

Abstract
Epigenetic mechanisms have been proposed to contribute to persistent aspects of addiction-related behaviors. One family of epigenetic molecules that may regulate maladaptive behavioral changes produced by cocaine use are the histone deacetylases (HDACs)—key regulators of chromatin and gene expression. In particular, the class IIa HDACs (HDAC4, HDAC5, HDAC7 and HDAC9) respond to changes in neuronal activity by modulating their distribution between the nucleus and cytoplasm—a process controlled in large part by changes in phosphorylation of conserved residues. Cocaine triggers a transient nuclear accumulation of HDAC5 that functions to limit the development of cocaine reward behavior. However, the role and regulation of the close family member, HDAC4, in cocaine behaviors remain largely unknown. In this study, we report that cocaine and cAMP signaling in striatum produced differential phosphorylation and subcellular localization of HDAC4 and HDAC5. Unlike HDAC5, cocaine exposure induced a modest hyperphosphorylation and nuclear export of HDAC4. Genetic deletion of HDAC4 in the nucleus accumbens reduced acute cocaine-produced locomotion, maximum locomotor sensitization and cocaine reward-related behavior. Interestingly, overexpression of an HDAC4 cytoplasm-concentrated mutant (S266E) increased cocaine reward behavior in the cocaine conditioned place preference assay, suggesting that cocaine-induced nuclear export of HDAC4 might function to facilitate the development of cocaine reward behaviors through a role in the cell cytoplasm. Together, our findings suggest that, despite high sequence homology, HDAC4 and HDAC5 are oppositely regulated by cocaine-induced signaling in vivo and have distinct roles in regulating cocaine behaviors.
Keywords: addiction, cocaine, HDAC, striatum
INTRODUCTION
In recent years, increasing attention has been paid to the role of epigenetic mechanisms in regulating drug addiction (Hyman, Malenka & Nestler 2006; Renthal & Nestler 2009; Robison & Nestler 2011). Work in the last decade has described changes in chromatin state (e.g. histone and DNA modifications such as methylation and acetylation) produced by drug exposure in multiple brain regions including the nucleus accumbens (NAc). These modifications alter the accessibility of DNA to transcriptional complexes, providing a molecular mechanism for producing stable changes in gene expression. There is accumulating evidence that exposure to illicit drugs is able to alter the expression, localization, and function of a number of epigenetic enzymes responsible for regulating histone modifications that enhance or reduce transcription of different gene targets.
Histone deacetylases (HDACs) catalyze the removal of acetyl groups from multiple lysine residues on histone tails, which in turn promotes chromatin condensation and reduces access to transcription-promoting complexes (Kouzarides 1999; Bertos, Wang & Yang 2001; Jiang et al. 2008). Class IIa HDACs (HDAC4, 5, 7 and 9) are an HDAC subclass that shuttles between the nucleus and cytoplasm in response to cell signaling events (McKinsey et al. 2000a; Bertos et al. 2001; McKinsey, Zhang & Olson 2001; Chawla et al. 2003; Parra & Verdin 2010; Parra 2015). They do not appear to bind directly to genomic DNA, but regulate chromatin and gene expression through indirect association with DNA-bound transcription factors, such as the MEF2 family (Miska et al. 1999). Cell signaling pathways, such as those stimulated by intracellular calcium and cAMP levels, modulate class IIa HDAC nucleocytoplasmic shuttling by regulating conserved CaMK sensitive serine residues (HDAC4 S246/467 and HDAC5 259/498; Grozinger & Schreiber 2000; McKinsey et al. 2000b; Wang et al. 2000; Schlumm et al. 2013). When these residues are dephosphorylated, class IIa HDACs localize to the nucleus, where they are thought to repress gene expression. In addition to binding with MEF2, class IIa HDACs also interact with class I HDACs, and other proteins, as part of a large repressor complex (Miska et al. 1999; Grozinger & Schreiber 2000; Huang et al. 2000; Fischle et al. 2002). In contrast, phosphorylation of the conserved residues promotes nuclear export, disrupts binding to MEF2 transcription factors, and increases binding to the 14–3-3 family of cytoplasmic anchoring proteins, presumably resulting in de-repression of several target genes (Wang et al., 2000; Grozinger & Schreiber 2000). Classically, nuclear export of class IIa HDACs is viewed as a termination of HDAC action in the nucleus, but recent evidence indicates that class IIa HDACs may also have cytoplasmic targets and functions (Cho & Cavalli 2012; Cho et al. 2013).
HDAC4, a member of the class IIa family, is expressed in multiple brain regions, including the forebrain and striatum (Bolger & Yao 2005; Tsankova et al. 2006; Renthal et al. 2007), and in the forebrain, it is important for spatial learning and memory (McQuown et al. 2011; Kim et al. 2012). Viral-mediated expression of HDAC4 or HDAC5 in the adult NAc of wild-type mice reduces cocaine reward behaviors (Kumar et al. 2005; Wang et al. 2010; Taniguchi et al. 2012), and compared with wild-type animals, HDAC5-deficient mice display increased cocaine reward sensitivity after repeated cocaine administration—a process that is rescued by NAc expression of HDAC5 during the prior cocaine administration (Renthal et al. 2007). In a prior study, we observed that non-contingent cocaine administration in mice triggers a transient dephosphorylation and nuclear accumulation of endogenous HDAC5 in the striatum, and expression of a mutant form of HDAC5 that cannot be phosphorylated at Ser279 (S279A) reduced the development, but not expression, of cocaine reward in the conditioned place preference assay (CPP, Taniguchi et al. 2012). HDAC4 has an analogous serine residue (S266) that has been shown to be a PKA-sensitive site that regulates nucleocytoplasmic localization in non-neuronal cells (Paroni et al. 2008; Walkinshaw et al. 2013), but the role of this phosphorylation site in regulating HDAC4 neuronal localization and its response to cocaine stimulation are unknown. Despite published evidence that overexpression of HDAC4 in the NAc can modulate cocaine reward, little is known about how or whether cocaine administration regulates HDAC4 levels and subcellular distribution in the NAc, and whether altering HDAC4 localization can function, like HDAC5, to influence drug reward behavior.
In the present study, we show that despite high sequence conservation, HDAC4 is regulated differently than HDAC5 in response to cocaine administration or following elevation of cAMP in striatal neurons. Given the opposite regulation, we predicted that loss of HDAC4 might oppositely influence cocaine-related behaviors compared with loss of HDAC5. We find that loss of HDAC4 in the NAc reduces cocaine reward and acute and sensitized locomotor responses to cocaine. Moreover, we find that overexpression of cytoplasmically localized HDAC4 mutant (S279E) significantly increases cocaine CPP, suggesting that the cocaine-induced nuclear export of HDAC4 might serve to regulate the development of cocaine reward-context learning. Our findings suggest that HDAC4 and HDAC5 have distinct regulation and functional roles following cocaine administration.
METHODS
Animals
All Hdac4loxP/loxP and C57Bl/6 mice (Charles River, Wilmington, MA) used in this study were adult males tested between 9 and 12 weeks old. C57Bl/6 mice were acclimated to the colony for at least 1 week following delivery. Hdac4loxP/loxP contained loxP sites flanking exon 5 of the Hdac4 gene and have been previously characterized to generate an out-of-frame mutation in the presence of Cre recombinase (Potthoff et al. 2007). Experimental littermate animals were generated from homozygous crosses of Hdac4loxP/loxP animals. All animals were housed on a 12-hour light–dark cycle with access to food and water ad libitum. All procedures were in accordance with the McLean Hospital Institutional Care and Use (IACUC) guidelines.
