

Abstract
Multidrug resistance and toxic side effects are the major challenges in cancer treatment with microtubule-targeting agents (MTAs), and thus, there is an urgent clinical need for new therapies. Chalcone, a common simple scaffold found in many natural products, is widely used as a privileged structure in medicinal chemistry. We have previously validated tubulin as the anticancer target for chalcone derivatives. In this study, an α-methyl-substituted indole-chalcone (FC77) was synthesized and found to exhibit an excellent cytotoxicity against the NCI-60 cell lines (average concentration causing 50% growth inhibition = 6 nM). More importantly, several multidrug-resistant cancer cell lines showed no resistance to FC77, and the compound demonstrated good selective toxicity against cancer cells versus normal CD34+ blood progenitor cells. A further mechanistic study demonstrated that FC77 could arrest cells which relates to the binding to tubulin, and inhibit the microtubule dynamics. The National Cancer Institute COMPARE analysis and molecular modeling indicated that FC77 had a mechanism of action similar to that of colchicine. Overall, our data demonstrate that this indole-chalcone represents a novel MTA template for further development of potential drug candidates for the treatment of multidrug-resistant cancers.
Keywords: Indole-chalcone, Multidrug resistance, Microtubule-targeting agent, Cancer
Graphical Abstract
1. Introduction
Microtubules, composed of α- and β-tubulin heterodimers, are a validated druggable target for a number of marketed antitumor drugs (Fig. 1). The drugs are classified into two categories, microtubule-stabilizing agents (MSAs) and microtubule-destabilizing agents (MDAs)1–3. MSAs, such as paclitaxel (Taxol®) and ixabepilone, prevent microtubules disassembly, whereas MDAs, such as vinblastine and vincristine, inhibit microtubule polymerization. A new MDA from marine sponges, eribulin, has recently been approved for the treatment of breast cancer and liposarcoma4. Indeed, microtubule-targeting agents (MTAs) represent one of the most commonly used classes of chemotherapies in the treatment of cancer5. However, there is a high risk for cancer cells to acquire drug resistance during MTA treatment; consequently, tumors often reveal a multidrug-resistant (MDR) phenotype6. Cancer cells can acquire multidrug resistance via different mechanisms, including the overexpression of ATP-binding cassette (ABC) transporters. ABC transporters are overexpressed in ~40% of breast cancer patients, and the proportion can increase to ~70% upon chemotherapy treatment7. Toxic side effects are another challenge that limits the clinical use of MTAs8. Therefore, there is an urgent clinical need for new MTA anticancer agents, which would ideally be able to selectively target MDR malignancies while showing low toxicity for normal tissues.
Fig. 1.

Representative antimicrotubule cancer therapeutic drugs.
Since substrates for ABC transporters are typically structurally complex, such as the clinical MTAs, anticancer agents with a simpler structure may offer a better opportunity to mitigate the risk of multidrug resistance. Chalcone is a simple and privileged structure in medicinal chemistry, with diverse biological activities, including anticancer activity9–11. Recently, our group has demonstrated, via a whole-cell-based photoaffinity labeling approach, that tubulin is the direct cellular target of chalcones, responsible for their anticancer effects. A mass spectrometry-based approach has revealed that a positive chalcone probe modified the peptide N337-K350 (NSSYFVEWIPNNVK), suggesting targeting of the colchicine-binding site (Supplementary Fig. S1), which is different from the paclitaxel- or vinblastine-binding sites12. Molecular modeling of this probe showed that its azide group was within 2.2 Å and 2.5 Å of the labeled peptide (Supplementary Fig. S2). To date, no clinical MTA agents have been reported to target the colchicine-binding site as anticancer agents.
Based on these results and our earlier structure–activity relationship (SAR) data13, seven indole-chalcones were developed and characterized in this study (Supplementary Table S1), and the lead compound (FC77; Fig. 2) was evaluated for its in vitro anticancer activity and the mechanism of action.
Fig. 2.

Chemical structure of the novel indole-chalcone FC77.
2. Materials and methods
2.1. Chemicals and reagents
All purchased reagents and solvents were used without further purification. Silica gel chromatography was performed on Whatman silica gel 60 Å (230–400 mesh). Nuclear magnetic resonance (NMR) spectra (1H and 13C) were recorded on a spectrometer (Bruker Ascend 400) and calibrated using the deuterated solvent residual as an internal reference. High-resolution mass spectrometry (HRMS) was performed using a Q-TOF micro mass spectrometer. Compound 4 was analyzed by high-performance liquid chromatography (HPLC; Agilent 1100) using an Agilent Eclipse Plus C18 column (4.6 × 100 mm, 3.5 μm) and a 20-min linear gradient from 100% A (20 mM ammonium acetate, pH 6.8, in 10% CH3CN) to 100% B (CH3CN) at a flow rate of 1 mL/min. Purities of the other compounds were analyzed by HPLC (Agilent 1100) using an ODS-A column (YMC Pack; 10 × 250 mm, 5 μm) with methanol:H2O (100:0 to 80:20 over 20 min and 80:20 thereafter) as the mobile phase with a flow rate of 2 mL/min. The separation was monitored at wavelengths of 254 and 365 nm. The purities of all final compounds were higher than 95%.
