

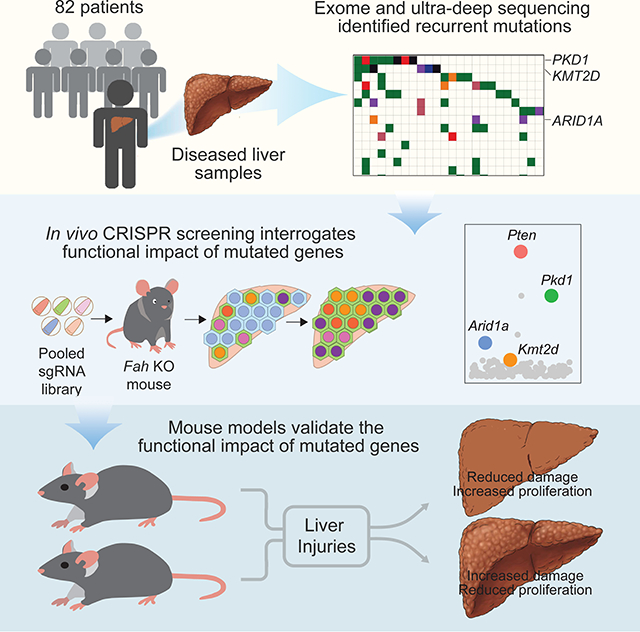
Summary
Normal tissues accumulate genetic changes with age, but it is unknown if somatic mutations promote clonal expansion of non-malignant cells in the setting of chronic degenerative diseases. Exome sequencing of diseased liver samples from 82 patients revealed a complex mutational landscape in cirrhosis. Additional ultra-deep sequencing identified recurrent mutations in PKD1, PPARGC1B, KMT2D, and ARID1A. The number and size of mutant clones increased as a function of fibrosis stage and tissue damage. To interrogate the functional impact of mutated genes, a pooled in vivo CRISPR screening approach was established. In agreement with sequencing results, examination of 147 genes again revealed that loss of Pkd1, Kmt2d, and Arid1a promoted clonal expansion. Conditional heterozygous deletion of these genes in mice was also hepatoprotective in injury assays. Pre-malignant somatic alterations are often viewed through the lens of cancer, but we show that mutations can promote regeneration, likely independent of carcinogenesis.
In Brief
Deep sequencing reveals the mutational landscape of human cirrhosis and in vivo CRISPR screening identifies functional roles for recurrently mutated genes in hepatocyte expansion and liver regenerative fitness.
Graphical Abstract
Introduction
Emerging evidence from multiple tissues clearly indicate that aging is associated with extensive somatic mutagenesis. Somatic mutations promote the clonal expansion of hematopoietic cells with age, a phenomenon termed clonal hematopoiesis (Genovese et al., 2014; Jaiswal et al., 2014). Mutations undergoing positive selection have also been detected in the eyelids of four individuals (Martincorena et al., 2015) and the esophagi of over 100 individuals (Martincorena et al., 2018; Yokoyama et al., 2019). In the cases of skin and esophagus, it is unknown if detected mutations have a physiological impact, and whether or not they contribute to the early phases of cancer development. These tissues were chosen for study partly because of the lack of spatial limits for cellular expansion at these sites. The extent of somatic genetic diversity within most other solid organs is not known, in part due to the fact that nested tissue structures such as intestinal crypts and hepatic lobules limit the expansion and thus detection of mutant clones in bulk sequencing.
An easier setting to detect mutations in tissues with nested architecture is in the context of chronic diseases, where environmental insults drive the expansion of regenerating clones. Liver disease is commonly caused by hepatitis C virus (HCV), hepatitis B virus (HBV), alcohol abuse, and non-alcoholic steatohepatitis (NASH) (Singal and El-Serag, 2015; White et al., 2012). The common pathological endpoint for these diverse processes is end-stage liver fibrosis, also known as cirrhosis, which affects up to 1% of the population and is a critical risk factor for hepatocellular carcinoma (HCC) (Kulik and El-Serag, 2019). It is known that mRNA expression changes in cirrhotic tissues are associated with disease progression (Hoshida et al., 2008), but it is less clear if recurrent mutations are selected, in part due to limited efforts to sequence liver tissues with the requisite depth and breadth.
