

Abstract
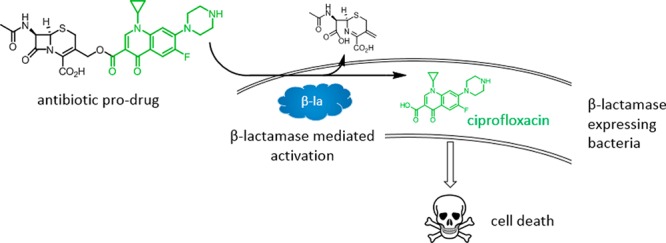
Expression of β-lactamase is the single most prevalent determinant of antibiotic resistance, rendering bacteria resistant to β-lactam antibiotics. In this article, we describe the development of an antibiotic prodrug that combines ciprofloxacin with a β-lactamase-cleavable motif. The prodrug is only bactericidal after activation by β-lactamase. Bactericidal activity comparable to ciprofloxacin is demonstrated against clinically relevant E. coli isolates expressing diverse β-lactamases; bactericidal activity was not observed in strains without β-lactamase. These findings demonstrate that it is possible to exploit antibiotic resistance to selectively target β-lactamase-producing bacteria using our prodrug approach, without adversely affecting bacteria that do not produce β-lactamase. This paves the way for selective targeting of drug-resistant pathogens without disrupting or selecting for resistance within the microbiota, reducing the rate of secondary infections and subsequent antibiotic use.
Introduction
Antimicrobial drug resistance is a global health emergency, threatening advances in many areas of medicine including surgery, cancer chemotherapy, organ transplantation, and survival of preterm infants.1,2 The most prevalent and important resistance determinant is the β-lactamase enzyme, which hydrolyzes members of the β-lactam class of antibiotic (e.g., penicillin, cephalosporins, and carbapenems) and thereby prevents engagement with their therapeutic targets the penicillin-binding proteins (PBPs).3,4 Of particular concern are the extended-spectrum β-lactamases (ESBLs) such as the CTX-M class, which are able to cleave a wide range of clinically relevant β-lactam antibiotics.5−7
Urinary tract infections (UTIs) are the most prevalent type of bacterial infection globally. These infections have a high rate of recurrence and can also lead to serious invasive infections such as sepsis, particularly in the elderly.8,9E. coli is the most common causative organism (∼75% cases), of which ∼50% are resistant to β-lactam antibiotics due to β-lactamase expression.8,10 As a consequence of the high rate of β-lactam resistance in UTI pathogens, second-line, broad-spectrum antibiotics such as ciprofloxacin are increasingly used therapeutically.11,12 Unfortunately, these broad-spectrum antibiotics are associated with disruption to the beneficial bacteria that colonize the gastrointestinal tract and other surfaces, known as the microbiota.13−17 This disruption can lead to serious secondary infections by antibiotic-resistant bacteria such as Clostridium difficle or fungi such as Candida albicans, leading to colitis and thrush, respectively.13,18 This is because antibiotics target conserved processes in bacteria such as cell wall, protein, DNA or RNA biosynthesis, which not only occur in the pathogens that cause infection but also in the members of the microbiota.19,20
An additional complication associated with some second-line therapeutics such as ciprofloxacin is host toxicity. Ciprofloxacin holds two black box warnings, one for increased risk of tendinitis and tendon rupture and one for exacerbation of muscle weakness in myasthenia gravis sufferers.21 Additionally, in 2015, the FDA officially recognized fluoroquinolone-associated disability (FQAD) as a syndrome. FQAD describes a range of disabling and potentially permanent side effects including disturbances of tendons, joints, muscles, nerves, the nervous system, and induction of type 2 diabetes.22,23 As a result, strategies with the potential to mitigate host toxicity by reducing exposure to ciprofloxacin are needed.
Given the drawbacks associated with broad-spectrum antibiotics, efforts have been made to limit their use.11,23−25 However, these efforts have had limited success with usage rates increasing globally, particularly in low- and middle-income countries.26 In part, this is due to a lack of access to fast and efficient diagnostic techniques and the need to respond quickly to serious bacterial infections with effective and cost-efficient treatment regimens that target a wide range of different bacterial pathogens.27 There is, therefore, a pressing need to develop new therapeutics that kill a broad range of different pathogens without damaging the host microbiota.
Since β-lactamase enzymes are not found in mammalian cells, we hypothesized that we could exploit this enzyme as a novel antibacterial target. Furthermore, β-lactamase expression is prevalent among UTI pathogens, which can both colonize the gut and cause infection of the GU tract.8,10 Consequently, this represents an opportunity to selectively target disease-causing bacteria without causing significant disruption to the microbiota or select for drug resistance as has been reported for broad-spectrum antibiotics such as ciprofloxacin.28−30 Therefore, the aim of this work was to develop a small molecule antibacterial agent that is selectively active against bacteria that express β-lactamase. To do this, we employed a prodrug strategy that utilized a β-lactam cleavable motif linked to the broad-spectrum antibiotic ciprofloxacin.
In support of our approach, the use of β-lactams as prodrug modifiers in antibody-directed enzyme prodrug therapy approaches has been explored in disease areas such as cancer (1–3, Figure 1).31−35 Additionally, β-lactam–fluoroquinolone conjugates have been proposed as a co-drug strategy to treat bacterial infections (4 and 5, Figure 1).36−39 However, our approach is different in that it is designed to selectively deliver a broad-spectrum, bactericidal antibiotic to only bacteria that express β-lactamase, while having minimal effect on bacteria that do not express the resistance determinant. By contrast, previous dual activity co-drug approaches were designed to have broad-spectrum activity against both drug-sensitive bacteria and those that express β-lactamase.
Figure 1.

Selected representative examples of cephalosporin prodrugs (PROTAX 1,332,34 and BMY-46633 3(35)) and co-drugs (MCO 4(38) and Ro 23-9424 5(39)).
Herein we describe the design and development of the prodrug, including optimization of the β-lactam motif to reduce the antibacterial activity of the intact molecule and increase the efficiency of β-lactamase mediated ciprofloxacin release. This is, to our knowledge, the first example of a β-lactam-fluoroquinolone prodrug with selective activity against drug-resistant bacteria.
Results and Discussion
Prodrug Design
In order to create a prodrug molecule that is selectively activated in β-lactamase producing bacteria, it was important to select a β-lactamase cleavable motif, linkage strategy, and active antibiotic that gave a stable nonbactericidal intact molecule and enabled the rapid and efficient release of the antibiotic upon activation by β-lactamase. The success of this strategy required a prodrug motif that would enable efficient substrate turnover rather than inhibition of the β-lactamase enzyme. The cephalosporin class of β-lactams are efficiently hydrolyzed by β-lactamases and have been previously employed as a prodrug motif as cleavage of the β-lactam ring is associated with the loss of the functional group at the 3′-position (Figure 2A).40 In addition, the chemistry associated with changing the 3′-substituent of cephalosporins is well-established and a wide variety of substituents at the C-3′ position are tolerated by β-lactamases.37,41,42 Consequently, a cephalosporin core was selected as the β-lactam component. To achieve the desired selectivity profile, ciprofloxacin was attached via the carboxylic acid to give the 3′-cephem ester 6 (Figure 2B). Derivatization of the carboxylic acid group of fluoroquinolone antibiotics is associated with a significant decrease in antibacterial activity due to a decreased ability to bind to bacterial DNA–enzyme complexes.43 While this choice of attachment site was selected to remove the ciprofloxacin activity from the intact prodrug, it remained likely that the prodrug molecule would retain antibacterial activity as a result of the ability of the cephem portion of the molecule to interact with PBPs. Therefore, to further increase selectivity, it was essential to undertake a program of optimization of the β-lactam motif to reduce PBP activity and increase or maintain β-lactamase activity. Initial optimization was performed on the cephalosporin portion of the prodrug to enable the rapid generation of analogues and evaluation of biological activity. The cephalosporin analogues with the most desirable activity profile were then selected for preparation as the full prodrug.
Figure 2.

(A) Mechanism of β-lactamase triggered cephalothin hydrolysis. (B) General structure of proposed cephalosporin–ciprofloxacin prodrug 6.
β-Lactam Analogue Preparation and Biological Evaluation
Analysis of the literature identified the amide functionality at C-7 of the cephem ring as central to PBP and β-lactamase activity,44−51 and therefore structural changes at this position provided the initial focus of investigation. By use of cephalothin 7 (Table 1) as the starting point, analogues were prepared to explore bioisosteric replacement,52,53 functionalities present in early generation β-lactam antibiotics, and to probe steric and electronic tolerance.54,55 All compounds were synthesized according to the previously reported methods (Figure S1 in Supporting Information).55,56 Antibacterial activity was assessed by determining the minimal concentration required to inhibit bacterial growth, known as the minimal inhibitory concentration (MIC), against the E. coli strain DH5α ± expression of the ESBL TEM-116.57 The susceptibility to β-lactamase mediated hydrolysis was assessed by determining the physiological efficiency (kcat/Km) of hydrolysis by recombinant AmpC protein.3,58,59
Table 1. Antibacterial Activities (MIC) and AmpC Hydrolytic Efficiency of Synthesized Compounds and Reference Compound Cephalothin (Ceph) 7.
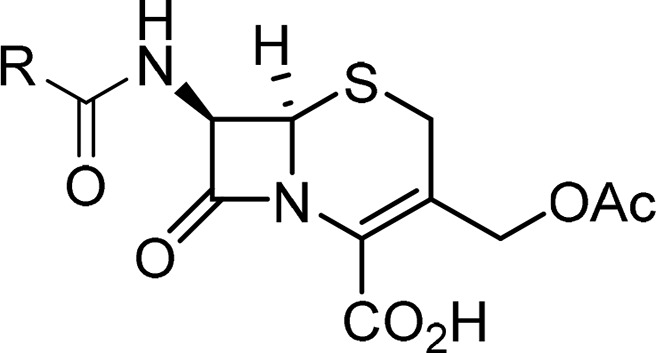

For all compounds (Table 1), a higher MIC value was determined for the E. coli strain expressing TEM-116 than the strain not expressing β-lactamase, indicating hydrolytic activity by the β-lactamase. Introduction of a substituent to the thiophene ring (8) or switching from a C-2 to a C-3 substitution (9) gave a modest increase in MIC values and a small decrease in kcat/Km compared to cephalothin 7. Although no measurable MIC value could be determined for any of the phenyl analogues (20–23), this was accompanied by a >3-fold decrease in kcat/Km. In general, a quaternary carbon (20–24) or tertiary carbon (10 and 25) at the α-position relative to the amide carbonyl was not well tolerated by AmpC. This finding is consistent with prior reports and has previously been exploited to reduce β-lactamase activity in the development of later-generation β-lactams. Compounds containing straight-chain aliphatic groups (26–28) retained some antibacterial activity; an increase in kcat/Km was observed with increasing chain length.
Examination of the tested analogues (Table 1) immediately revealed the importance of bulky benzylic substituents (11–19). Thiophene rings are frequently used as a bioisosteric replacement for a phenyl groups, and it is therefore perhaps unsurprising that there was only a modest 4-fold increase in MIC value against E. coli DH5α and a slight decrease in kcat/Km for 11 compared to cephalothin.52,53 However, introduction of substituents at the para-position (12–17) gave a further 2- to 4-fold increase in MIC against E. coli DH5α compared to unsubstituted benzyl 11. Substitution at the para-position also affected hydrolysis by AmpC with the following order of activity observed: F < Me = H < Cl = Br. Movement of the methyl substituent from the para- (12) to the meta-position (18) gave a 5-fold increase in kcat/Km and a 3-fold increase compared to cephalothin. High kcat/Km values were determined for bisaryl 16 and the para- and meta-substituted biphenyl ethers 17 and 19 (3.99 ± 0.88, 7.33 ± 1.72, and 31.14 ± 2.67, respectively). In addition, no measurable MIC values could be determined for 16, 17, or 19. This led us to question if the results were indicative of no antibacterial activity or simply a result of increased efflux activity out of, or a lack of permeability into, the bacterial cell.
β-Lactamase Hydrolytic Activity in Whole-Cell NMR Assay
To address the question of compound permeability/efflux, a whole-cell β-lactamase hydrolysis assay was used to detect the penetration of compounds into the periplasm.60,61 Hydrolytic decomposition of β-lactam rings is associated with changes in 1H NMR signals, which can be detected using whole bacterial cells in real time by 1H NMR spectroscopy (Figure S2). As hydrolysis occurred within the bacterial periplasm, only compounds with sufficient intracellular accumulation were hydrolyzed. Compounds 16 and 17 were selected as representative examples of high lipophilicity compounds with no measurable antibacterial activity and moderate in vitro β-lactamase hydrolysis. We evaluated the hydrolysis of bisaryl 16, biaryl ether 17, and cephalothin 7 in DH5α ± TEM-116 (Table 2). After 90 min, 16 was 69% hydrolyzed in DH5α + TEM-116 compared to 14% hydrolyzed in DH5α – TEM-116 and 17 was 53% hydrolyzed in DH5α + TEM-116 compared to 13% hydrolyzed in DH5α – TEM-116. These results indicated a high degree of in vivo β-lactamase mediated hydrolysis and that 16 and 17 accumulated in the bacterial cell. We therefore concluded that the lack of antibacterial activity of this compound against E. coli DH5α without β-lactamase was due to an absence of PBP engagement and not due to poor permeability or efflux activity.
