

Abstract
Traumatic brain injury (TBI) has been linked to the development of numerous psychiatric diseases, including substance use disorder. However, it can be difficult to ascertain from clinical data whether the TBI is cause or consequence of increased addiction vulnerability. Surprisingly few studies have taken advantage of animal models to investigate the causal nature of this relationship. In terms of a plausible neurobiological mechanism through which TBI could magnify the risk of substance dependence, numerous studies indicate that TBI can cause widespread disruption to monoaminergic signaling in striatal regions, and also increases neuroinflammation. In the current study, male Long-Evans rats received either a mild or severe TBI centered over the frontal cortex via controlled cortical impact, and were subsequently trained to self-administer cocaine over ten six-hour sessions. At the end of the study, markers of striatal dopaminergic function, and levels of inflammatory cytokine levels in the frontal lobes, were assessed via western blot and multiplex ELISA, respectively. There was significantly higher cocaine intake in a subset of animals with either mild or severe TBI. However, many animals within both TBI groups failed to acquire self-administration. Principal components analysis suggested that both dopaminergic and neuroinflammatory proteins were associated with overall cocaine intake, yet only an inflammatory component was associated with acquisition of self-administration, suggesting neuroinflammation may make a more substantial contribution to the likelihood of drug-taking. Should neuroinflammation play a causal role in mediating TBI-induced addiction risk, anti-inflammatory therapy may reduce the likelihood of substance abuse in TBI populations.
Keywords: Cytokine, dopamine, psychostimulant, brain injury, rat
Graphical Abstract
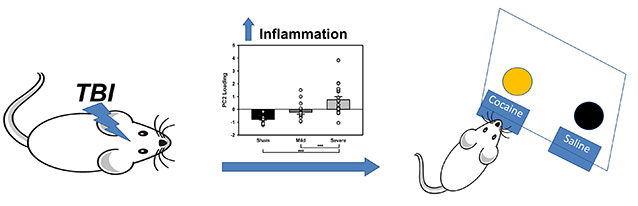
Both mild and severe traumatic brain injury caused an increase in cocaine self-administration in a subset of animals. Overall neuroinflammation (as measured by PCA) accounted for likelihood of acquisition of self-administration, while dopamine-related proteins did not, suggesting a role for inflammation in the development of addiction.
Introduction
Traumatic brain injury (TBI) is a major health problem, affecting over 2.5 million people in the United States annually (Center for Disease Control, 2017). Until recently, TBI was largely considered an acute problem. However, recent data on the long-term complications that arise from brain injuries suggest that TBI is best considered as a chronic condition (Draper & Ponsford, 2008; Corrigan & Hammond, 2013). Brain injury increases the risk for numerous psychiatric diseases, such as depression, anxiety, and addiction (Vaishnavi et al., 2009; Reeves & Panguluri, 2011; Zgaljardic et al., 2015), which suggests a priming effect wherein TBI induces or exacerbates susceptibility to these disorders. Further compounding this problem is the heterogeneous nature of brain injury: adverse outcomes, particularly those related to psychiatric disease, are difficult to predict from the initial clinical presentation (Rosenbaum & Lipton, 2012).
Addiction is one of the largest clinical and social challenges currently facing the world, resulting in 52,000 deaths per year across all substances of abuse in the United States alone (National Institute of Drug Abuse, 2015), while illicit substance abuse disorders affect approximately 2.3% of the adult population (Center for Behavioral Health Statistics and Quality, 2015). Addiction as a consequence of TBI is of particular concern due to the widespread prevalence of brain injury. While increased addiction risk has been established in specific populations following TBI (e.g. military, Miller et al., 2013), relatively little is known about the mechanisms by which this may occur, and the extent to which this is a generalized phenomenon across all brain injury and substances of abuse. Recently, multiple researchers have observed increased self-administration of alcohol after TBI (Lim et al., 2015; Mayeux et al., 2015; Weil et al., 2016). Furthermore, recent studies of brain injury in rats have demonstrated long-lasting increases in impulsive action and impulsive choice, (Vonder Haar et al., 2016; Vonder Haar et al., 2017), both of which are strongly associated with addiction vulnerability in humans, and are predictive of an addiction-like pattern of psychostimulant intake in animals (Perry et al., 2005; Belin et al., 2008).
Many clinical reports present a classic question of correlation versus causation with regard to TBI and addiction, as large-scale prospective studies are lacking. Moreover, relatively little research has explored the role of psychostimulant addiction after TBI relative to other substances such as opioids or alcohol. However, recent research suggests that TBI is significantly more prevalent in cocaine-dependent populations, and TBIs preceded onset of drug use while exacerbating white matter damage (Ma et al., 2015; Ramesh et al., 2015). Demonstrating a directional link between brain injury and increased cocaine self-administration would strengthen the argument that the injured brain is more susceptible to the addictive properties of psychostimulants, and enable investigations into the underlying neurobiological mechanisms which may enable addiction and impair cognitive function. If large-scale pathological changes as a result of TBI generate a pro-substance abuse phenotype, then these models may also shed light on the brain changes permissive of addiction. In particular, two pathophysiological features of TBI are also prominent in addiction: alterations to dopaminergic signaling, and neuroinflammation (Najjar et al., 2013; Ashok et al., 2017).
In brain-injured populations, the scale of changes to the dopamine system has been suggested to be a major mediator of dysfunction after injury (Bales et al., 2009). Detrimental outcomes are associated with variants of the human dopamine transporter (DAT) and dopamine D2 receptor genes (Wagner et al., 2014; Treble-Barna et al., 2017). Experimental injuries result in lower levels of DAT, and consequently, reduced dopamine release and firing rates (Wagner et al., 2005; Shimada et al., 2014; Chen et al., 2015). Moreover, dopaminergic therapies may improve symptoms in brain-injured populations, including response disinhibition, executive function, and motivation (Powell et al., 1996; Beers et al., 2005; Kraus et al., 2005; Moreno-López et al., 2016). In the addiction field, long-term dopaminergic changes such as reduced signaling, and aberrant plasticity are associated with a variety of drugs, including psychostimulants (Perez et al., 2010; Ashok et al., 2017), and dopaminergic connections between the dorsal and ventral striatum are essential for the development of addiction (Belin & Everitt, 2008). Despite this, it has not been determined if the changes observed in the dopamine system following a TBI also yield an increased vulnerability to stimulant addiction.
With regard to neuroinflammation, TBI can cause upregulation of inflammatory factors, lasting up to years in human patients (Johnson et al., 2013; Scott et al., 2015). This includes pro- and anti-inflammatory cytokines, as well as microglia activation (Holmin & Mathiesen, 1999; Shultz et al., 2012; Morganti et al., 2015; Morganti et al., 2016; Vonder Haar et al., 2016). In animal models, inflammatory factors are associated with impulsive behavioral deficits (Morganti et al., 2015; Vonder Haar et al., 2016; Vonder Haar et al., 2017). Recent research in psychiatry has also identified changes in neuroinflammation associated with abuse of a variety of substances, including psychostimulants (Dhillon et al., 2008; Fox et al., 2012; Najjar et al., 2013; Ray et al., 2014; Rodrigues et al., 2014). However, similar to aspects of the dopamine hypothesis of drug dependence, it is unclear whether these changes precede addiction, arise as a consequence thereof, or represent an interaction of these two alternatives. Further, while dopamine and inflammatory hypotheses are often discussed as separate mechanisms underlying cognitive changes, there is emerging evidence for interactions between dopamine signaling and neuroinflammation. In particular, immune activation can potentiate dopamine responses in the striatum, but lead to reduced motivation due to a downregulation of dopaminergic signaling (Felger et al., 2013; Petrulli et al., 2017). The current study aimed to characterize cocaine self-administration in TBI, and determine whether changes in dopamine or neuroinflammation were associated with elevated drug-taking.
