta, Biomembr. 2013, 1828, 715–723. 10.1016/j.bbamem.2012.09.002. [DOI] [PubMed] [Google Scholar]
- Ortiz-Suarez M. L.; Samsudin F.; Piggot T. J.; Bond P. J.; Khalid S. Full-Length Ompa: Structure, Function, and Membrane Interactions Predicted by Molecular Dynamics Simulations. Biophys. J. 2016, 111, 1692–1702. 10.1016/j.bpj.2016.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng G.; Geme J. W. S.; Waksman G. Repetitive Architecture of the Haemophilus Influenzae Hia Trimeric Autotransporter. J. Mol. Biol. 2008, 384, 824–836. 10.1016/j.jmb.2008.09.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balusek C.; Gumbart J. C. Role of the Native Outer-Membrane Environment on the Transporter Btub. Biophys. J. 2016, 111, 1409–1417. 10.1016/j.bpj.2016.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearer J.; Khalid S. Communication between the Leaflets of Asymmetric Membranes Revealed from Coarse-Grain Molecular Dynamics Simulations. Sci. Rep. 2018, 8, 1805. 10.1038/s41598-018-20227-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel D. S.; Re S.; Wu E. L.; Qi Y.; Klebba P. E.; Widmalm G.; Yeom M. S.; Sugita Y.; Im W. Dynamics and Interactions of Ompf and Lps: Influence on Pore Accessibility and Ion Permeability. Biophys. J. 2016, 110, 930–938. 10.1016/j.bpj.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.; Patel D. S.; Kucharska I.; Tamm L. K.; Im W. Refinement of Oprh-Lps Interactions by Molecular Simulations. Biophys. J. 2017, 112, 346–355. 10.1016/j.bpj.2016.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H. H.; Tang X. D.; Zhang Z. Y.; Dong C. J. Structural Insight into Lipopolysaccharide Transport from the Gram-Negative Bacterial Inner Membrane to the Outer Membrane. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2017, 1862, 1461–1467. 10.1016/j.bbalip.2017.08.003. [DOI] [PubMed] [Google Scholar]
- Gu Y. H.; Stansfeld P. J.; Zeng Y.; Dong H. H.; Wang W. J.; Dong C. J. Lipopolysaccharide Is Inserted into the Outer Membrane through an Intramembrane Hole, a Lumen Gate, and the Lateral Opening of Lptd. Structure 2015, 23, 496–504. 10.1016/j.str.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botos I.; Majdalani N.; Mayclin S. J.; McCarthy J. G.; Lundquist K.; Wojtowicz D.; Barnard T. J.; Gumbart J. C.; Buchanan S. K. Structural and Functional Characterization of the Lps Transporter Lptde from Gram-Negative Pathogens. Structure 2016, 24, 965–976. 10.1016/j.str.2016.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noinaj N.; Rollauer S. E.; Buchanan S. K. The Beta-Barrel Membrane Protein Insertase Machinery from Gram-Negative Bacteria. Curr. Opin. Struct. Biol. 2015, 31, 35–42. 10.1016/j.sbi.2015.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni D.; Wang Y.; Yang X.; Zhou H.; Hou X.; Cao B.; Lu Z.; Zhao X.; Yang K.; Huang Y. Structural and Functional Analysis of the Beta-Barrel Domain of Bama from Escherichia Coli. FASEB J. 2014, 28, 2677–2685. 10.1096/fj.13-248450. [DOI] [PubMed] [Google Scholar]
- Noinaj N.; Kuszak A. J.; Balusek C.; Gumbart J. C.; Buchanan S. K. Lateral Opening and Exit Pore Formation Are Required for Bama Function. Structure 2014, 22, 1055–1062. 10.1016/j.str.2014.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundquist K.; Bakelar J.; Noinaj N.; Gumbart J. C. C-Terminal Kink Formation Is Required for Lateral Gating in Bama. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E7942–E7949. 10.1073/pnas.1722530115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K. H.; Aulakh S.; Paetzel M. The Bacterial Outer Membrane Beta-Barrel Assembly Machinery. Protein Sci. 2012, 21, 751–768. 10.1002/pro.2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlova A.; Hwang H.; Lundquist K.; Balusek C.; Gumbart J. C. Living on the Edge: Simulations of Bacterial Outer-Membrane Proteins. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 1753–1759. 10.1016/j.bbamem.2016.01.020. [DOI] [PubMed] [Google Scholar]
- Gu Y.; Li H.; Dong H.; Zeng Y.; Zhang Z.; Paterson N. G.; Stansfeld P. J.; Wang Z.; Zhang Y.; Wang W.; et al. Structural Basis of Outer Membrane Protein Insertion by the Bam Complex. Nature 2016, 531, 64–69. 10.1038/nature17199. [DOI] [PubMed] [Google Scholar]
- Fleming P. J.; Patel D. S.; Wu E. L.; Qi Y.; Yeom M. S.; Sousa M. C.; Fleming K. G.; Im W. Bama Potra Domain Interacts with a Native Lipid Membrane Surface. Biophys. J. 2016, 110, 2698–2709. 10.1016/j.bpj.2016.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessen H. J.; Fleming P. J.; Fleming K. G.; Sodt A. J. Building Blocks of the Outer Membrane: Calculating a General Elastic Energy Model for Beta-Barrel Membrane Proteins. J. Chem. Theory Comput. 2018, 14, 4487–4497. 10.1021/acs.jctc.8b00377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang H.; Paracini N.; Parks J. M.; Lakey J. H.; Gumbart J. C. Distribution of Mechanical Stress in the Escherichia Coli Cell Envelope. Biochim. Biophys. Acta, Biomembr. 2018, 1860, 2566–2575. 10.1016/j.bbamem.2018.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montigny C.; Lyons J.; Champeil P.; Nissen P.; Lenoir G. On the Molecular Mechanism of Flippase- and Scramblase-Mediated Phospholipid Transport. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2016, 1861, 767–783. 10.1016/j.bbalip.2015.12.020. [DOI] [PubMed] [Google Scholar]
- Brunner J. D.; Schenck S.; Dutzler R. Structural Basis for Phospholipid Scrambling in the Tmem16 Family. Curr. Opin. Struct. Biol. 2016, 39, 61–70. 10.1016/j.sbi.2016.05.020. [DOI] [PubMed] [Google Scholar]
- Brunner J. D.; Lim N. K.; Schenck S.; Duerst A.; Dutzler R. X-Ray Structure of a Calcium-Activated Tmem16 Lipid Scramblase. Nature 2014, 516, 207–212. 10.1038/nature13984. [DOI] [PubMed] [Google Scholar]
- Lee B. C.; Menon A. K.; Accardi A. The Nhtmem16 Scramblase Is Also a Nonselective Ion Channel. Biophys. J. 2016, 111, 1919–1924. 10.1016/j.bpj.2016.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlock J. M.; Hartzell H. C. A Pore Idea: The Ion Conduction Pathway of Tmem16/Ano Proteins Is Composed Partly of Lipid. Pfluegers Arch. 2016, 468, 455–473. 10.1007/s00424-015-1777-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falzone M. E.; Malvezzi M.; Lee B. C.; Accardi A. Known Structures and Unknown Mechanisms of Tmem16 Scramblases and Channels. J. Gen. Physiol. 2018, 150, 933–947. 10.1085/jgp.201711957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bethel N. P.; Grabe M. Atomistic Insight into Lipid Translocation by a Tmem16 Scramblase. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 14049–14054. 10.1073/pnas.1607574113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang T.; Yu K.; Hartzell H. C.; Tajkhorshid E. Lipids and Ions Traverse the Membrane by the Same Physical Pathway in the Nhtmem16 Scramblase. eLife 2017, 6, e28671 10.7554/eLife.28671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B. C.; Khelashvili G.; Falzone M.; Menon A. K.; Weinstein H.; Accardi A. Gating Mechanism of the Extracellular Entry to the Lipid Pathway in a Tmem16 Scramblase. Nat. Commun. 2018, 9, 3251. 10.1038/s41467-018-05724-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekkers D. W. C.; Comfurius P.; Bevers E. M.; Zwaal R. F. A. Comparison between Ca2+-Induced Scrambling of Various Fluorescently Labelled Lipid Analogues in Red Blood Cells. Biochem. J. 2002, 362, 741–747. 10.1042/0264-6021:3620741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki J.; Fujii T.; Imao T.; Ishihara K.; Kuba H.; Nagata S. Calcium-Dependent Phospholipid Scramblase Activity of Tmem16 Protein Family Members. J. Biol. Chem. 2013, 288, 13305–13316. 10.1074/jbc.M113.457937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malvezzi M.; Chalat M.; Janjusevic R.; Picollo A.; Terashima H.; Menon A. K.; Accardi A.. Ca2+-Dependent Phospholipid Scrambling by a Reconstituted Tmem16 Ion Channel. Nat. Commun. 2013, 4. 10.1038/ncomms3367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe R.; Sakuragi T.; Noji H.; Nagata S. Single-Molecule Analysis of Phospholipid Scrambling by Tmem16f. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 3066–3071. 10.1073/pnas.1717956115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malvezzi M.; Andra K. K.; Pandey K.; Lee B. C.; Falzone M. E.; Brown A.; Iqbal R.; Menon A. K.; Accardi A. Out-of-the-Groove Transport of Lipids by Tmem16 and Gpcr Scramblases. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E7033–E7042. 10.1073/pnas.1806721115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulino C.; Neldner Y.; Lam A. K.; Kalienkova V.; Brunner J. D.; Schenck S.; Dutzler R. Structural Basis for Anion Conduction in the Calcium-Activated Chloride Channel Tmem16a. eLife 2017, 6, e26232 10.7554/eLife.26232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abellon-Ruiz J.; Kaptan S. S.; Basle A.; Claudi B.; Bumann D.; Kleinekathofer U.; van den Berg B. Structural Basis for Maintenance of Bacterial Outer Membrane Lipid Asymmetry. Nat. Microbiol 2017, 2, 1616–1623. 10.1038/s41564-017-0046-x. [DOI] [PubMed] [Google Scholar]
- Venken T.; Schillinger A. S.; Fuglebakk E.; Reuter N. Interactions Stabilizing the C-Terminal Helix of Human Phospholipid Scramblase 1 in Lipid Bilayers: A Computational Study. Biochim. Biophys. Acta, Biomembr. 2017, 1859, 1200–1210. 10.1016/j.bbamem.2017.03.019. [DOI] [PubMed] [Google Scholar]
- Palmgren M. G.; Nissen P. P-Type Atpases. Annu. Rev. Biophys. 2011, 40, 243–266. 10.1146/annurev.biophys.093008.131331. [DOI] [PubMed] [Google Scholar]
- Bublitz M.; Poulsen H.; Morth J. P.; Nissen P. In and out of the Cation Pumps: P-Type Atpase Structure Revisited. Curr. Opin. Struct. Biol. 2010, 20, 431–439. 10.1016/j.sbi.2010.06.007. [DOI] [PubMed] [Google Scholar]
- Bublitz M.; Musgaard M.; Poulsen H.; Thogersen L.; Olesen C.; Schiott B.; Morth J. P.; Moller J. V.; Nissen P. Ion Pathways in the Sarcoplasmic Reticulum Ca2+-Atpase. J. Biol. Chem. 2013, 288, 10759–10765. 10.1074/jbc.R112.436550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinoza-Fonseca L. M. The Ca2+-Atpase Pump Facilitates Bidirectional Proton Transport across the Sarco/Endoplasmic Reticulum. Mol. BioSyst. 2017, 13, 633–637. 10.1039/C7MB00065K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhekova H. R.; Ngo V.; da Silva M. C.; Salahub D.; Noskov S. Selective Ion Binding and Transport by Membrane Proteins - a Computational Perspective. Coord. Chem. Rev. 2017, 345, 108–136. 10.1016/j.ccr.2017.03.019. [DOI] [Google Scholar]
- Thogersen L.; Nissen P. Flexible P-Type Atpases Interacting with the Membrane. Curr. Opin. Struct. Biol. 2012, 22, 491–499. 10.1016/j.sbi.2012.05.009. [DOI] [PubMed] [Google Scholar]
- Sonntag Y.; Musgaard M.; Olesen C.; Schiott B.; Moller J. V.; Nissen P.; Thogersen L. Mutual Adaptation of a Membrane Protein and Its Lipid Bilayer During Conformational Changes. Nat. Commun. 2011, 2, 304. 10.1038/ncomms1307. [DOI] [PubMed] [Google Scholar]
- Gustavsson M.; Traaseth N. J.; Veglia G. Activating and Deactivating Roles of Lipid Bilayers on the Ca(2+)-Atpase/Phospholamban Complex. Biochemistry 2011, 50, 10367–10374. 10.1021/bi200759y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannsson A.; Keightley C. A.; Smith G. A.; Richards C. D.; Hesketh T. R.; Metcalfe J. C. The Effect of Bilayer Thickness and N-Alkanes on the Activity of the (Ca2+ + Mg2+)-Dependent Atpase of Sarcoplasmic Reticulum. J. Biol. Chem. 1981, 256, 1643–1650. [PubMed] [Google Scholar]
- Starling A. P.; East J. M.; Lee A. G. Effects of Phosphatidylcholine Fatty Acyl Chain Length on Calcium Binding and Other Functions of the (Ca(2+)-Mg2+)-Atpase. Biochemistry 1993, 32, 1593–1600. 10.1021/bi00057a025. [DOI] [PubMed] [Google Scholar]
- Lervik A.; Bresme F.; Kjelstrup S. Molecular Dynamics Simulations of the Ca2+-Pump: A Structural Analysis. Phys. Chem. Chem. Phys. 2012, 14, 3543–3553. 10.1039/c2cp23002j. [DOI] [PubMed] [Google Scholar]
- Autzen H. E.; Siuda I.; Sonntag Y.; Nissen P.; Moller J. V.; Thogersen L. Regulation of the Ca2+-Atpase by Cholesterol: A Specific or Non-Specific Effect?. Mol. Membr. Biol. 2015, 32, 75–87. 10.3109/09687688.2015.1073382. [DOI] [PubMed] [Google Scholar]