Table 2. Percentage Hydrolysis of Cephalothin (Ceph) 7 and Compounds 16 and 17 by DH5α Cells ± β-Lactamase in Whole-Cell NMR Hydrolysis Assay.
| % hydrolysis by
NMR |
|||||
|---|---|---|---|---|---|
| compd | concn (μM) | incubation time (min) | strain | –βla | +βla |
| Ceph 7 | 50 | 60 | DH5a ± TEM-116 | 61 | |
| 16 | 100 | 90 | 14 | 69 | |
| 17 | 100 | 90 | 13 | 53 | |
| Ceph 7 | 100 | 60 | DH5a ± CTX-M-1 | 0 | 100 |
| 16 | 100 | 60 | 0 | 100 | |
| 17 | 100 | 60 | 0 | 95 | |
Biological Evaluation in Uropathogenic E. coli
Initial assessment of compound activity was performed in the laboratory E. coli strain DH5α. To assess the activity of the β-lactams against a clinically relevant pathogenic strain of E. coli, we selected the uropathogenic strain CFT073. This bacterium was isolated from the blood of a patient with acute pyelonephritis, is devoid of all virulence plasmids commonly associated with uropathogenic strains, and proved tractable for genetic manipulation.62,63 The plasmid pSU18, without the coding sequence for β-lactamase (referred to here as pEMP) or encoding for the β-lactamase CTX-M-1, was introduced into CFT073, enabling comparison of compound activity in CFT073 and CFT073 + pSU18 ± β-lactamase. The primary β-lactamase used in this work was CTX-M-1 because CTX-M enzymes are the most prevalent β-lactamases among enterobacteria such as E. coli. As part of a class of extended-spectrum β-lactamases (ESBL) it confers resistance to most β-lactam antibiotics, with the exception of carbapenems.6
In the first instance, MIC values were determined for selected compounds against CFT073 + pSU18 ± CTX-M-1 (Table 3). For all the compounds tested, the MIC values for CFT073 + pSU18 were within 2-fold of those determined against DH5α. Next the hydrolytic activity of these compounds was assessed in the whole cell NMR assay (Table 3). For the majority of the compounds tested, a low level of hydrolysis, <20% after 60 min, was detected. However, levels of hydrolysis comparable to that observed for cephalothin (68%) were observed only for 24 and 26 (64% and 67%, respectively).
Table 3. Antibacterial Activities (MIC) against CFT073 ± CTX-M-1 and Percentage Hydrolysis in CFT073 + CTX-M-1 Cells for Selected Compounds and Reference Compound Cephalothin (Ceph) 7b.
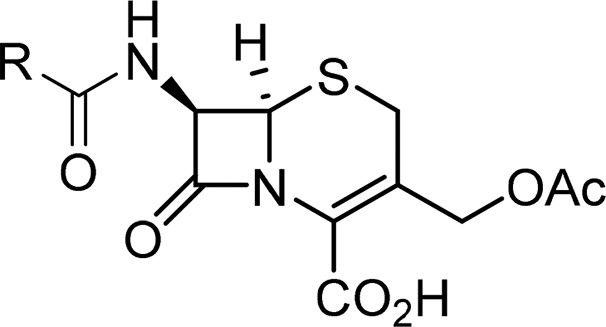

Percentage hydrolysis determined for 100 μM compound at 1 h by whole-cell NMR assay.
ND = not done.
A clear feature of the SAR was that CTX-M-1 mediated hydrolytic activity in whole CFT073 cells correlated with lipophilicity. Plotting the calculated log P (cLogP) values for compounds against the log of percentage hydrolysis revealed that moderate–high levels of hydrolysis (>30%) were only observed for compounds with cLogP values below 0.1 (Figure 3). Linear regression analysis revealed moderate correlation (R2 = 0.60), despite the degree of hydrolysis reflecting both cellular penetration and β-lactamase activity, which are both sensitive to compound lipophilicity.
Figure 3.

Plot of log of percentage hydrolysis in CFT073 + CTX-M-1 cells against calculated log P (cLogP) for synthesized compounds (filled circle) and cephalothin 7 (open circle): linear regression (dashed line), R2 = 0.60 (GraphPad Prism 7).
Interestingly, compounds 16 and 17 were hydrolyzed rapidly (69% and 54% after 90 min, respectively) in DH5α expressing the TEM-116 β-lactamase (Table 2) but only 4% hydrolyzed after 60 min in CFT073 expressing the CTX-M-1 β-lactamase. We hypothesized that the low level of hydrolytic activity observed for many of the compounds could be a result of poor intracellular accumulation in CFT073 E. coli since clinical isolates often have reduced permeability to antibiotics.64 To test this hypothesis, hydrolysis in DH5α expressing CTX-M-1 was determined for cepaholothin 7, 16, and 17 (Table 2). After 60 min complete hydrolysis for cepaholothin 7 and 16 and 94% hydrolysis for 17 were observed, suggesting that the low level of hydrolysis observed in CFT073 + CTX-M-1 was not due to the inability of CTX-M-1 to hydrolyze this chemotype. Instead it is likely that due to poor membrane permeability or increased efflux activity, lipophilic analogues were unable to engage with CTX-M-1 in CFT073. For compounds with low hydrolytic activity in the whole cell NMR assay with CFT073 we were unable to discern if a high MIC value in the absence of CTX-M-1 truly reflected a lack of antibacterial activity or a lack of permeability/high efflux activity. Therefore, compound 26, with its high MIC value in both CFT073 ± CTX-M-1 (≥400 μM) and high hydrolytic activity in whole CFT073 cells (67% after 60 min), was selected for incorporation into the full prodrug molecule. Compound 24, which also possessed a high MIC value in both CFT073 ± CTX-M-1 (≥400 μM) and high hydrolytic activity in whole CFT073 cells (64% after 60 min), was not progressed at this time as we wished to avoid the potential for toxicity problems arising from the furan ring, which has been identified as common toxicophore due to metabolic instability.65
Prodrug Preparation
Preparation of the prodrug derived from compound 26 required coupling of an activated 26 derivative, iodocephalosporin 30, to ciprofloxacin derivative 33 (Scheme 1). The iodocephalosporin 30 was prepared in three steps from commercially available 7-aminocephalosporanic acid (7-ACA). First, 7-ACA was reacted with acetic anhydride to give N-acetyl 26.55 Protection of the carboxylic acid as the tert-butyl ester was then performed using tert-butyl 2,2,2-trichloroacetimidate (TBTA) enabling formation of the tert-butyl ester 29 in the absence of base,66 which has previously been reported to be associated with isomerization from the Δ3-cephem to the biologically inactive Δ2-cephem.67−69 Iodination at the 3′-position with TMSI gave the activated iodocephalosporin 30 ready for coupling.36 The ciprofloxacin component was prepared by BOC protection of the piperazine NH to give 32 and subsequent conversion of the carboxylic acid to the sodium salt 33.70 Coupling of compounds 30 and 33 was performed in 3:1 1,4-dioxane/DMF to give the protected cephalosporin–ciprofloxacin conjugate 34.36,67 Finally, global deprotection with TFA to remove the BOC and tert-butyl ester afforded the final prodrug 35.71 Synthesis of 35 was achieved in seven steps from commercially available materials without the requirement for toxic metal reagents.
Scheme 1. Synthesis of Prodrug 35.

Reagents and conditions: (i) acetic anhydride, NaHCO3, H2O, acetone, 0 °C, 30 min; (ii) TBTA, DCM, 60 °C, 16 h; (iii) TMSI, DCM, rt, 2 h; (iv) Boc2O, 1 M NaOH, THF, rt, 16 h; (v) 0.1 M NaOH, MeOH, rt, 30 min; (vi) 3:1 1,4-dioxane/DMF, rt, 4 h; (vii) 1:1 TFA/DCM, anisole, 0 °C to rt.
In Vitro DNA Gyrase Activity
Members of the fluoroquinolone antibiotic family, including ciprofloxacin 31, target the type II topoisomerase enzymes, DNA gyrase, and topoisomerase IV. Inhibition of these enzymes results in the arrest of DNA replication and transcription preventing bacterial cell growth.43,72 Having successfully prepared prodrug 35, we moved to testing our hypothesis that the intact prodrug would not inhibit DNA gyrase or topoisomerase IV but β-lactamase triggered hydrolysis would result in the release of free ciprofloxacin capable of engaging these targets. To test this hypothesis, we evaluated the ability of prodrug 35 and ciprofloxacin to inhibit recombinant DNA gyrase enzyme activity in the absence and presence of the purified recombinant β-lactamase CTX-M-15 (Figure 4). Compounds were incubated with relaxed pBR322 plasmid DNA with and without recombinant CTX-M-15 and recombinant DNA gyrase. As predicted, inhibition of DNA gyrase by prodrug 35 was not observed in the absence of CTX-M-15. However, in the presence of CTX-M-15, 1 μM 35 was capable of reducing DNA gyrase activity by >50%. Ciprofloxacin 31 activity was not affected by CTX-M-15. These results confirmed that β-lactamase-specific hydrolysis of 35 results in liberation of active antibiotic capable of DNA gyrase inhibition in vitro.
Figure 4.

Activity of prodrug 35 and ciprofloxacin 31 against recombinant DNA gyrase ± CTX-M-15. (A) DNA was separated by agrose gel electrophoresis with 2 log DNA ladder: oc, open circle DNA; rel, relaxed DNA; sc, supercoiled DNA. (B) Quantification of gel bands corresponding to supercoiled DNA and normalized to no gyrase and gyrase only activity (ImageJ 1.52a): Gyr, DNA gyrase; cip, ciprofloxacin 31; PD, prodrug 35; CTX, CTX-M-15. Error bars represent SEM (n = 4); prodrug vs prodrug + CTX-M-15 was analyzed by unpaired t-test, p = 0.0004 (GraphPad Prism 7.03).
Selective Prodrug Activity against Uropathogenic E. coli Expressing β-Lactamase
Next, the activity of prodrug 35 was evaluated using whole bacterial cells. MIC values for 35 and ciprofloxacin 31 were determined in E. coli CFT073 expressing the disease-relevant β-lactamases CTX-M-1, New Delhi metallo-β-lactamase 1 (NDM-1), and Klebsiella pneumoniae carbapenemase (KPC-3) β-lactamase (Figure 5). NDM-1 is an example of an increasingly prevalent β-lactamase that is capable of hydrolyzing carbapenems, usually considered the last line of defense against β-lactamase expressing bacteria,73 while KPC enzymes are class A β-lactamases and the most common carbapenemases globally.74,75
Figure 5.

Antibacterial activities for prodrug 35 (blue) and ciprofloxacin 31 (red) against CFT073 E. coli cells WT and expressing empty plasmid (pEMP), CTX-M-15 (pCTX), NDM1 (pNMD1), and KPC (pKPC): (A) dose–response curves, where each point represents the mean ± SEM, n = 3; (B) summary of MIC values.
As expected, the MIC value determined for ciprofloxacin was consistent across all tested strains at 31 nM. The MIC determined for prodrug 35 in E. coli CFT073 WT and expressing empty plasmid (pEMP) was 310 nM, representing a 10-fold decrease in activity compared to ciprofloxacin 31 in the absence of β-lactamase. By contrast to bacteria without β-lactamase, the MIC value for 35 was 63 nM for E. coli CFT073 strains expressing CTX-M-1, NDM1, or KPC, only 2-fold higher than ciprofloxacin 31. These data demonstrate efficient and selective β-lactamase mediated prodrug cleavage and active antibiotic release, resulting in arrest of bacterial cell growth at concentrations comparable to that with free ciprofloxacin.
The activity of prodrug 35 compared to ciprofloxacin was profiled further in six independently isolated uropathogenic E. coli clinical isolates expressing the CTX-M-15 β-lactamase, which were obtained from Charing Cross Hospital, Imperial College NHS Trust. Three of the strains were ciprofloxacin sensitive (EC11, EC16, and EC17), and three were ciprofloxacin resistant (EC12, EC13, and EC19) as determined by diagnostic susceptibility testing. Activity of prodrug 35 was confirmed against the three ciprofloxacin sensitive bacterial strains, while no arrest in bacterial growth was observed for either ciprofloxacin 31 or prodrug 35 for strain EC12, EC13, or EC19 (Figure 6 and Table S1). These results demonstrate that the antibacterial activity of 35 observed in β-lactamase expressing strains is mediated through liberated ciprofloxacin and provide evidence for the clinical utility of 35. The gut microbiota includes both Gram-negative bacteria such as E. coli and Gram-positive organisms such as E. faecalis. Since we had shown that 35 was inactive against E. coli, we decided to further examine the potential clinical value of the prodrug by testing its activity against two representative E. faecalis strains that did not express β-lactamase. Prodrug 35 showed reduced activity compared to ciprofloxacin, indicating that our approach could minimize undesirable damage to the microbiota caused by fluoroquinolones (Figure S3). We also assessed the activity of 35 against CFT073 pEMP or pCTX-M-1 in the presence of human serum, which can modulate drug activity via protein-binding and also contains esterases that have the potential to activate the prodrug by cleaving the ester linkage. However, data from MIC assays performed in the presence of human serum (Figure S4) were equivalent to those obtained in the absence of serum (Figure 5). Combined, these findings provided further confidence in the selectivity of prodrug 35 and its stability in the host environment.
Figure 6.

Effect of prodrug 35 (blue) or ciprofloxacin 31 (red) against six uropathogenic E. coli clinical isolates. Each point represents the mean ± SEM, n = 3.
