

Abstract
Background
Decreased uterine blood flow is known to contribute to pregnancy complications such as gestational hypertension and preeclampsia. Previously, we showed that the loss of regulator of G protein signaling 2 (RGS2), a GTPase activating protein for Gq/11 and Gi/o class G proteins, decreases uterine blood flow in the nonpregnant state in mice. Here, we examined the effects of the absence of RGS2 and 5 on uterine blood flow and uterine vascular structure and function at early, mid, and late gestation, as well as peripartum period in mice.
Methods and Results
Abdominal Doppler ultrasonography was performed on adult female wild‐type, Rgs2−/−, and Rgs5−/− mice at pre‐pregnancy, gestational days 10, 15, and 18, and postpartum day 3. Uterine artery structure and function were also assessed by vessel myograph studies. At mid‐pregnancy, uterine blood flow decreased in both Rgs2−/− and Rgs5−/− mice, whereas resistive index increased only in Rgs2−/− mice. In uterine arteries from wild‐type mice, mRNA expression of RGS2 and 4 increased, whereas RGS5 expression remained elevated at mid‐pregnancy. These changes in gene expression were unique to uterine arteries because they were absent in mesenteric arteries and the aorta of wild‐type mice. In Rgs2−/− mice, uterine artery medial cross‐sectional area and G protein–coupled receptor‐mediated vasoconstriction increased in mid‐pregnancy, implicating a role for RGS2 in structural and functional remodeling of uterine arteries during pregnancy. In contrast, RGS5 absence increased vasoconstriction only in the peripartum period.
Conclusions
These data together indicate that RGS2 plays a critical role in the structural and functional remodeling of uterine arteries to impact uterine blood flow during pregnancy. Targeting the signaling pathway regulated by RGS2 may therefore be a therapeutic strategy for ameliorating utero‐placental perfusion disorders during pregnancy.
Keywords: animal model, G protein–coupled receptor signaling, knockout mice, pregnancy and postpartum, RGS proteins, uterine blood flow, vascular reactivity
Subject Categories: Basic Science Research, Vascular Biology, Physiology, Cell Signalling/Signal Transduction, Pathophysiology
Clinical Perspective
What Is New?
Absence of regulator of G protein signaling 2 (RGS2), a GTPase activating protein for Gq/11 class G proteins, causes augmented vascular reactivity and decreased uterine blood flow at mid‐pregnancy in mice; this study thus provides new evidence that RGS2 is necessary for normal uterine vascular adaptation to facilitate uteroplacental blood flow during pregnancy.
What Are the Clinical Implications?
Loss of fine‐tuning of G protein signaling by RGS2 protein attributed to loss‐of‐function mutations or protein degradation in the vasculature could predispose to pregnancy complications, including uteroplacental insufficiency and gestational hypertension.
Further studies focusing on vascular signaling pathways regulated by RGS2, using pharmacology and/or therapeutic genetics, could lead to identification of novel treatment for decreased uterine blood flow and uteroplacental insufficiency in pregnancy complications.
Introduction
The uterine artery is a primary feed vessel that conducts oxygenated blood to the uterine vascular bed and the placenta during pregnancy. Perfusion of the uterine wall is facilitated by a network of arcuate arteries that branch off from the main uterine artery, giving rise to the radial arteries in the myometrium, which, in turn, perfuse the endometrium as spiral arteries. Once placentation occurs during pregnancy, endovascular trophoblasts invade the radial arteries, converting them from resistance arterioles to capacitance vessels with increased diameter and decreased tone, allowing for increased blood flow to the placenta.1, 2 Blood flow to the uterus and the placenta therefore is greatly affected by changes in vascular tone and/or reactivity of the uterine artery to vasoactive substances during pregnancy. Indeed, proper function of the uterine artery has been shown to be critical for the progression of normal pregnancy, and impaired uterine blood flow has been associated with miscarriages3, 4, 5, 6 and pregnancy complications such as preeclampsia7, 8, 9
Increased uterine blood flow during pregnancy involves a decrease in vascular resistance of the uterine artery. The mechanisms that mediate decreased vascular resistance are not completely understood; however, several studies have shown that normal pregnancy triggers increased production of endothelium‐derived relaxing factors.2, 10 In addition, the vasculature becomes more sensitive to vasodilators, while developing refractoriness or decreased sensitivity to contractile agonists such as angiotensin II (Ang II).11 Several vasoactive substances act on G protein–coupled receptors (GPCRs) to reduce or increase vessel diameter via constriction or dilation, respectively, thereby affecting blood flow. Signaling downstream of GPCRs that influences vessel diameter is tightly regulated by regulators of G protein signaling (RGS) proteins. RGS proteins control the speed and amplitude of GPCR signaling by functioning as GTPase activating proteins that increase the rate of GTP hydrolysis by the intrinsic GTPase activity of the heterotrimeric G protein alpha subunit.12 Members of the R4/B family of RGS proteins have been shown to play key roles in vascular tone regulation. Whereas all members of R4/B RGS proteins can act as GTPase activating proteins for Gi/o and Gq/11 class G proteins,12, 13 RGS2 is known to be highly potent and selective towards Gq/11 class G proteins.14 Among the members of this family of RGS proteins, RGS2 and RGS5 have been most strongly linked to gestational hypertension and preeclampsia.15, 16 Meta‐analytical studies have shown that a single‐nucleotide polymorphism of the human Rgs2 gene (r4606) is a risk factor for the development of preeclampsia.15, 17 Previously, we examined the role of RGS2 in uterine blood flow and myogenic tone. We reported that the loss of even 1 copy of the Rgs2 gene impairs G protein regulation, causing increased uterine artery myogenic tone and decreased uterine blood flow in nonpregnant mice.18 However, the role of G protein regulation by RGS proteins in the uterine vascular bed and how it is involved in physiological adaptation of the vasculature to increase placental blood flow during pregnancy are unknown. In this study, we have profiled uterine blood flow and uterine artery vascular reactivity before, during, and after pregnancy in wild‐type (WT) mice and those null for Rgs2 or 5. Specifically, we examined how the absence of these R4 family members affects uterine blood flow and the reactivity of the uterine artery to GPCR activation at mid‐pregnancy and during peripartum period in first‐time pregnant mice. We report that loss of RGS2 impairs G protein regulation and is detrimental to uterine blood flow at mid‐pregnancy, a gestational period when many complications are reported to manifest.
Methods
The authors declare that data supporting all the findings in this study are available within the article. All materials and animal models used in the study are either commercially or publicly available.
Animals
Animal studies were performed according to protocols approved by the Animal Studies Committee of Drexel University College of Medicine heeding the National Institutes of Health guidance for the care and use of laboratory animals. All experiments were performed using 2‐ to 6‐month‐old female mice that have been backcrossed extensively into the C57BL/6 genetic background (Charles River Laboratories, Wilmington, MA). Generation of Rgs2−/− and Rgs5−/− mice has been described previously.19, 20 WT mice were generated in‐house from Rgs2 and Rgs5 het × het crosses. Mice were provided access to food and water ad libitum in our institution's animal facility at 22°C and a 12‐hour light/dark cycle.
Uterine Blood Flow Assessment by Doppler Ultrasound
Female mice at nonpregnant (NP), gestation day 10 (D10), gestation day 15 (D15), gestation day 18 (D18), and postpartum day 3 (PPD3) stages were anesthetized with isoflurane (1.0–1.5% isoflurane; Baxter Healthcare Corporation, Deerfield, IL; plus 1.5 L/min of O2), placed on a heating platform of a Vevo 2100 Imaging Station (Visual Sonics Inc, Toronto, Ontario, Canada), and gently secured with adhesive tape in order to remove hair from the abdomen with hair removal gel. During this process, body temperature was maintained at 37°C, and heart rate was recorded for the entirety of the ultrasound to ensure that mice remained within safe physiological limits. Uterine blood flow was measured by Doppler waveforms recorded using a 400‐MHz probe placed over the lower abdomen with coupling gel as previously described.18, 21 Ultrasound recordings were taken from the right and left uterine arteries close to the bladder or fetuses at a 30‐degree angle of insonation. After acquisition of waveforms, mice were removed from the isoflurane anesthesia, returned to their home cages, and allowed to recover. From each acquired waveform, peak systolic velocity (PSV), least diastolic velocity (LDV), and mean velocity were calculated. As previously described,18 average PSV, LDV, and mean velocity for each mouse was calculated and used to derive the following indices: Resistive Index=(Vmax−Vmin)/Vmax; Pulsatile Index=(Vmax−Vmin)/Vmean; and PSV/LDV ratio=Vmax/Vmin, where Vmax=PSV, Vmin=LDV and Vmean=mean velocity.
Quantitative Real‐Time PCR Analysis of RGS2, RGS4, and RGS5 Expression Levels in Uterine Arteries of WT, Rgs2−/−, and Rgs5−/− Mice
Expression levels of RGS2, RGS4, and RGS5 were determined by real‐time PCR using RNA extracted from uterine arteries of NP, D10, D15, D18, and PPD3 WT, Rgs2−/−, and Rgs5−/− mice and from mesenteric arteries and aorta of NP, D15, and PPD3 WT, Rgs2−/−, and Rgs5−/− mice. Total RNA was isolated using the TRIzol extraction method (Thermo Fisher Scientific, Waltham, MA), with tissue homogenizer tubes and an Omni Bead Ruptor 24 homogenizer (Omni International, Kennesaw, GA). RNA was purified using the Purelink RNA Mini Kit (Thermo Fisher Scientific). RNA was then reverse‐transcribed into cDNA using a Maxima First Strand cDNA Synthesis Kit (Thermo Fisher Scientific), according to the manufacturer's instructions. The following primer probes were used in the real‐time PCR assays with TaqMan gene expression master mix (Thermo Fisher Scientific), as directed by the manufacturer: Rgs2, Mm01292909_g1; Rgs5, Mm00654112_m1; Rgs4, Mm00501389_m1; and Gapdh, Mm99999915_g1. The ∆Ct method (where Ct is threshold cycle) was used to calculate RGS2, RGS4, and RGS5 mRNA expression after normalization to Gapdh expression.
Baseline Blood Pressure and Heart Rate Measurements
Blood pressure and heart rate were measured in WT, Rgs2−/−, and Rgs5−/− female mice at NP, D10, D15, D18, and PPD3 under isoflurane anesthesia, as previously described.22 Briefly, a fluid‐filled catheter was inserted into the right carotid artery, after which animals were allowed to stabilize for 15 minutes. Continuous recordings of mean blood pressure and heart rate were acquired using Lab Chart software (version 8.0; ADInstruments, Colorado Springs, CO) for 5 minutes.
