

Abstract
Exposure to toxic levels of hydrogen sulfide (H2S) produces an acute cardiac depression that can be rapidly fatal. We sought to characterize the time course of the cardiac effects produced by the toxicity of H2S in sheep, a human sized mammal, and to describe the in vivo and in vitro antidotal properties of methylene blue (MB), which has shown efficacy in sulfide intoxicated rats. Infusing NaHS (720 mg) in anesthetized adult sheep produced a rapid dilation of the left ventricular with a decrease in contractility, which was lethal within about 10 min by pulseless electrical activity. MB (7 mg/kg), administered during sulfide exposure, maintained cardiac contractility and allowed all of the treated animals to recover. At a dose of 350 mg NaHS, we were able to produce an intoxication, which led to a persistent decrease in ventricular function for at least 1 h in nontreated animals. Administration of MB, 3 or 30 min after the end of exposure, whereas all free H2S had already vanished, restored cardiac contractility and the pyruvate/lactate (P/L) ratio. We found that MB exerts its antidotal effects through at least 4 different mechanisms: (1) a direct oxidation of free sulfide; (2) an increase in the pool of “trapped” H2S in red cells; (3) a restoration of the mitochondrial substrate-level phosphorylation; and (4) a rescue of the mitochondrial electron chain. In conclusion, H2S intoxication produces acute and long persisting alteration in cardiac function in large mammals even after all free H2S has vanished. MB exerts its antidotal effects against life-threatening sulfide intoxication via multifarious properties, some of them unrelated to any direct interaction with free H2S.
Keywords: Hydrogen Sulfide Intoxication, Methylene Blue, Toxic Cardiac Failure
Hydrogen sulfide (H2S), which has been in the spotlight (Olson, 2013) as a potential physiological gaseous signaling molecule, remains a persistent public health risk (Guidotti, 1994) in farming (Chenard et al., 2003), wastewater (Austigard et al., 2018), gas (oil) (Hendrickson et al., 2004), and fishing industry (Dalgaard et al., 1972). H2S has also been used as a “method” of suicide (Kamijo et al., 2013; Reedy et al., 2011; Sams et al., 2013; Truscott, 2008).
In contrast to “light” sulfide intoxications during which clinical symptoms rapidly and spontaneously resolve, due to the extremely fast disappearance of free H2S from the body (Haggard, 1921; Haouzi et al., 2014; Klingerman et al., 2013; Toombs et al., 2010), the most severe cases of sulfide intoxication result in a persistent coma, with a life-threatening shock and breathing depression (Almeida et al., 2008; Almeida and Guidotti, 1999; Guidotti, 2010; Judenherc-Haouzi et al., 2016; Struve et al., 2001). Of note, the profound and rapid cardiac depression produced during H2S intoxication is also certainly a major contributor to the development of long-term neurological deficit in victims surviving the most severe forms of intoxication (Baldelli et al., 1993). There is no current standard of care (Haouzi et al., 2016) available to first responders (Judenherc-Haouzi et al., 2016; Sonobe and Haouzi, 2016a).
The mechanisms of H2S toxicity are quite complex (Beauchamp et al., 1984; Bouillaud and Blachier, 2011; Cheung et al., 2007; Guidotti, 2010; Haouzi et al., 2016). Although sulfide inhibits the activity of the cytochrome C oxidase (Cooper and Brown, 2008; Dorman et al., 2002), akin to the effects of CN, HS-/H2S can also directly alter the activity of critical ion channels (Tang et al., 2010) on neurons (Greer et al., 1995) or cardiac cells (Cheung et al., 2018; Judenherc-Haouzi et al., 2016). Despite the extremely fast disappearance of the pool of soluble/free sulfide in the blood and tissues (Haggard, 1921; Haouzi et al., 2014; Klingerman et al., 2013), ie, within a few minutes after the end of an acute exposure (Sonobe and Haouzi, 2016b), in severe intoxications, the deleterious effects of H2S persist well beyond the period of exposure potentially leading to a delayed death or long-term neurological sequelae (Tvedt et al., 1991). These persisting effects of a sulfide intoxication could be accounted for by the presence of a pool of sulfide combined with metalloproteins (Smith and Gosselin, 1966; Van de Louw and Haouzi, 2013) or cysteine residues of various proteins (Mustafa et al., 2009; Paul and Snyder, 2012) and/or could result from the production of reactive oxygen species (Eghbal et al., 2004).
The very rapid disappearance of free H2S represents the major challenge to treat severe forms of sulfide intoxication (Haouzi et al., 2016), because antidotes able to combine free HS-/H2S and catalyze its oxidation in blood or tissues (Beck et al., 1981; Brenner et al., 2014; Hall and Rumack, 1997; Haouzi, 2011; Haouzi et al., 2014, 2015, 2016; Mihajlovic, 1999; Smith, 1967, 1981; Smith and Gosselin, 1966; Van de Louw and Haouzi, 2013) become rapidly ineffective when used after removing victims from a toxic exposure, ie, when left with a pool of sulfide not accessible to trapping. We have recently found that methylene blue (MB), an old redox dye, counteracts the acute effects of lethal forms of H2S intoxication in sedated and unsedated rats as well as in isolated contracting cardiomyocytes (Cheung et al., 2018; Judenherc-Haouzi et al., 2016; Sonobe and Haouzi, 2015). This effect is still present after sulfide exposure, when all free H2S has vanished (Judenherc-Haouzi et al., 2016), restoring cardiac and metabolic function acutely depressed by H2S (Cheung et al., 2018; Judenherc-Haouzi et al., 2016; Sonobe and Haouzi, 2015). As a result, these antidotal effects of MB increased the immediate survival of rats severely intoxicated with sulfide (Judenherc-Haouzi et al., 2016; Sonobe et al., 2015). A clear beneficial effect was also observed in intoxicated cardiac myocytes, wherein MB, added 3 min after H2S exposure, restored contractility, intracellular Ca2+ homeostasis and potassium exchange profoundly altered by toxic levels of H2S (Cheung et al., 2018; Judenherc-Haouzi et al., 2016). One of the main interests of MB is therefore that it seems to possess antidotal properties against H2S toxicity regardless of the presence of a pool of free H2S. Also, MB has been shown to exert a protection against the toxic effects of cyanide (CN) (Brooks, 1933; Cheung et al., 2018; Eddy, 1930; Haouzi et al., 2018; Sahlin, 1926), and could therefore be used against other mitochondrial poisons. Finally, MB is already used for the emergency treatment of methemoglobinemia (Ash-Bernal et al., 2004; Clifton and Leikin, 2003; Ginimuge and Jyothi, 2010; Wendel, 1939; Wright et al., 1999).
The first issue that needs to be resolved pertains to the translation to human intoxication of observations obtained either in rats or in vitro. Indeed, rodents have long been shown to be able to reduce their metabolism in response to hypoxia (Frappell et al., 1992; Mortola, 1993), a protective mechanism that is also triggered by H2S (Haouzi et al., 2009), which includes a decrease in cardiac function (Blackstone et al., 2005; Rincon, 2005) and in breathing (Haouzi et al., 2009). Such a mechanism of defense is absent in larger mammals (Haouzi et al., 2008), challenging the relevance to humans of the cardiac and metabolic effects of H2S and MB reported in rodents. The size-dependent physiological response to sulfide requires therefore studies to be performed in large species, to test both the cardiac toxicity of H2S and its potential reversal by MB.
The second fundamental question that must be clarified relates to the mechanisms accounting for the counteracting effects of MB on H2S toxicity. We have postulated that this effect is related to MB potent redox properties (Cheung et al., 2018; Judenherc-Haouzi et al., 2016; Sonobe et al., 2015; Sonobe and Haouzi, 2015). Indeed MB is readily reduced into leuco-MB (LMB) by molecules such as NADH or NAPDH as soon as it is administered (Kelner and Alexander, 1985; Sevcik and Dunford, 1991). LMB is therefore a reducing agent capable of transferring the electron(s) gained from NA(P)DH to molecules with higher redox potentials such as oxidized metals—or O2. As LMB is oxidized back into MB in the process, a new hemi-cycle of reduction of MB can take place. A very simple mechanism to explain the antidotal effects of MB would be to assume that there is a direct redox interaction between MB and the pool of free H2S, that could act as a reducing agent (Li and Lancaster, 2013), this interaction remains, however, relevant as long as free H2S is present in the blood or tissue. Second, as recently proposed for CN (Haouzi et al., 2018), the couple MB/LMB, although producing a reduction of the atoms of iron in red cells, was hypothesized to lead to the cyclic and transient formation of oxidized forms of iron, increasing in turn the capacity of hemoglobin to trap CN and possibly H2S. Third, in contrast to NADH, the couple MB/LMB can diffuse in all cells including neurons and cardiomyocytes, we have previously proposed that MB/LMB could restore the redox environment of the mitochondria altered by the presence of sulfide (Cheung et al., 2018; Judenherc-Haouzi et al., 2016). For instance, the couple LMB/MB has been shown to provide electrons to oxidized complexes, when complex I is impeded (Riha et al., 2011; Rojas et al., 2009; 2012; Wen et al., 2011; Zhang et al., 2006). The blockade of complex IV by H2S (Cooper and Brown, 2008; Nicholls et al., 2013) makes however this explanation unlikely as the electron chain is already reduced (Kim et al., 2012), so complexes need to be first re-oxidized before allowing electrons to flow. Finally a mechanism has recently be offered suggesting that MB could allow the Krebs cycle to resume, by restoring the NADH/NAD, ratio due to the cyclic oxidation of NADH (Komlodi and Tretter, 2017), which accumulation during H2S intoxication inhibits the mitochondrial substrate-level phosphorylation (TCA cycle) (LaNoue et al., 1972; Liu et al., 2018).
Our first objective was to address the question of efficacy in a large mammal by describing in adult sheep the effects of MB during or following a lethal or sublethal exposure to H2S. In addition, we sought to determine the potential mechanisms of the antidotal properties of MB in vivo and in vitro. We focused our studies on the 4 potential mechanisms of action described in the above paragraph: (1) is there a direct redox interaction between MB and free H2S? (2) Does MB increase the capacity of hemoglobin to “trap” H2S? (3) Can MB restore the ratio NAD/NADH of animals intoxicated with sulfide? (4) Is the couple MB/LMB able to rescue the mitochondrial membrane potential during a sulfide intoxication, and if so how?
MATERIALS AND METHODS
In Vivo Experiments
The Sheep Model
Twenty-five adult female Dorset sheep (Ovis aries, ewes, 1–2 years old) weighing 44.9 ± 11.5 kg were used for these experiments. Out of these 25 sheep, 4 animals were studied for the determination of the doses required to get a model of sublethal intoxication. The data obtained in these 4 sheep are not included in the overall comparison, but are still presented in the Results section. This study was approved by the Pennsylvania State University College of Medicine Institutional Animal Care and Use Committee; all experiments were conducted in accordance with the Guideline for the Care and Use of Laboratory Animals published by the United States National Institutes of Health. The choice of using female sheep was not dictated by any specific scientific concerns but by practical considerations (cost, easiness to handle in the animal facility, weight, and age).