- Drachmann N. D.; Olesen C.; Moller J. V.; Guo Z.; Nissen P.; Bublitz M. Comparing Crystal Structures of Ca2+-Atpase in the Presence of Different Lipids. FEBS J. 2014, 281, 4249–4262. 10.1111/febs.12957. [DOI] [PubMed] [Google Scholar]
- Norimatsu Y.; Hasegawa K.; Shimizu N.; Toyoshima C. Protein-Phospholipid Interplay Revealed with Crystals of a Calcium Pump. Nature 2017, 545, 193–198. 10.1038/nature22357. [DOI] [PubMed] [Google Scholar]
- Sweadner K. J. An Ion-Transport Enzyme That Rocks. Nature 2017, 545, 162. 10.1038/nature22492. [DOI] [PubMed] [Google Scholar]
- Cornelius F.; Habeck M.; Kanai R.; Toyoshima C.; Karlish S. J. General and Specific Lipid-Protein Interactions in Na,K-Atpase. Biochim. Biophys. Acta, Biomembr. 2015, 1848, 1729–1743. 10.1016/j.bbamem.2015.03.012. [DOI] [PubMed] [Google Scholar]
- Morth J. P.; Pedersen B. P.; Buch-Pedersen M. J.; Andersen J. P.; Vilsen B.; Palmgren M. G.; Nissen P. A Structural Overview of the Plasma Membrane Na+,K+-Atpase and H+-Atpase Ion Pumps. Nat. Rev. Mol. Cell Biol. 2011, 12, 60–70. 10.1038/nrm3031. [DOI] [PubMed] [Google Scholar]
- Clausen M. V.; Hilbers F.; Poulsen H. The Structure and Function of the Na,K-Atpase Isoforms in Health and Disease. Front. Physiol. 2017, 8, 371. 10.3389/fphys.2017.00371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haviv H.; Cohen E.; Lifshitz Y.; Tal D. M.; Goldshleger R.; Karlish S. J. Stabilization of Na(+),K(+)-Atpase Purified from Pichia Pastoris Membranes by Specific Interactions with Lipids. Biochemistry 2007, 46, 12855–12867. 10.1021/bi701248y. [DOI] [PubMed] [Google Scholar]
- Haviv H.; Habeck M.; Kanai R.; Toyoshima C.; Karlish S. J. Neutral Phospholipids Stimulate Na,K-Atpase Activity: A Specific Lipid-Protein Interaction. J. Biol. Chem. 2013, 288, 10073–10081. 10.1074/jbc.M112.446997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habeck M.; Haviv H.; Katz A.; Kapri-Pardes E.; Ayciriex S.; Shevchenko A.; Ogawa H.; Toyoshima C.; Karlish S. J. Stimulation, Inhibition, or Stabilization of Na,K-Atpase Caused by Specific Lipid Interactions at Distinct Sites. J. Biol. Chem. 2015, 290, 4829–4842. 10.1074/jbc.M114.611384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapri-Pardes E.; Katz A.; Haviv H.; Mahmmoud Y.; Ilan M.; Khalfin-Penigel I.; Carmeli S.; Yarden O.; Karlish S. J. Stabilization of the Alpha2 Isoform of Na,K-Atpase by Mutations in a Phospholipid Binding Pocket. J. Biol. Chem. 2011, 286, 42888–42899. 10.1074/jbc.M111.293852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra N. K.; Peleg Y.; Cirri E.; Belogus T.; Lifshitz Y.; Voelker D. R.; Apell H. J.; Garty H.; Karlish S. J. Fxyd Proteins Stabilize Na,K-Atpase: Amplification of Specific Phosphatidylserine-Protein Interactions. J. Biol. Chem. 2011, 286, 9699–9712. 10.1074/jbc.M110.184234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habeck M.; Kapri-Pardes E.; Sharon M.; Karlish S. J. Specific Phospholipid Binding to Na,K-Atpase at Two Distinct Sites. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 2904–2909. 10.1073/pnas.1620799114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia A.; Pratap P. R.; Lupfert C.; Cornelius F.; Jacquemin D.; Lev B.; Allen T. W.; Clarke R. J. The Voltage-Sensitive Dye Rh421 Detects a Na+,K+-Atpase Conformational Change at the Membrane Surface. Biochim. Biophys. Acta, Biomembr. 2017, 1859, 813–823. 10.1016/j.bbamem.2017.01.022. [DOI] [PubMed] [Google Scholar]
- Jiang Q.; Garcia A.; Han M.; Cornelius F.; Apell H. J.; Khandelia H.; Clarke R. J. Electrostatic Stabilization Plays a Central Role in Autoinhibitory Regulation of the Na(+),K(+)-Atpase. Biophys. J. 2017, 112, 288–299. 10.1016/j.bpj.2016.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen K.; Garcia A.; Sani M. A.; Diaz D.; Dubey V.; Clayton D.; Dal Poggetto G.; Cornelius F.; Payne R. J.; Separovic F.; et al. Interaction of N-Terminal Peptide Analogues of the Na(+),K(+)-Atpase with Membranes. Biochim. Biophys. Acta, Biomembr. 2018, 1860, 1282–1291. 10.1016/j.bbamem.2018.03.002. [DOI] [PubMed] [Google Scholar]
- Levi V.; Rossi J. P.; Echarte M. M.; Castello P. R.; Gonzalez Flecha F. L. Thermal Stability of the Plasma Membrane Calcium Pump. Quantitative Analysis of Its Dependence on Lipid-Protein Interactions. J. Membr. Biol. 2000, 173, 215–225. 10.1007/s002320001021. [DOI] [PubMed] [Google Scholar]
- Lopreiato R.; Giacomello M.; Carafoli E. The Plasma Membrane Calcium Pump: New Ways to Look at an Old Enzyme. J. Biol. Chem. 2014, 289, 10261–10268. 10.1074/jbc.O114.555565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pignataro M. F.; Dodes-Traian M. M.; Gonzalez-Flecha F. L.; Sica M.; Mangialavori I. C.; Rossi J. P. Modulation of Plasma Membrane Ca2+-Atpase by Neutral Phospholipids: Effect of the Micelle-Vesicle Transition and the Bilayer Thickness. J. Biol. Chem. 2015, 290, 6179–6190. 10.1074/jbc.M114.585828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen J. P.; Vestergaard A. L.; Mikkelsen S. A.; Mogensen L. S.; Chalat M.; Molday R. S.. P4-Atpases as Phospholipid Flippases-Structure, Function, and Enigmas. Front. Physiol. 2016, 7. 10.3389/fphys.2016.00275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vestergaard A. L.; Coleman J. A.; Lemmin T.; Mikkelsen S. A.; Molday L. L.; Vilsen B.; Molday R. S.; Dal Peraro M.; Andersen J. P. Critical Roles of Isoleucine-364 and Adjacent Residues in a Hydrophobic Gate Control of Phospholipid Transport by the Mammalian P4-Atpase Atp8a2. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, E1334–1343. 10.1073/pnas.1321165111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourdon P.; Liu X. Y.; Skjorringe T.; Morth J. P.; Moller L. B.; Pedersen B. P.; Nissen P. Crystal Structure of a Copper-Transporting Pib-Type Atpase. Nature 2011, 475, 59–64. 10.1038/nature10191. [DOI] [PubMed] [Google Scholar]
- Autzen H. E.; Koldso H.; Stansfeld P. J.; Gourdon P.; Sansom M. S. P.; Nissen P. Interactions of a Bacterial Cu(I)-Atpase with a Complex Lipid Environment. Biochemistry 2018, 57, 4063–4073. 10.1021/acs.biochem.8b00326. [DOI] [PubMed] [Google Scholar]
- Ueno H.; Suzuki K.; Murata T. Structure and Dynamics of Rotary V1Motor. Cell. Mol. Life Sci. 2018, 75, 1789–1802. 10.1007/s00018-018-2758-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junge W.; Nelson N. Atp Synthase. Annu. Rev. Biochem. 2015, 84, 631–657. 10.1146/annurev-biochem-060614-034124. [DOI] [PubMed] [Google Scholar]
- Gruber G.; Manimekalai M. S.; Mayer F.; Muller V. Atp Synthases from Archaea: The Beauty of a Molecular Motor. Biochim. Biophys. Acta, Bioenerg. 2014, 1837, 940–952. 10.1016/j.bbabio.2014.03.004. [DOI] [PubMed] [Google Scholar]
- Srivastava A. P.; Luo M.; Zhou W.; Symersky J.; Bai D.; Chambers M. G.; Faraldo-Gomez J. D.; Liao M.; Mueller D. M. High-Resolution Cryo-Em Analysis of the Yeast Atp Synthase in a Lipid Membrane. Science 2018, 360, eaas9699 10.1126/science.aas9699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn A.; Vonck J.; Mills D. J.; Meier T.; Kuhlbrandt W. Structure, Mechanism, and Regulation of the Chloroplast Atp Synthase. Science 2018, 360, eaat4318 10.1126/science.aat4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekimoto T.; Ikeguchi M. Multiscale Molecular Dynamics Simulations of Rotary Motor Proteins. Biophys. Rev. 2018, 10, 605–615. 10.1007/s12551-017-0373-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S.; Warshel A. The Fof1 Atp Synthase: From Atomistic Three-Dimensional Structure to the Rotary-Chemical Function. Photosynth. Res. 2017, 134, 1–15. 10.1007/s11120-017-0411-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies K. M.; Anselmi C.; Wittig I.; Faraldo-Gomez J. D.; Kuhlbrandt W. Structure of the Yeast F1fo-Atp Synthase Dimer and Its Role in Shaping the Mitochondrial Cristae. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 13602–13607. 10.1073/pnas.1204593109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies K. M.; Strauss M.; Daum B.; Kief J. H.; Osiewacz H. D.; Rycovska A.; Zickermann V.; Kuhlbrandt W. Macromolecular Organization of Atp Synthase and Complex I in Whole Mitochondria. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 14121–14126. 10.1073/pnas.1103621108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss M.; Hofhaus G.; Schroder R. R.; Kuhlbrandt W. Dimer Ribbons of Atp Synthase Shape the Inner Mitochondrial Membrane. EMBO J. 2008, 27, 1154–1160. 10.1038/emboj.2008.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anselmi C.; Davies K. M.; Faraldo-Gómez J. D. Mitochondrial Atp Synthase Dimers Spontaneously Associate Due to a Long-Range Membrane-Induced Force. J. Gen. Physiol. 2018, 150, 763. 10.1085/jgp.201812033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laage S.; Tao Y.; McDermott A. E. Cardiolipin Interaction with Subunit C of Atp Synthase: Solid-State Nmr Characterization. Biochim. Biophys. Acta, Biomembr. 2015, 1848, 260–265. 10.1016/j.bbamem.2014.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acehan D.; Malhotra A.; Xu Y.; Ren M.; Stokes D. L.; Schlame M. Cardiolipin Affects the Supramolecular Organization of Atp Synthase in Mitochondria. Biophys. J. 2011, 100, 2184–2192. 10.1016/j.bpj.2011.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eble K. S.; Coleman W. B.; Hantgan R. R.; Cunningham C. C. Tightly Associated Cardiolipin in the Bovine Heart Mitochondrial Atp Synthase as Analyzed by 31p Nuclear Magnetic Resonance Spectroscopy. J. Biol. Chem. 1990, 265, 19434–19440. [PubMed] [Google Scholar]
- Santiago E.; Lopezmor N.; Segovia J. L. Correlation between Losses of Mitochondrial Atpase Activity and Cardiolipin Degradation. Biochem. Biophys. Res. Commun. 1973, 53, 439–445. 10.1016/0006-291X(73)90681-5. [DOI] [PubMed] [Google Scholar]
- Hsu P. C.; Samsudin F.; Shearer J.; Khalid S. It Is Complicated: Curvature, Diffusion, and Lipid Sorting within the Two Membranes of Escherichia Coli. J. Phys. Chem. Lett. 2017, 8, 5513–5518. 10.1021/acs.jpclett.7b02432. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Shaikh S. A.; Tajkhorshid E. Exploring Transmembrane Diffusion Pathways with Molecular Dynamics. Physiology 2010, 25, 142–154. 10.1152/physiol.00046.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stansfeld P. J.; Jefferys E. E.; Sansom M. S. Multiscale Simulations Reveal Conserved Patterns of Lipid Interactions with Aquaporins. Structure 2013, 21, 810–819. 10.1016/j.str.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonen T.; Cheng Y.; Sliz P.; Hiroaki Y.; Fujiyoshi Y.; Harrison S. C.; Walz T. Lipid-Protein Interactions in Double-Layered Two-Dimensional Aqp0 Crystals. Nature 2005, 438, 633–638. 10.1038/nature04321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aponte-Santamaria C.; Briones R.; Schenk A. D.; Walz T.; de Groot B. L. Molecular Driving Forces Defining Lipid Positions around Aquaporin-0. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 9887–9892. 10.1073/pnas.1121054109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briones R.; Aponte-Santamaria C.; de Groot B. L. Localization and Ordering of Lipids around Aquaporin-0: Protein and Lipid Mobility Effects. Front. Physiol. 2017, 8, 124. 10.3389/fphys.2017.00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. B.; Chen L. Y. In Silico Study of Aquaporin V: Effects and Affinity of the Central Pore-Occluding Lipid. Biophys. Chem. 2013, 171, 24–30. 10.1016/j.bpc.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong X.; Qin S. S.; Lu D. N.; Liu Z. Surface Tension Effects on the Phase Transition of a Dppc Bilayer with and without Protein: A Molecular Dynamics Simulation. Phys. Chem. Chem. Phys. 2014, 16, 8434–8440. 10.1039/C3CP55524K. [DOI] [PubMed] [Google Scholar]
- O’Connor J. W.; Klauda J. B. Lipid Membranes with a Majority of Cholesterol: Applications to the Ocular Lens and Aquaporin 0. J. Phys. Chem. B 2011, 115, 6455–6464. 10.1021/jp108650u. [DOI] [PubMed] [Google Scholar]