Determination of Uterine and Mesenteric Artery Reactivity
Reactivity of uterine and mesenteric arteries to vasoactive agents was studied using previously described methods, with modifications.18, 23 Briefly, mice were euthanized by deep anesthesia with the injection of ketamine/xylazine (ketamine; 43 mg/kg, IP, and xylazine; 6 mg/kg, IP), followed by cervical dislocation. The uterus and intestines were harvested and placed in a cooling chamber with PSS made up of: (in mmol/L) 140 NaCl, 5 KC1, 1.2 MgSO4, 2.0 CaC12, 10 NaAcetate, 10 HEPES, 1.2 Na2H2PO4, 5 glucose, and pH adjusted to 7.4 with NaOH. Segments of the main uterine arteries and second‐order mesenteric arteries were isolated, excised, and cannulated at both ends with glass pipettes and secured with nylon ligatures in a vessel chamber. Intraluminal pressure and vessel bath temperature were maintained in a static flow condition at 60 mm Hg and 37°C by a servo‐controlled pressure pump and a temperature control system, respectively, using a Living systems vessel chamber (Catamount, Burlington, VT) and IonOptix vessel dimensions analysis software as previously described.24, 25 The vessel lumen and chamber were filled with PSS of the same composition described above. After a 30‐minute equilibration period, vessels were treated with increasing concentrations of KCl‐PSS to assess vessel viability and examine non‐GPCR‐mediated contractility. Any vessel with <30% reduction in lumen diameter at the highest K+ concentration (80 mmol/L) was discarded. Viable vessels were exposed to a battery of contractile stimulation protocols, including electrical field stimulation (EFS) with increasing frequencies (2, 5, 10, 15, 20, and 30 Hz) of alternating current at 40 volts for 30 seconds per frequency, increasing concentrations of Ang II (10 pmol/L–1 μmol/L), phenylephrine (PE; 10 nmol/L–100 μmol/L), and endothelin‐1 (ET‐1; 0.1 nmol/L–1 μmol/L). To assess endothelium‐dependent vasodilation, vessels were preconstricted with 10 μmol/L of PE followed by increasing concentrations of acetylcholine (ACh; 1 nmol/L–100 μmol/L). To determine the effects of nitric oxide (NO) on vascular reactivity, uterine artery segments were preincubated with the NO synthase inhibitor, Nω‐Nitro‐l‐arginine methyl ester (1 mmol/L), for 20 minutes before the start of the contractility and vasodilatation protocols described above.
Statistical Analysis
For ex vivo, isolated vessel prep experiments, we used 1 to 2 vessel segments from each animal for data acquisition. We expressed vasoconstriction of each vessel segment as the percent change in lumen diameter from baseline according to the formula: [(Di−D)/Di]×100, where Di is the initial lumen diameter before application of the stimulus, and D is the measured lumen diameter in the presence of the stimulus, as previously described.23 Percent relaxation was calculated as the change in lumen diameter after preconstriction with PE using the formula: [(D−DPE)/(Di−DPE)]×100, where DPE is the steady‐state diameter after constriction with PE. EC50 and maximum values were calculated by determining nonlinear fit using GraphPad Prism software (GraphPad Software Inc, La Jolla, CA). Results are presented as means ± SEM. For in vivo, whole‐animal experiments, data from multiple animals at the same pregnancy state were averaged. For ex vivo studies, we averaged vascular reactivity data from replicate experiments using multiple vessel segments isolated from the same animal such that the unit of analysis was the number of animals. To assess the effects of the absence of RGS2 or RGS5 on GPCR‐induced vasoconstriction and NO‐dependent and NO‐independent vasodilation of preconstricted mesenteric and uterine arteries, Kolmogorov–Smirnov tests of normality were conducted. Data that passed the normality test were subjected to 2‐way ANOVA with repeated measures, followed by Newman–Keuls post‐hoc tests to determine between‐group differences. For data that did not pass the normality test, we used Mann–Whitney U tests to determine between‐group differences for each concentration of drug. These statistical tests were also used to determine any effect of the absence of RGS2 or RGS5 on uterine blood flow assessed by Doppler ultrasonography and on blood pressure at different pregnancy states. One‐way ANOVA and Dunnett's multiple comparisons tests were used to determine any significant differences from the NP state in mRNA expression. One‐way ANOVA, followed by Dunnett's multiple comparisons tests, was used to determine any significant differences from the NP state within genotypes for morphometric parameters, whereas a 2‐way ANOVA and Newman–Keuls post‐hoc tests were used to determine any significant differences between genotypes for morphometric parameters. P<0.05 were considered statistically significant.
Results
Deficiencies in RGS2 and 5 Increase Impedance to Uterine Blood Flow in Mid‐Pregnancy
Previously, we reported that the loss of 1 or both copies of the Rgs2 gene (Rgs2+/− or Rgs2−/−) impedes uterine blood flow in nonpregnant mice.18 To determine whether this phenomenon persists during pregnancy and the postpartum period, and whether it was unique to the absence of RGS2, we used Doppler ultrasonography to assess uterine vascular hemodynamics and waveforms before pregnancy (NP) and at gestational days 10 (D10), 15 (D15), and 18 (D18) and postpartum day 3 (PPD3) in WT, Rgs2−/−, and Rgs5−/− female mice. Similar to our previous report,18 uterine blood flow velocity before pregnancy tended to decrease in Rgs2−/− mice and was even further reduced in Rgs5−/− mice relative to WT controls (Figure 1A and 1B). In WT mice, peak systolic velocity, least diastolic velocity, and mean velocity tended to increase during pregnancy, plateauing at mid‐pregnancy (D15), and returning to levels below pre‐pregnancy velocities at PPD3 (Figure 1B and 1D). This pattern in uterine blood flow was inversely related to changes in uterine blood flow impedance, though there was a trend toward increased resistive and pulsatile index and PSV/LDV ratio at D10. In contrast to the patterns in WT mice, uterine blood flow velocity in Rgs2−/− and Rgs5−/− mice was relatively unchanged during pregnancy and appeared to be suppressed compared with that in WT mice. In Rgs2−/− mice, resistive index remained significantly elevated at D15, compared with WT controls. There was, however, no difference between the 3 genotypes at PPD3. These data indicated that the loss of RGS2, but not RGS5, impairs uterine blood flow hemodynamics during pregnancy. Moreover, although the loss of RGS2 significantly impeded uterine blood flow before pregnancy and also up to mid‐pregnancy, the effect of the absence of RGS5 on impedance remained at mid‐pregnancy (Figure 1E through 1G).
Figure 1.
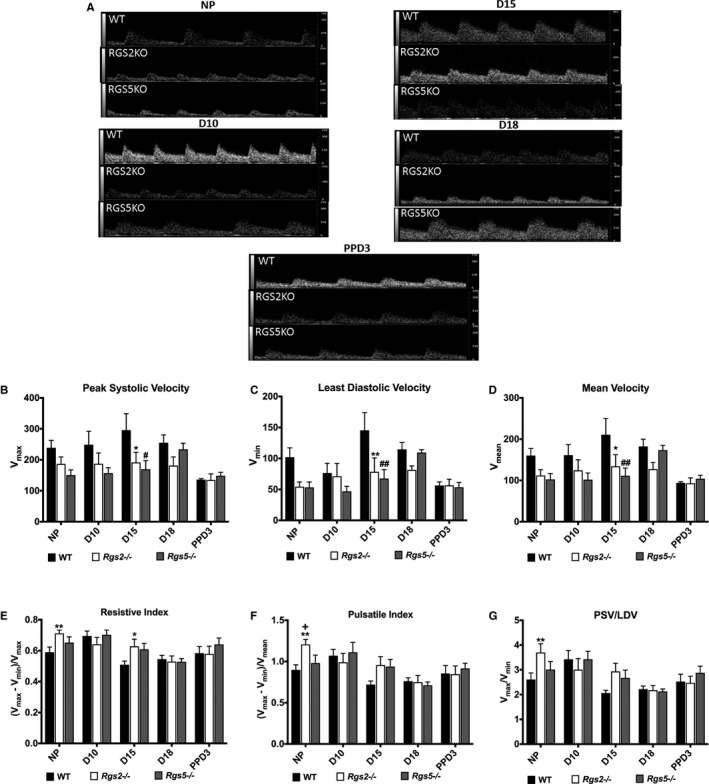
RGS2 and RGS5 deficiency increases impedance to uterine blood flow before pregnancy and at pregnancy day 15. A, Representative uterine artery Doppler ultrasonography waveforms from nonpregnant (NP), gestation day 10 (D10), gestation day 15 (D15), gestation day 18 (D18), and postpartum day 3 (PPD3) of wild‐type (WT), Rgs2−/−, and Rgs5−/− mice obtained under isoflurane anesthesia. Pulse wave velocities were acquired at an angle of 30° and frequency of 24 Hz. B through G, Uterine artery waveforms from WT (NP n=9, D10 n=5, D15 n=6, D18 n=5, and PPD3 n=5), Rgs2−/− (NP n=10, D10 n=5, D15 n=7, D18 n=5, and PPD3 n=4), and Rgs5−/− (NP n=5, D10 n=3, D15 n=4, D18 n=3, and PPD3 n=4) were analyzed to obtain (B) peak systolic velocity (PSV or Vmax), (C) least diastolic velocity (LDV or Vmin), and (D) mean velocity (MV or Vmean), which were used to derive (E) resistive index, (F) pulsatile index, and (G) PSV‐to‐LDV ratio for each genotype. Values are mean±SE. *,**P<0.05, 0.01 WT vs Rgs2−/−; #,## P<0.05, 0.01 WT vs Rgs5−/−; + P<0.05 Rgs2−/− vs Rgs5−/−.
Pregnancy Increases the Expression of RGS2 and 4 in WT Uterine Arteries
To determine the mechanism by which the loss of RGS2 and 5 decreases uterine blood flow during pregnancy, we examined expression profiles of these RGS proteins during pregnancy and postpartum period in uterine arteries from WT as well as Rgs2−/− and Rgs5−/− mice. Using RNA extracted from uterine arteries of NP, D10, D15, D18, and PPD3 mice, we performed gene expression analysis by RT‐qPCR. In WT mice, we found that RGS2 level markedly increased at D15, whereas it was severely suppressed at D18, returning to pre‐pregnancy level at PPD3 (Figure 2A). In contrast, RGS5 levels remained relatively elevated, except at D10 and PPD3, where they were markedly suppressed (Figure 2A). Next, we determined how pregnancy affects the expression profile of RGS4, also a member of the B/R4 RGS family and well expressed in the vasculature.13, 26 We found that similar to RGS2, the expression of RGS4 tended to decrease at D10 and D18, but appeared to increase at D15 (Figure 2A). We also noted that RGS2 and 4 were expressed at similar levels, whereas the expression of RGS5 was ≈30‐fold higher in the uterine vascular bed (Figure 2A).To determine whether there was any compensatory change in the expression of B/R4 RGS family members during pregnancy, we profiled the expression of RGS4 and 5 at the mRNA level in uterine arteries from Rgs2−/− mice and RGS2 and 4 in arteries from Rgs5−/− mice. In uterine arteries from Rgs5−/− mice, RGS2 mRNA levels remained unchanged and fairly low compared to RGS4 at all time points measured, except at PPD3, where the levels of RGS2 and 4 appeared similar (Figure 2C). In uterine arteries from Rgs2−/− mice, however, the expression profiles of RGS4 and RGS5 mRNA differed. Whereas RGS4 expression tended to increase at D10 and D15, RGS5 expression tended to decrease at D10 and D18 and returned to pre‐pregnancy level at D15 and PPD3 (Figure 2B). To determine whether the effect of pregnancy on mRNA expression of RGS2 was specific to the uterine vascular bed, we examined gene expression in the mesenteric vascular bed and aorta. In contrast to the uterine vascular bed expression profile, the expression of RGS2 and 4 tended to decrease at mid‐pregnancy, whereas RGS5 expression trended upward. In Rgs2−/− mesenteric arteries, RGS4 expression decreased slightly at D15 and PPD3, whereas the expression of RGS5 remained relatively unchanged (Figure 2E). In Rgs5−/− mesenteric arteries, however, RGS2 expression was unchanged at D15, but decreased markedly at PPD3, whereas RGS4 expression remained the same at all time points (Figure 2F). In the nonpregnant state, the expression of RGS5 was lower compared with RGS2 and 4, which had similar expression levels (Figure 2D through 2F). As shown in Figure 2G, mRNA expression of all 3 RGS proteins in the aorta remained unchanged at D15 and PPD3 relative to NP levels. In Rgs2−/− aorta, the expression of RGS5 was markedly reduced at D15 and tended to return to NP levels at PPD3; in contrast, RGS4 expression was unchanged (Figure 2H). In Rgs5−/− aorta, the expression of both RGS2 and 4 was suppressed at D15 and PPD3 (Figure 2I). We also noted that RGS4 expression was relatively low compared with RGS2 and 5 in the nonpregnant state (Figure 2G). These data, together, indicate that pregnancy increases mRNA expression of RGS2, and perhaps RGS4, at mid‐gestation, coinciding with the augmented uterine blood flow and decreased impedance. The data also indicate that, in the nonpregnant state, the absence of RGS2 induces a decrease in mRNA expression of both RGS4 and 5, whereas the loss of RGS5 triggers a compensatory increase in mRNA expression of RGS4, but has no effect on the expression of RGS2 in the uterine vasculature.