Cirrhotic livers have a grossly bumpy appearance due to their defining histologic feature: regenerative nodules separated by fibrous bands. Analysis of microdissected cirrhotic nodules by SNP, CAG repeat, array CGH analysis, or mitochondrial mutations revealed that half or more of all nodules have a clonal origin (Aihara et al., 1994; Fellous et al., 2009; Gong et al., 2010; Lin et al., 2010; Paradis et al., 1998). The prevalence of large nodules (0.5–5mm diameter) that likely arise from single cells suggests that if mutations existed in cirrhosis, they could be easily detected using next generation sequencing techniques, even in bulk samples comprised of heterogeneous nodules. These nodules also suggest selection for mutations that confer a selective advantage; however, the paucity of sequencing performed in cirrhotic livers means that few specific mutations or mechanisms for clonal expansion have been identified. Tert promoter mutations have been identified in dysplastic, likely pre-malignant, nodules (Nault et al., 2013) and Leptin receptor mutations have been identified in four cirrhotic patients (Ikeda et al., 2014). However, a broad landscape analysis of mutations in cirrhosis has not yet been performed in large numbers of patients. More generally, the impact of mutations found in most normal tissue sequencing studies have not been functionally validated.
In this study, we analyzed non-dysplastic liver tissues from 82 patients using both broad and deep sequencing approaches and identified a high density of mutations with allelic frequencies suggesting clonal expansion. To test whether recurrently mutated genes regulate hepatocyte clonal expansion, we performed in vivo CRISPR screens in the context of liver regeneration. This revealed a cohort of genes previously unknown to regulate regeneration, and which were also the most recurrently mutated within cirrhotic nodules, suggesting selection for mutations in these genes during liver disease. Importantly, many of the recurrently mutated genes that had the largest effects in regeneration are not frequently detected in liver cancer, and thus do not appear to promote carcinogenesis. Somatic mutations in non-malignant tissues are often viewed through the lens of progression to cancer, but our results suggest that recurrent mutations can confer adaptive changes that promote fitness and regeneration in response to chronic damage, potentially independent of cancer formation.
Results
Uncovering the burden of somatic mutations in diseased liver tissues
To survey mutations in liver disease in an unbiased fashion, we analyzed a total of 82 patients. We performed whole exome sequencing of non-malignant samples and paired blood from 53 patients that underwent surgical resection for early stage liver cancers. An additional 22 patients who had liver tissue and paired blood sequenced by the TCGA liver cancer project were reanalyzed (HCC TCGA, 2017). Six cirrhotic livers that were explanted during organ transplantation and one completely normal liver were also included.
Genomic DNA was extracted from tissue pieces that were histologically analyzed by a clinical pathologist with expertise in liver cancer and found to contain only normal liver cells or regenerative nodules (Figure 1A). No samples contained dysplastic, potentially malignant (hepatic adenoma), or malignant (HCC or cholangiocarcinoma) nodules, an important criterion because more advanced pathologic entities could bias the mutation spectrum towards cancer (Supplemental Table 1). The etiology of liver disease was predominantly HCV (72% in the UT Southwestern cohort), but patients also had HBV, NASH, and cryptogenic cirrhosis (Supplemental Table 1 and Figure S1A). Fibrosis stage in the samples ranged from no fibrosis (F0) to established cirrhosis (F4) (Figure S1A and S1B). The 60 exomes from our institution were sequenced to a mean depth of 86x (after duplicate removal), and 80% of exonic regions had greater than 100x coverage. The 22 TCGA cases that had paired blood and non-malignant liver were sequenced to a similar depth of 84x.
Figure 1. Whole exome sequencing reveals mutational burden within diseased livers.

A. Schema for liver tissue sampling. Fresh frozen non-malignant liver tissues from HCC surgical resections or livers removed during transplant surgeries were obtained for genomic DNA. The adjacent tissue was sectioned for histology. Sequenced tissue weights are shown on the right.
B. Number of mutations found in each patient sample. Classifications in the pie chart: among 389 total mutations, 260 are missense, 13 are nonsense, 4 are splice-site, 1 is a frameshift deletion and 111 are synonymous mutations.
C. Mutation count and rate in liver samples compared with cancer types. Mutation count equals the absolute number of mutations per patient sample with VAF > 5%. Mutation rate equals the sums of 2 times the VAFs for each mutation. The Wilcoxon Rank-Sum Test was used to compare mutation counts and rates between tissue types.
D. Classes of missense mutations or SNVs in liver tissues and HCC samples from TCGA.
E. The 4 mutation signatures with the highest cosine similarity are shown.
F. Correlation between mutation count and fibrosis stage. The p-value is calculated based on the one-way Jonckheere trend test.
G. Correlation between mutation count and ALT or AST, which are serum markers of hepatic damage.
H. VAF distribution for whole exome data. Mean VAF is 10.5% (+/− 0.514% SEM, with 95% confidence interval) and median VAF is 8.7%.
All data are presented as mean ± SEM. *, p<0.05, **, p<0.01,***, p<0.001,****, p<0.0001.
