

ABSTRACT
Monoclonal antibodies are among the fastest growing therapeutics in the pharmaceutical industry. Detecting higher-order structure changes of antibodies upon storage or mishandling, however, is a challenging problem. In this study, we describe the use of diethylpyrocarbonate (DEPC)-based covalent labeling (CL) – mass spectrometry (MS) to detect conformational changes caused by heat stress, using rituximab as a model system. The structural resolution obtained from DEPC CL-MS is high enough to probe subtle conformation changes that are not detectable by common biophysical techniques. Results demonstrate that DEPC CL-MS can detect and identify sites of conformational changes at the temperatures below the antibody melting temperature (e.g., 55 ᴼC). The observed labeling changes at lower temperatures are validated by activity assays that indicate changes in the Fab region. At higher temperatures (e.g., 65 ᴼC), conformational changes and aggregation sites are identified from changes in CL levels, and these results are confirmed by complementary biophysical and activity measurements. Given the sensitivity and simplicity of DEPC CL-MS, this method should be amenable to the structural investigations of other antibody therapeutics.
KEYWORDS: Antibody therapeutics, biopharmaceutical characterization, biophysical characterization, covalent labeling, diethylpyrocarbonate, mass spectrometry, liquid chromatography, protein conformation, protein higher-order structure, rituximab
Introduction
Monoclonal antibodies (mAbs) are among the fastest growing categories of therapeutics in the pharmaceutical industry.1–3 By 2020, around 70 mAb products are anticipated to be available on the market and their global sales are predicted to be nearly $125 billion, representing 15% of total pharmaceutical sales.1,4 Unlike small molecule drugs, the higher-order structure (HOS) of mAbs contributes to the greater binding specificity towards drug targets, resulting in higher therapeutic efficacy and less adverse effects. Changes in HOS upon storage or mishandling, e.g., protein misfolding and aggregation, however, can lead to reduced stability, loss of efficacy, unwanted actions, or possible immunogenicity.5,6 Monitoring HOS is thus essential to ensure efficacy and safety of mAb therapeutics throughout a product life cycle – from drug manufacturing to dose administration.6-9 Any new, structurally-informative method could be useful for biologics license applications (BLAs) because HOS characterization is required in the stability, lot-to-lot comparability, and biosimilar studies of antibody therapeutics.6,9,10
Detecting HOS changes of mAbs is challenging given their size and the multidomain nature. The current toolbox for HOS analysis of protein therapeutics has limitations.5,6,8 X-ray crystallography and nuclear magnetic resonance (NMR) spectroscopy can provide atomic-level resolution of protein structure, but these methods are time- and sample-consuming, and not amenable to all proteins. In contrast, biophysical techniques such as differential scanning calorimetry (DSC), dynamic light scattering (DLS), fluorescence spectroscopy, infrared (IR) spectroscopy, and circular dichroism (CD) spectroscopy are rapid, but provide only low-resolution ensemble averages of protein global conformation. These low-resolution methods still are commonly used to characterize HOS of mAb therapeutics,11-19 even though they do not provide information about small localized conformational changes, some of which may be potentially significant for drug efficacy and safety. Hence, there is a growing need for rapid and sample-efficient analytical tools with moderate resolution that can characterize HOS of therapeutic mAbs at the amino acid level. Current regulatory guidelines do not specify what method(s) should be used in characterizing HOS of biologics,20-26 allowing for the development of novel analytical techniques.
Mass spectrometry (MS) has become one of the most powerful methods for the analysis of proteins. From a recent evaluation of BLAs, the use of MS to characterize primary structure and HOS of protein therapeutics has increased in recent years.27 Characterizing HOS using MS requires that a protein’s structural information is encoded into the mass of that protein. Commonly-used MS-based approaches for studying protein structure include hydrogen/deuterium exchange (HDX) and covalent labeling.6,8,10 In HDX-MS, information regarding solvent accessibility and dynamics of backbone amides can be obtained from the exchange of hydrogens by deuteriums, thereby increasing the mass in a structurally informative manner. HDX-MS has been successfully used to investigate structural changes and identify aggregation sites in mAbs obtained from different storage or stress conditions.28-30 Although this technique has been commonly used in HOS analysis of mAb therapeutics,31-33 an analytical challenge is the accuracy of HDX measurements due to the transient nature of deuterium labeling that can lead to back exchange and scrambling. In addition, specialized robotic equipment and software are required to obtain optimal results.
Covalent labeling (CL) can also be used with MS to study protein HOS, but unlike HDX, CL is generally not subject to back exchange and scrambling. CL approaches use reagents to irreversibly modify solvent-exposed amino acid side chains, encoding structural information into the mass of the protein. CL along with MS detection (CL-MS), especially when used with bottom-up tandem MS (MS/MS), provides information about solvent accessibility of amino acid side chains, making it complementary to HDX.34-38 As an example, site-specific carboxyl group footprinting has been applied for the structural characterization of glycosylated therapeutic mAbs39,40 and for epitope-paratope mapping.41,42 A localized conformation change of mAbs as induced by deglycosylation was also identified using this technique.43 However, the reagent pair of 1-ethyl-3-(3-(dimethylamino)propyl)-carbodiimide hydrochloride (EDC) and glycine ethyl ester (GEE) that was used in the study can monitor only Glu and Asp residues, limiting its structural resolution. Using a more non-specific reagent capable of modifying a range of amino acid side chains simultaneously can allow protein structure to be probed with greater resolution.38 Hydroxyl radical footprinting (HRF), in which hydroxyl radicals (●OH) are generated and non-selectively label solvent accessible residues,44 has been used for the structural analysis of mAbs.40 HRF with MS detection has been used to identify epitopes and paratopes for antigen-antibody interactions41,45-47 and to determine dimer interfaces in formulated mAbs.48 However, specialized instrumentation such as a laser or a synchrotron source is needed for ●OH radical production. Moreover, data interpretation is complicated because ●OH radicals can generate over 50 different modification types on proteins.44 For day-to-day structural analysis of biotherapeutics, a less expensive and simpler experimental design and workflow is needed.
Diethylpyrocarbonate (DEPC) is a commercially-available reagent that can react with a range of nucleophilic side chains and N-termini of proteins, resulting in excellent protein structural coverage.38,49 Unlike radical reagents, no specialized instrumentation is required because DEPC can readily modify proteins once added to solution. DEPC labeling reactions also result in only one type of product, allowing very low levels of labeling to be straightforwardly identified and further increasing analytical sensitivity and structural coverage.34,38 CL-MS methods based on DEPC have been extensively developed by our group,35,50,51 and have been used to obtain insight into protein-ligand and protein-protein interactions as changes in side chain solvent accessibility at binding sites can be probed.35,52-56
Here, we extend the applicability of DEPC-based CL-MS to study protein conformational changes that occur upon heating. Using rituximab as a model mAb therapeutic, we demonstrate the ability to site-specifically detect subtle HOS changes at the temperatures far below the melting point of the mAb therapeutic, changes that are not detected by common biophysical methods. Sites of these structural changes are revealed at the amino-acid level using MS/MS and are validated by activity assays. Overall, given the simplicity, sensitivity, and straightforwardness of DEPC CL-MS, we predict that this method will be amenable to the structural investigations of other antibody therapeutics.
Results
Biophysical characterization of rituximab HOS after storage at mild to moderate stress
Before studying heat-stressed rituximab by covalent labeling, we used CD spectroscopy, fluorescence spectroscopy, and dynamic light scattering (DLS) to identify any structural perturbations upon heating. When heating rituximab for 4 h at temperatures below its melting point, we find that the three techniques are not able to detect any significant structural changes at 45°C or 55°C and reveal only mild changes at 65°C (Figure 1). Upon heating at 65°C, rituximab undergoes changes in its HOS, as indicated by intrinsic fluorescence and CD spectroscopy (Figure 1(a and b)), and unfolds to some extent as indicated by DLS (Figure 1(c)). These classical biophysical techniques provide only the weighted average structure of a global conformation, so the locations of any structural changes are unknown.
Figure 1.
