

Abstract
MYH9-related disease (MYH9-RD) is a rare, autosomal dominant disorder caused by mutations in MYH9, the gene encoding the actin-activated motor protein non-muscle myosin IIA (NMIIA). MYH9-RD patients suffer from bleeding syndromes, progressive kidney disease, deafness, and/or cataracts, but the impact of MYH9 mutations on other NMIIA-expressing tissues remains unknown. In human red blood cells (RBCs), NMIIA assembles into bipolar filaments and binds to actin filaments (F-actin) in the spectrin-F-actin membrane skeleton to control RBC biconcave disk shape and deformability. Here, we tested the effects of MYH9 mutations in different NMIIA domains (motor, coiled-coil rod, or non-helical tail) on RBC NMIIA function. We found that MYH9-RD does not cause clinically significant anemia and that patient RBCs have normal osmotic deformability as well as normal membrane skeleton composition and micron-scale distribution. However, analysis of complete blood count data and peripheral blood smears revealed reduced hemoglobin content and elongated shapes, respectively, of MYH9-RD RBCs. Patients with mutations in the NMIIA motor domain had the highest numbers of elongated RBCs. Patients with mutations in the motor domain also had elevated association of NMIIA with F-actin at the RBC membrane. Our findings support a central role for motor domain activity in NMIIA regulation of RBC shape and define a new sub-clinical phenotype of MYH9-RD.
Keywords: erythrocyte shape, actomyosin contractility, nonmuscle myosin IIA, cytoskeleton, spectrin-actin membrane skeleton
Introduction
Mutations in MYH9 – the gene encoding the heavy chain of non-muscle myosin IIA (NMIIA) – lead to a rare, autosomal dominant disorder known as MYH9-related disease (MYH9-RD, MIM #160775) 1–3. All MYH9-RD patients have reduced platelet counts and enlarged platelets (macrothrombocytopenia), as well as NMIIA-containing inclusions in leukocytes (Döhle bodies). Some patients also develop bleeding tendencies, nephropathy, sensorineural deafness, and/or pre-senile cataracts 4–6. Although NMIIA is widely expressed 7, no additional major hematological or extra-hematological pathologies have been identified.
MYH9-RD mutations (over 80 identified to date) occur in both the force-generating NMIIA motor domain and the filament-forming rod/tail domain 8. Motor domain mutations commonly affect two subdomains: the SH3/motor domain interface, which stabilizes motor domain folding and promotes ATPase activity, or the SH1 helix, which coordinates the NMIIA power stroke 9–12. Rod/tail domain mutations affect either the coiled-coil rod subdomain or the regulatory non-helical tail domain (Fig. 1, 13,14). Genotype-phenotype correlation studies have demonstrated that motor domain mutations cause the most severe disease with the highest probability of extra-hematological symptoms, while mutations in the non-helical tail domain cause the mildest disease 13,15. Different mutations within the same domain or affecting the same residue can also have varying effects on disease severity and progression 13.
Figure 1.
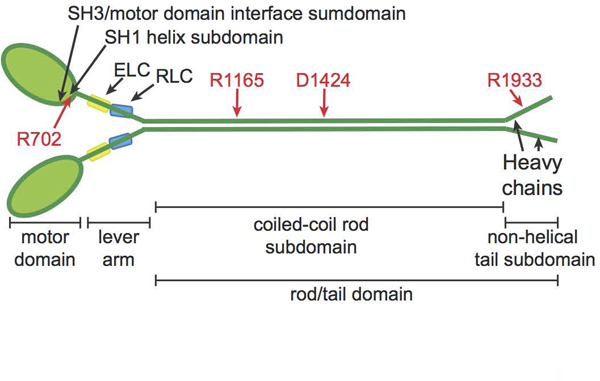
NMIIA molecule structure with domains and residues used in MYH9-RD genotype-phenotype comparisons. NMIIA heterohexameric molecules contain two heavy chains (green). Each heavy chain consists of an N-terminal motor domain with F-actin-activated ATPase activity, a flexible lever arm, a rod domain that mediates coiled-coil formation of the two heavy chains, and a non-helical tail with regulatory functions. MYH9-RD mutations occur in all domains, with motor domain mutations commonly in two subdomains: the SH3/motor domain interface and the SH1 helix. Each lever arm binds an essential light chain (ELC, yellow) and a regulatory light chain (RLC, blue). Four residues commonly affected by MYH9-RD mutations are denoted in red.
The major cell types affected by MYH9-RD (platelets and kidney podocytes) exclusively express NMIIA over the NMIIB and NMIIC isoforms and rely on specialized actin cytoskeletal structures to support complex cellular morphologies and functions 16–18. Similar to these cell types, human red blood cells (RBCs) predominantly express NMIIA 17,19,20. RBCs rely on the membrane skeleton, a quasi-hexagonal network of short (~37 nm) actin filament (F-actin) nodes interconnected by long, flexible (α1β1)2-spectrin tetramers. This network underlies the plasma membrane and supports RBC biconcave disk shape and deformability 21–23.
In RBCs, as in other cells, NMIIA forms heterohexamers (termed NMIIA molecules) of two heavy chains, two regulatory light chains, and two essential light chains (Fig. 1) 19,24. These NMIIA molecules form bipolar filaments that associate with membrane skeleton F-actin via their motor domains 20. Inhibition of NMIIA motor activity with blebbistatin reduces NMIIA-membrane skeleton association, leads to a loss of RBC biconcave disk shape (with cell elongation and a reduced dimple), and increases RBC deformability 20. This indicates a role for NMIIA motor activity in maintaining tension in the membrane skeleton. The impact of MYH9-RD mutations – which impair specific aspects of NMIIA activity and filament assembly 25,26 – on RBC shape and deformability remains unknown.
In this study, we investigate the relationship between MYH9 genotype and RBC NMIIA function in a cohort of 113 MYH9-RD patients (Table S1). These patients do not present clinically significant anemia and have normal membrane skeleton composition and organization, heavy chain and regulatory light chain phosphorylation, and osmotic deformability. However, patient RBCs exhibit abnormal sizes and elongated morphologies, based on RBC indices in the complete blood count (CBC) and light microscopy data from blood smears and confocal microscopy. Mutations in the motor domain cause the most pronounced RBC shape changes, agreeing with genotype-phenotype correlations in other cell types. Motor domain mutations also increase NMIIA association with the Triton-insoluble membrane skeleton, suggesting that enhanced NMIIA motor domain binding to F-actin underlies the elongated RBC morphology. Our results provide insights into the role of each domain in NMIIA function and define a new sub-clinical phenotype that could be important when MYH9-RD co-occurs with other conditions.
Materials and Methods
Blood collection, hematology, and shipping.
Whole blood was collected from healthy human donors or MYH9-RD patients into EDTA tubes by Dr. Remi Favier at Armand Trousseau Children’s Hospital in Paris, France or by Dr. Alessandro Pecci at the University of Pavia in Pavia, Italy. Complete blood counts (CBCs) were determined with automated hematology analyzers at the point of collection, and peripheral blood smears were prepared and stained with Wright-Giemsa (Sigma-Aldrich). Additional EDTA tubes containing whole blood were collected at the same time and shipped overnight at 4 degrees to the Fowler laboratory at The Scripps Research Institute (TSRI) in La Jolla, California, where osmotic fragility assays 27, ghost preparation, or fixation for confocal or TIRF imaging (see below) were performed upon arrival. The blood smear slides were shipped separately to the Fowler laboratory for image analysis. Osmotic ektacytometry assays 28 were performed on RBCs from blood shipped to Dr. Lydie Da Costa’s Hematology laboratory at Debré hospital in Paris, France.
