

ABSTRACT
Most strategies used to prepare homogeneous site-specific antibody-drug conjugates (ADCs) result in ADCs with a drug-to-antibody ratio (DAR) of two. Here, we report a disulfide re-bridging strategy to prepare homogeneous ADCs with DAR of one using a dual-maleimide pyrrolobenzodiazepine (PBD) dimer (SG3710) and an engineered antibody (Flexmab), which has only one intrachain disulfide bridge at the hinge. We demonstrate that SG3710 efficiently re-bridge a Flexmab targeting human epidermal growth factor receptor 2 (HER2), and the resulting ADC was highly resistant to payload loss in serum and exhibited potent anti-tumor activity in a HER2-positive gastric carcinoma xenograft model. Moreover, this ADC was tolerated in rats at twice the dose compared to a site-specific ADC with DAR of two prepared using a single-maleimide PBD dimer (SG3249). Flexmab technologies, in combination with SG3710, provide a platform for generating site-specific homogenous PBD-based ADCs with DAR of one, which have improved biophysical properties and tolerability compared to conventional site-specific PBD-based ADCs with DAR of two.
KEYWORDS: Antibody drug conjugates, engineered antibodies, site-specific conjugation, pyrrolobenzodiazepine dimers, cytotoxic payloads, drug to antibody ratio
Introduction
Although pyrrolobenzodiazepines (PBDs) such as anthramycin and sibiromycin were shown to be potent antitumor agents over 50 years ago, they have only recently emerged as cytotoxic warheads for antibody-drug conjugates (ADCs),1-3 which are a class of targeted therapeutics for the treatment of cancers.4-6 In their monomeric form, PBDs derive their cytotoxic activity by selectively binding to the minor groove of DNA and forming covalent bonds with the C2-NH2 group of guanine bases.7-10 Efforts to enhance this cytotoxic activity led to the synthesis of dimeric PBDs,11,12 which have two alkylating imine functions that allow the formation of minor groove DNA adducts, resulting in greater interference with DNA replication than monomeric PBDs.13-15
Early-generation dimeric PBD warheads (e.g., SG2000) have evolved over time to incorporate more desirable properties, such as enhanced solubility and improved DNA cross-linking ability, while retaining their potency.16 For example, Gregson et al.17 demonstrated that SG2057, which includes a 5-carbon linker between the two PBD monomers, had more than 3400-fold higher cytotoxicity and more than 10-fold DNA cross-linking ability compared with its 3-carbon linked PBD counterpart. These characteristics were incorporated into the SG3199 warhead by Tiberghien and colleagues,18 who also replaced the C2 aryls with methyl groups to increase the warhead’s solubility. The SG3199 warhead was subsequently linked to a maleimidocaproyl-polyethylene glycol (PEG8) linker via a self-immolative valine-alanine dipeptide at the N10 position to generate the SG3249 payload, which was used to prepare ADCs.19-24
Drawing upon the success of SG3249 as an ADC payload, we hypothesized that functionalization of the symmetrical N10 nitrogen with an additional cathepsin-B cleavable valine-alanine dipeptide and PEG8-maleimide linker would result in a symmetrical dual-maleimide PBD dimer, SG3710 (Figure 1), which could be used to re-bridge two adjacent cysteines, such as the ones at the immunoglobulin G1 (IgG1) hinge. The symmetrical nature of the SG3710 payload offers several key structural features that not only expand the applicability of PBD dimers, but also offer enhancement of the biochemical properties of previous versions of PBD dimers, such as SG3249, which is composed of a single chemical linker (Figure 1). The second PEG8 linker decreases the overall hydrophobicity of the SG3710 payload, a key parameter that has significant effects on ADC pharmacokinetics, biodistribution, and tumor efficacy.25-29 In addition, the symmetrical structure of SG3710 expands the utility of dual-linker PBDs and opens the door to developing other ADC warhead classes with a drug-to-antibody ratio (DAR) of one based on a site-specific disulfide re-bridging.
Figure 1.
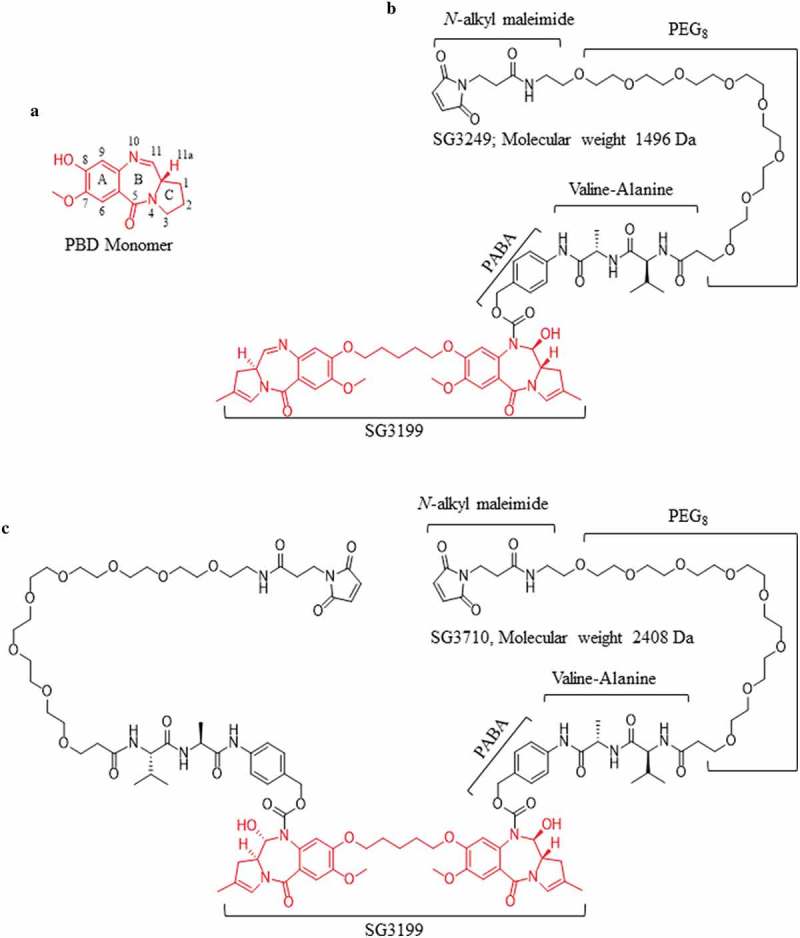
Chemical structures of the PBD monomer, SG3249, and SG3710. (a) Structure and numbering of the PBD monomer. (b) Structure of SG3249. Red denotes the released warhead, SG3199. The N-alkyl maleimide, PEG8 spacer, dipeptide valine-alanine (the cathepsin B cleavage site), and the self-immolative spacer (para-aminobenzyl alcohol, PABA) are schematically labeled. The molecular weight of SG3249 is 1496 Da. (c) Structure of the dual-maleimide SG3710. SG3710 has two linkers designed to bridge two adjacent cysteines, such as those at the IgG1 hinge. The molecular weight of SG3710 is 2408 Da.
The development of ADCs with a DAR of one could offer advantages over conventional site-specific ADCs prepared with DAR of two, including more favorable pharmacokinetic and biodistribution profiles, as well as improved tolerability. To test this hypothesis, we engineered an antibody, Flexmab, that enabled site-specific disulfide re-bridging with SG3710 to create an ADC with a DAR of one. The Flexmab antibody was engineered such that all but one pair of cysteines (C226, Kabat numbering)30 were mutated to valine. Additionally, an inter-chain disulfide was introduced at a buried position between the light and heavy chains to restore their covalent linkage. We site-specifically conjugated SG3710 to an anti-human epidermal growth factor receptor 2 (HER2) Flexmab derived from trastuzumab at the C226 thiols in the heavy chains, and we compared the resulting ADC in vitro and in vivo to a previously described site-specific anti-HER2 trastuzumab ADC with DAR of two, prepared using SG3249.20 We demonstrated that the combination of Flexmab and SG3710 technologies result in a platform for the preparation of site-specific PBD-based ADCs with DAR of one with improved properties compared to site-specific PBD-based ADCs with DAR of two.
Results
Engineering and characterization of Flexmab antibodies to enable site-specific re-bridging
Flexmab engineering was applied to trastuzumab, which selectively targets HER2, and to an isotype negative control antibody, NIP228. Both antibodies are kappa light chain isotype. Trastuzumab has been used extensively to treat patients with HER2-positive breast, colorectal, lung, and ovarian cancers, and it is the antibody component of ado-trastuzumab emtansine (Kadcyla), an ADC approved by the U.S. Food and Drug Administration in 2013.31-33 The engineered Flexmab contains five mutations that were selected based on rational design and in silico structural analysis (Figure S1). The light chain includes two mutations consisting of an F118C mutation that was used to generate a buried inter-chain disulfide bond with the heavy chain cysteine mutation L128C and a C214V mutation to eliminate the cysteine that forms the canonical inter-chain disulfide bridge with the cystine at position 220 in the heavy chain. The heavy chain contains three mutations including a L128C mutation, which forms a buried disulfide with F118C of the light chain, and C220V and C229V mutations to remove the canonical cysteines that form the inter-chain disulfide bridge with the light chain and the lower inter-chain disulfide bridge at the hinge, respectively. This approach resulted in the engineered Flexmab antibody, which has only one interchain disulfide bridge at the hinge formed by the cysteines at position 226 (Figure S1). We used the same mutagenesis strategy to engineer NIP228 as the isotype control antibody.
Trastuzumab-Flexmab, NIP228-Flexmab and their respective parental antibodies were transiently expressed in Chinese hamster ovary cells, which resulted in expression levels of 400 mg/L. Flexmabs and parental antibodies were purified using protein A affinity chromatography. Analytical size-exclusion chromatography after protein A purification revealed high monomeric content for both Flexmab antibodies (98%) that was similar to the monomeric content for their respective parental antibodies (98%) (Figure S4).
Next, we sought to determine whether the Flexmabs maintained thermostability in the same way as the parental antibodies. To this end, we used differential scanning calorimetry (DSC) to determine transition temperatures (TM) in degrees Celsius. Trastuzumab and trastuzumab-Flexmab DSCs were characterized by two transitions and had similar TMs of 68ºC and 82ºC for the first and second transitions, respectively (Figure S5A, B). Temperatures at the first and second transitions were 74ºC and 82ºC, respectively, for NIP228 and 66ºC and 82ºC, respectively, for NIP228-Flexmab (Figure S5C, D). Unlike trastuzumab and trastuzumab-Flexmab, which maintained their temperatures at each transition, NIP228-Flexmab’s temperature decreased by 8ºC compared to NIP228 for the first transition. Despite this decrease in TM, NIP228-Flexmab was very stable (lowest TM was 68ºC).
The BIAcore T100 was used to determine the kinetics of trastuzumab and trastuzumab-Flexmab binding to recombinant extracellular HER2. As shown in Table 1, trastuzumab and trastuzumab-Flexmab have similar equilibrium dissociation constant (Kd) for binding to HER2. The 1.7-fold difference in Kd for trastuzumab-Flexmab versus trastuzumab could be due to conformational changes occurring within the Fab binding arms as a result of Flexmab engineering and subsequent conjugation with SG3710; however, it is within the experimental error of this assay.
Table 1.
