

Abstract
Background
This is an updated version of the original Cochrane Review published in September 2014. The most common primary brain tumours in adults are gliomas. Gliomas span a spectrum from low to high grade and are graded pathologically on a scale of one to four according to the World Health Organization (WHO) classification. High‐grade glioma (HGG) carries a poor prognosis. Grade IV glioma is known as glioblastoma and carries a median survival in treated patients of about 15 months. Glioblastomas are rich in blood vessels (i.e. highly vascular) and also rich in a protein known as vascular endothelial growth factor (VEGF) that promotes new blood vessel formation (the process of angiogenesis). Anti‐angiogenic agents inhibit the process of new blood vessel formation and promote regression of existing vessels. Several anti‐angiogenic agents have been investigated in clinical trials, both in newly diagnosed and recurrent HGG, showing preliminary promising results. This review was undertaken to report on the benefits and harms associated with the use of anti‐angiogenic agents in the treatment of HGGs.
Objectives
To evaluate the efficacy and toxicity of anti‐angiogenic therapy in people with high‐grade glioma (HGG). The intervention can be used in two broad groups: at first diagnosis as part of 'adjuvant' therapy, or in the setting of recurrent disease.
Search methods
We conducted updated searches to identify published and unpublished randomised controlled trials (RCTs), including the Cochrane Central Register of Controlled Trials (CENTRAL; 2018, Issue 9), MEDLINE and Embase to October 2018. We handsearched proceedings of relevant oncology conferences up to 2018. We also searched trial registries for ongoing studies.
Selection criteria
RCTs evaluating the use of anti‐angiogenic therapy to treat HGG versus the same therapy without anti‐angiogenic therapy.
Data collection and analysis
Review authors screened the search results and reviewed the abstracts of potentially relevant articles before retrieving the full text of eligible articles.
Main results
After a comprehensive literature search, we identified 11 eligible RCTs (3743 participants), of which 7 were included in the original review (2987 participants). There was significant design heterogeneity in the included studies, especially in the response assessment criteria used. All eligible studies were restricted to glioblastomas and there were no eligible studies evaluating other HGGs. Ten studies were available as fully published peer‐reviewed manuscripts, and one study was available in abstract form. The overall risk of bias in included studies was low. This risk was based upon low rates of selection bias, detection bias, attrition bias and reporting bias. The 11 studies included in this review did not show an improvement in overall survival with the addition of anti‐angiogenic therapy (pooled hazard ratio (HR) of 0.95, 95% confidence interval (CI) 0.88 to 1.02; P = 0.16; 11 studies, 3743 participants; high‐certainty evidence). However, pooled analysis from 10 studies (3595 participants) showed improvement in progression‐free survival with the addition of anti‐angiogenic therapy (HR 0.73, 95% CI 0.68 to 0.79; P < 0.00001; high‐certainty evidence).
We carried out additional analyses of overall survival and progression‐free survival according to treatment setting and for anti‐angiogenic therapy combined with chemotherapy compared to chemotherapy alone. Pooled analysis of overall survival in either the adjuvant or recurrent setting did not show an improvement (HR 0.93, 95% CI 0.86 to 1.02; P = 0.12; 8 studies, 2833 participants; high‐certainty evidence and HR 0.99, 95% CI 0.85 to 1.16; P = 0.90; 3 studies, 910 participants; moderate‐certainty evidence, respectively). Pooled analysis of overall survival for anti‐angiogenic therapy combined with chemotherapy compared to chemotherapy also did not clearly show an improvement (HR 0.92, 95% CI 0.85 to 1.00; P = 0.05; 11 studies, 3506 participants; low‐certainty evidence). The progression‐free survival in the subgroups all showed findings that demonstrated improvements in progression‐free survival with the addition of anti‐angiogenic therapy. Pooled analysis of progression‐free survival in both the adjuvant and recurrent setting showed an improvement (HR 0.75, 95% CI 0.69 to 0.82; P < 0.00001; 8 studies, 2833 participants; high‐certainty evidence and HR 0.64, 95% CI 0.54 to 0.76; P < 0.00001; 2 studies, 762 participants; moderate‐certainty evidence, respectively). Pooled analysis of progression‐free survival for anti‐angiogenic therapy combined with chemotherapy compared to chemotherapy alone showed an improvement (HR 0.72, 95% CI 0.66 to 0.77; P < 0.00001; 10 studies, 3464 participants). Similar to trials of anti‐angiogenic therapies in other solid tumours, adverse events related to this class of therapy included hypertension and proteinuria, poor wound healing, and the potential for thromboembolic events, although generally, the rate of grade 3 and 4 adverse events was low (< 14.1%) and in keeping with the literature. The impact of anti‐angiogenic therapy on quality of life varied between studies.
Authors' conclusions
The use of anti‐angiogenic therapy does not significantly improve overall survival in newly diagnosed people with glioblastoma. Thus, there is insufficient evidence to support the use of anti‐angiogenic therapy for people with newly diagnosed glioblastoma at this time. Overall there is a lack of evidence of a survival advantage for anti‐angiogenic therapy over chemotherapy in recurrent glioblastoma. When considering the combination anti‐angiogenic therapy with chemotherapy compared with the same chemotherapy alone, there may possibly be a small improvement in overall survival. While there is strong evidence that bevacizumab (an anti‐angiogenic drug) prolongs progression‐free survival in newly diagnosed and recurrent glioblastoma, the impact of this on quality of life and net clinical benefit for patients remains unclear. Not addressed here is whether subsets of people with glioblastoma may benefit from anti‐angiogenic therapies, nor their utility in other HGG histologies.
Keywords: Humans; Angiogenesis Inhibitors; Angiogenesis Inhibitors/adverse effects; Angiogenesis Inhibitors/therapeutic use; Antibodies, Monoclonal, Humanized; Antibodies, Monoclonal, Humanized/adverse effects; Antibodies, Monoclonal, Humanized/therapeutic use; Antineoplastic Agents; Antineoplastic Agents/therapeutic use; Bevacizumab; Bevacizumab/therapeutic use; Brain Neoplasms; Brain Neoplasms/blood supply; Brain Neoplasms/drug therapy; Brain Neoplasms/mortality; Camptothecin; Camptothecin/analogs & derivatives; Camptothecin/therapeutic use; Dacarbazine; Dacarbazine/analogs & derivatives; Dacarbazine/therapeutic use; Glioblastoma; Glioblastoma/blood supply; Glioblastoma/drug therapy; Glioblastoma/mortality; Hypertension; Hypertension/chemically induced; Irinotecan; Irinotecan/therapeutic use; Lomustine; Lomustine/therapeutic use; Neoplasm Recurrence, Local; Neoplasm Recurrence, Local/drug therapy; ; /drug therapy; /mortality; Progression-Free Survival; Proteinuria; Proteinuria/chemically induced; Randomized Controlled Trials as Topic; Snake Venoms; Snake Venoms/therapeutic use; Temozolomide; Temozolomide/therapeutic use
Plain language summary
Drugs that target blood vessels in malignant brain tumours
Background The commonest primary brain tumours of adults are gliomas which comprise about two‐fifths of all primary brain tumours. Gliomas span a spectrum from low to high grade, and are graded pathologically on a scale of one to four, according to a classification by the World Health Organization (WHO) that was last updated in 2016. High‐grade glioma, including glioblastoma, are difficult to treat and carry a poor prognosis.
These tumours produce a protein that promotes the formation of new blood vessels (angiogenesis) to help them grow. Drugs have been developed to reduce the formation of new blood vessels and slow tumour growth. Bevacizumab, cediranib and cilengitide are anti‐angiogenic drugs that directly or indirectly target blood vessel formation and have been studied in glioblastoma in randomised clinical trials.
Study characteristics After a comprehensive search up to October 2018, we identified 11 eligible randomised clinical trials (totaling 3743 participants). All eligible studies were restricted to glioblastomas; there were no eligible studies that included other brain tumour types. The largest trials were conducted in newly diagnosed people with glioblastoma, treated with anti‐angiogenic therapy. Overall, the trials included in this systematic review did not show improvement in overall survival with the use of anti‐angiogenic therapy. Overall, the clinical trials in bevacizumab‐treated glioblastoma did prolong the time until tumour growth (progression‐free survival). Key results Anti‐angiogenic therapy does not significantly prolong life in newly diagnosed people with glioblastoma. In recurrent glioblastoma although there is no evidence of prolonging life over chemotherapy, when anti‐angiogenic therapies are used in combination with certain chemotherapy regimes there may be a small improvement in survival. Anti‐angiogenic agents delay tumour progression on magnetic resonance imaging (MRI) scans but is commonly associated with side effects, such as high blood pressure and protein in the urine.
Summary of findings
Summary of findings for the main comparison. Anti‐angiogenic therapy compared to no anti‐angiogenic therapy for high‐grade glioma (HGG).
| Anti‐angiogenic therapy compared to no anti‐angiogenic therapy for HGG | ||||
| Patient or population: people with HGG Setting: outpatient Intervention: anti‐angiogenic therapy Comparison: no anti‐angiogenic therapy | ||||
| Outcomes | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments |
| Overall survival (assessed with HR) | HR 0.95 (0.88 to 1.02) | 3743 (11 RCTs) | ⊕⊕⊕⊕ High | Strong, high‐certainty evidence that there is no or little overall survival benefit of anti‐angiogenic therapy for treatment of HGG, across treatment settings |
| Progression‐free survival (assessed with HR) | HR 0.73 (0.68 to 0.79) | 3595 (10 RCTs) | ⊕⊕⊕⊕ High | Strong, high‐certainty evidence that there is a progression‐free survival benefit of anti‐angiogenic therapy in HGG. The strength of evidence and size of the effect on progression‐free survival in relation to the overall survival results, questions the validity of progression‐free survival as a surrogate endpoint for progression‐free survival in this setting. |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; HGG: high‐grade glioma; HR: hazard ratio; RCT: randomised controlled trial | ||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect Very low certainty: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect | ||||
Summary of findings 2. Anti‐angiogenic therapy compared to no anti‐angiogenic therapy for high‐grade glioma (HGG), subgroups.
| Anti‐angiogenic therapy compared to no anti‐angiogenic therapy for HGG, subgroups | ||||
| Patient or population: people with HGG Setting: outpatient Intervention: anti‐angiogenic therapy Comparison: no anti‐angiogenic therapy (subgroup analysis) | ||||
| Outcomes | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments |
| Overall survival
(assessed with HR) Treatment setting: adjuvant (primary) |
HR 0.93 (0.86 to 1.02) | 2833 (8 RCTs) | ⊕⊕⊕⊕ High | Strong, high‐certainty evidence that there is no overall survival benefit of anti‐angiogenic therapy for treatment of HGG, in the adjuvant treatment setting |
| Overall survival
(assessed with HR) Treatment setting: recurrent |
HR 0.99 (0.85 to 1.16) | 910 (3 RCTs) |
⊕⊕⊕⊝ Moderate a | Moderate‐certainty evidence that anti‐angiogenic therapy does not confer a survival advantage compared to chemotherapy in recurrent HGG |
| Overall survival (assessed with HR) Subgroup: anti‐angiogenic therapy with chemotherapy |
HR 0.92 (0.85 to 1.00) | 3506 (11 RCTs) |
⊕⊕⊝⊝ Lowb |
Evidence that anti‐angiogenic therapy combined with chemotherapy does not improve overall survival compared to chemotherapy. Given the borderline statistical significance, a small survival benefit (of questionable clinical significance) cannot be excluded |
| Progression‐free survival (assessed with HR) Treatment setting: adjuvant (primary) |
HR 0.75 (0.69 to 0.82) | 2833 (8 RCTs) |
⊕⊕⊕⊕ High | Strong, high‐certainty evidence that there is a progression‐free survival benefit of anti‐angiogenic therapy in HGG in the adjuvant setting. The strength of evidence and size of the effect on progression‐free survival in relation to the overall survival results, questions the validity of progression‐free survival as a surrogate endpoint for overall survival in this setting. |
| Progression‐free survival
(assessed with HR) Treatment setting: recurrent |
HR 0.64 (0.54 to 0.76) | 762 (2 RCTs) | ⊕⊕⊕⊝ Moderate c,d | Some evidence that anti‐angiogenic therapy improves progression‐free survival in the recurrent setting. The strength of evidence and size of the effect on progression‐free survival in relation to the overall survival results, questions the validity of progression‐free survival as a surrogate endpoint for overall survival in this setting. |
| Progression‐free survival (assessed with HR) Subgroup: anti‐angiogenic therapy with chemotherapy |
HR 0.72 (0.66 to 0.77) | 3464 (10 RCTs) | ⊕⊕⊕⊕ High | Strong, high‐certainty evidence that there is a progression‐free survival benefit of anti‐angiogenic therapy combined with chemotherapy compared to chemotherapy in HGG. The strength of evidence and size of the effect on progression‐free survival in relation to the overall survival results, questions the validity of progression‐free survival as a surrogate endpoint for overall survival in this setting. |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; HGG: high‐grade glioma; HR: hazard ratio; RCT: randomised controlled trial | ||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect Very low certainty: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect | ||||
a The inconsistency of combining data of cediranib and bevacizumab in the recurrent setting has to be noted. In particular, the REGAL studies of cediranib tended to have inferior survival outcomes compared to the control, so there is the possibility that cediranib (but not all anti‐angiogenic therapy), does not have an impact on overall survival in the recurrent setting.
b Given a hazard ratio bordering on statistical significance, we have downgraded the certainty of this outcome to low.
c Given the small number of studies reporting PFS in the recurrent setting, the effect size is largely determined by the effect of the as yet unpublished 26101 clinical trial (Wick 2017)
d The inconsistency of combining data of cediranib and bevacizumab in the recurrent setting has to be noted. In particular, the REGAL studies of cediranib tended to have inferior survival outcomes compared to the control, so there is the possibility that cediranib (but not all anti‐angiogenic therapy), does not have an impact on progression‐free survival in the recurrent setting.
Background
Description of the condition
High‐grade gliomas (HGGs), comprising World Health Organization (WHO) grade III tumours (e.g. anaplastic astrocytoma, anaplastic oligodendroglioma, or anaplastic oligoastrocytoma) and grade IV tumours (e.g. glioblastoma), represent 75% of primary brain tumours in adults (CBTRUS 2015; Louis 2016). Standard initial treatment for glioblastoma, a WHO grade IV tumour and the most common glioma variant, involves maximal surgical resection followed by radiotherapy with concurrent and then adjuvant chemotherapy with the DNA, alkylator temozolomide. The five‐year survival of glioblastoma is approximately 10% (Stupp 2009). WHO grade III tumours have a better prognosis than glioblastoma but they are likely to progress and follow a similar clinical course. Traditional prognostic factors include age, performance status, histology, symptom severity, and extent of resection (Stupp 2009). The recursive partitioning analysis classification utilises a composite of these prognostic factors to define prognostic groupings (Curran 1993; Mirimanoff 2006). Molecular prognostic factors have been identified, including MGMT, 1p/19q LOH and IDH mutations. MGMT is the promoter hypermethylation of the methylguanine methyltransferase (MGMT) gene in glioblastoma. 1p/19q co‐deletion or loss of heterozygosity (LOH) involves loss of the short arm of chromosome 1 and the long arm of chromosome 19 in oligodendroglial tumours (1p/19q co‐deletion) and predicts a better prognosis (in grade 3 tumours). Mutations of prognostic importance include the genes encoding the enzyme isocitrate dehydrogenase (IDH) 1 and 2 which plays an important role in glucose metabolism (Yan 2009). These molecular markers are prognostic but MGMT methylation has also been associated with sensitivity to chemotherapy and radiotherapy (Stupp 2007).