Figure 2.
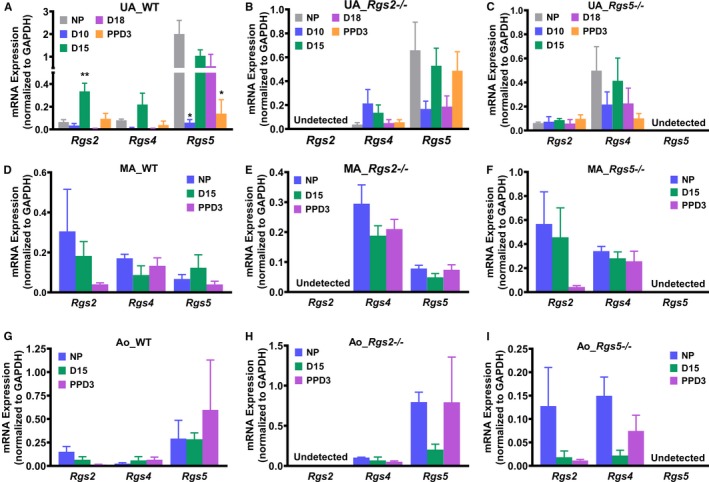
mRNA expression levels of RGS2 and RGS5 during pregnancy in wild‐type (WT) and knockout mouse uterine arteries, mesenteric arteries, and aorta. mRNA expression was determined from isolated uterine arteries, mesenteric arteries, and aorta by RT‐qPCR. A, mRNA expression of RGS2, RGS4, and RGS5 in uterine arteries from WT mice at nonpregnant (NP), gestation day 10 (D10), gestation day 15 (D15), gestation day 18 (D18), and postpartum day 3 (PPD3; RGS2: NP n=3, D10 n=5, D15 n=6, D18 n=3, and PPD3 n=5; RGS4: NP n=4, D10 n=4, D15 n=4, D18 n=4, and PPD3 n=3; RGS5: NP n=7, D10 n=4, D15 n=9, D18 n=5, and PPD3 n=4). B, mRNA expression of RGS2, RGS4, and RGS5 in uterine arteries from Rgs2−/− mice at NP, D10, D15, D18, and PPD3 (RGS4: NP n=4, D10 n=5, D15 n=4, D18 n=5, and PPD3 n=5; RGS5: NP n=6, D10 n=5, D15 n=4, D18 n=5, and PPD3 n=5). C, mRNA expression of RGS2, RGS4, and RGS5 in uterine arteries from Rgs5−/− mice at NP, D10, D15, D18, and PPD3 (RGS2: NP n=6, D10 n=3, D15 n=5, D18 n=5, and PPD3 n=10; RGS4: NP n=5, D10 n=3, D15 n=5, D18 n=5, and PPD3 n=5). D, mRNA expression of RGS2, RGS4, and RGS5 in mesenteric arteries from WT mice at NP, D15, and PPD3 (RGS2: NP n=3, D15 n=3, and PPD3 n=4; RGS4: NP n=4, D15 n=3, and PPD3 n=4; RGS5: n=4, D15 n=3, and PPD3 n=4). E, mRNA expression of RGS2, RGS4, and RGS5 in mesenteric arteries from Rgs2−/− mice at NP, D15, and PPD3 (RGS4: NP n=3, D15 n=3, and PPD3 n=3; RGS5: NP n=3, D15 n=3, and PPD3 n=3). F, mRNA expression of RGS2, RGS4, and RGS5 in mesenteric arteries from Rgs5−/− mice at NP, D15, and PPD3 (RGS2: NP n=3, D15 n=3, and PPD3 n=3; RGS4: NP n=3, D15 n=3, and PPD3 n=3). G, mRNA expression of RGS2, RGS4, and RGS5 in aorta from WT mice at NP, D15, and PPD3 (RGS2: NP n=3, D15 n=3, and PPD3 n=3; RGS4: NP n=3, D15 n=3, and PPD3 n=3; RGS5: NP n=3, D15 n=3, and PPD3 n=3). H, mRNA expression of RGS2, RGS4, and RGS5 in aorta from Rgs2−/− mice at NP, D15, and PPD3 (RGS4: NP n=3, D15 n=3, and PPD3 n=3; RGS5: NP n=3, D15 n=3, and PPD3 n=3). I, mRNA expression of RGS2, RGS4, and RGS5 in aorta from Rgs5−/− mice at NP, D15, and PPD3 (RGS2: NP n=3, D15 n=3, and PPD3 n=3; RGS4: NP n=3, D15 n=3, and PPD3 n=3). Values are mean±SE. *,**P<0.05, 0.01 vs NP.
Impaired Uterine Blood Flow in Rgs2−/− and Rgs5−/− Mice During Pregnancy Is Not Attributed to Gestational Hypertension
Because hypertension has been associated with augmented vascular tone, and because the absence of either RGS2 or RGS5 in mice is reported to cause hypertension, we tested whether impaired uterine blood flow in Rgs2−/− and Rgs5−/− mice was attributed to gestational hypertension. Baseline blood pressure, determined by carotid artery catheterization under anesthesia before pregnancy, at D10, D15, D18, and PPD3 was not significantly different between WT, Rgs2−/−, and Rgs5−/− mice at any time point (Figure 3). Similarly, heart rate was similar among all groups, except at D18, where heart rate was elevated in Rgs5−/− mice compared with WT mice (Figure 3D). This result led us to conclude that increased impedance to uterine blood flow in Rgs2−/− and Rgs5−/− mice was not attributed to chronic hypertension during pregnancy.
Figure 3.
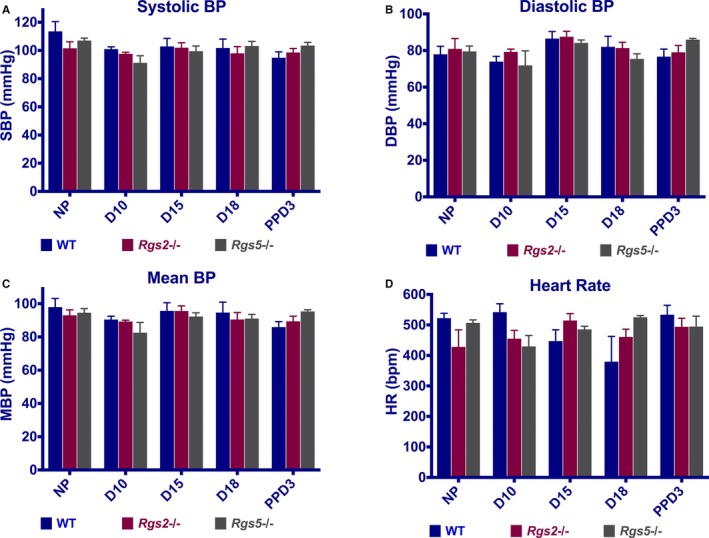
Systemic blood pressure of wild‐type (WT), Rgs2−/−, and Rgs5−/− mice before, during, and after pregnancy. Blood pressure (BP) was measured under isoflurane anesthesia by carotid artery catheterization. A, Systolic blood pressure (SBP), (B) diastolic blood pressure (DBP), (C) mean blood pressure (MBP), and (D) heart rate (HR) of WT, Rgs2−/−, and Rgs5−/− mice at nonpregnant (NP), gestation day 10 (D10), gestation day 15 (D15), gestation day 18 (D18), and postpartum day 3 (PPD3) stages (WT: NP n=5, D10 n=5, D15 n=5, D18 n=5, and PPD3 n=4; Rgs2−/−: NP n=4, D10 n=4, D15 n=6, D18 n=6, and PPD3 n=5; Rgs5−/−: NP n=5, D10 n=2, D15 n=3, D18 n=5, and PPD3 n=6). Values are mean±SE.
Decreased Uterine Blood Flow in Rgs2−/− Mice Is Accompanied by Increased Vascular Wall Thickening of the Uterine Artery at Mid‐Pregnancy
Physical properties of uterine arteries before pregnancy, at mid‐pregnancy, and postpartum period were examined to determine whether pregnancy‐induced structural remodeling was altered to contribute to decreased uterine blood flow in the absence of RGS2 or RGS5. As shown in Table 1, there was no difference in the physical properties of uterine arteries from WT, Rgs2−/−, and Rgs5−/− mice in the nonpregnant state. At mid‐pregnancy, there was a significant increase in lumen diameter of uterine arteries from WT and Rgs2−/−, but not Rgs5−/− mice. In addition, only Rgs2−/− uterine arteries showed a significant increase in vessel cross‐sectional area at mid‐pregnancy relative to the nonpregnant state. Moreover, whereas media/lumen ratio trended lower in WT and Rgs5−/− arteries, the ratio trended upward in Rgs2−/− arteries, suggesting a narrowing of effective luminal diameter at mid‐pregnancy. These data suggested that abnormal structural remodeling of the uterine vasculature, at least in the absence of RGS2, is involved in reduced uterine blood flow at mid‐pregnancy.
Table 1.
Morphometric Data of Pressurized Uterine Arteries From Rgs2−/−, Rgs5−/−, and WT Mice at 60 mm Hg in Physiological Saline Solution
| Genotype | WT | Rgs2−/− | Rgs5−/− | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Pregnancy day | NP | D15 | PPD3 | NP | D15 | PPD3 | NP | D15 | PPD3 |
| No. of animals | 6 | 6 | 9 | 8 | 6 | 6 | 4 | 6 | 6 |
| Average wall thickness, μm | 18.8±2.6 | 20.2±4.8 | 27.7±1.9 | 19.5±1.6 | 29.2±5.1 | 27.0±1.8 | 26.1±4.9 | 25.1±4.3 | 31.5±3.9 |
| Lumen diameter, μm | 193±9 | 254±22* | 289±16** | 197±6 | 277±15* | 242±27 | 211±11 | 252± 21 | 281±23 |
| Cross‐sectional area, ×1000 μm2 | 12.5±1.8 | 17.5±5.3 | 27.8±2.7* | 13.3±1.1 | 27.7±4.9** | 23.1±3.1 | 19.9± 4.4 | 22.8±5.2 | 30.7± 4.1 |
| Media/lumen ratio | 0.20±0.03 | 0.17±0.04 | 0.19±0.01 | 0.20±0.02 | 0.22±0.04 | 0.23±0.02 | 0.25±0.04 | 0.20±0.03 | 0.24±0.05 |
Values are mean±SE. D15 indicates gestation day 15; NP, nonpregnant; PPD3, postpartum day 3; WT, wild type.