Cell culture
Primary embryonic (E18) striatal cultures were generated from Long Evans rats (Charles River, Wilmington, MA) as previously described (Pulipparacharuvil et al. 2008; Taniguchi et al. 2012). For biochemistry experiments, crude striatal cells (grossly dissected whole striatum) were plated at 1.3 × 106 on 5 μg/ml poly-D-lysine coated 6-well plates. For transfection and immunocytochemistry experiments, enriched striatal cultures (subdissected from coronal slice) were plated at 1 × 105 on etched-glass coverslips coated with 5 μg/ml poly-D-lysine/0.5 μg/ml laminin solution in a 24-well plate. HEK293T cells were cultured on 6-well plates at 8 × 105−1 × 106 cells per well. Cells were transfected using Lipofectamine LTX (Life Technologies, Carlsbad, CA), and cell lysate was collected 48 hours after transfection.
Immunocytochemistry
Enriched striatal cultures were transfected at 8 days in vitro using a calcium phosphate method (Renthal et al. 2007; Pulipparacharuvil et al. 2008). For forskolin localization experiments, at 10 days in vitro cells were stimulated for 3 hours with DMSO or 10 μM forskolin prior to fixation. For nuclear export inhibition studies, at 10 days in vitro cells are treated for 3 hours with leptomycin B (3 ng/ml; Sigma-Aldrich, St. Louis, MO) or vehicle prior to fixation. Immunocytochemistry was conducted as described in Taniguchi et al. 2012. Briefly coverslips were incubated in anti-GFP antibody (1:10 000), anti-Flag (1:500) or anti-HDAC4 antibody (1:1000) followed by anti-mouse FITC (1:500) diluted in 1X TBS. After secondary incubation, coverslips were counterstained with Hoechst 33342 (1:50 000) to visualize the nucleus. The localization of HDAC4-GFP, HDAC4-Flag, HDAC5-GFP or HDAC4-Flag mutants was categorized as cytoplasmic mixed (not displayed), or nuclear using the GFP or Flag signal for each neuron under experimenter-blind conditions. The percentage for each category was calculated from the total number of transfected neurons counted in each condition, and the average across experiments is shown.
Plasmids and viruses
p1005+ GFP HDAC4-Flag WT was generated by PCR amplification of hHDAC4-Flag from pBJ-hHDAC4-Flag (Dr. Eric Olson, UTSW). PCR primers were designed to add XbaI restriction sites on 5’ and 3’ end of full-length hHDAC4-Flag. p1005+ GFP (Dr. Rachael Neve, MIT) backbone and PCR product were digested with XbaI, and compatible cohesive ends were ligated to produce p1005+ GFP HDAC4-Flag WT. p1005+ GFP HDAC4-Flag 3SA was generated by PPuMI and PflMI digestion of pcDNA-HDAC4.3SA-Flag (Addgene, Cambridge, MA), p1005+ GFP HDAC4-Flag WT was identically digested to excise the wild-type portion of the HDAC4 coding sequence and compatible cohesive end ligations generated p1005+ GFP HDAC4-Flag 3SA. p1005+ GFP HDAC4-Flag S266E and S266A was generated by site-directed mutagenesis of pBS-hHDAC4-Flag using primers that produce a serine to glutamine substitution at S266. pBS-hHDAC4-S266E/A-Flag was digested with PPuMI and PflMI digestion, and p1005+ GFP HDAC4-Flag S266E/A was generated using the same digestion/ligation as p1005+ GFP HDAC4-Flag 3SA. Diagnostic digests and sequencing were performed to confirm proper orientation and sequence. pEGFP-N1-HDAC5-GFP (Dr. Eric Olson, UTSW) and pcDNA3.1-GFP-HDAC4 (Addgene) were used to examine in vitro localization.
Sample preparation from cocaine-injected mice
C57BL/6 mice (Charles River) were injected once per day for seven consecutive days (intraperitoneal; i.p.) with saline or 20 mg/kg cocaine (Sigma-Aldrich) and sacrificed 4 hours after the last injection. Striata were rapidly dissected from 1 mm coronal sections prepared using a mouse brain matrix. To probe the phosphorylation status of HDAC4, samples are prepared as previously described (Taniguchi et al. 2012). Briefly, striata are dissolved in tissue lysis buffer with inhibitors and diluted with ninefold volume of radioimmunoprecipitation assay (RIPA, without SDS) buffer prior to sequential immunoprecipitation of HDAC5 and HDAC4, using anti-HDAC5 monoclonal antibody (Abcam, #50001) and anti-HDAC5 polyclonal antibody (cell signaling, #2082) followed by anti-HDAC4 polyclonal antibody (cell signaling, #2072). Cytosolic and nuclear extracts were prepared with NE-PER nuclear and cytoplasmic extraction kit (Pierce Biotechnology, Waltham, MA) according to manufacturer’s instructions. Samples were collected for protein quantification using detergent compatible Lowry colorimetric assay. Respectively, 80 and 50 μg of nuclear and cytoplasmic amounts were used for western blot analysis. Ratios are calculated relative to the saline condition and normalized to β-tubulin in the cytoplasmic and lamin A/C in the nuclear fraction.
Western blotting
Lysates from stimulated cultured neurons and cocaine-treated animals were first immunoprecipitated to isolate HDAC5. Subsequently, HDAC4 was immunoprecipitated from the HDAC5-immunodepIeted lysates. Protein extracts were subjected to SDS-PAGE, transferred to PVDF membrane, blocked with 10 percent (w/v) non-fat dry milk or LI-COR blocking buffer diluted in 1X PBS and incubated with antibody solutions. Primary antibodies were diluted in 1X TBS with 0.05 percent Tween-20 containing 3 percent (w/v) bovine serum albumin or LI-COR blocking buffer with 0.01 percent Tween-20 as follows: anti-HDAC4 (1:500), anti-P-S266 (1:500, lab made), anti-Flag (1:500), anti-β-tubulin (1:10 000) or anti-lamin A/C (1:500). Secondary antibodies were diluted in 1X TBS with 0.05 percent Tween-20 containing 10 percent (w/v) non-fat dry milk as follows or LI-COR blocking buffer with 0.01 percent Tween-20 and 0.1 percent SDS: anti-mouse HRP conjugated IgG (1:10 000), anti-rabbit HRP conjugated IgG (1:10 000), IRDye 680CW anti-mouse IgG (1:20 000) and IRDye 680RD anti-rabbit IgG (1:20 000). Blots were developed by enhanced chemiluminescence (ECL-Prime, Amersham Pharmacia Biotech, Little Chalfont, UK)) or imaged on LI-COR Odyssey Clx (LI-COR Biosciences, Lincoln, NE). Densitometry analysis was performed with NIH ImageJ (https://imagej.nih.gov/ij/) or LI-COR Image Studio (LI-COR Biosciences).
Viral-mediated gene transfer
Herpes simplex viruses (HSV) expression plasmids for the HDAC4 S266E mutant were packaged into high-titer viral particles as described previously (MIT viral core, Neve & Lim 2013). High-titer HSV-GFP was purchased from the MIT viral vector core. High titer AAV2/2-GFP and AAV2/2-Cre-GFP were purchased from Vector BiolLabs (Malvern, PA). Stereotaxic surgery was performed on anesthetized mice as previously described (Penrod et al. 2015). Virus was delivered bilaterally to the NAc (+1.6 mm anterior, +1.5 mm lateral and −4.4 mm ventral from bregma) at a 10° angle. Virus was delivered using Hamilton syringes at a rate of 0.1 μl/minute for a total of 0.5–0.7 μl ofvirus. Behavioral testing began after a recovery period (72 hours for HSV experiments for peak expression and 3 weeks for AAV experiments for peak expression and recombination). Viral placements were confirmed using GFP signal, which was co-expressed by all viruses.