Blood smear imaging and analysis.
Wright-Giemsa-stained peripheral blood smears were mounted in Permount (Fisher Scientific) and imaged using a Keyence BZ-X700 microscope with a 100x Plan Apochromat oil objective (NA 1.45). Images of smears were analyzed using the Hybrid Cell Count module of the BZ-X Analyzer software. The “Fill holes” feature was used to fill in the RBC central pallor regions, which were initially excluded by the Hybrid Cell Count. RBCs close to the image edge or touching other RBCs were automatically excluded based on size. RBCs whose shapes were distorted due to close proximity to other RBCs were then manually eliminated. The major axis and minor axes of the remaining cells were then recorded, and the aspect ratios were computed by dividing the major axis by the minor axis.
Immunofluorescence staining of RBCs.
Whole blood was collected from healthy human donors or MYH9-RD patients and shipped overnight to TSRI as explained above. RBC membranes were stained with fluorescent antibodies to glycophorin A (GPA) as follows. 25 μL of whole blood was added to 1 mL of 4% paraformaldehyde (PFA, Electron Microscopy Sciences) in Dulbecco’s PBS (DPBS – Gibco [2.67 mM KCl, 1.47 mM KH2PO4, 137.93 mM NaCl, 8.06 mM Na2HPO4-7H2O]), mixed, and incubated at room temperature overnight. Fixed RBCs were washed three times in DPBS by centrifuging for 5 minutes at 1000 x g and then blocked in 4% BSA (MP Biomedicals), 1% normal goat serum (Gibco) in DPBS (blocking buffer) at room temperature for one hour. Blocked RBCs were stained with a FITC-conjugated mouse anti-GPA antibody (BD Pharmingen 559943) in blocking buffer for one hour at room temperature and then washed three times in DPBS. GPA-stained RBCs were deposited on glass slides at 1000 rpm for 3 minutes using a Cytospin 4 machine (Thermo Scientific) and mounted with ProLong™ Gold mounting media and coverslipped prior to imaging.
In some experiments, RBC membranes were labeled with wheat germ agglutinin (WGA), which binds predominantly to GPA, the major sialic acid containing glycoprotein in RBCs. For WGA staining, shipped whole blood was washed three times in DPBS. Packed RBCs were then diluted (1:500) in DPBS and incubated in Alexa 555-conjugated WGA (Life Technologies W32464 at a final concentration of 1μg/ml) for 30 minutes at room temperature. The WGA-stained RBCs were then washed one time in DPBS and re-suspended in 4% PFA and fixed overnight at room temperature in the dark. RBCs were then washed three times in DPBS by centrifugation as above, cytospun onto glass slides and mounted with ProLong™ Gold mounting media and coverslipped prior to imaging.
For TIRF imaging of RBCs, the glass bottoms of 35mm tissue culture dishes with 0.17mm-thick cover glass bottoms (FluoroDish FD35–100) were coated with a 0.1% (w/v) solution of poly-L-lysine (Sigma-Aldrich P8920) for 30 minutes at room temperature. The poly-L-lysine solution was then removed and the cover glass was allowed to dry. 25μl whole blood was washed three times in DPBS. The RBC count in a 1:1000 dilution of whole blood in DPBS was determined using the Bio-Rad TC20™ Automated Cell Counter. This cell count was used to make a dilution of approximately one million RBCs in 500 μl of DPBS, which was pipetted onto the coated cover glass. RBCs were allowed to adhere to the poly-L-lysine for 30 minutes at room temperature. Non-adherent RBCs were then removed by aspirating the DPBS, and the adherent RBCs were fixed in 1 ml 4% PFA in DPBS for 30 minutes at room temperature. RBCs were then washed three times by pipetting and aspiration of 1 ml DPBS, permeabilized in 1 ml DPBS + 0.3% TX-100 for ten minutes, then incubated in blocking buffer for two hours. Permeabilized and blocked RBCs were incubated with mouse anti-α1-spectrin antibody (Abcam ab11751) in blocking buffer overnight at 4°C. RBCs were then washed two times in DPBS and incubated in Alexa-488-conjugated goat anti-mouse secondary antibody (Life Technologies A11001) in blocking buffer mixed with rhodamine phalloidin (Life Technologies R415) at a final concentration of 130nM at room temperature for one hour followed by two washes in DPBS. 2 mL of DPBS containing 0.02% sodium azide were added to the dishes to preserve the cells. Dishes were stored at 4°C and imaged within 1–2 days of staining.
Fluorescence microscopy.
RBC shape measurements of GPA- or WGA-stained RBCs were obtained from confocal Z-stacks acquired on a Zeiss LSM780 laser scanning confocal microscope using a 100x Plan Apochromat objective with 1.4 NA. Z-stacks were acquired at a digital zoom of 1.0 and a Z-step size of 0.25 μm.
RBCs immunostained for α1-spectrin and rhodamine phalloidin for F-actin were imaged using a Nikon Eclipse Ti inverted microscope with a 100x Apochromat oil objective (NA 1.49) using TIRF illumination with 488 and 561 laser lines and an ORCA-Flash 4.0 V2 Digital CMOS camera (Hamamatsu). Images were acquired using NIS-Elements 4.1 software.
Fluorescence image analysis.
RBC shape measurements were as previously described 20. Aspect ratios were measured from maximum intensity projections of Z-stacks using ImageJ and height measurements were acquired manually from XZ views of the center of each RBC in Volocity (version 6.3) by using the line function to measure the distance between the edges of fluorescent signal at the widest (rim) and narrowest (dimple) regions of each RBC. To correct for Z-stretch, the distance between Z-steps was set to 0.18 μm prior to recording height measurements. Stomatocytic RBCs, which were likely an artifact of shipping and occurred at similar frequencies in normal control and MYH9-RD samples, were excluded from 2D and 3D shape analysis.
Preparation of Mg++ ghosts for biochemical analyses.
Whole blood was collected from healthy human donors or MYH9-RD patients and shipped overnight as above. As previously described 20, blood was added to 5 volumes of ice-cold PBS/Dextran (150 mM NaCl, 5 mM NaHPO4 pH 7.4, 0.75% dextran [Sigma 31392], 0.02% NaN3), then settled at 1 × g to separate the RBCs from white blood cells and platelets 29. The sedimented RBCs were washed four times in ice-cold PBS (150 mM NaCl, 10 mM NaHPO4, pH 7.4) by centrifuging for 5 min at 600 × g and resuspending in PBS. Packed RBCs were then lysed in 40 volumes of ice-cold hypotonic lysis buffer containing magnesium (5 mM NaHPO4 pH 7.4, 2 mM MgCl2, 1 mM DTT, 20 μg/ml PMSF) while vortexing vigorously, followed by centrifuging for 10 min at 39,000 × g at 4°C to sediment the RBC membranes (Mg++ghosts) 30. The pellet was resuspended in ice-cold lysis buffer while vortexing, and membranes sedimented as above, for a total of 4 washes, to remove hemoglobin and other cytosolic proteins. Ghosts were then aliquoted, flash-frozen in liquid nitrogen, and stored at −80°C until use.
Preparation of membrane skeletons by TX-100 extraction of Mg++ ghosts.