*,**P<0.05, 0.01, NP vs D15 or PPD3.
Loss of RGS2 Sustains the Elevation of GPCR‐Evoked Vasoconstriction at Mid‐Pregnancy, Whereas Absence of RGS5 Augments Contractility of Uterine Arteries During Peripartum Period
Normal pregnancy induces refractoriness to contractile ligands, such as Ang II, norepinephrine, and ET‐1, but increases the level of, and sensitivity to, vasodilatory substances such as NO, endothelium‐derived hyperpolarizing factor, and prostaglandins to promote uterine blood flow and placental perfusion.10, 27 Because RGS2 and 5 potently regulate GPCR‐mediated contractile and vasodilatory responses to vasoactive ligands,28, 29, 30 we tested whether increased vasoconstriction attributed to the absence of either of these key B/R4 RGS proteins is involved in the augmented uterine blood flow impedance at mid‐pregnancy, noticeably in Rgs2−/− mice. Non‐G‐protein‐mediated constriction evoked with high‐potassium solution was unchanged between genotypes before, during, and after pregnancy within each genotype, with the exception of uterine arteries from WT mice, which had markedly decreased constriction at PPD3 compared with NP and D15 (Figure 4). Next, we examined vessel reactivity further using EFS to trigger the release of vasoactive substances, predominantly norepinephrine, from sympathetic nerve terminals innervating the uterine vasculature. In nonpregnant vessels, EFS‐induced constriction was augmented in uterine arteries from Rgs2−/− mice (Figure 5A). EFS‐induced constriction continued to trend higher in Rgs2−/− arteries at mid‐pregnancy, though there were no significant differences between genotypes (Figure 5B). At PPD3, uterine arteries from Rgs5−/− mice showed a robust increase in EFS‐induced constriction relative to the response in vessels from WT and Rgs2−/− mice (Figure 5C and 5F). In WT mice, uterine artery sensitivity at mid‐pregnancy appeared to be less sensitive to EFS, whereas such a trend was absent in vessels from Rgs2−/− and Rgs5−/− mice (Figure 5D through 5F). We then determined whether increased EFS‐induced constriction in Rgs2−/− arteries at NP and in Rgs5−/− arteries at PPD3 was attributed to augmented postsynaptic GPCR activity. As shown in Figure 6, uterine arteries from nonpregnant Rgs2−/− mice showed the highest contractile response to the α1‐adrenergic receptor agonist, PE (Figure 6A). Augmented constriction in Rgs2−/− arteries was sustained at mid‐pregnancy (Figure 6B). Similar to EFS‐induced constriction of uterine arteries at PPD3, constriction of uterine arteries from Rgs5−/− mice was markedly enhanced relative to vessels from WT and Rgs2−/− mice at this time point (Figure 6C). In the presence of the NO synthase inhibitor, Nω‐Nitro‐l‐arginine methyl ester, PE‐induced constriction during the nonpregnant state was augmented in WT and Rgs5−/−, but not in Rgs2−/− arteries (Figure 6G). At mid‐pregnancy, however, there was no difference in the contractile response to PE among the 3 groups (Figure 6H). At PPD3, uterine arteries from Rgs5−/− mice showed the strongest contractile response to PE, whereas the response was attenuated in WT and Rgs2−/− mice (Figure 6I).
Figure 4.
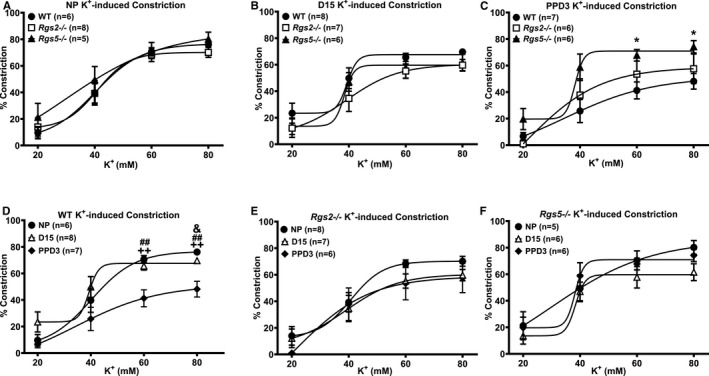
Contractile response of uterine arteries from wild‐type (WT), Rgs2−/−, and Rgs5−/− mice to high‐potassium (KCl) solution at nonpregnant (NP), gestation day 15 (D15), and postpartum day 3 (PPD3). A, Vasoconstriction of uterine arteries from WT, Rgs2−/−, and Rgs5−/− mice at NP expressed as percent decreases in vessel lumen diameter in response to KCl (WT n=6, Rgs2−/− n=8, and Rgs5−/− n=5 mice). B, Vasoconstriction of uterine arteries from WT, Rgs2−/−, and Rgs5−/− mice at D15 (WT n=8, Rgs2−/− n=7, and Rgs5−/− n=6 mice). C, Vasoconstriction of uterine arteries from WT, Rgs2−/−, and Rgs5−/− mice at PPD3 (WT n=7, Rgs2−/− n=6, and Rgs5−/− n=6 mice). D, Vasoconstriction of uterine arteries from WT mice at NP, D15, and PPD3 (NP n=6, D15 n=8, and PPD3 n=7 mice). E, Vasoconstriction of uterine arteries from Rgs2−/− mice at NP, D15, and PPD3 (NP n=8, D15 n=7, and PPD3 n=6 mice). F, Vasoconstriction of uterine arteries from Rgs5−/− mice at NP, D15, and PPD3 (NP n=5, D15 n=6, and PPD3 n=6 mice). Values are mean±SE. *P<0.05 WT vs Rgs5−/−; & P<0.05 NP vs D1; ## P<0.01 D15 vs PPD3; ++ P<0.01 NP vs PPD3.
Figure 5.
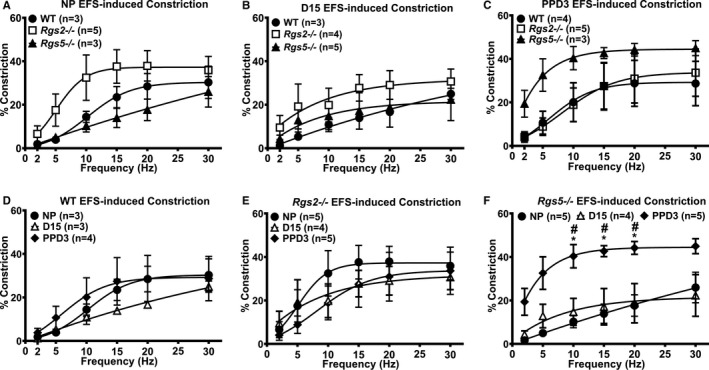
Electrical field stimulation (EFS) of uterine arteries from wild‐type (WT), Rgs2−/−, and Rgs5−/− mice at nonpregnant (NP), gestation day 15 (D15), and postpartum day 3 (PPD3). A, Contractile responses of WT, Rgs2−/−, and Rgs5−/− uterine arteries to EFS at NP (WT n=3, Rgs2−/− n=5, and Rgs5−/− n=3 mice). B, Contractile responses of WT, Rgs2−/−, and Rgs5−/− uterine arteries to EFS at D15 (WT n=4, Rgs2−/− n=5, and Rgs5−/− n=3 mice). C, Contractile responses of WT, Rgs2−/−, and Rgs5−/− uterine arteries at PPD3 (WT n=3, Rgs2−/− n=3, and Rgs5−/− n=4 mice). D, Contractile responses of WT uterine arteries at NP, D15, and PPD3 (NP n=3, D15 n=4, and PPD3 n=3 mice). E, Contractile responses of Rgs2−/− uterine arteries at NP, D15, and PPD3 (NP n=5, D15 n=4, and PPD3 n=4 mice). F, Contractile responses of Rgs5−/− uterine arteries at NP, D15, and PPD3 (NP n=3, D15 n=5, and PPD3 n=3 mice). Values are expressed as mean±SE of percent decreases in vessel lumen diameter. *P<0.05 NP vs PPD3; # P<0.05 D15 vs PPD3.
Figure 6.
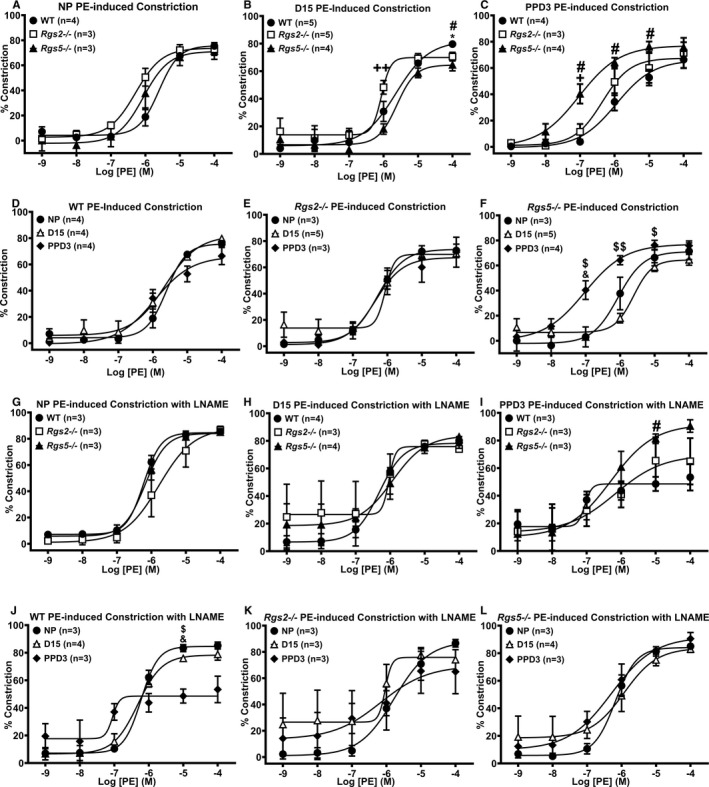
Contractile response of uterine arteries from wild‐type (WT), Rgs2−/−, and Rgs5−/− mice to phenylephrine (PE) in the absence (A through F) or presence (G through L) of Nω‐Nitro‐l‐arginine methyl ester (L‐NAME) at nonpregnant (NP), gestation day 15 (D15), and postpartum day 3 (PPD3). A and G, Contractile responses of WT, Rgs2−/−, and Rgs5−/− uterine arteries to PE at NP period (WT n=3, Rgs2−/− n=3, and Rgs5−/− n=4 mice). B and H, Contractile responses of WT, Rgs2−/−, and Rgs5−/− uterine arteries to PE at D15 (WT n=5, Rgs2−/− n=5, and Rgs5−/− n=4 mice). C and I, Contractile responses of WT, Rgs2−/−, and Rgs5−/− uterine arteries to PE at PPD3 (WT n=4, Rgs2−/− n=3, and Rgs5−/− n=4 mice). D and J, Contractile responses of WT uterine arteries at NP, D15, and PPD3 to PE (NP n=4, D15 n=4, and PPD3 n=4 mice). E and K, Contractile responses of Rgs2−/− uterine arteries at NP, D15, and PPD3 uterine arteries to PE (NP n=3, D15 n=5, and PPD3 n=3 mice). F and L, Contractile responses of Rgs5−/− uterine arteries at NP, D15, and PPD3 to PE (NP n=3, D15 n=5, and PPD3 n=4 mice). Values are expressed as mean±SE of percent decreases in vessel lumen diameter. *P<0.05 WT vs Rgs2−/−; # P<0.05 WT vs Rgs5−/−; +,++ P<0.05, 0.01 Rgs2−/− vs Rgs5−/−; $,$$ P<0.05, 0.01 D15 vs PPD3; & P<0.05 NP vs PPD3.