Binding kinetics of trastuzumab and trastuzumab-Flexmab to recombinant HER2.
| kon (1/Ms) | koff (1/s) | Kd, M | |
|---|---|---|---|
| Trastuzumab | 2.24 × 105 | 4.81 × 10−4 | 2.15 × 10−9 |
| Trastuzumab-Flexmab | 3.22 × 105 | 1.21 × 10−4 | 3.75 × 10−10 |
Kinetic measurements were carried out using BIAcore T100 biosensor. The equilibrium dissociation constants, Kd, were calculated as the ratio of the two rate constants, koff/kon.
Antibody-dependent cell-mediated cytotoxicity (ADCC), which can be an additional mechanism of action to augment cytotoxicity against HER2-expressing cancer cells, was similar in trastuzumab and trastuzumab-Flexmab (Figure 2(a)). As expected, the isotype controls NIP228 and NIP228-Flexmab did not show cytotoxicity against HER2-expressing BT-474 breast cancer cells (Figure 2(b)). Similar internalization between trastuzumab and trastuzumab-Flexmab was confirmed using a cell-binding assay with HER2-expressing SKBR-3 cells (data not shown).
Figure 2.
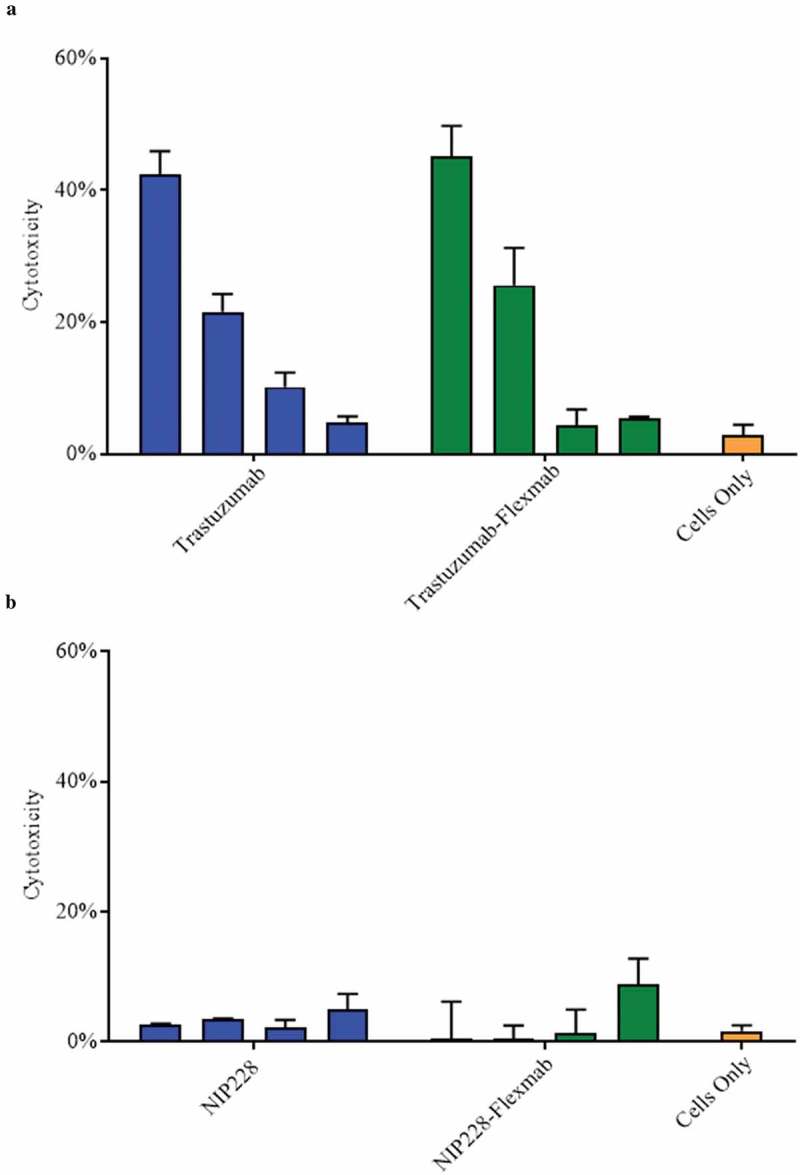
ADCC activity of antibodies and Flexmabs targeting BT-474 breast cancer cells. (a) ADCC of trastuzumab and trastuzumab-Flexmab. (b) ADCC of NIP228 and NIP-228-Flexmab. Error bars indicate mean ± standard deviation (n = 3). Antibodies and Flexmabs were tested at 1, 0.1, 0.01 and 0.001 μg/mL.
Synthesis of the dual maleimide PBD dimer SG3710
The dual-maleimide PBD dimer SG3710 was synthesized using starting material from SG3249.18 Generation of the dual-maleimide PBD dimer SG3710 (Figure S2 and supplementary appendix) was carried out in seven steps. The overall synthesis yield for the dual-maleimide PBD dimer SG3710 was 6.35%, a significant improvement over the 30-step route used to generate SG3249 that, despite being scalable to produce clinical-grade material,22-24 had an overall yield of 0.54%.18 Semi-preparative HPLC purification followed by lyophilization yielded SG3710 with a purity higher than 95%. SG3249 was prepared as described by Tiberghien et al.18
Preparation of site-specific re-bridged ADCs with a DAR of one using SG3710
Site-specific conjugation conditions were optimized to produce a disulfide re-bridged site-specific ADC with DAR of one using SG3710 (Figure 3). The re-bridging efficiency of the Flexmabs was optimized sequentially by experimentally monitoring of both the TCEP reduction and SG3710 conjugation efficiency using reduced and native liquid chromatography mass spectrometry (LCMS) (data not shown). The re-bridging efficiency of the trastuzumab-Flexmab and NIP228-Flexmab using SG3710 was maximized by first reducing the single interchain disulfide bond at the hinge using three equivalents of TCEP for one hour at room temperature, followed by the addition of three equivalents of SG3710 in 10% dimethyl sulfoxide (DMSO) for three hours at room temperature (Figure 3). Site-specific and efficiency of disulfide re-bridging using the SG3710 was demonstrated using orthogonal analytical methods.
Figure 3.
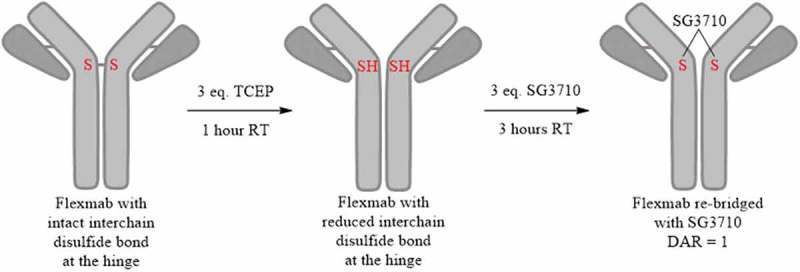
Schematic process for the site-specific re-bridging of Flexmab with SG3710. The site-specific re-bridging resulted in ADCs with a DAR of one.
Analytical size-exclusion chromatography confirmed high monomeric content (98%) for both trastuzumab-Flexmab-SG3710 and NIP228-Flexmab-SG3710 (Figure S6). To confirm that disulfide re-bridging with SG3710 was specific to the Flexmab heavy chains, the ADCs were analyzed using rRP-HPLC (Figure 4). In this analysis, in addition to using an absorbance of 280 nanometers, the ADCs were also detected using a PBD-specific absorbance of 330 nanometers.34 Overlays of the rRP-HPLC chromatograms of the parental and re-bridged Flexmabs revealed co-alignment of the retention times of the light chains, which indicates absence of conjugation at the light chains of the Flexmabs (Figure 4). Also, retention times shifted only for the re-bridged heavy chains of the Flexmabs (Figure 4). The trastuzumab-Flexmab heavy chain retention time was 19.75 minutes, whereas the trastuzumab-Flexmab-SG3710 re-bridged heavy chain retention time was 21.31 min (Figure 3(a)). The NIP228-Flexmab heavy chain retention time was 20.09 minutes, and the NIP228-Flexmab-SG3710 re-bridged heavy chain retention time was 21.57 min (Figure 4(b)). The shift in retention time for both Flexmab heavy chains indicated that conjugation of SG3710 is specific to the heavy chains.
Figure 4.
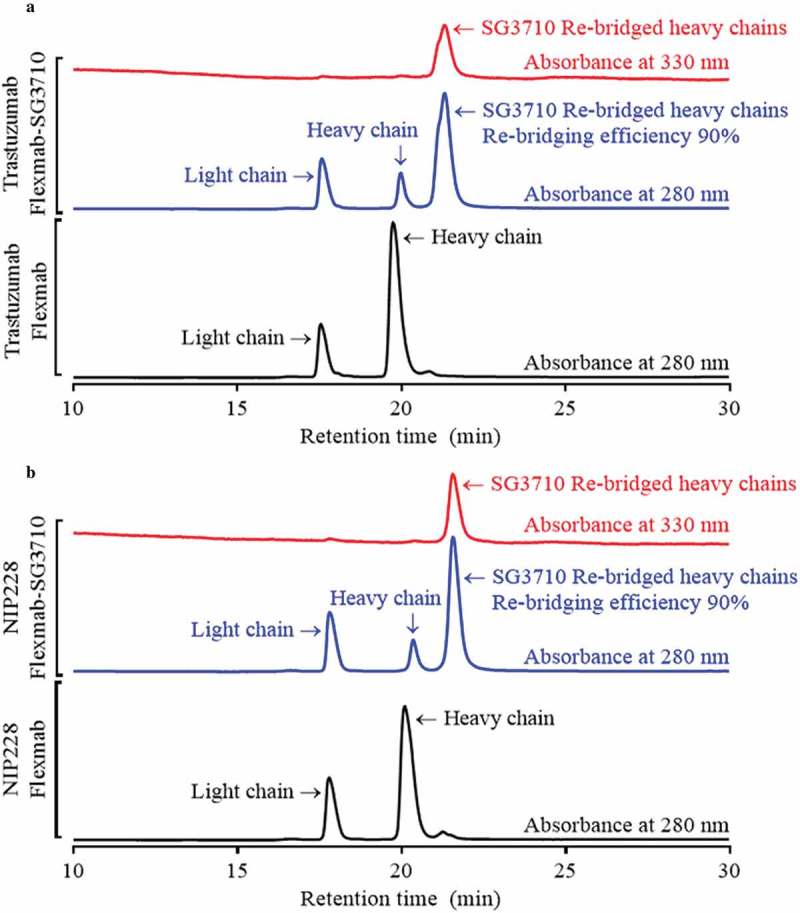
rRP-HPLC results for Flexmabs and Flexmabs re-bridged with SG3710. (a) rRP-HPLC results for trastuzumab and site-specific re-bridged trastuzumab-Flexmab-SG3710. (b) rRP-HPLC results of NIP228 and site-specific re-bridged NIP228-Flexmab-SG3710. Manual integration of the re-bridged versus parental heavy chains resulted in a re-bridging efficiency for both Flexmab ADCs of 90%. The analysis was carried out using absorbances of 280 and 330 nanometers.
