

ABSTRACT
Voltage-gated Ca2+ (CaV) channels couple membrane depolarization to Ca2+ influx, triggering a range of Ca2+-dependent cellular processes. CaV channels are, therefore, crucial in shaping neuronal activity and function, depending on their individual temporal and spatial properties. Furthermore, many neurotransmitters and drugs that act through G protein coupled receptors (GPCRs), modulate neuronal activity by altering the expression, trafficking, or function of CaV channels. GPCR-dependent mechanisms that downregulate CaV channel expression levels are observed in many neurons but are, by comparison, less studied. Here we show that the growth hormone secretagogue receptor type 1a (GHSR), a GPCR, can inhibit the forwarding trafficking of several CaV subtypes, even in the absence of agonist. This constitutive form of GPCR inhibition of CaV channels depends on the presence of a CaVβ subunit. CaVβ subunits displace CaVα1 subunits from the endoplasmic reticulum. The actions of GHSR on CaV channels trafficking suggest a role for this signaling pathway in brain areas that control food intake, reward, and learning and memory.
KEY WORDS: Voltage-gated calcium (Ca2+) channels, GPCR, CaVβ
Summary: A new mechanism that modulates the trafficking of several CaV channel subtypes to the plasma membrane through the constitutive activity of the exceptional G-protein-coupled receptor GHSR.
INTRODUCTION
Voltage-gated Ca2+ (CaV) channels are instrumental in coupling a change in transmembrane voltage to Ca2+ influx that, in turn, regulates numerous critical neuronal functions. Depending on cell type, developmental stage and subcellular location, different CaV subtypes are involved in orchestrating Ca2+-dependent signaling; CaV1 and CaV2 channels trigger transcription-dependent forms of synaptic plasticity (CaV1.2 and CaV1.3) (Wheeler et al., 2008) and fast neurotransmitter release (CaV2.1-3) (Dunlap et al., 1995; Catterall and Few, 2008; Pan and Zucker, 2009), whereas, CaV3 and CaV1.3 channels, that activate closer to the resting membrane potential, are involved in regulating cell excitability and shaping neuronal firing patterns (Molineux et al., 2006; Perez-Reyes, 2003; McKay et al., 2006; Xu and Lipscombe, 2001).
The temporal features and spatial distribution of each CaV channel subtype are strongly related to the specific roles of CaV channels in different cells (Dolphin, 2012). CaV channels associate with various proteins and these auxiliary subunits influence their trafficking to the plasma membrane (Dolphin, 2016; Felix et al., 2013; Simms and Zamponi, 2012). CaVβ and CaVα2δ are important auxiliary subunits that promote displacement of CaVα1 subunits from the endoplasmic reticulum (ER), and influence forward trafficking as well as stability at the plasma membrane (Felix et al., 2013; Dolphin, 2012). The influence of auxiliary subunits on CaV3 channels is less clear (Bichet et al., 2000; Fang and Colecraft, 2011; De Waard et al., 1994). Post-translational modifications, including asparagine-linked glycosylation, are known to promote cell surface expression of CaV3.2 (Weiss et al., 2013; Orestes et al., 2013) and indicate differences between the basic mechanisms controlling the plasma membrane density of different CaV channel subtypes.
An understanding of the mechanism that control CaV channel surface density in neurons is essential for a complete view of how cellular Ca2+ signals are regulated. Studies that explore CaV channel trafficking have started to contribute to knowledge in this field but there is much that we still do not know (Marangoudakis et al., 2012; Erickson et al., 2007). Activated GPCRs can promote CaV channel removal from the plasma membrane by internalization (Simms and Zamponi, 2012) and, in some cases, GPCR and CaV channels are internalized together (Kisilevsky and Zamponi, 2008). Our group has shown that constitutively active growth hormone secretagogue receptor type 1a (GHSR), reduces the density of CaV2.1 and CaV2.2 channel currents in both a heterologous expression system and in hypothalamic neurons (Lopez Soto et al., 2015). Here, we show that constitutively active GHSR regulates surface expression of several CaV channel subtypes. We present evidence that GHSR-mediated reduction in the current of CaV channels depends on the presence of an auxiliary CaVβ subunit, and functions by downregulating forward trafficking of CaV channels to the plasma membrane. Our data reveal a new link between CaVβ subunits and GHSR-dependent control of CaV channel activity.
RESULTS
We have shown previously that constitutively active GHSR targets presynaptic CaV2.1 and CaV2.2 in hypothalamic neurons and reduces their surface density (Lopez Soto et al., 2015). Here, we investigated whether constitutively active GHSR influences surface densities of other subtypes of CaV channels. We first assessed whether GHSR influences the size of CaV1.2 and CaV1.3 channel current densities. We used whole-cell patch clamp recording with 2 mM Ca2+ as the charge carrier. We recorded CaV channel currents from tsA201 cells co-transfected with CaV1.2 or CaV1.3, together with auxiliary subunits CaVα2δ1 and CaVβ3, either with GHSR or an empty plasmid. Compared to control cells, we found only very small CaV1.2 and CaV1.3 currents in cells that express GHSR (Fig. 1A), but this effect of GHSR was blocked by pre-incubation with the inverse GHSR agonist SPA (Fig. 1A). This suggests that the inhibitory actions of GHSR in tsA201 cells depend on constitutive activity of GHSR. Others have shown that GHSR is expressed at relatively high levels in hypothalamic neurons (Zigman et al., 2006). Furthermore, we have shown that constitutively active GHSR inhibits CaV2.1 and CaV2.2 currents in hypothalamic neurons (Lopez Soto et al., 2015). We, therefore, tested if GHSR also inhibits endogenous CaV1 channels in hypothalamic neurons by comparing CaV1 currents in hypothalamic neuronal cultures derived from wild type and GHSR-deficient (GHSR-null) mice. Total CaV currents in neurons isolated from GHSR-null mice were higher than those from wild type (Lopez Soto et al., 2015). To assess the relative size of CaV1 currents in hypothalamic neurons, in the presence and absence of GHSR, we used the dihydropyridine agonist Bay K 8644. The use of Bay K 8644 to induce increase in CaV currents is a robust method to isolate CaV1 from CaV2 channel currents in neurons (Thomas et al., 1985; Hess et al., 1984). The augmentation of CaV currents in response to Bay K 8644 was twofold greater in GHSR-null neurons as in those from wild type (Fig. 1B). Therefore, constitutive active GHSR also inhibits the activity of CaV1 channels.
Fig. 1.

GHSR constitutive activity reduces CaV1.2 and CaV1.3 currents in tsA201 cells, and reduces native CaV1 currents in cultured hypothalamic neurons. (A) Representative CaV current traces from tsA201 cells co-transfected with CaV1.2, CaVα1δ2, CaVβ3 and GHSR (+GHSR, n=8) or from controls transfected with empty plasmid (-GHSR, n=12), and average ICa for each condition (left). Representative CaV current traces from tsA201 cells co-transfected with CaV1.3, CaVα1δ2, CaVβ3 and GHSR (+GHSR, n=13) pre-incubated or not with SPA 1 µM (+SPA, n=9) and from controls transfected with empty plasmid (-GHSR, n=19), and average ICa for each condition (right). (B) Representative traces of the Bay K 8644 (BayK) effect (5 µM) on the Ba2+ current from GHSR-deficient (GHSR null, n=5) and wild-type (Wild type, n=7) hypothalamic neurons (left), and average IBa increase (right). Error bars represent mean±s.e.m., individual points represents current registered (A) or current increase for each cell (B). Kruskal–Wallis with Dunn's post-test (CaV1.3), Mann–Whitney test (CaV1.2) (A), and Student's t-test (B).
We next tested if constitutively active GHSR affects CaV3 channel currents. CaV3 channels are functionally and structurally more distant to CaV1 and CaV2 and, unlike the latter, are not retained in the ER but traffic to the plasma membrane in the absence of a CaVβ subunit. Fig. 2 shows that CaV3.2 currents and membrane surface distribution are unaffected by GHSR coexpression.
Fig. 2.

