

Abstract
Carbohydrates are essential moieties of many bioactive molecules in nature. However, efforts to elucidate their modes of action are often impeded by limitations in synthetic access to well-defined oligosaccharides. Most of the current methods rely on the design of specialized coupling partners to control selectivity during formation of glycosidic bonds. Herein, we report a commercially available phenanthroline to catalyze stereoretentive glycosylation with glycosyl bromides. The method provides efficient access to α−1,2-cis glycosides. This protocol has been performed for the large-scale synthesis of an octasaccharide adjuvant. Density functional theory calculations, together with kinetic studies, suggest that the reaction proceeds via double SN2 mechanism.
Keywords: organocatalysis, glycosylation, stereoselectivity, oligosaccharides, mechanism
Graphical Abstract
Catalytic Glycosylations: The commercially available phenanthroline serves as the efficient catalyst to promote the stereoselective formation of -1,2-cis glycosides synthesis. This newly developed catalyst system is not confined to the nature of the protecting groups bound on carbohydrate coupling partners to effect the selectivity.
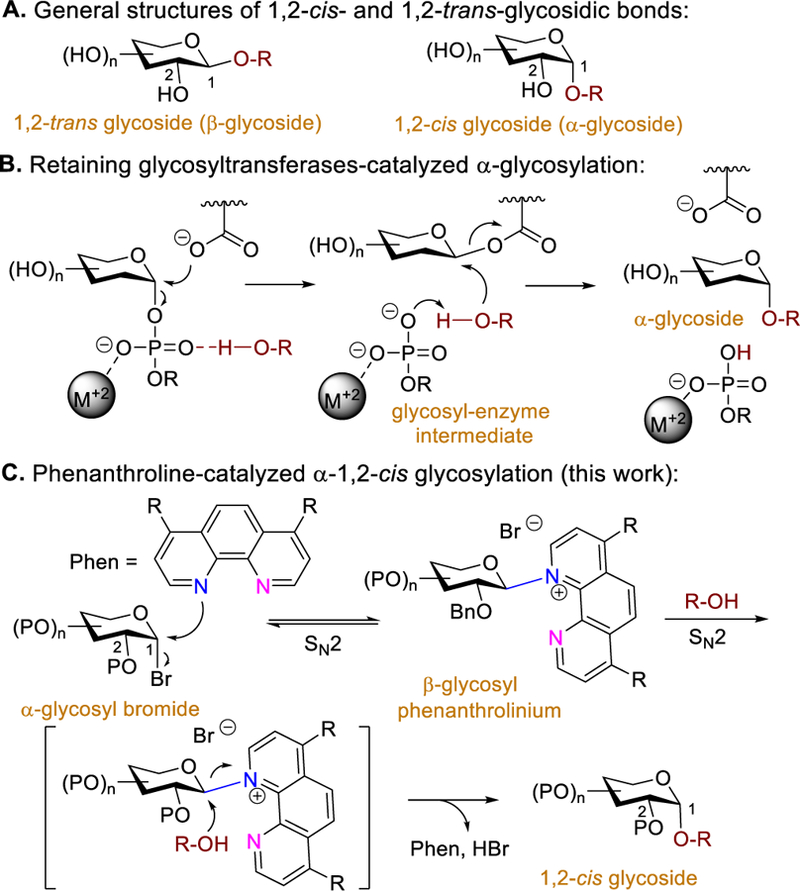
Glycosylations are fundamental methods for constructing complex carbohydrates. Key reactions involve glycosidic bond formation that connects glycosyl electrophiles to glycosyl nucleophiles to generate oligosaccharides, which play a critical role in cellular functions and disease processes.[1] As a result, the efficient preparation of well-defined oligosaccharides has been a major focus in carbohydrate synthesis. Despite recent advances,[2] the ability to forge α−1,2-cis glycosidic bonds in a stereoselective fashion is not easily predictable (Scheme 1A), due to the reaction’s high degree of variables and shifting SN1-SN2 mechanistic paradigm.[3] Most established methods have focused on tuning the steric and electronic nature of the protecting groups bound on electrophilic partners to achieve α−1,2-cis selectivity.[4] Recently, catalyst-controlled methods have emerged as a way to eliminate the need for the design of specific glycosyl electrophiles.[5] Although catalytic glycosylations concentrate on the use of catalysts to control the desired stereochemsitry, only limited examples for forming α−1,2-cis glycosides are known.[5d]
Scheme 1.
