

Abstract
Clustered regularly interspaced short palindromic repeat (CRISPR) machineries are prokaryotic immune systems that have been adapted as versatile gene editing and manipulation tools. We found that CRISPR-Cas nucleases Cpf1 (also known as Cas12a) and Cas9 exhibit differential guide RNA sequence requirements for cleavage of the two strands of target DNA in vitro. As a consequence of the differential guide RNA requirements, both Cas9 and Cpf1 enzymes can exhibit potent nickase activities on an extensive class of mismatched dsDNA targets. These properties allow the production of efficient nickases for a chosen dsDNA target sequence, without modification of the nuclease protein, using guide RNAs with a variety of patterns of mismatch to the intended DNA target. In parallel to the nicking activities observed with purified Cas9 in vitro, we observed sequence-dependent nicking for both perfectly matched and partially mismatched target sequences in a Sacchromyces cerevisae system. Our findings have implications for CRISPR spacer acquisition, off-target potential of CRISPR gene editing/manipulation, and tool development using homology directed nicking.
Introduction
Bacteria and archaea are constantly challenged by invasive genetic elements (e.g. bacteriophage, transposons, and plasmids). To combat these threats, prokaryotes evolved an adaptive immune system known as CRISPR-Cas (clustered regularly interspaced short palindromic repeats and CRISPR-associated proteins)1–4. This immune system is able to capture foreign genetic elements into repeats in the CRISPR loci as short DNA segments. These captured “spacer” sequences are expressed in the context of precursor CRISPR RNAs (pre-crRNAs), which are processed into small CRISPR RNAs (referred to as guide RNAs or gRNAs)1–4. CRISPR-Cas proteins use the gRNA complementarity and often a protospacer adjacent motif (PAM) to recognize and cleave the target thus conferring immunity to the invading elements1–4. The CRISPR-Cas systems have been adapted as versatile genome editing tools that are ubiquitously used in many disciplines5.
Strepotococcus pyogenes Cas9 is a type II-A CRISPR-Cas nuclease that is widely studied and used in genome editing and epigenome manipulation6,7. Cas9 is a blunt cutting nuclease that can target specific DNA sequences using a NGG PAM and a gRNA8. Many variants of Cas9 have been developed (e.g. transcription activating/repressing, nicking, nuclease-dead, base editing etc.)9–11. One set of observations of particular note in reference to such variants are experiments in which nicking of targets can be used to guide homology-dependent gene editing, with apparent advantages in specificity12 and efficiency13 over double stranded break strategies. These assessments were carried out with mutant variants of Cas9 in which one of the nuclease domains has been inactivated. Sternberg et al.14 describe a conformational shift mechanism which would arguably coordinate the two nuclease domains to provide concerted double strand cleavage without substantial accumulation of nicked intermediates, while Szczelkun et al.15 (Figure 6Sb) describe one case of a truncated gRNA:target match that leads to slower coordination and accumulation in vitro of a nicked substrate. In addition, Strohkendl et al.16 reported temporary nicking with Cpf1 due to mismatches influencing the timescale of cleavage of each DNA strand.
Cpf1 is a minimal type V CRISPR-Cas nuclease that uses a single Cas endonuclease paired with a gRNA to cleave complementary DNA targets17–20. Cpf1 homologs from three species have been successfully adapted for genome editing: Acidaminococcus sp. (AsCpf1), Francisella novicida (FnCpf1), and Lachnospiraceae bacterium (LbCpf1)18. Cpf1 homologs recognizes T-rich PAMs (reported as TTTN/TTTV for As/Lb-Cpf1 and TTV for FnCpf1, theoretically opening a wide array of sites not available for Cas9 editing17,21. Cpf1 processes its own pre-CRISPR RNA, allowing this to be a potential platform for multi-gene functional analysis21,22. Despite numerous possible advantages, Cpf1 homologs have yet to be used as extensively in genome editing applications as Cas9.
Examining the consequences of Cpf1 and Cas9 activities on DNA topology for a broad set of potential substrates, we observed efficient nicking activities with both nucleases on specific classes of mismatched DNA targets. Given that either nicking or double stranded cleavage of DNA is sufficient to induce a variety of repair and replacement mechanisms in vivo13,23–27, the resulting profiles illuminate a dual capability of CRISPR-Cas nucleases to initiate genetic change through both types of interaction. These observations challenge binary models in which CRISPR-Cas9 nucleases either cleave or fail to cleave individual targets. Instead, our data indicate a scenario in which cleaving and nicking targets can and will coexist in a single experimental or natural condition.
Results
Sequence dependence for CRISPR-Cas nuclease activity was first addressed by building variant libraries of plasmids with a diversity of perfectly matched (“wild-type”) and mismatched (“mutant”) target sequences. Each sequence (wild-type or mutant) is represented by several barcoded species. We challenged the variant library with CRISPR-Cas nuclease protein programmed with single gRNA, and used sequencing to determine which templates remained following nuclease treatment. Additional information on the nature of the cleaved substrates comes from assays with and without a whole-library (backbone) linearization step after CRISPR-Cas nuclease cleavage of substrates and before PCR (Figure 1a). In particular, we note that backbone-intact assays provide a potential to distinguish nicked from closed circles due to the higher PCR yields observed from molecules where a nicking event has allowed the two plasmid strands to become topologically unlinked28.
Figure 1: High throughput assay for nicking and cleavage by CRISPR-Cas nucleases.

a) Schematic of in vitro high throughput plasmid libraries and subsequent steps used to assess representation or each sequence before and after CRISPR-Cas interaction. Each library consists of a uniform backbone into which a variety of potential target sites are inserted at a single location. Potential targets match one of several gRNA or control sequences (panel b); with each library including a diversity of both matched and mismatched sequences. To assess representation for each sequence before and after CRISPR-Cas interaction, samples were amplified with primers flanking the potential cleavage site and subjected to high throughput sequencing. Amplification and sequencing of an unreacted sample (no CRISPR-Cas interaction) provides a baseline fraction of normalization of each variant; this reference amplification and sequencing is carried out for every experiment and used for subsequent normalization. To assess topological state before and after CRISPR-Cas interaction, samples were split into two pools that are treated identically with the exception that one of the two subpools is cleaved at a site outside of the region to be amplified (“Method 1”), while the other pool is not subjected an outside cleavage (“Method 2”). Amplification of uncleaved circular templates (Method 2) is known to enrich for nicked over closed circular templates28.
b) Depiction of target sequences synthesized for the Cpf1 and Cas9 variant libraries. Regions in red indicate the PAM, blue indicates seed region (positions 1–10), green indicates the distal region (positions 11–20), and magenta is the barcode region. For each target, the following variants were synthesized: wild-type, single variants, single deletions, and double consecutive transversion variants.