2.2. Synthesis of indole-chalcones
The synthetic route is presented in Supplementary Scheme S1 using two patents as references.14, 15 To a solution of indole-3-carboxaldehyde (1 mmol) in ethanol (4 mL), piperidine (1.2 mmol) and the corresponding acetophenone (0.5 mmol) were added. After the mixture was stirred at 95 °C for 48 h, the reaction was quenched with hydrochloric acid diluted to pH 6 and then extracted with ethyl acetate. The organic layer was washed with aqueous NaHCO3, water, and brine, then dried over anhydrous Na2SO4, and finally concentrated. The residue was recrystallized in ethanol at −20 °C for 24 h to afford the target compound FC77. Recrystallization yield: 8.2%. 1H NMR (400MHz, CDCl3): δ 8.71 (1H, br, NH), 7.69 (1H, s, Ar-H), 7.64 (1H, s, =CH), 7.59 (1H, d, J = 8.0 Hz, Ar-H), 7.45 (1H, d, J = 8.0 Hz, Ar-H), 7.30 (1H, d, J = 7.2 Hz, Ar-H), 7.22 (1H, t, J = 7.5 Hz, Ar-H), 7.03 (2H, s, Ar-H), 3.96 (3H, s, OCH3), 3.89 (6H, s, 2OCH3), 2.32 (3H, s, CH3). 13C NMR (100MHz, CDCl3): δ 197.98, 152.78, 135.46, 134.52, 131.76, 127.64, 125.93, 123.37, 121.04, 118.44, 113.31, 111.45, 106.94, 60.97, 56.26, 15.39. HRMS (ESI+) m/z Calculated for C21H22NO4 352.1543; Observed 352.1543 (M+H+). HPLC Purity: 97.4%, Rt = 35.70 min, UV 254 nm. The NMR, HRMS, and purity spectra are included in the Supplemental Information.
2.3. Cell lines and cell culture
Human A549, A549/T, A549/DDP, HCT-116, HCT-116/L, HL60, HL60/DOX, K562, K562/HHT300, CCRF-CEM, and CCRF-CEM/VLB100 cells were authenticated via DNA analysis by Genetica DNA Laboratories (Cincinnati, OH, USA) or by the University of Arizona Genomics Core. Cells were cultured following our standard protocols12, 13, 16–20 and tested monthly for Mycoplasma contamination. De-identified mobilized peripheral blood (MPB) was obtained after informed consent according to protocols approved by the University of Minnesota Institutional Review Board.
K562 and K562/HHT300 cell lines were provided by Dr. Tang.21 K562/HHT300 was developed from K562 upon chronic exposure to homoharringtonine, a protein translation inhibitor. HL60 and HL60/DOX cell lines were provided by Dr. Ganapathi.22 HL60/DOX was developed from HL60 upon chronic exposure to doxorubicin (a topoisomerase inhibitor). CCRF-CEM and CCRF-CEM/VLB100 were provided by Dr. Beck23, 24. CCRF-CEM/VLB100 was developed from CCRF-CEM upon chronic exposure to vinblastine, an antimicrotubule agent. A549, A549/T, A549/DDP, HCT-116 and HCT-116/L were obtained from State Key Laboratory of Oncogenes and Related Genes, Cancer Institute of Shanghai Jiaotong University. A549/T and A549/DDP were developed from A549 upon chronic exposure to paclitaxel and cisplatin, respectively. HCT-116/L was developed from HCT-116 upon chronic exposure to oxaliplatin. We have evaluated all the parental cell lines and the MDR cell lines, using the names as in their original reports. Comparison has been made only between the MDR cell line with its corresponding parental cell line.
2.4. Cell viability measurement
In vitro cytotoxicity of the compounds was assayed by determining their ability to inhibit the growth of tumor cells. In brief, cells were plated in a 96-well plate (at a density of ~4,000 cells/well for adherent cells and ~10,000 cells/well for suspension cells). The cells were treated with a series of dilutions of the test compounds, with the final concentration of dimethyl sulfoxide (DMSO) of 1% in the cell medium; cells treated with medium containing 1% DMSO served as a control. After a 48- or 72-h treatment, the relative cell viability in each well was determined using a CellTiter-Blue cell viability assay kit. The concentration causing 50% inhibition of the cell growth (GI50) of each candidate compound was determined by fitting the relative cell viability to the drug concentration using a dose-response model in the GraphPad Prism program (GraphPad Software, San Diego, CA, USA). For the National Cancer Institute (NCI)-60 screening panel, the treatment lasted for 72 h.
2.5. Colony formation assay
Cells (100 cells per condition for leukemia cell lines or 500 cells per condition for MPB) were treated with doxorubicin, vincristine, or FC77 for 24 h in a standard culture medium and then harvested and plated in MethoCult H4034 optimum CFC medium (Stem Cell Technologies) according to the manufacturer’s recommendations. Colonies were scored after 7–14 days under an inverted microscope.
2.6. Analysis of cell-cycle distribution by flow cytometry
The cell cycle was analyzed by flow cytometry based on the DNA content following our established procedures13, 25, with slight modifications. Briefly, HL60 cells (106 cells/mL) or HCT116 cells (5*105 cells/mL) were treated with compounds at their corresponding GI50 concentrations with 1% DMSO for different time points (1–12 h). The cells were then washed twice with ice-cold phosphate-buffered saline (PBS) and resuspended in 1 mL of PBS. The suspension was mixed with cold 70% ethanol (9 mL) and fixed at 4 °C overnight. Cell pellets were collected by centrifugation, washed twice with PBS, then resuspended in 1 mL of a propidium iodide (PI) staining solution (50 μg/mL PI, 200 μg/mL RNase A, and 0.1% Triton X-100 in PBS), and incubated at room temperature for 30 min in the dark. The cell-cycle distribution was then analyzed using a BD FACSCalibur flow cytometer.
