

Abstract

Novel anthraquinone compounds that induce ER stress and paraptosis-like cell death were designed and synthesized. Compound 4a is the first organic micromolecule to kill tumor cells by only paraptosis, and its mechanism of action has been further explored. Paraptosis does not appear to involve either phosphatidylserine translocation associated with apoptosis or cell cycle arrest. The bisbenzyloxy and N-(2-hydroxyethyl)formamide structures may be two critical pharmacophores for paraptosis. Bisbenzyloxy can induce ER stress, and the N-(2-hydroxyethyl)formamide structure can increase the ratio of LC3II/I and cytoplasmic vacuolization and facilitates paraptosis. Some antitumor drugs fail to eradicate malignant cell lines with impaired apoptotic pathways; paraptosis may be a route to kill such cells and provides a new potential strategy for cancer chemotherapy.
Keywords: Anthraquinone, paraptosis, ER stress, noncaspase-dependent cell death, vacuolation
Programmed cell death is divided into two general types: caspase-dependent and noncaspase-dependent cell death. Apoptosis is one of the most typical forms of caspase-dependent programmed cell death. Paraptosis is a new noncaspase-dependent type of programmed cell death that appears to differ from apoptosis, necrosis, and autophagy and is characterized by ER stress, extensive cytoplasmic vacuolization, and swelling of the endoplasmic reticulum and/or mitochondria. Little is known at present regarding the molecular basis of paraptosis.1−3 Some antitumor drugs fail to eradicate malignant cell lines with impaired apoptotic pathways; paraptosis may be a route to kill such cells and provides a new potential strategy for cancer chemotherapy.
Factors that induce paraptosis include organometallic complexes, viruses,4 natural products, and their analogues. Compounds that induce paraptosis are predominantly organometallic complexes such as copper,5−7 ruthenium,8,9 titanium,10 and rhenium11 complexes. Some natural products can also induce tumor cell death through paraptosis such as taxol12,13 (Figure 1A), curcumin,14 and celastrol.15 Although organometallic complexes can induce cell death through the paraptotic pathway, it is easy to cause heavy metal poisoning during treatment. However, few natural product derivatives or organics have been reported to induce paraptosis.
Figure 1.
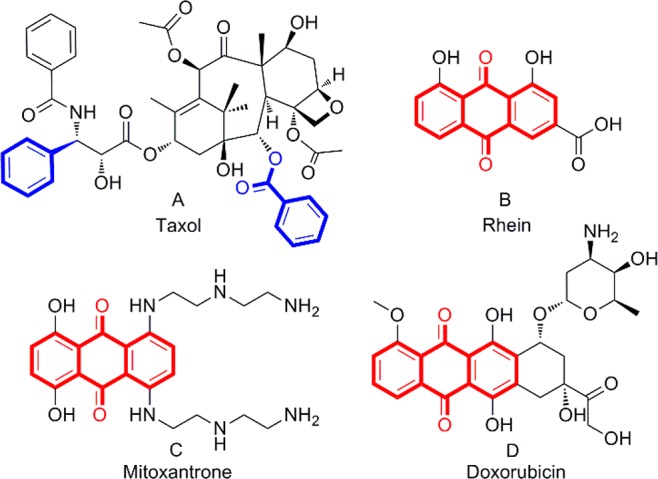
Taxol, rhein, and anthraquinone-based drugs that have been used clinically to treat tumors.
Rhein (Figure 1B) is a natural product isolated from Rheum palmatum that has moderate anticancer activities in different human tumor cells and is very well tolerated by the human body.16,17 There are several anthraquinone-based drugs, such as mitoxantrone and doxorubicin (Figure 1C,D), used for the treatment of various cancers in the clinic, and their antitumor effect is exerted through the apoptotic pathway.18,19
It has been reported in the literature that bisbenzyloxy may be an essential group to induce ER stress in cells.20 Here, we used rhein as a lead compound, introduced a bisbenzyloxy group at its 1,8-OH position to ensure ER stress, and then modified and structurally optimized it at the C-3 position and designed and synthesized a series of novel anthraquinone compounds that can induce cell death by paraptosis. The general structure of the compound is shown in Figure 2. Optimized compound 4a (Figure 2) is the first organic micromolecule to kill tumor cells by only paraptosis without other cell death mechanisms and has excellent activity in vitro. The morphological characteristics of paraptotic cells and the expression of related proteins were further examined. Along with analyzing structure–activity relationships (SARs) and mechanisms of action, the mechanism by which paraptosis is induced by the compounds was elucidated.