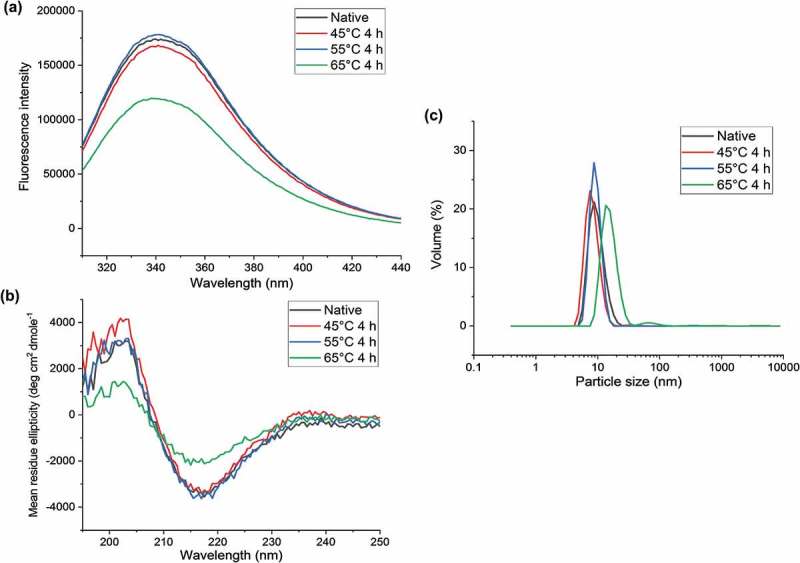
Biophysical characterization of rituximab at 37°C (native, black) and thermally-stressed conditions, after incubation of the rituximab formulation at 45°C for 4 h (red), 55°C for 4 h (blue), and 65°C for 4 h (green). Techniques used in structural characterization were (a) tryptophan fluorescence spectroscopy, (b) far-UV circular dichroism spectroscopy, and (c) dynamic light scattering. The essentially identical overlap of the spectra of the native and 45°C and 55°C heat-stressed samples indicates that these techniques do not detect any structural changes to rituximab after preheating to these temperatures. Upon heating at 65°C, however, structural changes and aggregation occur.
DEPC labeling with MS detection as a tool for HOS analysis of rituximab
DEPC-based covalent labeling together with MS detection was used to identify any structural changes undergone by rituximab that could not be detected by CD, fluorescence, or DLS. DEPC is a reactive electrophile that can modify solvent accessible nucleophilic side chains (Cys, His, Lys, Thr, Tyr, Ser) and N-termini of proteins (Figures S1a and S1b in the Supplemental Material). The resulting carbethoxylated products of these residues have a mass shift of +72.021 Da, and the specific protein modification sites can be identified and semi-quantified after proteolytic digestion, liquid chromatographic separation of the resulting peptides, and tandem MS analysis (Figure 2). Any changes to the extent of covalent labeling at particular residues can be used to probe HOS changes to proteins upon comparing one condition (e.g. native) to another (e.g., heated). During the labeling conditions, the DEPC to protein molar ratio is limited at 4 to 1 to minimize labeling-induced structural perturbations to a protein during the labeling reaction, while at the same time providing sufficient labeling extents to identify the modified sites.35 CD and intrinsic fluorescence spectroscopy confirm that the DEPC-modified rituximab undergoes no significant structural changes, as compared to the spectra of unlabeled rituximab (Figure S2).
Figure 2.

Scheme showing DEPC labeling with MS detection for the structural analysis of antibody therapeutics. DEPC reagent modifies solvent accessible amino acids. The modified protein is subjected to proteolytic digestion and the modified peptides are analyzed using LC–MS/MS. Sites of protein conformational changes can be revealed by changes in the extent of labeling at specific residues.
To obtain the extent of labeling at a residue level, the DEPC-labeled rituximab was subjected to “bottom-up” analysis via proteolytic digestion and LC-MS/MS (Figures S1c and S3). Peptide identification and peak area determination were performed using custom-designed software described previously,56 as described in the Materials and Methods section. A rituximab sequence coverage of over 90% was obtained in the tryptic peptide mapping, allowing almost the entire structure of the protein to be probed. Upon DEPC labeling, up to 47 residues are found to be modified in each light chain (LC) while 107 residues are modified in each heavy chain (HC), which together corresponds to 23% of the residues in rituximab. Considering the average distance between adjacent DEPC modification sites,50 the effective resolution for probing rituximab’s structure using DEPC is ~ 8 Å. Some DEPC-modified residues could be reliably detected down to levels as low as 0.001%, however a more conservative labeling threshold of 0.01% was used for all peptides. Detection of peptides at these labeling levels together with the excellent structural coverage allows us to monitor subtle structure changes to the protein upon heat treatment. The limits of quantitation (LOQ) were experimentally determined by spiking experiments of a model synthetic tryptic peptide (rituximab HC peptide with amino acids 306–321; DEPC-labelled at different sites), into a digest of unlabeled rituximab at varying concentrations (Figure S4).
To identify regions of rituximab that undergo subtle structural changes upon heating, DEPC modification levels at individual amino acid residues of heat-treated rituximab were compared to the modification levels of the un-treated protein. For each stress condition, unpaired student t-tests were used to determine if the labeling levels are significantly different at a 95% confidence level. For strongly nucleophilic residues, like His and Lys, changes in labeling extent are determined by the absolute difference in DEPC label levels (Equation 1).
where and are the average % DEPC modification of a given residue in the stressed and native proteins, respectively. (See Equation 3 in the Materials and Methods for a calculation of % DEPC modification.) For weakly nucleophilic residues, such as Tyr, Ser, and Thr, modification extents are much smaller than for His and Lys residues due to their lower intrinsic reactivity with DEPC. Thus, a ratio difference, which is more sensitive to a small modification change, is used to determine labeling changes for Tyr, Ser, and Thr residues (Equation 2).
DEPC CL-MS for probing subtle structural changes of rituximab
In Table 1 the number of DEPC-modifiable residues in each rituximab domain that undergo significant changes in modification extent after heating for 4 h at 45°C is graphically depicted. The sites that undergo statistically significant labeling changes are binned into three groups – low, medium, and high – based on the extent of the labeling change. These bins were obtained upon plotting the DEPC modification extents, from all stress temperatures, on a single histogram and finding that they distribute into three ranges (see Table 1 for details). Relatively few changes in DEPC labeling levels are found in rituximab samples stressed at 45°C. Most of these changes are increases in labeling, as might be expected when the protein is heated and undergoes unfolding. In addition, the variable regions have far fewer changes than the constant domains. More than 70% of the labeling changes occur at Tyr, Ser, and Thr residues, and the labeling changes that occur at His and Lys residues are always less than 20%. While the extent of labeling of both sets of residues are influenced by changes in solvent accessibility, labeling of the weakly nucleophilic Tyr, Ser, and Thr residues is more sensitive to changes in local microenvironment. Thus, labeling changes predominantly in these residues suggests that the structural changes at 45°C are primarily changes in the local microenvironment instead of large structural changes that lead to significant differences in solvent accessibility. Consistent with this idea is the fact that the labeling changes are scattered throughout the protein structure rather than clustered in certain regions of the protein (Figure 3(a)). DEPC labeling sites and levels for rituximab under native and the lower temperature thermally-stressed conditions (45°C and 55°C) are listed in full in Tables S1 and S2, respectively.
Table 1.
Changes in DEPC modification extents after the heat stress at 45°C for 4 h. The pie charts indicate the fraction of modified residues that undergo statistically significant labeling changes within each domain of rituximab. Red represents labeling increases while blue represents decreases. The bar charts for each domain indicate the number of residues whose extent of covalent labeling (CL) change falls within low (L), medium (M), and high (H) bins.
 |
Figure 3.