GHSR constitutive activity fails to reduce CaV3.2 density on the plasma membrane. (A) Representative CaV current traces (left) from tsA201 cells co-transfected with CaV3.2 and GHSR (+GHSR, n=10) or empty plasmid (-GHSR, n=15), and average ICa for each condition (right). (B) Photomicrographs (left) and average of GFP plasma membrane signal (in percent) (right) of tsA201 cells co-transfected with CaV3.2-GFP and GHSR (+GHSR, n=88) and from controls transfected with empty plasmid (-GHSR, n=94). Green and red signals correspond to the eGFP tag on CaV3.2 and the CellMask membrane marker, respectively. Scale bar: 10 µm. Error bars represent mean±s.e.m., individual points represent each cell analyzed.
CaV3 channels do not require CaVβ subunits to traffic to the plasma membrane but they do interact with CaVβ and CaVα2δ subunits when coexpressed in the same cell (Wyatt et al., 1998; Dolphin et al., 1999; Arias et al., 2005; Dubel et al., 2004). When we coexpressed GHSR with CaVβ3, GHSR reduces CaV3.2 current density, and the inhibitory effect of GHSR was blocked by the inverse agonist SPA (Fig. 3A). In order to visualize CaV3 channels in living cells, we used GFP-tagged CaV3.2 (CaV3.2-GFP) and analyzed its surface expression with and without expression of GHSR. We found reduced levels of CaV3.2-GFP fluorescence at the cell surface only when GHSR was coexpressed with CaVβ3, and this effect of GHSR was blocked by application of SPA (Fig. 3B). By contrast, CaVα2δ1 did not influence the ability of GHSR to reduce the density of CaV3 currents (CaV3.2+CaVα2δ1=20.6±4.2 pA/pF versus CaV3.2+CaVα2δ1+GHSR=12.1±2.9 pA/pF, n=5 and 7, P=0.11). Our results suggest that the CaVβ auxiliary subunit is necessary to reconstitute the inhibitory effect constitutively active GHSR has on CaV3 currents.
Fig. 3.

GHSR constitutive activity reduces CaV3.2 density on plasma membrane and, consequently, the CaV3.2 current in a CaVβ3-dependent manner. (A) Representative CaV current traces (left) from tsA201 cells co-transfected with CaV3.2, CaVβ3 and GHSR (+GHSR) pre-incubated or not with SPA 1 µM (+GHSR+SPA), and from controls transfected with empty plasmid (-GHSR) and average ICa in presence of increasing CaVβ3/CaV3.2 molar ratios (right). (B) Photomicrographs (left) and average of GFP plasma membrane signal (in percent) (right) of tsA201 cells co-transfected with CaV3.2-GFP, CaVβ3 and GHSR (+GHSR, n=21) pre-incubated or not with SPA 1 µM (+GHSR+SPA, n=14) and from controls transfected with empty plasmid (-GHSR, n=27). Green and red signals correspond to the eGFP tag on CaV3.2 and the membrane marker CellMask, respectively. Scale bar: 10 µm. Dots represent mean±s.e.m., numbers in brackets represent the number of analyzed cells in A. Error bars represent mean±s.e.m. and individual points represent each cell analyzed in B. Mann–Whitney test and Kruskal–Wallis with Dunn's post-test (A). Kruskal–Wallis with Dunn's post hoc test (B).
We next tested if the inhibitory effects of constitutively active GHSR on CaV2 channels also depend on the presence of CaVβ subunits. In the absence of CaVβ, CaV2.2 currents were small but measurable, and coexpression of GHSR did not have an effect on CaV2.2 current levels (Fig. 4A). This result is in contrast to control conditions under which GHSR reduces CaV2.2 current density in cells that also express CaVβ and CaVα2δ (Fig. 4B). In addition, we found no differences in CaV2.2-GFP plasma membrane signal in cells expressing GHSR in the absence of CaVβ when compared to control cells (Fig. 4C, top and bottom panels, respectively). We also tried to record CaV2.2 currents in cells in the absence of CaVα2δ but we were unable to detect CaV2.2 currents above baseline (3.33±1.50 pA/pF, n=5, P=0.091). However, we did observe a plasma membrane CaV2.2-GFP signal in cells not expressing CaVα2δ (Fig. 4C, middle panel) and this signal was reduced by GHSR coexpression by an amount that was similar to the reduction observed in cells expressing GHSR, CaV2.2, CaVβ and CaVα2δ (Fig. 4C, bottom panel). Our results suggest that CaVβ, but not CaVα2δ, is required for the inhibitory actions of constitutively active GHSR on CaV channels, and that the actions of GHSR involve reduced expression of CaV2.2 at the plasma membrane.
Fig. 4.

Presence of CaVβ3 but not that of CaVα2δ1 is required for GHSR constitutive activity to reduce CaV2.2 current by decreasing CaV2.2 density on plasma membrane. (A) Representative CaV currents from tsA201 cells co-transfected with CaV2.2, CaVα2δ1 and GHSR (+GHSR, n=44) or from controls transfected with empty plasmid (-GHSR, n=51) and average ICa for each condition. (B) Representative CaV currents from tsA201 cells co-transfected with CaV2.2, CaVβ3, CaVα2δ1 and GHSR (+GHSR, n=22) or empty plasmid (-GHSR, 48) and average ICa for each condition. (C) Photomicrographs and averaged GFP plasma membrane signal (in percent) of tsA201 cells co-transfected with CaV2.2-GFP, CaVα2δ1 and GHSR (+GHSR, n=87) and from controls transfected with empty plasmid (-GHSR, n=135) (top). Photomicrographs and average of GFP plasma membrane signal (in percent) of tsA201 cells co-transfected with CaV2.2-GFP, CaVβ3 and GHSR (+GHSR, n=51) or from controls transfected with empty plasmid (-GHSR, n=42) (middle). Photomicrographs and average of GFP plasma membrane signal (in percent) of tsA201 cells co-transfected with CaV2.2-GFP, CaVβ3, CaVα2δ1 and GHSR (+GHSR, n=29) or from controls transfected with empty plasmid (-GHSR, n=27) (bottom). Green and red signals correspond to the eGFP tag on CaV2.2 and the membrane marker CellMask, respectively. Scale bars: 10 µm. Error bars represent mean±s.e.m. and individual points represent each cell analyzed. Mann–Whitney test.
The effects of GHSR on CaV current density might involve impaired forward trafficking from the ER and Golgi and/or enhanced internalization from the plasma membrane to recycling endosomes (RE) (Simms and Zamponi, 2012). To distinguish between these possibilities, we analyzed the subcellular localization of CaV channels in the presence or absence of GHSR. We used eGFP-tagged CaVβ3 and CaVβ2a (CaVβ3-eGFP and CaVβ2a-eGFP, respectively), as well as genetically encoded plasma membrane, ER, Golgi and RE markers to study CaV2.2 channels distribution among these different compartments in tsA201 cells. We know that the inhibitory effects of GHSR on CaV2.2 are observed in cells that express either the CaVβ3 or the CaVβ2a subtype (Lopez Soto et al., 2015). CaVβ2a can be palmitoylated and this modification increases its interaction with the plasma membrane (Chien et al., 1995; Chien et al., 1996; Chien et al., 1998), whereas CaVβ3 is a soluble protein and only migrates to the plasma membrane when in complex with CaVα1 (Bichet et al., 2000). Consistent with our functional studies, we observed a reduced CaVβ2a-eGFP signal at the plasma membrane when GHSR is coexpressed and this effect was blocked by SPA pre-incubation (Fig. 5A). We also found a concomitant increase in the CaVβ2a-eGFP signal at the ER, accompanied by a mild decrease in the proportional amount of CaVβ2a-eGFP located at Golgi complex. Under these conditions, the CaVβ2a-eGFP signal in recycling endosomes was unchanged across different experimental conditions (Fig. 5B). We also assayed changes in the distribution CaVβ3-eGFP that were due to the presence of GHSR and found they were similar to the distribution of CaVβ2a-GFP (Fig. 6). In agreement with this result, we found that dominant-negative version of Rab11b, a protein that controls internalization of CaV1.2 to endosomes, does not alter the reduction of the CaV current density caused by GHSR [CaV2.2+CaVα1δ2+CaVβ3+Rab11bGDP=7.4±1.7 pA/pF (n=10) versus CaV2.2+CaVα1δ2+CaVβ3+Rab11bGDP+GHSR=0.0±0.0 pA/pF (n=4), P=0.0208]. We also found a similar result when assaying the small GTPase RhoA. This protein is involved in modifying the surface densities of CaV2.1, CaV2.2 and CaV2.3 by promoting an internalization-dependent mechanism (Rousset et al., 2015); and we did not find an effect of a RhoA inhibitor (C3 toxin) on reduction of CaV2.2 currents caused by GHSR [CaV2.2+CaVα1δ2+CaVβ3+C3=19.6±3.6 pA/pF (n=4) versus CaV2.2+CaVα1δ2+CaVβ3+C3+GHSR=0.0±0.0 pA/pF (n=4), P=0.0017].