Figure 2.
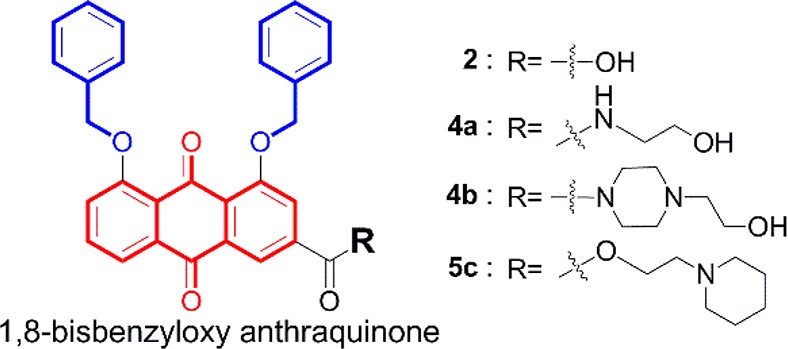
Examples of bioactive compounds with paraptotic activity.
The general procedure for anthraquinone compound synthesis and the total yield are shown in the Supporting Information (Scheme S1 and Table S1). The effect of all compounds on cancer cell proliferation was investigated by MTT and CCK-8 assay, and their IC50 values and cell morphology are listed in Table S2 and Figure S1. Compounds 4a, 4b, and 5c inhibited tumor cell proliferation and induced cell vacuoles (Figure S1). It is worth noting that 4a has an IC50 value (1.5 ± 0.3 μM) at the single digit micromolar level and is more sensitive in hepatocellular carcinoma SMMC-7721 cells than in the other cells tested (Figure S2). Therefore, 4a was selected for further investigation.
Inverted phase contrast microscopy showed that a large number of vacuoles appeared in the cells after 4a treatment for 48 h (Figures 3A and S3). As the concentration of 4a increased, the number of vacuoles in the cells gradually increased, but the volume of the vacuoles slowly shrank. At the same time, morphological changes such as cell shrinkage, cytoplasmic loss, and nuclear chromatin pyknosis were observed (Figures 3A and S4). Since vacuoles were found in the cytosol, to determine if the vacuoles were derived from the ER, SMMC-7721 cells were stained with ER-Tracker Red and Hoechst 33342. It was clearly seen under a laser scanning confocal microscope that the nuclei shrank after 4a treatment. In addition, vacuoles were observed and were surrounded by positive ER-specific markers, indicating that the vacuoles originated from the ER. With an increase in drug concentration, the number of vacuoles gradually increased, the entire endoplasmic reticulum cytoplasm was occupied by vacuoles, and cell contents began to be lost in large quantities (Figures 3B and S5).
Figure 3.
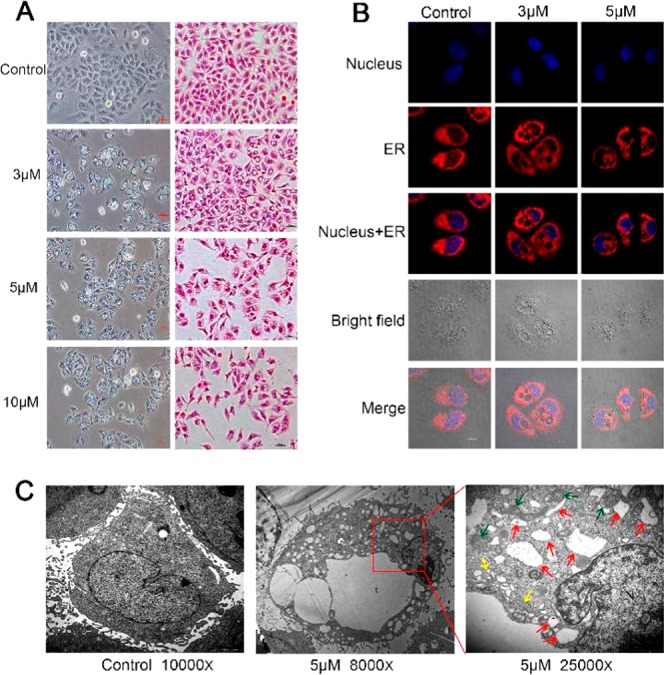
(A) Phase contrast images and HE staining after SMMC-7721 cells were treated with 4a for 48 h. (B) SMMC-7721 cells were incubated with 4a for 48 h and analyzed using ER-Tracker Red and Hoechst 33342 assays by laser scanning confocal microscopy. ER-Tracker Red (red) and Hoechst 33342 (blue) were used to visualize ER structures and nuclei, respectively. (C) TEM imaging of control and treated (4a, 5 μM) SMMC-7721 cells. The abnormal ER, mitochondria, and Golgi apparatus are indicated by red arrowheads, green arrowheads, and yellow arrowheads, respectively.