2.7. Differential scanning fluorimetry assay
Briefly, a tubulin monomer was mixed with colchicine, paclitaxel, vinblastine or FC77 (50 μM) in the presence of GDP (0.1 mg/mL), and the melting temperature of tubulin was measured using BioRad CFX96 Real-Time PCR System following established procedures26, 27.
2.8. Microtubule dynamics
The individual microtubule dynamics were measured in the presence of MTAs and analyzed as described previously28. Briefly, LLC-PK1 porcine kidney cells, stably expressing enhanced green fluorescent protein (EGFP)–tubulin, were treated with Taxol (Sigma–Aldrich, St. Louis, MO, USA), vincristine sulfate (Sigma), or FC77 for 30 min prior to imaging. A total of 121 frames were collected every 0.5 s for 60 s, while the lengths of individual microtubules were determined using the TipTracker software without modification29.
2.9. Statistical analysis
The in vitro cytotoxicity assay was performed at least three times in triplicate. Comparisons were made using a two-tailed Student’s t-test. A value of p ≤ 0.05 was considered statistically significant.
3. Results
3.1. FC77 shows strong cytotoxicity against NCI-60 cancer cell lines
The NCI-60 human cancer cell line screening has been widely used to identify potential anticancer drug candidates, to help elucidate the mechanism of action, and to help prioritize the types of malignancies for the development30. Through this screening, FC77 was found to be a low-nanomolar inhibitor of the growth of the majority of the NCI-60 human cancer cell lines (average GI50: ~6 nM; Table 1 and Supplementary Fig. S4), with some preference toward leukemia, colon, and central nervous system cancers (GI50 < 4 nM for all 19 cell lines), as well as toward non-small-cell lung cancers, with only one exception (HOP-92).
Table 1.
The 72-h cytotoxicity profile of FC77 against the NCI-60 cancer cell line panel.
| Cell line | GI50 (nM) | Cell line | GI50 (nM) | Cell line | GI50 (nM) | Cell line | GI50 (nM) |
|---|---|---|---|---|---|---|---|
| Leukemia | Colon cancer | CNS cancer | A498 | 2.5 | |||
| CCRF-CEM | 5.1 | COLO 205 | 3.5 | SF-268 | 5.5 | ACHN | 6.2 |
| Hl-60 (TB) | 2.3 | HCC-2998 | 4.3 | SF-295 | 3.0 | CAKI-1 | 4.2 |
| K562 | 3.5 | HCT-116 | 3.6 | SF-539 | 2.8 | RXF 393 | N.D. |
| MOLT-4 | 5.5 | HCT-15 | 3.4 | SNB-19 | 4.1 | TK-10 | > 100 |
| PRMI-8226 | 4.8 | HT29 | 4.0 | SNB-75 | 3.3 | UO-31 | 7.9 |
| SR | 3.3 | KM12 | 3.6 | U251 | 4.4 | Prostate cancer | |
| Non-small-cell lung cancer | SW-620 | 4.2 | Ovarian cancer | PC-3 | 7.9 | ||
| A549/ATCC | 4.1 | Melanoma | IGROV1 | 5.6 | DU-145 | 3.8 | |
| EKVX | 5.2 | LOX IMVI | 4.7 | OVCAR-3 | 3.3 | Breast cancer | |
| HOP-62 | 5.4 | MALME-3M | > 100 | OVCAR-4 | 25.1 | MCF-7 | 3.5 |
| HOP-92 | 74.1 | M14 | 3.4 | OVCAR-5 | 85.1 | MDA-MB-231/ATCC | 3.9 |
| NCI-H226 | 6.2 | MDA-MB-435 | 2.3 | OVCAR-8 | 3.5 | HS 578T | 4.8 |
| NCI-H23 | 5.1 | SK-MEL-2 | 12.3 | NCI/ADR-RES | 3.2 | BT-549 | 5.9 |
| NCI-H460 | 3.7 | SK-MEL-28 | 17.4 | SK-OV-3 | 7.8 | T-47D | N.D. |
| NCI-H522 | 2.8 | SK-MEL-5 | 4.3 | Renal cancer | MDA-MB-468 | 3.3 | |
| UACC-257 | > 100 | 786–0 | 6.5 |
CNS, central nervous system; GI50, concentration causing 50% inhibition of the cell growth; NCI, National Cancer Institute; N.D., not determined.
3.2. FC77 inhibits the growth of multiple parental and corresponding MDR cancer cells
We further explored the anticancer potential of FC77 against leukemia (HL60 vs. HL60/DOX, K562 vs. K562/HHT300, CCRF-CEM vs. CCRF-CEM/VLB100), non-small-cell lung cancer (A549 vs. A549/T and A549/DDP), and colon cancer (HCT-116 vs. HCT-116/L) cell lines and their corresponding MDR counterparts.
The drug-resistant cell lines all demonstrated MDR phenotypes, as shown for paclitaxel and vinblastine (Supplementary Fig. S5), potentially via upregulation of ABC transporters18. Unlike paclitaxel and vinblastine, FC77 retained its cytotoxicity against the MDR cancer cell lines, with the GI50 values of 1–53.4 nM (Fig. 3). Although FC77 did not reveal selectivity toward A549/T cells (GI50 = 53.4 ± 1.1 nM), it was much more potent than paclitaxel (GI50 = 2,090 ± 64 nM; Supplementary Fig. S5) against this cell line. For parental CCRF-CEM and HCT-116 cells and their resistant derivatives, FC77 showed less selectivity; however, the GI50 values were in the low nanomolar range (1–8 nM). Moreover, K562/HHT300 and, particularly, HL60/DOX and A549/DDP cells were more sensitive to FC77 than their parental cells, suggesting the potential of FC77 to treat MDR malignancies as a complementary treatment to the current standard chemotherapies.