As previously described 20, five volumes of ice-cold TX-100 extraction buffer (0.2% TX-100 [Sigma], 2 mM MgCl2, 1 mM EGTA, 10 mM NaHPO4 pH 7.5, 1 mM DTT, 1:2000 protease inhibitor cocktail [Sigma P8340], and phosphatase inhibitor cocktail [Thermo Scientific product #88667]) was added to a 50 μL aliquot of Mg++ ghosts. The sample was then vortexed and incubated on ice for five minutes. A small volume was removed to make the “Total” gel sample. The remainder was centrifuged at 21,000 × g at 4°C in an Eppendorf 5424 microcentrifuge to sediment the TX-100-insoluble membrane skeletons. A small volume of the supernatant was removed to make the “Supe” gel sample, and the rest of the supernatant was discarded. The TX-100-insoluble pellet was resuspended by vortexing and sonication in sufficient TX-100 extraction buffer to obtain the volume of the original sample after removal of the aliquot for the “Total” gel sample. A small volume of the resuspended pellet was then used to make the “Pellet” gel sample. Gel samples were prepared by mixing with equal volumes of 2× SDS sample buffer (210 mM Tris-Cl, pH 6.8, 3 mM EDTA, 20% sucrose, 0.01% bromophenol blue, 6% SDS, 85 mM DTT) and boiling for five minutes.
SDS-PAGE and Immunoblotting.
For protein stains of Mg++ ghosts, ghost gel samples were run on Novex™ WedgeWell™ 4–20% Tris-glycine gels with a 4% stacking gel (Invitrogen) at 150 V for 75 minutes. The gels were then stained with One-Step Blue protein gel stain (Biotium). For immunoblotting, ghost and membrane skeleton gel samples were run on BioRad 4–20% Criterion-TGX midi-gels. For myosin heavy chain blots, proteins were transferred onto 0.2 μm nitrocellulose (Genesee Scientific) at 120V for 1 hr at 4°C. The transfer buffer contained 20% methanol, 125 mM Tris, 96 mM glycine, and 0.1% SDS and was precooled to 4°C before transfer. For myosin light chain blots, proteins were transferred onto 0.45 μm Immobilon-FL PVDF (Millipore) at 120V for 1 hr at 4°C. The transfer buffer contained 20% methanol, 125 mM Tris, and 96 mM glycine and was precooled to 4°C before transfer. After transfer, membranes were blocked in 1% fish gelatin in 1xTBS (50 mM Tris-Cl, pH 7.6; 150 mM NaCl) for 1 hr at room temperature and then incubated with primary antibodies overnight at 4°C. Membranes were washed four times in 1xTBS + 0.1% Tween20, incubated with secondary antibody at room temperature for 1 hr, and then washed four times in 1xTBS + 0.1% Tween20. Primary and secondary antibodies were diluted into 1xTBS with 1% fish gelatin and 0.1% Tween20. Primary antibodies used were: mouse anti-NMIIA tail domain antibody (Abcam ab55456), rabbit anti-pS1943 NMIIA (Cell Signaling Technologies 5026), rabbit anti-myosin light chain (Cell Signaling Technologies 8505), and mouse anti-pS19 myosin light chain (Cell Signaling Technologies 3675), each diluted 1:1000. The mouse anti-actin (Chemicon MAB1501) antibody was diluted 1:10000. Secondary antibodies used were: goat anti-mouse IRDye 680LT (LI-COR 926–68020) and goat anti-rabbit IRDye 800CW (LI-COR 926–32211) at a dilution of 1:10000. Labeled bands on blots were imaged on a LI-COR Odyssey Infrared Imaging System, and background-corrected intensity measurements of protein bands were measured using ImageJ.
Statistical analysis.
Data are presented in dot plots or bar graphs as mean ± standard deviation (SD). Means were compared between two samples using unpaired t-tests. If the samples did not have equal SDs, Welch’s t-test was used. If one or both samples were not normally distributed, the Mann-Whitney test was used. Means were compared between three or more samples using one-way ANOVAs followed by Tukey’s multiple comparisons test. If one or more samples were not normally distributed, the Kuskal-Wallis test was performed, followed by Dunn’s multiple comparisons test. Differences between variances were detected using F-tests for two samples and the Brown-Forsythe test for three or more samples. Statistical significance was defined as p < 0.05. Statistical analysis was performed using GraphPad Prism 7.03 software.
Results
MYH9-RD mutations reduce RBC hemoglobin content and size in complete blood count measurements
Mutations in membrane skeleton proteins can reduce RBC survival in the circulation, leading to anemia 21,23,31. To determine if MYH9-RD mutations cause anemia, we analyzed complete blood count (CBC) data from 12 normal donor controls and 113 MYH9-RD patients (Table S1). We did not control for patient age or sex, which impact RBC deformability and CBC indices 32–34. However, due to the low prevalence of MYH9-RD 35, an age and sex-controlled study may not have sufficient power to detect differences in RBC phenotypes. We mitigated age and sex effects by including a large number of patients. Patients had reduced platelet counts, as expected (Fig. 2A). Patients do not exhibit anemia, based on normal RBC count, hemoglobin level (HGB), and hematocrit (HCT), nor compensated hemolysis based on reticulocyte percentage. MYH9-RD clinical presentations such as elevated WHO bleeding score (Table S2) or chronic renal failure (Table S3) did not affect patient RBC indices.
Figure 2.
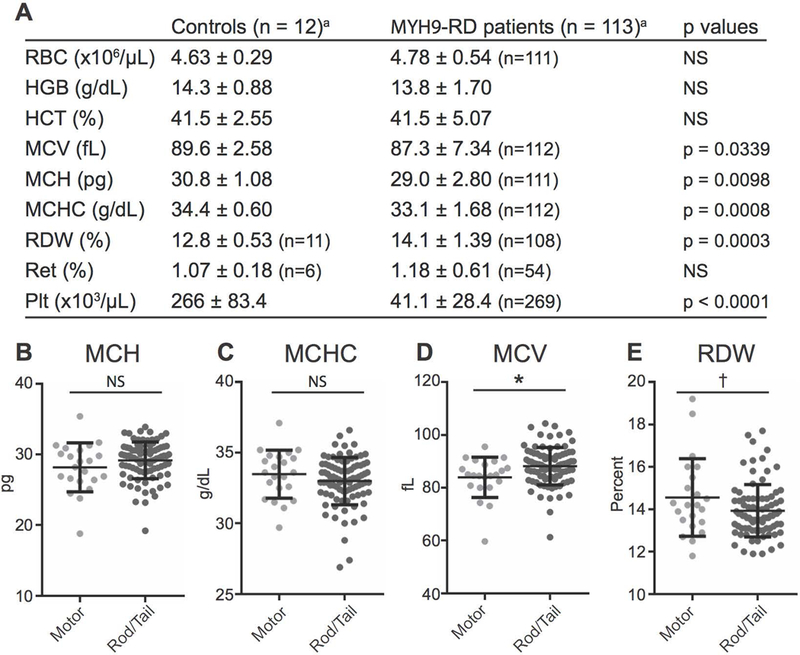
The NMIIA domain affected by MYH9-RD mutations determines differences in RBC volume and size but not RBC hemoglobin content. (A) Hematological analysis of normal controls and MYH9-RD patients. Values are mean ± SD. RBC indicates red blood cell count; HBG, hemoglobin; HCT, hematocrit; MCV, mean corpuscular volume; MCH, mean corpuscular hemoglobin; MCHC, mean corpuscular hemoglobin concentration; RDW, red cell distribution width; Ret, reticulocytes; Plt, automatic platelet count. NS indicates not statistically significant (p > 0.05). Comparisons of RBC, HGB, HCT, and MCV made with Welch’s t test. Comparisons of MCH, MCHC, RDW, Ret, and Plt made with Mann-Whitney test,a unless otherwise noted. Patients with mutations in the motor and tail domains do not have significant differences in mean corpuscular hemoglobin (MCH) (B) or mean corpuscular hemoglobin concentration (MCHC) (C) values. (D) Patients with motor domain mutations have significantly lower mean corpuscular volume (MCV) values, p = 0.0149. (E) Mean values for red cell distribution width (RDW) are not significantly different between motor domain and tail domain patients. However, the variance in RDW is significantly larger for motor domain patients by F-test (p = 0.0123). Lines in dot plots represent mean ± S.D. Motor, n = 22; Tail, n = 90, with the exception of RDW measurements: Motor, n = 22; Tail, n = 86. All comparisons between means were made by unpaired t-test.