Although many normal tissue sequencing studies use a single high sensitivity mutation calling algorithm, we elected to employ multiple algorithms to reduce false positive calls. For exome sequencing analyses, we assessed the results of 6 mutation callers: Mutect, Strelka2, Shimmer, Speedseq, Varscan, and Manta. We called mutations that were recognized by at least 3 of 6 algorithms, that had variant allele frequencies (VAFs) of 5–25% in liver specimens, and <5% in the blood. We applied these criteria (except that any VAFs above 5% were included) to public HCC data from 363 patients and found that our pipeline was able to identify the recurrent mutations in HCC (Figure S1C).
The diseased liver samples harbored extensive mutational burden and heterogeneity. Each tissue sample weighed between 4 and 28mg (mean = 16.5mg) and was approximately 2.5mm in each dimension (Figure 1A). A total of 389 somatic mutations were identified (Supplemental Table 2) with a mean of 4.74 (+/−0.94 SEM, range of 0–57) mutations per sample (Figure 1B). Each ~16mg liver sample contained approximately 5 out of the 500,000 lobules that comprise an average liver. Since the average human liver is approximately 1,500g and contains 90,000 of these tissue fragments, a conservative estimate would predict 400,000 mutations across an entire cirrhotic organ. Missense mutations were the most common, followed by synonymous, nonsense, splice-site, and frameshift mutations (Figure 1B). Next, we compared the mutation count and rate between liver tissues and cancers sequenced from other sources after reanalyzing all of the raw data with our pipeline. The absolute mutation count per sample in liver tissues was similar to gastric cancer and hepatoblastoma cohorts, but lower than Ewing’s sarcoma, rhabdomyosarcoma, kidney clear cell carcinoma, HCC, and lung adenocarcinoma cohorts (Figure 1C). When VAF was used to calculate mutation rate per base pair of genomic DNA, livers had a lower mutation rate than all of the analyzed cancers (Figure 1C). One confounding factor of this analysis is that liver samples are likely polyclonal, while cancer samples have undergone expansion of a dominant clone. Although mutation counts for HCCs were higher, the single-nucleotide variant (SNV) pattern was similar, supporting parallel mechanisms of mutagenesis (Figure 1D). As with other cancers, transition mutations were generally more frequent than transversion mutations (exchanges of purine for pyrimidine bases). In particular, C to T transition mutations were high in liver tissues, a phenomenon also seen in HCCs. Mutational signature analysis identified the defective DNA mismatch repair related signatures 6 and 15 (http://cancer.sanger.ac.uk/cosmic/signatures), the exposure to chewing tobacco mutagens related signature 29, and the exposure to smoking tobacco related mutagens related signature 4 (Figure 1E). Of these signatures, signature 6 and signature 4 were previously implicated in HCC, showing that the mutational processes identified within liver cancers began long before tumor initiation.
We then examined clinical correlates of mutation burden. We first noted that the patients without fibrosis (F0 fibrosis patients) had few mutations. Overall, we found a highly significant association between fibrosis stage and mutation count (Figure 1F and Supplemental Table 2; p = 0.0001). There was also a significant association between serum liver enzyme ALT and AST levels, used clinically to assess the extent of liver damage, and mutation count (Figure 1G). Together these indicated that higher levels of hepatic fibrosis, inflammation, and damage were associated with increasing numbers of clones carrying mutations. No associations were found between mutation count and age, sex, smoking, or disease etiology (HCV, HBV, or NASH). The presence of HCC was also not associated with mutation count, since the six noncancer bearing livers removed during organ transplantations harbored a similar mutation count as livers resected for HCC (p-value = 0.73).
Ultra-deep targeted sequencing identified recurrent somatic mutations
Since whole exome data yielded mainly 5–15% VAF mutations (Figure 1H), it was likely that a large proportion of somatic mutations with <5% VAF were left undetected. Therefore, we performed ultra-deep targeted resequencing of 136 genes (29 genes that were frequently mutated in HCC and 107 genes that were mutated in diseased liver exomes, listed in Supplemental Table 3) in all of the above 60 samples and an additional completely normal sample from a liver transplantation donor using the Agilent SureSelect platform with molecular barcoding, which afforded a higher sensitivity for mutations with VAFs under 5%. For ultra-deep sequencing, we used a modified set of mutation calling criteria. First, we exploited the molecular barcodes by using a preprocessing step to exclude sequencing artifacts. We then assessed the results of 7 mutation callers. LoFreq-star was added to increase sensitivity for low VAF mutations (Wilm et al., 2012). We identified mutations that were recognized by at least 3 of these 7 algorithms, that had VAFs of 0.5–25% in liver specimens, and <5% in the blood. Ultra-deep sequencing of paired blood samples allowed us to exclude low VAF clonal hematopoietic mutations that might have contaminated the liver. For 11 of the 61 patients, 4–7 biopsies per liver were sequenced to examine distinct and shared mutations across a larger swath of liver. The average sequencing depth was 1784x (also after duplicate removal) for blood and 1617x for liver.