Re-bridging of the Flexmab heavy chains by SG3710 was also demonstrated by monitoring the elution at 330 nanometers, which is a PBD-specific absorbance (red signals in Figure 4).34 The rRP-HPLC also demonstrated the presence of parental heavy chains for both Flexmab ADCs (Figure 4). Manual integration of the peaks representing parental and re-bridged Flexmab heavy chains produced a nearly identical conjugation efficiency of 90% for the trastuzumab-Flexmab-SG3710 and NIP228-Flexmab-SG3710 ADCs (Figure 4). Hydrophobic interaction chromatography (HIC) was used to confirm conjugation efficiency of the intact re-bridged ADCs and to assess the hydrophobicity change compared with that of the parental Flexmabs (Figure 5). As with the rRP-HPLC,34 an absorbance of 280 nm and a PBD-specific absorbance of 330 nm was used to identify re-bridged Flexmabs ADCs. Manual integration of the HIC peaks confirmed that the efficiency of re-bridging for both trastuzumab-Flexmab-SG3710 and NIP228-Flexmab-SG3710 was 90% (Figure 5), which is similar to the efficiency of conjugation determined using rRP-HPLC (Figure 4).
Figure 5.
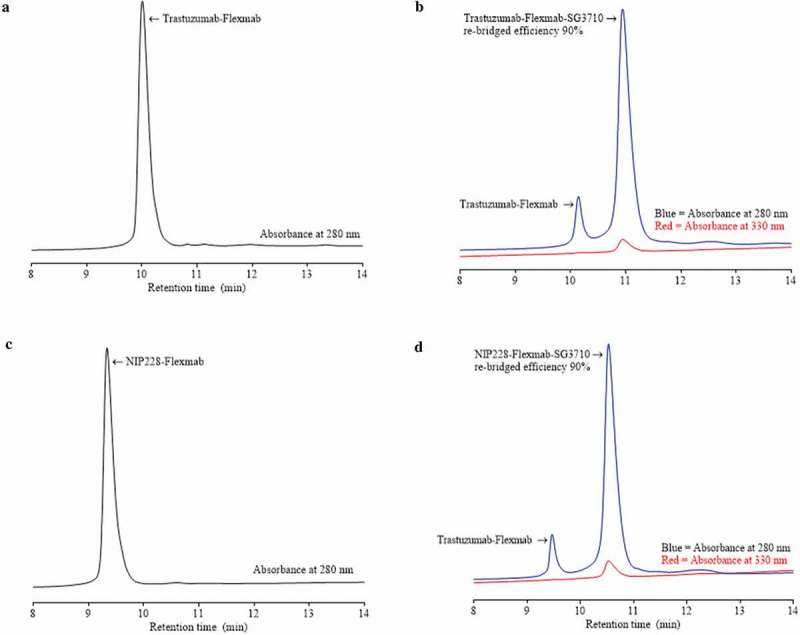
HIC of Flexmabs and Flexmab re-bridged with SG3710. (a) HIC results for trastuzumab-Flexmab. (b) HIC results for trastuzumab-Flexmab-SG3710. (c) HIC results for NIP228-Flexmab. (d) HIC results for NIP228-Flexmab-SG3710. Efficiency of conjugation, determined by manual integration of the re-bridged versus the parental peaks, was 90% for both ADCs. An absorbance of 280 nanometers was used for both the antibodies and the Flexmab-ADCs; a PBD-specific absorbance of 330 nanometers was also used for the ADCs.
Lastly, LCMS in non-reducing conditions was used as a complementary method to confirm the re-bridging of the heavy chains in the Flexmabs and to calculate DAR by determining the ratio of the peak height intensity of re-bridged versus parental species. This analysis was carried out using glycosylated Flexmabs and Flexmab ADCs. The molecular weight of trastuzumab-Flexmab G0f glycoform was 147986 Da (Figure 6(a)), whereas that of re-bridged trastuzumab-Flexmab G0f glycoform molecular weight was 150399 Da (Figure 6(b)). The mass difference between parental and re-bridged trastuzumab-Flexmab was 2413 Da, which corresponds to the mass of one SG3710 (Figure 1). The molecular weight of NIP228-Flexmab G0f glycoform was 146770 Da (Figure 6(c)), whereas that of re-bridged NIP228-Flexmab G0f glycoform molecular mass was 149199 Da (Figure 6(d)). The mass difference between parental and re-bridged NIP228-Flexmab is 2429 Da, which corresponds to the mass of one SG3710 with one of the maleimide hydrolized (Figure 1). These data confirm the re-bridging of the two Flexmabs at C226 on the heavy chains with one SG3710. Based on the ratio of the re-bridged peak height intensity to that of the parental peak height intensity of the G0f glycoform, the DARs were 0.9 for trastuzumab-Flexmab-SG3710 and 0.85 for NIP228-Flexmab-SG3710, respectively (Figure 6(b,d)). These DARs value are compatible with the efficiency of conjugation determined using rRP-HPLC and HIC.
Figure 6.
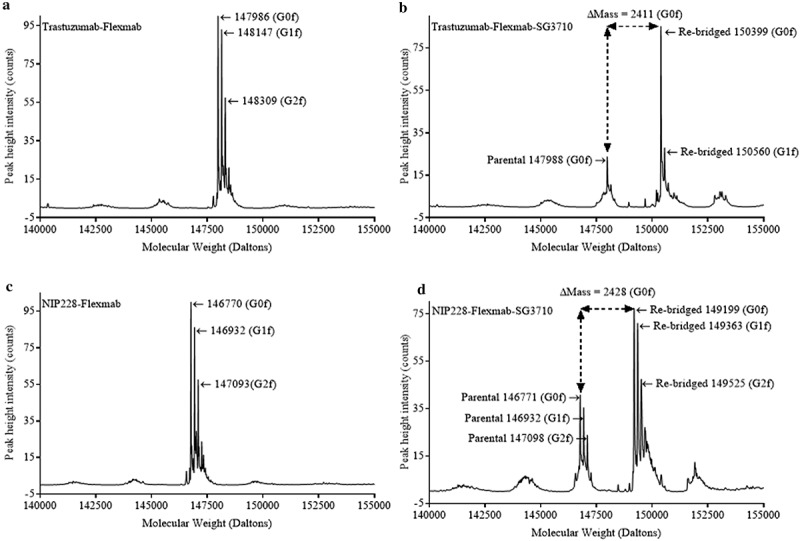
LCMS in non-reducing conditions of glycosylated Flexmab and glycosylated Flexmab-SG3710. (a) LCMS results for trastuzumab-Flexmab showing the molecular weights of the three major glycoforms. (b) LCMS results for trastuzuman-Flexmab-SG3710. G0f and G1f glycoforms and their respective molecular masses are shown. Those two glycoforms correspond to re-bridged ADCs with one SG3710. The molecular mass of the G0f parental is also shown. The mass difference between re-bridged and parental is 2411 Da, which correspond to the mass of one SG3710. (c) LCMS results for NIP228-Flexmab. The molecular weights of the three major glycoforms are shown. (d) LCMS results for NIP228-Flexmab-SG3710. G0f, G1f and G2f glycoforms and their respective molecular masses are shown. Those three glycoforms correspond to re-bridged ADC with one SG3710. The molecular mass of the G0f, G1f and G2f parental glycoforms are shown. The mass difference between re-bridged and parental G0f glycoforms is 2428 Da, which correspond to the mass of one SG3710.
Preparation of site-specific ADCs with DAR of two using SG3249
To compare in vitro and in vivo potencies and rat tolerability of the ADCs with DAR of one prepared using SG3710, we prepared site-specific ADCs with a DAR of two for both trastuzumab and NIP228 using SG3249 as described previously.20 The conjugation and analytical characterization of these conjugates are shown in Figure S3.
Mouse serum stability of the ADCs re-bridged with SG3710
To evaluate the ex vivo stability of the re-bridged ADCs, trastuzumab-Flexmab-SG3710 and NIP228-Flexmab-SG3710 were incubated in mouse serum for seven days at 37°C. The stability of the re-bridged ADCs after one, three, and seven days was compared to that of the ADCs when the incubation started by monitoring the changes in re-bridged versus parental ADCs using affinity capture non-reducing LCMS (Figure S7). After seven days of incubation, affinity capture non-reducing LCMS revealed that less than 1% of the SG3710 payload was lost from both ADCs (Figure S7). This finding is noticeably different from results reported for other ADCs generated using stochastic methods for conjugation at the hinge cysteines, for which conventional payloads bearing a single linker resulted in significant drug losses.35-41 Even though the C226 position in the Flexmabs was exposed to solvent, we believe that the dual-linker design of SG3710 contributed to the high stability we observed in serum.
ADC linker designs have evolved to include a single protease-specific dipeptide such as valine-citrulline or valine-alanine for cathepsin B, acid pH sensitivity, or a disulfide-linkage to facilitate primary release of the warhead after ADC internalization and lysosomal trafficking.36,42,43 Currently, linkers using enzymatic cleavage contain only a single dipeptide cleavage site to enable subsequent release of the warhead, the speed of which can be fine-tuned by altering the selection of dipeptide residues.43,44 In addition to requiring two retro-Michael reactions to prematurely release the payload, SG3710 incorporates two cathepsin-B cleavable valine-alanine sites, and thus requires two enzymatic cleavage events to release the SG3199 warhead. The combination of these two features likely contributed to the high stability of the SG3710 ADCs in mouse serum observed in this study.
Cell binding and in vitro cytotoxicity of the re-bridged ADCs with a DAR of one versus site-specific ADCs with DAR of two
BIAcore data demonstrated similar (within the experimental error) Kd between trastuzumab and trastuzumab-Flexmab for binding to recombinant HER2 (Table 1). In vitro assessment of the target affinity and specificity of trastuzumab-Flexmab-SG3710 was conducted using flow cytometry with SKBR-3 (HER2-positive, 1.5 × 106 HER2 receptors/cell) and MCF-7 (HER2-negative) cell lines. Trastuzumab-Flexmab-SG3710 showed high affinity (EC50 = 15.7 nM) and selectivity to the SKBR-3 cell line, whereas no binding occurred to the MCF-7 cell line (Figure S8A, B). No binding was observed for the NIP228-Flexmab-SG3710 control with either SKBR-3 or MCF-7 cells (Figure S8A, B). These cell lines were also used to carry out cytotoxicity assays to compare the potency of the trastuzumab-Flexmab-SG3710 with that of trastuzumab engineered for site-specific conjugation of two SG3249 payloads (herein referred as trastuzumab-C239i-SG3249 Figure S8C, D).20 As expected, the potency of trastuzumab-Flexmab-SG3710 (IC50 = 35.5 pM, 5% cell viability) was slightly lower than that of trastuzumab-C239i-SG3249 (IC50 = 14.4 pM, 2% cell viability) on the SKBR-3 cell line after five days of incubation. The trastuzumab-C239i-SG3249 IC50 data agreed with the data previously reported for this ADC (IC50 ~ 11.6 pM) using the SKBR-3 cell line.18 The two-fold difference in IC50 between trastuzumab-Flexmab-SG3710 and trastuzumab-C239i-SG3249 is due to different drug loads of the ADCs (i.e., trastuzumab-Flexmab-SG3710 has a DAR of one, while trastuzumab-C239i-SG3249 has a DAR of two). However, two-fold difference in IC50 is within the margin of experimental error and thus the potency of the two ADCs, despite their difference in drug load, is similar. Specificity towards HER2 was maintained for the trastuzumab-Flexmab-SG3710 as demonstrated by minimal cell killing (93% viability) on the MCF-7 cell line (Figure S8C, D). No cytotoxicity was observed for the NIP228-Flexmab-SG3710 negative control with either SKBR-3 or MCF-7 cells (Figure S8A, B).