Changes in cell organelles were observed by transmission electron microscopy (TEM). After treatment with 4a at 5 μM for 48 h, several large vacuoles and many small vacuoles were present in the cytoplasm, the cellular membrane structure remained intact, the nucleus was concentrated and smaller, and autophagosomes and apoptotic bodies were not found (Figures 3C and S6, S8, and S11). Further observation showed that the vacuoles originated from the expansion and swelling of the rough ER, and the vacuoles were wrapped in monolayer membranes rather than bilayer membranes, similar to autophagy (Figures 3C and S6, S8, and S9). We also observed that mitochondrial swelling, vacuoles, and ridges disappeared in some cells (Figures S11 and S12). These results indicate that 4a treatment induced the formation of massive cytoplasmic vacuoles and damaged the integrity of the ER, mitochondrial, and Golgi apparatus structures in human SMMC-7721 cells. Compound 4a triggered ER vacuolation with the specific characteristics of paraptosis.
Next, we further examined the effect of vacuoles on cell viability. A CCK-8 assay was performed after the treatment of SMMC-7721 cells with 3, 5, or 10 μM (which can induce cell vacuole formation) 4a for 48 h. With an increase in the 4a concentration, the cell viability was highly decreased (Figures 4C and S25). To investigate whether growth inhibition induced by 4a was associated with regulation of the cell cycle and the induction of apoptosis, the DNA content of the cellular nuclei and the percentages of apoptotic cells were detected by flow cytometry. Given that early changes in apoptosis occur at the cell membrane surface and that PS is a negatively charged phospholipid normally present inside the cell membrane, as the cell undergoes apoptosis and the distribution of this asymmetrical phospholipid is disrupted, PS leaks outside the cell membrane.21 Flow cytometry revealed that apoptotic cells were not detected during 4a-induced cytoplasmic vacuolation-mediated cell death (Figures 4A,D and S13–S18). Except in the 10 μM treatment group, in which the cell viability was 22.42 ± 1.42%, the cell membranes remained normal and did not appear to indicate PS eversion in early apoptosis (Figures 4A,D and S17). Interestingly, the cell cycle was not altered by treatment with 4a at any concentration (Figures 4B,E and S19–S24), which may be a new biological characteristic of paraptosis. However, we found that treatment with 5c not only induced paraptosis-like cell death but also induced apoptosis. Vacuolar cells and a few apoptotic cells were clearly observed (Figure S26), and flow cytometry also detected apoptotic cells with PS translocation (Figures S27–S31).
Figure 4.

(A) Flow cytometric analysis of apoptosis and the cell cycle. Cells were collected and stained with annexin V-fluorescein isothiocyanate (FITC) and PI. (B) Flow cytometric analysis of cell cycle populations. (C) CCK-8 assays were performed after the treatment of SMMC-7721 cells with 4a for 48 h. (D) Frequency distribution graph of apoptotic cells. (E) Frequency distribution graph of the cell cycle.
Caspase-3 is activated in the apoptotic cell both by extrinsic (death ligand) and intrinsic (mitochondrial) pathways. As an executioner caspase, the caspase-3 zymogen has virtually no activity until it is cleaved by an initiator caspase.22,23 However, we did not detect cleaved caspase-3 in 4a-treated cells (Figure 5). The above results indicated that 4a-induced cell death, which does not cause apoptosis or affect the cell cycle, is a noncaspase-dependent method of cell death.
Figure 5.

Immunoblot analysis of caspase-3. SMMC-7721 cells were treated for 12, 24, and 48 h with 4a (IC60) and cisplatin (IC60), and cellular extracts were analyzed to detect both the inactive (procaspase-3) and active (cleaved caspase-3) forms of caspase-3. β-Actin was shown as a loading control, and cisplatin was used as a positive control for apoptosis.