Fig. 3.

Comparison of cytotoxicity of FC77 against multidrug-resistant cancer cell lines and their parental cancer cell lines. *p < 0.05, ***p < 0.001.
3.3. FC77 shows a safer therapeutic window and low toxic side effects
The differential anticancer activity of FC77 against HL60 and HL60/DOX cells was further evaluated by a colony formation assay (Fig. 4), which reflects their leukemic stem cell activity. The clonogenic potential of HL60 and HL60/DOX cells was significantly and dose-dependently reduced upon treatment with this indole-chalcone. FC77 was more active against drug-resistant HL60/DOX cells than against HL60 cells in the colony formation assay. A significant difference was observed at a low drug concentration (0.01 μM). As expected, conventional chemotherapeutic agents (doxorubicin and vincristine) were highly active against HL60 cells but much less effective against the resistant HL60/DOX cell line. To evaluate the potential therapeutic window of FC77, we also evaluated its effects on colony formation by normal CD34+ blood progenitor cells derived from MPB of healthy donors31. While doxorubicin and vincristine showed a greater inhibitory effect on the colony formation by normal MPB cells than on that by MDR HL60/DOX cells, FC77 was markedly less toxic to normal MPB cells than to MDR HL60/DOX leukemia cells (Fig. 4), which suggests its safer therapeutic window and translational potential.
Fig. 4.

Preferential cytotoxicity of FC77 to HL60/DOX cells over HL60 cells, and its therapeutic window for CD34+ MPB cells, assessed via a leukemia colony-forming cell (L-CFC) assay. Cells (100 cells per condition for leukemia cell lines or 500 cells per condition for MPB) were treated with doxorubicin, vincristine, or FC77 for 24 h in a standard culture medium and then harvested and plated in MethoCult H4034 optimum CFC medium (Stem Cell Technologies) according to the manufacturer’s recommendations. Colonies were scored after 7–14 days under an inverted microscope. *p < 0.05.
3.4. FC77 significantly induces cell cycle arrest
We then characterized the impact of FC77 and three standard drugs (doxorubicin, paclitaxel, and vinblastine) on the HL60 cell-cycle distribution. The concentrations of the compounds were equal to their corresponding 48-h GI50s. The cells treated with FC77 showed a significantly larger population in the G2/M phase than the control cells (Fig. 5A). Vinblastine also significantly induced cell cycle arrest, while paclitaxel slightly increased the G2/M population, consistent with their antimicrotubule mechanisms of action. Doxorubicin showed no induction of cell cycle arrest. Given that FC77 showed the impact on cell cycle, we further evaluated its effects toward HL60 and HCT116 using different time points (Fig. 5B and C). FC77 can arrest these two cells in a significant time-dependent manner. The HL60 cells showed the population of ~14–25% in the G2/M phase. HCT116 cells can be arrested in the G2/M phase from ~25% at 1h to ~60 % at 12 h.
Fig. 5.

(A) Effects of FC77 and standard therapeutic drugs on the HL60 cell cycle upon a 6-h treatment. DOX, doxorubicin; TAX, paclitaxel; VIN, vinblastine. **p < 0.01, ***p < 0.001 versus control group. Effect of FC77 on cell cycle distribution towards HL-60 (B) and HCT-116 cells (C) at different time points (1 h, 3 h, 6 h, 9 h, 12 h) with concentrations of corresponding GI50s (HL60, 2.8 nM; HCT-116, 1 nM). *p<0.05, **p<0.01, ***p<0.001 versus G2/M phase of control group. All the experiments are triplicated.
3.5. FC77 directly interacts with tubulin
We next explored the ability of FC77 to directly interact with tubulin by measuring its effect on the melting temperature of tubulin by differential scanning fluorimetry27. As shown in Fig. 6, colchicine and vinblastine increased the melting temperature by 3 and 2 °C, respectively, while paclitaxel increased the temperature by only 1 °C. We speculated that the weak effect of paclitaxel could be due to its preferential interaction with microtubules, instead of tubulin. FC77 increased the melting temperature by 4 °C, i.e., more than the three standard MTAs, indicating its direct interaction with tubulin.
Fig. 6.

Thermal shifts in the tubulin monomer (in the presence of GDP, 0.1 mg/mL) with colchicine, paclitaxel, vinblastine or FC77 (50 μM), as determined by differential scanning fluorimetry. Control: General tubulin buffer (PEM) containing 80 mM PIPES pH 6.9, 2 mM MgCl2 and 0.5 mM EGTA. The melting temperatures of tubulin (°C) with different treatments are presented in the table.
3.6. FC77 significantly blocks the dynamics of individual microtubules
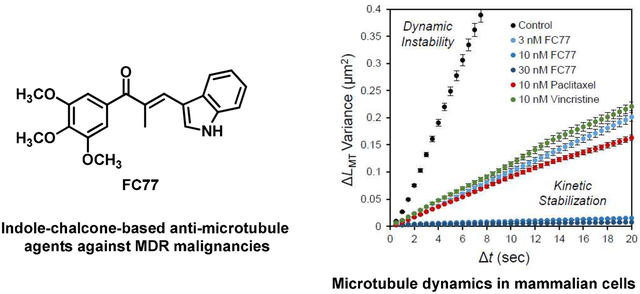
The effect of FC77 on the microtubule dynamics was characterized via direct imaging of microtubule dynamics in intact LLC-PK1 porcine kidney cells (Fig. 7). Normally, microtubules in intact cells undergo dynamic changes via polymerization and depolymerization, exhibiting a high assembly variance over time (DMSO control in Fig. 7A). As expected, paclitaxel and vincristine (10 nM) significantly reduced the dynamics of individual microtubules. A similar reduction was achieved with FC77 when its concentration was 3 nM (Fig. 7A). However, FC77 completely blocked the microtubule dynamics when its concentration reached 10 nM (Fig. 7A, B, and D). A dose-response quantitative analysis showed that FC77 was a more potent inhibitor than paclitaxel and vincristine. The half-maximal inhibitory concentration (IC50) was approximately 3 nM for FC77 compared to 14 nM and 37 nM for paclitaxel and vincristine, respectively (Fig. 7C).