Materials and Methods
Subjects
Subjects were 68 Long-Evans male rats purchased from Charles River (Saint-Constant, QC), approximately 3 months old at the time of surgery. Rats were single-housed on a reverse light cycle in standard cages with a plastic hut and paper towel available as enrichment. Food and water were available ad libitum. Housing and testing were performed in accordance with the Canadian Council on Animal Care and all procedures were approved by the University of British Columbia Animal Care Committee.
Apparatus
Cocaine self-administration took place in a bank of eight standard operant chambers (Med Associates, St. Albans, VT) equipped with two retractable levers, two cue lights above the levers, a tone generator, an infusion pump, a centrally located food hopper and a houselight. Only the levers, cue lights, tone generator and infusion pump were used in the current study.
Drugs and Reagents
Buprenorphine (0.3 mg/mL) was purchased from McGill University (Montreal, QC). Heparin (10,000/mL) and bupivacaine was purchased from Associated Veterinary Purchasing Company (Langley, BC) and mixed into sterile saline to create a 20% heparin solution. Cocaine hydrochloride (Medisca, Richmond, BC) was dissolved in sterile saline at a concentration of 6 mg/mL.
TBI Surgery
Rats were anesthetized with 5% isoflurane in 0.8 L/min oxygen and placed in a stereotaxic frame with 2% isoflurane maintenance via nosecone. Buprenorphine (0.01 mg/kg, s.c.), lactated ringer solution (5 ml, s.c.) and bupivacaine (0.1 mL of 0.5% solution, s.c. at incision site) were administered. Under aseptic conditions, a midline incision was made in the scalp and the fascia retracted. A 6.0 mm diameter circular craniotomy was performed centered at AP +3.0, ML 0.0 mm from bregma. Animals received either a bilateral frontal controlled cortical impact (CCI) surgery using an electromagnetic CCI device (Leica Biosystems, Buffalo Grove, IL) or sham procedure as previously described (Vonder Haar et al., 2016; Vonder Haar & Winstanley, 2016). Injuries were performed with a 5 mm diameter flat-faced tip centered over the medial prefrontal cortex (AP +3.0, ML 0.0 from bregma). Rats were randomly assigned to one of three surgical conditions as per previous work (Vonder Haar et al., 2016; Vonder Haar et al., 2017): Severe TBI (n = 28), Mild TBI (n = 23), or Sham (n = 17). Severe impact parameters used an impact depth (DV) of −2.5 mm @ 3 m/s for 0.5 s; mild impact settings were considerably less (force = 11.1% of severe), DV −0.8 @ 1 m/s for 0.5 s; sham surgeries followed an ‘intact sham’ procedure (Martens et al., 2012; Vonder Haar & Winstanley, 2016), with no craniotomy. Following injury, bleeding was stopped with sterile gauze and the incision sutured. Buprenorphine (0.01 mg/kg) was administered for pain management 10 and 24 hours post-surgery. Animals were monitored daily for weight gain, hydration, and pain signs and if needed, given softened chow, subcutaneous saline, or buprenorphine respectively.
Jugular Vein Catheterization Surgery
After a one week recovery period following the TBI procedure, rats underwent jugular vein catheter implantation as previously described (Ferland & Winstanley, 2016). Rats were anesthetized with isoflurane as above, and buprenorphine (0.01 mg/kg, s.c.), lactated ringer solution (8 ml, s.c.) and bupivacaine (0.2 mL of 0.5% solution, s.c. at each incision site) were administered. Under aseptic conditions, an incision was made between the scapulae and another on the right side of the chest. The backport was mounted between the scapulae and a Silastic silicone tube was sutured into the right jugular vein. Catheter patency was maintained with daily flushes of 0.1 mL 20% heparin solution and animal health monitored as above. Buprenorphine was administered for pain management 10 and 24 hours post-surgery.
Self-Administration Protocol
Following one week recovery from catheter surgery, cocaine self-administration began. Rats were placed in operant chambers with cocaine (0.5 mg/kg/infusion), or saline available on a fixed ratio-1 schedule. The active lever (counterbalanced left/right) had an illuminated cue light above it. Lever presses to the active lever resulted in a cocaine infusion over 4.5 s, during which a tone (5 Hz) played and the cue light flashed (2 Hz) (Calu et al., 2007). Presses to the inactive lever had no programmed consequences. There was a 40 s time out after an infusion in which active lever presses had no consequences. Following the time out, the cue light was illuminated to signal cocaine availability. Rats were given 10 sessions in a row for 6 hours each day with a maximum of 30 infusions per hour.
Rats that demonstrated a minimum of five infusions per day for the last three days of self-administration were considered to have acquired self-administration, and this was observed in 69% of Sham animals (9/13), 63% of Mild TBI animals (12/19), but only 42% of Severe TBI animals (10/24). Only these animals were included in the analysis of behavioral outcomes. Non-acquisition animals displayed an average of approximately one press on each lever over the last three sessions. All animals were included in tissue analyses. Blood draws from the catheter confirmed catheter patency at the conclusion of the study.
Tissue Collection and Preparation
One day after completion of cocaine self administration (post-injury day 25, post-cocaine day 11), animals (approx. 450g) were restrained in DecapiCone plastic restraints (Braintree Scientific) and rapidly decapitated without anesthesia, brains placed on frozen CO2 (‘dry ice’), and stored at −80° C. Brains were then sliced on a Leica cryostat at 150 μm, and tissue punch samples (2 mm) taken from the medial prefrontal cortex (12 slices), and orbitofrontal cortex (10 slices) for ELISA analyses, while punches were taken from the nucleus accumbens (6 slices) and dorsal striatum (12 slices) for Western blot analyses. Tissue punches were stored at −80° C until preparation for analysis. Tissue was then ultrasonically lysed in radioimmunoprecipitation assay buffer (RIPA) buffer and spun at 15,500 RCF, supernatant extracted and measured for protein content using absorbance at 280 nm wavelength light. Samples were then assayed via ELISA or Western blot as described below.
ELISA Analysis of Cytokine Levels
ELISA analyses were conducted as previously described (Vonder Haar et al., 2016; Vonder Haar et al., 2017). Protein was analyzed using a multiplex kit for IL-1α, IL-1β, IL-2, IL-4, IL-6, IL-10, IL12, TNFα, and IFNγ (Quansys Q-plex, Logan, UT) according to manufacturer protocols. The optical density was read using a Q-view imager. Protein concentrations were calculated using the standard curve.
Western Blot Analyses of Dopamine-related Proteins
Protein was analyzed for levels of dopamine receptor D1 (DRD1; Santa Cruz Biotechnology sc-14001, 1:200, 75 kDa band), dopamine receptor D2 (DRD2; sc-9113, 1:200 50 kDa band), and dopamine transporter (DAT; Millipore AB2231, 1:1000, 75 kDa band). For each region, and protein measurement, 75 μg protein was boiled for five minutes in Laemmli buffer with 5% 2-mercaptoethanol and loaded into 10% polyacrylamide gels, run at 110V for 1.5 hours, then transferred onto a PVDF membrane, blocked for one hour in PBS-based blocking buffer (LI-COR 927–40000), and incubated in primary antibody in blocking buffer overnight at 4° on an orbital shaker. Blots were washed (3X PBST) and then incubated with secondary antibody in blocking buffer (LI-COR 926–68021 goat anti-rabbit IgG, 1:10,000) at room temperature for one hour under agitation. Samples were then incubated in β-tubulin in blocking buffer (Millipore 05–661, mouse monoclonal 1:10,000, 50 kDa band) overnight at 4° on an orbital shaker, and incubated in secondary antibody in blocking buffer (LI-COR 925–32210 anti-mouse IgG, 1:15,000) for one hour at room temperature under agitation. Samples were then visualized on an Odyssey Infrared Imaging System (LI-COR). On each blot, β-tubulin was normalized to the maximum value (lane normalization factor), and each sample was normalized to its normalized β-tubulin value (normalized signal) (LI-COR, 2016). Because multiple blots were performed, blot number was included as a covariate in all analyses.
