

SUMMARY
Mitochondrial Ca2+ uniporter (MCU)-mediated Ca2+ uptake promotes the buildup of reducing equivalents that fuel oxidative phosphorylation for cellular metabolism. Although MCU modulates mitochondrial bioenergetics, its function in energy homeostasis in vivo remains elusive. Here we demonstrate that deletion of the Mcu gene in mouse liver (MCUΔhep) and in Danio rerio by CRISPR/Cas9 inhibits mitochondrial Ca2+ (mCa2+) uptake, delays cytosolic Ca2+ (cCa2+) clearance, reduces oxidative phosphorylation, and leads to increased lipid accumulation. Elevated hepatic lipids in MCUΔhep were a direct result of extramitochondrial Ca2+-dependent protein phosphatase-4 (PP4) activity, which dephosphorylates AMPK. Loss of AMPK recapitulates hepatic lipid accumulation without changes in MCU-mediated Ca2+ uptake. Furthermore, reconstitution of active AMPK, or PP4 knockdown, enhances lipid clearance in MCUΔhep hepatocytes. Conversely, gain-of-function MCU promotes rapid mCa2+ uptake, decreases PP4 levels, and reduces hepatic lipid accumulation. Thus, our work uncovers an MCU/PP4/AMPK molecular cascade that links Ca2+ dynamics to hepatic lipid metabolism.
Graphical Abstract
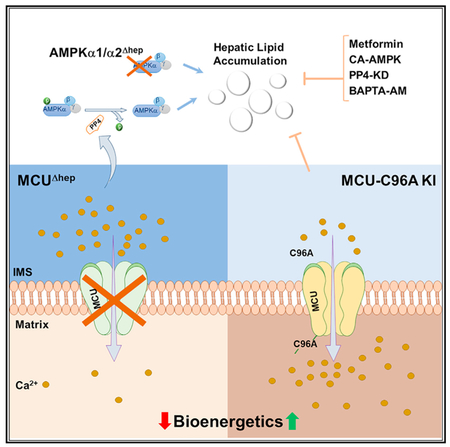
In Brief
Hepatic mitochondrial Ca2+ shapes bioenergetics and lipid homeostasis. Tomar et al. demonstrate that MCU-mediated cCa2+ buffering serves as a crucial step in controlling hepatic fuel metabolism through an MCU/PP4/AMPK molecular cascade. Identification of these molecular signaling events aids in understanding how perturbation of mitochondrial ion homeostasis may contribute to the etiology of metabolic disorders.
INTRODUCTION
Mitochondrial Ca2+ (mCa2+) uptake plays a fundamental role in the spatiotemporal regulation of cytosolic Ca2+ (cCa2+) and cellular bioenergetics in most cell types (Balaban, 2009; Baughman et al., 2011; Cárdenas et al., 2010; De Stefani et al., 2011; Denton and McCormack, 1980; Drago et al., 2011; Hajnóczky et al., 2000; Mallilankaraman et al., 2012a, 2012b). Because of the highly selective permeability of the inner mitochondrial membrane (IMM), mCa2+ uptake is tightly regulated by the mitochondrial electrochemical gradient (ΔΨm), generated by the electron transport chain and facilitated by the mitochondrial Ca2+ uni-porter (MCU) complex (Baughman et al., 2011; De Stefani et al., 2011; Kirichok et al., 2004). The core component of the complex, the MCU, and its regulators MICU1, MICU2, MCUR1, essential MCU regulator (EMRE), and MCUb have been identified and actively studied in recent years (Baughman et al., 2011; De Stefani et al., 2011, 2016; Kamer and Mootha, 2014; Mallilankaraman et al., 2012a, 2012b; Patron et al., 2014; Payne et al., 2017; Perocchi et al., 2010; Plovanich et al., 2013; Raffaello et al., 2013; Sancak et al., 2013; Tomar et al., 2016). The MCU-regulated mCa2+ fine-tunes mitochondrial bioenergetics through activation of Ca2+-dependent dehydrogenases and promotion of ATP synthesis (Balaban, 2009; Glancy and Balaban, 2012; Glancy et al., 2013). However, MCU-mediated mCa2+ overload favors opening of the mitochondrial permeability transition pore (mPTP), leading to a cascade of cell death events (Bernardi, 1999; Biasutto et al., 2016; Izzo et al., 2016; Shanmughapriya et al., 2015a). Despite evidence that MCU functions as a multi-faceted oligomeric complex, the molecular mechanisms underlying the requirement of this complex for cellular metabolism in vivo remain highly debated (Kwong et al., 2015; Liu et al., 2017; Luongo et al., 2015; Pan et al., 2013; Tomar et al., 2016). The liver plays a central role in carbohydrate and lipid metabolism, and metabolic dysfunction in this organ has a significant effect on whole-body energy homeostasis. An imbalance between lipid catabolism and anabolism, resulting in hepatic lipid accumulation, is observed in several metabolic disorders, including diabetes, obesity, and nonalcoholic fatty liver disease (Fu et al., 2011; Mayes and Felts, 1967; Perry et al., 2014; van den Berghe, 1991). Mitochondria comprise around 18% of the hepatocyte volume and form a very efficient cCa2+ regulatory system in hepatocytes (Weibel et al., 1969). Hepatic mitochondria have a high Ca2+ threshold for the opening of MCU channels and are directly coupled to oxidative phosphorylation (OXPHOS) (Paillard et al., 2017). Therefore, in hepatic tissue, mCa2+ may play a critical role in shaping metabolic outcomes. However, the mechanistic link between Ca2+ dynamics and lipid metabolism, if any, remains unknown.
Because the MCU is the primary driver of changes in mCa2+ and is responsible for Ca2+-induced remodeling of mitochondrial function, we generated loss- and gain-of-function mouse models for this protein. Genetic ablation of Mcu from hepatocytes results in depletion of matrix Ca2+ and impaired OXPHOS activity. Hepatic lipid accumulation occurs as a result of delayed cCa2+ clearance and protein phosphatase 4-dependent AMP-activated protein kinase (AMPK) dephosphorylation. In parallel, acute deletion of hepatic AMPKα1α2 isoforms was sufficient to raise hepatic triglyceride levels, supporting a model where impaired AMPK activity is the mechanistic link between mCa2+ buffering and steatosis. Importantly, cCa2+ chelation or metformin treatment restored AMPK phosphorylation and lipid clearance in MCUΔhep hepatocytes. These results demonstrate that MCU-mediated cCa2+ buffering serves as a crucial step in controlling hepatic fuel metabolism.
RESULTS
Loss of Mcu Lowers Fatty Acid Oxidation-Coupled Mitochondrial Oxygen Consumption in Hepatocytes
To decipher the role of mCa2+ in liver metabolism, we generated a liver-specific knockout mouse model of Mcu (MCUfl/flAlb-Cre+; hereafter referred to as MCUΔhep) by crossing the MCUfl/fl C57BL/6 mouse with the hepatocyte-specific Cre-recombinase line (Figures 1A and S1A). MCUΔhep mitochondria showed a striking deficit in their MCU current (IMCU), which further establishes that MCU is the core component of the uniporter complex necessary for hepatic mCa2+ uptake (Figures 1B–1D). Consistently, permeabilized hepatocytes from MCUfl/fl but not MCUΔhep exposed to a bolus of 10 03B1μM Ca2+ exhibited rapid mCa2+ uptake (Figures 1E and 1F) without any effect on ΔΨm(Figures 1E, S1B, and S1C). Although mitochondria are known to sequester inositol 1,4,5-trisphosphate receptor (IP3R) and calcium-release activated channel (CRAC)-mediated cCa2+ (Cárdenas et al., 2010; Gilabert and Parekh, 2000; Hajnóczky et al., 1999, 2000; Hawkins et al., 2010; Pacher et al., 2000), it was unknown whether the basal mitochondrial matrix Ca2+ is altered in MCUΔhep mitochondria. Using permeabilized primary hepatocytes (Mallilankaraman et al., 2012a, 2012b; Shanmughapriya et al., 2015b; Tomar et al., 2016), we found that MCUΔhep hepatocytes have 0.5 μM mitochondrial matrix calcium, which is ~8-fold less than Mcufl/fl hepatocytes (4.0 μM) (Figures 1G and 1H).To further validate the specificity of Mcu loss in MCUΔhep mice, we reconstituted the FLAG-tagged MCU wild-type (WT) in MCUΔhep mice through adenoviral delivery (Figure 1I). The adenoviral expression of MCU-FLAG restored both MCU-mediated mCa2+ uptake and basal matrix Ca2+ (Figures 1J–1M). We next sought to determine whether deletion of MCU lowers mitochondrial reactive oxygen species (mROS) production in hepatocytes. Primary hepatocytes isolated from MCUfl/fl and MCUΔhep mice were incubated with MitoSOX Red to visualize mROS levels, and basal mROS is greatly suppressed in MCUΔhep (Figures S1D and S1E), indicating a role for MCU-dependent mCa2+ uptake in mROS production.
Figure 1. Hepatocyte MCU Activity Regulates FAO-Coupled Mitochondrial Respiration.

(A) Generation and confirmation of MCUΔhep mice by PCR genotyping (left) and western blotting (right).
(B) Schematic representation of the patch-clamp technique for measuring MCU channel activity.
(C) Representative IMCU traces derived from MCUfl/fl and MCUΔhep mitoplasts.
(D) IMCU densities (picoamperes/picofarads) in mitoplasts. n = 6.
(E) Representative traces of extramitochondrial Ca2+ ([Ca2+]out) clearance and DJm in permeabilized hepatocytes from MCUfl/fl and MCUΔhep. n = 3.
(F) mCa2+ uptake rate calculated from (E). n = 3.
(G) Representative traces of [Ca2+]out rise from MCUfl/fl and MCUΔhep. n = 3.
(H) Quantification of mCa2+ after addition of carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) from (G). n = 3.
(I) Immunoblot for the reconstitution of MCU in MCUΔhep mice using adenovirus-mediated delivery. n = 2.
(J) Reconstitution of MCU restores [Ca2+]out clearance ability. n = 4.
(K) mCa2+ uptake rate calculated from (J). n = 4.
(L) MCU reconstitution restores the matrix Ca2+ in MCUΔhep. Shown are representative traces of [Ca2+]out rise in response to FCCP. n = 4.
(M) Quantification of mCa2+ calculated after addition of FCCP from (L). n = 4.
(N) MCU reconstitution restores the OCR in MCU KO hepatocytes. n = 3.
(O) Quantification of basal, ATP-coupled, and maximal OCRs in hepatocytes from (N). n = 3.
(P) The FAO OCR was measured in MCUfl/fl and MCUΔhep hepatocytes using palmitate as a substrate with or without etomoxir (40 mM). n = 3.
(Q) Quantification of basal, ATP-coupled, and maximal FAO-coupled OCRs from (P). n = 3.
(R) Measurement of hepatocyte ATP. n = 3.
(S) Measurement of AMP:ATP ratio in hepatocytes. n = 3. Statistical analysis: mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001; ns, non-significant.
(T) Schematic of hepatocyte deletion of MCU, showing reduction in mCa2+ and bioenergetic parameters.
Next, we found no changes in OXPHOS protein complex expression (Figure S1H), despite a greatly diminished basal mitochondrial oxygen consumption rate (OCR) in MCUΔhep hepatocytes (Figures S1F and 1G). Maximal uncoupled and ATP-coupled OCRs were also found to be significantly decreased (Figures S1F and 1G), suggesting a lack of optimal production of Ca2+-dependent reducing equivalents like nicotinamide adenine dinucleotide (NADH) in the matrix (Glancy and Balaban, 2012; Glancy et al., 2013; White et al., 2005). Adenoviral reconstitution of MCU-FLAG in MCUΔhep mice restored the basal, maximal, and ATP-coupled OCRs in primary hepatocytes (Figures 1N and 1O). Because mCa2+ occupies a central role in mitochondrial bioenergetics, and alterations in MCU-mediated mCa2+ uptake may account for the differences observed in glucose and fat oxidation, we speculated that the fatty acid oxidation (FAO)-coupled OCR was compromised in MCUΔhep hepatocytes. To examine the FAO-dependent OCR, hepatocytes were supplemented with exogenous palmitate in glucose-free medium. To confirm endogenous FAO involvement, etomoxir was used as an inhibitor of carnitine palmitoyltransferase-1 (CPT-1). Basal and maximal FAO-coupled OCRs were significantly reduced in MCUΔhep hepatocytes compared with the control (Figures 1P and 1Q). CPT-1 expression was unchanged, indicating that the reduction in FAO-coupled OCR was not due to the alteration in mitochondrial fatty acid transport (Figures S1I and S1J). MCUΔhep hepatocytes did not display changes in the mitochondrial biogenesis-related transcription factors PGC1a, mitochondrial transcription factor A (mtTFA), and NRF1 (Figures S1K and S1L) but did show significant changes in the AMP:ATP ratio, consistent with the decreased OXPHOS activity (Figures 1R and 1S). These data clearly indicate that both glucose- and FAO-dependent OCRs and ATP production in hepatocytes are Ca2+-dependent (Figure 1T).