Selective Bactericidal Activity against β-Lactamase Expressing Bacteria
Finally, the ability of prodrug 35 to kill bacteria rather than arrest growth was evaluated. Survival of E. coli CFT073 pEMP or pCTX-M-1 with no treatment or exposed to ciprofloxacin 31 or prodrug 35 was determined over time by CFU counts (Figure 7). After 6 h incubation with prodrug 35 there was >100-fold greater killing of E. coli expressing CTX-M-1, compared with bacteria that did not express the enzyme. Killing activity of 35 in E. coli expressing CTX-M-1 was almost identical to free ciprofloxacin, while growth comparable to no treatment controls was detected for CFT073 expressing empty plasmid incubated with 35. These findings demonstrate that it is possible to selectively kill β-lactamase-producing bacteria using our prodrug approach, without adversely affecting bacteria that do not produce β-lactamase.
Figure 7.
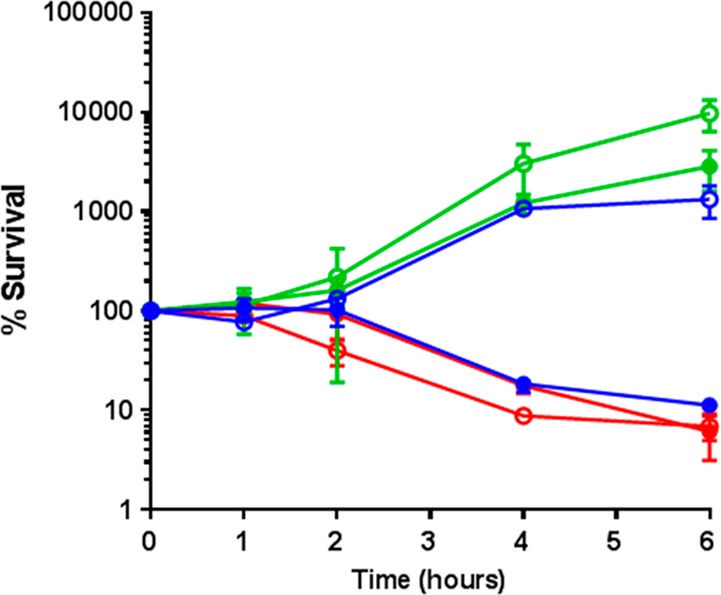
Survival of CFT073 pEMP (open circle) and pCTX (filled circle) with no treatment (green), ciprofloxacin 31 (78 nM) (red), or prodrug 35 (78 nM) (blue): Cipro, ciprofloxacin; PD, prodrug 35.
Conclusions
A novel cephalosporin–fluoroquinolone antibiotic prodrug has been designed, synthesized, and evaluated for biological activity. A program of optimization was successfully undertaken to reduce the antibacterial activity of the intact prodrug though modification to the cephalosporin component. Prodrug 35 exhibits similar growth inhibitory activity to ciprofloxacin against uropathogenic E. coli expressing the diverse ESBLs CTX-M-1, NDM-1, and KPC but little activity against strains that did not express β-lactamases. The selectively observed for bactericidal activity was even greater, with prodrug 35 killing β-lactamase expressing bacteria at the same rate as free ciprofloxacin while not affecting the growth of bacteria that did not express β-lactamases.
Overall, the activity of prodrug 35 is consistent with (1) permeability to pathogenic Gram-negative bacteria, (2) a low-level of antibacterial activity for the intact prodrug, (3) β-lactamase mediated intracellular release of ciprofloxacin upon cleavage of the cephalosporin, and (4) activation of the prodrug by a broad range of β-lactamases.
Together, these studies demonstrate that our prodrug approach can harness resistance as a therapeutic opportunity to selectively kill antibiotic-resistant bacteria. Since fluoroquinolones are a clinically useful, broad-spectrum antibiotic, we envisage that increasing the selectivity profile will have two major advantages. First, increased selectivity of fluoroquinolones will enable maintenance of the microbiota leading to reduced secondary infection rate and subsequent antibiotic use. Second, there is a decreased side-effect profile due to minimized exposure of host cells to fluoroquinolone antibiotic.
The focus of this work was uropathogenic E. coli (UPEC), which is a major cause of UTIs and frequently expresses β-lactamase. Our approach is expected to result in high concentrations of active ciprofloxacin at the site of infection (bladder and kidneys), without causing disruption to the host microbiota. However, it is important to consider that the primary reservoir of UPEC is the gut, and it is envisaged that our prodrug approach would also enable the selective decolonization of ESBL-expressing E. coli from the GI tract of people who suffer from recurrent UTI. An additional use could be the treatment of lung infections caused by P. aeruginosa in patients with cystic fibrosis, for which fluoroquinolones are the only available oral antibiotics.76,77 Since >65% P. aeruginosa isolates express the AmpC β-lactamase,78 it is possible that our prodrug approach could be used treat the infection without the associated damage to the microbiota.
In summary, this study paves the way for the development and use of small molecule therapeutics that selectively target drug-resistant pathogens using broad-spectrum antibiotics while minimizing selection for resistance and without collateral damage to the microbiota. This complements ongoing efforts to alter the spectrum of activity of existing antibiotics to enable them to be used in new ways. For example, recent work from Liu and co-workers79 described an approach to broaden the spectrum of activity of the otherwise Gram-positive restricted oxazolidinone antibiotics to confer activity against Gram-negative bacteria. By contrast, our approach restricts the activity of the normally broad-spectrum agent ciprofloxacin to only those bacteria that express the β-lactamase resistance determinant. We anticipate that the modification of existing antibiotics will prolong and expand their clinical utility, while efforts to discover new antibiotic classes are underway.
Experimental Section
Experimental Procedures (Chemistry)
Unless otherwise stated, reactions were conducted in oven-dried glassware under an atmosphere of argon using anhydrous solvents. All commercially obtained reagents and solvents were used as received. TLC analysis was performed on precoated aluminum sheets of silica (60 F254 nm, Merck) and visualized using short-wave UV light. Column chromatography was also performed on an Isolera Spektra Four purification system using Biotage Flash silica cartridges (SNAP KPSil, SNAP Ultra, or SNAP KP-C18-HS). 1H NMR spectra were recorded on Bruker Av-400 spectrometers at 400 MHz using an internal deuterium lock. Chemical shifts are quoted in parts per million (ppm) using the following internal references: CDCl3 (δH 7.26), D2O (δH 4.79), and DMSO-d6 (δH 2.50). Signal multiplicities are recorded as singlet (s), doublet (d), triplet (t), quartet (q), multiplet (m), doublet of doublets (dd), triplet of triplets (tt), apparent (app), broad (br), or obscured (obs). Coupling constants, J, were measured to the nearest 0.1 Hz. 13C NMR spectra were recorded on Bruker Av-400 spectrometers at 101 MHz using an internal deuterium lock. Chemical shifts are quoted to 0.1 ppm, unless greater accuracy was required, using the following internal references: CDCl3 (δC 77.0) and DMSO-d6 (δC 39.5). High resolution mass spectra were recorded on a Waters LCT with a Waters Aquilty UPLC I-class system operating in ES+ or ES– mode. Analytical separation was performed using a Waters BEH Acquity C18, 50 mm × 2.1 mm column using a flow rate of 0.5 mL/min in a 4 min gradient elution at 40 °C. The mobile phase was a mixture of 99.9% water and 0.1% formic acid (solvent A) and 99.9% acetonitrile and 0.1% formic acid (solvent B). Gradient elution was as follows: 95:5 (A/B) to 5:95 (A/B) over 3.2 min and then reversion back to 95:5 (A/B) over 0.3 min, finally 95:5 (A/B) for 0.5 min. For accurate mass determination, samples were referenced against leucine enkaphalin or sulfadimethoxine. All tested compounds were ≥95% pure by LCMS analysis. All final compounds were screened through computational PAINS and aggregator filters and gave no structural alerts as potential assay interference compounds or aggregators.80,81 All compounds were soluble at the concentrations used for biological evaluation.
General Synthetic Procedures
Method A. 7-Aminocephalosporanic acid (1 equiv) was dissolved in sat. NaHCO3 (aq) and acetone added, followed by acid chloride (1.2 or 2 equiv). The reaction was stirred at room temperature for 30 min, then washed with EtOAc. The aqueous layer was acidified to pH 2 with 1 M HCl and extracted with DCM (×3).55 The organic extracts were combined, dried over Na2SO4, evaporated and the resulting solid was triturated with ice-cold Et2O (unless otherwise stated) to afford the product.
Method B. 7-Aminocephalosporanic acid (1 equiv) and acid chloride (2 equiv) were dissolved in EtOAc and heated to reflux for 30 min. After cooling to room temperature, aniline (1.3–3 equiv) was added and stirred for 1 h before the reaction mixture was diluted with 3% NaHCO3 (aq). The aqueous layer was separated and the organic layer washed with 3% NaHCO3 (aq) (×2). The aqueous layers were combined, washed with EtOAc, and acidified to pH 2 with 1 M HCl.55 The desired product was isolated as described.
Method C. Carboxylic acid (1 equiv) was dissolved in DCM and oxalyl chloride (1.2 equiv) added followed by DMF (1 drop) and the reaction stirred for 16 h.56 The solvent was removed under reduced pressure to afford the acyl chloride, which was used without further purification.
Preparation of Compounds. (6R,7R)-3-(Acetoxymethyl)-7-(2-(4-bromothiophen-2-yl)acetamido)-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic Acid (8)
2-(4-Bromothiophen-2-yl)acetic acid (203 mg, 0.92 mmol), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (193 mg, 1.01 mmol), and 7-aminocephalosporanic acid (250 mg, 0.92 mmol) were suspended in DMF (8 mL) and stirred at room temperature for 48 h.82 The resulting mixture was filtered and the filtrate diluted with H2O and extracted with EtOAc (×3). The organic extracts were combined, washed with 1 M LiCl (aq) and brine, and dried over Na2SO4. Solvent was removed under reduced pressure and the resulting oil triturated with Et2O. The precipitate was collected by vacuum filtration and washed with DCM to afford the product as beige amorphous solid (36 mg, 8%). IR (solid): υmax 3273, 3101, 2837, 1774, 1748, 1707, 1662, 1539, 1223 cm–1. 1H NMR (400 MHz, DMSO-d6) δ 9.15 (d, J = 8.1 Hz, 1H), 7.51 (d, J = 1.2 Hz, 1H), 6.93 (s, 1H), 5.68–5.59 (m, 1H), 5.06 (d, J = 4.8 Hz, 1H), 5.00 (d, J = 12.6 Hz, 1H), 4.70 (d, J = 12.6 Hz, 1H), 3.78 (d, J = 2.6 Hz, 2H), 3.58 (d, J = 18.0 Hz, 1H), 3.42 (d, J = 18.7 Hz, 1H), 2.02 (s, 3H). 13C NMR (101 MHz, DMSO) δ 170.3, 169.4, 162.8, 139.1, 128.5, 122.8, 107.7, 59.0, 57.2, 35.6, 25.4, 20.6. HRMS (ESI+): calcd for C16H17BrN2O6S2 (M + H)+ 496.9453, found 496.9479.
(6R,7R)-3-(Acetoxymethyl)-8-oxo-7-(2-(thiophen-3-yl)acetamido)-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic Acid (9)
3-Thiopheneacetic acid (104 mg, 0.74 mmol), oxalyl chloride (76 μL, 0.88 mmol), and DMF (1 drop) were reacted in DCM (3 mL) according to method C. The resulting acid chloride and 7-aminocephalosporanic acid (100 mg, 0.37 mmol) were reacted in EtOAc (5 mL) prior to the addition of aniline (100 μL) according to method B. The aqueous layer was extracted with DCM (×3), and the organic layers were combined, dried over Na2SO4, and evaporated. The resulting solid was triturated with ice-cold DCM to afford the product as an off-white amorphous solid (48 mg, 33%). IR (solid): υmax 3284, 1751, 1730, 1651, 1621, 1536, 1241 cm–1. 1H NMR (400 MHz, DMSO-d6) δ 8.95 (d, J = 8.3 Hz, 1H), 7.46 (dd, J = 4.9, 3.0 Hz, 1H), 7.26 (dd, J = 2.9, 1.0 Hz, 1H), 7.03 (dd, J = 4.9, 1.2 Hz, 1H), 5.46 (dd, J = 8.3, 4.8 Hz, 1H), 4.99 (d, J = 11.9 Hz, 1H), 4.93 (d, J = 4.8 Hz, 1H), 4.73 (d, J = 11.9 Hz, 1H), 3.61–3.49 (m, 2H), 3.45 (d, J = 17.2 Hz, 1H), 3.19 (d, J = 17.3 Hz, 1H), 2.00 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 170.6, 170.5, 163.3, 162.8, 135.8, 135.6, 128.6, 125.7, 122.3, 64.7, 58.5, 57.2, 36.4, 25.1, 20.8. HRMS (ESI+): calcd for C16H16N2O6S2 (M + H)+ 397.0528, found 397.0540.