Data Analysis
Data were analyzed using the R statistical package (http://www.r-project.org/). Transformations were applied as appropriate to normalize distributions. Log transformations were applied to data bounded on the lower end (Infusions, Presses, some ELISA variables, some WB variables), square root transformations were applied to right-skewed data (some ELISA variables, some WB variables), arcsine-square root transformations were applied to percent data (discrimination indices). The repeated measures data (self-administration) were analyzed using linear regression. Models were constructed using all interactions, and dropping nonsignificant predictors, and then using the Akaike Information Criterion (AIC) to compare among them (Bozdogan, 1987). Principal components analysis (PCA) was carried out on cytokine and dopamine-related protein data to reduce complex datasets into simpler components for analysis. For PCAs, subjects with fewer than half the observations from cytokine or dopamine protein measurement were removed from analysis (N = 10, Sham = 2, Mild = 2, Severe = 6), while other missing values were filled in with group means (n = 23/318 instances). These corrections were only carried out for PCA as a full set of observations is required. Cytokine, dopamine-related proteins, and principal components data were analyzed by one-way ANOVA, and Tukey’s HSD posthoc tests followed any significant effects. A p-value of 0.05 or less was considered significant.
Results
Cocaine Self-Administration
A linear regression was performed to look at the number of infusions taken (Infusions ~ Injury + Session; R2 = 0.09). The mild TBI group took significantly more infusions than the Sham group (β = 0.59, t = 4.44, p < 0.001), as did the Severe TBI group (β = 0.44, t = 3.05, p = 0.003), and infusions taken increased across sessions, (β = 0.15, t = 2.66, p = 0.008; Figure 1A). A linear regression examining the total number of active lever presses (Active Presses ~ Injury + Session; R2 = 0.10) revealed significantly increased presses in Mild TBI animals compared to Sham (β = 0.58, t = 4.33, p < 0.001), as well as more responses in the Severe TBI group (β = 0.62, t = 4.29, p < 0.001), plus a general increase in responding across sessions, (β = 0.12, t = 2.13, p = 0.034; Figure 1B). A linear regression for inactive lever presses (Inactive Presses ~ Injury + Session; R2 = 0.23) demonstrated no difference between Sham and Mild TBI groups (β = 0.05, t = 0.39, p = 0.701), while the Severe TBI group made significantly more unreinforced responses (β = 1.01, t = 7.51, p < 0.001). Overall, inactive lever presses declined across Session, as expected (β = −0.19, t = 3.62, p < 0.001; Figure 1C). A linear regression was also performed to look at the discrimination index (Active Presses / [Active + Inactive Presses]; Lever Index ~ Injury + Session; R2 = 0.16). Across sessions, all groups increased their preference for the active over the inactive lever (Session: β = 0.25, t = 4.33, p < 0.001; Figure 1D), but while there was no difference between Sham and Mild TBI groups (β = 0.22, t = 1.64, p = 0.103), the Severe TBI group were markedly worse at discriminating the active from inactive lever relative to Sham animals (β = −0.54, t = 3.62, p < 0.001). A linear regression also examined the discrimination index for presses occurring during the cue light signaling cocaine availability (Active Presses During Cue / Total Active Presses; Cue Index ~ Injury*Session; R2 = 0.20). Again, there was no difference between Sham and Mild TBI groups (β = −0.22, t = −1.59, p = 0.113), with rats responding almost exclusively during the cue, and no effect of session (β = −0.01, t = 0.06, p = 0.949). However, the Severe TBI group again showed signs of failing to appropriately discriminate the contingencies of drug availability, as indicated by a significant difference from the Sham group (β = −1.07, t = 7.13, p < 0.001; Figure 1E). An analysis of within-session dynamics examining cocaine infusions during the first hour (First Hour Infusion ~ Injury*Session; R2 = 0.12) showed no significant difference in escalation between the Mild or Severe TBI versus Sham groups over sessions (Mild: β = 0.02, t = 0.45, p = 0.656; Severe: β = −0.03, t = 0.50, p = 0.619; data not shown).
Figure 1.

Behavioral outcomes in cocaine self-administration. A) Both Mild and Severe animals took more infusions overall (p < 0.001, p = 0.003, respectively). B) Increased active lever presses were also observed in Mild and Severe animals relative to Sham (p’s < 0.001). C) Severe TBI animals pressed the inactive lever more than Shams (p < 0.001). D) Severe TBI animals also showed weaker discrimination of the active lever relative to Sham (p < 0.001). E) Severe TBI animals were also worse at discriminating the cue that signaled cocaine availability (p < 0.001). *** = p < 0.001; ** = p < 0.01.
Saline self-administration
Similar to the analysis of cocaine self-administration, a linear regression was performed to look at the number of infusions taken by animals (Infusions ~ Injury + Session; R2 = 0.19). There were no significant differences between Sham and Mild (β = −0.04, t = 0.20, p = 0.839) or Severe (β = −0.06, t = 0.30, p = 0.764) TBI groups, with response rates declining across sessions, (β = −0.44, t = 5.20, p < 0.001; Figure 2A). Similarly, a linear regression examining the total number of active lever presses (Active Presses ~ Injury + Session; R2 = 0.13) indicated no significant differences between Sham and Mild (β = −0.07, t = 0.32, p = 0.753) or Severe (β = 0.15, t = 0.68, p = 0.498) TBI groups, with a decline in presses across sessions, (β = −0.36, t = 4.09, p < 0.001; Figure 2B). A separate regression evaluated total number of inactive lever presses (Inactive Presses ~ Injury + Session; R2 = 0.26). Surprisingly, there was a small, but significant decrease in responding to the inactive lever in both Mild and Severe TBI groups relative to sham controls (Mild vs. Sham: β = −0.81, t = 4.16, p < 0.001; Severe vs. Sham: β = −0.49, t = 2.54, p = 0.013), with response rates generally declining over subsequent sessions, (β = −0.38, t = 4.76, p < 0.001; Figure 2C). Discrimination indices were not analyzed for these animals because no reinforcer was available.
Figure 2.

Behavioral outcomes in saline self-administration. A) There were no group differences in number of infusions. B) There were no group differences in active lever presses. C) Both Mild and Severe TBI animals pressed the inactive lever less than Shams (p < 0.001, p = 0.013, respectively). *** = p < 0.001; * = p < 0.05.
Cytokine Levels
An ANOVA was conducted on the levels of each cytokine (Cytokine ~ Injury*Drug) dropping nonsignificant interactions and factors, and a Tukey’s HSD posthoc followed significant omnibus tests. There were no significant group or drug differences for IL-1α, IL-1β, IL-2, IL-4, TNFα (p’s > 0.16). There were significant injury related differences in IL-6, IL-10, and IL-12 levels (F(2, 53) = 4.91, p = 0.011; F(2, 59) = 3.18, p = 0.049; F(2, 57) = 27.42, p < 0.001, respectively; Figure 3, non-significant results are not shown). The Severe group had significantly lower IL-6 levels (p = 0.008) and lower IL-10 levels (p = 0.038) than the Mild group. IL-12 levels were stratified by injury severity, with Mild TBI having higher levels than Sham (p < 0.001), and Severe having higher levels than both Sham (p < 0.001) and Mild TBI (p = 0.009). There were no significant drug differences on cytokine levels.
Figure 3.

Levels of inflammatory cytokines. A) The Severe group had lower IL-6 levels than the Mild group (p = 0.008). B) Severely-injured animals also had lower IL-10 relative to Mild (p = 0.038). C) Mild animals had significantly higher levels of IL-12 relative to Sham (p < 0.001), and Severe TBI animals had higher levels than both Sham and Mild TBI groups (p < 0.001, p = 0.009, respectively). Open circles represent individual data points; *** = p < 0.001; ** = p < 0.01; * = p < 0.05.