Fig. 7.

Effects of paclitaxel, vincristine, and FC77 on the microtubule dynamics in LLC-PK1 cells. (A) Representative plots of the changes in the microtubule length over time under different conditions. Each trace represents an individual microtubule. (B) Mean-squared displacement of the microtubule dynamic end per unit of time. Microtubules undergoing dynamic instability exhibit large displacements (DMSO), while those that are kinetically stabilized or have lost the dynamics exhibit small displacements over time (10 nM FC77). Data are presented as the mean ± standard error of the mean. (C) Effective diffusion coefficients of the microtubule dynamic end as a function of the concentration of paclitaxel (red), vincristine (blue), and FC77 (black). Effective diffusion coefficients were estimated from the mean-squared displacements of the dynamic end shown in (B). The IC50 values were approximately 3 nM for FC77, 14 nM for paclitaxel, and 37 nM for vincristine. The best-fit sigmoid curves were obtained for all estimated diffusion coefficients. The diffusion coefficient data for paclitaxel is from our previous study22. (D) Microtubule dynamics in the presence of paclitaxel, vincristine, and FC77. Representative kymographs of the EGFP–tubulin signal are shown for individual microtubules under the indicated conditions. For each kymograph, the microtubule is aligned to the vertical axis, while time is plotted on the horizontal axis.
3.7. FC77 shows a similar microtubule-depolymerizing mechanism with that of colchicine
To confirm the antimicrotubule mechanism of FC77, we used the NCI COMPARE algorithm to compare the FC77 activity profile with those of other anticancer therapeutics. The similarity relative to FC77 was expressed using a Pearson’s correlation coefficient (PCC). A PCC value of > 0.5 is generally considered significant32, 33. Our COMPARE analysis results showed that FC77 was similar in its mechanism of action to five representative antimicrotubule agents (Table 2). Colchicine, in particular, showed a PCC value of 0.658 relative to FC77, indicating a similar microtubule-depolymerizing mechanism, while FC77 was approximately 10-fold more potent than colchicine (average GI50 = 57.5 nM in the NCI database)34. Additionally, colchicine was not highly active against HL60/DOX cells (GI50 = 10.3 μM), which showed a 25-fold higher resistance than that of parental HL60 cells (Supplementary Fig. S7).
Table 2.
The top antimicrotubule agents with cancer cell growth inhibitory patterns similar to that of FC77.
| Name | PCC |
|---|---|
| Colchicine | 0.658 |
| Epothilone A | 0.567 |
| Epothilone D | 0.564 |
| Docetaxel | 0.555 |
| Vincristine | 0.547 |
PCC, Pearson’s correlation coefficient.
4. Discussion
Chalcone is a privileged structure in medicinal chemistry because it shows diverse biological activities, including anticancer activity9. Nevertheless, there is not enough convincing evidence to confirm its anticancer binding targets. In our previous study, we have unambiguously confirmed β-tubulin as a direct cellular target of chalcones via whole-cell-based photoaffinity12. The results prompted us to investigate drug candidates based on the chalcone scaffold and their molecular mechanisms.
FC77, an indole-chalcone derivative, was demonstrated to show marked potency against multiple cancer cell lines by targeting microtubules. Since multidrug resistance is a major challenge for clinical MTA cancer treatment, our findings suggest the potential of FC77 to mitigate this clinical issue. The potential advantage of FC77 is its simple structure compared with those of complex clinical antimicrotubule agents. Combretastatin A-4, a natural product from Combretum caffrum35 and an MTA with a simple structure similar to that of FC77, were evaluated in parallel and found to have a similar selectivity profile and potencies (Supplementary Fig. S6), indicating that MTAs with simple structures may be useful for treating MDR malignancies. Moreover, it is reported that tubulin binding agents that specifically target the colchicine binding site may circumvent the MDR36, 37. Thus, several experiments were then applied to support its colchicine-binding property.
The cell cycle arrest is a key feature of antimicrotubule agent-induced cytotoxicity38, 39. Apoptosis, on the other hand, can be induced by therapeutic agents with various mechanisms of action, making it difficult to understand the upstream mechanisms. Thus, the increased G2/M phase population in two cell lines after treatment with FC77 suggested that FC77 induced the arrest at G2 and/or M phase. Additional data, including the melting temperature (biochemical binding), COMPARE (correlative analysis), and molecular modeling (Supplementary Figs. S1–S3) data, as well as our previous whole-cell-based photoaffinity labeling data12, collectively support this mechanism of action. A whole-cell microtubule dynamics assay was used, instead of a tubulin polymerization/depolymerization biochemical assay, to confirm the direct interaction of FC77 with tubulin, mainly because the concentrations needed to induce polymerization or depolymerization could be more than 1,000-fold higher than cytotoxic concentrations and because there might be a limited correlation, if any, between the biochemical potency and in vitro cytotoxicity, as has been reported for tubulysin by Fecik et al.40. Besides, molecular modeling showed that the compound could be well docked into the colchicine-binding site, and a good superposition with colchicine was observed. A clear SAR (Supplemental Information), with a decent correlation (R2 = 0.70) between the GI50 values of the compounds and their activities predicted by an atom-based QSAR model, was also obtained (Supplementary Fig. S3C). The 5-methoxyl and 1-amino functional groups of FC77 interacted with Cys241 and Asn101 of tubulin via hydrogen bonding (Supplementary Fig. S3A and B), consistent with the potent cytotoxicity of the compound.