HDAC4 recombination validation
Hdac4loxP/loxP received AAV2/2-GFP or AAV2/2-Cre-GFP as described previously. Three weeks after viral infusion, animals were rapidly decapitated; brains were extracted, frozen on dry ice and stored at −80° C until ready for dissection. Brains were transferred from −80° C and placed into a cryostat at −25° C. Brains were mounted onto the cryostat chuck with tissuetek and fast frozen with cryospray (Freeze-It, Thermo Fisher Scientific). Forty-micometer sections were taken until the NAc was revealed. Bilateral punches were taken using a 1-mm stainless steel punch tool and placed in cold microcentrifuge tubes for later RNA extraction. Brains were sectioned through the depth of the punch to confirm that punches were taken from NAc. NAc punches were lysed in QIAzol lysis reagent (Qiagen Germantown, MD), and RNA was isolated using miRNAeasy collection kit (Qiagen). mRNA was converted to cDNA using SuperScriptIII-RT (Invitrogen Carlsbad, CA) and random hexamer primers. HDAC4 expression was quantified using RT-qPCR with probes Hdac4 5’-CGAGCACAGAGGTGAAGATG-3’ (forward) and 5-GGTGGAGAGCTCTGGTCAA-3’ (reverse) and GAPDH 5’-AGGTCGGTGTGAACGGATTTG-3’ (forward) and 5’-TGTAGACCATGTAGTTGAGGTCA-3’ (reverse). Fold changes relative to GAPDH were determined using the ΔΔCt method, in which mean fold change (2-ΔΔCtAVE) and SEM (abs(((2-ΔΔCtAVE × 2-ΔΔCtSEM) − (2-ΔΔCtAVE/2-ΔΔCtSEM))/2)) were determined and expressed relative to GFP control condition.
Locomotor sensitization
Animals were sensitized to repeated cocaine injections, as previously described (Smith et al. 2016). Briefly, following a 1-hour habituation session in the test chamber, the mice receive saline or cocaine (i.p.) injections and are returned to the chamber for an additional hour. All mice received four daily saline injection sessions followed by daily cocaine injections (15 mg/kg) for the next 7 days. Hdac4 cKONAc cocaine locomotor sensitization was assessed 2–3 weeks after low dose (5 mg/kg) CPP. Hdac4 cKONAc acute cocaine locomotor responding in drug-naïve mice was assessed at 3 weeks following initial viral delivery. For each locomotor session, the sum of the first 30 or 60 minutes of locomotor activity is displayed.
Conditioned place preference
Mice were conditioned to cocaine using a balanced, accelerated paradigm to accommodate the timing of transient HSV expression (Pliakas et al. 2001; Smith et al. 2016). Briefly, after recovery from viral-mediated gene transfer (i.e. 72 hours for HSV studies), the mice freely explore a three-chambered apparatus, and time in each chamber is recorded. On the next 2 days, animals received two daily pairings with either side of the chamber: i.p. saline in the morning and i.p. cocaine (5 or 10 mg/kg) in the afternoon. Twenty-four hours after the last conditioning sessions, animals were returned to the chamber and allowed to freely explore both sides. Time spent in each chamber was recorded, and time spent in paired or unpaired (middle and unpaired) sides on pre-test and post-test is displayed. Only animals bilaterally expressing virus (as determined by GFP expression) in the NAc were included in final analysis.
Luciferase assay
3X MRE-luciferase and TK-Renilla luciferase plasmids have been previously described (Flavell et al. 2006). Primary striatal cultures were transfected using calcium phosphate method at 8 days in vitro, and 48 hours later, the cells were stimulated with or without depolarization buffer (isotonic, 60 mM KCl final; Pulipparacharuvil et al. 2008) for 6 hours, and dual luciferase activity levels were measured as recommended by the manufacturer (Promega, Madison, WI).
Immunohistochemistry
To confirm placement and viral-mediated gene transfer, mouse brains were rapidly extracted and processed for immunohistochemistry as previously described (Penrod et al. 2015). Briefly, brains were dissected, fixed, and cryoprotected. Fifty-micrometer sections from the olfactory bulbs to the dorsal hippocampus were generated using a freezing microtome. Free-floating sections were blocked in 3 percent (w/v) bovine serum albumin and incubated in chicken anti-GFP (1:1000, Aves) for 3–16 hours followed by anti-chicken 488 conjugated IgY (1:200). Sections were then counterstained with the nuclear dye, Hoechst 33342 (1:50,000 Thermo Fisher Scientific, Waltham, MA), dehydrated and mounted for fluorescent microscopy visualization.
Data analysis
Student’s t-tests and one-way, two-way or repeated measures ANOVAs with Sidak’s multiple comparison post hoc tests were used, as appropriate. Grubb’s outlier analysis was used on final datasets. Statistical analysis and graphical depictions were prepared using GRAPHPAD PRISM software (GraphPad, La Jolla, CA).
RESULTS
HDAC4 and HDAC5 phosphorylation are differentially regulated by cAMP and cocaine
Previous work has demonstrated that elevation of cAMP levels stimulates dephosphorylation at multiple HDAC5 residues, which results in a robust accumulation of HDAC5 protein in the cell nucleus (McKinsey et al. 2000b; McKinsey et al. 2001; Chawla et al. 2003; Vega et al. 2004; Taniguchi et al. 2012; Walkinshaw et al. 2013). The HDAC5 S279 phosphorylation site, which resides within the nuclear localization sequence, regulates the nuclear export rate, and dephosphorylation of this site is induced by cocaine or elevated cAMP signaling in striatal neurons (Taniguchi et al. 2012). Interestingly, HDAC4 and HDAC5 show full sequence conservation of the residues surrounding this site (Fig. 1a), and as predicted, we found that the HDAC5 P-S279 antibody (Taniguchi et al. 2012) also site-specifically recognizes HDAC4 P-S266 (Fig. 1b). To test whether HDAC4 P-S266 has a similar effect on subcellular localization as HDAC5 P-S279, we generated a phospho-mimetic mutant (S266E) of HDAC4 and tested its distribution in striatal neurons. Similar to the HDAC5 S2 79E mutant (Taniguchi et al. 2012), the HDAC4 S266E protein concentrated in the cytoplasm of striatal neurons (Fig. 1c) and reduced nuclear accumulation under conditions of nuclear export inhibition (leptomycin B treatment, Fig. 1d; FInteraction(3, 16) = 4.895, P = 0.0134; Sidak’s post hoc, cytoplasmic vervus nuclear P < 0.01 WT, S266A, 3SA, n.s. S266E), suggesting that P-S266/S279 has a similar effect on subcellular distribution of these closely related proteins.
Figure 1.