A total of 214 somatic mutations were identified using the criteria described above. Within one liver sample, a mean of 1.66 (+/−0.32 SEM, range 0 to 11) mutations were identified even with stringent mutation calling (Figure S2A and Supplemental Table 3). As seen in exomes, missense mutations were the most common, followed by synonymous, frameshift, nonsense, and splice-site mutations (Figure S2B). Again, the proportions of mutation types (missense, nonsense, etc.) and SNVs were similar to what was found with exome sequencing of liver (Figure S2B and S2C) and HCC, but the genes in which these mutations occurred were often different than that of liver cancers. As with exomes, the ultra-deep data identified a significant correlation between mutational burden and fibrosis (Figure S2D and Supplemental Table 3; p = 0.00418). Targeted ultra-deep sequencing of two normal liver samples from a young, healthy transplant donor and a patient without liver disease yielded only 0 and 1 mutation, respectively. As expected, the mean VAF of mutations was lower in the ultra-deep sequencing as compared to exome sequencing (Figure S2E).
There was gene level recurrency between patients in PKD1 (13% of patients), KMT2D (10%), STARD9 (10%), APOB (10%), PPARGC1B (10%), ALMS1 (8%), ALB (8%), ARID1A (6%), TRP53 (6%), and PKHD1 (6%) (Figure 2). Many of these mutations were predicted to be deleterious by both Poylphen and SIFT (Supplemental Table 3) and due to the locations of the mutations across the genes (Figure S2F). The recurrency is likely underestimated because in most patients only one piece of tissue was sampled from the entire liver. Many of the most frequently mutated genes are not observed in liver cancers (PKD1, PPARGC1B, KMT2D, ALMS1, PKHD1), but a subset was prominent in TCGA HCC studies (ALB, APOB, ARID1A, ARID2, TP53). Only 5 of the 29 most mutated genes in HCC were detected in more than two patients and did not include CTNNB1, RB1, AXIN1, and KEAP1, some of the most established drivers of HCC (Figure 2). This suggested distinct selection pressures between HCC and background liver. Both PKD1 and PKHD1 are involved in monogenic polycystic liver disease in addition to their known roles in kidney disease (Tahvanainen et al., 2005). Multiple chromatin remodeling genes such as ARID1A, SUZ12, ARID2, ARID1B, SMARCA4, and BRD9 were also identified. Some of the mutations in recurrently altered genes were validated by Sanger sequencing (Figure S3).
Figure 2. Ultra-deep targeted sequencing reveals recurrently mutated genes within diseased liver tissues.

Sequencing of 136 genes was performed in 129 liver samples and paired blood from 61 patients. The waterfall plot for 26 genes with recurrent mutations is shown here. Genes previously found to be recurrently mutated in the HCC TCGA study are labeled in red.
Clonal expansion of mutations within nodules
The fundamental histological unit of the liver is the lobule, a collection of cells consisting of portal triads at the periphery and a central vein in the middle connected by hepatocytes and capillaries arranged in radial cords. Hepatocytes, or at least subsets of hepatocytes, have the capacity to expand in number in response to liver injury (Font-Burgada et al., 2015; Lin et al., 2018; Wang et al., 2015). During regeneration, lobules expand and develop into nodules, but it is unlikely that hepatocytes move beyond nodular boundaries. Because of this restrictive architecture, we did not expect mutations with VAFs above 25%, especially with bulk sequencing of samples that contained multiple nodules (see histologic measurement approach shown in Figure 3A). The fact that most whole exome and ultra-deep identified mutations had VAFs between 1–15% supported this hypothesis (Figure 1H and Figure S2E).
Figure 3. A comparison of mutant clone and nodule volumes indicates clonal expansion with increasing liver fibrosis.

A. The formula for nodule volume calculations based on measured nodule dimensions.
B. Individual mutant clone volumes calculated from ultra-deep sequencing VAFs (upper). Mutant clone volumes from F0–3 and F4 livers were compared (lower). There are 15 clones in F0–3 and 85 clones in F4 samples and each clone is represented by a blue circle.
C. Individual nodule volumes calculated using the measurements obtained in Figure 3A (upper). Nodule volumes from F0–3 and F4 livers were compared (lower). We measured 22 nodules in 11 F0–3 samples and 100 nodules in 53 F4 samples. Each nodule is a red circle.
D. Ratios of individual mutant clone volumes / average nodule volume of each sample (upper). The ratios from F0–3 and F4 livers were compared (lower). Each ratio is a green circle.