- Gu R. X.; Ingolfsson H. I.; de Vries A. H.; Marrink S. J.; Tieleman D. P. Ganglioside-Lipid and Ganglioside-Protein Interactions Revealed by Coarse-Grained and Atomistic Molecular Dynamics Simulations. J. Phys. Chem. B 2017, 121, 3262–3275. 10.1021/acs.jpcb.6b07142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon M. A.; Schlessinger J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2010, 141, 1117–1134. 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broughton S. E.; Hercus T. R.; Lopez A. F.; Parker M. W. Cytokine Receptor Activation at the Cell Surface. Curr. Opin. Struct. Biol. 2012, 22, 350–359. 10.1016/j.sbi.2012.03.015. [DOI] [PubMed] [Google Scholar]
- Bocharov E. V.; Mineev K. S.; Pavlov K. V.; Akimov S. A.; Kuznetsov A. S.; Efremov R. G.; Arseniev A. S. Helix-Helix Interactions in Membrane Domains of Bitopic Proteins: Specificity and Role of Lipid Environment. Biochim. Biophys. Acta, Biomembr. 2017, 1859, 561–576. 10.1016/j.bbamem.2016.10.024. [DOI] [PubMed] [Google Scholar]
- Pawar A. B.; Sengupta D. Effect of Membrane Composition on Receptor Association: Implications of Cancer Lipidomics on Erbb Receptors. J. Membr. Biol. 2018, 251, 359–368. 10.1007/s00232-018-0015-1. [DOI] [PubMed] [Google Scholar]
- Polyansky A. A.; Volynsky P. E.; Efremov R. G. Multistate Organization of Transmembrane Helical Protein Dimers Governed by the Host Membrane. J. Am. Chem. Soc. 2012, 134, 14390–14400. 10.1021/ja303483k. [DOI] [PubMed] [Google Scholar]
- Lelimousin M.; Limongelli V.; Sansom M. S. Conformational Changes in the Epidermal Growth Factor Receptor: Role of the Transmembrane Domain Investigated by Coarse-Grained Metadynamics Free Energy Calculations. J. Am. Chem. Soc. 2016, 138, 10611–10622. 10.1021/jacs.6b05602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkhipov A.; Shan Y.; Das R.; Endres N. F.; Eastwood M. P.; Wemmer D. E.; Kuriyan J.; Shaw D. E. Architecture and Membrane Interactions of the Egf Receptor. Cell 2013, 152, 557–569. 10.1016/j.cell.2012.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkhipov A.; Shan Y.; Kim E. T.; Shaw D. E. Membrane Interaction of Bound Ligands Contributes to the Negative Binding Cooperativity of the Egf Receptor. PLoS Comput. Biol. 2014, 10, e1003742 10.1371/journal.pcbi.1003742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koland J. G. Coarse-Grained Molecular Simulation of Epidermal Growth Factor Receptor Protein Tyrosine Kinase Multi-Site Self-Phosphorylation. PLoS Comput. Biol. 2014, 10, e1003435 10.1371/journal.pcbi.1003435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abd Halim K. B.; Koldso H.; Sansom M. S. Interactions of the Egfr Juxtamembrane Domain with Pip2-Containing Lipid Bilayers: Insights from Multiscale Molecular Dynamics Simulations. Biochim. Biophys. Acta, Gen. Subj. 2015, 1850, 1017–1025. 10.1016/j.bbagen.2014.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedger G.; Sansom M. S.; Koldso H. The Juxtamembrane Regions of Human Receptor Tyrosine Kinases Exhibit Conserved Interaction Sites with Anionic Lipids. Sci. Rep. 2015, 5, 9198. 10.1038/srep09198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coskun U.; Grzybek M.; Drechsel D.; Simons K. Regulation of Human Egf Receptor by Lipids. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 9044–9048. 10.1073/pnas.1105666108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedger G.; Shorthouse D.; Koldso H.; Sansom M. S. Free Energy Landscape of Lipid Interactions with Regulatory Binding Sites on the Transmembrane Domain of the Egf Receptor. J. Phys. Chem. B 2016, 120, 8154–8163. 10.1021/acs.jpcb.6b01387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavent M.; Seiradake E.; Jones E. Y.; Sansom M. S. P. Structures of the Epha2 Receptor at the Membrane: Role of Lipid Interactions. Structure 2016, 24, 337–347. 10.1016/j.str.2015.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaszuba K.; Grzybek M.; Orlowski A.; Danne R.; Rog T.; Simons K.; Coskun U.; Vattulainen I. N-Glycosylation as Determinant of Epidermal Growth Factor Receptor Conformation in Membranes. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 4334–4339. 10.1073/pnas.1503262112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavent M.; Karia D.; Kalli A. C.; Domanski J.; Duncan A. L.; Hedger G.; Stansfeld P. J.; Seiradake E.; Jones E. Y.; Sansom M. S. P. Interactions of the Epha2 Kinase Domain with Pips in Membranes: Implications for Receptor Function. Structure 2018, 26, 1025–1034. 10.1016/j.str.2018.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koldso H.; Shorthouse D.; Helie J.; Sansom M. S. Lipid Clustering Correlates with Membrane Curvature as Revealed by Molecular Simulations of Complex Lipid Bilayers. PLoS Comput. Biol. 2014, 10, e1003911 10.1371/journal.pcbi.1003911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthis N. J.; Campbell I. D. The Tail of Integrin Activation. Trends Biochem. Sci. 2011, 36, 191–198. 10.1016/j.tibs.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesner S.; Legate K. R.; Fassler R. Integrin-Actin Interactions. Cell. Mol. Life Sci. 2005, 62, 1081–1099. 10.1007/s00018-005-4522-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalli A. C.; Hall B. A.; Campbell I. D.; Sansom M. S. P. A Helix Heterodimer in a Lipid Bilayer: Prediction of the Structure of an Integrin Transmembrane Domain Via Multiscale Simulations. Structure 2011, 19, 1477–1484. 10.1016/j.str.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chng C. P.; Tan S. M. Leukocyte Integrin Alpha L Beta 2 Transmembrane Association Dynamics Revealed by Coarse-Grained Molecular Dynamics Simulations. Proteins: Struct., Funct., Genet. 2011, 79, 2203–2213. 10.1002/prot.23044. [DOI] [PubMed] [Google Scholar]
- Vararattanavech A.; Chng C. P.; Parthasarathy K.; Tang X. Y.; Torres J.; Tan S. M. A Transmembrane Polar Interaction Is Involved in the Functional Regulation of Integrin Alpha L Beta 2. J. Mol. Biol. 2010, 398, 569–583. 10.1016/j.jmb.2010.03.027. [DOI] [PubMed] [Google Scholar]
- Pagani G.; Gohlke H. On the Contributing Role of the Transmembrane Domain for Subunit-Specific Sensitivity of Integrin Activation. Sci. Rep. 2018, 8, 5733. 10.1038/s41598-018-23778-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderwood D. A.; Campbell I. D.; Critchley D. R. Talins and Kindlins: Partners in Integrin-Mediated Adhesion. Nat. Rev. Mol. Cell Biol. 2013, 14, 503–517. 10.1038/nrm3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt T.; Suk J. E.; Ye F.; Situ A. J.; Mazumder P.; Ginsberg M. H.; Ulmer T. S. Annular Anionic Lipids Stabilize the Integrin Alphaiibbeta3 Transmembrane Complex. J. Biol. Chem. 2015, 290, 8283–8293. 10.1074/jbc.M114.623504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X.; McLean M. A.; Sligar S. G. Phosphatidylinositol 4,5-Bisphosphate Modulates the Affinity of Talin-1 for Phospholipid Bilayers and Activates Its Autoinhibited Form. Biochemistry 2016, 55, 5038–5048. 10.1021/acs.biochem.6b00497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X.; McLean M. A.; Sligar S. G. Conformational Equilibrium of Talin Is Regulated by Anionic Lipids. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 1833–1840. 10.1016/j.bbamem.2016.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalli A. C.; Wegener K. L.; Goult B. T.; Anthis N. J.; Campbell I. D.; Sansom M. S. P. The Structure of the Talin/Integrin Complex at a Lipid Bilayer: An Nmr and Md Simulation Study. Structure 2010, 18, 1280–1288. 10.1016/j.str.2010.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalli A. C.; Campbell I. D.; Sansom M. S. Conformational Changes in Talin on Binding to Anionic Phospholipid Membranes Facilitate Signaling by Integrin Transmembrane Helices. PLoS Comput. Biol. 2013, 9, e1003316 10.1371/journal.pcbi.1003316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arcario M. J.; Tajkhorshid E. Membrane-Induced Structural Rearrangement and Identification of a Novel Membrane Anchor in Talin F2f3. Biophys. J. 2014, 107, 2059–2069. 10.1016/j.bpj.2014.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehrbod M.; Trisno S.; Mofrad M. R. On the Activation of Integrin Alphaiibbeta3: Outside-in and inside-out Pathways. Biophys. J. 2013, 105, 1304–1315. 10.1016/j.bpj.2013.07.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalli A. C.; Campbell I. D.; Sansom M. S. Multiscale Simulations Suggest a Mechanism for Integrin inside-out Activation. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 11890–11895. 10.1073/pnas.1104505108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlowski A.; Kukkurainen S.; Poyry A.; Rissanen S.; Vattulainen I.; Hytonen V. P.; Rog T. Pip2 and Talin Join Forces to Activate Integrin. J. Phys. Chem. B 2015, 119, 12381–12389. 10.1021/acs.jpcb.5b06457. [DOI] [PubMed] [Google Scholar]
- Hughes P. E.; Diaz-Gonzalez F.; Leong L.; Wu C.; McDonald J. A.; Shattil S. J.; Ginsberg M. H. Breaking the Integrin Hinge. A Defined Structural Constraint Regulates Integrin Signaling. J. Biol. Chem. 1996, 271, 6571–6574. 10.1074/jbc.271.12.6571. [DOI] [PubMed] [Google Scholar]
- Lau T. L.; Kim C.; Ginsberg M. H.; Ulmer T. S. The Structure of the Integrin Alphaiibbeta3 Transmembrane Complex Explains Integrin Transmembrane Signalling. EMBO J. 2009, 28, 1351–1361. 10.1038/emboj.2009.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provasi D.; Negri A.; Coller B. S.; Filizola M. Talin-Driven inside-out Activation Mechanism of Platelet Alphaiibbeta3 Integrin Probed by Multimicrosecond, All-Atom Molecular Dynamics Simulations. Proteins: Struct., Funct., Genet. 2014, 82, 3231–3240. 10.1002/prot.24540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalli A. C.; Rog T.; Vattulainen I.; Campbell I. D.; Sansom M. S. P. The Integrin Receptor in Biologically Relevant Bilayers: Insights from Molecular Dynamics Simulations. J. Membr. Biol. 2017, 250, 337–351. 10.1007/s00232-016-9908-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shattil S. J.; Kim C.; Ginsberg M. H. The Final Steps of Integrin Activation: The End Game. Nat. Rev. Mol. Cell Biol. 2010, 11, 288–300. 10.1038/nrm2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehrbod M.; Mofrad M. R. Localized Lipid Packing of Transmembrane Domains Impedes Integrin Clustering. PLoS Comput. Biol. 2013, 9, e1002948 10.1371/journal.pcbi.1002948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEver R. P.; Zhu C. Rolling Cell Adhesion. Annu. Rev. Cell Dev. Biol. 2010, 26, 363–396. 10.1146/annurev.cellbio.042308.113238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun F.; Schroer C. F. E.; Xu L.; Yin H.; Marrink S. J.; Luo S. Z. Molecular Dynamics of the Association of L-Selectin and Ferm Regulated by Pip2. Biophys. J. 2018, 114, 1858–1868. 10.1016/j.bpj.2018.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai X.; Moraes T. F.; Reithmeier R. A. F. Structural Biology of Solute Carrier (Slc) Membrane Transport Proteins. Mol. Membr. Biol. 2017, 34, 1–32. 10.1080/09687688.2018.1448123. [DOI] [PubMed] [Google Scholar]
- Lin L.; Yee S. W.; Kim R. B.; Giacomini K. M. Slc Transporters as Therapeutic Targets: Emerging Opportunities. Nat. Rev. Drug Discovery 2015, 14, 543–560. 10.1038/nrd4626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta K.; Donlan J. A. C.; Hopper J. T. S.; Uzdavinys P.; Landreh M.; Struwe W. B.; Drew D.; Baldwin A. J.; Stansfeld P. J.; Robinson C. V. The Role of Interfacial Lipids in Stabilizing Membrane Protein Oligomers. Nature 2017, 541, 421–424. 10.1038/nature20820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlessinger A.; Yee S. W.; Sali A.; Giacomini K. M. Slc Classification: An Update. Clin. Pharmacol. Ther. 2013, 94, 19–23. 10.1038/clpt.2013.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perland E.; Fredriksson R. Classification Systems of Secondary Active Transporters. Trends Pharmacol. Sci. 2017, 38, 305–315. 10.1016/j.tips.2016.11.008. [DOI] [PubMed] [Google Scholar]