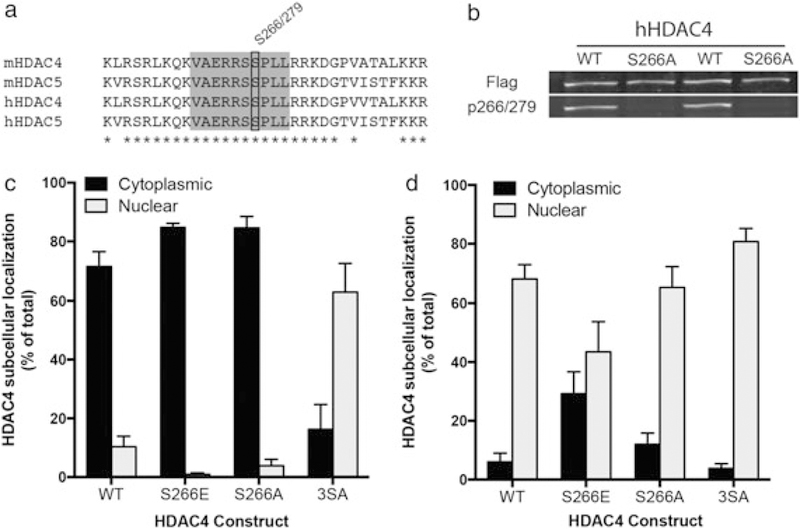
HDAC4 S266 drives cytoplasmic localization when phosphorylated. (a) Residue conservation comparison between mouse and human HDAC4 and HDAC5 near the S266/S279 residue (black box). Gray box encompasses the epitope used for antibody production; asterisks indicate 100 percent identity sites. (b) HDAC4 WT or HDAC S266A overexpressed in HEK293 cells show equivalent total HDAC expression (top panel) but the absence of p266 signal (bottom panel) in the alanine mutant condition. (c) Phosphomimetic mutation of S266 (S266E) promotes HDAC4 cytoplasmic accumulation. Mutation of critical serine residues to alanine (S246A/S467A/S632A) shifts HDAC4 subcellular localization to the nucleus. (d) Under conditions where nuclear export is blocked (3 hour of 10 ng/ml leptomycin B treatment), phosphomimetic mutation of S266 (S266E) causes enhanced cytoplasmic localization, indicating reduced nuclear import. In (c) and (d), transfected neurons were identified by GFP expression (driven by a second promoter), and subcellular localization was determined by Flag signal (C-terminal tag on all HDAC4 constructs)
Because elevation of cAMP signaling (10 μM forskolin) in striatal neurons triggers the rapid and robust (~75 percent) dephosphorylation of HDAC5 P-S279 (Taniguchi et al. 2012), we asked whether HDAC4 P-S266 is similarly regulated. To examine HDAC4 phosphorylation specifically, a sequential immunoprecipitation strategy was employed. HDAC5 was immunodepleted from the tissue lysate using a HDAC5-specific antibody, and subsequently, HDAC4 was immunoprecipitated using a HDAC4-specific antibody (or vice versa, Supporting Information Fig S1a). Surprisingly, we observed that forskolin treatment did not significantly decrease P-S266 levels (Fig. 2a), suggesting distinct regulation of these two closely related HDACs. Consistent with this observation, elevation of cAMP levels promoted accumulation of GFP-tagged HDAC5 into the cell nucleus (Fig. 2b, left; FInteraction(2, 12) = 6.385, P = 0.0129; Sidak’s post hoc, nuclear compartment P < 0.05), whereas cAMP elevation did not alter GFP-tagged HDAC4’s cytoplasmic distribution (Fig. 2b, right; FCompartment(2, 12) = 0.3519, P = 0.0057) in cultured primary striatal neurons. Like forskolin treatment, cocaine administration (i.p.) in mice produces a transient dephosphorylation of HDAC5 S279 and enrichment of HDAC5 in the nuclear fraction from isolated striatal tissues (Taniguchi et al. 2012). To test whether chronic cocaine administration regulates HDAC4 in a similar fashion, we repeatedly injected wild-type mice with cocaine (20 mg/kg; i.p.) or saline and then analyzed P-S266 and the nuclear/cytoplasmic ratio of HDAC4. At 4 hours following the last cocaine injection, a time point where HDAC5 S279 is significantly dephosphorylated (~35 percent; Taniguchi et al. 2012) and enriched in the nucleus, we observed an approximately fourfold increase in P-S266 HDAC4 levels (Fig. 2c, right; P = 0.0525). Consistent with the role of this residue in regulating HDAC4 and HDAC5 subcellular localization, we detected a significant decrease in HDAC4 nuclear levels (Fig. 2d, right; t = 2.623, df = 9, P = 0.0277). These data suggest that HDAC4 and HDAC5, despite high sequence homology, are distinctly regulated by cAMP signaling and cocaine in striatum.
Figure 2.
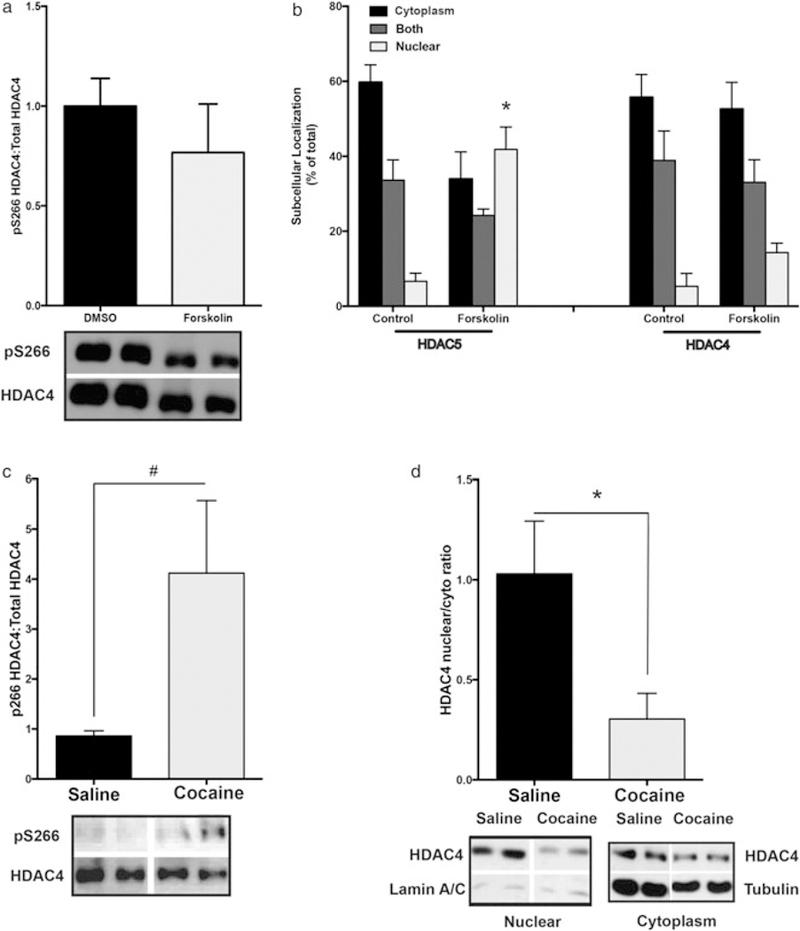
HDAC4 phosphorylation is differentially regulated by cAMP and cocaine. (a) Forskolin treatment (10 μM, 3 hours) does not alter HDAC4 S266 phosphorylation (from HDAC5 immunodepleted, HDAC4 immunoprecipitated lysates) compared with DMSO-treated cultures (mean ± SEM, n = 6/condition, three independent experiments). (b) Forskolin treatment (10 μM, 3 hours) promotes nuclear accumulation of HDAC5-GFP but does not alter HDAC4-GFP subcellular distribution (mean ± SEM, n = 3 wells/condition, ~80 cells per well, two to three independent experiments). (c) Chronic cocaine (20 mg/kg; i.p.; 7 days) weakly increases S266 phosphorylation of HDAC4 (from HDAC5-immunodepleted, HDAC4 immunoprecipitated lysates) compared with animals treated with repeated saline injections (mean ± SEM, n = 9 mice/condition). (d) Cocaine promotes cytoplasmic accumulation of HDAC4. Nuclear/cytoplasmic ratios of HDAC4, relative to saline, normalized to lamin A/C in nuclear fraction and to β-tubulin in cytoplasmic fraction (mean ± SEM, n = 5–6 mice/condition). Asterisks indicate significant difference from control group in pairwise comparison; *P < 0.05, # near significant result P =.0525
HDAC4 in the NAc is required for multiple cocaine behavioral responses
Because cocaine administration regulates the subcellular localization of HDAC4, we tested the importance of this gene in two cocaine-induced behaviors—cocaine reward, which measures an animal’s preference to explore a chamber previously associated with drug experience, and locomotor sensitization, which measures a naïve and adaptive motor response that develops with repeated cocaine administration. Because total Hdac4 KO mice do not survive past weaning (Vega et al. 2004), we investigated the role of HDAC4 in cocaine behaviors using a conditional gene deletion approach. Young adult Hdac4fl/fl mice (Potthoff & Olson 2007) received bilateral infusions of AAV2-Cre-GFP into the NAc to generate conditional Hdac4 KO mice (Hdac4 cKONAc) or littermate controls (AAV2-GFP infusions). Three weeks after virus infusion to allow for recombination (Fig. 3a; t = 2.236, df = 10, P = 0.0494), all mice were tested sequentially in CPP (5 or 10 mg/kg cocaine; Pliakis et al., 2001; Smith et al. 2016) and locomotor sensitization tests. Hdac4 cKONAc mice failed to condition to a low dose of cocaine (5 mg/kg; FConditioning(1, 29) = 10.1, P = 0.0035; Sidak’s post hoc pre versus post, GFP P < 0.01, Hdac4 cKONAc n.s.), but conditioned like wild-type controls to high-dose cocaine in the CPP assay (10 mg/kg, Fig. 3b; FConditioning(1, 37) = 35.49, P < 0.0001; Sidak’s post hoc pre versus post, GFP and Hdac4 cKONAc P < 0.001), suggesting that HDAC4 in the NAc enhances an animal’s sensitivity to the rewarding effects of cocaine, particularly at low doses. The mice were then left undisturbed in their home cages for 2–3 weeks before locomotor sensitization testing. All mice were subjected to four daily injections of saline and then seven consecutive daily injections of cocaine (15 mg/kg; i.p.), and locomotion was monitored for 1 hour (Smith et al. 2016). Compared with control mice, Hdac4 cKONAc mice displayed normal locomotion following the saline injections (Fig. 3c). There was a small, but non-significant, reduction in locomotion after the first cocaine injection (Fig. 3c, Coc1), but a significant reduction in sensitized locomotion developed after repeated cocaine administration (Fig. 3c; FSession (3, 87) = 115, P < 0.001; FInteraction(3, 87) = 3.402, P = 0.0212; Sidak’s post hoc, Coc7 P < 0.01). Because all of the mice had received prior, low-dose cocaine in the preceding CPP test, we next tested drug-naïve Hdac4 cKONAc and control mice to examine possible differences in naïve cocaine locomotor responses to a range of cocaine doses. We observed a significant reduction in drug-induced locomotion in the drug-naïve Hdac4 cKONAc mice at both 15 and 30 mg/kg cocaine (Fig 3c; FGenotype(3, 98) = 11.45, P = 0.0010; FDose(3, 98) = 11.24, P < 0.001; FInteraction(3, 98) = 3.78, P = 0.0129; Sidak’s post hoc, 15 mg/kg P < 0.05, 30 mg/kg P < 0.01). Together, these findings indicate that HDAC4 in the NAc is required for normal cocaine reward behavior and both naïve and sensitized locomotor responses to cocaine.