Fig. 5.

GHSR constitutive activity reduces CaVβ2a density on the plasma membrane while increasing it on the ER. (A) Confocal images of tsA201 cells co-transfected with CaV2.2, CaVβ2a-eGFP, CaVα2δ1, plasma membrane marker (PM-marker), endoplasmic reticulum-marker (ER-marker) and GHSR pre-incubated with SPA (1 µM) (+GHSR+SPA, n=18) or not (+GHSR, n=18) and controls expressing empty plasmid (-GHSR, n=19). (B) Confocal images of tsA201 cells co-transfected with CaV2.2, CaVβ2a-eGFP, CaVα2δ1, Golgi complex marker (Golgi-marker), recycling endosome marker (RE-marker) and GHSR pre-incubated with SPA (1 µM) (+GHSR+SPA, n=15) or not (+GHSR, n=20) and controls expressing empty plasmid (-GHSR, n=16). Bar graphs show the colocalization of green signal from CaVβ2a-eGFP with blue signal (from PM-marker or Golgi-marker) or with red signal (from ER-marker or RE-marker). Error bars represent mean±s.e.m. and individual points represent each quantified cell. Scale bars: 10 µm. One way-ANOVA with Tukey's post hoc test (F2, 52=10.28) (A) and Kruskal–Wallis with Dunn's post-test (B).
Fig. 6.

GHSR constitutive activity reduces CaVβ3 density on the plasma membrane while increasing it on the ER. (A) Confocal images of tsA201 cells co-transfected with CaV2.2, CaVβ3-eGFP, CaVα2δ1, plasma membrane marker (PM-marker), endoplasmic reticulum-marker (ER-marker) and GHSR pre-incubated with SPA (1 µM) (+GHSR+SPA, n=14) or not (+GHSR, n=21) and of controls transfected with empty plasmid (-GHSR, n=25). (B) Confocal images of tsA201 cells co-transfected with CaV2.2, CaVβ3-eGFP, CaVα2δ1, Golgi marker (Golgi-marker), recycling endosome marker (RE-marker) and GHSR pre-incubated with SPA (1 µM) (+GHSR+SPA, n=16) or not (+GHSR, n=16) and of controls transfected with empty plasmid (-GHSR, n=7). Bar graphs show the colocalization of green signal from CaVβ3-eGFP with blue signal (from PM-marker or Golgi-marker) or with red signal (from ER-marker or RE-marker). Scale bars:10 µm. Error bars represent mean±s.e.m., individual points represent each quantified cell. One way-ANOVA with Turkey's post-test (F2, 57=16.43) (CaVβ3-eGFP/PM-marker overlap) and Kruskal–Wallis with Dunn's post-test.
Next we asked if GHSR can modify the subcellular location of CaVβ subunits independently of the CaV channel, by repeating the above experimental series in the absence of CaVα1. Under these conditions, without CaV2.2, GHSR coexpression failed to modify the distribution of CaVβ3 and CaVβ2a. As reported by others (Bichet et al., 2000), CaVβ3 alone does not traffic to the plasma membrane (Fig. 7). We also tested if the CaVα1 and CaVβ interaction is required for the inhibitory effect of constitutive active GHSR. We assayed the Trp391Ala mutant of CaV2.2 mutant (CaV2.2W391A) that has impaired affinity for CaVβ (Van Petegem et al., 2008; Leroy et al., 2005). As shown in Fig. 8A, this mutant failed to block the GHSR inhibitory effect when CaVβ3 or CaVβ2a were present. Moreover, we also assayed a C-terminally truncated form of CaVβ2a (CaVβ2aTF8n) that has been reported to fail to increase CaV2.1 currents and to change activation parameters (Leyris et al., 2009), suggesting it is unable to interact with the CaVα1 subunit. As we show in Fig. 8B, GHSR impairs the CaV2.2 current in the presence of CaVβ2aTF8n. Taking together, these experiments indicate that a Trp391Ala change in CaVα1 or truncation of CaVβ are not sufficient to block the inhibitory effect of GHSR.
Fig. 7.

GHSR constitutive activity fails to reduce CaVβ density on plasma membrane in the absence of CaVα1. (A) Confocal images of tsA201 cells co-transfected with CaVβ2a-eGFP, CaVα2δ1, plasma membrane marker (PM-marker), endoplasmic reticulum-marker (ER-marker) and GHSR (+GHSR, n=9) or of controls transfected with empty plasmid (-GHSR, n=9). (B) Confocal images of tsA201 cells co-transfected with CaVβ3-eGFP, CaVα2δ1, plasma membrane marker (PM-marker), endoplasmic reticulum-marker (ER-marker) and GHSR (+GHSR, n=7) or of controls transfected with empty plasmid (-GHSR, n=8). Bar graphs show the colocalization of green signal from CaVβ3-eGFP or CaVβ2a-eGFP with blue signal (from PM-marker) or with red signal (from ER-marker). Scale bars: 10 µm. Error bars represent mean±s.e.m., individual points represent each quantified cell. Student's t-test (A) and Mann–Whitney test (B).
Fig. 8.
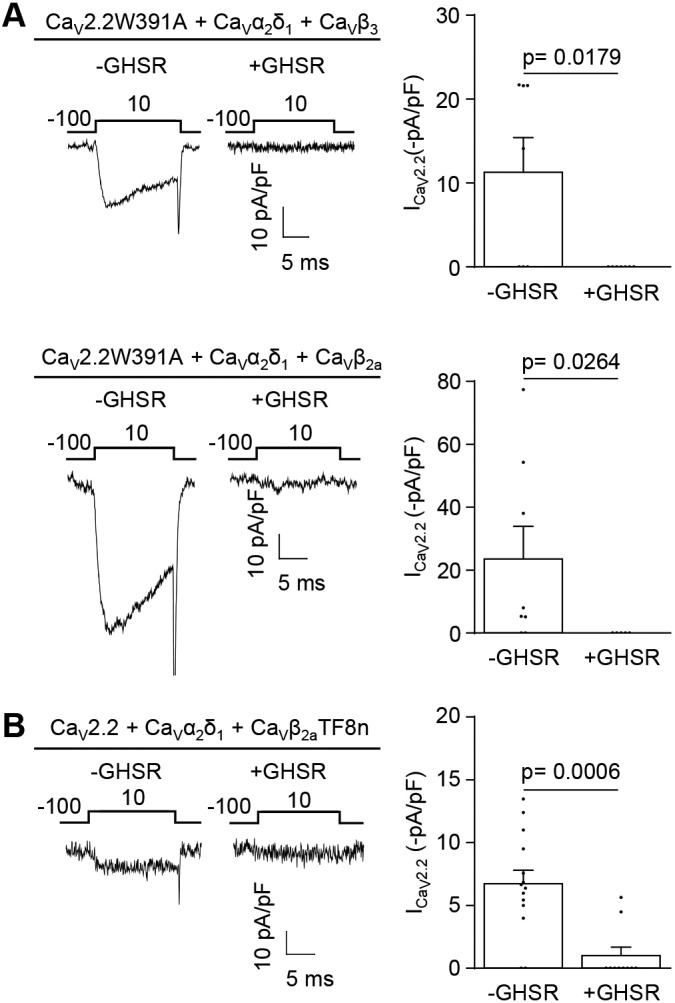
A W391A mutation of CaVα1 or truncation of CaVβ are not sufficient to block the inhibitory effect of GHSR on CaV2.2 activity. (A) Representative CaV current traces from tsA201 cells co-transfected with CaV2.2W391A, CaVα1δ2, CaVβ3 and GHSR (+GHSR, n=7) or from controls transfected with empty plasmid (-GHSR, n=7), and average ICa for each condition (top). Representative CaV current traces from tsA201 cells co-transfected with CaV2.2W391A, CaVα1δ2, CaVβ2a and GHSR (+GHSR, n=5) or from controls transfected with empty plasmid (-GHSR, n=8), and average ICa for each condition (bottom). Error bars represent mean±s.e.m., individual points represents current registered. Student's t-test (top) and Mann–Whitney test (bottom). (B) Representative CaV current traces from tsA201 cells co-transfected with CaV2.2, CaVα1δ2, CaVβ2aTF8n and GHSR (+GHSR, n=10) or from controls transfected with empty plasmid (-GHSR, n=14), and average ICa for each condition. Error bars represent mean±s.e.m., individual points represents current registered. Mann–Whitney test.