The generation of ER vacuoles is usually associated with persistent ER stress. Therefore, we reasoned that 4a-induced vacuolation might result in the induction of persistent ER stress. The vacuolization of cells gradually increased (Figures 6B and S32), the expression of GRP78, a key hallmark of the ER stress response,24 significantly increased, and this process was time-dependent after treatment with 4a (Figures 6A and S33). Vacuolation is also a feature of autophagy, and these results prompted us to explore the role of autophagy in 4a-induced vacuole formation and cell death. Therefore, we examined whether 4a activated autophagy in SMMC-7721 cells. The essential proteins LC3 and p62 in the autophagic flow pathway were detected by Western blotting. When autophagosomes are formed, LC3 is synthesized as proLC3 in the cytosol and cleaved by Atg4 to produce LC3-I. LC3-I is activated by Atg7, transferred to Atg3, and conjugated to PE. The LC3-PE conjugate is LC3-II, a membrane-bound form of LC3. The Atg16 conjugate functions in LC3 lipidation.25,26 LC3-II is transported in an Atg5-dependent manner to the inner layer of the autophagosome and binds to receptors (e.g., p62, etc.) that recognize the substance to be degraded, encapsulating the substance to form the autophagosome.27,28 P62 trapped by LC3 is selectively transported into the autophagosome, which results in the downregulation of p62 expression in the cells. LC3-II located in the outer membrane of the autophagosome is cleaved by Atg4 to LC3-I, PE is separated, and LC3-I is recycled for repeated use29,30 (Figure 7D). The treatment of cells with 4a increased the ratio of LC3-II/I. The higher the LC3-II/I ratio, the more vacuoles were observed in the cells (Figures 6A,B, and S32–S34). Because p62 protein levels also increased, autophagosome formation was inhibited following 4a treatment. The evidence strongly suggests that 4a triggers ER vacuolation death, which does not depend on the typical autophagy pathway but proceeds through a paraptotic pathway.
Figure 6.
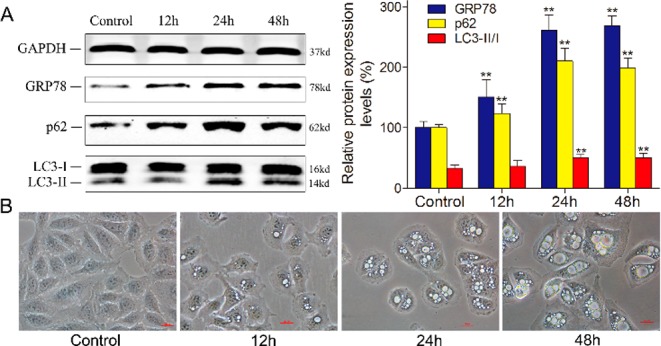
(A) Western blotting analysis of the expression of GRP78, p62, and LC3 after 4a (3 μM) treatment at different time points in SMMC-7721 cells. GAPDH is shown as a loading control; ** = P < 0.01. (B) Phase contrast images of SMMC-7721 cells treated with 3 μM 4a at the indicated time points. All images were acquired at the same magnification.
Figure 7.

Summary of the SAR and the mechanism of paraptosis. (A) Western blotting analysis of the expression of GRP78, p62, and LC3 after the treatment of SMMC-7721 cells with the same concentration of rhein, 2, 4a, and cisplatin. GAPDH is shown as a loading control; ** = P < 0.01. (B) Phase contrast images of SMMC-7721 cells treated with 10 μM rhein, 2, 4a, and cisplatin for 48 h; all images were acquired at the same magnification. (C) Summary of the SAR and mechanism of paraptosis. (D) LC3-PE binding system during mammalian autophagy.
To study the structure–activity relationship (SAR) between 4a and the induction of paraptosis, we tested the expression levels of related proteins using Western blotting.