Figure 3.
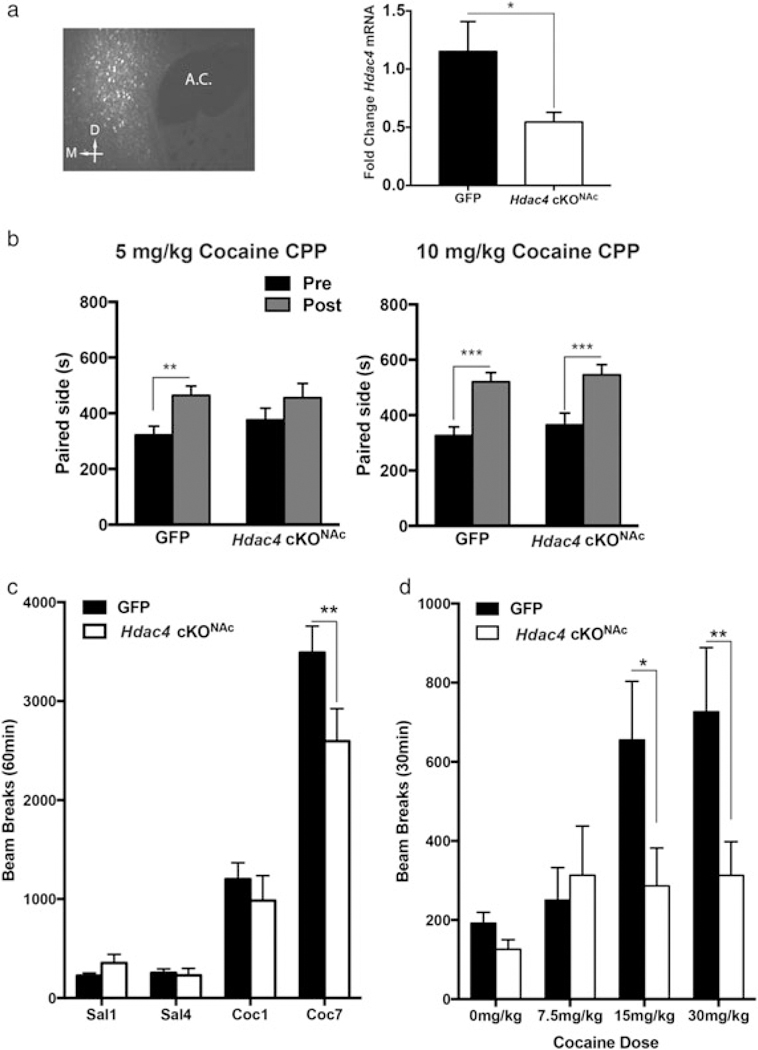
Loss of HDAC4 limits locomotor responses to acute and sensitizing regimens of cocaine. (a) Representative targeted expression in the NAc >21 days post-infection. Viral-mediated Cre expression significantly reduces HDAC4 mRNA. NAc punches from AAV-GFP or AAV-Cre-GFP Hdac4ffl/fl mice 3 weeks post-infection. HDAC4 mRNA is normalized to GAPDH and expressed as fold change compared with GFP control group (mean ± SEM, n = 6–7 NAc punches/condition). (b) Hdac4 NAc cKO disrupts low-dose (5 mg/kg) cocaine-conditioned place preference (left) but does not alter high-dose (10 mg/kg) cocaine CPP (right, mean ± SEM, n = 12–19 mice/condition). (c) Hdac4 NAc cKO decreases locomotor sensitization magnitude (mean ± SEM, n = 12–19 mice/condition). (d) Hdac4 NAc cKO decreases acute locomotor responses to cocaine (mean ± SEM, n = 8–10 mice/condition). Asterisks indicate significant difference from pre-test (b) or control group (a, c and d) in pairwise comparison, *P < 0.05,**P < 0.01, ***P < 0.001. AC, anterior commissure; D, dorsal; M, medial
Cytoplasmic HDAC4 enhances cocaine reward
Chronic cocaine administration increases the phosphorylation and cytoplasmic localization of HDAC4 (Fig. 2c and d), and loss of HDAC4 reduces cocaine reward behavior (Fig. 3a), particularly at a threshold dose of drug (5 mg/kg). We therefore speculated that the cocaine-induced HDAC4 nuclear export might function to facilitate cocaine reward behavior. To test this idea, we generated recombinant HSV that overexpress a cytoplasm-localized HDAC4 mutant (S266E) or GFP alone (Fig. 1c and d). Animals bilaterally overexpressing HDAC4 S266E, but not GFP, in the adult NAc displayed significant cocaine conditioned place preference at a threshold dose (Fig. 4a; FConditioning(1, 26) = 10.3, P = 0.0035; Sidak’s post hoc, pre versus post, SE P < 0.05). Unlike overexpression of the nuclear mutant, HDAC4–3SA, which dramatically reduced depolarization-induced MEF2-dependent transcription, overexpression of the HDAC4 S266E mutant had no significant effect on basal or depolarization-induced MEF2-dependent transcription activity in striatal cultures (Fig. 4b; FConstruct(1, 48) = 27.84, P < 0.0001, FInteraction(2, 48) = 7.222, P = 0.0018; Sidak’s post hoc, basal versus KCl, GFP and S266E P < 0.001, 3SA ns), suggesting that overexpression of HDAC4 S266E does not function as a dominant negative protein in striatal neurons. Together, these findings suggest that phosphorylated HDAC4 can enhance cocaine reward-like behavior, possibly through a role in the cytoplasm.
Figure 4.

