

Abstract
Skeletal muscle satellite cells (SCs) are adult muscle stem cells responsible for muscle regeneration after acute or chronic injuries. The lineage progression of quiescent SC toward activation, proliferation, and differentiation during the regeneration is orchestrated by cascades of transcription factors (TFs). Here, we elucidate the function of TF Yin Yang1 (YY1) in muscle regeneration. Muscle‐specific deletion of YY1 in embryonic muscle progenitors leads to severe deformity of diaphragm muscle formation, thus neonatal death. Inducible deletion of YY1 in SC almost completely blocks the acute damage‐induced muscle repair and exacerbates the chronic injury‐induced dystrophic phenotype. Examination of SC revealed that YY1 loss results in cell‐autonomous defect in activation and proliferation. Mechanistic search revealed that YY1 binds and represses mitochondrial gene expression. Simultaneously, it also stabilizes Hif1α protein and activates Hif1α‐mediated glycolytic genes to facilitate a metabolic reprogramming toward glycolysis which is needed for SC proliferation. Altogether, our findings have identified YY1 as a key regulator of SC metabolic reprogramming through its dual roles in modulating both mitochondrial and glycolytic pathways.
Keywords: Hif1α, metabolic reprogramming, muscle satellite cell, skeletal muscle regeneration, YY1
Subject Categories: Metabolism, Stem Cells, Transcription
Introduction
Skeletal muscle has a robust regenerative capacity, with rapid reestablishment of full power occurring even after severe damage that causes widespread myofiber necrosis. The cells responsible for muscle regeneration are resident muscle stem cells, also called satellite cells (SCs) which are located in a niche beneath the ensheathing basal lamina on the surface of the myofibers and maintained in a quiescent stage (Aziz et al, 2012; Bentzinger et al, 2012; Relaix & Zammit, 2012). Most satellite cells in postnatal muscles originate from a population of embryonic precursors that express paired box protein 7 (Pax7) and/or the related Pax3. These cells are of mesodermal origin and arise from a dorsal structure of the developing somite (known as the dermomyotome). Under the control of Pax3, the cells delaminate from the ventrolateral lips of the dermomyotome and migrate to the sites of limb and diaphragm to provide myogenic progenitor cells at these sites. In mouse, during fetal stages E12.5–E17.5, the level of Pax3 protein is reduced, accompanied by the appearance of Pax7+ progenitors contributing to the formation of fetal myofibers and establishment of the major source of postnatal satellite cells (Buckingham, 2007; Buckingham & Relaix, 2007; Bentzinger et al, 2012). Upon injury in adult muscle, SCs are rapidly activated, undergo proliferative expansion, and eventually differentiate and fuse to form new myofibers (Aziz et al, 2012; Bentzinger et al, 2012; Relaix & Zammit, 2012). A subset of SCs undergoes self‐renewal and return to quiescent state to replenish the adult stem cell pool. SCs are inextricably linked to Pax7 which is expressed and maintained in virtually all quiescent SCs in adult mouse muscles (Seale et al, 2000; Buckingham, 2007; Buckingham & Relaix, 2007), thus provides a valuable target locus to facilitate genetic manipulation of the SC genome.
In the process of regeneration, activation of SCs following muscle injury results in the expansion of myogenic cell pool and leads to the initiation of myogenic program. This program, orchestrated by a regulated cascade of myogenic regulatory factors (MRFs) including MyoD, Myf5, myogenin, and MRF4, together with Pax3 and Pax7, drives the myogenic lineage progression (Aziz et al, 2012; Bentzinger et al, 2012). Besides MRFs, the intrinsic complexity of myogenesis arises also from hierarchical interactions between transcriptional regulators, regulatory RNAs, and chromatin‐remodeling factors. Yin Yang1, YY1, is a ubiquitously expressed TF which regulates various processes of development and differentiation (Gordon et al, 2006). Previous work from our group and others identified YY1 as an epigenetic repressor of multiple muscle genes and muscle relevant miRNAs/lincRNAs during myoblast differentiation into myotube (Wang et al, 2007, 2008; Lu et al, 2012, 2013; Zhou et al, 2012a) Deregulation of the YY1‐regulated molecular circuitries leads to aberrant myogenic differentiation, which contributes to pathogenesis of muscle diseases including rhabdomyosarcoma and Duchenne muscular dystrophy (DMD; Wang et al, 2008, 2012; Zhou et al, 2012a). Altogether, these studies performed mainly on C2C12 myoblast cell line highlight the importance of YY1 in myoblast differentiation and underscore a need to determine the full spectrum of YY1 function in SC lineage progression and in vivo.
Beside the transcriptional control, metabolic control is emerging as a key regulatory mechanism of SC fate transition. Increasing evidence demonstrates that SC functions are largely controlled by two major metabolic states of the cell: oxidative (occurring in the mitochondria) and glycolytic (occurring in the cytoplasm; Garcia‐Prat et al, 2017). Glycolysis is the enzymatic conversion of glucose to pyruvate, which generates two net ATP molecules per molecule of glucose. In contrast, cells in oxygen‐rich environments can use oxidative phosphorylation (OXPHOS) for more efficient ATP production, which nets an average of 34 additional ATP molecules per glucose by oxidizing pyruvate to acetyl coenzyme A (Acetyl‐CoA), then to carbon dioxide and water in the mitochondrial tricarboxylic acid (TCA) cycle. As a major location of energy production, mitochondria play a fundamental role in cell metabolism, and its function is therefore subject to tight quality control. Outstanding questions relevant to SCs include the metabolic status of quiescent satellite cells (QSCs) and how metabolism controls the transition of QSC to activated and differentiated states. Recent evidence indicates that QSCs have mitochondrial activity probably due to the aerobic SC niche, and their activation into more committed progenitors is accompanied by an increased usage of glycolytic pathway (Ryall et al, 2015). This so‐called Warburg metabolism (aerobic glycolysis) originally discovered in cancer cells is believed to allow SCs to respond to the rapid increase in energy demand that characterizes the activated state; it also provides glycolytic intermediates which are chemical building blocks that are critical for synthesis of amino acids, lipids, and nucleic acids, which is important for SCs that need to continually replicate and divide. The metabolic reprogramming has been observed in many stem cells (Ryall et al, 2015). For example, emerging lines of evidence suggest that metabolic reprogramming is required at each step of the conversion of differentiated cells such as fibroblasts to iPSCs and the subsequent conversion to differentiated cells such as neurons (Gibson & Thakkar, 2018). Thus, metabolic reprogramming is not just passive bystanders but may modulate stemness, cell differentiation, and reprogramming. Nevertheless, our knowledge of metabolic programming in SC is incomplete; how SCs achieve the successful use of glycolysis during their activation remains unclear and becomes a focus of this investigation.
In this study, utilizing several genetic mouse models, we initiated a thorough analysis of YY1 function in skeletal muscle development and regeneration. Conditional deletion of YY1 in Pax7 expressing cells led to failure of diaphragm muscle development, thus the neonatal death. Inducible deletion of YY1 in SCs severely impaired the process of acute injury‐induced muscle regeneration. Moreover, deletion of YY1 in a dystrophic mdx mouse exacerbated the chronic injury‐induced dystrophic phenotype. Further examination revealed the YY1 deletion resulted in cell‐autonomous defect in SC activation and proliferation, pointing to YY1 as a key regulator of SC expansion. In search for molecular mechanisms underlying the above phenotypes, global profiling of YY1 regulated transcriptome and binding events revealed that during SC activation YY1 binds and directly represses mitochondrial gene expression; simultaneously, it also stabilizes Hif1α protein to activate Hif1α‐mediated glycolytic genes. Loss of YY1 led to up‐regulation of mitochondrial genes and inhibited glycolysis, thus causing the defect in SC activation. Altogether, our findings have identified YY1 as a key regulator of SC metabolic reprogramming through its dual roles in modulating both mitochondrial and glycolytic pathways.
Results
Conditional deletion of YY1 in muscle progenitor cells causes defect in muscle development
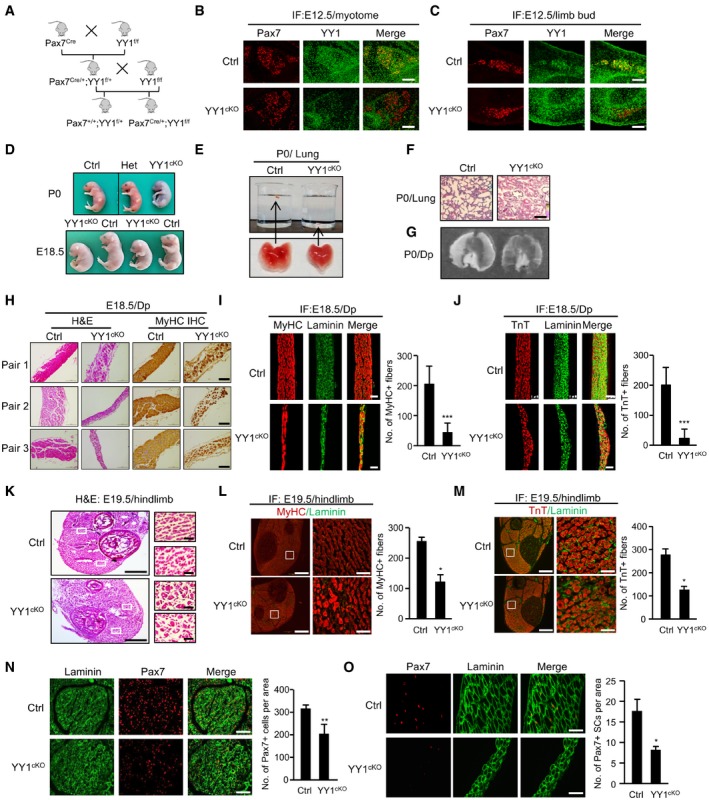
Constitutive ablation of YY1 in mice results in peri‐implantation lethality (Donohoe et al, 1999). To investigate the roles of YY1 in SC function and muscle regeneration, YY1 was selectively ablated in Pax7‐derived cells by crossing Pax7Cre knock‐in mice expressing Cre recombinase from the Pax7 locus (Pax7Cre; Keller et al, 2004) with mice bearing floxed YY1 alleles (Fig EV1A, exon 1 is flanked by loxP sites; YY1f/f; Affar el et al, 2006). Conditional knock out allele, Pax7Cre/+; YY1f/f (thereafter referred to as YY1cKO), and control (Ctrl), Pax7+/+; YY1f/+ littermates, were produced in F2 generation and used for the study (Fig 1A). We first confirmed the successful deletion of YY1 in YY1cKO by staining for both Pax7 and YY1 in the embryos. As reported before (Donohoe et al, 1999), YY1 is ubiquitously expressed and relatively elevated in somite, limb bud, and tail tip. At E12.5 day, there was already a complete deletion of YY1 in Pax7+ cells in myotome by immunofluorescence (IF) staining (merging of Pax7/red with YY1/green in Ctrl yielded yellow cells, which were rarely found in YY1cKO; Fig 1B). When examining limb buds (Fig 1C), the majority of cells in YY1cKO appeared red; a few were yellow probably because Pax7 was not fully activated in these cells yet (Pax7 does not express in limb bud until ~ E11.5; Hutcheson et al, 2009). Unexpectedly, YY1cKO mice died soon after birth even though they were only slightly smaller than the Ctrl and heterozygous (Het, Pax7Cre/+; YY1flox/+) mice and showed no overt morphological deformity (Figs 1D and EV1B). These pups appeared unable to breathe despite frequent gasping and died within an hour after birth, which prompted us to examine their lungs. Indeed, postmortem analysis indicated that the lungs were not able to fill with air, thus sank in water (Fig 1E); histological examination revealed collapsed alveoli (Fig 1F), confirming that the lungs failed to inflate at birth. Since diaphragm (Dp) muscle is critical for the opening of newborn lung (Borensztein et al, 2013; Merrell & Kardon, 2013) and Pax7+ muscle progenitors are important for Dp muscle development (Babiuk et al, 2003; Merrell & Kardon, 2013), we suspected there may be a defect in Dp muscle formation resulting from the YY1 deletion in Pax7+ progenitors. Indeed, the YY1cKO Dp muscle appeared much thinner (Fig 1G), containing markedly reduced MyHC+ muscle fibers visualized by hematoxylin and eosin (H&E) and immunohistochemistry (IHC) staining on multiple pairs of littermate embryos (E18.5 day; Fig 1H). IF staining with MyHC and troponin T proteins also confirmed the massive loss of muscle fibers in the YY1cKO Dp (Fig 1I and J). Further examining the limb muscles which are also developed from Pax7+ progenitors, we found the basic pattern of hindlimb muscles was present in P0 YY1cKO mice, but each muscle appeared slightly smaller and the number of MyHC+ or TnT+ fibers was also decreased (52 and 54%) compared to Ctrl littermates (Fig 1K–M) while the size of myofibers (measured by cross‐sectional area, CSA) was increased (Fig EV1C). These results suggested the deletion of YY1 in Pax7‐expressing muscle progenitors causes a defect in body/limb muscle formation, highlighting a critical need for YY1 during embryonic/fetal muscle development. To further test this notion, we examined the progenitor cells along developmental stages. At the early embryonic stage (E12.5 day), staining for Pax7 or MyoD did not reveal obvious reduction in the number of progenitors in myotome, nor was there any evident defect in the myotome structure (Fig EV1D). However, when examined at days E14.5 (Fig EV1E) and E18.5 (Figs 1N and EV1F), a marked decrease in the number of Pax7+ (35%) or MyoD+ (39%) cells was found in the limb muscle. Similarly, the number of Pax7+ cells was also reduced in the Dp muscle of iKO vs. Ctrl mouse (Fig 1O). Altogether, the above results suggested YY1 plays a critical role in muscle development and the expansion of Pax7+ progenitor pool during embryonic/fetal myogenesis.
Figure EV1. Conditional deletion of YY1 in muscle progenitor cells causes defect in cell expansion.
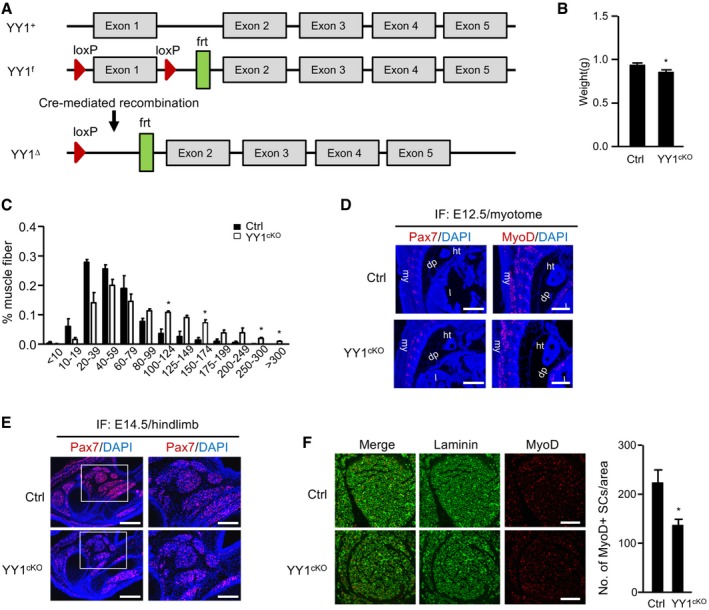
- Schematic illustration of YY1 locus from wild‐type (YY1+), YY1 floxed (YY1f) with the exon 1 flanked by two loxP sites and YY1 Cre‐mediated (YY1∆) mice.
- Quantification of body weight from Ctrl or YY1cKO littermate mice (n = 4, each).
- Cross‐section area (CSA) of individual myofibers in the above harvested limb muscles was measured. The percentage of myofibers with a defined range of CSA over the total myofibers was calculated (n = 3 mice, each).
- Pax7 (red) or Myod (red) staining was performed on cryosections of myotome from Ctrl or YY1cKO mice at E12.5. ht: heart; my: myotome; dp: diaphragm; l: liver; Scar bar = 400 μm.
- Pax7 (red) staining was performed on cryosections of hindlimbs from Ctrl or YY1cKO mice at E14.5. Scale bar (left) = 400 μm. Scale bar (right) = 200 μm.
- Limb muscles (E18.5) were subject to IF staining for MyoD and laminin. Scale bar = 100 μm. Quantification of the numbers of MyoD+ muscle progenitor cells per area is shown on the right (n = 3 mice, each).
Figure 1. Conditional deletion of YY1 in muscle progenitor cells causes defect in cell expansion.
-
ABreeding scheme for generating conditional knock out (YY1cKO), Pax7Cre/+; YY1f/f, and control (Ctrl), Pax7+/+; YY1f/+ littermates.
-
B, CImmunofluorescent (IF) staining of Pax7 and YY1 was performed on cryosections of myotome or limb bud from Ctrl or YY1cKO mice to confirm the ablation of YY1 in Pax7‐expressing progenitors. Scale bar = 100 μm.
-
DRepresentative images of Ctrl, heterozygous (Het), and YY1cKO newborn (P0) mice or embryos at E18.5 day.
-
EExcised lungs from P0 Ctrl or YY1cKO pups were placed in a beaker for sinking test. The lung from YY1cKO pup sank whereas Ctrl floated.