The animals were pre-medicated with Ketamine (20 mg/kg) IM, then a catheter (Medtronic 3F) was placed in the right external jugular vein for anesthesia induction (Propofol). Anesthesia was subsequently maintained by urethane (80 mg/kg) and alphachloralose (15 mg/kg) as described previously (Haouzi et al., 2015; Haouzi and Chenuel, 2005). The animals were tracheostomized and an inflatable-cuff cannula (Shiley no. 7) was inserted. A catheter was placed in the right femoral artery for sampling arterial blood and for monitoring arterial blood pressure.
Additional venous catheters (Medtronic 3F) were placed in the left jugular vein and right femoral vein for injection of NaHS and antidotes, respectively. To prevent H2S-induced apnea, the animals were mechanically ventilated throughout the experiments, in SIMV mode (servo Siemens 900C) with a mandatory ventilation of 7–8 l/min to keep PaCO2 around 38 Torr. The animals were therefore able to trigger the ventilator increasing their ventilation as needed.
Measurements
Inspiratory and expiratory flows were recorded through the tracheostomy using a Hans-Rudolph pneumotachograph (Hans-Rudolph, Kansas City, Missouri) connected to a differential pressure transducer. The expiratory circuit of the ventilator was connected to a home-made mixing chamber, where mixed expired O2, CO2, and H2S fractions were continuously measured using an O2 (Oxystar-100), CWE, and CO2 (model 17630, VacuMed, Ventura, California) analyzers and 2 sulfide analyzers (Interscan RM series, Simi Valley, California; 1–200 ppm and 0.001–1 ppm). Each gas analyzer was calibrated with different gas mixtures containing a known concentration of CO2 and O2 in air, and H2S in N2. The time constant of the response of the circuit, including the sulfide analyzers, was determined as the time needed to drop H2S concentration by 64% and was found to average 123 s. The femoral arterial catheter was connected to a pressure transducer (TA-100, CWE Inc., Ardmore, Pennsylvania).
The analog output signals from the sensors were digitized at 200 Hz (Power Lab 16/35, ADInstruments, Inc., Colorado Springs, Colorado) and were visualized online. Data were stored for further analysis by LabChart 7 (ADInstruments, Inc., Colorado Springs, Colorado).
Mean blood pressure was computed from the instantaneous blood pressure signal. The respiratory flow signal was integrated for breath-by-breath calculation of tidal volume and minute ventilation (Haouzi et al., 2014; Klingerman et al., 2013). Oxygen consumption (O2) and CO2 production were computed under STPD conditions using VE, the inspiratory and mixed expiratory fraction of O2 and CO2 (Haouzi and Chenuel, 2005).
Echocardiographic Evaluation of Cardiac Function
The left hemithorax was shaved once the animal was sedated and 3 subcutaneous electrodes were placed on the chest for continuous acquisition of the ECG signal. Two-dimensional (2D) and M-mode studies were realized with a Philips ATL HDI 5000 ultrasound system, using a P4-2 (ie, 4.0–2.0 MHz broadband) phased array transducer. 2D left longitudinal parasternal views were obtained, then M-mode was used, due to its superior time resolution, to monitor the acute changes in left ventricular (LV) size and contractility with the cursor placed at the tip of the mitral leaflets (approximately mid-ventricle). Images were optimized to allow clear visualization of the LV antero-septal and posterior wall endocardial borders, avoiding papillary muscles and mitral chordae as much as possible. Simultaneous 2D and M-mode LV imaging was continuously monitored throughout the experimentation. Ten-second clips were recorded every 30–60 s and stored on magnetic optical disks for subsequent analysis.
Protocol
NaHS (sodium hydrosulphide hydrate, Sigma Aldrich, St. Louis, Missouri) was dissolved in sterile saline at a concentration of 20 mg/ml assuming an equimolar conversion of NaHS into H2S, after taking into account 25% hydration of NaHS, as reported previously (Haouzi et al., 2014; Klingerman et al., 2013). NaHS solutions were prepared a few minutes before the infusion and kept in airtight syringes. The solution was infused intravenously using an infusion pump (Fusion 100; Chemyx Inc; Stafford, Texas) at a rate that is described below. As a first approach, we relied on the study of Burrows (1984) to determine the dose of MB to be used. This study showed that MB could be safely used up to at least 15 mg/kg to treat methemoglobinemia (DL 50 ranged from 30 to 40 mg/kg). The dose of 7 mg/kg MB was previously tested in 3 sheep and was found to provoke no side effects on blood pressure or cardiac function, whereas increasing O2 by 2–3-fold, akin to the effects produced in the rats (Haouzi et al., 2018). This dose of MB was infused over 10 min. Two different protocols were used:
Protocol 1: lethal H2S exposure
Eight sheep received a total dose of 720 mg H2S (16 mg/kg) over a period of 10 min, which we had previously determined to be lethal. This protocol was developed to allow enough time (10 min) to study the effects of sulfide on circulation and cardiac function, the exposure consisting in a stepwise increment of the dose of H2S from 60 to 96 mg/min. Half of the animals received saline and half of the animals received MB at a dose of 7 mg/kg for 10 min during H2S exposure.
Protocol 2: sublethal intoxication
Protocol 1 was modified to be able to produce a significant reduction in cardiac function, blood pressure, and O2during H2S exposure, whereas allowing the animals to survive. We first used a dose of H2S reduced by ∼25% (590 mg) in 4 sheep, allowing all the animals to survive the period of infusion, but they all deceased between 3.23 and 5 min after the end of the infusion. The protocol was therefore modified and at a total dose of ∼350 mg over 4.5 min: 75 mg/min for 3 min then 85 mg/min for 1.5 min, the animals remained alive for at least the entire duration of the experiments, although displaying a severe depression in cardiac function during sulfide exposure. MB was infused at the dose of 7 mg/kg for 10 min as in protocol 1, but infusion was started 3 min after the end of H2S infusion, at a time when expired H2S was back to zero.
Arterial blood was sampled before the intoxication, then at 1, 5, 15, and 30 min into recovery from sulfide exposure in each surviving animal. Arterial blood gases partial pressure of O2 (PaO2), CO2 (PaCO2), and lactate concentration were measured using i-STAT1 blood gas analyzer (ABAXIS, Union City, California). In addition, a spectrophotometry analysis of the blood (Eppendorf BioSpectrometer basic, Eppendorf AG, Hamburg, Germany) was performed for determination of the presence of hemoglobin, methemoglobin, and MB. Aliquots of blood were also centrifuged and the supernatant (plasma) was used for determination of pyruvate concentrations.
As we found out that cardiac function continued to decline in the group that received saline (see Result section), MB was administered 30 min after the end of sulfide exposure in 4 of the control animals, but over 5 instead of 10 min, following the protocol described above.
At the end of the experiments, animals were euthanatized using a bolus injection of barbiturate (Euthasol, 12 ml, 400 mg/ml) in the jugular catheter.
In Vitro Studies
Interactions Between H2S and MB in PBS, in Blood and With Hemoglobin
Aliquots of the arterial blood sampled from each animal were heparinized and immediately combined with deionized water (dH2O) and phosphate-buffered saline (PBS) for determination of the level of hemoglobin and methemoglobin as well as the concentration of MB in the blood. Fifty microliters of blood were added to a tube containing 4.65 ml of dH2O and 300 μl of PBS, before transferring this solution into a cuvette for spectrophotometry analysis. A calibration curve was established by adding known concentration of methemoglobin (human vvhemoglobin, Sigma no. 7379) as described previously (Haouzi et al., 2018). The concentration of MB in the blood was determined by mixing 200 μl of whole blood to 4.65 ml of distilled H2O and 150 μl of PBS. Tubes were gently mixed and 2 ml of the sample was added to a 10 × 10 mm cuvette for analysis (Eppendorf Macro Vis). The spectrum of absorbance in each solution was analyzed between 300 and 800 nm (Eppendorf Basic Biospectrophotometer).
In addition, a solution of 100 μM of H2S (final concentration) was mixed with a solution containing 100 μM MB (final concentration) and blood. Blood was obtained from sheep and the protocol was intended to mimic the in vivo conditions of MB exposure in protocol 1. After incubation for 10 min, blood was analyzed by spectrophotometry (Eppendorf BioSpectrometer basic, Eppendorf AG, Hamburg, Germany), following the protocol described above for the determination of MB and hemoglobin/methemoglobin.
Finally, NaHS, at different concentrations, was mixed with a solution containing MB in PBS then transferred to a 2-ml cuvette, entirely filed with the solution, that was hermetically closed preventing any O2 from air to diffuse into the solution. The samples were analyzed over time by spectrophotometry to determine the potential accumulation of LMB in solution, a marker of sulfide oxidation.
The interaction between hemoglobin, MB (100 μM), and NADH (β-nicotinamide adenine dinucleotide, Sigma no. N8129, 200 μM) was investigated with known concentrations of methemoglobin (100%, 50%, or 0%).
Determination of lactate and pyruvate concentrations
A pyruvate Assay Kit (Abcan, Cambridge, Massachusetts) was used according to manufacturer’s instruction. As this assay is based on the determination of a redox reaction, involving NADH, we determined any potential direct reaction between MB and the assay. The effects of MB were determined by reacting different concentrations with the assay and a calibration curve was established with and without MB using known concentration of pyruvate. We found no direct effect of the presence of MB on the determination of pyruvate. MB did not produce any artifact in the determination of pyruvate that could interfere with its determination. When known concentrations of pyruvate ranging from 20 to 100 μM were added to a solution of MB before determination of pyruvate concentrations, no artifact was created by MB besides the upward shift of the absorbance produced by the presence of MB itself, which was corrected in all our measurements.
In addition, the presence of MB did not affect the determination of lactate by the i-STAT1 blood gas analyzer (ABAXIS, Union City, California). The ratio pyruvate/lactate (P/L) was used as a marker of cytoplasmic NADH/NAD ratio.
Interactions Between MB, NADH, and Cytochrome C (CC), Effects of H2O2
As the spectra of absorbance of MB, NADH, and CC do not have significant overlap, we studied the interactions, at neutral pH and ambient temperature of MB (100 μM) and NADH (200 μM) alone or mixed with cytochrome C (50 μM). Absorbance was determined every 5 min over time up to 90 min. The interactions between MB and NADH were studied in the cuvette either open to the air or capped.
Finally, we tested the effects of H2O2 (5, 50, or 500 μM) on reduced cytochrome C (50 μM) in PBS at ambient temperature.