Loss of Mcu Promotes Hepatic Lipid Accumulation and Lowers Ketone Body Production
We next investigated the broader metabolic effects of hepatocyte-specific Mcu ablation in mice. Although the body weight was approximately equivalent, the total body fat content of MCUΔhep mice was significantly increased during both the fed and fasting states (Figures 2A and S3A). The ratios of liver:body weight and white adipose tissue (WAT):body weight were also significantly increased in MCUΔhep mice (Figures S3B and S3C). Biochemical analysis of livers from MCUΔhep mouse livers revealed significant increases in fed and fasted triglycerides (TAGs) despite an increase in plasma TAGs being observed only in the fasting state (Figures 2B and 2C). During fasting, plasma ketones were significantly decreased in MCUΔhep mice (Figure 2D), suggesting a link between MCU-dependent mCa2+ and acetyl-coenzyme A (CoA) oxidation. Ten-week-old mice were subjected to three different states (fed, fasted, and re-fed) for assessment of food intake, locomotor activity, heat, fat utilization, O2 consumption, and respiratory exchange ratio (RER). These whole-body metabolic parameters were unchanged between control and MCUΔhep mice (Figures S2A–S2F).
Figure 2. Loss of MCU Promotes Hepatic Lipid Accumulation and Increases Total Body Fat.

(A) Body mass was monitored using NMR and DEXA before and after 24 h fasting. n = 11–12 mice per group.
(B) Liver triglycerides were measured using an enzymatic assay. n = 4–6 mice per group.
(C) Hepatic deletion of MCU results in increased plasma TAG in the fasting state, as measured by enzymatic assay. n = 5–6 mice per group.
(D) MCUΔhep mice showed decreased plasma ketones under the fasting state, as measured by enzymatic assay. n = 5 mice per group.
(E) Primary hepatocytes were isolated from MCUfl/fl and MCUΔhep mice and cultured in low glucose, high glucose, and high glucose followed by starvation. Lipid droplets were visualized by confocal microscopy. n = 3.
(F) Quantification of the number of lipid droplets from (E). n = 3.
(G) Quantification of the size of lipid droplets from (E). n = 15–30 cells from each isolation. n = 3.
(H) Representative electron micrograph showing a large number of lipid droplets in MCU KO hepatocytes. n = 3 mice per group.
(I) Representative histological section image showing massive oil red O staining in MCU KO liver. n 40 images. n = 3 mice.
(J) Schematic of hepatic ablation of MCU, showing increased lipid deposition in the liver, which subsequently increased the whole-body fat content. Statistical analysis: mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
Hepatic lipid metabolism is tightly linked with glucose metabolism. Therefore, we assessed glucose homeostasis in MCUΔhep mice. Fasting and fed blood glucose levels, as well as glucose tolerance, were unchanged between MCUfl/fl and MCUΔhep mice (Figures S3D and S3E). Stored hepatic glycogen is the primary energy source during early fasting, and hepatocytes generate glucose from glycogen through glycogenolysis (Petersen et al., 2017). Interestingly, MCUΔhep mice showed increased glycogen during the fed state, with the same level of clearance in the fasting state (Figure S3F). MCUfl/fl and MCUΔhep hepatocytes had comparable glucose output, which suggests that deletion of hepatic Mcu does not compromise glycogenolysis (Figure S3G). MCUΔhep hepatocytes had normal glucose up-take (Figures S3H and S3I). This suggests that the elevated TAG levels and reduced ketone bodies are possibly due to lower mitochondrial matrix Ca2+ in MCUΔhep mice. We next investigated whether deletion of Mcu in hepatocytes led to lipid accumulation in the liver. Hepatocytes isolated from MCUfl/fl and MCUΔhep animals were cultured in the presence of low (5 mM) and high (25 mM) glucose, followed by starvation. MCUΔhep hepatocytes retained significantly higher levels of lipid vesicles compared with the control (Figures 2E–2G). Upon starvation, lipids were depleted in control but not in MCUΔhep hepatocytes, suggesting a lack of fat mobilization (Figures 2F and 2G). In addition to the number of lipid droplets, the size of the lipid droplets was significantly larger in MCUΔhep hepatocytes (Figure 2G). Consistent with confocal imaging analysis, ultrastructural studies revealed increased accumulation of lipid droplets in MCUΔhep mice under fed and fasting conditions (Figure 2H). Oil red O staining of liver sections recapitulated this and revealed extensive lipid staining in MCUΔhep liver sections (Figure 2I). Hepatic lipid accumulation has been linked to increased endoplasmic reticulum (ER) stress; however, MCUΔhep hepatocytes have an intact ER with no sign of stress (Figures S3J and S3K).
We asked whether Mcu deletion induces acute hepatic inflammation. Plasma alanine aminotransferase (ALT) levels were found to be unchanged between MCUfl/fl and MCUΔhep mice (Figure S3L). H&E staining of both MCUfl/fl and MCUΔhep mouse livers revealed no infiltration of inflammatory cells in MCUΔhep tissue (Figure S3M). We next investigated whether loss of Mcu has any effect on liver TAG secretion and de novo lipogenesis. Analysis of liver TAG secretion and hepatic and white adipose lipo-genesis were found to be unchanged between MCUfl/fl and MCUΔhep mice (Figures S3N–S3P), indicating that hepatic lipid accumulation in MCUΔhep mice could be due to decreased fatty acid utilization as a result of MCU deletion (Figure 2J).
Genetic Ablation of Mcu Limits mCa2+ Uptake and Elevates Total Body Fat in Zebrafish
Because liver-specific deletion of Mcu in mice with the C57BL/6 genetic background caused aberrant lipid accumulation, the zebrafish model system could address the conservation of this mechanism across species. Adaptation of the CRISPR/Cas9 gene editing strategy eliminates morpholino-induced false positive and negative genetic mutant phenotypes (Figure 3A; Clark et al., 2011). We targeted a region proximal to the Mcu translation initiation codon and confirmed the 268-bp deletion allele by DNA sequencing, qRT-PCR, and western blotting (Figures 3B–3D). Isolated primary zebrafish cells were used to assess the mCa2+ dynamics for both control and MCU knockout (KO) zebrafish. As expected, mCa2+ uptake was significantly reduced in MCU KO zebrafish without altering ΔΨm (Figures 3E, 3F, S4A, and S4B). Similar to MCUΔhep hepatocytes, cellular ATP levels were lower in MCU KO zebrafish and, to a lesser extent, in MCU+/−cells (Figure 3G). During zebrafish cell isolation, we found that supernatants collected from MCU KO zebrafish cells appeared to have a strikingly higher fat content that was visually apparent (Figure 3H). We therefore extended the in vitro observation to the in vivo imaging approach of fat deposition in intact live zebrafish. The fluorescence intensity of lipids in the dorsal fin and tail fin were considerably increased (Figures 3I–3K), further supporting our results in MCUΔhep mice showing that MCU deletion results in higher lipid accumulation.
Figure 3. Loss of MCU Limits mCa2+ Uptake and Elevates Total Body Fat in Zebrafish.

(A) Schematic of the generation of global MCU KO zebrafish using CRISPR/Cas9.
(B) Genotyping for MCU deletion.
(C) Bar graph showing MCU mRNA abundance in WT, MCU+/−, and MCU−/− zebrafish.
(D) Western blot for MCU expression. n = 3.
(E) Measurement of mCa2+ uptake. n = 5–10.
(F) Quantification of ionomycin-induced peak mCa2+ levels from (E). n = 5–10.
(G) Bar graph representing cellular ATP levels in cells isolated from MCU+/+, MCU+/−, and MCU−/− zebrafish. n = 4.
(H) Adult WT and MCU−/−zebrafish were homogenized and centrifuged. MCU−/−zebrafish samples show a clear yellow color lipid layer on top of the protein lysate. n = 6.
(I) Adult WT and MCU−/−zebrafish were stained with Lipid Green to monitor the distribution of lipids in the whole body. n = 3. (J and K) Bar graphs represent the quantification of Lipid Green staining from the dorsal (J) or tail (K) fin. n = 3.
Statistical analysis: mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
MCU Deletion in Hepatocytes Restrains Nutrient Sensing through Altered cCa2+ Signaling
MCU-mediated mCa2+ plays an essential role in mitochondrial bioenergetics and is required for maintenance of hepatic ATP content (Figure 1). Having observed elevated TAGs and decreased ketone body levels in MCUΔhep mice, we speculated that the decreased ATP levels might activate signaling via the energy-sensing AMPK (Hardie, 2015; Hardie et al., 2012; Kahn et al., 2005; Mihaylova and Shaw, 2011). In contrast to our expectations, MCU deletion resulted in a large reduction in T172-phosphorylated (active) AMPK-α, whereas total AMPK protein and mRNA levels remained unchanged (Figures 4A, 4B, and S5A). Expression of the upstream kinases LKB1 and calcium-regulated kinase calcium/calmodulin dependent protein kinase kinase-II (CaMKK-II) remained unchanged (Figures 4A and 4C). However, downstream inhibitory phosphorylation of acetyl-CoA carboxylase (ACC) was decreased (Figure 4A). Expression of two other key proteins involved in lipid synthesis, fatty acid synthase and SREBP1, remained unchanged (Figure 4A). Adenoviral reconstitution of MCU in MCUΔhep hepatocytes restored phosphorylation of AMPK (Figure 4A). To rule out a global dephosphorylation event, we also monitored extracellular signal-regulated kinase (ERK) phosphorylation and found normal ERK phosphorylation in MCU KO hepatocytes (Figure S5B). To confirm the potential of impaired AMPK signaling to drive hepatic lipid accumulation in MCUΔhep mice, we next generated hepatic AMPK-KO (AMPKΔhep) mice by injecting adenovirus-associated virus 8-Cre (AAV8-Cre) into AMPKα1/α2fl/fl mice, which have LoxP sites flanking both the AMPKα1 and AMPKα2 loci. The loss of AMPK protein was validated by western blotting (Figure 4D). Biochemical analysis of hepatic tissue collected from overnight-fasted mice showed significant hepatic lipid accumulation in AMPKΔhep mice (Figure 4E). This observation was further validated by oil red O staining of liver tissue section and boron-dipyrromethene (BODIPY) staining of primary hepatocytes (Figures 4F, S5C, and S5D). However, the loss of AMPK did not affect mCa2+ uptake or ΔΨm (Figures S5E and S5F), indicating that AMPK is downstream of MCU in lipid accumulation. The involvement of AMPK was further confirmed by using consti tutively active AMPK (AMPKα1 T172D, hereafter called CAAMPK) (Hardie, 2011; King et al., 2009). Primary hepatocytes isolated from MCUfl/fl and MCUΔhep mice were infected with an CA-AMPK-expressing adenovirus and monitored for lipid accumulation (Figures 4G–4J and S5G).
Figure 4. Ablation of mCa2+ Uptake Augments the cCa2+ Rise That Enhances AMPK Dephosphorylation.

(A) Hepatocytes isolated from MCUfl/fl and MCUΔhep mice were lysed in radioimmunoprecipitation assay (RIPA) buffer and immunoblotted for the indicated antibodies. n = 6.
(B) Bar graph representing the pAMPK:AMPK ratio from (A). n = 6.
(C) Immunoblot analysis for phospho and total CaMKK-II. n = 6.(D) Hepatocyte lysates from AMPKα1/α2fl/fl and AMPKα1/α2fl/flCre+ mice were probed for AMPKa protein abundance. n = 3.