(6R,7R)-3-(Acetoxymethyl)-8-oxo-7-((R)-2-phenylpropanamido)-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic Acid (10)
(R)-(−)-2-Phenylpropionic acid (101 μL, 0.74 mmol), oxalyl chloride (76 μL, 0.88 mmol), and DMF (1 drop) were reacted in DCM (3 mL) according to method C. The resulting acid chloride and 7-aminocephalosporanic acid (100 mg, 0.37 mmol) were reacted in EtOAc (5 mL) prior to the addition of aniline (100 μL) according to method B. The aqueous layer was extracted with DCM (×3), and the organic layers were combined, dried over Na2SO4 and evaporated. The resulting solid was triturated with ice-cold DCM to afford the product as a white amorphous solid (42 mg, 28%). IR (solid): υmax 3571, 3317, 1785, 1759, 1718, 1662, 1517, 1238 cm–1. 1H NMR (400 MHz, CDCl3) δ 8.94 (d, J = 8.4 Hz, 1H), 7.34–7.26 (m, 4H), 7.23–7.18 (m, 1H), 5.57 (dd, J = 8.4, 4.8 Hz, 1H), 4.99–4.91 (m, 2H), 4.68 (d, J = 12.3 Hz, 1H), 3.82 (q, J = 7.1 Hz, 1H), 3.44 (d, J = 17.6 Hz, 1H), 3.24 (d, J = 17.7 Hz, 1H), 1.99 (s, 3H), 1.34 (d, J = 7.0 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 174.1, 170.3, 163.7, 163.1, 141.6, 128.1, 127.2, 126.6, 63.7, 58.5, 57.4, 44.2, 25.2, 20.7, 17.8. HRMS (ESI+): calcd for C24H24N2O6S (M + H)+ 405.1120, found 405.1131.
(6R,7R)-3-(Acetoxymethyl)-8-oxo-7-(2-phenylacetamido)-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic Acid (11)
7-Aminocephalosporanic acid (100 mg, 0.35 mmol) and phenylacetyl chloride (97 μL, 0.73 mmol) were reacted in sat. NaHCO3(aq) (10 mL) and acetone (5 mL) according to method A to afford the product as a white amorphous solid (45 mg, 31%). IR (solid): υmax 3254, 3034, 1778, 1737, 1707, 1654, 1536, 1223 cm–1. 1H NMR (400 MHz, DMSO-d6) δ 13.71 (br s, 1H), 9.10 (d, J = 8.3 Hz, 1H), 7.35–7.18 (m, 5H), 5.68 (dd, J = 8.3, 4.8 Hz, 1H), 5.08 (d, J = 4.8 Hz, 1H), 5.00 (d, J = 12.8 Hz, 1H), 4.68 (d, J = 12.8 Hz, 1H), 3.66–3.47 (m, 4H), 2.03 (s, 3H). 13C NMR (101 MHz, DMSO) δ 170.9, 170.2, 164.7, 162.8, 135.8, 129.0, 128.2, 126.54, 126.48, 123.1, 62.7, 59.1, 57.4, 41.6, 25.5, 20.6. HRMS (ESI+): calcd for C18H19N2O6S (M + H)+ 391.0964, found 391.0972.
(6R,7R)-3-(Acetoxymethyl)-8-oxo-7-(2-(p-tolyl)acetamido)-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic Acid (12)
4-Methylphenylacetic acid (111 mg, 0.74 mmol), oxalyl chloride (76 μL, 0.88 mmol), and DMF (1 drop) were reacted in DCM (3 mL) according to method C. The resulting acid chloride and 7-aminocephalosporanic acid (100 mg, 0.37 mmol) were reacted in EtOAc (5 mL) prior to the addition of aniline (100 μL) according to method B. The resulting precipitate was collected by vacuum filtration and washed with DCM to afford the product as a white amorphous solid (63 mg, 42%). IR (solid): υmax 3261, 3045, 1778, 1752, 1707, 1655, 1536, 1223 cm–1. 1H NMR (400 MHz, DMSO-d6) δ 13.66 (br s, 1H), 9.04 (d, J = 8.3 Hz, 1H), 7.15 (d, J = 8.1 Hz, 2H), 7.10 (d, J = 8.0 Hz, 2H), 5.67 (dd, J = 8.3, 4.8 Hz, 1H), 5.08 (d, J = 4.8 Hz, 1H), 5.00 (d, J = 12.8 Hz, 1H), 4.69 (d, J = 12.8 Hz, 1H), 3.62 (d, J = 18.1 Hz, 1H), 3.54–3.41 (m, 3H), 2.26 (s, 3H), 2.03 (s, 3H). 13C NMR (101 MHz, DMSO) δ 171.1, 170.2, 164.8, 162.8, 135.5, 132.7, 128.9, 128.8, 62.7, 59.1, 57.4, 41.2, 25.5, 20.63, 20.57. HRMS (ESI+): calcd for C19H21N2O6S (M + H)+ 405.1120, found 405.1119.
(6R,7R)-3-(Acetoxymethyl)-7-(2-(4-fluorophenyl)acetamido)-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic Acid (13)
4-Fluorophenylacetyl chloride (98 μL, 0.74 mmol) and 7-aminocephalosporanic acid (100 mg, 0.37 mmol) were reacted in EtOAc (5 mL) prior to the addition of aniline (100 μL) according to method B. The resulting mixture was cooled to 4 °C and the precipitate collected by vacuum filtration and washed with ice-cold DCM to afford the product as a white amorphous solid (61 mg, 41%). IR (solid): υmax 3273, 1763, 1736, 1659, 1532, 1215 cm–1. 1H NMR (400 MHz, DMSO-d6) δ 13.67 (br s, 1H), 9.10 (d, J = 8.2 Hz, 1H), 7.30 (dd, J = 8.4, 5.6 Hz, 2H), 7.12 (app t, J = 8.8 Hz, 2H), 5.67 (dd, J = 8.1, 4.8 Hz, 1H), 5.08 (d, J = 4.8 Hz, 1H), 5.00 (d, J = 12.8 Hz, 1H), 4.69 (d, J = 12.8 Hz, 1H), 3.65–3.46 (m, 4H), 2.03 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 170.8, 170.1, 164.6, 162.8, 131.9, 130.9, 130.8, 126.4, 123.3, 115.0, 114.8, 62.7, 59.1, 57.4, 40.6, 25.5, 20.5. HRMS (ESI+): calcd for C18H18FN2O6S (M + H)+ 409.0870, found 409.0864.
(6R,7R)-3-(Acetoxymethyl)-7-(2-(4-chlorophenyl)acetamido)-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic Acid (14)
Hexanoyl chloride (139 mg, 0.74 mmol) and 7-aminocephalosporanic acid (100 mg, 0.37 mmol) were reacted in EtOAc (5 mL) prior to the addition of aniline (75 μL) according to method B. The resulting precipitate was collected by vacuum filtration and washed with ice-cold DCM to afford the product as a cream amorphous solid (104 mg, 67%). IR (solid): υmax 3265, 3056, 1778, 1748, 1707, 1643, 1536, 1223 cm–1. 1H NMR (400 MHz, DMSO-d6) δ 13.69 (br s, 1H), 9.13 (d, J = 8.2 Hz, 1H), 7.36 (d, J = 8.5 Hz, 2H), 7.29 (d, J = 8.5 Hz, 2H), 5.67 (dd, J = 8.2, 4.8 Hz, 1H), 5.08 (d, J = 4.8 Hz, 1H), 5.00 (d, J = 12.8 Hz, 1H), 4.68 (d, J = 12.8 Hz, 1H), 3.65–3.45 (m, 4H), 2.03 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 170.6, 170.2, 164.6, 162.8, 134.8, 131.3, 130.9, 128.2, 62.7, 59.1, 57.4, 40.8, 25.5, 20.6. HRMS (ESI+): calcd for C18H18ClN2O6S (M + H)+ 447.0394, found 447.0414.
(6R,7R)-3-(Acetoxymethyl)-7-(2-(4-bromophenyl)acetamido)-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic Acid (15)
3-Bromophenylacetyl chloride (171 mg, 0.74 mmol) and 7-aminocephalosporanic acid (100 mg, 0.37 mmol) were reacted in EtOAc (5 mL) prior to the addition of aniline (75 μL) according to method B. The resulting precipitate was collected by vacuum filtration and washed with ice-cold DCM to afford the product as a cream amorphous solid (131 mg, 76%). IR (solid): υmax 3265, 3060, 1782, 1748, 1707, 1651, 1543, 1223 cm–1. 1H NMR (400 MHz, DMSO-d6) δ 13.66 (br s, 1H), 9.14 (d, J = 8.2 Hz, 1H), 7.50 (d, J = 8.4 Hz, 2H), 7.23 (d, J = 8.4 Hz, 2H), 5.67 (dd, J = 8.2, 4.8 Hz, 1H), 5.08 (d, J = 4.8 Hz, 1H), 5.00 (d, J = 12.8 Hz, 1H), 4.68 (d, J = 12.8 Hz, 1H), 3.65–3.45 (m, 4H), 2.03 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 170.6, 170.2, 164.7, 162.8, 135.2, 131.3, 131.1, 126.3, 123.4, 119.7, 62.7, 59.1, 57.4, 40.8, 25.5, 20.6. HRMS (ESI+): calcd for C18H18BrN2O6S (M + H)+ 469.0069, found 469.0076.
(6R,7R)-7-(2-([1,1′-Biphenyl]-4-yl)acetamido)-3-(acetoxymethyl)-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic Acid (16)
4-Biphenylacetic acid (156 mg, 0.736 mmol), oxalyl chloride (76 μL, 0.88 mmol), and DMF (1 drop) were reacted in DCM (3 mL) according to method C. The resulting acid chloride and 7-aminocephalosporanic acid (100 mg, 0.37 mmol) were reacted in EtOAc (5 mL) prior to the addition of aniline (100 μL) according to method B. The resulting precipitate was collected by vacuum filtration and washed with ice-cold Et2O to afford the product as a white amorphous solid (112 mg, 65%). IR (solid): υmax 3302, 1756, 1737, 1654, 1621, 1536, 1237 cm–1. 1H NMR (400 MHz, DMSO-d6) δ 9.02 (d, J = 8.3 Hz, 1H), 7.65 (d, J = 7.2 Hz, 2H), 7.60 (d, J = 8.2 Hz, 2H), 7.45 (app t, J = 7.6 Hz, 2H), 7.41–7.30 (m, 3H), 5.48 (dd, J = 8.3, 4.8 Hz, 1H), 5.00 (d, J = 11.9 Hz, 1H), 4.94 (d, J = 4.8 Hz, 1H), 4.75 (d, J = 12.0 Hz, 1H), 3.62 (d, J = 13.9 Hz, 1H), 3.54 (d, J = 13.9 Hz, 1H), 3.46 (d, J = 17.2 Hz, 1H), 3.20 (d, J = 17.2 Hz, 1H), 2.00 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 170.9, 170.4, 163.4, 162.8, 139.9, 138.3, 135.7, 135.2, 129.6, 128.8, 127.2, 126.5, 126.5, 64.7, 58.5, 57.2, 41.2, 25.1, 20.7. HRMS (ESI+): calcd for C24H23N2O6S (M + H)+ 467.1277, found 467.1287.
(6R,7R)-3-(Acetoxymethyl)-8-oxo-7-(2-(4-phenoxyphenyl)acetamido)-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic Acid (17)
4-Phenoxyphenylacetic acid (170 mg, 0.74 mmol), oxalyl chloride (76 μL, 0.88 mmol), and DMF (1 drop) were reacted in DCM (3 mL) according to method C. The resulting acid chloride and 7-aminocephalosporanic acid (100 mg, 0.37 mmol) were reacted in EtOAc (5 mL) prior to the addition of aniline (75 μL) according to method B. The aqueous layer was extracted with DCM (×3), and the organic layers were combined, dried over Na2SO4, and evaporated. The resulting solid was precipitated from hot DCM to afford the product as a cream amorphous solid (24 mg, 14%). IR (solid): υmax 3280, 1774, 1730, 1655, 1532, 1223 cm–1. 1H NMR (400 MHz, DMSO-d6) δ 9.05 (d, J = 8.3 Hz, 1H), 7.37 (app t, J = 7.9 Hz, 2H), 7.29 (d, J = 8.5 Hz, 2H), 7.12 (t, J = 7.4 Hz, 1H), 7.04–6.89 (m, 5H), 5.57 (dd, J = 8.2, 4.8 Hz, 1H), 5.03–4.95 (m, 2H), 4.72 (d, J = 12.4 Hz, 1H), 3.59–3.46 (m, 3H), 3.33 (d, J = 17.7 Hz, 1H), 2.01 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 171.0, 170.3, 163.7, 163.2, 156.9, 155.2, 131.0, 130.6, 130.0, 123.2, 118.6, 118.3, 63.7, 58.8, 57.3, 40.7, 25.3, 20.6. HRMS (ESI+): calcd for C24H23N2O7S (M + H)+ 483.1226, found 483.1212.