Criteria Used for Determination of Reduced and Oxidized Hemoglobin and Cytochrome C
In keeping with previous spectra published in the literature, the following criteria were used to identify the reduced and oxidized form of hemoglobin (Horecker, 1943; von Kompen, 1983; Zijlstra and Buursma, 1997), cytochrome C (Appaix et al., 2000; Koch and Schneider, 2007; Stotz et al., 1938), MB (Bergmann and O’Konski, 1963), and NADH (Albani, 2007): reduced oxyhemoglobin was defined by the presence of 2 peaks of absorbance at 545 and 575 nm (Horecker, 1943; von Kompen, 1983; Zijlstra and Buursma, 1997) with positive ratio of these 2 peaks A575/A545; based on this ratio, it is possible to identify the concentrations of methemoglobinemia in solution above 4% (Haouzi et al., 2018). For cytochrome C (Appaix et al., 2000; Koch and Schneider, 2007; Stotz et al., 1938), the change from the oxidized to the reduced form can be characterized by a shift of the Soral band from 410 to 415 nm with a decrease in the peak magnitude along with the apparition of 2 peaks at 520 and 550 instead of one at 530 nm (Appaix et al., 2000; Koch and Schneider, 2007; Stotz et al., 1938). The change in concentrations in NAD were evaluated by the peak of absorption at 340 nm, whereas the oxidized form has virtually no absorption at this wavelength (Albani, 2007); the concentrations of oxidized (blue) form of MB were determined by the maximal peak at 660 nm (with a smaller peak at 615 nm) and 290 nm in the ultraviolet (Bergmann and O’Konski, 1963) (see Result section for representative spectra of absorbance).
Mitochondrial Redox Potential in Isolated Cardiomyocytes
Cardiac myocytes were isolated from the LV free wall and septum from adult C57BL6 mice (10–12 weeks old), as reported previously (Cheung et al., 2018, 2018). In all experiments, myocytes were used within 2–6 h of isolation. All protocols applied to the mice in this study were approved and supervised by the Institutional Animal Care and Use Committee at Temple University.
Myocytes were seeded on laminin-coated coverslips. They were loaded with mitochondrial membrane potential indicator rhodamine 123 (13 µM; Invitrogen) and mitochondrial superoxide-sensitive fluorophore MitoSOX Red (22 µM; Invitrogen) in the extracellular media containing 2% BSA, 0.06% pluronic acid, and 20 µM sulfinpyrazone at 37°C for 30 min. Cells were washed, and exposed to PBS, NaHS (100 µM), MB (20 µg/ml), and NaHS + MB (MB added 3 min after NaHS) for 10 min.
Data Analysis
All data were expressed as mean ± SD. The variables of interest were compared within each group, before NaHS infusion, then at 1, 5, 15, and 30 min after the end of NaHS infusion or up to the moment when the animal died, using repeated ANOVA, followed by a post hoc multiple comparison procedure (Turkey’s multiple comparison).
An experienced echocardiography reader analyzed the TTE images and videos in a blinded manner using Synapse Cardiovascular software (Fujifilm). LV end-diastolic (Dd) and end-systolic (Sd) diameters were measured from M-mode views, guided by 2D images to position the sampling line at mid ventricle, through the largest diameter, just above the posterior papillary muscle tip. Clips failing to show a clear delineation of the endocardial borders were discarded. For each evaluation, 3–5 consecutive cycles were measured and averaged. Fractional shortening (FS) was computed as (Dd − Sd)/Dd, the ejection fraction (EF) was calculated using the Teichholz formula (Teichholz et al., 1976) based on the determination of the end-diastolic volume (Dv), end-systolic volume (Sv), and systolic ejection volume (SEV). Cardiac output was calculated as SEV × HR.
For the cardiomyocyte studies, cells were washed and then imaged using a Carl Zeiss Meta 510 Meta confocal microscope with a 40× oil objective with 1.7× digital zoom at 488 and 561 nm for rhodamine 123 and MitoSOX Red, respectively. Results were expressed as means ± SE and data were compared using 2-way ANOVA.
RESULTS
In Vivo Studies
Effects of Lethal Infusion of NAHS on Cardiac Function, Interaction With MB (Protocol 1)
This study was performed in 8 sheep. Four received H2S alone, whereas 4 received MB at the same time (7 mg/kg over 10 min). All the nontreated animals displayed a very similar pattern of response to H2S exposure. After an initial phase, wherein blood pressure increased for about 2 min, ABP, O2, and CO2 decreased very rapidly leading to a complete circulatory arrest within 10 min. The decline of the circulatory parameters and gas exchange rate was very fast as displayed in Figure 1.
Figure 1.

Mean ± SD values of oxygen uptake (O2), CO2 output (CO2), and mean arterial blood pressure (ABP) in 8 sheep exposed to a lethal level of H2S (protocol 1). Left panels (closed symbols) display the time course of the effects of H2S alone, whereas the right panels (open symbols) show the effects of H2S along with MB. The red-doted lines in the right panels correspond to the average data of the untreated animals shown here for comparison. MB increased O2 and CO2, which remained above the levels in nontreated animals during the entire period of exposure to sulfide. Note that (1) O2 and CO2 were still above 100 ml/min at a time when all of the nontreated animals were dead, (2) blood pressure was maintained around baseline levels even when the cardiac function was very much depressed suggesting that a significant peripheral vasoconstriction occurred.
Echocardiography studies (Figure 2) showed that after a brief period of increased contractility, LV dilation and decreased contractility occurred starting 2–3 min into H2S infusion, together with a sinus bradycardia. Cardiac output also decreased rapidly (Figure 3). In 3 out of the 4 untreated intoxicated animals, PEA was the cause of asystole at 10 min, as the ECG still showed a sinus rhythm with a frequency of ∼45 beats/min, whereas no ventricular contraction or pulse pressure could be identified. In the remaining animal, a cardiac arrest occurred secondary to an episode of ventricular tachycardia (VT) at 9 min, associated with the disappearance of the pulse pressure, O2, and cardiac output.
Figure 2.

A, Example in 1 sheep of a 2D echocardiography study during a lethal sulfide infusion. Note the very rapid distension of the heart and increase in left ventricular volume after few minutes of H2S exposure, leading to a suppression of the cardiac systole (PEA) in less than 10 min. B, Effects of lethal levels of H2S (protocol 1) on cardiac function determined by echocardiography in 4 untreated animals (saline, left panels), and in 4 animals treated with MB (right panels). From top to bottom, mean ± SD values of end-diastolic and end-systolic diameter, cardiac output (CO), left ventricular ejection fraction (LVEF), and heart rate (determined from the ECG signal) are displayed. In the nontreated intoxicated group, note that a rapid depression in cardiac contractility led to a cardiogenic shock followed by a cardiac asystole. Note that when cardiac contractions ceased, heart rate (ECG signal) is maintained at an average of 50 QRS/min, defining a PEA. MB administered during H2S infusion maintained the cardiac function within a range compatible with survival; for instance, cardiac output still averaged 3 l/min at a time when all the control sheep where in cardiac asystole. C, Temporal profile of the changes (mean ± SD) in LVEF), O2, and blood pressure following sulfide exposure (protocol 1, lethal administration) in the MB-treated sheep (open symbols), whereas the red-doted lines correspond to the average data obtained in the untreated animals. Time zero is the time of exposure to sulfide. Note that in the MB-treated group, all the parameters retuned to baseline within a few minutes after the cessation of the exposure to sulfide and remained stable, at a time when all the nontreated animals had already died. Lactate also returned progressively to normal reaching 2.8 mM at 30 min into recovery. Concentrations of MB remained present in the blood, but decreased form 121 μM at 1 min to 20 μM at 30 min.
Figure 3.

A and B, Absorbance of the solution of hemoglobin obtained from the blood sampled 1 min after the end of exposure in a sheep that received H2S only (red lines, before cardiac asystole occurred) and in a sheep that received H2S plus MB (blue line). Note that a peak of absorbance at 620 nm was present when both MB and H2S were administered, for comparison the spectrum of absorbance of MB in blood in also shown (black line). C, effects of mixing blood to NaHS (100 μM), MB (60 μM), and NaHS (60 μM) plus MB (60 μM). Note that only in the latter was a peak of absorbance produced at 620 nm.
In the MB-treated animals, blood pressure followed an initial pattern similar to that of the untreated animals associated with a progressive dilation of the heart, but by the 5–6th minute, the changes in circulation and cardiac function departed from the nontreated animals, as contractility remained steady while the levels of cardiac output and blood pressure were maintained until the end of sulfide infusion. O2 and CO2 increased during MB infusion opposing the depression produced by H2S, for instance O2 was still almost half of its baseline value (0.112 ± 0.08 l/min) at the end of exposure in MB-treated animals (p < .01 vs. nontreated animals), whereas cardiac output averaged 3.2 ± 0.9 l/min (p < .001) (Figures 1 and 2). All variables—including O2, blood pressure, or LV EF—returned to baseline during the recovery period from sulfide exposure (Figure 2), whereas lactate reached 6.1 mM at 1 min (p < .01), subsiding over time reaching 2.8 mM at 30 min. The concentration of MB was found to average 121 μM, 1 min into recovery decreasing progressively over time but was still present in the blood 30 min after the end of MB administration (Figure 2). The study of the hemoglobin absorbance did not reveal the presence of any methemoglobinemia (Figure 3); however, in contrast to nontreated intoxicated animals in which blood could be sampled in animals in asystole within 1 min after exposure, a new peak of absorbance, different from the peak of absorbance of MB, was observed at 620 nm in the MB-treated group (Figure 3).
Finally, the level of mixed expired H2S fraction (Figure 4) was found to be significantly lower in the MB-treated group than in the nontreated animals, reaching half of its expected value during MB infusion (p < .01). As cardiac output was higher in the treated group, the product of cardiac output and the mean alveolar fraction of H2S (a surrogate for the arterial concentration of gaseous H2S [Haouzi et al., 2014; Klingerman et al., 2013] was computed as a marker of the flow of H2S circulation). This product was not different between the 2 groups of animals (Figure 4), reflecting the fact that the lower level of alveolar H2S, and thus in the blood, was in large part the result of a “dilution” by a higher cardiac output in the MB-treated animals.
Figure 4.

A, Temporal profile of the changes (mean ± SD) in mixed expired fraction of H2S (expressed in ppm during lethal sulfide administration (protocol 1) in the non-MB-treated group (closed symbols) and in the MB-treated sheep (open symbols). Note that the level of sulfide in the expired gas was lower in the MB treated group. On note, (B) is illustrating the actual flow of H2S reaching the central circulation, ie, cardiac output times the concentration of gaseous H2S extrapolated from the estimation of alveolar fraction of H2S. Indeed, as H2S was infused at a constant rate, the concentration of H2S in the blood reaching the central circulation was dictated by the level of venous return/cardiac output (see text for further comments). C, Shows the in vitro effects of mixing MB and H2S in the absence of O2, clearly MB is reduced by the presence of H2S, which is being oxidized in the process.