Introduction and synopsis of current work
In an effort to identify an effective strategy for a stereoselective synthesis of α−1,2-cis glycosides that would obviate the necessity for substrate prefunctionalization, we consider whether the anomeric selectivity could be controlled by a simple catalyst. We recognized that retaining glycosyltransferases are known to catalyze α-glycosidic bond formation with the net retention of anomeric configuration (Scheme 1B).[6] As such, we envisioned that a catalyst capable of acting as a glycosyltransferase to provide 1,2-cis glycosides with predictable α-selectivity and in high yields would likely find broad applications. Pyridine has been reported by Lemieux and Morgan to serve as a nucleophilic catalyst to displace the anomeric leaving group of a glycosyl electrophile.[7] The glycosyl pyridinium ion formed in the reaction prefers the equatorial position to avoid the steric and electrostatic interactions associated with positioning that group in the axial orientation.[7] Invertive substitution by a nucleophile would then afford α−1,2-cis glycoside. However, it was apparent from the outset of our studies that the adaption of the pyridine system could present challenges. As an axial pyridinium ion can also be generated, from the reaction of an electrophile with pyridine, to compete for access to β−1,2-trans glycoside,[7,8] pyridine-mediated reaction proceeds with marginal bias for the α-selectivity. Herein, we report the discovery of a commercially available phenanthroline to stereoselectively catalyze formation of α−1,2-cis glycosides (Scheme 1C). Phenanthroline is a rigid and planar structure with two fused pyridine rings whose nitrogen atoms are positioned to act cooperatively. We postulated that the first nitrogen atom serves as a catalytic nucleophile to react with an electrophile to generate a covalent β-glycosyl phenanthrolium ion preferentially as phenanthroline is more sterically demanding than pyridine. The second nitrogen atom could non-covalently interact with carbohydrate moiety or form a hydrogen bond with the alcohol nucleophile to facilitate invertive substitution. These features should effectively promote a double SN2 mechanism.
To initiate our investigation, α-glycosyl bromide 1 was chosen as a model electrophilic partner and galactopyranoside 2 as a nucleophile to simplify the analysis of coupling product mixtures (Table 1). Previous reports have documented the ability of glycosyl bromides to function as the effective electrophiles under various conditions.[9] The reaction of 2 with 1, having a C(2)-benzyl (Bn) group,[2] often proceeds via an SN1-like pathway. As expected, use of Lewis acid, silver triflate (AgOTf), provided a 4:1 (α:β) mixture of disaccharide 3. Upon exploring a wide range of reaction parameters (Figures S1 - S6), we discovered that coupling of 2 with 1 in 15 mol% of 4,7-diphenyl-1,10-phennathroline (4) as a catalyst and isobutylene oxide (IBO) as a hydrogen bromide scavenger in tert-butyl methyl ether (MTBE) at 50 oC for 24 h provides the highest yield and selectivity of 3 (73%, α:β > 20:1). In the absence of catalyst 4, no reaction was apparent after 24 h. We also conducted the reaction with catalysts 5 - 8, and three trends are observed. First, the yield of 3 is correlated with the ability of the catalyst to displace the C(1)-bromide. The C(2)- and C(9)-methyl groups of 5 reduce the accessibility of the pyridine nitrogen for displacing the anomeric bromide. Second, the catalyst’s conformation can influence the efficiency and selectivity. For instance, 2,2’-bipyridine (6) is less α-selective than 4 potentially due the two nitrogen atoms being disrupted by the free-rotation about the bond linking the pyridine rings. Third, the selectivity is correlated with the efficiency of the catalyst. As expected, pyridine (7) is not as selective as catalyst 4. Since 4-(dimethylamino)pyridine (8) is a more effective catalyst than 7,[10] disaccharide 3 was obtained in higher yield. To explore if this catalytic system could be suitable for a large scale synthesis, we examined the reaction on a 4 mmol scale of 1 and 4.4 mmol of 2. At high concentration (2 M), use of 5 mol% of catalyst 4 proved sufficient to provide 3 (70%, α:β >20:1) (Figure S7).