(6R,7R)-3-(acetoxymethyl)-8-oxo-7-(2-(m-tolyl)acetamido)-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic Acid (18)
3-Methylphenylacetic acid (111 mg, 0.74 mmol), oxalyl chloride (76 μL, 0.88 mmol), and DMF (1 drop) were reacted in DCM (3 mL) according to method C. The resulting acid chloride and 7-aminocephalosporanic acid (100 mg, 0.37 mmol) were reacted in EtOAc (5 mL) prior to the addition of aniline (75 μL) according to method B. The aqueous layer was extracted with DCM (×3), and the organic layers were combined, dried over Na2SO4, and evaporated. The resulting solid was triturated with DCM to afford the product as a cream amorphous solid (36 mg, 24%). IR (solid): υmax 3288, 1726, 1662, 1625, 1526, 1223 cm–1. 1H NMR (400 MHz, DMSO-d6) δ 9.00 (d, J = 8.3 Hz, 1H), 7.17 (app t, J = 7.5 Hz, 1H), 7.12–6.98 (m, 3H), 5.51 (dd, J = 8.1, 4.7 Hz, 1H), 5.05–4.91 (m, 2H), 4.74 (d, J = 12.1 Hz, 1H), 3.56–3.41 (m, 3H), 3.26 (d, J = 17.5 Hz, 1H), 2.27 (s, 3H), 2.01 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 171.0, 170.5, 163.5, 163.3, 137.2, 135.8, 129.7, 128.1, 127.1, 126.1, 64.3, 58.6, 57.3, 41.5, 25.2, 21.0, 20.7. HRMS (ESI+): calcd for C19H21N2O6S (M + H)+ 405.1120, found 405.1126.
(6R,7R)-3-(Acetoxymethyl)-8-oxo-7-(2-(3-phenoxyphenyl)acetamido)-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic Acid (19)
3-Phenoxyphenylacetic acid (170 mg, 0.74 mmol), oxalyl chloride (76 μL, 0.88 mmol), and DMF (1 drop) were reacted in DCM (3 mL) according to method C. The resulting acid chloride and 7-aminocephalosporanic acid (100 mg, 0.37 mmol) were reacted in EtOAc (5 mL) prior to the addition of aniline (75 μL) according to method B. The aqueous layer was extracted with DCM (×3), and the organic layers were combined, dried over Na2SO4, and evaporated. The resulting solid was precipitated from hot DCM to afford the product as a cream amorphous solid (22 mg, 12%). IR (solid): υmax 3280, 3042, 1771, 1726, 1659, 1528, 1226 cm–1. 1H NMR (400 MHz, DMSO-d6) δ 9.04 (d, J = 8.2 Hz, 1H), 7.39 (app t, J = 7.9 Hz, 2H), 7.31 (t, J = 7.9 Hz, 1H), 7.13 (t, J = 7.4 Hz, 1H), 7.07–6.95 (m, 4H), 6.87 (dd, J = 8.1, 2.2 Hz, 1H), 5.55 (dd, J = 8.1, 4.8 Hz, 1H), 5.02–4.94 (m, 2H), 4.71 (d, J = 12.3 Hz, 1H), 3.59–3.45 (m, 3H), 3.30 (d, J = 17.7 Hz, 1H), 2.01 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 170.6, 170.3, 163.6, 163.1, 156.6, 156.5, 138.0, 130.0, 129.69, 124.2, 123.3, 119.2, 118.6, 116.7, 63.7, 58.8, 57.2, 41.4, 25.3, 20.6. HRMS (ESI+): calcd for C24H23N2O7S (M + H)+ 483.1226, found 483.1233.
(6R,7R)-3-(Acetoxymethyl)-7-benzamido-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic Acid (20)
7-Aminocephalosporanic acid (100 mg, 0.37 mmol) and benzoyl chloride (51 μL, 0.44 mmol) were reacted in sat. NaHCO3 (aq) (10 mL) and acetone (5 mL) according to method A to afford the product as a white amorphous solid (75 mg, 55%). IR (solid): υmax 3250, 1774, 1752, 1710, 1651, 1520, 1223 cm–1. 1H NMR (400 MHz, DMSO-d6) δ 13.69 (br s, 1H), 9.41 (d, J = 8.0 Hz, 1H), 7.91 (d, J = 7.7 Hz, 2H), 7.57 (t, J = 7.3 Hz, 1H), 7.48 (app t, J = 7.5 Hz, 2H), 5.88 (dd, J = 8.1, 4.8 Hz, 1H), 5.19 (d, J = 4.8 Hz, 1H), 4.99 (d, J = 12.8 Hz, 1H), 4.70 (d, J = 12.7 Hz, 1H), 3.64 (d, J = 18.0 Hz, 1H), 3.50 (d, J = 18.0 Hz, 1H), 2.03 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 170.2, 166.9, 164.0, 162.8, 133.0, 131.8, 128.3, 127.7, 123.1, 62.7, 59.8, 57.6, 25.5, 20.5. HRMS (ESI+): calcd for C17H17N2O6S (M + H)+ 377.0807, found 377.0807.
(6R,7R)-3-(Acetoxymethyl)-7-(4-methylbenzamido)-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic Acid (21)
7-Aminocephalosporanic acid (100 mg, 0.37 mmol) and 4-methylbenzoyl chloride (97 μL, 0.74 mmol) were reacted in sat. NaHCO3 (aq) (10 mL) and acetone (5 mL) according to method A to afford the product as a white amorphous solid (39 mg, 26%). IR (solid): υmax 3258, 1774, 1730, 1648, 1525, 1223 cm–1. 1H NMR (400 MHz, DMSO-d6) δ 9.29 (d, J = 8.1 Hz, 1H), 7.83 (d, J = 7.8 Hz, 2H), 7.29 (d, J = 7.9 Hz, 2H), 5.81 (dd, J = 8.1, 4.7 Hz, 1H), 5.14 (d, J = 4.8 Hz, 1H), 4.99 (d, J = 12.5 Hz, 1H), 4.72 (d, J = 12.5 Hz, 1H), 3.59 (d, J = 17.7 Hz, 1H), 3.43 (d, J = 17.7 Hz, 1H), 2.37 (s, 3H), 2.03 (s, 3H). 13C NMR (101 MHz, DMSO) δ 170.3, 166.8, 163.5, 163.0, 141.8, 130.3, 128.8, 127.8, 63.4, 59.5, 57.6, 25.4, 21.0, 20.6. HRMS (ESI+): calcd for C18H19N2O6S (M + H)+ 391.0964, found 391.0972.
(6R,7R)-3-(Acetoxymethyl)-7-(4-methoxybenzamido)-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic Acid (22)
7-Aminocephalosporanic acid (100 mg, 0.37 mmol) and 4-methoxybenzoyl chloride (100 μL, 0.74 mmol) were reacted in EtOAc (5 mL) prior to the addition of aniline (100 μL) according to method B. The aqueous layer was extracted with DCM (×3), and the organic layers were combined, dried over Na2SO4, and evaporated. The resulting solid was triturated with ice-cold Et2O to afford the product as a white amorphous solid (36 mg, 24%). IR (solid): υmax 3254, 1774, 1752, 1705, 1640, 1528, 1223 cm–1. 1H NMR (400 MHz, DMSO-d6) δ 9.22 (d, J = 8.1 Hz, 1H), 7.92 (d, J = 8.9 Hz, 2H), 7.01 (d, J = 8.9 Hz, 2H), 5.84 (dd, J = 8.1, 4.8 Hz, 1H), 5.16 (d, J = 4.8 Hz, 1H), 4.98 (d, J = 12.7 Hz, 1H), 4.70 (d, J = 12.7 Hz, 1H), 3.82 (s, 3H), 3.62 (d, J = 17.9 Hz, 1H), 3.46 (d, J = 17.9 Hz, 1H), 2.03 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 170.2, 166.3, 164.1, 162.9, 162.1, 129.7, 125.1, 113.6, 62.9, 59.7, 57.7, 55.4, 25.5, 20.6. HRMS (ESI+): calcd for C18H19N2O7S (M + H)+ 407.0913, found 407.0919.
(6R,7R)-3-(Acetoxymethyl)-7-(4-nitrobenzamido)-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic Acid (23)
7-Aminocephalosporanic acid (100 mg, 0.37 mmol) and 4-nitrobenzoyl chloride (136 mg, 0.74 mmol) were reacted in sat. NaHCO3 (aq) (10 mL) and acetone (5 mL) according to method A. Several drops of MeOH were added prior to the addition of ice-cold Et2O to afford the product as a white amorphous solid (54 mg, 35%). IR (solid): υmax 3265, 3064, 2971, 1785, 1748, 1711, 1640, 1595, 1524, 1223 cm–1. 1H NMR (400 MHz, DMSO-d6) δ 13.72 (s, 1H), 9.80 (d, J = 7.8 Hz, 1H), 8.34 (d, J = 8.8 Hz, 2H), 8.13 (d, J = 8.8 Hz, 2H), 5.88 (dd, J = 7.8, 4.7 Hz, 1H), 5.22 (d, J = 4.8 Hz, 1H), 5.00 (d, J = 12.8 Hz, 1H), 4.71 (d, J = 12.8 Hz, 1H), 3.66 (d, J = 17.9 Hz, 1H), 3.51 (d, J = 18.0 Hz, 1H), 2.03 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 170.7, 166.0, 164.0, 163.3, 149.9, 139.0, 129.8, 124.1, 63.2, 60.4, 58.0, 26.0, 21.0, 15.6. HRMS (ESI+): calcd for C17H16N3O8S (M + H)+ 422.0658, found 422.0660.
(6R,7R)-3-(Acetoxymethyl)-7-(furan-2-carboxamido)-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic Acid (24)
7-Aminocephalosporanic acid (100 mg, 0.37 mmol) and 2-furoyl chloride (73 μL, 0.74 mmol) were reacted in sat. NaHCO3 (aq) (10 mL) and acetone (5 mL) according to method A to afford the product as a white amorphous solid (30 mg, 22%). IR (solid): υmax 3243, 1793, 1718, 1710, 1632, 1595, 1223 cm–1. 1H NMR (400 MHz, DMSO-d6) δ 13.71 (br s, 1H), 9.29 (d, J = 8.2 Hz, 1H), 7.90 (d, J = 1.7 Hz, 1H), 7.36 (d, J = 3.5 Hz, 1H), 6.65 (dd, J = 3.5, 1.7 Hz, 1H), 5.81 (dd, J = 8.2, 4.8 Hz, 1H), 5.16 (d, J = 4.8 Hz, 1H), 4.99 (d, J = 12.8 Hz, 1H), 4.70 (d, J = 12.8 Hz, 1H), 3.64 (d, J = 18.0 Hz, 1H), 3.50 (d, J = 18.0 Hz, 1H), 2.03 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 170.7, 164.4, 163.3, 158.4, 146.8, 146.5, 115.4, 112.4, 63.2, 59.6, 58.1, 26.0, 21.0. HRMS (ESI+): calcd for C15H15N2O7S (M + H)+ 367.0600, found 367.0609.
(6R,7R)-3-(Acetoxymethyl)-7-(cyclohexanecarboxamido)-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic Acid (25)
7-Aminocephalosporanic acid (100 mg, 0.37 mmol) and cyclohexanecarbonyl chloride (60 μL, 0.44 mmol) were reacted in sat. NaHCO3(aq) (10 mL) and acetone (5 mL) according to method A to afford the product as a white amorphous solid (15 mg, 11%). IR (solid): υmax 3261, 2926, 2851, 1778, 1737, 1711, 1648, 1532, 1215 cm–1. 1H NMR (400 MHz, DMSO-d6) δ 13.67 (br s, 1H), 8.71 (d, J = 8.2 Hz, 1H), 5.63 (dd, J = 8.2, 4.8 Hz, 1H), 5.07 (d, J = 4.8 Hz, 1H), 4.99 (d, J = 12.8 Hz, 1H), 4.67 (d, J = 12.8 Hz, 1H), 3.61 (d, J = 18.1 Hz, 1H), 3.46 (d, J = 18.0 Hz, 1H), 2.34–2.24 (m, 1H), 2.03 (s, 3H), 1.76–1.57 (m, 5H), 1.39–1.14 (m, 5H). 13C NMR (101 MHz, DMSO) δ 176.1, 170.2, 164.8, 162.9, 62.8, 59.0, 57.6, 43.3, 29.8, 28.6, 25.4, 25.4, 25.2, 25.0, 20.6. HRMS (ESI+): calcd for C17H23N2O6S (M + H)+ 383.1277, found 383.1264.
(6R,7R)-7-Acetamido-3-(acetoxymethyl)-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic Acid (26)
7-Aminocephalosporanic acid (500 mg, 1.84 mmol) was suspended in H2O (8 mL), NaHCO3 (387 mg, 4.60 mmol) was added and the resulting mixture stirred at room temperature for 10 min before being cooled to 0 °C. Acetic anhydride (347 μL, 0.368 mmol) in acetone (10 mL) was added and the reaction stirred at 0 °C for 30 min. Acetone was removed under reduced pressure, and the resulting material was diluted in H2O and neutralized with sat. NaHCO3 (aq). The aqueous solution was washed with EtOAc, acidified to pH 2 with 1 M HCl and extracted with EtOAc (×3). The organic layers were combined, washed with brine, dried over Na2SO4, and evaporated to afford the product as a colorless foam (471 mg, 81%). IR (solid): υmax 3317, 2937, 1771, 1718, 1755, 1625, 1528, 1219 cm–1. 1H NMR (400 MHz, DMSO-d6) δ 13.67 (br s, 1H), 8.84 (d, J = 8.4 Hz, 1H), 5.68 (dd, J = 8.3, 4.9 Hz, 1H), 5.08 (d, J = 4.9 Hz, 1H), 5.00 (d, J = 12.8 Hz, 1H), 4.68 (d, J = 12.8 Hz, 1H), 3.63 (d, J = 18.0 Hz, 1H), 3.48 (d, J = 18.1 Hz, 1H), 2.03 (s, 3H), 1.91 (s, 3H). 13C NMR (101 MHz, DMSO) δ 170.2, 170.1, 165.0, 162.9, 126.4, 123.4, 62.7, 59.0, 57.4, 22.1, 20.6. HRMS (ESI+): calcd for C12H15N2O6S (M + Na)+ 337.0470, found 337.0479.
