

Abstract
Accidental or intentional misuse of acetaminophen (APAP) is the leading cause of acute liver failure in the Western world. Although mechanisms that trigger APAP-induced liver injury (AILI) are well known, those that halt the progression of APAP liver disease and facilitate liver recovery are less understood. Heparan sulfate proteoglycans (HSPGs) bind to and regulate various tissue injury factors through their heparan sulfate (HS) chains, but the importance of HSPGs in liver injury in vivo remains unknown. Here, we examined the role of syndecan-1, the major cell-surface HSPG of hepatocytes, in AILI. Ablation of syndecan-1 in mice led to unopposed progression of liver injury upon APAP overdose. However, direct APAP hepatoxicity and liver injury at early times post-APAP overdose were unaffected by syndecan-1, suggesting that syndecan-1 influences later mechanisms that lead to liver repair. The exuberant liver injury phenotypes in syndecan-1 null (Sdc1–/–) mice were traced to a deficiency in protein kinase B (Akt) activation in hepatocytes, which led to a delayed increase in glycogen synthase kinase-3β (GSK-3β)-mediated hepatocyte apoptosis. Inhibition of Akt worsened, whereas inhibition of GSK-3β and caspases protected mice from AILI. Moreover, administration of purified syndecan-1, HS, or engineered heparan compounds containing 2-O-sulfate groups rescued Sdc1–/– mice from AILI by potentiating Akt signaling and inhibiting GSK-3β-mediated apoptosis in hepatocytes. In addition, HS showed a significantly prolonged therapeutic efficacy as compared to N-acetylcysteine.
Conclusion:
These results demonstrate that 2-O-sulfated domains in syndecan-1 HS halt disease progression and promote liver repair by enhancing hepatocyte survival in AILI. We propose that syndecan-1 is a critical endogenous factor that controls the balance between prosurvival signaling and apoptosis in hepatocytes in APAP liver disease.
Heparan sulfate (HS) regulates many factors implicated in tissue injury, such as growth factors, cytokines, and matrix components(1,2) HS binds specifically to these molecules and regulates their function by affecting their activity, stability, conformation, or destination. These observations suggest that HS is an important modulator of tissue injury, but precisely how HS regulates injury and repair mechanisms in vivo remains ill defined. HS is ubiquitously expressed inside, on, and in the surrounding environment of all adherent cells. HS in vivo is found covalently conjugated to specific core proteins as HS proteoglycans (HSPGs).(1) HS is comprised of un-branched, repeating disaccharide units of hexuronic acid, either glucuronic acid (GlcA) or iduronic acid (IdoA), alternating with an unsubstituted or N-substituted glucosamine on which the substituents are either acetate (GlcNAc) or sulfate (GlcNS). In HS biosynthesis, a nonsulfated HS precursor is polymerized on specific serine residues of HSPG core proteins and then extensively modified in the Golgi by N-deacetylase N-sulfotransferases, C5 epimerase, 2-O-sulfotransferase (2OST), 6OSTs, and 3OSTs.(3) These reactions proceed largely in a sequential manner, but do not go to completion, resulting in highly heterogeneous mature HS chains. This unique and complex modification pattern of HS enables HS to interact with many molecules and regulate diverse biological processes(4) but the contribution of specific HS modifications in biological processes in vivo has yet to be clearly determined.
The syndecan family of type I transmembrane cell-surface HSPGs, comprised of four members in mammals (syndecan-1 through-4), is the major source of HS.(1) Syndecans function primarily as receptors for HS-binding molecules at the cell surface and as soluble modulators of molecular interactions in the extracellular environment when released from the cell surface by ectodomain shedding. Surprisingly, mice lacking syndecan-1 or −4 do not show major developmental abnormalities, but do show striking pathological phenotypes when injured or infected.(1,2,5) suggesting that certain postdevelopmental functions of syndecans are specific and cannot be compensated for by other syndecans or other HSPGs. Reinforcing this view, accumulating evidence suggests that syndecan-1 is an important modulator of both infectious and sterile tissue injury.(1,5) In humans, syndecan-1 ectodomains are elevated in blood of patients with sepsis, hemorrhagic shock, and ischemia-reperfusion injury, among other fluids from infected or injured tissues.(5) In mice, ablation of syndecan-1 leads to drastically altered responses to infectious or inflammatory stimuli.(5) Syndecan-1 null (Sdc1–/–) mice show significantly increased tissue damage in several models of sterile inflammation(6–8) but are markedly protected from bacterial infection in the lung, skin, and cornea.(5,9,10) These results suggest that syndecan-1 is an important factor that orchestrates tissue injury responses, but that certain pathogens have either adapted or evolved to subvert these functions of syndecan-1 to promote their survival.
Syndecan-1 is expressed abundantly in the liver on the sinusoidal surface of hepatocytes,(7,8,11) suggesting that syndecan-1 plays an important role in hepatic responses to agents that are metabolized by the liver and those that cause liver disease. Indeed, syndecan-1 mediates the hepatic clearance of triglyceride-rich lipoproteins(11) and is up-regulated in hepatocytes infected with hepatitis C virus.(12) Acetaminophen (APAP) is a widely used analgesic that is considered safe at therapeutic doses, but provokes various degrees of liver damage, which can lead to liver failure and even death, when ingested at supratherapeutic doses. In fact, APAP overdose is the leading cause of acute liver failure (ALF) of any etiology in the Western world. In the United States alone, APAP overdose accounts for more than 56,000 emergency room visits and 2,600 hospitalizations annually and carries a 26% mortality rate without liver transplantation.(13–15) Although it is well known that the generation of N-acetyl-p-benzoquinone imine (NAPQI), the hepatotoxic metabolite of APAP, instigates liver injury,(16) the biological programs that result in disease progression and perpetuation of liver injury versus healing are less understood. Here, we show that syndecan-1 is a critical endogenous factor that protects against APAP liver disease by promoting hepatocyte survival and liver repair.
Materials and Methods
A detailed description of reagents is provided in the Supporting Information.
MOUSE MODEL OF APAP-AND CONCANAVALIN A-INDUCED LIVER INJURY
Wild-type (Wt) littermates and Sdc1–/– mice on the C57BL/6J background were used (6- to 8-week-old females and males). Unchallenged Sdc1–/– mice are fertile and healthy with normal tissue morphology, complete blood counts, and serum chemistry parameters(7,8) Mice were housed in microisolator cages in a specific pathogen-free facility accredited by the American Association for Laboratory and Animal Care in a 12-hour light/dark cycle and fed a basal rodent chow ad libitum. All animal experiments were approved by the Institutional Animal Care and Use Committee of Boston Children’s Hospital (Boston, MA) and complied with U.S. federal Guidelines for the Care and Use of Laboratory Animals. For Concanavalin A (ConA)-induced liver injury, mice were injected intravenously with 25 mg/kg of ConA. At 24 hours postinjection, mice were euthanized, blood was collected, and serum levels of alanine aminotransferase (ALT) and aspartase aminotransferase (AST) were measured using an automated Roche Cobas 6000 serum chemistry analyzer (Department of Laboratory Medicine, Boston Children’s Hospital). For APAP-induced liver injury, mice were fasted overnight (12–15 hours) to reduce hepatic glutathione (GSH) levels before APAP treatment. APAP was dissolved in warm (55°C) pyrogen-free phosphate-buffered saline (PBS) immediately before each experiment, cooled to 37°C, and injected intraperitoneally at 500 or 625 mg/kg. In some experiments, mice were injected intraperitoneally with 0.5 mg/kg of purified syndecan-1 ectodomain, HS, heparin, or modified heparan compounds, 200 mg/kg of lithium, 1 mg/ kg of triciribine, 25 mg/kg of Ac-DEVD-CMK, or 50 mg/kg of N-acetylcysteine (NAC) before or after APAP administration as indicated. Doses were determined as those minimally required in our preliminary experiments. All reagents were prepared in PBS except triciribine and Ac-DEVD-CMK, which were dissolved in ethanol and diluted to the indicated dose in PBS. At the indicated times, mice were euthanized and serum levels of ALT and AST were measured. Liver lobes were also isolated and processed for immunoblotting, cell-based assays, fluorescence-activated cell sorting (FACS), and histopathological and immunohistochemical (IHC) analyses as described.