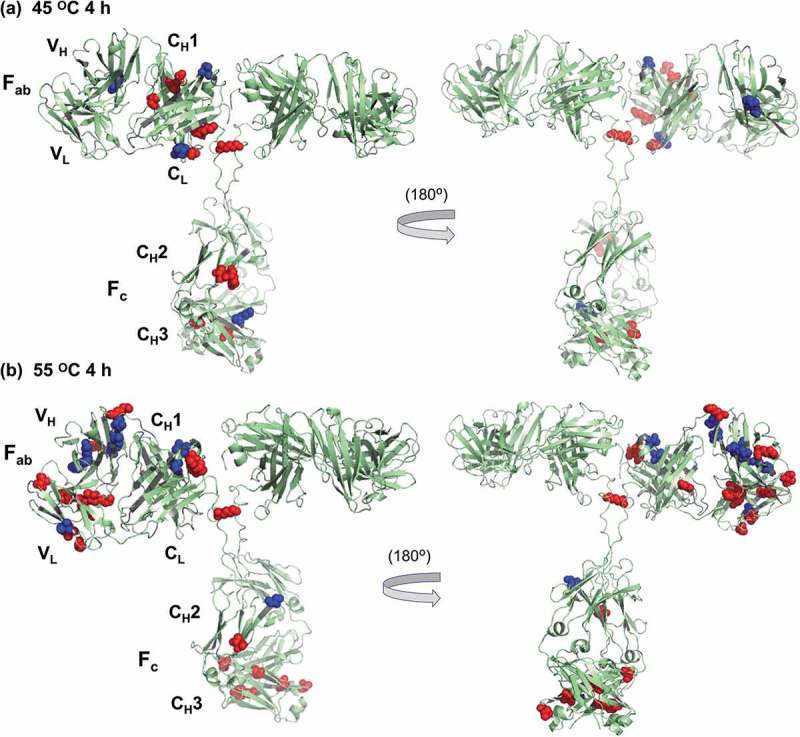
Sites on rituximab that undergo significant labeling changes after heat stress at (a) 45°C for 4 h and (b) 55°C for 4 h, as compared to non-stressed rituximab. Spheres represent residues that undergo significant changes in label levels (p < 0.05). Red represents labeling increases while blue represents decreases. Note that, for clarity, only one asymmetric unit of rituximab structure is labeled in this figure. As no full-length structures of rituximab are available in a protein data bank (PDB), we used existing Fab and Fc crystal structures of rituximab to generate a molecular model for the entire rituximab molecule. A full-length human IgG1 model, from which atomic coordinates were generated using PDBs 2IG2 (Fc) and 1FC2 (Fab) with a hinge region and other details theoretically modeled57,58, was used as a template. Fc (PDB 4W4N) and Fab (PDB 4KAQ) structures of rituximab were then aligned to the template, using the molecular visualization system PyMOL.
Activity assays were conducted to determine if the DEPC labeling changes reflect significant enough structural changes to affect rituximab activity and thus validate the DEPC labeling results. Three types of activity assays were performed: 1) rituximab bridging ELISA, 2) Alamar blue assay, and 3) Raji cell pull-down assay (Figure S5). The bridging ELISA assay was used to evaluate binding of the Fc region, thereby reporting on the structural integrity of Fc region. In addition, complement-dependent cytotoxicity (CDC) activity of the Fc region was assessed using an Alamar blue assay.59,60 The structural integrity of Fab region was evaluated using CD20-positive Raji B cells as part of a pull-down assay. These activity assays were conducted using rituximab preheated at different temperatures spanning the range of temperatures studied by DEPC labeling. Results from the bridging ELISA show that there is no significant change in Fc binding activity of rituximab after preheating to 45°C (Figure 4(a)). CDC activity of Fc region is also estimated to remain unchanged after heat stress at 45°C (Figure 4(b)). Similarly, the Fab binding activity of rituximab stressed at 45°C is estimated not to differ from control samples (Figure 4(c)). Together, these activity assays are consistent with the idea that the protein does not undergo significant structural changes at 45°C, as indicated by the DEPC labeling experiments. The number of residues undergoing labeling changes is relatively small in both the Fab and Fc regions and consists primarily of Tyr, Ser, and Thr residues that are more sensitive to local microenvironment changes. Moreover, the modification sites are scattered throughout the protein (Figure 3(a)), suggesting little effect on the conformation of the Fab and Fc regions, as confirmed by the activity assays.
Figure 4.

Structural changes revealed from DEPC CL-MS experiments are validated using rituximab activity assays. The structural integrity of the Fc region is evaluated by (a) a rituximab bridging ELISA that measures Fc binding to a capture antibody and (b) an Alamar blue assay that measures complement dependent cytotoxicity (CDC). The structural integrity of the Fab region is assessed by (c) a Raji cell pull-down assay that measures Fab binding to CD20 antigen on B cells. For each temperature condition, the means of two groups i.e. stressed vs. control (37°C) were compared using student t-test at 95% confidence level. Each assay was performed in triplicate (n = 3).
More residues undergo significant changes in modification extent after heating at 55°C for 4 h, and these changes can be found in all rituximab domains, particularly in the VH and VL domains of the Fab region (Table 2). Most of the changes in each domain are increases in labeling, with an exception of the VH and CL domains where there is a similar or higher number of labeling decreases. Almost twice as many labeling changes occur at His and Lys residues upon heating to 55°C compared to heating at 45°C, although most of the changes are characterized as low or medium changes. Most of the labeling changes at Tyr, Ser, and Thr are characterized as medium or high changes. Overall, these results indicate likely changes in protein topology, although the moderate labeling changes at His and Lys residues suggest somewhat modest structural changes. In contrast to the heat stress at 45°C, after heating at 55°C, the sites whose label levels undergo significant changes are found to cluster in the Fab region of the protein, especially in VH, VL, and CL domains (Figure 3(b)). Such clustering suggests localized structural changes in the Fab region of rituximab. Similar clustering of residues undergoing labeling changes are not observed in the Fc region (Figure 3(b)).
Table 2.
Changes in DEPC modification extents after the heat stress at 55°C for 4 h. The pie charts indicate the fraction of modified residues that undergo statistically significant labeling changes within each domain of rituximab. Red represents labeling increases while blue represents decreases. The bar charts for each domain indicate the number of residues whose extent of covalent labeling (CL) change falls within low (L), medium (M), and high (H) bins.
 |
Further insight into the meaning of the labeling data can be obtained via comparison to the activity assays. A small, but statistically significant, change in Fc binding activity from the bridging ELISA is measured at 55°C (Figure 4(a)), but the CDC activity of the Fc region does not change significantly after heating at 55°C (Figure 4(b)). The lack of consistency in the Fc-related assays might correlate with the lack of clustering of labeled residues in the Fc as seen in the DEPC labeling experiments (Figure 3(b)). One possible explanation is that the conformational changes in the Fc region are significant enough to influence the ELISA but too subtle to affect the Alamar blue assay. A significant change in Fab binding activity is observed from the Raji cell pull-down assay for rituximab preheated at 55°C (Figure 4(c)). This result is very consistent with the observed labeling changes in the Fab region that show significant clustering of residues that undergo labeling changes (Figure 3(b)).
Investigation of conformation change upon higher heat stress
As indicated earlier, heating at 65°C leads to greater changes in the fluorescence, CD, and DLS data. In fact, heating at this temperature for 4 h results in a cloudy sample, and DLS data suggest the formation of small protein aggregates, as indicated by a small percentage of species at 100 nm (Figure 1(c)). Additional support for this conclusion is also found from size-exclusion chromatography (SEC) measurements (Figure S6), which reveal the presence of high molecular weight species upon heating at 65°C and not upon heating at 45°C or 55°C. Upon applying our DEPC CL-MS method to rituximab thermally-stressed at 65°C, we find significant labeling changes in all rituximab domains (Table 3 and Table S3). Most notably, many more decreases in labeling are observed than increases in labeling, which is likely explained by protein aggregation. Significant decreases in labeling are found in both the Fab and Fc regions, while only a few residues in the Fab region undergo increases in labeling. Around 30% of the labeling changes at 65°C are found at His and Lys residues, and most of these can be characterized as medium or high changes in labeling extent. For Tyr, Ser, and Thr residues, most sites undergo medium or high extents of labeling changes. Overall, these results suggest more profound changes in the solvent accessibility and the local microenvironment for many sites in the protein upon heating at 65°C. When the residues that undergo changes in labeling are mapped on the rituximab structure, clusters of residues are found in the CH3 domain of Fc region and the variable domains of the Fab region (Figure 5).
Table 3.
Changes in DEPC modification extents after the heat stress at 65°C for 4 h. The pie charts indicate the fraction of modified residues that undergo statistically significant labeling changes within each domain of rituximab. Red represents labeling increases while blue represents decreases. The bar charts for each domain indicate the number of residues whose extent of covalent labeling (CL) change falls within low (L), medium (M), and high (H) bins.
 |
Figure 5.

Sites on rituximab that undergo significant (a) increases and (b) decreases in DEPC modification after heat stress at 65°C for 4 hr, as compared to non-stressed rituximab. Spheres represent residues that undergo significant changes in label levels (p < 0.05). Red represents labeling increases while blue represents decreases. Note that, for clarity, only one asymmetric unit of rituximab structure is labeled in this figure. (See Figure 3 for more details about rituximab’s molecular model.).
The labeling changes are consistent with activity assays that indicate changes in the Fab and Fc regions. Results from the bridging ELISA show that there is a significant change in Fc binding after heating to 65°C (Figure 4(a)). CDC activity of the Fc region (Figure 4(b)) and Fab binding activity (Figure 4(c)) are also significantly different from control samples. The activity assay results are consistent with the DEPC labeling results, indicating significant structural changes to rituximab in both Fab and Fc regions.