(6R,7R)-3-(Acetoxymethyl)-7-butyramido-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic Acid (27)
Butyryl chloride (76 μL, 0.74 mmol) and 7-aminocephalosporanic acid (100 mg, 0.37 mmol) were reacted in EtOAc (5 mL) prior to the addition of aniline (100 μL) according to method B. The aqueous layer was extracted with DCM (×3), and the organic layers were combined, dried over Na2SO4, and evaporated. The resulting solid was triturated with ice-cold DCM to afford the product as a white amorphous solid (15 mg, 12%). IR (solid): υmax 3265, 2960, 1774, 1751, 1715, 1654, 1539, 1223 cm–1. 1H NMR (400 MHz, DMSO-d6) δ 8.74 (d, J = 8.2 Hz, 1H), 5.61 (dd, J = 7.9, 4.8 Hz, 1H), 5.04 (d, J = 4.7 Hz, 1H), 4.99 (d, J = 12.6 Hz, 1H), 4.69 (d, J = 12.6 Hz, 1H), 3.57 (d, J = 17.8 Hz, 1H), 3.39 (d, J = 17.7 Hz, 1H), 2.21–2.13 (m, 2H), 2.02 (s, 3H), 1.52 (app h, J = 7.2 Hz, 2H), 0.86 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, DMSO) δ 172.9, 170.3, 164.4, 163.1, 63.3, 58.9, 57.4, 36.6, 25.4, 20.6, 18.7, 13.5. HRMS (ESI+): calcd for C14H19N2O6S (M + H)+ 343.0964, found 343.0959.
(6R,7R)-3-(Acetoxymethyl)-7-hexanamido-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic Acid (28)
Hexanoyl chloride (103 μL, 0.74 mmol) and 7-aminocephalosporanic acid (100 mg, 0.37 mmol) were reacted in EtOAc (5 mL) prior to the addition of aniline (100 μL) according to method B. The resulting precipitate was collected by vacuum filtration and washed with ice-cold DCM to afford the product as a white amorphous solid (78 mg, 57%). IR (solid): υmax 3283, 3183 2930, 1774, 1752, 1711, 1651, 1536, 1223 cm–1. 1H NMR (400 MHz, DMSO-d6) δ 13.65 (br s, 1H), 8.78 (d, J = 8.2 Hz, 1H), 5.67 (dd, J = 8.2, 4.8 Hz, 1H), 5.08 (d, J = 4.8 Hz, 1H), 4.99 (d, J = 12.8 Hz, 1H), 4.68 (d, J = 12.8 Hz, 1H), 3.62 (d, J = 18.1 Hz, 1H), 3.48 (d, J = 18.1 Hz, 1H), 2.23–2.13 (m, 2H), 2.03 (s, 3H), 1.56–1.46 (m, 2H), 1.32–1.18 (m, 4H), 0.86 (t, J = 7.0 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 173.0, 170.1, 164.9, 162.8, 126.4, 123.2, 62.7, 59.0, 57.5, 34.6, 30.7, 25.5, 24.8, 21.8, 20.5, 13.8. HRMS (ESI+): calcd for C16H23N2O6S (M + H)+ 371.1277, found 371.1290.
tert-Butyl (6R,7R)-7-Acetamido-3-(acetoxymethyl)-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylate (29)
Compound 26 (75 mg, 0.24 mmol) was dissolved in DCM (2 mL), tert-butyl 2,2,2-trichloroacetimidate (170 μL, 0.96 mmol) was added, and the reaction was heated to 60 °C for 24 h. After cooling to room temperature the reaction was diluted with MeOH. Solvent was removed under reduced pressure and the resulting solid triturated with cold DCM. The solute was loaded directly onto a 10 g SNAP KPSil column and purified by column chromatography (0–10% MeOH in DCM) to afford the product as a cream glassy solid (82 mg, 93%). IR (thin film): υmax 3291, 2982, 1774, 1718, 1670, 1528, 1368, 1223 cm–1. 1H NMR (400 MHz, CDCl3) δ 6.39 (d, J = 9.0 Hz, 1H), 5.84 (dd, J = 9.0, 4.9 Hz, 1H), 5.09 (d, J = 13.3 Hz, 1H), 4.95 (d, J = 4.9 Hz, 1H), 4.80 (d, J = 13.2 Hz, 1H), 3.55 (d, J = 18.4 Hz, 1H), 3.36 (d, J = 18.4 Hz, 1H), 2.08 (s, 3H), 2.06 (s, 3H), 1.53 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 170.7, 170.4, 164.9, 160.5, 127.5, 123.8, 84.0, 63.3, 59.4, 57.5, 27.9, 26.6, 23.0, 20.9. HRMS (ESI+): calcd for C16H23N2O6S (M + H)+ 371.1277, found 371.1276.
7-(4-(tert-Butoxycarbonyl)piperazin-1-yl)-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic Acid (32)
Ciprofloxacin 31 (500 mg, 1.51 mmol) was dissolved in 1 M NaOH (aq) (5 mL) and THF (10 mL) added, followed by the dropwise addition of Boc2O (360 mg, 1.66 mmol) in THF (10 mL) and stirred at room temperature for 16 h. Solvent was removed under reduced pressure and the resulting material diluted in H2O and neutralized with sat. NH4Cl (aq). The precipitate was collected by vacuum filtration and washed with H2O to afford the product as a white amorphous solid (502 mg, 77%). IR (solid): υmax 2971, 1733, 1688, 1629, 1249 cm–1. 1H NMR (400 MHz, CDCl3) δ 14.95 (s, 1H), 8.78 (s, 1H), 8.05 (d, J = 12.9 Hz, 1H), 7.37 (d, J = 7.1 Hz, 1H), 3.73–3.62 (m, 4H), 3.53 (tt, J = 7.3, 4.0 Hz, 1H), 3.34–3.25 (m, 4H), 1.50 (s, 9H), 1.43–1.37 (m, 2H), 1.24–1.17 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 167.1, 154.7, 153.1, 147.7, 113.0, 112.7, 108.5, 105.1, 80.5, 35.4, 28.6, 8.4. HRMS (ESI+): calcd for C22H27N3O5F (M+H)+ 432.1935, found 432.1951.
Sodium 7-(4-(tert-Butoxycarbonyl)piperazin-1-yl)-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylate (33)
Compound 32 (105 mg, 0.240 mmol) was suspended in MeOH (2.44 mL), 0.1 M NaOH (aq) (2.44 mL) was added, and the reaction mixture was stirred at 30 °C for 30 min. Solvent was removed under reduced pressure and resulting material suspended in H2O (5 μL) and EtOH (5 mL) and evaporated to dryness (×3). Then, the solid was suspended in DCM and evaporated to afford the product as a cream amorphous solid (111 mg, quant.). IR (solid): υmax 1617, 1478, 1242 cm–1.
tert-Butyl (6R,7R)-7-Acetamido-3-(((7-(4-(tert-butoxycarbonyl)piperazin-1-yl)-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carbonyl)oxy)methyl)-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylate (34)
Compound 29 (198 mg, 0.54 mmol) was dissolved in DCM (8 mL) and TMSI (117 μL, 0.82 mmol) added dropwise.83 The reaction mixture was stirred in the dark for 2 h at room temperature, then diluted with DCM and washed with 10% (wt/v) Na2SO3 (aq). The organic layer was dried over Na2SO4 and evaporated to a give compound 30 as a yellow glassy solid. Compound 30 (120 mg, 0.27 mmol) and compound 33 (100 mg, 0.26 mmol) were suspended in anhydrous 1,4-dioxane (3.5 mL), and DMF (1.15 mL) was added dropwise. The reaction mixture was stirred in the dark for 2 h before the solvent was removed under a stream of N2. The resulting material was dissolved in minimal DCM and loaded directly onto a 10 g SNAP Ultra cartridge and purified by column chromatography (0–6% MeOH in DCM) to afford the product as a pale yellow glassy solid (108 mg, 52%). IR (solid): υmax 2974, 1782, 1685, 1618, 1250 cm–1. 1H NMR (400 MHz, DMSO-d6) δ 8.86 (d, J = 8.5 Hz, 1H), 8.44 (s, 1H), 7.80 (d, J = 13.3 Hz, 1H), 7.47 (d, J = 7.4 Hz, 1H), 5.71 (dd, J = 8.4, 4.9 Hz, 1H), 5.15–5.09 (m, 2H), 4.86 (d, J = 13.1 Hz, 1H), 3.72–3.62 (m, 3H), 3.58–3.50 (m, 4H), 3.25–3.16 (m, 4H), 1.91 (s, 3H), 1.49 (s, 9H), 1.43 (s, 9H), 1.31–1.25 (m, 2H), 1.13–1.05 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 171.5, 170.0, 164.9, 164.2, 160.5, 153.7, 152.6 (d, 1JC–F = 247.5 Hz), 148.4, 143.8 (d, 2JC–F = 10.1 Hz), 138.0, 126.2, 123.5, 122.1 (d, 3JC–F = 7.1 Hz), 111.6 (d, 2JC–F = 22.2 Hz), 108.6, 106.7 (d, 3JC–F = 3.0 Hz), 82.8, 79.2, 62.6, 59.0, 57.4, 54.9, 49.50, 49.46, 34.9, 28.1, 27.5, 25.6, 22.1, 7.6, 7.5. HRMS (ESI+): calcd for C36H44FN5O9S (M + H)+ 742.2922, found 742.2930.
(6R,7R)-7-Acetamido-3-(((1-cyclopropyl-6-fluoro-4-oxo-7-(piperazin-1-yl)-1,4-dihydroquinoline-3-carbonyl)oxy)methyl)-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic Acid (35)
Compound 34 (15 mg, 0.02 mmol) was dissolved in anhydrous DCM (0.3 mL) and cooled to 0 °C. Anhydrous anisole (3 drops) was added followed by the dropwise addition of TFA (0.3 mL). The reaction mixture was stirred at 0 °C for 30 min, warmed to room temperature, and stirred for a further 40 min. Solvent was removed under a stream of N2 and the resulting gum triturated with ice-cold EtOAc. The precipitate was collected, diluted in H2O and DCM, and basified to pH 9 with 3% NaHCO3 (aq). The aqueous phase was separated and loaded directly onto a 12 g SNAP KP-C18-HS cartridge and purified by reverse-phase column chromatography (0–100% MeCN in H2O). Fractions containing product were freeze-dried to afford the product as a white solid (2.5 mg, 23%). 1H NMR (500 MHz, D2O) δ 8.58 (s, 1H), 7.73 (d, J = 13.0 Hz, 1H), 7.46 (d, J = 7.2 Hz, 1H), 5.58 (d, J = 4.6 Hz, 1H), 5.11 (d, J = 12.6 Hz, 1H), 5.07 (d, J = 4.7 Hz, 1H), 4.81 (d, J = 12.6 Hz, 1H), 3.68–3.31 (m, 11H), 2.00 (s, 3H), 1.27 (d, J = 6.9 Hz, 2H), 1.06 (d, J = 4.3 Hz, 2H). HRMS (ESI+): calcd for C27H29FN5O7S (M + H)+ 586.1772, found 586.1794.
Acknowledgments
We thank the Imperial Confidence in Concept scheme and the Wellcome Trust (Pathfinder Award 204337/Z/16/Z) for funding this work. A.M.E. also acknowledges support from the National Institute for Health Research (NIHR) Imperial Biomedical Research Centre (BRC). J.S. acknowledges funding the Engineering and Physical Sciences Research Council (Grant EP/M027546/1) and the Biotechnology and Biological Sciences Research Council-funded South West Biosciences Doctoral Training Partnership (Training Grant Reference BB/J014400/1, studentship to C.L.T.). T.B.C. is a Sir Henry Dale Fellow jointly funded by the Wellcome Trust and Royal Society (Grant 107660/Z/15Z). We thank Peter Haycock from the Imperial College Department of Chemistry NMR Facility and Lisa Haigh from the Imperial College Department of Chemistry Mass Spec Facility for their support with NMR and mass spectrometry data collection, respectively. We thank Prof. Matthew B. Avison, University of Bristol, for providing the pSU18 vectors used in this study. We also thank Ali Abdolrasouli, Charing Cross Hospital, for providing the clinical isolates used in this study.
Glossary
Abbreviations Used
- E. coli
Escherichia coli
- FDA
Food and Drug Administration
- GI
gastrointestinal
- GU
genitourinary
- kcat
catalytic constant for the conversion of substrate to product
- P. aeruginosa
Pseudomonas aeruginosa
- TMSI
trimethylsilyl iodide.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.8b01923.