Cpf1 nicking and cleavage specificity
Sequence dependence for LbCpf1 activity was investigated using a variant library of plasmids for four different canonical guide sequences: EGFP-1, EGFP-2, unc-22A, and rol-6 (Figure 1b). Initial assays were carried out with the backbone-linearization step (Method 1, Figure 1a). For each target, the libraries contained unmodified sequences, single-base variants, double-adjacent transversions, and deletions. Using loss-of-amplification across the cleavage junction as an assay, LbCpf1 showed a similar specificity profile to previous characterizations of Cas9, less tolerance of mutations in the seed region (positions 1–10, with position 1 being PAM-proximal) while sequence requirements in the distal region (positions 11–20) were more lax (left panel of Figure 2–3; left panel of Supplementary Figure 1–6). This trend is consistent for all three classes of variants, for all Cpf1s, and on all tested targets (left panel of Supplementary Figure 1–6).
Figure 2: Library-based assessment of nicking and cleavage activities of LbCpf1 on single mutant target variants.

Line graphs on left show retention profiles for whole-library assays with a post-reaction backbone cleavage step that avoid preferential recovery of nicked substrates. Line graphs on right show retention profiles without such a cleavage step (i.e., with preferential recovery of nicked sequences). For the assays shown in this figure, nuclease LbCpf1 was programmed with an EGFP-1 gRNA and interacted with a mixed target library as described in Figure 1a. Retention scores are shown for single base transversions at each indicated position in the EGFP-1 target, and have been normalized using median retention for a set of unrelated target sequences included in the library [unc-22A, rol-6, and EGFP-2]) (Sequencing Data: AF_SOL_820; For details on retention score calculation, see “Materials and Methods”.).
Figure 3: Library-based assessment of nicking and cleavage activities of LbCpf1 on double consecutive transversion and deletion target variants.
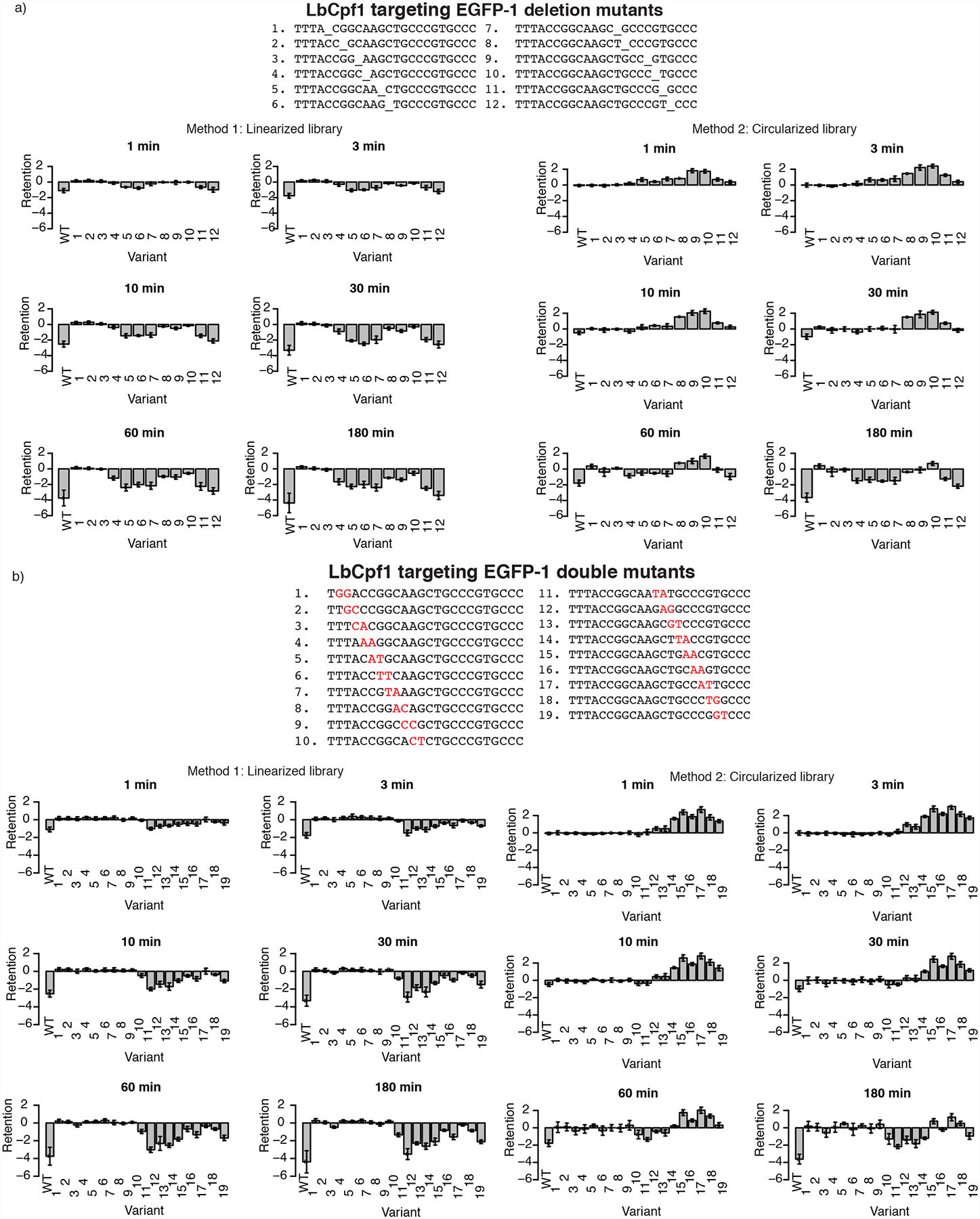
Bar graphs on left show retention profiles for whole-library assays with a post-reaction backbone cleavage step that avoid preferential recovery of nicked substrates. Bar graphs on right show retention profiles without such a cleavage step (i.e., with preferential recovery of nicked sequences). a) Single deletion variants, b) Double consecutive transversion variants. Bar graphs show median retention of indicated variants at each base, with error bars representing the standard deviation of observed retention among distinct barcoded instances of each variant target sequence. (Left: linearized library; Right: circular library) (Sequence Data: AF_SOL_820; For details on retention score calculation, see “Materials and Methods”.). Number of distinct barcoded instances for each variant assessed (n for standard deviation calculation) were as follows: Left: WT: 26, Deletions 1–12: 7, 9, 8, 9, 6, 3, 10, 2, 6, 6, 3, 6. Consecutive Transversions 1–19: 19, 24, 11, 4, 5, 8, 7, 6, 6, 9, 9, 5, 8, 11, 7, 8, 8, 5, 5. Right: WT: 24, Deletions 1–12: 6, 8, 8, 9, 6, 4, 6,1, 5, 6, 2, 5, Consecutive Transversions 1–19: 15, 20, 9, 5, 3, 8, 5, 5, 8, 9, 9, 6, 9, 11, 6, 6, 5, 5, 4.