Sublethal Exposure (Protocol 2)
The protocol consisted in infusing 350 mg over 4.5 min: 75 mg/min for 3 min then 85 mg/min for 1.5 min, and allowed all the animals to remain alive after the exposure for the entire duration of the protocol (Figure 5). This protocol was applied in 13 animals. The changes in cardiac function and hemodynamics produced during the infusion of sulfide were not very different from those produced by the protocol 1. LV dilation developed, starting as early as the 1st minute of exposure. Although global LV hypokinesis was observed in all animals, 2 of them also showed evidence of regional wall motion abnormality with either posterior wall or septal akinesis. When NAHS infusion was stopped, all animals displayed a transient phase of “recovery” with a hyperdynamic left ventricle for 3–7 min before worsening again. One animal died at 3 min during this phase and was excluded from the study. All of the other 12 sheep survived and were therefore included in the protocol. Six sheep received saline 3 min into recovery and remained alive until the end of the protocol (up to 1 h). They all displayed a significant depression in cardiac contractility that persisted until the end of the study. Significant cardiac conduction abnormalities and arrhythmia were also present, consisting in changes in ventricular repolarization, ie, short QT and wide and ample T waves. These abnormalities of ventricular repolarization occurred very early following the end of H2S infusion. More significant arrhythmias such as supra-VT lasting several minutes were noticed in 2 sheep, whereas a self-terminating 3-min VT was observed just at the cessation of H2S infusion in 1 sheep. Wide QRS, suggesting bundle branch block, developed at 6 min in 1 animal. Transient ectopies were observed (premature atrial and/or ventricular contractions) followed by a brief and reversible episode of VT in 1 sheep.
Figure 5.

Temporal profile of the changes (mean ± SD) in left ventricle ejection fraction (EF) and cardiac output (A), expired H2S, blood pressure, O2 and CO2 (B) following sulfide exposure in protocol 2 (sublethal intoxication). The closed symbols represent the untreated animals while the open symbols represent the animals treated by MB (3 min after the end of sulfide exposure) whereas the red-dotted line corresponds to the average data of the untreated animals. Note the drop in EF during H2S exposure in both groups and that in the nontreated animals, a continuous decrease in EF developed over time reaching −50% of the baseline value at 30 min along with a significant reduction in cardiac output. MB in all instance restored the cardiac function. Also note that (1) H2S in expired gas was undetectable, when MB was administered, (2) blood pressure was not different between the 2 groups of animals, reflecting a rise in peripheral vascular resistance in the non-treated sheep, and (3) O2 consumption and CO2 production increased in response to MB (see text for discussion). C, PL ratio also increased after MB and returned to baseline values. Concentrations of MB decreased form 121 μM at 1 min to 20 μM at 30 min.
Blood lactate concentrations increased from 0.9 to 5.3 mM (p < .01) at 1 min after the end of H2S infusion (Figure 5). Lactate remained elevated throughout the 30-min period of recovery averaging 3.2 mM at 30 min (p < .01). This increase in lactate was associated with a marked decrease in PL ratio by more than 10-fold at 1 min, remaining below baseline even at 30 min (Figure 5).
MB infusion had 3 major impacts on the recovery outcome. First, the persistent decrease in cardiac contractility was suppressed following MB infusion; LVEF or cardiac output returned to or even above baseline reaching 98 ± 7% of baseline versus 54 ± 8% for the nontreated animals (p < .05) and 6.4 ± 0.9 l/min versus 3.8 l ± 0.5/min for the nontreated animals (p < .01) respectively. Second, CO2 and O2 increased following MB infusion (Figure 5); as a result, the O2 deficit was “paid” much more rapidly in the MB-treated group (615 s) than in the saline group (885 s, p < .05). Third, the P/L ratio doubled as soon as MB was administered (p < .01, Figure 5) and returned to baseline level for the rest of the experiment.
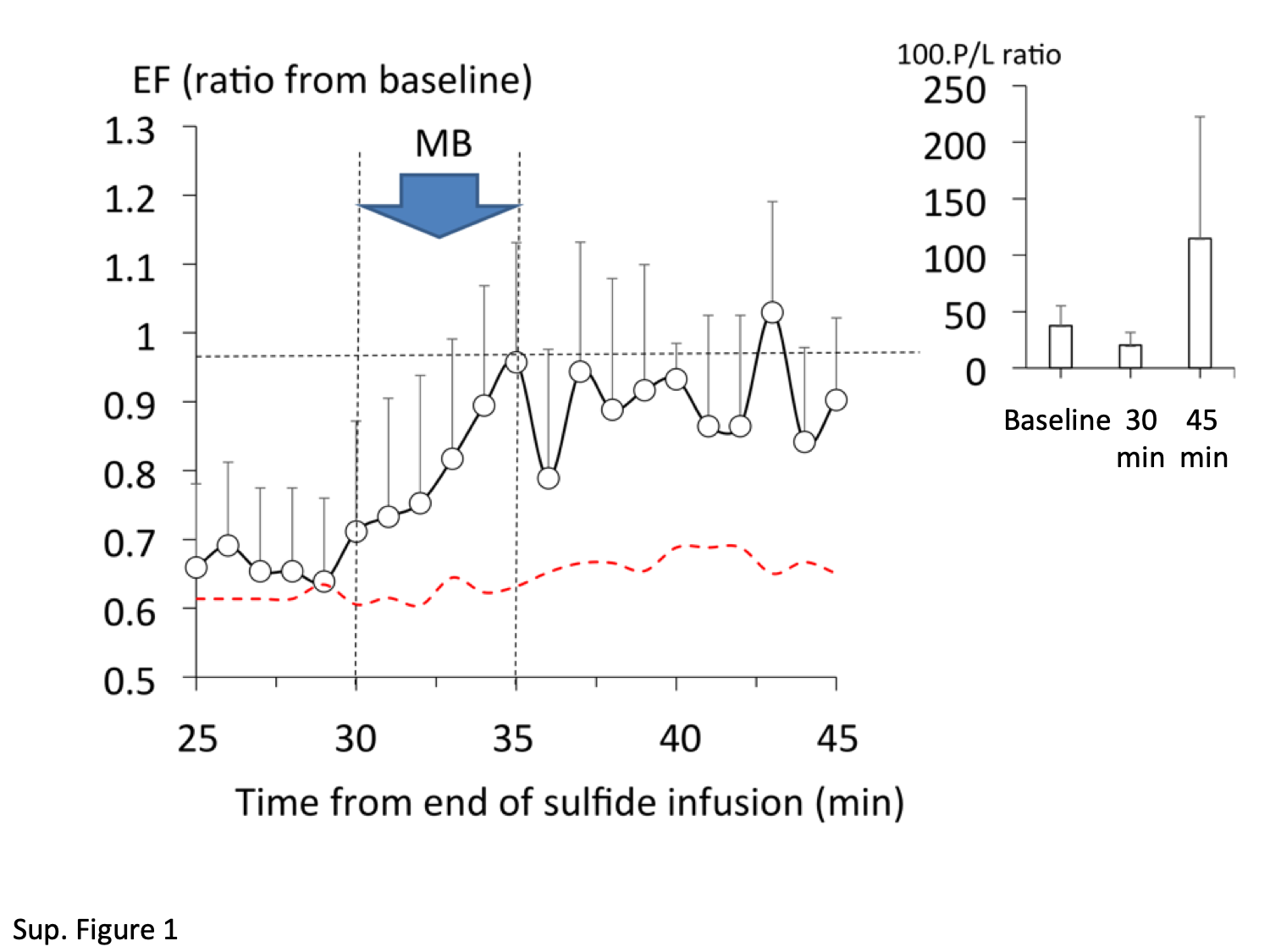
In 4 out of the 6 control animals (saline), MB was administered 30 min after the end of sulfide exposure. In all instances, a severe reduction in EF, ie, less than 60% of baseline was present at the time of MB administration. MB produced a rapid rise in EF and in P/L ratio in every treated animal (Supplementary Figure 1).
In Vitro Studies
Interaction Between H2S and MB: Is Free H2S Spontaneously Oxidized by MB?
A reduction of MB (oxidation of H2S) was observed when MB and H2S were mixed in PBS, while preventing O2 to diffuse into the cuvette. However, this rapid drop in MB absorbance was only observed when high concentrations of H2S were used (from 200 μM to 5 mM) (Figure 4).
Interaction Between MB, H2S, and Hemoglobin: Does MB Increase the Trapping of Free H2S by Hemoglobin?
In order to clarify the nature of the peak of absorbance of hemoglobin at 620 nm present in the blood of the animals intoxicated by H2S but that received MB, we first tried to reproduce this peak by mixing MB (100 μM) or H2S (100 μM) alone or in combination with a hemoglobin solution as well as with a methemoglobin solution (100% or 50%). We were only able to create this new peak of absorbance when MB was mixed in the blood with H2S, akin to the peak found in vivo, as demonstrated in Figures 3 and 6. Neither MB nor H2S alone (100 μM) produced such a change.
Figure 6.

Absorbance of solution of containing 100% methemoglobin mixed with PBS (black lines) or mixed with a solution containing 100 μM NaHS in PBS (red lines) (A). The same reactions were studied with a solution containing 50% hemoglobin-50% methemoglobin (B). 100% hemoglobin (C), and 100% hemoglobin with MB alone (D, green line) or with H2S (100 μM) plus MB (60 μM) (D, blue line). Note that the addition of MB allowed the formation of a new peak of absorbance, reflecting the creation of a combined pool of H2S on hemoglobin.
Interactions Between MB, NADH, and Cytochrome C
Mixing MB and NADH (neutral pH at ambient temperature) produced a rapid disappearance of the peak at 335–340 nm specific of NADH (Figure 7), the reaction started immediately as soon as the 2 molecules were in contact. In our experimental conditions, almost all the 200 μM NADH in 2 ml (400 nmol of NADH) were consumed by 100 μM of MB (200 nmol) in about 40 min. Conversely, when NADH was present in excess (2 mM) and O2 was prevented to diffuse into the solution (by capping the cuvette filled with the solution), there was a rapid decrease in absorbance of MB, reduced into LMB, but unable to be re-oxidized after having consumed the volume of O2 present in the PBS solution (Figure 7). The re-oxygenation of the solution led to the immediate restoration of the blue color.
Figure 7.

A, Absorbance of a solution of MB (100 μM), NADH (200 μM), and of a solution where MB was mixed with NADH (neutral pH, ambient temperature). When mixed in the presence O2, there was a drop in NADH concentrations. In the absence of O2, ie, as soon as the O2 present in solution is consumed, the re-oxidation of LMB into MB is rendered impossible, LMB replaces MB in the solution, demonstrating the restoration of MB from LMB via an increase in O2 utilization outside the electron chain. B, schematic representation of the potential effects of NADH oxidation by MB in condition wherein NADH is not used anymore by the complex I, ie, when the electron chain remains in reduced state during H2S inhibition of the cytochrome C oxidase. Any inhibition of the TCA cycle, resulting from the increase in NADH/NAD ratio, will be alleviated by MB allowing the TCA cycle to resume, also note the effects of MB on P/L ratio.