Discussion
In this study, we have shown the applicability of DEPC CL-MS as an analytical tool to provide site-specific information about changes in the HOS of protein therapeutics. Sites of the structural changes are revealed from differences in DEPC modification levels compared to a control sample. The full meaning of the quantitative changes in labeling is not fully understood at this stage, but labeling increases often suggest greater unfolding in a given region and labeling decreases imply burial of side chains usually from aggregation. Changes in the solvent-accessible surface area (SASA) of a given residue is a primary factor that governs side chain reactivity,36-38 but changes in reactivity can also be caused by changes in local tertiary structure around a given residue (e.g., microenvironment).38
Previous studies have identified the melting temperatures (Tm) for different domains of rituximab to be 71°C, 74°C, and 83°C for the CH2, Fab, and CH3 domains, respectively.61 We stressed the rituximab samples at temperatures far below these Tm values as we were interested in studying the protein under mild to moderate thermal stress to see if DEPC CL-MS could report on any subtle structural changes. These lower temperatures more realistically mimic stresses that protein therapeutics may undergo.62 Results from DEPC CL-MS after heating at 45°C suggest that no significant structural changes occur at this temperature, which is consistent with fluorescence, CD, and DLS measurements as well as the activity assays. Even though a few residues undergo statistically significant labeling changes, the number of labeling changes that occur at His and Lys residues, which are the better reporters of SASA changes, is relatively small. Most of the labeling changes occur at Tyr, Ser, and Thr residues (Table 1), which are sites that are more sensitive to local microenvironment changes such as changes in H-bonding or nearby electrostatic interactions. While the extent of DEPC modification of both sets of residues are influenced by SASA, the labeling changes that occur at the weakly nucleophilic Tyr, Ser, and Thr residues are much less correlated with the changes in SASA.38 Similar observations are made in HRF experiments, where the primary structure and HOS more significantly influence residues with poor intrinsic reactivity.36 The predominance of labeling changes at Tyr, Ser, and Thr residues suggests that any HOS changes at 45°C are changes in the local microenvironment instead of significant unfolding that would dramatically change the SASA of residues. Moreover, the labeling changes are scattered throughout the protein structure rather than clustered at certain regions of the protein (Figure 3), again implying very local effects. The data from the activity assays (Figure 4) are in agreement with this idea.
A greater number of labeling changes occur upon heating rituximab to 55°C than at 45°C, and these labeling changes are clustered in the Fab domain of the protein. This extent and localized nature of labeling changes indicate that CL-MS is revealing structural perturbations, changes that are not measurable by fluorescence, CD, and DLS. Around 30% of the labeling changes at 55°C occur at His and Lys residues, and these changes are mostly moderate or high increases in labeling (Table 2), indicating that the protein is unfolding in these regions. As alluded to earlier, the reactivity of the more nucleophilic His and Lys residues is better correlated with SASA, and thus the labeling of these residues are more sensitive to local unfolding events that would expose them to a greater extent.35 It is important to note that most of the His and Lys labeling changes at 55°C are classified as moderate or low increases, meaning there is less than a 20% increase in labeling. While we have not established a quantitative relationship between labeling extents and SASA, the fact that the biophysical techniques do not detect any structural changes suggests that any structural changes that do occur after heating to 55°C are subtle. A larger number of Tyr, Ser, and Thr residues also undergo labeling changes after stressing at 55°C than at 45°C, and most are moderate or high increases (Table 2). Again, these residues are less correlated with SASA changes, but the significant number of labeling changes to these residues also suggests a structural change.
The locations of the structural changes after heating to 55°C are worth noting. The most significant and clustered labeling changes are found in the Fab region, which is reported to be the most sensitive region to heat stress in IgG1 molecules.63 Our results would seem to contrast with DSC data for rituximab obtained by Andersen et al. in which the CH2 domain is found to have the lowest Tm of 71°C;61 however, this difference is almost certainly due to differences in the nature of our labeling experiments and the DSC experiments. In our experiments, the protein reaches a thermal equilibrium after 4 h of heating at 55°C before being cooled and analyzed, whereas the DSC experiment involves a temperature ramp where the protein is exposed to a given temperature for only a minute without associated cooling.61 Because different protein domains can change structures at different rates and re-fold to different extents upon cooling, one might not expect there to be a strong correlation between our heat-stress experiments and DSC experiments. Very telling is the fact that several residues that undergo significant labeling changes cluster in the variable domains, VH and VL, where the complementarity-determining regions (CDRs) for CD20 are located (Figure 3 and Table 2).64 Among the residues undergoing significant labeling changes at 55°C are Thr91 on the LC, located in the middle of CDR L3, His33 on the LC and His35 and Lys74 on the HC, which are located close to CDRs L1, H1, and H2, respectively (Figure S7b). Structural changes near these residues, as indicated by the labeling changes, are nicely consistent with the decreased Fab binding that was found in the activity assay (Figure 4(c)). To the best of our knowledge, our CL-MS technique is the first to report a subtle HOS change of a therapeutic mAb at the temperature as low as 55°C.
Heating at 65°C more significantly alters the conformation of rituximab, as revealed by biophysical measurements, activity assays, and CL-MS. Indeed, some fraction of the protein aggregates upon heating for 4 h at this temperature, which is consistent with the propensity of rituximab to transiently unfold at temperatures below its Tm.62 Aggregation is also indicated by the fact that most of the measured changes in CL-MS are moderate or high labeling decreases (Table 3). It may seem surprising that the 65°C data does not have a more even mix of labeling increases and decreases because the protein probably unfolds to some degree in addition to aggregating. It should be noted, though, that our technique is inherently much more sensitive to aggregation events than unfolding events. The amino acids that are labeled by DEPC are primarily polar residues that are more likely to reside on the protein surface anyway. Consequently, protein unfolding events only mildly increase their SASA and DEPC reactivity, while aggregation events completely bury these normally exposed residues, dramatically changing their SASA and DEPC reactivity. If a given residue is more exposed in some protein molecules but buried in other protein molecules, the decrease in labeling due to residue burial will likely counterbalance any labeling increase due to increased solvent exposure.
This greater sensitivity to aggregation events gives us excellent insight into regions of the protein that self-associate, as we have shown previously for other proteins.52,53,56 Clusters of residues that undergo decreases in labeling are found in both Fab and Fc regions (Figure 5). Structural changes in the Fab region are consistent with a reduction in Fab binding to CD20 (Figure 4(c)), and many labeling changes are found near the CDRs. Among the labeling changes are Tyr70 on the LC and His35, Lys67, and Lys74 on the HC, which sit close to CDRs L2, H1, H2 and H2, respectively (Figure S8a). Labeling changes in the Fc region, particularly the CH3 domain are also confirmed by a reduction in Fc binding and CDC activity (Figure 4). Clusters of residues that undergo decreases in DEPC labeling in the CH3 domain of Fc (Ser258, Ser428, His439, and Tyr440) and the variable domains of Fab (Ser5, Tyr70, and Tyr101 on the LC and Thr118 and Ser120 on the HC) suggest these regions as the likely aggregation interfaces (Figure S8b).
Our DEPC CL-MS technique compares favorably to other MS-based structural techniques (e.g., HDX,28-30 HRF,48 and dimethyl labeling65) that have been used recently to study HOS changes or identify aggregation interfaces of mAbs. Most of the previous studies used harsh stress conditions (freeze-thaw, UV light, or temperature around the Tm) to induce conformation changes to the mAbs under study. Consistent with the results reported in our work here, most aggregation sites in mAbs are found to be in the Fab region, particularly in the variable domains,28,30,48,56,65 although there is at least one example of aggregation involving the Fc region.29 To our knowledge, there are no published experimental studies that report the aggregation interface for rituximab upon heating. Nevertheless, computational tools have predicted aggregation prone regions66-68 in Fab and Fc regions, of which many are located close to the sites found to decrease in labeling, such as Ser5 (LC), Thr118 and Ser120 (HC, Fab) and Ser258 (HC, Fc).
From a methodological perspective, CL-MS using DEPC has some advantages over other MS-based techniques, especially for revealing aggregation interfaces. In HDX-MS, reduced deuterium exchange along a protein backbone is used to identify aggregation sites, even though protein-protein interactions are primarily mediated by side chain interactions. DEPC labeling reports on the SASA of side chains, thereby providing a more direct indication of aggregation sites. CL-MS also does not require specialized equipment like HDX-MS, thereby simplifying the overall approach. HRF is another side-chain labeling technique that has been used for studying mAb interactions and structure, but HRF techniques also require specialized lasers41,45,46,69,70 or synchrotron40,48 sources to generate the radicals. In addition, radicals generate multiple reaction products, as opposed to a single one with DEPC, which can decrease the sensitivity of the technique.