In order to understand the extent of and potential restrictions on clonal expansion, we calculated the volumes of mutant clones and measured the volumes of liver nodules. Specifically, we used the masses of sequenced tissues multiplied by the VAFs of the 214 mutations detected in ultra-deep sequencing. The resulting mean and median mutant clone volumes were 1.14mm3 and 0.59mm3 (Figure 3B). To compare these volumes with actual nodular volumes, we measured nodular dimensions from histology (Figure 3A). The calculated mean and median nodular volumes were 1.02mm3 and 0.73mm3 (Figure 3C and Supplemental Table 4). Since we could not precisely identify which mutations arose from which nodules, we could not determine if nodules came from single mutant cells using this data, but we could conclude that mutant clone and nodule volumes were similar (Figure 3B,C). This supported the idea that clone sizes could not expand far beyond nodule sizes and that spatial restrictions on clone size exist. To determine if mutant clones expand over time within nodules, we examined the ratio of individual mutant clone volumes to the average nodule volume per patient. This metric significantly increased in F4 livers compared with F0–3 livers (Figure 3D; p=0.0014). Thus, with time and chronic liver damage, nodules are more likely to contain increasingly clonal populations of mutant cells, suggesting clonal dominance. Altogether, our data showed that both the number and volume of mutant clones increased over time as a function of liver fibrosis and damage.
To test the idea that nodule architecture can physically restrain clonal outgrowths, we analyzed the multi-site ultra-deep sequencing performed on 4–7 tissue pieces from the same livers in 11 patients (Figure 4). We did not know which pieces were adjacent and which were distant because the original positions were not mapped. Out of a total of 119 mutations found in these patients, only 7 pairs were shared. The small number of overlapping mutations supported the concept that nodular architecture restrains expansive clonal outgrowths, and thus most mutations within nodules are private. This also confirmed that we were not merely detecting mutations from clonal hematopoiesis, which would more likely be present in all pieces from the same patient. The shared mutations likely indicated that the tissue pieces were directly adjacent and shared a mutant nodule.
Figure 4. Venn diagrams representing livers sequenced at multiple locations.

In these diagrams, each box represents one patient, each circle represents one piece of liver, and each number represents the mutation count within a piece. Intersecting circles depict mutations that are shared. Circle size scales with mutation number, not tissue size. A table of mutation counts per piece of liver is at the bottom.
Chromosome level copy number variations can be observed in diseased livers
Besides mutations, chromosome level aneuploidy is an important class of somatic variation that is hypothesized to accumulate during liver damage (Duncan et al., 2010, 2012a, 2012b). Chromosome level variations can be called with exome sequencing (Zare et al., 2017), so we examined somatic copy-number alterations in cancer and liver specimens using CNVkit. To first test if the CNVkit pipeline could identify recurrent CNVs previously confirmed by Affymetrix 6.0 SNP arrays, we called CNVs in 317 published TCGA HCC samples (Figure S4A). In agreement with TCGA, we also identified recurrent copy number gains in 1q and 8q as well as losses in 8p and 17p within HCCs. To further support the validity of CNV calls, we correlated mRNA expression with CNVs. Figure S4B showed that across all genes in the human genome, gene expression is more likely to be positively associated with their CNVs than negatively associated. We also examined mutations in association with CNVs, and found that genes with copy number loss harbored fewer somatic mutations (Figure S4C). In most diseased livers, our results showed few clear examples of chromosome level CNV changes (representative plot for one sample shown in Figure S4D). This lack of evidence for aneuploidy is consistent with single cell CNV sequencing performed in mouse livers (Knouse et al., 2014). However, two samples without any histologic evidence of dysplasia or cancer showed clear gains of 1q and 8q (Figure S4E). When CNVs from all 82 patients were averaged, disparate regions in chromosome 19 showed recurrent gains, but it is unclear if these represented chromosome level ploidy changes or more localized duplications (Figure S4F). This data showed that aneuploidy, though rare, can be detected in bulk liver samples and are potentially drivers of clonal expansion.