After treating cells with the same concentration of rhein, 2, 4a, and cisplatin, rhein failed to activate the expression of GRP78, 2, and 4a treatment significantly activated GRP78 expression, and cisplatin treatment strongly inhibited GRP78 protein expression compared to their expression in the control group (Figures 7A and S36–S37). It has been shown that bisbenzyloxy is an important group that produces a sustained ER stress response. Our experimental data validate speculation in the literature reported by Zheng et al.20 In addition, compared with that in the control, the expression level of p62 did not decrease in any of the treatment groups, and the LC3-II/I ratio did not change except in the 4a treatment group (Figures 7A and S36–S37). When SMMC-7721 cells were treated with the same concentration of 2 and 4a for 48 h, the formation of vacuoles was clearly observed in cells treated with 4a, and little vacuolation was observed in cells treated with compound 2 (Figures 6B and S35), although 2 can produce ER stress. The above results show that an increase in the ratio of LC3-II/I may be related to the presence of N-(2-hydroxyethyl)formamide at the C-3 position of the 1,8-dibenzyloxy-9,10-anthraquinone ring and indicate that the occurrence of cellular vacuoles may be related to the upregulation of LC3-II (Figure 7C). LC3-II is the terminal protein in the autophagy signaling pathway. Activated LC3-II binds to p62 to form autophagosomes and participates in autophagy (Figure 7D), and the expression level of p62 decreases significantly. However, we found that activated LC3-II did not bind to p62 to form the double membranes associated with autophagy but may be related to cytoplasmic vacuolization and the formation of single membrane vacuoles in paraptosis (Figure 7C,D) after 4a treatment.
In summary, the novel anthraquinone compound 4a is the first organic micromolecule to kill tumor cells by only paraptosis in vitro. In paraptosis, we found no translocation of PS and no cell cycle arrest, which is different from the traditional apoptosis model and is also a new biological characteristic of paraptosis. Here, with the study of its mechanism of action and SAR, we confirmed that bisbenzyloxy is an important pharmacophore that produces a sustained ER stress response. N-(2-Hydroxyethyl)formamide may be related to the upregulation of LC3-II. Activated LC3-II may promote cytoplasmic vacuolization and accelerate this process in paraptosis. These findings provide new insights for us to determine paraptosis and may be useful for designing new effective induced paraptosis drugs.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81460561) and the Key Laboratory of Early Prevention and Treatment for Regional High-Frequency Tumor, Guangxi Medical University, Ministry of Education.
Glossary
ABBREVIATIONS
- ER
endoplasmic reticulum
- CCK-8
CellCountingKit-8
- MTT
3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide
- IC50
50% inhibitory concentration
- GRP78
glucose-regulated protein
- LC3
microtubule-associated protein light chain 3
- p62
Sequestosome 1
- PE
phosphatidyl ethanolamine
- PS
phosphatidylserine
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00624.
Experimental details for chemical synthesis, product analysis, NMR spectroscopy, mass spectrometry, drug screening, protein expression, cell culture, FACS analysis, cell viability assay, laser scanning confocal microscopy, transmission electron microscopy, and Figures S1–S37 (PDF)
Author Contributions
§ W.T. and J.L. contributed equally.
Financial support was from the National Natural Science Foundation of China (81460561).
The authors declare no competing financial interest.
Supplementary Material
References