Angiogenesis refers to the development of new blood vessels from pre‐existing vessels, and abnormal angiogenesis has been implicated in disease processes including malignant tumours (Fidler 1994; Folkman 1990). The dependence of tumour growth on the development of new blood vessels is now a well‐established aspect of cancer biology (Folkman 1971). Anti‐angiogenic therapy, the targeting of tumour blood vessels, interferes with tumour growth and spread in HGG (as described in Description of the intervention section below), is a novel anti‐cancer strategy that has been the subject of various clinical trials in HGG and other cancers. Angiogenesis inhibitors have been developed that block new tumour blood vessel formation and also induce regression of existing tumour blood vessels.
Description of the intervention
Vascular endothelial growth factor (VEGF) is a protein and a key regulator of new blood vessel formation. Activation of the VEGF receptor starts a number of processes that promote cell growth and survival of tumour blood vessels. In addition, VEGF mediates leakiness of blood vessels and is involved in promoting the circulating progenitor cells from the bone marrow to distant sites of new vessel formation (Hicklin 2005; Zebrowski 1999). The anti‐VEGF antibody (bevacizumab) is an antibody that binds circulating angiogenic factor VEGF‐A and prevents the formation of new blood vessels, and has activity in recurrent glioblastoma (Friedman 2009; Khasraw 2010; Kreisl 2009). Other anti‐angiogenic agents have also demonstrated favourable activity in glioblastoma. Anti‐angiogenic agents may be classified as direct, indirect or mixed depending on their mechanism of action and target (Gasparini 2005), as blood vessel formation can be targeted by several mechanisms, as follows (Sivakumar 2005).
Binding of circulating angiogenic factors, such as VEGF‐A, e.g. anti‐VEGF antibodies (bevacizumab) or decoys such as VEGF‐trap (aflibercept).
Blockade of angiogenic factor cell surface receptors (R), e.g. with anti‐VEGF‐R antibodies or with small molecular intracellular tyrosine kinase inhibitors (TKIs) (cediranib).
Imitators of endogenous angiogenesis inhibitors, e.g. angiostatin, endostatin, thrombospondin.
Inhibition of tumour and endothelial cell adhesion and migration by integrin inhibitors (cilengitide).
Certain side effects of anti‐angiogenesis have been observed consistently in clinical studies of these agents (e.g. bleeding, hypertension, delayed wound healing, gastrointestinal perforation and thromboses) and thus are considered a 'class' effect (Batchelor 2013; Chinot 2014; Gilbert 2014).
How the intervention might work
Anti‐angiogenic therapy has been shown to 'normalise' tumour‐associated blood vessel structure and function, and as such may improve vessel leakiness and pressures within tumours. The effectiveness of chemotherapy can be improved by this 'normalisation' of tumour blood vessels, leading to a reduction in the surrounding tumour interstitial oedema and pressure in a way that enhances the delivery of the cytotoxic agent into the tumour (Khasraw 2010). Also, continued therapy may eventually lead to vessel regression, thus depriving cancers of their nutrient source. Anti‐angiogenic therapy may also paradoxically improve tumour oxygenation, thus enhance the effectiveness of radiation therapy (Scaringi 2013). There is preclinical evidence indicating that anti‐angiogenic agents may enhance the effectiveness of chemotherapy on established tumours through a variety of other mechanisms and inhibit new tumour growth by inhibiting the vital process of angiogenesis (Kerbel 2008).
Why it is important to do this review
The purpose of this review is to find, organise and summarise high‐level evidence in terms of benefit and harms of anti‐angiogenic therapy in people with HGGs in order to provide meaningful conclusions for clinical practice and further research.
Anti‐angiogenic therapy has become a cornerstone of therapy for relapsed HGG, despite an absence of high‐level clinical evidence of benefit. Our earlier review did not find clear evidence supporting the use of anti‐angiogenic therapy in either the adjuvant or relapsed setting (Khasraw 2014). As more trials have been completed and updated results are available for several studies, it is important to update this review so that we can interpret clinical data in the face of our improved understanding of the biology, so that the evidence is applied in a way that delivers the most effective treatment to each patient.
Objectives
To evaluate the efficacy and toxicity of anti‐angiogenic therapy in people with high‐grade glioma (HGG). The intervention can be used in two broad groups: at first diagnosis as part of 'adjuvant' therapy, or in the setting of recurrence or progressive disease.
Methods
Criteria for considering studies for this review
Types of studies
We only examined randomised controlled trials (RCTs) comparing therapy with anti‐angiogenic therapy to a control treatment without anti‐angiogenic therapy in people with high‐grade glioma (HGG).
Types of participants
Adults with a histologic diagnosis of World Health Organization (WHO) grade III glioma or grade IV glioma (glioblastoma).
Types of interventions
We included studies when the agent evaluated was mechanistically described as an angiogenesis inhibitor. Therefore, we included agents targeting multiple molecular pathways only when the primary mechanism of action was an important angiogenesis pathway. We included studies which included a secondary intervention in the intervention group (such as chemotherapy or radiotherapy), as long as this secondary intervention was the same in the control group. The control group could be placebo/best supportive care or could have an active intervention (such as chemotherapy), as long as anti‐angiogenic therapy was not included. We included studies from the primary setting (early diagnosis) and the relapsed setting.
Comparisons will be as follows:
Treatment with anti‐angiogenic therapy versus standard care (all patients)
-
Treatment with anti‐angiogenic therapy versus standard care (subgroups):
Primary (adjuvant) setting
Recurrent setting
Anti‐angiogenic therapy plus chemotherapy versus chemotherapy alone.
When the protocol was designed, numerous other possibilities for comparison were considered, however, these have been discarded to simplify the analysis and avoid confusion, these can be found in Appendix 1.
Types of outcome measures
We considered studies including at least one of the following outcomes for evaluation.
Primary outcomes
Overall survival, defined as time from randomisation to death.
Secondary outcomes
Progression‐free survival, defined as time from randomisation to either death from any cause or disease progression.
Disease progression, as defined according to the widely used RECIST criteria used for solid tumours is not used for brain tumours (Therasse 2000). Therefore, we assessed progression‐free survival according to the Macdonald criteria (Macdonald 1990), or the new international working party criteria (response assessment in neuro‐oncology (RANO)) (Wen 2010). There is some debate in the literature regarding the best method of assessing progression radiologically (Wen 2010). Although the Macdonald criteria have been accepted as the standard of care for assessment of progression, the RANO criteria have been developed by consensus to specifically deal with some of the issues associated with the Macdonald criteria. In particular, chemoradiotherapy is well recognised to cause 'pseudoprogression' in 10% to 30% of all patients (Brandsma 2009; Wen 2010). Nevertheless, we used accepted radiological guidelines for response rates in this study. The Macdonald and RANO criteria are listed in Table 3 and Table 4.
1. MacDonald Criteria.
| Response | Criteria |
| Complete response | Requires all of the following: complete disappearance of all enhancing measurable and non‐measurable disease sustained for at least 4 weeks; no new lesions; no corticosteroids; and stable or improved clinically |
| Partial response | Requires all of the following: ≥ 50% decrease compared with baseline in the sum of products of perpendicular diameters of all measurable enhancing lesions sustained for at least 4 weeks; no new lesions; stable or reduced corticosteroid dose; and stable or improved clinically |
| Stable disease | Requires all of the following: does not qualify for complete response, partial response, or progression; and stable clinically |
| Progression | Defined by any of the following: ≥ 25% increase in sum of the products of perpendicular diameters of enhancing lesions; any new lesion; or clinical deterioration |
2. RANO criteria.
| Response | Criteria |
| Complete response | Requires all of the following: complete disappearance of all enhancing measurable and non‐measurable disease sustained for at least 4 weeks; no new lesions; stable or improved non‐enhancing (T2/FLAIR) lesions; and patient must be off corticosteroids or on physiologic replacement doses only, and stable or improved clinically. In the absence of a confirming scan 4 weeks later, this response will be considered only stable disease. |
| Partial response | Requires all of the following: ≥ 50% decrease, compared with baseline, in the sum of products of perpendicular diameters of all measurable enhancing lesions sustained for at least 4 weeks; no progression of non‐measurable disease; no new lesions; stable or improved non‐enhancing (T2/ FLAIR) lesions on same or lower dose of corticosteroids compared with baseline scan; and patient must be on a corticosteroid dose not greater than the dose at time of baseline scan and is stable or improved clinically. In the absence of a confirming scan 4 weeks later, this response will be considered only stable disease. |
| Stable disease | Stable disease occurs if the patient does not qualify for complete response, partial response, or progression (see next section) and requires the following: stable non‐enhancing (T2/FLAIR) lesions on same or lower dose of corticosteroids compared with baseline scan and clinically stable status. In the event that the corticosteroid dose was increased for new symptoms and signs without confirmation of disease progression on neuroimaging, and subsequent follow‐up imaging shows that this increase in corticosteroids was required because of disease progression, the last scan considered to show stable disease will be the scan obtained when the corticosteroid dose was equivalent to the baseline dose. |
| Progression | Progression is defined by any of the following: ≥ 25% increase in sum of the products of perpendicular diameters of enhancing lesions (compared with baseline if no decrease) on stable or increasing doses of corticosteroids; a significant increase in T2/FLAIR non‐enhancing lesions on stable or increasing doses of corticosteroids compared with baseline scan or best response after initiation of therapy, not due to comorbid events; the appearance of any new lesions; clear progression of non‐measurable lesions; or definite clinical deterioration not attributable to other causes apart from the tumour, or to decrease in corticosteroid dose. Failure to return for evaluation as a result of death or deteriorating condition should also be considered as progression. |
Quality of life, where assessed using an objective grading measure, such as those described in Mauer 2008.
Adverse events, classified according to WHO or National Cancer Institute Common Terminology Criteria (NCI‐CTCAE (CTCAE 2017)), including the percentage of treatment‐related deaths.
Search methods for identification of studies
We did not apply any language restrictions to the searches.
Electronic searches
We conducted searches to identify published and unpublished RCTs. Due to the relatively recent availability of targeted anti‐angiogenic drugs, we considered a literature search starting in 2000 sufficient for the purpose of this review. We searched the following databases.
The Cochrane Central Register of Controlled Trials (CENTRAL; 2018, Issue 9).
MEDLINE Ovid (up to September week 4, 2018).
Embase Ovid (up week 41, 2018).
The CENTRAL, MEDLINE and Embase search strategies are listed in Appendix 2, Appendix 3, Appendix 4.
Searching other resources
We also searched databases of ongoing trials.
We handsearched reference lists from trials selected by electronic searching to identify further relevant trials. We handsearched published abstracts between the years 2000 to 2018 from conference proceedings from the European Society for Medical Oncology (published in the Annals of Oncology), the European Council for Clinical Oncology (published in the European Journal of Cancer), proceedings of the conferences of the Society of Neuro‐oncology (SNO), the European Association of Neuro‐oncology (EANO) and the World Federation of Neuro‐oncology (WFNO), as well as the American Society for Clinical Oncology (ASCO). In addition, we asked members of the relevant Cancer Groups (EANO, SNO), experts in the field and manufacturers of relevant drugs to provide details of outstanding clinical trials and any relevant unpublished material.
Data collection and analysis
Selection of studies
Two review authors (MA, MK) independently assessed the titles and abstracts retrieved by the search strategy for potential eligibility. Full text articles were obtained, where possible, of potentially eligible studies (see criteria in section Types of interventions). These were assessed for eligibility independently and in a blinded fashion (to authors, journal, drug company, institutions and results) by two of the review authors (MA, MK). It was agreed that disagreement will be resolved by consensus with a third review author (any of the authors).
Abstracts or unpublished data were agreed to be included only if sufficient information on the study design, characteristics of participants, interventions and outcomes was available. Further information or final results were sought from the primary author.
Please see Figure 1 for PRISMA flow diagram of study selection.
1.

Study flow diagram.
Data extraction and management
Data extraction was performed independently by two review authors (MA, MK). Data were entered into Review Manager 5 (Review Manager 2014) for analysis. We recorded the following for each eligible trial: study design, participants, setting, interventions, and quality components, duration of follow‐up, efficacy outcomes, biomarker analyses and side effects. For studies with more than one publication, we extracted data on all outcomes from the most recent publication. Short‐term adverse events were to be highlighted if considered significant.
Two review authors (MA, MK) independently extracted details of study population, interventions and outcomes by using a standardised data extraction form. Blinding of study participants and investigators was also assessed. Differences in data extraction were resolved by consensus with a third review author (any 2 authors), referring back to the original article. There was no disagreement regarding selection of the studies among the authors.
The data extraction form included the following items.
General information: title, authors, source, contact address, country, published/unpublished, language and year of publication, sponsor of trial.
Trial characteristics, including study design, duration/follow‐up, quality assessment (as specified above).
Participants: inclusion and exclusion criteria, sample size, baseline characteristics, similarity of groups at baseline, withdrawals and losses to follow‐up.
Interventions: dose, route and timing of chemotherapy, anti‐angiogenic therapy and comparison intervention.
-
Outcomes
For time‐to‐event (survival and disease progression), we extracted hazard ratio (HR) and 95% confidence interval (CI), log rank Chi2, log rank P values, number of events, number of participants per group, median‐, one‐, two‐, three‐ and five‐year survival rates.
For dichotomous outcomes (radiological response and adverse events), we extracted the number of participants in each group who experienced the outcome of interest and the number of participants assessed at endpoint in order to estimate the risk ratio (RR).
For continuous outcomes (quality of life measures), we extracted the final or change value and standard deviation of the outcome of interest and the number of participants analysed at endpoint for each treatment group in order to estimate the difference in means and 95% CI between treatment arms.
For time‐to‐event (survival and disease progression), we extracted hazard ratio (HR) and 95% confidence interval (CI), log rank Chi2, log rank P values, number of events, number of participants per group, median‐, one‐, two‐, three‐ and five‐year survival rates.
For dichotomous outcomes (radiological response and adverse events), we extracted the number of participants in each group who experienced the outcome of interest and the number of participants assessed at endpoint in order to estimate the risk ratio (RR).
For continuous outcomes (quality of life measures), we extracted the final or change value and standard deviation of the outcome of interest and the number of participants analysed at endpoint for each treatment group in order to estimate the difference in means and 95% CI between treatment arms.
Where possible, we extracted data for intention‐to‐treat analysis for all outcomes.
We estimated HRs and their 95% CIs directly or indirectly from the published data (Altman 2001). HRs can be estimated (under some assumptions) from log rank Chi2, log rank P values, observed to expected event ratios, from ratios of median survival times or time point survival rates (Machin 1997; Parmar 1998).
We recorded the time points at which outcomes were collected and reported.
Assessment of risk of bias in included studies
Two review authors (MK and MA) independently assessed for quality all studies that met the inclusion criteria, with disagreement resolved by a third review author if required. We assessed the risk of bias for every included study using the Cochrane 'Risk of bias' tool (Higgins 2011a).
Each study was assessed independently by two review authors, for the use of random allocation to the comparison groups. We also included trials which permitted a cross‐over for participants after disease progression.