The Absence of RGS2 Augments Sensitivity to α‐Adrenergic Receptor Stimulation in the Resistance Vasculature at Mid‐Pregnancy
Global inactivation of the Rgs2 gene has been reported to prolong the contractile response evoked by the activation of Gq/11‐coupled GPCRs.31 Therefore, we determined whether augmented sensitivity to α‐adrenergic receptor stimulation at mid‐pregnancy was unique to the uterine vascular bed. As observed in uterine arteries, the absence of RGS2 or 5 did not affect G‐protein–independent constriction of mesenteric arteries stimulated with high‐potassium PSS before, during, or after pregnancy (Figure 7A through 7C). Similarly, PE‐induced constriction of mesenteric arteries from nonpregnant WT, Rgs2−/−, and Rgs5−/− mice was not different at NP (Figure 7D). However, mesenteric arteries from Rgs2−/− mice showed a robust increase in sensitivity to PE at mid‐pregnancy (Figure 7E and Table 2). At PPD3, sensitivity to PE appeared to increase (Table 2), whereas submaximal constriction was augmented in mesenteric arteries from Rgs2−/− and Rgs5−/− mice (Figure 7F and Table 2). Interestingly, EFS‐induced constriction of mesenteric arteries from Rgs5−/− mice was augmented at mid‐pregnancy (Figure 7H); however, it appeared to decrease in Rgs2−/− and Rgs5−/− mice at PPD3 (Figure 7I). Taken together, these data indicate that the loss of RGS2 increases sensitivity of small resistance arteries to α‐adrenergic receptor stimulation at mid‐pregnancy.
Figure 7.
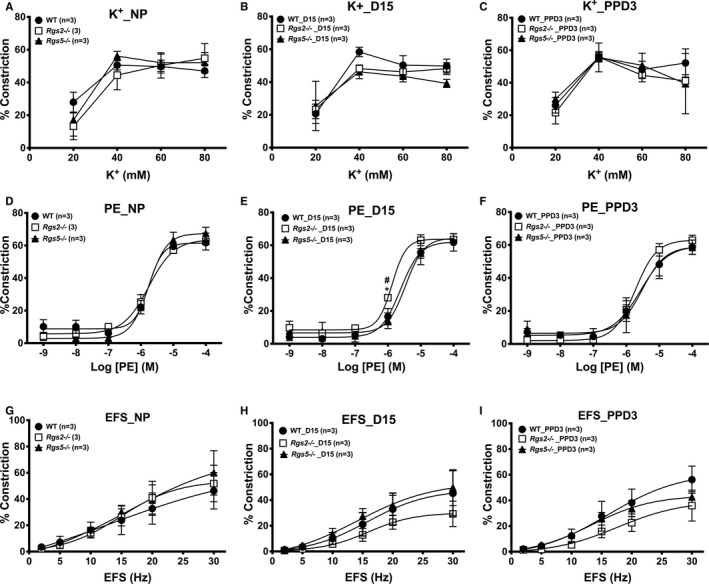
Contractile response of mesenteric arteries from wild type (WT), Rgs2−/−, and Rgs5−/− mice to high‐potassium (K+) solution, phenylephrine (PE), and electrical field stimulation (EFS) at nonpregnant (NP), gestation day 15 (D15), and postpartum day 3 (PPD3). A through C, Vasoconstriction of mesenteric arteries from WT, Rgs2−/−, and Rgs5−/− mice at NP (A), D15 (B), and PPD3 (C) expressed as percent decreases in vessel lumen diameter in response to high K+ (WT n=6, Rgs2−/− n=8, and Rgs5−/− n=8 mice). D through F, Vasoconstriction of mesenteric arteries from WT, Rgs2−/−, and Rgs5−/− mice at NP (D), D15 (E), and PPD3 (F) expressed as percent decreases in vessel lumen diameter in response to PE (WT n=7, Rgs2−/− n=8, and Rgs5−/− n=8 mice). G through I, Vasoconstriction of mesenteric arteries from WT, Rgs2−/−, and Rgs5−/− mice at NP (D), D15 (E), and PPD3 (F) expressed as percent decreases in vessel lumen diameter in response to EFS (WT n=5, Rgs2−/− n=6, and Rgs5−/− n=6 mice). Values are expressed as mean±SE of percent decreases in vessel lumen diameter. *P<0.05, WT vs Rgs2−/−; # P<0.05, Rgs2−/− vs Rgs5−/−.
Table 2.
Maximal Contractile Response and Potency (EC50) of Phenylephrine on Uterine and Mesenteric Arteries From WT, Rgs2−/−, and Rgs5−/− Mice at Nonpregnant, Mid‐Pregnant, and Peripartum States
| No. of Animals | Maximal Response | EC50 (μmol/L) | |
|---|---|---|---|
| NP | |||
| UA | |||
| WT | 4 | 75.86±2.70 | 2.60±0.66 |
| Rgs2−/− | 3 | 73.10±4.19 | 0.63±0.13 |
| Rgs5−/− | 3 | 72.32±7.15 | 0.96±0.32 |
| MA | |||
| WT | 3 | 61.43±1.58 | 1.55±0.34 |
| Rgs2−/− | 3 | 63.91±6.60 | 1.90±0.64 |
| Rgs5−/− | 3 | 67.39±3.50 | 1.74±0.09 |
| D15 | |||
| UA | |||
| WT | 5 | 84.33±0.67 | 2.37±0.55 |
| Rgs2−/− | 5 | 70.98±3.24* | 0.75±0.21* |
| Rgs5−/− | 4 | 63.44±4.30** | 2.38±0.48 |
| MA | |||
| WT | 3 | 62.15±6.27 | 3.55±0.87 |
| Rgs2−/− | 3 | 64.65±1.77 | 1.47±0.27* |
| Rgs5−/− | 3 | 67.12±5.99 | 4.42±1.78 |
| PPD3 | |||
| UA | |||
| WT | 4 | 61.70±6.66 | 0.71±0.20 |
| Rgs2−/− | 3 | 75.14±11.11 | 0.95±0.48 |
| Rgs5−/− | 4 | 79.34±4.34 | 0.15±0.09 |
| MA | |||
| WT | 3 | 60.79±7.29 | 4.52±0.86 |
| Rgs2−/− | 3 | 64.14±2.02 | 1.84±0.08* |
| Rgs5−/− | 3 | 62.96±2.83 | 1.47±0.44* |
Values are mean±SE. D15 indicates gestation day 15; MA, mesenteric arteries; NP, nonpregnant; PPD3, postpartum day 3; UA, uterine arteries; WT, wild type.
*,**P<0.05, 0.01, WT vs Rgs2−/− or Rgs5−/−.
The Absence of RGS2 or 5 Does Not Alter Sensitivity or Maximal Contractile Response of Uterine Arteries to Ang II or ET‐1 During Pregnancy
To test whether the absence of RGS2 or RGS5 generally augments G‐protein–dependent vasoconstriction in uterine arteries during pregnancy, we examined contractile response to other vasoactive agents. In NP and D15 mice, Ang II–induced constriction was similar among WT, Rgs2−/−, and Rgs5−/− uterine arteries (Figure 8A and 8B). However, Ang II–induced constriction was markedly increased in Rgs2−/− and Rgs5−/− uterine arteries at PPD3 (Figure 8C); when compared with constriction at NP and D15 within each group, the contractile response remained elevated at PPD3 in Rgs2−/− and Rgs5−/− uterine arteries, indicating that the increase was not attributed to apparent decrease in WT uterine artery constriction (Figure 8D through 8F). Contractile response to ET‐1 was slightly decreased in Rgs5−/− arteries at NP (Figure 9A). However, sensitivity of Rgs2−/− arteries to ET‐1 was markedly decreased at D15 relative to WT and Rgs5−/− (Figure 9B), or to the NP and PPD3 states (Figure 9B and 9E). In contrast to Ang II–induced constriction, contractile response to ET‐1 was decreased in WT uterine arteries at PPD3 (Figure 9D), whereas the response in Rgs2−/− and Rgs5−/− uterine arteries appeared to increase relative to WT (Figure 9C).
Figure 8.
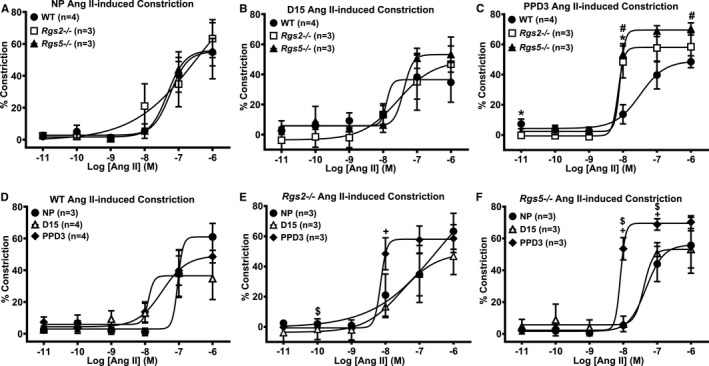
Contractile response of uterine arteries from wild‐type (WT), Rgs2−/−, and Rgs5−/− mice to angiotensin (ANG) II at nonpregnant (NP), gestation day 15 (D15), and postpartum day 3 (PPD3). A, Contractile responses of WT, Rgs2−/−, and Rgs5−/− uterine arteries to ANG II at NP period (WT n=4, Rgs2−/− n=3, and Rgs5−/− n=3 mice). B, Contractile responses of WT, Rgs2−/−, and Rgs5−/− uterine arteries to ANG II at D15 (WT n=4, Rgs2−/− n=3, and Rgs5−/− n=3 mice). C, Contractile responses of WT, Rgs2−/−, and Rgs5−/− uterine arteries to ANG II at PPD3 (WT n=4, Rgs2−/− n=3, and Rgs5−/− n=3 mice). D, Vasoconstrictor responses of WT uterine arteries at NP, D15, and PPD3 to ANG II (NP n=3, D15 n=4, and PPD3 n=4 mice). E, Contractile responses of Rgs2−/− uterine arteries at NP, D15, and PPD3 uterine arteries to ANG II (NP n=3, D15 n=3, and PPD3 n=3 mice). F, Vasoconstrictor responses of Rgs5−/− uterine arteries at NP, D15, and PPD3 to ANG II (NP n=3, D15 n=3, and PPD3 n=3 mice). Values are expressed as mean±SE of percent decreases in vessel lumen diameter. Data were analyzed using the nonparametric Mann–Whitney U test. *P<0.05 WT vs Rgs2−/−; # P<0.05 WT vs Rgs5−/−; $ P<0.05 NP vs PPD3; + P<0.05 D15 vs PPD3.
Figure 9.