Author Present Address
⊥ D.C.M: Critical Care Research Group, Nuffield Department of Clinical Neurosciences, University of Oxford, OX3 9DU Oxford, United Kingdom.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Notes
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health
Supplementary Material
References
- Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations, Review on Antimicrobial Resistance; O’Neill J., Chair; HM Government, Wellcome Trust, 2014. [Google Scholar]
- Rossolini G. M.; Arena F.; Pecile P.; Pollini S. Update on the Antibiotic Resistance Crisis. Curr. Opin. Pharmacol. 2014, 18, 56–60. 10.1016/j.coph.2014.09.006. [DOI] [PubMed] [Google Scholar]
- Kong K.-F.; Schneper L.; Mathee K. Beta-Lactam Antibiotics: From Antibiosis to Resistance and Bacteriology. APMIS. 2010, 118 (1), 1–36. 10.1111/j.1600-0463.2009.02563.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes R.; Amador P.; Prudêncio C. β-Lactams. Rev. Med. Microbiol. 2013, 24 (1), 7–17. 10.1097/MRM.0b013e3283587727. [DOI] [Google Scholar]
- Bush K. Proliferation and Significance of Clinically Relevant β-Lactamases. Ann. N. Y. Acad. Sci. 2013, 1277 (1), 84–90. 10.1111/nyas.12023. [DOI] [PubMed] [Google Scholar]
- Cantón R.; González-Alba J. M.; Galán J. C. CTX-M Enzymes: Origin and Diffusion. Front. Microbiol. 2012, 3, 110. 10.3389/fmicb.2012.00110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaikh S.; Fatima J.; Shakil S.; Rizvi S. M. D.; Kamal M. A. Antibiotic Resistance and Extended Spectrum Beta-Lactamases: Types, Epidemiology and Treatment. Saudi J. Biol. Sci. 2015, 22 (1), 90–101. 10.1016/j.sjbs.2014.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores-Mireles A. L.; Walker J. N.; Caparon M.; Hultgren S. J. Urinary Tract Infections: Epidemiology, Mechanisms of Infection and Treatment Options. Nat. Rev. Microbiol. 2015, 13 (5), 269–284. 10.1038/nrmicro3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peach B. C.; Garvan G. J.; Garvan C. S.; Cimiotti J. P. Risk Factors for Urosepsis in Older Adults: A Systematic Review. Gerontol. Geriatr. Med. 2016, 2, 2333721416638980. 10.1177/2333721416638980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tandogdu Z.; Wagenlehner F. M. E. Global Epidemiology of Urinary Tract Infections. Curr. Opin. Infect. Dis. 2016, 29 (1), 73–79. 10.1097/QCO.0000000000000228. [DOI] [PubMed] [Google Scholar]
- Kabbani S.; Hersh A. L.; Shapiro D. J.; Fleming-Dutra K. E.; Pavia A. T.; Hicks L. A. Opportunities to Improve Fluoroquinolone Prescribing in the United States for Adult Ambulatory Care Visits. Clin. Infect. Dis. 2018, 67 (1), 134–136. 10.1093/cid/ciy035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheld W. M. Maintaining Fluoroquinolone Class Efficacy: Review of Influencing Factors. Emerging Infect. Dis. 2003, 9 (1), 1–9. 10.3201/eid0901.020277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becattini S.; Taur Y.; Pamer E. G. Antibiotic-Induced Changes in the Intestinal Microbiota and Disease. Trends Mol. Med. 2016, 22 (6), 458–478. 10.1016/j.molmed.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewardson A. J.; Gaïa N.; Francois P.; Malhotra-Kumar S.; Delémont C.; Martinez de Tejada B.; Schrenzel J.; Harbarth S.; Lazarevic V. Collateral Damage from Oral Ciprofloxacin versus Nitrofurantoin in Outpatients with Urinary Tract Infections: A Culture-Free Analysis of Gut Microbiota. Clin. Microbiol. Infect. 2015, 21 (4), 344.e1–344.e11. 10.1016/j.cmi.2014.11.016. [DOI] [PubMed] [Google Scholar]
- Dethlefsen L.; Relman D. A. Incomplete Recovery and Individualized Responses of the Human Distal Gut Microbiota to Repeated Antibiotic Perturbation. Proc. Natl. Acad. Sci. U. S. A. 2011, 108 (Suppl. 1), 4554–4561. 10.1073/pnas.1000087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan Å.; Edlund C.; Nord C. E. Effect of Antimicrobial Agents on the Ecological Balance of Human Microflora. Lancet Infect. Dis. 2001, 1 (2), 101–114. 10.1016/S1473-3099(01)00066-4. [DOI] [PubMed] [Google Scholar]
- Jernberg C.; Lofmark S.; Edlund C.; Jansson J. K. Long-Term Impacts of Antibiotic Exposure on the Human Intestinal Microbiota. Microbiology 2010, 156 (11), 3216–3223. 10.1099/mic.0.040618-0. [DOI] [PubMed] [Google Scholar]
- Brown K. A.; Khanafer N.; Daneman N.; Fisman D. N. Meta-Analysis of Antibiotics and the Risk of Community-Associated Clostridium Difficile Infection. Antimicrob. Agents Chemother. 2013, 57 (5), 2326–2332. 10.1128/AAC.02176-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chellat M. F.; Raguž L.; Riedl R. Targeting Antibiotic Resistance. Angew. Chem., Int. Ed. 2016, 55 (23), 6600–6626. 10.1002/anie.201506818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis K. Platforms for Antibiotic Discovery. Nat. Rev. Drug Discovery 2013, 12 (5), 371–387. 10.1038/nrd3975. [DOI] [PubMed] [Google Scholar]
- FDA. Highlights of prescribing information (ciprofloxacin hydrochloride). www.fda.gov/medwatch (accessed Oct 25, 2018).
- Press Announcements. FDA updates warnings for fluoroquinolone antibiotics. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm513183.htm (accessed Oct 24, 2018).
- Marchant J. When Antibiotics Turn Toxic. Nature 2018, 555 (7697), 431–433. 10.1038/d41586-018-03267-5. [DOI] [PubMed] [Google Scholar]
- Dingle K. E.; Didelot X.; Quan T. P.; Eyre D. W.; Stoesser N.; Golubchik T.; Harding R. M.; Wilson D. J.; Griffiths D.; Vaughan A.; Finney J. M.; Wyllie D. H.; Oakley S. J.; Fawley W. N.; Freeman J.; Morris K.; Martin J.; Howard P.; Gorbach S.; Goldstein E. J. C.; Citron D. M.; Hopkins S.; Hope R.; Johnson A. P.; Wilcox M. H.; Peto T. E. A.; Walker A. S.; Crook D. W.; Del Ojo Elias C.; Crichton C.; Kostiou V.; Giess A.; Davies J. Effects of Control Interventions on Clostridium Difficile Infection in England: An Observational Study. Lancet Infect. Dis. 2017, 17 (4), 411–421. 10.1016/S1473-3099(16)30514-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewardson A. J.; Vervoort J.; Adriaenssens N.; Coenen S.; Godycki-Cwirko M.; Kowalczyk A.; Huttner B. D.; Lammens C.; Malhotra-Kumar S.; Goossens H.; Harbarth S.; Vervoort J.; Lammens C.; Malhotra-Kumar S.; Goossens H.; Adriaenssens N.; Coenen S.; Kowalczyk A.; Godycki-Cwirko M.; Stewardson A. J.; Huttner B.; Harbarth S.; Brossier C.; Delemont C.; de Tejada B. M.; Renzi G.; Schrenzel J.; Van Bylen S.; Vanbergen J.; Koeck P.; Leysen P.; Vandercam K.; Kluijtmans J.; Borkiewicz A.; Heyvaert F.; Michels N.; Deswaef G.; Beckx T.; Declerck H.; Embrechts K.; Verheyen N.; Bauwens T.; Beghin J.; Verpooten L.; Bombeke K.; Vandenabeele T.; Muras M.; Swistak J.; Wesolowska A.; Sterniczuk E.; Brzozowska L.; Cichowska K.; Szewczyk J.; Krupinska G.; Blaszczyk H.; Stawinska U.; Szyler M.; Kasielski M.; Rydz R.; Myszkowska A.; Zebrowska L. Effect of Outpatient Antibiotics for Urinary Tract Infections on Antimicrobial Resistance among Commensal Enterobacteriaceae: A Multinational Prospective Cohort Study. Clin. Microbiol. Infect. 2018, 24 (9), 972–979. 10.1016/j.cmi.2017.12.026. [DOI] [PubMed] [Google Scholar]
- Klein E. Y.; Van Boeckel T. P.; Martinez E. M.; Pant S.; Gandra S.; Levin S. A.; Goossens H.; Laxminarayan R. Global Increase and Geographic Convergence in Antibiotic Consumption between 2000 and 2015. Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (15), E3463–E3470. 10.1073/pnas.1717295115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapid Diagnostics: Stopping Unnecessary Use of Antibiotics, Review on Antimicrobial Resistance; O’Neill J., Chair; HM Government, Wellcome Trust, 2015. [Google Scholar]
- Mota R.; Pinto M.; Palmeira J.; Gonçalves D.; Ferreira H. Intestinal Microbiota as a Reservoir of Extended-Spectrum β-Lactamase-Producing Escherichia Coli: An Exploratory Study in Healthy University Students. J. Glob. Antimicrob. Resist. 2018, 14, 10–11. 10.1016/j.jgar.2018.05.023. [DOI] [PubMed] [Google Scholar]
- de Lastours V.; Goulenok T.; Guérin F.; Jacquier H.; Eyma C.; Chau F.; Cattoir V.; Fantin B. Ceftriaxone Promotes the Emergence of AmpC-Overproducing Enterobacteriaceae in Gut Microbiota from Hospitalized Patients. Eur. J. Clin. Microbiol. Infect. Dis. 2018, 37 (3), 417–421. 10.1007/s10096-018-3186-x. [DOI] [PubMed] [Google Scholar]
- Ríos E.; López M. C.; Rodríguez-Avial I.; Culebras E.; Picazo J. J. Detection of Escherichia Coli ST131 Clonal Complex (ST705) and Klebsiella Pneumoniae ST15 among Faecal Carriage of Extended-Spectrum β-Lactamase- and Carbapenemase-Producing Enterobacteriaceae. J. Med. Microbiol. 2017, 66 (2), 169–174. 10.1099/jmm.0.000399. [DOI] [PubMed] [Google Scholar]
- Shepherd T. A.; Jungheim L. N.; Meyer D. L.; Starling J. J. A Novel Targeted Delivery System Utilizing a Cephalosporin-Oncolytic Prodrug Activated by an Antibody β-Lactamase Conjugate for the Treatment of Cancer. Bioorg. Med. Chem. Lett. 1991, 1 (1), 21–26. 10.1016/S0960-894X(01)81083-0. [DOI] [Google Scholar]
- Jungheim L. N.; Shepherd T. A.; Meyer D. L. Synthesis of Acylhydrazido-Substituted Cephems. Design of Cephalosporin-Vinca Alkaloid Prodrugs: Substrates for an Antibody Targeted Enzyme. J. Org. Chem. 1992, 57 (8), 2334–2340. 10.1021/jo00034a027. [DOI] [Google Scholar]
- Rodrigues M. L.; Carter P.; Wirth C.; Mullins S.; Lee A.; Blackburn B. K. Synthesis and β-Lactamase-Mediated Activation of a Cephalosporin-Taxol Prodrug. Chem. Biol. 1995, 2 (4), 223–227. 10.1016/1074-5521(95)90272-4. [DOI] [PubMed] [Google Scholar]
- Phelan R. M.; Ostermeier M.; Townsend C. A. Design and Synthesis of a β-Lactamase Activated 5-Fluorouracil Prodrug. Bioorg. Med. Chem. Lett. 2009, 19 (4), 1261–1263. 10.1016/j.bmcl.2008.12.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudyma T. W.; Bush K.; Colson K. L.; Firestone R. A.; King H. D. Synthesis and Release of Doxorubicin from a Cephalosporin Based Prodrug by a β-Lactamase-Immunoconjugate. Bioorg. Med. Chem. Lett. 1993, 3 (2), 323–328. 10.1016/S0960-894X(01)80902-1. [DOI] [Google Scholar]
- Demuth T. P.; White R. E.; Tietjen R. A.; Storrin R. J.; Skuster J. R.; Andersen J. A.; McOsker C. C.; Freedman R.; Rourke F. J. Synthesis and Antibacterial Activity of New C-10 Quinolonyl-Cephem Esters. J. Antibiot. 1991, 44 (2), 200–209. 10.7164/antibiotics.44.200. [DOI] [PubMed] [Google Scholar]
- Bryskier A. Dual β-Lactam-Fluoroquinolone Compounds: A Novel Approach to Antibacterial Treatment. Expert Opin. Invest. Drugs 1997, 6 (10), 1479–1499. 10.1517/13543784.6.10.1479. [DOI] [PubMed] [Google Scholar]
- O’Callaghan C. H.; Sykes R. B.; Staniforth S. E. A New Cephalosporin with a Dual Mode of Action. Antimicrob. Agents Chemother. 1976, 10 (2), 245–248. 10.1128/AAC.10.2.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgopapadakou N. H.; Bertasso A.; Chan K. K.; Chapman J. S.; Cleeland R.; Cummings L. M.; Dix B. A.; Keith D. D. Mode of Action of the Dual-Action Cephalosporin Ro 23-9424. Antimicrob. Agents Chemother. 1989, 33 (7), 1067–1071. 10.1128/AAC.33.7.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Callaghan C. H.; Kirby S. M.; Morris A.; Waller R. E.; Duncombe R. E. Correlation between Hydrolysis of the β-Lactam Bond of the Cephalosporin Nucleus and Expulsion of the 3-Substituent. J. Bacteriol. 1972, 110 (3), 988–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth T. P.; O’Donnell M. E.; O’Connor M. J.; St Ledger J. O. β-Lactamase-Dependent Prodrugs—Recent Developments. Tetrahedron 2000, 56 (31), 5699–5707. 10.1016/S0040-4020(00)00419-1. [DOI] [Google Scholar]