In vivo efficacy of the re-bridged ADCs with DAR of one versus the site-specific ADCs with DAR of two
The in vivo efficacy of trastuzumab-Flexmab-SG3710 and trastuzumab-C239i-SG3249 ADCs was investigated after single-dose injections (1 mg/kg) in female athymic mice bearing NCI-N87 HER2-positive subcutaneous xenografts, and tumor growth was monitored for 85 days (Figure 7(a)). Despite having half the drug load, trastuzumab-Flexmab-SG3710 exhibited equivalent efficacy compared to trastuzumab-C239i-SG3249 with complete tumor regression maintained throughout the study (Figure 7(a)). These results prompted us to conduct a dose de-escalation study to evaluate the minimum effective dose necessary to achieve anti-tumor activity. A single dose of 0.6 mg/kg of trastuzumab-Flexmab-SG3710 yielded sustained tumor regression for 60 days (Figure 7(b)). A similar tumor regression was observed with trastuzumab-C239i-SG3249 dosed at 0.3 mg/kg (Figure 7(b)). A single dose of 0.3 mg/kg of trastuzumab-Flexmab-SG3710 yielded sustained tumor regression for approximately 45 days with slight tumor regrowth observed between days 45 and 60 (Figure 7(b))
Figure 7.
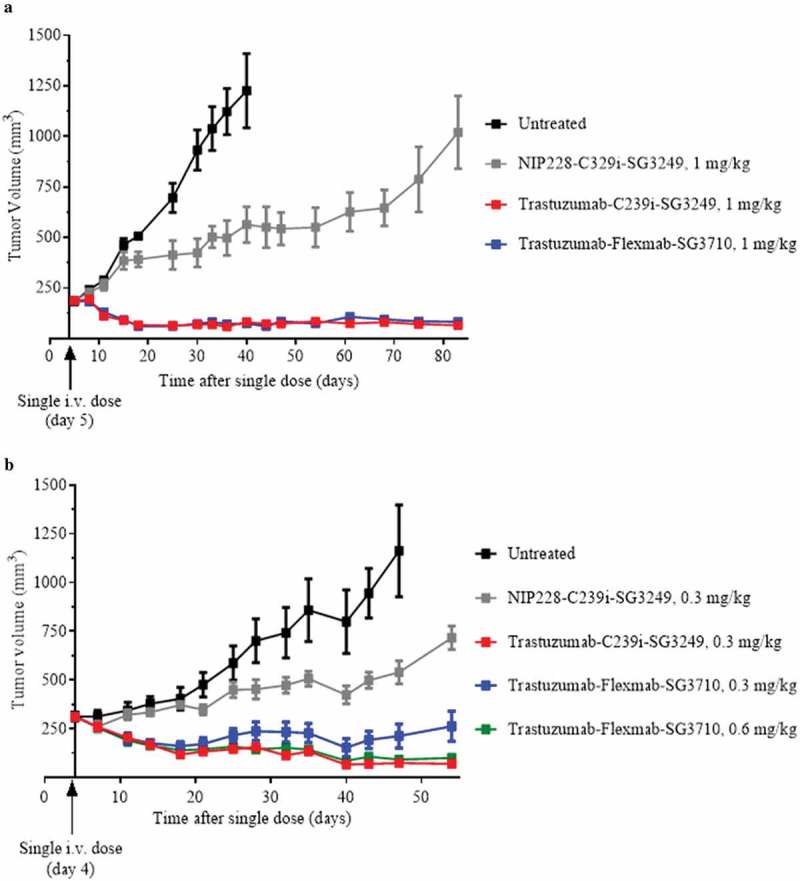
In vivo efficacy results in female athymic mice bearing NCI-N87 HER2-positive subcutaneous xenografts. (a) Results for 1 mg/kg dose of NIP228-C239i-SG3249 (grey), trastuzumab-C239i-SG3249 (red) and trastuzumab-Flexmab-SG3710 (blue). Five mice per group were dosed intravenously five days after their tumors reached a volume of 200 mm3. (b) In this efficacy study, the intravenous dose of NIP228-C239i-SG3249 (grey) and trastuzumab-C239i-SG3249 (red) were 0.3 mg/kg, while trastuzumab-Flexmab-SG3710 was dosed at 0.3 mg/kg (blue) and 0.6 mg/kg (green). The mice were treated on day four after tumors reached a volume of 200 mm3. Untreated mice were used as negative controls (black).
Total antibody analysis in mice
Serum concentration of total antibody for each of the molecules was measured in nude mice administered with a single intravenous dose of 1 mg/kg (Figure 8). Due to insufficient timepoints, pharmacokinetic analysis could not be performed, and thus half-lives could not be calculated. However, at later timepoints, trastuzumab-C239i-SG3249, had significantly faster clearance than trastuzumab-Flexmab-SG3710. Moreover, trastuzumab-Flexmab-SG3710 had similar clearance of trastuzumab and trastuzumab-Flexmab (Figure 8). Taken together, these data indicated that neither Flexmab engineering nor SG3710 re-bridging had adverse effects on clearance, and that trastuzumab-Flexmab-SG3710 had an improved pharmacokinetic profile compared to trastuzumab-C239i-SG3249.
Figure 8.
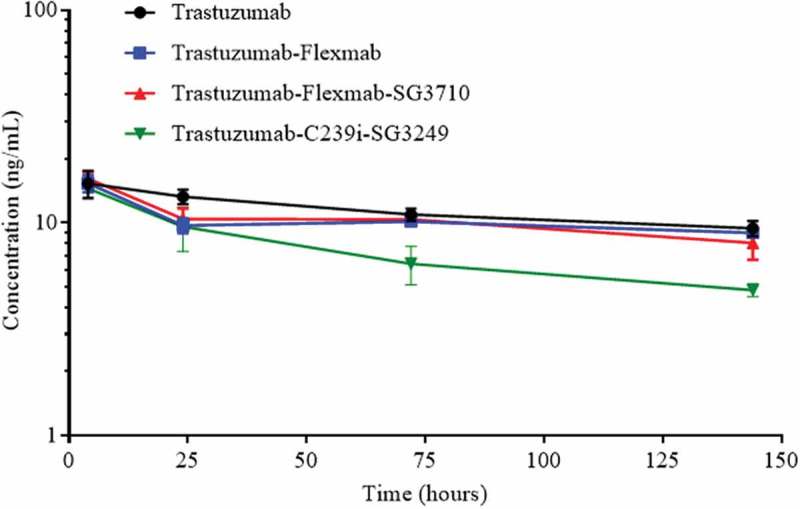
Serum concentration of total antibody after a single intravenous administration of 1 mg/kg of trastuzumab, trastuzumab-Flexmab, trastuzumab-Flexmab-SG3710 and trastuzumab-C239i-SG3249 in nude mice (n = 5 mice/group).
Exploratory toxicology study in rat
Single-dose tolerability studies were carried out over 22 days in naïve male Sprague-Dawley rats revealed that trastuzumab-Flexmab-SG3710 could be dosed at 4 mg/kg, twice the dose of trastuzumab-C239i-SG3249, which elicited dose-limiting toxicities at 2 mg/kg (Figure S9). In general, the predominant toxicity in rats treated with PBD ADCs is dose-dependent bone marrow suppression.19 Similarly, in this study, we observed dose-dependent decreases in white blood cell and platelet counts that peaked on day 15 post-dose. Importantly, the degree of bone marrow suppression was similar in animals treated with 2 mg/kg of trastuzumab-C239i-SG3249 and those treated with 4 mg/kg of trastuzumab-Flexmab-SG3710 (Figure S9). In addition, body weight measurements supported the greater tolerability of trastuzumab-Flexmab-SG3710 ADC at 4 mg/kg, with body weight changes for these animals after 22 days being nearly identical to those from animals treated with trastuzumab-C239i-SG3249 at 2 mg/kg (Figure S9).
Discussion
Previous studies have demonstrated a correlation between drug loading and in vitro cytotoxicity with ADCs containing higher DAR values exhibiting greater potency. However, an inverse relationship is often observed between ADCs with higher drug loads and their efficacy and tolerability. For example, Hamblett and colleagues generated anti-CD30 ADCs using the mc-vc-PAB-MMAE payload in which the number of payloads ranged from two, four, and eight drugs per antibody.25 When the authors compared the biophysical properties of these ADCs in tumor-bearing mice, they observed that the ADCs with two or four drugs per antibody had slower clearance rates and longer half-lives and were tolerated better than ADCs with eight drugs. Similarly, in our study the ADC with the higher drug load, trastuzumab-C239i-SG3249, was slightly more potent in vitro and in vivo than trastuzumab-Flexmab-SG3710. In line with Hamblett’s observations,25 the ADC with greater hydrophobicity, trastuzumab-C239i-SG3249, showed more rapid clearance in vivo than trastuzumab-Flexmab-SG3710. In addition, the lower drug loading of trastuzumab-Flexmab-SG3710 improved tolerability in rats, confirming that a balance must be struck between improving efficacy while preserving tolerability.
Hinrichs and colleagues have further elucidated the importance of balancing anti-tumor activity with tolerability when advancing PBD-based ADCs through the preclinical pipeline.19 After previous fractionated dosing regimens of ADCs based on tubulin-inhibitors, the authors used fractionated dosing with the PBD-based ADC 1C1-C239i-SG3249 (anti-EphA2, DAR = 2) at 0.5 mg/kg and compared the anti-tumor effects to those of single-dose administrations of 1C1-C239i-SG3249 ADC at 1 mg/kg in preclinical models of breast, gastric, and pancreatic cancer.19 They observed that, although dose-fractionation effectively lowers the highest peak serum drug concentrations, equivalent total exposure was maintained, which improved the tolerability of the PBD-based ADCs while maintaining their anti-tumor activity. Fractionated dosing could further enhance the tolerability of trastuzumab-Flexmab-SG3710 by reducing organ-specific toxicities observed with PBDs, such as lower peripheral blood counts and bone marrow suppression.45 Although this approach was not tested herein, such experiments could provide an additional rationale for evaluating PBD-based ADCs with lower drug load to enhance their therapeutic window.
In summary, we demonstrated that the combination of the engineered anti-HER2 Flexmab and SG3710 technologies results in a stable, potent ADC with reduced hydrophobicity and improved tolerability. The benefits of the Flexmab are extensive and include its applicability to any IgG1 using straight-forward molecular biology techniques, high expression yields, retention of binding affinity and specificity of the parental antibody, and the elimination of disulfide scrambling during conjugation. The SG3710 payload further contributes its own set of highly desirable characteristics, including a scalable synthesis and a symmetrical chemical structure that enables highly efficient disulfide-bridging. Perhaps the most significant property of SG3710 is the modularity that this symmetrical PBD dimer affords. One could easily envision replacing the maleimide chemical handles with other functional groups such as alkynes for site-specific disulfide bridging of azide-containing biomolecules using Cu-catalyzed chemistry. SG3710 could also be chemically tuned such that a single payload requires two unique events (e.g., cathepsin-B cleavage and pH sensitivity) to enable warhead release, which could increase the payload’s tumor selectivity. Because ADC tolerability is a major translational hurdle to overcome,46,47 the data presented here suggest that a combination of Flexmab and SG3710 technologies could improve the safety of future PBD-based ADCs.