An unexpected feature of Cpf1 cleavage was observed in assays with the circular variant library in which no linearization was carried out before assessing retention (Method 2). Certain target site variants had highly positive retention scores, which suggest these variants increase (rather than decrease) their representation during a short-time cleavage reaction. Log-retention scores were observed as high as ~3 (i.e., 23 or 8-fold enrichment) for double mutants from time points 1,3, 10, and 30 minutes (right panels of LbCpf1: Figure 2–3; AsCpf1 and FnCpf1: Supplementary Figures 7–27). For some targets, these effects disappeared at longer reaction times, as expected if there was eventual cleavage of the circular targets (Figure 2–3). The enhanced recovery led to the hypothesis of a rapid nicking of the initial substrate followed by a relatively slow cleavage on the opposite strand (nicked plasmids enhance PCR amplification compared to linear and supercoiled substrates28). This would explain the observed over-representation after addition of Cpf1 and the highly positive retention score followed by much slower loss of the observed retention.
The nicking hypothesis was tested by reacting Cpf1::gRNA complexes with individual plasmid substrates and examining topology using ethidium-containing native agarose DNA gels. Sequence variants with highly positive retentions were identified in the high throughput data and either cloned into a DNA plasmid or synthesized as a gRNA. Using a DNA target with double-consecutive transversion mutations in positions 12–13 and 14–15 in wild-type EGFP-1 gRNA, we found LbCpf1 was rapidly capable of converting closed circular to nicked circular substrates in these assays (Figure 4a–b). In addition, the nicking ability was found to be specific, with no observed nicking of substrates lacking homology to the gRNA (Figure 4c). The nicking ability was confirmed in AsCpf1 and FnCpf1 with their respective EGFP-1 gRNAs (Supplementary Figure 28–29).
Figure 4: Gel-based assessments of nicking and cleavage by LbCpf1 and Cas9.
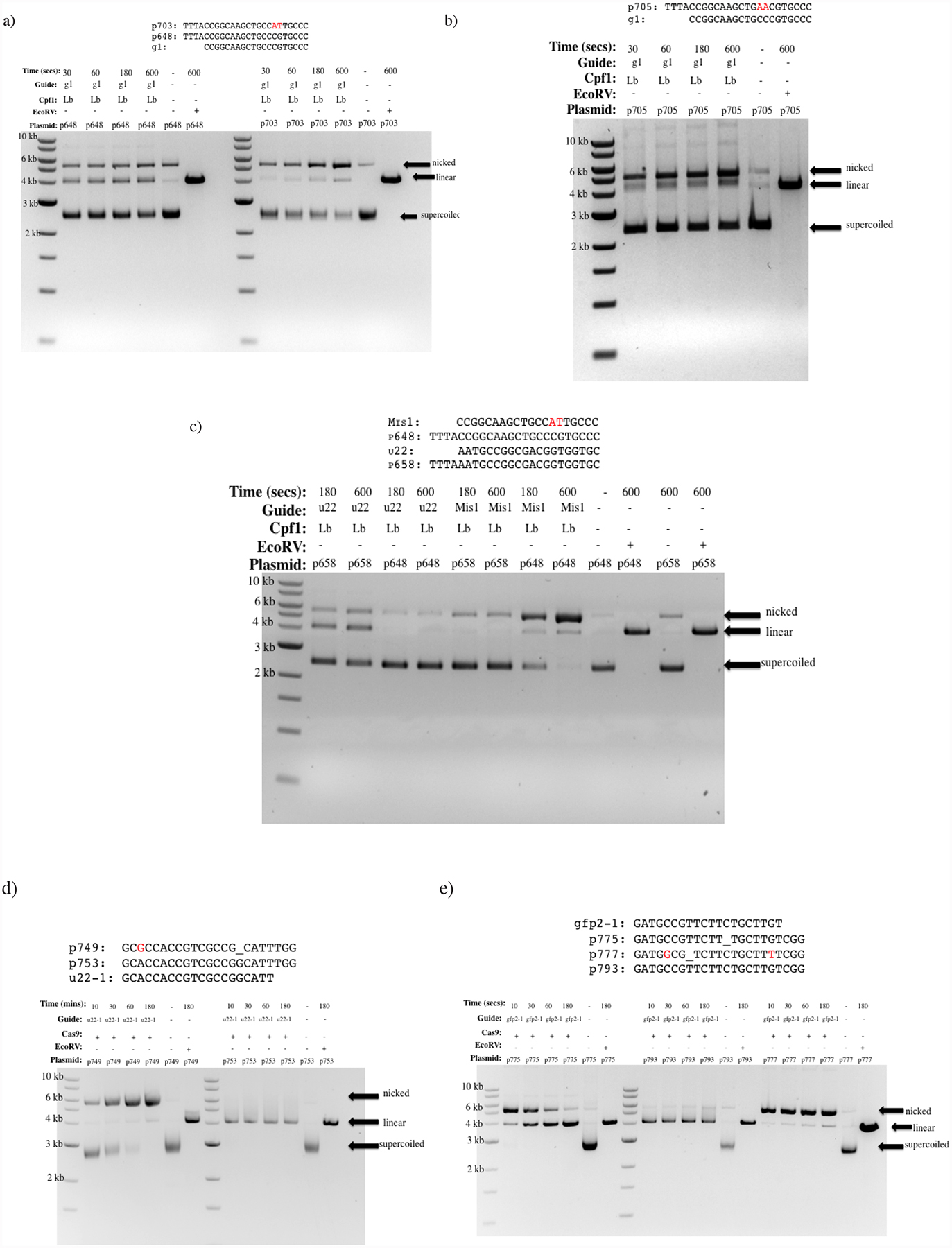
a-c) Gel-based assays for nicking by LbCpf1. a) Left: Wild-type EGFP-1 target (p648) reacted with wild-type EGFP-1 gRNA. Gels show a mixture of linear, nicked, and uncut target plasmids, Right: a mutated EGFP-1 target (p703) with wild-type EGFP-1 gRNA showed preferential accumulation of nicked plasmid. b) Gel assay with mutated EGFP-1 target (p705) and wild-type EGFP-1 gRNA. Preferential nicking with some linearization is observed. c) Specificity assessment using nicking gel assay. Tested gRNAs are wild-type unc-22A (u22) and mismatched EGFP-1 (Mis1). Tested targets are wild-type unc-22A (p658) and EGFP-1 (p648). The u22 gRNA linearizes the wild-type unc-22A (p658) target while having no effect on the EGFP-1 (p648) target. The mismatched EGFP-1 gRNA (Mis1) promotes nicking when paired with the EGFP-1 target but not with unc-22A. Nicking is thus shown to be RNA-guided and specific. d), e) Specificity and mismatch effects on Cas9 nicking and cleavage activities. d) Cas9 activity wild-type unc-22A gRNA and mutated unc-22A target (left), and with wild-type unc-22A gRNA and wild-type unc-22A target (right). Cas9 can be seen to efficiently nick at the mismatched target. e) Wild-type EGFP-2 gRNA with mutated EGFP-2 DNA targets (p775 and p777) and wild-type EGFP-2 with wild-type EGFP-2 DNA (middle). Both mutated EGFP-2 targets are nicked efficiently. The p775 mutation if given enough time eventually linearizes the plasmid (left). The p777 mutation remains nicked through the time course (right).