Table 1:
Reaction development.[a]
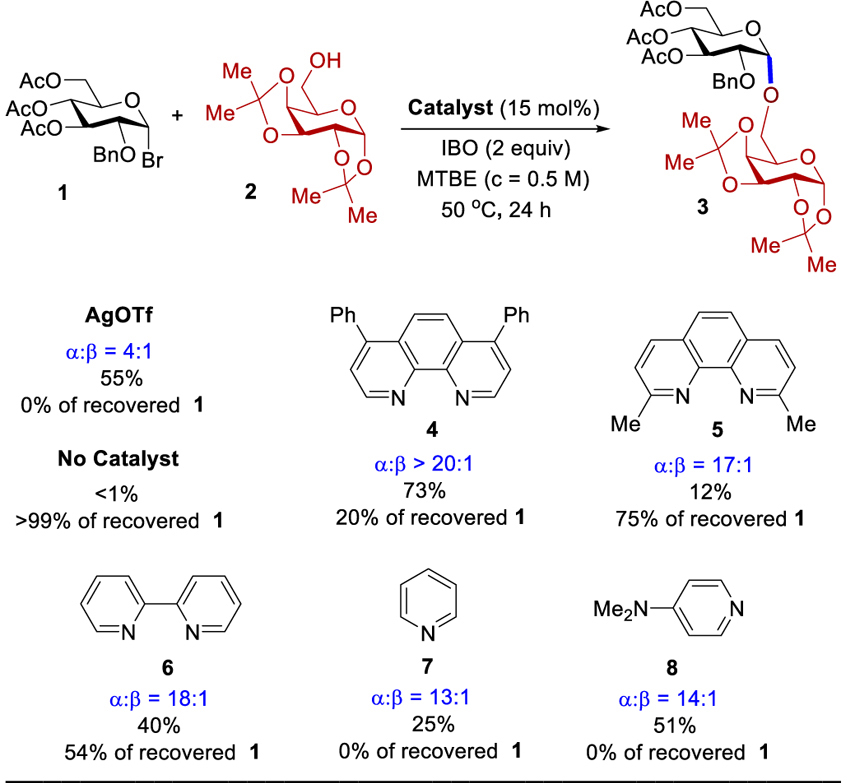 |
The reaction was conducted with 0.2 mmol of 1 and 0.4 mmol of 2. Isolated yields averaged three runs. The (α/β) ratios were determined by 1H NMR analysis.
There are several underlying factors that potentially influence the efficiency and the selectivity of the coupling products, including the protecting group nature of glycosyl electrophiles,[2–4] the reactivity of nucleophiles, and the reaction conditions. As a result, a number of α−1-bromo electrophiles were investigated (Table 2). To validate that catalyst 4 could overturn the “remote” participation of the C(3)-, C(4)-, and C(6)-acyl protecting groups, we first explored the coupling with glucosyl bromides having non-participatory benzyl protecting groups. No significant compromise to the α-selectivity was observed with disaccharides 9 and 10 with use of tetrabenzyl glucosyl bromide. This protocol is more α-selective than other approaches. For example, while our system provided 10 with α:β = 14:1, TMSOTf-mediated coupling with imidate electrophiles provided 10 with marginal α- selectivity (α:β = 1:1.2 – 4:1).[11] In addition, these glucosyl bromides effectively coupled secondary alcohols to afford 11-13 with high α-selectivity. For the challenging C(4)-hydroxy, the SN1-SN2 reaction paradigm was slightly shifted (14: α:β = 7:1). A protected serine nucleophile also exhibited high α-selectivity (15: α:β = 20:1).