In vivo CRISPR screening identified genes that mediate clonal hepatic expansion
To identify genes that regulate hepatic regeneration in the context of chronic liver disease, we designed a CRISPR/Cas9 screen in Fumarylacetoacetate hydrolase (Fah) knockout (KO) mice. Fah KO mice model a monogenic degenerative liver disease called hereditary tyrosinemia, which if left untreated eventually leads to cirrhosis. This disease and model are effectively “cured” by treatment with a drug called nitisinone (NTBC), which inhibits accumulation of hepatotoxic metabolites associated with impaired tyrosine catabolism (Figure S5) (Grompe et al., 1995; Overturf et al., 1996). Thus, KO livers exert a selection pressure for wild-type or FAH producing hepatocytes. We generated a plasmid designed to transiently express a transposon that co-expressed Cas9, an sgRNA against any gene of choice, and Fah (used as a selection marker) (Figure 5A,B). First, we showed that mice hydrodynamically injected only with the transposon plasmid died after NTBC withdrawal, whereas mice receiving both transposon and Sleeping Beauty transposase (SB100) plasmids remained healthy, as would be expected if liver function had been rescued by FAH expression (Figure 5C). Within one week, livers receiving sgRNAs targeting Pten harbored PTEN-deficient, FAH-positive hepatocytes and after one month, most hepatocytes were FAH positive and Pten deficient (Figure 5D,E). In these Pten targeted mice, whole liver steatohepatitis was also observed on gross and microscopic inspection (Figure 5F), recapitulating the phenotype of classical liver-specific Pten knockout mice (Qiu et al., 2008). In contrast, livers receiving a nontargeting sgGal4 did not perturb Pten but did generate FAH-positive cells, and these livers appeared completely normal (Figure 5D, Supplemental Table 5). These experiments confirmed the expected behavior of individual sgRNAs in this in vivo system.
Figure 5. In-vivo CRISPR screening identified genes that increase clonal expansion.

A. Sleeping Beauty transposon used for stable hepatocyte expression of FAH, Cas9, and sgRNA. The plasmid was injected intravenously.
B. Schema of in vivo loss-of-function screen to identify genes that regulate liver regeneration using the Fah KO hereditary tyrosinemia model.
C. Body weights of Fah KO mice undergoing liver repopulation after plasmids were delivered +/− SB100 transposase plasmid by hydrodynamic transfection (HDT). NTBC was withdrawn immediately after HDT. Data are represented as mean ± standard deviation; n = 3 mice per group.
D. FAH IHC staining shows FAH+, PTEN negative hepatocytes one week after HDT of plasmids carrying Fah, Cas9, and a Pten sgRNA (scale bar = 50 μm).
E. Representative IHC staining one month after transposon HDT (scale bar = 100μm).
F. The appearance of livers 1 month after transposon HDT.
G. Scatterplot showing average enrichment of individual sgRNAs after liver repopulation from 5 independent replicates. The sgRNA count was defined as the number of sequencing reads that perfectly match the sgRNA target sequence (see Supplemental Table 5).
H. Identification of candidate genes using positive robust rank aggregation (RRA) score as assessed by MAGeCK. The RRA score reflected whether or not the distribution of sgRNAs targeting a gene were significantly skewed within a ranked list of all sgRNAs. Assuming that if a gene had no biological effect, then sgRNAs targeting this gene should be uniformly distributed (Li et al., 2014).
I. Top 10 gene candidates in the repopulated liver based on RRA scores. The screen was performed in 5 independent mouse replicates. A red square means that the gene was found within the top 10 enriched genes in that screened mouse.
To broadly assess the genes implicated in liver sequencing, we generated an sgRNA library with 882 guides targeting 147 mutated genes identified in our sequencing studies (Figure 5B, Supplemental Table 5). This pool of transposon plasmids encoding sgRNAs along with FAH was hydrodynamically delivered into Fah KO mice such that each of five mice received the entire library, then NTBC was withdrawn. In this way, many clones of transfected and FAH-rescued hepatocytes bearing different sgRNAs would compete with each other during liver regeneration and sgRNAs that conferred a selective advantage in regenerating hepatocyte clones would become over-represented relative to other clones. One month later, repopulated sgRNAs were quantified by deep sequencing. Distinct sgRNAs against Arid1a, Pten, Pkd1, and Kmt2d were consistently enriched among individual mice subjected to the screen (Figure 5G-I and Supplemental Table 5). Pten and Arid1a encode well characterized tumor suppressors known to influence liver growth. We previously showed that Arid1a-deficient hepatocytes have enhanced regenerative capacity in the liver (Sun et al. 2016). Kmt2d and Pkd1 had not previously been identified as regulators of liver regeneration or HCC. The most enriched genes in the screen (Pkd1, Kmt2d, and Arid1a) corresponded to the most recurrently mutated genes in the sequencing studies (Figure 2 and Figure 5G-I), demonstrating that the mouse screen recapitulated key aspects of human liver disease progression.