- Guan L.; Mirza O.; Verner G.; Iwata S.; Kaback H. R. Structural Determination of Wild-Type Lactose Permease. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 15294–15298. 10.1073/pnas.0707688104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita A.; Singh S. K.; Kawate T.; Jin Y.; Gouaux E. Crystal Structure of a Bacterial Homologue of Na+/Cl--Dependent Neurotransmitter Transporters. Nature 2005, 437, 215–223. 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- Quistgaard E. M.; Low C.; Guettou F.; Nordlund P. Understanding Transport by the Major Facilitator Superfamily (Mfs): Structures Pave the Way. Nat. Rev. Mol. Cell Biol. 2016, 17, 123–132. 10.1038/nrm.2015.25. [DOI] [PubMed] [Google Scholar]
- Lensink M. F.; Govaerts C.; Ruysschaert J. M. Identification of Specific Lipid-Binding Sites in Integral Membrane Proteins. J. Biol. Chem. 2010, 285, 10519–10526. 10.1074/jbc.M109.068890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez-Germa C.; Loura L. M.; Domenech O.; Montero M. T.; Vazquez-Ibar J. L.; Hernandez-Borrell J. Phosphatidylethanolamine-Lactose Permease Interaction: A Comparative Study Based on Fret. J. Phys. Chem. B 2012, 116, 14023–14028. 10.1021/jp309726v. [DOI] [PubMed] [Google Scholar]
- Martens C.; Shekhar M.; Borysik A. J.; Lau A. M.; Reading E.; Tajkhorshid E.; Booth P. J.; Politis A. Direct Protein-Lipid Interactions Shape the Conformational Landscape of Secondary Transporters. Nat. Commun. 2018, 9, 4151. 10.1038/s41467-018-06704-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimanius D.; Lindahl E.; Andersson M.. Uptake Dynamics in the Lactose Permease (Lacy) Membrane Protein Transporter. Sci. Rep. 2018, 8. 10.1038/s41598-018-32624-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan N. A Glimpse of Membrane Transport through Structures-Advances in the Structural Biology of the Glut Glucose Transporters. J. Mol. Biol. 2017, 429, 2710–2725. 10.1016/j.jmb.2017.07.009. [DOI] [PubMed] [Google Scholar]
- Mueckler M.; Thorens B. The Slc2 (Glut) Family of Membrane Transporters. Mol. Aspects Med. 2013, 34, 121–138. 10.1016/j.mam.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iglesias-Fernandez J.; Quinn P. J.; Naftalin R. J.; Domene C. Membrane Phase-Dependent Occlusion of Intramolecular Glut1 Cavities Demonstrated by Simulations. Biophys. J. 2017, 112, 1176–1184. 10.1016/j.bpj.2017.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hresko R. C.; Kraft T. E.; Quigley A.; Carpenter E. P.; Hruz P. W. Mammalian Glucose Transporter Activity Is Dependent Upon Anionic and Conical Phospholipids. J. Biol. Chem. 2016, 291, 17271–17282. 10.1074/jbc.M116.730168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazmier K.; Claxton D. P.; McHaourab H. S. Alternating Access Mechanisms of Leut-Fold Transporters: Trailblazing Towards the Promised Energy Landscapes. Curr. Opin. Struct. Biol. 2017, 45, 100–108. 10.1016/j.sbi.2016.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loland C. J. The Use of Leut as a Model in Elucidating Binding Sites for Substrates and Inhibitors in Neurotransmitter Transporters. Biochim. Biophys. Acta, Gen. Subj. 2015, 1850, 500–510. 10.1016/j.bbagen.2014.04.011. [DOI] [PubMed] [Google Scholar]
- Khelashvili G.; LeVine M. V.; Shi L.; Quick M.; Javitch J. A.; Weinstein H. The Membrane Protein Leut in Micellar Systems: Aggregation Dynamics and Detergent Binding to the S2 Site. J. Am. Chem. Soc. 2013, 135, 14266–14275. 10.1021/ja405984v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeVine M. V.; Khelashvili G.; Shi L.; Quick M.; Javitch J. A.; Weinstein H. Role of Annular Lipids in the Functional Properties of Leucine Transporter Leut Proteomicelles. Biochemistry 2016, 55, 850–859. 10.1021/acs.biochem.5b01268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick M.; Winther A. M.; Shi L.; Nissen P.; Weinstein H.; Javitch J. A. Binding of an Octylglucoside Detergent Molecule in the Second Substrate (S2) Site of Leut Establishes an Inhibitor-Bound Conformation. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 5563–5568. 10.1073/pnas.0811322106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal S.; Khelashvili G.; Shi L.; Weinstein H. The Cost of Living in the Membrane: A Case Study of Hydrophobic Mismatch for the Multi-Segment Protein Leut. Chem. Phys. Lipids 2013, 169, 27–38. 10.1016/j.chemphyslip.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohail A.; Jayaraman K.; Venkatesan S.; Gotfryd K.; Daerr M.; Gether U.; Loland C. J.; Wanner K. T.; Freissmuth M.; Sitte H. H.; et al. The Environment Shapes the Inner Vestibule of Leut. PLoS Comput. Biol. 2016, 12, e1005197 10.1371/journal.pcbi.1005197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest L. R.; Zhang Y. W.; Jacobs M. T.; Gesmonde J.; Xie L.; Honig B. H.; Rudnick G. Mechanism for Alternating Access in Neurotransmitter Transporters. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 10338–10343. 10.1073/pnas.0804659105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton P. J.; Belovich A. N.; Khelashvili G.; Saunders C.; Erreger K.; Javitch J. A.; Sitte H. H.; Weinstein H.; Matthies H. J. G.; Galli A. Pip2 Regulates Psychostimulant Behaviors through Its Interaction with a Membrane Protein. Nat. Chem. Biol. 2014, 10, 582–589. 10.1038/nchembio.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchmayer F.; Schicker K.; Steinkellner T.; Geier P.; Stubiger G.; Hamilton P. J.; Jurik A.; Stockner T.; Yang J. W.; Montgomery T.; et al. Amphetamine Actions at the Serotonin Transporter Rely on the Availability of Phosphatidylinositol-4,5-Bisphosphate. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 11642–11647. 10.1073/pnas.1220552110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong W. C.; Amara S. G. Membrane Cholesterol Modulates the Outward Facing Conformation of the Dopamine Transporter and Alters Cocaine Binding. J. Biol. Chem. 2010, 285, 32616–32626. 10.1074/jbc.M110.150565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanlon S. M.; Williams D. C.; Schloss P. Membrane Cholesterol Modulates Serotonin Transporter Activity. Biochemistry 2001, 40, 10507–10513. 10.1021/bi010730z. [DOI] [PubMed] [Google Scholar]
- Magnani F.; Tate C. G.; Wynne S.; Williams C.; Haase J. Partitioning of the Serotonin Transporter into Lipid Microdomains Modulates Transport of Serotonin. J. Biol. Chem. 2004, 279, 38770–38778. 10.1074/jbc.M400831200. [DOI] [PubMed] [Google Scholar]
- Khelashvili G.; Doktorova M.; Sahai M. A.; Johner N.; Shi L.; Weinstein H. Computational Modeling of the N-Terminus of the Human Dopamine Transporter and Its Interaction with Pip2 -Containing Membranes. Proteins: Struct., Funct., Genet. 2015, 83, 952–969. 10.1002/prot.24792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khelashvili G.; Stanley N.; Sahai M. A.; Medina J.; LeVine M. V.; Shi L.; De Fabritiis G.; Weinstein H. Spontaneous Inward Opening of the Dopamine Transporter Is Triggered by Pip2-Regulated Dynamics of the N-Terminus. ACS Chem. Neurosci. 2015, 6, 1825–1837. 10.1021/acschemneuro.5b00179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razavi A. M.; Khelashvili G.; Weinstein H. A Markov State-Based Quantitative Kinetic Model of Sodium Release from the Dopamine Transporter. Sci. Rep. 2017, 7, 40076. 10.1038/srep40076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razavi A. M.; Khelashvili G.; Weinstein H. How Structural Elements Evolving from Bacterial to Human Slc6 Transporters Enabled New Functional Properties. BMC Biol. 2018, 16, 31. 10.1186/s12915-018-0495-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khelashvili G.; Weinstein H. Functional Mechanisms of Neurotransmitter Transporters Regulated by Lipid-Protein Interactions of Their Terminal Loops. Biochim. Biophys. Acta, Biomembr. 2015, 1848, 1765–1774. 10.1016/j.bbamem.2015.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adkins E. M.; Samuvel D. J.; Fog J. U.; Eriksen J.; Jayanthi L. D.; Vaegter C. B.; Ramamoorthy S.; Gether U. Membrane Mobility and Microdomain Association of the Dopamine Transporter Studied with Fluorescence Correlation Spectroscopy and Fluorescence Recovery after Photobleaching. Biochemistry 2007, 46, 10484–10497. 10.1021/bi700429z. [DOI] [PubMed] [Google Scholar]
- Foster J. D.; Adkins S. D.; Lever J. R.; Vaughan R. A. Phorbol Ester Induced Trafficking-Independent Regulation and Enhanced Phosphorylation of the Dopamine Transporter Associated with Membrane Rafts and Cholesterol. J. Neurochem. 2008, 105, 1683–1699. 10.1111/j.1471-4159.2008.05262.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremona M. L.; Matthies H. J. G.; Pau K.; Bowton E.; Speed N.; Lute B. J.; Anderson M.; Sen N.; Robertson S. D.; Vaughan R. A.; et al. Flotillin-1 Is Essential for Pkc-Triggered Endocytosis and Membrane Microdomain Localization of Dat. Nat. Neurosci. 2011, 14, 469–U103. 10.1038/nn.2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones K. T.; Zhen J.; Reith M. E. Importance of Cholesterol in Dopamine Transporter Function. J. Neurochem. 2012, 123, 700–715. 10.1111/jnc.12007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahbek-Clemmensen T.; Lycas M. D.; Erlendsson S.; Eriksen J.; Apuschkin M.; Vilhardt F.; Jorgensen T. N.; Hansen F. H.; Gether U. Super-Resolution Microscopy Reveals Functional Organization of Dopamine Transporters into Cholesterol and Neuronal Activity-Dependent Nanodomains. Nat. Commun. 2017, 8, 740. 10.1038/s41467-017-00790-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penmatsa A.; Wang K. H.; Gouaux E. X-Ray Structure of Dopamine Transporter Elucidates Antidepressant Mechanism. Nature 2013, 503, 85–90. 10.1038/nature12533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K. H.; Penmatsa A.; Gouaux E. Neurotransmitter and Psychostimulant Recognition by the Dopamine Transporter. Nature 2015, 521, 322–327. 10.1038/nature14431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penmatsa A.; Wang K. H.; Gouaux E. X-Ray Structures of Drosophila Dopamine Transporter in Complex with Nisoxetine and Reboxetine. Nat. Struct. Mol. Biol. 2015, 22, 506–U101. 10.1038/nsmb.3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laursen L.; Severinsen K.; Kristensen K. B.; Periole X.; Overby M.; Muller H. K.; Schiott B.; Sinning S. Cholesterol Binding to a Conserved Site Modulates the Conformation, Pharmacology, and Transport Kinetics of the Human Serotonin Transporter. J. Biol. Chem. 2018, 293, 3510–3523. 10.1074/jbc.M117.809046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman J. A.; Green E. M.; Gouaux E. X-Ray Structures and Mechanism of the Human Serotonin Transporter. Nature 2016, 532, 334–339. 10.1038/nature17629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjerregaard H.; Severinsen K.; Said S.; Wiborg O.; Sinning S. A Dualistic Conformational Response to Substrate Binding in the Human Serotonin Transporter Reveals a High Affinity State for Serotonin. J. Biol. Chem. 2015, 290, 7747–7755. 10.1074/jbc.M114.573477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuvel D. J.; Jayanthi L. D.; Bhat N. R.; Ramamoorthy S. A Role for P38 Mitogen-Activated Protein Kinase in the Regulation of the Serotonin Transporter: Evidence for Distinct Cellular Mechanisms Involved in Transporter Surface Expression. J. Neurosci. 2005, 25, 29–41. 10.1523/JNEUROSCI.3754-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraro M.; Masetti M.; Recanatini M.; Cavalli A.; Bottegoni G. Mapping Cholesterol Interaction Sites on Serotonin Transporter through Coarse Grained Molecular Dynamics. PLoS One 2016, 11, 24. 10.1371/journal.pone.0166196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeppelin T.; Ladefoged L. K.; Sinning S.; Periole X.; Schiott B. A Direct Interaction of Cholesterol with the Dopamine Transporter Prevents Its out-to-Inward Transition. PLoS Comput. Biol. 2018, 14, e1005907 10.1371/journal.pcbi.1005907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Periole X.; Zeppelin T.; Schiott B.. Dimer Interface of the Human Serotonin Transporter and Effect of the Membrane Composition. Sci. Rep. 2018, 8. 10.1038/s41598-018-22912-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderluh A.; Hofmaier T.; Klotzsch E.; Kudlacek O.; Stockner T.; Sitte H. H.; Schutz G. J. Direct Pip2 Binding Mediates Stable Oligomer Formation of the Serotonin Transporter. Nat. Commun. 2017, 8, 14089. 10.1038/ncomms14089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coincon M.; Uzdavinys P.; Nji E.; Dotson D. L.; Winkelmann I.; Abdul-Hussein S.; Cameron A. D.; Beckstein O.; Drew D. Crystal Structures Reveal the Molecular Basis of Ion Translocation in Sodium/Proton Antiporters. Nat. Struct. Mol. Biol. 2016, 23, 248–255. 10.1038/nsmb.3164. [DOI] [PubMed] [Google Scholar]