Table 2:
Scope with respect to glycosyl electrophiles.[a]
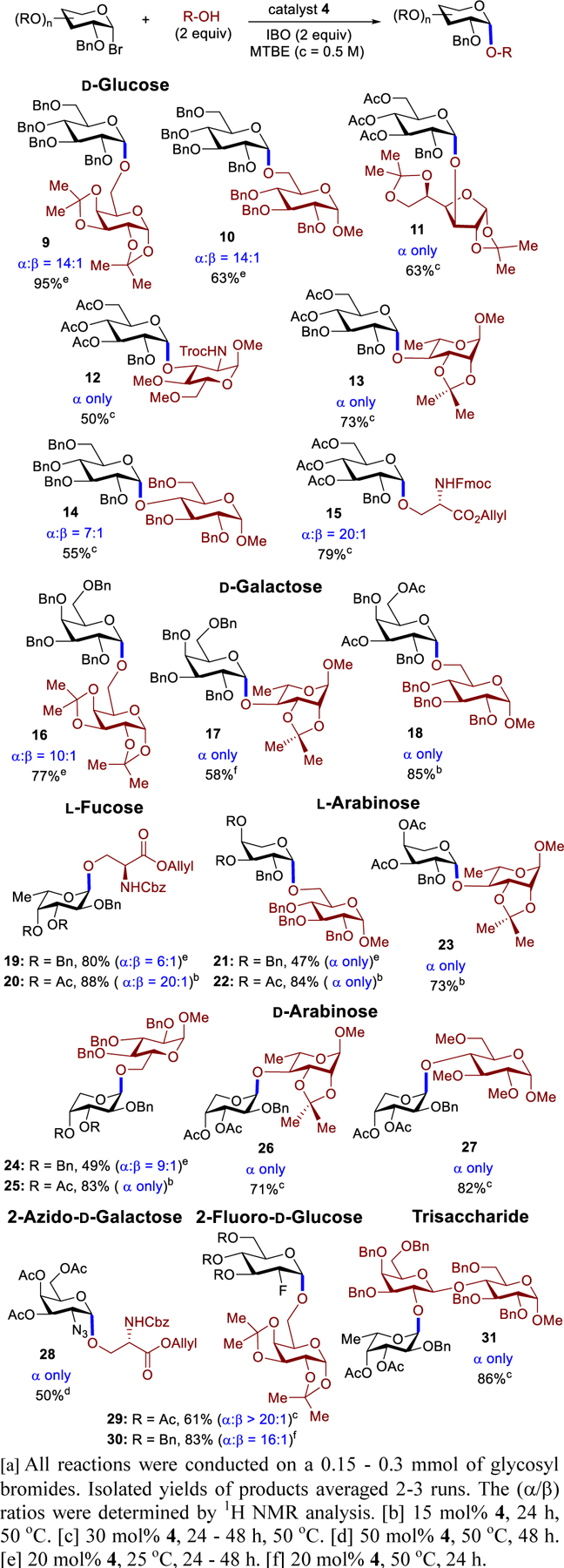 |
Next, we investigated the ability of catalyst 4 to overturn the inherent bias of D-galactose whose axial C(4)-benzyl group has been reported to favor β-product formation.[12] Under our catalytic system, galactosyl bromides served as effective electrophiles to deliver 16-18 (Table 2) with high α-selectivity. In contrast, the amide-promoted reaction provided 16 with α:β = 3:1.[13] We discovered that while tribenzyl L-fucosyl bromide afforded 19 with α:β = 6:1, use of triacetyl L-fucose provided exclusively α−20. Both 19 and 20 are key units of a thrombospondin type 1 associated an autosomal recessive disorder.[14] Use of tribenzyl L-arabinosyl bromide also provided exclusively α−21, albeit with 47% yield due to decomposition during the course of the reaction. The yield could be improved with use of the acetyl protecting groups (22: 84%). The diacetyl L-arabinose was compatible with C(4)-hydroxy to afford α−23.[15] A similar trend was observed with D-arabinose (24 - 27). For comparison, this protocol to produce 24 (α:β = 9:1) is more α-selective than the thioglycoside method with use of NIS/AgOTf as the activating agent (α:β = 3:1).[15]
We also sought to explore the selectivity trends with electrophiles bearing C(2)-azido and C(2)-fluoro groups (Table 2). Use of C(2)-azido galactose substrate provided exclusively α−28, a tumor-associated mucin TN antigen precursor.[16] To compare, 28 had been previously prepared as a 4:1 (α:β) mixture with use of AgClO4 as the activating reagent.[17] Next, we turned our attention to 2-fluoro-D-glucose substrate. The ability of C(2)-F bond to have an impact on the stereochemistry of the product has been reported.[18] While the 2-fluoro-glucose having benzyl protecting groups is highly β-selective under TMSOTf-mediated reactions, the analogous acetyl electrophile affords a 1:1 (α:β) mixture. In contrast, both substrates are α-selective under phenanthroline conditions (29, α:β > 20:1; 30, α:β = 16:1). This catalyst system is also amendable to the synthesis of a protected human milk α-trisaccharide 31 (86%).[19]