Materials and methods
Cell lines and culture conditions
We obtained SKBR-3 (HER2+), NCI-N87 (HER2+), and MCF-7 (HER2-) cell lines from ATCC and maintained them in T175 tissue culture flasks (Corning) using the manufacturer’s recommended media (McCoys 5A + 10% fetal bovine serum (FBS) for SKBR-3 cells; RPMI-1640 medium for NCI-N87 cells; Dulbecco’s modified Eagle medium (DMEM) + 10% FBS) for MCF-7 cells. 293F cells (Invitrogen) used for transient transfection of antibodies were maintained in 293F FreeStyle media (Invitrogen). SKBR-3 and MCF-7 cells were cultured in a 37°C incubator with 5% CO2. The 293F cells were cultured in shake flasks (2 L, Corning) at 37°C with 8% CO2 and rotation at 120 rpm.
Reagents and chemicals
All chemical reagents were purchased from Sigma Aldrich, VWR, or JT Baker unless otherwise specified and all were used without additional purification.
Design of the anti-Her2 and isotype control NIP228 Flexmabs
The trastuzumab antibody48 (Figure S1) and the MedImmune proprietary isotype control antibody (NIP228) were used to engineer the Flexmabs. The kappa light chain sequence contained two mutations, F118C and C214V (Kabat antibody numbering),30 whereas the heavy chain contained three mutations, L124C, C220V, and C228V (Figure S1). The light chain and heavy chain DNA sequences were codon-optimized for mammalian expression and procured from GeneArt (Life Technologies). The optimized Flexmab constructs were sub-cloned using standard molecular biology techniques into a MedImmune proprietary mammalian expression vector.48
Antibodies
The design, cloning, expression, purification, and characterization of trastuzumab, NIP228, trastuzumab-C239i, and NIP228-C239i have been described previously.20
Expression and purification of antibodies and Flexmabs
Expression and purification were carried out using previously published methods.20,48,49 After transient 293F expression and protein A purification, the antibodies were dialyzed into conjugation buffer (1X phosphate-buffered saline (PBS), 0.1 mM ethylenediaminetetraacetic acid (EDTA), pH 7.2) using Slide-A-Lyzer dialysis cassettes at 4°C (10 kDa molecular weight cutoff (MWCO), Thermo Fisher Scientific) and concentrated to 8 mg/mL using Vivaspin concentrators (10 kDa MWCO, GE Healthcare). Concentrations were determined using a Nanodrop spectrophotometer (A280, Thermo Fisher Scientific) with an extinction coefficient of 1.4. Transient expression yields after 10 days were 400 mg/L.
Binding to recombinant HER2
The Biacore T-100 biosensor, CM5 sensor chip, and related reagents were purchased from GE Healthcare. Anti-human Fc antibody (Thermo Fisher Scientific, cat. 31125) as the active surface (10000 response units) and blank reference surface chip were immobilized using the Amine Coupling Kit (GE Healthcare). Binding experiments were carried out at room temperature. 3 nM solutions of trastuzumab and trastuzumab-Flexmab in 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffered saline (HBS) (0.01 mol/L HEPES pH 7.4, 0.15 mol/L NaCl, 3 mmol/L EDTA, 0.05% v/v surfactant P20) were injected over the active sensor surface at a flow rate of 10 μL/minute for 1 minute. Next, serial dilutions (0.78–400 nM) of recombinant human HER2 (MedImmune) in HBS buffer were injected over both active and references surfaces at a flow rate of 40 μL/minute for 1.5 minutes. After completion of the association phase, dissociation was monitored for 15 minutes at the same flow rate. At the end of each cycle, the surfaces were regenerated using glycine pH 1.5 at a flow rate of 30 μL/minute for 0.5 minutes. Before the kinetics analysis, the control curves obtained with samples injected over reference surfaces were subtracted from the biosensor curves obtained after injection of the samples over active surfaces. The results were analyzed using BIAcore T-100 evaluation software.
Antibody-dependent cell-mediated cytotoxicity
The ADCC assay was performed as described previously.20,50 Human HER2-positive BT-474 breast cancer cells (ATCC HTB-20™), which express HER2 on their surfaces, were used as target cells. Cells were washed once in PBS and then suspended at 0.2 × 106 cell/mL in phenol-red free Roswell Park Memorial Institute medium supplemented with 5% FBS (Invitrogen; assay medium). Peripheral blood mononuclear cells from healthy donors were used as effector cells. Donor blood was collected into 10 mL EDTA tubes and pooled. Lymphocyte separation medium (15 mL) was added into each SepMate™-50 tube (StemCell). Donor blood was diluted with an equal volume of Hanks’ balanced salt solution (StemCell) and added to each SepMate™-50 tube. Tubes were centrifuged at 1200 × g for 10 minutes at room temperature. The top layer (platelets) of each tube was carefully removed. The new top layer, which contained the mononuclear cells, was poured into a new 50 mL conical tube and centrifuged at 400 × g for 10 minutes at room temperature. The supernatant was poured off and cells were washed once with Hanks’ balanced salt solution using new 50 mL conical tubes. Cells were then centrifuged at 300 × g for 10 minutes at room temperature, after which the supernatant was poured off, and cells were suspended in assay medium at 2.5 × 106 cell/mL. Antibodies were diluted to a concentration of 3 μg/mL in assay medium. Ten-fold serial dilutions were prepared for each set of antibodies in the assay medium to a concentration of 1 μg/mL to 1 ng/mL. On day 1, antibodies, target cells, and effector cells were combined by the addition of 50 μL of the serially diluted antibodies to 50 μL of BT-474 target cells, along with 50 μL of effector cells into wells of a 96-well plate (12.5:1 ratio of effector and target cells). To correct for non-antibody-mediated lactate dehydrogenase (LDH) release in target cells, antibodies and target cells were combined in triplicate by the adding of 50 μL of the serially diluted antibodies to 50 μL of BT-474 cells along with 50 μL of assay medium into wells. Three control wells with 150 μL of assay medium for lysis solution correction were included on each plate. In addition, 12 control wells with 50 μL of target cells and 100 μL of assay medium (six wells for target cell basal LDH release and six wells for target cell maximum LDH release) were included on each plate. Assay plates were centrifuged at 1,100 rpm for three minutes to ensure effector and target cell contact. Plates were incubated at 37°C in 5% CO2 for 24 h. The CytoTox 96 Non-radioactive Cytotoxicity Assay kit (Promega), which measures LDH released in culture media from cells undergoing lysis, was used to quantify cell lysis. On day 2, 15 μL of 10 × lysis solution (Promega) was added to the lysis solution correction and target cell maximum LDH release control wells and incubated for five minutes. Plates were centrifuged at 1,100 rpm for five minutes to pellet cells. Supernatant (50 μL) from each well was pipetted into a new white wall clear-bottom plate. CytoTox 96® reagent (50 μL) was added to generate red formazan product in the dark for 15–30 minutes at room temperature. Stop solution (50 μL; Promega) was added to each well, and the absorbance at OD490 nm was measured within one hour.
ADCC activity was calculated using the following formula:
% Cytotoxicity = (Experimental – Effector spontaneous – Target spontaneous)/(Target maximum – Target spontaneous) × 100.
Target maximum LDH released was calculated using the following formula:
Target cell maximum LDH released = target cell maximum – lysis solution
The data were plotted and analyzed to fit a sigmoidal dose-dependent curve using Prism software (GraphPad).
Differential scanning calorimetry (DSC) of antibodies and Flexmabs
DSC experiments were carried out using a MicroCal VP-DSC calorimeter (Malvern). Native and Flexmab antibodies were extensively dialyzed into 25 mM histidine-HCl at pH 6 and 4°C and formulated at 0.5 mg/mL. The raw data were normalized for concentration and scan rate (1°C/minute). Data analysis and deconvolution were carried out using the Origin 7 software (Malvern). Deconvolution analysis was conducted using a non-two-state model and the best fits were obtained using 15 iteration cycles. The denaturation temperatures, Tm °C, corresponding to the maximum of the transition peaks were determined for each construct.
Synthesis of SG3710 and SG3249 payloads
Synthesis of SG3710 is shown in Figure S2, and experimental details are provided in the supplementary information. The synthesis of SG3249 has been previously described.18
Site-specific conjugation of SG3710 to Flexmabs
Flexmabs (15 mg, 100 nmoles) in conjugation buffer (1X PBS, 1 mM EDTA, pH 7.2, 3 mL) were reduced using tris(2-carboxyethyl)phosphine hydrochloride (TCEP, 3 eq., 300 nmoles, Thermo Fisher Scientific) for one hour at room temperature. After reduction, DMSO (10% v/v, 300 μL) was added to the reduced Flexmabs, followed by the addition of SG3710 (3 eq., 300 nmoles, in DMSO). After three hours incubation at room temperature, the reaction was quenched using N-acetyl cysteine (5 eq. over SG3710, 1.5 μmoles, Sigma Aldrich) and the Flexmab-ADCs were dialyzed against three buffer exchanges of conjugation buffer at 4ºC using a Slide-A-Lyzer dialysis cassette (10 kDa MWCO, Thermo Fisher Scientific). The Flexmab-ADCs were diluted at a 1:5 ratio with deionized H2O and loaded onto a type II ceramic hydroxyapatite (CHT) column (Bio-Rad) at 5 mL/min using an AKTA Pure (GE Healthcare). The column was washed with 20 column volumes of CHT buffer A (10 mM NaPO3, pH 7.0) to remove excess of SG3710 and conjugation reagents. Elution of the Flexmab-ADCs was performed using a linear gradient of CHT buffer B (2 M NaCl in 10 mM NaPO3, pH 7.0) for 20 minutes. The eluted ADCs were dialyzed overnight at 4ºC into conjugation buffer using a Slide-A-Lyzer dialysis cassette (10 kDa MWCO) and diluted at a 1:5 ratio with HIC buffer A (25 mM Tris-HCl, 1.5M (NH4)2 SO4, pH 8.0). The ADCs were loaded onto a semi-preparative HIC column (HiTrap Butyl-S FF, GE Healthcare) at 1 mL/min using an AKTA Pure (GE Healthcare) and washed with 20 column volumes of HIC buffer A. The ADCs were eluted using a linear gradient of HIC buffer B (25 mM Tris-HCl, 5% isopropyl alcohol, pH 7.0) for 45 minutes at 1 mL/minute. Purified ADCs were dialyzed into 25 mM histidine-HCl, pH 6.0 overnight at 4ºC, concentrated using a Vivaspin concentrator (10 kDa MWCO) to 2 mg/mL, and filtered through a 0.2 μm syringe filter (Pall Corporation). To ensure removal of endotoxin, the ADCs were filtered using Mustang E syringe filters (0.2 μm; VWR).