- Lu F.; Li S.; Jiang Y.; Jiang J.; Fan H.; Lu G.; Deng D.; Dang S.; Zhang X.; Wang J.; et al. Structure and Mechanism of the Uracil Transporter Uraa. Nature 2011, 472, 243–246. 10.1038/nature09885. [DOI] [PubMed] [Google Scholar]
- Arakawa T.; Kobayashi-Yurugi T.; Alguel Y.; Iwanari H.; Hatae H.; Iwata M.; Abe Y.; Hino T.; Ikeda-Suno C.; Kuma H.; et al. Crystal Structure of the Anion Exchanger Domain of Human Erythrocyte Band 3. Science 2015, 350, 680–684. 10.1126/science.aaa4335. [DOI] [PubMed] [Google Scholar]
- Lee C.; Kang H. J.; von Ballmoos C.; Newstead S.; Uzdavinys P.; Dotson D. L.; Iwata S.; Beckstein O.; Cameron A. D.; Drew D. A Two-Domain Elevator Mechanism for Sodium/Proton Antiport. Nature 2013, 501, 573–577. 10.1038/nature12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landreh M.; Marklund E. G.; Uzdavinys P.; Degiacomi M. T.; Coincon M.; Gault J.; Gupta K.; Liko I.; Benesch J. L.; Drew D.; et al. Integrating Mass Spectrometry with Md Simulations Reveals the Role of Lipids in Na(+)/H(+) Antiporters. Nat. Commun. 2017, 8, 13993. 10.1038/ncomms13993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gournas C.; Papageorgiou I.; Diallinas G. The Nucleobase-Ascorbate Transporter (Nat) Family: Genomics, Evolution, Structure-Function Relationships and Physiological Role. Mol. BioSyst. 2008, 4, 404–416. 10.1039/b719777b. [DOI] [PubMed] [Google Scholar]
- Kalli A. C.; Sansom M. S.; Reithmeier R. A. Molecular Dynamics Simulations of the Bacterial Uraa H+-Uracil Symporter in Lipid Bilayers Reveal a Closed State and a Selective Interaction with Cardiolipin. PLoS Comput. Biol. 2015, 11, e1004123 10.1371/journal.pcbi.1004123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alguel Y.; Amillis S.; Leung J.; Lambrinidis G.; Capaldi S.; Scull N. J.; Craven G.; Iwata S.; Armstrong A.; Mikros E.; et al. Structure of Eukaryotic Purine/H(+) Symporter Uapa Suggests a Role for Homodimerization in Transport Activity. Nat. Commun. 2016, 7, 11336. 10.1038/ncomms11336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martzoukou O.; Karachaliou M.; Yalelis V.; Leung J.; Byrne B.; Amillis S.; Diallinas G. Oligomerization of the Uapa Purine Transporter Is Critical for Er-Exit, Plasma Membrane Localization and Turnover. J. Mol. Biol. 2015, 427, 2679–2696. 10.1016/j.jmb.2015.05.021. [DOI] [PubMed] [Google Scholar]
- Pyle E.; Kalli A. C.; Amillis S.; Hall Z.; Lau A. M.; Hanyaloglu A. C.; Diallinas G.; Byrne B.; Politis A. Structural Lipids Enable the Formation of Functional Oligomers of the Eukaryotic Purine Symporter Uapa. Cell Chem. Biol. 2018, 25, 840–848. 10.1016/j.chembiol.2018.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reithmeier R. A. F.; Casey J. R.; Kalli A. C.; Sansom M. S. P.; Alguel Y.; Iwata S. Band 3, the Human Red Cell Chloride/Bicarbonate Anion Exchanger (Ae1, Slc4a1), in a Structural Context. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 1507–1532. 10.1016/j.bbamem.2016.03.030. [DOI] [PubMed] [Google Scholar]
- Abbas Y. M.; Toye A. M.; Rubinstein J. L.; Reithmeier R. A. F. Band 3 Function and Dysfunction in a Structural Context. Curr. Opin. Hematol. 2018, 25, 163–170. 10.1097/MOH.0000000000000418. [DOI] [PubMed] [Google Scholar]
- Schubert D.; Boss K. Band-3 Protein Cholesterol Interactions in Erythrocyte-Membranes - Possible Role in Anion Transport and Dependency on Membrane Phospholipid. FEBS Lett. 1982, 150, 4–8. 10.1016/0014-5793(82)81295-7. [DOI] [PubMed] [Google Scholar]
- Kohne W.; Deuticke B.; Haest C. W. M. Phospholipid Dependence of the Anion Transport-System of the Human-Erythrocyte Membrane - Studies on Reconstituted Band-3 Lipid Vesicles. Biochim. Biophys. Acta, Biomembr. 1983, 730, 139–150. 10.1016/0005-2736(83)90326-7. [DOI] [PubMed] [Google Scholar]
- Klugerman A. H.; Gaarn A.; Parkes J. G. Effect of Cholesterol Upon the Conformation of Band-3 and Its Transmembrane Fragment. Can. J. Biochem. Cell Biol. 1984, 62, 1033–1040. 10.1139/o84-132. [DOI] [PubMed] [Google Scholar]
- Maneri L. R.; Low P. S. Structural Stability of the Erythrocyte Anion Transporter, Band-3, in Different Lipid Environments - a Differential Scanning Calorimetric Study. J. Biol. Chem. 1988, 263, 16170–16178. [PubMed] [Google Scholar]
- Rodgers W.; Glaser M. Distributions of Proteins and Lipids in the Erythrocyte-Membrane. Biochemistry 1993, 32, 12591–12598. 10.1021/bi00210a007. [DOI] [PubMed] [Google Scholar]
- Wu J.; McNicholas C. M.; Bevensee M. O. Phosphatidylinositol 4,5-Bisphosphate (Pip2) Stimulates the Electrogenic Na/Hco3 Cotransporter Nbce1-a Expressed in Xenopus Oocytes. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 14150–14155. 10.1073/pnas.0906303106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalli A. C.; Reithmeier R. A. F. Interaction of the Human Erythrocyte Band 3 Anion Exchanger 1 (Ae1, Slc4a1) with Lipids and Glycophorin A: Molecular Organization of the Wright (Wr) Blood Group Antigen. PLoS Comput. Biol. 2018, 14, e1006284. 10.1371/journal.pcbi.1006284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crichton P. G.; Lee Y.; Kunji E. R. The Molecular Features of Uncoupling Protein 1 Support a Conventional Mitochondrial Carrier-Like Mechanism. Biochimie 2017, 134, 35–50. 10.1016/j.biochi.2016.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L.; Wang S.; Zhu Q.; Wu B.; Liu Z.; OuYang B.; Chou J. J. Specific Interaction of the Human Mitochondrial Uncoupling Protein 1 with Free Long-Chain Fatty Acid. Structure 2017, 25, 1371–1379. 10.1016/j.str.2017.07.005. [DOI] [PubMed] [Google Scholar]
- Khademi S.; O’Connell J. 3rd; Remis J.; Robles-Colmenares Y.; Miercke L. J.; Stroud R. M. Mechanism of Ammonia Transport by Amt/Mep/Rh: Structure of Amtb at 1.35 A. Science 2004, 305, 1587–1594. 10.1126/science.1101952. [DOI] [PubMed] [Google Scholar]
- Khademi S.; Stroud R. M. The Amt/Mep/Rh Family: Structure of Amtb and the Mechanism of Ammonia Gas Conduction. Physiology 2006, 21, 419–429. 10.1152/physiol.00051.2005. [DOI] [PubMed] [Google Scholar]
- Mirandela G. D.; Tamburrino G.; Hoskisson P. A.; Zachariae U.; Javelle A. The Lipid Environment Determines the Activity of the Escherichia Coli Ammonium Transporter Amtb. FASEB J. 2019, 33, 1989. 10.1096/fj.201800782R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrick J. W.; Boone C. D.; Liu W.; Conover G. M.; Liu Y.; Cong X.; Laganowsky A. Allostery Revealed within Lipid Binding Events to Membrane Proteins. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 2976–2981. 10.1073/pnas.1719813115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey F. A.; Lok S. M. Common Features of Enveloped Viruses and Implications for Immunogen Design for Next-Generation Vaccines. Cell 2018, 172, 1319–1334. 10.1016/j.cell.2018.02.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baylon J. L.; Tajkhorshid E. Capturing Spontaneous Membrane Insertion of the Influenza Virus Hemagglutinin Fusion Peptide. J. Phys. Chem. B 2015, 119, 7882–7893. 10.1021/acs.jpcb.5b02135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeevan B. G. C.; Gerstman B. S.; Chapagain P. P. Membrane Association and Localization Dynamics of the Ebola Virus Matrix Protein Vp40. Biochim. Biophys. Acta, Biomembr. 2017, 1859, 2012–2020. 10.1016/j.bbamem.2017.07.007. [DOI] [PubMed] [Google Scholar]
- Olson M. A.; Lee M. S.; Yeh I. C. Membrane Insertion of Fusion Peptides from Ebola and Marburg Viruses Studied by Replica-Exchange Molecular Dynamics Simulations. J. Comput. Chem. 2017, 38, 1342–1352. 10.1002/jcc.24717. [DOI] [PubMed] [Google Scholar]
- Kamath S.; Wong T. C. Membrane Structure of the Human Immunodeficiency Virus Gp41 Fusion Domain by Molecular Dynamics Simulation. Biophys. J. 2002, 83, 135–143. 10.1016/S0006-3495(02)75155-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattarai N.; Gc J. B.; Gerstman B. S.; Stahelin R. V.; Chapagain P. P. Plasma Membrane Association Facilitates Conformational Changes in the Marburg Virus Protein Vp40 Dimer. RSC Adv. 2017, 7, 22741–22748. 10.1039/C7RA02940C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrmans M.; Marrink S. J. Molecular View of the Role of Fusion Peptides in Promoting Positive Membrane Curvature. J. Am. Chem. Soc. 2012, 134, 1543–1552. 10.1021/ja207290b. [DOI] [PubMed] [Google Scholar]
- Chernomordik L. V.; Kozlov M. M. Mechanics of Membrane Fusion. Nat. Struct. Mol. Biol. 2008, 15, 675–683. 10.1038/nsmb.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín-Acebes M. A.; Vázquez-Calvo Á.; Caridi F.; Saiz J.-C.; Sobrino F.. Lipid Involvement in Viral Infections: Present and Future Perspectives for the Design of Antiviral Strategies; IntechOpen: Rijeka, Croatia, 2013; pp 291–321. [Google Scholar]
- Lorizate M.; Krausslich H. G. Role of Lipids in Virus Replication. Cold Spring Harbor Perspect. Biol. 2011, 3, a004820. 10.1101/cshperspect.a004820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teissier E.; Pecheur E. I. Lipids as Modulators of Membrane Fusion Mediated by Viral Fusion Proteins. Eur. Biophys. J. 2007, 36, 887–899. 10.1007/s00249-007-0201-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chlanda P.; Zimmerberg J. Protein-Lipid Interactions Critical to Replication of the Influenza a Virus. FEBS Lett. 2016, 590, 1940–1954. 10.1002/1873-3468.12118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown R. S.; Wan J. J.; Kielian M. The Alphavirus Exit Pathway: What We Know and What We Wish We Knew. Viruses 2018, 10, 89. 10.3390/v10020089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Votteler J.; Sundquist W. I. Virus Budding and the Escrt Pathway. Cell Host Microbe 2013, 14, 232–241. 10.1016/j.chom.2013.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waheed A. A.; Freed E. O. The Role of Lipids in Retrovirus Replication. Viruses 2010, 2, 1146–1180. 10.3390/v2051146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan R. B.; Tanner L.; Wenk M. R. Implications for Lipids During Replication of Enveloped Viruses. Chem. Phys. Lipids 2010, 163, 449–459. 10.1016/j.chemphyslip.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T.; Suzuki T. Function of Membrane Rafts in Viral Lifecycles and Host Cellular Response. Biochem. Res. Int. 2011, 2011, 245090. 10.1155/2011/245090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugger B.; Glass B.; Haberkant P.; Leibrecht I.; Wieland F. T.; Krausslich H. G. The Hiv Lipidome: A Raft with an Unusual Composition. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 2641–2646. 10.1073/pnas.0511136103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorizate M.; Sachsenheimer T.; Glass B.; Habermann A.; Gerl M. J.; Krausslich H. G.; Brugger B. Comparative Lipidomics Analysis of Hiv-1 Particles and Their Producer Cell Membrane in Different Cell Lines. Cell. Microbiol. 2013, 15, 292–304. 10.1111/cmi.12101. [DOI] [PubMed] [Google Scholar]
- Gerl M. J.; Sampaio J. L.; Urban S.; Kalvodova L.; Verbavatz J. M.; Binnington B.; Lindemann D.; Lingwood C. A.; Shevchenko A.; Schroeder C.; et al. Quantitative Analysis of the Lipidomes of the Influenza Virus Envelope and Mdck Cell Apical Membrane. J. Cell Biol. 2012, 196, 213–221. 10.1083/jcb.201108175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanova P. T.; Myers D. S.; Milne S. B.; McClaren J. L.; Thomas P. G.; Brown H. A. Lipid Composition of Viral Envelope of Three Strains of Influenza Virus - Not All Viruses Are Created Equal. ACS Infect. Dis. 2015, 1, 399–452. 10.1021/acsinfecdis.5b00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerviel A.; Thomas A.; Chaloin L.; Favard C.; Muriaux D. Virus Assembly and Plasma Membrane Domains: Which Came First?. Virus Res. 2013, 171, 332–340. 10.1016/j.virusres.2012.08.014. [DOI] [PubMed] [Google Scholar]