-
FP0 lungs were stained with hematoxylin and eosin (H&E), and representative images are shown. Scale bar = 100 μm.
-
GRepresentative images of diaphragms (Dps) isolated from P0 Ctrl or YY1cKO littermates.
-
HThe above Dps were used for H&E or immunohistochemistry (IHC) staining for MyHC. Representative images from three pairs of littermates are shown. Scale bar = 100 μm.
-
I, JThe above harvested Dps were subjected to IF staining for MyHC or troponin T (TnT) together with laminin. Scale bar = 50 μm. Quantifications of positively stained fibers per area are shown on the right (n = 3 mice, each).
-
KRepresentative images of H&E staining of hindlimb muscles isolated from Ctrl or YY1cKO embryos at E18.5 day. Scale bar = 400 μm (left) or 50 μm (right).
-
L, MThe above harvested limb muscles were subjected to IF staining for MyHC or TnT together with laminin. Scale bar = 400 μm (left) or 50 μm (right). Quantifications of the numbers of MyHC+ or TnT+ fibers per area are shown on the right (n = 3 mice, each).
-
N, OThe above harvested limb (N) or diaphragm (O) muscles were subjected to IF staining for Pax7 and laminin. Scale bar = 100 μm (N) or 50 μm (O). Quantification of the numbers of Pax7+ muscle progenitor cells per area is shown on the right (n = 3 mice, each).
Inducible ablation of YY1 in adult muscle severely hampered injury‐induced muscle regeneration
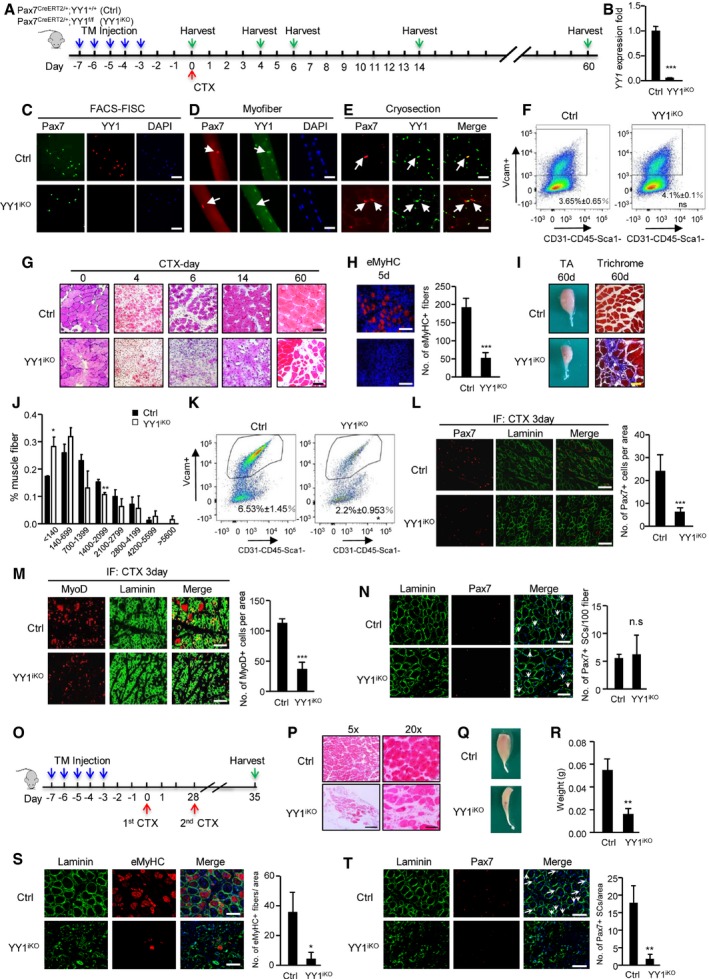
To circumvent the developmental defect and neonatal death, we next employed an inducible Cre driver in which tamoxifen (TM)‐inducible CreER protein is expressed from a modified Pax7 locus, Pax7CreERT2 (Lepper et al, 2009). Mice with the Pax7CreERT2/+allele were crossed to YY1f/f mice, to generate Pax7CreERT2/+;YY1f/f mice (termed YY1iKO) to permanently disrupt YY1 function in adult Pax7+ SCs upon the administration of TM. Pax7CreERT2/+; YY1+/+mice treated with TM were used as control (Ctrl). Following the administration of five consecutive doses of TM (Fig 2A), SCs were FACS‐isolated and cultured; YY1 mRNA was efficiently depleted (94%, Figs 2B and EV2A). Consistently, DNA analysis also showed lack of YY1 gene in the YY1iKO genome (Fig EV2B). IF staining for YY1 protein also revealed it was eliminated from freshly isolated Pax7+ SCs (FISCs) whereas readily detected in the Ctrl (Fig 2C). Furthermore, when examining freshly isolated extensor digitorum longus (EDL) myofibers from Ctrl or YY1iKO mice, loss of YY1 in SCs was also readily seen with an ablation efficacy of 94% (Figs 2D and EV2C). Lastly, on muscle cryosections YY1 protein was not detected in the majority of Pax7‐expressing QSCs from YY1iKOmice, where only 6% SCs escaped recombination and remained YY1+ (Fig 2E). Importantly, TM‐treated YY1iKO mice remained viable and displayed no obvious phenotype or changes in body weight under physiological conditions up to 1 year after TM injection. In addition, 3 weeks after TM injection, we found no obvious change in the number of SCs isolated by FACS between Ctrl and YY1iKO littermates (Fig 2F). Consistently, the number of SCs on single myofibers did not differ between the littermates 3 days or 2 months after the TM treatment (Fig EV2D and E), indicating YY1 loss did not have impact on SC maintenance.
Figure 2. Inducible ablation of YY1 in adult mouse muscle blocks injury‐induced muscle regeneration.
-
ASchematic outline of the tamoxifen (TM) administration used in the study and experimental design for testing the effect of YY1 deletion on cardiotoxin (CTX)‐induced muscle regeneration process for control (Ctrl), Pax7CreERT2/+; YY1+/+ and inducible knock out (YY1iKO), Pax7CreERT2/+; YY1f/f mice.
-
BSCs were FACS‐sorted 3 days after the last TM injection and cultured for 1.5 days; RT–qPCR detection of YY1 mRNA shows the ablation in YY1iKO cells.
-
C–EIF staining for Pax7 and YY1 on (C) freshly isolated FISCs or (D) single myofibers from EDL muscles or (E) cryosections from TA muscles showing the deletion of YY1 protein from YY1iKO cells. Scale bar = 100 μm in (C) or 50 μm in (D, E).
-
FRepresentative FACS plots. About 100,000 cells from 2‐month‐old Ctrl and YY1iKO mice were sorted by FACS 3 weeks post‐TM injection. The percentage of SCs was shown. (n = 3 mice, each).
-
GH&E staining was performed on the injured TA muscles collected from the designated times post‐CTX injection to visualize the degree of regeneration. Scale bar = 100 μm.
-
HIF staining for eMyHC on the TA muscles 5 days post‐CTX injury. Quantifications of the number of eMyHC+ fibers are shown on the right (n = 3, each). Scale bar = 50 μm.
-
ILeft: representative images of TA muscles isolated from Ctrl or YY1iKO mice 60 days post‐CTX injury. Right: Masson's trichrome staining of the above muscles to visualize the degree of fibrosis. Scale bar = 100 μm.
-
JCross‐section area (CSA) of individual myofibers in TA muscles 4 weeks after injury was measured. The distribution for CSA was calculated (n = 3 mice).
-
KRepresentative FACS plots showing the percentage of SCs sorted from TA muscles 3 days after CTX injury of Ctrl and YY1iKO mice.
-
L, MImmunostaining for Pax7 or MyoD together with laminin was performed on the TA muscles 3 days post‐CTX injury. Scale bar = 100 μm. Quantifications of the numbers of Pax7+ or MyoD+ SCs per area are shown on the right (n = 3 mice, each).
-
NIF staining for Pax7 and laminin on TA muscles 4 weeks after CTX injury of Ctrl or YY1iKO mice. Quantifications of the numbers of Pax7+ SCs per 100 fibers are shown on the right (n = 3 mice, each). Scale bar = 100 μm.
-
OSchematic outline of the second CTX injury 28 days after the first injury.
-
PH&E staining was performed on TA muscles of Ctrl or YY1iKO mice 7 days after second CTX injury. Scale bar = 400 μm (left) or 100 μm (right).
-
QRepresentative images of above TA muscles 7 days after second injury.
-
RQuantifications of the weight of above TA muscles from three pairs of littermate mice.
-
SIF staining for eMyHC and laminin on TA muscles 7 days after the second CTX injury of Ctrl or YY1iKO mice. Quantifications of the number of eMyHC+ fibers per area are shown on the right (n = 3 mice, each). Scale bar = 100 μm.
-
TIF staining for Pax7 and laminin on the above muscles and the quantifications of the number of Pax7+ SCs per area are shown on the right (n = 3 mice, each). Scale bar = 100 μm.
Figure EV2. Inducible deletion of YY1 in satellite cells impairs acute injury‐induced muscle regeneration.
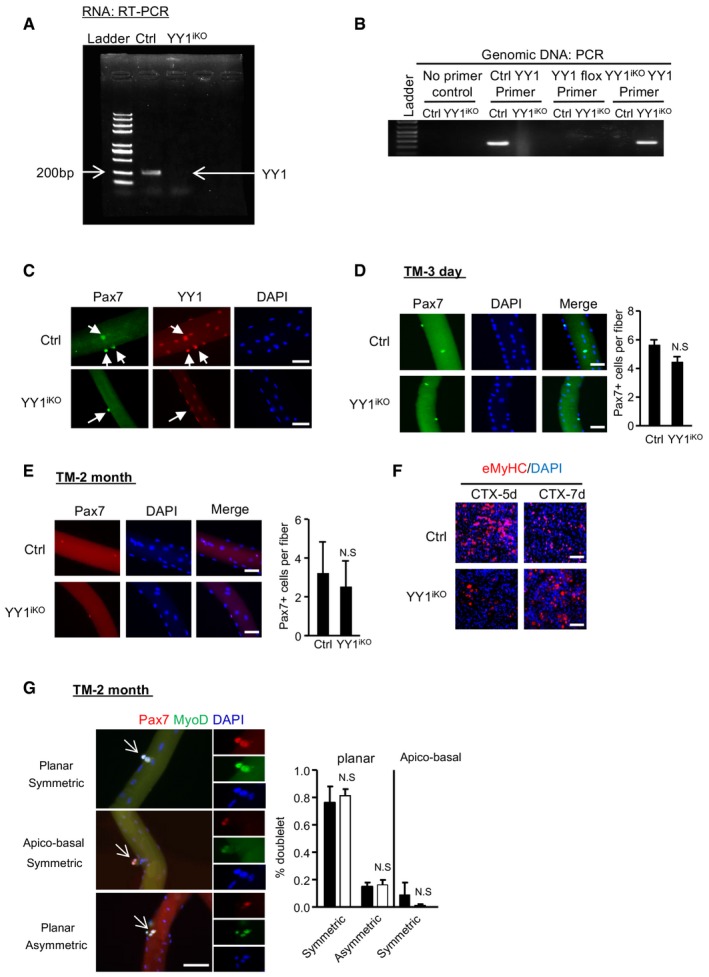
-
ASemi‐quantitative RT–PCR detection of YY1 mRNA expression in SCs harvested 3 days after the last dose of tamoxifen (TM) injection to confirm the ablation of YY1 in YY1iKO mice.
-
BPCR validation of YY1 ablation from the genomic DNA of FACS‐sorted SCs.
-
CSingle myofibers were isolated from EDL muscles from Ctrl or YY1iKO mice and cultured for 2 days before immunofluorescence (IF) staining for Pax7 and YY1; YY1 was not detected from the YY1iKO cells (arrows). Scale bar = 50 μm.
-
D, EIF staining for Pax7 on freshly isolated single myofibers from Ctrl or YY1iKO mice 3 days (D) or 2 months (E) post‐TM injection. Quantifications of Pax7+ SCs per fiber are shown on the right (n = 3, each). Scale bar = 50 μm.
-
FIF staining for eMyHC on the TA muscles 5 or 7 days post‐CTX injury. Scale bar = 100 μm.
-
GCtrl and YY1iKO cells were cultured for 48 h and IF‐stained for Pax7 (red) and MyoD (green). White arrows indicate dividing cell doublets. Scale bar = 50 μm. Right: quantification of symmetrical or asymmetrical doublets based on the symmetry of Pax7 expression in the two dividing cells at either planar or apico‐basal orientation (n = 3, each).
Next, to study the role for YY1 in muscle regeneration, we employed a commonly used cardiotoxin (CTX)‐induced muscle injury‐regeneration model (Chen et al, 2017). Tibialis anterior (TA) muscles of Ctrl or YY1iKO animals were subjected to a single CTX injury and then allowed to recover for 3–60 days before analysis of the regenerated tissue (Fig 2G). YY1iKO mice exhibited normal muscle size and morphology in the absence of injury (Fig 2G, day 0). The CTX injection quickly induced extensive muscle damage and infiltration of inflammatory cells in both Ctrl and YY1iKO muscles (Fig 2G). Strikingly, muscle regeneration was severely disrupted in YY1iKO mice. In Ctrl muscle, during the acute phase of regeneration (1–7 days after injury), SC descendants fused to form small new myofibers expressing embryonic myosin heavy chain (eMyHC) characterized by centrally localized nuclei; these eMyHC fibers were readily seen at day 5 (Fig 2H). In contrast, YY1iKO muscle was composed of degenerating myofibers, fibrotic tissues, and inflammatory cells at this phase; eMyHC+ regenerating fibers were rarely seen until day 7 when the Ctrl muscle was composed of regenerated myofibers with larger size that had down‐regulated eMyHC (Fig EV2F). Fourteen days after injury, muscle damage and inflammatory cells in Ctrl muscle were largely cleared, and the regenerated myofibers continued to grow and mature, as their sizes became homogenous (Fig 2G). In contrast, YY1iKO muscle was still occupied by damaged fibers and inflammatory cells, with only traces of regenerating myofibers (Fig 2G). If left to 60 days after injury, Ctrl muscle had fully regenerated, and muscle architecture was largely restored, with the normal myofiber hypertrophy seen after injury (Fig 2I). In contrast, YY1iKO muscle nearly failed to regenerate and reconstitute muscle structure, instead it displayed severe atrophy with markedly reduced fiber size (Fig 2I and J) and evident fibrosis (Fig 2I). Collectively, these results suggested that YY1 deletion leads to severe loss of regenerative capacity, and thus, the expression of YY1 in SCs is essential for timely and proper repair of damaged skeletal muscle tissue after acute injury.
Muscle regeneration requires activation and proliferation of satellite cells and subsequent differentiation of myoblasts into myotubes. Considering embryonic deletion of YY1 caused a defect in the expansion of muscle progenitors (Fig 1N and O), we asked whether the expansion of SC pool in YY1iKO mice had been compromised. Indeed, 3 days after CTX injection, the number of SCs sorted by FASC from YY1iKO vs. Ctrl was largely reduced (66%; Fig 2K); consistently, staining of the above injured muscles for Pax7 or MyoD also revealed a significant reduction in Pax7+ or MyoD+ cells in YY1iKO muscle (Fig 2L and M, 73 and 67%, respectively). Interestingly, staining of Pax7 4 weeks after CTX injury revealed no difference in the number of Pax7+ cells per 100 fibers between Ctrl and YY1iKO mice (Fig 2N), suggesting YY1 loss may not impact the ability of SC to self‐renew and return to quiescent stage. Consistently, no significant change was found on the number of asymmetrically dividing Pax7+MyoD+ cells when monitoring the planar and apico‐basal cell divisions and recording daughter cells in doublets that were identical (symmetric) or non‐identical (asymmetric cell division; Fig EV2G). Nevertheless, when the mice were subject to the second CTX injury 28 days after the first injury (Fig 2O), compared to the first injury, YY1iKO mice suffered from an even more severely hampered regeneration (Fig 2P–S, 88% reduction in eMyHC+ fibers in iKO vs. Ctrl) and nearly complete loss of SC expansion (Fig 2T, 91% loss of Pax7+ cells in iKO vs. Ctrl). Altogether, the above results confirmed the key role of YY1 in SC expansion during acute injury‐induced muscle regeneration.