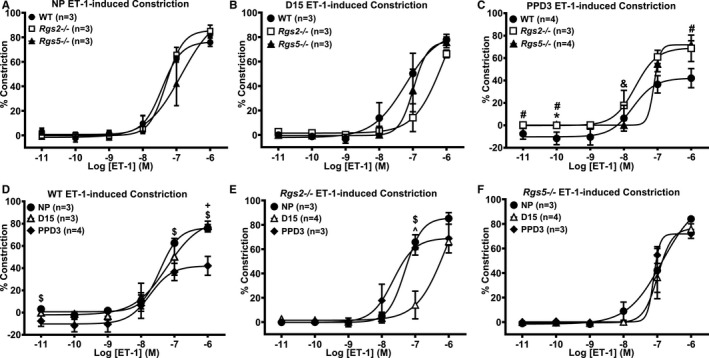
Contractile response of uterine arteries from wild‐type (WT), Rgs2−/−, and Rgs5−/− mice to endothelin (ET)‐1 at nonpregnant (NP), gestation day 15 (D15), and postpartum day 3 (PPD3). A, Contractile responses of WT, Rgs2−/−, and Rgs5−/− uterine arteries to ET‐1 at NP period (WT n=3, Rgs2−/− n=3, and Rgs5−/− n=3 mice). B, Contractile responses of WT, Rgs2−/−, and Rgs5−/− uterine arteries to ET‐1 at D15 (WT n=3, Rgs2−/− n=4, and Rgs5−/− n=3 mice). C, Contractile responses of WT, Rgs2−/−, and Rgs5−/− uterine arteries to ET‐1 at PPD3 (WT n=4, Rgs2−/− n=3, and Rgs5−/− n=4 mice). D, Contractile responses of WT uterine arteries at NP, D15, and PPD3 to ET‐1 (NP n=3, D15 n=3, and PPD3 n=4 mice). E, Contractile responses of Rgs2−/− uterine arteries at NP, D15, and PPD3 uterine arteries to ET‐1 (NP n=4, D15 n=3, and PPD3 n=3 mice). F, Contractile responses of Rgs5−/− uterine arteries at NP, D15, and PPD3 to ET‐1 (NP n=3, D15 n=3, and PPD3 n=4 mice). Values are expressed as mean±SE of percent decreases in vessel lumen diameter. Data were analyzed using the nonparametric Mann–Whitney U test. *P<0.05 WT vs Rgs2−/−; # P<0.05 WT vs Rgs5−/−; & P<0.05 Rgs2−/− vs Rgs5−/−; ^ P<0.05 NP vs D15; $ P<0.05 NP vs PPD3; + P<0.05 D15 vs PPD3.
The Absence of RGS2 Increases Endothelium‐Dependent Vasodilatory Response in Uterine Arteries During Pregnancy
Last, we assessed effects of the absence of RGS2 or 5 on GPCR‐induced vasodilatation during pregnancy, in preconstricted uterine arteries from WT, Rgs2−/−, and Rgs5−/− mice stimulated with ACh in the absence or presence of Nω‐Nitro‐l‐arginine methyl ester to induce endothelium‐dependent and endothelium‐independent vasodilation, respectively. In the nonpregnant and postpartum states, endothelium‐dependent vasodilation was similar among groups (Figure 10A and 10C). However, Rgs2−/− vessels were more sensitive to ACh at mid‐pregnancy (Figure 10B). Increased sensitivity to ACh was abolished by the presence of Nω‐Nitro‐l‐arginine methyl ester; however, under this condition, vasodilation was augmented in WT vessels at mid‐pregnancy (Figure 10H). In mesenteric arteries, ACh‐induced vasodilatation trended lower in vessels from Rgs2−/− mice in the nonpregnant state (Figure 11A). At mid‐pregnancy, there was a leftward shift in ACh‐evoked vasodilatory response in vessels from Rgs2−/− and Rgs5−/− mice; however, such trends disappeared at PPD3 (Figure 11B versus 11C). These results together suggested a compensatory increase in vasodilatation of uterine and mesenteric arteries at mid‐pregnancy in the absence of RGS2, at least partly, through the NO‐dependent mechanism.
Figure 10.
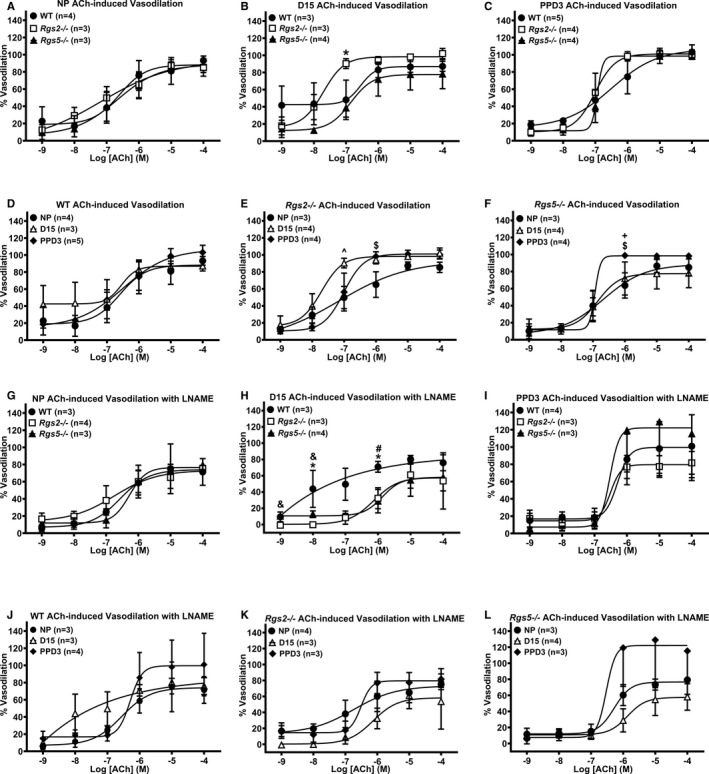
Vasodilatory response of uterine arteries from wild‐type (WT), Rgs2−/−, and Rgs5−/− mice to acetylcholine (ACh) in the absence (A through F) or presence (G through L) of Nω‐Nitro‐L‐arginine methyl ester (L‐NAME) at nonpregnant (NP), gestation day 15 (D15), and postpartum day 3 (PPD3). All the vessels were preconstricted with 10 μmol/L of phenylephrine before the application of increasing concentrations of ACh. A and G, Vasodilatory responses of WT, Rgs2−/−, and Rgs5−/− uterine arteries to ACh at NP period (WT n=4, Rgs2−/− n=3, and Rgs5−/− n=3 mice). B and H, Vasodilatory responses of WT, Rgs2−/−, and Rgs5−/− uterine arteries to ACh at D15 (WT n=3, Rgs2−/− n=3, and Rgs5−/− n=4 mice). C and I, Vasodilatory responses of WT, Rgs2−/−, and Rgs5−/− uterine arteries to ACh at PPD3 (WT n=5, Rgs2−/− n=3, and Rgs5−/− n=4 mice). D and J, Vasodilatory responses of WT uterine arteries at NP, D15, and PPD3 to ACh (NP n=3, D15 n=5, and PPD3 n=4 mice). E and K, Vasodilatory responses of Rgs2−/− uterine arteries at NP, D15, and PPD3 uterine arteries to ACh (NP n=4, D15 n=3, and PPD3 n=3 mice). F and L, Vasodilatory responses of Rgs5−/− uterine arteries at NP, D15, and PPD3 to ACh (NP n=3, D15 n=4, and PPD3 n=4 mice). Values are expressed as mean±SE of percent increases in vessel lumen diameter. Data were analyzed using the nonparametric Mann–Whitney U test. *P<0.05 WT vs Rgs2−/−; # P<0.05 WT vs Rgs5−/−; & P<0.05 Rgs2−/− vs Rgs5−/−; ^ P<0.05 NP vs D15; $ P<0.05 NP vs PPD3; + P<0.05 D15 vs PPD3.
Figure 11.
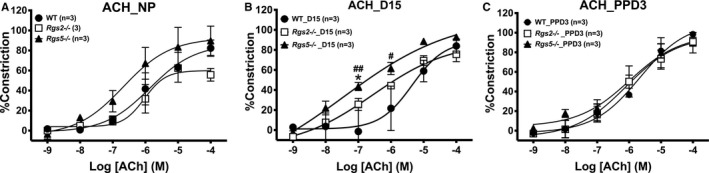
Vasodilatory response of mesenteric arteries from wild‐type (WT), Rgs2−/−, and Rgs5−/− mice to acetylcholine (ACh) at nonpregnant (NP), gestation day 15 (D15), and postpartum day 3 (PPD3). All vessels were preconstricted with 10 μmol/L of phenylephrine before the application of increasing concentrations of ACh. A through C, Vasodilatory responses of WT, Rgs2−/−, and Rgs5−/− uterine arteries to ACh at NP (A), D15 (B), and PPD3 (C) period (WT n=6, Rgs2−/− n=6, and Rgs5−/− n=6 mice). Values are expressed as mean±SE of percent increases in vessel lumen diameter. *P<0.05 Rgs5−/− vs Rgs2−/−; #,## P<0.05, 0.01 WT vs Rgs5−/− NP vs D15.