(E) Hepatic TAG levels. n = 5.
(F) Assessment of hepatic lipid accumulation by Oil-Red-O staining. n = 5.
(G) Primary hepatocytes isolated from MCUfl/fl and MCUΔhep mice were infected with a CA-AMPK-expressing adenovirus. Hepatocytes were stained with oil red O and measured. n = 6–12 replicates from 3 mice.
(H) Visualization of lipid droplets from CA-AMPK reconstituted MCUΔhep hepatocytes. n = 3.
(I) Quantification of the number of lipid droplets from (H). n = 20–30 cells. n = 3.
(J) Schematic depicting a link between MCU and AMPK phosphorylation.
(K) GCaMP6-expressing hepatocytes from MCUfl/fl and MCUΔhep were stimulated with thapsigargin (Tg) or Vasopressin, and cCa2+ dynamics were monitored. n = 3.
(L) Quantification of the cCa2+ clearance rate from (K). n = 15–25 cells. n = 3.
(M) GCaMP6-expressing hepatocytes from MCUfl/fl and MCUΔhep were stimulated with Vasopressin and cCa2+ dynamics were monitored. n = 3.
(N) Quantification of the cCa2+ clearance rate from (M). n = 15–25 cells. n = 3.
(O) Hepatocytes isolated from MCUfl/fl and MCUΔhep adult mice were transduced with an adenovirus expressing the cCa2+ sensor GCaMP6, and basal cCa2+ fluorescence was quantified. n = 74–91 cells from 3 mice in each group.
(P) Hepatocytes were treated with the intracellular Ca2+ chelator BAPTA-AM for various times. Cell lysates were immunoblotted for the indicated antibodies. n = 4.
(Q) Hepatocytes were treated with BAPTA-AM overnight, and oil red O stain was quantified. 12 replicates from 3 mice per group.
(R) Visualization of lipid droplets from control and BAPTA-AM-treated hepatocytes. n = 3 mice per group.
(S) Quantification of lipid droplets from control and BAPTA-AM-treated hepatocytes. n = 20–30 cells per group. n = 3 mice per group.
(T) Schematic depicting how cCa2+ determines lipid clearance, possibly through AMPK phosphorylation.
Statistical analysis: mean ± SEM. **p < 0.01, ***p < 0.001.
Blockade of mCa2+ Uptake Augments a cCa2+ Rise that Enhances AMPK Dephosphorylation
To decipher the underlying mechanism of AMPK dephosphorylation in MCUΔhep mice, we investigated whether loss of MCU has any effect on cCa2+ dynamics. Hepatocytes were infected with an adenovirus expressing the cCa2+-reporter GCaMP6, and cCa2+ dynamics were monitored in response to thapsigargin and vasopressin. The MCUΔhep hepatocytes exhibited delayed cCa2+ clearance after stimulation with thapsigargin and vasopressin (Figures 4K–4N). Remarkably, basal cCa2+ was elevated in MCUΔhep hepatocytes (Figure 4O). Because loss of MCU caused persistent elevation of cCa2+, we investigated whether chelation of cCa2+ restores the phosphorylated form of AMPK in MCUΔhep hepatocytes. When hepatocytes were pretreated with 1,2-Bis (2-aminophenoxy)ethane-N,N,N’,N’-tetraacetic acid tetrakis (acetoxymethyl ester) (BAPTA-AM; 25 μM), a significant amount of phosphorylated AMPK was detected, even as early as 30 min (Figure 4P). Next, we investigated whether BAPTA-AM-mediated chelation of Ca2+ c improves lipid clearance in MCUΔhep hepatocytes. Interestingly, chelation of cCa2+ reduced the lipid content in MCUΔhep hepatocytes, as indicated by oil red O and BODIPY staining (Figures 4Q–4S). Collectively, these data indicate that blockade of mCa2+ buffering results in AMPK dephosphorylation through aberrant cCa2+ elevation, leading to hepatic lipid accumulation in MCUΔhep mice (Figure 4T).
Cytosolic Ca2+ Rise Augments AMPK Dephosphorylation through Protein Phosphatase-4
Having determined that AMPK dephosphorylation is a cCa2+-dependent signal, we used pharmacologic and genetic tools to identify the phosphatase responsible for AMPK dephosphorylation. The pan protein phosphatase inhibitor (PP1, PP2A, PP2B, and PP4 inhibitor) okadaic acid restored AMPK phosphorylation, suggesting a phosphatase dependent-event (Figure 5A). Because AMPK dephosphorylation is a cCa2+-dependent protein phosphatase and calcineurin is a Ca2+-calmodulin-activated protein phosphatase, we tested whether FK506 would prevent AMPK dephosphorylation. FK506 pre-treatment did not restore AMPK phosphorylation in MCUΔhep mice (Figure 5B). We asked whether the Ca2+-dependent, ubiquitously expressed PP2A subfamily phosphatase PP4 could be responsible for AMPK dephosphorylation (Shui et al., 2007). Pre-treating MCUΔhep hepatocytes with the PP4 inhibitor cantharidin restored the phosphorylated form of AMPK (Figure 5C). We also found that PP4 protein expression was increased in MCUΔhep hepatocytes (Figures 5D and 5E), indicating a cCa2+-regulated increased PP4 protein abundance. To verify the involvement of PP4 in AMPK dephosphorylation, we silenced PP4 expression in MCUΔhep hepatocytes using small interfering RNA (siRNA) (Figure 5F). PP4 knockdown resulted in restoration of AMPK phosphorylation (Figure 5F). Next we used the reverse approach to confirm the involvement of PP4 in AMPK dephosphorylation by ectopic expression of PP4. Overexpression of PP4-FLAG resulted in dephosphorylation of AMPK in MCUfl/fl hepatocytes that was further enhanced in MCUΔhep hepatocytes, demonstrating PP4 to be a major phosphatase of AMPK in the liver (Figure 5G). To identify whether AMPK and PP4 exist as a Ca2+-dependent protein complex, we performed protein-protein interaction studies, which demonstrated that AMPK and PP4 bind one another (Figure 5H) and that this binding is regulated by cCa2+ (Figures 5I and 5J). Silencing of PP4 in MCUΔhep hepatocytes decreased the lipid content, as indicated by oil red O and BODIPY staining (Figures 5K–5M). These findings reveal that the blockade of rapid sequestration of Ca2+ into the mitochondrial matrix by MCU deletion results in activation of Ca2+-dependent PP4, which dephosphorylates AMPK (Figure 5N).
Figure 5. AMPK Dephosphorylation in MCU KO Hepatocytes Is Likely Due to PP4.

(A) Hepatocytes were treated with okadaic acid (OA) (50 nM) for 2 h. Cell lysates were immunoblotted for the indicated antibodies. n = 4.
(B) MCUfl/fl and MCUΔhep hepatocytes were treated with FK506 (2 μM) for 2 h and immunoblotted for the indicated antibodies. n = 4.
(C) MCUfl/fl and MCUΔhep hepatocytes were treated with cantharidin (50 μM) for 2 h and immunoblotted for the indicated antibodies. n = 4.
(D) Hepatocytes isolated from MCUfl/fl and MCUΔhep mice were lysed and immunoblotted for the indicated antibodies. n = 4.
(E) Densitometric analysis of PP4 protein abundance. n = 4.
(F) Hepatocytes were transfected with PP4 siRNA for 72 h. Cell lysates were immunoblotted for the indicated antibodies. n = 4.
(G) Hepatocytes were transfected with PP4-FLAG for 48 h. Cell lysates were immunoblotted for the indicated antibodies. n = 4.
(H–J) HepG2 cells were transfected with AMPK-hemagglutinin (HA) and PP4-FLAG plasmids for 48 h. Cell lysates were immunoprecipitated with an HA antibody and probed with the indicated antibodies (H). Under a similar condition (I and J), cells were treated with BAPTA-AM in the presence or absence of ionomycin (50 nM) stimulation. The reciprocal immunoprecipitation was performed with HA or FLAG antibodies and probed with the indicated antibodies. n = 3.
(K) Visualization of lipid droplets from scrambled (Scr) siRNA- and PP4 siRNA-treated hepatocytes. n = 3.
(L) Quantification of lipid droplets from Scr siRNA- and PP4 siRNA-treated hepatocytes. n = 20–30 cells. n = 3.
(M) Hepatocytes isolated from MCUfl/fl and MCUΔhep adult mice were transfected with PP4 siRNA for 72 h. Cells were stained with oil red O and quantified. n = 3.
(N) Schematic depicting aberrant cCa2+ clearance in MCU KO hepatocytes exhibiting AMPK dephosphorylation through elevated PP4 activity.
Statistical analysis: mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
The Gain-of-Function MCU Controls PP4, which Enhances Hepatic Lipid Clearance during Fasting
We sought to understand whether MCU hyperactivation has the potential for higher lipid mobilization and increased FAO. A recent report revealed that the human MCU C97A mutation results in hyperactivation of MCU channel activity (Figure S6A; Dong et al., 2017). Therefore, we generated a gain-of-function MCU mouse model (C96A-knockin [KI]) with a global MCU C96A (corresponding to human MCU C97) KI using the CRISPR/Cas9 approach (Figures 6A and S6B), which was confirmed by restriction fragment length polymorphism (RFLP) and Sanger sequencing (Figures 6A and S6C). The C96A-KI mice were viable (Figure 6B) but had reduced body weight compared with the WT control (Figures 6C and S6D). We investigated the mCa2+ dynamics and noted that C96A-KI hepatocytes exhibited an increased mCa2+ uptake rate as well as higher basal matrix mCa2+ under normal ΔΨm (Figures 6D–6G, S6E, and S6F). Although western blot analysis of OXPHOS proteins revealed no differences between WT and C96A-KI hepatocytes (Figure 6H), C96A-KI hepatocytes exhibited higher basal and maximal OCRs (Figures 6I and 6J).
Figure 6. Hepatocytes Harboring MCU-C96A Display Reduced PP4 and Augmented Hepatic Lipid Clearance during Fasting.

(A) Schematic depicting the strategy used to generate MCU-C96A KI mice. Top right: genotyping of MCU-C96A KI mice. Bottom: MCU protein abundance.
(B) 12-week-old MCU-C96A KI mice are viable.
(C) MCUC96A-KI mice have reduced body weight at 12 weeks. n = 5.
(D) MCU-mediated extramitochondrial Ca2+ clearance and mCa2+ uptake. n = 3.
(E) MCU-mediated mCa2+ uptake rate was calculated from (D). n = 3.
(F) Measurement of basal mCa2+ after addition of FCCP. n = 3.
(G) Quantification of basal mCa2+ after addition of FCCP from (F). n = 3.
(H) Western blot analysis of mitochondrial respiratory chain subunits of complex I (NDUFB8), complex II (succinate dehydrogenase complex iron sulfur subunit B [SDHB]), complex III (UQCRC2), complex IV (MTCO1), and ATP synthase subunit ATP5A. MCU and TOM20 were used as loading controls. n = 3.
(I) Measurement of mitochondrial OCR. n = 3.
(J) Measurement of basal, ATP coupled, and maximal OCR from (I). n = 3.
(K) Hepatocytes isolated from WT and C96A-KI mice were lysed and probed with the indicated antibodies. n = 3.
(L) Densitometric analysis of pAMPK/AMPK ratio from (K). n = 3.
(M) Densitometric analysis of PP4 protein expression from (K). n = 3.
(N) Measurement of liver and triglycerides (TAGs) under fasting conditions. Measurement of these parameters is described in Figure 2. n = 5.
(O) Measurement of plasma triglycerides (TAG) under fasting conditions. n = 5.
(P) Measurement of plasma ketone bodies under fasting conditions. n = 5.
(Q) Visualization of lipid droplets from WT and C96A KI hepatocytes. n = 3.
(R) Quantification of lipid droplets from (Q). n = 20–30 cells. n = 3.