YY1 deletion aggravates dystrophic phenotype in mdx mice
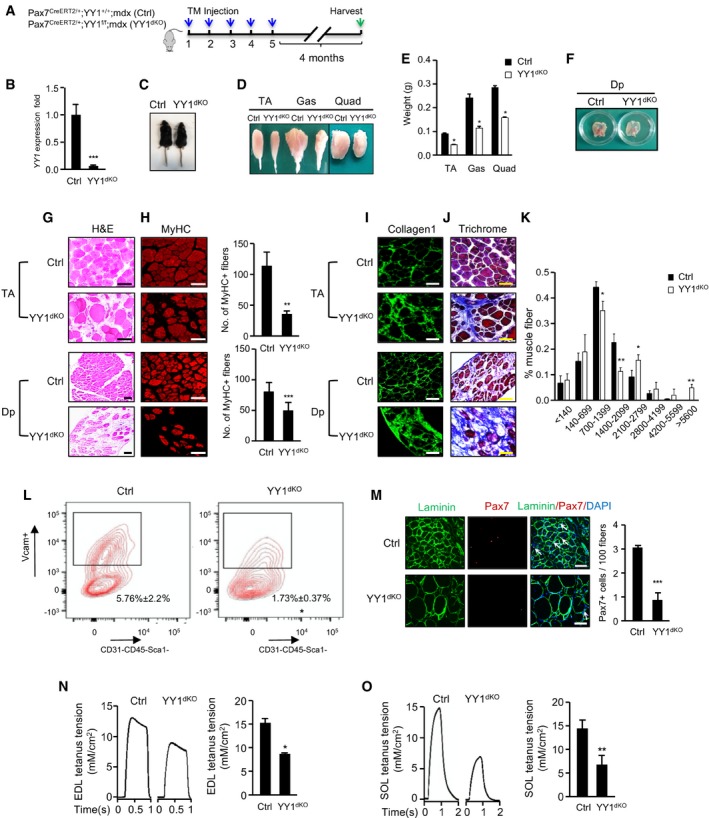
To further solidify the impact of YY1 deletion on injury‐induced muscle regeneration, we next employed a mdx mouse, in which mutation in dystrophin gene leads to continuous degeneration and regeneration of myofibers accompanied by repeated activation and enhanced turnover of SCs, mimicking the phenotype of human DMD (Bulfield et al, 1984). YY1iKO mice were crossed with mdx mice to generate double KO (dKO) mice. Two‐month‐old Pax7CreERT2/+; YY1f/f; mdx mice were treated with five consecutive doses of TM to delete YY1 in SCs; Pax7CreERT2/+; YY1+/+; mdx mice treated with TM were used as control (Ctrl; Fig 3A and B). After 4 months, YY1dKO had a much smaller body size than Ctrl littermates (Fig 3C). The size and weight of limb muscles including TA, gastrocnemius (GAS), and quadruple (Quad) were all markedly reduced (50, 53, and 45%; Fig 3D and E). Likewise, Dp muscle was markedly thinner (Fig 3F). Further histological examination revealed a reduced number of muscle fibers (Fig 3G and H, MyHC staining) accompanied by an exacerbation of fibrosis (Fig 3I and J, collagen and trichrome staining) in both limb (TA) and Dp muscles. Moreover, a higher number of hypertrophic myofibers was found in YY1dKO vs. Ctrl mice (Fig 3K). Altogether, the above findings suggested that the deletion of YY1 in SCs of mdx aggravates dystrophic phenotypes. Expectedly, a largely reduced number of SCs (70%) was sorted out by FACS from TA muscles of YY1dKO vs. Ctrl mice (Fig 3L); consistently, the number of Pax7+ cells was also largely reduced by 71% (Fig 3M), suggesting YY1 deletion led to a defect in SC expansion during the regeneration in a chronic muscle injury setting. Lastly, to confirm the functional consequence of the weakened muscles in YY1dKO, the mice were subjected to muscle functional test by detecting the maximal isometric tetanic force on EDL and soleus (SOL) muscles. As expected, the muscles from YY1dKO showed impaired tetanus force (Fig 3N and O, reduced by 43 and 54%, respectively) compared to the Ctrl. Altogether, the above findings led us to conclude that YY1 plays an essential role in maintaining SC pool and enables continuous muscle regeneration under chronic injury.
Figure 3. YY1 deletion aggravates dystrophic phenotype in mdx mice.
-
AOutline of the TM administration scheme to obtain Ctrl or YY1dKO mice.
-
BSCs were FACS‐sorted 4 months after the last TM injection; RT–qPCR detection of YY1 mRNA shows the ablation in YY1dKO cells.
-
CRepresentative images of Ctrl or YY1dKO mice 4 months after the TM administration.
-
DRepresentative images of limb muscles (tibialis anterior, TA; gastrocnemius, Gas; quadruple, Quad) from Ctrl or YY1dKO mice.
-
EQuantifications of muscle weight from three pairs of littermate mice.
-
FRepresentative images of Dp muscles isolated from Ctrl or YY1dKO mice 4 months after the TM injection.
-
G, H(G) H&E or (H) MyHC staining was performed on TA or Dp muscles from Ctrl or YY1dKO mice. Quantifications of the numbers of MyHC+ fibers per area of TA or Dp muscles are shown on the right (n = 3 mice, each). Scale bar = 100 μm.
-
I, J(I) Collagen 1 or (J) Masson's trichrome staining was performed on TA or Dp muscles from Ctrl or YY1dKO mice. Scale bar = 100 μm.
-
KThe distribution of fiber size measured by CSA in TA muscles from three pairs of 6‐month‐old Ctrl and YY1dKO mice was calculated (n = 3 mice, each).
-
LRepresentative FACS plots showing the percentage of SCs sorted from 6‐month‐old Ctrl and YY1dKO mice (n = 3 mice, each).
-
MIF staining for Pax7 and laminin on TA muscles of Ctrl or YY1dKO mice. Quantifications of the numbers of Pax7+ SCs per 100 fibers are shown on the right (n = 3 mice, each).
-
N, OEDL or SOL muscles from Ctrl or YY1dKO mice were subjected to measurement of maximal isometric tetanic force. Left: representative trace images of normalized tetanic force. (Right): quantification of the force by normalizing with cross‐sectional area (CSA; n = 3 mice, each).
YY1 deletion leads to cell‐autonomous defect in SC activation and proliferation
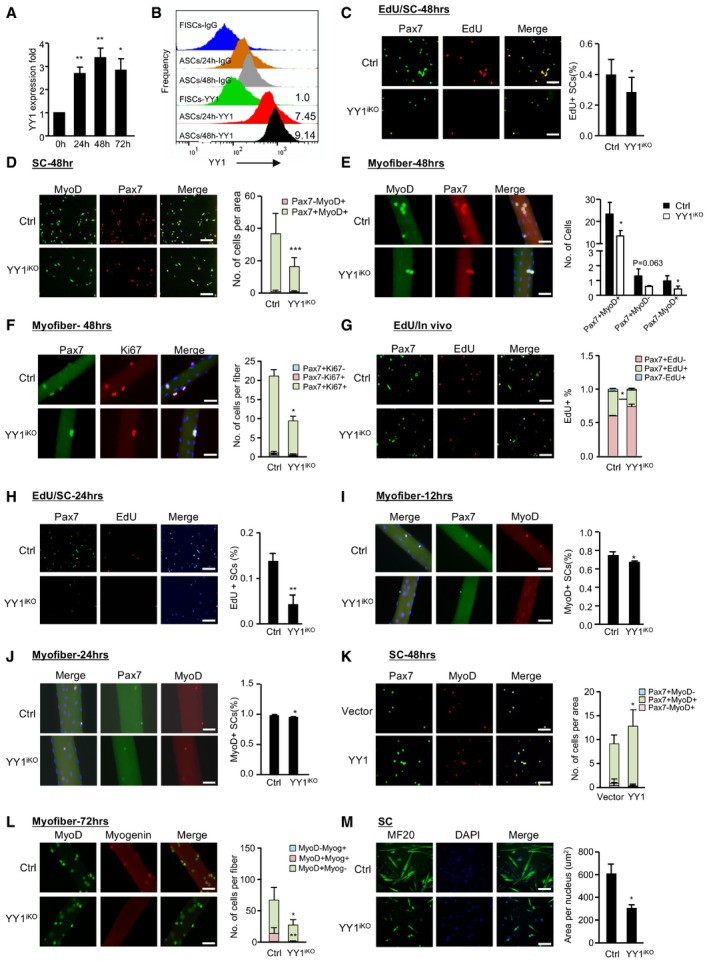
To further investigate the cellular mechanism underlying the hampered SC pool expansion in YY1iKO mice, we first examined YY1 expression dynamics in SCs and found it was lowly expressed in FISCs but quickly induced upon SC activation and its level continued to increase into 48 h as SCs fully activated and proliferated (Figs 4A and B, and EV3A); this pattern was concomitant with a sharp increase in YY1 mRNA level in the acute SC activation/proliferation phase after CTX injection (Fig EV3B). The above data led us to speculate that YY1 may promote the proliferative expansion of SCs. To test this notion, freshly isolated SCs (FISCs) by FACS sorting were cultured and labeled by 5‐ethynyl‐2′‐deoxyuridine (EdU; Fig EV3C). YY1‐depleted SCs showed a marked reduction in the number of EdU+ cells at 48 h in culture (Figs 4C, and EV3D and E), reflecting a proliferative delay; this was also confirmed by a marked reduction in the number of Pax7+MyoD+ cells (Figs 4D and EV3F). Furthermore, when freshly isolated EDL myofiber explants were cultured for 2 days to allow SC activation and proliferation on the fibers, the number of Pax7+MyoD+ cells from YY1iKO myofibers was also significantly reduced compared with the Ctrl (Fig 4E, 42%). The proportion of Pax7+MyoD+ cells (Fig EV3G), on the other hand, showed no significant difference, suggesting that although the cell proliferation was delayed the lineage progression was not blocked. Consistently, when stained for Ki67 and Pax7, a much lower number of double positive cells was found on the YY1iKO myofibers (Fig 4F, 56.24%). When further examined in vivo (Fig EV3H), EdU labeling in regenerating muscles for 12 h also revealed a reduced (12%) number of proliferating SCs at 2.5 days after CTX injury (Fig 4G). Altogether, the above results solidified the delayed proliferation caused by YY1 loss. To pinpoint the possible defect in the earlier stage, i.e., activation of quiescent SCs to enter the first cell division, we found that at 24 h before SCs entered the first cell cycle (Tang & Rando, 2014), the number of EdU+ cells was reduced (10%) in YY1iKO SCs (Fig 4H), indicating a possible defect at the very early activation stage. To solidify the notion, we found the percentage of MyoD+ cells was significantly decreased when examining the single myofibers cultured in growth medium for 12 or 24 h (Fig 4I and J) but not 48 h (Fig EV3I). Lastly, lentiviral re‐expression of YY1 in YY1iKO cells (Fig EV3J) indeed restored the proliferative capacity of these cells (Fig 4K), pinpointing YY1 loss as the cause of the defect. Interestingly, YY1iKO SCs did not show an increased propensity for differentiation as seen in other cases (Zhu et al, 2016); the number of Myog+MyoD+ cells on YY1iKO myofibers at day 3 in culture was instead largely reduced (97% of Ctrl level, Fig 4L). In addition, SCs from YY1iKO mice did not form MyHC+ myotubes efficiently when differentiated (50% of Ctrl level, Fig 4M). Furthermore, TUNNEL assay revealed no evident apoptosis (Fig EV3K). Altogether, the above results demonstrated that inducible YY1 deletion in Pax7‐expressing SCs causes cell‐autonomous defect in their activation and proliferation capacity.
Figure 4. YY1 deletion leads to cell‐autonomous defect in SC pool expansion.
-
AFACS‐isolated SCs from Pax7‐nGFP mice were cultured for the designated time (0, 24, 48, or 72 h). YY1 expression was quantified by RT–qPCR. 18S was used as the normalization control.
-
BFlow cytometric analysis of YY1 protein or IgG control in FACS‐isolated SCs cultured for the designated time (0, 24, or 48 h). YY1 protein level is normalized to IgG levels. The numbers indicate the normalized level of YY1 protein relative to that of FISCs.
-
CAn equal number of FACS‐isolated SCs from Ctrl or YY1iKO mice were cultured for 48 h and EdU‐labeled for 8 h, followed by immunostaining for Pax7 (green) and EdU (red). Quantifications of the percentage of EdU+ cells are shown on the right (n = 3 mice, each). Scale bar = 100 μm.
-
DThe above cells were also IF‐stained for Pax7 (red) and MyoD (green). Quantifications of each population, Pax7+MyoD+ or Pax7−MyoD+, are shown on the right (n = 3, each). Scale bar = 100 μm.
-
ESingle EDL myofibers were cultured for 48 h before staining for Pax7 (red) and MyoD (green). Quantifications of the numbers of each population, Pax7+MyoD−, Pax7−MyoD+, or Pax7+MyoD+, are shown on the right (n = 3 mice, each). Scale bar = 50 μm.
-
FPax7 (green) and Ki67 (red) staining was performed on the above single EDL myofibers. Scale bar = 50 μm. Quantifications of the numbers of each population, Pax7+Ki67−, Pax7−Ki67+, or Pax7+Ki67+, are shown on the right (n = 3 mice, each).
-
GSCs expansion in vivo was determined by EdU labeling for 12 h 2.5 days after CTX injury. Quantifications of the numbers of EdU+ cells are shown on the right (n = 3 mice, each). Scale bar = 100 μm.
-
HAn equal number of FACS‐isolated SCs from Ctrl or YY1iKO mice were cultured for 24 h and EdU‐labeled for 12 h, followed by immunostaining for Pax7 (green) and EdU (red) was performed. Quantifications of the percentage of EdU+ cells are shown on the right (n = 3 mice, each). Scale bar = 100 μm.
-
I, JSingle EDL myofibers were cultured for 12 (I) or 24 (J) h in growth media before staining for Pax7 (green) and MyoD (red). Scale bar = 50 μm. Quantifications of the proportion of MyoD+ cells are shown on the right (n = 3 mice, each).
-
KFACS‐isolated SCs from YY1iKO mice were infected with YY1‐expressing or control viruses, followed by immunostaining for Pax7 (green) and MyoD (red) 36 h post‐transfection. Scale bar = 100 μm. Quantifications of the numbers of each population, Pax7+MyoD−, Pax7−MyoD+, or Pax7+MyoD+, are shown on the right (n = 3 mice, each).
-
LSingle EDL myofibers were cultured for 3 days, followed by immunostaining for myogenin (red) and MyoD (green). Scale bar = 50 μm. Quantifications of the numbers of each population, MyoD+Myog+, MyoD−Myog+, or MyoD+Myog−, are shown on the right (n = 3 mice, each).
-
MFACS‐isolated SCs were cultured for 2 days in proliferation medium followed by 2 days in differentiation medium; the degree of differentiation was assessed by IF staining of MF20+. Scale bar = 100 μm. Quantifications of the numbers of MF20+ area per nucleus are shown on the right (n = 3 mice, each).
Figure EV3. Inducible deletion of YY1 in satellite cells engenders a cell‐autonomous defect in activation and proliferation.
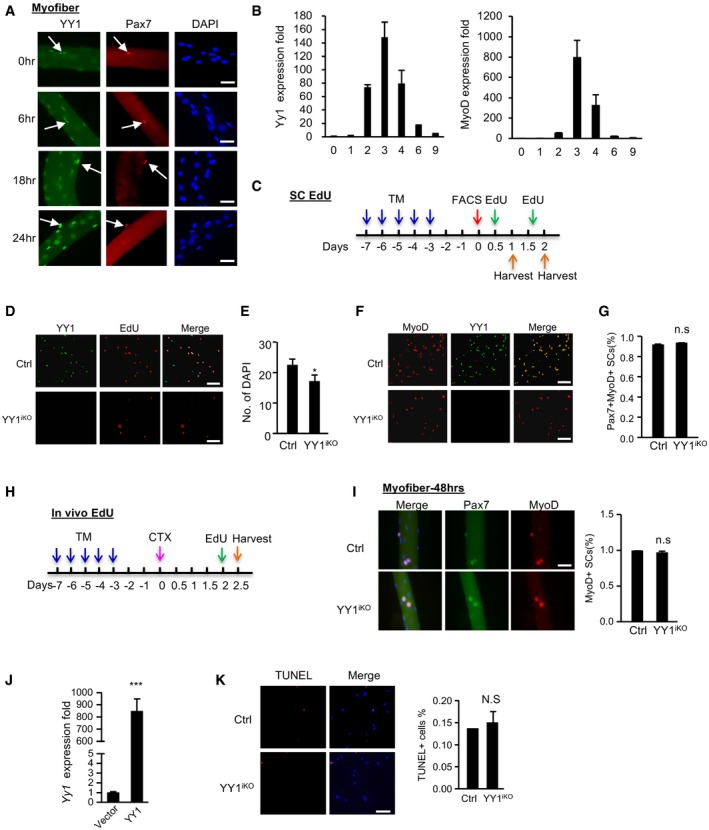
- Single myofibers were isolated from EDL muscles and cultured for the designated time (0, 6, 18, or 24 h), followed by immunostaining for Pax7 (red) and YY1 (green). Scale bar = 50 μm. White arrows indicate Pax7+YY1+ cells.
- qRT–PCR detection of Yy1 and MyoD mRNA expression level during CTX‐induced muscle regeneration.
- Schematic illustration of the in vitro EdU labeling assays. To assess SCs activation, cells were cultured for 12 h and EdU‐labeled for 12 h. To assess the proliferation, cells were cultured for 40 h and labeled for 8 h before harvesting for EdU staining.
- As shown in Fig 4C, FACS‐isolated SCs from Ctrl or YY1iKO mice were cultured for 48 h and EdU‐labeled for 8 h, followed by immunostaining for YY1 (green) and EdU (red) to show. Scale bar = 100 μm.