In conclusion, we demonstrate here the ability of DEPC CL-MS to investigate HOS of antibody therapeutics. DEPC directly probes solvent accessibility and the microenvironment of side chains, and any changes in DEPC reactivity are indicative of changes in protein conformation. With a broad spectrum of modifiable residues coupled with the sensitivity of MS, the structural resolution and sensitivity of our DEPC labeling technique is high enough for probing subtle protein conformational changes. We have shown that DEPC CL-MS can reveal subtle HOS changes in rituximab at the temperatures below the Tm (e.g., 55°C), temperatures at which classical biophysical techniques are not sensitive enough to detect any changes. The results from DEPC labeling are validated via activity assays. At higher heat stress (e.g., 65°C), HOS changes and aggregation are apparent from clusters of residues that undergo decreases in labeling in both Fab and Fc regions. These results are confirmed by complementary biophysical and activity measurements. With its structural resolution, sensitivity, and simplicity, DEPC CL-MS should be amenable to the structural investigations of other antibody therapeutics. Future work will demonstrate its applicability to HOS characterization of other therapeutic antibodies and antibody-antigen complexes.
Materials and methods
Materials
Rituximab formulation (Rituxan® 100 mg/10 mL vial, lot# 3209283, Genentech) was ordered from Myoderm. Diethylpyrocarbonate (DEPC) (#D5758), imidazole (#I5513), iodoacetamide (#I6125), tris(2-carboxyethyl)phosphine (TCEP) (#C4706), and trypsin (#T1426) were obtained from Sigma-Aldrich. Urea (#AC424581000) was purchased from Acros Organics. Sodium phosphate monobasic monohydrate (#S0710-1) was obtained from EM Science. Sodium phosphate dibasic anhydrous (#S374-500), LC/MS-grade formic acid (#A117-50), acetonitrile (#A998-4), and water (#W7-4) were purchased from Fisher Scientific.
Heat treatments
Samples were aliquoted (5 μL) from a rituximab formulation stored at 4°C. Control samples were incubated at 37°C for 5 min prior to DEPC labeling. For thermal treatments, the samples were incubated at 45°C, 55°C, and 65°C for 4 h in a temperature-controlled water bath. The samples were cooled to 37°C prior to labeling.
DEPC labeling reactions
Rituximab samples (10 mg/mL, 69.5 μM) in formulation with only minor dilution (to 58 μM) were reacted with DEPC. The reagent was first diluted in acetonitrile to make an intermediate solution, and the final solution of DEPC was then prepared in water. Labeling of rituximab was performed at 37°C for 5 min at a DEPC to protein molar ratio of 4 to 1. The reaction was quenched by the addition of imidazole at a 1:50 DEPC to imidazole molar ratio. For each thermal treatment, at least three replicates were performed on the rituximab samples.
Proteolytic digestion
Following quenching the labeled samples were added into a urea-containing tube and diluted in 50 mM phosphate buffer at pH 7.4. The resulting mixture contained 8 M urea for protein denaturation. L-methionine was added at the final concentration of 2 mg/mL to minimize oxidation during protein digestion. To achieve complete digestion of rituximab, TCEP was added at the final concentration of 25 mM to reduce the disulfide bonds. Iodoacetamide (25 mM) was simultaneously added to alkylate the reduced Cys residues. The samples were kept in the dark at room temperature for 20 min. Tween® 80 was then removed from the samples using DetergentOUT™ Tween® Micro spin columns (#786–214, G-Biosciences). Subsequently, trypsin was added to the resulting samples at a 1:10 (w/w) enzyme to substrate, and the labeled protein was digested overnight at 37°C. Following digestion, an Amicon® centrifugal filter with a 10 kDa molecular weight cutoff (#UFC501096, Millipore) was used to remove trypsin from the resulting rituximab peptides. The flow-through was collected, flash-frozen in liquid nitrogen, and stored at −20°C until LC-MS/MS analysis.
HPLC separation
Online LC-MS/MS analyses were performed on all rituximab digests. A sample containing approximately 2 μg rituximab peptides was loaded on an Easy-NanoLC 1000 system (Thermo Scientific). A flow rate of 300 nL/min was used. Samples were first trapped and desalted on an Acclaim™ PepMap™ C18 trap column (2 cm x 75 μm ID, 3 μm; Thermo Scientific). Separation of rituximab peptides was then performed using a FortisBIO C18 nanocolumn (15 cm x 75 μm ID, 1.7 μm; Fortis Technologies). LC/MS-grade water (solvent A) and acetonitrile (solvent B), each containing 0.1% formic acid, were used as mobile phases. A shallow gradient was utilized to achieve sufficient separation of peptides on analytical column. A linear gradient of solvent B was increased from 0% B to 50% B over 90 min. The LC column was then flushed by elevating the mobile phase composition to 95% B over 15 min and holding at 95% B for another 20 min.
Mass spectrometry
Mass spectra were acquired on a Thermo Scientific Orbitrap Fusion mass spectrometer. The nano-electrospray ionization source was operated in the positive mode using a needle voltage of 2,000 V. The ion transfer tube temperature was set to 300°C. The resolution of Orbitrap was set to 60,000 and the MS1 AGC target and maximum injection time were optimized and set to 1 × 106 ions and 100 msec, respectively. Tandem mass spectrometry (MS/MS) was performed on linear quadrupole ion trap for the most abundant peptide ions, with ion abundances above 5,000. The precursor ions were selected using a quadrupole mass filter at an isolation width of 2.0, and the MS2 AGC target and maximum injection time were set to 5 × 104 ions and 100 msec, respectively. Tandem mass spectra were generated using collisional-induced dissociation with a normalized collision energy of 35%. To avoid a biased selection of high-abundance ions, a dynamic exclusion of 60 sec was activated after 5 spectra were acquired for any given precursor ion within 5 sec. Mass detection during MS and MS/MS was done in centroid mode to ease the data analysis.
Peptide identification and peak quantification
A custom software pipeline described previously56 and specifically designed for protein CL-MS studies was used to identify and quantify labeling sites. Raw mass spectral data files from the LC-MS/MS analyses were firstly converted to .mgf format using MSConvertGUI software.71,72 SearchGUI was used to analyze the .mgf files for the peptide identification.73 The sequence of rituximab’s light and heavy chains were added to a sequence database constructed from the common Repository of Adventitious Proteins (cRAP database).74 Tandem mass spectra were searched against the custom database and its reverse, the decoy database. Several search engines (X!Tandem,75,76 MS-GF+,77 OMSSA,78 and MyriMatch79) were all used. The search parameters were set as follows: a precursor mass tolerance of 10 ppm, carbamidomethylation of Cys as a fixed modification, oxidation of Met as a variable modification, and DEPC modification of His, Lys, Ser, Thr, Tyr, and N-terminus as a user variable modification (mass addition of 72.0211). Nonspecific enzyme cleavage was selected to improve the sequence coverage of searches, accounting for nonspecific proteolytic digestion and degradation of rituximab peptides during sample preparation. Next, PeptideShaker was used to visualize the results from multiple search engines.80 False discovery rates were set at 1% and post-translational modifications were scored using the PhosphoRS algorithm. The peptide identification results were reformatted and exported to a .csv file and processed to contain only rituximab features, which were to be used as a custom database for LC-MS peak identification. MZmine was used to analyze the raw LC-MS data files for peptide peak quantification.81,82 Mass detection was performed at the MS1 level. Chromatograms were reconstructed and deconvoluted using m/z and ion abundances data, and peak areas were quantified. Deconvoluted chromatograms from multiple samples were aligned using the Join Aligner algorithm. The quantified, identified, and aligned data were finally exported in .csv format. DEPC modification levels (L) of each labeled residue were calculated as follows (Equation 3).
where is the peak area of unmodified peptide that has a sequence (i) containing the residue of interest and possesses a certain charge state (z) in the mass spectrum, and is the peak area of peptide in which the residue of interest is modified. A pictorial representation of this calculation can be found in Figure S3.
Note that the DEPC modification extents calculated by Equation 3 are only used for the relative quantitation, i.e., comparing the protein of interest under different conditions (control vs. stressed). The label levels do not reflect the absolute quantitation of modified species as the addition of a carbethoxyl group to the modified peptide and different LC solvent conditions during elution of peptides lead to different ionization efficiency of the unmodified and modified peptides.