(S) Representative histological section image shows reduced lipid accumulation in C96A-KI mice. n = 3 mice per group. Statistical analysis: mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
Next we monitored the AMPK and ACC phosphorylation status and the expression of PP4 in C96A-KI hepatocytes. C96A-KI he patocytes had higher levels of phosphorylated AMPK and ACC and a significant reduction in PP4 protein abundance (Figures 6K–6M). Biochemical analysis of hepatic tissue from C96A-KI mice clearly showed a reduction in liver and plasma TAG levels along with a significant increase in plasma ketone levels (Figures 6N–6P). We next investigated the effect of C96A-KI on high-glucose-induced lipid accumulation in primary hepatocytes. C96A-KI hepatocytes showed a clear reduction in lipid droplet accumulation under high-glucose conditions (Figures 6Q and 6R). In support of these data, oil red O staining confirmed enhanced lipid clearance by C96A-KI mice during fasting (Figure 6S). This newly generated mouse model further validates the involvement of MCU in the regulation of hepatic lipid homeostasis.
Restoration of AMPK Phosphorylation in MCUΔhep Mice Improves Lipid Clearance
It has been shown that cells treated with metformin exhibit increased AMPK phosphorylation (Hawley et al., 2002; Madiraju et al., 2014; Shaw et al., 2005; Zhou et al., 2001). Metformin treatment did not alter mCa2+ uptake (Figure S7A). We next tested whether supplementing metformin would promote AMPK phosphorylation and lipid clearance in MCUΔhep. MCUΔhep hepatocytes treated with metformin had significantly increased AMPK phosphorylation, similar to the effect of 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) (Figures 7A and 7B). Given the effect of metformin on AMPK phosphorylation and fatty acid metabolism, we examined whether metformin administration could deplete lipid accumulation in MCUΔhep mice. After 3 weeks of oral metformin administration (1.25 mg/mL), we noted a marked reduction in lipid content in MCUΔhep livers, as evidenced by oil red O staining (Figure 7C). Similarly, acute metformin treatment markedly reduced hepatocyte lipid content, as evidenced by BODIPY-positive lipid vesicle numbers (Figures 7D and 7E), but did not normalize the remaining lipid droplet size (Figure S7B).
Figure 7. Activation of AMPK by Metformin Corrects Hepatic Lipidome Remodeling in MCU KO Mice.

(A) Hepatocytes isolated from MCUfl/fl and MCUΔhep treated with metformin (1 mM), AICAR (100 μM) for 16 hours. After treatment, cell lysates were probed with indicated antibodies. n = 3.
(B) Bar graph showing the quantification of pAMPK in Figure 7A. n = 3.
(C) Representative histological section image shows clearance of lipids in MCU KO metformin-administered mice. n = 3.
(D) Visualization of lipid droplets from MCUfl/fl and MCUΔhep mice with or without metformin treatment. n = 3.
(E) Quantification of lipid droplets from (D). n = 15–30 cells per mice. n = 3.
(F) Lipids were analyzed as described in STAR Methods, and the identified features were subjected to a hierarchical cluster analysis. n = 4.
(G) Significant top 15 lipids (false discovery rate [FDR] = 0.001) representing the relative lipid modulation among the groups. The y axis represents normalized values. *, significantly different from MCUΔhep; #, significantly different from metformin-administrated MCUΔhep. n = 4 per group.
Statistical analysis: mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
Finally, we conducted a comprehensive lipidomics analysis and characterized the lipid species in control, MCUΔhep, and metformin-treated MCUΔhep mice. Lipidomics analysis of MCUΔhep liver tissue showed a significant accumulation of TAG and diacylglycerol (DAG) lipid species (Figures 7F, 7G, S7C, and S7D). Notably, administration of metformin not only restored AMPK phosphorylation but also significantly lowered several TAG and DAG species (Figures 7F and 7G). Collectively, these data indicate that loss of MCU results in altered hepatic lipid metabolism, leading to hepatic lipid accumulation and lipidomic remodeling.
DISCUSSION
We report that hepatic deletion of MCU lowers hepatic mCa2+, reduces FAO-coupled mitochondrial respiration, and increases hepatic lipid accumulation, linking mCa2+ handling to hepatic lipid metabolism. Our characterization of this mouse model reveals that, following MCU ablation, there is an accumulation of cCa2+ because of a lack of mCa2+ sequestration via MCU into the matrix. This observation is in contrast to earlier reports heart- and endothelium-specific MCU KO models, in which no apparent changes were observed in mCa2+ (Luongo et al., 2015; Pan et al., 2013; Tomar et al., 2016). Depletion of mCa2+ and reduced cCa2+ clearance ability in MCUΔhep hepatocytes indicates that MCU could be a major player in shaping metabolic outcomes in the liver. Although mCa2+ overload is necessary for PTP opening and mitochondrial dysfunction, MCU KO failed to protect against cell death under ionomycin and oxidative stress conditions, signifying the physiological importance of mCa2+ buffering and cell survival (Nemani et al., 2018; Pan et al., 2013). Recently, it has been demonstrated that prevention of MCU-mediated Ca2+ up-take exhibited enhanced mitochondrial shape transition through Miro-1 and triggered cell death in a PTP-independent pathway (Nemani et al., 2018) Alternatively, MCU-dependent Ca2+ enhances mitochondrial constriction, which is Drp1-independent, suggesting that Ca2+ dynamics may be an initiator of preconstruction (Chakrabarti et al., 2018). Nevertheless, altering mCa2+ may contribute significantly to changes in the mitochondrial phenotype and cellular metabolism.
It has been established that the cCa2+ clearance mechanisms operate differentially between tissues (Fieni et al., 2012; Paillard et al., 2017; Pan et al., 2013). For example, there are several reasons why one could speculate that mCa2+ remained unchanged in myocytes after MCU deletion. The extracellularly and intracellularly mediated cCa2+ rise is rapidly cleared by the high-Ca2+ affinity sarcoplasmic or endoplasmic reticulum Ca2+ ATPase (SERCA) and plasma membrane pumps (plasma membrane Ca2+ ATPase [PMCA]) and sodium Ca2+ exchanger (NCX) in cardiomyocytes (Fearnley et al., 2011), where ΔΨm and mitochondrial buffering capacity is weaker because of excitation-contraction (E-C) coupling. In non-excitable cell types like hepatocytes, MCU clears cCa2+ faster than SERCA and PMCA pumps. Because mCa2+ uptake is primarily driven by the electro-chemical gradient across the mitochondrial inner membrane, it is possible that hepatocyte mitochondria are maintained in a hyperpolarized state specifically to facilitate cCa2+ clearance and maintain heterogeneity (Collins et al., 2002; Kuznetsov et al., 2004). Another striking difference we observed in hepatocytes was the reduction of both glucose and fatty acid-dependent mitochondrial OXPHOS. This further strengthens our finding of lower basal mCa2+ in MCU KO hepatocytes.
The most striking phenotype of MCUΔhep mice is dyslipidemia and ectopic accumulation of lipid in the liver, which was associated with reduced fasting plasma ketones. These data suggest an impairment in FAO and, consequently, ketone body production in MCU KO hepatocytes. The lipid accumulation phenotype was observed in the mouse as well as zebrafish model systems, indicating conservation of MCU-regulated lipid metabolism across species. During starvation, AMPK promotes phosphorylation and inactivation of ACC, which results in fatty acid uptake into mitochondria for β-oxidation (Hardie et al., 2012). Remarkably, loss of MCU in the liver caused impairment of FAO and massive accumulation of total lipids under fasting conditions, suggesting the possibility that AMPK activity is impaired by the dysregulated cellular Ca2+ dynamics (Figures 4K–4O). As predicted, deletion of MCU in hepatocytes caused delayed cCa2+ clearance under basal and hormonal stimulation. Activation of AMPK by increased AMP:ATP is well-established in the literature, but our findings demonstrate that, despite greatly decreased ATP and increased AMP in MCU KO hepatocytes, there was decreased AMPK activation (Figures 1S and 4A). In follow-up studies, we tested whether loss of AMPK alone was sufficient to confer a fatty liver phenotype. Interestingly, liver-specific AMPK KO mice displayed similar hepatic lipid accumulation, confirming the central role of AMPK in MCU KO-induced hepatic lipid accumulation. Chelation of cCa2+ restored phospho-AMPK levels in MCU KO hepatocytes, confirming the effect of Ca2+-dependent regulation (Figure 4P).
It has been demonstrated that, in addition to its regulation by AMP, AMPK is activated by the upstream kinases LKB1 and CaMKK-II, which phosphorylate a conserved Thr172 in the a subunit (Hawley et al., 2005; Hurley et al., 2005; Woods et al., 2003). CaMKK-II activates AMPK via phosphorylation of the a subunit in the presence of increased cCa2+ (Hawley et al., 2005; Hurley et al., 2005). Because we propose a model of Ca2+-mediated regulation of AMPK, it is pertinent to question whether CaMKK-II plays a role in this mechanism because of its known Ca2+-induced activation.
However, we did not observe any significant alteration in CaMKK-II phosphorylation in MCUΔhep mice. This finding is also consistent with studies that have demonstrated that CaMKK-II has limited distribution in hepatic tissue in comparison with neurons and T cells (Anderson et al., 1998; Mihaylova and Shaw, 2011). Surprisingly, our biochemical analysis found significant levels of AMPK dephosphorylation without any effect on the upstream kinases LKB1 and CaMKK-II in MCU KO hepatocytes, suggesting a mechanism of AMPK regulation independent of the canonical signaling cascade.
Although ATP antagonizes AMPK activation, dephosphorylation-dependent inactivation of AMPK is mediated by serine-threonine phosphatase proteins, including PP2A and PP2Cα (Hardie, 2011). Our search for a Ca2+-dependent AMPK dephosphorylation mechanism revealed that pharmacologic inhibition of cCa2+ regulated PP4 by cantharidin and knockdown of PP4 restored AMPK phosphorylation and reduced hepatic lipid accumulation in MCU KO hepatocytes (Figure 5). Our study demonstrates direct regulation of AMPK through Ca2+-induced PP4 activity. To further test the physiological role of MCU, we created a unique gain-of-function MCU mouse model that displays higher mCa2+ uptake and rapidly clears cCa2+ (Figure 6). Interestingly, MCU C96A-KI mice not only exhibited higher MCU-mediated Ca2+ uptake but also reduced PP4 levels and lower circulating and hepatic TAGs in the fasted state.
The CA form of double ACC KI mice exhibits elevated lipogenesis and lower FAO, implying that AMPK activation is crucial for lipid clearance (Fullerton et al., 2013). Our data provide evidence that sustained cCa2+ elevation in MCUΔhep mice results in activation of ACC because of AMPK dephosphorylation and, consequently, lipid accumulation. Strikingly, our MCU C96A-KI mice restored AMPK and ACC phosphorylation states and rapid clearance of hepatic lipid during fasting. The insulin-sensitizing role of metformin has been linked to AMPK phosphorylation in the setting of obesity and insulin resistance (Howell et al., 2017; Li et al., 2011). Administration of metformin in MCUΔhep mice improved lipid clearance through MCU-independent AMPK phosphorylation, supporting a therapeutic intervention for disorders of hepatic insulin resistance and lipid accumulation.
STAR⋆METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to and will be fulfilled by the Lead Contact, Muniswamy Madesh (muniswamy@uthscsa.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse models
Hepatocyte-specific Mcu knockouts (MCUΔhep) were generated by crossing MCUfl/fl mice (Luongo et al., 2015) with hepatocyte-specific Cre recombinase transgenic mice (B6.Cg-Tg(Alb-cre)21Mgn/J, The Jackson Laboratory, USA). Hepatic AMPK-KO (AMPKΔhep) mice were generated by retro orbital injection of the 1×1011 viral particles of adenovirus associated virus 8-Cre (AAV8-Cre under the control of the TGB promoter) in the AMPKα1/α2fl/fl mice which have the Lox-P site on both AMPKα1 and AMPKα2 loci. Mice were allowed to rest for 2 weeks before the experiment. To generate MCU-C96A knock in mice, gRNA1, gRNA2, ssODN containing BssHII site along with Cas9 protein was microinjected in single-celled embryo and transferred to surrogate mother. Details of oligo’s sequence used for C96A-KI are given in Figure S5B. The C96A-KI is confirmed by restriction fragment analysis using BssHII restriction enzyme. All mice were grouped according to same sex and age for all the experiments. For studies utilizing primary hepatocytes in vitro, both male and female mice were used. However, male mice were used for all in vivo experiments. All animal experiments were approved by Temple University’s IACUC and followed AAALAC guidelines.