To evaluate the determinants for nicking and cleavage, we carried out a number of reciprocal experiments in which mutated gRNAs were used in assays with wild-type targets. These assays showed nicking and some linearization, as expected if the ability to form a nicking enzyme is a general feature of certain classes of gRNA::target mismatch. We note that there was some non-equivalence in the target-mutated versus gRNA-mutated assays, depending on the individual gRNAs (Supplementary Figure 30). These data indicate an interaction in which specific sequence along with the pattern of mismatches determine the balance between nicking and cleavage activities.
The above observations raise the possibility that an appropriately designed gRNA might produce effective single-strand nicking activity on an arbitrary substrate. Some possible guidelines for such design are suggested from the patterns of mismatch effects on initial sequence recovery in Figures 2–3 and Supplementary Figure 1–6. In particular, we see a strong tendency to nick for templates with a combination of transversion point mutations in the distal region (positions 9–15). Taking consecutive double mutation at positions 12 and 13 as a provisional lead for such assays, we first tested the ability to produce nicking activities towards additional targets where no high throughput analysis of target specificity had been carried out (targets dm22085 (DNMT1), fc596 (FANCF) and wp1058 (WTAP)). While these three sequences show different degrees of cleavage for matched gRNA:target combinations, all three bias toward nicking with the indicated double mutant target (Supplementary Figure 31–33).
Cas9 nicking and cleavage specificity
The type II CRISPR effector enzyme Cas9 has been a workhorse tool for genome editing and for a wide variety of experimental applications in vitro and in vivo. We tested S. pyogenes Cas9 for nicking activity in assays and libraries that (as with the tested Cpf1 libraries) contained wild-type, single variants, single deletions, and double consecutive mutations in four targets: EGFP-1, EGFP-2, unc-22A, and rol-6 (Figure 1b). While yielding some evidence of preferential nicking of specific substrates, these assays yielded a less dramatic distinction between nicking and double strand cleavage for individual template sequences than had been seen with Cpf1 (Supplementary Figure 34–45). Encouraged by the differences (but with the knowledge that an optimal nicking activity might require more extensive mutational analysis), we repeated the Cas9 assays using an unc-22A library obtained through random oligonucleotide synthesis with a broader set of multiple mutations (Fu et al.29,30; Supplementary Figure 46). These assays showed a strong preferential nicking for a variety of double mutants (Supplementary Figure 47). Examining an extended list of the randomly mutagenized targets from this library for which positive retention scores indicated a strong nicking activity (Supplementary Table 1), we chose a design with two mutations, a single deletion in the seed region at position 5 and a mismatch (A to G) in the distal region at position 18 for the unc-22A target. DNA templates with this double variant reacted with Cas9 complexed to wild-type unc-22A gRNA confirmed robust target specific nicking (Figure 4d). EGFP-2 variant targets that had a highly positive retention score in the high throughput assays were likewise confirmed as nicking substrates in agarose gel assays (Figure 4e). Conversely, wild-type unc-22A and EGFP-2 targets were assayed for nicking with the equivalently mismatched (mutant) gRNA. Similar to Cpf1, the equivalent mutations in the gRNA with wild-type targets showed nicking and some linearization that varied depending on gRNAs (Supplementary Figure 48–49). Finally, we observed that some non-intended targets were nicked (e.g. a variant target sequence of unc-22A nicking with wild-type EGFP-2 gRNA). We hypothesized that targets with certain minimal and/or broken homologies can induce nicking. We confirmed this by testing non-targeting DNA with gRNAs that showed positive retention scores and confirmed the nicking observed in the high throughput results. For example, we observed in the high throughput assays a variant of the unc-22A target that evidently nicks with EGFP-2 gRNA (Supplementary Figure 50). This variant brings the unc-22A sequence somewhat closer to the EGFP-2 sequence, although still providing rather limited homology (contiguous complementarity confined to 6 consecutive matches in the seed region). While demonstrating a less stringent sequence requirement, nicking remained a specific process, with only residual nicking for unrelated targets observed in the gel assay (e.g., >16-hour time point, Supplementary Figure 51).
Tuning of relative nicking activity
Analysis of complex libraries at multiple time points or with differing enzyme:substrate ratios provides a combined view that allows selection of substrates with a high preference for nicking over cleavage, with little or no cleavage even on extended incubation. Such substrates are exemplified for Cas9 by unc-22A double transversion (positions 5,12) (Supplementary Figure 47). For LbCpf1 maximal nicking is seen at certain time points with double consecutive transversion mutations in positions (12/13 and 14/15; Figure 3, Supplementary Figure 1–6). These examples illustrate the value of an optimization round in identifying the most specific nicking reagents for a given target, and of the value of a broad variant survey in characterizing potential off-target nicking consequences for a given gRNA.
Nicking in vivo in S. cerevisiae
Assessments of Cas9 activity in vivo were carried out in S. cerevisiae, using methodologies of Cas9 and gRNA expression and of target library production described in Fu et al.30. These high throughput assays allow tracking of topology as a function of sequence for a target library, allowing determination of which templates in a complex pool are cut, which are nicked, and which are not cut. Assays in yeast allow assessment of nuclease activities in a system with rapid cell division and where the DNA targets are present in a chromatinized context (in contrast to the supercoiled plasmid context of the above assays, which may be much more representative of bacterial than of eukaryotic chromosomes).