Biophysical characterizations and activity assays
The technical details of these methods can be found in the Supplementary Material.
Funding Statement
This work was supported by the National Institutes of Health (NIH) for Small Business Innovation Research (SBIR) under Grant R43 GM116211. The data described herein were acquired on an Orbitrap Fusion mass spectrometer funded by NIH grant S10OD010645. The content is solely the responsibility of the authors and does not necessarily represent the official views of NIH; National Institute of General Medical Sciences [R43 GM116211]; National Institutes of Health [S10OD010645].
Abbreviations
- CD
circular dichroism
- CDC
complement-dependent cytotoxicity
- CDR
complementarity-determining region
- CL
covalent labeling
- CL, CH1, CH2, CH3
constant domains
- DEPC
diethylpyrocarbonate
- DLS
dynamic light scattering
- ELISA
enzyme-linked immunosorbent assay
- Fab
antigen-binding fragments
- Fc
crystallizable fragments
- HC/H
heavy chain
- HOS
higher-order structure
- IgG1
immunoglobulin gamma subtype 1
- LC
liquid chromatography
- LC/L
light chain
- mAb
monoclonal antibody
- MS
mass spectrometry
- MS/MS
tandem mass spectrometry
- OD
optical density
- SASA
solvent-accessible surface area
- Tm
melting temperature
- UV
ultraviolet radiation
- VL, VH
variable domains
- TIC
total ion chromatogram
- XIC
extracted ion chromatogram
Acknowledgments
The authors acknowledge Prof. Stephen J. Eyles for his help with the Thermo Scientific Orbitrap Fusion mass spectrometer. Dr. Lizz Bartlett, Dr. Mahalia A.C. Serrano, Kingshuk Dutta, Hsin-Ting Huang, Tyler M. Marcinko, and Bo Zhao are thanked for their assistance with some of the biophysical characterization. The authors gratefully acknowledge Akshada Abhyankar, Padmini Deosthale, and Ann Feil for their preliminary involvement in the work involving biophysical and activity assays. Meizhe Wang is thanked for her comments on some figures of this manuscript. P.L. also acknowledges his doctoral fellowship from the Faculty of Pharmaceutical Sciences, Chulalongkorn University (Bangkok, Thailand).
Disclosure of Potential Conflicts of Interest
Eric M. Graban and John E. Hale are employees of QuarryBio Inc, which is commercializing DEPC CL-MS methods to characterize the higher order structure of protein therapeutics.
Supplemental material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Ecker DM, Jones SD, Levine HL.. The therapeutic monoclonal antibody market. mAbs. 2015;7:9–14. doi: 10.4161/19420862.2015.989042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lindsley CW. New 2017 data and statistics for pharmaceutical products. ACS Chem Neurosci. 2018;9:1518–19. doi: 10.1021/acschemneuro.8b00320. [DOI] [PubMed] [Google Scholar]
- 3.Kaplon H, Reichert JM. Antibodies to watch in 2019. mAbs. 2018. doi: 10.1080/19420862.2018.1556465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.EvaluatePharma® World Preview 2017, Outlook to 2022. London (England, United Kingdom): Evaluate Ltd; 2017. p. 8. [Google Scholar]
- 5.Frokjaer S, Otzen DE. Protein drug stability: a formulation challenge. Nat Rev Drug Discov. 2005;4:298–306. doi: 10.1038/nrd1695. [DOI] [PubMed] [Google Scholar]
- 6.Berkowitz SA, Engen JR, Mazzeo JR, Jones GB. Analytical tools for characterizing biopharmaceuticals and the implications for biosimilars. Nat Rev Drug Discov. 2012;11:527–40. doi: 10.1038/nrd3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weiss WF, Gabrielson JP, Al-Azzam W, Chen G, Davis DL, Das TK, Hayes DB, Houde D, Singh SK. Technical decision making with higher order structure data: perspectives on higher order structure characterization from the biopharmaceutical industry. J Pharm Sci. 2016;105:3465–70. doi: 10.1016/j.xphs.2016.09.003. [DOI] [PubMed] [Google Scholar]
- 8.Kaltashov IA, Bobst CE, Abzalimov RR, Wang G, Baykal B, Wang S. Advances and challenges in analytical characterization of biotechnology products: mass spectrometry-based approaches to study properties and behavior of protein therapeutics. Biotechnol Adv. 2012;30:210–22. doi: 10.1016/j.biotechadv.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaltashov IA, Bobst CE, Abzalimov RR, Berkowitz SA, Houde D. Conformation and dynamics of biopharmaceuticals: transition of mass spectrometry-based tools from academe to industry. J Am Soc Mass Spectrom. 2010;21:323–37. doi: 10.1016/j.jasms.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beck A, Diemer H, Ayoub D, Debaene F, Wagner-Rousset E, Carapito C, Van Dorsselaer A, Sanglier-Cianférani S. Analytical characterization of biosimilar antibodies and Fc-fusion proteins. TrAC Trends Analyt Chem. 2013;48:81–95. doi: 10.1016/j.trac.2013.02.014. [DOI] [Google Scholar]
- 11.Jung SK, Lee KH, Jeon JW, Lee JW, Kwon BO, Kim YJ, Bae JS, Kim D-I, Lee SY, Chang SJ. Physicochemical characterization of Remsima®. mAbs. 2014;6:1163–77. doi: 10.4161/mabs.32221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Magnenat L, Palmese A, Fremaux C, D’Amici F, Terlizzese M, Rossi M, Chevalet L. Demonstration of physicochemical and functional similarity between the proposed biosimilar adalimumab MSB11022 and Humira®. mAbs. 2017;9:127–39. doi: 10.1080/19420862.2016.1259046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seo N, Polozova A, Zhang M, Yates Z, Cao S, Li H, Kuhns S, Maher G, McBride HJ, Liu J. Analytical and functional similarity of Amgen biosimilar ABP 215 to bevacizumab. mAbs. 2018;10:678–91. doi: 10.1080/19420862.2018.1452580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee KH, Lee J, Bae JS, Kim YJ, Kang HA, Kim SH, Lee SJ, Lim KJ, Lee JW, Jung SK, et al. Analytical similarity assessment of rituximab biosimilar CT-P10 to reference medicinal product. mAbs. 2018;10:380–96. doi: 10.1080/19420862.2018.1433976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nupur N, Chhabra N, Dash R, Rathore AS. Assessment of structural and functional similarity of biosimilar products: rituximab as a case study. mAbs. 2018;10:143–58. doi: 10.1080/19420862.2017.1402996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ambrogelly A, Gozo S, Katiyar A, Dellatore S, Kune Y, Bhat R, Sun J, Li N, Wang D, Nowak C, et al. Analytical comparability study of recombinant monoclonal antibody therapeutics. mAbs. 2018;10:513–38. doi: 10.1080/19420862.2018.1438797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Flores-Ortiz LF, Campos-García VR, Perdomo-Abúndez FC, Pérez NO, Medina-Rivero E. Physicochemical properties of rituximab. J Liq Chromatogr Relat Technol. 2014;37:1438–52. doi: 10.1080/10826076.2013.794738. [DOI] [Google Scholar]
- 18.Hickey JM, Toprani VM, Kaur K, Mishra RPN, Goel A, Oganesyan N, Lees A, Sitrin R, Joshi SB, Volkin DB. Analytical comparability assessments of 5 recombinant CRM197 proteins from different manufacturers and expression systems. J Pharm Sci. 2018;107:1806–19. doi: 10.1016/j.xphs.2018.03.002. [DOI] [PubMed] [Google Scholar]
- 19.Thiagarajan G, Semple A, James JK, Cheung JK, Shameem M. A comparison of biophysical characterization techniques in predicting monoclonal antibody stability. mAbs. 2016;8:1088–97. doi: 10.1080/19420862.2016.1189048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.ICH Harmonised Tripartite Guideline Q5E comparability of biotechnological/biological products subject to changes in their manufacturing process; 2004. Nov [accessed 2016 Mar 12] http://www.ich.org/products/guidelines/quality/quality-single/article/comparability-of-biotechnologicalbiological-products-subject-to-changes-in-their-manufacturing-proc.html.
- 21.ICH Harmonised Tripartite Guideline Q6B specifications: test procedures and acceptance criteria for biotechnological/biological products; 1999. Mar [accessed 2016 Mar 12] http://www.ich.org/products/guidelines/quality/quality-single/article/specifications-test-procedures-and-acceptance-criteria-for-biotechnologicalbiological-products.html.