Our data indicate that constitutively active GHSR interferes with the surface expression of CaV1, CaV2 and CaV3 channels by promoting the retention of CaVα1 subunits in the ER when CaVβ subunits are present.
DISCUSSION
GHSR is crucially important in the regulation of appetite and bodyweight, and in learning and memory. Here, we extend previous studies of the effect constitutively active GHSR exerts on CaV2 channels, and show that GHSR also downregulates the activity of CaV1 and CaV3 channels. GHSR prevents CaV channels from leaving the ER, but only when CaVβ subunits are present, resulting in reduced density of surface channel.
GPCR-dependent regulation of neuronal CaV channels alter receptor and channel trafficking in many cases, including that of the mu 1 opioid receptor (OPRM1), opioid-receptor receptor-like 1 (ORL1, also known as OPRL1), dopamine receptors (DRDs) and γ-aminobutyric acid (GABA) type B receptor subunits (GABBR1 and GABBR2). This form of modulation is long lasting and independent of neuronal activity (Gray et al., 2007). Several of these receptors reduce the surface density of CaV channels by internalizing the channel or co-internalizing a GPCR–channel complex. ORL1 reduces CaV channel density at the plasma membrane after long exposures to the agonist, and this process implicates increasing trafficking to early endosomes or to lysosomes (Altier et al., 2006). Removal of channels from the surface is unlikely to participate in the constitutively active GHSR mechanism since we failed to observe any increase of channel complexes on the recycling endosome. However, as we report for GHSR, other GPCRs can regulate CaV channel activity and surface density in the absence of agonist. For instance, ORL1 exerts a negative modulation of CaV2.2 channel activity in absence of nociceptin, by forming a complex between itself and the CaV2.2 channel (Beedle et al., 2004). Moreover, DRD1 and DRD2 interact directly with CaV2.2 to increase channel surface expression levels under basal conditions, whereas agonist-activated DRD1 and DRD2 internalize together with CaV channels (Kisilevsky and Zamponi, 2008; Kisilevsky et al., 2008). These reports have shown that GPCRs are able to impact on channel trafficking by directly interacting with the channel itself. In the case of GHSR, we know that the effect depends on the degree of GHSR gene expression (Lopez Soto et al., 2015), which opens the possibility that chronic inhibition of CaV trafficking needs direct interaction of GPCR and the CaV channel.
We have shown here that constitutive active GHSR reduces forward trafficking of CaV channels only when the CaVβ subunit is present. Indeed, the ratio of CaVβ to CaVα1 influences how much CaV3 currents are downregulated by GHSR. As this effect depends on the stoichiometry of channel subunits, we suggest that the interaction of channels subunits is needed such that GHSR is able to exert its inhibitory effect. Consistent with this, we found that CaVα1 must be present for GHSR to modify the subcellular localization of eGFP-tagged CaVβ. However, we also found that the inhibitory effect of GHSR was not changed by CaV2.2W391A or CaVβ2aTF8n. These results suggest that the interaction between CaVα1 and CaVβ, mediated by W391 in the α-interaction domain (AID) and/or the segment lacking in CaVβ2aTF8n, is not required for the GHSR inhibitory effect. More experiments are, therefore, required to conclude whether the presence of CaVβ alone is sufficient to mediate the inhibitory effect of GHSR.
Considering that the chronic inhibition of CaV channels by GHSR relies on retention of CaV channels at the ER − during which we observed a mild decrease in the proportional amount of channels located at Golgi complex (CaVβ2a only) − we postulate that CaVβ acts as an inhibitor for forward trafficking when GHSR is active in addition to its established stimulatory role. In this regard, previous studies have shown that CaVβ controls forward trafficking of CaV channels (Simms and Zamponi, 2012) by preventing channel ubiquitylation and posterior degradation through the proteasome, by masking a putative ER-retention domain (Altier et al., 2011; Fang and Colecraft, 2011). However, several reports suggest a dual function of CaVβ, as (1) stimulator of forward trafficking and (2) mediator of trafficking to endosomes. Hidalgo's group has postulated a mechanism in which small GTPases and dynamin simultaneously interact with CaVβ dimers and, as a consequence, stimulate the endocytosis of channel complexes (Gonzalez-Gutierrez et al., 2007; Miranda-Laferte et al., 2011). We found that two different small GTPases, Rab11b and RhoA, are not involved in the mechanism that underlie GHSR basal inhibition of CaV, supporting the idea that an internalization process is unlikely to mediate this effect. If CaV channels are retained in intracellular compartments, an open question is what happens to them. There are several reports demonstrating that reduced CaV trafficking is followed by increased channel degradation through the proteasome (Waithe et al., 2011; Marangoudakis et al., 2012; Altier et al., 2011). Interestingly, it has been shown that CaVβ is necessary for the increase of NeDD4-1-mediated CaV channel degradation through proteasomes and lysosomes (Rougier et al., 2011). One key difference between the findings described by Rougier et al. and us is that, according to Rougier and colleagues, CaV3 channels are not affected by NeDD4-1 (officially known as NEDD4) − even in presence of CaVβ, indicating that distinct mechanisms are involved in both processes. More research is needed to conclude which molecular players execute the effect of GHSR basal activation on CaV trafficking.
We have shown previously that constitutively active GHSR reduces the surface density of CaV2 channels (Lopez Soto et al., 2015). Here, we extended our study to other CaV channel subtypes to show that this chronic basal inhibition by GHSR is common to all CaV subtypes, including neuronal CaV1 currents. CaV1 channels control Ca2+-modulated transcription by coupling voltage changes to Ca2+ influx at dendrites and soma of neurons (Dolmetsch et al., 2001; West et al., 2001). The best-studied effect of GPCR activity on CaV1 is the enhanced activity through acute activation of GPCRs that are coupled to Gs proteins (Olson et al., 2005). In our case, the number of CaV1 channels is chronically reduced − an effect that would compete with Ca2+ release from internal compartments that has been described in response to GHSR activation in neurons (Cabral et al., 2012; Cowley et al., 2003; Andrews et al., 2009) − indicating that GHSR exert a fine control of Ca2+ dynamics and, consequently, Ca2+-dependent gene activation in neurons.
We also demonstrated that GHSR inhibits CaV3.2 currents. In neurons, CaV3 channels control the shape and frequency of action potentials (Perez-Reyes, 2003; Zhang et al., 2013), and changes in channel activity due to alternative splicing (Murbartian et al., 2004; Latour et al., 2004) or nonsense mutations (Powell et al., 2009) are responsible for pathophysiological states, such as epilepsy (Hamed, 2008). Yet, the mechanisms that control CaV3 trafficking and surface membrane stability are largely unknown (Zhang et al., 2013). What is known, however, is that hormonal changes during epilepsy can alter the surface expression of CaV3.1 (Qiu et al., 2006). Our current data suggest that neurons that express GHSR at high levels negatively modulate the participation of CaV3 in waveform and frequency of action potentials. A crucial finding is that the presence of CaVβ is mandatory for GHSR-mediated negative modulation of CaV3 currents. In this regard, the interaction between CaVβ and CaV3 is not clearly established. Some reports that support this interaction have shown that CaV3 currents are enhanced following coexpression of CaVβ (Dolphin et al., 1999; Dubel et al., 2004; Dolphin, 2003). By contrast other studies have failed to demonstrate a direct impact of CaVβ on CaV3 current levels (Leuranguer et al., 1998; Bae et al., 2010). Our data indicate for the first time that CaVβ is required for impairment of CaV3.2 trafficking mediated by constitutive active GPCR, thereby uncovering a new inhibitory function of CaVβ on CaV3 currents.
GHSRs are widely expressed in the brain (Zigman et al., 2006; Mani et al., 2014). On the basis of our current and previous data; we propose that this receptor controls CaV density in neurons. Moreover, we propose that impairment of CaV trafficking by GHSR can be overcome by activation of other GPCRs that counteract the signaling cascade of constitutively active GHSRs. This would lead to a time- and place-specific fine-tuning of CaV membrane insertion. Since each neuronal CaV channel subtype has fundamental functions, constitutively active GHSRs would have great impact on the activity of GHSR-expressing neurons. This new form of neuronal activity modulation might, therefore, be relevant in the regulation of neuronal excitability, synaptic plasticity and neurotransmitter release that depends on channel complexes t formed with CaVβ in GHSR-expressing neurons.