While sequencing provides a valuable assay for determining the population of molecules present before and after exposure in vivo to a Cas9+gRNA pair, it remains important to approach such data with considerable care. One concern is that the in vivo situation in yeast will represent a dynamic equilibrium between any cleavage or nicking by the Cas9 enzyme and repair processes (that might fix a nick or break) or other cellular processes (e.g. replication across a nick, generating a break) that might interconvert the various states. To investigate in vivo activities, we evaluated the incidence of nicked substrates in vivo rather than the much more complicated kinetic question of determining the rate of new nicks in the presence of repair and other activities.
In developing assays to test nicking of DNA in vivo, the simple detection of relaxed circles among extracted DNAs is not sufficient to infer whether nicking had occurred in vivo, or nicking occurred during extraction and/or analysis of DNA. To ensure definitive resolution of this issue, we asked a number of additional questions. Is the nicking signal (enhanced PCR yield observed in DNA pools containing close matches to the guide) is reproducible in parallel samples and in different biological backgrounds? Is the nicking signal specific to molecules in the target pool with homology to the Cas9 gRNA and PAM site? For partially-matched targets, the nicking signal decrease for target sequences with substantial numbers of gRNA mismatches? Does the nicking signal increase with induction of Cas9 and with longer exposure times in vivo? Does the in vivo nicking signal for different target variants correlate with measured in vitro nicking?
Our analysis found all the criteria fulfilled. The nicking signal (preferential retention of gRNA-matched targets in samples processed from uncut yeast DNA) is consistent between replicates of induced yeast (Figure 5a), depends on both PAM sequence and gRNA homology (Figure 5a–c), is lost with multiple mutations in the target (Figure 5b–c 4–7 mismatch lane), increases with longer exposure times (Figure 5b–c, comparing single-generation and 2.5 generation samples), and is correlated (R=0.47, p-value=0.00015; n=66) with nicking observed in vitro (Figure 5d–f). These data thus provide support for the hypothesis that the state of extracted DNA reflects the configuration in vivo and that a fraction of this DNA is indeed in a nicked form. Based on the observed differential retention (a maximum of ~3-fold in vivo) and the maximum enhancement for fully nicked DNA (similarly found in Lin et al.28 and our observations), we estimate that between 1/6 and 1/3 of susceptible targets are nicked at any time in vivo. We note a modest difference among in vitro conditions (two different buffers) and the in vivo yeast observations. All show nicking of mismatched (and to some extent, fully matched) targets, while differences are seen in the relative proportions for different sequences. Of interest, a substantial proportion of nicked targets is observed to persist even for fully matched Cas9 targets in the yeast assays (Figure 5b–c, black dots).
Figure 5. Assays for target-match-dependent nicking of precise and imprecise targets by Cas9 in vivo.

a) Reproducible nicking signal for matched targets following Cas9 induction in yeast. This plot compares differential retention
[log2(circular_assay_retention) - log2(linear_assay_retention)] for induced libraries from two different but functionally equivalent yeast strains, BY4741 and ΔKU70. Individual dots represent different target sequences, each assessed as a median over multiple barcoded instances in each library. Targets unrelated to unc-22A (grey hollow circles; “Protospacer 4” variants29) and with perfect unc-22A match in the gRNA homology but mismatches in the GG pam (blue squares) show no substantial differential retention in either sample. Perfectly matched targets with canonical “NGG” PAM sites (black circle) show substantial differential retention in the two yeast populations. Targets with single mismatch (red dots) show a spectrum of different retention differentials, ranging from no difference to differences comparable to the perfect target match. The two yeast populations give highly similar results with calculated sample-to-sample correlations between the two yeast populations of 0.81 (Pearson; two-tailed p-value=9.6E-17, n=66), and 0.76 (Spearman; two-tailed p-value=1.6E-13, n=66). For details on retention score calculation, see “Materials and Methods”. b-e) Similar sequence requirements for nicked substrate accumulation under diverse in vitro and in vivo conditions. Top graphs show circular and linear assay retention scores for yeast (in vivo) experiments in the BY4741 and ΔKU70 genetic backgrounds. Each plot shows median retention for multiple (barcoded) transversion variants at each position (averaged where duplicate sample are available for the relevant conditions). Retentions are calculated using the unrelated PS4 sequence as an internal standard, with initial library abundance obtained from measurement of target species incidence in parallel libraries with yeast that have not been induced. Below are equivalent retention score plots for in vitro analysis in which an equivalent library of targets was interacted with purified Cas9 in one of two buffer conditions (a relatively active “Thermo-Pol” buffer condition and a less active “Cas9 buffer” condition; see methods for buffer details). f) Differential retention comparison between in vitro and in vivo samples. The in vivo sample shown here is from BY4741 (at 2.5 generations) compared to the initial rate (1 minute) of nicking in vitro (Cas9 buffer). Correlations are Pearson: 0.54, two-tailed p-value=3.6E-6 and Spearman 0.49, two-tailed p-value=3.2E-5 (n=66). Each point on the plot represents a single gRNA homology+PAM sequence class, showing a median of differential retentions derived from independent flanking-barcoded instances of each sequence.
Discussion
We show that two families of RNA-programmed CRISPR-Cas nucleases, As/Fn/Lb-Cpf1 and Cas9 can efficiently nick DNA targets with perfect and/or mismatched homology to the programming gRNA. The ability to program native CRISPR-Cas nucleases to nick has uses in technology and implications in the biology of CRISPR immunity.
The ability to tailor some CRISPR-Cas nucleases to nick at specific sites “on demand” provides a potential alternative to mutated CRISPR nuclease variants for gene editing/manipulation. While mutation of defined cleavage domains8,31,32 provides a capability for nicking activities for some CRISPR nucleases, the approach is not available for all CRISPR-Cas systems (particularly Cpf1, where a single domain may execute both cleavage reactions22,33,34). In addition, such approaches limit the multifunctional applications of CRISPR in a single system, since co-expression of nickase and wild-type enzymes will generate cleavage at intended nickase sites as well as nicking at intended cleavage sites. Use of a single wild-type enzyme with gRNAs for nicking and cleavage would surmount this challenge, with tuning of gRNAs a likely requirement in making such an approach effective. While we have not investigated such applications in microbial or other systems (e.g. mammalian cells), the use of mismatched gRNAs with wild-type CRISPR enzyme to direct nicking could be valuable for gene editing or replace nickases in gene manipulation. As an example, base editors composed of a catalytically dead Cas9 fused to a deaminase could potentially be redesigned to involve a fully functional Cas9 fused to a deaminase that can be guided to specific areas via mismatched gRNAs.