Measures of treatment effect
Data analysis
We presented summary statistics for the primary endpoints (time‐to‐event data) as HRs (Cox 1972).
Unit of analysis issues
Measurement of progression‐free survival has been done under different response criteria. Nevertheless, utilising the HR for the comparisons minimises the significance of this issue, as the control group is subject to the same response criteria. Nevertheless, consideration of the different response criteria must be made when interpreting the pooled analysis of the progression‐free survival data.
Dealing with missing data
We contacted the first author of the most recent publication in cases of missing data. Specifically, we contacted the first and senior authors of the BELOB study to try to obtain a HR for progression‐free survival, but this was unavailable (Taal 2014).
Assessment of heterogeneity
We assessed heterogeneity between studies using Cochranes Q‐test, with a significance threshold of alpha = 0.1, and by estimation of the percentage heterogeneity between trials which cannot be ascribed to sampling variation (Higgins 2003).
In case of substantial heterogeneity, the extra variation was incorporated in the analysis by using a random‐effects model.
We considered the following factors as possible sources of heterogeneity.
Differing clinical settings (primary versus recurrent disease).
Different applications of anti‐angiogenic treatment (scheduling etc.).
Different types of angiogenesis inhibitors (as classified above).
Differences in prognostic factors between studies.
Study quality.
We considered these factors in the sensitivity and subgroup analyses, except in the case of differing prognostic factors as there were limited data available to analyse this (apart from analysis by setting: primary versus recurrent).
Assessment of reporting biases
Given the overall small number of studies included in this meta‐analysis, we did not construct funnel plots to assess reporting bias. We performed qualitative assessment of reporting bias for individual studies, denoting whether registered endpoints at ClinicalTrials.gov and other trial registries were reported in the final publication.
Data synthesis
We pooled results in a meta‐analysis.
For time‐to‐event data, we pooled HRs using the generic inverse variance facility of Review Manager 5.
In the trials with multiple treatment groups, we compared the anti‐angiogenic intervention to the specific control comparator. Specifically, when dealing with multiple interventions, we split the shared control group across the two interventions. We felt that this approach was more advantageous than combining all experimental groups, as this allowed investigations of heterogeneity across intervention arms (Higgins 2011a).
We used fixed‐effect models for all meta‐analyses and random‐effects models with inverse variance weighting in cases of substantial heterogeneity for all meta‐analyses (DerSimonian 1968).
Summary of Findings
We presented the overall certainty of the evidence for each outcome according to the GRADE approach, which takes into account issues not only related to internal validity (risk of bias, inconsistency, imprecision, publication bias) but also to external validity such as directness of results (Langendam 2013). We created 'Summary of findings' tables based on the methods described the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a), and used GRADEpro GDT. We used the GRADE checklist and GRADE Working Group certainty of evidence definitions (Meader 2014). We downgraded the evidence from 'high' certainty by one level for serious (or by two for very serious) concerns for each limitation.
High‐certainty: we are very confident that the true effect lies close to that of the estimate of the effect.
Moderate‐certainty: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different.
Low‐certainty: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect.
Very low‐certainty: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect.
The outcomes specified included overall survival and progression‐free survival.
Subgroup analysis and investigation of heterogeneity
In this update we re‐analysed subgroups analysed in the initial review.. We did not perform any additional subgroup analyses at this time.
Sensitivity analysis
We performed a sensitivity analysis after the main meta‐analysis for both primary and secondary outcomes. We analysed several subgroups to deal with the sources of heterogeneity listed above. Specifically, we conducted a separate analysis for bevacizumab‐containing anti‐angiogenic therapy, apart from the main analysis to account for the different types of anti‐angiogenic therapy used across studies, with bevacizumab appearing most frequently. We excluded the vandetanib study as part of the sensitivity analysis for progression‐free survival due to its higher risk of bias and the outlying nature of its results (Lee 2015); however this did not change results of the analysis significantly.
Review update
Out of the 11 studies analysed in this review, only one was available in abstract form and with details restricted to what was presented at scientific conferences and additional details from clinical trials.gov. We will undertake updates of this review as soon as all eligible studies are published in full in the peer‐reviewed literature. With future reviews, we will consider the mechanism of action, whether the drug is an antibody or a small molecule, and a direct, indirect or mixed inhibitor for classification, as well as different types of chemotherapy, schedules of anti‐angiogenic agents, and combinations of different anti‐angiogenic strategies. We will classify studies using anti‐angiogenic therapies as second‐line according to their prior use in first‐line (primary) as well. We will adopt this classification depending on the availability of relevant studies in the future. As soon as studies using combinations of anti‐angiogenic and other targeted drugs become available, we will add further comparisons.
Results
Description of studies
Results of the search
The initial search performed in 2014 yielded 252 references from CENTRAL, 33 references through MEDLINE and 624 references from Embase. Conference searches yielded an additional 10 references. We deleted duplicates.
For the update, we reran the search from 2016 to October 2018 and found 310 additional references in CENTRAL, 177 in MEDLINE and 220 in Embase. Once we removed duplicates, we collated a total of 590 references. We utilised a machine learning algorithm, the Cochrane RCT classifier on the updated reference list to identify records that were likely to be randomised controlled trials (RCTs), and we reduced the collated list to 305 references.
Two review authors (MK and MA) independently reviewed abstracts, and excluded articles that obviously did not meet the inclusion criteria at this stage. We subsequently evaluated the eligibility of 55 full text records.
The study selection process is demonstrated in Figure 1.
Included studies
We identified 11 eligible RCTs, all of which were peer‐reviewed published journal articles.
Randomised trials in newly diagnosed glioblastoma
Avastin in glioblastoma (AVAglio) study (Chinot 2014)
This was a phase III double‐blind placebo‐controlled trial evaluating bevacizumab in people with newly diagnosed glioblastoma (Chinot 2014).
In this study, four to seven weeks after surgical resection of glioblastoma, 921 participants were randomised to standard temozolomide (75 mg/m2/ day) and radiotherapy (2 Gy 5 days a week; maximum, 60 Gy) with bevacizumab (10 mg/kg intravenously every 2 weeks) or placebo. Four weeks after completion of radiotherapy, participants were commenced on 6 cycles of standard maintenance temozolomide (150 mg/m2 to 200 mg/m2 orally days 1 to 5, every 28 days). Bevacizumab or placebo was continued every two weeks during the maintenance phase and then every three weeks at a dose of 15 mg/kg until disease progression or toxicity. Both overall survival and progression‐free survival (investigator assessed) were co‐primary endpoints. Secondary endpoints included progression‐free survival assessed by an independent central review, one‐year survival rates, health‐related quality of life (EORTC QLQ‐C30 and BN20 scales Fayers 2001; Taphoorn 2010), and safety. Exploratory endpoints were performance status and the use of corticosteroids.
One of the secondary endpoints of the AVAglio study was to compare quality of life between treatment arms, as measured by the EORTC QLQ‐C30 scale and its companion brain tumour module BN20, and 78% to 91% of evaluable people without disease progression completed each assessment in the first year. Baseline scores were comparable between arms. Although there was no difference between treatment arms in health‐related quality of life score changes over time, the addition of bevacizumab did not worsen or improve participant response over time compared with placebo. However, bevacizumab‐treated participants experienced a longer time to health‐related quality of life deterioration compared with the control arm (Chinot 2014).
In AVAglio, radiologic progression was defined as a 25% or more increase in the size of enhancing lesions, unequivocal progression of existing non‐enhancing lesions or any new lesions. Clinical progression was defined as worsening neurologic symptoms. Stratification included age, performance status, MGMT status (MGMT is the promoter hypermethylation of the methylguanine methyltransferase), extent of surgical resection, Mini Mental State Examination (MMSE), corticosteroid and antiepileptic use.
Radiation Therapy Oncology Group (RTOG) 0825 study (Gilbert 2014)
This was a phase III double‐blind placebo‐controlled trial evaluating bevacizumab in people with newly diagnosed glioblastoma (Gilbert 2014). It enrolled 621 participants with newly diagnosed glioblastoma stratified based on MGMT methylation status and molecular profile. Participants then received three weeks of radiation therapy and daily temozolomide plus bevacizumab or a continuation of their standard therapy plus placebo. The primary objectives were overall survival and progression‐free survival. Secondary endpoints included toxicity, symptom burden, health‐related quality of life, neurocognitive function and identification of participant subsets more likely to benefit from bevacizumab.
As part of this study, net clinical benefit was measured using longitudinal measures of patient reported outcomes including the MD Anderson Symptom Inventory‐Brain Tumor Module (MDASI‐BT) and the EORTC Quality of Life Questionnaire/Brain Tumor Module (EORTC QLQ‐C30/BN20). These were completed at baseline and longitudinally (weeks 6, 10, 22, 34 and 46).
Participants also completed neurocognitive testing with the Hopkins Verbal Learning Test‐Revised (HVLT‐R), Trail Making Test (TMT) and Controlled Oral Word Association (COWA) at baseline and longitudinally (week 6, 10, 22, 34, and 46). Six neurocognitive test scores as well as the Clinical Trial Composite (CT COMP) score (i.e. the average performance across all neurocognitive tests) were examined over time.
Bevacizumab plus irinotecan versus temozolomide in newly diagnosed O6‐methylguanine‐DNA methyltransferase non‐methylated glioblastoma: the GLARIUS study (Herrlinger 2016)
This was a randomised phase II study in people with MGMT non‐methylated newly diagnosed glioblastoma (Herrlinger 2016). One hundred and seventy participants were randomised in a 2:1 ratio to bevacizumab and irinotecan or temozolomide. All participants received radiation therapy at a standard dose for six weeks. Participants in the bevacizumab/irinotecan arm received four cycles of bevacizumab over the course of radiation and then received bevacizumab/irinotecan every two weeks until disease progression. The primary endpoint of the study was six‐month progression‐free survival. Secondary endpoints included overall survival, steroid use and toxicity. Cross‐over to bevacizumab and irinotecan following progression on temozolomide was permitted. Of the 54 subjects in the temozolomide arm, 29 crossed over to bevacizumab and irinotecan.
Randomised phase II trial of irinotecan and bevacizumab as neo‐adjuvant and adjuvant to temozolomide‐based chemoradiation compared with temozolomide‐chemoradiation for unresectable glioblastoma (TEMAVIR study) (Chauffert 2014)
This was a randomised two‐arm phase II study of bevacizumab in combination with irinotecan in the upfront setting (Chauffert 2014). The predefined primary outcome was six‐month progression‐free survival. The experimental arm of the study consisted of neo‐adjuvant bevacizumab, 10 mg/kg intravenously and irinotecan, 125 mg/m2 intravenously every two weeks for four cycles, before chemoradiotherapy with temozolomide and bevacizumab 10 mg/kg intravenously two‐weekly. Adjuvant bevacizumab and irinotecan were given for six months in the experimental arm. The control group consisted of standard chemoradiation according to the Stupp protocol (Stupp 2014.
Neo‐adjuvant treatment with temozolomide and bevacizumab previous to temozolomide plus radiation plus bevacizumab therapy in unresectable glioblastoma (Genom‐009 study) (Balana 2016)
This was a randomised phase II study comparing neoadjuvant temozolomide followed by concurrent chemoradiation versus the same protocol with the addition of intravenous bevacizumab at 10 mg/kg on days one and 15 of each cycle in the neoadjuvant stage and day one, 15 and 30 of the concurrent stage (Balana 2016). The study was specifically looking at poor prognosis unresectable glioblastoma.
The cilengitide, temozolomide, and radiation therapy in treating patients with newly diagnosed glioblastoma and methylated gene promoter status (CENTRIC) study (Stupp 2014)
The CENTRIC study combined cilengitide with standard treatment for people with newly diagnosed glioblastoma and methylated MGMT gene promoter (CENTRIC; Stupp 2014). It was an international phase III trial that enrolled 545 participants with glioblastoma harbouring MGMT promoter methylation, adding cilengitide (2000 mg twice weekly intravenously) to standard radiotherapy/temozolomide compared with radiotherapy/temozolomide without cilengitide. The primary endpoint was overall survival and secondary endpoints included progression‐free survival, safety and tolerability, pharmacokinetics, quality of life, general health and work status.
Cilengitide, temozolomide, and radiation therapy in treating patients with newly diagnosed glioblastoma and unmethylated gene promoter status (CORE study) (Nabors 2015)
This was a randomised three‐arm open‐label phase II study of cilengitide in MGMT non‐methylated newly diagnosed glioblastoma (Nabors 2015). The first intervention arm of the study included cilengitide at standard dosing (2000 mg intravenously twice weekly) through chemoradiation (6 weeks) and as maintenance until week 34. The second intervention arm of the study included cilengitide at intensive dosing (2000 mg intravenously five times weekly) throughout chemoradiation and then 2000 mg intravenously twice weekly as maintenance until week 34. The control arm and both intervention arms included standard chemoradiotherapy with temozolomide 75 mg/m2 followed by six cycles of maintenance temozolomide at 150 mg/m2 to 200 mg/m2 days 1 to 5, every 28 days.
Vandetanib study (Lee 2015)
This was a randomised non‐comparative phase II study of standard Stupp 2005 chemoradiation protocol, with or without vandetanib (100 mg daily) starting up to one week prior to commencement of chemoradiation (Lee 2015). Participants were randomised in a 2:1 ratio to receive vandetanib in addition to standard treatment versus standard chemoradiation. The study was terminated early after an interim futility analysis was performed.
Randomised trials in recurrent glioblastoma
Cediranib in glioblastoma alone and with lomustine (REGAL) study (Batchelor 2013)
A phase III, randomised, multicenter study comparing cediranib, as monotherapy and in combination with lomustine versus lomustine alone (Batchelor 2013). Three hundred and twenty‐five participants with disease progression after receiving only one prior systemic temozolomide‐containing chemotherapy regimen were randomly assigned in a 2:2:1 ratio to one of three arms: cediranib monotherapy 30 mg orally daily; cediranib 20 mg orally daily in combination with oral lomustine 110 mg/m2 once every six weeks; or lomustine 110 mg/m2 once every six weeks in combination with a placebo. The primary endpoint was to assess the relative efficacy of cediranib ‐ either alone or in combination with lomustine ‐ versus lomustine alone by independent central radiographic assessment of progression‐free survival.
Single‐agent bevacizumab or lomustine versus a combination of bevacizumab plus lomustine in patients with recurrent glioblastoma (Dutch BELOB) study with bevacizumab (Taal 2014)
This was a randomised three arm phase II study of 153 participants with glioblastoma at first recurrence treated with: 1) bevacizumab 10 mg/kg every two weeks versus 2) bevacizumab every two weeks plus lomustine 110/m2 every six weeks for six cycles capped at 200 mg or 3) single agent lomustine. The primary endpoint was overall survival at nine months. Secondary endpoints included median progression‐free survival, six‐month progression‐free survival, quality of life, deterioration free survival and safety in the form of safety monitoring in the first 10 participants. For the purposes of this analysis, the single agent lomustine arm was considered the control group and the two other groups were intervention groups.
EORTC 26101 study with bevacizumab (Wick 2017)
This was a randomised phase III trial evaluating bevacizumab plus lomustine versus intravenous lomustine in recurrent glioblastoma (Wick 2017). This was the follow‐on study evaluating the schedules first determined in the BELOB trial. The control arm consisted of single agent lomustine at 110 mg/m² every six weeks (maximum dose 200 mg) and the experimental arm consisted of lomustine 90 mg/m² every six weeks (maximum dose 160 mg) with bevacizumab 10 mg/kg every two weeks. The primary endpoint was overall survival.