CELL-BASED ASSAYS
Primary Wt and Sdc1–/– hepatocytes were isolated from perfused livers by collagenase digestion.(11) Briefly, mice were cannulated through the portal vein and after nicking the inferior vena cava, perfused with 20 mL of Hank’s balanced salt solution (HBSS) containing 5 mM of ethylenediaminetetraacetic acid followed by 20 mL of HBSS containing 1 mg/mL of collagenase and 1 mM of CaCl2. Livers were isolated, torn, shaken loose in hepatocyte culture medium (Dulbecco’s modified Eagle’s medium with 25 mM of glucose, 20% fetal bovine serum, 100 IU/mL of penicillin G, and 100 μg/ mL of streptomycin), and filtered through a 70-μm cell strainer. Cells were washed once with culture medium, overlaid with 70% Percoll, and centrifuged at 800g for 10 minutes. Hepatocyte pellets were collected, washed, and counted, and viability was determined by trypan blue. Hepatocyte preparations with a viability >90% were plated onto type I collagen precoated 24-well plates at 5 × 105 cells/well. The culture medium was changed at 6 hours postplating to remove detached hepatocytes and cultured for an additional 18 hours before use. Primary hepatocytes were incubated with 40 mM of APAP in hepatocyte culture medium, and cell viability was measured with the nonradioactive cytotoxicity assay kit at the indicated times. For measurement of GSH consumption, primary hepatocytes plated on six-well plates precoated with type I collagen were incubated with 40 mM of APAP for 0–24 hours, washed, trypsinized, and transferred to a microfoge tube. Trypsinized cells were counted, centrifuged, lysed by freeze-thawing, and the GSH concentration in cell lysates was determined with the GSH assay kit.
HISTOPATHOLOGICAL ANALYSES
Isolated livers were fixed in 4% paraformaldehyde/ PBS for 2 days at 4°C, paraffin-embedded, and sectioned. Liver sections (5 μm) were stained with hematoxylin-eosin (H& E) or immunostained with anticleaved caspase-3, anti-phospho-GSK-3β (glycogen synthase kinase-3β) or anti-phospho-Akt (protein kinase B) and Alexa 594 secondary antibodies. Brightfield images were captured using the Zeiss Axio Imager Z1 upright microscope (Carl Zeiss Microscopy, Peabody, MA, USA), and fluorescence images were captured using the Apotome 3-D optical sectioning technology.
STATISTICAL ANALYSIS
All data are expressed as mean ± SEM. Statistical significance between the experimental and control groups was analyzed by two-tailed unpaired Student t test, between multiple groups by one-way analysis of variance (ANOVA) followed by Dunnett’s post-hoc test, and Kaplan-Meier survival curves by log-rank test using GraphPad Prism software (version 5.0b; Graph-Pad Software Inc.), where P values <0.05 were determined to be significant.
Results
APAP CAUSES SEVERE LIVER INJURY IN SYNDECAN-1-DEFICIENT MICE
We initially examined the hypothesis that if syndecan-1 is indeed important in the pathogenesis of liver injury, then hosts without syndecan-1 will respond differently to agents that cause liver damage, such as APAP and ConA. The hepatotoxic effects of APAP are primarily attributed to the inactivation of mitochondrial and cytoplasmic proteins by NAPQI conjugation and NAPQI-mediated depletion of cellular GSH,(17) whereas ConA causes hepatic damage through membrane-bound tumor necrosis factor alpha (TNFα) on T cells.(18) Wt and Sdc1–/– mice were injected intraperitoneally with 500 mg/kg of APAP or injected intravenously with 25 mg/kg of ConA, and liver injury was assessed by measuring serum levels of ALT and AST and by liver histopathology at 24 hours postinjection. ALT and AST levels were significantly increased by approximately 4- and 5-fold, respectively, in Sdc1–/– mice injected with APAP compared to Wt mice treated identically (Fig. 1A). Histological evaluation revealed that necrosis emanating from the central veins was more severe and widespread in Sdc1–/– livers compared to Wt livers (Fig. 1B). At a higher dose of APAP (625 mg/kg), Sdc1–/– mice also showed significantly elevated serum transaminase levels (Supporting Fig. S1) and increased mortality compared to Wt mice (Fig. 1C). However, liver damage was similar in Wt and Sdc1–/– mice administered Con A, and even slightly reduced in Sdc1–/– mice (Fig. 1A), suggesting that syndecan-1 does not affect hepatic damage caused by membrane-bound TNFα. These observations suggest that syndecan-1 prominently protects against APAP-induced liver injury (AILI).
FIG. 1.

Sdc1–/–mice show increased susceptibility to AILI. (A) Wt and Sdc1–/–mice were injected intraperitoneally with APAP (500 mg/kg) or injected intravenously with Con A (25 mg/kg), and serum levels of ALT and AST were measured at 24 hours post-injection. Data shown are mean ± SEM (APAP group: n = 23 for Wt, n = 17 for Sdc1–/–; Con A group: n = 5 for Wt, n = 7 for Sdc1–/–; *P values vs. Wt group, Student t test). (B) Representative H&E-stained liver sections of mice at 24 hours post-APAP (500 mg/kg; original magnification, ×l00). (C) Survival curve of Wt and Sdc1–/– mice injected with 625 mg/kg of APAP (*P vs. Wt, log-rank test).
SYNDECAN-1 ATTENUATES APAP-INDUCED LIVER INJURY IN AN HS-DEPENDENT MANNER
To pursue how syndecan-1 protects against APAP liver disease, we first examined whether administration of purified syndecan-1 could rescue Sdc1–/– mice from severe liver injury and whether the effect is mediated by a specific structural component of syndecan-1. Sdc1–/– mice were pretreated with purified syndecan-1 ectodomain, HS, chondroitin sulfate (CS), or syndecan-1 core protein lacking both HS and CS, and liver damage was assessed at 24 hours post-APAP. Serum ALT levels were significantly reduced by 99% and 86% in mice given syndecan-1 ectodomain or HS, respectively, compared to control mice that were given APAP only, whereas CS and core protein virtually had no protective effect (Fig. 2A). Although some centrilobular necrosis was still evident in livers of mice given syndecan-1 ectodomain or HS, the necrotic area was substantially smaller compared to mice given APAP only or APAP and CS (Fig. 2B). These results establish that HS chains of syndecan-1 protect against AILI.
FIG. 2.
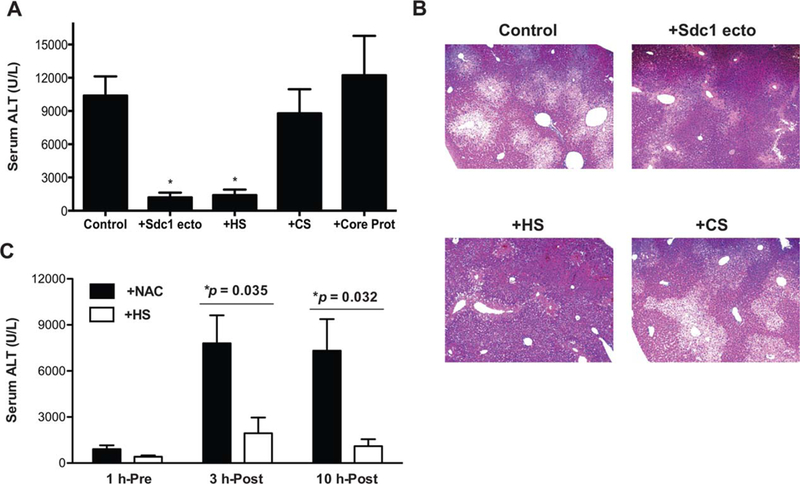
Exogenous syndecan-1 rescues Sdc1–/– mice from APAP-induced liver damage in an HS-dependent manner. (A) Sdc1–/– mice were injected intraperitoneally with PBS (control) or 0.5 mg/kg of purified syndecan-1 ectodomain, HS, CS, or core protein (Core Prot) 1 hour pre-APAP injection (500 mg/kg), and serum ALT levels were measured at 24 hours post-APAP (mean ± SEM; n = 8 for all groups; *P < 0.05, one-way ANOVA followed by Dunnett’s test). (B) Representative H&E-stained liver sections from APAP+PBS (control), APAP+Sdc1 ecto, APAP+HS, and APAP+CS groups (original magnification, X100). (C) Sdc1–/– mice were injected with 0.5 mg/kg of HS or 50 mg/kg of NAC at 1 hour before, 3 hours after, or 10 hours after APAP, and serum ALT levels were determined at 24 hours post-APAP (mean ± SEM; n = 7 in all groups; *P values between NAC and HS groups).