Gene-targeted mice confirm genes that regulate liver regeneration
To more rigorously assess the function of new regulators of liver regeneration, we conditionally deleted Arid1a, Kmt2d, or Pkd1 from hepatocytes in mice. As mentioned previously, the mutation pattern and computational predictions suggested loss of gene function (Figure S2F). Because the mutations in human diseased livers likely impact only one of two alleles in most clones, we conditionally deleted one copy of Arid1a in the adult liver using Albumin-Cre to achieve a more realistic partial loss-of-function. We then treated mice with classic liver injury assays using hepatotoxic chemicals such as CCl4 (a centrilobular toxin) and DDC (a biliary toxin that causes cholestasis) (Figure 6A). Arid1a heterozygous livers showed modestly reduced liver injury after CCl4 treatment (Figure 6B), but were substantially protected from DDC as indicated by lower bilirubin levels and higher liver/body mass ratios (Figure 6C). Consistent with the results of the CRISPR screen, these data demonstrated that even a partial loss-of-function in Arid1a conferred a selective advantage upon hepatocytes in response to liver injury.
Figure 6. Arid1a and Kmt2d heterozygosity protect against chemical liver injuries.

A. Schema for Aridla experiments. Arid1afl/+ mice or Arid1afl/+; Alb-Cre mice were injected with a single dose of CCl4 to induce acute liver injury. In a separate experiment, DDC diet was given for 2 weeks and then a normal diet for 3 days to evaluate liver injury and recovery.
B. Serum alkaline phosphatase (ALKP) from Arid1afl/+ mice or Arid1afl/+; Alb-Cre mice at baseline and 24 hours after a single dose of CCl4 (n = 7 and 6 for baseline and 7 and 5 for CCl4).
C. Serum total bilirubin after 2 weeks of DDC diet and liver/body mass ratios after 2 weeks of DDC diet and 3 days of normal diet (n = 10 and 8).
D. Schema for Kmt2d experiments. AAV-TBG-Cre (5×1010) was injected intravenously into Kmt2dfl/+ mice to delete one allele in hepatocytes. AAV-TBG-GFP was injected as control. Two weeks later, mice were injected with one dose of CCl4. In a separate set of experiments, mice were put on T3 diet for 1 month to enforce hepatocyte proliferation.
E. Serum ALT or AST measured 24 hours after CCl4 (n = 4 and 5).
F. H&E staining showing hepatic necrosis 48 hours after CCl4. Scale bar: 2000μm.
G. Quantification of necrosis on H&E and liver/body mass ratios, assessed 48 hours after CCl4 (n = 4 and 5).
H. Proliferation as assessed by Ki-67 one month after T3 diet. Scale bar: 100μm.
I. Quantification of Ki-67 positive cells and liver/body mass ratios after T3 diet (n = 3 and 4).
All data are presented as mean ± SEM. *, p<0.05, **, p<0.01.
We next examined Kmt2d loss by delivering hepatocyte targeting adeno-associated virus (AAV)-TBG-GFP or AAV-TBG-Cre to heterozygous Kmt2dfl/+ mice. We first showed that neither virus had an independent effect on the CCl4 liver injury assay (Figure S6A). After a single dose of CCl4 (Figure 6D), Kmt2d heterozygosity protected from liver damage, as indicated by reduced serum transaminases (Figure 6E) and reduced hepatocyte necrosis (Figure 6F,G). To determine if hepatocytes with just one copy of Kmt2d also showed altered proliferative capacity, we subjected mice to a diet with increased Triiodothyronine (T3) thyroid hormone to induce hepatocyte proliferation ((Fanti et al., 2014); Figure 6D). Kmt2d heterozygous mice showed a significantly increased proliferative response as quantified by Ki-67 staining (Figure 6H,I). The combination of decreased cell death and increased proliferation indicated that a partial loss-of-function in Kmt2d conferred a selective advantage upon hepatocytes in response to liver injury.
We also asked if heterozygous loss of Pkd1 could influence hepatocyte fitness (Figure 7A). After delivering control AAV-TBG-GFP or AAV-TBG-Cre to Pkd1+/+ or Pkd1fl/+ mice (Piontek et al., 2004), we observed that Pkd1 heterozygous vs. control livers had reduced serum transaminases and hepatocyte necrosis after CCl4 (Figure 7B-D and Figure S6B). Interestingly, there was no concomitant increase in Ki-67 staining (data not shown), suggesting that Pkd1 heterozygosity impacted hepatocyte survival but not proliferation. We further assessed Pkd1 heterozygous livers after multiple doses of CCl4 and found that heterozygous mice had significantly reduced fibrosis than did control mice (Figure 7E,F). This demonstrated that loss of one Pkd1 allele could decrease chronic hepatic fibrosis. These data confirmed that a number of recurrent genetic changes conferred an adaptive advantage on hepatocytes in the face of chronic liver injury.
Figure 7. Pkd1 heterozygosity protects against CCl4 induced necrosis and fibrosis.