As shown in Figure 8, oxidized cytochrome C in solution (50 μM) could only be reduced when both NADH (200 μM) and MB (100 μM) were present at the same time suggesting that the reduction of MB by NADH represents the first step of re-oxydation of ferric iron, therefore primarily produced by LMB and not by NADH.
Figure 8.

A, Absorbance of a solution of oxidized cytochrome C (CC, 50 μM) mixed with MB (100 μM), NADH (200 μM), or both MB + NADH at neutral pH and ambient temperature. Note that only in the presence of both MB and NADH was cytochrome C reduced (shift of the Soral band to the left and apparition of 2 peaks of absorbance at 520 and 550 nm—specific of reduced CC—with a disappearance of the peak at 525 nm, specific to oxidized CC). Neither NADH nor MB alone can reduce the oxidized form of CC. The bottom panel shows how that this reaction in vitro was already present at 5 min. B, Effect of H2O2 (500 μM in PBS) versus PBS of the absorbance of a solution of cyctochrome C (CC, 50 μM), previously reduced by MB (100 μM) and NADH (200 μM). H2O2 or PBS were applied at time zero, note that in the nontreated (PBS) solution, at 55 min cytochrome C is still under reduced form, whereas a significant amount of NADH has already been consumed. The presence of H2O2 led to a rapid re-oxidation of cytochrome C. These observations are the basis for our proposed hypothesis, displayed in (C), of a potential cyclic re-oxidation of complexes I to III of the electron chain (via H2O2 production during LMB re-oxidation by O2) and their direct reduction by LMB (see Discussion section for further details).
Effects of H2O2 of Oxidized Cytochrome C
Concentration of H2O2 in the high millimolar range produced a modification of the absorbance of cytochrome C with a progressive disappearance of the Soral band reflecting H2O2 toxicity. However, lower concentrations of H2O2 (500 μM) led to a re-oxidation of the cytochrome C previously reduced by NADH and MB (Figure 8), this effect was maximal at 5 min (Figure 8).
Effects of MB on NAHS Toxicity on Mitochondrial Superoxide (O2−) Levels and Mitochondrial Membrane Potential (ΔΨm) in Isolated Cardiomyocytes: Can MB Restore ΔΨm?
Fluorescence signal of MitoSOX Red which detects mitochondrial superoxide anion, colocalized with the mitochondrial membrane potential marker rhodamine 123, confirming MitoSOX Red fluorescence was mitochondrial in origin (Figure 9). NAHS (100 µM) decreased Δψm and increased O2−, whereas MB (20 µg/ml added 3 min NAHS) reversed the deleterious changes brought on by NAHS (Figure 9). MB by itself had no effects on Δψm or O2− (Figure 9). Of note, we have previously reported that in this preparation, there was no difference in the concentration of H2S in the dish within the time frame of these experiments with or without MB (Judenherc-Haouzi et al., 2016) and confirmed this observation by measuring the amount of H2S present in solution which was similar in both conditions (with or without MB).
Figure 9.

Isolated LV myocytes from adult C57BL6 mice loaded with mitochondrial indicator rhodamine 123 (123 μM, Invitrogen) and mitochondrial superoxide-sensitive fluorophore MitoSOX red (22 μM; Invitrogen). Cells were exposed for 10 min to PBS, NaSH (100 μM), MB (20 μg/ml), and NaHS + MB (MB added 3 min after NaHS). Cells were imaged using a Carl Zeiss Meta 510 Meta confocal microscope was used with 1.7× digital zoom at 488 and 561 nm for rhodamine 123 and MitoSOX Red, respectively. H2S decreased the mitochondrial proton gradient, a marker of an impaired electron transport chain activity, in intoxicated cells and increased the production of reactive O2 species levels. MB rescued both responses. **p < .01; ***p < .001. Note that MB alone had no effect on the either signal.
DISCUSSION
In adult sheep, a lethal sulfide exposure caused a very rapid distension of the left ventricle and a profound depression in cardiac contractility, which led to a pulseless electrical activity within 10 min. In less severe exposure, ie, conditions during which animals were alive at the end of exposure to H2S, a persistent cardiac failure potentially lethal was present after the cessation of exposure. This long-lasting depression in cardiac contractility consisted, after a brief period of improvement, in a linear decrease in cardiac output with time along with persistent hyperlactacidemia and decrease in P/L ratio. MB opposed the effects of H2S when administered at the same time as a lethal sulfide exposure allowing all animals to survive. In a more clinically relevant scenario, ie, when MB was administered following a sublethal exposure, MB was able to significantly improve cardiac contractility and to restore the P/L ratio. Interestingly, when MB was administered 30 min after the end of sulfide infusion, MB could still significantly improve the persistent depressed cardiac contractility. Of note, whether administered at 3 or 30 min, these responses were observed while free sulfide had disappeared from the blood (based on determination of sulfide concentrations in the expired gas).
Depression in Cardiac Contractility During H2S Intoxication in a Large Mammal
We have previously established that as soon as H2S diffuses into the blood and cells, the vast majority of free H2S almost instantly disappears due to its immediate “combination” with various proteins and its rapid oxidation into polysulfide, sulfite, sulfate, and thiosulfate—a reaction catalyzed in cells by a quinone oxido-reductase (Bouillaud and Blachier, 2011; Lagoutte et al., 2010; Leschelle et al., 2005; Modis et al., 2013). A small portion of free sulfide is still present in the blood, as long as the exposure is maintained, comprising of gaseous H2S (Barrett et al., 1988; Carroll and Mather, 1989; De Bruyn et al., 1995; Douabul and Riley, 1979) and the sulfhydryl anion HS− (Almgren et al., 1976; Millero, 1986). However, as soon as exposure ceases, the rate of oxidation of H2S is so rapid that this remaining pool of “free/soluble” H2S almost immediately vanishes from the blood and tissues (Haggard, 1921; Haouzi et al., 2014; Klingerman et al., 2013; Toombs et al., 2010). The present study confirms this finding, as H2S disappears from the expired gas of the animals almost immediately after the cessation of infusion (Figure 5). In contrast to “free” H2S, the pool of combined sulfide can persist at low concentrations for relatively long periods of time (Haouzi et al., 2014) and has been put forward to explain the persistent symptoms of toxicity.
We have also previously found that as soon as the concentrations of H2S reached about 10 μM in the blood during sulfide exposure, toxic symptoms develop in the form of a rapid decrease in cardiac contractility in the rat (Judenherc-Haouzi et al., 2016; Sonobe and Haouzi, 2015; Sonobe et al., 2015; Haouzi and Sonobe, 2015). This circulatory failure constitutes one of the main life-threatening manifestations of sulfide toxicity. These cardiac alterations are therefore similar in large and small mammals and can be accounted by various alteration in cardiac ion channels that we have previously characterized, including a profound inhibition of L-type Ca2+ channels (Judenherc-Haouzi et al., 2016). Of note, the risk of PEA (or much more rarely ventricular fibrillation) still persisted after the exposure. On a practical standpoint, sheep alive 5 min after exposure remained alive for at least an hour without any treatment. In most of these animals, the cardiac function remained however seriously depressed, deteriorating even more over time.
Effects of MB on the Pool of Free Sulfide
Two situations must be clearly distinguished, as they do not account for the same effect of MB. During H2S exposure, because there is a pool of free/gaseous H2S present in the blood and tissues, the effects of MB could therefore be mediated by a direct interaction with H2S (Resch et al., 1989). H2S could, as a reducing agent, be reacting with the oxidized form of MB (Li and Lancaster, 2013) as demonstrated in the present study at high concentration of H2S (millimolar range). However, at concentrations consistent with those observed in H2S intoxication, ie, micromolar range, we have previously found that mixing MB and H2S did not affect in a measurable manner the concentrations of gaseous H2S at least in the time frame used for our measurements (Judenherc-Haouzi et al., 2016).
We also observed that a new peak of absorbance of hemoglobin, at 620 nm, developed but only in the animals that received MB and H2S at the same time, in contrast to those that received H2S only. A very similar effect was produced in vitro (Figures 3 and 6): a peak of absorbance at 620 nm was produced when both MB and H2S (100 μM) were mixed with blood. This peak is traditionally considered as a marker of the presence of “sulfhemoglobin” (Bagarinao and Vetter, 1992), but then what is the role of MB in “potentiating” this reaction? As previously suggested (Stossel and Jennings, 1966), methemoglobinemia is not typically present following MB administration (Burrows, 1984; Stossel and Jennings, 1966), at least not at the doses that were used in the present study and within the resolution of methemoglobinemia determination. After all, MB is a treatment of methemoglobinemia. However, although MB by itself is not able to oxidize Fe2+ to Fe3+, a cyclic conversion of hemoglobin into methemoglobin, due to the decrease in NADPH (and possibly reduced glutathione in the red cells) could be produced. This reaction will be opposed by the reduction by LMB of ferric iron (Fe3+) and its cyclic transformation into a ferrous form (Wendel, 1934). Such enhanced cyclic reaction could increase the chance for molecules of free sulfide to be trapped without any significant increase in mean concentration of methemoglobin.
The role of H2O2 produced during the re-oxidation of LMB by O2 should be considered, as the formation of ferryl iron (Fe4+), produced by H2O2, has been already shown to lead to a peak of absorbance at 620 nm, when H2S is present (Michel, 1938). Incidentally, adding H2S to a solution of hemoglobin and looking for a peak of absorbance at 620 nm is a method used to identify the presence of ferryl iron (HBFe4+) (Chintagari et al., 2016). In other words, the presence of a peak at 620 nm in our studies may well reflect the presence of ferryl iron reacting with sulfide. One could propose that molecules of hydrogen peroxide produced during the re-oxidation of LMB by O2 (Schirmer et al., 2011; Tretter et al., 2014; Wainwright and Amaral, 2005), before being “transformed” into H2O by the catalase presents in red cells, could change the redox state of iron into a ferryl state (Patel et al., 1996), “fossilizing” H2S and in turn impeding sulfide diffusion to the tissues. This could certainly account for the lessening of H2S toxicity without creating a permanent methemoglobinemia.
Finally, it should be pointed out that the artificial situation consisting in administering MB and H2S (intravenously) at the same time can create a virtuous circle as well as a vicious circle wherein any improvement in cardiac function and thus cardiac output by MB will decrease the concentration of free H2S in the blood by a simple phenomenon of dilution (Figure 4), whereas for the same rate of NaHS infusion, as cardiac output decreases, sulfide concentrations will rise with an inverse relation to systemic blood flow.