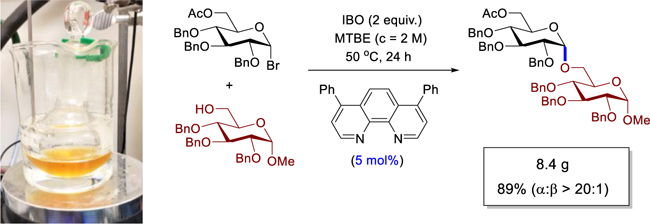
The critical question remains whether the phenanthronline system could be applicable for construction of larger oligosaccharides. To illustrate this potential, we examined the synthesis of octasaccharide 40 (Figure 1), a backbone of the natural α-glucan polysaccharides,[20] the potential vaccine adjuvants. These α-glucans are heterogeneous in size and composition. As such, well-defined oligosaccharides are required to study bioactive fragments. In our approach, a catalyst loading of 5 mol% proved efficient to promote the coupling of commercially available 33 with glycosyl bromide 32 to provide 8.4 g of product 34 in 89% yield with α:β >20:1. Disaccharide 34 was then converted into glycosyl bromide 36,21 which was used in the coupling to 35 to afford tetrasaccharide 37 (86%, α:β >20:1). Another coupling iteration afforded octasaccharide 40 (77%, α:β >20:1).
Figure 1.

Mechanistic studies.
Having obtained 1,2-cis product in high yield and α-selectivity, we sought to investigate the mechanism of this catalytic glycosylation. We first attempted to detect a transient β-covalent phenanthrolinium ion using mass spectrometry. In the event, α-bromide 1 was treated with stoichiometric amount of 4 in MTBE (0.5 M) for 24 h at 50 oC. Formation of phenanthrolinium ion 41 (Figure 1A) was confirmed using electrospray ionization (ESI) with an m/z ratio of 711.2710.[22] We next evaluated if the stereochemistry of the α−1,2-cis product would be dictated by the anomeric configuration of glycosyl bromide (Figure 1B).[23] We observed that β-bromide 42[24] only slowly anomerized to α-bromide 1 without catalyst 4 (Figure S10). However, β-bromide 42 rapidly converted into α-bromide 1 in the presence of 15 mol% of 4 within 1 h at 25 oC (Figure 1B).[25] We also performed the reaction of β-anomer 42 with nucleophile 2 in the presence of 4, and we observed that isomerization of β−42 to α−1 is faster than formation of product 3.[26,27] Collectively, these results suggest that β-bromide 42 is not the reacting partner in the phenanthroline system.