In total, 11 studies with 3743 participants reported data for overall survival. Hazard ratios for overall survival from all studies were eligible to be pooled. In one study (BELOB), the hazard ratio was estimated using the Parmar method (Higgins 2011b; Parmar 1998). Although the BELOB study was a three‐arm study of lomustine (control), lomustine and bevacizumab, and bevacizumab monotherapy (Taal 2014), the hazard ratios were calculated for both data sets of bevacizumab‐containing regimens compared to lomustine to allow inclusion in this analysis. Although the GLARIUS (Herrlinger 2016), and TEMAVIR (Chauffert 2014), studies had different chemotherapy arms (irinotecan versus temozolomide), the authors felt pooling the data from this study was appropriate as both groups had a chemotherapy backbone, albeit different. We accounted for the limitations of the data from the GLARIUS study in the sensitivity analysis.
Eight of the 11 studies compared the addition of anti‐angiogenic therapy in the primary or adjuvant setting, whereas the other three studies were in the relapsed setting. There was additional variability in the interventions across the studies. Seven of the 11 studies utilised bevacizumab as the anti‐angiogenic treatment, with two studies of cilengitide, one of vandetanib and one of cediranib. In the primary studies, the GLARIUS study used a different cytotoxic chemotherapy (irinotecan) of uncertain efficacy in gliomas, compared to all the other studies, which used standard chemoradiation with temozolomide. The TEMAVIR study used a different backbone (bevacizumab/irinotecan) instead of temozolomide in the adjuvant and neoadjuvant components of the study, whilst utilising temozolomide during radiation. The relapsed studies used lomustine as the comparator.
Excluded studies
We excluded all non‐randomised, single‐arm studies and studies that had an anti‐angiogenic agent but no control arm or a historic controlled arm. One randomised study was excluded (Duerinck 2016), as in the control arm, 20 out of 22 cases received bevacizumab, an anti‐angiogenic therapy. We did not include another study, AVAreg in the meta‐analysis as it was a non‐comparative randomised study, for which there was inadequate data to assess the endpoints of this meta‐analysis (Brandes 2016). We contacted the primary investigator of the study to evaluate whether adequate data were available for analysis prior to this study's exclusion. We excluded the randomised TAVAREC clinical trial (Van Den Bent 2017), as currently available data are only in the form of conference proceedings and are a mix of low‐grade and high‐grade gliomas (grade 2 and grade 3) and would be therefore excluded from this study protocol. Additionally, no specific results were reported of axitinib versus non‐bevacizumab‐containing control therapy for this reason. Of note, the BRAIN study (Friedman 2009), and National Cancer Institute study (Kreisl 2009), used to support Food and Drug Administration approval of bevacizumab in the USA for recurrent glioblastoma, were not eligible for inclusion in this analysis because neither study had a control arm not containing anti‐angiogenic therapy. Finally we excluded the REGOMA clinical study which has been presented in abstract form, as given regorafenib is a multi‐kinase inhibitor, its predominant mechanism of action may not be anti‐angiogenic (Lombardi 2018).
Risk of bias in included studies
Overall, the risk of bias in included studies was low. Ten of the 11 studies had an overall low risk of bias (see Figure 2). The vandetanib study had a moderate risk of bias due to the fact that participants and investigators were not blinded to treatment assignment (Lee 2015); it is unclear as to whether investigators were blinded to outcome assessment and insufficient information regarding allocation concealment was not present. Figure 3 demonstrates a summary 'Risk of bias' figure.
2.

'Risk of bias' graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.

'Risk of bias' summary: review authors' judgements about each risk of bias item for each included study.
Allocation
All but three studies demonstrated a central allocation system, reducing risk of allocation concealment bias. The CORE, TEMAVIR and vandetanib studies did not report allocation concealment measures (Chauffert 2014; Lee 2015; Nabors 2015).
Blinding
Three studies were completely blinded. For the REGAL (cediranib) study, participants and investigators were blinded (Batchelor 2013). The placebo and cediranib packaging were identical and the participants could only be unblinded in the event of a medical emergency. For the RTOG 0825 study, both participants and investigators were blinded (Gilbert 2014). Investigators were blinded to the MGMT status of the patient. Salvage bevacizumab was available on progression resulting in unblinding once a code‐breaking form was submitted. For AVAglio both participants and investigators were blinded (Chinot 2014). Unblinding was permitted on progression only if determining subsequent therapy. In the intervention group 31% per cent of participants continued bevacizumab beyond progression and 48% crossed over to bevacizumab in the control group. All other studies were unblinded. All of the studies had centralised radiological review, except the BELOB and TEMAVIR studies, which were assessed by local investigators, thereby increasing risk of outcome assessment bias (Chauffert 2014; Taal 2014). The vandetanib study did not report whether outcome assessment was analysed locally or centrally (Lee 2015).
Incomplete outcome data
For most of the studies, outcome data capture was near complete, thereby minimising the risk of attrition bias. In REGAL, 10 participants evenly balanced across treatment arms were lost to follow‐up (Batchelor 2013). In the AVAglio study, of the 921 participants, 26 participants dropped out before the study commenced, with a further 23 in the second phase of the study and a further 28 in the third phase (Chinot 2014). The intervention and control group was well matched in dropout rate. In the CENTRIC study, only one participant was lost to follow‐up, and the rate of participants not receiving the assigned intervention was low in both groups (9 and 15 respectively) (Stupp 2014). In the RTOG 0825 study, 16 participants dropped out, with eight in each arm (Gilbert 2014). In the REGAL cediranib study, 10 participants were lost to follow‐up and 12 participants did not receive their allocated intervention (5, 6 and 1 patient) respectively (Batchelor 2013). The BELOB study had a similarly low dropout rate with only three participants dropping out across the entire study (Taal 2014). In the CORE study only two participants were lost to follow‐up and most participants received the allocated intervention (Nabors 2015). Eight participants were lost to follow‐up in the GLARIUS study (Herrlinger 2016). There were no reported participants lost to follow‐up in the TEMAVIR or Genom‐009 clinical trials (Balana 2016; Chauffert 2014). In the vandetanib study, seven participants in the control arm withdrew consent prior to commencing treatment and were not analysed and 10 participants withdrew consent on the intervention arm after commencing treatment. No participants were reported as lost to follow‐up (Lee 2015). Moreover, nine studies used intention‐to‐treat analysis, further limiting the risk of attrition bias. The vandetanib study used a per protocol analysis (Lee 2015), and the Genom‐009 study did not report whether an intention‐to‐treat or per protocol analysis was performed (Balana 2016). In the EORTC 26101 study, only 22 participants were lost to follow‐up across both treatment arms (Wick 2017).
Selective reporting
All trials were registered at either ClinicalTrials.gov or Eudra‐CT, and reported the preplanned outcomes, limiting the risk of selective reporting. The AVAglio study did include an unplanned analysis of 'deterioration‐free survival' as a surrogate for the quality of life data which was not prespecified in the trial protocol, although quality of life was a planned endpoint (Chinot 2014).
Other potential sources of bias
Most studies used an intention‐to‐treat analysis, and where information was available, all had similar schedules for follow‐up between the control and experimental arms. The RTOG 0825 study had additional scans (such as dynamic MRI), which were performed periodically and did not relate to the study's outcome assessments (Gilbert 2014). Most studies had pharmaceutical sponsorship, which has previously been noted to result in favourable outcomes for pharmaceuticals (Lexchin 2003). However, most studies reported unfavourable results, thereby negating the risk of this bias.
Effects of interventions
Anti‐angiogenic therapy compared to no anti‐angiogenic therapy for high‐grade glioma (HGG) (all patients)
Overall survival
Meta‐analysis of all 11 studies of anti‐angiogenic therapy (n = 3743 participants) found no observed difference in overall survival: fixed‐effect pooled hazard ratio (HR) 0.95, 95% confidence interval (CI) 0.88 to 1.02; P = 0.16; high‐certainty evidence; (Analysis 1.1; Figure 4; Table 1). We did not observe significant heterogeneity (I2 = 0.42) in the results.
1.1. Analysis.

Comparison 1 Anti‐angiogenic therapy compared to no anti‐angiogenic therapy for high‐grade glioma (all patients, overall survival), Outcome 1 Hazard for OS.
4.

Forest plot of meta‐analysis of overall survival, categorized by setting (Analysis 1.1)
In terms of participant population, although there were some minor differences in molecular profile (for instance the use of the better prognosis MGMT methylated participants in the REGAL study (Batchelor 2013), compared to the worse prognosis MGMT unmethylated participants in GLARIUS and CORE studies (Herrlinger 2016; Nabors 2015), the major participant‐related difference was in the treatment setting (see Included studies section above).
Progression‐free survival
A meta‐analysis of 10 studies (3595 participants) reported data for progression‐free survival. The BELOB study did not report progression‐free survival and we were unable to obtain this data from the study authors (Taal 2014). A meta‐analysis of all trials of anti‐angiogenic therapy using a fixed‐effect model found an observed difference in progression‐free survival: HR 0.73, 95% CI 0.68 to 0.79; P < 0.00001; high‐certainty evidence; Analysis 2.1; Figure 5; Table 1). Due to the significant heterogeneity (I2 = 65%), we performed a random‐effects meta‐analysis to confirm this finding (HR 0.75, 95% CI 0.66 to 0.87; P < 0.00001, analysis not shown). This heterogeneity is most likely due to differences between the studies in terms of both the patient population and clinical setting and the intervention applied (as described in overall survival section above).
2.1. Analysis.
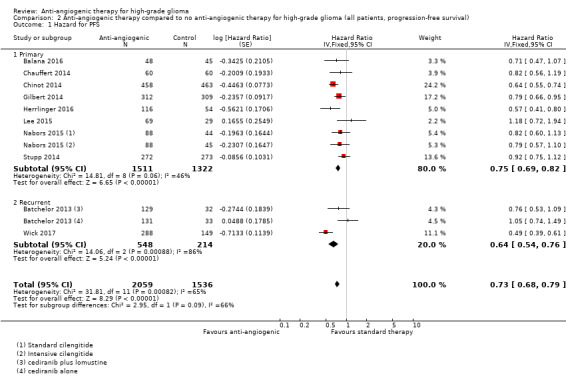
Comparison 2 Anti‐angiogenic therapy compared to no anti‐angiogenic therapy for high‐grade glioma (all patients, progression‐free survival), Outcome 1 Hazard for PFS.
5.
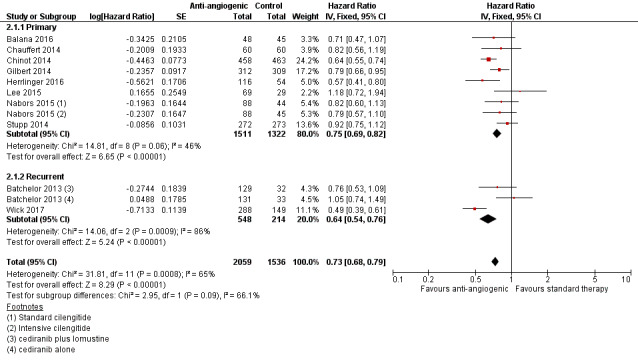
Forest plot of meta‐analysis of progression‐free survival, categorized by setting (Analysis 2.1)
Quality of life
There is inadequate published data to formally assess and pool quality of life endpoints. However, the two primary studies that have been published in full differ in the reported impact of anti‐angiogenic therapy on patient quality of life (Gilbert 2014; Taphoorn 2015, in: Chinot 2014). The AVAglio study protocol secondary endpoint was EORTC QLQ‐C30. All participants were required to fill in the quality of life questionnaires. There was no difference between treatment arms in health‐related quality of life score changes over time. The addition of bevacizumab did not worsen or improve participant response over time compared with placebo. Bevacizumab‐treated participants experienced a longer time to health‐related quality of life deterioration compared with the control arm (Chinot 2014). The paper reports a measure of "deterioration‐free survival" that includes survival. Deterioration was classified as a sustained 10‐point deterioration from baseline or death. This outcome measure was not included in the trial protocol but was reported in the full paper.
The RTOG 0825 study showed that over time an increased symptom burden, a worse quality of life, and a decline in neurocognitive function were more frequent in the bevacizumab group (Gilbert 2014). This has only been reported in abstract form (Armstrong 2013; Wefel 2013, in: Gilbert 2014). The trial had an optional net clinical benefits study, of which 80% of trial participants initially participated. It is important to note that participants were not required to fill in forms upon progression, and as a result of the consistent improvement in progression‐free survival in the bevacizumab arm, more participants completed the various questionnaires throughout the study in the intervention arm. For example, in week 34, 107 participants in the bevacizumab arm completed the forms compared to 72 in the control group. Consequently, there was a significant deterioration in the quality of life for participants in the bevacizumab‐containing regimen across many domains. One possibility is that progression of non‐contrast enhancing disease resulted in the bevacizumab‐containing regimen was responsible for the discrepancies in form completion. Conversely, unlike the AVAglio study, this study did prespecify the nature of quality of life analyses in the original protocol.
The EORTC 26101 study did demonstrate that the combination of bevacizumab and lomustine resulted in decreased quality of life in the social functioning domain, which was considered clinically significant (Wick 2017). However, there were no significant differences in global health status, physical functioning, motor dysfunction or communication deficit between the two arms. Moreover, if progression was not included as an event, overall there was no significant difference in time to deterioration in health‐related quality of life between the two arms. Finally, there was no significant difference observed between the two groups in the time to starting glucocorticoids.
The only other study to formally present quality of life data was the GLARIUS trial, which was confounded by the use of a different chemotherapy backbone (Herrlinger 2016). Neverthless, there was no observed difference in quality of life between the two groups on QLQ‐C30 and other quality of life domains.
In summary, the impact of anti‐angiogenic therapy including bevacizumab on quality of life remains unclear.
Adverse events
There were some differences in the toxicity profiles of the different regimens, particularly dependent upon whether anti‐angiogenic therapy was combined with cytotoxic therapy. The toxicity data from the published studies are presented in Table 5. The studies of bevacizumab reported similar rates of common and serious toxicities including wound complications (0.8% to 3.3%), hypertension (4.2% to 27%), thromboembolic complications (2.5% to 7.7%), and gastrointestinal perforation (0.8% to 5.3%). These findings mainly differed due to different combination regimens and different populations of people (e.g. higher rates of complications in the poor prognosis TEMAVIR group (Chauffert 2014)). Overall the other anti‐angiogenic therapies were well tolerated, although in the REGAL study, an excess of haematological toxicity was seen in the cediranib combined with lomustine groups (38.3% thrombocytopaenia) (Batchelor 2013).