We next explored the therapeutic effects of HS in AILI by comparing its effects with those of N-acetyl-cysteine (NAC), a clinical antidote for APAP overdose.(19) However, NAC has a limited time window of efficacy, and delayed or prolonged treatment can have a negative impact on the clinical outcome.(20) For instance, the mortality rate can be increased up to 15-fold in patients who receive NAC >12 hours post-APAP ingestion.(21) Furthermore, unintentional APAP overdose accounts for nearly half of the cases in the United States and these patients tend to be treated late with NAC, leading to greater morbidity and mortality.(15) NAC effectively reduced ALT levels in Sdc1–/– mice when given 1 hour pre-APAP, but not when administered 3 or 10 hours post-APAP (Fig. 2C), consistent with its limited window of efficacy. On the other hand, HS significantly reduced ALT levels not only when mice were pretreated, but also when given at 3 or 10 hours post-APAP (Fig. 2C). These results indicate that HS has a significantly prolonged therapeutic efficacy, as compared to NAC, and that HS attenuates AILI through a mechanism distinct from that of NAC. Furthermore, these findings suggest that syndecan-1 and HS, either alone or in combination with NAC, could provide a new therapeutic strategy to mitigate AILI, especially in treating patients admitted after NAC treatment is no longer effective.
2-O-SULFATED DOMAINS IN HS ATTENUATE AILI
Having demonstrated that HS chains of syndecan-1 protect against AILI, we next investigated the impact of HS sulfation because sulfate modifications are thought to govern HS functions.(22) We first tested the effects of chemically desulfated heparin compounds, each selectively lacking sulfate groups at the N-, 2-O-, and 6-O-positions. As expected, unmodified heparin, which is a structural and functional pharmaceutical analog of HS, significantly reduced APAP-induced liver damage as assessed by serum ALT (Fig. 3A) and histological analyses (Fig. 3C). However, whereas N-and 6-O-desulfated heparin compounds were similarly protective as unmodified heparin, 2-O-desulfated heparin did not mitigate AILI (Fig. 3A,C).
FIG. 3.
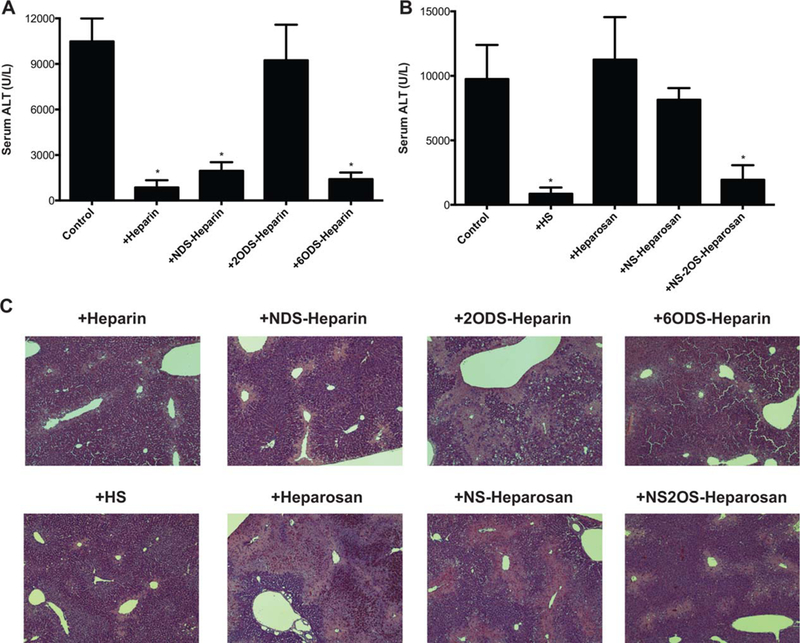
2-O-sulfated heparan compounds inhibit AILI. (A) Sdc1–/–mice were injected intraperitoneally with PBS (control) or 0.5 mg/kg of heparin, N-desulfated heparin (NDS-heparin), 2-O-desulfated heparin (2ODS-heparin), or 6-O-desulfated heparin (6ODS-heparin) 1 hour pre-APAP injection (500 mg/kg), and serum ALT was determined at 24 hours post-APAP. Data shown are mean ± SEM (n = 5 for PBS and heparin groups, n = 6 for all modified heparin groups; *P < 0.05, one-way ANOVA followed by Dunnett’s test). (B) Sdc1–/– mice were injected intraperitoneally with PBS (control) or 0.5 mg/kg of HS, heparosan, N-sulfated heparosan (NS-heparosan), or N- and 2-O-sulfated heparosan (NS2OS-heparosan) 1 hour pre-APAP injection, and serum ALT was measured at 24 hours post-APAP (mean ± SEM; n = 6 for all groups; *P < 0.05, ANOVA followed by Dunnett’s test). (C) Representative H&E-stained liver sections at 24 hours post-APAP from mice treated without or with heparin, HS, or modified heparan compounds (original magnification, ×100).
To confirm the results from the chemically desulfated heparin studies, we next examined the effects of engineered heparosan on AILI. Unmodified heparosan is a capsular polysaccharide from Escherichia coli that has an identical structure to unmodified HS. Two different sulfated heparosan derivatives, N-sulfated and N- and 2-O-sulfated heparosan, were synthesized for this study using a chemoenzymatic approach.(23) Administration of heparosan or N-sulfated heparosan did not reduce ALT levels (Fig. 3B) or liver necrosis (Fig. 3C) in Sdc1–/– mice subjected to APAP overdose. However, similar to HS, N- and 2-O-sulfated heparosan significantly reduced ALT levels (Fig. 3B) and markedly diminished liver necrosis (Fig. 3C). These data indicate that 2-O-sulfated domains are critical in the ability of HS to suppress AILI.
SYNDECAN-1 HALTS THE PROGRESSION OF APAP LIVER DISEASE
To determine how syndecan-1 inhibits AILI, we next assessed whether syndecan-1 ablation affects hepatic GSH depletion as a measure of excess NAPQI formation and activity. The rate and extent of GSH depletion and resynthesis were similar in Wt and Sdc1–/– livers, with maximal depletion by 4 hours post-APAP and almost complete repletion by 12 hours post-APAP (Fig. 4A). Furthermore, isolated primary Wt and Sdc1–/– hepatocytes were similarly susceptible to APAP-induced cell death (Fig. 4B), and GSH consumption in primary Wt and Sdc1–/– hepatocytes incubated with APAP was similar (Fig. 4C). These data indicate that syndecan-1 does not affect the formation and toxicity of NAPQI or its inactivation by GSH.
FIG. 4.