Despite the absence of anemia, MYH9-RD patient RBCs had small but significant reductions in hemoglobin content (lower MCH and MCHC) and abnormal RBC size (lower MCV and higher RDW) (Fig. 2A). To investigate whether the location of the MYH9 mutation affects CBC parameters, we compared: (i) patients with either motor or coiled-coil rod/non-helical tail domain mutations, (ii) patients with mutations in an NMIIA subdomain (SH3/motor domain interface, SH1 helix, coiled-coil rod, or non-helical tail), and (iii) patients with mutations in frequently affected residues (R702, R1165, D1424, or R1933) 8. This analysis – based on a previous genotype-phenotype correlation study 13 – included groups for which we analyzed at least six unique patients (Table S1, Fig. 1). While patients with motor or rod/tail domain mutations had similar RBC hemoglobin contents (Fig. 2B, C), motor domain patients had significantly greater changes in RBC size, with lower MCVs and larger RDW variation (Fig. 2D, E). However, we observed no relationship between NMIIA subdomain or specific residue affected and RBC hemoglobin content or cell size (Fig. S1).
MYH9-RD mutations affect RBC morphology
Given that MYH9-RD patient RBCs have abnormal sizes, we next examined RBC shapes in Wright-Giemsa-stained peripheral blood smears (Fig. 3). While most control RBCs were round (Fig. 3Ai), we observed elongated RBCs in MYH9-RD patient smears (Fig. 3Aii-xii, black arrowheads), including many elongated cells with crenated edges similar to those in echinocytes (not seen in controls). Elongated RBCs were most frequent in patients with motor domain mutations (Fig. 3Aii-vi) and least common in patients with non-helical tail domain mutations (Fig. 3Axi-xii). However, the number of elongated RBCs varied considerably between patients, with some patients (e.g. the F41Ib patient in Fig. 3Aiii) having very few.
Figure 3.
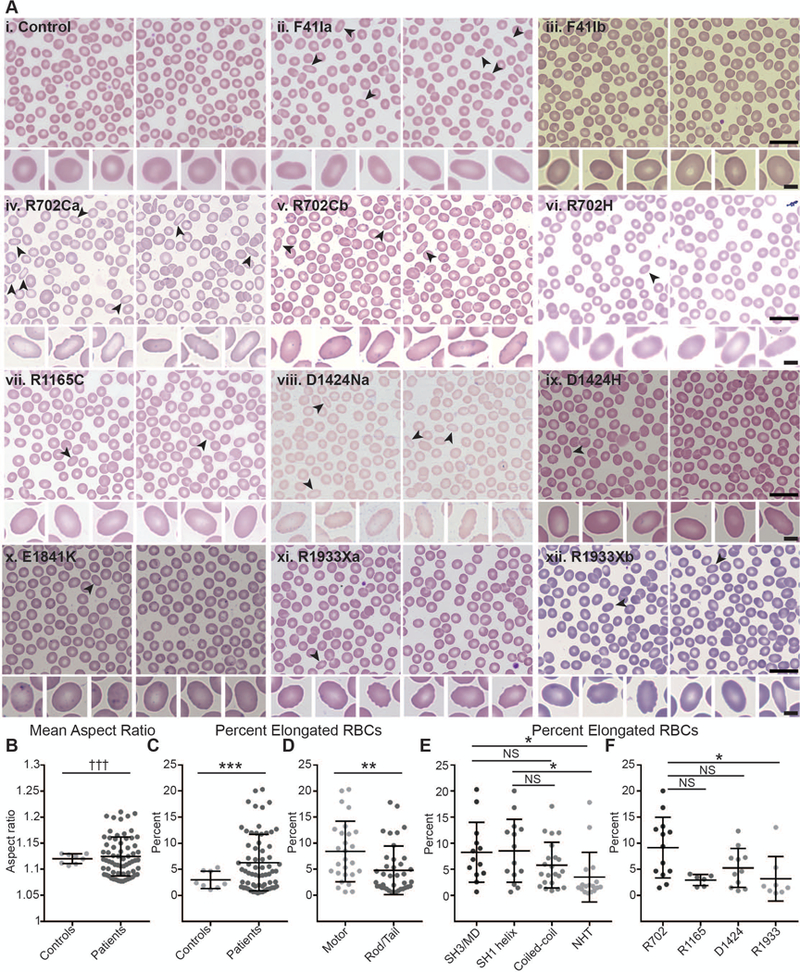
Wright-Giemsa-stained blood smears from MYH9-RD patients reveal elongated RBCs. (A) Representative images of smears from a normal donor control with no MYH9 mutations (i) and MYH9-RD patients with F41I mutations (ii-iii), R702C mutations (iv-v), a R702H mutation (vi), a R1165C mutation (vii), a D1424N mutation (viii), a D1424H mutation (ix), an E1841K mutation (x), or R1933X mutations (xi-xii) (scale = 20 μm). Elongated RBCs in top rows are denoted by black arrowheads. Bottom rows show typical round RBCs from the normal control (i) and the six most elongated RBCs from each patient (ii-xii) (scale = 4 μm). Lowercase letters following mutations are used to distinguish unique patients with the same mutation. (B) Mean aspect ratios (maximum/minimum axis) measured from images of smears from normal controls or MYH9-RD patients. Welch’s t-test showed no significant differences, but variances were significantly different by F-test (p = 0.0009). (C) Percentage of elongated RBCs (aspect ratios greater than or equal to 1.3) in images of smears from normal controls or MYH9-RD patients. Controls vs. Patients, p = 0.0008 by Welch’s t-test. (D) Percentage of elongated RBCs (aspect ratios greater than or equal to 1.3) in images of smears from MYH9-RD patients with motor or tail domain mutations. Motor vs. Tail, p = 0.0054 by Mann-Whitney test. (E) Percentage of elongated RBCs (aspect ratios greater than or equal to 1.3) in images of smears from MYH9-RD patients with mutations in the SH3/motor domain interface, the SH1 helix, the coiled-coil rod domain, or the non-helical tail (NHT). Kruskal-Wallis test followed by Dunn’s multiple comparisons test showed significant differences between the SH3/MD and the NHT (p = 0.0227) and between the SH1 helix and the NHT (p = 0.0225). (F) Percentage of elongated RBCs (aspect ratios greater than or equal to 1.3) in images of smears from MYH9-RD patients with mutations in R702, R1165, D1424, or R1933 residues of NMIIA. Kruskal-Wallis test followed by Dunn’s multiple comparisons test showed a significant difference between R702 and R1933 patients (p = 0.0227). Lines in dot plots represent mean ± S.D. Controls, n = 8; Patients, n = 66; Motor, n = 27; Tail, n = 39; SH3/MD, n = 13; SH1 helix, n = 14; Coiled-coil, n = 2; NHT, n = 17; R702, n = 13; R1165, n = 6; D1424, n = 11; R1933, n = 8.