- Sperandio S.; de Belle I.; Bredesen D. E. An alternative, nonapoptotic form of programmed cell death. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 14376–14381. 10.1073/pnas.97.26.14376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon M. J.; Kim E. H.; Kwon T. K.; Park S. A.; Choi K. S. Simultaneous mitochondrial Ca2+ overload and proteasomal inhibition are responsible for the induction of paraptosis in malignant breast cancer cells. Cancer Lett. 2012, 324, 197–209. 10.1016/j.canlet.2012.05.018. [DOI] [PubMed] [Google Scholar]
- Sperandio S.; Poksay K.; de Belle I.; Lafuente M. J.; Liu B.; Nasir J.; Bredesen D. E. Paraptosis: mediation by MAP kinases and inhibition by AIP-1/Alix. Cell Death Differ. 2004, 11, 1066–1075. 10.1038/sj.cdd.4401465. [DOI] [PubMed] [Google Scholar]
- Monel B.; Compton A. A.; Bruel T.; Amraoui S.; Burlaud-Gaillard J.; Roy N.; Guivel-Benhassine F.; Porrot F.; Génin P.; Meertens L. Zika virus induces massive cytoplasmic vacuolization and paraptosis-like death in infected cells. EMBO J. 2017, 36, 1653–1668. 10.15252/embj.201695597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardito S.; Bassanetti I.; Bignardi C.; Elviri L.; Tegoni M.; Mucchino C.; Bussolati O.; Franchi-Gazzola R.; Marchiò L. Copper binding agents acting as copper ionophores lead to caspase inhibition and paraptotic cell death in human cancer cells. J. Am. Chem. Soc. 2011, 133, 6235–6242. 10.1021/ja109413c. [DOI] [PubMed] [Google Scholar]
- Gandin V.; Tisato F.; Dolmella A.; Pellei M.; Santini C.; Giorgetti M.; Marzano C.; Porchia M. In vitro and in vivo anticancer activity of copper (I) complexes with homoscorpionate tridentate tris (pyrazolyl) borate and auxiliary monodentate phosphine ligands. J. Med. Chem. 2014, 57, 4745–4760. 10.1021/jm500279x. [DOI] [PubMed] [Google Scholar]
- Marzano C.; Gandin V.; Pellei M.; Colavito D.; Papini G.; Lobbia G. G.; Del Giudice E.; Porchia M.; Tisato F.; Santini C. In vitro antitumor activity of the water soluble copper (I) complexes bearing the tris (hydroxymethyl) phosphine ligand. J. Med. Chem. 2008, 51, 798–808. 10.1021/jm701146c. [DOI] [PubMed] [Google Scholar]
- Li C.; Ip K. W.; Man W. L.; Song D.; He M. L.; Yiu S. M.; Lau T. C.; Zhu G. Cytotoxic (salen) ruthenium (III) anticancer complexes exhibit different modes of cell death directed by axial ligands. Chem. Sci. 2017, 8, 6865–6870. 10.1039/C7SC02205K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierroz V.; Rubbiani R.; Gentili C.; Patra M.; Mari C.; Gasser G.; Ferrari S. Dual mode of cell death upon the photo-irradiation of a Ru II polypyridyl complex in interphase or mitosis. Chem. Sci. 2016, 7, 6115–6124. 10.1039/C6SC00387G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cini M.; Williams H.; Fay M. W.; Searle M. S.; Woodward S.; Bradshaw T. D. Enantiopure titanocene complexes–direct evidence for paraptosis in cancer cells. Metallomics. Metallomics. 2016, 8, 286–297. 10.1039/C5MT00297D. [DOI] [PubMed] [Google Scholar]
- Ye R. R.; Tan C. P.; Chen M. H.; Hao L.; Ji L. N.; Mao Z. W. Mono-and Dinuclear Phosphorescent Rhenium (I) Complexes: Impact of Subcellular Localization on Anticancer Mechanisms. Chem. - Eur. J. 2016, 22, 7800–7809. 10.1002/chem.201505160. [DOI] [PubMed] [Google Scholar]
- Chen T. S.; Wang X. P.; Sun L.; Wang L. X.; Xing D.; Mok M. Taxol induces caspase-independent cytoplasmic vacuolization and cell death through endoplasmic reticulum (ER) swelling in ASTC-a-1 cells. Cancer Lett. 2008, 270, 164–172. 10.1016/j.canlet.2008.05.008. [DOI] [PubMed] [Google Scholar]
- Sun Q.; Chen T.; Wang X.; Wei X. Taxol induces paraptosis independent of both protein synthesis and MAPK pathway. J. Cell. Physiol. 2010, 222, 421–432. 10.1002/jcp.21982. [DOI] [PubMed] [Google Scholar]
- Yoon M. J.; Kang Y. J.; Lee J. A.; I Kim Y. M.; Kim A.; Lee Y. S.; Park J. H.; Lee B. Y.; Kim I. A.; Kim H. S.; Kim S. A.; Yoon A. R.; Yun C. O.; Kim E. Y.; Lee K.; Choi K. S. Stronger proteasomal inhibition and higher CHOP induction are responsible for more effective induction of paraptosis by dimethoxycurcumin than curcumin. Cell Death Dis. 2017, 5, e1112 10.1038/cddis.2014.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W. B.; Feng L. X.; Yue Q. X.; Wu W. Y.; Guan S. H.; Jiang B. H.; Yang M.; Liu X.; Guo D. A. Paraptosis accompanied by autophagy and apoptosis was induced by celastrol, a natural compound with influence on proteasome, ER stress and Hsp90. J. Cell. Physiol. 2012, 227, 2196–2206. 10.1002/jcp.22956. [DOI] [PubMed] [Google Scholar]