Zebrafish
To generate zebrafish mcu mutant, three different sgRNA target sequences were designed (with PAM sequences in bold, #1: GCC GGGTTTCACTTCAGAGATGG (+strand), #2: GGCTGCGAAAGTGTGTAGATCGG (+ strand), and #3: TGTGCCCTGATGCCTCTGT GAGG (−strand). sgRNAs were in vitro transcribed and co-injected with mRNA coding for nCas9n into 1-cell zebrafish embryos. Activity of sgRNAs was tested by loss of underlined restriction enzyme sites Hpy118I (TCNGA), Sau3AI (GATC) and EcoNI (#3, CCTN5AGG). Primers mcu-F1 (TAGAGACCGTGGTATTTCAGTCTAC) and mcu-R1 (TGCACTGCACAGTGATGATCAGCA) were used for genotyping. A deletion allele tpl124, generated using sgRNA#2, containing a 268 base pair deletion encompassing parts of exon 1 and intron 1, was selected for further study. All Zebrafish experiments were approved by Temple University’s IACUC and followed AAALAC guidelines.
Cell culture
Primary hepatocytes were grown in Williams E medium containing 1% (v/v) antibiotic-antimycotic solution (GIBCO), 1% (v/v) 200mM L-glutamine, and 1% (v/v) non-essential amino acids under standard culture condition. HepG2 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM)/10% FBS, supplemented with 1% (v/v) antibiotic-antimycotic solution (GIBCO) at 5% CO2 and 37 C.
METHOD DETAILS
Isolation of mouse primary hepatocytes
Primary adult mouse hepatocytes were isolated from 10–12 week old male and female animals using a two-step collagenase perfusion technique with slight modifications (Li et al., 2010). In brief, mice liver was sequentially perfused with perfusion medium-I (DPBS containing 10mM HEPES, 0.05% w/v KCl, 5mM Glucose, 200 μM EDTA, pH 7.4) and perfusion medium-II (DPBS containing 30mM HEPES, 0.05% w/v KCL, 5mM Glucose, 1mM CaCl2, pH 7.4) containing collagenase D (400 μg/ml). Liver lobes were dissected, dissociated, and crude hepatocyte preparation passed through the gauze mesh filter (100 μM diameter). The crude hepatocyte preparation was centrifuged at the speed of 50 g for 2 min to pellet down parenchymal hepatocytes. The hepatocytes were washed five times with perfusion medium-II using the above-mentioned centrifuge conditions. Following the wash, cells were plated in culture dishes in hepatocyte attachment medium (Williams E medium containing 1% (v/v) antibiotic-antimycotic solution (GIBCO), 1% (v/v) 200mM L-glutamine, 1% (v/v) non-essential amino acids, and 10% heat inactivated fetal bovine serum). The next day, attachment media was replaced with hepatocyte culture medium (Williams E medium containing 1% (v/v) antibiotic-antimycotic solution (GIBCO), 1% (v/v) 200mM L-glutamine, and 1% (v/v) non-essential amino acids).
Mitoplast IMCU patch-clamp recording
Mitoplast patch-clamp recordings for IMCU current were performed at 30 C as reported earlier (Chaudhuri et al., 2013; Hoffman et al., 2013; Joiner et al., 2012; Kirichok et al., 2004; Tomar et al., 2016) with slight modifications. In brief, mitoplasts isolated from hepatocytes of MCUfl/fl and MCUΔhep mice were bathed in a solution containing 150mM sodium gluconate, 5.4mM KCl, 5mM CaCl2, and 10mM HEPES (pH 7.2). The pipette solution contained 150mM sodium gluconate, 5mM NaCl, 135mM sucrose, 10mM HEPES, and 1.5mM EGTA (pH 7.2). IMCU currents were recorded using an Axon200B patch-clamp amplifier with a Digidata 1320A acquisition board (pCLAMP 10.0 software; Axon Instruments). The external/bath solution (5 mM Ca2+) was chosen on the basis of previous measurements.
Measurement of mitochondrial Ca2+ uptake, ΔΨm, and mitochondrial matrix Ca2+
Mitochondrial Ca2+ uptake and ΔΨm were measured as described earlier using dual-wavelength emission fluorimeter with slight modifications (Mallilankaraman et al., 2012a; Mallilankaraman et al., 2012b; Shanmughapriya et al., 2015a; Tomar et al., 2016). Freshly isolated mouse primary hepatocytes were washed with Ca2+/Mg2+ free DPBS (GIBCO). An equal number of cells (5×106 cells) were permeabilized with 40 μg/ml digitonin in 1.5 mL of intracellular medium (ICM-120 mM KCl, 10 mM NaCl, 1 mM KH2PO4, 20 mM HEPES-Tris, pH 7.2) supplemented with 2 μM thapsigargin to block the SERCA pump and 5mM succinate to energize the mitochondria. Fura-FF (0.5 mM) was loaded at 0 s time point for the measurement of extramitochondrial Ca2+ ([Ca2+]out). Fluorescence emissions were monitored in a multi-wavelength excitation dual-wavelength emission fluorimeter (Delta RAM, PTI). The [Ca2+]out is shown as the ratio of Fura-FF/FA fluorescence (excitation 340 nm/380 nm) and ΔΨm as the ratio of the JC-1 fluorescence of J-aggregate (570 nm excitation/595 nm emission) and monomer (490 nm excitation/535 nm emission) forms of JC-1. The ΔΨm indicator, JC-1 (800 nM), a bolus of 10 μM Ca2+, and the mitochondrial uncoupler, FCCP (3 μM), were added at the indicated time points with constant stirring at 37 C. To measure the mitochondrial matrix Ca2+, hepatocytes were permeabilized in 40 mg/ml digitonin in 1.5 mL of intracellular medium supplemented with thapsigargin, Ru360 (1 μM, to block mitochondrial Ca2+ uptake), CGP37157 (1 μM, to block mitochondrial Ca2+ efflux), and Fura-FF (0.5 μM). Mitochondrial uncoupler, FCCP (10 μM) was added at indicated time points without adding any Ca2+ bolus to release the mitochondrial Ca2+ store.
Mitochondrial and cytosolic Ca2+ measurements using confocal live cell imaging system
Primary zebrafish cells were grown on Poly-L-lysine coated glass coverslips and loaded with mCa2+ sensor Rhod-2 (2 μM) for 45 min in extracellular media (ECM-120mM NaCl, 5mM KCl, 1mM KH2PO4, 0.2mM MgCl2, 0.1mM EGTA, 20mM HEPES, pH 7.4). Cells were washed and imaged in ECM using Carl Zeiss LSM510 confocal live cell imaging system using the 560 nm excitation (Ex) laser and 580 nm emission (Em) spectra is collected. Ionomycin was added at indicated time points to increase cCa2+ levels. For cCa2+ measurements, primary hepatocytes were grown on 25-mm glass coverslips and transduced with GCaMP6 encoding adenovirus. Coverslips were mounted in an open perfusion micro-incubator (PDMI-2; Harvard Apparatus) at 37 C and imaged using the Carl Zeiss LSM510 confocal live cell imaging system using the Ex-488 nm and Em-510 nm. After 1 min of baseline recording, vasopressin (100nM) or thapsigargin (2 μM) was added, and confocal images were obtained every 3 s at 488-nm excitation using a 40x oil objective. Images were analyzed and quantitated using ZEN 2010 and ImageJ software.
Mitochondrial respiration measurements
Primary hepatocytes isolated from MCUfl/fl and MCUΔhep mice were plated in Poly-L-lysine coated 96 well Seahorse culture plates. Oxygen consumption rate (OCR) was measured at 37 C in an XF96 extracellular flux analyzer (Seahorse Bioscience). Respiratory chain inhibitors were added sequentially at indicated time points. For the measurement of FAO coupled OCR, cells were glucose depleted overnight and treated with carnitine palmitoyl transferase-1 inhibitor Etomoxir (40 μM) for 1 hour to monitor endogenous fatty acid utilization. Exogenous palmitate is used as fatty acid substrate for the OCR measurement using XF96 extracellular flux analyzer (Seahorse Bioscience).
Cellular AMP/ATP measurement
Mouse hepatocyte and zebrafish total cellular ATP levels were measured using CellTiter-Glo luminescent assay kit (Promega, Madison, WI, USA) as per the manufacturer’s protocol. AMP levels were measured using the AMP-Glo luminescent assay kit (Promega, Madison, WI, USA). Luminescence was measured using a microplate reader (Infinite M1000 PRO, Tecan).
Mitochondrial reactive oxygen species measurement
Primary hepatocytes isolated from MCUfl/fl and MCUΔhep mice were plated on Poly-L-lysine coated glass coverslips. Next day, cells were loaded with the mitochondrial superoxide sensitive fluorophore MitoSOX Red (Life Technologies; 2 μM) and Dihydrorhodamine 123 (Rhod123; 2.5 μg/ml) in William’s E medium (without serum) at 37 C for 30 min. Cells were then washed and imaged using a Carl Zeiss 510 confocal microscope with a 40 3 oil immersion objective at 561 nm as described earlier (Mukhopadhyay et al., 2007). MitoSOX fluorescence was quantified using ImageJ software and plotted as arbitrary units in Graphpad Prism 6 software.
TMRM staining for ΔΨm
Mouse primary hepatocytes and zebrafish primary cells were placed on Poly-L-lysine coated glass coverslips. The next day, cells were stained with Tetramethylrhodamine, methyl ester (TMRM; 50 nM) and Dihydrorhodamine 123 (Rhod123; 2.5 μg/ml) for 30 min at 37 C. Images were acquired using a Carl Zeiss 510 confocal microscope using a 40 × oil objective at excitations of 561 nm and 488 nm, respectively. Images were quantified for TMRM fluorescence using ImageJ software and plotted as arb unit in Graphpad Prism 6 software.
Metabolic assessment of mice
The metabolic status of MCUfl/fl and MCUΔhep male mice were assessed using a Comprehensive Laboratory Animal Monitoring System (CLAMS; Columbus Instruments, Columbus, OH) as described earlier (Tomar et al., 2016). Mice were single housed in 12 hour light/dark cycles with a normal chow diet and water ad libitum. Mice were allowed to acclimate for 3 days and then oxygen consumption, carbon dioxide production, food intake and locomotor activity were measured continuously for 3 days (24 hr Fed, 24 hr Fasted and 24hrs Re-fed state) at an ambient temperature of 22 C. During the fasted state, the chow diet is restricted for 24 hours. The respiratory exchange ratio is the ratio of carbon dioxide production and oxygen consumption. For the oxidation of carbohydrate six molecules of oxygen is utilized with generation of six molecules of CO2, resulting a RER of 1.0. However, for the oxidation of one fatty acid molecule 23 oxygen molecules are utilized with a generation of 16 CO2 molecules, resulting in a RER of 0.7. Therefore, we can calculate the percentage of fat utilization by using following equation:
Animal body composition, liver/plasma biochemistry, glucose tolerance test and glucose output assay
Normal chow diet fed 10 weeks old male mice used for body fat composition analysis by nuclear magnetic resonance (NMR) and dual emission X-ray absorptiometry (DEXA) (Echo Medical Systems, Houston, TX). Enzymatic colorimetric assay was performed to monitor liver/plasma triglyceride, plasma ketones, liver glycogen, and blood glucose as described earlier (Alenghat et al., 2008; Tomar et al., 2016). For the glucose tolerance test, 12 week old male mice were fasted overnight for approximately 16 hours by transferring mice to clean cages. Next day, mouse body weight (wt) was measured and 20% (w/v) glucose solution (2g of glucose/kg body mass) was injected intraperitoneally. The blood glucose levels were measured at the indicated time points through the tail puncture using the OneTouch Ultra Glucose Meter. The glucose output assay was performed on primary hepatocytes isolated from 12-weeks old MCUfl/fl and MCUΔhep mice. The freshly isolated hepatocytes were maintained in Williams E medium containing 1% (v/v) antibiotic-antimycotic solution (GIBCO), 1% (v/v) 200mM L-glutamine, 1% (v/v) non-essential amino acids, and 10% heat inactivated fetal bovine serum overnight. The following day, the media was changed to glucose output media (GOM: 118 mM NaCl, 4.7 mM KCl, 1.2 mM MgSO4, 1.2 mM KH2PO4, 1.2mM CaCl2, 20mM NaHCO3, 20mM HEPES pH 7.4, and BSA 0.025%) for 2h before induction of glycogenolysis. The cells were stimulated with 5nM Glucagon in GOM for 2h, and culture supernatants were collected and glucose content was measured by Glucose (HK) Assay Kit.