- Rossmann M. G. Structure of Viruses: A Short History. Q. Rev. Biophys. 2013, 46, 133–180. 10.1017/S0033583513000012. [DOI] [PubMed] [Google Scholar]
- Reddy T.; Sansom M. S. Computational Virology: From the inside Out. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 1610–1618. 10.1016/j.bbamem.2016.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber R. G.; Marzinek J. K.; Holdbrook D. A.; Bond P. J. Multiscale Molecular Dynamics Simulation Approaches to the Structure and Dynamics of Viruses. Prog. Biophys. Mol. Biol. 2017, 128, 121–132. 10.1016/j.pbiomolbio.2016.09.010. [DOI] [PubMed] [Google Scholar]
- Kasson P.; DiMaio F.; Yu X.; Lucas-Staat S.; Krupovic M.; Schouten S.; Prangishvili D.; Egelman E. H.. Model for a Novel Membrane Envelope in a Filamentous Hyperthermophilic Virus. eLife 2017, 6. 10.7554/eLife.26268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrant J. D.; Bush R. M.; Amaro R. E. Microsecond Molecular Dynamics Simulations of Influenza Neuraminidase Suggest a Mechanism for the Increased Virulence of Stalk-Deletion Mutants. J. Phys. Chem. B 2016, 120, 8590–8599. 10.1021/acs.jpcb.6b02655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy T.; Shorthouse D.; Parton D. L.; Jefferys E.; Fowler P. W.; Chavent M.; Baaden M.; Sansom M. S. P. Nothing to Sneeze At: A Dynamic and Integrative Computational Model of an Influenza a Virion. Structure 2015, 23, 584–597. 10.1016/j.str.2014.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parton D. L.; Tek A.; Baaden M.; Sansom M. S. P. Formation of Raft-Like Assemblies within Clusters of Influenza Hemagglutinin Observed by Md Simulations. PLoS Comput. Biol. 2013, 9, e1003034. 10.1371/journal.pcbi.1003034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risselada H. J.; Marelli G.; Fuhrmans M.; Smirnova Y. G.; Grubmuller H.; Marrink S. J.; Muller M. Line-Tension Controlled Mechanism for Influenza Fusion. PLoS One 2012, 7, e38302. 10.1371/journal.pone.0038302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossman J. S.; Lamb R. A. Influenza Virus Assembly and Budding. Virology 2011, 411, 229–236. 10.1016/j.virol.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boonstra S.; Blijleven J. S.; Roos W. H.; Onck P. R.; van der Giessen E.; van Oijen A. M. Hemagglutinin-Mediated Membrane Fusion: A Biophysical Perspective. Annu. Rev. Biophys. 2018, 47, 153–173. 10.1146/annurev-biophys-070317-033018. [DOI] [PubMed] [Google Scholar]
- Goronzy I. N.; Rawle R. J.; Boxer S. G.; Kasson P. M. Cholesterol Enhances Influenza Binding Avidity by Controlling Nanoscale Receptor Clustering. Chem. Sci. 2018, 9, 2340–2347. 10.1039/C7SC03236F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzinek J. K.; Holdbrook D. A.; Huber R. G.; Verma C.; Bond P. J. Pushing the Envelope: Dengue Viral Membrane Coaxed into Shape by Molecular Simulations. Structure 2016, 24, 1410–1420. 10.1016/j.str.2016.05.014. [DOI] [PubMed] [Google Scholar]
- Vanegas J. M.; Heinrich F.; Rogers D. M.; Carson B. D.; La Bauve S.; Vernon B. C.; Akgun B.; Satija S.; Zheng A. H.; Kielian M.; et al. Insertion of Dengue E into Lipid Bilayers Studied by Neutron Reflectivity and Molecular Dynamics Simulations. Biochim. Biophys. Acta, Biomembr. 2018, 1860, 1216–1230. 10.1016/j.bbamem.2018.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma K. K.; Marzinek J. K.; Tantirimudalige S. N.; Bond P. J.; Wohland T. Single-Molecule Studies of Flavivirus Envelope Dynamics: Experiment and Computation. Prog. Biophys. Mol. Biol. 2018, 10.1016/j.pbiomolbio.2018.09.001. [DOI] [PubMed] [Google Scholar]
- Kuhn R. J.; Zhang W.; Rossmann M. G.; Pletnev S. V.; Corver J.; Lenches E.; Jones C. T.; Mukhopadhyay S.; Chipman P. R.; Strauss E. G.; et al. Structure of Dengue Virus: Implications for Flavivirus Organization, Maturation, and Fusion. Cell 2002, 108, 717–725. 10.1016/S0092-8674(02)00660-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy T.; Sansom M. S. P. The Role of the Membrane in the Structure and Biophysical Robustness of the Dengue Virion Envelope. Structure 2016, 24, 375–382. 10.1016/j.str.2015.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wewer C. R.; Khandelia H. Different Footprints of the Zika and Dengue Surface Proteins on Viral Membranes. Soft Matter 2018, 14, 5615–5621. 10.1039/C8SM00223A. [DOI] [PubMed] [Google Scholar]
- Fajardo-Sanchez E.; Galiano V.; Villalain J. Spontaneous Membrane Insertion of a Dengue Virus Ns2a Peptide. Arch. Biochem. Biophys. 2017, 627, 56–66. 10.1016/j.abb.2017.06.016. [DOI] [PubMed] [Google Scholar]
- Oliveira E. R.; de Alencastro R. B.; Horta B. A. The Mechanism by Which P250l Mutation Impairs Flavivirus-Ns1 Dimerization: An Investigation Based on Molecular Dynamics Simulations. Eur. Biophys. J. 2016, 45, 573–580. 10.1007/s00249-016-1147-9. [DOI] [PubMed] [Google Scholar]
- Lin M. H.; Hsu H. J.; Bartenschlager R.; Fischer W. B. Membrane Undulation Induced by Ns4a of Dengue Virus: A Molecular Dynamics Simulation Study. J. Biomol. Struct. Dyn. 2014, 32, 1552–1562. 10.1080/07391102.2013.826599. [DOI] [PubMed] [Google Scholar]
- Fajardo-Sanchez E.; Galiano V.; Villalain J. Molecular Dynamics Study of the Membrane Interaction of a Membranotropic Dengue Virus C Protein-Derived Peptide. J. Biomol. Struct. Dyn. 2017, 35, 1283–1294. 10.1080/07391102.2016.1179595. [DOI] [PubMed] [Google Scholar]
- Briggs J. A. G.; Wilk T.; Welker R.; Krausslich H. G.; Fuller S. D. Structural Organization of Authentic, Mature Hiv-1 Virions and Cores. EMBO J. 2003, 22, 1707–1715. 10.1093/emboj/cdg143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayton G. S.; Voth G. A. Multiscale Computer Simulation of the Immature Hiv-1 Virion. Biophys. J. 2010, 99, 2757–2765. 10.1016/j.bpj.2010.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin M. H.; Chen C. P.; Fischer W. B. Patch Formation of a Viral Channel Forming Protein within a Lipid Membrane - Vpu of Hiv-1. Mol. BioSyst. 2016, 12, 1118–1127. 10.1039/C5MB00798D. [DOI] [PubMed] [Google Scholar]
- Charlier L.; Louet M.; Chaloin L.; Fuchs P.; Martinez J.; Muriaux D.; Favard C.; Floquet N. Coarse-Grained Simulations of the Hiv-1 Matrix Protein Anchoring: Revisiting Its Assembly on Membrane Domains. Biophys. J. 2014, 106, 577–585. 10.1016/j.bpj.2013.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzmann N.; Chipot C.; Penin F.; Dehez F. Assessing the Physiological Relevance of Alternate Architectures of the P7 Protein of Hepatitis C Virus in Different Environments. Bioorg. Med. Chem. 2016, 24, 4920–4927. 10.1016/j.bmc.2016.07.063. [DOI] [PubMed] [Google Scholar]
- Shukla A.; Dey D.; Banerjee K.; Nain A.; Banerjee M. The C-Terminal Region of the Non-Structural Protein 2b from Hepatitis a Virus Demonstrates Lipid-Specific Viroporin-Like Activity. Sci. Rep. 2015, 5, 15884. 10.1038/srep15884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs E. L. A.; Gomes R. G. B.; Elhussein M.; Collier W.; Findlow I. S.; Khalid S.; McCormick C. J.; Williamson P. T. F. Interaction between the Ns4b Amphipathic Helix, Ah2, and Charged Lipid Headgroups Alters Membrane Morphology and Ah2 Oligomeric State - Implications for the Hepatitis C Virus Life Cycle. Biochim. Biophys. Acta, Biomembr. 2015, 1848, 1671–1677. 10.1016/j.bbamem.2015.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler D. E.; Penin F.; Schulten K.; Chipot C. The P7 Protein of Hepatitis C Virus Forms Structurally Plastic, Minimalist Ion Channels. PLoS Comput. Biol. 2012, 8, e1002702 10.1371/journal.pcbi.1002702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker M. K.; Gangupomu V. K.; Abrams C. F. Characterization of the Water Defect at the Hiv-1 Gp41 Membrane Spanning Domain in Bilayers with and without Cholesterol Using Molecular Simulations. Biochim. Biophys. Acta, Biomembr. 2014, 1838, 1396–1405. 10.1016/j.bbamem.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akabori K.; Huang K.; Treece B. W.; Jablin M. S.; Maranville B.; Woll A.; Nagle J. F.; Garcia A. E.; Tristram-Nagle S. Hiv-1 Tat Membrane Interactions Probed Using X-Ray and Neutron Scattering, Cd Spectroscopy and Md Simulations. Biochim. Biophys. Acta, Biomembr. 2014, 1838, 3078–3087. 10.1016/j.bbamem.2014.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitiello G.; Falanga A.; Petruk A. A.; Merlino A.; Fragneto G.; Paduano L.; Galdiero S.; D’Errico G. Fusion of Raft-Like Lipid Bilayers Operated by a Membranotropic Domain of the Hsv-Type I Glycoprotein Gh Occurs through a Cholesterol-Dependent Mechanism. Soft Matter 2015, 11, 3003–3016. 10.1039/C4SM02769H. [DOI] [PubMed] [Google Scholar]
- Guardado-Calvo P.; Atkovska K.; Jeffers S. A.; Grau N.; Backovic M.; Perez-Vargas J.; de Boer S. M.; Tortorici M. A.; Pehau-Arnaudet G.; Lepault J.; et al. A Glycerophospholipid-Specific Pocket in the Rvfv Class Ii Fusion Protein Drives Target Membrane Insertion. Science 2017, 358, 663–667. 10.1126/science.aal2712. [DOI] [PubMed] [Google Scholar]
- Del Vecchio K.; Frick C. T.; Gc J. B.; Oda S. I.; Gerstman B. S.; Saphire E. O.; Chapagain P. P.; Stahelin R. V. A Cationic, C-Terminal Patch and Structural Rearrangements in Ebola Virus Matrix Vp40 Protein Control Its Interactions with Phosphatidylserine. J. Biol. Chem. 2018, 293, 3335–3349. 10.1074/jbc.M117.816280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla A.; Padhi A. K.; Gomes J.; Banerjee M. The Vp4 Peptide of Hepatitis a Virus Ruptures Membranes through Formation of Discrete Pores. J. Virol. 2014, 88, 12409–12421. 10.1128/JVI.01896-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Q.; Chen C. L.; Herrmann A. Bilayer Conformation of Fusion Peptide of Influenza Virus Hemagglutinin: A Molecular Dynamics Simulation Study. Biophys. J. 2004, 87, 14–22. 10.1529/biophysj.103.024562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lague P.; Roux B.; Pastor R. W. Molecular Dynamics Simulations of the Influenza Hemagglutinin Fusion Peptide in Micelles and Bilayers: Conformational Analysis of Peptide and Lipids. J. Mol. Biol. 2005, 354, 1129–1141. 10.1016/j.jmb.2005.10.038. [DOI] [PubMed] [Google Scholar]
- Hung H. M.; Hang T. D.; Nguyen M. T. Multiscale Simulations on Conformational Dynamics and Membrane Interactions of the Non-Structural 2 (Ns2) Transmembrane Domain. Biochem. Biophys. Res. Commun. 2016, 478, 193–198. 10.1016/j.bbrc.2016.07.069. [DOI] [PubMed] [Google Scholar]
- Hu Y.; Patel S. Thermodynamics of Cell-Penetrating Hiv1 Tat Peptide Insertion into Pc/Ps/Chol Model Bilayers through Transmembrane Pores: The Roles of Cholesterol and Anionic Lipids. Soft Matter 2016, 12, 6716–6727. 10.1039/C5SM01696G. [DOI] [PubMed] [Google Scholar]
- Apellaniz B.; Rujas E.; Carravilla P.; Requejo-Isidro J.; Huarte N.; Domene C.; Nieva J. L. Cholesterol-Dependent Membrane Fusion Induced by the Gp41 Membrane-Proximal External Region-Transmembrane Domain Connection Suggests a Mechanism for Broad Hiv-1 Neutralization. J. Virol. 2014, 88, 13367–13377. 10.1128/JVI.02151-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worch R.; Krupa J.; Filipek A.; Szymaniec A.; Setny P. Three Conserved C-Terminal Residues of Influenza Fusion Peptide Alter Its Behavior at the Membrane Interface. Biochim. Biophys. Acta, Gen. Subj. 2017, 1861, 97–105. 10.1016/j.bbagen.2016.11.004. [DOI] [PubMed] [Google Scholar]
- Mavioso I. C. V. C.; de Andrade V. C. R.; Carvalho A. J. P.; do Canto A. M. T. M. Molecular Dynamics Simulations of T-2410 and T-2429 Hiv Fusion Inhibitors Interacting with Model Membranes: Insight into Peptide Behavior, Structure and Dynamics. Biophys. Chem. 2017, 228, 69–80. 10.1016/j.bpc.2017.06.012. [DOI] [PubMed] [Google Scholar]
- Risselada H. J. Membrane Fusion Stalks and Lipid Rafts: A Love-Hate Relationship. Biophys. J. 2017, 112, 2475–2478. 10.1016/j.bpj.2017.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victor B. L.; Lousa D.; Antunes J. M.; Soares C. M. Self-Assembly Molecular Dynamics Simulations Shed Light into the Interaction of the Influenza Fusion Peptide with a Membrane Bilayer. J. Chem. Inf. Model. 2015, 55, 795–805. 10.1021/ci500756v. [DOI] [PubMed] [Google Scholar]