Effects of MB on H2S Inhibition of TCA Cycle: Interactions Between MB and NADH
The fixation of H2S on CCO (Cooper and Brown, 2008) prevents its re-oxidation by O2. As a consequence, (1) the mitochondrial complexes upstream to CCO are maintained in a reduced state and become unable to transfer protons across the inner mitochondrial membrane leading to an inhibition of mitochondrial ATP synthesis (Kim et al., 2012); (2) NADH is not oxidized anymore by an already reduced complex I. The increase in NADH/NAD ratio impedes the TCA cycle (LaNoue et al., 1972; Liu et al., 2018), which in turn suppresses the formation of ATP via the mitochondrial substrate-level phosphorylation (TCA cycle) and catalyzes the transformation of pyruvate into lactate in the cytoplasm (Burgner and Ray, 1984). Based on our previous data obtained on cell cultures and showing that in the high micromolar range, MB increased O2 even when the electron chain was blocked, we proposed (Haouzi et al., 2018) that the increase of O2 consumption produced by MB is mainly the result of the re-oxidation of LMB by O2 and thus does not necessarily reflect a recovery of the electron chain (Atamna et al., 2008; Daudt et al., 2012; Poteet et al., 2012; Wen et al., 2011; Zhang et al., 2006). In this context the increase in CO2 that we observed is extremely relevant and could be used as a marker of the TCA cycle activity. The antidotal effect of MB after exposure could therefore be explained by the consequences of the oxidation of NADH allowing the Krebs cycle to resume its activity at a rate corresponding to the rate of MB reduction (or NADH oxidation). The restoration by MB of the P/L ratio (Albrecht et al., 1971; Levine, 1977), a marker of NAD/NADH ratio, supports this hypothesis.
MB Does Rescue the Mitochondrial Membrane Potential
The results illustrated in Figure 9 suggest that MB could rescue the mitochondrial membrane potential altered by H2S. Series of studies have suggested that MB (in the form LMB) could support the transfer of protons through the mitochondrial membrane against a concentration gradient, essential for the production of adenosine triphosphate (ATP), by allowing electrons to “flow” between complexes (Lindahl and Oberg, 1961; Scott and Hunter, 1966; Zhang et al., 2006). These effects, although not very well understood, have been put forward to account for MB-LMB beneficial effects in various conditions associated with altered mitochondrial functions (Atamna et al., 2008; Atamna and Kumar, 2010; Daudt et al., 2012; Lin et al., 2012; Poteet et al., 2012). In all these studies, the rationale was that LMB provides electrons to the complex III or IV, restoring in turn part of the electron chain activity and allowing ATPase activity to resume (Riha et al., 2011; Rojas et al., 2009, 2012; Wen et al., 2011; Zhang et al., 2006). As mentioned in the Introduction, the blockade of complex IV by H2S (Cooper and Brown, 2008; Nicholls et al., 2013) makes such a hypothetical mechanism immaterial, as the electron chain is already in a reduced state. To try to reconcile the effects of MB during sulfide intoxication with the data obtained in Figure 9, we are speculating that, akin to the effects of H2S in red cells, the presence of hydrogen peroxide produced during the re-oxidation of LMB could first re-oxidize the reduced complex III (Jancura et al., 2014; Jünemann et al., 2000), as illustrated in Figure 8. The question is whether such a re-oxidation will be able to restore the capacity of the electron chain to be reduced again by LMB or NADH in a cyclic manner, transferring electrons between the complexes I and III, whereas complex IV remains “blocked.” As summarized in Figure 8, a local production of small concentrations of H2O2 during LMB re-oxidation may represent an interesting rescue mechanism to be explored. Finally, the meaning of a decrease in the production of reactive O2 species by MB in cells intoxicated by H2S (Figure 9) should be understood in the light on recent data published by Olson et al. (2018). Indeed, because the methodology used to identify ROS in vitro may not be able to differentiate between oxygen or sulfur reactive species (DeLeon et al., 2016), the effects of MB on the production and oxidation of polysulfide and reactive sulfur species (rather than or in addition to ROS) remains to be characterized.
In conclusion, MB exerts its antidotal properties during and following H2S intoxication in a large mammal through mechanisms that are not only dependent on the pool of free H2S. Indeed, MB also appears to counteract the deleterious metabolic consequences produced by H2S at the mitochondrial level.
SUPPLEMENTARY DATA
Supplementary data are available at Toxicological Sciences online.
FUNDING
This work was supported by the National Institutes of Health Office of the Director (NIH OD), and the National Institute of Neurological Disorders and Stroke (NINDS) (U01 NS097162-03).
Supplementary Material
{kind=link}
REFERENCES
- Albani J. R. (2007). Principles and Applications of Fluorescence Spectroscopy. Blackwell Science, Oxford; Ames, IA, viii, 255 pp., 4 pp. of plates. [Google Scholar]
- Albrecht V., Roigas H., Schultze M., Jacobasch G., Rapoport S. (1971). The influence of pH and methylene blue on the pathways of glucose utilization and lactate formation in erythrocytes of man. Eur. J. Biochem. 20, 44–50. [DOI] [PubMed] [Google Scholar]
- Almeida A. F., Guidotti T. L. (1999). Differential sensitivity of lung and brain to sulfide exposure: A peripheral mechanism for apnea. Toxicol. Sci. 50, 287–293. [DOI] [PubMed] [Google Scholar]
- Almeida A. F., Nation P. N., Guidotti T. L. (2008). Mechanism and treatment of sulfide-induced coma: A rat model. Int. J. Toxicol. 27, 287–293. [DOI] [PubMed] [Google Scholar]
- Almgren T., Dyrssen D., Elgquist B., Johansson O. (1976). Dissociation of hydrogen sulfide in seawater and comparison of pH scales. Mar. Chem. 4, 289–297. [Google Scholar]
- Appaix F., Minatchy M., Riva-Lavieille C., Olivares J., Antonsson B., Saks V. A. (2000). Rapid spectrophotometric method for quantitation of cytochrome c release from isolated mitochondria or permeabilized cells revisited. Biochim. Biophys. Acta 1457, 175–181. [DOI] [PubMed] [Google Scholar]
- Ash-Bernal R., Wise R., Wright S. M. (2004). Acquired methemoglobinemia: A retrospective series of 138 cases at 2 teaching hospitals. Medicine 83, 265–273. [DOI] [PubMed] [Google Scholar]
- Atamna H., Kumar R. (2010). Protective role of methylene blue in Alzheimer’s disease via mitochondria and cytochrome c oxidase. J. Alzheimers Dis. 20(Suppl. 2), S439–S452. [DOI] [PubMed] [Google Scholar]
- Atamna H., Nguyen A., Schultz C., Boyle K., Newberry J., Kato H., Ames B. N. (2008). Methylene blue delays cellular senescence and enhances key mitochondrial biochemical pathways. FASEB J. 22, 703–712. [DOI] [PubMed] [Google Scholar]
- Austigard A. D., Svendsen K., Heldal K. K. (2018). Hydrogen sulphide exposure in waste water treatment. J. Occup. Med. Toxicol. 13, 10.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagarinao T., Vetter R. D. (1992). Sulfide-hemoglobin interactions in the sulfide-tolerant salt marsh resident in the California killifish Fundulus parvipinnis. J. Comp. Physiol. B 162, 614–624. [Google Scholar]
- Baldelli R. J., Green F. H., Auer R. N. (1993). Sulfide toxicity: Mechanical ventilation and hypotension determine survival rate and brain necrosis. J. Appl. Physiol. 75, 1348–1353. [DOI] [PubMed] [Google Scholar]
- Barrett T. J., Anderson G. M., Lugowski J. T. (1988). The solubility of hydrogen sulphide in 0-5 m NaCl solutions at 25-95 C and one atmosphere. Geochim. Cosmochim. Acta 52, 807–811. [Google Scholar]
- Beauchamp R. O. Jr, Bus J. S., Popp J. A., Boreiko C. J., Andjelkovich D. A. (1984). A critical review of the literature on hydrogen sulfide toxicity. Crit. Rev. Toxicol. 13, 25–97. [DOI] [PubMed] [Google Scholar]
- Beck J. F., Bradbury C. M., Connors A. J., Donini J. C. (1981). Nitrite as antidote for acute hydrogen sulfide intoxication? Am. Ind. Hyg. Assoc. J. 42, 805–809. [DOI] [PubMed] [Google Scholar]
- Bergmann K., O’Konski C. T. (1963). A spectroscopic study of methylene blue monomer, dimer, and complexes with montmorillonite. J. Phys. Chem. 67, 2169–2177. [Google Scholar]
- Blackstone E., Morrison M., Roth M. B. (2005). H2S induces a suspended animation-like state in mice. Science 308, 518–15845845. [DOI] [PubMed] [Google Scholar]
- Bouillaud F., Blachier F. (2011). Mitochondria and sulfide: A very old story of poisoning, feeding, and signaling? Antioxid. Redox. Signal. 15, 379–391. [DOI] [PubMed] [Google Scholar]
- Brenner M., Benavides S., Mahon S. B., Lee J., Yoon D., Mukai D., Viseroi M., Chan A., Jiang J., Narula N., et al. (2014). The vitamin B12 analog cobinamide is an effective hydrogen sulfide antidote in a lethal rabbit model. Clin. Toxicol. 52, 490–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks M. M. (1933). Methylene blue as antidote for cyanide and carbon monoxide poisoning. J. Am. Med. Assoc. 100, 59. [Google Scholar]
- Burgner J. W. 2nd, Ray W. J. Jr. (1984). On the origin of the lactate dehydrogenase induced rate effect. Biochemistry 23, 3636–3648. [DOI] [PubMed] [Google Scholar]
- Burrows G. E. (1984). Methylene blue: Effects and disposition in sheep. J. Vet. Pharmacol. Ther. 7, 225–231. [DOI] [PubMed] [Google Scholar]
- Carroll J. J., Mather A. E. (1989). The solubility of hydrogen sulfide in water from 0 to 90°C and pressures to 1 MPa. Geochim. Cosmochim. Acta 53, 1163–1170. [Google Scholar]
- Chenard L., Lemay S. P., Lague C. (2003). Hydrogen sulfide assessment in shallow-pit swine housing and outside manure storage. J. Agric. Saf. Health 9, 285–302. [DOI] [PubMed] [Google Scholar]
- Cheung J. Y., Wang J., Zhang X. Q., Song J., Davidyock J. M., Prado F. J. (2018). Methylene blue counteracts H2S-induced cardiac ion channel dysfunction and ATP reduction. Cardiovasc. Toxicol. 18, 407–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung J. Y., Wang J. F., Zhang X.-Q., Song J., Tomar D., Madesh M., Judenherc-Haouzi A., Haouzi P. (2018). Methylene blue counteracts cyanide cardiotoxicity: Cellular mechanisms. J. Appl. Physiol. 124, 1164–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung N. S., Peng Z. F., Chen M. J., Moore P. K., Whiteman M. (2007). Hydrogen sulfide induced neuronal death occurs via glutamate receptor and is associated with calpain activation and lysosomal rupture in mouse primary cortical neurons. Neuropharmacology 53, 505–514. [DOI] [PubMed] [Google Scholar]