To further understand the relationship between cytokines, a principal component analysis was conducted. Three principal components were extracted, accounting for 89.87% of the variance (Figure 4A). Component loadings, representing the degree to which each cytokine contributed, were examined to understand potential relations between cytokines. PC1 (60.0% variance) had relatively equal, negative loadings across all cytokines, with the exception of IL-12. In contrast, PC2 (16.84% variance) was dominated by a large, positive IL-12 loading. PC3 (13.03% variance) had large (opposite) loadings for IL-1β, and IL-4 (Figure 4B–C). Each component was compared in a two-factor ANOVA to assess differences across the Injury and Drug conditions (PC ~ Injury*Drug). There were no TBI-related differences in PC1 (F(2,49) = 2.16, p = 0.126) and PC3 (F(2,49) = 2.06, p = 0.139) or cocaine-related differences in PC1 (F(1,49) = 0.01, p = 0.933), PC2 (F(1,49) = 0.16, p = 0.694) and PC3 (F(1,49) = 0.69, p =0.409). However, there was a difference between injury groups in PC2 (F(2,49) = 15.83, p < 0.001; Figure 4D). Specifically, the Severe TBI group had significantly higher levels than the Sham (p < 0.001) and Mild TBI (p = 0.001) groups, but the Sham and Mild TBI were not significantly different from each other (p = 0.133).
Figure 4.

Principal components analysis of cytokine data. A) Scree plot of extracted components. The first three were selected for further analysis. B) Relative loading of different cytokines in PC1 vs. PC2, illustrating the degree to which each cytokine contributes to its component. PC2 displayed relatively common profiles, with the exception of IL-12. C) Relative loading of different cytokines in PC2 vs. PC3. PC2 revealed orthogonally opposed cytokine loadings, with IL-12 again showing the largest, and most independent effect. D) Relative PC2 loading (group mean +/− SEM; open circles represent individual data points), stratified by group. The Severe TBI group had significantly higher levels than both Sham and Mild TBI (p < 0.001, p = 0.001, respectively), which is likely driven by the strong group differences in IL-12. The Sham and Mild TBI were not significantly different (p = 0.133). *** = p < 0.001.
Dopamine Receptor and Transporter Levels
An ANOVA was conducted on the protein levels from the dorsal striatum and accumbens, with blot as a covariate (Protein ~ Injury*Drug + Blot) dropping nonsignificant interactions and factors, and Tukey’s HSD posthoc followed significant omnibus tests. There were no injury-related differences in striatum D2 receptors, striatum DAT, accumbens D2 receptors, or accumbens DAT (p’s > 0.265). There were no drug-related differences in striatum D1 receptors, striatum DAT, or accumbens D2 receptors (p’s > 0.725). There were injury-related differences in striatum and accumbens D1 receptors (F(2,53) = 4.91, p = 0.011; F(2,53) = 4.84, p = 0.012; Figure 5A–B, non-significant results are not shown); both Mild and Severe TBI groups had significantly lower levels in the striatum (p = 0.020; p = 0.019), and the Mild TBI group had significantly higher levels in the accumbens relative to both Sham and Severe TBI (p = 0.042; p = 0.017). There were drug-related differences in striatum D2 receptors (F(1,52) = 6.04, p = 0.017; Figure 5C), accumbens D1 receptors (F(1,53) = 4.76, p = 0.034; Figure 5D), and accumbens DAT (F(1,53) = 5.29, p = 0.025; Figure 5E); cocaine self-administering animals had higher D2 receptor levels in the striatum (p = 0.014), lower D1 in the accumbens (p = 0.035), and higher levels of DAT in the accumbens (p = 0.026).
Figure 5.

Levels of markers of dopaminergic signaling. A) Both Mild and Severe TBI groups had lower levels of D1 receptors in the striatum (p = 0.020, p = 0.019, respectively). B) However, the Mild TBI had higher D1 receptor levels in the nucleus accumbens compared to Sham or Severe TBI (p = 0.042, p = 0.017, respectively). C) Cocaine animals had increased D2 receptor levels in the striatum (p = 0.014). D) Cocaine animals demonstrated lower D1 receptor levels in the accumbens (p = 0.035). E) Cocaine animals displayed higher levels of DAT in the accumbens (p = 0.026). Open circles represent individual data points; * = p < 0.05.
To evaluate holistic changes in dopaminergic signaling, a principal components analysis was conducted on normalized protein levels. Four principal components were extracted, accounting for 80.63% of the variance (Figure 6A). Component loadings, representing the degree to which each protein contributed, were examined to understand potential relations between proteins. PC1 (27.48% variance) strongly loaded on the protein levels from the accumbens, while PC3 (16.86%) was heavily dominated by striatal protein levels. PC2 (22.15%) loaded heavily on DAT in the striatum opposite D2 receptors in the accumbens, while PC4 (14.14%) was dominated by D2 receptors in the accumbens and DAT in the striatum (Figure 6B–D). Each component was then compared in a two-factor ANOVA to evaluate any differences in injury or drug conditions (PC~Injury*Drug). There were no TBI-related differences in PC1 (F(2,57) = 0.10, p = 0.908), PC2 (F(2,57) = 0.01, p = 0.987), or PC3 (F(2,57) = 1.82, p = 0.171). There were no drug-related differences in PC1 (F(1,57) = 0.018, p = 0.892), PC2 (F(1,57) = 0.13, p = 0.714), PC3 (F(1,57) = 0.99, p = 0.323), or PC4 (F(1,57) = 0.12, p = 0.725). There was a significant difference between injury groups in PC4 (F(2,57) = 3.71, p = 0.031; Figure 6E). Specifically, the Severe TBI group had lower levels than the Sham group (p = 0.028).
Figure 6.

Principal components analysis of markers of dopaminergic function. A) Scree plot of extracted components. The first four were selected for further analysis. B) Relative loading of different dopamine markers in PC1 vs. PC4 illustrating the degree to which each protein contributes to its component. C) Relative loading of different dopamine markers in PC2 vs. PC4. D) Relative loading of different dopamine markers in PC3 vs. PC4. E) Relative PC4 loading, stratified by injury group (group mean +/− SEM; open circles represent individual data points). The Severe TBI group had significantly lower levels than the Sham group (p = 0.028). Comparison of PC4 across these cases shows relatively opposing loadings for the accumbens versus striatum, and D1 versus D2, highlighting the importance of these relationships in TBI. * = p < 0.05.
Association of Cellular Markers with Infusions & Active Presses
The principal components extracted from the Western blot and ELISA were used to evaluate the relationship between cytokines, dopaminergic signaling, and number of infusions obtained and total active presses. Model selection was carried out by starting with the full model (Infusion or Presses ~ All components + Injury + Session; Infusion R2 = 0.19; Press R2 =0.19), and removing insignificant predictors, then comparing the AIC of all models. For total infusions, a model with the significant predictors Injury (Mild: β = −0.21, t = 0.86, p = 0.392; Severe: β = 0.67, t = 2.75, p = 0.007), inflammation PC3 (β = −0.48, t = 3.74, p < 0.001), dopamine PC1 (β = −0.42, t = 4.42, p < 0.001), and dopamine PC4 (β = −0.71, t = 5.22, p < 0.001) accounted for 19.34% of the variance (Figure 7A–B). For active presses, a model with the significant predictors Injury (Mild: β = −0.22, t = 0.82, p = 0.415; Severe: β = 0.97, t = 3.60, p < 0.001), inflammation PC3 (β = −0.53, t = 3.74, p < 0.001), dopamine PC1 (β = −0.47, t = 4.49, p < 0.001), and dopamine PC4 (β = −0.71, t = 4.72, p < 0.001) accounted for 18.91% of the variance.
Figure 7.

Regression predictions of behavior as a function of extracted principal components. A) Number of infusions as predicted by a regression including injury, session, inflammation PC3, dopamine PC1, and dopamine PC4. Specifically shown is the relationship of high levels of PC3 to higher cocaine intake. B) The same function shown at the end of cocaine self-administration, demonstrating that PC3 interacts with session, but that higher levels are still associated with relatively low cocaine intake. C) Logistic regression function, in which inflammation PC3 was the only significant predictor. This data demonstrates the ability of PC3 to account for acquisition of cocaine self-administration, suggesting that inflammation precedes high levels of cocaine intake, rather than being caused by it.