To compare RBC elongation between samples, we measured RBC aspect ratios (major/minor axis) in 8 normal controls and 66 MYH9-RD patients (Table S1). We excluded RBCs with morphologies affected by close proximity to other RBCs or by artifacts of the smear preparation process (Fig. S2). Patients and controls had similar mean aspect ratios, although patient values had a larger variance (Fig. 3B, Fig. S3). Because many patients had low numbers of highly elongated RBCs (see Fig. S4 for examples), we also determined the percentage of RBCs with aspect ratios greater than or equal to 1.3 in each sample. Patients displayed a higher percentage of elongated RBCs than controls (Fig. 3C), and patients with motor domain mutations had higher percentages of elongated RBCs at the domain (Fig. 3D), subdomain (Fig. 3E), and residue (Fig. 3F) level. We observed similar differences in the coefficients of variation (standard deviation/mean) of aspect ratios (Fig. S3).
We showed previously that NMIIA activity maintains the biconcave disk shape of RBCs 20. To evaluate whether MYH9 mutations affect RBC biconcavity, we used confocal fluorescence microscopy to obtain 3D images of RBCs stained for the membrane marker glycophorin A (GPA) (Fig. S5A). For these and subsequent analyses, we controlled for shipping artifacts by comparing shipped patient samples to shipped normal controls. Consistent with the elongated RBC shapes in peripheral blood smears (Fig. 3), RBCs from some MYH9-RD patients (N93K, R702Cc, and D1424Na [lowercase letters distinguish unique patients with the same mutation]) had increased mean aspect ratios compared to the control (Fig. S5B). However, the patients examined had normal biconcavity, as reflected in the ratio of dimple versus rim height measured in XZ slices through the center of each RBC (Fig. S5C). Thus, while blebbistatin treatment causes both RBC elongation and loss of biconcavity, MYH9-RD mutations cause only RBC elongation.
MYH9-RD patients have normal RBC osmotic fragility and deformability by ektacytometry
Because MYH9-RD mutations cause reduced RBC hemoglobin content and RBC elongation, we hypothesized that MYH9-RD may also affect RBC hydration, surface area to volume ratio, or membrane mechanical properties. To test this hypothesis, we measured osmotic fragility in RBCs from shipped controls, MYH9-RD patients, and a patient with mild hereditary spherocytosis (Fig. S6, Table S1). While the hereditary spherocytosis patient had increased osmotic fragility, as expected 31, MYH9-RD patient osmotic fragility curves overlapped with curves from the controls (Fig. S6A, B).
We next performed osmotic gradient ektacytometry, which measures RBC deformability at varying suspending medium osmolalities and under a defined shear stress 28, on three patients with mutations in the motor domain (S96Lb, F41Ia, and F41L [lowercase letters distinguish unique patients with the same mutation]) (Table S1). The ektacytometry curve from the patient with an F41I mutation (images of a smear from this patient are shown in Fig. 3Aii) overlapped with curves from fresh and shipped normal controls (Fig. S7A). All three patients tested had similar values as controls for: maximum elongation index (EI max), which measures the maximum ability of RBCs to deform under isotonic conditions; the osmolality at which RBCs are least deformable under hypotonic conditions (O min), which measures RBC surface area-to-volume ratio and corresponds to the osmolarity of 50% RBC hemolysis in the osmotic fragility test; and the osmolality at which RBCs exhibit half-maximal deformability under hypertonic conditions (O hyper), which measures RBC membrane mechanical properties and reflects the mean corpuscular hemoglobin concentration and the RBC hydration state (Fig. S7B) 28,36. Together with the osmotic fragility data, this suggests that MYH9-RD mutations do not cause significant changes in RBC hydration, surface area to volume ratio, or membrane mechanical properties detected by ektacytometry.
The RBC membrane skeleton is unaffected in MYH9-RD patients
Mutations in membrane skeleton proteins can compromise skeleton connectivity, affecting its composition and/or organization 23,31,37. Compared to shipped controls and published data 20,36,38, RBC membranes (Mg++ ghosts) from multiple MYH9-RD patients contained normal levels of major membrane skeleton components such as α1/β1-spectrin, band 3, protein 4.1R, and actin (Fig. S8A). Total internal reflection microscopy (TIRF) images of RBCs spread on poly-L-lysine show actin and α1-spectrin localized in a dense, reticular pattern at the membrane with no detectable differences between the shipped controls and the motor domain patients examined (Fig. S8B, C). Thus, MYH9-RD mutations do not lead to global rearrangements or disassembly of the membrane skeleton.
MYH9-RD patients have normal RBC NMIIA phosphorylation but increased NMIIA association with the RBC membrane
MYH9-RD mutations may potentially compromise RBC NMIIA activity by affecting NMIIA phosphorylation. Phosphorylation at serine 1943 of the NMIIA heavy chain (HC) inhibits NMIIA filament assembly and promotes filament turnover; phosphorylation at serine 19 of the NMIIA regulatory light chain (RLC) makes NMIIA molecules competent for F-actin-activated MgATPase activity and force generation 14,39–41. Both the HC and RLC are phosphorylated in RBCs 20, indicating active regulation of motor activity and filament assembly. To determine if MYH9-RD affects RBC NMIIA phosphorylation, we measured phosphorylation at these residues in shipped control and patient Mg++ ghosts using immunoblotting (Fig. S9A, Table S1). The majority of patients had HC and RLC phosphorylation levels similar to shipped controls (Fig. S9B-E), suggesting that MYH9-RD mutations do not affect the regulation of RBC NMIIA contractility or filament assembly by HC or RLC phosphorylation.
MYH9-RD mutations may also affect RBC NMIIA activity by altering the ability of NMIIA to bind membrane skeleton F-actin or form bipolar filaments and other higher-order structures. To test this hypothesis, we isolated membrane skeletons by extracting RBC membrane ghosts with Triton X-100 to solubilize lipids and transmembrane proteins and remove any remaining cytosolic components (Fig. 4A-C, Table S1, 20,36,38). Compared to shipped controls and patients with coiled-coil rod or non-helical tail domain mutations, patients with motor domain mutations had double the percentage of NMIIA associated with the Triton-insoluble membrane skeleton pellet (Fig. 4D, E). This suggests that motor domain mutations may increase the binding of NMIIIA motor domains to membrane skeleton F-actin. This increased F-actin binding may be related to the high percentages of elongated RBCs we observe in patients with motor domain mutations.
Figure 4.