- 22.U.S. Food and Drug Administration Quality considerations in demonstrating biosimilarity of a therapeutic protein product to a reference product to a reference product; 2015. Apr [accessed 2017 Sep 25]. https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291134.pdf.
- 23.U.S. Food and Drug Administration Points to consider in the manufacture and testing of monoclonal antibody products for human use; 1997. Feb 28 [accessed 2017 Sep 23]. https://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/OtherRecommendationsforManufacturers/UCM153182.pdf.FDA. [DOI] [PubMed]
- 24.European Medicines Agency Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: quality issues (revision 1); 2014. Jun 3 [accessed 2017 Sep 25]. http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000886.jsp&mid=WC0b01ac058002956b.
- 25.European Medicines Agency Guideline on development, production, characterisation and specification for monoclonal antibodies and related products; 2016. Aug 4 [accessed 2017 Sep 25]. http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000867.jsp&mid=WC0b01ac058002956b.
- 26.U.S. Food and Drug Administration Guidance concerning demonstration of comparability of human biological products, including therapeutic biotechnologyderived products; 1996. Apr [accessed 2018 Jul 21]. https://www.fda.gov/drugs/guidancecomplianceregulatoryinformation/guidances/ucm122879.htm.
- 27.Rogstad S, Faustino A, Ruth A, Keire D, Boyne M, Park J. A retrospective evaluation of the use of mass spectrometry in FDA biologics license applications. J Am Soc Mass Spectrom. 2017;28:786–94. doi: 10.1007/s13361-016-1531-9. [DOI] [PubMed] [Google Scholar]
- 28.Zhang A, Singh SK, Shirts MR, Kumar S, Fernandez EJ. Distinct aggregation mechanisms of monoclonal antibody under thermal and freeze-thaw stresses revealed by hydrogen exchange. Pharm Res. 2012;29:236–50. doi: 10.1007/s11095-011-0538-y. [DOI] [PubMed] [Google Scholar]
- 29.Iacob RE, Bou-Assaf GM, Makowski L, Engen JR, Berkowitz SA, Houde D. Investigating monoclonal antibody aggregation using a combination of H/DX-MS and other biophysical measurements. J Pharm Sci. 2013;102:4315–29. doi: 10.1002/jps.23754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bommana R, Chai Q, Schöneich C, Weiss WF, Majumdar R. Understanding the increased aggregation propensity of a light-exposed IgG1 monoclonal antibody using hydrogen exchange mass spectrometry, biophysical characterization, and structural analysis. J Pharm Sci. 2018;107:1498–511. doi: 10.1016/j.xphs.2018.01.017. [DOI] [PubMed] [Google Scholar]
- 31.Wei H, Mo J, Tao L, Russell RJ, Tymiak AA, Chen G, Iacob RE, Engen JR. Hydrogen/deuterium exchange mass spectrometry for probing higher order structure of protein therapeutics: methodology and applications. Drug Discov Today. 2014;19:95–102. doi: 10.1016/j.drudis.2013.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Majumdar R, Middaugh CR, Weis DD, Volkin DB. Hydrogen-deuterium exchange mass spectrometry as an emerging analytical tool for stabilization and formulation development of therapeutic monoclonal antibodies. J Pharm Sci. 2015;104:327–45. doi: 10.1002/jps.24224. [DOI] [PubMed] [Google Scholar]
- 33.Bonnington L, Lindner I, Gilles U, Kailich T, Reusch D, Bulau P. Application of hydrogen/deuterium exchange-mass spectrometry to biopharmaceutical development requirements: improved sensitivity to detection of conformational changes. Anal Chem. 2017;89:8233–37. doi: 10.1021/acs.analchem.7b01670. [DOI] [PubMed] [Google Scholar]
- 34.Mendoza VL, Vachet RW. Probing protein structure by amino acid-specific covalent labeling and mass spectrometry. Mass Spectrom Rev. 2009;28:785–815. doi: 10.1002/mas.20203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mendoza VL, Vachet RW. Protein surface mapping using diethylpyrocarbonate with mass spectrometric detection. Anal Chem. 2008;80:2895–904. doi: 10.1021/ac701999b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xie B, Sood A, Woods RJ, Sharp JS. Quantitative protein topography measurements by high resolution hydroxyl radical protein footprinting enable accurate molecular model selection. Sci Rep. 2017;7:4552. doi: 10.1038/s41598-017-04689-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang W, Ravikumar Krishnakumar M, Chance Mark R, Yang S. Quantitative mapping of protein structure by hydroxyl radical footprinting-mediated structural mass spectrometry: a protection factor analysis. Biophys J. 2015;108:107–15. doi: 10.1016/j.bpj.2014.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Limpikirati P, Liu T, Vachet RW. Covalent labeling-mass spectrometry with non-specific reagents for studying protein structure and interactions. Methods. 2018;144:79–93. doi: 10.1016/j.ymeth.2018.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaur P, Tomechko SE, Kiselar J, Shi W, Deperalta G, Wecksler AT, Gokulrangan G, Ling V, Chance MR. Characterizing monoclonal antibody structure by carboxyl group footprinting. mAbs. 2015;7:540–52. doi: 10.1080/19420862.2015.1023683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kaur P, Kiselar J, Shi W, Yang S, Chance MR. Covalent labeling techniques for characterizing higher order structure of monoclonal antibodies. State-of-the-Art and Emerging Technologies for Therapeutic Monoclonal Antibody Characterization Volume 3 Defining the Next Generation of Analytical and Biophysical Techniques. American Chemical Society; 2015. p. 45–73. [Google Scholar]
- 41.Li KS, Chen G, Mo J, Huang RYC, Deyanova EG, Beno BR, O’Neil SR, Tymiak AA, Gross ML. Orthogonal mass spectrometry-based footprinting for epitope mapping and structural characterization: the IL-6 receptor upon binding of protein therapeutics. Anal Chem. 2017;89:7742–49. doi: 10.1021/acs.analchem.7b01748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wecksler AT, Kalo MS, Deperalta G. Mapping of Fab-1: vEGFInterface using carboxyl group footprinting mass spectrometry. J Am Soc Mass Spectrom. 2015;26:2077–80. doi: 10.1007/s13361-015-1273-0. [DOI] [PubMed] [Google Scholar]
- 43.Pan LY, Salas-Solano O, Valliere-Douglass JF. Localized conformational interrogation of antibody and antibody-drug conjugates by site-specific carboxyl group footprinting. mAbs. 2017;9:307–18. doi: 10.1080/19420862.2016.1268306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu G, Chance MR. Hydroxyl radical-mediated modification of proteins as probes for structural proteomics. Chem Rev. 2007;107:3514–43. doi: 10.1021/cr0682047. [DOI] [PubMed] [Google Scholar]
- 45.Li J, Wei H, Krystek SR, Bond D, Brender TM, Cohen D, Feiner J, Hamacher N, Harshman J, Huang RY-C, et al. Mapping the energetic epitope of an antibody/interleukin-23 interaction with hydrogen/deuterium exchange, fast photochemical oxidation of proteins mass spectrometry, and alanine shave mutagenesis. Anal Chem. 2017;89:2250–58. doi: 10.1021/acs.analchem.6b03058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang Y, Wecksler AT, Molina P, Deperalta G, Gross ML. Mapping the binding interface of VEGF and a monoclonal antibody Fab-1 fragment with fast photochemical oxidation of proteins (FPOP) and mass spectrometry. J Am Soc Mass Spectrom. 2017;28:850–58. doi: 10.1007/s13361-017-1601-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Opuni Kwabena FM, Al‐Majdoub M, Yefremova Y, El‐Kased Reham F, Koy C, Glocker Michael O. Mass spectrometric epitope mapping. Mass Spectrom Rev. 2016;37:229–41. doi: 10.1002/mas.21516. [DOI] [PubMed] [Google Scholar]
- 48.Deperalta G, Alvarez M, Bechtel C, Dong K, McDonald R, Ling V. Structural analysis of a therapeutic monoclonal antibody dimer by hydroxyl radical footprinting. mAbs. 2013;5:86–101. doi: 10.4161/mabs.22964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Trinquier G, Sanejouand YH. Which effective property of amino acids is best preserved by the genetic code? Protein Eng Des Sel. 1998;11:153–69. doi: 10.1093/protein/11.3.153. [DOI] [PubMed] [Google Scholar]
- 50.Zhou Y, Vachet RW. Increased protein structural resolution from diethylpyrocarbonate-based covalent labeling and mass spectrometric detection. J Am Soc Mass Spectrom. 2012;23:708–17. doi: 10.1007/s13361-011-0332-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhou Y, Vachet RW. Diethylpyrocarbonate labeling for the structural analysis of proteins: label scrambling in solution and how to avoid it. J Am Soc Mass Spectrom. 2012;23:899–907. doi: 10.1007/s13361-012-0349-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mendoza VL, Barón-Rodríguez MA, Blanco C, Vachet RW. Structural Insights into the pre-amyloid tetramer of β-2-microglobulin from covalent labeling and mass spectrometry. Biochemistry. 2011;50:6711–22. doi: 10.1021/bi2004894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mendoza VL, Antwi K, Barón-Rodríguez MA, Blanco C, Vachet RW. Structure of the preamyloid dimer of β-2-microglobulin from covalent labeling and mass spectrometry. Biochemistry. 2010;49:1522–32. doi: 10.1021/bi901748h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Srikanth R, Mendoza VL, Bridgewater JD, Zhang G, Vachet RW. Copper Binding to β-2-microglobulin and its pre-amyloid oligomers. Biochemistry. 2009;48:9871–81. doi: 10.1021/bi901172y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu T, Marcinko TM, Kiefer PA, Vachet RW. Using covalent labeling and mass spectrometry to study protein binding sites of amyloid inhibiting molecules. Anal Chem. 2017;89:11583–91. doi: 10.1021/acs.analchem.7b02915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Borotto NB, Zhou Y, Hollingsworth SR, Hale JE, Graban EM, Vaughan RC, Vachet RW. Investigating therapeutic protein structure with diethylpyrocarbonate labeling and mass spectrometry. Anal Chem. 2015;87:10627–34. doi: 10.1021/acs.analchem.5b03180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Padlan EA. Anatomy of the antibody molecule. Mol Immunol. 1994;31:169–217. [DOI] [PubMed] [Google Scholar]
- 58.Martz E. Atomic coordinate files for whole IgG1; 1996. Feb 7 [accessed 2018 Aug 4]. https://www.umass.edu/microbio/rasmol/padlan.htm.