Association of Cellular Markers with Acquisition of Self-Administration
A Fisher’s Exact test revealed no significant overall group differences in acquisition (p = 0.215). To better understand why some rats, particularly in the TBI condition, were less likely to acquire self-administration of cocaine, the components extracted from the ELISA and Western blots were compared in a logistic regression with acquisition (acquired/not acquired) as the binary outcome variable. Model selection was performed on a regression containing injury, the three inflammation components and three dopamine components (Acquisition ~ All components + Injury), dropping insignificant predictors, and selecting the model with the lowest AIC. A model with only inflammation PC3 was associated with acquisition (β = −1.05, t = 2.89, p = 0.004; Figure 7C).
Discussion
In this study, we demonstrated increased levels of cocaine self-administration in a subset of animals following frontal TBI, regardless of injury severity. While there is increasing evidence for a link between addiction and brain injury in patients (Graham & Cardon, 2008; Miller et al., 2013; Zgaljardic et al., 2015), these are, to our knowledge, the first data to demonstrate a causal relationship between TBI and elevated cocaine-taking. However, it should be noted that this effect was not uniform across the TBI groups, and many animals failed to acquire cocaine self-administration. Because an unbiased view of acquisition was desired, no sucrose pre-training was performed as is common in many drug self-administration studies. The current study also sought to determine whether frontal TBI increased local neuroinflammation and altered levels of dopamine-related proteins in the striatum, in order to probe potential mechanisms driving TBI-induced addiction risk. In keeping with previous data, we observed increased levels of IL-12 that were stratified by injury severity, as well as changes in other inflammatory cytokines. By condensing all neuroinflammatory data in a PCA, a strong relationship was observed between an inflammatory component (PC3) and number of cocaine infusions taken. We also observed signs of disrupted dopamine signaling as a result of TBI, with western blot analyses detecting lower D1 receptor levels in the dorsal striatum of both injury groups, but increased levels of D1 in the accumbens of mild-injured animals. A similar PCA data analysis reduced the dopamine data into four components, and found that two of them (PC1, PC4) were related to cocaine intake. However, only the neuroinflammation component (PC3) associated with cocaine intake also predicted acquisition of cocaine self-administration, potentially suggesting a more pronounced role for inflammatory factors within the frontal cortex in determining addiction susceptibility.
The fact that not all animals, in either the severe or mild TBI groups, showed robust drug-taking after injury may, at first glance, appear problematic. However, we are not the first to report this phenomenon in preclinical studies: only a subset of animals exhibited elevated consumption of alcohol after blast TBI (Lim et al., 2015). Furthermore, this data pattern strongly mimics the human condition, wherein not all individuals become susceptible to addiction after a brain injury. Although only a subpopulation of severely or mildly injured animals went on to acquire self-administration, these rats took more infusions than the subgroup of sham animals that likewise acquired drug-taking behavior under this reinforcement schedule. Such individual differences may also prove useful, in that selective changes in pathology may be identified which confer vulnerability to addiction, and these can inform therapeutics development or screening protocols to identify at-risk individuals. These findings also suggest a need to strongly consider methodology when studying the interaction of brain injury and addiction. The use of within-subjects paradigms, where biological samples can be directly correlated with the performance of animals, will be essential to elucidate potential relationships between physiological variables and behavioral alterations.
A particularly interesting finding of the current study is that even milder injuries can lead to substantial changes in the profile of addiction. A similar finding was also observed by Weil et al. (Weil et al., 2016), in a model of concussive brain injury, albeit limited to female subjects. It has been suggested that inflammation may play a mediating role in behavioral deficits following both milder and more severe injuries (Haber et al., 2013; Yang et al., 2013), and may therefore have some degree of ‘priming’ effect. If true, injured animals may display more rapid sensitization, and larger drug effects at baseline. Multiple studies have demonstrated increased conditioned place preference to cocaine in models of TBI, supporting this hypothesis (Merkel et al., 2017a; Merkel et al., 2017b), and in the current data, injured animals had a large initial increase in cocaine intake relative to shams. Despite this, a large portion of the injured group failed to acquire self-administration. Findings have suggested that alterations to dopamine receptors and transporter can lead to cocaine aversion (O’Neill et al., 2013; Hasbi et al., 2017), potentially due to high levels of sensitization to an otherwise effective dose. Findings of drug sensitization after TBI have been reported before (Lowing et al., 2014), however, this was with regard to the sedative effects of ethanol. Further, it has been shown that inflammatory challenge, outside of injury events, can lead to sensitization to the effects of cocaine (Tortorelli et al., 2015). These data, taken together, suggest that a high level of sensitization to the effects of cocaine could actually lead to aversion, and may potentially explain some of the differences in acquisition in the current study. This may also explain the considerable heterogeneity in the profile of addiction after TBI in human subjects: some individuals may display increased responsivity to a drug, while others may be so sensitive that the drug becomes aversive.
It should be noted that severely-injured animals also had difficulty discriminating the reinforcement contingencies in play, similar to previous reports (Martens et al., 2012; Vonder Haar et al., 2014b; Vonder Haar et al., 2014c). However, even though inactive lever presses were consistently higher in this group as compared to mild TBI and sham-injured controls, the proportion of responses on the active lever hovered around 80% for the last three sessions of self-administration training – well over chance responding. It is therefore unlikely that the elevated drug-taking observed in this cohort was not goal-directed. Previous data does not suggest that this form of severe TBI increases rats’ tendency to make lever-press responses in general; if anything, at low response requirements such as those used in the current study, injured rats may display lower pressing rates (Vonder Haar & Winstanley, 2016). The elevated inactive lever press responses may therefore reflect increased stimulus/response generalization, or potentially an increased locomotor response to cocaine, resulting in greater levels of general activity. However, in one case, locomotor sensitization to other psychostimulants such as amphetamine was actually reduced after TBI (Karelina et al., 2017), thus poor discrimination or increased generalization is more likely.
Despite a small corpus of literature on experimental TBI and addiction, a substantial amount of consensus can be found between the few existing studies and the current research. This is particularly interesting because these studies were conducted in a variety of models of head injury (concussive, focal, blast) and used multiple drugs of abuse (alcohol-previous work, cocaine-current study). It should be noted that differences in injury model and drug of abuse may limit comparisons for these studies, but common results are then suggestive of general actions as opposed to drug- or injury-specific findings. Across nearly all experiments, increased self-administration of the addictive substance was observed in TBI groups (Lim et al., 2015; Mayeux et al., 2015; Weil et al., 2016), although one study did report the opposite, with reduced alcohol intake in the acute period after a moderate closed-head injury (Lowing et al., 2014). In the current study, we were able to assess the contributions of multiple dopamine-related proteins in the striatum, as well as a host of inflammatory signaling molecules within frontal regions, to explore potential mechanisms influencing drug-taking after TBI. Brain injury caused a reduction in D1 receptor density in the dorsal striatum, which is reminiscent of observations after chronic cocaine use, although in those cases the accumbens was typically affected as well (Moore et al., 1998; Ramôa et al., 2014). However, it should be noted that these measurements were taken after self-administration, and changes in D1 and DAT in the accumbens, as well as D2 receptor levels in the striatum, could be directly attributed to the effects of cocaine (Figure 5). The current data suggest that neuroinflammatory factors may play a more causal role; not only was PC3 from the cytokine ELISA data analysis associated with higher levels of self-administration, but this was the only component that was predictive of whether self-administration was acquired. Inflammation was also identified as a potential contributor to addiction after injury by Mayeux and colleagues (Mayeux et al., 2015), who found higher glial activity with increased self-administration.