- Quantification of the number of DAPI+ cells for Fig 4C is shown (n = 3 mice, each).
- FISCs were cultured for 1.5 days and IF‐stained MyoD with YY1. Scale bar = 100 μm.
- Quantification of the percentage of Pax7+MyoD+ cells in Fig 4E is shown (n = 3 mice, each).
- Schematic illustration of the in vivo EdU labeling assay. Two days after CTX injection, EdU injection via i.p was performed followed by FACS isolation of SCs 12 h later.
- Single EDL myofibers were cultured for 48 h in growth media before staining for Pax7 (green) and MyoD (red). Scale bar = 50 μm. The percentage of MyoD+ cells was shown (n = 3 mice, each).
- SCs isolated from YY1iKO mice were transfected with a lentiviral YY1‐expressing or empty viruses. The overexpression of YY1 mRNA was detected by RT–qPCR (n = 3, each).
- SCs were sorted and cultured for 36 h, followed by TUNEL assay. Quantifications of TUNEL+ cells% are shown on the right (n = 3, each). Scale bar = 100 μm.
Transcriptomic and global binding profiling reveals YY1 repression of mitochondrial genes in SCs
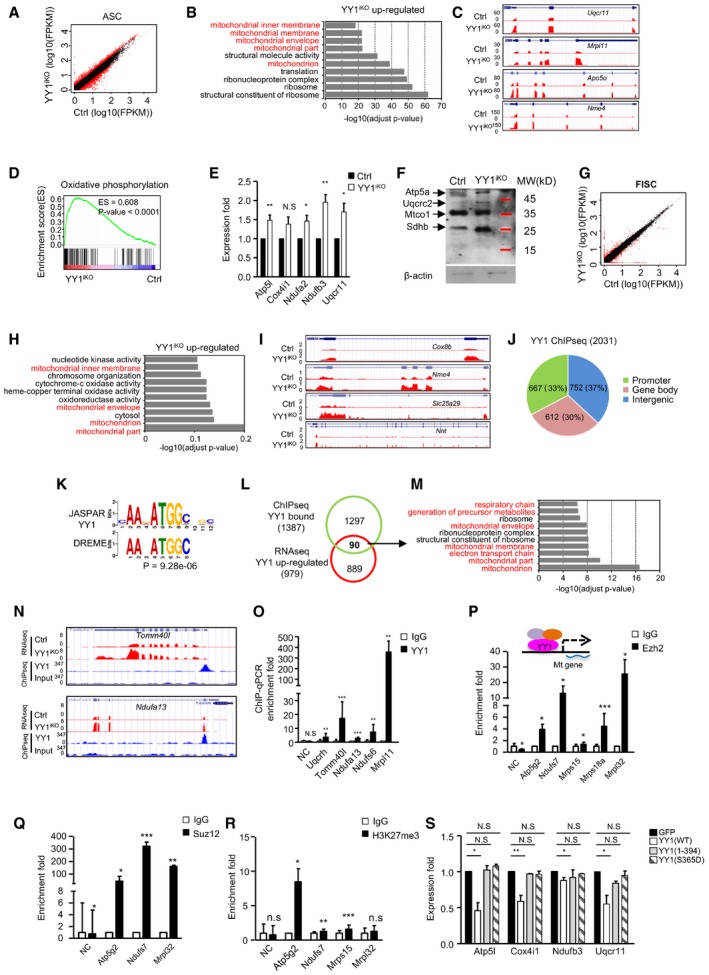
To probe into the molecular mechanisms underlying YY1 influence on SCs activation/proliferation, we performed RNA sequencing (RNAseq) to interrogate the transcriptomic changes caused by YY1 loss. Total RNAs from ASCs (cultured for 36 h) were extracted and subject to PolyA+ RNAseq. A total of 979 genes were up‐regulated whereas 924 down‐regulated in YY1iKO vs. Ctrl (Fig 5A; Table EV1). Forty‐nine lincRNAs were found differentially expressed, which is in line with our prior finding that YY1 regulates dozens of lincRNAs in C2C12 myoblasts (Lu et al, 2013). Gene Ontology (GO) analysis revealed terms such as “cell cycle” and “cell division” are prominent among the down‐regulated transcripts (Fig EV4B; Table EV1), consistent with the observed delay in cell activation/proliferation (Fig 4). Interestingly, the up‐regulated genes were dominant by mitochondrion‐related GO terms such as “mitochondrial membrane”, “mitochondrial envelope”, and “mitochondria part” (Fig 5B and C; Table EV1). Gene set enrichment analysis (GSEA) also disclosed that the up‐regulated genes were enriched in oxidative phosphorylation (OXPHOS) pathway (Fig 5D). Specifically, among the enriched mitochondrial genes, genes related to (i) electron transport chain, including Ndufa2, Ndufb3, Uqcr11, and Cox7a2; (ii) mitochondrial translation, including Mrpl11, Mrpl52, Mrpl13, and Chchd1; (iii) purine nucleoside triphosphate metabolic process, including ATP5j2, ATP5f1, ATP5o, and Nme1; and (iv) mitochondrial transmembrane transporter, including Tomm5, Timm9, and Slc25a33, were found on the top of the list (Table EV1). Additionally, an array of genes such as Sod1, Nox1, and Txn1, correlated to regulate cellular redox homeostasis, were also up‐regulated (Table EV1). The above RNAseq results were further confirmed by RT–qPCR on several selected genes including Atp5l, Cox4i1, Ndufa2, Ndufb3, and Uqcr11 (Fig 5E). Examining more closely into the expression of Complexes I–V, we found that the majority of subunits in Complexes I, III, IV, and V were up‐regulated by RNAseq (Fig EV4A), which was substantiated by the elevated protein expression levels of Sdhb (Complex II subunit, 30 kDa), Uqcrc2 (Complex III subunit, 48 kDa), and Atp5a (Complex V subunit, 55 kDa) using a mitoprofile antibody cocktail for Western detection (Fig 5F). Curious about the potential expression changes in myogenic genes, by GSEA, we found that down‐regulated genes were not enriched in myogenic genes, further supporting a role for YY1 in cell proliferation but not lineage progression (Fig EV4C and D). Furthermore, when total RNAs from FISCs were subject to transcriptomic profiling (Fig 5G; Table EV2), mitochondrion‐related genes were also markedly enriched in the up‐regulated panel of genes (Fig 5H; Table EV2), including Cox8b, Nme4, Slc25a29, Nnt1, Cox7a1, Rars2, Lars2, and Hscb (Figs 5I, and EV4E and F), despite the down‐regulated genes were enriched for “extracellular region part” etc. (Fig EV4G; Table EV2), distinct from those identified in ASCs. Together, the above findings led us to speculate that YY1 functions to repress mitochondrial genes and oxidative phosphorylation.
Figure 5. Transcriptomic and binding profiling reveals YY1 repression of mitochondrial genes in SCs.
-
ARNAseq was performed with RNAs extracted from ASCs (FACS purified and cultured for 36 h) of YY1iKO or Ctrl mice; scatter plot shows differentially expressed genes with a fold change ≥ 2 (red dots) in YY1iKO vs. Ctrl.
-
BGene ontology (GO) analyses of the above up‐regulated genes. The top 10 enriched GO terms are displayed on the Y axis and adjusted P values on the X axis.
-
CGenomic snapshots showing the examples of mitochondrial genes (Uqcr11, Mrpl11, Apo5o, and Nme4) up‐regulated in YY1iKO vs. Ctrl cells.
-
DGSEA analysis shows oxidative phosphorylation gene set is enriched in YY1iKO vs. Ctrl cells (P‐value < 0.0001).
-
ERT–qPCR validation of the expression of the selected up‐regulated mitochondrial genes in YY1iKO vs. Ctrl.
-
FProtein levels of the respiratory chain complexes I–V components in YY1iKO vs. Ctrl ASCs were detected using a mitoprofile antibody cocktail. β‐actin was used as the loading control.
-
GRNAseq was performed using RNAs from freshly isolated SCs (FISCs); scatter plot shows differentially expressed genes with a fold change ≥ 2 (red dots) in YY1iKO vs. Ctrl.
-
HGO analysis for the up‐regulated genes in YY1iKO vs. Ctrl.
-
IGenomic snapshots show the selected mitochondrial genes (Cox8b, Nme4, Slc25a29, Nnt) up‐regulated in YY1iKO vs. Ctrl FISCs.
-
JChIPseq was performed using chromatins from WT ASCs, and the genomic distribution of 2,031 YY1 binding peaks is shown.
-
KDe novo motif prediction by DREME revealed the enrichment of canonical YY1 motifs in the above binding regions.
-
LVenn diagrams show the overlapping (90 genes) between the above identified binding target genes (1,387) and the up‐regulated genes from ASC RNAseq (979).
-
MGO analysis of the above 90 genes revealed an extreme enrichment of mitochondrial‐related terms.
-
NGenomic snapshots of two of the above identified mitochondrial genes (Tomm40I and Ndufa13) that are bound by YY1 in their TSSs (ChIPseq tracks) and up‐regulated by YY1 deletion (RNAseq tracks).
-
OChIP–qPCR validation of YY1 binding on some mitochondrial genes. Negative control (NC) represents a genomic region on Chromosome 11 with no YY1 binding peak identified. Enrichment fold was calculated as the amount of amplified DNA from YY1 binding sites normalized to values obtained from IgG control.
-
P–RChIP–qPCR was performed to show the enrichment of Ezh2 (P), Suz12 (Q), and H3K27 me3 (R) binding on selected mitochondrial genes.
-
SA construct to express wild‐type YY1(WT) or two mutants lacking DNA‐binding activity, YY1(1–394) and YY1(S365D), were transfected into YY1iKO cells; the expression of mitochondrial gene was measured.
Figure EV4. Transcriptomic and ChIPseq profiling reveals YY1 repression of mitochondrial genes in SCs.
-
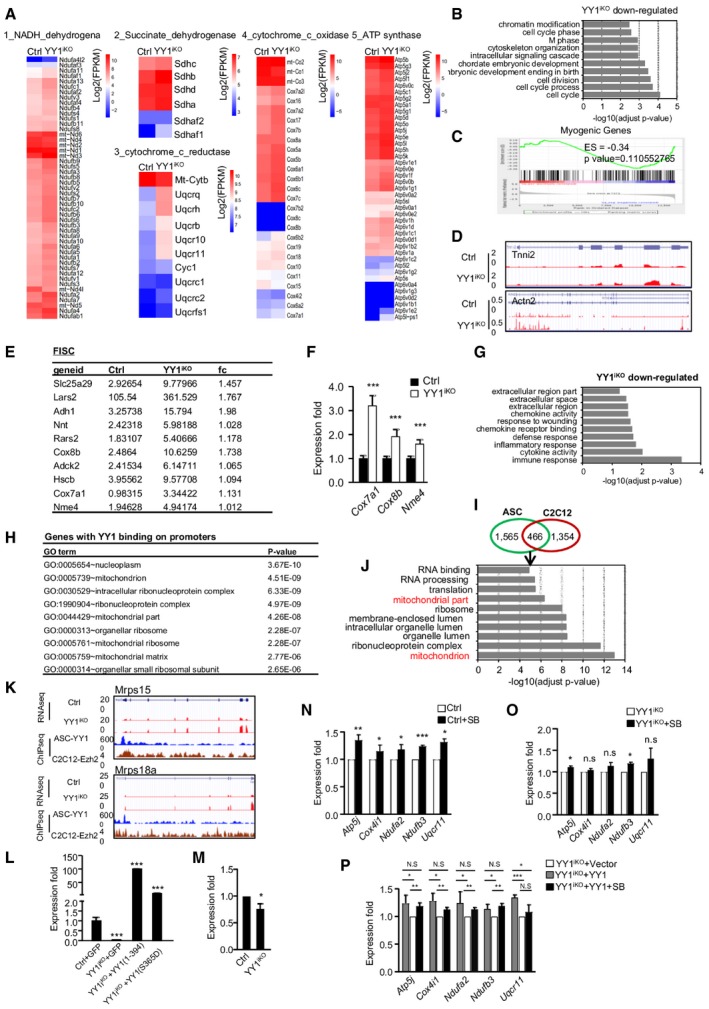
AHeat maps indicating gene expression (Log2[FPKM]) of complex I–V in Ctrl vs. YY1iKO ASCs.
-
BGene ontology (GO) analysis of the down‐regulated genes in YY1iKO vs. Ctrl ASCs. The top 10 enriched GO terms are displayed on the Y axis and adjusted P values on the X axis.
-
CGSEA analysis shows myogenic genes were not enriched in YY1iKO vs. Ctrl ASCs (P‐value = 0.11).
-
DGenomic snapshots showing the selected myogenic genes, Tnni2 and Actn2, in YY1iKO vs. Ctrl ASCs.
-
EList of up‐regulated mitochondrial genes in YY1iKO FISCs measured by RNAseq (in FPKM).
-
FRT–qPCR validation of the expression of selected mitochondrial genes up‐regulated in YY1iKO FISCs (n = 3 mice, each).
-
GGO analysis for the down‐regulated genes in YY1iKO vs. Ctrl FISCs.
-
HGO analysis of all genes with YY1 binding in their promoters. The top 10 enriched GO terms are displayed with relevant adjusted P values.
-
IVenn diagrams showing the overlapping of the YY1 ChIPseq from ASCs and C2C12 myoblasts. A total of 466 peaks were found in both datasets.
-
JGO analysis of genes associated with the above 466 peaks. The top 10 enriched GO terms are displayed on Y axis and adjusted P values on the X axis.
-
KEzh2 ChIPseq data from C2C12 myoblasts and YY1 ChIPseq data from ASCs were compared. Genomic snapshots showing the co‐binding of YY1 and Ezh2 on selected mitochondrial genes, Mrps15 and Mrps18a.
-
LCtrl or YY1iKO ASCs were infected with a pRlenti‐GFP control, pRlenti‐YY1(1–394), or pRlenti‐YY1(S365D) virus to overexpress the mutants of YY1. RT–qPCR detection showing the overexpressed YY1 mutant mRNAs (n = 3, each).
-
MRT–qPCR analysis of Pax7 expression in YY1iKO vs. Ctrl ASCs (n = 3 mice, each).
-
N, OASCs from Ctrl or YY1iKO were treated with SB202190 (SB) and analyzed for the expression of selected mitochondrial genes (n = 3 mice, each).
-
PYY1iKO ASCs were transfected with YY1 and treated with SB to further examine YY1‐dependent p38 repression on mitochondrial gene expression (n = 3 mice, each).
To further pinpoint whether YY1 represses the mitochondrial genes through direct binding and transcriptional regulation, genome‐wide chromatin immunoprecipitation coupled with massive parallel sequencing (ChIPseq) was performed on chromatins from ASCs isolated from C57B/L wild‐type mice, which led to the identification of a total of 2,031 binding peaks (Table EV3). Expectedly, these peaks distributing in intergenic, gene body, and promoter regions (Fig 5J) were enriched for a bona fide YY1 binding motif (Fig 5K). Gene Ontology (GO) analysis revealed terms such as “nucleoplasm” and “mitochondrial small ribosomal subunit” were prominent among genes with YY1 binding at promoters (Fig EV4H). When intersecting with the 979 genes up‐regulated from RNAseq, 90 were found to be likely direct targets of YY1 with binding in promoter regions (Fig 5L), whereas the majority of genes may be indirectly activated upon YY1 deletion. Strikingly, these genes were extremely enriched for mitochondrion/OXPHOS‐related terms, such as “respiratory chain”, “mitochondrial part”, and “electron transport chain”, but “cell cycle” or “cell division” related terms no longer appeared (Fig 5M; Table EV3). From both the ChIPseq (Fig 5N) and ChIP–qPCR validation (Fig 5O), these mitochondrial genes were bound by YY1 in their transcriptional start site (TSS) regions, suggesting a direct transcriptional regulation. Of note, the majority of the above YY1 ChIPseq peaks in ASCs did not overlap with those previously identified in C2C12 myoblasts (Fig EV4I), reflecting the intrinsic difference between the isolated satellite cells and the myoblast cell line. Nevertheless, the 466 peaks shared between the ASC and C2C12 datasets were associated with genes enriched high in “mitochondrion part” “mitochondria” (Fig EV4J), again pointing to YY1 as a key regulator of mitochondria function in myoblast cells. Altogether, our findings suggested that YY1 acts to bind and repress mitochondrial genes upon SC activation. Since it is known that polycomb repressive complex 2 (PRC2) can interact with YY1 to deposit H3K27me3 marks and repress multiple myogenic loci in myoblasts (Caretti et al, 2004; Wang et al, 2008), we asked whether this synergism also occurs to repress mitochondrial loci in SCs. Indeed, examining available Ezh2 ChIPseq data (Mousavi et al, 2012), we found that co‐binding of Ezh2 with YY1 is evident on a list of mitochondrial genes that were up‐regulated in YY1iKO cells (Fig EV4K). Further validation by ChIP–PCR using antibodies against Ezh2 or Suz12 confirmed the PRC2 binding on several mitochondrial genes (Fig 5P and Q) as well as the enrichment of H3K27me3 on the same loci (Fig 5R). In line with this, Ehz2 loss in mouse was shown to cause a defect in SC proliferation and muscle regeneration (Juan et al, 2011), phenocopying YY1iKO. Moreover, in order to substantiate that YY1 represses mitochondrial genes through its direct DNA‐binding activity, re‐expression of a wild‐type (WT) YY1 restored the elevated mitochondrial genes in iKO SCs, but a mutant YY1 (S365D) lacking DNA‐binding activity failed to do so (Alexander & Rizkallah, 2017); similarly, a second mutant, YY1 (1–394), lacking DNA‐binding domain also did not restore the mitochondrial expression in iKO SCs (Figs 5S and EV4L). Lastly, as YY1/PRC2 was previously reported to repress Pax7 expression, upon p38 activation (Palacios et al, 2010), we tested whether Pax7 expression is increased by YY1 loss and surprisingly found Pax7 was decreased in iKO vs. Ctrl SCs (Fig EV4M); this is probably because the YY1/PRC2 repression on Pax7 occurs upon myoblast fusion to myotube but not in actively proliferating myoblast cells (Palacios et al, 2010). In addition, treatment of ASCs with SB202190 (SB), a p38 inhibitor, led to up‐regulation of mitochondrial genes in Ctrl but not YY1iKO cells; overexpressing YY1 restored mitochondrial gene expression in iKO when the cells were treated with SB, altogether suggesting that YY1/PRC2‐mediated repression of mitochondrial genes is p38‐dependent (Fig EV4N–P).