- Ruiz-Herrero T.; Hagan M. F. Simulations Show That Virus Assembly and Budding Are Facilitated by Membrane Microdomains. Biophys. J. 2015, 108, 585–595. 10.1016/j.bpj.2014.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingsworth L. R.; Lemkul J. A.; Bevan D. R.; Brown A. M. Hiv-1 Env Gp41 Transmembrane Domain Dynamics Are Modulated by Lipid, Water, and Ion Interactions. Biophys. J. 2018, 115, 84–94. 10.1016/j.bpj.2018.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Oliveira Dos Santos Soares R.; Bortot L. O.; van der Spoel D.; Caliri A. Membrane Vesiculation Induced by Proteins of the Dengue Virus Envelope Studied by Molecular Dynamics Simulations. J. Phys.: Condens. Matter 2017, 29, 504002. 10.1088/1361-648X/aa99c6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers D. M.; Kent M. S.; Rempe S. B. Molecular Basis of Endosomal-Membrane Association for the Dengue Virus Envelope Protein. Biochim. Biophys. Acta, Biomembr. 2015, 1848, 1041–1052. 10.1016/j.bbamem.2014.12.018. [DOI] [PubMed] [Google Scholar]
- Chen G. F.; Xu T. H.; Yan Y.; Zhou Y. R.; Jiang Y.; Melcher K.; Xu H. E. Amyloid Beta: Structure, Biology and Structure-Based Therapeutic Development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. 10.1038/aps.2017.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett P. J.; Song Y. L.; Van Horn W. D.; Hustedt E. J.; Schafer J. M.; Hadziselimovic A.; Beel A. J.; Sanders C. R. The Amyloid Precursor Protein Has a Flexible Transmembrane Domain and Binds Cholesterol. Science 2012, 336, 1168–1171. 10.1126/science.1219988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panahi A.; Bandara A.; Pantelopulos G. A.; Dominguez L.; Straub J. E. Specific Binding of Cholesterol to C99 Domain of Amyloid Precursor Protein Depends Critically on Charge State of Protein. J. Phys. Chem. Lett. 2016, 7, 3535–3541. 10.1021/acs.jpclett.6b01624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nierzwicki L.; Czub J. Specific Binding of Cholesterol to the Amyloid Precursor Protein: Structure of the Complex and Driving Forces Characterized in Molecular Detail. J. Phys. Chem. Lett. 2015, 6, 784–790. 10.1021/acs.jpclett.5b00197. [DOI] [PubMed] [Google Scholar]
- Di Scala C.; Troadec J. D.; Lelievre C.; Garmy N.; Fantini J.; Chahinian H. Mechanism of Cholesterol-Assisted Oligomeric Channel Formation by a Short Alzheimer Beta-Amyloid Peptide. J. Neurochem. 2014, 128, 186–195. 10.1111/jnc.12390. [DOI] [PubMed] [Google Scholar]
- Di Scala C.; Yahi N.; Lelievre C.; Garmy N.; Chahinian H.; Fantini J. Biochemical Identification of a Linear Cholesterol-Binding Domain within Alzheimer’s Beta Amyloid Peptide. ACS Chem. Neurosci. 2013, 4, 509–517. 10.1021/cn300203a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockhart C.; Klimov D. K. Cholesterol Changes the Mechanisms of a Beta Peptide Binding to the Dmpc Bilayer. J. Chem. Inf. Model. 2017, 57, 2554–2565. 10.1021/acs.jcim.7b00431. [DOI] [PubMed] [Google Scholar]
- Song Y. L.; Hustedt E. J.; Brandon S.; Sanders C. R. Competition between Homodimerization and Cholesterol Binding to the C99 Domain of the Amyloid Precursor Protein. Biochemistry 2013, 52, 5051–5064. 10.1021/bi400735x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y.; Xu T. H.; Harikumar K. G.; Miller L. J.; Melcher K.; Xu H. E. Dimerization of the Transmembrane Domain of Amyloid Precursor Protein Is Determined by Residues around the Gamma - Secretase Cleavage Sites. J. Biol. Chem. 2017, 292, 15826–15837. 10.1074/jbc.M117.789669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J. X.; Yau W. M.; Tycko R. Evidence from Solid-State Nmr for Nonhelical Conformations in the Transmembrane Domain of the Amyloid Precursor Protein. Biophys. J. 2011, 100, 711–719. 10.1016/j.bpj.2010.12.3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y.; Mittendorf K. F.; Lu Z.; Sanders C. R. Impact of Bilayer Lipid Composition on the Structure and Topology of the Transmembrane Amyloid Precursor C99 Protein. J. Am. Chem. Soc. 2014, 136, 4093–4096. 10.1021/ja4114374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez L.; Foster L.; Meredith S. C.; Straub J. E.; Thirumalai D. Structural Heterogeneity in Transmembrane Amyloid Precursor Protein Homodimer Is a Consequence of Environmental Selection. J. Am. Chem. Soc. 2014, 136, 9619–9626. 10.1021/ja503150x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez L.; Meredith S. C.; Straub J. E.; Thirumalai D. Transmembrane Fragment Structures of Amyloid Precursor Protein Depend on Membrane Surface Curvature. J. Am. Chem. Soc. 2014, 136, 854–857. 10.1021/ja410958j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmin T.; Dimitrov M.; Fraering P. C.; Dal Peraro M. Perturbations of the Straight Transmembrane Alpha-Helical Structure of the Amyloid Precursor Protein Affect Its Processing by Gamma-Secretase. J. Biol. Chem. 2014, 289, 6763–6774. 10.1074/jbc.M113.470781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez L.; Foster L.; Straub J. E.; Thirumalai D. Impact of Membrane Lipid Composition on the Structure and Stability of the Transmembrane Domain of Amyloid Precursor Protein. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, E5281–E5287. 10.1073/pnas.1606482113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantelopulos G. A.; Straub J. E.; Thirumalai D.; Sugita Y. Structure of App-C991–99 and Implications for Role of Extra-Membrane Domains in Function and Oligomerization. Biochim. Biophys. Acta, Biomembr. 2018, 1860, 1698–1708. 10.1016/j.bbamem.2018.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun F.; Chen L.; Wei P.; Chai M.; Ding X.; Xu L.; Luo S. Z. Dimerization and Structural Stability of Amyloid Precursor Proteins Affected by the Membrane Microenvironments. J. Chem. Inf. Model. 2017, 57, 1375–1387. 10.1021/acs.jcim.7b00196. [DOI] [PubMed] [Google Scholar]
- Audagnotto M.; Lemmin T.; Barducci A.; Dal Peraro M. Effect of the Synaptic Plasma Membrane on the Stability of the Amyloid Precursor Protein Homodimer. J. Phys. Chem. Lett. 2016, 7, 3572–3578. 10.1021/acs.jpclett.6b01721. [DOI] [PubMed] [Google Scholar]
- Audagnotto M.; Lorkowski A. K.; Dal Peraro M. Recruitment of the Amyloid Precursor Protein by Gamma-Secretase at the Synaptic Plasma Membrane. Biochem. Biophys. Res. Commun. 2018, 498, 334–341. 10.1016/j.bbrc.2017.10.164. [DOI] [PubMed] [Google Scholar]
- Bai X. C.; Yan C.; Yang G.; Lu P.; Ma D.; Sun L.; Zhou R.; Scheres S. H. W.; Shi Y. An Atomic Structure of Human Gamma-Secretase. Nature 2015, 525, 212–217. 10.1038/nature14892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong R.; Chang S.; Xia W. M.; Wong S. T. C. Molecular Dynamics Simulation Study Reveals Potential Substrate Entry Path into Gamma-Secretase/Presenilin-1. J. Struct. Biol. 2015, 191, 120–129. 10.1016/j.jsb.2015.07.001. [DOI] [PubMed] [Google Scholar]
- MacKenzie K. R.; Prestegard J. H.; Engelman D. M. A Transmembrane Helix Dimer: Structure and Implications. Science 1997, 276, 131–133. 10.1126/science.276.5309.131. [DOI] [PubMed] [Google Scholar]
- Trenker R.; Call M. E.; Call M. J. Crystal Structure of the Glycophorin a Transmembrane Dimer in Lipidic Cubic Phase. J. Am. Chem. Soc. 2015, 137, 15676–15679. 10.1021/jacs.5b11354. [DOI] [PubMed] [Google Scholar]
- Smith S. O.; Eilers M.; Song D.; Crocker E.; Ying W. W.; Groesbeek M.; Metz G.; Ziliox M.; Aimoto S. Implications of Threonine Hydrogen Bonding in the Glycophorin a Transmembrane Helix Dimer. Biophys. J. 2002, 82, 2476–2486. 10.1016/S0006-3495(02)75590-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming K. G.; Engelman D. M. Specificity in Transmembrane Helix-Helix Interactions Can Define a Hierarchy of Stability for Sequence Variants. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 14340–14344. 10.1073/pnas.251367498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassenaar T. A.; Pluhackova K.; Moussatova A.; Sengupta D.; Marrink S. J.; Tieleman D. P.; Bockmann R. A. High-Throughput Simulations of Dimer and Trimer Assembly of Membrane Proteins. The Daft Approach. J. Chem. Theory Comput. 2015, 11, 2278–2291. 10.1021/ct5010092. [DOI] [PubMed] [Google Scholar]
- Domanski J.; Sansom M. S. P.; Stansfeld P. J.; Best R. B. Balancing Force Field Protein-Lipid Interactions to Capture Transmembrane Helix-Helix Association. J. Chem. Theory Comput. 2018, 14, 1706–1715. 10.1021/acs.jctc.7b00983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cybulski L. E.; Ballering J.; Moussatova A.; Inda M. E.; Vazquez D. B.; Wassenaar T. A.; de Mendoza D.; Tieleman D. P.; Killian J. A. Activation of the Bacterial Thermosensor Desk Involves a Serine Zipper Dimerization Motif That Is Modulated by Bilayer Thickness. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 6353–6358. 10.1073/pnas.1422446112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covino R.; Ballweg S.; Stordeur C.; Michaelis J. B.; Puth K.; Wernig F.; Bahrami A.; Ernst A. M.; Hummer G.; Ernst R. A Eukaryotic Sensor for Membrane Lipid Saturation. Mol. Cell 2016, 63, 49–59. 10.1016/j.molcel.2016.05.015. [DOI] [PubMed] [Google Scholar]
- Zhou K.; Dichlberger A.; Martinez-Seara H.; Nyholm T. K. M.; Li S.; Kim Y. A.; Vattulainen I.; Ikonen E.; Blom T. A Ceramide-Regulated Element in the Late Endosomal Protein Laptm4b Controls Amino Acid Transporter Interaction. ACS Cent. Sci. 2018, 4, 548–558. 10.1021/acscentsci.7b00582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blom T.; Li S. Q.; Dichlberger A.; Back N.; Kim Y. A.; Loizides-Mangold U.; Riezman H.; Bittman R.; Ikonen E. Laptm4b Facilitates Late Endosomal Ceramide Export to Control Cell Death Pathways. Nat. Chem. Biol. 2015, 11, 799–806. 10.1038/nchembio.1889. [DOI] [PubMed] [Google Scholar]
- Venter H.; Mowla R.; Ohene-Agyei T.; Ma S. T. Rnd-Type Drug Efflux Pumps from Gram-Negative Bacteria: Molecular Mechanism and Inhibition. Front. Microbiol. 2015, 6, 377. 10.3389/fmicb.2015.00377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travers T.; Wang K. J.; Lopez C. A.; Gnanakaran S. Sequence- and Structure-Based Computational Analyses of Gram-Negative Tripartite Efflux Pumps in the Context of Bacterial Membranes. Res. Microbiol. 2018, 169, 414–424. 10.1016/j.resmic.2018.01.002. [DOI] [PubMed] [Google Scholar]
- Du D.; Wang Z.; James N. R.; Voss J. E.; Klimont E.; Ohene-Agyei T.; Venter H.; Chiu W.; Luisi B. F. Structure of the Acrab-Tolc Multidrug Efflux Pump. Nature 2014, 509, 512–515. 10.1038/nature13205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargiu A. V.; Ramaswamy V. K.; Malloci G.; Malvacio I.; Atzori A.; Ruggerone P. Computer Simulations of the Activity of Rnd Efflux Pumps. Res. Microbiol. 2018, 169, 384–392. 10.1016/j.resmic.2017.12.001. [DOI] [PubMed] [Google Scholar]
- Schuldiner S. Emre, a Model for Studying Evolution and Mechanism of Ion-Coupled Transporters. Biochim. Biophys. Acta, Proteins Proteomics 2009, 1794, 748–762. 10.1016/j.bbapap.2008.12.018. [DOI] [PubMed] [Google Scholar]
- Nathoo S.; Litzenberger J. K.; Bay D. C.; Turner R. J.; Prenner E. J. Visualizing a Multidrug Resistance Protein, Emre, with Major Bacterial Lipids Using Brewster Angle Microscopy. Chem. Phys. Lipids 2013, 167, 33–42. 10.1016/j.chemphyslip.2013.01.007. [DOI] [PubMed] [Google Scholar]
- Dutta S.; Morrison E. A.; Henzler-Wildman K. A. Emre Dimerization Depends on Membrane Environment. Biochim. Biophys. Acta, Biomembr. 2014, 1838, 1817–1822. 10.1016/j.bbamem.2014.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banigan J. R.; Leninger M.; Her A. S.; Traaseth N. J. Assessing Interactions between a Polytopic Membrane Protein and Lipid Bilayers Using Differential Scanning Calorimetry and Solid-State Nmr. J. Phys. Chem. B 2018, 122, 2314–2322. 10.1021/acs.jpcb.8b00479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padariya M.; Kalathiya U.; Baginski M. Structural and Dynamic Insights on the Emre Protein with Tpp(+) and Related Substrates through Molecular Dynamics Simulations. Chem. Phys. Lipids 2018, 212, 1–11. 10.1016/j.chemphyslip.2017.12.004. [DOI] [PubMed] [Google Scholar]