MATERIALS AND METHODS
Cell culture and transient transfection
tsA201 cells were grown in Dulbecco's modified Eagle's medium (DMEM; Gibco) with 10% fetal bovine serum (FBS; Internegocios) and subcultured when 80% confluent. For patch-clamp experiments, tsA201 cells were co-transfected with plasmids containing GHSR (GHSR, GenBank accession no. AY429112) and voltage-gated Ca2+ channel subunits CaV1.2 (Cacna1c, GenBank accession no. AY728090), or CaV1.3-GFP (Cacna1d, GenBank accession no. AF370009), or CaV2.2 (Cacna1b, GenBank accession no. AF055477) or CaV3.2-GFP (Cacna1 h, GenBank accession no. NM021098) with or without auxiliary subunits CaVβ3 (Cacnb3, GenBank accession no. M88751) and CaVα2δ1 (Cacna2d1, GenBank accession no. AF286488), using Lipofectamine 2000 (Invitrogen). Some experiments were performed using different mutants, such as CaV2.2W391A (Addgene no. 58734) (Leroy et al., 2005) or CaVβ2aTF8n (Leyris et al., 2009). For some experiments, which lacked one or more plasmids, cells were transfected with empty plasmid pcDNA3.1 (+) to maintain the total cDNA amount in the transfection mix and eGFP-containing plasmid to identify the cells transfected. Other clones used in this paper were Rab11bS25N (Rab11bGDP) and pCDNA3-C3-toxin (C3-toxin). For live imaging experiments green fluorescent protein (GFP)-tagged CaV2.2 (CaV2.2-GFP) was used. For confocal imaging experiments, enhanced (e)GFP-tagged-auxiliary subunits CaVβ (CaVβ2a-eGFP, Cacnb2, GenBank accession no. M80545 and CaVβ3-eGFP, Cacnb3, GenBank accession no. M88751), and plasmids that encode for different intracellular compartments markers: pPalmitoyl-mTurquoise2 (plasma membrane marker, Addgene no. 36209), mCh-Sec61β (ER marker, Addgene catalog no. 49155), pmTurquoise2-Golgi (Golgi marker, Addgene catalog no. 36205) and DsRed-Rab11WT (recycling endosome marker, Addgene catalog no. 12679) were used. After transfection, cells were kept in culture to allow the expression of Ca2+ channels for 24-48 h, depending on the CaV channel subtype. Then, cells were dispersed with 0.25 mg/ml trypsin, rinsed twice and kept at room temperature (23°C) in DMEM during the patch-clamp experimental day.
Clones of Ca2+ channel subunits (except CaV3.2-GFP and CaV2.2-GFP) were generated in D.L.’s lab. CaV3.2-GFP clone was a gift from Dr E. Bourinet (Institut de Génomique Fonctionnelle, Université de Montpellier, France). CaV2.2-GFP clone was provided by Dr G. W. Zamponi (Department of Physiology and Pharmacology, University of Calgary, Canada). Auxiliary subunits CaVβ2a-eGFP and CaVβ3-eGFP were a gift from Dr Byung Chang Suh (Department of Brain Science, Daegu Gyeongbuk Institute of Science and Technology, South Korea). The GHSR clone was provided by Dr J. Marie (Université de Montpellier, Montpellier, France). The CaVβ2aTF8n clone was a gift from Dr P. Charnet (CRBM, CNRS, Université de Montpellier, France), the Rab11bS25N (Rab11bGDP) clone was provided by Dr M. V. Khvotchev (Department of Neurology, University of California, San Francisco, CA) and the pCDNA3-C3-toxin clone was provided by Dr C. Davio (University of Buenos Aires, Argentina).
Drugs
For patch-clamp and imaging experiments on tsA201 cells, the inverse GHSR agonist, [D-Arg1,D-Phe5,D-Trp7,9,Leu11]-substance P (SPA; Santa Cruz Biotechnology, Inc.) was used. For patch-clamp on mouse neuronal primary culture CaV1 agonist Bay K 8644 (Sigma-Aldrich) was used.
Animals
Wild type and GHSR1a-deficient (GHSR-null) mice (3–5-month-old females) were bred at the IMBICE animal facility. Wild-type mice, on a pure C57BL/6 background. GHSR-null mice, which fail to express GHSR1a, were derived from crosses between heterozygous animals that had been back-crossed >10 generations onto a C57BL/6 genetic background. All animals were housed in a 12 h light–dark cycle in a climate-controlled room (22°C) with ad libitum access to water and food. This study was performed in strict accordance with the recommendations of the Guide for the Care and Use of Laboratory Animals of the National Research Council and all efforts were made to minimize animal suffering. All experimentation received approval from the Institutional Animal Care and Use Committee of the IMBICE (ID:10-0112).
Mouse primary neuron culture
Neuron cultures were obtained from GHSR-null mice at embryonic days 16–18. The protocol used was similar to the one described by Raingo et al. (2012). In brief, the necks of pregnant mice were dislocated and embryos quickly removed. The brain of the embryo was exposed, placed on its dorsal side and the the hypothalamus was removed with forceps. Brains were placed in sterile Hank's solution and rinsed twice. Then, cells were dissociated at 37°C for 20 min with 0.25 mg/ml trypsin (Microvet). Enzyme digestion was stopped by addition of 300 µl FBS, and 0.28 mg/ml deoxyribonuclease I from bovine pancreas (Sigma-Aldrich) was added. Cells were mechanically dissociated using several glass pipettes with consecutive smaller-tip diameters. We plated about 50,000 cells on 12-mm diameter glass coverslips that had previously been treated with poly-L-lysine (Sigma-Aldrich) and were laid over 24-well plates. We incubated cells at 37°C in a 95% air and 5% CO2 atmosphere with DMEM (Microvet)/F12 (1:1), supplemented with B27 (1:50, Gibco), 10% FBS, 0.25% glucose, 2 mM glutamine (Gibco), 3.3 µg/ml insulin (Novo Nordisk Pharmaceutical Industries, Inc.), 40 µg/ml gentamicin sulfate (Richet) and a 1% vitamin solution (Microvet). At day 4 of culture, half of the medium was replaced by fresh medium containing cytosine β-d-arabinofuranoside (Sigma-Aldrich) to reach a final concentration of 5 µM.
Electrophysiology
Ion channel currents were recorded by using an Axopatch 200 amplifier (Molecular Devices). Data were sampled at 20 kHz and filtered at 10 kHz (−3 dB) using PCLAMP8.2 software (Molecular Devices). Recording electrodes with resistances between 2 and 4 MΩ were used and filled with internal solution. Series resistances of <6 MΩ were admitted and compensated to 80% with a 10 µs lag time. Current leak was subtracted on-line using a P/−4 protocol. All recordings were obtained at room temperature (23°C).
Ca2+ currents of transiently transfected tsA201 cells
Whole-cell patch-clamp recordings were performed on transfected (GFP-positive) tsA201 cells. Internal pipette solution contained (in mM): 134 CsCl, 10 EGTA, 1 EDTA, 10 HEPES pH 7.2 and 4 MgATP, with CsOH. External solution contained (in mM): 2 CaCl2, 1 MgCl2, 10 HEPES pH 7.4 and 140 choline chloride, with CsOH. Some experiments were made using BaCl2 (10 mM or 20 mM) instead of CaCl2 to amplify CaV current amplitude. Cells were held at −100 mV to remove closed-state inactivation (Thaler et al., 2004).
The test-pulse protocol consisted of voltage square pulses that were applied every 10 s; CaV1.2: −100 mV to +10 mV for 15 ms, CaV2.2: −100 mV to +10 mV 25ms, CaV1.3: −100 mV to −10 mV for 15 ms, and CaV3.2: −100 mV to −20 mV for 200 ms.