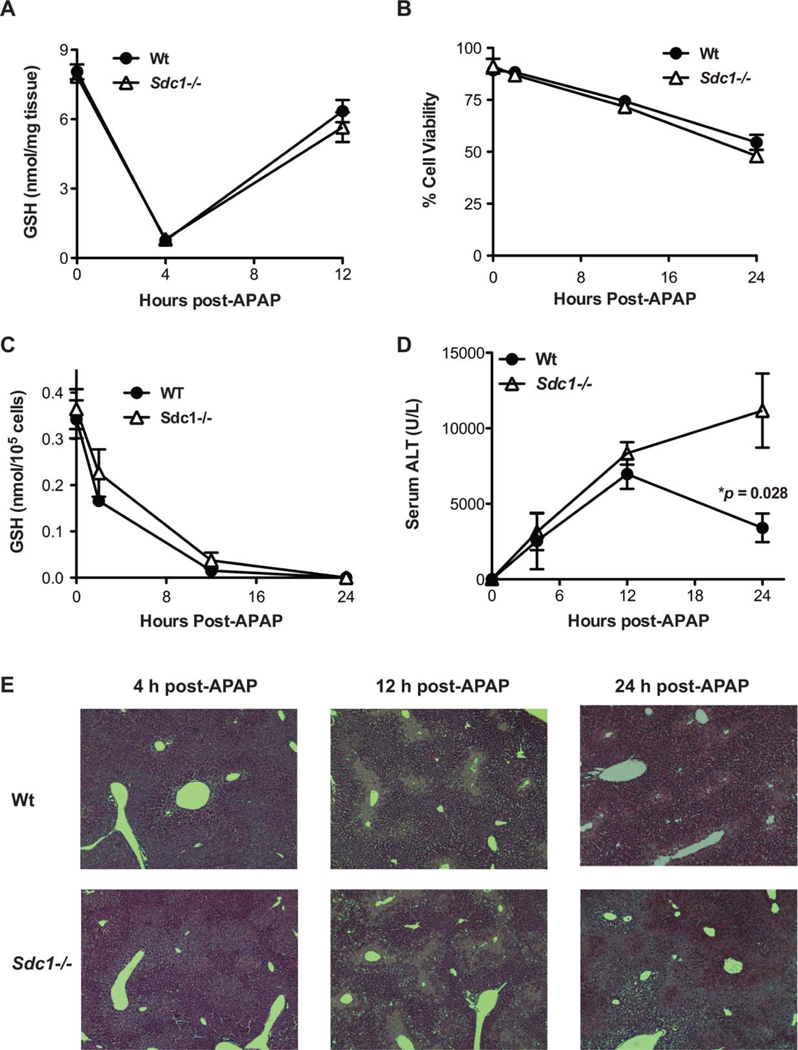
Syndecan-1 deficiency does not affect direct APAP hepatotoxicity, but interferes with hepatic repair in AILI. (A) Wt and Sdc1–/– mice were injected with APAP (500 mg/kg) and total liver GSH levels were measured at the indicated times (mean ± SEM; n = 4 in both Wt and Sdc1–/– groups). (B) Primary hepatocytes isolated from Wt or Sdc1–/– livers were incubated with 40 mM of APAP and cell viability was determined (n = 4). (C) Wt and Sdc1–/– primary hepatocytes were incubated with 40 mM of APAP and GSH levels were measured (n = 4). (D) Serum ALT levels in Wt and Sdc1–/– mice were measured at the indicated times post-APAP injection (n = 5; *P between Wt and Sdc1–/– at 24 hours post-APAP). (E) Representative H&E-stained Wt and Sdc1–/– liver sections at the indicated times post-APAP (original magnification, ×100).
We next evaluated the time course of liver injury by APAP. The pathogenesis of AILI in mice closely resembles that of the human disease, except that peak toxicity is observed earlier in mice (6–12 hours) than in humans (24–48 hours).(17) In mice, NAPQI formation and GSH depletion begin immediately post-APAP dosing and continue for 3–4 hours.(24) Hepatocyte necrosis begins during this time and continues until around 12 hours post-APAP overdose, around which time a tissue repair response starts and continues until approximately 72 hours post-APAP.(24) ALT levels similarly increased in Wt and Sdc1–/– mice up to 12 hours post-APAP, at which time ALT levels started to decline in Wt mice (Fig. 4D). In contrast, ALT levels continued to increase and high levels were sustained in Sdc1–/– mice at 24 hours post-APAP (Fig. 4D). Histological analyses also showed a similarly progressive liver injury up to 12 hours post-APAP in both backgrounds, but markedly worsened injury in Sdc1–/– livers, characterized by hepatocyte necrosis and inflammatory cell infiltrates surrounding centrilobular regions, was observed at 24 hours post-APAP (Fig. 4E). These results indicate that syndecan-1 does not play a critical role in the onset of AILI. Instead, these data suggest that syndecan-1 protects against APAP-induced liver disease by halting the progression of liver injury and facilitating liver repair. Consistent with this idea, proliferating cell nuclear antigen expression, a marker of cell proliferation, was markedly reduced in Sdc1–/– livers compared to Wt livers at 12 and 24 hours post-APAP (Supporting Fig. S2). Furthermore, in preliminary studies examining the role of syndecan-1 in furosemide-induced liver injury where significant liver toxicity is observed by 5 hours after furosemide injection, but liver damage is most severe at 24 hours after furosemide,(25,26) ALT levels were significantly increased by 4-fold in Sdc1–/– mice injected with furosemide compared to Wt mice treated identically (Supporting Fig. S3). These results suggest that syndecan-1 may also halt the perpetuation of liver injury in acute liver diseases other than AILI.
SYNDECAN-1 DEFICIENCY INHIBITS Akt ACTIVATION AND ENHANCES GSK-3β-MEDIATED HEPATOCYTE APOPTOSIS IN APAP LIVER DISEASE
Tissue repair is strongly regulated by growth factors. Removal of growth factors or inhibition of signaling can result in cell death and progression of tissue damage.(27) Activation of phosphoinositide 3-kinase-Akt signaling by growth factors is an important pathway that facilitates tissue repair by enhancing cell proliferation and survival. Akt stimulation by growth factors facilitates liver regeneration,(28) and cell-surface HSPGs serve as coreceptors for various heparinbinding growth factors that promote tissue repair, such as hepatocyte growth factor (HGF) and fibroblast growth factors (FGFs).(1,22) We therefore examined whether syndecan-1 influences Akt signaling in APAP-overdosed mice. At 12 hours post-APAP, the time point where ALT levels begin to decline in Wt mice but continue to increase in Sdc1–/– mice, IHC analysis revealed widespread expression of Ser473-phosphorylated Akt (pAkt) in Wt, but not in Sdc1–/– livers (Fig. 5A), which was confirmed by immunoblotting of liver extracts (Fig. 5B). Interestingly, levels of both FGF-2 and HGF in total liver extracts were similar in Wt and Sdc1–/– mice (Supporting Fig. S4), suggesting that syndecan-1 increases Akt activation by potentiating growth factor signaling, but not its expression levels.
FIG. 5.