Elevations in inflammatory cytokines after TBI have also been linked with impulsive behavior, which is a strong predictor of addictive phenotypes (Perry et al., 2005; Belin et al., 2008). Perhaps the most consistent relationship after TBI has been increases in levels of IL-12, which have been linked with impulsive action and impulsive decision-making (Vonder Haar et al., 2016; Vonder Haar et al., 2017). In the current study, we also observed that frontal TBI increased levels of IL-12 in areas of frontal cortex important for the regulation of impulse control, and PCA again confirmed that IL-12 levels seem to be regulated somewhat independently from the majority of other cytokines. Both preclinical and clinical studies have established that high impulsivity can potentiate vulnerability for addiction. Despite this, PC2, which was largely dominated by IL-12, was not the best predictor of the total amount of cocaine taken or the acquisition of cocaine self-administration. However, deficits in impulse control are only one pathway to addiction. While it is still possible that TBI-induced increases in impulsivity, potentially driven by IL-12, may amplify addiction risk, the current data suggest that other neuroinflammatory mechanisms may play a more potent role in facilitating drug-taking behavior. These differences in principal component makeup between prior studies (Vonder Haar et al., 2016; Vonder Haar et al., 2017) and the current data warrant further investigation to determine specific contributions of any given cytokine or group of cytokines to the effects of brain injury.
Specifically, PC3 was heavily dominated by IL-1β. This cytokine is a product of toll-like receptor 4 activation, which has been shown to be crucial to the development of cocaine self-administration in the ventral tegmental area (VTA) (Northcutt et al., 2015). Furthermore, antagonism of IL-1β within the VTA can prevent resumption of cocaine-seeking in the extinction-reinstatement model of relapse, further pointing to an important role for this cytokine in mediating drug dependency (Brown et al., 2018). Elevated levels of IL-1β, as measured in the periphery and centrally, have been repeatedly associated with psychiatric conditions often comorbid with addictions, such as bipolar disorder and schizophrenia (see Goldsmith et al., 2016 for a recent meta-analysis). IL-1β can also modulate neuronal function through a variety of mechanisms, including altering both GABAergic and glutamatergic signaling (Viviani et al., 2003; Bajo et al., 2015). Although a role for frontal IL-1β in cocaine self-administration has yet to be investigated, modulating output from both medial prefrontal and orbitofrontal regions can impact drug-taking and drug-seeking in rodents (Winstanley et al., 2009; Martín-García et al., 2014), and neuroimaging data also implicate these brain areas in the maintenance of the addicted state (Jasinska et al., 2014). It is therefore possible that cytokine-mediated alterations in frontal function directly contribute to a pro-addiction phenotype following TBI.
In sum, these data support the hypothesis that TBI can directly increase risk for drug dependence, and that modulating inflammation may be able to reduce the likelihood of developing substance use disorder following TBI. This could be a particularly promising hypothesis to test, as anti-inflammatory therapies also reduce other symptoms of TBI (Haber et al., 2013; Vonder Haar et al., 2014a; Anderson et al., 2015). However, both TBI and addiction are heterogeneous phenomena, which pose significant challenges for resolving the factors driving TBI-induced elevations in addiction risk using animal models or clinical data. While the current study represents an important first step, additional research is undoubtedly needed to parse this complex relationship. Public awareness of the negative association between TBI and mental health outcomes continues to grow, increasing pressure to identify the critical mediators and develop targeted interventions, yet this phenomenon is worryingly understudied by both the clinical and behavioral neuroscience community. These data support the need to establish a more complete understanding of the neurobiological pathways through which immune signaling molecules impact brain function and behavior, information that may critically enable target validation and therapeutic ligand development for the psychiatric complications associated with TBI.
Acknowledgements
We would like to thank the Anthony Phillips laboratory at UBC for generously providing additional self-administration chambers and expert advice for this project. Funding for this project was provided by operating grants from the Canadian Institutes of Health Research (CIHR; PJT-148631) awarded to CAW, and the National Institute of Neurological Disorders and Stroke (R21NS087458) awarded to SR. CV was supported by a (CIHR) post-doctoral fellowship, and WVU startup funds. In the past three years, CAW has received salary support from the CIHR New Investigator Award program and the Michael Smith Foundation for Health Research.
Abbreviations
- ANOVA
Analysis of variance
- AIC
Akaike information criterion
- CCI
controlled cortical impact
- DAT
dopamine transporter
- ELISA
Enzyme-linked immunosorbent assay
- IL
interleukin
- PCA
Principal components analysis
- TBI
Traumatic brain injury
- TNFα
Tumor necrosis factor-α
- VTA
ventral tegmental area
- WB
Western Blot
Footnotes
Conflict of Interest
In the past three years, CAW has been a member of an Advisory Board for Shire Pharmaceuticals, and has been retained as an expert witness by Hogan Lovells LLP, with reference to unrelated matters. The authors have no other financial disclosures or potential conflicts of interest to declare.
Data Accessibility
Primary data may be made accessible on a case-by-case basis by contacting the corresponding author.
References
- Anderson GD, Peterson TC, Vonder Haar C, Farin FM, Bammler TK, MacDonald JW, Kantor ED & Hoane MR (2015) Effect of traumatic brain injury, erythropoietin, and anakinra on hepatic metabolizing enzymes and transporters in an experimental rat model. The AAPS Journal, 17, 1255–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashok AH, Mizuno Y, Volkow ND & Howes OD (2017) Association of stimulant use with dopaminergic alterations in users of cocaine, amphetamine, or methamphetamine: A systematic review and meta-analysis. JAMA Psychiatry, 74, 511–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajo M, Varodayan FP, Madamba SG, Robert AJ, Casal LM, Oleata CS, Siggins GR & Roberto M (2015) IL-1 interacts with ethanol effects on GABAergic transmission in the mouse central amygdala. Front. Pharmacol, 6, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bales JW, Wagner AK, Kline AE & Dixon CE (2009) Persistent cognitive dysfunction after traumatic brain injury: A dopamine hypothesis. Neurosci. Biobehav. Rev, 33, 981–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beers SR, Skold A, Dixon CE & Adelson PD (2005) Neurobehavioral effects of amantadine after pediatric traumatic brain injury: A preliminary report. J. Head Trauma Rehabil, 20, 450–463. [DOI] [PubMed] [Google Scholar]
- Belin D & Everitt BJ (2008) Cocaine seeking habits depend upon dopamine-dependent serial connectivity linking the ventral with the dorsal striatum. Neuron, 57, 432–441. [DOI] [PubMed] [Google Scholar]
- Belin D, Mar AC, Dalley JW, Robbins TW & Everitt BJ (2008) High impulsivity predicts the switch to compulsive cocaine-taking. Science, 320, 1352–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozdogan H (1987) Model selection and Akaike’s information criterion (AIC): The general theory and its analytical extensions. Psychometrika, 52, 345–370. [Google Scholar]
- Brown KT, Levis SC, O’Neill CE, Northcutt AL, Fabisiak TJ, Watkins LR & Bachtell RK (2018) Innate immune signaling in the ventral tegmental area contributes to drug-primed reinstatement of cocaine seeking. Brain, Behav., Immun, 67, 130–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calu DJ, Stalnaker TA, Franz TM, Singh T, Shaham Y & Schoenbaum G (2007) Withdrawal from cocaine self-administration produces long-lasting deficits in orbitofrontal-dependent reversal learning in rats. Learn. Memory, 14, 325–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Center for Behavioral Health Statistics and Quality (2015) Behavioral health trends in the United States: Results from the 2014 Survey on Drug Use and Health. Retrieved from http://www.samhsa.gov/data/. [Google Scholar]
- Center for Disease Control (2017) Injury prevention and control: Traumatic brain injury. Retrieved from www.cdc.gov/TraumaticBrainInjury/, 2018.