Ba2+ currents of primary neuronal cultures
Mouse neurons that had been cultured for 7–15 days were patched in voltage-clamp whole-cell mode at a holding potential of -80 mV applying squared test pulses to 0 mV for 20 ms every 10 s (Raingo et al., 2007). Internal pipette solution contained (mM): 134 CsCl, 10 EGTA, 1 EDTA, 10 HEPES pH 7.2 and 4 MgATP, with CsOH. Neurons were bathed with high [Na+] external solution containing (mM): 135 NaCl, 4.7 KCl, 1.2 MgCl2, 2.5 CaCl2, 10 HEPES pH 7.4 and 10 glucose, with NaOH. After getting the whole cell configuration, CaV currents were recorded replacing the external solution by a high [Ba2+] solution containing (mM): 10 BaCl2, 110 choline chloride, 20 tetraethylammonium chloride, 1 MgCl2, 10 HEPES pH 7.4, 10 glucose and 0.001 tetrodotoxin (TTX; Sigma-Aldrich), with CsOH.
Imaging
In experiments presented in Figs 2–4, tsA201 cells had been co-transfected with CaV2.2-GFP or CaV3.2-GFP, with or without its auxiliary subunits, GHSR or pcDNA3.1 (+). At 48 or 24 h after transfection of CaV3.2-GFP or CaV2.2-GFP, respectively, culture medium was replaced by 1 ml of 1 μg/ml of membrane marker solution (CellMask orange plasma membrane stain; Molecular probes) and cells were kept at 37°C for 1 min. After that, cells were rinsed three times with 1× phosphate-buffered saline (PBS). To finish, PBS was removed and a clean coverslip was placed over the cell layer.
Fluorescence photomicrographs were obtained using an optical fluorescence microscope (Eclipse 50i; Nikon), equipped with B2A and G2A filters and a camera (DS-Ri1; Nikon). Photomicrographs were analyzed with FIJI free software, using the CellMask red signal to mark out the plasma membrane and quantify green fluorescence intensity in both the internal area (excluding plasma membrane) and the total area of each cell as integrated density. The fluorescence intensity corresponding to the membrane (membrane fluorescence) was calculated as the difference between the fluorescence corresponding to the total (total fluorescence) and the internal area. Finally, the CaV2.2-GFP or CaV3.2-GFP membrane fluorescence (in percent) was calculated for each cell, by using: (membrane fluorescence/total fluorescence)×100.
In experiments presented in Figs 5–7 tsA201 cells had been plated on glass coverslips treated previously with poly-L-lysine and laid over 12-well plates. Cells were co-transfected 24 h later with CaVβ2a-eGFP or CaVβ3-eGFP and with different plasmid combinations by using Lipofectamine. Further 24 h later, cells were rinsed with 1×PBS. After that, cells were fixed with 4% paraformaldehyde in PBS for 20 min, followed by a rinse with 1×PBS twice and were mounted on glass slides using VECTASHIELD® mounting medium.
Confocal images were collected using a Zeiss LSM 800 confocal microscope and ZEN software. Quantification of colocalization of CaVβ2a-eGFP or CaVβ3-eGFP with the different intracellular compartment markers was performed using Just another Colocalization Plugin (JaCoP) from FIJI, to calculate Manders' overlap coefficient for each marker (CaVβ-eGFP/marker overlap).
Statistics
Data were analyzed and visualized by using the OriginPro 8 (Origin-Lab) and GraphPad Prism 5 (GraphPad Software, Inc.) software. We used the Kolmogorov–Smirnov to test for conformity to a normal distribution; variance homogeneity was examined by using Bartlett's (normal distributed data) and Brown-Forsythe′s (no normal distributed data) test. P values were calculated from one- or two-sample t-tests (normally distributed data) or Mann–Whitney test (no normally distributed data), and multiple comparison one way-ANOVA with Tukey's post hoc test (normal distributed data) or non-parametric Kruskal–Wallis test with Dunn's post-test (no normal distributed data). P values were calculated and included in the figures. Specific statistical test used is indicated for each data set. Data were expressed as mean±s.e.m.
Acknowledgements
We thank Dr Jeffrey Zigman (The University of Texas Southwestern Medical Center, Dallas, TX) for providing GHSR-null mice and Dr Mario Perello and Guadalupe Garcia Romero (both Multidisciplinary Institute of Cellular Biology (IMBICE), Universidad Nacional de La Plata, Buenos Aires, Argentina) for help and advice in housing and caring for the mouse colony. We thank Sylvia Denome for her excellent technical assistance and Daniel Dubreuil (both Department of Neuroscience, Brown University, Providence, RI) for kindly helping us with confocal microscopy.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: E.R.M., E.J.L.S., J.R.; Methodology: E.R.M., E.J.L.S., V.M.D., S.S.R., J.R.; Formal analysis: E.R.M., E.J.L.S., V.M.D., J.R.; Investigation: E.R.M., E.J.L.S., D.L., J.R.; Resources: D.L., J.R.; Writing - original draft: E.R.M., J.R.; Writing - review & editing: E.R.M., E.J.L.S., V.M.D., D.L., J.R.; Supervision: J.R.; Project administration: J.R.; Funding acquisition: J.R.
Funding
This work was supported by grants from the Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET) (PICT2013-1145 and PICT2015-3330 to J.R.). E.R.M., V.M.D., S.S.R. and J.R. were supported by Consejo Nacional de Investigaciones Científicas y Técnicas and Comisión de Investigaciones de la Provincia de Buenos Aires (CIC).
References
- Altier C., Khosravani H., Evans R. M., Hameed S., Peloquin J. B., Vartian B. A., Chen L., Beedle A. M., Ferguson S. S., Mezghrani A. et al. (2006). ORL1 receptor-mediated internalization of N-type calcium channels. Nat. Neurosci. 9, 31-40. 10.1038/nn1605 [DOI] [PubMed] [Google Scholar]
- Altier C., Garcia-Caballero A., Simms B., You H., Chen L., Walcher J., Tedford H. W., Hermosilla T. and Zamponi G. W. (2011). The Cavbeta subunit prevents RFP2-mediated ubiquitination and proteasomal degradation of L-type channels. Nat. Neurosci. 14, 173-180. 10.1038/nn.2712 [DOI] [PubMed] [Google Scholar]
- Andrews Z. B., Erion D., Beiler R., Liu Z.-W., Abizaid A., Zigman J., Elsworth J. D., Savitt J. M., Dimarchi R., Tschoep M. et al. (2009). Ghrelin promotes and protects nigrostriatal dopamine function via a UCP2-dependent mitochondrial mechanism. J. Neurosci. 29, 14057-14065. 10.1523/JNEUROSCI.3890-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias J. M., Murbartián J., Vitko I., Lee J.-H. and Perez-Reyes E. (2005). Transfer of beta subunit regulation from high to low voltage-gated Ca2+ channels. FEBS Lett. 579, 3907-3912. 10.1016/j.febslet.2005.06.008 [DOI] [PubMed] [Google Scholar]
- Bae J., Suh E. J. and Lee C. (2010). Interaction of T-type calcium channel Ca(V)3.3 with the beta-subunit. Mol. Cells 30, 185-191. 10.1007/s10059-010-0106-z [DOI] [PubMed] [Google Scholar]
- Beedle A. M., Mcrory J. E., Poirot O., Doering C. J., Altier C., Barrere C., Hamid J., Nargeot J., Bourinet E. and Zamponi G. W. (2004). Agonist-independent modulation of N-type calcium channels by ORL1 receptors. Nat. Neurosci. 7, 118-125. 10.1038/nn1180 [DOI] [PubMed] [Google Scholar]
- Bichet D., Cornet V., Geib S., Carlier E., Volsen S., Hoshi T., Mori Y. and De Waard M. (2000). The I-II loop of the Ca2+ channel alpha1 subunit contains an endoplasmic reticulum retention signal antagonized by the beta subunit. Neuron 25, 177-190. 10.1016/S0896-6273(00)80881-8 [DOI] [PubMed] [Google Scholar]
- Cabral A., Suescun O., Zigman J. M. and Perello M. (2012). Ghrelin indirectly activates hypophysiotropic CRF neurons in rodents. PLoS ONE 7, e31462 10.1371/journal.pone.0031462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall W. A. and Few A. P. (2008). Calcium channel regulation and presynaptic plasticity. Neuron 59, 882-901. 10.1016/j.neuron.2008.09.005 [DOI] [PubMed] [Google Scholar]