Site-specific conjugation of SG3249 to antibodies
Site-specific conjugation of SG3249 to antibodies with the C239i insertion was carried out as previously described.20,33 Briefly, C239i antibodies (20 mg, 133 nmoles) in conjugation buffer (3.5 mL) were reduced using TCEP (40 eq., 5.33 µmoles) for three hours at 37°C. After reduction, the antibodies were dialyzed through two buffer exchanges of conjugation buffer overnight at 4°C, then filtered through 0.2 µm syringe filters. The next day, (L)-dehydroascorbic acid (20 eq., 2.67 μmoles, in DMSO) was added to the reduced antibodies for four hours at room temperature. The SG3249 payload (8 eq., 0.96 μmoles, in DMSO) was added and the conjugation reaction proceeded for one hour at room temperature. Excess of SG3249 was quenched using N-acetyl cysteine (4 eq. over SG3249, 3.84 μmoles, in deionized H2O) for 15 minutes at room temperature and the ADCs were dialyzed against three exchanges of conjugation buffer at 4ºC using a Slide-A-Lyzer dialysis cassette. The ADCs were diluted at a 1:5 ratio with deionized H2O and loaded onto a type II ceramic hydroxyapatite column at 5 mL/min using an AKTA Pure (GE Healthcare). The column was washed with 20 column volumes of CHT buffer A(10 mM NaPO3, pH 7.0) to remove excess payload and conjugation reagents. The ADCs were eluted using a linear gradient of CHT buffer B(2 M NaCl, 10 mM NaPO3, pH 7.0) for 20 minutes. The purified ADCs were then formulated in the same way as the Flexmab-ADCs.
Analytical characterization of the ADCs
The C239i ADCs were analytically characterized as described previously.20,50 A summary of their analytical characterization is shown in Figure S3.
Size-exclusion high-performance liquid chromatography (HPLC) was used to analyze purity and aggregation with an Agilent 1200 series HPLC system. Samples (100 µg in 100 µL) were injected onto a TSK Gel column (G3000SW, 8 mm I.D. × 30 cm × 5 µm, Tosoh Biosciences) using 0.1 M NaPO4, 0.1 M NaSO4, and 10% isopropanol, at pH 6.8 as the mobile phase at a flow rate of 1 mL/minute. Absorbance of eluted peaks was measured at 280 nm followed by manual integration to determine purity and percent aggregation.
Analytical HIC-HPLC was used to analyze hydrophobicity. Flexmabs and Flexmabs-ADCs (50 µg in 50 µL) were loaded onto a Proteomix HIC Butyl-NP5 column (4.6 mm I.D. × 3.5 cm × 5 µm, Sepax Technologies) using HIC buffer A (25 mM Tris-HCl, 1. 5M (NH4)2 SO4, pH 8) and eluted using a linear gradient of HIC buffer B (25 mM Tris-HCl, 5% isopropyl alcohol, pH 7, 5–100%) for 13 minutes at 0.8 mL/minute. Absorbance was measured at 280 nm and at 330 nm,34 and eluted peaks were manually integrated to determine conjugation efficiency.
Reduced reverse-phase (rRP-HPLC) was used to confirm site-specific re-bridging of SG3710 onto the heavy chain of the Flexmabs. Flexmabs and Flexmabs-ADCs were treated with dithiothreitol (50 mM) for 30 minutes at room temperature. After reduction, the Flexmabs and Flexmab-ADCs were injected onto a PLRP-S column (1000Å, 2.1 mm × 50 mm, Agilent) and eluted using a gradient mobile phase of solvent A (0.1% trifluoroacetic acid in water) and solvent B (0.1% trifluoroacetic acid in acetonitrile) consisting of 5% to 100% solvent B for 25 minutes. rRP-HPLC was carried out at 80°C using a flow rate of 1 mL/minute. Absorbance was measured at 280 nm and 330 nm.
Intact and reduced reverse-phase LCMS was used to confirm the molecular weights of the Flexmabs and Flexmab-ADCs. Two µg (4 µL) of Flexmabs or Flexmab-ADCs were injected onto an Agilent 1200 series HPLC system connected in series to an Agilent 6520 Accurate-Mass Time-of-Flight LCMS system. Flexmabs or Flexmab-ADCs were loaded onto a Zorbax 300 Diphenyl Rapid Resolution HD column (2.1 mm × 50 mm × 1.8 µm) and eluted using a flow rate of 0.5 mL/minute consisting of a step gradient of 1–80% solvent B (0.1% formic acid in acetonitrile) after two minutes (solvent A: 0.1 formic acid in water). Data was acquired and analyzed using MassHunter software (Agilent) and DARs and the efficiency of conjugation were calculated based on deconvoluted peak analysis.
Cell binding by flow cytometry
Binding affinities and specificities of the trastuzumab-Flexmab and NIP228-Flexmab ADCs were assessed using flow cytometry. On the day of the cell binding experiments, SKBR-3 (HER2+) and MCF-7 (HER2-) cells were dissociated from their flask with TrypLE trypsin (Thermo Fisher Scientific) and resuspended in their respective growth media. Cells were counted on a ViCell cell viability analyzer (Beckman Coulter) and brought to a concentration of 1 × 106 cells/mL. Cells were transferred in duplicate to wells (5 × 104 cells/well) of a 96-well plate (Falcon) and centrifuged at 1,200 rpm and 4ºC. Pelleted cells were resuspended in 180 µL of flow cytometry buffer (PBS pH 7.2, 2% FBS, on ice) and antibodies or ADCs were added to cells (20 µL of serial dilution: 200 µg/ml-0.01 µg/mL; final concentration 20–0.001 µg/mL). Unconjugated antibodies or ADCs and cells were incubated at 4ºC for one hour and were then washed with flow cytometry buffer and pelleted by centrifugation (twice, 1,200 rpm). After the final centrifugation, cell pellets were resuspended in AlexaFluor 647-conjugated anti-human secondary antibody (Thermo Fisher Scientific Cat. A-21445;150 µL, 8 µg/mL, pH 7.2 in PBS, 2% FBS) and incubated at 4ºC for one hour. Cells were washed with flow cytometry buffer and centrifuged (twice, 1200 rpm), followed by resuspension in 135 µL of flow cytometry buffer. 4ʹ,6-diamidino-2-phenylindole hydrochloride was added (15 µL from 10X stock, 1 µM final, Sigma Aldrich) to each cell suspension to act as a live/dead stain. Fluorescence data from the cells were collected using a LSRII flow cytometer (Beckton Dickson) and data were analyzed using FlowJo analysis software. Binding curves were generated using Prism (GraphPad).
Mouse serum stability studies
Mouse serum (Jackson ImmunoResearch Laboratories) was filtered through a 0.2 μm syringe filter (Pall Corporation) into sterile polypropylene tubes and kept on ice. The ADCs (200 μg) were added to mouse serum for a final concentration of 200 μg/mL and samples were incubated at 37°C. Aliquots (200 μL) were taken from each sample at 0, 24, 72, and 148 hours of incubation. Samples collected at baseline (i.e., time zero), which were used as controls, were placed on dry ice within the first minutes after the addition of ADCs to serum. Samples were stored at −80°C until they were subjected to affinity capture and analysis by non-reduced LCMS. Anti-human IgG (Fc-specific) agarose beads (Sigma Aldrich) was used for affinity capture of the ADCs from mouse serum. At each time point, 50 μL of anti-human Fc agarose beads were mixed with 300 μL of PBS and 100 μL of thawed serum sample for 30 min at room temperature under continuous rotation. The beads were washed three times with 1× PBS to remove unbound serum proteins and the ADCs were eluted using 100 μL IgG elution buffer (Thermo Fisher Scientific) and neutralized with 20 μL 1 M Tris-HCl pH 8. To facilitate further analysis, the eluted ADCs were deglycosylated using PNGase F (New England BioLabs) in accordance with the manufacturer’s instructions. The deglycosylated ADCs (20 μL) were analyzed by non-reduced LCMS (as described above), and the raw data was analyzed using MassHunter (Agilent).
Cytotoxity assay
On the day of the cytotoxicity assay, SKBR-3 and MCF-7 cells were dissociated from their flask with 0.25% trypsin/EDTA (Thermo Fisher Scientific), then 1:1 10% media was added to neutralize the trypsin/EDTA. Cells were centrifuged at 1,100 RPM’s for 3 minutes and were resuspended in their growth media. Cells were counted on a ViCell cell viability analyzer (Beckman Coulter) and brought to a concentration of 1.0 × 106 cells/mL in their respective growth media. Cell suspensions (100 μL, 1.0 × 104 cells/well) were transferred to white/white-bottomed 96-well plates (Corning). Cells were allowed to rest at room temperature for 20 min and then incubated to further adhere for 2 hours in a 37ºC incubator with 5% CO2. After a two-hour incubation, 2X, 3-fold serial dilutions of antibodies and ADCs were prepared with a range of 20 µg/mL – 3 ng/mL and 100 µL of each dilution was added to the wells in triplicate (10 µg/mL – 1.5 ng/mL final ADC concentrations, 100 μL total volume/well). Media (100 μL) was added to untreated wells of each plate to serve as negative controls. Five days after treatment, plates were removed from the incubator and allowed to equilibrate to room temperature for about 25 minutes. Plates were centrifuged (1,100 rpm, three minutes) and supernatants were carefully flicked off the plates. RPMI without phenol red or FBS was added to each well (50 μL), to all plates, followed by the addition of CellTiter-Glo (Promega) reagent (50 μL). Plates were shaken for one hour in the dark at room temperature and luminescence was measured using an Envision plate reader (PerkinElmer). Percent viability was calculated using the following equation:
Experimental data were plotted using GraphPad Prism to generate half maximal inhibitory concentration (IC50) curves.
Mouse tumor growth inhibition
All studies using animals were performed humanely using a protocol approved by the MedImmune Institutional Animal Care and Use Committee in a facility accredited by AAALAC International. NCI-N87 cells (5 × 106) in 50% Matrigel were inoculated subcutaneously into 4–6 week-old female athymic nude mice (Charles River Laboratories). When tumors reached a size of 200 mm3, five mice each were randomly assigned to groups. ADCs were administered intravenously via the lateral tail vein at the indicated doses on day 5 (1 mg/kg dose) and day 4 (all other doses) after cell inoculation. Tumor volumes were measured twice weekly with calipers. Untreated mice were used as controls. The tumor volumes were calculated using the formula:
Body weights were monitored daily to assess treatment tolerability. The tumor growth data were analyzed using Prism software (GraphPad). Tumor volumes were expressed as mean ± standard error of the mean.
Total antibody analysis in mice
Serum concentration of trastuzumab, trastuzumab-Flexmab, trastuzumab-Flexmab-SG3710 and trastuzumab-C239i-SG3249 were measured by immunoassay to determine pharmacokinetic profile of total antibody. We did not determine total ADC because those ADCs wehre previously shown to be highly stable in serum.20 Three athymic nude mice (Charles River Laboratories) per group were given a single intravenous dose of 1 mg/kg. Blood samples were collected into K2-EDTA tubes from each mouse through orbital bleeding at 4, 24, and 72 hours and through terminal bleeding at 144 hours post-injection; these tubes were processed into plasma. Pre-dose bleed samples were used as controls. Two μg/mL (30 μL/well) of the anti-human Fc antibody (Thermo Fisher Scientific, cat. 31125) were coated in 96-well plates overnight in PBS at 4ºC. The plates were washed five times with PBS containing Tween 80 (PBST) and blocked with 3% non-fat dry milk in PBS (blocking buffer) for one hour at room temperature. A two-fold dilution of antibodies or ADCs in blocking buffer, starting at 20 μg/mL, was added to each well (30 μL/well) and incubated at room temperature for one hour. The plates were washed five times with PBST and 1 μg/mL of biotinylated anti-trastuzumab antibody (MedImmune) was added (30 μl/well) in 3% bovine serum albumin (BSA, Thermo Fisher Scientific). The plates were washed again five times and a dilution of 1:3,000 of horseradish peroxidase–conjugated streptavidin (Thermo Fisher Scientific) in 3% BSA was added (30 μL/well). The plates were washed five times with PBST and developed with (3,3ʹ,5,5ʹ tetramethylbenzidine substrate (30 μL/well), followed by the addition of 0.2 N HCl (30 μL/well) to stop the reaction. Plates were read at 450 nm. Data were plotted using Prism5 (Graph Pad). Standard curves for each test article were prepared in 10% pooled normal mouse plasma.