YY1 loss leads to a defect in activating glycolytic pathway during SC activation via decreasing Hif1α protein
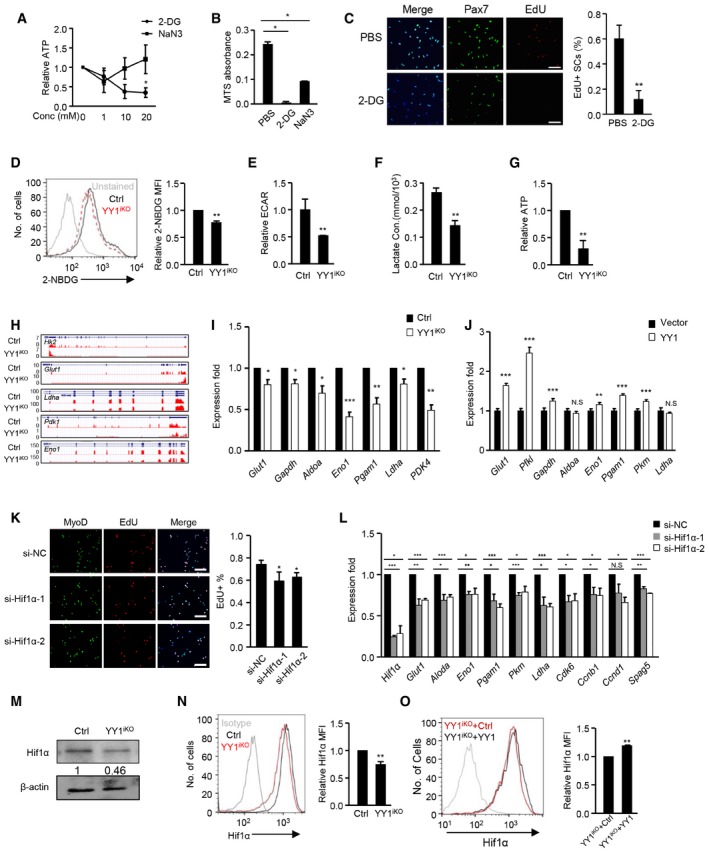
Altogether, the above genome‐wide interrogation revealed a key role of YY1 in repressing mitochondrial genes in ASCs, leading us to speculate that mitochondrial function needs to be inhibited during SCs activation and proliferation. Indeed, a role for the metabolic switch from fatty acid oxidation to aerobic glycolysis is thought to occur to support increased macromolecule biosynthesis during SC activation (Ryall et al, 2015). In line with this notion, we found that treatment of ASCs with a glycolytic inhibitor 2‐Deoxy‐D‐glucose (2‐DG) drastically reduced ATP production even at 1 mM concentration, whereas treatment with a respiration inhibitor sodium azide (NaN3) caused a decrease of ATP production at 1 mM but the level bounced back at higher concentrations (10 and 20 mM; Fig 6A). Consistently, 2‐DG treatment markedly inhibited SC proliferation (98% reduction) while NaN3 caused much milder effect (62% reduction) compared to PBS‐treated cells by MTS assay (Fig 6B); EdU labeling assay also confirmed the above result showing a 81% reduction in EdU+ SCs upon 2‐DG treatment (Fig 6C). The above hypersensitivity to the cytotoxicity of 2‐DG indicated ASCs are indeed more dependent on glycolysis than OXPHOS. We thus suspected that in addition to the elevated mitochondrial genes, the YY1iKO cells might also suffer from hindered activation of glycolytic metabolism; both defects contribute to the delayed activation/proliferation. Indeed, a reduced (22%) level of glucose uptaking was found in YY1iKO vs. Ctrl (Fig 6D). Also, measurement of the basal extracellular acidification rate (ECAR, a marker of glycolysis) by Seahorse extracellular flux bioanalyzer revealed a 48% decrease in glycolytic usage in YY1iKO cells (Fig 6E); this was further strengthened by a reduced (46%) level of lactate production (Fig 6F) as well as an evident decrease (70%) of ATP production (Fig 6G).
Figure 6. YY1 loss leads to a defect in glycolytic switch during SC activation via Hif1α destabilization.
-
AAn equal number of FACS‐isolated SCs from C57 mice were cultured for 24 h and treated with 0, 1, 10, or 20 mM 2‐DG (glycolytic inhibitor) or NaN3 (respiration inhibitor) for 3 h before measurement of ATP production. The relative ATP levels normalized to the 0 mM values are plotted. N = 3 mice.
-
BSCs from C57 mice were treated with 10 mM 2‐DG or 10 mM NaN3 for 36 h before measurement of proliferation rate by MTS assay (n = 3 mice, each).
-
CSCs from C57 mice were treated with 10 mM 2‐DG for 24 h and subject to EdU labeling for 6 h. The percentage of Pax7+/EdU+ cells over the total number of Pax7+ cells was quantified (n = 3 mice, each).
-
DCtrl and YY1iKO ASCs were stained with 60 μg/ml 2‐NBDG for 45 min, and the fluorescence intensity (MFI) of 2‐NBDG was measured by flow cytometry (n = 3 mice, each).
-
E–GAn equal number of Ctrl and YY1iKO cells were cultured for 36 h; basal extracellular acidification rate (ECAR) level (E), lactate concentration (F), and ATP production (G) were measured and normalized with cell numbers. N = 3 mice.
-
HGenomic snapshots depict RNAseq profiles of the selected glycolytic genes (Hk2, Glut1, Ldha, Pdk1, and Eno1) down‐regulated in YY1iKO vs. Ctrl ASCs.
-
IExpression of the selected glycolytic genes was quantified by RT–qPCR. Hsp90ab1 was used as the normalization control.
-
JRe‐expression of YY1 by transfecting YY1iKO ASCs with a YY1 WT expressing plasmid led to up‐regulation of glycolytic genes including Glut1, Pfkl, Gapdh, Eno1, Pgam1, and Pkm. Hsp90ab1 was used as the normalization control.
-
KFACS‐isolated SCs from Pax7‐nGFP mice were transfected with Hif1α or control siRNAs and EdU‐labeled for 5 h. The percentage of MyoD+EdU+ cells over the total number of MyoD+ cells was quantified. Scale bar = 100 μm. N = 3 mice.
-
LThe expression of glycolytic genes and cell cycle‐related genes was detected by RT–qPCR in the above transfected cells. β‐actin was used as the normalization control.
-
MHif1α protein level was detected by Western blotting in YY1iKO vs. Ctrl ASCs. β‐actin was used as the loading control.
-
NLeft: image of flow cytometric analysis of Hif1α protein or IgG control in YY1iKO vs. Ctrl ASCs. Right: relative mean fluorescence intensity (MFI) of Hif1α protein is shown. N = 3 mice.
-
OYY1iKO ASCs were transfected with empty YY1 WT viruses; the relative expression level of Hif1α protein was detected by the above described flow cytometric analysis N = 3 mice.
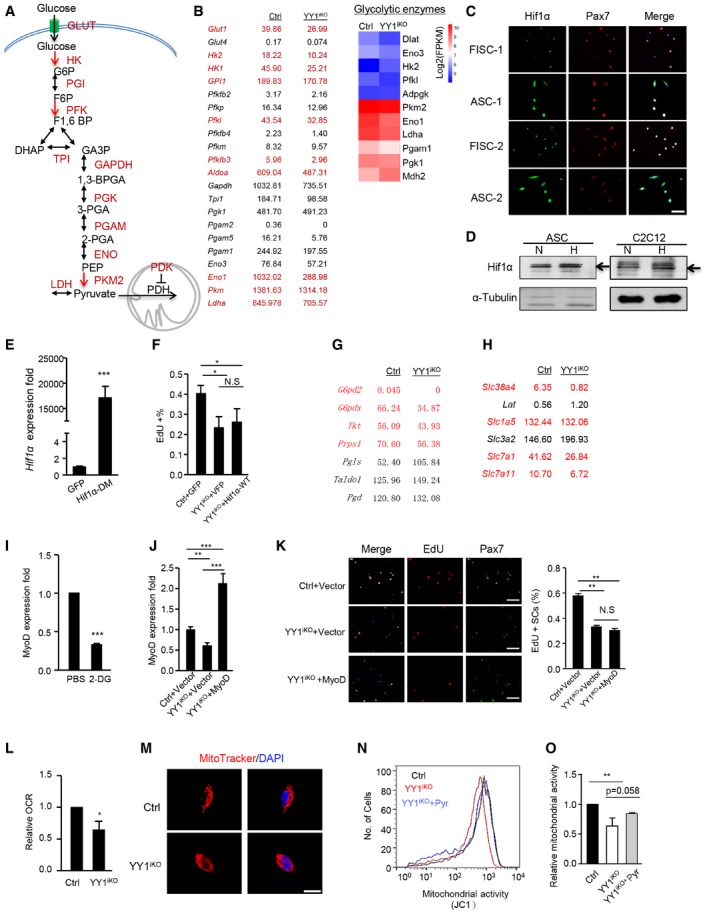
To answer what causes the reduced glycolysis in YY1iKO cells, analyses of RNAseq data revealed a striking down‐regulation of a panel of glycolytic genes even though glycolysis‐related terms were not enriched in GO analysis of the 924 down‐regulated genes; these genes encode glucose transporters and enzymes involved in virtually every step of glucose metabolism, including Glut1, Hk2, Eno1, Ldha, and Pdk1 (Figs 6H and I, and EV5A and B; Table EV1), suggesting YY1 may modulate the expression of these genes. Indeed, their expressions could be largely rescued by re‐expression of WT YY1 in YY1iKO cells (Fig 6J). Nevertheless, no direct binding of YY1 was identified on their TSS regions by ChIPseq (Table EV3). To search for plausible mechanisms explaining their down‐regulation, we noticed that the majority of these genes, including Glut1, Hk2, Gpi, Pfkl, Pfkfb3, Aldoa, Eno1, Pkm, Ldha, and Pdk1, are known trans‐activated targets of hypoxia‐inducible factor‐1 (Hif1α; Ito & Suda, 2014), raising an intriguing possibility that the compromised glycolysis in YY1iKO cells is mediated by Hif1α. Xie et al (2018) in fact recently demonstrated the possible induction of Hif1α in SC proliferation, which nevertheless needs to be confirmed and the exact mechanism behind its involvement in SC, remains unclear. Similar to YY1, an increase in Hif1α protein was observed in ASCs vs. FISCs (Fig EV5C), suggesting a potential promoting role during SC activation/proliferation. Expectedly, knock‐down of Hif1α reduced the EdU+ SCs (Fig 6K) and the expression of cell cycle genes as well as glycolytic genes (Fig 6L). Of note, despite the popular notion that hypoxia is a strong driver of Hif1α expression through its stabilizing effect, a steady level of Hif1α protein was detected in ASCs cultured in normoxic condition (21% O2; Fig 6M), which could be further stabilized in hypoxic condition (1% O2; Fig EV5D). Expectedly, Hif1α protein level was significantly decreased in YY1iKO cells vs. Ctrl by Western blotting (Fig 6M, 54%) and flow cytometric assay (Fig 6N, 26%), suggesting YY1 increases Hif1α protein levels in ASCs. Consistently, lentiviral re‐expression of YY1 in YY1iKO cells led to detectable restoration of Hif1α protein level by flow cytometric assay (Fig 6O).
Figure EV5. Hif1α mediates YY1 effect on glycolysis in ASCs.
-
AIllustration of the key enzymes on glycolytic pathway.
-
BLeft: expression of the enzymes or glucose transporters in Ctrl and YY1iKO ASCs measured by RNAseq (in FPKM). Highlighted are known Hif1α target genes. Right: heat maps indicating gene expression (Log2[FPKM]) of glycolytic enzymes in Ctrl and YY1iKO ASCs.
-
CIF staining for Pax7 (red) and Hif1α (green) on freshly isolated SCs or ASCs cultured for 48 h. An evident up‐regulation of Hif1α protein can be observed. Scale bar = 50 μm.
-
DWestern blot of Hif1α in ASCs or C2C12 myoblasts cultured under normoxic (N; 21% O2) or hypoxic (H; 1% O2) conditions. α‐Tubulin was used as a loading control. Black arrows indicate the location of Hif1α protein.
-
EYY1iKO ASCs were infected with pRlenti‐Hif1α (P402A/P564A; Hif1α‐DM) or a control pRlenti‐GFP virus to overexpress a constitutively active form of Hif1α. RT–qPCR detection of the overexpressed Hif1α DM mRNAs (n = 3, each).
-
FFACS‐sorted cells were infected with Ctrl or wt Hif1α and subjected to EdU labeling for 5.5 h. The percentage of MyoD+/EdU+ cells over the total number of MyoD+ cells was quantified (n = 3 mice, each).
-
G, HExpression levels of pentose cycle enzymes (G) and amino‐acid transporters (H) in Ctrl and YY1iKO ASCs measured by RNAseq (in FPKM). Highlighted are genes down‐regulated in YY1iKO ASCs.
-
ISCs from C57 mice were treated with 10 mM 2‐DG for 24 h. RT–qPCR detection of MyoD mRNAs was shown (n = 3 mice, each).
-
JSCs isolated from Ctrl and YY1iKO mice were transfected with a MyoD expressing or empty vector. RT–qPCR detection of the overexpressed MyoD mRNAs was shown (n = 3, each).
-
KThe above transfected SCs were labeled with EdU for 6 h. The percentage of Pax7+EdU+ cells over the total number of Pax7+ cells was quantified. Scale bar = 100 μm. N = 3 mice.
-
LAn equal number of Ctrl and YY1iKO cells were cultured for 36 h, and basal oxygen consumption rate (OCR) was measured and normalized with cell numbers (n = 3, each).
-
MMitoTracker staining of cultured SCs (48 h) isolated from Ctrl or YY1iKO mice to visualize the mitochondrial structures. Scale bar = 20 μm.
-
NCtrl, YY1iKO, or YY1iKO ASCs treated with 100 μM sodium pyruvate were stained with 2 μM 5,5′,6,6′‐tetrachloro‐1,1′,3,3′‐tetraethyl‐benzimidazolylcarbocyanine chloride (JC‐1) for 15 min to measure the mitochondrial membrane potential in living cells. The representative image of flow cytometry analyses is shown.
-
OQuantifications of the membrane potential levels are shown (n = 3, each).
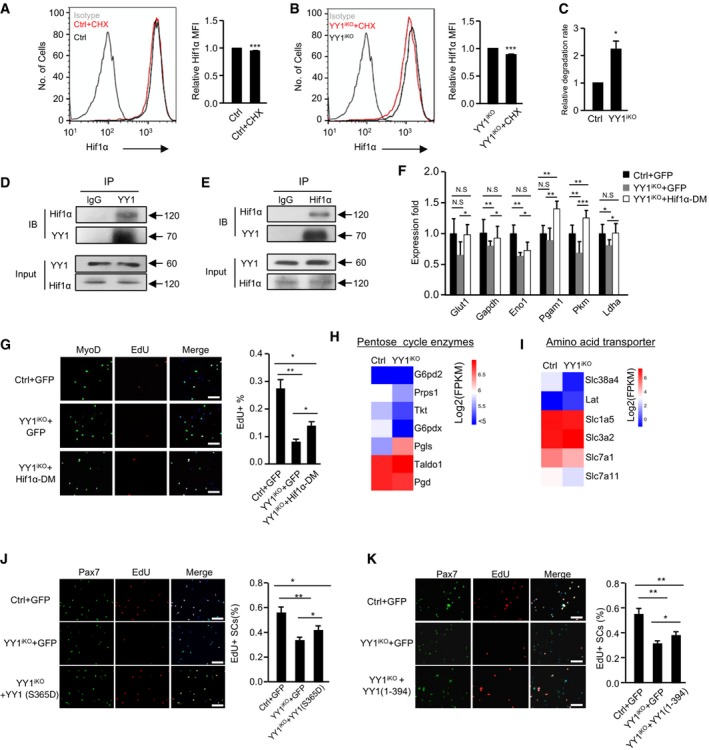
To answer how YY1 regulates Hif1α, since no direct binding of YY1 on the Hif1α promoter/enhancer was detected to suggest a transcriptional regulation, we considered the stabilizing effect of YY1 on Hif1α protein. Indeed, treatment with cycloheximide (CHX) indicated the degradation rate of Hif1α protein was much higher (2.23‐fold) in YY1iKO compared to Ctrl cells (Fig 7A–C). To further ask how YY1 stabilizes Hif1α protein, we noticed a prior study reported the physical interaction of both proteins in HCT116 cells (Wu et al, 2013). Indeed, by co‐immunoprecipitation (Co‐IP), a steady interaction was detected between endogenous YY1 and Hif1α proteins in C2C12 myoblasts in normoxic condition (Fig 7D and E), suggesting YY1 may stabilize Hif1α through physically interacting with it to prevent its degradation.