- Cowley M. A., Smith R. G., Diano S., Tschöp M., Pronchuk N., Grove K. L., Strasburger C. J., Bidlingmaier M., Esterman M., Heiman M. L. et al. (2003). The distribution and mechanism of action of ghrelin in the CNS demonstrates a novel hypothalamic circuit regulating energy homeostasis. Neuron 37, 649-661. 10.1016/S0896-6273(03)00063-1 [DOI] [PubMed] [Google Scholar]
- Chien A. J., Zhao X., Shirokov R. E., Puri T. S., Chang C. F., Sun D., Rios E. and Hosey M. M. (1995). Roles of a membrane-localized beta subunit in the formation and targeting of functional L-type Ca2+ channels. J. Biol. Chem. 270, 30036-30044. 10.1074/jbc.270.50.30036 [DOI] [PubMed] [Google Scholar]
- Chien A. J., Carr K. M., Shirokov R. E., Rios E. and Hosey M. M. (1996). Identification of palmitoylation sites within the L-type calcium channel beta2a subunit and effects on channel function. J. Biol. Chem. 271, 26465-26468. 10.1074/jbc.271.43.26465 [DOI] [PubMed] [Google Scholar]
- Chien A. J., Gao T., Perez-Reyes E. and Hosey M. M. (1998). Membrane targeting of L-type calcium channels. Role of palmitoylation in the subcellular localization of the beta2a subunit. J. Biol. Chem. 273, 23590-23597. [DOI] [PubMed] [Google Scholar]
- De Waard M., Pragnell M. and Campbell K. P. (1994). Ca2+ channel regulation by a conserved beta subunit domain. Neuron 13, 495-503. 10.1016/0896-6273(94)90363-8 [DOI] [PubMed] [Google Scholar]
- Dolmetsch R. E., Pajvani U., Fife K., Spotts J. M. and Greenberg M. E. (2001). Signaling to the nucleus by an L-type calcium channel-calmodulin complex through the MAP kinase pathway. Science 294, 333-339. 10.1126/science.1063395 [DOI] [PubMed] [Google Scholar]
- Dolphin A. C. (2003). Beta subunits of voltage-gated calcium channels. J. Bioenerg. Biomembr. 35, 599-620. 10.1023/B:JOBB.0000008026.37790.5a [DOI] [PubMed] [Google Scholar]
- Dolphin A. C. (2012). Calcium channel auxiliary alpha2delta and beta subunits: trafficking and one step beyond. Nat. Rev. Neurosci. 13, 542-555. 10.1038/nrn3317 [DOI] [PubMed] [Google Scholar]
- Dolphin A. C. (2016). Voltage-gated calcium channels and their auxiliary subunits: physiology and pathophysiology and pharmacology. J. Physiol. 594, 5369-5390. 10.1113/JP272262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolphin A. C., Wyatt C. N., Richards J., Beattie R. E., Craig P., Lee J.-H., Cribbs L. L., Volsen S. G. and Perez-Reyes E. (1999). The effect of alpha2-delta and other accessory subunits on expression and properties of the calcium channel alpha1G. J. Physiol. 519, 35-45. 10.1111/j.1469-7793.1999.0035o.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubel S. J., Altier C., Chaumont S., Lory P., Bourinet E. and Nargeot J. (2004). Plasma membrane expression of T-type calcium channel alpha(1) subunits is modulated by high voltage-activated auxiliary subunits. J. Biol. Chem. 279, 29263-29269. 10.1074/jbc.M313450200 [DOI] [PubMed] [Google Scholar]
- Dunlap K., Luebke J. I. and Turner T. J. (1995). Exocytotic Ca2+ channels in mammalian central neurons. Trends Neurosci. 18, 89-98. 10.1016/0166-2236(95)80030-6 [DOI] [PubMed] [Google Scholar]
- Erickson M. A., Haburćák M., Smukler L. and Dunlap K. (2007). Altered functional expression of Purkinje cell calcium channels precedes motor dysfunction in tottering mice. Neuroscience 150, 547-555. 10.1016/j.neuroscience.2007.09.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang K. and Colecraft H. M. (2011). Mechanism of auxiliary beta-subunit-mediated membrane targeting of L-type (Ca(V)1.2) channels. J. Physiol. 589, 4437-4455. 10.1113/jphysiol.2011.214247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felix R., Calderón-Rivera A. and Andrade A. (2013). Regulation of high-voltage-activated Ca2+ channel function, trafficking, and membrane stability by auxiliary subunits. Wiley Interdiscip. Rev. Membr. Transp. Signal. 2, 207-220. 10.1002/wmts.93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Gutierrez G., Miranda-Laferte E., Neely A. and Hidalgo P. (2007). The Src homology 3 domain of the beta-subunit of voltage-gated calcium channels promotes endocytosis via dynamin interaction. J. Biol. Chem. 282, 2156-2162. 10.1074/jbc.M609071200 [DOI] [PubMed] [Google Scholar]
- Gray A. C., Raingo J. and Lipscombe D. (2007). Neuronal calcium channels: splicing for optimal performance. Cell Calcium. 42, 409-417. 10.1016/j.ceca.2007.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamed S. A. (2008). Neuroendocrine hormonal conditions in epilepsy: relationship to reproductive and sexual functions. Neurologist 14, 157-169. 10.1097/NRL.0b013e3181618ada [DOI] [PubMed] [Google Scholar]
- Hess P., Lansman J. B. and Tsien R. W. (1984). Different modes of Ca channel gating behaviour favoured by dihydropyridine Ca agonists and antagonists. Nature 311, 538-544. 10.1038/311538a0 [DOI] [PubMed] [Google Scholar]
- Kisilevsky A. E. and Zamponi G. W. (2008). D2 dopamine receptors interact directly with N-type calcium channels and regulate channel surface expression levels. Channels (Austin) 2, 269-277. 10.4161/chan.2.4.6402 [DOI] [PubMed] [Google Scholar]
- Kisilevsky A. E., Mulligan S. J., Altier C., Iftinca M. C., Varela D., Tai C., Chen L., Hameed S., Hamid J., Macvicar B. A. et al. (2008). D1 receptors physically interact with N-type calcium channels to regulate channel distribution and dendritic calcium entry. Neuron 58, 557-570. 10.1016/j.neuron.2008.03.002 [DOI] [PubMed] [Google Scholar]
- Latour I., Louw D. F., Beedle A. M., Hamid J., Sutherland G. R. and Zamponi G. W. (2004). Expression of T-type calcium channel splice variants in human glioma. Glia 48, 112-119. 10.1002/glia.20063 [DOI] [PubMed] [Google Scholar]
- Leroy J., Richards M. W., Butcher A. J., Nieto-Rostro M., Pratt W. S., Davies A. and Dolphin A. C. (2005). Interaction via a key tryptophan in the I-II linker of N-type calcium channels is required for beta1 but not for palmitoylated beta2, implicating an additional binding site in the regulation of channel voltage-dependent properties. J. Neurosci. 25, 6984-6996. 10.1523/JNEUROSCI.1137-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuranguer V., Bourinet E., Lory P. and Nargeot J. (1998). Antisense depletion of beta-subunits fails to affect T-type calcium channels properties in a neuroblastoma cell line. Neuropharmacology 37, 701-708. 10.1016/S0028-3908(98)00060-4 [DOI] [PubMed] [Google Scholar]
- Leyris J.-P., Gondeau C., Charnet A., Delattre C., Rousset M., Cens T. and Charnet P. (2009). RGK GTPase-dependent CaV2.1 Ca2+ channel inhibition is independent of CaVbeta-subunit-induced current potentiation. FASEB J. 23, 2627-2638. 10.1096/fj.08-122135 [DOI] [PubMed] [Google Scholar]
- López Soto E. J., Agosti F., Cabral A., Mustafa E. R., Damonte V. M., Gandini M. A., Rodriguez S., Castrogiovanni D., Felix R., Perello M. et al. (2015). Constitutive and ghrelin-dependent GHSR1a activation impairs CaV2.1 and CaV2.2 currents in hypothalamic neurons. J. Gen. Physiol. 146, 205-219. 10.1085/jgp.201511383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani B. K., Walker A. K., Lopez Soto E. J., Raingo J., Lee C. E., Perelló M., Andrews Z. B. and Zigman J. M. (2014). Neuroanatomical characterization of a growth hormone secretagogue receptor-green fluorescent protein reporter mouse. J. Comp. Neurol. 522, 3644-3666. 10.1002/cne.23627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marangoudakis S., Andrade A., Helton T. D., Denome S., Castiglioni A. J. and Lipscombe D. (2012). Differential ubiquitination and proteasome regulation of Ca(V)2.2 N-type channel splice isoforms. J. Neurosci. 32, 10365-10369. 10.1523/JNEUROSCI.0851-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mckay B. E., Mcrory J. E., Molineux M. L., Hamid J., Snutch T. P., Zamponi G. W. and Turner R. W. (2006). Ca(V)3 T-type calcium channel isoforms differentially distribute to somatic and dendritic compartments in rat central neurons. Eur. J. Neurosci. 24, 2581-2594. 10.1111/j.1460-9568.2006.05136.x [DOI] [PubMed] [Google Scholar]