The study described here also highlights the fact that a fraction of Cas9’s observed activities in vivo either in native systems or in engineering applications may reflect nicking rather than cleavage activities, with the balance likely dependent on in vivo conditions as well as the sequences of gRNAs and targets. Of particular importance here, observations that nicking activities can provide advantages in genome editing13,23,24,26,27 indicate that such conditions could prove advantageous. We note that the consequences of nicking will depend in each system on the kinetic balances between nick ligation, single stranded exo- and endonuclease activities that might extend or convert nicks35, other modes of DNA repair, and DNA replication/division rates. These kinetic parameters will vary substantially based on intrinsic cellular properties, on specific genomic positions, and on stochastic ordering of events. Of particular interest, we note recent work of Davis and Maizels13 provide assays for nick-induced repair and editing in mammalian systems.
An understanding of sequence requirements for nicking as well as full cleavage of targets will be critical for identification and assessment of potential off-target effects of CRISPR-Cas nucleases in vitro and in vivo as these are applied in experimental, biotechnological, and clinical settings. In particular, the existence of a guide-specific nicking repertoire impacts the selection of gRNA targets to avoid off-target consequences during genome editing. Although nicked double stranded DNA in vivo is less detrimental than a double stranded break, nicks lead to downstream repair events that can cause unexpected mutations (e.g. Kuzminov et al.25). Of the various algorithms that score and/or pick gRNAs based on potential off-target effects36,37, many entail a user-selected threshold for candidate gRNA mismatch and/or leave the user to select candidate gRNAs based a mismatch-count-based estimate of off-target cleavage potential. Understanding the nicking abilities of CRISPR-Cas nucleases may thus offer considerable value in gRNA design and selection.
Imperfect homology-dependent nicking of CRISPR-Cas nucleases has implications on the mechanism of spacer acquisition in CRISPR immunity. Spacer acquisition is the process in which nucleic acids from foreign genetic elements (e.g. plasmids, bacteriophages, etc.) are integrated in the CRISPR loci. The integrated spacers are later transcribed and processed and used for host defense1,38. Cas1 and Cas2 catalyze spacer acquisition39,40. It has been shown that the effector CRISPR-Cas nucleases (nuclease active and inactive) in type II CRISPR systems are necessary for spacer acquisition41,42. In addition, there is evidence that type I CRISPR-Cas systems in the presence of non-targeting gRNAs can increase spacer acquisition with evident strand bias, a phenomenon that is called “primed adaptation”43–45. Although primed adaptation has not been reported for the type II systems to this date, it is possible that spacer acquisition is conserved among CRISPR-Cas systems. These observations in conjunction with knowledge that nicking can be induced with mismatch gRNAs could be relevant in the increased ability to acquire spacers in the presence of a non-targeting gRNAs. In particular, the nicked product could provide a strand specific advantage for the integration of nucleic acids in the CRISPR loci. An additional possibility is that the minimal homology between the gRNA and DNA target produces a nick that serves as an anchor for the CRISPR-Cas nuclease to recruit Cas1 and Cas2.
Methods
Variant plasmid library
The Cpf1 library was created using pooled oligonucleotide synthesis and was inserted into a plasmid vector population as described Fu et al.29. A Cas9 unc-22A variant library created using degenerate oligonucleotide synthesis and a library created using pooled oligonucleotide synthesis were previously characterized in Fu et al.30 and were used in this study. The library created by pooled oligonucleotide synthesis was retransformed and re-grown from Fu et al.30.
High throughput in vitro target specificity assays
Cpf1 in vitro target specificity assays were performed as reported with Fu et al.29, in 100 mM NaCl, 50 mM Tris-HCl, 10 mM MgCl2, 100 μg/ml BSA, pH 7.9. Cpf1 gRNAs were synthesized by Integrated DNA Technologies. Each reaction contained 50 ng/ul of Cpf1 protein and gRNA at a 1:1 ratio and was incubated at 37 °C.
For Cas9, we used the modified single (chimeric) gRNA structure of Jinek et al.8. Target specificity in vitro assays for Cas9 were performed as detailed in Fu et al.29 (Thermo-pol buffer: 20 mM Tris-HCl, 10 mM (NH4)2SO4, 10 mM KCl, 2 mM MgSO4, 0.1% Triton X-100, pH 8.8). In addition to the buffer used for these studies, we have performed a set of parallel assays using an alternative buffer similar to that of Jinek et al.8 (Cas9 buffer: reaction buffer: 20 mM HEPES, 100 mM NaCl, 5 mM MgCl2, 0.1 mM EDTA, pH 6.5). Similar results were obtained in the two buffers, albeit (as previously observed; Fu et al.30) with differences in cleavage kinetics and completion.
Our initial assessment nicking and cleavage relies on the known stimulation of PCR yields from nicked plasmid as compared to circular (supercoiled) plasmids, while linearized plasmids decrease skew in PCR yields28. As an initial assay to assess target nicking and cleavage after Cas9/Cpf1 interaction, we start with circular plasmid libraries that carry a wide variety of target variants between defined PCR primers. Aliquots of the library are interacted with Cas9/Cpf1 and DNA extracted, along with control aliquots of the circular (unreacted) library. Each extracted DNA sample is then split into two pools, one of which is amplified and sequenced after digestion outside of the target region with a restriction enzyme that linearizes the population of molecules outside of the amplified region (so there is no longer a preference for nicked circles in overall PCR yield). The second pool is amplified without linearization, resulting in counts that would retain a preference for nicked over closed circular or double-strand cleaved molecules. We note that nicking by Cpf1/Cas9 in the amplified (target homology) region resulted in an increase of PCR yields and that both gel and PCR enhancement assays would see nicked molecules with a single cleaved phosphodiester bond in additional to molecules in which other processes had extended an initial phosphodiester break in vitro35 or in vivo. In practice, the observed net stimulations of PCR yields from nicking are on the order of 23.
Supplementary Table 3 lists gRNA and target plasmid sequences.
Calculation of retention scores
For each sequence ‘X’:
Retention[X]= log2(ReprestentationX[Library with addition of CRISPR-Cas Nuclease]/RepresentationX[(Uncleaved Library) or (All Variants of a specified target)]
We normalize first to total reads from each experimental condition that have the expected length (35–36 bps) and barcode (scaling to total library counts). As an alternative normalization for comparison, we scale different libraries by counting tags matching a non-targeted gRNA sequence or all non-targeted gRNA sequences. For example, experiments with gRNAs targeting EGFP-1, EGFP-2, and unc-22A was normalized to the sum all variants of the rol-6 target sequence or normalized to the sum of all non-targeted variants. For experiments where the rol-6 was the intended target, the library was normalized to total sum of unc-22A variants. As expected, the different normalization approaches yield highly comparable results. Normalizations used for display are noted for each figure.