There have been some concerns about the chalcone template because it contains a pan-assay-interference-compounds (PAINS) fragment41, 42. However, the use of PAINS as a filter has been challenged43, 44. Compounds with a potentially promiscuous template must be carefully evaluated before they are excluded. In this study, the effect of FC77 on microtubule dynamics in intact cells was characterized, and the compound was demonstrated to completely inhibit the microtubule dynamics when its concentration reached 10 nM, which is much lower than the typical concentrations for PAINS effects. A dose-response quantitative analysis showed that FC77 is a more potent inhibitor of microtubule dynamics than are paclitaxel and vincristine. Besides, our previous data also support β-tubulin as a direct cellular target of chalcones12. Collectively, these studies demonstrated that the reported compounds possess specific activities and their apparent activity is not an artifact due to the PAINS potential.
In summary, we synthesized a series of chalcone compounds, for which a clear SAR was demonstrated, with a good correlation with their modeled binding interactions with tubulin. FC77 exhibited excellent cytotoxicity against the NCI-60 human cancer cell lines, with the GI50 values in the low nanomolar range. A mechanistic study demonstrated that FC77 arrested the cell cycle which might relate to the binding to tubulin, and inhibited the microtubule dynamics. The COMPARE analysis indicated that FC77 had a mechanism of action similar to that of colchicine and other MTAs. These data consistently support the antimicrotubule mechanism of action of FC77. More importantly, MDR cancer cell lines showed no resistance to FC77, and this compound demonstrated selective cytotoxicity against cancer cells, with less toxicity against normal CD34+ blood progenitor cells. Overall, our data show that FC77 represents a novel MTA template for drug development to treat MDR cancers.
Supplementary Material
Acknowledgements
This research was funded by grants from the Shanghai Municipal Commission of Health and Family Planning (2017YQ052 to C.Z.); the Young Elite Scientists Sponsorship Program by the China Association for Science and Technology (2017QNRC061 to C.Z.); the Key Research and Development Program of Ningxia (2018BFH02001 to W. Z. and 2018BFH02001–01 to C.Z.); the Shanghai “ChenGuang” Project (16CG42 to C.Z.); the National Natural Science Foundation of China (81502978 to C.Z.); the National Cancer Institute, National Institutes of Health, USA (R01CA163864 to C.X.); Ningxia Medical University (XT2017022 to C.X.); the University of Minnesota Masonic Cancer Center and Academic Health Center Heme Malignancy Tissue Bank; and the National Heart, Lung, and Blood Institute, National Institutes of Health, USA (T32HL007062 to C.E.E.). We also thank Dr. Li Su for the cell cycle analysis.
Footnotes
Conflicts of interest
The authors declare that they have no competing interests.
Supporting Information
Supplementary data related to this article can be found at http://dx.doi.org/###, including Structure-activity relationship, chalcone synthesis and characterization, docking studies, NCI60 panel data, spectra of synthesized compounds and references.
References
- 1.Chen SM; Meng LH; Ding J New microtubule-inhibiting anticancer agents. Expert Opin. Investig. Drugs 2010, 19, (3), 329–343. [DOI] [PubMed] [Google Scholar]
- 2.Gigant B; Wang C; Ravelli RB; Roussi F; Steinmetz MO; Curmi PA; Sobel A; Knossow M Structural basis for the regulation of tubulin by vinblastine. Nature 2005, 435, (7041), 519–522. [DOI] [PubMed] [Google Scholar]
- 3.Li W Drugs targeting tubulin polymerization. Pharm. Res 2012, 29, (11), 2939–2942. [DOI] [PubMed] [Google Scholar]
- 4. https://www.clinicaltrials.gov/ct2/results?term=eribulin+OR+E7389.
- 5.Dumontet C; Jordan MA Microtubule-binding agents: a dynamic field of cancer therapeutics. Nat. Rev. Drug Discov 2010, 9, (10), 790–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kavallaris M Microtubules and resistance to tubulin-binding agents. Nat. Rev. Cancer 2010, 10, (3), 194–204. [DOI] [PubMed] [Google Scholar]
- 7.Fletcher JI; Haber M; Henderson MJ; Norris MD ABC transporters in cancer: more than just drug efflux pumps. Nat. Rev. Cancer 2010, 10, (2), 147–156. [DOI] [PubMed] [Google Scholar]
- 8.Wieczorek M; Tcherkezian J; Bernier C; Prota AE; Chaaban S; Rolland Y; Godbout C; Hancock MA; Arezzo JC; Ocal O; Rocha C; Olieric N; Hall A; Ding H; Bramoulle A; Annis MG; Zogopoulos G; Harran PG; Wilkie TM; Brekken RA; Siegel PM; Steinmetz MO; Shore GC; Brouhard GJ; Roulston A The synthetic diazonamide DZ-2384 has distinct effects on microtubule curvature and dynamics without neurotoxicity. Sci. Transl. Med 2016, 8, (365), 365ra159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhuang C; Zhang W; Sheng C; Zhang W; Xing C; Miao Z Chalcone: A Privileged Structure in Medicinal Chemistry. Chem Rev 2017, 117, (12), 7762–7810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Singh P; Anand A; Kumar V Recent developments in biological activities of chalcones: a mini review. Eur. J. Med. Chem 2014, 85, 758–777. [DOI] [PubMed] [Google Scholar]
- 11.Zhou B; Xing C Diverse Molecular Targets for Chalcones with Varied Bioactivities. Med. Chem. (Los Angeles) 2015, 5, (8), 388–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou B; Yu X; Zhuang C; Villalta P; Lin Y; Lu J; Xing C Unambiguous Identification of beta-Tubulin as the Direct Cellular Target Responsible for the Cytotoxicity of Chalcone by Photoaffinity Labeling. ChemMedChem 2016, 11, (13), 1436–1445. [DOI] [PubMed] [Google Scholar]
- 13.Zhou B; Jiang P; Lu J; Xing C Characterization of the Fluorescence Properties of 4-Dialkylaminochalcones and Investigation of the Cytotoxic Mechanism of Chalcones. Arch. Pharm. (Weinheim) 2016, 349, (7), 539–552. [DOI] [PubMed] [Google Scholar]
- 14.Kyowa H; K.K. K Propenone derivatives Europe Patent 0680950A1, 1994.