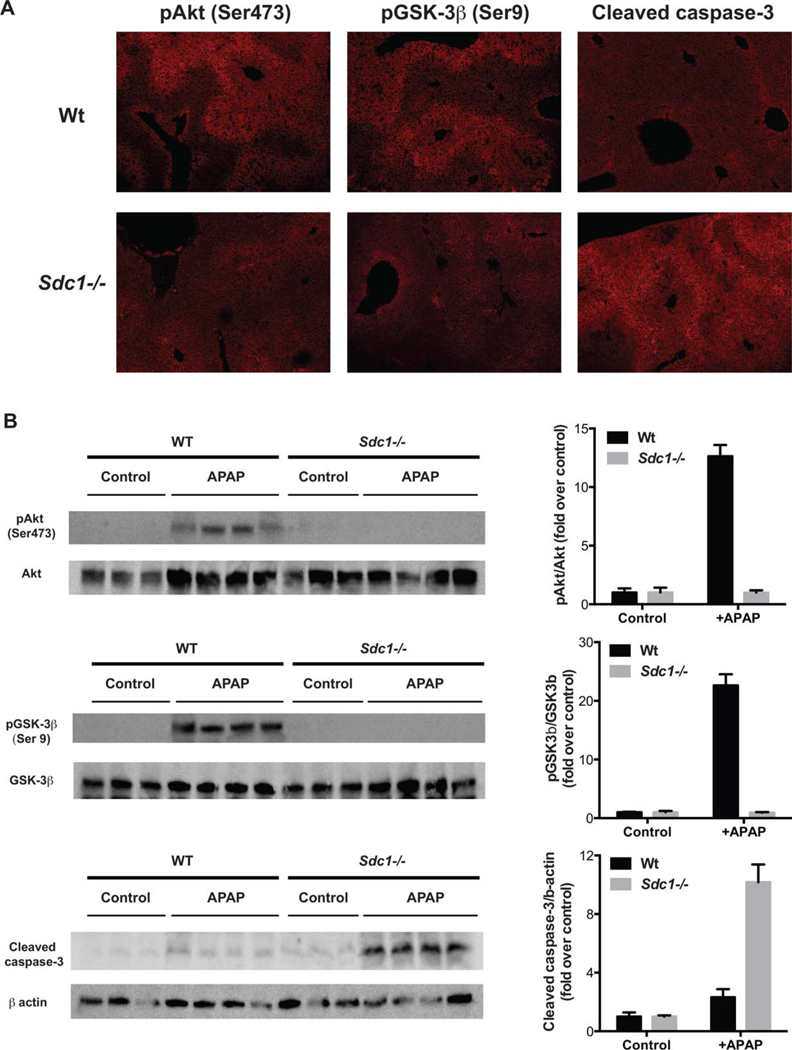
Syndecan-1 ablation inhibits Akt signaling and GSK-3β inactivation and enhances hepatocyte apoptosis in AILI. Wt and Sdc1–/– mice were injected with APAP (500 mg/kg) and livers were isolated at 12 hours post-APAP. (A) Liver sections were immunostained with anti-pAkt (Ser473), anti-pGSK-3β (Ser9), or anticleaved caspase-3 antibodies (original magnification, ×100), and (B) liver lysates were immunoblotted for pAkt, total Akt, pGSK-3β, total GSK-3β, cleaved caspase-3, and β-actin.
We next assessed whether syndecan-1 regulates inactivation of GSK-3β by Akt. GSK-3β is a Ser/Thr protein kinase and is one of the principal physiological targets of Akt.(29) Unlike most kinases where phosphorylation leads to activation, phosphorylation of GSK-3β (Ser9) by Akt results in its inactivation.(29) Furthermore, silencing of GSK-3β, but not GSK-3α, using antisense oligonucleotides has been shown to attenuate APAP-induced liver damage,(30) suggesting that GSK-3β is an important mediator. At 12 hours post-APAP, similar to pAkt, the signal for pGSK-3β was markedly increased in Wt, but not in Sdc1–/– livers by IHC (Fig. 5A) and immunoblotting (Fig. 5B).
GSK-3β mediates apoptosis induced by many apoptotic factors,(31) and hepatocyte apoptosis is a common feature of many liver diseases, including AILI.(32–36) Akt signaling promotes cell survival, in part, by inhibiting GSK-3β-mediated apoptosis.(37) We therefore examined whether syndecan-1 modulates hepatocyte apoptosis in APAP liver disease. Expression of cleaved caspase-3, a marker of apoptosis, was markedly increased in Sdc1–/– livers compared to Wt livers at 12 hours post-APAP (Fig. 5A), which was confirmed by immunoblotting for cleaved caspase-3 (Fig. 5B) and FACS analysis of Annexin V binding to primary hepatocytes (Supporting Fig. S5). Apoptosis was apparently triggered by signals derived from neighboring necrotic hepatocytes given that apoptotic hepatocytes in Sdc1–/– livers were especially evident in areas surrounding the centrilobular necrotic area (Fig. 5A). At a higher dose of APAP (625 mg/kg), delayed apoptosis starting at around 12 hours and continuing at 24 hours post-APAP was also observed in Wt livers by both Annexin V binding and cleaved caspase-3 immunoblotting (Supporting Fig. S6), indicating that the observed apoptotic phenotypes are not restricted to the Sdc1–/– background. Together, these data suggest that syndecan-1 halts the progression of AILI and facilitates liver repair by enhancing Akt activation and inhibiting GSK-3β-mediated hepatocyte apoptosis.
To pursue this hypothesis, we examined the effects of inhibitors of Akt (triciribine), GSK-3β (lithium), and caspase-3 (Ac-DEVD-CMK) on AILI. ALT levels were significantly increased in Wt mice treated with APAP and triciribine relative to those given APAP only (Fig. 6A), demonstrating that Akt inhibition worsens liver damage. In contrast, AILI was significantly attenuated by lithium (Fig. 6B) and Ac-DEVD-CMK (Fig. 6C) in susceptible Sdc1–/– mice, indicating that inhibition of GSK-3β or caspase-3 activity mitigates liver damage. Furthermore, because the inhibitors were given several hours post-APAP challenge, these results indicate that Akt facilitates, whereas GSK-3β and caspase-3 impede, recovery from APAP-induced liver damage. Delayed administration of LiCl and Ac-DEVD-CMK, and also purified syndecan-1 ectodomain and HS, similarly impeded liver injury in Wt mice injected with a higher dose of APAP (625 mg/kg; Supporting Fig. S7), indicating that the phenotypes observed in Sdc1–/– mice are not restricted to this background. Akt inhibition by triciribine was confirmed by immunoblotting for pAkt in liver extracts, whereas GSK-3β inhibition by lithium was confirmed by immunoblotting for β-catenin (Supporting Fig. S8). Triciribine and Ac-DEVD-CMK were dissolved in ethanol, and not dimethyl sulfoxide (DMSO), and diluted to the indicated dose in PBS to rule out the inhibitory effect of DMSO on cytochrome P450 (CYP) enzymes.(38) Ethanol can also induce CYP enzymes and potentially exacerbate AILI, but at the low concentration used in this study, it did not affect ALT levels in both Wt and Sdc1–/– mice. These data indicate that unopposed GSK-3β-mediated hepatocyte apoptosis leads to progressive liver injury in APAP-injected Sdc1–/– mice. Interestingly, GSK-3β ablation in mice results in extensive hepatocyte apoptosis and embryonic lethality.(39) Thus, too little or too much GSK-3β activity can promote hepatocyte apoptosis, suggesting that GSK-3β has dual functions in hepatocyte apoptosis depending on the triggering signal. Alternately, the role of GSK3β inactivation may be to promote liver repair, and the delayed apoptosis observed may be a default pathway related to the failure to regenerate.
FIG. 6.
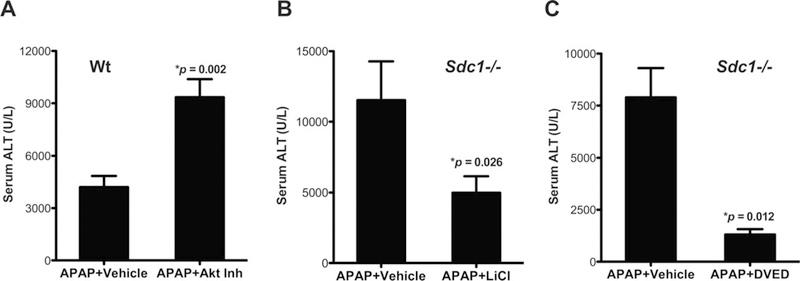
Inhibition of Akt aggravates, whereas inhibition of GSK-3β or caspases protects against, AILI. (A) Wt mice were injected with vehicle (2% EtOH/PBS) or Akt inhibitor (triciribine at 1 mg/kg) at 5 hours post-APAP, and ALT was measured at 24 hours post-APAP (mean ± SEM; n = 6; *P vs. APAP 1 vehicle). (B) Sdc1–/– mice were injected with vehicle (PBS) or LiCl (200 mg/kg) at 5 hours post-APAP, and ALT was determined at 24 hours post-APAP (n = 5; *P vs. APAP 1 vehicle). (C) Sdc1–/– mice were administered vehicle (EtOH/PBS) or Ac-DEVD-CMK (25 mg/kg) at 3 hours post-APAP, and ALT was determined at 24 hours post-APAP (mean ± SEM; n = 5; *P vs. APAP 1 vehicle).