Figure 7. YY1 stabilizes Hif1α protein through direct interaction.
-
A, BCtrl or YY1iKO ASCs were treated with CHX (cycloheximide) for 90 min, and Hif1α protein was measured by flow cytometry. The numbers indicate the degradation rate after CHX treatment. N = 3 mice.
-
CThe relative degradation rate was calculated by the ratio of degradation rate in YY1iKO (11.14% ± 1.71%) vs. Ctrl (5.08% ± 0.796%). N = 3 mice.
-
DLysates from C2C12 myoblasts (GM) were subject to co‐immunoprecipitation (Co‐IP) assays with anti‐YY1 or anti‐IgG antibodies and blotted with anti‐Hif1α or anti‐YY1 antibodies.
-
EThe above lysates were also immunoprecipitated with anti‐Hif1α or anti‐IgG antibodies and blotted with anti‐YY1 or anti‐Hif1α antibodies.
-
FASCs from YY1iKO mice were infected with pRlenti‐Hif1α (P402A/P564A; Hif1α‐DM) viruses to overexpress a non‐degradable Hif1α protein or GFP‐expressing viruses. ASCs from Ctrl mice were also infected with the GFP viruses. Thirty‐six hours after infection, the expression of target glycolytic genes was detected by RT–qPCR. Hsp90ab1 was used as the normalization control.
-
GSCs were isolated from Ctrl or YY1iKO mice and infected with the above viruses. EdU labeling was then performed for 5.5 h. The percentage of MyoD+EdU+ cells over the total number of MyoD+ cells was quantified. Scale bar = 100 μm. N = 3 mice.
-
H, IHeat maps indicating gene expression levels (Log2[FPKM]) of pentose cycle enzymes (H) and amino acid transporter (I) in YY1iKO vs. Ctrl.
-
J, KYY1 (S365D) (J) or YY1 (1‐394) (K) mutant was expressed in YY1iKO ASCs and cells were then EdU‐labeled for 6 h. A GFP‐expressing plasmid was used as the negative control. The percentage of Pax7+/EdU+ cells over the total number of Pax7+ cells was quantified.
To further demonstrate the causative involvement of Hif1α in the phenotypes of YY1iKO, we found that overexpression of a constitutively active form of Hif1α, HIF1α (P402A/P564A; Esteban et al, 2006; with double mutations on two propyl residues, P402A and P564A, to prevent degradation, thus named Hif1α‐DM) in YY1iKO cells (Fig EV5E) fully rescued the abundance of glycolytic genes, including Glut1, Gapdh, Eno1, Pgam1, Pkm, and Ldha (Fig 7F). Consequently, cell proliferation was also rescued by EdU labeling assay (Fig 7G). Supportively, overexpression of a wild‐type Hif1α that could not be stabilized failed to rescue the proliferative defect in YY1iKO cells (Fig EV5F). Altogether, the above results thus suggested that Hif1α protein destabilization upon YY1 loss might lead to comprised glycolysis, which partially mediates the proliferation defect seen in YY1iKO cells. As suggested by a prior report (Ryall et al, 2015), the glycolytic pathway is central to cell proliferation because of its ability to provide building blocks for macromolecule synthesis. Consistently, when examining the RNAseq data closely, we indeed found some pentose cycle enzymes key to nucleotide synthesis were down‐regulated (Figs 7H and EV5G). In addition, several genes regulating amino‐acid transporters and key to protein synthesis were also down‐regulated (Figs 7I and EV5H). To connect the glycolysis with MyoD function, interestingly, we found treatment of 2DG significantly lowered MyoD level (Fig EV5I), indicating glycolysis controls MyoD level. However, re‐expression of MyoD in iKO cells failed to rescue the defective proliferation (Fig EV5J and K), suggesting MyoD alone does not mediate YY1 and glycolytic control of cell activation. Altogether, our findings led us to believe that during SCs activation YY1 orchestrates metabolic rewiring through up‐regulating Hif1α which activates many glycolytic genes and simultaneously repressing mitochondrial genes transcriptionally; the dual functions allow it to contribute to the full expansion of SCs. In line with this idea, we found the two YY1 mutant constructs lacking transcriptionally activities but possessing protein interacting domain only partially rescued the defective proliferation of YY1iKO SCs (Fig 7J and K).
Lastly, it is interesting to mention that despite the marked increase in many mitochondrial genes, measurement of the basal oxygen consumption rate (OCR, an indicator of mitochondrial oxidative activity) by the Seahorse surprisingly revealed a 36% reduction in YY1iKO vs. Ctrl ASCs (Fig EV5L). Consistently, MitoTracker labeling of mitochondria in the fixed YY1iKO ASCs showed reduced formation of tubular structures characterizing mature mitochondria (Fig EV5M). Quantitative measurement of the mitochondrial membrane potential by staining with JC‐1 in living cells also showed a mild (37%) but reproducible decrease (Fig EV5N and O). It is possible that the compromised OXPHOS in YY1iKO cells probably resulted from a decreased substrate (pyruvate) flow into mitochondria as a consequence of glycolytic suppression, thus reducing respirational rate despite the elevated mitochondrial gene levels. In line with this notion, we found treatment of YY1iKO cells with sodium pyruvate could partially rescue the decreased mitochondrial activity (Fig EV5N and O).
Discussion
In this study, we elucidated YY1 function in injury‐induced muscle regeneration as well as embryonic muscle development via the use of multiple genetic mouse models. The phenotypic characterization uncovered that ablation of YY1 profoundly impacts the process of regeneration in both acute and chronic injuries as well as the embryonic muscle formation, underscoring the key functions of YY1 in regulating both skeletal muscle regeneration and muscle development. Loss of YY1 caused an intrinsic delay in SC activation and proliferation, which contributes to the severely disrupted regenerative ability in the mutant mouse. Further genome‐wide interrogation led to the revelation of YY1 as a crucial factor in controlling metabolic reprogramming during SC proliferation through direct repression of mitochondrial genes transcriptionally and simultaneously enhancing glycolytic pathway via Hif1α stabilization (Fig 8).
Figure 8. Schematic model of the role of YY1 during adult muscle regeneration.
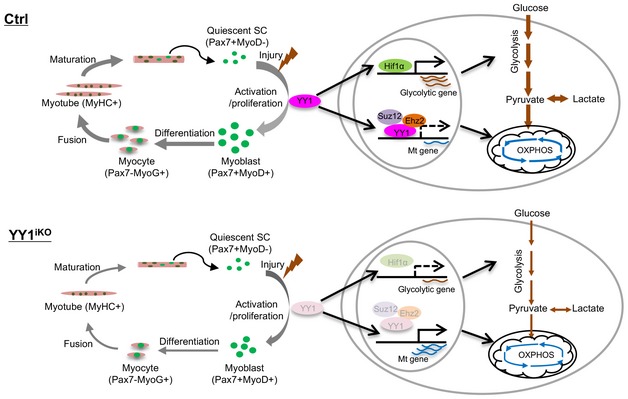
YY1 controls the activation of SCs by acting as a switch of metabolic reprogramming. In Ctrl SCs, its expression is increased during SC activation upon injury‐induced muscle damage and it transcriptionally suppresses mitochondrial gene expression through direct binding to their TSS regions together with PRC2 repressive complex. Meanwhile, it also stabilizes Hif1α protein to facilitate its trans‐activating of glycolytic genes. As a result, successful activation of glycolytic pathways occurs, allowing SCs to activate and proliferation to drive the repairing of the damaged muscle. When YY1 is deleted in the YY1iKO mouse, Hif1α level is decreased due to protein destabilization, leading to a reduction in glycolytic gene expression and glycolytic rate. Meanwhile, although the expression of mitochondrial genes is up‐regulated, OXPHOS could not be increased possibly because of reduced flow of pyruvate substrate into mitochondria. As a result of the inhibited metabolic reprogramming, SC activation is repressed and the regeneration is compromised. Quiescent SCs are defined by expression of Pax7 and lack of MyoD expression (Pax7+MyoD−), activated and proliferating myoblasts by the concomitant expression of Pax7 and MyoD (Pax7+MyoD+), differentiating myocytes by expression of MyoG (Pax7−MyoG+), and differentiated myotubes are marked by expression of Myosin heavy chain (MyHC). Error bars represent SD's of the mean. Student's t‐test (two‐sided): *P < 0.05, **P < 0.01, ***P < 0.001.
As a TF discovered more than two decades ago, roles of YY1 have been studied in many cellular processes (Gordon et al, 2006; He & Casaccia‐Bonnefil, 2008). Here, we for the first time illuminated its function in regulating adult satellite cell activity in the process of in vivo muscle regeneration. The iKO cells in culture displayed an evident reduction in their proliferative rate; the activation step also appeared to be inhibited (Fig 4H–J), underscoring its key function in the very early steps of SC activities. Nonetheless, the lineage progression of FISCs grown in culture did not seem to be blocked as the percentage of Pax7+MyoD+ cells did not differ in Ctrl vs. iKO cells; by 48 h, almost all iKO SCs were MyoD+ (Fig EV3G). When analyzing the in vivo regenerative process, YY1iKO mutant mouse, on the other hand, suffered from severe block of regenerative process upon acute injury, suggesting that in addition to the intrinsic delay of SC pool expansion, YY1 deletion may have caused other defects, for example, in SC cross‐talking with the niche environment; this warrants further investigation in the future.
To probe into the mechanisms underlying the delayed proliferation in YY1iKO cells, we have uncovered a role for YY1 in regulating dual pathways in metabolism. First, mitochondrial gene loci are directly bound and repressed by YY1 genes in a complex with PRC2; this action of YY1 is thus dependent on its transcriptional function, and the mutants lacking DNA‐binding activity could not restore the mitochondrial expression in iKO cells (Fig 5S). Notably, several prior studies have shed light on the important regulatory roles of YY1 in mitochondrial function and oxidative phosphorylation in various cells. For example, Blattler et al (2012) demonstrated that skeletal muscle tissue‐specific YY1 KO mice have severely defective mitochondria morphology and oxidative function associated with exercise intolerance. Perekatt et al (2014) also disclosed that YY1 is essential for intestinal stem cell renewal; loss of YY1 promotes the intestinal stem cells to exist from their niche and proliferation of the cells is unaffected. Very interestingly, in contrast to our findings, YY1 was found to be an activating factor for mitochondrial genes and functions in these studies. For example, in intestinal stem cells YY1 binds to mitochondrial complex I genes and is required for their expression; mitochondrial structure and function are compromised upon YY1 loss (Perekatt et al, 2014). These rather opposing findings suggest that YY1 binding to mitochondrial genes can cause pleiotropic regulatory effects depending on the cellular context. Considering the ubiquitous expression of YY1 in many types of stem cells, we suspect it can act as a key TF regulating mitochondrial function in linking stem cell metabolism to their lineage progression.
In addition to directly controlling the expression of mitochondrial genes, we for the first time showed that YY1 could also modulate glycolytic pathway through stabilizing Hif1α protein, thus underpinning YY1 as a metabolic controller with dual functions simultaneously modulating two main bioenergetic pathways to facilitate the proliferative growth of SCs. Hif1α is emerging as a key regulator of glycolysis via direct trans‐activation of many glycolytic genes (Ito & Suda, 2014); here, we for the first elucidated its functionality and mechanism in regulating SC metabolism and extended findings from two prior reports (Yang et al, 2017; Xie et al, 2018). In the study by Yang et al, when both Hif1α and Hif2α genes were deleted by Pax7CreER, an obvious delay was observed during CTX injury‐induced muscle regeneration (Fig 4); nevertheless, no single mutant was made to further investigate their separate roles. In the more recent report by Xie et al studying Hif2α functionality, overexpression of Hif1α in SCs was shown to promote cell proliferation although no further mechanistic insight was provided. We showed that YY1 regulation of Hif1α most likely occurs through the physical interaction of both proteins, which is independent of its transcriptional activity (Fig 7D and E). It is also interesting to point out that our dissection of YY1‐Hif1α connection in SCs was mainly conducted in a normoxic condition, leading to the unexpected finding that YY1 can stabilize Hif1α protein in normoxia. In contrast to the common belief that Hif1α function is dependent on hypoxia, the normoxic Hif1α protein appears to exert an important function in regulating SC activation. During the course of the preparation of this manuscript, Wu et al (2018) recently reported that YY1 alters tumor cell metabolism by activating glucose‐6‐phosphate dehydrogenase (G6PD), which is in line with our finding; nonetheless, the YY1 activation of G6PD was shown to be directly at transcriptional level, distinct from the mechanism presented in our study.
Taken together, our study demonstrates that YY1 plays dual roles in suppressing mitochondrial genes and increasing Hif1α to facilitate a smooth activation of glycolytic pathway that is needed for SC activation/proliferation. Interestingly, although its function in the proliferative stage appears to repress mitochondrial activation and to enhance glycolysis, it becomes an activator of mitochondrial genes and oxidative phosphorylation in the differentiation stage according to Blattler et al (2012). This is reminiscent of a recent report showing YY1 activates genes involved in mitochondrial bioenergetics in B cells (Kleiman et al, 2016). This duality of YY1 function thus renders YY1 a key regulator of metabolic remodeling driving the entire SCs lineage progression from activation to differentiation. This seemingly opposite regulatory abilities on mitochondrial genes stem from its competence to form distinct co‐repressive or co‐activating complexes at different stages. Specifically, we found that YY1 and PRC2 synergistically repress mitochondrial loci in ASCs, while PGC1α was shown to be recruited by YY1 to increase mitochondrial genes in skeletal muscle tissue (Cunningham et al, 2007). To summarize, our findings demonstrated that this metabolic reprogramming orchestrated by YY1 plays a direct role in controlling stem cell fate. Better deciphering the interplay between bioenergy and stem cell differentiation should improve our ability to manipulate stem cells in vitro, which is of great interest for the development of regenerative medicine (Chen & Chan, 2017; Schell et al, 2017).
Materials and Methods
Animal studies
Pax7Cre (Keller et al, 2004), Pax7CreERT2/+ (Lepper et al, 2009), and Tg: Pax7‐nGFP mouse strains (Sambasivan et al, 2009) were kindly provided by Dr. Shahragim Tajbakhsh. YY1f/f (Affar el et al, 2006) and C57BL/10 ScSn DMDmdx (mdx) mouse strains were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). The YY1 conditional KO (YY1cKO) strain (Ctrl: Pax7+/+; YY1f/+, YY1cKO: Pax7Cre/+; YY1f/f) was generated by crossing Pax7Cre mice with YY1f/f mice. The YY1‐inducible conditional KO (YY1iKO) strain (Ctrl: Pax7CreERT2/+; YY1+/+, YYiKO: Pax7CreERT2/+; YY1f/fmice) was generated by crossing Pax7CreERT2/+ mice with YY1f/fmice. The YY1/mdx double KO (YY1dKO) strain (Ctrl: Pax7CreERT2/+; YY1+/+; Mdx, YY1dKO: Pax7CreERT2/+; YY1f/f; Mdx) was generated by crossing YY1iKO with mdx mice. Primers used for genotyping are shown in Table EV4. Inducible conditional deletion of YY1 was administered by tamoxifen (TM; T5648, Sigma) intraperitoneally (IP) at 2 mg per 20 g body weight. For cardiotoxin (CTX) studies, approximately 2‐month‐old mice were injected with 50 μl of 10 μg/ml CTX (Latoxan, France) solution into TA muscles. Muscles were harvested at designated time points for further analysis. All animal handling procedures and protocols were approved by the Animal Ethics Committee at Chinese University of Hong Kong (CUHK). For EdU incorporation assay in vivo, 2 days after CTX injection, EdU injection via intraperitoneally (IP) at 0.25 mg per 20 g body weight was performed, followed by FACS isolation of SCs 12 h later. Cells were then collected and fixed with 4% PFA. EdU‐labeled cells were visualized using “click” chemistry with an Alexa Fluor® 594‐conjugated azide. Pictures were captured with a fluorescence microscope (Leica).