- Yang X.; Sun G.; Yang C.; Wang B. Novel rhein analogues as potential anticancer agents. ChemMedChem 2011, 6, 2294–2301. 10.1002/cmdc.201100384. [DOI] [PubMed] [Google Scholar]
- Su Z. Y.; Tian W.; Li J.; Wang C. M.; Pan Z. Y.; Li D. R.; Hou H. X. Biological evaluation and molecular docking of Rhein as a multi-targeted radiotherapy sensitization agent of nasopharyngeal carcinoma. J. Mol. Struct. 2017, 1147, 462–468. 10.1016/j.molstruc.2017.06.123. [DOI] [Google Scholar]
- Koceva-Chyła A.; Jedrzejczak M.; Skierski J.; Kania K.; Jóźwiak Z. Mechanisms of induction of apoptosis by anthraquinone anticancer drugs aclarubicin and mitoxantrone in comparison with doxorubicin: relation to drug cytotoxicity and caspase-3 activation. Apoptosis 2005, 10, 1497–1514. 10.1007/s10495-005-1540-9. [DOI] [PubMed] [Google Scholar]
- Kluza J. M.; Marchetti P.; Gallego M. A.; Lancel S.; Fournier C.; Loyens A.; Beauvillain J. C.; Bailly C. Mitochondrial proliferation during apoptosis induced by anticancer agents: effects of doxorubicin and mitoxantrone on cancer and cardiac cells. Oncogene 2004, 23, 7018–7030. 10.1038/sj.onc.1207936. [DOI] [PubMed] [Google Scholar]
- Zheng H.; Dong Y.; Li L.; Sun B.; Liu L.; Yuan H.; Lou H. Novel benzo[α]quinolizidine analogs induce cancer cell death through paraptosis and apoptosis. J. Med. Chem. 2016, 59, 5063–5076. 10.1021/acs.jmedchem.6b00484. [DOI] [PubMed] [Google Scholar]
- Segawa K.; Kurata S.; Yanagihashi Y.; Brummelkamp T. R.; Matsuda F.; Nagata S. Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science 2014, 344, 1164–1168. 10.1126/science.1252809. [DOI] [PubMed] [Google Scholar]
- Kothakota S.; Azuma T.; Reinhard C.; Klippel A.; Tang J.; Chu K.; McGarry T. J.; Kirschner M. W.; Koths K.; Kwiatkowski D. J. Caspase-3-generated fragment of gelsolin: effector of morphological change in apoptosis. Science 1997, 278, 294–298. 10.1126/science.278.5336.294. [DOI] [PubMed] [Google Scholar]
- Liu X.; Zou H.; Slaughter C.; Wang X. DFF, a heterodimeric protein that functions downstream of caspase-3 to trigger DNA fragmentation during apoptosis. Cell 1997, 89, 175–184. 10.1016/S0092-8674(00)80197-X. [DOI] [PubMed] [Google Scholar]
- Lee A. S. The ER chaperone and signaling regulator GRP78/BiP as a monitor of endoplasmic reticulum stress. Methods 2005, 35, 373–381. 10.1016/j.ymeth.2004.10.010. [DOI] [PubMed] [Google Scholar]
- Ichimura Y.; Kirisako T.; Takao T.; Satomi Y.; Shimonishi Y.; Ishihara N.; Mizushima N.; Tanida I.; Kominami E.; Ohsumi M. A ubiquitin-like system mediates protein lipidation. Nature 2000, 408, 488–492. 10.1038/35044114. [DOI] [PubMed] [Google Scholar]
- Xie Z.; Klionsky D. J. Autophagosome formation: core machinery and adaptations. Nat. Cell Biol. 2007, 9, 1102. 10.1038/ncb1007-1102. [DOI] [PubMed] [Google Scholar]
- Mathew R.; Karp C. M.; Beaudoin B.; Vuong N.; Chen G.; Chen H. Y.; Bray K.; Reddy A.; Bhanot G.; Gelinas C. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009, 137, 1062–1075. 10.1016/j.cell.2009.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuragi Y.; Ichimura Y.; Komatsu M. p62/SQSTM 1 functions as a signaling hub and an autophagy adaptor. FEBS J. 2015, 282, 4672–4678. 10.1111/febs.13540. [DOI] [PubMed] [Google Scholar]
- Pankiv S.; Clausen T. H.; Lamark T.; Brech A.; Bruun J. A.; Outzen H.; Øvervatn A.; Bjørkøy G.; Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- Ichimura Y.; Kumanomidou T.; Sou Y. S.; Mizushima T.; Ezaki J.; Ueno T.; Kominami E.; Yamane T.; Tanaka K.; Komatsu M. Structural basis for sorting mechanism of p62 in selective autophagy. J. Biol. Chem. 2008, 283, 22847–22857. 10.1074/jbc.M802182200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.