3. Adverse events.
| Grade ≥ 3 adverse event | REGAL (%) C = cediranib, L = cediranib + lomustine) |
AVAglio (%) | RTOG 0825 (%, CRT = chemo radiotherapy, A = adjuvant chemotherapy) |
BELOB (%, B = bev L = bev + lomustine |
CENTRIC (%) | CORE (%) S = standard cilengitide I = intensive cilengitide |
TEMAVIR (%) | Genom‐009 (%) N = neoadjuvant C = concurrent |
Vandetanib (%) | GLARIUS (%) | 26101 B = bev + lomustine L = lomustine alone |
| Haemorrhage | C: 0 L: 0.8 |
3.3 | CRT: 0 A: 1.6 |
NR | 2 | NR | 5.3 | N: 1 C: 0 |
NR | 1.6 | NR |
| Wound‐healing complications | NR | 3.3 | CRT: 1 A: 1.6 |
NR | NR | NR | NR | NR | 0 | 0.8 | |
| Thromboembolic events (arterial ATE, venous VTE) |
C: 3.1 L: 4.9 |
ATE: 5 VTE: 7.6 |
CRT: 4.6 A: 7.7 |
B: 0 L: 5.8 |
4 | S: 7.9 I: 2.5 |
ATE: 1.7 VTE: 8.8 |
N: 4.2 C: 0 |
4.3 | ATE: 0 VTE: 7.6 | B: 4.9 L: 0 |
| Hypertension | C: 14.1 L: 6.5 |
11.3 | CRT: 1.3 A: 4.2 |
B: 26 L: 27 |
NR | NR | 0 | N: 4.2 C: 2.1 |
1.4 | 8.4 | B: 23.7 L: 0.7 |
| Proteinuria | NR | 5.4 | NR | B: 0 L: 5.8 |
NR | NR | NR | N: 0 C: 0 |
NR | NR | NR |
| GI perforation | NR | 1.1 | CRT: 0 A: 0.8 |
NR | NR | NR | 5.3 | N: 2.1 C: 0 |
1.4 | 0.8 | NR |
| Thrombocytopaenia | C: 1.6 L: 38.3 |
15 | CRT: 10.2 A: 11.1 |
B: 2 L: 17 |
10.6 | S: 2.2 I: 0 |
3.5 | N: 2.1 C: 6.1 |
7.2 | NR | *all haem B: 53.7 L: 49.7 |
| Fatigue | C: 16,4 L: 15.4 |
7.4 | CRT: 0 A: 13.1 |
B: 4 L: 15 |
5 | S: 5.6 I: 2.5 |
NR | N: 4.2 C: 0 |
5.8 | NR | NR |
| Diarrhoea | C: 6.3 L: 5.7 |
NR | NR | NR | 1 | S: 0 I: 1.2 |
7 | NR | 1.4 | 5.9 | NR |
ATE: arterial thromboembolic event GI: gastrointestinal NR: not reported VTE: venous thromboembolic event
Anti‐angiogenic therapy compared to no anti‐angiogenic therapy for high‐grade glioma (HGG) (subgroups)
Overall survival
Pooled data: overall survival ‐ primary setting
A meta‐analysis of eight studies of anti‐angiogenic therapy in the primary setting (2833 participants) found no observed difference in overall survival: fixed‐effect HR 0.93, 95% CI 0.86 to 1.02; P = 0.12; high‐certainty evidence (Analysis 1.1; Figure 4; Table 2). There was no significant heterogeneity (I2 = 24%), and as such, we did not perform a random‐effects meta‐analysis.
Pooled data: overall survival ‐ recurrent (relapsed) setting
In a meta‐analysis of three studies (910 participants) of anti‐angiogenic therapy in the recurrent setting, the fixed‐effect HR for overall survival was 0.99 (95% CI 0.85 to 1.16; P = 0.90; moderate‐certainty evidence; Analysis 1.1). However, there was significant statistical heterogeneity (I2= 0.65) and a random‐effects meta‐analysis confirms this (overall survival: HR 1.00, 95% CI 0.76 to 1.31, P = 0.93; I2 = 0.65).
Pooled data: overall survival ‐ anti‐angiogenic therapy versus no anti‐angiogenic therapy
In a meta‐analysis of two studies (237 participants) of anti‐angiogenic therapy without chemotherapy, the HR for overall survival was 1.26 (95% CI 0.96 to 1.65; P = 0.10). There was no observed heterogeneity (I2 = 0).
Pooled data: overall survival ‐ anti‐angiogenic therapy with chemotherapy versus chemotherapy
A meta‐analysis of 11 studies of anti‐angiogenic therapy with chemotherapy (3506 participants) found no significant improvement in overall survival with a HR of 0.92 (95% CI 0.85 to 1.00; P = 0.05; low‐certainty evidence; Analysis 3.2). There was no significant observed heterogeneity (I2 = 35%).
3.2. Analysis.

Comparison 3 Anti‐angiogenic therapy compared to no anti‐angiogenic therapy for high‐grade glioma (subgroups, overall survival), Outcome 2 Hazard for OS anti‐angiogenic plus chemotherapy.
Pooled data: overall survival ‐ bevacizumab versus no bevacizumab
Given the larger number of studies with bevacizumab, we performed a separate analysis of all the bevacizumab‐containing regimens (see Differences between protocol and review). A meta‐analysis of seven studies of bevacizumab (2502 participants) found no observed difference in overall survival: fixed‐effect HR 0.94, 95% CI 0.85 to 1.02; P = 0.15; Analysis 3.3). There was no significant heterogeneity (I2 = 47%).
3.3. Analysis.

Comparison 3 Anti‐angiogenic therapy compared to no anti‐angiogenic therapy for high‐grade glioma (subgroups, overall survival), Outcome 3 Hazard for OS bevacizumab.
Progression‐free survival
Pooled data: progression‐free survival (primary and relapsed setting)
A meta‐analysis of eight studies for progression‐free survival in the primary setting (2833 participants) found a difference in the observed progression‐free survival: HR 0.75, 95% CI 0.69 to 0.82; P < 0.00001; high‐certainty evidence (Analysis 2.1; Table 2). There was no significant heterogeneity (I2 = 46%). Although data have been pooled for the recurrent setting, as only two trials were reported with two substantially different regimens (bevacizumab and cediranib), with substantial consequent heterogeneity (I2 = 86%), it is not possible to infer any meaningful conclusions from this data. An observed difference in progression‐free survival (HR 0.64, 95% CI 0.54 to 0.76; P < 0.00001) was nevertheless noted.
Pooled data: progression‐free survival (anti‐angiogenic therapy with chemotherapy versus chemotherapy)
A meta‐analysis of 10 studies of anti‐angiogenic therapy with chemotherapy (3464 participants) found an observed difference in progression‐free survival: HR 0.72, 95% CI 0.66 to 0.77; P < 0.00001; high‐certainty (Analysis 4.1). There was significant heterogeneity (I2 = 64%). A random‐effects model meta‐analysis confirmed similar results (HR 0.73, 95% CI 0.64 to 0.84; P < 0.00001).
4.1. Analysis.

Comparison 4 Anti‐angiogenic therapy compared to no anti‐angiogenic therapy for high‐grade glioma (subgroups, progression‐free survival), Outcome 1 Hazard for PFS anti‐angiogenic therapy plus chemotherapy.
Pooled data: progression‐free survival (bevacizumab versus no bevacizumab)
A meta‐analysis of six studies of bevacizumab (2362 participants) found a significant observed difference in progression‐free survival: HR 0.65, 95% CI 0.60 to 0.72; P < 0.00001 (Analysis 4.2). There was significant heterogeneity (I2 = 61%) in these studies, as for the greater pooled analysis of progression‐free survival. As such, we performed a random‐effects meta‐analysis with similar results (HR 0.65, 95% CI 0.55 to 0.77; P < 0.00001).
4.2. Analysis.

Comparison 4 Anti‐angiogenic therapy compared to no anti‐angiogenic therapy for high‐grade glioma (subgroups, progression‐free survival), Outcome 2 Hazard for PFS bevacizumab.
Sensitivity analysis
We did not perform a sensitivity analysis according to study quality, removing the only moderate‐certainty study (Lee 2015), which did not result in different results. We further evaluated sensitivity to differing classes of agents and treatment regimens as described above (Analysis 3.1: Analysis 3.2; Analysis 3.3; Analysis 4.1; Analysis 4.2). There were insufficient data to conduct a pooled analysis on the basis of MGMT status or molecular profile at this stage. In general, the subgroup analysis showed that overall survival and progression‐free survival largely remain the same when accounting for studies with differing interventions and settings.
3.1. Analysis.

Comparison 3 Anti‐angiogenic therapy compared to no anti‐angiogenic therapy for high‐grade glioma (subgroups, overall survival), Outcome 1 Hazard for OS anti‐angiogenic therapy alone.
Discussion
Summary of main results
The trials included in this systematic review did not show an overall improvement in overall survival with anti‐angiogenic therapy. They did however, demonstrate a significant overall improvement in progression‐free survival. These divergent results reflect the uncertainty regarding the efficacy of anti‐angiogenic therapy in high‐grade glioma (HGG) and also further question the correlation between radiological progression‐free survival and overall survival in HGG. There is concern that assessment of progression‐free survival was based largely on the appearance on magnetic resonance imaging (MRI) scans and that this may or may not have a relationship to the underlying biologic progression of the malignancy.
Given the large number of high‐certainty evidence studies performed in the primary setting with limited heterogeneity in results, this meta‐analysis presents strong data arguing against the routine use of anti‐angiogenic therapy in this setting. If anti‐angiogenic therapy is to be used in the primary setting in the future, careful consideration should be made to patient selection, in order to optimise patient groups most likely to benefit from these therapies.
Retrospective analysis of the AVAglio study suggests that people with IDH1 wild‐type proneural glioblastoma may derive an overall survival benefit from primary bevacizumab treatment (Chinot 2014). However, this has not been validated in an independent data set and it is unclear if it will be evaluated in a randomised controlled trial (RCT) (Sandmann 2015, in: Chinot 2014).
Despite widespread use of bevacizumab in the recurrent setting, there is still insufficient data to support either its use alone or in combination with chemotherapy over chemotherapy alone. Previous studies have suggested a role for anti‐angiogenic therapy in the relapsed setting for quality of life reasons. In particular, there is evidence that anti‐angiogenic therapy may have quality of life benefits in the relapsed setting (Vredenburgh 2010). Moreover, the CABARET study demonstrated that a proportion of participants in both treatment arms (both bevacizumab‐containing arms) had an improved quality of life (Field 2017). In the main efficacy analysis of this study, there is also evidence to suggest that bevacizumab combined with carboplatin is not superior to bevacizumab alone and that single agent bevacizumab should be used instead of that particular combination (Field 2015). Nevertheless, the results presented here suggest that bevacizumab, either alone or in combination with chemotherapy, are not superior to chemotherapy alone in terms of efficacy or quality of life.
It should also be noted that some of the agents evaluated are more direct angiogenic pathway inhibitors and are arguably more potent as anti‐angiogenic agents than others and this may explain the greater observed effects. For example, bevacizumab and cediranib are inhibitory of vascular endothelial growth factor (VEGF) or VEGF‐R signalling respectively, whereas cilengitide is thought to mediate its anti‐angiogenic effects through inhibition of tumour and endothelial cell adhesion and migration. These differences may also account for the progression‐free survival prolongation observed with bevacizumab that was not seen with cilengitide.
The quality of life data from the largest studies with bevacizumab in HGG that have been presented are not consistent. The AVAglio results suggest that quality of life improves in participants receiving bevacizumab compared with placebo, but they used a measure of deterioration‐free survival which was not prespecified in the trial protocol (Chinot 2014). However, the RTOG 0825 study suggests the opposite, i.e. that quality of life of people with HGG on bevacizumab may be worse than people who have not received bevacizumab (Gilbert 2014). However, the RTOG 0825 study had optional participation in the net clinical benefits component of the study and did not require completion of the form on progression. As such, a larger number of participants in the bevacizumab‐containing regimen completed the questionnaires and this may be responsible for the divergence in results. This may be related to failure to capture progression by non‐contrast enhancing disease (Radbruch 2011), or differences in radiological assessment criteria between the two studies (Chinot 2013). The EORTC study has demonstrated that bevacizumab, in combination with lomustine, does not have health‐related quality of life benefits compared to lomustine alone in recurrent disease (Wick 2017). As bevacizumab was given with lomustine, this study does not provide direct evidence for or against bevacizumab's quality of life benefit if given alone in the relapsed setting.
Gliomas and HGGs are an especially important group of tumours because of their disproportionate impact on people's well‐being and quality of life, in addition to longevity. However, the impact of anti‐angiogenic therapy, including bevacizumab on quality of life remains unclear.
Overall completeness and applicability of evidence
The analysed studies were heterogeneous in terms of the interventions applied, the clinical settings and in their study designs. This review included different pharmacologic interventions that directly or indirectly target angiogenesis. Bevacizumab, cilengitide, cediranib and vandetanib are different agents in their mechanism of action, in their anti‐angiogenic effects and in terms of pharmacodynamic and pharmacokinetic effects. The studies also differed in their clinical settings (some were primary and others were recurrent), but they all were restricted to glioblastoma and were largely high‐quality randomised studies. As such, the evidence presented here is relatively complete, particularly for drawing conclusions in the primary setting.
Quality of the evidence
We demonstrate the overall risk of bias in the studies included in the meta‐analysis as low (Figure 2 and Figure 3). Of the 11 studies, we judged 10 to be of high‐certainty as they were randomised studies with low overall risk of bias (see Risk of bias in included studies). We considered the other study (vandetanib) to be of moderate‐certainty, due to lack of information regarding allocation concealment, and lack of blinding, but was nevertheless a randomised non‐comparative study (Lee 2015).
Table 1 demonstrates the results of the primary analyses for anti‐angiogenic therapy compared with no anti‐angiogenic therapy for overall survival and progression‐free survival. Overall, there is consistent, high‐certainty evidence that demonstrates no improvement in overall survival across treatment settings. Paradoxically, there is consistent, high‐certainty evidence, demonstrating improvement in progression‐free survival across treatment settings. Given the validity of overall survival as an endpoint, the combination of these findings suggests that progression‐free survival may not be an appropriate surrogate endpoint in glioblastoma. Table 2 demonstrates the results of various subgroup analyses. In general, the subgroups are in agreement with the primary analysis. However, a small survival benefit of anti‐angiogenic therapy combined with chemotherapy compared with chemotherapy alone cannot be definitively excluded.
Potential biases in the review process
There was no prespecified subset analysis of trials of anti‐angiogenic therapy alone, anti‐angiogenic therapy with chemotherapy and bevacizumab alone. However, at the time of writing of the review, it was noted that the differing classes of agents would form the basis of the sensitivity analysis.
Agreements and disagreements with other studies or reviews
The results of the updated meta‐analysis are broadly similar to those found in our original review in 2014 (Khasraw 2014). Compared to the previous review, there are additional studies and most studies have been published in full, meriting a more complete assessment of the data. An additional meta‐analysis was reported in 2017 (Lombardi 2017), with broadly similar findings of an improved progression‐free survival with no overall survival benefit. The authors of this meta‐analysis included studies of temsirolimus, enzastaurin and dasatanib, which were not included in this analysis as they were not deemed to be mechanistically primarily anti‐angiogenic therapy inhibitors, as predefined in our protocol. Nevertheless, the results are broadly in keeping with the findings we have observed in our study.
Authors' conclusions
Implications for practice.
High quality evidence is available and does not support a survival advantage for anti‐angiogenics in primary disease either alone or in combination with therapy.
Anti‐angiogenic therapy in relapsed glioblastoma does not confer an overall survival advantage over chemotherapy.
Implications for research.
Further research needs to be done to evaluate whether specific subgroups benefit from anti‐angiogenic therapy and to identify biomarkers of response to anti‐angiogenic therapy. Further research regarding optimal imaging assessment for anti‐angiogenic therapies may also be of help in determining the utility of anti‐angiogenic therapy.