- 59.Gazzano-Santoro H, Ralph P, Ryskamp TC, Chen AB, Mukku VR. A non-radioactive complement-dependent cytotoxicity assay for anti-CD20 monoclonal antibody. J Immunol Methods. 1997;202:163–71. [DOI] [PubMed] [Google Scholar]
- 60.Zhang H, Song L, Ye H, Hu L, Liang W, Liu D. Characterization of a novel humanized anti-CD20 antibody with potent anti-tumor activity against Non-Hodgkin’s Lymphoma. Cell Physiol Biochem. 2013;32:645–54. doi: 10.1159/000354468. [DOI] [PubMed] [Google Scholar]
- 61.Andersen Christian B, Manno M, Rischel C, Thórólfsson M, Martorana V. Aggregation of a multidomain protein: A coagulation mechanism governs aggregation of a model IgG1 antibody under weak thermal stress. Protein Sci. 2009;19:279–90. doi: 10.1002/pro.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nejadnik MR, Randolph TW, Volkin DB, Schöneich C, Carpenter JF, Crommelin DJA, Jiskoot W. Postproduction handling and administration of protein pharmaceuticals and potential instability issues. J Pharm Sci. 2018;107:2013–19. doi: 10.1016/j.xphs.2018.04.005. [DOI] [PubMed] [Google Scholar]
- 63.Vermeer AW, Norde W. The thermal stability of immunoglobulin: unfolding and aggregation of a multi-domain protein. Biophys J. 2000;78:394–404. doi: 10.1016/S0006-3495(00)76602-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Du J, Wang H, Zhong C, Peng B, Zhang M, Li B, Huo S, Guo Y, Ding J. Structural Basis for Recognition of CD20 by Therapeutic Antibody Rituximab. J Biolo Chem. 2007;282:15073–80. doi: 10.1074/jbc.M701654200. [DOI] [PubMed] [Google Scholar]
- 65.Jhan S-Y, Huang L-J, Wang T-F, Chou -H-H, Chen S-H. Dimethyl labeling coupled with mass spectrometry for topographical characterization of primary amines on monoclonal antibodies. Anal Chem. 2017;89:4255–63. doi: 10.1021/acs.analchem.7b00320. [DOI] [PubMed] [Google Scholar]
- 66.Courtois F, Schneider CP, Agrawal NJ, Trout BL. Rational design of biobetters with enhanced stability. J Pharm Sci. 2015;104:2433–40. doi: 10.1002/jps.24520. [DOI] [PubMed] [Google Scholar]
- 67.Wang X, Das TK, Singh SK, Kumar S. Potential aggregation prone regions in biotherapeutics: A survey of commercial monoclonal antibodies. mAbs. 2009;1:254–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.van der Kant R, Karow-Zwick AR, Van Durme J, Blech M, Gallardo R, Seeliger D, Aßfalg K, Baatsen P, Compernolle G, Gils A, et al. Prediction and reduction of the aggregation of monoclonal antibodies. J Mol Biol. 2017;429:1244–61. doi: 10.1016/j.jmb.2017.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Watson C, Sharp JS. Conformational analysis of therapeutic proteins by hydroxyl radical protein footprinting. Aaps J. 2012;14:206–17. doi: 10.1208/s12248-012-9336-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jones LM, B. Sperry J, A. Carroll J, Ml G. Fast photochemical oxidation of proteins for epitope mapping. Anal Chem. 2011;83:7657–61. doi: 10.1021/ac2007366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chambers MC, Maclean B, Burke R, Amodei D, Ruderman DL, Neumann S, Gatto L, Fischer B, Pratt B, Egertson J, et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat Biotech. 2012;30:918–20. doi: 10.1038/nbt.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Holman JD, Tabb DL, Mallick P. 2002. Employing proteowizard to convert raw mass spectrometry data. Current Protocols Bioinf. John Wiley & Sons, Inc.; Suppl. 46;13.24.1–13.24.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vaudel M, Barsnes H, Berven FS, Sickmann A, Martens L. SearchGUI: an open-source graphical user interface for simultaneous OMSSA and X!Tandem searches. Proteomics. 2011;11:996–99. doi: 10.1002/pmic.201000595. [DOI] [PubMed] [Google Scholar]
- 74.The Global Proteome Machine Organization cRAP protein sequences; 2012. Jan 1 [accessed 2017 Oct 18]. http://www.thegpm.org/crap/index.html.
- 75.Craig R, Beavis RC. A method for reducing the time required to match protein sequences with tandem mass spectra. Rapid Commun Mass Spectrom. 2003;17:2310–16. doi: 10.1002/rcm.1198. [DOI] [PubMed] [Google Scholar]
- 76.Craig R, Beavis RC. TANDEM: matching proteins with tandem mass spectra. Bioinformatics. 2004;20:1466–67. doi: 10.1093/bioinformatics/bth092. [DOI] [PubMed] [Google Scholar]
- 77.Kim S, Pevzner PA. MS-GF+ makes progress towards a universal database search tool for proteomics. Nat Commun. 2014;5:5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Geer LY, Markey SP, Kowalak JA, Wagner L, Xu M, Maynard DM, Yang X, Shi W, Bryant SH. Open Mass Spectrometry Search Algorithm. J Proteome Res. 2004;3:958–64. doi: 10.1021/pr0499491. [DOI] [PubMed] [Google Scholar]
- 79.Tabb DL, Fernando CG, Chambers MC. MyriMatch: highly accurate tandem mass spectral peptide identification by multivariate hypergeometric analysis. J Proteome Res. 2007;6:654–61. doi: 10.1021/pr0604054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vaudel M, Burkhart JM, Zahedi RP, Oveland E, Berven FS, Sickmann A, Martens L, Barsnes H. PeptideShaker enables reanalysis of MS-derived proteomics data sets. Nat Biotech. 2015;33:22–24. doi: 10.1038/nbt.3109. [DOI] [PubMed] [Google Scholar]
- 81.Katajamaa M, Miettinen J, Orešič M. MZmine: toolbox for processing and visualization of mass spectrometry based molecular profile data. Bioinformatics. 2006;22:634–36. doi: 10.1093/bioinformatics/btk039. [DOI] [PubMed] [Google Scholar]
- 82.Pluskal T, Castillo S, Villar-Briones A, Orešič M. MZmine 2: modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinformatics. 2010;11:395. doi: 10.1186/1471-2105-11-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.