2-O-SULFATED DOMAINS IN SYNDECAN-1 HS ENHANCE Akt ACTIVATION AND INHIBIT HEPATOCYTE APOPTOSIS IN AILI
Having shown that activation of Akt and subsequent inhibition of GSK-3b-mediated hepatocyte apoptosis are deficient in Sdc1–/– livers and that these aberrant signaling responses underlie the increased susceptibility of Sdc1–/– mice to AILI, we next tested whether purified syndecan-1 ectodomains and 2-O-sulfated heparan compounds can rescue these deficiencies. Treatment of Sdc1–/– mice with purified ectodomain, HS, or N- and 2-O-sulfated heparosan markedly increased pAkt and phosphorylated GSK-3β (pGSK-3β) levels in livers, whereas 2-O-desulfated heparin and heparosan had minimal effects compared to mice given APAP only (Fig. 7A,B). Furthermore, mice given syndecan-1 ectodomain, HS, or N- and 2-O-sulfated heparosan, but not 2-O-desulfated heparin or heparosan, showed substantially reduced signal for cleaved caspase-3 compared to mice given APAP only (Fig. 7C). Immunostaining of liver sections confirmed that syndecan-1 ectodomain and heparan compounds containing 2-O-sulfate groups enhance Akt activation and inhibit GSK-3β-mediated hepatocyte apoptosis (Supporting Fig. S9). However, few apoptotic hepatocytes were still evident in livers of mice administered N- and 2-O-sulfated heparosan (Supporting Fig. S9), suggesting that intact ectodomain and HS are more effective at inhibiting hepatocyte apoptosis than the engineered heparosan compound. Cumulatively, these data indicate that 2-O-sulfated domains in syndecan-1 HS attenuate the progression of AILI by enhancing Akt signaling and inhibiting GSK-3β-mediated hepatocyte apoptosis.
FIG. 7.
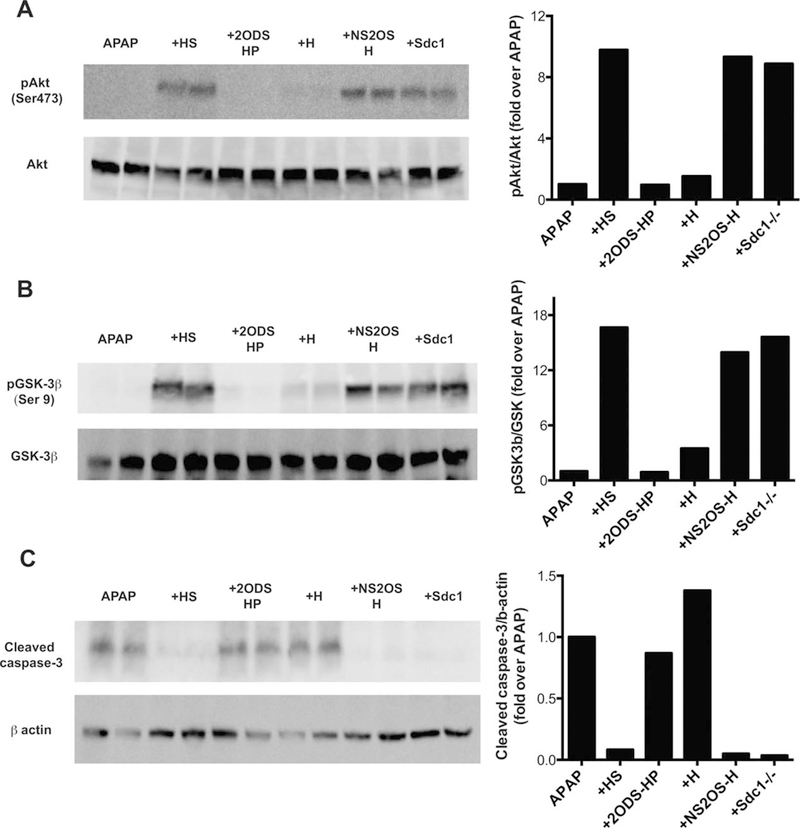
2-O-sulfated heparan compounds enhance Akt activation and GSK-3β inactivation and inhibit caspase-3 activation in AILI. Sdc1–/– mice were injected with 0.5 mg/kg of HS, 2-O-desulfated heparin (2ODS-HP), heparosan (H), N- and 2-O-suflated heparosan (NS2OS-H), or purified syndecan-1 ectodomain (Sdcl) at 1 hour pre-APAP, and livers isolated at 12 hours post-APAP were homogenized and liver lysates were immunoblotted for (A) pAkt and total Akt, (B) pGSK-3β and total GSK-3β, or (C) cleaved caspase-3 and β-actin.
Discussion
In this study, we identified a function of syndecan-1 in potently inhibiting AILI. Sdc1–/– mice were significantly more susceptible to AILI compared to Wt mice as demonstrated by increased transaminase levels and centrilobular necrosis, and decreased survival. These pathological phenotypes were rescued by administration of purified syndecan-1 ectodomain or HS, but not core protein, indicating that syndecan-1 protects against AILI through its HS chains. Remarkably, we also found that this protective activity is dependent on 2-O-sulfate groups of HS. This strict requirement for a particular HS modification was surprising, because HS activities in general are thought to depend more on the overall organization of HS domains and net negative charge than on the fine structure.(40) Our study also showed that syndecan-1 attenuates AILI independent of NAPQI activity and after GSH has already recovered. Instead, syndecan-1 facilitates later events that lead to liver repair, which were traced to a potentiation of prosurvival Akt signaling and inhibition of delayed GSK-3β-mediated hepatocyte apoptosis, which apparently is the default pathway when syndecan-1 is absent. Syndecan-1 is therefore a critical endogenous factor that shifts the balance from a detrimental, injury-perpetuating apoptotic pathway to a beneficial prosurvival pathway in APAP liver disease.
The observed specificity of syndecan-1 HS and 2-O-sulfated heparan compounds indicates the importance of specific molecular interactions rather than nonspecific electrostatic effects. Thus, a key to fully understanding this unprecedented liver repair mechanism in AILI will be future studies to identify the molecular targets regulated by 2-O-sulfated domains in syndecan-1 HS. HS positively regulates several heparin-binding molecules that activate Akt signaling, promote cell survival, and facilitate liver repair and regeneration, such as HGF, epidermal growth factors, FGFs, and Wnts.(41,42) Furthermore, 2-O-sulfate groups are required for HS to potentiate prosurvival signaling by many heparin-binding growth factors.(22) Hence, the requirement for 2-O-sulfate groups and Akt signaling points to the possibility that syndecan-1 HS accentuates prosurvival signaling by several heparin-binding growth factors to protect against AILI. However, most growth factors require other sulfate groups in addition to 2-O-sulfates for full activation.(22) Moreover, the observation that 2-O-sulfated heparosan, which contains GlcA but no IdoA, enhances Akt signaling and attenuates APAP-induced liver damage suggests that 2-O-sulfated glucuronic acid (GlcA2S) groups may be essential. Although 2-O-sulfation occurs mostly on IdoA and rarely on GlcA in most naturally occurring HS, higher levels of GlcA2S are found in liver HS.(43) These data suggest an intriguing possibility where GlcA2S-containing domains in syndecan-1 HS promote hepatocyte survival and liver recovery in APAP liver disease. However, our results do not completely exclude the possibility that 2-O-sulfated iduronic acid–containing domains can function similarly. Regardless, the strict requirement for 2-O-sulfate groups revealed in this study suggests that syndecan-1 HS may regulate a previously unidentified factor that potentiates Akt signaling and facilitate liver repair in APAP liver disease. A recent study showed that syndecan-1 HS binds to and inhibits the antimicrobial peptide, CRAMP, in a 2-O-sulfate–dependent manner(10) suggesting that unique biological targets specifically regulated by 2-O-sulfate motifs exist in vivo.