Next, the kinetic studies were conducted at 50 oC, using C6D6 as the NMR solvent and toluene as a quantitative internal standard, with α-bromide 1 and 2-propanol as coupling partners in the presence of IBO and catalyst 4. The rates of reaction were plotted as functions of the concentrations of catalyst 4 (Figure 1C-1) and 2-propanol (Figure 1C-2). Overall, the kinetic data suggested that the reaction proceeds via double SN2-like mechanism (Figures 1C and S11-S15), as the rate of the reaction is both catalyst and nucleophile dependent. The biphasic kinetic in Figure 1C-2 suggests a shift in the rate-determining step (RDS) at different concentrations of 2-propanol. At high concentration of 2-propanol, the RDS is formation of the phenanthrolinium ion (first step in Scheme 1C), which is further supported by the linear dependence of rate on catalyst concentration (Figure 1C-1). At low concentration of 2-propanol, nucleophilic attack (second step in Scheme 1C) is the RDS.
Finally, to understand the role of the phenanthroline catalyst in controlling α−1,2-cis glycosylation, the transition structures and intermediates for nucleophilic addition of phenanthroline or pyridine to α-bromide 43 have been optimized utilizing density functional theory (DFT) calculations at the B3LYP/6–31+G(d,p) level with the SMD implicit solvent model (Figures 1D and S16). Non-covalent interaction (NCI) analysis[28] (Figure S18) indicates that the β-phenanthrolinium ion is stabilized by interactions between the C-1 axial hydrogen of glycosyl moiety and the nitrogen of phenanthroline (the H1–N2 distance is 1.964 Å and the C1–H1---N2 angle is 137°).[29] NCI analysis, however, does not show corresponding interactions for the β-pyridinium ion. DFT calculations find that β-phenanthrolinium ion is more stable than the α-complex (ΔGβ → α = +6.7 kcal/mol, Figure S19), thereby shielding the β-face and favoring SN2 attack on the α-face to yield α−1,2-cis glycoside.
In conclusion, the phenanthroline-catalyzed glycosylation strategy provides a general platform for α-selective formation of 1,2-cis glycosides under mild and operationally simple conditions. This system is not confined to the predetermined nature of the coupling partners and mimics the glycosyltransferase-catalyzed mechanism. Our computational and experimental studies indicate a double SN2 pathway involving phenanthroline-catalyzed glycosylation with α-glycosyl bromide. Efforts to utilize this method to enable other glycosidic bonds are underway.
Supplementary Material
Scheme 2.
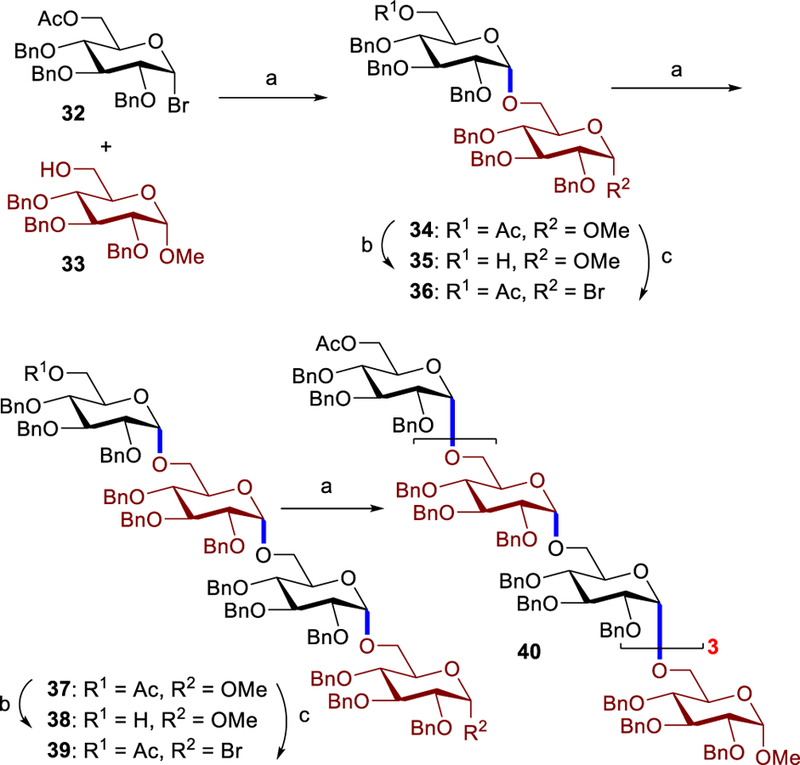
Synthesis of octasaccharide 40. [a] 5 – 15 mol% of 4, IBO (2 equiv.), MTBE (2 M), 50 oC, 24 h, 34: 89%, α:β > 20:1; 37: 86%, α:β > 20:1; 40: 77%, α:β > 20:1. [b] NaOMe, MeOH, CH2Cl2, 25 oC, 35: 99%, 38: 70%. [c] PTSA, Ac2O, 70 oC, 2 h (glycosyl acetates: 36S: 61%, 39S: 51%, see SI); then HBr/AcOH, CH2Cl2, 0 oC, 15 min, and 36 and 39 were used in the next step without further purification.