- Chintagari N. R., Jana S., Alayash A. I. (2016). Oxidized ferric and ferryl forms of hemoglobin trigger mitochondrial dysfunction and injury in alveolar type I cells. Am. J. Respir. Cell. Mol. Biol. 55, 288–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clifton J. 2nd, Leikin J. B. (2003). Methylene blue. Am. J. Ther. 10, 289–291. [DOI] [PubMed] [Google Scholar]
- Cooper C. E., Brown G. C. (2008). The inhibition of mitochondrial cytochrome oxidase by the gases carbon monoxide, nitric oxide, hydrogen cyanide and hydrogen sulfide: Chemical mechanism and physiological significance. J. Bioenerg. Biomembr. 40, 533–539. [DOI] [PubMed] [Google Scholar]
- Dalgaard J. B., Dencker F., Fallentin B., Hansen P., Kaempe B., Steensberg J. (1972). Fatal poisoning and other health hazards connected with industrial fishing. Br. J. Ind. Med. 29, 307–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daudt D. R. 3rd, Mueller B., Park Y. H., Wen Y., Yorio T. (2012). Methylene blue protects primary rat retinal ganglion cells from cellular senescence. Invest. Ophthalmol. Vis. Sci. 53, 4657–4667. [DOI] [PubMed] [Google Scholar]
- De Bruyn W. J., Swartz E., Hu J. H., Shorter J. A., Davidovits P., Worsnop D. R., Zahniser M. S., Kolb C. E. (1995). Henry’s law solubilities and Setchenow coefficients for biogenic reduced sulfur species obtained from gas-liquid uptake measurements. J. Geophys. Res. 100, 7245–7251. [Google Scholar]
- DeLeon E. R., Gao Y., Huang E., Arif M., Arora N., Divietro A., Patel S., Olson K. R. (2016). A case of mistaken identity: Are reactive oxygen species actually reactive sulfide species? Am. J. Physiol. Regul. Integr. Comp. Physiol. 310, R549–R560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorman D. C., Moulin F. J., McManus B. E., Mahle K. C., James R. A., Struve M. F. (2002). Cytochrome oxidase inhibition induced by acute hydrogen sulfide inhalation: Correlation with tissue sulfide concentrations in the rat brain, liver, lung, and nasal epithelium. Toxicol. Sci. 65, 18–25. [DOI] [PubMed] [Google Scholar]
- Douabul A. A., Riley J. P. (1979). The solubility of gases in distilled water and seawater - V. Hydrogen sulphide. Deep Sea Res. 26A, 259–268. [Google Scholar]
- Eddy N. B. (1930). Antagonism between methylene blue and sodium cyanide. J. Pharm. Exp. Ther. 39, 271. [Google Scholar]
- Eghbal M. A., Pennefather P. S., O’Brien P. J. (2004). H2S cytotoxicity mechanism involves reactive oxygen species formation and mitochondrial depolarisation. Toxicology 203, 69–76. [DOI] [PubMed] [Google Scholar]
- Frappell P., Lanthier C., Baudinette R. V., Mortola J. P. (1992). Metabolism and ventilation in acute hypoxia: A comparative analysis in small mammalian species. Am. J. Physiol. 262, R1040–R1046. [DOI] [PubMed] [Google Scholar]
- Ginimuge P. R., Jyothi S. D. (2010). Methylene blue: Revisited. J. Anaesth. Clin. Pharmacol. 26, 517–520. [PMC free article] [PubMed] [Google Scholar]
- Greer J. J., Reiffenstein R. J., Almeida A. F., Carter J. E. (1995). Sulfide-induced perturbations of the neuronal mechanisms controlling breathing in rats. J. Appl. Physiol. 78, 433–440. [DOI] [PubMed] [Google Scholar]
- Guidotti T. L. (1994). Occupational exposure to hydrogen sulfide in the sour gas industry: Some unresolved issues. Int. Arch. Occup. Environ. Health 66, 153–160. [DOI] [PubMed] [Google Scholar]
- Guidotti T. L. (2010). Hydrogen sulfide: Advances in understanding human toxicity. Int. J. Toxicol. 29, 569–581. [DOI] [PubMed] [Google Scholar]
- Haggard H. W. (1921). The fate of sulfides in the blood. J. Biol. Chem. 49, 519–529. [Google Scholar]
- Hall A. H., Rumack B. H. (1997). Hydrogen sulfide poisoning: An antidotal role for sodium nitrite? Vet. Hum. Toxicol. 39, 152–154. [PubMed] [Google Scholar]
- Haouzi P. (2011). Sulfide and methemoglobinemia. Respir. Physiol. Neurobiol. 179, 119–120. [Google Scholar]
- Haouzi P., Chenuel B. (2005). Control of arterial PCO2 by somatic afferents in sheep. J. Physiol. 569, 975–987. [ 10.1113/jphysiol.2005.089649] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haouzi P., Sonobe T. (2015). Cardiogenic shock induced reduction in cellular O2 delivery as a hallmark of acute H2S intoxication. Clin. Toxicol. 53, 416–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haouzi P., Bell H. J., Notet V., Bihain B. (2009). Comparison of the metabolic and ventilatory response to hypoxia and H2S in unsedated mice and rats. Respir. Physiol. Neurobiol. 167, 316–322. [DOI] [PubMed] [Google Scholar]
- Haouzi P., Chenuel B., Sonobe T. (2015). High-dose hydroxocobalamin administered after H2S exposure counteracts sulfide-poisoning-induced cardiac depression in sheep. Clin. Toxicol. 53, 28–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haouzi P., Chenuel B., Sonobe T., Klingerman C. M. (2014). Are H2S-trapping compounds pertinent to the treatment of sulfide poisoning? Clin. Toxicol. 52, 566.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haouzi P., Gueguinou M., Sonobe T., Judenherc-Haouzi A., Tubbs N., Trebak M.. 2018. Revisiting the physiological effects of methylene blue as a treatment of cyanide intoxication. Clin. Toxicol. 56828–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haouzi P., Notet V., Chenuel B., Chalon B., Sponne I., Ogier V., Bihain B. (2008). H2S induced hypometabolism in mice is missing in sedated sheep. Respir. Physiol. Neurobiol. 160, 109–115. [DOI] [PubMed] [Google Scholar]
- Haouzi P., Sonobe T., Judenherc-Haouzi A. (2016). Developing effective countermeasures against acute hydrogen sulfide intoxication: Challenges and limitations. Ann. N.Y. Acad. Sci. 1374, 29–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haouzi P., Sonobe T., Torsell-Tubbs N., Prokopczyk B., Chenuel B., Klingerman C. M. (2014). In vivo interactions between cobalt or ferric compounds and the pools of sulphide in the blood during and after H2S poisoning. Toxicol. Sci. 141, 493–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickson R. G., Chang A., Hamilton R. J. (2004). Co-worker fatalities from hydrogen sulfide. Am. J. Ind. Med. 45, 346–350. [DOI] [PubMed] [Google Scholar]
- Horecker B. L. (1943). The absorption spectra of hemoglobin and its derivatives in the visible and near infra-red regions. J. Biol. Chem. 148, 173–183. [Google Scholar]
- Jancura D., Stanicova J., Palmer G., Fabian M. (2014). How hydrogen peroxide is metabolized by oxidized cytochrome c oxidase. Biochemistry 53, 3564–3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judenherc-Haouzi A., Zhang X.-Q., Sonobe T., Song J., Rannals M. D., Wang J. F., Tubbs N., Cheung J. Y., Haouzi P. (2016). Methylene blue counteracts H2S toxicity-induced cardiac depression by restoring L-type Ca channel activity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 310, R1030–R1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jünemann S., Heathcote P., Rich P. R. (2000). The reactions of hydrogen peroxide with bovine cytochrome c oxidase. Biochim. Biophys. Acta 1456, 56–66. [DOI] [PubMed] [Google Scholar]
- Kamijo Y., Takai M., Fujita Y., Hirose Y., Iwasaki Y., Ishihara S. (2013). A multicenter retrospective survey on a suicide trend using hydrogen sulfide in Japan. Clin. Toxicol. 51, 425–428. [DOI] [PubMed] [Google Scholar]
- Kelner M. J., Alexander N. M. (1985). Methylene blue directly oxidizes glutathione without the intermediate formation of hydrogen peroxide. J. Biol. Chem. 260, 15168–15171. [PubMed] [Google Scholar]
- Kim H. J., Khalimonchuk O., Smith P. M., Winge D. R. (2012). Structure, function, and assembly of heme centers in mitochondrial respiratory complexes. Biochim. Biophys. Acta. 1823, 1604–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingerman C. M., Trushin N., Prokopczyk B., Haouzi P. (2013). H2S concentrations in the arterial blood during H2S administration in relation to its toxicity and effects on breathing. Am. J. Physiol. Regul. Integr. Comp. Physiol. 305, R630–R638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch H., Schneider D.. 2007. Folding, assembly, and stability of transmembrane cytochromes. Curr. Chem. Biol. 1, 59–74. [Google Scholar]
- Komlodi T., Tretter L. (2017). Methylene blue stimulates substrate-level phosphorylation catalysed by succinyl-CoA ligase in the citric acid cycle. Neuropharmacology 123, 287–298. [DOI] [PubMed] [Google Scholar]
- Lagoutte E., Mimoun S., Andriamihaja M., Chaumontet C., Blachier F., Bouillaud F. (2010). Oxidation of hydrogen sulfide remains a priority in mammalian cells and causes reverse electron transfer in colonocytes. Biochim. Biophys. Acta 1797, 1500–1511. [DOI] [PubMed] [Google Scholar]
- LaNoue K. F., Bryla J., Williamson J. R. (1972). Feedback interactions in the control of citric acid cycle activity in rat heart mitochondria. J. Biol. Chem. 247, 667–679. [PubMed] [Google Scholar]
- Leschelle X., Goubern M., Andriamihaja M., Blottière H. M., Couplan E., Gonzalez-Barroso M-d-M., Petit C., Pagniez A., Chaumontet C., Mignotte B., et al. (2005). Adaptative metabolic response of human colonic epithelial cells to the adverse effects of the luminal compound sulfide. Biochim. Biophys. Acta 1725, 201–212. [DOI] [PubMed] [Google Scholar]
- Levine S. (1977). Interaction between ethyl methylene blue and cyanide-induced increases in blood lactate. J. Lab. Clin. Med. 89, 632–639. [PubMed] [Google Scholar]
- Li Q., Lancaster J. R. Jr. (2013). Chemical foundations of hydrogen sulfide biology. Nitric Oxide 35, 21–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin A.-L., Poteet E., Du F., Gourav R. C., Liu R., Wen Y., Bresnen A., Huang S., Fox P. T., Yang S.-H., et al. (2012). Methylene blue as a cerebral metabolic and hemodynamic enhancer. PLoS One 7, e46585.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl P. E., Oberg K. E. (1961). The effect of rotenone on respiration and its point of attack. Exp. Cell Res. 23, 228–237. [DOI] [PubMed] [Google Scholar]
- Liu Y., Hu L., Ma T., Yang J., Ding J. (2018). Insights into the inhibitory mechanisms of NADH on the alphagamma heterodimer of human NAD-dependent isocitrate dehydrogenase. Sci. Rep. 8, 3146.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel H. (1938). A study of Sulfhemoglobin. J. Biol. Chem. 126, 323–348. [Google Scholar]
- Mihajlovic A. (1999). Antidotal mechanisms for hydrogen sulfide toxicity. PhD thesis, Department of Pharmaceutical Sciences, University of Toronto, Toronto.