Discussion
Dysregulation of G protein signaling is implicated in the pathogenesis of cardiovascular disorders during pregnancy. In this study, we define the role of RGS2 and 5 in the uterine vascular bed and how their presence is critical to proper structural and functional adaptation of the uterine artery during various stages of pregnancy and the peripartum period. In normal pregnancy, the uterine vasculature undergoes a number of adaptive changes that together ensure that the placenta is well perfused by the maternal circulation for delivery of oxygen and nutrients. The adaptive changes include desensitization of uterine vessels to endogenous vasoactive agents that can decrease vessel diameter, and hence blood flow, by evoking vasoconstriction.1, 2 Many of these endogenous vasoactive substances, such as Ang II and norepinephrine, elicit their contractile effects by activating Gq/11‐ and Gi/o‐coupled receptors in the medial layer of the uterine artery. The level of these endogenous substances increases because of pregnancy‐induced activation of many hormonal systems, such as the renin‐angiotensin‐aldosterone system.32, 33, 34 Thus, increases in the activity and/or expression of the signaling molecules that serve as brakes in the contractile signaling pathway are likely a component of the functional adaptation rendering the uterine artery less sensitive to contractile agents during pregnancy. By acting as GAPs, RGS2 and RGS5 proteins serve as brakes for vascular Gq/11 and Gi/o protein signaling involved in vasoconstriction. Indeed, our previous study demonstrated that mice lacking RGS2 show impaired uterine blood flow accompanied by augmented myogenic tone in the nonpregnant state, and that these are attributed, at least partly, to increased signaling by Gq/11 and Gi/o class G proteins.18 The results in this study indicate that impaired uterine blood flow attributed to increased impedance in the absence of RGS2 persists during pregnancy and is notably evident in mid‐pregnancy. As would be expected in normal pregnancy, impedance to uterine blood flow decreases to facilitate a rise in utero‐placental perfusion in pregnant WT mice.2 In contrast, uterine blood flow remains low with elevated impedance in Rgs2−/− mice and, to some extent, also in Rgs5−/− mice. These findings suggest that, perhaps, a more stringent control of vascular G protein signaling by RGS proteins is part of the adaptive mechanisms that facilitate increased uterine perfusion and hence increased maternal blood flow to the placenta during pregnancy. Several lines of evidence from this study lend support to this hypothesis. First, in WT mice, vascular expression of RGS2 and RGS4 at the mRNA level robustly increases several‐fold above pre‐pregnancy levels by mid‐pregnancy. Second, the expression of RGS2 or RGS4 at mid‐pregnancy is maintained at pre‐pregnancy levels in the absence of RGS5, and, similarly, mRNA expression of RGS4 and 5 remains unchanged at mid‐pregnancy in the absence of RGS2. These findings suggest a potential compensatory mechanism, at least at mRNA expression level. These changes in gene expression profiles appear to be unique to the uterine vascular bed, given that they were not observed in vessels of similar or higher caliber from different vascular beds. However, such compensatory mechanisms could not depend entirely on gene expression, given that RGS5 expression in the nonpregnant state and at mid‐pregnancy is more than 30‐fold higher than RGS2 and RGS4 in the uterine vascular bed. Third, vascular medial cross‐sectional area increases in uterine arteries from Rgs2−/− mice, thereby decreasing the lumen‐to‐media ratio. This could be attributed to smooth muscle proliferation resulting from hyperactivation of mitogenic GPCR signaling in the absence of RGS2, as previously reported.35, 36
Several studies have indicated that gestational hypertension is a potential cause of decreased uterine blood flow during pregnancy.37, 38, 39, 40 High blood pressure increases transmural pressure that, in turn, causes an increase in myogenic tone because of stretch‐evoked smooth muscle constriction.2, 41 Such an action causes a decrease in lumen diameter, thereby decreasing placental perfusion by the maternal circulatory system.18, 42 Because the loss of RGS2 (and RGS5 in 1 study) elevates blood pressure in the nonpregnant state,28, 31 we presumed that decreases in uterine blood flow in female Rgs2−/− mice during pregnancy, was likely attributed to gestational hypertension. However, both Rgs2−/− and Rgs5−/− mice are normotensive, and Rgs5−/− mice actually show a mild decrease in systolic blood pressure at mid‐pregnancy. This suggests a couple of pathophysiological mechanisms that possibly mediate impaired uterine blood flow in Rgs2−/− mice during pregnancy: The mechanism could be intrinsic to the uterine vascular bed and/or activated by some yet unknown factor(s) acting on this vascular bed during pregnancy. The uterine artery‐intrinsic mechanism is supported by our previous study, in which we showed a decrease in uterine blood flow accompanied by augmented myogenic tone in uterine arteries from both Rgs2+/− and Rgs2−/− mice.18 The extrinsic factor hypothesis is supported by the vascular reactivity data shown in this study. Specifically, uterine arteries from both knockout mice are more sensitive to direct or indirect α‐adrenergic receptor stimulation before pregnancy, as previously reported.31, 43 In addition, the enhanced constriction is sustained in uterine arteries from Rgs2−/− mice at mid‐pregnancy, and in Rgs5−/− mice during the peripartum period. Because adrenergic receptor‐mediated constriction is dependent on the activation of Gq/11 class G proteins, this finding suggests that Gq/11 regulation by RGS2 is more critical to reducing uterine vascular sensitivity to contractile vasoactive agents and is part of the adaptive remodeling of the uterine vascular bed to facilitate uteroplacental blood flow during pregnancy. The observation that increased RGS4 expression and restoration of RGS5 expression to pre‐pregnancy level at mid‐pregnancy in Rgs2−/− uterine arteries were not sufficient to reduce α‐adrenergic receptor‐mediated constriction is an additional line of evidence for a separate role for RGS2 and 5 during pregnancy. Indeed, our results further indicate that the absence of RGS2 also causes a general increase in sensitivity of resistance arteries to contractile adrenergic agonists. Moreover, it is noteworthy that inhibition of NO synthase elevates constriction in WT and Rgs5−/− arteries to the same level as Rgs2−/− arteries. This suggests that augmented constriction of uterine arteries from Rgs2−/− mice is partly attributed to endothelial dysfunction, which is known to be a consequence of the absence of RGS2 in the vasculature.43 In contrast to α‐adrenergic receptor agonists, sensitivity to ET‐1 is rather decreased in Rgs2−/− and Rgs5−/− arteries and is more pronounced in Rgs5−/− relative to WT arteries at mid‐pregnancy. This could be attributed to enhanced vasodilatory action of ET‐1 in the vascular endothelium that may be unique to the absence of RGS5 facilitating G‐protein–mediated production of endothelium‐derived relaxing factors during pregnancy. Contrary to ET‐1 and PE, increased uterine artery sensitivity to Ang II stimulation is observed in Rgs2−/− and Rgs5−/− mice only during the postpartum period, suggesting that the mechanisms mediating the insensitivity of the uterine vasculature to angiotensin receptor stimulation during pregnancy is not affected by the absence of either RGS proteins. However, just as for adrenergic receptor stimulation during the peripartum period, the absence of either RGS2 or 5 also markedly augments sensitivity to Ang II in this period. Because the G proteins that are controlled by RGS2 and 5 are less regulated in knockout mice, this augmented constriction in the peripartum period could be attributed to exacerbation of receptor supersensitivity resulting from a decline in the level of several hormones, including Ang II down to pre‐pregnancy levels. Such impaired reversal of vascular adaptation to pregnancy could be indicative of future cardiovascular disorders or the development of complications in subsequent pregnancies, though this hypothesis was not tested in the current study.
A hallmark of vascular adaptation to facilitate uteroplacental blood flow during pregnancy is the increase in the production of endothelium‐derived relaxing factors and increased sensitivity of the circulatory system to these endogenous agents.1, 2 This mechanism is not affected by the absence of either RGS2 or 5. Indeed, the absence of RGS2 appears to augment endothelium‐dependent vasodilation in both the uterine and mesenteric vascular beds at mid‐pregnancy, which is contrary to a previous study showing that RGS2 deficiency in the vasculature impairs acetylcholine‐induced, endothelium‐derived hyperpolarizing factor–dependent vasodilatation.43 However, augmented vasodilation in Rgs2−/− mice during pregnancy is largely mediated by NO, because it is abolished by inhibition of NO synthase. Because the absence of RGS2 increases vascular constriction and decreases uterine blood flow at mid‐pregnancy, augmented vasodilation could be a compensatory response, albeit inadequate to counter the increased impedance to blood flow, but sufficient to prevent the elevation of systemic blood pressure resulting from increased contractility of the resistance vasculature.
Several findings in this study contradict those in a recent report about the role of RGS5 in pregnancy.16 First, contrary to the extreme hypertensive phenotype previously reported, the female Rgs5−/− mouse model used in this study is normotensive and does not develop hypertension during pregnancy. Second, there is no effect of the absence of RGS2 or 5 on litter size or body weight of the newborns. Third, expression of RGS5 decreases throughout pregnancy and the peripartum state, whereas vascular reactivity of uterine arteries from Rgs5−/− mice is augmented in the nonpregnant state. The basis for these differences is unclear, though 2 independent groups created the same model, both targeting exon 1 of the Rgs5 gene.20, 28 That genetic background can mask or enhance (patho)‐physiological phenotype is well established.44, 45, 46, 47, 48 Thus, it is likely that the predicted cardiovascular phenotypes resulting from the loss of RGS5 are dampened by a compensatory mechanism involving increased expression and/or function of RGS2, and that the activation of such a compensatory mechanism may depend on the genetic background of the knockout mouse model.
Our study adds to accumulating evidence of the critical role that G protein signaling regulation by RGS proteins plays in the physiological adaptation of the circulatory system in normal pregnancy. The loss of RGS5 is implicated in pregnancy complications, including gestational hypertension and preeclampsia,16 whereas certain polymorphisms in the Rgs2 gene are also associated with preeclampsia and the development of chronic hypertension following preeclampsia in overweight women.15, 49, 50 In this study, we have shown how the expression and function of RGS2, 4, and 5 change in the course of pregnancy and during the peripartum period. More importantly, we establish decreased uterine blood flow, which is yet another serious pregnancy complication that can result from a deficiency in, or the absence of, RGS2 or 5, without the hallmark gestational hypertension. In addition, our findings show how the function of these RGS proteins is intrinsic to the uterine vascular bed, and that their absence, individually and at a specific period in the reproductive cycle, is causally involved in uterine vascular dysfunction. Further studies of G protein signaling regulation by RGS proteins is critical to fully unravel the etiology of pregnancy complications and the ensuing cardiovascular and metabolic disorders. Acquiring such knowledge is imperative in the effort to develop novel therapies for pregnancy‐related disorders.
Author contributions
PO‐O conceived and designed the study; JNK, SAD, EAO, and PO‐O performed experiments and analyzed data; PO‐O and JNK drafted, edited, and revised the manuscript. PO‐O approved the final version of the manuscript.
Sources of Funding
The study is funded, in part, by grants from the American Heart Association (16SDG27260276), NIH‐NHLBI (R01 HL139754), Pennsylvania Department of Health (CURE), and Margaret Q. Landenberger Foundation to Patrick Osei‐Owusu. The funding agencies had no role in the study design or execution.
Disclosures
None.
Acknowledgments
We are grateful to Dr John Kehrl at the NIH‐NIAID for generously providing the Rgs5−/− mice. We thank all members of the Osei‐Owusu laboratory for technical assistance and for comments on the manuscript.
(J Am Heart Assoc. 2019;8:e010917 DOI: 10.1161/JAHA.118.010917.)
Parts of this study were presented as an abstract at the American Heart Association Council on Hypertension Scientific Sessions, September 14 to 17, 2017, in San Francisco, CA.