Exploratory toxicology studies in rats
Male Sprague Dawley rats (aged 8–12 weeks, n = 5 per group) were given a single intravenous injection on day 1 of 0.75, 1.5, 3, or 4 mg/kg of trastuzumab-Flexmab-SG3710 or 0.75, 1.5, or 2 mg/kg of trastuzumab-C239i-SG3249 and monitored for 21 days. Control rats (five per group) were given a single intravenous injection of vehicle control on day one. In all main study animals, changes in body weight, clinical pathology, and gross pathology with organ weights were assessed, and microscopic observations were made. Hematology and serum chemistry samples were collected and analyzed using standard methodologies on days 8 and 15. Additional samples for coagulation analysis were collected and analyzed on day 22 only. The toxicology studies and clinical chemistry data analysis were carried out at Charles River Laboratories (Reno, Nevada). The data were plotted using Prism (Graph Pad).
Funding Statement
These studies were supported by MedImmune, the global biologics R&D arm of AstraZeneca.
Abbreviations
- ADC
antibody-drug conjugate
- ADCC
antibody-dependent cell-mediated cytotoxicity
- BT-474
breast cancer cells
- CHT
type II ceramic hydroxyapatite
- DAR
drug-to-antibody ratio
- DMEM
Dulbecco’s modified Eagle medium
- DMSO
dimethyl sulfoxide
- DSC
differential scanning calorimetry
- EC50
half maximal effective concentration
- EDTA
ethylenediaminetetraacetic acid
- FBS
fetal bovine serum
- HIC
hydrophobic interaction chromatography
- HER2
human epidermal growth factor receptor 2
- HPLC
high performance liquid chromatography
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- Kd
equilibrium dissociation constant
- IgG1
immunoglobulin G1
- LDH
lactate dehydrogenase
- LCMS
liquid chromatography mass spectrometry
- MCF-7
breast cancer cells
- MED
minimum efficacious dose
- MTD
maximum tolerated dose
- MWCO
molecular weight cutoff
- mc-vc-PAB-MMAE
monomethyl auristatin E
- PABA
para-aminobenzyl alcohol
- PBS
phosphate-buffered saline
- PBD
pyrrolobenzodiazepine dimers
- PC-3
prostate cancer cells
- PEG
polyethylene glycol
- rLCMS
reduced liquid chromatography mass spectrometry
- rRP-HPLC
reduced reverse phase high performance liquid chromatography
- RPMI
Roswell Park Memorial Institute
- SKBR-3
breast cancer cells
- TCEP
tris(2-carboxyethyl)phosphine hydrochloride
- TI
therapeutic index
- TM
transition temperatures
Acknowledgments
The authors would like to thank the members of the MedImmune animal facility for the help with the mouse studies. The authors thank Song Cho for help with the ADCC studies; Amit Kumar and Frank Comer for critically reviewing the manuscript. The pharmacokinetics samples processing and assays validation were carried out at BioAgilytix. Editorial support was provided by Deborah Berlyne (funded by MedImmune).
Contributions
All author contributed to the research, and all reviewed and approved the manuscript.
Disclosure of potential conflicts of interest
The authors are employees of Medimmune and may have ownership interest in AstraZeneca.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
References
- 1.Leimgruber W, Stefanović V, Schenker F, Karr A, Berger J.. Isolation and characterization of anthramycin, a new antitumor antibiotic. J Am Chem Soc. 1965;3:5791–93.doi: 10.1021/ja00952a050 PMID:5845427. [DOI] [PubMed] [Google Scholar]
- 2.Hurley LH. Pyrrolo(1,4)benzodiazepine antitumor antibiotics, comparative aspects of anthramycin, tomaymycin and sibiromycin. J Antibiot. 1977;30:349–70. PMID:328469. [DOI] [PubMed] [Google Scholar]
- 3.Mantaj J, Jackson PJ, Rahman KM, Thurston DE.. From anthramycin to pyrrolobenzodiazepine (PBD)-containing antibody-drug conjugates (ADCs). Angew Chem Int Ed Engl. 2017;56:462–88. doi: 10.1002/anie.201510610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peters C, Brown S. Antibody-drug conjugates as novel anti-cancer chemotherapeutics. Biosci Rep. 2015;35:e00225. doi: 10.1042/BSR20150089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vezina HE, Cotreau M, Han TH, Gupta M. Antibody-drug conjugates as cancer therapeutics: past, present, and future. J Clin Pharmacol. 2017;10:S11–S25. doi: 10.1007/s11864-016-0418-0. [DOI] [PubMed] [Google Scholar]
- 6.Sievers EL, Senter PD. Antibody-drug conjugates in cancer therapy. Annu Rev Med. 2013;64:15–29. doi: 10.1146/annurev-med-050311-201823. [DOI] [PubMed] [Google Scholar]
- 7.Hurley LH, Reck T, Thurston DE, Langley DR, Holden KG, Hertzberg RP, Hoover JR, Gallagher G, Faucette LF, Mong SM. Pyrrolo[1,4]benzodiazepine antitumor antibiotics: relationship of DNA alkylation and sequence specificity to the biological activity of natural and synthetic compounds. Chem Res Toxicol. 1988;1:258–68. PMID:2979741. [DOI] [PubMed] [Google Scholar]
- 8.Hartley JA. The development of pyrrolobenzodiazepines as antitumour agents. Expert Opin Investig Drugs. 2011:20733–44. doi: 10.1517/13543784.2011.573477. [DOI] [PubMed] [Google Scholar]
- 9.Hertzberg RP, Hecht SM, Reynolds VL, Molineux IJ, Hurley LH. DNA sequence specificity of the pyrrolo[1,4]benzodiazepine antitumor antibiotics. Methidiumpropyl-EDTA-iron(II) footprinting analysis of DNA binding sites for anthramycin and related drugs. Biochemistry. 1986;25:1249–58. PMID:3008824. [DOI] [PubMed] [Google Scholar]
- 10.Wells G, Martin CR, Howard PW, Sands ZA, Laughton CA, Tiberghien A, Woo CK, Masterson LA, Stephenson MJ, Hartley JA, et al. Design, synthesis, and biophysical and biological evaluation of a series of pyrrolobenzodiazepine-poly(N-methylpyrrole) conjugates. J Med Chem. 2006;49:5442–61. doi: 10.1021/jm051199z. [DOI] [PubMed] [Google Scholar]
- 11.Rahman KM, Thompson AS, James CH, Narayanaswamy M, Thurston DE. The pyrrolobenzodiazepine dimer SJG-136 forms sequence-dependent intrastrand DNA cross-links and monoalkylated adducts in addition to interstrand cross-links. J Am Chem Soc. 2009;131:13756–66. doi: 10.1021/ja902986x. [DOI] [PubMed] [Google Scholar]
- 12.Smellie M, Bose DS, Thompson AS, Jenkins TC, Hartley JA, Thurston DE. Sequence-selective recognition of duplex DNA through covalent interstrand cross-linking: kinetic and molecular modeling studies with pyrrolobenzodiazepine dimers. Biochemistry. 2003;42:8232–39. doi: 10.1021/bi034313t. [DOI] [PubMed] [Google Scholar]
- 13.Gregson SJ, Howard PW, Hartley JA, Brooks NA, Adams LJ, Jenkins TC, Kelland LR, Thurston DE. Design, synthesis, and evaluation of a novel pyrrolobenzodiazepine DNA-interactive agent with highly efficient cross-linking ability and potent cytotoxicity. J Med Chem. 2001;44:737–48. PMID:11262084. [DOI] [PubMed] [Google Scholar]
- 14.Kamal A, Prabhakar S, Shankaraiah N, Ratna Reddy C, Venkat Reddy P. Synthesis of C8–C8/C2–C8-linked triazolo pyrrolobenzodiazepine dimers by employing ‘click’ chemistry and their DNA-binding affinity. Tetrahedron Lett. 2008;49:3620–24. doi: 10.1016/j.tetlet.2008.04.006. [DOI] [Google Scholar]
- 15.Kopka ML, Goodsell DS, Baikalov I, Grzeskowiak K, Cascio D, Dickerson RE. Crystal structure of a covalent DNA-drug adduct: anthramycin bound to C-C-A-A-C-G-T-T-G-G and a molecular explanation of specificity. Biochemistry. 1994;33:13593–610. PMID:7947769. [DOI] [PubMed] [Google Scholar]
- 16.Gregson JS, Howard WP, Thurston ED, Jenkins CT, Kelland RL. Synthesis of a novel C2/C2ʹ-exo unsaturated pyrrolobenzodiazepine cross-linking agent with remarkable DNA binding affinity and cytotoxicity. Chem Commun. 1999:797–98. doi: 10.1039/A809791G. [DOI] [Google Scholar]
- 17.Gregson SJ, Howard PW, Gullick DR, Hamaguchi A, Corcoran KE, Brooks NA, Hartley JA, Jenkins TC, Patel S, Guille MJ, et al. Linker length modulates DNA cross-linking reactivity and cytotoxic potency of C8/C8ʹ ether-linked C2-exo-unsaturated pyrrolo[2,1-c][1,4]benzodiazepine (PBD) dimers. J Med Chem. 2004;47:1161–74. doi: 10.1021/jm030897l. [DOI] [PubMed] [Google Scholar]
- 18.Tiberghien AC, Levy JN, Masterson LA, Patel NV, Adams LR, Corbett S, Williams DG, Hartley JA, Howard PW. Design and synthesis of Tesirine, a clinical antibod-drug conjugate pyrrolobenzodiazepine dimer payload. ACS Med Chem Lett. 2016;24:983–87. doi: 10.1021/acsmedchemlett.6b00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hinrichs JM, Ryan PM, Zheng B, Afif-Rider S, Yu XQ, Gunsior M, Zhong H, Harper J, Bezabeh B, Vashisht K, et al. Fractionated dosing improves preclinical therapeutic index of pyrrolobenzodiazepine-containing antibody drug conjugates. Clin Cancer Res. 2017;23:5858–68. doi: 10.1158/1078-0432.CCR-17-0219. [DOI] [PubMed] [Google Scholar]
- 20.Dimasi N, Fleming R, Zhong H, Bezabeh B, Kinneer K, Christie RJ, Fazenbaker C, Wu H, Gao C. Efficient preparation of site-specific antibody-drug conjugates using cysteine insertion. Mol Pharm. 2017;14:1501– 16. doi: 10.1021/acs.molpharmaceut.6b00995. [DOI] [PubMed] [Google Scholar]