Acknowledgements
We are grateful for financial support from the National Institutes of Health (U01 GM120293). We thank Profs. David Crich (Wayne State) and Daniel Quinn (Iowa) for suggestions with kinetic studies as well as the Lumigen Center for instrumental assistance.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Supporting information and the ORCID identification number(s) for the author(s) of this article can be found under: http://doi.org/10.1002/anie.
References
- [1].(a) Ohtsubo K, Marth JD, Cell 2016, 126, 855–867; [DOI] [PubMed] [Google Scholar]; (b) van Kooyk Y, Rabinovich G, Nat. Immunol 2008, 9, 593–601. [DOI] [PubMed] [Google Scholar]
- [2].(a) McKay MJ, Nguyen HM, ACS Catal 2012, 2, 1563–1595; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nielsen MM, Pedersen CM, Chem. Rev 2018, 118, 8285–8358. [DOI] [PubMed] [Google Scholar]
- [3].Leng W-L, Yao H, He J-X, Liu X-W Acc. Chem. Res 2018, 51, 628–639. [DOI] [PubMed] [Google Scholar]
- [4].(a) Kim J-H, Yang H, Boons G-J, Angew. Chem. Int 2005, 44, 947–949; [DOI] [PubMed] [Google Scholar]; (b) Kim J-H, Yang H, Park J, Boons G-J, J. Am. Chem. Soc 2005, 127, 12090–12097; [DOI] [PubMed] [Google Scholar]; (c) Yasomanee JP, Demchenko AV, J. Am. Chem. Soc 2012, 134, 20097–20102; [DOI] [PubMed] [Google Scholar]; (d) Yasomanee JP, Demchenko AV, Angew. Chem. Int. Ed 2014, 53, 10453–10456. [DOI] [PubMed] [Google Scholar]
- [5].(a) Geng Y, Kumar A, Faidallah HM, Albar HA, Mhkalid IA, Schmidt RR; Angew. Chem. Int. Ed 2013, 52, 10089–10092; [DOI] [PubMed] [Google Scholar]; (b) Buda S, Mawoj M, Golebiowska P, Dyduch K, Michalak A, Mlynarski J, J. Org. Chem 2015, 80, 770–780; [DOI] [PubMed] [Google Scholar]; (c) Peng P, Schmidt RR, J. Am. Chem. Soc 2015, 137, 12653–12659; [DOI] [PubMed] [Google Scholar]; (d) Kimura T, Eto T, Takahashi D, Toshima K, Org. Lett 2016, 18, 3190–3193; [DOI] [PubMed] [Google Scholar]; (e) Sun L, Wu X, Xiong D-C, Ye X-S, Angew. Chem. Int. Ed 2016, 55, 8041–8044; [DOI] [PubMed] [Google Scholar]; (f) Park Y, Harper KC, Kuhl N, Kwan EE, Liu RY, Jacobsen EN, Science 2017, 355, 162–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lairson LL, Henrissat B, Davies GJ, Withers SG, Annu. Rev. Biochem 2008, 77, 521–555. [DOI] [PubMed] [Google Scholar]
- [7].(a) Lemieux RU, Morgan AR, J. Am. Chem. Soc 1963, 85, 1889–1890. [Google Scholar]; (b) Lemieux RU, Morgan AR, Can. J. Chem 1965, 43, 2205–2213. [Google Scholar]
- [8].Garcia BA, Gin DY, J. Am. Chem. Soc 2000, 122, 4269–4279. [Google Scholar]
- [9].Lanz G, Madsen R, Eur. J. Org. Chem 2016, 3119–3125.