- Miranda-Laferte E., Gonzalez-Gutierrez G., Schmidt S., Zeug A., Ponimaskin E. G., Neely A. and Hidalgo P. (2011). Homodimerization of the Src homology 3 domain of the calcium channel beta-subunit drives dynamin-dependent endocytosis. J. Biol. Chem. 286, 22203-22210. 10.1074/jbc.M110.201871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molineux M. L., Mcrory J. E., Mckay B. E., Hamid J., Mehaffey W. H., Rehak R., Snutch T. P., Zamponi G. W. and Turner R. W. (2006). Specific T-type calcium channel isoforms are associated with distinct burst phenotypes in deep cerebellar nuclear neurons. Proc. Natl. Acad. Sci. USA 103, 5555-5560. 10.1073/pnas.0601261103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murbartian J., Arias J. M. and Perez-Reyes E. (2004). Functional impact of alternative splicing of human T-type Cav3.3 calcium channels. J. Neurophysiol. 92, 3399-3407. 10.1152/jn.00498.2004 [DOI] [PubMed] [Google Scholar]
- Olson P. A., Tkatch T., Hernandez-Lopez S., Ulrich S., Ilijic E., Mugnaini E., Zhang H., Bezprozvanny I. and Surmeier D. J. (2005). G-protein-coupled receptor modulation of striatal CaV1.3 L-type Ca2+ channels is dependent on a Shank-binding domain. J. Neurosci. 25, 1050-1062. 10.1523/JNEUROSCI.3327-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orestes P., Osuru H. P., Mcintire W. E., Jacus M. O., Salajegheh R., Jagodic M. M., Choe W., Lee J., Lee S. S., Rose K. E. et al. (2013). Reversal of neuropathic pain in diabetes by targeting glycosylation of Ca(V)3.2 T-type calcium channels. Diabetes 62, 3828-3838. 10.2337/db13-0813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan B. and Zucker R. S. (2009). A general model of synaptic transmission and short-term plasticity. Neuron 62, 539-554. 10.1016/j.neuron.2009.03.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Reyes E. (2003). Molecular physiology of low-voltage-activated t-type calcium channels. Physiol. Rev. 83, 117-161. 10.1152/physrev.00018.2002 [DOI] [PubMed] [Google Scholar]
- Powell K. L., Cain S. M., Ng C., Sirdesai S., David L. S., Kyi M., Garcia E., Tyson J. R., Reid C. A., Bahlo M. et al. (2009). A Cav3.2 T-type calcium channel point mutation has splice-variant-specific effects on function and segregates with seizure expression in a polygenic rat model of absence epilepsy. J. Neurosci. 29, 371-380. 10.1523/JNEUROSCI.5295-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J., Bosch M. A., Jamali K., Xue C., Kelly M. J. and Ronnekleiv O. K. (2006). Estrogen upregulates T-type calcium channels in the hypothalamus and pituitary. J. Neurosci. 26, 11072-11082. 10.1523/JNEUROSCI.3229-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raingo J., Castiglioni A. J. and Lipscombe D. (2007). Alternative splicing controls G protein-dependent inhibition of N-type calcium channels in nociceptors. Nat. Neurosci. 10, 285-292. 10.1038/nn1848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raingo J., Khvotchev M., Liu P., Darios F., Li Y. C., Ramirez D. M. O., Adachi M., Lemieux P., Toth K., Davletov B. et al. (2012). VAMP4 directs synaptic vesicles to a pool that selectively maintains asynchronous neurotransmission. Nat. Neurosci. 15, 738-745. 10.1038/nn.3067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rougier J.-S., Albesa M., Abriel H. and Viard P. (2011). Neuronal precursor cell-expressed developmentally down-regulated 4-1 (NEDD4-1) controls the sorting of newly synthesized Ca(V)1.2 calcium channels. J. Biol. Chem. 286, 8829-8838. 10.1074/jbc.M110.166520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousset M., Cens T., Menard C., Bowerman M., Bellis M., Brusés J., Raoul C., Scamps F. and Charnet P. (2015). Regulation of neuronal high-voltage activated Ca(V)2 Ca(2+) channels by the small GTPase RhoA. Neuropharmacology 97, 201-209. 10.1016/j.neuropharm.2015.05.019 [DOI] [PubMed] [Google Scholar]
- Simms B. A. and Zamponi G. W. (2012). Trafficking and stability of voltage-gated calcium channels. Cell Mol. Life Sci. 69, 843-856. 10.1007/s00018-011-0843-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaler C., Gray A. C. and Lipscombe D. (2004). Cumulative inactivation of N-type CaV2.2 calcium channels modified by alternative splicing. Proc. Natl. Acad. Sci. USA 101, 5675-5679. 10.1073/pnas.0303402101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas G., Chung M. and Cohen C. J. (1985). A dihydropyridine (Bay k 8644) that enhances calcium currents in guinea pig and calf myocardial cells. A new type of positive inotropic agent. Circ. Res. 56, 87-96. 10.1161/01.RES.56.1.87 [DOI] [PubMed] [Google Scholar]
- Van Petegem F., Duderstadt K. E., Clark K. A., Wang M. and Minor D. L. Jr (2008). Alanine-scanning mutagenesis defines a conserved energetic hotspot in the CaValpha1 AID-CaVbeta interaction site that is critical for channel modulation. Structure 16, 280-294. 10.1016/j.str.2007.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waithe D., Ferron L., Page K. M., Chaggar K. and Dolphin A. C. (2011). Beta-subunits promote the expression of Ca(V)2.2 channels by reducing their proteasomal degradation. J. Biol. Chem. 286, 9598-9611. 10.1074/jbc.M110.195909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss N., Black S. A. G., Bladen C., Chen L. and Zamponi G. W. (2013). Surface expression and function of Cav3.2 T-type calcium channels are controlled by asparagine-linked glycosylation. Pflugers Arch. 465, 1159-1170. 10.1007/s00424-013-1259-3 [DOI] [PubMed] [Google Scholar]
- West A. E., Chen W. G., Dalva M. B., Dolmetsch R. E., Kornhauser J. M., Shaywitz A. J., Takasu M. A., Tao X. and Greenberg M. E. (2001). Calcium regulation of neuronal gene expression. Proc. Natl. Acad. Sci. USA 98, 11024-11031. 10.1073/pnas.191352298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler D. G., Barrett C. F., Groth R. D., Safa P. and Tsien R. W. (2008). CaMKII locally encodes L-type channel activity to signal to nuclear CREB in excitation-transcription coupling. J. Cell Biol. 183, 849-863. 10.1083/jcb.200805048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt C. N., Page K. M., Berrow N. S., Brice N. L. and Dolphin A. C. (1998). The effect of overexpression of auxiliary Ca2+ channel subunits on native Ca2+ channel currents in undifferentiated mammalian NG108-15 cells. J. Physiol. 510, 347-360. 10.1111/j.1469-7793.1998.347bk.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W. and Lipscombe D. (2001). Neuronal Ca(V)1.3alpha(1) L-type channels activate at relatively hyperpolarized membrane potentials and are incompletely inhibited by dihydropyridines. J. Neurosci. 21, 5944-5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Jiang X., Snutch T. P. and Tao J. (2013). Modulation of low-voltage-activated T-type Ca(2)(+) channels. Biochim. Biophys. Acta 1828, 1550-1559. 10.1016/j.bbamem.2012.08.032 [DOI] [PubMed] [Google Scholar]
- Zigman J. M., Jones J. E., Lee C. E., Saper C. B. and Elmquist J. K. (2006). Expression of ghrelin receptor mRNA in the rat and the mouse brain. J. Comp. Neurol. 494, 528-548. 10.1002/cne.20823 [DOI] [PMC free article] [PubMed] [Google Scholar]