Cases of positive retention (as noted above and in [reference to increased yield for nicked circles in PCR]) are indicative of potential target nicking rather than cleavage. We note a maximum positive retention in our experiments of approximately 3 log-2 units, yielding a comparable estimate. A set of assays using Cas9 D10A nickase also allows us a more quantitative conversion between degree of enhancement in PCR yield and quantitative fraction of nicked templates (100% nicking yields a retention ~2.5–3, again in the same range; Supplementary Figure 52–54).
Nicking agarose gel assays
Target DNA were cloned into a parent vector (pHRL-TK, Promega) using NotI and Acc65I restriction sites. Cpf1 or Cas9 reactions were set up as above for the library assays. For time points, samples were stopped with 100 mM EDTA, 2% SDS, and 80 U/μL Proteinase K and/or flash frozen with dry ice. For visualization, frozen samples were thawed and immediately loaded on ~1–1.5% agarose gel (TAE buffer, Ethidium Bromide Concentration [0.3 mg/L]). Supplementary Table 3 lists gRNA and target plasmid sequences.
Protein components
As/Fn/Lb-Cpf1 constructs were generated by assembly of synthetic gene fragments into an E. coli expression plasmid using the NEBuilder HiFi DNA Assembly Cloning Kit (NEB #E5520S). As/Fn/Lb-Cpf1 expression vectors contained N-terminal 6xHis-tag and SV40 NLS, and C-terminal SV40 NLS. Recombinant proteins were expressed in modified E. coli NiCo21 (DE3) cells (NEB C2925H) harboring the Cpf1 expression plasmid by growing in LB at 23°C for 16 hr in presence of IPTG at 0.4mM. Cells were disrupted by sonication prior to chromatographic purification.
As/Fn/Lb-Cpf1 was purified using HiTrap DEAE FF (GE Healthcare), HisTrap HP (Ni-NTA) (GE Healthcare) and HiTrapSP HP (GE Healthcare) columns. Recombinant proteins were dialyzed and concentrated into 20mM Tris-HCl (pH7.4), 500mM NaCl, 1mM DTT, 0.1mM EDTA and 50% glycerol.
Cas9 and LbCpf1 protein preparations were NEB M0386S and M0653S respectively.
Cas9 in vivo assays
Two yeast strains were transfected with a pooled library cloned into a plasmid carrying a Cas9 expression cassette (driven by the GalL promoter), a gRNA expression construct (driven by a tetracyline inducible RPR1 promoter), and a target or non-target sequence adjacent to a potential “PAM” site (‘NGG’) (plasmid map available here: https://benchling.com/s/O5VobNjd). Target sequences are inserted into the plasmid at a defined location, with the majority deriving from a library of variants of the unc-22A, while variants of a sequence of unrelated origin (PS4) provide a number of internal controls29. The two yeast populations analyzed were from strains BY4741 (BY) and KU70 deletion strain (KU) from MATa collection. BY is an auxotrophic wild-type S288C lab strain, while KU was chosen for analysis based on its intrinsic defects in non-homologous double strand break repair. We found no major difference between the two strains. Each strain was analyzed without specific induction of Cas9 (“baseline levels”). Additionally, each strain was analyzed after one division and at 2.5 generations as assessed by optical density (these were ~90 and ~230 minutes after an initial 4-hour metabolic adaptation to galactose media from dextrose) following induction with galactose and 250 ng/ml anhydrotetracycline (ATc). Control, single-generation, and 2.5 generation time points in yeast were analyzed in duplicate for KU70, with control and single generation time points analyzed in duplicate for BY4741.
In our analysis (Figure 5; Supplementary Table 1), we use the difference in log retention score for any given target (circular-linear), as the provisional metric for evaluation, validating this measurement as described. Retention scores for these comparisons are calculated from individual DNA pools that have been subdivided following extraction. One aliquot is analyzed following an experimental template-linearization cleavage step (which we call a “linear” assay a.k.a. Method 1), while a second aliquot is analyzed without such a step (designated a “circular” assay a.k.a. Method 2). As noted above, previous biochemical analysis28 and our further analysis in vitro confirm that this assessment indeed provides a metric that assesses nicking of the DNA.
Statistics and Reproducibility:
Several features of experimental design with complex libraries are key to consistent assessment of nicking activities. First, we ensure that each potential target sequence (wild-type or mutant) is present in several barcoded version in each library, allowing independent measurements for each sequence based on the individual barcodes (the number of such sequences that are used for the analysis are indicated in each figure with a value n). Second, each library contains multiple internal reference standards—plasmids with a parallel structure but with target sequences unrelated to the gRNA being assayed. For the Cas9 libraries described here, the unrelated reference sequences were from several distinct unrelated targets. As shown on the left of each line-plot figure, the relative representation of these unrelated targets was constant following interaction with a specific gRNA, while representation of targeted sequences was, as noted, consistently modulated. Additional internal control support comes from a subset of gRNA-homologous targets with central PAM variants, which also show little or no change relative to reference sequences (shown for Cpf1 targets as positions −2 and −3). Third, we carried out assessments of retention at multiple time points, and with more than one target for each enzyme, with comparable results. Cpf1 assays on each target were carried out with several different cognate enzymes and time points with similar results, Cas9 in vitro assays were each performed in several independent time series with comparable results, and Cas9 in vivo assays were performed in two different yeast strains with comparable results.
For nicking assessments using electrophoresis, all main figures (Figure 4a–e) gRNA::target combination was tested at multiple time points, with multiple experimental trials; for Supplemental Figures 28–33 and 48–51, a single series with multiple time points was carried out. In each case, an expected migration shift on nicking was observed.
Data availability
The raw data that support our findings is available in Short Reads Archive: PRJNA503740 (see Supplementary Table 2 for information on data for each corresponding figure).
Supplementary Material
Acknowledgements:
We thank the Daniel Herschlag, Peter Fineran, Gaelen Hess, Nimit Jain, and colleagues in our laboratories for their input and discussion. We are grateful to Joseph A. Meacham for advice on reagents and Cameron Lee for lending reagents.