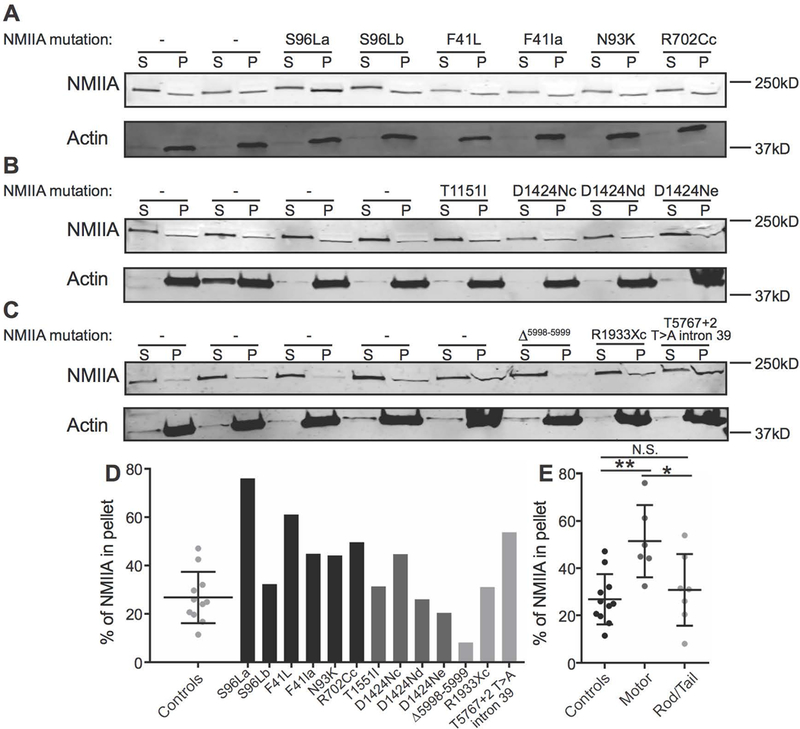
Association of NMIIA with the RBC membrane skeleton in MYH9-RD patients. (A-C) RBC Mg++ ghosts from normal donor shipped controls and MYH9-RD motor (A), coiled-coil rod (B), and non-helical tail (C) domain patients were extracted in Triton X-100 buffer followed by SDS-PAGE and immunoblotting for NMIIA heavy chain (top) and actin (bottom) in the supernatant (S) and Triton-insoluble membrane skeleton pellet (P) fractions. Each pair of lanes represents a unique normal donor or patient, in the same order as in Figure S9. (D) Quantification of % NMIIA in the membrane skeleton pellet fraction, comparing normal shipped controls (dot plot) with individual motor domain patients (black bars), coiled-coil rod domain patients (dark gray bars), and non-helical tail domain patients (light gray bars). Each dot in the dot plot and each bar in the bar graph represents one technical replicate of each sample. (E) Quantification in (D) grouped by normal donor shipped controls, motor domain patients, and coiled-coil rod/non-helical tail domain patients. One-way ANOVA followed by Tukey’s multiple comparisons test, Controls vs. Motor, p = 0.0039; Motor vs. Rod/Tail, p = 0.0276. In dot plots, lines represent mean ± S.D.
Discussion
NMIIA contractility controls RBC biconcave shape and deformability through interactions between the NMIIA motor domain and membrane skeleton F-actin 20. Here, we tested the impact of MYH9-RD mutations on NMIIA function in RBCs. We found that MYH9-RD patients do not have anemia and have normal RBC membrane skeletons and osmotic deformability. However, MYH9-RD RBCs exhibit elongated morphology, reduced size and hemoglobin content, and increased size variability. The extent of these abnormal RBC phenotypes varied, with motor domain mutations causing the most severe effects, as seen in other cells and tissues from MYH9-RD patients 8,13,15. Patients with motor domain mutations also had increased NMIIA association with RBC membrane skeleton F-actin. These results highlight the central role of the NMIIA motor domain in maintaining tension in the membrane skeleton to control RBC morphology.
In a previous study 20, we determined the function of NMIIA contractility in RBCs using blebbistatin, which locks NMIIA motor domains in a weak actin-binding state 42. Blebbistatin treatment caused RBC elongation in 2D and decreased biconcavity in 3D. MYH9-RD mutations also cause 2D RBC elongation but do not affect RBC biconcavity. Blebbistatin treatment likely has a larger impact on NMIIA contractility, as blebbistatin can bind all NMIIA molecules present. Individuals with MYH9-RD mutations, on the other hand, are heterozygous 4–6, and mutant NMIIA molecules have varying degrees of normal activity 25,26. Thus, MYH9-RD RBCs may have sufficient NMIIA activity to support biconcavity but not to prevent elongation.
Blebbistatin treatment decreases NMIIA-membrane skeleton association as measured by TIRF images of RBCs immunostained for NMIIA 20. By contrast, MYH9-RD motor domain mutations increase NMIIA-membrane skeleton association in Triton X-100 assays. Purified NMIIs with motor domain mutations have reduced MgATPase activity but bind F-actin for longer time periods, resulting in increased tension generation 25,43,44. Weakening NMIIA-F-actin binding by blebbistatin treatment can rescue impaired proplatelet formation in MYH9-RD patient megakaryocytes 45. These data suggest that increased tension may underlie MYH9-RD disease phenotypes, including in RBCs from patients with motor domain mutations. Both reduced (blebbistatin) and increased (MYH9-RD) NMIIA-F-actin association lead to RBC elongation, suggesting that normal RBC morphology requires an optimal amount of membrane skeleton tension.
Given that blebbistatin inhibition of NMIIA contractility increases RBC deformability in microfluidics assays 20, we were surprised that MYH9-RD patients had normal osmotic deformability in osmotic fragility and ektacytometry assays. NMIIA likely interacts with a small subset of membrane skeleton F-actins, as discussed previously 20. Thus, increased NMIIA-membrane skeleton association in MYH9-RD may not increase RBC membrane tension enough to affect osmotic deformability. Alternatively, a small percentage of patient RBCs could display large changes in deformability that we failed to detect in our ektacytometry assays. Future studies using microfluidics assays will test this possibility by measuring deformability in individual RBCs and allowing a more direct comparison with our blebbistatin data.
In common human RBC membrane disorders, including hereditary spherocytosis (HS) and hereditary elliptocytosis (HE), many patients have a small percentage of affected RBCs, as in blebbistatin-treated and MYH9-RD RBCs. HS and HE patients with low numbers of affected RBCs also have small changes in hemoglobin content and other CBC indices 31, as seen in MYH9-RD. However, both HS and HE affect RBC deformability in ektacytometry, while MYH9-RD does not. Many RBC membrane disorders cause changes in membrane protein levels or protein modifications 31,46. While we did not detect changes in major membrane proteins or in phosphorylation of the NMIIA heavy or light chains, future proteomic or phospho-proteomic studies may detect changes that contribute to the changes in RBC shape and CBC indices in MYH9-RD.
MYH9-RD may affect low numbers of RBCs because changes in NMIIA contractility may have different effects on RBC membrane properties at different stages in RBC lifetimes. Previous studies suggest a role for NMII in erythroblast cell division prior to enucleation and in reticulocyte maturation 47–49. Future studies using mouse models of MYH9-RD 50 could test whether MYH9-RD affects erythroblast terminal differentiation. Over the course of the RBC lifespan, RBCs gradually decrease in membrane surface area, volume, and deformability 32,33,51. Younger RBCs with higher deformability may thus be more sensitive to changes in NMIIA activity than older, stiffer RBCs. Alternatively, MYH9 mutations may make RBCs more vulnerable to repeated deformation in the circulation, leading to gradual elongation. Future studies could test this hypothesis by repeating the experiments in this study with age-separated RBCs 51,52.