Lipid staining in primary hepatocytes, liver sections and whole zebrafish
Primary mice hepatocytes were isolated from MCUfl/fl and MCUΔhep and cultured in Poly-L-lysine coated glass coverslips with Williams E medium containing 5mM Glucose (low glucose condition) or 25mM Glucose (high glucose condition) for 24 hours. The next day, the medium for the hepatocytes was replaced with Earle’s Balanced Salt Solution (EBSS) to induce starvation for 2 hours. After incubation, cells were loaded with BODIPY® 493/503 (1 μg/ml) in serum-free medium for 30 min. Cells were washed and images were acquired using a Carl Zeiss 510 confocal microscope using a 40 × oil objective at 488 nm excitation. Images were quantified for the size and number of lipid droplets using ImageJ software. For the histological evaluation of hepatic lipid accumulation, mice liver was perfused with PBS and subsequently with 4% paraformaldehyde to fix the tissue. Liver tissue was embedded in paraffin to cut the 5 μM thin tissue sections and subsequently stained for Oil Red O and hematoxylin and eosin (H&E). Histological images were acquired in bright field light microscope (Nikon, USA). Zebrafish lipid content was visualized by staining with Lipid Green. Wild-type and MCU-KO zebrafish were incubated in 100 μM of Lipid Green for 15 min (Lee et al., 2011). After incubation, zebrafish were anesthetized in tricaine containing water and in vivo confocal microscopy was performed using the Carl Zeiss LSM710 two-photon confocal system.
Electron microscopy to visualize hepatic ultrastructure
MCUfl/fl and MCUΔhep mice were anesthetized, and the liver was perfused with PBS for 2 minutes at 35 C followed by 0.15 M cacodylate buffer (Ted Pella Inc., Redding, CA) pH7.4 containing 2.5% glutaraldehyde (Electron Microscopy Sciences, Hartfield, PA), 2% formaldehyde (fresh from paraformaldehyde (EMS)) with 2mM CaCl2 and 20 mM KCl at 35 C for 5 minutes. Liver lobes from the animal were removed and fixed for an additional 2–3 h on ice in the same tissue fixing solution. Liver tissue was sectioned, and images were acquired using the Zeiss LIBRA120 TEM equipped with Gatan UltraScan, 1000 2k × 2k CCD EFTEM, and energy filtering.
Immunoblotting and co-immunoprecipitation assay
Immunoblotting for the detection of protein expression and phosphorylation was performed using the equal amount of total cell lysate prepared from primary mice hepatocytes or zebrafish. Proteins were extracted from primary hepatocytes isolated from mice liver or zebrafish using RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.25% deoxycholic acid, 1 mM EDTA, 1% NP-40 supplemented with protease inhibitor cocktail (Roche) and PhosSTOP Phosphatase Inhibitor Cocktail (Roche)). Protein concentrations were quantified using Pierce™ 660nm Protein Assay (Thermo Scientific Inc.) and equal amounts of protein were separated on 4%–12% Bis-Tris polyacrylamide gel. Proteins were transferred to a PVDF membrane, and probed with indicated antibodies. For co-immunoprecipitation, proteins were extracted from HepG2 cells transiently transfected with AMPK-HA and/or PP4-Flag using RIPA buffer. Indicated antibodies were used to pull-down the specific protein. The input (10% of total cell lysates) and immunoprecipitates were probed with specified antibody.
Global lipidomic profiling
Preparation of samples for lipidomic analysis
To monitor global lipidomics alterations in MCUΔhep mice, 12-week-old mice were used. MCUΔhep mice were fed with Metformin (1.25mg/ml metformin in drinking water) for three weeks. Mice liver were harvested and frozen in liquid nitrogen until the lipids were extracted. Liver sections were subjected to homogenization to uniformity in ice cold phosphate buffered saline (PBS) such that a final suspension of 10% w/v liver homogenate in PBS were obtained. 50 μL of this suspension was used in the extraction and analysis of liver lipids.
Extraction of lipids from liver tissues
50 μL of liver homogenate was diluted with 150 μL of PBS. To the mixture thus obtained, a mixture of internal standards were added followed by vortexing and incubating on ice for 10 minutes for equilibration of the IS with the sample. Following incubation, 1 mL of methanol was added to the sample followed by sonication to mix the samples. Thereafter 0.5 mL of chloroform was added and the samples were sonicated again to mix the solvents to obtain a well dispersed monophasic extraction mixture. This mixture was then incubated at 48 C for one hour to optimize the extraction of highly non polar lipids. Following incubation, a phase break implemented out by the addition of 1 mL of chloroform followed by 2mL of water. The resultant mixture was vortexed for 30 s and then centrifuged at 3500 g to enable phase separation. Following centrifugation, the bottom organic layer containing the lipids was transferred to a fresh tube. The top aqueous layer was washed with an additional 1 mL of CHCl3, vortexed, centrifuged and the separated organic layer was combined with the previous organic fraction. The lipid extracts thus obtained was dried via vacuum centrifugation and re-suspended in 200 mL of methanol. The re-suspended solution was incubated at 48 C for 10 minutes vortexed, and 50 μL transferred to 200 μL autosampler vials for analysis via ultra-performance liquid chromatography high resolution mass spectrometry.
Analysis of lipids by untargeted mass spectrometry
5 μL of the lipid extract prepared as detailed above was analyzed following separation on a Acquity CSH C18 reverse phase column (100Å, 3.5 μm, 3 mm × 150 mm, Waters) using a Shimadzu Nexera UPLC chromatographic system coupled to Sciex TripleTOF 5600+ quadrupole time of flight mass analyzer. Data was acquired in the positive mode in an untargeted data dependent manner in the mass range of 50–1200 Da. A 28 minute chromatographic separation utilizing a linear gradient of 60:40 acetonitrile:water with 10mM ammonium formate and 0.1% formic acid as mobile phase A and 90:10 isopropanol:acetonitrile with 10mM ammonium formate and 0.1% formic acid as mobile phase B.
Data Analysis
The acquired data was subjected to post processing via MarkerView software (Sciex LLC). Following normalization to total ion current and alignment of retention times, features were extracted as m/z retention time pairs. The extracted features were subjected statistical analysis using Metaboanalyst. The data was first subjected to log transformation followed by centering using Pareto scaling (mean-centered and divided by the square of the standard deviation of each variable). Cluster analysis was performed using Euclidean distance measure and Ward clustering algorithm. ANOVA and Tukey’s HSD post hoc analysis with an FDR = 0.001 threshold was used to identify the top feature differences between the three groups. Those features identified as being significantly different were then identified via comparison against Lipidmaps database as well as the elution retention time window for that lipid class.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data were expressed as the mean ± SE and statistical significance was evaluated via Student’s unpaired t test, one-way and twoway ANOVA with Tukey’s HSD post hoc analysis, where appropriate. p < 0.05 was considered statistically significant. All experiments were conducted at least three times unless specified. Replicates and statistical analysis information is reported in the figure legends. Data were plotted either with Sigma Plot 11.0 software or GraphPad Prism version 6 software.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit Polyclonal anti- ACC | Cell Signaling Technology | Cat# 3662 |
| Rabbit Polyclonal anti- AMPKα | Cell Signaling Technology | Cat# 2532 |
| Rabbit Polyclonal anti- ATF-6α | Santa Cruz Biotechnology Inc. | Cat# sc-166659 |
| Mouse monoclonal anti- ATP5A | Abeam | Cat# ab14748 |
| Rabbit monoclonal anti- CaMKII-α (D10C11) | Cell Signaling Technology | Cat# 11945 |
| Rabbit Polyclonal anti- Carnitine Palmitoyl Transferase-1 | Alpha Diagnostic Intl. Inc. | Cat# CPT1M11-A |
| Mouse monoclonal anti- ERK1/ERK2 | Thermo Scientific Pierce | Cat# MA5–15605 |
| Rabbit monoclonal anti- Fatty Acid Synthase (C20G5) | Cell Signaling Technology | Cat# 3180 |
| Mouse monoclonal anti- FLAG M2-Peroxidase (HRP) | Sigma-Aid rich | Cat# A8592–1MG |
| Mouse monoclonal anti- FLAG M2 | Sigma-Aid rich | Cat# F1804–1MG |
| Rat monoclonal anti- HA-Peroxidase | Sigma-Aid rich | Cat# 12013819001 |
| Rabbit monoclonal anti- LKB1 (D60C5) | Cell Signaling Technology | Cat# 3047 |
| Rabbit Polyclonal anti- MCU | Sigma-Aid rich | Cat# HPA016480 |
| Rabbit Polyclonal anti- MCUR1 | Tomar et al., 2016 | N/A |
| Rabbit Polyclonal anti- MICU1 | Tomar et al., 2016 | N/A |
| Rabbit Polyclonal anti- mtTFA | Abeam | Cat# ab138351 |
| Rabbit Polyclonal anti- NRF1 | Cell Signaling Technology | Cat# 12381 |
| Mouse monoclonal Total OXPHOS Rodent WB Antibody Cocktail | Abeam | Cat# ab110413 |
| Rabbit Polyclonal anti- Phospho- ACC (Ser79) | Cell Signaling Technology | Cat# 3661 |
| Rabbit Polyclonal anti- Phospho-AMPKα (Thr172) | Cell Signaling Technology | Cat# 2531 |
| Rabbit monoclonal anti- Phospho-CaMKII (Thr286) (D21E4) | Cell Signaling Technology | Cat# 12716S |
| Rabbit Polyclonal anti- Phospho-p44/42 MAPK (Erk½) (Thr202/Tyr204) | Cell Signaling Technology | Cat# 9101 |
| Rabbit monoclonal anti- Phospho-PERK (Thr980) (16F8) | Cell Signaling Technology | Cat# 3179 |
| Rabbit Polyclonal anti- PGC1 alpha | Abeam | Cat# ab54481 |
| Mouse monoclonal anti- PPX (C-6) | Santa Cruz Biotechnology Inc. | Cat# sc-374106 |
| Rabbit Polyclonal anti- SREBP-1 (C-20) | Santa Cruz Biotechnology Inc. | Cat# sc-366 |
| Rabbit Polyclonal anti- Tom20 (FL-145) | Santa Cruz Biotechnology Inc. | Cat# sc11415 |
| Rabbit monoclonal anti- XBP-1 s (D2C1 F) | Cell Signaling Technology | Cat# 12782 |
| Mouse monoclonal anti- β-Actin (C4) | Santa Cruz Biotechnology Inc. | Cat# sc47778 |
| Goat anti-Mouse IgG (H+L)-HRP Conjugate | Bio-Rad Laboratories | Cat# 1721011 |
| Goat anti-rabbit IgG, HRP-linked | Cell Signaling Technology | Cat# 7074S |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Acetonitrile | Sigma-Aid rich | Cat# 271004 |
| Ammonium formate | Sigma-Aid rich | Cat# 516961 |
| Antibiotic-antimycotic solution | Thermo Fisher Scientific | Cat# 15240112 |
| Antimycin A | Sigma-Aid rich | Cat# A8674 |
| BAPTA-AM | Thermo Fisher Scientific | Cat# B6769 |
| Bis-Tris polyacrylamide gel (4–12%) | Thermo Fisher Scientific | Cat # WG1402BOX |
| BODIPY® 493/503 | Thermo Fisher Scientific | Cat# D3922 |
| CaCI2 | Sigma-Aldrich | Cat# 21115 |
| Cacodylate buffer | Electron Microscopy Sciences | Cat# 11652 |
| CGP37157 | Sigma-Aldrich | Cat# C8874 |
| Chloroform | Sigma-Aldrich | Cat# CX1055 |
| Collagenase D | Sigma-Aldrich | Cat# 11088866001 |
| Digitonin | Sigma-Aldrich | Cat# D141 |
| Dihydrorhodamine 123 | Thermo Fisher Scientific | Cat# D632 |
| DPBS Ca2+/Mg2+ free | Thermo Fisher Scientific | Cat# 14190136 |
| DPBS with Ca2+/Mg2+ | Thermo Fisher Scientific | Cat# 14040117 |
| Dulbecco’s modified Eagle’s medium (DMEM) | Thermo Fisher Scientific | Cat# 11885092 |
| Earie’s Balanced Salt Solution (EBSS) | Thermo Fisher Scientific | Cat# 24010043 |
| EDTA | Thermo Fisher Scientific | Cat# AM9260G |
| EGTA | Sigma-Aldrich | Cat# 03777 |
| Etomoxir | Sigma-Aldrich | Cat# 236020 |