Cytoplasmic accumulation of HDAC4 promotes cocaine reward. (a) Overexpression (inset shows representative targeted expression in the NAc ~7 days post-infection and viral-mediated expression in cultured striatal neurons) of a cytoplasmically localized HDAC4 mutant enhances a subthreshold conditioning experience (mean ± SEM, n = 7–12 mice/condition). (b) Overexpression of a nuclear localized, but not cytoplasmically localized, HDAC4 mutant represses MRE-luciferase activity by KCl-induced depolarization. Asterisks indicate significant difference from pre-test or control, *P < 0.05, ***P < 0.001. AC, anterior commissure
DISCUSSION
In this study, we report that, unlike HDAC5 (Belfield et al. 2006; Ha 2010; Mihaylova et al. 2011; Taniguchi et al. 2012) and HDAC4 in other cell types (Du et al. 2008; Kozhemyakina et al. 2009; Mihaylova et al. 2011; Liu & Schneider 2013; Walkinshaw et al. 2013), elevation of cAMP signaling in striatal neurons fails to trigger robust dephosphorylation and accumulation of HDAC4 in the cell nucleus, suggesting distinct and cell type-specific regulatory mechanisms between these closely related class IIa HDACs. Moreover, we find that cocaine administration produces an increase in HDAC4 phosphorylation (statistical trend) and a significant reduction in HDAC4 nuclear levels in striatal tissues, which is opposite of the effects of cocaine on HDAC5 (Fig. 2, Taniguchi et al. 2012). While HDAC4 in the adult NAc appears to be dispensable for cocaine reward-like behavior in the conditioned place preference test at higher doses (10 mg/kg), it is required for the development of preference to low doses (5 mg/kg), perhaps indicating that novel mechanisms recruited by higher doses of cocaine are able to compensate for the loss of HDAC4. We also find that HDAC4 is required in the adult NAc for acute locomotor responses to cocaine —particularly at moderate to high doses—and for cocaine locomotor sensitization (Fig. 3), suggesting a NAc-specific role for HDAC4 in naïve and adaptive motor responses to cocaine. We show that a mutant form of HDAC4 with a phosphomimetic mutation at a key regulatory serine residue (S266E) promotes cytoplasmic accumulation of HDAC4 and cocaine reward, the opposite localization and behavioral phenotype observed for HDAC5 (Taniguchi et al. 2012). Taken together with previous work, our findings add to the growing evidence that despite high homology and often overlapping expression patterns, there are cell type-specific and/or brain region-specific roles and regulation for the different class IIa HDAC family members.
Our current observations raise a number of interesting questions for future study. Our analysis of the phospho-site mutants on HDAC4 and HDAC5 suggests strongly that phosphorylation at the conserved residues serves a similar role in regulating subcellular localization. However, while cAMP signaling triggers a robust dephosphorylation and nuclear accumulation of HDAC5 (Fig. 2; Belfield et al. 2006; Ha 2010; Mihaylova et al. 2011; Taniguchi et al. 2012), HDAC4 appears to be largely insensitive to cAMP signaling in cultured striatal neurons. Consistent with these observations, HDAC4 and HDAC5 phosphorylation and subcellular localization were distinctly regulated by in vivo cocaine exposure. Although somewhat surprising given the high homology between HDAC4 and HDAC5, multiple studies in non-neuronal cells report differential sensitivity to signaling events that regulate phosphorylation/nuclear export as well as interdependence between closely related HDACs to promote coordinated export events (Chang et al. 2005; Backs et al. 2006; Zhang et al. 2007; Backs et al. 2008; Métrich et al. 2010; Liu et al. 2012). Similar to our current findings that cocaine triggers nuclear export of HDAC4 (Fig. 2d), a previous study (Renthal et al. 2007) reported that chronic cocaine administration increased the levels of P-S259—another highly conserved phosphorylation site that promotes cytoplasmic localization of the class IIa HDACs (McKinsey et al. 2000a, b). This suggests that chronic cocaine administration might trigger hyperphosphorylation at multiple HDAC4 residues that regulate subcellular localization.
Because Hdac5 KO mice display enhanced cocaine reward behavior (Renthal et al. 2007), particularly after prior cocaine experience, and overexpression of HDAC4 in the NAc reduces cocaine reward behavior and drug self-administration (Kumar et al. 2005; Wang et al. 2010), we predicted that genetic disruption of HDAC4 in the adult NAc might enhance behavioral responses to cocaine. However, we noted that Hdac4 cKONAc mice failed to condition to a near-threshold dose of cocaine (5 mg/kg), which suggests that endogenous HDAC4 increases the sensitivity to cocaine reward, albeit to a modest extent. Moreover, we detected reductions in both naïve and sensitized cocaine locomotor responses in Hdac4 cKONAc mice, suggesting that HDAC4 enhances the sensitivity to cocaine in multiple behavioral readouts.
Because repeated cocaine exposure promoted HDAC4 hyperphosphorylation and increased cytoplasmic localization, we examined the behavioral consequence of overexpression of a cytoplasmically accumulated HDAC4 mutant. We reasoned that cytoplasmic localization on HDAC4 might reflect the activation of gene expression and removal of its gene repressor functions in the cell nucleus. In this case, like expression of HDAC5 S279E (Taniguchi et al. 2012), we would predict that overexpression of HDAC4 S266E would have no effect on cocaine CPP because it is not retained in the nucleus and unable to repress transcription. However, if cocaine-induced relocalization of HDAC4 serves a cytoplasmic role, then overexpression of the HDAC4 S266E mutant might alter cocaine CPP. Interestingly, we observed that overexpression of HDAC4 in the cytoplasm actually enhanced cocaine reward behavior at a near-threshold conditioning experience. In the two 5 mg/kg conditioning experiments (Figs 3b & 4a), the behavior of the control groups is variable. We find that a number of variables contributed to the conditioning effect, particularly at a near-threshold dose, including experimenter sex, background strain, and surgical or other prior experience. In these experiment, animals differed in strain (Hdac4Fl/Fl versus C57Bl/6 J), housing experience (breed in colony versus ordered from vendor) and surgical experience (AAV infusion with 2–3 weeks of recovery versus HSV infusion with 2–3 days of recovery). Despite the difficulty in comparing the experiments directly, the concordance between the two experiments (loss of function reduces while overexpression enhances conditioning) supports the interpretation that cocaine-induced cytoplasmic localization of HDAC4 promotes cocaine responding.
The finding of a cytoplasmic role for HDAC4 is surprising, but not unprecedented. For example, previous work has found that cytoplasmically localized HDAC5 can enhance tubulin deacetylation (Cho & Cavalli 2012; Cho et al. 2013). In the future, it will be interesting to test whether cytoplasmic HDAC4 promotes cocaine CPP independent of its ability to regulate nuclear gene expression and/or whether the cytoplasmic mutant of HDAC4 functions as a dominant negative by associating with nuclear factors and titrating them away from the nucleus. Regarding the latter possibility, HDAC4 can interact with multiple nuclear proteins, including other HDACs, repressor complex members and transcription factors (Miska et al. 1999; Huang et al. 2000; Fischle et al. 2002; Backs et al. 2008) whose removal from the nucleus could promote cocaine reward. However, arguing against this possibility, we did not observe a significant repression or enhancement of MRE-luciferase activity of HDAC4 S266E on basal or depolarization-induced MEF2-dependent transcription (Fig. 4b).
Overall, our current findings indicate that the same cocaine regimen that promotes HDAC5 dephosphorylation and nuclear accumulation in striatum enhances HDAC4 phosphorylation and cytoplasmic localization. Our previous work suggested that cocaine and D1 dopamine receptor stimulation of cAMP signaling activates a PP2A phosphatase-dependent dephosphorylation of HDAC5, which promotes nuclear accumulation (Taniguchi et al. 2012). Because neither elevation of cAMP in striatal neurons nor cocaine administration produces HDAC4 dephosphorylation and nuclear accumulation, we speculate that HDAC4 is a poor substrate for PP2A or that there are competing kinase reactions, such as PKA, that enhance HDAC4 phosphorylation at S266. This differential regulation of HDAC4 and HDAC5 is possibly mediated by differential affinity for the numerous phosphatase and kinase cascades downstream of the coordinated glutamatergic and dopaminergic activity produced by cocaine exposure, and future research will be required to determine the underlying signaling mechanisms that differentially regulate these closely related HDACs in the striatum. Our behavioral findings also suggest distinct, and possibly opposite, roles for HDAC4 and HDAC5 in multiple cocaine behaviors. In the future, understanding the relevant signaling cascades and downstream target genes or cell processes that ultimately mediate HDAC4’s and HDAC5”s effects on cocaine behavior will be essential for understanding their roles in drug addiction-related behaviors.