There is a need to clarify the impact of anti‐angiogenic therapy on quality of life in people with glioblastoma.
What's new
| Date | Event | Description |
|---|---|---|
| 19 November 2018 | Amended | Minor correction to Abstract and typo in CoI corrected. |
| 12 November 2018 | New citation required but conclusions have not changed | We identified new studies, but our conclusions remain unchanged. |
| 12 November 2018 | New search has been performed | We updated the literature searches. |
Acknowledgements
We thank Jo Platt for designing and running the searches and Gail Quinn, Clare Jess and Tracey Harrison for their contribution to the editorial process.
This project was supported by the National Institute for Health Research (NIHR), via Cochrane Infrastructure funding to the Cochrane Gynaecological, Neuro‐oncology and Orphan Cancer Group. The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, NHS or the Department of Health.
Appendices
Appendix 1. Comparisons included in review protocol
Primary chemotherapy for newly diagnosed HGG in combination with direct angiogenesis inhibitor (e.g. bevacizumab, aflibercept, or cedirinib), compared to the same chemotherapy without direct angiogenesis inhibitor.
Primary chemotherapy in combination with indirect angiogenesis inhibitor (e.g. cilengitide), compared to the same chemotherapy without indirect angiogenesis inhibitors.
Second‐line chemotherapy in combination with direct angiogenesis inhibitor, compared to the same chemotherapy without direct angiogenesis inhibitors.
First progression following initial treatment of newly diagnosed HGG treated with angiogenesis inhibitor compared to therapy not involving angiogenesis inhibitors.
Second‐line chemotherapy in combination with indirect angiogenesis inhibitor, compared to the same chemotherapy without indirect angiogenesis inhibitor.
Appendix 2. CENTRAL search strategy
#1 MeSH descriptor Glioma explode all trees #2 glioma* or glioblastoma* or astrocytoma* or oligodendroglioma* or oligoastrocytoma* or GBM #3 (#1 OR #2) #4 MeSH descriptor Angiogenesis Inhibitors explode all trees #5 angiogenesis and inhibit* #6 antiangiogenic or anti‐angiogenic #7 MeSH descriptor Vascular Endothelial Growth Factors explode all trees #8 vascular endothelial growth factor* #9 VEGF #10 VEGFR or VEGF‐R #11 bevacizumab or avastin #12 cilengitide #13 cediranib or recentin or azd2171 #14 aflibercept or ave0005 #15 (#4 OR #5 OR #6 OR # OR #8 OR #9 OR #10 OR #11 OR #12 OR #13 OR #14) #16 (#3 AND #15)
Appendix 3. MEDLINE Ovid search strategy
1 exp Glioma/ 2 (glioma* or glioblastoma* or astrocytoma* or oligodendroglioma* or oligoastrocytoma* or GBM).mp. 3 1 or 2 4 exp Angiogenesis Inhibitors/ 5 (angiogenesis and inhibit*).mp. 6 (antiangiogenic or anti‐angiogenic).mp. 7 exp Vascular Endothelial Growth Factors/ 8 vascular endothelial growth factor*.mp. 9 VEGF.mp. 10 (VEGFR or VEGF‐R).mp. 11 (bevacizumab or avastin).mp. 12 cilengitide.mp. 13 (cediranib or recentin or azd2171).mp. 14 (aflibercept or ave0005).mp. 15 4 or 5 or 6 or 7 or 8 or 9 or 10 or 11 or 12 or 13 or 14 16 3 and 15 17 randomised controlled trial.pt. 18 controlled clinical trial.pt. 19 randomized.ab. 20 placebo.ab. 21 clinical trials as topic.sh. 22 randomly.ab. 23 trial.ti. 24 17 or 18 or 19 or 20 or 21 or 22 or 23 25 16 and 24
key:pt=publication type, ab=abstract, ti=title, mp=title, abstract, original title, name of substance word, subject heading word, protocol supplementary concept, rare disease supplementary concept, unique identifier
Appendix 4. Embase Ovid search strategy
1 exp glioma/ 2 (glioma* or glioblastoma* or astrocytoma* or oligodendroglioma* or oligoastrocytoma* or GBM).mp. 3 1 or 2 4 exp angiogenesis inhibitor/ 5 (angiogenesis and inhibit*).mp. 6 (antiangiogenic or anti‐angiogenic).mp. 7 exp vasculotropin/ 8 vascular endothelial growth factor*.mp. 9 VEGF.mp. 10 (VEGFR or VEGF‐R).mp. 11 (bevacizumab or avastin).mp. 12 cilengitide.mp. 13 (cediranib or recentin or azd2171).mp. 14 (aflibercept or ave0005).mp. 15 or/4‐14 16 3 and 15 17 crossover procedure/ 18 double‐blind procedure/ 19 randomized controlled trial/ 20 single‐blind procedure/ 21 random*.mp. 22 factorial*.mp. 23 (crossover* or cross over* or cross‐over*).mp. 24 placebo*.mp. 25 (double* adj blind*).mp. 26 (singl* adj blind*).mp. 27 assign*.mp. 28 allocat*.mp. 29 volunteer*.mp. 30 17 or 18 or 19 or 20 or 21 or 22 or 23 or 24 or 25 or 26 or 27 or 28 or 29 31 16 and 30
key: mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer, device trade name, keyword
Data and analyses
Comparison 1. Anti‐angiogenic therapy compared to no anti‐angiogenic therapy for high‐grade glioma (all patients, overall survival).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Hazard for OS | 11 | 3743 | Hazard Ratio (Fixed, 95% CI) | 0.95 [0.88, 1.02] |
| 1.1 Primary | 8 | 2833 | Hazard Ratio (Fixed, 95% CI) | 0.93 [0.86, 1.02] |
| 1.2 Recurrent | 3 | 910 | Hazard Ratio (Fixed, 95% CI) | 0.99 [0.85, 1.16] |
Comparison 2. Anti‐angiogenic therapy compared to no anti‐angiogenic therapy for high‐grade glioma (all patients, progression‐free survival).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Hazard for PFS | 10 | 3595 | Hazard Ratio (Fixed, 95% CI) | 0.73 [0.68, 0.79] |
| 1.1 Primary | 8 | 2833 | Hazard Ratio (Fixed, 95% CI) | 0.75 [0.69, 0.82] |
| 1.2 Recurrent | 2 | 762 | Hazard Ratio (Fixed, 95% CI) | 0.64 [0.54, 0.76] |
Comparison 3. Anti‐angiogenic therapy compared to no anti‐angiogenic therapy for high‐grade glioma (subgroups, overall survival).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Hazard for OS anti‐angiogenic therapy alone | 2 | 237 | Hazard Ratio (Fixed, 95% CI) | 1.26 [0.96, 1.65] |
| 2 Hazard for OS anti‐angiogenic plus chemotherapy | 11 | 3506 | Hazard Ratio (Fixed, 95% CI) | 0.92 [0.85, 1.00] |
| 3 Hazard for OS bevacizumab | 7 | 2502 | Hazard Ratio (Fixed, 95% CI) | 0.94 [0.85, 1.02] |
Comparison 4. Anti‐angiogenic therapy compared to no anti‐angiogenic therapy for high‐grade glioma (subgroups, progression‐free survival).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Hazard for PFS anti‐angiogenic therapy plus chemotherapy | 10 | 3464 | Hazard Ratio (Fixed, 95% CI) | 0.72 [0.66, 0.77] |
| 2 Hazard for PFS bevacizumab | 6 | 2362 | Hazard Ratio (Fixed, 95% CI) | 0.65 [0.60, 0.72] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Balana 2016.
| Methods | 2‐arm RCT | |
| Participants | People with de novo unresectable glioblastoma Inclusion criteria
Exclusion criteria
|
|
| Interventions |
TMZ arm Neoadjuvant treatment consisted of:
cycles) Concurrent treatment consisted of:
(60 Gy in 2 Gy fractions, for 42 days) plus temozolomide (75 mg/m2/d) for a maximum of 49 days. The experimental arm included the same therapy as the control arm with the addition of bevacizumab 10 mg/kg on days 1, 15 of each cycle in the neoadjuvant stage and on days 1, 15, 30 of the concurrent stage. |
|
| Outcomes |
Primary endpoint
Secondary endpoints
|
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Central block randomisation without stratification |
| Allocation concealment (selection bias) | Low risk | Central randomisation; allocation concealed |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | It is not indicated that any specific blinding procedures were undertaken. As the intervention arm required an intravenous infusion, it is assumed that this was an open‐label study and at high risk of performance bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The primary endpoint was also assessed by central blinded radiological review, reducing risk of detection bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Eight participants in the control arm did not receive the allocated intervention, whereas one patient was excluded from analysis in the intervention arm. |
| Selective reporting (reporting bias) | Unclear risk | Endpoints were not included in initial registration information on ClinicalTrials.gov. |
| Other bias | Low risk | Roche Spain provided funds for data management and centralised review of radiological assessments. |
Batchelor 2013.
| Methods | 3‐arm RCT comparing:
|
|
| Participants | People with glioblastoma, recurrent Inclusion criteria
Exclusion criteria
|
|
| Interventions |
Experimental 1. Cediranib alone
2. Cediranib + lomustine
Control
|
|
| Outcomes |
Response assessment by modified Macdonald criteria |
|
| Notes | Median survival
Some minor differences in the baseline characteristics of groups: 62% (intervention) versus 50% (control) had better Karnofsky performance. Also baseline steroid use differed between the arms (40% in intervention arm versus 50% in control arm). Study sponsored by AstraZeneca |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomised according to full publication (JCO article). From trial protocol method of randomisation was computer program (IWRS) |
| Allocation concealment (selection bias) | Low risk | Allocation was strictly concealed, with a unique ID tracking system |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Both the participants and investigators were blinded (double‐blinded). |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Outcomes were assessed with centralised radiographic review, with masking to study arm. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There was a very low dropout rate (10 participants total), making attrition bias less significant. Follow‐up was similar across all study groups. |
| Selective reporting (reporting bias) | Low risk | Preplanned endpoints were assessed in the final publication, confirmed on registration at ClinicalTrials.gov |
| Other bias | Low risk | No significant other bias observed |
Chauffert 2014.
| Methods | 2‐arm RCT | |
| Participants | People with de novo unresectable supratentorial glioblastoma Inclusion criteria
Exclusion criteria
|
|
| Interventions |
Experimental arm The experimental arm consisted of neo‐adjuvant bevacizumab, 10 mg/kg IV and IRI, 125 mg/m2 IV, every 2 weeks for 4 cycles, before RT. RT, 60 Gy in 30 fractions, was given 5 days/week. The CTV was the enhanced T1‐weighted abnormality GTV plus a 2 cm margin. Oral TMZ, 75 mg/m2 /day, and bevacizumab, 10 mg/kg IV every 2 weeks, were given concomitantly to RT. Adjuvant bevacizumab and IRI were given every 2 weeks for 6 months |
|
| Outcomes |
Primary endpoint
Secondary endpoints
|
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomised study, method not specified |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not specified |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | This was designed as an open‐label study, thereby making it at high risk of performance bias due to inability to blind investigators or participants. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | The primary endpoint, progression‐free survival, was subject to performance bias as local investigators assessed for disease progression and there was no centralised review. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The study has not reported number of participants lost to follow‐up. |
| Selective reporting (reporting bias) | Low risk | The study reported all preplanned endpoints registered on ClinicalTrials.gov. |
| Other bias | Unclear risk | Only 66.7% of participants in the intervention arm completed radiotherapy, which is a proven effective intervention, due to the study design. This compares to 93% in the control arm and may be a source of biased results. |
Chinot 2014.
| Methods | 2‐arm RCT | |
| Participants | People with glioblastoma Inclusion criteria
Exclusion criteria
|
|
| Interventions |
Experimental
RT/TMZ + Bev (N = 458) 6 weeks radiation plus temozolomide 75 mg/m2 daily orally and bevacizumab 10 mg/kg every 2 weeks, 4‐week break, then 6 cycles (of 4 weeks) of bevacizumab 10 mg/kg every 2 weeks + temozolomide 150 mg ‐ 200 mg/m2 orally. Day 1‐5, then bevacizumab 15 mg/kg every 3 weeks until progression |
|
| Outcomes |
Progression‐free survival by modified Macdonald criteria (described in detail in Chinot 2013) |
|
| Notes |
The trial was sponsored by Hoffman La Roche. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation via stratified block, centrally with interactive voice‐response system |
| Allocation concealment (selection bias) | Low risk | Allocation concealed via interactive voice‐response system |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Both participants and investigators were blinded. Unblinding was only allowed in the case of an emergency pre‐progression and every effort was made to maintain the blind post‐progression. The packaging and storage of the placebo and bevacizumab was identical. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Outcomes were assessed with centralised radiographic review, with masking to study arm. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There was a very low dropout rate (9 participants in total lost to follow‐up), making attrition bias less significant. Follow‐up was similar across all study groups. |
| Selective reporting (reporting bias) | Low risk | The endpoints registered at ClinicalTrials.gov were reported as intended in the final publication, making it a low risk of selective reporting. |
| Other bias | Low risk | No significant other bias observed |
Gilbert 2014.
| Methods | 2‐arm RCT | |
| Participants | People with glioblastoma Inclusion criteria
|
|
| Interventions |
Radiotherapy (60 Gy) and daily temozolomide with bevacizumab or placebo beginning during week 4 of radiotherapy and was continued for up to 12 cycles of adjuvant temozolomide. Participants undergo chemoradiotherapy and receive adjuvant temozolomide as in arm 1. Participants also receive bevacizumab IV over 30‐90 minutes once every 2 weeks beginning in week 4 of chemoradiotherapy and continuing until the completion of adjuvant temozolomide. |
|
| Outcomes |
RECIST used for response assessment, but took into account pseudoprogression (i.e. 1st post‐treatment scan not adequate to declare progression) |
|
| Notes | Sponsored by National Cancer Institute | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | From the trial protocol accompanying the publication, randomisation was computer assisted. The Zelen method was used. The first 60 participants had 2:1 chance of intervention, the next 60 participants had 1:2 of intervention with equal chance of intervention or placebo thereafter |
| Allocation concealment (selection bias) | Low risk | Allocation was concealed via an online data management centre |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Both participants and investigators were blinded. Unblinding was only allowed in the case of an emergency pre‐progression. Unblinding was allowed post‐progression. The packaging and storage of the placebo and bevacizumab was identical. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Outcomes were assessed by a blinded centralised radiological review under the auspices of ACRIN. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 8 participants lost to follow‐up in each arm |
| Selective reporting (reporting bias) | Low risk | Have reported on endpoints as planned on trial protocol |
| Other bias | Low risk | No significant other bias observed |
Herrlinger 2016.
| Methods | 2‐arm RCT | |
| Participants | People with glioblastoma, MGMT non‐methylated
Exclusion criteria
|
|
| Interventions | All participants received involved‐field radiotherapy (RT; 30x2 Gy)
starting day 22 to 35 after surgery Experimental arm
Control arm
|
|
| Outcomes |
Primary endpoint
Secondary endpoints
|
|
| Notes | Sponsored by Roche | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐based randomised minimisation method |
| Allocation concealment (selection bias) | Low risk | Computer‐based randomised minimisation method |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | It is not indicated that any specific blinding procedures were undertaken. As the intervention arm required an intravenous infusion, it is assumed that this was an open‐label study and at high risk of performance bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Outcomes were assessed by centralised review. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Six participants in the intervention arm and two participants in the control arm were not assessable due to various reasons. |
| Selective reporting (reporting bias) | Low risk | The study reported all preplanned endpoints registered on ClinicalTrials.gov. |
| Other bias | Unclear risk | Sponsored by Roche |
Lee 2015.