In vitro muscle functional test
The test was performed according to previous method (Zhang et al, 2016). Dissected EDL/SOL muscles with intact tendons were mounted into warmed 95%O2/5%CO2‐bubbled Krebs solution. One tendon was attached to a force transducer (In Vitro Muscle Test System 1200, Aurora Scientific Inc., Aurora, ON, Canada); the other end was tied to a hook connected to the lever arm of a position feedback motor. The optimum length (L0) was adjusted based on the maximal twitch force. Muscles were stimulated with supramaximal intensity at 100 Hz for 400 ms for tetanic force measurements. Specific tetanic force values were calculated by peak tetanic force normalized to the muscle physiological CSAs (PCSAs).
Cell culture
Mouse C2C12 myoblast cells (CRL‐1772) and 293 T cells were obtained from ATCC and cultured in DMEM medium (12800‐017, Gibco) with 10% fetal bovine serum (10270‐106, Gibco), penicillin/streptomycin (1×; 15140‐122, Gibco) at 37°C in 5% CO2 incubator. SCs were cultured in F10 medium (F0723, Merck Millipore) with 20% fetal bovine serum, penicillin/streptomycin (1×), and 2.5 ng/ml basic fibroblast growth factor (13256, Life Technologies). To induce SCs differentiation, cells were switched to F10 medium with 2% horse serum (126050088, Invitrogen). To inhibit protein degradation, SCs were treated with 6 μM MG132 (M8699, Sigma) for 2 h. To inhibit protein synthesis, SCs were treated with 100 μg/ml cycloheximide for 90 min. To provide alternate carbon substrate to YY1iKO cells, SCs were treated with 100 μM sodium pyruvate (11360070, Thermo Fisher) for 24 h. For hypoxic treatment, cells were exposed to hypoxia in vitro in New Brunswick™ Galaxy® 48 S incubator (MG Scientific, USA) with 1% oxygen for indicated time. Transient transfection of plasmids or siRNA was conducted with Lipofectamine 2000 (11668‐019, Invitrogen) following manufacturer's protocol. For viral infection, pRlenti‐GFP or pRlenti‐Hif1α (P402A/P564A; Hif1α double mutant or Hif1α‐DM), pSL5 or pSL5/YY1 along with the packaging plasmids was transfected into HEK293T cells. Forty‐eight hours after transfection, culture supernatant was collected and used for infecting SCs. To investigate the effect of p38 signaling on mitochondrial genes, cells were treated with 10 μM SB202190 (S7076, Sigma) for 24 h (Wang et al, 2018).
FACS isolation of SCs
SCs were sorted based on established methods (Cheung et al, 2012). Briefly, hindlimb muscles from mice were digested with collagenase II (800 units/ml) for 90 min at 37°C, and then digested muscles were triturated and washed in washing medium [Hams F‐10 media (Gibco), 10% HIHS (Gibco), penicillin/streptomycin (1×, Gibco)] before SCs were liberated by treating with collagenase II (800 units/ml)/dispase (11 unit/ml) for 30 min. Mononuclear cells were filtered with a 45‐mm cell strainer and incubated with primary antibodies (Vcam1‐biotin, CD31‐fluorescein isothiocyanate [FITC], CD45‐FITC, and Sca1‐Alxa647). The Vcam1 signal was amplified with streptavidin‐PE‐cy7. All antibodies were used at a dilution of 1:75. The BD FACSAria IV cell sorter (BD Biosciences) was used for SC sorting following the manufacturer's instructions.
Myofiber isolation
As described before (Zhou et al, 2015; Chen et al, 2017), extensor digitorum longus (EDL) muscles were dissected and digested with Collagenase II (800 units/ml) in DMEM media (Gibco) at 37°C for 75 min. Single myofibers were released by gentle trituration with washing medium (mentioned above). Fibers were cultured in Ham's F10 medium with 10% HS and P/S (1×) or Ham's F10 medium supplemented with 10% FBS and 2.5 ng/ml basic fibroblast growth factor at designated time points. Fixation was carried out in 4% paraformaldehyde at room temp for 15 min.
Cell proliferation assay
EdU assay was conducted following manufacturer's instructions (C10339, Thermo Scientific). Growing cells on coverslips were incubated in culture medium with 10 μM EdU for designated times. Cells were then washed for three times in PBS and fixed with 4% PFA for 20 min. EdU‐labeled cells were visualized using “click” chemistry with an Alexa Fluor® 594‐conjugated azide. Pictures were captured with a fluorescence microscope (Leica). MTS assay was conducted using CellTiter 96® Aqueous One Solution Reagent (Promega, Madison, WI) following the manufacturer's instructions. Briefly, cells were seeded at a density of 10 × 103 cells/well (SCs) in a 96‐well plate and cultured for 12 h, followed by treatment of 10 mM 2‐DG/NaN3 for 36 h. The proliferation was measured with absorbance at 490 nm with background subtraction at 650 nm.
TUNEL assay
TUNEL assays to monitor apoptosis were carried out with the In Situ Cell Death Detection Kit (Roche) according to the manufacturer's protocol. Briefly, freshly isolated cells on coverslips were cultured for 36 h, followed by washing twice in PBS and fixing with 4% PFA for 15 min. TUNEL staining was carried out by adding reaction mixture of label solution and enzyme solution for 60 min at dark. Fluorescent images were captured with a fluorescence microscope (Leica).
Cellular bioenergetics
Cellular bioenergetics was analyzed on a Seahorse extracellular flux bioanalyzer (XF24) based on previously described methods (Ryall et al, 2015). Briefly, cells were seeded at a density of 50 × 103 cells/well (SCs) in a Seahorse XF24 plate. Cells were cultured for 36 h in growth media. Immediately prior to the assay, cultured cells were washed twice in minimal assay media (Seahorse Biosciences, 37°C, pH 7.40) supplemented with 25 mM glucose and 1 mM sodium pyruvate. All cells were equilibrated in minimal assay media for 1 h in a non‐CO2 incubator immediately prior to the assay. The basal rate of extracellular acidification rate (ECAR) was measured in triplicate for 2 min (over a total of 15 min) from the rate of decline in O2 partial pressure (OCR) and rate of change in assay pH (ECAR).
Lactate assay
The lactate level in cell supernatant was measured using the Lactate Assay Kit (1200011002, Eton Bioscience, USA) according to the manufacturer's instructions. Briefly, cells were seeded at a density of 20 × 103 cells/well (SCs) in a 96‐well plate and cultured for 40 h, and the cell culture supernatant was then collected and mixed with L‐Lactate assay solution for 30 min at 37°C incubator. The concentration of L‐Lactate was measured with an absorbance at 490 nm.
Measurement of glucose uptake
Briefly, cells were seeded at a density of 20 × 104 cells/well (SCs) in a 12‐well plate and cultured for 36 h. Cells were then rinsed twice with NO‐glucose medium with 20% FBS and incubated with 60 μg/ml 2‐NBDG for 45 min. Trypsinized cells were then analyzed by flow cytometer BD FACSAria Fusion (BD Biosciences). Raw data were analyzed by FlowJo software.
MitoTracker fluorescence staining
MitoTracker®Red CMXRos (M7512, Invitrogen) was added into growth media of SCs at a final concentration of 10 nM for 3 min, followed by washing with PBS and fixing the cells with 4% paraformaldehyde. Images were visualized by 100× oil lens of a fluorescence microscopy (Leica).
Measurement of mitochondrial membrane potential
Briefly, SCs were seeded at a density of 20 × 104 cells/well in a 12‐well plate and cultured for 36 h. Trypsinized cells were stained with 2 μM 5,5′,6,6′‐tetrachloro‐1,1′,3,3′‐tetraethyl‐benzimidazolylcarbocyanine chloride (JC‐1; Invitrogen) and analyzed by flow cytometer FACSCanto II Analyzer (BD Biosciences). Raw data were analyzed by FlowJo software.
ATP measurement
ATP concentrations in cells were determined luminometrically using an ApoSENSOR™ Cell Viability Assay Kit (K254, Biovision, USA) according to the manufacturer's instructions. Briefly, cells were seeded at a density of 10 × 103 cells/well (SCs) in a 96‐well plate and cultured for 36 h. Cells were lysed with Nuclear Releasing Buffer for 5 min; 10 μl ATP Monitoring Enzyme was added to the cell lysate, and the concentration of ATP was measured with a luminometer (PerkinElmer).
Plasmids
The full‐length YY1 expression plasmid was described before (Wang et al, 2008). The full‐length Myod expression plasmid was described before (Skapek et al, 1996). pRlenti‐GFP or pRlenti‐Hif1α (P402A/P564A; carrying the following substitutions: P402A and P564A) was a kind gift from Prof. Miguel Esteban (GIBH, Guangzhou, China; Esteban et al, 2006). pSL5 or pSL5/YY1 was a kind gift from Prof. Guangchao Sui (NEFU, Harbin, China; Wan et al, 2012). pRlenti‐YY1 (1–394) and pRlenti‐YY1 (S365D) expressing vector were constructed by PCR amplification from pCEP4F‐YY1(1–399) or pCS2(+)Flag‐YY1(S365D), following by T–A cloning into pRlenti‐GFP using BamH I and EcoR I sites.
Real‐time PCR
Total RNAs from satellite cells were extracted using TRIzol reagent (Life Technologies) according to the manufacturer's protocol. Real‐time PCR was performed by using SYBR Green Master Mix (Applied Biosystem) on ABI PRISM 7900HT (Applied Biosystem). HSP90 was used for normalization. All the experiments were designed in triplicates. Primers used for RT–qPCR are shown in Table EV4.
Immunoblotting, immunofluorescence, and immunohistochemistry
For Western blot assays, in vitro cultured cells were harvested, washed with ice‐cold PBS, and lysed in cell lysis buffer. Whole‐cell lysates were subjected to SDS–PAGE, and protein expression was visualized using an enhanced chemiluminescence detection system (GE Healthcare, Little Chalfont, UK) as described before (Wang et al, 2007). The following dilutions were used for each antibody: YY1 (Santa Cruz Biotechnology; 1:1,000), α‐tubulin (Sigma; 1:5,000), Hif1α (Abcam; 1:1,000), β‐actin (ImmunoWay; 1:5,000), and OXPHOS Rodent WB Antibody Cocktail (Abcam, 1:250). For immunofluorescence staining, cultured cells and myofibers were fixed in 4% PFA for 15 min and blocked with 3% BSA within 1 h. Primary antibodies were applied to samples with indicated dilution below, and the samples were kept at 4°C overnight. For Pax7 staining on frozen muscle sections, an antigen retrieval step was performed before blocking through boiling samples in 0.01 M citric acid (pH 6.0) for 5 min in a microwave. After 4% BBBSA (4% IgG‐free BSA in PBS; Jackson, ref: 001‐000‐162) blocking, the sections were further blocked with the donkey anti‐mouse IgG (H + L; 1/100 in PBS; Jackson, ref: 115‐007‐003) for 30 min. The biotin‐conjugated anti‐mouse IgG (1:500 in 4% BBBSA, Jackson, ref: 115‐065‐205) and Cy3‐Streptavidin (1:1,250 in 4% BBBSA, Jackson, ref: 016‐160‐084) were used as secondary antibodies. H&E staining on frozen muscle sections was performed as previously described (Diao et al, 2012). Masson's trichrome staining was performed according to the manufacturer's instructions (ScyTek Laboratories, Logan, UT). All fluorescent images were captured with a fluorescence microscope (Leica). Primary antibodies used include the following: YY1 (1:100) for staining of isolated SCs (Santa Cruz Biotechnology, Inc), MyoD (1:100; Santa Cruz Biotechnology, Inc), and myogenin (1:200; Santa Cruz Biotechnology, Inc); Pax7 (1:100; Developmental Studies Hybridoma Bank) and MF20 (1:50; Developmental Studies Hybridoma Bank); troponin (1:200; Sigma‐Aldrich), MyHC (1:200; Sigma‐Aldrich), and laminin (1:800; Sigma‐Aldrich); Ki67 (1:200; Thermo); collagen I (1:200; Southern Biotech); MyoD (1:200; Dako); and Hif1α (1:200; Abcam) and YY1 (1:200; Abcam) for staining of muscle cryosections.
Flow cytometric analysis of protein levels
SCs were collected by incubation with 0.5% trypsin, fixed by using 4% PFA for 15 min at RT, and then permeabilized with treatment of 90% ice‐cold methanol. After blocking with 1% BSA for 30 min at RT, cells were stained with a rabbit anti‐Hif1α (Abcam, ab2185) or a rabbit anti‐YY1 (Santa Cruz Biotechnology, sc‐1703) or anti‐rabbit IgG control (Jackson ImmunoResearch Laboratories) for 45 min at 4°C. Cells were then stained with Alexa Fluor® 594‐conjugated goat anti‐rabbit secondary antibody (Thermo Fisher Scientific, R37117) for 15 min at 4°C and analyzed by flow cytometer BD FACSAria Fusion (BD Biosciences). Raw data were analyzed by FlowJo software.
Co‐immunoprecipitation assay
C2C12 cells were cross‐linked with 200 μg/ml of 3,3′‐Dithiodipropionic acid di(N‐hydroxysuccinimide ester) (Sigma) for 15 min and then harvested and lysed in RIPA buffer (25 mM HEPES pH 7.4, 1% nonidet P‐40, 0.1% SDS, 0.5% sodium deoxycholate, 1 × PIC). 5 μg of antibodies against YY1 (Santa Cruz Biotechnology, Cat# SC‐1703, rabbit polyclonal), Hif1α (Santa Cruz Biotechnology, Cat# SC‐10790, rabbit polyclonal), or normal mouse IgG (Santa Cruz Biotechnology, Cat# SC‐2025) was incubated with above cell lysate overnight at 4°C with rotation. After extensive washing with RIPA buffer, the bound proteins were eluted by boiling in 50 μl of 1 × loading buffer (125 mM Tris–HCl pH 6.8, with 4% SDS, 20% (v/v) glycerol) and subjected to Western blotting analysis.
ChIP assays
Ten million SCs were cultured for 36 h and subsequently cross‐linked in 1% formaldehyde and processed according to previously described (Lu et al, 2012; Zhou et al, 2012b; Peng et al, 2017). In all, 5 μg of antibodies against YY1 (Santa Cruz Biotechnology, Cat# SC‐1703, rabbit polyclonal), Ezh2 (Active Motif, Cat# 61642, rabbit polyclonal), Suz12 (Abcam, Cat# 12073, rabbit polyclonal), H3K27me3 (Millipore, Cat# 07‐449, rabbit polyclonal), or normal mouse IgG (Santa Cruz Biotechnology, Cat# SC‐2025) was used for one immunoprecipitation. Immunoprecipitated genomic DNA was resuspended in 20 μl of water. PCRs were performed with 1 μl of immunoprecipitated material as a template and products were analyzed by qRT–PCR on a 7900HT system (Life Technologies). Relative enrichment is calculated as the amount of amplified DNA from ChIP group relative to values from IgG group. Primers used for ChIP–qPCR are shown in Table EV4.
ChIP sequencing and data analysis
For library construction, we used a protocol as described before (Lu et al, 2013; Zhou et al, 2015; Peng et al, 2017). Briefly, the purified DNA (100 ng) was end‐repaired, and A‐nucleotide overhangs were added by incubation with the Taq Klenow fragment lacking exonuclease activity. After the attachment of anchor sequences, fragments were PCR‐amplified using Illumina‐supplied primers. The purified DNA library products were evaluated using Bioanalyzer (Agilent) and SYBR qPCR and diluted to 10 nM for sequencing on Illumina HiSeq 2000 sequencer (YY1; pair‐end with 50 bp). A data analysis pipeline CASAVA 1.8 (Illumina) was employed to perform the initial bioinformatics analysis including base calling and converting the results into raw reads in FASTQ format. For peak defining, the sequenced reads were mapped to the mouse reference genome (mm9) using bowtie2 (Langmead & Salzberg, 2012). The alignment was performed, and only the uniquely aligned reads were kept. The protein DNA‐binding peaks (sites) were identified using model‐based analysis for ChIPseq (MACS, version 2.1.1; Zhang et al, 2008) with input sample as the background. During the peak calling, the P‐value cutoff was set to under 0.0001 for YY1 ChIPseq experiment.
RNAseq and data analysis
For library construction, we used a protocol as described before (Lu et al, 2013; Zhou et al, 2015). The purified library products were evaluated using a Bioanalyzer (Agilent) and SYBR qPCR and sequenced on an Illumina HiSeq 2000 sequencer (pair‐end with 50 bp). Sequenced fragments were mapped to reference mouse genome (mm9) using TopHat2 (Kim et al, 2013). Cufflinks (Trapnell et al, 2012) were then used to estimate the relative abundance of transcripts in RNAseq experiments. Abundances were reported in fragments per kilobase per million (FPKM), which is conceptually analogous to the reads per kilobase per million (RPKM) used for single‐end RNAseq. Differentially expressed genes were identified if the fold change ≥ 2 by comparing YY1iKO and Ctrl samples.
Statistical test
The statistical significance was assessed by the Student's t‐test (two‐sided). *P < 0.05, **P < 0.01, ***P < 0.001 and n.s.: not significant (P ≥ 0.05).