| FCCP | Sigma-Aldrich | Cat# C2920 |
| Fetal bovine serum | HyClone-GE | Cat# SH30071.04 |
| Formic acid | Sigma-Aldrich | Cat# 33015 |
| Fura-FF | Sigma-Aldrich | Cat# 17085 |
| Glucose | Sigma-Aldrich | Cat# G8644–100ML |
| Glucagon | Sigma-Aldrich | Cat# G2044 |
| Glutaraldehyde | Electron Microscopy Sciences | Cat# 16020 |
| HEPES | Thermo Fisher Scientific | Cat# 15630080 |
| lonomycin | Thermo Fisher Scientific | Cat# I24222 |
| JC-1 | Thermo Fisher Scientific | Cat# T3168 |
| KCl | Sigma-Aldrich | Cat# P9333 |
| KH2PO4 | Sigma-Aldrich | Cat# P5655 |
| L-glutamine | Thermo Fisher Scientific | Cat# 25030164 |
| MgCl2 | Sigma-Aldrich | Cat# 449172 |
| MitoSOX Red | Thermo Fisher Scientific | Cat# M 36008 |
| NaCI | Sigma-Aldrich | Cat# 746398 |
| Non-essential amino acids | Thermo Fisher Scientific | Cat# 11140050 |
| Oil Red O | Sigma-Aldrich | Cat# O0625 |
| Oligomycin | Sigma-Aldrich | Cat# 75351 |
| Paraformaldehyde | Electron Microscopy Sciences | Cat# 15700 |
| PhosSTOP Phosphatase Inhibitor Cocktail | Sigma-Aldrich | Cat# 4906845001 |
| Poly-L-lysine | Sigma-Aldrich | Cat# P4707–50ML |
| complete Protease Inhibitor Cocktail | Sigma-Aldrich | Cat# 11697498001 |
| Rhod-2-AM | Thermo Fisher Scientific | Cat# R1245MP |
| RIPA buffer | EMD-Millipore | Cat# 20–188 |
| Rotenone | Sigma-Aldrich | Cat# 557368 |
| Ru360 | Sigma-Aldrich | Cat# 557440 |
| Sodium gluconate | Sigma-Aldrich | Cat# S2054 |
| Sucrose | Sigma-Aldrich | Cat# 84097 |
| Tetramethylrhodamine methyl ester | Thermo Fisher Scientific | Cat# T668 |
| Thapsigargin | Thermo Fisher Scientific | Cat# T7458 |
| Tricaine | Sigma-Aldrich | Cat# E10521 |
| Vasopressin | Sigma-Aldrich | Cat# 1711100 |
| William’s E medium | Thermo Fisher Scientific | Cat# 32551020 |
| Critical Commercial Assays | ||
| Pierce™ 660nm Protein Assay | Thermo Fisher Scientific | Cat# 22662 |
| CellTiter-Glo Assay | Promega | Cat# G7571 |
| AMP Glo Assay | Promega | Cat# V5011 |
| ALT Activity Assay | Sigma-Aid rich | Cat# MAK052 |
| β-hydroxybutyrate Assay | Cayman | Cat# 700190 |
| Glucose (HK) Assay Kit | Sigma-Aid rich | Cat# GAHK20 |
| Triglyceride Assay | Cayman | Cat# 10010303 |
| Experimental Models: Cell Lines | ||
| HepG2 | ATCC | Cat# HB-8065 |
| Primary Mouse Hepatocytes | This Study | N/A |
| Experimental Models: Organisms/Strains | ||
| C57BL/6J Mice | The Jackson Laboratory, USA | Stock# 000664 |
| MCUfl/fl mice | Luongo et al., 2015 | N/A |
| B6.Cg-Tg(Alb-cre)21 Mgn/J | The Jackson Laboratory, USA | Stock# 003574 |
| MCUΔhep mice | This Study | N/A |
| AMPKα1/α2fl/fl mice | Foretz et al., 2010 | N/A |
| AMPK α1/α2Δhepmice | This Study | N/A |
| MCU-C96A-KI mice | This Study | N/A |
| MCU-KO zebrafish | This Study | N/A |
| Oligonucleotides | ||
| PP4-siRNA (Smart Pool) | Dharm aeon | Cat# L-040058–02-0005 |
| Recombinant DNA | ||
| Ad-T172DAMPKα1 (CA-AMPK) | This Study | N/A |
| Ad-MCU-Flag | This Study | N/A |
| Ad-GCaMP6.m | This Study | N/A |
| pECE HA AMPKα1 AS | Addgene | Plasmid # 69831 |
| pEBG-T172D AMPKaα1 | King et al., 2009 | N/A |
| PP4-Flag | OriGene Technologies Inc. | Cat# MR2 04327 |
| Software and Algorithms | ||
| Fiji ImageJ software | Fiji ImageJ | https://fiji.sc/ |
| Sigma Plot 11.0 | Systat Software Inc. | https://systatsoftware.com/products/sigmaplot/ |
| Zen 2010 | Carl Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software/zen.html |
| Graphpad Prism 6 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Metaboanalyst | Xia Lab at McGill Univeristy | https://www.metaboanalyst.ca/ |
| MarkerView software | Sciex LLC | https://sciex.com/products/software/markerview-software |
| Canvas 11.0 | ACD Systems | https://www.canvasgfx.com/en/support/canvas-11/ |
Highlights.
Mitochondrial Ca2+ powers FAO-dependent hepatocyte mitochondrial respiration
Hepatic MCU deletion promotes lipid accumulation and lowers ketone bodies
Blockade of mCa2+ buffering enhances AMPK dephosphorylation through PP4
Restoration of AMPK activity in MCUΔhep model improves lipid clearance
ACKNOWLEDGMENTS
We thank Anne Brunet for sharing the AMPK-HA construct. The AMPKα1 T172D-expressing plasmid was provided by Dr. Kenneth R. Hallows (University of Southern California Keck School of Medicine, Los Angeles, CA, USA). We thank Dr. John W. Elrod for sharing the MCUfl/fl mice. We thank Dr. Mitchell A. Lazar and Dr. Kevin J. Foskett (University of Pennsylvania, Philadelphia, PA, USA) for critically reading the manuscript, helpful thoughts, and suggestions. We also thank Jean L. Ross and Shannon Modla for EM sample preparation and image acquisition. This research was funded by the NIH (R01GM109882, R01HL086699, R01HL142673, and 1S10RR027327 to M. Madesh and U01HD087198 to D.S.W.). D.T. is supported by American Heart Association postdoctoral fellowship grant 17POST33660251. This work also received support via a Young Investigator Award from SCIEX for clinical lipidomic research (to D.S.W.). Services and products in support of the research project were generated by the VCU Massey Cancer Center Lipidomics Shared Resource (Developing Core) supported in part NIH-NCI Cancer Center support grant P30 CA016059. Metabolic profiling was performed by the University of Pennsylvania Diabetes Research Center Mouse Phenotyping, Physiology and Metabolism Core (NIH grant P30-DK19525).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found with this article online at https://doi.org/10.1016/j.celrep.2019.02.107.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Alenghat T, Meyers K, Mullican SE, Leitner K, Adeniji-Adele A, Avila J, Bućan M, Ahima RS, Kaestner KH, and Lazar MA (2008). Nuclear receptor corepressor and histone deacetylase 3 govern circadian metabolic physiology. Nature 456, 997–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson KA, Means RL, Huang Q-H, Kemp BE, Goldstein EG, Selbert MA, Edelman AM, Fremeau RT, and Means AR (1998). Components of a calmodulin-dependent protein kinase cascade. Molecular cloning, functional characterization and cellular localization of Ca2+/calmodulin-dependent protein kinase kinase β. J. Biol. Chem 273, 31880–31889. [DOI] [PubMed] [Google Scholar]
- Balaban RS (2009). The role of Ca(2+) signaling in the coordination of mitochondrial ATP production with cardiac work. Biochim. Biophys. Acta 1787, 1334–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, et al. (2011). Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476, 341–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi P (1999). Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol. Rev 79, 1127–1155. [DOI] [PubMed] [Google Scholar]
- Biasutto L, Azzolini M, Szabò I, and Zoratti M (2016). The mitochondrial permeability transition pore in AD 2016: An update. Biochim. Biophys. Acta 1863, 2515–2530. [DOI] [PubMed] [Google Scholar]
- Cárdenas C, Miller RA, Smith I, Bui T, Molgó J, Müller M, Vais H, Cheung KH, Yang J, Parker I, et al. (2010). Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 142, 270–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti R, Ji WK, Stan RV, de Juan Sanz J, Ryan TA, and Higgs HN (2018). INF2-mediated actin polymerization at the ER stimulates mitochondrial calcium uptake, inner membrane constriction, and division. J. Cell Biol 217, 251–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri D, Sancak Y, Mootha VK, and Clapham DE (2013). MCU encodes the pore conducting mitochondrial calcium currents. eLife 2, e00704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark KJ, Balciunas D, Pogoda HM, Ding Y, Westcot SE, Bedell VM, Greenwood TM, Urban MD, Skuster KJ, Petzold AM, et al. (2011). In vivo protein trapping produces a functional expression codex of the vertebrate proteome. Nat. Methods 8, 506–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins TJ, Berridge MJ, Lipp P, and Bootman MD (2002). Mitochondria are morphologically and functionally heterogeneous within cells. EMBO J. 21, 1616–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Stefani D, Raffaello A, Teardo E, Szabò I, and Rizzuto R (2011). A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476, 336–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Stefani D, Rizzuto R, and Pozzan T (2016). Enjoy the Trip: Calcium in Mitochondria Back and Forth. Annu. Rev. Biochem 85, 161–192. [DOI] [PubMed] [Google Scholar]
- Denton RM, and McCormack JG (1980). The role of calcium in the regulation of mitochondrial metabolism. Biochem. Soc. Trans 8, 266–268. [DOI] [PubMed] [Google Scholar]
- Dong Z, Shanmughapriya S, Tomar D, Siddiqui N, Lynch S, Nemani N, Breves SL, Zhang X, Tripathi A, Palaniappan P, et al. (2017). Mitochondrial Ca2+ Uniporter Is a Mitochondrial Luminal Redox Sensor that Augments MCU Channel Activity. Mol. Cell 65, 1014–1028.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drago I, Pizzo P, and Pozzan T (2011). After half a century mitochondrial calcium in- and efflux machineries reveal themselves. EMBO J. 30, 4119–4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearnley CJ, Roderick HL, and Bootman MD (2011). Calcium signaling in cardiac myocytes. Cold Spring Harb. Perspect. Biol 3, a004242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fieni F, Lee SB, Jan YN, and Kirichok Y (2012). Activity of the mitochondrial calcium uniporter varies greatly between tissues. Nat. Commun 3, 1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foretz M, Hébrard S, Leclerc J, Zarrinpashneh E, Soty M, Mithieux G, Sakamoto K, Andreelli F, and Viollet B (2010). Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J. Clin. Invest 120, 2355–2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu S, Yang L, Li P, Hofmann O, Dicker L, Hide W, Lin X, Watkins SM, Ivanov AR, and Hotamisligil GS (2011). Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 473, 528–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullerton MD, Galic S, Marcinko K, Sikkema S, Pulinilkunnil T, Chen ZP, O’Neill HM, Ford RJ, Palanivel R, O’Brien M, et al. (2013). Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat. Med 19, 1649–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilabert JA, and Parekh AB (2000). Respiring mitochondria determine the pattern of activation and inactivation of the store-operated Ca(2+) current I(CRAC). EMBO J. 19, 6401–6407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glancy B, and Balaban RS (2012). Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry 51, 2959–2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glancy B, Willis WT, Chess DJ, and Balaban RS (2013). Effect of calcium on the oxidative phosphorylation cascade in skeletal muscle mitochondria. Biochemistry 52, 2793–2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajnóczky G, Hager R, and Thomas AP (1999). Mitochondria suppress local feedback activation of inositol 1,4, 5-trisphosphate receptors by Ca2+. J. Biol. Chem 274, 14157–14162. [DOI] [PubMed] [Google Scholar]
- Hajnóczky G, Csordás G, Madesh M, and Pacher P (2000). The machinery of local Ca2+ signalling between sarco-endoplasmic reticulum and mitochondria. J. Physiol 529, 69–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG (2011). AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev. 25, 1895–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG (2015). AMPK: positive and negative regulation, and its role in whole-body energy homeostasis. Curr. Opin. Cell Biol 33, 1–7. [DOI] [PubMed] [Google Scholar]