| Methods | 2‐arm RCT | |
| Participants | People with de novo glioblastoma Inclusion
Exclusion
|
|
| Interventions |
Control arm
Intervention arm
|
|
| Outcomes |
Primary endpoint
Secondary endpoints
|
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomised study, method not specified |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not specified |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | This was designed as an open‐label study, thereby making it at high risk of performance bias due to inability to blind investigators or participants. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It is unclear as to whether radiological assessment was performed centrally or by investigators. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | One patient in the intervention arm and seven participants in the control arm did not receive assigned intervention. |
| Selective reporting (reporting bias) | Low risk | The study reported all preplanned endpoints registered on ClinicalTrials.gov. |
| Other bias | Unclear risk | Sponsored by Investigator‐Sponsored Study Program of Astra‐Zeneca. |
Nabors 2015.
| Methods | 3‐arm RCT comparing:
|
|
| Participants | People with glioblastoma, MGMT unmethylated Inclusion criteria
Exclusion criteria
|
|
| Interventions |
Experimental: standard cilengitide (N = 88) or intensive cilengitide (N = 88)
Control: chemoradiation alone
|
|
| Outcomes |
|
|
| Notes | Minor difference in baseline groups: 55% were performance status 1 in control arm versus 45% in intervention arm Study sponsored by Merck |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomised, method unknown |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment status unknown/not reported |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | This was designed as an open‐label study, thereby making it at high risk of performance bias due to inability to blind investigators or participants. However this is less subject to bias with overall survival as the primary endpoint. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The independent review committee assessing progression‐free survival was masked to treatment allocation. As such, the secondary endpoint, progression‐free survival, was not subject to significant detection bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Only 2 participants were lost‐to‐follow‐up, both in the control group. 13 participants withdrew consent for the study. The study follow‐up schedule was the same across treatment groups. |
| Selective reporting (reporting bias) | Low risk | Have currently reported intended endpoints |
| Other bias | Unclear risk | Study sponsored by Merck |
Stupp 2014.
| Methods | 2‐arm RCT comparing:
|
|
| Participants | People with glioblastoma Inclusion criteria
Exclusion criteria
|
|
| Interventions |
|
|
| Outcomes |
Primary endpoint
|
|
| Notes | Sponsored by Merck. Statistics however ran in parallel between Merck and the EORTC, reducing risk of this bias | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation via stratified block, centrally with interactive voice‐response system |
| Allocation concealment (selection bias) | Low risk | Allocation concealed via interactive voice‐response system |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | This was designed as an open‐label study, thereby making it at high risk of performance bias due to inability to blind investigators or participants. However this is less subject to bias with overall survival as the primary endpoint. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The independent review committee assessing progression‐free survival was masked to treatment allocation and the database remained masked to primary outcome variables for all parties prior to final analysis. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Only one patient was lost to follow‐up across the study. Twenty‐four participants in the intervention arm withdrew consent for the study and 17 in the control arm withdrew consent. |
| Selective reporting (reporting bias) | Low risk | The study reported all preplanned endpoints registered on ClinicalTrials.gov. |
| Other bias | Unclear risk | The study was funded by Merck. The principal investigators had full access to and reviewed all data, and had final responsibility for the decision to submit for publication. |
Taal 2014.
| Methods | 3‐arm RCT comparing:
|
|
| Participants | People with glioblastoma, recurrent Inclusion/exclusion criteria
|
|
| Interventions |
Experimental:bevacizumab (N = 51) or bevacizumab/lomustine (N = 55)
Amendment 3: restart combination arm on 90 mg/m2 lomustine (cap at 160 mg) (bevacizumab/lomustine 90) Control: lomustine alone (N = 47)
|
|
| Outcomes |
Progression‐free survival by RANO criteria |
|
| Notes | Unrestricted grant from Roche NL but independent statistics | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Central stratified block randomisation procedure |
| Allocation concealment (selection bias) | Low risk | Web‐based allocation programme |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | This was designed as an open‐label study, thereby making it at high risk of performance bias due to inability to blind investigators or participants. However this is less subject to bias with overall survival as the primary endpoint. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Radiographic disease progression was assessed by local investigators, who were also aware of the treatment assignment. Consequently, a secondary endpoint, progression‐free survival, is at high risk of bias. This is somewhat mitigated by the fact the primary endpoint of the study was overall survival at 9 months. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Five participants dropped out across the whole study, therefore unlikely to have attrition bias |
| Selective reporting (reporting bias) | Low risk | Preplanned endpoints were assessed in the final publication based upon registration information on the EU clinical trials database (EUDRA‐CT). Of note, the primary endpoint was changed from progression‐free survival to 6‐month overall survival based upon emerging data from other clinical trials. As the initial endpoint, progression‐free survival, was still reported, we consider this to be at low risk of reporting bias. |
| Other bias | Low risk | No significant other bias was observed. |
Wick 2017.
| Methods | 2‐arm randomised phase III study | |
| Participants | Recurrent glioblastoma
|
|
| Interventions |
Control arm
Experimental arm
|
|
| Outcomes |
Primary endpoint
|
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Central adaptive stratified sampling (minimisation) procedure |
| Allocation concealment (selection bias) | Low risk | Central allocation (EORTC) |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | This was designed as an open‐label study, thereby making it at high risk of performance bias due to inability to blind investigators or participants. However this is less subject to bias with overall survival as the primary endpoint. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Outcomes were assessed with centralised radiographic review, with masking to study arm. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There was a very low dropout rate (22 participants total), making attrition bias less significant. |
| Selective reporting (reporting bias) | Low risk | The study reported all preplanned endpoints as confirmed at ClinicalTrials.gov and in the accompanying publication of the trial protocol with the final publication. |
| Other bias | Low risk | Staff at the EORTC and the first author reviewed all the data. The EORTC was the trial sponsor and vouches for the integrity, accuracy, and completeness of the data. F. Hoffmann–La Roche supported EORTC 26101 through an educational grant and provided bevacizumab free of charge but had no role in analysing the data or writing the manuscript. |
ACRIN: American College of Radiology Imaging Network. ANC: absolute neutrophil count CTV: clinical target volume ECOG: Eastern Cooperative Oncology Group EORTC: European Organisation for Research and Treatment of Cancer Gd‐MRI: gadolinium‐magnetic resonance imaging GTV: gross tumour volume IRI: immunoreactive insulin IV: intravenous MGMT: methylguanine‐DNA methyltransferase MRI: magnetic resonance imaging PS: performance scale RCT: randomised controlled trial RPA: recursive partitioning analysis RT: radiotherapy RTX: radiotherapy SGOT: serum glutamic‐oxaloacetic transaminase SGPT: serum glutamic‐pyruvic transaminase ULN: upper limit of normal QT: QT interval TMZ: temozolomide WHO: World Health Organisation WBC: white blood cells
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Brandes 2016 | Non‐comparative randomised controlled trial for which inadequate data exists to assess endpoints of this meta‐analysis. The primary investigator of this study was contacted for this data prior to exclusion of this study from the meta‐analysis. |
| Duerinck 2016 | In the control arm, 20 out of 22 cases received bevacizumab, an anti‐angiogenic therapy. |
| Friedman 2009 | No control arm not containing anti‐angiogenic therapy |
| Kreisl 2009 | No control arm not containing anti‐angiogenic therapy |
| Lombardi 2018 | Predominant mode of action multi‐kinase inhibition. |
| Van Den Bent 2017 | Currently available data are only in the form of conference proceedings and is a mix of low‐grade and high‐grade gliomas (grade 2 and grade 3) and would be therefore excluded from this study protocol. |
Characteristics of studies awaiting assessment [ordered by study ID]
Wirsching 2018.
| Methods | 2 arm RCT (arm A, N=50) or without bevacizumab (arm B, N=25) in patients with newly diagnosed glioblastoma aged greater than65 years. The primary objective was to obtain evidence for prolongation of median OS by the addition of bevacizumab to RT. Response was assessed by RANO criteria. |
| Participants | Patients with newly diagnosed glioblastoma, aged > 65 years. |
| Interventions | Arm A: hypofractionated RT (40 Gy in 15 fractions) with bevacizumab (10 mg/kg every 2 weeks) (N=50) Arm B: hypofractionated RT (40 Gy in 15 fractions) alone (N=25) |
| Outcomes | Overall survival, progression‐free survival |
| Notes |
Differences between protocol and review
The background has been shortened in the review but the methods and analysis plan has been adhered to. Funnel plots corresponding to meta‐analysis of the primary outcome were planned to be used to exclude publication bias. However, due to the small number of total studies involved, this was not undertaken. For data analysis it was planned to use risk ratio (RR) for dichotomous outcomes and standardised mean differences for continuous outcomes. However, as summary statistics in the form of hazard ratios were used in the trials included in the meta‐analysis, these data analyses were not performed.
The following subgroups were planned to be analysed, but were not included in final analysis as there was inadequate data.
Specific a priori subgroups included
Histology: HGG (WHO: grades III versus IV), astrocytoma versus oligodendroglioma.
Extent of resection (resection versus biopsy).
Known patient‐related prognostic factors, such as age and performance status.
Anti‐angiogenics as monotherapy, or in combination with chemotherapy (or chemoradiotherapy).
Drug class (monoclonal antibodies versus tyrosine kinase inhibitors (TKIs), versus indirect angiogenesis inhibitor, versus other (unspecified or not definitely known, e.g. thalidomide).
Status of promoter hypermethylation of the MGMT gene.
Recursive partitioning analysis classification.
Loss of heterozygosity for 1p and 19q.
Contributions of authors
MA: writing of the text and analysis MK: design of protocol, analysis and writing of the text NP: design of protocol and final approval of the protocol All authors prepared the review and approved the final version of the review.
Sources of support
Internal sources
None, Other.
External sources
None, Other.
Declarations of interest
Nick Pavlakis ‐ none known
Helen Wheeler ‐ the analysis for this Cochrane Review is based on peer‐reviewed data which was prepared by an independent steering trials committee. My involvement in the Australian Roche Advisory Board was to discuss completed trial results and how the drug may be introduced into the clinic in Australian centres. My participation on the merck serono centric steering committee was to review ongoing trial recruitment and serious adverse events. None of these activities influenced the analysis of the review data or contributed to any presented/published conclusions.
Robin Grant ‐ no conflict of interest related to this review
John Simes ‐ I have no relevant conflicts of interest to declare. My institution has received research funding support from Merck KGa and Roche.
Malaka Ameratunga‐ none known
Mustafa Khasraw ‐ none known. My institution has received research funding support from Merck KGa and I have served on glioblastoma advisory boards of Roche.
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Balana 2016 {published data only}
- Balana C, Las Penas R, Sepulveda JM, Gil‐Gil MJ, Luque R, Gallego O, et al. Bevacizumab and temozolomide versus temozolomide alone as neoadjuvant treatment in unresected glioblastoma:the GENOM 009 randomized phase II trial. Journal of Neurooncology 2016;127:569‐79. [DOI: 10.1007/s11060-016-2065-5] [DOI] [PubMed] [Google Scholar]
Batchelor 2013 {published data only}
- Batchelor TT, Mulholland P, Neyns B, Nabors LB, Campone M, Wick A, et al. Phase III randomised trial comparing the efficacy of cediranib as monotherapy, and in combination with lomustine, versus lomustine alone in patients with recurrent glioblastoma. Journal of Clinical Oncology 2013;31(26):3212‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Chauffert 2014 {published data only}
- Chauffert B, Feuvret L, Bonnetain F, Taillandier L, Frappaz D, Taillia H, et al. Randomized phase II trial of irinotecan and bevacizumab as neo‐adjuvant and adjuvant to temozolomide‐based chemoradiation compared with temozolomide‐chemoradiation for unresectable glioblastoma: final results of the TEMAVIR study from ANOCEF. Annals of Oncology 2014;25:1442‐7. [DOI: 10.1093/annonc/mdu148] [DOI] [PubMed] [Google Scholar]
Chinot 2014 {published data only}
- Chinot O, Wick W, Mason W, Henriksson R, Saran F, Nishikawa R, et al. Bevacizumab plus radiotherapy‐temozolomide for newly diagnosed glioblastoma. New England Journal of Medicine 2014;370(8):709‐22. [DOI: 10.1056/NEJMoa1308345] [DOI] [PubMed] [Google Scholar]
- Sandmann T, Bourgon R, Garcia J, Li C, Cloughesy T, Chinot Ol, et al. Patients with proneural glioblastoma may derive overall survival benefit from the addition of bevacizumab to first‐line radiotherapy and temozolomide: retrospective analysis of the AVAglio Trial. Journal of Clinical Oncology 2015;33(25):2735‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taphoorn MJ, Henriksson R, Bottomley A, Cloughesy T, Wick W, Mason WP, et al. Health‐related quality of life in a randomized phase III study of bevacizumab, temozolomide, and radiotherapy in newly diagnosed glioblastoma.. Journal of Clinical Oncology 2015;33(19):2166‐75. [DOI] [PubMed] [Google Scholar]
Gilbert 2014 {published data only}
- Armstrong TS, Won M, Wefel JS, Gilbert MR, Pugh SL, Brachman D, et al. Comparative impact of tumor and treatment on patient reported outcomes (PROs) in patients with glioblastoma (GBM) enrolled In RTOG 0825. Journal of Clinical Oncology 2013; Vol. 31 Suppl 15.
- Gilbert MR, Dignam J, Armstrong TS, Wefel JS, Blumenthal D, Vogelbaum MA, et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. New England Journal of Medicine 2014;370(8):699‐708. [DOI: 10.1056/NEJMoa1308573] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wefel J, Pugh S, Armstrong T, Gilbert M, Won M, Wendland M, et al. Neurocognitive function outcomes in patients with glioblastoma (GBM) enrolled in RTOG 0825. Journal of Clinical Oncology 2013; Vol. 31 Suppl 15.