- Chen Y-H, Huang EY-K, Kuo T-T, Ma H-I, Hoffer BJ, Tsui P-F, Tsai J Jr, Chou Y-C & Chiang Y-H (2015) Dopamine release impairment in striatum after different levels of cerebral cortical fluid percussion injury. Cell Transplant, 24, 2113–2128. [DOI] [PubMed] [Google Scholar]
- Corrigan JD & Hammond FM (2013) Traumatic Brain Injury as a Chronic Health Condition. Arch. Phys. Med. Rehabil, 94, 1199–1201. [DOI] [PubMed] [Google Scholar]
- Dhillon NK, Peng F, Bokhari S, Callen S, Shin S-H, Zhu X, Kim K-J & Buch SJ (2008) Cocaine-mediated alteration in tight junction protein expression and modulation of CCL2/CCR2 axis across the blood-brain barrier: Implications for HIV-dementia. J. Neuroimmune Pharmacol, 3, 52–56. [DOI] [PubMed] [Google Scholar]
- Draper K & Ponsford J (2008) Cognitive functioning ten years following traumatic brain injury and rehabilitation. Neuropsychology, 22, 618–625. [DOI] [PubMed] [Google Scholar]
- Felger JC, Mun J, Kimmel HL, Nye JA, Drake DF, Hernandez CR, Freeman AA, Rye DB, Goodman MM & Howell LL (2013) Chronic interferon-α decreases dopamine 2 receptor binding and striatal dopamine release in association with anhedonia-like behavior in nonhuman primates. Neuropsychopharmacology, 38, 2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferland JMN & Winstanley CA (2016) Risk-preferring rats make worse decisions and show increased incubation of craving after cocaine self-administration. Addict. Biol, 22, 991–1001. [DOI] [PubMed] [Google Scholar]
- Fox HC, D’sa C, Kimmerling A, Siedlarz KM, Tuit KL, Stowe R & Sinha R (2012) Immune system inflammation in cocaine dependent individuals: Implications for medications development. Hum. Psychopharmacol. Clin. Exp, 27, 156–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldsmith D, Rapaport M & Miller B (2016) A meta-analysis of blood cytokine network alterations in psychiatric patients: Comparisons between schizophrenia, bipolar disorder and depression. Mol. Psychiatry, 21, 1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham DP & Cardon AL (2008) An update on substance use and treatment following traumatic brain injury. Ann. N. Y. Acad. Sci, 1141, 148–162. [DOI] [PubMed] [Google Scholar]
- Haber M, Abdel Baki SG, Grin’kina NM, Irizarry R, Ershova A, Orsi S, Grill RJ, Dash P & Bergold PJ (2013) Minocycline plus N-acetylcysteine synergize to modulate inflammation and prevent cognitive and memory deficits in a rat model of mild traumatic brain injury. Exp. Neurol, 249, 169–177. [DOI] [PubMed] [Google Scholar]
- Hasbi A, Perreault ML, Shen MY, Fan T, Nguyen T, Alijaniaram M, Banasikowski TJ, Grace AA, O’Dowd BF & Fletcher PJ (2017) Activation of dopamine D1–D2 receptor complex attenuates cocaine reward and reinstatement of cocaine-seeking through inhibition of DARPP-32, ERK, and ΔFosB. Front. Pharmacol, 8, 924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmin S & Mathiesen T (1999) Long-term intracerebral inflammatory response after experimental focal brain injury in rat. Neuroreport, 10, 1889–1891. [DOI] [PubMed] [Google Scholar]
- Jasinska AJ, Stein EA, Kaiser J, Naumer MJ & Yalachkov Y (2014) Factors modulating neural reactivity to drug cues in addiction: a survey of human neuroimaging studies. Neurosci. Biobehav. Rev, 38, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Stewart JE, Begbie FD, Trojanowski JQ, Smith DH & Stewart W (2013) Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain, 136, 28–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karelina K, Gaier KR & Weil ZM (2017) Traumatic brain injuries during development disrupt dopaminergic signaling. Exp. Neurol, 297, 110–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus MF, Smith GS, Butters M, Donnell AJ, Dixon E, Yilong C & Marion D (2005) Effects of the dopaminergic agent and NMDA receptor antagonist amantadine on cognitive function, cerebral glucose metabolism and D2 receptor availability in chronic traumatic brain injury: A study using positron emission tomography (PET). Brain Inj, 19, 471–479. [DOI] [PubMed] [Google Scholar]
- LI-COR (2016) Western blot normalization handbook. Retrieved from https://www.licor.com/documents/gtk1a4mrphretj1gvmzld0f920g7h8sh, 2016.
- Lim YW, Meyer NP, Shah AS, Budde MD, Stemper BD & Olsen CM (2015) Voluntary alcohol intake following blast exposure in a rat model of mild traumatic brain injury. PLoS One, 10, e0125130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowing JL, Susick LL, Caruso JP, Provenzano AM, Raghupathi R & Conti AC (2014) Experimental traumatic brain injury alters ethanol consumption and sensitivity. J. Neurotrauma, 31, 1700–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Steinberg JL, Keyser-Marcus L, Ramesh D, Narayana PA, Merchant RE, Moeller FG & Cifu DX (2015) Altered white matter in cocaine-dependent subjects with traumatic brain injury: A diffusion tensor imaging study. Drug Alcohol Depend, 151, 128–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens KM, Vonder Haar C, Hutsell BA & Hoane MR (2012) A discrimination task used as a novel method of testing decision-making behavior following traumatic brain injury . J Neurotrauma, 29, 2505–2512. [DOI] [PubMed] [Google Scholar]
- Martín-García E, Courtin J, Renault P, Fiancette J-F, Wurtz H, Simonnet A, Levet F, Herry C & Deroche-Gamonet V (2014) Frequency of cocaine self-administration influences drug seeking in the rat: Optogenetic evidence for a role of the prelimbic cortex. Neuropsychopharmacology, 39, 2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayeux JP, Teng SX, Katz PS, Gilpin NW & Molina PE (2015) Traumatic brain injury induces neuroinflammation and neuronal degeneration that is associated with escalated alcohol self-administration in rats. Behav. Brain Res, 279, 22–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkel SF, Andrews AM, Lutton EM, Razmpour R, Cannella LA & Ramirez SH (2017a) Dexamethasone attenuates the enhanced rewarding effects of cocaine following experimental traumatic brain injury. Cell Transplant, 26, 1178–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkel SF, Razmpour R, Lutton EM, Tallarida CS, Heldt NA, Cannella LA, Persidsky Y, Rawls SM & Ramirez SH (2017b) Adolescent traumatic brain injury induces chronic mesolimbic neuroinflammation with concurrent enhancement in the rewarding effects of cocaine in mice during adulthood. J. Neurotrauma, 34, 165–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller SC, Baktash SH, Webb TS, Whitehead CR, Maynard C, Wells TS, Otte CN & Gore RK (2013) Risk for addiction-related disorders following mild traumatic brain injury in a large cohort of active-duty U.S. airmen. Am. J. Psychiatry, 170, 383–390. [DOI] [PubMed] [Google Scholar]
- Moore RJ, Vinsant SL, Nader MA, Porrino LJ & Friedman DP (1998) Effect of cocaine self-administration on striatal dopamine D1 receptors in rhesus monkeys. Synapse, 28, 1–9. [DOI] [PubMed] [Google Scholar]
- Moreno-López L, Manktelow AE, Sahakian BJ, Menon DK & Stamatakis EA (2016) Anything goes? Regulation of the neural processes underlying response inhibition in TBI patients. Eur. Neuropsychopharmacol, 27, 159–169. [DOI] [PubMed] [Google Scholar]
- Morganti JM, Jopson TD, Liu S, Riparip L-K, Guandique CK, Gupta N, Ferguson AR & Rosi S (2015) CCR2 antagonism alters brain macrophage polarization and ameliorates cognitive dysfunction induced by traumatic brain injury. J. Neurosci, 35, 748–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morganti JM, Riparip L-K & Rosi S (2016) Call off the dog (ma): M1/M2 polarization is concurrent following traumatic brain injury. PLoS One, 11, e0148001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najjar S, Pearlman DM, Alper K, Najjar A & Devinsky O (2013) Neuroinflammation and psychiatric illness. J. Neuroinflammation, 10, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Institute of Drug Abuse (2015) Overdose Death Rates. Retrieved from https://www.drugabuse.gov/related-topics/trends-statistics/overdose-death-rates, 2017.