- [10].Spivey AC, Arseniyadis S, Angew. Chem. Int. Ed 2004, 43, 5436–5441. [DOI] [PubMed] [Google Scholar]
- [11].Nigudkar SS, Stine KJ, Demchenko AV, J. Am. Chem. Soc 2014, 136, 921–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Chatterjee S, Moon S, Hentschel F, Gilmore K, Seeberger PH, J. Am. Chem. Soc 2018, 140, 11942–11953. [DOI] [PubMed] [Google Scholar]
- [13].Lu SR, Lai YH, Chen JH, Liu CY, Mong KKT, Angew. Chem. Int. Ed 2011, 50, 7315–7320. [DOI] [PubMed] [Google Scholar]
- [14].Vasudevan D, Takeuchi H, Johar SS, Majerus E, Haltiwanger RS, Curr. Biol 2015, 25, 286–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Gao PC, Zhu SY, Cao H, Yang JS, J. Am. Chem. Soc 2016, 138, 1684–1688. [DOI] [PubMed] [Google Scholar]
- [16].Pratt MR, Bertozzi CR, Chem. Soc. Rev 2005, 34, 58–68. [DOI] [PubMed] [Google Scholar]
- [17].Kuduk SD, Schwarz JB, Chen X-T, Glunz PW, Sames D, Ragupathi G, Livingston PO, Danishefsky SJ, J. Am. Chem. Soc 1998, 120, 12474–12485. [Google Scholar]
- [18].(a) Bucher C, Gilmour R, Angew. Chem. Int. Ed 2010, 49, 8724–8728; [DOI] [PubMed] [Google Scholar]; (b) Durantie E, Bucher C, Gilmour R, Chem. Eur. J 2012, 18, 8208–8215. [DOI] [PubMed] [Google Scholar]
- [19].Chen X, Adv. Carbohydr. Chem. Biochem 2015, 72, 113–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].(a) van Bueren AL, Higgins M, Wang D, Burke RD, Boraston AB, Nat. Struct. Mol. Biol 2007, 14, 76–84; [DOI] [PubMed] [Google Scholar]; (b) Bittencourt VCB, Figueiredo RT, da Silva RB, Mourao-Sa DS, Fernandez PL, Sassaki GL, Mulloy B, Bozza MT, Barreto-Bergter EB, J. Biol. Chem 2006, 281, 22614–22623. [DOI] [PubMed] [Google Scholar]
- [21].Cao Y, Okada Y, Yamada H, H. Carbohydr. Res 2006, 341, 2219–2223. [DOI] [PubMed] [Google Scholar]
- [22]. Fragmentation of 41 using collision induced dissociation led to formation of phenanthroline with an m/z ratio of 333.1396 (Figure S8).
- [23].Singh Y, Wang TH, Geringer SA, Stine KJ, Demchenko AV, J. Org. Chem 2018, 83, 374–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lemieux RU, Hendriks KB, Stick RV, James K J. Am. Chem. Soc 1975, 97, 4056–4062. [Google Scholar]
- [25]. β−42 rapidly converted into α−1 in the presence of catalyst 4 within 15 min at 50 oC.
- [26]. Coupling of 2 with β-bromide 42 under standard conditions provided 3 in comparable yield and α-selectivity to that obtained with α-bromide 1.
- [27]. The α:β ratio of product 3 is kinetically-derived and is not reflective of a thermodynamic distribution arising from post-coupling isomerization (Figure S9).
- [28].Johnson ER, Keinan S, Mori-Sanchez P, Contreras-Garcia J, Cohen AJ, Yang W, J. Am. Chem. Soc 2010, 132, 6498–6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29]. The optimized geometry of the transition state shows that because of the back-side displacement nature of the SN2 transition state, the distance is too large for any stabilizing interaction between the hydrogen of the alcohol and the nitrogen of phenanthroline (Figure S17).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.