Herrlinger 2016 {published data only}
- Herrlinger U, Schaefer N, Steinbach JP, Weyerbrock A, Hau P, Goldbrunner R, et al. Bevacizumab plus irinotecan versus temozolomide in newly diagnosed O6‐methylguanine–DNA methyltransferase nonmethylated glioblastoma: The Randomized GLARIUS Trial.. Journal of Clinical Oncology 2016;34(14):1611‐19. [DOI] [PubMed] [Google Scholar]
Lee 2015 {published data only}
- Lee EQ, Kaley TJ, Duda DG, Schiff D, Lassman AB, Wong ET, et al. A multicenter, phase II, randomized, noncomparative clinical trial of radiation and temozolomide with or without vandetanib in newly diagnosed glioblastoma patients. Clinical Cancer Research 2015;21(16):3610‐8. [DOI: 10.1158/1078-0432.CCR-14-3220] [DOI] [PMC free article] [PubMed] [Google Scholar]
Nabors 2015 {published data only}
- Nabors LB, Fink KL, Mikkelsen T, Grujicic D, Tarnawski R, Nam DH, et al. Two cilengitide regimens in combination with standard treatment for patients with newly diagnosed glioblastoma and unmethylated MGMT gene promoter: results of the open‐label, controlled, randomized phase II CORE study. Neuro‐Oncology 2015;17(5):708‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Stupp 2014 {published data only}
- Stupp R, Hegi ME, Gorlia T, Erridge S, Perry J, Hong Y‐K, et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071‐22072 study): a multicentre, randomised, open‐label, phase 3 trial. The Lancet Oncology 2014;15:1100‐8. [DOI] [PubMed] [Google Scholar]
Taal 2014 {published data only}
- Taal W, Oosterkamp HM, Walenkamp A, Dubbink H, Beerepoot LV, Hanse M, et al. Single‐agent bevacizumab or lomustine versus a combination of bevacizumab plus lomustine in patients with recurrent glioblastoma (BELOB trial): a randomised controlled phase 2 trial. The Lancet Oncology 2014;15(9):943‐53. [DOI] [PubMed] [Google Scholar]
Wick 2017 {published data only}
- Wick W, Gorlia T, Bendszus M, Taphoorn M, Sahm F, Harting I, et al. Lomustine and bevacizumab in progressive glioblastoma. New England Journal of Medicine 2017;377(20):1954‐63. [DOI: 10.1056/NEJMoa1707358] [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Brandes 2016 {published data only}
- Brandes AA, Finocchiaro G, Zagonel V, Reni M, Caserta C, Fabi A, et al. AVAREG: a phase 2, randomized, noncomparative study of fotemustine or bevacizumab for patients with recurrent glioblastoma. Neuro‐Oncology 2016;18(9):1304‐12. [DOI: 10.1093/neuonc/now035] [DOI] [PMC free article] [PubMed] [Google Scholar]
Duerinck 2016 {published data only}
- Duerinck J, Du Four S, Vandervorst S, D'Haene N, Mercier M, Michotte A, et al. Randomized phase II study of axitinib versus physicians best alternative choice of therapy in patients with recurrent glioblastoma. Journal of Neurooncology 2016;128:147‐55. [DOI: 10.1007/s11060-016-2092-2] [DOI] [PubMed] [Google Scholar]
Friedman 2009 {published data only}
- Friedman HS, Prados MD, Wen PY, Mikkelsen T, Schiff D, Abrey LE, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. Journal of Clinical Oncology 2009;27(28):4733‐40. [DOI] [PubMed] [Google Scholar]
Kreisl 2009 {published data only}
- Kreisl TM, Kim L, Moore K, Duic P, Royce C, Stroud I, et al. Phase II trial of single‐agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. Journal of Clinical Oncology 2009;27(5):740‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lombardi 2018 {published data only}
- Lombardi G, Salvo GL, Ruda R, Franceschi E, Eoli M, Faedi M, et al. Updated results of REGOMA: A randomized, multicenter, controlled open‐label phase II clinical trial evaluating regorafenib in relapsed glioblastoma (GBM) patients (PTS). Journal of Clinical Oncology. May 2018; Vol. 36, issue 15:2047.
Van Den Bent 2017 {published data only}
- Bent M, Klein M, Smits M, Reijneveld JC, Idbaih A, Clement P, et al. Final results of the EORTC Brain Tumor Group randomized phase II TAVAREC trial on temozolomide with or without bevacizumab in 1st recurrence grade II/III glioma without 1p/19q co‐deletion. Journal of Clinical Oncology 2017; Vol. 35 Suppl 15. [DOI: 10.1200/JCO.2017.35.15_suppl.2009] [DOI]
References to studies awaiting assessment
Wirsching 2018 {published data only}
- Wirsching HG, Tabatabai G, Roelcke U, Hottinger AF, Jörger F, Schmid A, et al. Bevacizumab plus hypofractionated radiotherapy versus radiotherapy alone in elderly patients with glioblastoma: the randomized, open‐label, phase II ARTE trial. Annals of Oncology 10 Apr 2018;29(6):1423‐30. [DOI] [PubMed] [Google Scholar]
Additional references
Altman 2001
- Altman DG. Systematic reviews of evaluations of prognostic variables. BMJ (Clinical Research edition) 2001;323(7306):224‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Brandsma 2009
- Brandsma D, Bent MJ. Pseudoprogression and pseudoresponse in the treatment of gliomas. Current Opinion in Neurology 2009;22(6):633‐8. [DOI] [PubMed] [Google Scholar]
CBTRUS 2015
- Ostrom QT, Gittleman H, Fulop J, Liu M, Blanda R, Kromer C, et al. CBTRUS Statistical Report: primary brain and central nervous system tumors diagnosed in the United States in 2008‐2012. Neuro‐Oncology 2015;Suppl 4:iv1‐iv62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Chinot 2013
- Chinot OL, Macdonald DR, Abrey LE, Zahlmann G, Kerloeguen Y, Cloughesy TF. Response assessment criteria for glioblastoma: practical adaptation and implementation in clinical trials of antiangiogenic therapy. Current Neurology Neurosciences Reports 2013;13:347. [DOI] [PMC free article] [PubMed] [Google Scholar]
Cox 1972
- Cox DR. Regression models and life tables. Journal of the Royal Statistical Society 1972;34(2):187‐220. [Google Scholar]
CTCAE 2017
- NIH‐NIC. Common terminology criteria for adverse events (CTCAE v5.0). National Cancer Institute 2017.
Curran 1993
- Curran WJ Jr, Scott CB, Horton J, Nelson JS, Weinstein AS, Fischbach AJ, et al. Recursive partitioning analysis of prognostic factors in three radiation therapy oncology group malignant glioma trials. Journal of the National Cancer Institute 1993;85(9):704‐10. [DOI] [PubMed] [Google Scholar]
DerSimonian 1968
- DerSimonian R, Laird N. Meta‐analysis in clinical trials. Controlled Clinical Trials 1986;7:177‐88. [DOI] [PubMed] [Google Scholar]
Fayers 2001
- Fayers PM, Aaronson NK, Bjordal K, Grønvold M, Curran D, Bottomley A, EORTC Quality of Life Group. EORTC QLQ‐C30 Scoring Manual. 3rd Edition. Brussels: European Organisation for Research and Treatment of Cancer, 2001. [ISBN: 2‐9300‐6416‐1] [Google Scholar]
Fidler 1994
- Fidler IJ, Ellis LM. The implications of angiogenesis for the biology and therapy of cancer metastasis . Cell 1994;79(2):185‐8. [DOI] [PubMed] [Google Scholar]
Field 2015
- Field KM, Simes J, Nowak AK, Cher L, Wheeler H, Hovey EJ, et al. Randomized phase 2 study of carboplatin and bevacizumab in recurrent glioblastoma. Neuro‐oncology 2015;17(11):1504‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Field 2017
- Field KM, King MT, Simes J, Espinoza D, Barnes EH, Sawkins K, et al. Health‐related quality of life outcomes from CABARET: a randomized phase 2 trial of carboplatin and bevacizumab in recurrent glioblastoma. Journal of Neuro‐oncology 2017;133(3):623‐31. [DOI] [PubMed] [Google Scholar]
Folkman 1971
- Folkman J, Bach M, Rowe JW, Davidoff F, Lambert P, Hirsch C, et al. Tumor angiogenesis ‐ therapeutic implications. New England Journal of Medicine 1971;285(21):1182‐6. [DOI] [PubMed] [Google Scholar]
Folkman 1990
- Folkman J. What is the evidence that tumors are angiogenesis dependent?. Journal of the National Cancer Institute 1990;82(1):4‐6. [DOI] [PubMed] [Google Scholar]
Gasparini 2005
- Gasparini G, Longo R, Fanielli M, Teicher BA. Combination of antiangiogenic therapy with other anticancer therapies: results, challenges and open questions. Journal of Clinical Oncology 2005;23(6):1295‐311. [DOI] [PubMed] [Google Scholar]
Hicklin 2005
- Hicklin DJ, Ellis LM. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. Journal of Clinical Oncology 2005;23(5):1011‐27. [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG . Measuring inconsistency in meta‐analyses. BMJ 2003;327:557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011a
- Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Higgins 2011b
- Higgins JP, Deeks JJ, editor(s). Chapter 7: Selecting studies and collecting data. In: Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Kerbel 2008
- Kerbel RS. Tumor angiogenesis. New England Journal of Medicine 2008;358:2039‐49. [DOI: 10.1056/NEJMra0706596] [DOI] [PMC free article] [PubMed] [Google Scholar]
Khasraw 2010
- Khasraw M, Lassman A. Advances in the treatment of malignant gliomas. Current Oncology Reports 2010;12(1):26‐33. [DOI] [PubMed] [Google Scholar]
Langendam 2013
- Langendam MW, Akl EA, Dahm P, Glasziou P, Guyatt G, Schunemann HJ. Assessing and presenting summaries of evidence in Cochrane Reviews. Systematic Reviews 2013;23(2):81. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lexchin 2003
- Lexchin J, Bero LA, Djulbegovic B, Clark O. Pharmaceutical industry sponsorship and research outcome and quality: systematic review. BMJ 2003;326:1167‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lombardi 2017
- Lombardi G, Pambuku A, Bellu L, Farina M, Della Puppa A, Denaro l, et al. Effectiveness of antiangiogenic drugs in glioblastoma patients: A systematic review and meta‐analysis of randomized clinical trials. Critical Reviews in Oncology/Hematology 2017;111:94‐102. [DOI] [PubMed] [Google Scholar]
Louis 2016
- Louis DN, Perry A, Reifenberger G, Deimling A, Figarella‑Branger D, Cavenee WK, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System. Acta Neuropathology 2016;131(6):803‐20. [DOI: 10.1007/s00401-016-1545-1] [DOI] [PubMed] [Google Scholar]
Macdonald 1990
- Macdonald DR, Cascino TL, Schold SC Jr, Cairncross JG. Response criteria for Phase II studies of supratentorial malignant gliomas. Journal of Clinical Oncology 1990;8(7):1277‐80. [DOI] [PubMed] [Google Scholar]
Machin 1997
- Machin D, Stenning SP, Parmar MKP. Thirty years of Medical Research Council randomized trials in solid tumors. Journal of Clinical Oncology 1997;9:100‐14. [DOI] [PubMed] [Google Scholar]
Mauer 2008
- Mauer ME, Bottomley A, Taphoorn MJ. Evaluating health‐related quality of life and symptom burden in brain tumour patients: instruments for use in clinical trials and clinical practice. Current Opinion in Neurology 2008;21:741‐53. [DOI] [PubMed] [Google Scholar]
Meader 2014
- Meader N, King K, Llewellyn A, Norman G, Brown J, Rodgers M, et al. A checklist designed to aid consistency and reproducibility of GRADE assessments: development and pilot validation. Systematic Reviews 2014;3:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
Mirimanoff 2006
- Mirimanoff R‐O, Gorlia T, Mason W, Bent MJ, Kortmann R‐D, Fisher B, et al. Radiotherapy and temozolomide for newly diagnosed glioblastoma: recursive partitioning analysis of the EORTC 26981/22981‐NCIC CE3 phase III randomized trial. Journal of Clinical Oncology 2006;24(16):2563‐9. [DOI] [PubMed] [Google Scholar]
Parmar 1998
- Parmar MKB, Torri V, Steward L. Extracting summary statistics to perform meta‐analysis of the published literature for survival endpoints. Statistics in Medicine 1998;17:2815‐34. [DOI] [PubMed] [Google Scholar]
Radbruch 2011
- Radbruch A, Lutz K, Wiestler B, Bäumer P, Heiland S, Wick W, et al. Relevance of T2 signal changes in the assessment of progression of glioblastoma according to the Response Assessment in Neurooncology criteria. Neuro‐oncology 2011;14(2):222‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Review Manager 2014 [Computer program]
- Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Scaringi 2013
- Scaringi C, Enrici RM, Minniti G. Combining molecular targeted agents with radiation therapy for malignant gliomas. Onco Targets and Therapy 2013;6:1079‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sivakumar 2005
- Sivakumar B, Harry LE, Paleolog EM. Modulating angiogenesis. JAMA 2005;292(8):972‐7. [DOI] [PubMed] [Google Scholar]
Stupp 2005
- Stupp R, Mason WP, Bent MJ, Weller M, Fisher B, Taphoorn MJB, et al: European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups and the National Cancer Institute of Canada Clinical Trials Group. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. New England Journal of Medicine 2005;352:987‐96. [DOI] [PubMed] [Google Scholar]
Stupp 2007
- Stupp R, Hegi ME, Gilbert MR, Chakravarti A. Chemoradiotherapy in malignant glioma: standard of care and future directions. Journal of Clinical Oncology 2007;25(26):4127‐36. [DOI] [PubMed] [Google Scholar]
Stupp 2009
- Stupp R, Hegi ME, Mason WP, Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5‐year analysis of the EORTC‐NCIC trial. Lancet Oncology 2009;10(5):459‐66. [DOI] [PubMed] [Google Scholar]
Taphoorn 2010
- Taphoorn MJB, Claassens L, Aaronson NK, Coens C, Mauer M, Osoba D, et al. An international validation study of the EORTC brain cancer module (EORTC QLQ‐BN20) for assessing health‐related quality of life and symptoms in brain cancer patients. European Journal of Cancer 2010;46(6):1033‐40. [DOI] [PubMed] [Google Scholar]
Therasse 2000
- Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, et al. New guidelines to evaluate the response to treatment in solid tumors. Journal of the National Cancer Institute 2000;92(3):205‐16. [DOI] [PubMed] [Google Scholar]
Vredenburgh 2010
- Vredenburgh J, Cloughesy T, Samant M, Prados M, Wen P, Mikkelson T, et al. Corticosteroid use in patients with glioblastoma at first or second relapse treated with bevacizumab in the BRAIN Study. The Oncologist 2010;15(12):1329‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wen 2010
- Wen PY, Macdonald DR, Reardon DA, Cloughesy TF, Sorensen AG, Galanis E, et al. Updated Response Assessment Criteria for High‐Grade Gliomas: Response Assessment in Neuro‐Oncology Working Group. Journal of Clinical Oncology 2010;28(11):1963‐72. [DOI] [PubMed] [Google Scholar]
Yan 2009
- Yan H, Parsons DW, Jin G, McLendon R, Ahmed Rasheed B, Yuan W, et al. IDH1 and IDH2 mutations in gliomas. New England Journal of Medicine 2009;360:765‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
Zebrowski 1999
- Zebrowski BK, Yano S, Liu WB, Shaheen RM, Hicklin DJ, Putnam JB, et al. Vascular endothelial growth factor levels and induction of permeability in malignant pleural effusions. Clinical Cancer Research 1999;5(11):3364‐68. [PubMed] [Google Scholar]
References to other published versions of this review
Khasraw 2010b
- Khasraw M, Lassman A, Wheeler H, Pavlakis N. Anti‐angiogenic therapy for high grade glioma. Cochrane Database of Systematic Reviews 2010, Issue 1. [DOI: 10.1002/14651858.CD008218] [DOI] [PMC free article] [PubMed] [Google Scholar]
Khasraw 2014
- Khasraw M, Ameratunga MS, Grant R, Wheeler H, Pavlakis N. Antiangiogenic therapy for high‐grade glioma. Cochrane Database of Systematic Reviews 2014, Issue 9. [DOI: 10.1002/14651858.CD008218.pub3] [DOI] [PubMed] [Google Scholar]