- Shoshan-Barmatz V.; Keinan N.; Zaid H. Uncovering the Role of Vdac in the Regulation of Cell Life and Death. J. Bioenerg. Biomembr. 2008, 40, 183–191. 10.1007/s10863-008-9147-9. [DOI] [PubMed] [Google Scholar]
- Zaid H.; Abu-Hamad S.; Israelson A.; Nathan I.; Shoshan-Barmatz V. The Voltage-Dependent Anion Channel-1 Modulates Apoptotic Cell Death. Cell Death Differ. 2005, 12, 751–760. 10.1038/sj.cdd.4401599. [DOI] [PubMed] [Google Scholar]
- Hiller S.; Garces R. G.; Malia T. J.; Orekhov V. Y.; Colombini M.; Wagner G. Solution Structure of the Integral Human Membrane Protein Vdac-1 in Detergent Micelles. Science 2008, 321, 1206–1210. 10.1126/science.1161302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ujwal R.; Cascio D.; Colletier J. P.; Faham S.; Zhang J.; Toro L.; Ping P. P.; Abramson J. The Crystal Structure of Mouse Vdac1 at 2.3 Angstrom Resolution Reveals Mechanistic Insights into Metabolite Gating. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 17742–17747. 10.1073/pnas.0809634105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayrhuber M.; Meins T.; Habeck M.; Becker S.; Giller K.; Villinger S.; Vonrhein C.; Griesinger C.; Zweckstetter M.; Zeth K. Structure of the Human Voltage-Dependent Anion Channel. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 15370–15375. 10.1073/pnas.0808115105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostovtseva T. K.; Bezrukov S. M. Vdac Regulation: Role of Cytosolic Proteins and Mitochondrial Lipids. J. Bioenerg. Biomembr. 2008, 40, 163–170. 10.1007/s10863-008-9145-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villinger S.; Briones R.; Giller K.; Zachariae U.; Lange A.; de Groot B. L.; Griesinger C.; Becker S.; Zweckstetter M. Functional Dynamics in the Voltage-Dependent Anion Channel. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 22546–22551. 10.1073/pnas.1012310108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiser B. P.; Salari R.; Eckenhoff R. G.; Brannigan G. Computational Investigation of Cholesterol Binding Sites on Mitochondrial Vdac. J. Phys. Chem. B 2014, 118, 9852–9860. 10.1021/jp504516a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy M. T.; Ong T. C.; Clark L.; Teijido O.; van der Wel P. C. A.; Garces R.; Wagner G.; Rostovtseva T. K.; Griffin R. G. Lipid Dynamics and Protein-Lipid Interactions in 2d Crystals Formed with the Beta-Barrel Integral Membrane Protein Vdac1. J. Am. Chem. Soc. 2012, 134, 6375–6387. 10.1021/ja300347v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mlayeh L.; Chatkaew S.; Leonetti M.; Homble F. Modulation of Plant Mitochondrial Vdac by Phytosterols. Biophys. J. 2010, 99, 2097–2106. 10.1016/j.bpj.2010.07.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mlayeh L.; Krammer E. M.; Leonetti M.; Prevost M.; Homble F. The Mitochondrial Vdac of Bean Seeds Recruits Phosphatidylethanolamine Lipids for Its Proper Functioning. Biochim. Biophys. Acta, Bioenerg. 2017, 1858, 786–794. 10.1016/j.bbabio.2017.06.005. [DOI] [PubMed] [Google Scholar]
- Young J. A. T.; Collier R. J. Anthrax Toxin: Receptor Binding, Internalization, Pore Formation, and Translocation. Annu. Rev. Biochem. 2007, 76, 243–265. 10.1146/annurev.biochem.75.103004.142728. [DOI] [PubMed] [Google Scholar]
- Kalu N.; Atsmon-Raz Y.; Momben Abolfath S.; Lucas L.; Kenney C.; Leppla S. H.; Tieleman D. P.; Nestorovich E. M. Effect of Late Endosomal Dobmp Lipid and Traditional Model Lipids of Electrophysiology on the Anthrax Toxin Channel Activity. Biochim. Biophys. Acta, Biomembr. 2018, 1860, 2192–2203. 10.1016/j.bbamem.2018.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berube B. J.; Wardenburg J. B. Staphylococcus Aureus Alpha-Toxin: Nearly a Century of Intrigue. Toxins 2013, 5, 1140–1166. 10.3390/toxins5061140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guros N. B.; Balijepalli A.; Klauda J. B. The Role of Lipid Interactions in Simulations of the Alpha-Hemolysin Ion-Channel-Forming Toxin. Biophys. J. 2018, 115, 1720–1730. 10.1016/j.bpj.2018.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K.; Caaveiro J. M. M.; Morante K.; Gonzalez-Manas J. M.; Tsumoto K.. Structural Basis for Self-Assembly of a Cytolytic Pore Lined by Protein and Lipid. Nat. Commun. 2015, 6. 10.1038/ncomms7337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesa-Galloso H.; Delgado-Magnero K. H.; Cabezas S.; Lopez-Castilla A.; Hernandez-Gonzalez J. E.; Pedrera L.; Alvarez C.; Tieleman D. P.; Garcia-Saez A. J.; Lanio M. E.; et al. Disrupting a Key Hydrophobic Pair in the Oligomerization Interface of the Actinoporins Impairs Their Pore-Forming Activity. Protein Sci. 2017, 26, 550–565. 10.1002/pro.3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ermakova E.; Kurbanov R.; Zuev Y. Coarse-Grained Molecular Dynamics of Membrane Semitoroidal Pore Formation in Model Lipid-Peptide Systems. J. Mol. Graphics Modell. 2019, 87, 1–10. 10.1016/j.jmgm.2018.11.003. [DOI] [PubMed] [Google Scholar]
- Zimmerman B.; Kelly B.; McMillan B. J.; Seegar T. C. M.; Dror R. O.; Kruse A. C.; Blacklow S. C. Crystal Structure of a Full-Length Human Tetraspanin Reveals a Cholesterol-Binding Pocket. Cell 2016, 167, 1041–1051. 10.1016/j.cell.2016.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manat G.; Roure S.; Auger R.; Bouhss A.; Barreteau H.; Mengin-Lecreulx D.; Touze T. Deciphering the Metabolism of Undecaprenyl-Phosphate: The Bacterial Cell-Wall Unit Carrier at the Membrane Frontier. Microb. Drug Resist. 2014, 20, 199–214. 10.1089/mdr.2014.0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H. Y.; Chou C. C.; Hsu M. F.; Wang A. H. Proposed Carrier Lipid-Binding Site of Undecaprenyl Pyrophosphate Phosphatase from Escherichia Coli. J. Biol. Chem. 2014, 289, 18719–18735. 10.1074/jbc.M114.575076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Eerden F. J.; Melo M. N.; Frederix P.; Marrink S. J. Prediction of Thylakoid Lipid Binding Sites on Photosystem Ii. Biophys. J. 2017, 113, 2669–2681. 10.1016/j.bpj.2017.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Eerden F. J.; Melo M. N.; Frederix P. W. J. M.; Periole X.; Marrink S. J. Exchange Pathways of Plastoquinone and Plastoquinol in the Photosystem Ii Complex. Nat. Commun. 2017, 8, 15214. 10.1038/ncomms15214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapoport T. A.; Li L.; Park E. Structural and Mechanistic Insights into Protein Translocation. Annu. Rev. Cell Dev. Biol. 2017, 33, 369–390. 10.1146/annurev-cellbio-100616-060439. [DOI] [PubMed] [Google Scholar]
- Knyazev D. G.; Kuttner R.; Zimmermann M.; Sobakinskaya E.; Pohl P. Driving Forces of Translocation through Bacterial Translocon Secyeg. J. Membr. Biol. 2018, 251, 329–343. 10.1007/s00232-017-0012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corey R. A.; Pyle E.; Allen W. J.; Watkins D. W.; Casiraghi M.; Miroux B.; Arechaga I.; Politis A.; Collinson I. Specific Cardiolipin-Secy Interactions Are Required for Proton-Motive Force Stimulation of Protein Secretion. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 7967–7972. 10.1073/pnas.1721536115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalbey R. E.; Kuhn A.; Zhu L.; Kiefer D. The Membrane Insertase Yidc. Biochim. Biophys. Acta, Mol. Cell Res. 2014, 1843, 1489–1496. 10.1016/j.bbamcr.2013.12.022. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Capponi S.; Zhu L.; Gellenbeck P.; Freites J. A.; White S. H.; Dalbey R. E. Yidc Insertase of Escherichia Coli: Water Accessibility and Membrane Shaping. Structure 2017, 25, 1403–1414. 10.1016/j.str.2017.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B.; Miller T. F. Long-Timescale Dynamics and Regulation of Sec-Facilitated Protein Translocation. Cell Rep. 2012, 2, 927–937. 10.1016/j.celrep.2012.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall S. S.; Niesen M. J. M.; Muller A.; Tiemann K.; Saladi S. M.; Galimidi R. P.; Zhang B.; Clemons W. M.; Miller T. F. A Link between Integral Membrane Protein Expression and Simulated Integration Efficiency. Cell Rep. 2016, 16, 2169–2177. 10.1016/j.celrep.2016.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Lehn R. C.; Zhang B.; Miller T. F. 3rd. Regulation of Multispanning Membrane Protein Topology Via Post-Translational Annealing. eLife 2015, 4, e08697 10.7554/eLife.08697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niesen M. J. M.; Wang C. Y.; Van Lehn R. C.; Miller TF I. I. I. Structurally Detailed Coarse-Grained Model for Sec-Facilitated Co-Translational Protein Translocation and Membrane Integration. PLoS Comput. Biol. 2017, 13, e1005427. 10.1371/journal.pcbi.1005427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fathizadeh A.; Elber R. A Mixed Alchemical and Equilibrium Dynamics to Simulate Heterogeneous Dense Fluids: Illustrations for Lennard-Jones Mixtures and Phospholipid Membranes. J. Chem. Phys. 2018, 149, 072325. 10.1063/1.5027078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan C. K.; Han W.; Wu Y. D. Parameterization of Pace Force Field for Membrane Environment and Simulation of Helical Peptides and Helix-Helix Association. J. Chem. Theory Comput. 2012, 8, 300–313. 10.1021/ct2004275. [DOI] [PubMed] [Google Scholar]
- Wassenaar T. A.; Ingolfsson H. I.; Priess M.; Marrink S. J.; Schafer L. V. Mixing Martini: Electrostatic Coupling in Hybrid Atomistic-Coarse-Grained Biomolecular Simulations. J. Phys. Chem. B 2013, 117, 3516–3530. 10.1021/jp311533p. [DOI] [PubMed] [Google Scholar]
- Mardt A.; Pasquali L.; Wu H.; Noe F. Vampnets for Deep Learning of Molecular Kinetics. Nat. Commun. 2018, 9, 5. 10.1038/s41467-017-02388-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J.; Rauscher S.; Nawrocki G.; Ran T.; Feig M.; de Groot B. L.; Grubmuller H.; MacKerell A. D. Charmm36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2017, 14, 71–73. 10.1038/nmeth.4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong H.; Blois T. M.; Cao Z.; Bowie J. U. Method to Measure Strong Protein-Protein Interactions in Lipid Bilayers Using a Steric Trap. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 19802–19807. 10.1073/pnas.1010348107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoval-Perez A.; Pluhackova K.; Bockmann R. A. Critical Comparison of Biomembrane Force Fields: Protein-Lipid Interactions at the Membrane Interface. J. Chem. Theory Comput. 2017, 13, 2310–2321. 10.1021/acs.jctc.7b00001. [DOI] [PubMed] [Google Scholar]
- Petrov D.; Zagrovic B. Are Current Atomistic Force Fields Accurate Enough to Study Proteins in Crowded Environments?. PLoS Comput. Biol. 2014, 10, e1003638. 10.1371/journal.pcbi.1003638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javanainen M.; Martinez-Seara H.; Vattulainen I. Excessive Aggregation of Membrane Proteins in the Martini Model. PLoS One 2017, 12, e0187936. 10.1371/journal.pone.0187936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming K. G. Energetics of Membrane Protein Folding. Annu. Rev. Biophys. 2014, 43, 233–255. 10.1146/annurev-biophys-051013-022926. [DOI] [PubMed] [Google Scholar]
- Frisz J. F.; Lou K. Y.; Klitzing H. A.; Hanafin W. P.; Lizunov V.; Wilson R. L.; Carpenter K. J.; Kim R.; Hutcheon I. D.; Zimmerberg J.; et al. Direct Chemical Evidence for Sphingolipid Domains in the Plasma Membranes of Fibroblasts. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, E613–E622. 10.1073/pnas.1216585110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyman E.; Hsieh C. L.; Eggeling C. From Dynamics to Membrane Organization: Experimental Breakthroughs Occasion a ″Modeling Manifesto’’. Biophys. J. 2018, 115, 595–604. 10.1016/j.bpj.2018.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorr J. M.; Scheidelaar S.; Koorengevel M. C.; Dominguez J. J.; Schafer M.; van Walree C. A.; Killian J. A. The Styrene-Maleic Acid Copolymer: A Versatile Tool in Membrane Research. Eur. Biophys. J. 2016, 45, 3–21. 10.1007/s00249-015-1093-y. [DOI] [PMC free article] [PubMed] [Google Scholar]