A hallmark of fulminant hepatic failure is often the rapid induction of hepatocyte apoptosis(44) However, the role of hepatocyte apoptosis in AILI is rather controversial. APAP overdose induces central features of apoptosis, such as release of mitochondrial cytochrome c and activation of caspases.(33,34) Furthermore, cleaved caspase products are increased in APAP-overdosed patients, caspase inhibitors protect against APAP-induced liver injury in rodent models(33,36) and a NAC/prostaglandin E2 combinatorial treatment mitigates APAP-induced liver damage in zebrafish, in part, by reducing caspase activation and hepatocyte apoptosis.(45) Despite these data strongly pointing to the importance of apoptotic cell death in AILI, several studies have also provided evidence that caspase activation is transient and limited post-APAP overdose and is insufficient to cause apoptotic hepatocyte death.(46,47) Our studies also showed that there are few apoptotic hepatocytes in Wt mice injected with 500 mg/kg of APAP. However, consistent with a report showing induction of hepatocyte apoptosis at a higher dose of APAP,(34) delayed apoptosis starting at around 12 hours post-APAP and continuing at 24 hours post-APAP was also observed in Wt mice injected with a higher dose of 625 mg/kg of APAP. Furthermore, liver injury can continue to occur even after levels of circulating APAP have declined to undetectable levels,(48) and NAC effectively suppresses AILI if given early, but is not as effective when given at later times post-APAP. These observations suggest an important role for propagation of preexisting injury rather than continuation of de novo direct injury.
Based on these findings, we propose that APAP overdose activates an apoptotic response in hepatocytes, which perpetuates liver injury, but the response is halted and fails to go to completion when hepatocytes express sufficient levels of syndecan-1 or when supplied with syndecan-1 or 2-O-sulfated heparan compounds. Furthermore, the discordant observations of hepatocyte apoptosis in AILI may be attributed to differences in the expression level of syndecan-1, especially in patients. In support of this idea, there are anecdotes of individuals consuming massive quantities of APAP without liver injury(49) whereas, in some cases, individuals develop ALF although they have not taken doses of APAP exceeding the recommended daily maximum of 4 g.(50) Examining whether low expression of syndecan-1 is a risk factor for AILI may be well worth trying. In addition, given that hepatocyte apoptosis is of major importance in many liver diseases,(44) the use of selectively 2-O-sulfated heparan compounds, capable of correcting the imbalance in prosurvival and apoptotic signaling in hepatocytes, may also have broader utility in treating liver disorders other than AILI.
Supplementary Material
Abbreviations:
- 2OST
2-O-sulfotransferase
- AILI
APAP-induced liver injury
- Akt
protein kinase B
- ALF
acute liver failure
- ALT
alanine transaminase
- ANOVA
analysis of variance
- APAP
acetaminophen
- AST
aspartate transaminase
- ConA
concanavalin A
- CS
chondroitin sulfate
- CYP
cytochrome P450
- DMSO
dimethyl sulfoxide
- FACS
fluorescence-activated cell sorting
- FGFs
fibroblast growth factors
- GlcA
glucuronic acid
- GlcA2S
2-O-sulfated glucuronic acid
- GlcNAc
N-acetylglucosamine
- GSH
glutathione
- GSK-3β
glycogen synthase kinase-3β
- HBSS
Hank’s balanced salt solution
- H&E
hematoxylin-eosin
- HGF
hepatocyte growth factor
- HS
heparan sulfate
- HSPG
heparan sulfate proteoglycan
- IdoA
iduronic acid
- IHC
immunohistochemical
- NAC
N-acetylcysteine
- NAPQI
N-acetyl-p-benzoquinone imine
- pAkt
phosphorylated Akt
- PBS
phosphate-buffered saline
- p-GSK-3β
phosphorylated GSK-3β
- Sdcl–/–
syndecan-1 null
- TNFα
tumor necrosis factor alpha
- Wt
wild type
Footnotes
Supporting Information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep.29265/suppinfo.
Potential conflict of interest: Nothing to report.
REFERENCES
- 1 ).Bernfield M, Götte M, Park PW, Reizes O, Fitzgerald ML, Lincecum J, Zako M. Functions of cell surface heparan sulfate proteoglycans. Annu Rev Biochem 1999;68:729–777. [DOI] [PubMed] [Google Scholar]
- 2 ).Bishop JR, Schuksz M, Esko JD. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature 2007;446: 1030–1037. [DOI] [PubMed] [Google Scholar]
- 3 ).Esko JD, Selleck SB. Order out of chaos: assembly of ligand binding sites in heparan sulfate. Annu Rev Biochem 2002;71: 435–471. [DOI] [PubMed] [Google Scholar]
- 4 ).Perrimon N, Bernfield M. Specificities of heparan sulphate proteoglycans in developmental processes. Nature 2000;404:725–728. [DOI] [PubMed] [Google Scholar]
- 5 ).Teng YH, Aquino RS, Park PW. Molecular functions of syndecan-1 in disease. Matrix Biol 2012;31:3–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6 ).Bode L, Salvestrini C, Park PW, Li JP, Esko JD, Yamaguchi Y, et al. Heparan sulfate and syndecan-1 are essential in maintaining murine and human intestinal epithelial barrier function. J Clin Invest 2008;118:229–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7 ).Hayashida K, Chen Y, Bartlett AH, Park PW. Syndecan-1 is an in vivo suppressor of Gram-positive toxic shock. J Biol Chem 2008;283:19895–19903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8 ).Hayashida K, Parks WC, Park PW. Syndecan-1 shedding facilitates the resolution of neutrophilic inflammation by removing sequestered CXC chemokines. Blood 2009;114:3033–3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9 ).Park PW, Pier GB, Hinkes MT, Bernfield M. Exploitation of syndecan-1 shedding by Pseudomonas aeruginosa enhances virulence. Nature 2001;411:98–102. [DOI] [PubMed] [Google Scholar]
- 10 ).Hayashida A, Amano S, Gallo RL, Linhardt RJ, Liu J, Park PW. 2-O-sulfated domains in syndecan-1 heparan sulfate inhibit neutrophil cathelicidin and promote Staphylococcus aureus corneal infection. J Biol Chem 2015;290:16157–16167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11 ).Stanford KI, Bishop JR, Foley EM, Gonzales JC, Niesman IR, Witztum JL, Esko JD. Syndecan-1 is the primary heparan sulfate proteoglycan mediating hepatic clearance of triglyceride-rich lipoproteins in mice. J Clin Invest 2009;119:3236–3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12 ).Zhang F, Sodroski C, Cha H, Li Q, Liang TJ. Infection of hepatocytes with HCV increases cell surface levels of heparan sulfate proteoglycans, uptake of cholesterol and lipoprotein, and virus entry by upregulating SMAD6 and SMAD7. Gastroenterology 2017;152:257–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13 ).Lee WM. Acetaminophen and the U.S. Acute Liver Failure Study Group: lowering the risks of hepatic failure. HEPATOLOGY 2004;40:6–9. [DOI] [PubMed] [Google Scholar]
- 14 ).Ostapowicz G, Fontana RJ, Schiodt FV, Larson A, Davern TJ, Han SH, et al. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med 2002;137:947–954. [DOI] [PubMed] [Google Scholar]
- 15 ).Lee WM, Squires RH Jr., Nyberg SL, Doo E, Hoofnagle JH. Acute liver failure: summary of a workshop. HEPATOLOGY 2008; 47:1401–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16 ).Dahlin DC, Miwa GT, Lu AY, Nelson SD. N-acetyl-p-benzo-quinone imine: a cytochrome P-450-mediated oxidation product of acetaminophen. Proc Natl Acad Sci USA 1984;81:1327–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17 ).Larson AM. Acetaminophen hepatotoxicity. Clin Liver Dis 2007;11:525–548, vi. [DOI] [PubMed] [Google Scholar]