References
- 1. Thaler I, Manor D, Itskovitz J, Rottem S, Levit N, Timor‐Tritsch I, Brandes JM. Changes in uterine blood flow during human pregnancy. Am J Obstet Gynecol. 1990;162:121–125. [DOI] [PubMed] [Google Scholar]
- 2. Osol G, Mandala M. Maternal uterine vascular remodeling during pregnancy. Physiology. 2009;24:58–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Steer CV, Tan SL, Dillon D, Mason BA, Campbell S. Vaginal color Doppler assessment of uterine artery impedance correlates with immunohistochemical markers of endometrial receptivity required for the implantation of an embryo. Fertil Steril. 1995;63:101–108. [DOI] [PubMed] [Google Scholar]
- 4. Salle B, Bied‐Damon V, Benchaib M, Desperes S, Gaucherand P, Rudigoz RC. Preliminary report of an ultrasonography and colour Doppler uterine score to predict uterine receptivity in an in‐vitro fertilization programme. Hum Reprod. 1998;13:1669–1673. [DOI] [PubMed] [Google Scholar]
- 5. Cacciatore B, Simberg N, Fusaro P, Tiitinen A. Transvaginal Doppler study of uterine artery blood flow in in vitro fertilization‐embryo transfer cycles. Fertil Steril. 1996;66:130–134. [DOI] [PubMed] [Google Scholar]
- 6. Mansour GM, Hussein SH, Abd El Hady RM, Mohammed HF, Abd El Gawad MM, Abou Gabal AI, Al‐Awadhy RM, Saied ME. Uterine artery flow velocity waveform (FVW) type and subednometrial vascularity in recurrent pregnancy loss. J Matern Fetal Neonatal Med. 2018;6 (issue 1‐6). doi: 10.1080/14767058.2018.1495190. [DOI] [PubMed] [Google Scholar]
- 7. Gallo DM, Poon LC, Akolekar R, Syngelaki A, Nicolaides KH. Prediction of preeclampsia by uterine artery Doppler at 20‐24 weeks’ gestation. Fetal Diagn Ther. 2013;34:241–247. [DOI] [PubMed] [Google Scholar]
- 8. Pedroso MA, Palmer KR, Hodges RJ, Costa FD, Rolnik DL. Uterine artery Doppler in screening for preeclampsia and fetal growth restriction. Rev Bras Ginecol Obstet. 2018;40:287–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rolnik DL, da Silva Costa F, Sahota D, Hyett J, McLennan A. Quality assessment of uterine artery Doppler measurement in first trimester combined screening for pre‐eclampsia. Ultrasound Obstet Gynecol. 2019;53:245–250. [DOI] [PubMed] [Google Scholar]
- 10. Chu ZM, Beilin LJ. Nitric oxide‐mediated changes in vascular reactivity in pregnancy in spontaneously hypertensive rats. Br J Pharmacol. 1993;110:1184–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Conrad KP, Morganelli PM, Brinck‐Johnsen T, Colpoys MC. The renin‐angiotensin system during pregnancy in chronically instrumented, conscious rats. Am J Obstet Gynecol. 1989;161:1065–1072. [DOI] [PubMed] [Google Scholar]
- 12. Watson N, Linder ME, Druey KM, Kehrl JH, Blumer KJ. RGS family members: GTPase‐activating proteins for heterotrimeric G‐protein alpha‐subunits. Nature. 1996;383:172–175. [DOI] [PubMed] [Google Scholar]
- 13. Chidiac P, Roy AA. Activity, regulation, and intracellular localization of RGS proteins. Receptors Channels. 2003;9:135–147. [PubMed] [Google Scholar]
- 14. Heximer SP, Watson N, Linder ME, Blumer KJ, Hepler JR. RGS2/G0S8 is a selective inhibitor of Gqalpha function. Proc Natl Acad Sci USA. 1997;94:14389–14393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kvehaugen AS, Melien O, Holmen OL, Laivuori H, Oian P, Andersgaard AB, Dechend R, Staff AC. Single nucleotide polymorphisms in G protein signaling pathway genes in preeclampsia. Hypertension. 2013;61:655–661. [DOI] [PubMed] [Google Scholar]
- 16. Holobotovskyy V, Chong YS, Burchell J, He B, Phillips M, Leader L, Murphy TV, Sandow SL, McKitrick DJ, Charles AK, Tare M, Arnolda LF, Ganss R. Regulator of G protein signaling 5 is a determinant of gestational hypertension and preeclampsia. Sci Transl Med. 2015;7:290ra88. [DOI] [PubMed] [Google Scholar]
- 17. Kvehaugen AS, Melien O, Holmen OL, Laivuori H, Dechend R, Staff AC. Hypertension after preeclampsia and relation to the C1114G polymorphism (rs4606) in RGS2: data from the Norwegian HUNT2 study. BMC Med Genet. 2014;15:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jie L, Owens EA, Plante LA, Fang Z, Rensing DT, Moeller KD, Osei‐Owusu P. RGS2 squelches vascular Gi/o and Gq signaling to modulate myogenic tone and promote uterine blood flow. Physiol Rep. 2016;4:e12692–e12704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Oliveira‐Dos‐Santos AJ, Matsumoto G, Snow BE, Bai D, Houston FP, Whishaw IQ, Mariathasan S, Sasaki T, Wakeham A, Ohashi PS, Roder JC, Barnes CA, Siderovski DP, Penninger JM. Regulation of T cell activation, anxiety, and male aggression by RGS2. Proc Natl Acad Sci USA. 2000;97:12272–12277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cho H, Park C, Hwang IY, Han SB, Schimel D, Despres D, Kehrl JH. Rgs5 targeting leads to chronic low blood pressure and a lean body habitus. Mol Cell Biol. 2008;28:2590–2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hernandez‐Andrade E, Ahn H, Szalai G, Korzeniewski SJ, Wang B, King M, Chaiworapongsa T, Than NG, Romero R. Evaluation of utero‐placental and fetal hemodynamic parameters throughout gestation in pregnant mice using high‐frequency ultrasound. Ultrasound Med Biol. 2014;40:351–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Osei‐Owusu P, Owens EA, Jie L, Reis JS, Forrester SJ, Kawai T, Eguchi S, Singh H, Blumer KJ. Regulation of renal hemodynamics and function by RGS2. PLoS One. 2015;10:e0132594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Osei‐Owusu P, Knutsen RH, Kozel BA, Dietrich HH, Blumer KJ, Mecham RP. Altered reactivity of resistance vasculature contributes to hypertension in elastin insufficiency. Am J Physiol Heart Circ Physiol. 2014;306:H654–H666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Owens EA, Jie L, Reyes BAS, Van Bockstaele EJ, Osei‐Owusu P. Elastin insufficiency causes hypertension, structural defects and abnormal remodeling of renal vascular signaling. Kidney Int. 2017;92:1100–1118. [DOI] [PubMed] [Google Scholar]
- 25. Sweazea KL, Walker BR. Impaired myogenic tone in mesenteric arteries from overweight rats. Nutr Metab (Lond). 2012;9:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Heximer SP, Blumer KJ. RGS proteins: Swiss army knives in seven‐transmembrane domain receptor signaling networks. Sci STKE. 2007;2007:pe2. [DOI] [PubMed] [Google Scholar]
- 27. Bird IM, Sullivan JA, Di T, Cale JM, Zhang L, Zheng J, Magness RR. Pregnancy‐dependent changes in cell signaling underlie changes in differential control of vasodilator production in uterine artery endothelial cells. Endocrinology. 2000;141:1107–1117. [DOI] [PubMed] [Google Scholar]
- 28. Holobotovskyy V, Manzur M, Tare M, Burchell J, Bolitho E, Viola H, Hool LC, Arnolda LF, McKitrick DJ, Ganss R. Regulator of G‐protein signaling 5 controls blood pressure homeostasis and vessel wall remodeling. Circ Res. 2013;112:781–791. [DOI] [PubMed] [Google Scholar]
- 29. Ganss R. Keeping the balance right: regulator of G protein signaling 5 in vascular physiology and pathology. Prog Mol Biol Transl Sci. 2015;133:93–121. [DOI] [PubMed] [Google Scholar]
- 30. Osei‐Owusu P, Blumer KJ. Regulator of G protein signaling 2: a versatile regulator of vascular function. Prog Mol Biol Transl Sci. 2015;133:77–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Heximer SP, Knutsen RH, Sun X, Kaltenbronn KM, Rhee MH, Peng N, Oliveira‐dos‐Santos A, Penninger JM, Muslin AJ, Steinberg TH, Wyss JM, Mecham RP, Blumer KJ. Hypertension and prolonged vasoconstrictor signaling in RGS2‐deficient mice. J Clin Invest. 2003;111:445–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Skinner SL, Lumbers ER, Symonds EM. Analysis of changes in the renin‐angiotensin system during pregnancy. Clin Sci. 1972;42:479–488. [DOI] [PubMed] [Google Scholar]
- 33. Alhenc‐Gelas F, Tache A, Saint‐Andre JP, Milliez J, Sureau C, Corvol P, Menard J. The renin‐angiotensin system in pregnancy and parturition. Adv Nephrol Necker Hosp. 1986;15:25–33. [PubMed] [Google Scholar]
- 34. Sastre E, Blanco‐Rivero J, Caracuel L, Callejo M, Balfagon G. Alterations in perivascular sympathetic and nitrergic innervation function induced by late pregnancy in rat mesenteric arteries. PLoS One. 2015;10:e0126017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mao Y, Su J, Lei L, Meng L, Qi Y, Huo Y, Tang C. Impaired regulator of G protein signaling 2 transcription facilitates vascular remodeling in injured rat aorta. J Cardiovasc Med (Hagerstown). 2014;15:572–578. [DOI] [PubMed] [Google Scholar]
- 36. Momen A, Afroze T, Sadi AM, Khoshbin A, Zhang H, Choi J, Gu S, Zaidi SH, Heximer SP, Husain M. Enhanced proliferation and altered calcium handling in RGS2‐deficient vascular smooth muscle cells. J Recept Signal Transduct Res. 2014;34:476–483. [DOI] [PubMed] [Google Scholar]
- 37. Woods LL, Brooks VL. Role of the renin‐angiotensin system in hypertension during reduced uteroplacental perfusion pressure. Am J Physiol. 1989;257(1 Pt 2):R204–R209. [DOI] [PubMed] [Google Scholar]
- 38. Tam KB, George E, Cockrell K, Arany M, Speed J, Martin JN Jr, Lamarca B, Granger JP. Endothelin type A receptor antagonist attenuates placental ischemia‐induced hypertension and uterine vascular resistance. Am J Obstet Gynecol. 2011;204:330.e1–330.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lang U, Baker RS, Braems G, Zygmunt M, Kunzel W, Clark KE. Uterine blood flow—a determinant of fetal growth. Eur J Obstet Gynecol Reprod Biol. 2003;110(suppl 1):S55–S61. [DOI] [PubMed] [Google Scholar]
- 40. Reho JJ, Peck J, Novak J, Ramirez RJ. Hypertension induced by episodic reductions in uteroplacental blood flow in gravid rat. Hypertens Pregnancy. 2011;30:208–220. [DOI] [PubMed] [Google Scholar]
- 41. Hill MA, Zou H, Potocnik SJ, Meininger GA, Davis MJ. Invited review: arteriolar smooth muscle mechanotransduction: Ca(2+) signaling pathways underlying myogenic reactivity. J Appl Physiol (1985). 2001;91:973–983. [DOI] [PubMed] [Google Scholar]
- 42. Hu XQ, Xiao D, Zhu R, Huang X, Yang S, Wilson S, Zhang L. Pregnancy upregulates large‐conductance Ca(2+)‐activated K(+) channel activity and attenuates myogenic tone in uterine arteries. Hypertension. 2011;58:1132–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Osei‐Owusu P, Sabharwal R, Kaltenbronn KM, Rhee MH, Chapleau MW, Dietrich HH, Blumer KJ. Regulator of G protein signaling 2 deficiency causes endothelial dysfunction and impaired endothelium‐derived hyperpolarizing factor‐mediated relaxation by dysregulating Gi/o signaling. J Biol Chem. 2012;287:12541–12549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kashyap S, Warner G, Hu Z, Gao F, Osman M, Al Saiegh Y, Lien KR, Nath K, Grande JP. Cardiovascular phenotype in Smad3 deficient mice with renovascular hypertension. PLoS One. 2017;12:e0187062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Arnold C, Demirel E, Feldner A, Genove G, Zhang H, Sticht C, Wieland T, Hecker M, Heximer S, Korff T. Hypertension‐evoked RhoA activity in vascular smooth muscle cells requires RGS5. FASEB J. 2018;32:2021–2035. [DOI] [PubMed] [Google Scholar]
- 46. Villar AJ, Kim J, De Blank P, Gillespie AM, Kozy HM, Ursell PC, Epstein CJ. Effects of genetic background on cardiovascular anomalies in the Ts16 mouse. Dev Dyn. 2005;232:131–139. [DOI] [PubMed] [Google Scholar]
- 47. Taddei I, Morishima M, Huynh T, Lindsay EA. Genetic factors are major determinants of phenotypic variability in a mouse model of the DiGeorge/del22q11 syndromes. Proc Natl Acad Sci USA. 2001;98:11428–11431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jelinek M, Wallach C, Ehmke H, Schwoerer AP. Genetic background dominates the susceptibility to ventricular arrhythmias in a murine model of beta‐adrenergic stimulation. Sci Rep. 2018;8:2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Karppanen T, Kaartokallio T, Klemetti MM, Heinonen S, Kajantie E, Kere J, Kivinen K, Pouta A, Staff AC, Laivuori H. An RGS2 3'UTR polymorphism is associated with preeclampsia in overweight women. BMC Genet. 2016;17:121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Perschbacher KJ, Deng G, Fisher RA, Gibson‐Corley KN, Santillan MK, Grobe JL. Regulators of G‐protein signaling in cardiovascular function during pregnancy. Physiol Genomics. 2018;50:590–604. [DOI] [PMC free article] [PubMed] [Google Scholar]