- 21.Harper J, Lloyd C, Dimasi N, Toader D, Marwood R, Lewis L, Bannister D, Jovanovic J, Fleming R, D’Hooge F, et al. Preclinical evaluation of MEDI0641, a pyrrolobenzodiazepine-conjugated antibody-drug conjugate targeting 5T4. Mol Cancer Ther. 2017;16:1576–87. doi: 10.1158/1535-7163.MCT-16-0825. [DOI] [PubMed] [Google Scholar]
- 22.Saunders LR, Bankovich AJ, Anderson WC, Aujay MA, Bheddah S, Black K, Desai R, Escarpe PA, Hampl J, Laysang A, et al. A DLL3-targeted antibody-drug conjugate eradicates high-grade pulmonary neuroendocrine tumor-initiating cells in vivo. Sci Transl Med. 2015;7:302ra136. doi: 10.1126/scitranslmed.aac9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Flynn MJ, Zammarchi F, Tyrer PC, Akarca AU, Janghra N, Britten CE, Havenith CEG, Levy J-N, Tiberghien A, Masterson LA, et al. ADCT-301, a pyrrolobenzodiazepine (PBD) dimer-containing antibody-drug conjugate (ADC) targeting CD25-expressing hematological malignancies. Mol Cancer Ther. 2016;15:2709–21. doi: 10.1158/1535-7163.MCT-16-0233. [DOI] [PubMed] [Google Scholar]
- 24.Cho S, Zammarchi F, Williams DG, Havenith CEG, Monks NR, Tyrer P, D'Hooge F, Fleming R, Vashisht K, Dimasi N , et al. Antitumor activity of MEDI3726 (ADCT-401), a pyrrolobenzodiazepine antibody-drug conjugate targeting PSMA, in pre-clinical models of prostate cancer. Mol Cancer Ther. 2018;10:2176–86. doi: 10.1158/1535-7163.MCT-17-0982. [DOI] [PubMed] [Google Scholar]
- 25.Hamblett KJ, Senter PD, Chace DF, Sun MM, Lenox J, Cerveny CG, Kissler KM, Bernhardt SX, Kopcha AK, Zabinski RF, et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin Cancer Res. 2004;10:7063–70. doi: 10.1158/1078-0432.CCR-04-0789. [DOI] [PubMed] [Google Scholar]
- 26.Boswell CA, Mundo EE, Zhang C, Bumbaca D, Valle NR, Kozak KR, Fourie A, Chuh J, Koppada N, Saad O, et al. Impact of drug conjugation on pharmacokinetics and tissue distribution of anti-STEAP1 antibody-drug conjugates in rats. Bioconjug Chem. 2011;22:1994–2004. doi: 10.1021/bc200212a. [DOI] [PubMed] [Google Scholar]
- 27.Herbertson RA, Tebbutt NC, Lee FT, MacFarlane DJ, Chappell B, Micallef N, Lee ST, Saunder T, Hopkins W, Smyth FE, et al. Phase I biodistribution and pharmacokinetic study of Lewis Y-targeting immunoconjugate CMD-193 in patients with advanced epithelial cancers. Clin Cancer Res. 2009;15:6709. doi: 10.1158/1078-0432.CCR-09-0536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burke PJ, Hamilton JZ, Jeffrey SC, Hunter JH, Doronina SO, Okeley NM, Miyamoto JB, Anderson ME, Stone IJ, Ulrich ML, et al. Optimization of a PEGylated glucuronide-monomethylauristatin E linker for antibody-drug conjugates. Mol Cancer Ther. 2017;16:116–23. doi: 10.1158/1535-7163.MCT-16-0343. [DOI] [PubMed] [Google Scholar]
- 29.Lyon RP, Bovee TD, Doronina SO, Burke PJ, Hunter JH, Neff-LaFord HD, Jonas M, Anderson ME, Setter JR, Senter PD. Reducing hydrophobicity of homogeneous antibody-drug conjugates improves pharmacokinetics and therapeutic index. Nat Biotechnol. 2015;33:733–35. doi: 10.1038/nbt.3212. [DOI] [PubMed] [Google Scholar]
- 30.Kabat EA, Wu TT, Perry H, Gottesman K, Foeller C. Sequences of proteins of immunological interest. 5th Bethesda (MD): NIH Publication No. 91–3242; 1991. [Google Scholar]
- 31.Vogel CL, Cobleigh MA, Tripathy D, Gutheil JC, Harris LN, Fehrenbacher L, Slamon DJ, Murphy M, Novotny WF, Burchmore M, et al. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J Clin Oncol. 2002;20:719–26. doi: 10.1200/JCO.2002.20.3.719. [DOI] [PubMed] [Google Scholar]
- 32.Yan M, Schwaederle M, Arguello D, Millis SZ, Gatalica Z. HER2 expression status in diverse cancers: review of results from 37,992 patients. Cancer Metastasis Rev. 2015;34:157–64. doi: 10.1007/s10555-015-9552-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J, Pegram M, Oh D-Y, Diéras V, Guardino E, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med. 2012;367:1783–91. doi: 10.1056/NEJMoa1209124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jeffrey SC, Burke PJ, Lyon RP, Meyer DW, Sussman D, Anderson M, Hunter JH, Leiske CI, Miyamoto JB, Nicholas ND, et al. A potent anti-CD70 antibody-drug conjugate combining a dimeric pyrrolobenzodiazepine drug with site-specific conjugation technology. Bioconjug Chem. 2013;7:1256–63. doi: 10.1021/bc400217g. [DOI] [PubMed] [Google Scholar]
- 35.Alley SC, Benjamin DR, Jeffrey SC, Okeley NM, Meyer DL, Sanderson RJ, Senter PD. Contribution of linker stability to the activities of anticancer immunoconjugates. Bioconjug Chem. 2008;19:759–65. doi: 10.1021/bc7004329. [DOI] [PubMed] [Google Scholar]
- 36.Trail PA, Willner D, Knipe J, Henderson AJ, Lasch SJ, Zoeckler ME, TrailSmith MD, Doyle TW, King HD, Casazza AM, et al. Effect of linker variation on the stability, potency, and efficacy of carcinoma-reactive BR64-doxorubicin immunoconjugates. Cancer Res. 1997;57:100–05. PMID:8988048. [PubMed] [Google Scholar]
- 37.Acchione M, Kwon H, Jochheim CM, Atkins WM. Impact of linker and conjugation chemistry on antigen binding, Fc receptor binding and thermal stability of model antibody-drug conjugates. mAbs. 2012;4:362–72. doi: 10.4161/mabs.19449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pillow TH, Tien J, Parsons-Reponte KL, Bhakta S, Li H, Staben LR, Li G, Chuh J, Fourie-O’Donohue A, Darwish M, et al. Site-specific trastuzumab maytansinoid antibody–drug conjugates with improved therapeutic activity through linker and antibody engineering. J Med Chem. 2014;57:890–7899. doi: 10.1021/jm500552c. [DOI] [PubMed] [Google Scholar]
- 39.Tumey LN, Charati M, He T, Sousa E, Ma D, Han X, Clark T, Casavant J, Loganzo F, Barletta F, et al. Mild method for succinimide hydrolysis on ADCs: impact on ADC potency, stability, exposure, and efficacy. Bioconjug Chem. 2014;25:1871–80. doi: 10.1021/bc500357n. [DOI] [PubMed] [Google Scholar]
- 40.Jackson D, Atkinson J, Guevara CI, Zhang C, Kery V, Moon S-J, Virata C, Yang P, Lowe C, Pinkstaff J, et al. In vitro and in vivo evaluation of cysteine and site specific conjugated herceptin antibody-drug conjugates. PLoS One. 2014;9:e83865. doi: 10.1371/journal.pone.0083865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Christie RJ, Fleming R, Bezabeh B, Woods R, Mao S, Harper J, Joseph A, Wang Q, Xu Z-Q, Wu H, et al. Stabilization of cysteine-linked antibody drug conjugates with N-aryl maleimides. J Control Release. 2015;220PtB:660–70. doi: 10.1016/j.jconrel.2015.09.032. [DOI] [PubMed] [Google Scholar]
- 42.Erickson HK, Widdison WC, Mayo MF, Whiteman K, Audette C, Wilhelm SD, Singh R. Tumor delivery and in vivo processing of disulfide-linked and thioether-linked antibody−maytansinoid conjugates. Bioconjug Chem. 2010;21:84–92. doi: 10.1021/bc900315y. [DOI] [PubMed] [Google Scholar]
- 43.Dubowchik GM, Firestone RA, Padilla L, Willner D, Hofstead SJ, Mosure K, Knipe JO, Lasch SJ, Trail PA. Cathepsin B-labile dipeptide linkers for lysosomal release of doxorubicin from internalizing immunoconjugates: model studies of enzymatic drug release and antigen-specific in vitro anticancer activity. Bioconjug Chem. 2002;13:855–69. PMID: 12121142. [DOI] [PubMed] [Google Scholar]
- 44.Cazzamalli S, Dal Corso A, Neri D. Linker stability influences the anti-tumor activity of acetazolamide-drug conjugates for the therapy of renal cell carcinoma. J Controlled Release. 2017;246:39–45. doi: 10.1016/j.jconrel.2016.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pillow TH, Schutten M, Yu SF, Ohri R, Sadowsky J, Poon KA, Solis W, Zhong F, Del Rosario G, Go MAT, et al. Modulating therapeutic activity and toxicity of pyrrolobenzodiazepine antibody-drug conjugates with self-immolative disulfide linkers. Mol Cancer Ther. 2017;16:871–78. doi: 10.1158/1535-7163.MCT-16-0641. [DOI] [PubMed] [Google Scholar]
- 46.Lowe D. More thoughts on AbbVie’s Rova-T implosion. Sci Transl Med. 2018. http://blogs.sciencemag.org/pipeline/archives/2018/06/05/more-thoughts-on-abbvies-rova-t-implosion. [Google Scholar]
- 47.ASH Clinical News All clinical trials of vadastuximab talirine suspended following patient deaths. 2017; https://www.ashclinicalnews.org/news/clinical-trials-vadastuximab-talirine-suspended-following-patient-deaths/
- 48.Dimasi N, Fleming R, Wu H, Gao C. Molecular engineering strategies and methods for the expression and purification of IgG1-based bispecific bivalent antibodies. Methods. 2018. doi: 10.1016/j.ymeth.2018.08.004. [DOI] [PubMed] [Google Scholar]
- 49.Dimasi N, Gao C, Fleming R, Woods RM, Yao XT, Shirinian L, Kiener PA, Wu H. The design and characterization of oligospecific antibodies for simultaneous targeting of multiple disease mediators. J Mol Biol. 2009;393:672–92. doi: 10.1016/j.jmb.2009.08.032. [DOI] [PubMed] [Google Scholar]
- 50.Thompson P, Fleming R, Bezabeh B, Huang F, Mao S, Chen C, Harper J, Zhong H, Gao X, Yu X-Q, et al. Rational design, biophysical and biological characterization of site-specific antibody-tubulysin conjugates with improved stability, efficacy and pharmacokinetics. J Control Release. 2016;236:100–16. doi: 10.1016/j.jconrel.2016.06.025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.