- Millero F. J. (1986). The thermodynamics and kinetics of hydrogen sulfide system in natural waters. Mar. Chem. 18, 121–147. [Google Scholar]
- Modis K., Coletta C., Erdelyi K., Papapetropoulos A., Szabo C. (2013). Intramitochondrial hydrogen sulfide production by 3-mercaptopyruvate sulfurtransferase maintains mitochondrial electron flow and supports cellular bioenergetics. FASEB J. 27, 601–611. [DOI] [PubMed] [Google Scholar]
- Mortola J. P. (1993). Hypoxic hypometabolism in mammals. News Physiol. Sci. 8, 79–82. [Google Scholar]
- Mustafa A. K., Gadalla M. M., Sen N., Kim S., Mu W., Gazi S. K., Barrow R. K., Yang G., Wang R., Snyder S. H. (2009). H2S signals through protein S-sulfhydration. Sci. Signal. 2, ra72.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls P., Marshall D. C., Cooper C. E., Wilson M. T. (2013). Sulfide inhibition of and metabolism by cytochrome c oxidase. Biochem. Soc. Trans. 41, 1312–1316. [DOI] [PubMed] [Google Scholar]
- Olson K. R. (2013). Hydrogen sulfide: Both feet on the gas and none on the brake? Front. Physiol. 4, 2.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson K. R., Gao Y., Arif F., Arora K., Patel S., DeLeon E. R., Sutton T. R., Feelisch M., Cortese-Krott M. M., Straub K. D. (2018). Metabolism of hydrogen sulfide (H2S) and production of reactive sulfur species (RSS) by superoxide dismutase. Redox. Biol. 15, 74–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel R. P., Svistunenko D. A., Darley-Usmar V. M., Symons M. C., Wilson M. T. (1996). Redox cycling of human methaemoglobin by H2O2 yields persistent ferryl iron and protein based radicals. Free Radic. Res. 25, 117–123. [DOI] [PubMed] [Google Scholar]
- Paul B. D., Snyder S. H. (2012). H(2)S signalling through protein sulfhydration and beyond. Nat. Rev. Mol. Cell Biol. 13, 499–507. [DOI] [PubMed] [Google Scholar]
- Poteet E., Winters A., Yan L.-J., Shufelt K., Green K. N., Simpkins J. W., Wen Y., Yang S.-H. (2012). Neuroprotective actions of methylene blue and its derivatives. PLoS One 7, e48279.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reedy S. J., Schwartz M. D., Morgan B. W. (2011). Suicide fads: Frequency and characteristics of hydrogen sulfide suicides in the United States. West J. Emerg. Med. 12, 300–304. [PMC free article] [PubMed] [Google Scholar]
- Resch P., Field R. J., Schneider F. W., Burger M. (1989). Reduction of methylene blue by sulfide ion in the presence and absence of oxygen: Simulation of the methylene blue-O2-HS- CSTR oscillations. J. Phys. Chem. 93, 8181–8186. [Google Scholar]
- Riha P. D., Rojas J. C., Gonzalez-Lima F. (2011). Beneficial network effects of methylene blue in an amnestic model. NeuroImage 54, 2623–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rincon P. (2005). Mice put in ‘suspended animation’. BBC News, April 21, 2005.
- Rojas J. C., Bruchey A. K., Gonzalez-Lima F. (2012). Neurometabolic mechanisms for memory enhancement and neuroprotection of methylene blue. Prog. Neurobiol. 96, 32–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas J. C., Simola N., Kermath B. A., Kane J. R., Schallert T., Gonzalez-Lima F. (2009). Striatal neuroprotection with methylene blue. Neuroscience 163, 877–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahlin B. (1926). The antagonism between methylene blue and cyan potassium. Skand. Arch. Physiol. 47, 284–291. [Google Scholar]
- Sams R. N., Carver H. W. 2nd, Catanese C., Gilson T. (2013). Suicide with hydrogen sulfide. Am. J. Forensic Med. Pathol. 34, 81–82. [DOI] [PubMed] [Google Scholar]
- Schirmer R. H., Adler H., Pickhardt M., Mandelkow E. (2011). “Lest we forget you–methylene blue…”. Neurobiol. Aging 32, 2325.e7-16. [DOI] [PubMed] [Google Scholar]
- Scott A., Hunter F. E. Jr (1966). Support of thyroxine-induced swelling of liver mitochondria by generation of high energy intermediates at any one of three sites in electron transport. J. Biol. Chem. 241, 1060–1066. [PubMed] [Google Scholar]
- Sevcik P., Dunford H. B. (1991). Kinetics of the oxidation of NADH by methylene blue in a closed system. J. Phys. Chem. 95, 2411–2415. [Google Scholar]
- Smith R. P. (1967). The oxygen and sulfide binding characteristics of hemoglobins generated from methemoglobin by two erythrocytic systems. Mol. Pharmacol. 3, 378–385. [PubMed] [Google Scholar]
- Smith R. P. (1981). Nitrite treatment for hydrogen sulfide poisoning. Ann. Intern. Med. 95, 782.. [DOI] [PubMed] [Google Scholar]
- Smith R. P., Gosselin R. E. (1966). On the mechanism of sulfide inactivation by methemoglobin. Toxicol. Appl. Pharmacol. 8, 159–172. [DOI] [PubMed] [Google Scholar]
- Sonobe T., Haouzi P. (2015). H2S induced coma and cardiogenic shock in the rat: Effects of phenothiazinium chromophores. Clin. Toxicol. 53, 525–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonobe T., Haouzi P. (2016a). Sulfide intoxication-induced circulatory failure is mediated by a depression in cardiac contractility. Cardiovas. Toxicol. 16, 67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonobe T., Haouzi P. (2016b). H2S concentrations in the heart after acute H2S administration: Methodological and physiological considerations. Am. J. Physiol. Heart Circ. Physiol. 311, H1445.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonobe T., Chenuel B., Cooper T. K., Haouzi P. (2015). Immediate and long-term outcome of acute H2S intoxication induced coma in unanesthetized rats: Effects of methylene blue. PLoS One 10, e0131340.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stossel T. P., Jennings R. B. (1966). Failure of methylene blue to produce methemoglobinemia in vivo. Am. J. Clin. Pathol. 45, 600–604. [DOI] [PubMed] [Google Scholar]
- Stotz E., Sidwell A. E., Hogness T. R. (1938). The spectrophotometric determination of equilibrium in the oxidation-reduction systems: The potential of cytochrome c. J. Biol. Chem. 124, 1–23. [Google Scholar]
- Struve M. F., Brisbois J. N., James R. A., Marshall M. W., Dorman D. C. (2001). Neurotoxicological effects associated with short-term exposure of Sprague-Dawley rats to hydrogen sulfide. Neurotoxicology 22, 375–385. [DOI] [PubMed] [Google Scholar]
- Tang G., Wu L., Wang R. (2010). Interaction of hydrogen sulfide with ion channels. Clin. Exp. Pharmacol. Physiol. 37, 753–763. [DOI] [PubMed] [Google Scholar]
- Teichholz L. E., Kreulen T., Herman M. V., Gorlin R. (1976). Problems in echocardiographic volume determinations: Echocardiographic-angiographic correlations in the presence of absence of asynergy. Am. J. Cardiol. 37, 7–11. [DOI] [PubMed] [Google Scholar]
- Toombs C. F., Insko M. A., Wintner E. A., Deckwerth T. L., Usansky H., Jamil K., Goldstein B., Cooreman M., Szabo C. (2010). Detection of exhaled hydrogen sulphide gas in healthy human volunteers during intravenous administration of sodium sulphide. Br. J. Clin. Pharmacol. 69, 626–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tretter L., Horvath G., Holgyesi A., Essek F., Adam-Vizi V. (2014). Enhanced hydrogen peroxide generation accompanies the beneficial bioenergetic effects of methylene blue in isolated brain mitochondria. Free Radic. Biol. Med. 77, 317–330. [DOI] [PubMed] [Google Scholar]
- Truscott A. (2008). Suicide fad threatens neighbours, rescuers. CMAJ 179, 312–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tvedt B., Skyberg K., Aaserud O., Hobbesland A., Mathiesen T. (1991). Brain damage caused by hydrogen sulfide: A follow-up study of six patients. Am. J. Ind. Med. 20, 91–101. [DOI] [PubMed] [Google Scholar]
- Van de Louw A., Haouzi P. (2013). Ferric iron and cobalt (III) compounds to safely decrease hydrogen sulfide in the body? Antioxid. Redox. Signal. 19, 510–516. [DOI] [PubMed] [Google Scholar]
- von Kompen E. J. (1983). Spectrophotometry of hemoglobin and hemoglobin derivatives In Advances in Clinical Chemistry 23 (Latner A. L., Schwartz M., Eds.), pp. 199–258. Academic Press, New York. [DOI] [PubMed] [Google Scholar]
- Wainwright M., Amaral L. (2005). The phenothiazinium chromophore and the evolution of antimalarial drugs. Trop. Med. Int. Health 10, 501–511. [DOI] [PubMed] [Google Scholar]
- Wen Y., Li W., Poteet E. C., Xie L., Tan C., Yan L.-J., Ju X., Liu R., Qian H., Marvin M. A., et al. (2011). Alternative mitochondrial electron transfer as a novel strategy for neuroprotection. J. Biol. Chem. 286, 16504–16515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendel W. B. (1934). The mechanism of antidotal action of methylene blue in cyanide poisoning. Science 80, 381–382. [Google Scholar]
- Wendel W. B. (1939). The control of methemoglobinemia with methylene blue. J. Clin. Invest. 18, 179–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright R. O., Lewander W. J., Woolf A. D. (1999). Methemoglobinemia: Etiology, pharmacology, and clinical management. Ann. Emerg. Med. 34, 646–656. [DOI] [PubMed] [Google Scholar]
- Zhang X., Rojas J. C., Gonzalez-Lima F. (2006). Methylene blue prevents neurodegeneration caused by rotenone in the retina. Neurotox. Res. 9, 47–57. [DOI] [PubMed] [Google Scholar]
- Zijlstra W. G., Buursma A. (1997). Spectrophotometry of hemoglobin: Absorption spectra of bovine oxyhemoglobin, deoxyhemoglobin, carboxyhemoglobin, and methemoglobin. Comp. Biochem. Physiol. 118, 743–749. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.