- 15.Kyowa H; K.K. K Propenone derivatives. U.S. Patent 5952355A, 1995.
- 16.Zhang Y; Srinivasan B; Xing C; Lu J A new chalcone derivative (E)-3-(4-methoxyphenyl)-2-methyl-1-(3,4,5-trimethoxyphenyl)prop-2-en-1-one suppresses prostate cancer involving p53-mediated cell cycle arrests and apoptosis. Anticancer Res. 2012, 32, (9), 3689–3698. [PubMed] [Google Scholar]
- 17.Srinivasan B; Johnson TE; Lad R; Xing C Structure-activity relationship studies of chalcone leading to 3-hydroxy-4,3’,4’,5’-tetramethoxychalcone and its analogues as potent nuclear factor kappaB inhibitors and their anticancer activities. J. Med. Chem 2009, 52, (22), 7228–7235. [DOI] [PubMed] [Google Scholar]
- 18.Das SG; Hermanson DL; Bleeker N; Lowman X; Li Y; Kelekar A; Xing C Ethyl 2-amino-6-(3,5-dimethoxyphenyl)-4-(2-ethoxy-2-oxoethyl)-4H-chromene-3-carboxylate (CXL017): a novel scaffold that resensitizes multidrug resistant leukemia cells to chemotherapy. ACS Chem. Biol 2013, 8, (2), 327–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Das SG; Srinivasan B; Hermanson DL; Bleeker NP; Doshi JM; Tang R; Beck WT; Xing C Structure-activity relationship and molecular mechanisms of ethyl 2-amino-6-(3,5-dimethoxyphenyl)-4-(2-ethoxy-2-oxoethyl)-4H-chromene-3-carboxylate (CXL017) and its analogues. J. Med. Chem 2011, 54, (16), 5937–5948. [DOI] [PubMed] [Google Scholar]
- 20.Aridoss G; Zhou B; Hermanson DL; Bleeker NP; Xing C Structure-activity relationship (SAR) study of ethyl 2-amino-6-(3,5-dimethoxyphenyl)-4-(2-ethoxy-2-oxoethyl)-4H-chromene-3-carboxylate (CXL017) and the potential of the lead against multidrug resistance in cancer treatment. J. Med. Chem 2012, 55, (11), 5566–5581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tang R; Cohen S; Perrot JY; Faussat AM; Zuany-Amorim C; Marjanovic Z; Morjani H; Fava F; Corre E; Legrand O; Marie JP P-gp activity is a critical resistance factor against AVE9633 and DM4 cytotoxicity in leukaemia cell lines, but not a major mechanism of chemoresistance in cells from acute myeloid leukaemia patients. BMC Cancer 2009, 9, 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vaziri SA; Grabowski DR; Tabata M; Holmes KA; Sterk J; Takigawa N; Bukowski RM; Ganapathi MK; Ganapathi R c-IAP1 is overexpressed in HL-60 cells selected for doxorubicin resistance: effects on etoposide-induced apoptosis. Anticancer Res. 2003, 23, (5A), 3657–3661. [PubMed] [Google Scholar]
- 23.Chen M; Beck WT DNA topoisomerase II expression, stability, and phosphorylation in two VM-26-resistant human leukemic CEM sublines. Oncol. Res 1995, 7, (2), 103–111. [PubMed] [Google Scholar]
- 24.Beck WT; Mueller TJ; Tanzer LR Altered surface membrane glycoproteins in Vinca alkaloid-resistant human leukemic lymphoblasts. Cancer Res. 1979, 39, (6 Pt 1), 2070–2076. [PubMed] [Google Scholar]
- 25.Cao D; Han X; Wang G; Yang Z; Peng F; Ma L; Zhang R; Ye H; Tang M; Wu W; Lei K; Wen J; Chen J; Qiu J; Liang X; Ran Y; Sang Y; Xiang M; Peng A; Chen L Synthesis and biological evaluation of novel pyranochalcone derivatives as a new class of microtubule stabilizing agents. Eur. J. Med. Chem 2013, 62, 579–589. [DOI] [PubMed] [Google Scholar]
- 26.Cala O; Remy MH; Guillet V; Merdes A; Mourey L; Milon A; Czaplicki G Virtual and biophysical screening targeting the gamma-tubulin complex--a new target for the inhibition of microtubule nucleation. PloS one 2013, 8, (5), e63908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhuang C; Narayanapillai S; Zhang W; Sham YY; Xing C Rapid identification of Keap1-Nrf2 small-molecule inhibitors through structure-based virtual screening and hit-based substructure search. J. Med. Chem 2014, 57, (3), 1121–1126. [DOI] [PubMed] [Google Scholar]
- 28.Castlea BT; McCubbinb S; Prahla LS; Bernensa JN; Septb D; Oddea DJ Mechanisms of kinetic stabilization by the drugs paclitaxel and vinblastine. Mol. Biol. Cell 2017, 28, (9), 1238–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prahl LS; Castle BT; Gardner MK; Odde DJ Quantitative analysis of microtubule self-assembly kinetics and tip structure. Methods Enzymol. 2014, 540, 35–52. [DOI] [PubMed] [Google Scholar]