Footnotes
Competing interests
Ryan T. Fuchs, Megumu Mabuchi, Jennifer Curcuru, and G. Brett Robb are employees of New England Biolabs. A provisional patent “Compositions and Methods for Nicking Target DNA Sequences” related to this work has been filed by Stanford University (Inventors: Becky Xu Hua Fu, Andrew Fire and Justin D. Smith)
References
- 1.Terns MP & Terns RM CRISPR-based adaptive immune systems. Curr. Opin. Microbiol 14, 321–7 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barrangou R et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–12 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Barrangou R & Marraffini LA CRISPR-Cas systems: Prokaryotes upgrade to adaptive immunity. Mol. Cell 54, 234–44 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heler R, Marraffini LA & Bikard D Adapting to new threats: the generation of memory by CRISPR-Cas immune systems. Mol. Microbiol 93, 1–9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carroll D Genome Editing: Past, Present, and Future . Yale J. Biol. Med 90, 653–659 (2017). [PMC free article] [PubMed] [Google Scholar]
- 6.Mali P, Esvelt KM & Church GM Cas9 as a versatile tool for engineering biology. Nat. Methods 10, 957–63 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Terns RM & Terns MP CRISPR-based technologies: prokaryotic defense weapons repurposed. Trends Genet. 30, 111–118 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jinek M et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–21 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Murovec J, Pirc Ž & Yang B New variants of CRISPR RNA-guided genome editing enzymes. Plant Biotechnol. J 15, 917–926 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cebrian-Serrano A & Davies B CRISPR-Cas orthologues and variants: optimizing the repertoire, specificity and delivery of genome engineering tools. Mamm. Genome 28, 247–261 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eid A, Alshareef S & Mahfouz MM CRISPR base editors: genome editing without double-stranded breaks. Biochem. J 475, 1955–1964 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shen B et al. Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat. Methods 11, 399–402 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Davis L & Maizels N Homology-directed repair of DNA nicks via pathways distinct from canonical double-strand break repair. Proc. Natl. Acad. Sci. U. S. A 111, E924–32 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sternberg SH, LaFrance B, Kaplan M & Doudna JA Conformational control of DNA target cleavage by CRISPR-Cas9. Nature 527, 110–3 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Szczelkun MD et al. Direct observation of R-loop formation by single RNA-guided Cas9 and Cascade effector complexes. Proc. Natl. Acad. Sci. U. S. A 111, 9798–803 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Strohkendl I, Saifuddin FA, Rybarski JR, Finkelstein IJ & Russell R Kinetic Basis for DNA Target Specificity of CRISPR-Cas12a. Mol. Cell 71, 816–824.e3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zetsche B et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163, 759–71 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bayat H, Modarressi MH & Rahimpour A The Conspicuity of CRISPR-Cpf1 System as a Significant Breakthrough in Genome Editing. Curr. Microbiol 75, 107–115 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Fernandes H, Pastor M & Bochtler M Type II and type V CRISPR effector nucleases from a structural biologist’s perspective. Postepy Biochem. 62, 315–326 [PubMed] [Google Scholar]
- 20.Fonfara I, Richter H, Bratovič M, Le Rhun A & Charpentier E The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature 532, 517–21 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Kim HK et al. In vivo high-throughput profiling of CRISPR–Cpf1 activity. Nat. Methods 14, 153–159 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Fonfara I, Richter H, Bratovič M, Le Rhun A & Charpentier E The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature 532, 517–21 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Vriend LEM & Krawczyk PM Nick-initiated homologous recombination: Protecting the genome, one strand at a time. DNA Repair (Amst). 50, 1–13 (2017). [DOI] [PubMed] [Google Scholar]
- 24.Vriend LEM et al. Distinct genetic control of homologous recombination repair of Cas9-induced double-strand breaks, nicks and paired nicks. Nucleic Acids Res. 44, 5204–17 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuzminov A Single-strand interruptions in replicating chromosomes cause double-strand breaks. Proc. Natl. Acad. Sci 98, 8241–8246 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gao Y et al. Single Cas9 nickase induced generation of NRAMP1 knockin cattle with reduced off-target effects. Genome Biol. 18, 13 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Satomura A et al. Precise genome-wide base editing by the CRISPR Nickase system in yeast. Sci. Rep 7, 2095 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin C-H, Chen Y-C & Pan T-M Quantification Bias Caused by Plasmid DNA Conformation in Quantitative Real-Time PCR Assay. PLoS One 6, e29101 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fu BXH, Hansen LL, Artiles KL, Nonet ML & Fire AZ Landscape of target: guide homology effects on Cas9-mediated cleavage. Nucleic Acids Res. 42, 13778–87 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fu BXH, St. Onge, R. P., Fire, A. Z. & Smith, J. D. Distinct patterns of Cas9 mismatch tolerance in vitro and in vivo. Nucleic Acids Res. 44, 5365–5377 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gasiunas G, Barrangou R, Horvath P & Siksnys V Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. U. S. A 109, E2579–86 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ran FA et al. Double Nicking by RNA-Guided CRISPR Cas9 for Enhanced Genome Editing Specificity. Cell 154, 1380–1389 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dong D et al. The crystal structure of Cpf1 in complex with CRISPR RNA. Nature 532, 522–6 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Yamano T et al. Crystal Structure of Cpf1 in Complex with Guide RNA and Target DNA. Cell 165, 949–962 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen JS et al. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science (80-.). eaar6245 (2018). doi: 10.1126/science.aar6245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koo T, Lee J & Kim J-S Measuring and Reducing Off-Target Activities of Programmable Nucleases Including CRISPR-Cas9. Mol. Cells 38, 475–481 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang X-H, Tee LY, Wang X-G, Huang Q-S & Yang S-H Off-target Effects in CRISPR/Cas9-mediated Genome Engineering. Mol. Ther. Nucleic Acids 4, e264 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bhaya D, Davison M & Barrangou R CRISPR-Cas systems in bacteria and archaea: versatile small RNAs for adaptive defense and regulation. Annu. Rev. Genet 45, 273–97 (2011). [DOI] [PubMed] [Google Scholar]
- 39.Sternberg SH, Richter H, Charpentier E & Qimron U Adaptation in CRISPR-Cas Systems. Mol. Cell 61, 797–808 (2016). [DOI] [PubMed] [Google Scholar]
- 40.Jackson SA et al. CRISPR-Cas: Adapting to change. Science (80-.). 356, eaal5056 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Wei Y, Terns RM & Terns MP Cas9 function and host genome sampling in Type II-A CRISPR-Cas adaptation. Genes Dev. 29, 356–61 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Heler R et al. Cas9 specifies functional viral targets during CRISPR–Cas adaptation. Nature 519, 199–202 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fineran PC et al. Degenerate target sites mediate rapid primed CRISPR adaptation. Proc. Natl. Acad. Sci. U. S. A 111, E1629–38 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Staals RHJ et al. Interference-driven spacer acquisition is dominant over naive and primed adaptation in a native CRISPR-Cas system. Nat. Commun 7, 12853 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Richter C et al. Priming in the Type I-F CRISPR-Cas system triggers strand-independent spacer acquisition, bi-directionally from the primed protospacer. Nucleic Acids Res. 42, 8516–26 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data that support our findings is available in Short Reads Archive: PRJNA503740 (see Supplementary Table 2 for information on data for each corresponding figure).