- Albrecht H. A.; Beskid G.; Chan K. K.; Christenson J. G.; Cleeland R.; Deitcher K. H.; Georgopapadakou N. H.; Keith D. D.; Pruess D. L. Cephalosporin 3′-Quinolone Esters with a Dual Mode of Action. J. Med. Chem. 1990, 33 (1), 77–86. 10.1021/jm00163a013. [DOI] [PubMed] [Google Scholar]
- Correia S.; Poeta P.; Hébraud M.; Capelo J. L.; Igrejas G. Mechanisms of Quinolone Action and Resistance: Where Do We Stand?. J. Med. Microbiol. 2017, 66 (5), 551–559. 10.1099/jmm.0.000475. [DOI] [PubMed] [Google Scholar]
- Dunn G. L. Ceftizoxime and Other Third-Generation Cephalosporins: Structure-Activity Relationships. J. Antimicrob. Chemother. 1982, 10 (Suppl. C), 1–10. 10.1093/jac/10.suppl_C.1. [DOI] [PubMed] [Google Scholar]
- Kaushik D.; Rathi S.; Jain A. Ceftaroline: A Comprehensive Update. Int. J. Antimicrob. Agents 2011, 37 (5), 389–395. 10.1016/j.ijantimicag.2011.01.017. [DOI] [PubMed] [Google Scholar]
- Fung-Tomc J. C. Fourth-Generation Cephalosporins. Clin. Microbiol. Newsl. 1997, 19 (17), 129–136. 10.1016/S0196-4399(97)82485-3. [DOI] [Google Scholar]
- Sader H. S.; Jones R. N. Historical Overview of the Cephalosporin Spectrum: Four Generations of Structural Evolution. Antimicrob. Newsl. 1992, 8 (12), 75–82. 10.1016/0738-1751(92)90022-3. [DOI] [Google Scholar]
- García-Rodríguez J. A.; Muñoz Bellido J. L.; García Sánchez J. E. Oral Cephalosporins: Current Perspectives. Int. J. Antimicrob. Agents 1995, 5 (4), 231–243. 10.1016/0924-8579(95)00015-Z. [DOI] [PubMed] [Google Scholar]
- Turck M. Cephalosporins and Related Antibiotics: An Overview. Clin. Infect. Dis. 1982, 4 (Suppl. 2), S281–S287. 10.1093/clinids/4.Supplement_2.S281. [DOI] [PubMed] [Google Scholar]
- Snyder N. J.; Tabas L. B.; Berry D. M.; Duckworth D. C.; Spry D. O.; Dantzig A. H. Structure-Activity Relationship of Carbacephalosporins and Cephalosporins: Antibacterial Activity and Interaction with the Intestinal Proton-Dependent Dipeptide Transport Carrier of Caco-2 Cells. Antimicrob. Agents Chemother. 1997, 41 (8), 1649–1657. 10.1128/AAC.41.8.1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonawane V. C. Enzymatic Modifications of Cephalosporins by Cephalosporin Acylase and Other Enzymes. Crit. Rev. Biotechnol. 2006, 26 (2), 95–120. 10.1080/07388550600718630. [DOI] [PubMed] [Google Scholar]
- Oberdorf C.; Schepmann D.; Vela J. M.; Diaz J. L.; Holenz J.; Wunsch B. Thiophene Bioisosteres of Spirocyclic σ Receptor Ligands. 1. N-Substituted Spiro[piperidine-4,4’-thieno[3,2-c ]pyrans]. J. Med. Chem. 2008, 51 (20), 6531–6537. 10.1021/jm8007739. [DOI] [PubMed] [Google Scholar]
- Brown N.Bioisosteres in Medicinal Chemistry; Wiley-VCH: Weinheim, Germany, 2012. [Google Scholar]
- Essack S. Y. The Development of β-Lactam Antibiotics in Response to the Evolution of β-Lactamases. Pharm. Res. 2001, 18 (10), 1391–1399. 10.1023/A:1012272403776. [DOI] [PubMed] [Google Scholar]
- Buckwell S. C.; Page M. I.; Waley S. G.; Longridge J. L. Hydrolysis of 7-Substituted Cephalosporins Catalysed by β-Lactamases I and II from Bacillus Cereus and by Hydroxide Ion. J. Chem. Soc., Perkin Trans. 2 1988, 0 (10), 1815–1821. 10.1039/P29880001815. [DOI] [Google Scholar]
- Rao W.-H.; Zhan B.-B.; Chen K.; Ling P.-X.; Zhang Z.-Z.; Shi B.-F. Pd(II)-Catalyzed Direct Sulfonylation of Unactivated C(Sp3)-H Bonds with Sodium Sulfinates. Org. Lett. 2015, 17 (14), 3552–3555. 10.1021/acs.orglett.5b01634. [DOI] [PubMed] [Google Scholar]
- Pai H.; Lyu S.; Lee J. H.; Kim J.; Kwon Y.; Kim J. W.; Choe K. W.; Jeong B. C.; Lee S. H. Survey of Extended-Spectrum Beta-Lactamases in Clinical Isolates of Escherichia coli and Klebsiella pneumoniae: Prevalence of TEM-52 in Korea. J. Clin. Microbiol. 1999, 37 (6), 1758–1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush K.; Sykes R. B. Methodology for the Study of Beta-Lactamases. Antimicrob. Agents Chemother. 1986, 30 (1), 6–10. 10.1128/AAC.30.1.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders C. C. Chromosomal Cephalosporinases Responsible for Multiple Resistance to Newer β-Lactam Antibiotics. Annu. Rev. Microbiol. 1987, 41 (1), 573–594. 10.1146/annurev.mi.41.100187.003041. [DOI] [PubMed] [Google Scholar]
- Ma J.; Cao Q.; McLeod S. M.; Ferguson K.; Gao N.; Breeze A. L.; Hu J. Target-Based Whole-Cell Screening by 1 H-NMR Spectroscopy. Angew. Chem., Int. Ed. 2015, 54 (16), 4764–4767. 10.1002/anie.201410701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J.; McLeod S.; MacCormack K.; Sriram S.; Gao N.; Breeze A. L.; Hu J. Real-Time Monitoring of New Delhi Metallo-β-Lactamase Activity in Living Bacterial Cells by 1 H-NMR Spectroscopy. Angew. Chem., Int. Ed. 2014, 53 (8), 2130–2133. 10.1002/anie.201308636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch R. A.; Burland V.; Plunkett G.; Redford P.; Roesch P.; Rasko D.; Buckles E. L.; Liou S.-R.; Boutin A.; Hackett J.; Stroud D.; Mayhew G. F.; Rose D. J.; Zhou S.; Schwartz D. C.; Perna N. T.; Mobley H. L. T.; Donnenberg M. S.; Blattner F. R. Extensive Mosaic Structure Revealed by the Complete Genome Sequence of Uropathogenic Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 2002, 99 (26), 17020–17024. 10.1073/pnas.252529799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mobley H. L.; Green D. M.; Trifillis A. L.; Johnson D. E.; Chippendale G. R.; Lockatell C. V.; Jones B. D.; Warren J. W. Pyelonephritogenic Escherichia coli and Killing of Cultured Human Renal Proximal Tubular Epithelial Cells: Role of Hemolysin in Some Strains. Infect. Immun. 1990, 58 (5), 1281–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghai I.; Ghai S. Understanding Antibiotic Resistance via Outer Membrane Permeability. Infect. Drug Resist. 2018, 11, 523–530. 10.2147/IDR.S156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson L. A. Reactive Metabolites in the Biotransformation of Molecules Containing a Furan Ring. Chem. Res. Toxicol. 2013, 26 (1), 6–25. 10.1021/tx3003824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Yuan H.; Wright S. C.; Wang H.; Larrick J. W. Synthesis and Preliminary Cytotoxicity Study of a Cephalosporin-CC-1065 Analogue Prodrug. BMC Chem. Biol. 2001, 1 (1), 4. 10.1186/1472-6769-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H. W.; Kang T. W.; Kim E. N.; Cha K. H.; Shin J.; Cho D. O.; Choi N. H.; Kim J. W.; Hong C. Preparation Of Ceph-3-Em Esters Unaccompanied By δ3 To δ2 Isomerization Of The Cephalosporin Derivatives. Synth. Commun. 1999, 29 (11), 1873–1887. 10.1080/00397919908086176. [DOI] [Google Scholar]
- Kaiser G. V.; Cooper R. D. G.; Koehler R. E.; Murphy C. F.; Webber J. A.; Wright I. G.; Van Heyningen E. M. Chemistry of Cephalosporin Antibiotics. XIX. Transformation of Δ2-Cephem to Δ3-Cephem by Oxidation-Reduction at Sulfur. J. Org. Chem. 1970, 35 (7), 2430–2433. 10.1021/jo00832a081. [DOI] [PubMed] [Google Scholar]
- Popa E.; Huang M.-J.; Brewstera M. E.; Bodora N. On the Mechanism of Cephalosporin Isomerization. J. Mol. Struct.: THEOCHEM 1994, 315, 1–7. 10.1016/0166-1280(94)03792-J. [DOI] [Google Scholar]
- Tanaka K. S. E.; Houghton T. J.; Kang T.; Dietrich E.; Delorme D.; Ferreira S. S.; Caron L.; Viens F.; Arhin F. F.; Sarmiento I.; Lehoux D.; Fadhil I.; Laquerre K.; Liu J.; Ostiguy V.; Poirier H.; Moeck G.; Parr T. R.; Rafai Far A. Bisphosphonated Fluoroquinolone Esters as Osteotropic Prodrugs for the Prevention of Osteomyelitis. Bioorg. Med. Chem. 2008, 16 (20), 9217–9229. 10.1016/j.bmc.2008.09.010. [DOI] [PubMed] [Google Scholar]
- Chan M. F.; Castillo R. S.; Li Q.; Doppalapudi V. R.; Hixton M. S.; Lobl T. J.. Improved Beta-Lactam Antibiotics. WO 01/83492 A1, 2001.
- Aldred K. J.; Kerns R. J.; Osheroff N. Mechanism of Quinolone Action and Resistance. Biochemistry 2014, 53, 1565–1574. 10.1021/bi5000564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A. U.; Maryam L.; Zarrilli R. Structure, Genetics and Worldwide Spread of New Delhi Metallo-β-Lactamase (NDM): A Threat to Public Health. BMC Microbiol. 2017, 17 (1), 101. 10.1186/s12866-017-1012-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yigit H.; Queenan A. M.; Anderson G. J.; Domenech-Sanchez A.; Biddle J. W.; Steward C. D.; Alberti S.; Bush K.; Tenover F. C. Novel Carbapenem-Hydrolyzing Beta-Lactamase, KPC-1, from a Carbapenem-Resistant Strain of Klebsiella Pneumoniae. Antimicrob. Agents Chemother. 2001, 45 (4), 1151–1161. 10.1128/AAC.45.4.1151-1161.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Queenan A. M.; Bush K. Carbapenemases: The Versatile Beta-Lactamases. Clin. Microbiol. Rev. 2007, 20 (3), 440–458. 10.1128/CMR.00001-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remmington T.; Jahnke N.; Harkensee C. Oral Anti-Pseudomonal Antibiotics for Cystic Fibrosis. Cochrane Database Syst. Rev. 2016, (10), CD005405. 10.1002/14651858.CD005405.pub4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langton Hewer S. C.; Smyth A. R. Antibiotic Strategies for Eradicating Pseudomonas aeruginosa in People with Cystic Fibrosis. Cochrane Database Syst. Rev. 2017, (11), CD004197. 10.1002/14651858.CD004197.pub5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkin S. D.; Abid S.; Foster M.; Bose M.; Keller A.; Hollaway R.; Sader H. S.; Greenberg D. E.; Finklea J. D.; Castanheira M.; Jain R. Multidrug-Resistant Pseudomonas aeruginosa from Sputum of Patients with Cystic Fibrosis Demonstrates a High Rate of Susceptibility to Ceftazidime-Avibactam. Infect. Drug Resist. 2018, 11, 1499–1510. 10.2147/IDR.S173804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R.; Miller P. A.; Vakulenko S. B.; Stewart N. K.; Boggess W. C.; Miller M. J. A Synthetic Dual Drug Sideromycin Induces Gram-Negative Bacteria To Commit Suicide with a Gram-Positive Antibiotic. J. Med. Chem. 2018, 61 (9), 3845–3854. 10.1021/acs.jmedchem.8b00218. [DOI] [PubMed] [Google Scholar]
- Baell J. B.; Holloway G. A. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 2010, 53 (7), 2719–2740. 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- Irwin J. J.; Duan D.; Torosyan H.; Doak A. K.; Ziebart K. T.; Sterling T.; Tumanian G.; Shoichet B. K. An Aggregation Advisor for Ligand Discovery. J. Med. Chem. 2015, 58 (17), 7076–7087. 10.1021/acs.jmedchem.5b01105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quotadamo A.; Linciano P.; Davoli P.; Tondi D.; Costi M.; Venturelli A. An Improved Synthesis of CENTA, a Chromogenic Substrate for β-Lactamases. Synlett 2016, 27 (17), 2447–2450. 10.1055/s-0035-1562454. [DOI] [Google Scholar]
- Reaction was performed under strict anhydrous conditions. Increasing the equivalents of TMSI or extending the reaction time led to decomposition of the product and a reduction in recovered material.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.