Supplementary Material
A. Representative blot demonstrating immunoprecipitation specificity. Cultured striatal neuron lysate is independently immunoprecipitated using HDAC5 or HDAC4 antibodies. Immunoprecipitation specificaly isolates the HDAC of interest as demonstrated by the absence of the alternate HDAC. Blot is displayed at short and long exposure times to demonstrate effectiveness of he immunoprecipitation. B. Time spent in the unpaired chambers for all cocaine CPP experiments. Hdac4 cKONAc 5 mg/kg (FConditioning (1,29) = 10.1, p = 0.0035; Sidak’s post-hoc pre v post, GFP p < 0.01, Hdac4 cKONAc n.s.), Hdac4 cKONAc 10 mg/kg (FConditioning (1,29) = 43.1, p < 0.001; Sidak’spost-hoc pre v post, GFP p < 0.001, Hdac4 cKONAc p< 0.001), HDAC4 HSV 5 mg/kg (FConditioning (1,14) = 7.029, p = 0.019; Sidak’s post-hoc pre v post,GFP n.s., S266E p < 0.05)
Acknowledgements
The authors thank Dr. David Potter, Brandon Hughes, Yuhong Guo, Ben Zirlin and Emilia Pulver for technical assistance and Dr. Rachael Neve (Viral Core facility at MIT) for HSV-HDAC4 viruses. pcDNA-HDAC4.3SA-FLAG was a gift from Tso-Pang Yao (Addgene plasmid #30486). pcDNA-HDAC4-FLAG was a gift from Eric Verdin (Addgene plasmid #13821). This work is supported by F32DA036319 (to R.D.P.), F31DA035073 (to M.B.C.), the Fugaku Trust for Medicinal Research and a NARSAD Young Investigator Award (to M.T.) and R01DA027664 and R01DA032078 (to C.W.C.). Funding agencies played no role in the design or interpretation of the data.
Footnotes
SUPPORTING INFORMATION
Additional Supporting Information may be found online in the supporting information tab for this article.
References
- Backs J, Song K, Bezprozvannaya S, Chang S, Olson EN (2006) CaM kinase II selectively signals to histone deacetylase 4 during cardiomyocyte hypertrophy. J Clin Invest 116:1853–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backs J, Backs T, Bezprozvannaya S, McKinsey TA, Olson EN (2008) Histone deacetylase 5 acquires calcium/calmodulin-dependent kinase II responsiveness by oligomerization with histone deacetylase 4. Mol Cell Biol 28:3437–3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belfield JL, Whittaker C, Cader MZ, Chawla S (2006) Differential effects of Ca2+ and cAMP on transcription mediated by MEF2D and cAMP-response element-binding protein in hippocampal neurons. J Biochem 281:27724–27732. [DOI] [PubMed] [Google Scholar]
- Bertos NR, Wang AH, Yang XJ (2001) Class II histone deacetylases: structure, function, and regulation. Biochem Cell Biol 79:243–252. [PubMed] [Google Scholar]
- Bolger TA, Yao T-P (2005) Intracellular trafficking of histone deacetylase 4 regulates neuronal cell death. J Neurosci 25:9544–9553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang S, Bezprozvannaya S, Li S, Olson EN (2005) An expression screen reveals modulators of class II histone deacetylase phosphorylation. Proc Natl Acad Sci U S A 102:8120–8125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawla S, Vanhoutte P, Arnold FJL, Huang CL-H, Bading H (2003) Neuronal activity-dependent nucleocytoplasmic shuttling of HDAC4 and HDAC5. J Neurochem 85:151–159. [DOI] [PubMed] [Google Scholar]
- Cho Y, Cavalli V (2012) HDAC5 is a novel injury-regulated tubulin deacetylase controlling axon regeneration. EMBO J 31:3063–3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho Y, Sloutsky R, Naegle KM, Cavalli V (2013) Injury-induced HDAC5 nuclear export is essential for axon regeneration. Cell 155:894–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du M, Perry RLS, Nowacki NB, Gordon JW, Salma J, Zhao J, Aziz A, Chan J, Siu KWM, McDermott JC (2008) Protein kinase A represses skeletal myogenesis by targeting myocyte enhancer factor 2D. Mol Cell Biol 28:2952–2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischle W, Dequiedt F, Hendzel MJ, Guenther MG, Lazar MA, Voelter W, Verdin E (2002) Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol Cell 9:45–57. [DOI] [PubMed] [Google Scholar]
- Flavell SW, Cowan CW, Kim T-K, Greer PL, Lin Y, Paradis S, Griffith EC, Hu LS, Chen C, Greenberg ME (2006) Activity-dependent regulation of MEF2 transcription factors suppresses excitatory synapse number. Science 311:1008–1012. [DOI] [PubMed] [Google Scholar]
- Grozinger CM, Schreiber SL (2000) Regulation of histone deacetylase 4 and 5 and transcriptional activity by 14–3-3-dependent cellular localization. Proc Natl Acad Sci U S A 97:7835–7840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha CH, Kim JY, Zhao J, Wang W, Jhun BS, Wong C, Jin ZG (2010) PKA phosphorylates histone deacetylase 5 and prevents its nuclear export, leading to the inhibition of gene transcription and cardiomyocyte hypertrophy. Proc Natl Acad Sci 107:15467–15472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang EY, Zhang J, Miska EA, Guenther MG, Kouzarides T, Lazar MA (2000) Nuclear receptor corepressors partner with class II histone deacetylases in a Sin3-independent repression pathway. Genes Dev 14:45–54. [PMC free article] [PubMed] [Google Scholar]
- Hyman SE, Malenka RC, Nestler EJ (2006) Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci 29:565–598. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Langley B, Lubin FD, Renthal W, Wood MA, Yasui DH, Kumar A, Nestler EJ, Akbarian S, Beckel-Mitchener AC (2008) Epigenetics in the nervous system. J Neurosci 28:11753–11759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, Akhtar MW, Adachi M, Mahgoub M, Bassel-Duby R, Kavalali ET, Olson EN, Monteggia LM (2012) An essential role for histone deacetylase 4 in synaptic plasticity and memory formation. J Neurosci 32:10879–10886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T (1999) Histone acetylases and deacetylases in cell proliferation. Curr Opin Genet Dev 9:40–48. [DOI] [PubMed] [Google Scholar]
- Kozhemyakina E, Cohen T, Yao T-P, Lassar AB (2009) Parathyroid hormone-related peptide represses chondrocyte hypertrophy through a protein phosphatase 2A/histone deacetylase 4/MEF2 pathway. Mol Cell Biol 29:5751–5762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Choi K-H, Renthal W, Tsankova NM, Theobald DEH, Truong H- T, Russo SJ, LaPlant Q, Sasaki TS, Whistler KN, Neve RL, Self DW, Nestler EJ (2005) Chromatin remodeling is a key mechanism underlying cocaine-induced plasticity in striatum. Neuron 48:303–314. [DOI] [PubMed] [Google Scholar]