Author contributions
HW and HS conceived the projects, designed the experiments, and wrote the manuscript. FC conducted most of the experiments. JZ analyzed RNAseq and ChIPseq data. YL, YC, LW, ZZ, CCW, and BZ helped to perform some of the mouse assays. CC‐LW helped to perform assays of measuring mitochondrial membrane potential. JY provided help on the analysis of fiber size. YZ helped to review and edit the manuscript. ZW and THC provided suggestions on some satellite cell experiments.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
We thank Prof. Miguel Esteban for sharing the Hif1α(P402A/P564A) plasmid; Prof. Guangchao Sui for sharing pSL5/YY1 plasmid; Prof. Raed Rizkallah for sharing pCS2(+)Flag‐YY1(S365D) plasmid; Dr. James G. Ryall for advice on cellular bioenergetics; Prof. Gabrielle Kardon for their technical assistance on handling mouse embryos; Dr. Han Zhu for his suggestions on fluorescence‐activated cell sorting (FACS). We thank General Research Funds (GRF) from the Research Grants Council (RGC) of the Hong Kong Special Administrative Region (Project Code: 14100415, 14133016, 14106117, and 14100018 to H.W.; 14102315 and 14116918 to H.S.); CUHK Focused Innovations Scheme: Scheme B to H.S. (Project Code: 1907307); National Natural Science Foundation (NSFC) of China (Grant No: 31871304 to H.W.); NSFC/RGC Joint Research Scheme from RGC (Project Code: N_CUHK413/18 to H.S.). Direct Grant from CUHK (Project Number: 2017.049 to H.W. and 2017.009 to H.S.). This work was also supported by the Hong Kong Epigenomics Project (EpiHK).
The EMBO Journal (2019) 38: e99727
Contributor Information
Hao Sun, Email: haosun@cuhk.edu.hk.
Huating Wang, Email: huating.wang@cuhk.edu.hk.
Data availability
RNAseq and ChIPseq data used in this study have been deposited in Gene Expression Omnibus (GEO) database under the accession codes GSM2893681, GSM2893682, GSM2893683, GSM2893684, GSM2893685, and GSM2893686.
References
- Affar el B, Gay F, Shi Y, Liu H, Huarte M, Wu S, Collins T, Li E (2006) Essential dosage‐dependent functions of the transcription factor yin yang 1 in late embryonic development and cell cycle progression. Mol Cell Biol 26: 3565–3581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander KE, Rizkallah R (2017) Aurora A phosphorylation of YY1 during mitosis inactivates its DNA binding activity. Sci Rep 7: 10084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aziz A, Sebastian S, Dilworth FJ (2012) The origin and fate of muscle satellite cells. Stem Cell Rev 8: 609–622 [DOI] [PubMed] [Google Scholar]
- Babiuk RP, Zhang W, Clugston R, Allan DW, Greer JJ (2003) Embryological origins and development of the rat diaphragm. J Comp Neurol 455: 477–487 [DOI] [PubMed] [Google Scholar]
- Bentzinger CF, Wang YX, Rudnicki MA (2012) Building muscle: molecular regulation of myogenesis. Cold Spring Harb Perspect Biol 4: a008342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blattler SM, Verdeguer F, Liesa M, Cunningham JT, Vogel RO, Chim H, Liu H, Romanino K, Shirihai OS, Vazquez F, Ruegg MA, Shi Y, Puigserver P (2012) Defective mitochondrial morphology and bioenergetic function in mice lacking the transcription factor Yin Yang 1 in skeletal muscle. Mol Cell Biol 32: 3333–3346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borensztein M, Monnier P, Court F, Louault Y, Ripoche MA, Tiret L, Yao Z, Tapscott SJ, Forne T, Montarras D, Dandolo L (2013) Myod and H19‐Igf2 locus interactions are required for diaphragm formation in the mouse. Development 140: 1231–1239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckingham M (2007) Skeletal muscle progenitor cells and the role of Pax genes. C R Biol 330: 530–533 [DOI] [PubMed] [Google Scholar]
- Buckingham M, Relaix F (2007) The role of Pax genes in the development of tissues and organs: Pax3 and Pax7 regulate muscle progenitor cell functions. Annu Rev Cell Dev Biol 23: 645–673 [DOI] [PubMed] [Google Scholar]
- Bulfield G, Siller WG, Wight PA, Moore KJ (1984) X chromosome‐linked muscular dystrophy (mdx) in the mouse. Proc Natl Acad Sci USA 81: 1189–1192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caretti G, Di Padova M, Micales B, Lyons GE, Sartorelli V (2004) The Polycomb Ezh2 methyltransferase regulates muscle gene expression and skeletal muscle differentiation. Genes Dev 18: 2627–2638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Chan DC (2017) Mitochondrial dynamics in regulating the unique phenotypes of cancer and stem cells. Cell Metab 26: 39–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, He L, Zhao Y, Li Y, Zhang S, Sun K, So K, Chen F, Zhou L, Lu L, Wang L, Zhu X, Bao X, Esteban MA, Nakagawa S, Prasanth KV, Wu Z, Sun H, Wang H (2017) Malat1 regulates myogenic differentiation and muscle regeneration through modulating MyoD transcriptional activity. Cell Discov 3: 17002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung TH, Quach NL, Charville GW, Liu L, Park L, Edalati A, Yoo B, Hoang P, Rando TA (2012) Maintenance of muscle stem‐cell quiescence by microRNA‐489. Nature 482: 524–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, Puigserver P (2007) mTOR controls mitochondrial oxidative function through a YY1‐PGC‐1alpha transcriptional complex. Nature 450: 736–740 [DOI] [PubMed] [Google Scholar]
- Diao Y, Guo X, Li Y, Sun K, Lu L, Jiang L, Fu X, Zhu H, Sun H, Wang H, Wu Z (2012) Pax3/7BP is a Pax7‐ and Pax3‐binding protein that regulates the proliferation of muscle precursor cells by an epigenetic mechanism. Cell Stem Cell 11: 231–241 [DOI] [PubMed] [Google Scholar]
- Donohoe ME, Zhang X, McGinnis L, Biggers J, Li E, Shi Y (1999) Targeted disruption of mouse Yin Yang 1 transcription factor results in peri‐implantation lethality. Mol Cell Biol 19: 7237–7244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteban MA, Tran MG, Harten SK, Hill P, Castellanos MC, Chandra A, Raval R, O'Brien TS, Maxwell PH (2006) Regulation of E‐cadherin expression by VHL and hypoxia‐inducible factor. Cancer Res 66: 3567–3575 [DOI] [PubMed] [Google Scholar]
- Garcia‐Prat L, Sousa‐Victor P, Munoz‐Canoves P (2017) Proteostatic and metabolic control of stemness. Cell Stem Cell 20: 593–608 [DOI] [PubMed] [Google Scholar]
- Gibson GE, Thakkar A (2018) Mitochondria/metabolic reprogramming in the formation of neurons from peripheral cells: cause or consequence and the implications to their utility. Neurochem Int 117: 65–76 [DOI] [PubMed] [Google Scholar]
- Gordon S, Akopyan G, Garban H, Bonavida B (2006) Transcription factor YY1: structure, function, and therapeutic implications in cancer biology. Oncogene 25: 1125–1142 [DOI] [PubMed] [Google Scholar]
- He Y, Casaccia‐Bonnefil P (2008) The Yin and Yang of YY1 in the nervous system. J Neurochem 106: 1493–1502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutcheson DA, Zhao J, Merrell A, Haldar M, Kardon G (2009) Embryonic and fetal limb myogenic cells are derived from developmentally distinct progenitors and have different requirements for beta‐catenin. Genes Dev 23: 997–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Suda T (2014) Metabolic requirements for the maintenance of self‐renewing stem cells. Nat Rev Mol Cell Biol 15: 243–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juan AH, Derfoul A, Feng X, Ryall JG, Dell'Orso S, Pasut A, Zare H, Simone JM, Rudnicki MA, Sartorelli V (2011) Polycomb EZH2 controls self‐renewal and safeguards the transcriptional identity of skeletal muscle stem cells. Genes Dev 25: 789–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller C, Hansen MS, Coffin CM, Capecchi MR (2004) Pax3: Fkhr interferes with embryonic Pax3 and Pax7 function: implications for alveolar rhabdomyosarcoma cell of origin. Genes Dev 18: 2608–2613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiman E, Jia H, Loguercio S, Su AI, Feeney AJ (2016) YY1 plays an essential role at all stages of B‐cell differentiation. Proc Natl Acad Sci USA 113: E3911–E3920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL (2012) Fast gapped‐read alignment with Bowtie 2. Nat Methods 9: 357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepper C, Conway SJ, Fan CM (2009) Adult satellite cells and embryonic muscle progenitors have distinct genetic requirements. Nature 460: 627–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Zhou L, Chen EZ, Sun K, Jiang P, Wang L, Su X, Sun H, Wang H (2012) A Novel YY1‐miR‐1 regulatory circuit in skeletal myogenesis revealed by genome‐wide prediction of YY1‐miRNA network. PLoS One 7: e27596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Sun K, Chen X, Zhao Y, Wang L, Zhou L, Sun H, Wang H (2013) Genome‐wide survey by ChIP‐seq reveals YY1 regulation of lincRNAs in skeletal myogenesis. EMBO J 32: 2575–2588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrell AJ, Kardon G (2013) Development of the diaphragm – a skeletal muscle essential for mammalian respiration. FEBS J 280: 4026–4035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mousavi K, Zare H, Wang AH, Sartorelli V (2012) Polycomb protein Ezh1 promotes RNA polymerase II elongation. Mol Cell 45: 255–262 [DOI] [PubMed] [Google Scholar]
- Palacios D, Mozzetta C, Consalvi S, Caretti G, Saccone V, Proserpio V, Marquez VE, Valente S, Mai A, Forcales SV, Sartorelli V, Puri PL (2010) TNF/p38alpha/polycomb signaling to Pax7 locus in satellite cells links inflammation to the epigenetic control of muscle regeneration. Cell Stem Cell 7: 455–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng XL, So KK, He L, Zhao Y, Zhou J, Li Y, Yao M, Xu B, Zhang S, Yao H, Hu P, Sun H, Wang H (2017) MyoD‐ and FoxO3‐mediated hotspot interaction orchestrates super‐enhancer activity during myogenic differentiation. Nucleic Acids Res 45: 8785–8805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perekatt AO, Valdez MJ, Davila M, Hoffman A, Bonder EM, Gao N, Verzi MP (2014) YY1 is indispensable for Lgr5 + intestinal stem cell renewal. Proc Natl Acad Sci USA 111: 7695–7700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Relaix F, Zammit PS (2012) Satellite cells are essential for skeletal muscle regeneration: the cell on the edge returns centre stage. Development 139: 2845–2856 [DOI] [PubMed] [Google Scholar]
- Ryall JG, Dell'Orso S, Derfoul A, Juan A, Zare H, Feng X, Clermont D, Koulnis M, Gutierrez‐Cruz G, Fulco M, Sartorelli V (2015) The NAD(+)‐dependent SIRT1 deacetylase translates a metabolic switch into regulatory epigenetics in skeletal muscle stem cells. Cell Stem Cell 16: 171–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambasivan R, Gayraud‐Morel B, Dumas G, Cimper C, Paisant S, Kelly RG, Tajbakhsh S (2009) Distinct regulatory cascades govern extraocular and pharyngeal arch muscle progenitor cell fates. Dev Cell 16: 810–821 [DOI] [PubMed] [Google Scholar]
- Schell JC, Wisidagama DR, Bensard C, Zhao H, Wei P, Tanner J, Flores A, Mohlman J, Sorensen LK, Earl CS, Olson KA, Miao R, Waller TC, Delker D, Kanth P, Jiang L, DeBerardinis RJ, Bronner MP, Li DY, Cox JE et al (2017) Control of intestinal stem cell function and proliferation by mitochondrial pyruvate metabolism. Nat Cell Biol 19: 1027–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seale P, Sabourin LA, Girgis‐Gabardo A, Mansouri A, Gruss P, Rudnicki MA (2000) Pax7 is required for the specification of myogenic satellite cells. Cell 102: 777–786 [DOI] [PubMed] [Google Scholar]
- Skapek SX, Rhee J, Kim PS, Novitch BG, Lassar AB (1996) Cyclin‐mediated inhibition of muscle gene expression via a mechanism that is independent of pRB hyperphosphorylation. Mol Cell Biol 16: 7043–7053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang AH, Rando TA (2014) Induction of autophagy supports the bioenergetic demands of quiescent muscle stem cell activation. EMBO J 33: 2782–2797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L (2012) Differential gene and transcript expression analysis of RNA‐seq experiments with TopHat and Cufflinks. Nat Protoc 7: 562–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan M, Huang W, Kute TE, Miller LD, Zhang Q, Hatcher H, Wang J, Stovall DB, Russell GB, Cao PD, Deng Z, Wang W, Zhang Q, Lei M, Torti SV, Akman SA, Sui G (2012) Yin Yang 1 plays an essential role in breast cancer and negatively regulates p27. Am J Pathol 180: 2120–2133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Hertlein E, Bakkar N, Sun H, Acharyya S, Wang J, Carathers M, Davuluri R, Guttridge DC (2007) NF‐kappaB regulation of YY1 inhibits skeletal myogenesis through transcriptional silencing of myofibrillar genes. Mol Cell Biol 27: 4374–4387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Garzon R, Sun H, Ladner KJ, Singh R, Dahlman J, Cheng A, Hall BM, Qualman SJ, Chandler DS, Croce CM, Guttridge DC (2008) NF‐kappaB‐YY1‐miR‐29 regulatory circuitry in skeletal myogenesis and rhabdomyosarcoma. Cancer Cell 14: 369–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Zhou L, Jiang P, Lu L, Chen X, Lan H, Guttridge DC, Sun H, Wang H (2012) Loss of miR‐29 in myoblasts contributes to dystrophic muscle pathogenesis. Mol Ther 20: 1222–1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Zhu H, Situ C, Han L, Yu Y, Cheung TH, Liu K, Wu Z (2018) p110alpha of PI3K is necessary and sufficient for quiescence exit in adult muscle satellite cells. EMBO J 37: e98239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Kasim V, Kano MR, Tanaka S, Ohba S, Miura Y, Miyata K, Liu X, Matsuhashi A, Chung UI, Yang L, Kataoka K, Nishiyama N, Miyagishi M (2013) Transcription factor YY1 contributes to tumor growth by stabilizing hypoxia factor HIF‐1alpha in a p53‐independent manner. Cancer Res 73: 1787–1799 [DOI] [PubMed] [Google Scholar]
- Wu S, Wang H, Li Y, Xie Y, Huang C, Zhao H, Miyagishi M, Kasim V (2018) Transcription factor YY1 promotes cell proliferation by directly activating the pentose phosphate pathway. Can Res 78: 4549–4562 [DOI] [PubMed] [Google Scholar]
- Xie L, Yin A, Nichenko AS, Beedle AM, Call JA, Yin H (2018) Transient HIF2A inhibition promotes satellite cell proliferation and muscle regeneration. J Clin Invest 128: 2339–2355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Yang S, Wang C, Kuang S (2017) The hypoxia‐inducible factors HIF1alpha and HIF2alpha are dispensable for embryonic muscle development but essential for postnatal muscle regeneration. J Biol Chem 292: 5981–5991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS (2008) Model‐based analysis of ChIP‐Seq (MACS). Genome Biol 9: R137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZK, Li J, Liu J, Guo B, Leung A, Zhang G, Zhang BT (2016) Icaritin requires Phosphatidylinositol 3 kinase (PI3K)/Akt signaling to counteract skeletal muscle atrophy following mechanical unloading. Sci Rep 6: 20300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Wang L, Lu L, Jiang P, Sun H, Wang H (2012a) Inhibition of miR‐29 by TGF‐beta‐Smad3 signaling through dual mechanisms promotes transdifferentiation of mouse myoblasts into myofibroblasts. PLoS One 7: e33766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Wang L, Lu L, Jiang P, Sun H, Wang H (2012b) A novel target of microRNA‐29, Ring1 and YY1‐binding protein (Rybp), negatively regulates skeletal myogenesis. J Biol Chem 287: 25255–25265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Sun K, Zhao Y, Zhang S, Wang X, Li Y, Lu L, Chen X, Chen F, Bao X, Zhu X, Wang L, Tang LY, Esteban MA, Wang CC, Jauch R, Sun H, Wang H (2015) Linc‐YY1 promotes myogenic differentiation and muscle regeneration through an interaction with the transcription factor YY1. Nat Commun 6: 10026 [DOI] [PubMed] [Google Scholar]
- Zhu H, Xiao F, Wang G, Wei X, Jiang L, Chen Y, Zhu L, Wang H, Diao Y, Wang H, Ip NY, Cheung TH, Wu Z (2016) STAT3 regulates self‐renewal of adult muscle satellite cells during injury‐induced muscle regeneration. Cell Rep 16: 2102–2115 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Data Availability Statement
RNAseq and ChIPseq data used in this study have been deposited in Gene Expression Omnibus (GEO) database under the accession codes GSM2893681, GSM2893682, GSM2893683, GSM2893684, GSM2893685, and GSM2893686.