- Northcutt A, Hutchinson M, Wang X, Baratta M, Hiranita T, Cochran T, Pomrenze M, Galer E, Kopajtic T & Li C (2015) DAT isn’t all that: cocaine reward and reinforcement requires Toll Like Receptor 4 signaling. Mol. Psychiatry, 20, 1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill B, Tilley MR & Gu HH (2013) Cocaine produces conditioned place aversion in mice with a cocaine-insensitive dopamine transporter. Genes, Brain and Behav., 12, 34–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez M, Gabach L, Almiron R, Carlini V, De Barioglio SR & Ramirez O (2010) Different chronic cocaine administration protocols induce changes on dentate gyrus plasticity and hippocampal dependent behavior. Synapse, 64, 742–753. [DOI] [PubMed] [Google Scholar]
- Perry JL, Larson EB, German JP, Madden GJ & Carroll ME (2005) Impulsivity (delay discounting) as a predictor of acquisition of IV cocaine self-administration in female rats. Psychopharmacology, 178, 193–201. [DOI] [PubMed] [Google Scholar]
- Petrulli J, Kalish B, Nabulsi N, Huang Y, Hannestad J & Morris E (2017) Systemic inflammation enhances stimulant-induced striatal dopamine elevation. Transl. Psychiatry, 7, e1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell JH, Al-Adawi S, Morgan J & Greenwood RJ (1996) Motivational deficits after brain injury: Effects of bromocriptine in 11 patients. J. Neurol. Neurosurg. Psychiatry, 60, 416–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramesh D, Keyser-Marcus LA, Ma L, Schmitz JM, Lane SD, Marwitz JH, Kreutzer JS & Moeller FG (2015) Prevalence of traumatic brain injury in cocaine-dependent research volunteers. Am J Addict, 24, 341–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramôa CP, Doyle SE, Lycas MD, Chernau AK & Lynch WJ (2014) Diminished role of dopamine D1-receptor signaling with the development of an addicted phenotype in rats. Biol. Psychiatry, 76, 8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray LA, Roche D, Heinzerling K & Shoptaw S (2014) Opportunities for the development of neuroimmune therapies in addiction. Int. Rev. Neurobiol, 118, 381–401. [DOI] [PubMed] [Google Scholar]
- Reeves RR & Panguluri RL (2011) Neuropsychiatric complications of traumatic brain injury. J. Psychosoc. Nurs. Ment. Health Serv, 49, 42–50. [DOI] [PubMed] [Google Scholar]
- Rodrigues L, Gobira PH, de Oliveira AC, Pelição R, Teixeira AL, Moreira FA & Campos AC (2014) Neuroinflammation as a possible link between cannabinoids and addiction. Acta Neuropsychiatrica, 26, 334–346. [DOI] [PubMed] [Google Scholar]
- Rosenbaum SB & Lipton ML (2012) Embracing chaos: The scope and importance of clinical and pathological heterogeneity in mTBI. Brain Imaging Behav., 6, 255–282. [DOI] [PubMed] [Google Scholar]
- Scott G, Hellyer PJ, Ramlackhansingh AF, Brooks DJ, Matthews PM & Sharp DJ (2015) Thalamic inflammation after brain trauma is associated with thalamo-cortical white matter damage. J. Neuroinflammation, 12, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada R, Abe K, Furutani R & Kibayashi K (2014) Changes in dopamine transporter expression in the midbrain following traumatic brain injury: An immunohistochemical and in situ hybridization study in a mouse model. Neurol. Res, 36, 239–246. [DOI] [PubMed] [Google Scholar]
- Shultz SR, MacFabe DF, Foley KA, Taylor R & Cain DP (2012) Sub-concussive brain injury in the Long-Evans rat induces acute neuroinflammation in the absence of behavioral impairments. Behav. Brain Res, 229, 145–152. [DOI] [PubMed] [Google Scholar]
- Tortorelli LS, Engelke DS, Lunardi P, Mello e Souza T, Santos-Junior JG & Goncalves C-A (2015) Cocaine counteracts LPS-induced hypolocomotion and triggers locomotor sensitization expression. Behav. Brain Res, 287, 226–229. [DOI] [PubMed] [Google Scholar]
- Treble-Barna A, Wade SL, Martin LJ, Pilipenko V, Yeates KO, Taylor HG & Kurowski BG (2017) Influence of dopamine-related genes on neurobehavioral recovery after traumatic brain injury during early childhood. J. Neurotrauma, 34, 1919–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaishnavi S, Rao V & Fann JR (2009) Neuropsychiatric problems after traumatic brain injury: Unraveling the silent epidemic. Psychosomatics, 50, 198–205. [DOI] [PubMed] [Google Scholar]
- Viviani B, Bartesaghi S, Gardoni F, Vezzani A, Behrens M, Bartfai T, Binaglia M, Corsini E, Di Luca M & Galli C (2003) Interleukin-1β enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J. Neurosci, 23, 8692–8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonder Haar C, Anderson GD, Elmore BE, Moore LH, Wright AM, Kantor ED, Farin FM, Bammler TK, MacDonald JW & Hoane MR (2014a) Comparison of the effect of minocycline and simvastatin on functional recovery and gene expression in a rat traumatic brain injury model. J. Neurotrauma, 31, 961–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonder Haar C, Lam FCW, Adams WA, Riparip L-K, Kaur S, Muthukrishna M, Rosi S & Winstanley CA (2016) Frontal traumatic brain injury in rats causes long-lasting impairments in impulse control that are differentially sensitive to pharmacotherapeutics and associated with chronic neuroinflammation. ACS Chem. Neurosci, 7, 1531–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonder Haar C, Maass WR, Jacobs EA & Hoane MR (2014b) Deficits in discrimination following experimental frontal brain injury are mediated by motivation and can be improved by nicotinamide administration. J. Neurotrauma, 31, 1711–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonder Haar C, Martens KM, Riparip L-K, Rosi S, Wellington CL & Winstanley CA (2017) Frontal traumatic brain injury increases impulsive decision making in rats: A potential role for the inflammatory cytokine interleukin-12. J. Neurotrauma, 34, 2790–2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonder Haar C, Smith TR, French EJ, Martens KM, Jacobs EA & Hoane MR (2014c) Simple tone discriminations are disrupted following experimental frontal traumatic brain injury in rats. Brain Inj., 28, 235–243. [DOI] [PubMed] [Google Scholar]
- Vonder Haar C & Winstanley CA (2016) Minor functional deficits in basic response patterns for reinforcement following frontal traumatic brain injury in rats. J. Neurotrauma, 33, 1892–1900. [DOI] [PubMed] [Google Scholar]
- Wagner AK, Scanion JM, Becker CR, Ritter AC, Niyonkuru C, Dixon CE, Conley YP & Price JC (2014) The influence of genetic variants on striatal dopamine transporter and D2 receptor binding after TBI. J. Cereb. Blood Flow Metab, 34, 1328–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner AK, Sokoloski JE, Ren D, Chen X, Khan AS, Zafonte RD, Michael AC & Dixon CE (2005) Controlled cortical impact injury affects dopaminergic transmission in the rat striatum. J. Neurochem, 95, 457–465. [DOI] [PubMed] [Google Scholar]
- Weil ZM, Karelina K, Gaier KR, Corrigan TE & Corrigan JD (2016) Juvenile traumatic brain injury increases alcohol consumption and reward in female mice. J. Neurotrauma, 33, 895–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winstanley CA, Bachtell RK, Theobald DEH, Laali S, Green TA, Kumar A, Chakravarty S, Self DW & Nestler EJ (2009) Increased impulsivity during withdrawal from cocaine self-administration: Role for ΔFosB in the orbitofrontal cortex. Cereb. Cortex, 19, 435–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SH, Gangidine M, Pritts TA, Goodman MD & Lentsch AB (2013) Interleukin 6 mediates neuroinflammation and motor coordination deficits after mild traumatic brain injury and brief hypoxia in mice. Shock, 40, 471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zgaljardic DJ, Seale GS, Schaefer LA, Temple RO, Foreman J & Elliott TR (2015) Psychiatric Disease and Post-Acute Traumatic Brain Injury. J. Neurotrauma, 32, 1911–1925. [DOI] [PubMed] [Google Scholar]