- Liu Y, Schneider MF (2013) Opposing HDAC4 nuclear fluxes due to phosphorylation by β adrenergic activated PKA or by activity or Epac activated CaMKII in skeletal muscle fibres. J Physiol (Lond). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Hernandez-Ochoa EO, Randall WR, Schneider MF (2012) AJP: Cell Physiology 303:C334–C347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinsey TA, Zhang CL, Lu J, Olson EN (2000a) Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature 408:106–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinsey TA, Zhang CL, Olson EN (2000b) Activation of the myocyte enhancer factor-2 transcription factor by calcium/calmodulin-dependent protein kinase-stimulated binding of 14–3-3 to histone deacetylase 5. Proc Natl Acad Sci U S A 97:14400–14405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinsey TA, Zhang CL, Olson EN (2001) Identification of a signal-responsive nuclear export sequence in class II histone deacetylases. Mol Cell Biol 21:6312–6321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuown SC, Barrett RM, Matheos DP, Post RJ, Rogge GA, Alenghat T, Mullican SE, Jones S, Rusche JR, Lazar MA, Wood MA (2011) HDAC3 is a critical negative regulator of long-term memory formation. J Neurosci 31:764–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Métrich M, Laurent A-C, Breckler M, Duquesnes N, Hmitou I, Courillau D, Blondeau J- P, Crozatier B, Lezoualc’h F, Morel E (2010) Epac activation induces histone deacetylase nuclear export via a Ras-dependent signalling pathway. Cell Signal 22:1459–1468. [DOI] [PubMed] [Google Scholar]
- Mihaylova MM, Vasquez DS, Ravnskjaer K, Denechaud P-D, Yu RT, Alvarez JG, Downes M, Evans RM, Montminy M, Shaw RJ (2011) Class IIa histone deacetylases are hormone-activated regulators of FOXO and mammalian glucose homeostasis. Cell 145:607–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miska EA, Karlsson C, Langley E, Nielsen SJ, Pines J, Kouzarides T (1999) HDAC4 deacetylase associates with and represses the MEF2 transcription factor. EMBO J 18:5099–5107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neve RL, Lim F (2013) Generation of high-titer defective HSV-1 vectors. Curr Protoc Neurosci Chapter 4:Unit4.13. [DOI] [PubMed] [Google Scholar]
- Paroni G, Cernotta N, Russo CD, Gallinari P, Pallaoro M, Foti C, Talamo F, Orsatti L, Steinkühler C, Brancolini C (2008) PP2A regulates HDAC4 nuclear import. Mol Biol Cell 19:655–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra M (2015) Class IIa HDACs—new insights into their functions in physiology and pathology. FEBS J 282:1736–1744. [DOI] [PubMed] [Google Scholar]
- Parra M, Verdin E (2010) Regulatory signal transduction pathways for class IIa histone deacetylases. Curr Opin Pharmacol 10:454–460. [DOI] [PubMed] [Google Scholar]
- Penrod RD, Wells AM, Carlezon WA, Cowan CW (2015) Use of adeno-associated and herpes simplex viral vectors for in vivo neuronal expression in mice. Curr Protoc Neurosci 73:4.37.1–4.37.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pliakas AM, Carlson RR, Neve RL, Konradi C, Nestler EJ, Carlezon WA (2001) Altered responsiveness to cocaine and increased immobility in the forced swim test associated with elevated cAMP response element-binding protein expression in nucleus accumbens. J Neurosci 21:7397–7403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potthoff MJ, Olson EN (2007) MEF2: a central regulator of diverse developmental programs. Development 134: 4131–4140. [DOI] [PubMed] [Google Scholar]
- Potthoff MJ, Wu H, Arnold MA, Shelton JM, Backs J, McAnally J, Richardson JA, Bassel-Duby R, Olson EN (2007) Histone deacetylase degradation and MEF2 activation promote the formation of slow-twitch myofibers. J Clin Invest 117:2459–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulipparacharuvil S, Renthal W, Hale CF, Taniguchi M, Xiao G, Kumar A, Russo SJ, Sikder D, Dewey CM, Davis MM, Greengard P Nairn AC, Nestler EJ, Cowan CW (2008) Cocaine regulates MEF2 to control synaptic and behavioral plasticity. Neuron 59:621–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renthal W, Nestler EJ (2009) Histone acetylation in drug addiction. Semin Cell Dev Biol 20:387–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renthal W, Maze I, Krishnan V, Covington HE, Xiao G, Kumar A, Russo SJ, Graham A, Tsankova N, Kippin TE, Kerstetter KA, Neve RL, Haggarty SJ, McKinsey TA, Bassel-Duby R, Olson EN, Nestler EJ (2007) Histone deacetylase 5 epigenetically controls behavioral adaptations to chronic emotional stimuli. Neuron 56:517–529. [DOI] [PubMed] [Google Scholar]
- Robison AJ, Nestler EJ (2011) Transcriptional and epigenetic mechanisms of addiction. Nat Rev Neurosci 12:623–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlumm F, Mauceri D, Freitag HE, Bading H (2013) Nuclear calcium signaling regulates nuclear export of a subset of class IIa histone deacetylases following synaptic activity. J Biochem 288:8074–8084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith LN, Penrod RD, Taniguchi M, Cowan CW (2016) Assessment of cocaine-induced behavioral sensitization and conditioned place preference in mice.J Vis Exp . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi M, Carreira MB, Smith LN, Zirlin BC, Neve RL, Cowan CW (2012) Histone deacetylase 5 limits cocaine reward through cAMP-induced nuclear import. Neuron 73:108–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsankova NM, Berton O, Renthal W, Kumar A, Neve RL, Nestler EJ (2006) Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci 9:519–525. [DOI] [PubMed] [Google Scholar]
- Vega RB, Matsuda K, Oh J, Barbosa AC, Yang X, Meadows E, McAnally J, Pomajzl C, Shelton JM, Richardson JA, Karsenty G, Olson EN (2004) Histone deacetylase 4 controls chondrocyte hypertrophy during skeletogenesis. Cell 119:555–566. [DOI] [PubMed] [Google Scholar]
- Walkinshaw DR, Weist R, Xiao L, Yan K, Kim GW, Yang XJ (2013) Dephosphorylation at a conserved SP motif governs cAMP sensitivity and nuclear localization of class IIa histone deacetylases. J Biochem 288:5591–5605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang AH, Kruhlak MJ, Wu J, Bertos NR, Vezmar M, Posner BI, Bazett-Jones DP, Yang YJ (2000) Regulation of histone deacetylase 4 by binding of 14–3-3 proteins. Mol Cell Biol 20:6904–6912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Lv Z, Hu Z, Sheng J, Hui B, Sun J, Ma L (2010) Chronic cocaine-induced H3 acetylation and transcriptional activation of CaMKIIalpha in the nucleus accumbens is critical for motivation for drug reinforcement. Neuropsychopharmacology 35:913–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Kohlhaas M, Backs J, Mishra S, Phillips W, Dybkova N, Chang S, Ling H, Bers DM, Maier LS, Olson EN, Brown JH (2007) CaMKIIdelta isoforms differentially affect calcium handling but similarly regulate HDAC/MEF2 transcriptional responses. J Biochem 282:35078–35087. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A. Representative blot demonstrating immunoprecipitation specificity. Cultured striatal neuron lysate is independently immunoprecipitated using HDAC5 or HDAC4 antibodies. Immunoprecipitation specificaly isolates the HDAC of interest as demonstrated by the absence of the alternate HDAC. Blot is displayed at short and long exposure times to demonstrate effectiveness of he immunoprecipitation. B. Time spent in the unpaired chambers for all cocaine CPP experiments. Hdac4 cKONAc 5 mg/kg (FConditioning (1,29) = 10.1, p = 0.0035; Sidak’s post-hoc pre v post, GFP p < 0.01, Hdac4 cKONAc n.s.), Hdac4 cKONAc 10 mg/kg (FConditioning (1,29) = 43.1, p < 0.001; Sidak’spost-hoc pre v post, GFP p < 0.001, Hdac4 cKONAc p< 0.001), HDAC4 HSV 5 mg/kg (FConditioning (1,14) = 7.029, p = 0.019; Sidak’s post-hoc pre v post,GFP n.s., S266E p < 0.05)