- 18 ).Kaufmann T, Jost PJ, Pellegrini M, Puthalakath H, Gugasyan R, Gerondakis S, et al. Fatal hepatitis mediated by tumor necrosis factor TNFalpha requires caspase-8 and involves the BH3-only proteins Bid and Bim. Immunity 2009;30:56–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19 ).Harrison PM, Keays R, Bray GP, Alexander GJ, Williams R. Improved outcome of paracetamol-induced fulminant hepatic failure by late administration of acetylcysteine. Lancet 1990;335: 1572–1573. [DOI] [PubMed] [Google Scholar]
- 20 ).Smilkstein MJ, Knapp GL, Kulig KW, Rumack BH. Efficacy of oral N-acetylcysteine in the treatment of acetaminophen overdose. Analysis of the national multicenter study (1976 to 1985). N Engl J Med 1988;319:1557–1562. [DOI] [PubMed] [Google Scholar]
- 21 ).Schmidt LE, Dalhoff K, Poulsen HE. Acute versus chronic alcohol consumption in acetaminophen-induced hepatotoxicity. Hepatology 2002;35:876–882. [DOI] [PubMed] [Google Scholar]
- 22 ).Whitelock JM, Iozzo RV. Heparan sulfate: a complex polymer charged with biological activity. Chem Rev 2005;105:2745–2764. [DOI] [PubMed] [Google Scholar]
- 23 ).Chen J, Jones CL, Liu J. Using an enzymatic combinatorial approach to identify anticoagulant heparan sulfate structures. Chem Biol 2007;14:986–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24 ).Jaeschke H, Williams CD, McGill MR, Xie Y, Ramachandran A. Models of drug-induced liver injury for evaluation of phytotherapeutics and other natural products. Food Chem Toxicol 2013;55:279–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25 ).Wong SG, Card JW, Racz WJ. The role of mitochondrial injury in bromobenzene and furosemide induced hepatotoxicity. Toxicol Lett 2000;116:171–181. [DOI] [PubMed] [Google Scholar]
- 26 ).Mitchell JR, Potter WZ, Hinson JA, Jollow DJ. Hepatic necrosis caused by furosemide. Nature 1974;251:508–511. [DOI] [PubMed] [Google Scholar]
- 27 ).Franke TF, Kaplan DR, Cantley LC. PI3K: downstream AKTion blocks apoptosis. Cell 1997;88:435–437. [DOI] [PubMed] [Google Scholar]
- 28 ).Haga S, ozaki M, Inoue H, okamoto Y, ogawa W, Takeda K, et al. The survival pathways phosphatidylinositol-3 kinase (PI3-K)/phosphoinositide-dependent protein kinase 1 (PDK1)/Akt modulate liver regeneration through hepatocyte size rather than proliferation. HEPATOLOGY 2009;49:204–214. [DOI] [PubMed] [Google Scholar]
- 29 ).Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995;378:785–789. [DOI] [PubMed] [Google Scholar]
- 30 ).Shinohara M, Ybanez MD, Win S, Than TA, Jain S, Gaarde WA, et al. Silencing glycogen synthase kinase-3beta inhibits acetaminophen hepatotoxicity and attenuates JNK activation and loss of glutamate cysteine ligase and myeloid cell leukemia sequence 1. J Biol Chem 2010;285:8244–8255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31 ).Jope RS, Johnson GV. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci 2004;29:95–102. [DOI] [PubMed] [Google Scholar]
- 32 ).Huebener P, Pradere JP, Hernandez C, Gwak GY, Caviglia JM, Mu X, et al. The HMGB1/RAGE axis triggers neutrophilmediated injury amplification following necrosis. J Clin Invest 2015;125:539–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33 ).El-Hassan H, Anwar K, Macanas-Pirard P, Crabtree M, Chow SC, Johnson VL, et al. Involvement of mitochondria in acetaminophen-induced apoptosis and hepatic injury: roles of cytochrome c, Bax, Bid, and caspases. Toxicol Appl Pharmacol 2003;191:118–129. [DOI] [PubMed] [Google Scholar]
- 34 ).Murthy A, Defamie V, Smookler DS, Di Grappa MA, Horiuchi K, Federici M, et al. Ectodomain shedding of EGFR ligands and TNFR1 dictates hepatocyte apoptosis during fulminant hepatitis in mice. J Clin Invest 2010;120:2731–2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35 ).Volkmann X, Anstaett M, Hadem J, Stiefel P, Bahr MJ, Lehner F, et al. Caspase activation is associated with spontaneous recovery from acute liver failure. HEPATOLOGY 2008;47:1624–1633. [DOI] [PubMed] [Google Scholar]
- 36 ).Hu B, Colletti LM. CXC receptor-2 knockout genotype increases X-linked inhibitor of apoptosis protein and protects mice from acetaminophen hepatotoxicity. HEPATOLOGY 2010;52: 691–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37 ).Pap M, Cooper GM. Role of glycogen synthase kinase-3 in the phosphatidylinositol 3-Kinase/Akt cell survival pathway. J Biol Chem 1998;273:19929–19932. [DOI] [PubMed] [Google Scholar]
- 38 ).Yoon MY, Kim SJ, Lee BH, Chung JH, Kim YC. Effects of dimethylsulfoxide on metabolism and toxicity of acetaminophen in mice. Biol Pharm Bull 2006;29:1618–1624. [DOI] [PubMed] [Google Scholar]
- 39 ).Hoeflich KP, Luo J, Rubie EA, Tsao MS, Jin O, Woodgett JR. Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature 2000;406:86–90. [DOI] [PubMed] [Google Scholar]
- 40 ).Kreuger J, Spillmann D, Li JP, Lindahl U. Interactions between heparan sulfate and proteins: the concept of specificity. J. Cell Biol 2006;174:323–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41 ).Riehle KJ, Dan YY, Campbell JS, Fausto N. New concepts in liver regeneration. J Gastroenterol Hepatol 2011;26(Suppl 1): 203–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42 ).Limaye PB, Bowen WC, Orr AV, Luo J, Tseng GC, Michalopoulos GK. Mechanisms of hepatocyte growth factormediated and epidermal growth factor-mediated signaling in transdifferentiation of rat hepatocytes to biliary epithelium. HEPATOLOGY 2008;47:1702–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43 ).Kovensky J, Cirelli AF. Occurrence of 2-O-sulphated D-glucuronic acid in rat liver heparan sulphate. Carbohydr Res 1993;245: 361–365. [DOI] [PubMed] [Google Scholar]
- 44 ).Malhi H, Guicciardi ME, Gores GJ. Hepatocyte death: a clear and present danger. Physiol Rev 2010;90:1165–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45 ).North TE, Babu IR, Vedder LM, Lord AM, Wishnok JS, Tannenbaum SR, et al. PGE2-regulated wnt signaling and N-acetylcysteine are synergistically hepatoprotective in zebrafish acetaminophen injury. Proc Natl Acad Sci USA 2010;107:17315–17320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46 ).Gujral JS, Knight TR, Farhood A, Bajt ML, Jaeschke H. Mode of cell death after acetaminophen overdose in mice: apoptosis or oncotic necrosis? Toxicol Sci 2002;67:322–328. [DOI] [PubMed] [Google Scholar]
- 47 ).McGill MR, Sharpe MR, Williams CD, Taha M, Curry SC, Jaeschke H. The mechanism underlying acetaminophen-induced hepatotoxicity in humans and mice involves mitochondrial damage and nuclear DNA fragmentation. J Clin Invest 2012;122: 1574–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48 ).Bartolone JB, Cohen SD, Khairallah EA. Immunohistochemical localization of acetaminophen-bound liver proteins. Fundam Appl Toxicol 1989;13:859–862. [DOI] [PubMed] [Google Scholar]
- 49 ).Shayiq RM, Roberts DW, Rothstein K, Snawder JE, Benson W, Ma X, Black M. Repeat exposure to incremental doses of acetaminophen provides protection against acetaminophen-induced lethality in mice: an explanation for high acetaminophen dosage in humans without hepatic injury. HEPATOLOGY 1999;29:451–463. [DOI] [PubMed] [Google Scholar]
- 50 ).Lee WM. Acetaminophen toxicity: changing perceptions on a social/medical issue. HEPATOLOGY 2007;46:966–970. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.