Taken together, our data support a central role for the NMIIA motor domain in binding to F-actin to maintain tension in the membrane skeleton and control RBC morphology. We found that MYH9-RD mutations increase binding of NMIIA to the RBC membrane skeleton and cause elongated RBC morphology and that these effects were strongest in patients with motor domain mutations. While these RBC phenotypes do not cause anemia, they may be exacerbated by co-occurring conditions that impact RBC membrane skeleton structure, cell shape, and deformability: RBC aging 32,33,51, hemolytic anemias 23,31,37, or sickle cell disease 53–55. Along with recent reports of elevated liver enzymes 56,57 and impaired organelle transport in natural killer cells 58,59, RBC elongation adds to a growing list of sub-clinical alterations of MYH9-RD.
Supplementary Material
Acknowledgements
We are very grateful to the MYH9-RD patients who volunteered to participate in this study. We thank Elly Jarvis for her assistance in measuring RBC shape in images of peripheral blood smears. We thank Catherine Cheng and Justin Parreno for helpful suggestions and comments. This work was supported by National Institutes of Health Grant HL083464 to V.M.F. and the Joseph and Lillian Pleitgen Trust (V.M.F.). A.S. was supported by NIH/NCATS CTSA Awards TL1 TR001113 and TL1 TR002551. The work of A.P. is supported by a grant from the IRCCS Policlinico San Matteo Foundation and the Telethon Foundation (grant number GGP17106). L.D.C was supported by the Laboratory of Excellence for Red Cells (LABEX GR-Ex)-ANR Avenir-11-LABX-005–02. K.P was supported by a postdoctoral fellowship from the National Hemophilia Foundation, U.S.A
References
- 1.Kelley MJ, Jawien W, Ortel TL, Korczak JF. Mutation of MYH9, encoding non-muscle myosin heavy chain A, in May-Hegglin anomaly. Nat Genet. 2000;26(1):106–108. [DOI] [PubMed] [Google Scholar]
- 2.Seri M, Cusano R, Gangarossa S, et al. Mutations in MYH9 result in the May-Hegglin anomaly, and Fechtner and Sebastian syndromes. The May-Heggllin/Fechtner Syndrome Consortium. Nat Genet. 2000;26(1):103–105. [DOI] [PubMed] [Google Scholar]
- 3.Kunishima S, Matsushita T, Kojima T, et al. Identification of six novel MYH9 mutations and genotype-phenotype relationships in autosomal dominant macrothrombocytopenia with leukocyte inclusions. J Hum Genet. 2001;46(12):722–729. [DOI] [PubMed] [Google Scholar]
- 4.Balduini CL, Savoia A, Seri M. Inherited thrombocytopenias frequently diagnosed in adults. J Thromb Haemost. 2013;11(6):1006–1019. [DOI] [PubMed] [Google Scholar]
- 5.Kunishima S, Saito H. Advances in the understanding of MYH9 disorders. Curr Opin Hematol. 2010;17(5):405–410. [DOI] [PubMed] [Google Scholar]
- 6.Althaus K, Greinacher A. MYH9-related platelet disorders. Semin Thromb Hemost. 2009;35(2):189–203. [DOI] [PubMed] [Google Scholar]
- 7.Golomb E, Ma X, Jana SS, et al. Identification and characterization of nonmuscle myosin II-C, a new member of the myosin II family. J Biol Chem. 2004;279(4):2800–2808. [DOI] [PubMed] [Google Scholar]
- 8.Pecci A, Ma X, Savoia A, Adelstein RS. MYH9: Structure, functions and role of non-muscle myosin IIA in human disease. Gene. 2018;664:152–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bauer CB, Holden HM, Thoden JB, Smith R, Rayment I. X-ray structures of the apo and MgATP-bound states of Dictyostelium discoideum myosin motor domain. J Biol Chem. 2000;275(49):38494–38499. [DOI] [PubMed] [Google Scholar]
- 10.Dominguez R, Freyzon Y, Trybus KM, Cohen C. Crystal structure of a vertebrate smooth muscle myosin motor domain and its complex with the essential light chain: visualization of the pre-power stroke state. Cell. 1998;94(5):559–571. [DOI] [PubMed] [Google Scholar]
- 11.Gourinath S, Himmel DM, Brown JH, Reshetnikova L, Szent-Gyorgyi AG, Cohen C. Crystal structure of scallop Myosin s1 in the pre-power stroke state to 2.6 a resolution: flexibility and function in the head. Structure. 2003;11(12):1621–1627. [DOI] [PubMed] [Google Scholar]
- 12.Risal D, Gourinath S, Himmel DM, Szent-Gyorgyi AG, Cohen C. Myosin subfragment 1 structures reveal a partially bound nucleotide and a complex salt bridge that helps couple nucleotide and actin binding. Proc Natl Acad Sci U S A. 2004;101(24):8930–8935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pecci A, Klersy C, Gresele P, et al. MYH9-related disease: a novel prognostic model to predict the clinical evolution of the disease based on genotype-phenotype correlations. Hum Mutat. 2014;35(2):236–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dulyaninova NG, Bresnick AR. The heavy chain has its day: regulation of myosin-II assembly. Bioarchitecture. 2013;3(4):77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saposnik B, Binard S, Fenneteau O, et al. Mutation spectrum and genotype-phenotype correlations in a large French cohort of MYH9-Related Disorders. Mol Genet Genomic Med. 2014;2(4):297–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Patel-Hett S, Wang H, Begonja AJ, et al. The spectrin-based membrane skeleton stabilizes mouse megakaryocyte membrane systems and is essential for proplatelet and platelet formation. Blood. 2011;118(6):1641–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maupin P, Phillips CL, Adelstein RS, Pollard TD. Differential localization of myosin-II isozymes in human cultured cells and blood cells. J Cell Sci. 1994;107 ( Pt 11):3077–3090. [DOI] [PubMed] [Google Scholar]
- 18.Faul C, Asanuma K, Yanagida-Asanuma E, Kim K, Mundel P. Actin up: regulation of podocyte structure and function by components of the actin cytoskeleton. Trends Cell Biol. 2007;17(9):428–437. [DOI] [PubMed] [Google Scholar]
- 19.Fowler VM, Davis JQ, Bennett V. Human erythrocyte myosin: identification and purification. J Cell Biol. 1985;100(1):47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith AS, Nowak RB, Zhou S, et al. Myosin IIA interacts with the spectrin-actin membrane skeleton to control red blood cell membrane curvature and deformability. Proc Natl Acad Sci U S A. 2018;115(19):E4377–E4385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fowler VM. The human erythrocyte plasma membrane: a Rosetta Stone for decoding membrane-cytoskeleton structure. Curr Top Membr. 2013;72:39–88. [DOI] [PubMed] [Google Scholar]
- 22.Gokhin DS, Fowler VM. Feisty filaments: actin dynamics in the red blood cell membrane skeleton. Curr Opin Hematol. 2016;23(3):206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mohandas N, Gallagher PG. Red cell membrane: past, present, and future. Blood. 2008;112(10):3939–3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wong AJ, Kiehart DP, Pollard TD. Myosin from human erythrocytes. J Biol Chem. 1985;260(1):46–49. [PubMed] [Google Scholar]
- 25.Hu A, Wang F, Sellers JR. Mutations in human nonmuscle myosin IIA found in patients with May-Hegglin anomaly and Fechtner syndrome result in impaired enzymatic function. J Biol Chem. 2002;277(48):46512–46517. [DOI] [PubMed] [Google Scholar]