- Hardie DG, Ross FA, and Hawley SA (2012). AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol 13, 251–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins BJ, Irrinki KM, Mallilankaraman K, Lien YC, Wang Y, Bhanumathy CD, Subbiah R, Ritchie MF, Soboloff J, Baba Y, et al. (2010). S-glutathionylation activates STIM1 and alters mitochondrial homeostasis. J. Cell Biol 190, 391–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley SA, Gadalla AE, Olsen GS, and Hardie DG (2002). The antidiabetic drug metformin activates the AMP-activated protein kinase cascade via an adenine nucleotide-independent mechanism. Diabetes 51, 2420–2425. [DOI] [PubMed] [Google Scholar]
- Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG, and Hardie DG (2005). Calmodulin-dependent protein kinase-b is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2, 9–19. [DOI] [PubMed] [Google Scholar]
- Hoffman NE, Chandramoorthy HC, Shamugapriya S, Zhang X, Rajan S, Mallilankaraman K, Gandhirajan RK, Vagnozzi RJ, Ferrer LM, Sreekrishnanilayam K, et al. (2013). MICU1 motifs define mitochondrial calcium uni-porter binding and activity. Cell Rep. 5, 1576–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell JJ, Hellberg K, Turner M, Talbott G, Kolar MJ, Ross DS, Hoxhaj G, Saghatelian A, Shaw RJ, and Manning BD (2017). Metformin Inhibits Hepatic mTORC1 Signaling via Dose-Dependent Mechanisms Involving AMPK and the TSC Complex. Cell Metab. 25, 463–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley RL, Anderson KA, Franzone JM, Kemp BE, Means AR, and Witters LA (2005). The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J. Biol. Chem 280, 29060–29066. [DOI] [PubMed] [Google Scholar]
- Izzo V, Bravo-San Pedro JM, Sica V, Kroemer G, and Galluzzi L (2016). Mitochondrial Permeability Transition: New Findings and Persisting Uncertainties. Trends Cell Biol. 26, 655–667. [DOI] [PubMed] [Google Scholar]
- Joiner ML, Koval OM, Li J, He BJ, Allamargot C, Gao Z, Luczak ED, Hall DD, Fink BD, Chen B, et al. (2012). CaMKII determines mitochondrial stress responses in heart. Nature 491, 269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn BB, Alquier T, Carling D, and Hardie DG (2005). AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 1, 15–25. [DOI] [PubMed] [Google Scholar]
- Kamer KJ, and Mootha VK (2014). MICU1 and MICU2 play nonredundant roles in the regulation of the mitochondrial calcium uniporter. EMBO Rep. 15, 299–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King JD Jr., Fitch AC, Lee JK, McCane JE, Mak DO, Foskett JK, and Hallows KR (2009). AMP-activated protein kinase phosphorylation of the R domain inhibits PKA stimulation of CFTR. Am. J. Physiol. Cell Physiol 297, C94–C101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirichok Y, Krapivinsky G, and Clapham DE (2004). The mitochondrial calcium uniporter is a highly selective ion channel. Nature 427, 360–364. [DOI] [PubMed] [Google Scholar]
- Kuznetsov AV, Usson Y, Leverve X, and Margreiter R (2004). Subcellular heterogeneity of mitochondrial function and dysfunction: evidence obtained by confocal imaging. Mol. Cell. Biochem 256–257, 359–365. [DOI] [PubMed] [Google Scholar]
- Kwong JQ, Lu X, Correll RN, Schwanekamp JA, Vagnozzi RJ, Sargent MA, York AJ, Zhang J, Bers DM, and Molkentin JD (2015). The Mitochondrial Calcium Uniporter Selectively Matches Metabolic Output to Acute Contractile Stress in the Heart. Cell Rep. 12, 15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, So JH, Jeon JH, Choi EB, Lee YR, Chang YT, Kim CH, Bae MA, and Ahn JH (2011). Synthesis of a new fluorescent small molecule probe and its use for in vivo lipid imaging. Chem. Commun. (Camb.) 47, 7500–7502. [DOI] [PubMed] [Google Scholar]
- Li WC, Ralphs KL, and Tosh D (2010). Isolation and culture of adult mouse hepatocytes. Methods Mol. Biol 633, 185–196. [DOI] [PubMed] [Google Scholar]
- Li Y, Xu S, Mihaylova MM, Zheng B, Hou X, Jiang B, Park O, Luo Z, Lefai E, Shyy JY, et al. (2011). AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 13, 376–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JC, Parks RJ, Liu J, Stares J, Rovira II, Murphy E, and Finkel T (2017). The In Vivo Biology of the Mitochondrial Calcium Uniporter. Adv. Exp. Med. Biol 982, 49–63. [DOI] [PubMed] [Google Scholar]
- Luongo TS, Lambert JP, Yuan A, Zhang X, Gross P, Song J, Shanmughapriya S, Gao E, Jain M, Houser SR, et al. (2015). The Mitochondrial Calcium Uniporter Matches Energetic Supply with Cardiac Workload during Stress and Modulates Permeability Transition. Cell Rep. 12, 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madiraju AK, Erion DM, Rahimi Y, Zhang XM, Braddock DT, Albright RA, Prigaro BJ, Wood JL, Bhanot S, MacDonald MJ, et al. (2014). Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature 510, 542–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallilankaraman K, Cárdenas C, Doonan PJ, Chandramoorthy HC, Irrinki KM, Golenár T, Csordás G, Madireddi P, Yang J, Müller M, et al. (2012a). MCUR1 is an essential component of mitochondrial Ca2+ up-take that regulates cellular metabolism. Nat. Cell Biol 14, 1336–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallilankaraman K, Doonan P, Cárdenas C, Chandramoorthy HC, Müller M, Miller R, Hoffman NE, Gandhirajan RK, Molgó J, Birnbaum MJ, et al. (2012b). MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca(2+) uptake that regulates cell survival. Cell 151, 630–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayes PA, and Felts JM (1967). Regulation of fat metabolism of the liver. Nature 215, 716–718. [DOI] [PubMed] [Google Scholar]
- Mihaylova MM, and Shaw RJ (2011). The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol 13, 1016–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay P, Rajesh M, Haskó G, Hawkins BJ, Madesh M, and Pacher P (2007). Simultaneous detection of apoptosis and mitochondrial su-peroxide production in live cells by flow cytometry and confocal microscopy. Nat. Protoc 2, 2295–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemani N, Carvalho E, Tomar D, Dong Z, Ketschek A, Breves SL, Jaña F, Worth AM, Heffler J, Palaniappan P, et al. (2018). MIRO-1 Determines Mitochondrial Shape Transition upon GPCR Activation and Ca2+ Stress. Cell Rep 23, 1005–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Csordás P, Schneider T, and Hajnóczky G (2000). Quantification of calcium signal transmission from sarco-endoplasmic reticulum to the mitochondria. J. Physiol 529, 553–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paillard M, Csordás G, Szanda G, Golenár T, Debattisti V, Bartok A, Wang N, Moffat C, Seifert EL, Spät A, and Hajnóczky G (2017). Tissue-Specific Mitochondrial Decoding of Cytoplasmic Ca2+ Signals Is Controlled by the Stoichiometry of MICU½ and MCU. Cell Rep. 18, 2291–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, Fergusson MM, Rovira II, Allen M, Springer DA, et al. (2013). The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat. Cell Biol 15, 1464–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patron M, Checchetto V, Raffaello A, Teardo E, Vecellio Reane D, Mantoan M, Granatiero V, Szabò I, De Stefani D, and Rizzuto R (2014). MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol. Cell 53, 726–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne R, Hoff H, Roskowski A, and Foskett JK (2017). MICU2 Restricts Spatial Crosstalk between InsP3R and MCU Channels by Regulating Threshold and Gain of MICU1-Mediated Inhibition and Activation of MCU. Cell Rep. 21, 3141–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perocchi F, Gohil VM, Girgis HS, Bao XR, McCombs JE, Palmer AE, and Mootha VK (2010). MICU1 encodes a mitochondrial EF hand protein required for Ca(2+) uptake. Nature 467, 291–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RJ, Samuel VT, Petersen KF, and Shulman GI (2014). The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature 510, 84–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen MC, Vatner DF, and Shulman GI (2017). Regulation of hepatic glucose metabolism in health and disease. Nat. Rev. Endocrinol 13, 572–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plovanich M, Bogorad RL, Sancak Y, Kamer KJ, Strittmatter L, Li AA, Girgis HS, Kuchimanchi S, De Groot J, Speciner L, et al. (2013). MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS ONE 8, e55785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffaello A, De Stefani D, Sabbadin D, Teardo E, Merli G, Picard A, Checchetto V, Moro S, Szabò I, and Rizzuto R (2013). The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 32, 2362–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Markhard AL, Kitami T, Kovács-Bogdán E, Kamer KJ, Udeshi ND, Carr SA, Chaudhuri D, Clapham DE, Li AA, et al. (2013). EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 342, 1379–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanmughapriya S, Rajan S, Hoffman NE, Higgins AM, Tomar D, Nemani N, Hines KJ, Smith DJ, Eguchi A, Vallem S, et al. (2015a). SPG7 Is an Essential and Conserved Component of the Mitochondrial Permeability Transition Pore. Mol. Cell 60, 47–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanmughapriya S, Rajan S, Hoffman NE, Zhang X, Guo S, Kolesar JE, Hines KJ, Ragheb J, Jog NR, Caricchio R, et al. (2015b). Ca2+ signals regulate mitochondrial metabolism by stimulating CREB-mediated expression of the mitochondrial Ca2+ uniporter gene MCU. Sci. Signal 8, ra23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, Montminy M, and Cantley LC (2005). The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 310, 1642–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shui JW, Hu MC, and Tan TH (2007). Conditional knockout mice reveal an essential role of protein phosphatase 4 in thymocyte development and pre-T-cell receptor signaling. Mol. Cell. Biol 27, 79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomar D, Dong Z, Shanmughapriya S, Koch DA, Thomas T, Hoffman NE, Timbalia SA, Goldman SJ, Breves SL, Corbally DP, et al. (2016). MCUR1 Is a Scaffold Factor for the MCU Complex Function and Promotes Mitochondrial Bioenergetics. Cell Rep. 15, 1673–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berghe G (1991). The role of the liver in metabolic homeostasis: implications for inborn errors of metabolism. J. Inherit. Metab. Dis 14, 407–420. [DOI] [PubMed] [Google Scholar]
- Weibel ER, Stäubli W, Gnägi HR, and Hess FA (1969). Correlated morphometric and biochemical studies on the liver cell. I. Morphometric model, stereologic methods, and normal morphometric data for rat liver.J. Cell Biol 42, 68–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White C, Li C, Yang J, Petrenko NB, Madesh M, Thompson CB, and Foskett JK (2005). The endoplasmic reticulum gateway to apoptosis by Bcl-X(L) modulation of the InsP3R. Nat. Cell Biol. 7, 1021–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LGD, Neumann D, Schlattner U, Wallimann T, Carlson M, and Carling D (2003). LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr. Biol 13, 2004–2008. [DOI] [PubMed] [Google Scholar]
- Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, et al. (2001). Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Invest 108, 1167–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.