- 30.Shoemaker RH The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 2006, 6, (10), 813–823. [DOI] [PubMed] [Google Scholar]
- 31.Bhatia S; Reister S; Mahotka C; Meisel R; Borkhardt A; Grinstein E Control of AC133/CD133 and impact on human hematopoietic progenitor cells through nucleolin. Leukemia 2015, 29, (11), 2208–2220. [DOI] [PubMed] [Google Scholar]
- 32.Naasani I; Seimiya H; Yamori T; Tsuruo T FJ5002: a potent telomerase inhibitor identified by exploiting the disease-oriented screening program with COMPARE analysis. Cancer Res. 1999, 59, (16), 4004–4011. [PubMed] [Google Scholar]
- 33.Yamori T; Matsunaga A; Sato S; Yamazaki K; Komi A; Ishizu K; Mita I; Edatsugi H; Matsuba Y; Takezawa K; Nakanishi O; Kohno H; Nakajima Y; Komatsu H; Andoh T; Tsuruo T Potent antitumor activity of MS-247, a novel DNA minor groove binder, evaluated by an in vitro and in vivo human cancer cell line panel. Cancer Res. 1999, 59, (16), 4042–4049. [PubMed] [Google Scholar]
- 34. https://dtp.cancer.gov/dtpstandard/servlet/MeanGraph?searchtype=NSC&searchlist=757&outputformat=HTML&outputmedium=page&chemnameboolean=AND&debugswitch=false&assaytype=&testshortname=&dataarraylength=75&endpt=GI50&button=Mean+Graph&highconc=−4.0.
- 35.Lee L; Robb LM; Lee M; Davis R; Mackay H; Chavda S; Babu B; O’Brien EL; Risinger AL; Mooberry SL; Lee M Design, synthesis, and biological evaluations of 2,5-diaryl-2,3-dihydro-1,3,4-oxadiazoline analogs of combretastatin-A4. J. Med. Chem 2010, 53, (1), 325–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arnst KE; Wang Y; Hwang DJ; Xue Y; Costello T; Hamilton D; Chen Q; Yang J; Park F; Dalton JT; Miller DD; Li W A Potent, Metabolically Stable Tubulin Inhibitor Targets the Colchicine Binding Site and Overcomes Taxane Resistance. Cancer Res. 2018, 78, (1), 265–277. [DOI] [PubMed] [Google Scholar]
- 37.Nguyen TL; Cera MR; Pinto A; Lo Presti L; Hamel E; Conti P; Gussio R; De Wulf P Evading Pgp activity in drug-resistant cancer cells: a structural and functional study of antitubulin furan metotica compounds. Mol. Cancer Ther 2012, 11, (5), 1103–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu C; Zuo Y; Wang R; Liang B; Yue X; Wen G; Shang N; Huang L; Chen Y; Du J; Bu X Discovery of potent cytotoxic ortho-aryl chalcones as new scaffold targeting tubulin and mitosis with affinity-based fluorescence. J. Med. Chem 2014, 57, (15), 6364–6382. [DOI] [PubMed] [Google Scholar]
- 39.Shen KH; Chang JK; Hsu YL; Kuo PL Chalcone arrests cell cycle progression and induces apoptosis through induction of mitochondrial pathway and inhibition of nuclear factor kappa B signalling in human bladder cancer cells. Basic & Clin. Pharmacol. Toxicol 2007, 101, (4), 254–261. [DOI] [PubMed] [Google Scholar]
- 40.Balasubramanian R; Raghavan B; Begaye A; Sackett DL; Fecik RA Total synthesis and biological evaluation of tubulysin U, tubulysin V, and their analogues. J. Med. Chem 2009, 52, (2), 238–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baell JB Feeling Nature’s PAINS: Natural Products, Natural Product Drugs, and Pan Assay Interference Compounds (PAINS). J. Nat. Prod 2016, 79, (3), 616–628. [DOI] [PubMed] [Google Scholar]
- 42.Gilberg E; Gutschow M; Bajorath J X-ray Structures of Target-Ligand Complexes Containing Compounds with Assay Interference Potential. J. Med. Chem 2018, 61, (3), 1276–1284. [DOI] [PubMed] [Google Scholar]
- 43.Senger MR; Fraga CA; Dantas RF; Silva FP Jr. Filtering promiscuous compounds in early drug discovery: is it a good idea? Drug Discov. Today 2016, 21, (6), 868–872. [DOI] [PubMed] [Google Scholar]
- 44.Meng N; Tang H; Zhang H; Jiang C; Su L; Min X; Zhang W; Zhang H; Miao Z; Zhang W; Zhuang C Fragment-growing guided design of Keap1-Nrf2 protein-protein interaction inhibitors for targeting myocarditis. Free Radic. Biol. Med 2018, 117, 228–237. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.