- 26.Franke JD, Dong F, Rickoll WL, Kelley MJ, Kiehart DP. Rod mutations associated with MYH9-related disorders disrupt nonmuscle myosin-IIA assembly. Blood. 2005;105(1):161–169. [DOI] [PubMed] [Google Scholar]
- 27.Parpart AK, Lorenz PB, et al. The osmotic resistance (fragility) of human red cells. J Clin Invest. 1947;26(4):636–640. [PubMed] [Google Scholar]
- 28.Da Costa L, Suner L, Galimand J, et al. Diagnostic tool for red blood cell membrane disorders: Assessment of a new generation ektacytometer. Blood Cells Mol Dis. 2016;56(1):9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hardwicke J, Ricketts CR, Squire JR. Effect of dextran of various molecular sizes on erythrocyte sedimentation rate. Nature. 1950;166(4232):988–989. [DOI] [PubMed] [Google Scholar]
- 30.Fowler VM, Bennett V. Erythrocyte membrane tropomyosin. Purification and properties. J Biol Chem. 1984;259(9):5978–5989. [PubMed] [Google Scholar]
- 31.Da Costa L, Galimand J, Fenneteau O, Mohandas N. Hereditary spherocytosis, elliptocytosis, and other red cell membrane disorders. Blood Rev. 2013;27(4):167–178. [DOI] [PubMed] [Google Scholar]
- 32.Canham PB. Difference in geometry of young and old human erythrocytes explained by a filtering mechanism. Circ Res. 1969;25(1):39–45. [DOI] [PubMed] [Google Scholar]
- 33.Bartosz G Erythrocyte aging: physical and chemical membrane changes. Gerontology. 1991;37(1–3):33–67. [DOI] [PubMed] [Google Scholar]
- 34.Cheng CK, Chan J, Cembrowski GS, van Assendelft OW. Complete blood count reference interval diagrams derived from NHANES III: stratification by age, sex, and race. Lab Hematol. 2004;10(1):42–53. [PubMed] [Google Scholar]
- 35.Savoia A, Pecci A. MYH9-Related Disorders. In: Adam MP, Ardinger HH, Pagon RA, et al. , eds. GeneReviews((R)) Seattle (WA)1993. [Google Scholar]
- 36.Moyer JD, Nowak RB, Kim NE, et al. Tropomodulin 1-null mice have a mild spherocytic elliptocytosis with appearance of tropomodulin 3 in red blood cells and disruption of the membrane skeleton. Blood. 2010;116(14):2590–2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mohandas N, Evans E. Mechanical properties of the red cell membrane in relation to molecular structure and genetic defects. Annu Rev Biophys Biomol Struct. 1994;23:787–818. [DOI] [PubMed] [Google Scholar]
- 38.Yu J, Fischman DA, Steck TL. Selective solubilization of proteins and phospholipids from red blood cell membranes by nonionic detergents. J Supramol Struct. 1973;1(3):233–248. [DOI] [PubMed] [Google Scholar]
- 39.Vicente-Manzanares M, Ma X, Adelstein RS, Horwitz AR. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat Rev Mol Cell Biol. 2009;10(11):778–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heissler SM, Manstein DJ. Nonmuscle myosin-2: mix and match. Cell Mol Life Sci. 2013;70(1):1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Conti MA, Adelstein RS. Nonmuscle myosin II moves in new directions. J Cell Sci. 2008;121(Pt 1):11–18. [DOI] [PubMed] [Google Scholar]
- 42.Kovacs M, Toth J, Hetenyi C, Malnasi-Csizmadia A, Sellers JR. Mechanism of blebbistatin inhibition of myosin II. J Biol Chem. 2004;279(34):35557–35563. [DOI] [PubMed] [Google Scholar]
- 43.Kim KY, Kovacs M, Kawamoto S, Sellers JR, Adelstein RS. Disease-associated mutations and alternative splicing alter the enzymatic and motile activity of nonmuscle myosins II-B and II-C. J Biol Chem. 2005;280(24):22769–22775. [DOI] [PubMed] [Google Scholar]
- 44.Ma X, Kovacs M, Conti MA, et al. Nonmuscle myosin II exerts tension but does not translocate actin in vertebrate cytokinesis. Proc Natl Acad Sci U S A. 2012;109(12):4509–4514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen Y, Boukour S, Milloud R, et al. The abnormal proplatelet formation in MYH9-related macrothrombocytopenia results from an increased actomyosin contractility and is rescued by myosin IIA inhibition. J Thromb Haemost. 2013;11(12):2163–2175. [DOI] [PubMed] [Google Scholar]
- 46.Huisjes R, Bogdanova A, van Solinge WW, Schiffelers RM, Kaestner L, van Wijk R. Squeezing for Life - Properties of Red Blood Cell Deformability. Front Physiol. 2018;9:656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ubukawa K, Guo YM, Takahashi M, et al. Enucleation of human erythroblasts involves non-muscle myosin IIB. Blood. 2012;119(4):1036–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moura PL, Hawley BR, Mankelow TJ, et al. Non-muscle Myosin II drives vesicle loss during human reticulocyte maturation. Haematologica. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nowak RB, Papoin J, Gokhin DS, et al. Tropomodulin 1 controls erythroblast enucleation via regulation of F-actin in the enucleosome. Blood. 2017;130(9):1144–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang Y, Conti MA, Malide D, et al. Mouse models of MYH9-related disease: mutations in nonmuscle myosin II-A. Blood. 2012;119(1):238–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Costa M, Ghiran I, Peng CK, Nicholson-Weller A, Goldberger AL. Complex dynamics of human red blood cell flickering: alterations with in vivo aging. Phys Rev E Stat Nonlin Soft Matter Phys. 2008;78(2 Pt 1):020901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rennie CM, Thompson S, Parker AC, Maddy A. Human erythrocyte fraction in “Percoll” density gradients. Clin Chim Acta. 1979;98(1–2):119–125. [DOI] [PubMed] [Google Scholar]
- 53.Kato GJ, Steinberg MH, Gladwin MT. Intravascular hemolysis and the pathophysiology of sickle cell disease. J Clin Invest. 2017;127(3):750–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu SC, Yi SJ, Mehta JR, et al. Red cell membrane remodeling in sickle cell anemia. Sequestration of membrane lipids and proteins in Heinz bodies. J Clin Invest. 1996;97(1):29–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Evans E, Mohandas N, Leung A. Static and dynamic rigidities of normal and sickle erythrocytes. Major influence of cell hemoglobin concentration. J Clin Invest. 1984;73(2):477–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pecci A, Biino G, Fierro T, et al. Alteration of liver enzymes is a feature of the MYH9-related disease syndrome. PLoS One. 2012;7(4):e35986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Favier R, DiFeo A, Hezard N, Fabre M, Bedossa P, Martignetti JA. A new feature of the MYH9-related syndrome: chronic transaminase elevation. Hepatology. 2013;57(3):1288–1289. [DOI] [PubMed] [Google Scholar]
- 58.Sanborn KB, Mace EM, Rak GD, et al. Phosphorylation of the myosin IIA tailpiece regulates single myosin IIA molecule association with lytic granules to promote NK-cell cytotoxicity. Blood. 2011;118(22):5862–5871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sanborn KB, Rak GD, Maru SY, et al. Myosin IIA associates with NK cell lytic granules to enable their interaction with F-actin and function at the immunological synapse. J Immunol. 2009;182(11):6969–6984. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.