A. Schema for Pkd1 experiments. AAV-TBG-Cre (5×1010) was injected intravenously into Pkdfl/+ mice to delete one Pkd1 allele in the liver. AAV-TBG-GFP was injected to generate control mice. Two weeks later, mice were injected with one dose of CCl4 to induce injury. In a separate experiment, 12 weeks of biweekly CCl4 was used to induce chronic damage and liver fibrosis.
B. Serum ALT and AST in Pkd1 het mice 24 hours after CCl4 injection (n = 11 and 11).
C. Necrotic cells (circled in yellow) in Pkd1 heterozygous livers 48 hours post CCl4 injection. Scale bar: 200μm.
D. Quantification of necrosis 48 hours post CCl4 injection (n = 11 and 11).
E. Sirius Red staining of liver sections after 12 weeks of biweekly CCl4 injections. Scale bar: 500pm.
F. Quantification of Sirius Red staining after chronic CCl4 injury (n = 9 and 9).
All data are presented as mean ± SEM. *, p<0.05, **, p<0.01.
Discussion
Decades of liver damage can result in cirrhosis, a pathologic state associated with tissue dysfunction and in some cases, multifocal tumorigenesis. This observation is attributed to the “field effect” within damaged livers, but the molecular characteristics of the field remain incompletely defined. Until now, DNA level genomic changes have not been successfully measured in the earliest phases of liver damage, potentially due to sequencing approaches focused only on selected cancer genes (Nault et al., 2013, 2014). Our results showed that mutations accumulate in liver tissues as they become more damaged and fibrotic. The average piece of diseased liver the size of a raindrop contains 1–2 mutations, indicative of a high level of somatic mutagenesis across the organ. Many normal tissue sequencing studies have used single callers with multifocal sampling to emphasize sensitivity. We intersected multiple callers in an attempt to increase specificity, potentially at the expense of sensitivity. Even so, it appears that the rate of mutation in diseased liver is lower than that of skin or esophagus, but probably higher than that of aged colonic crypts (Lee-Six et al., 2018a). It is possible that tissues restricted by architectural boundaries have a lower detectable mutation rate due to an inability for cells to proliferate without restrictions. Caveats of this comparison include differences in disease processes, sampling, sequencing breadth, sequencing depth, and mutation calling between studies.
Though previously untested, the biological relevance of recurrent somatic alterations occurring in normal tissues have generally been hypothesized to 1) drive clonal expansion, and 2) represent steps towards transformation. An in vivo screen allowed us to interrogate the first assumption and prioritize functional genes in an orthogonal fashion with respect to genomics. It was remarkable that ARID1A was one of the top hits in both the CRISPR screen and sequencing analyses, since we had previously shown a clear role for this gene in regeneration (Sun et al., 2016). Other recurrently mutated genes such as PKD1 and KMT2D also demonstrated unexpected but robust fitness promoting effects in the context of injury. Kmt2c and Kmt2d knockout mice were previously shown to protect against fatty liver disease (Kim et al., 2015, 2016), but had not been studied in liver regeneration. It is unknown if clonal expansion is dependent on mutations occurring in particular stem or progenitor cells marked by SOX9, TERT, or AXIN2 (Font-Burgada et al., 2015; Lin et al., 2018; Wang et al., 2015). While it is known that the TERT promoter is frequently mutated in cirrhosis and also likely functions to promote hepatocyte fitness, we did not focus on non-coding mutations in this study. Also, mutations in SOX9 or AXIN2 were not observed at an appreciable frequency. Overall, the mutational recurrency, in vivo CRISPR screening, and animal modeling experiments supported the idea that somatic mutational mechanisms impinge on pathways that promote cell proliferation and resistance to toxic insults, in addition to pathways that lead to transformation. Potential “benefits” of normal tissue mutations have not been highlighted in the context of clonal hematopoiesis or squamous tissue mutations (Lee-Six et al., 2018b; Martincorena et al., 2015, 2018), although it is likely that adaptive mutations also exist in those settings.
In the liver, many recurrently mutated genes are not known to be major drivers of cancer initiation or progression. For example, PKD1, PKDH1, and PPARGC1B mutations are not commonly detected in cancer. This is in line with recent findings in the esophagus, which show that NOTCH1 is recurrently mutated in normal but not as frequently in cancer samples (Martincorena et al., 2018). It is likely that these mutational events promote regeneration in normal cells without driving or even impairing transformation. Two recurrently mutated genes from our study could fit this model. PPARGC1B is considered one of the master regulators of mitochondrial biogenesis and de novo lipogenesis. Intriguingly, overexpression of PPARGC1B in the liver promoted hepatocyte injury, fibrogenesis, and HCC development while liver-specific deletion reduced injury and HCC development (Piccinin et al., 2018). Moreover, PPARGC1B mutations are observed in less than 1% of HCCs. Thus, the deleterious PPARGC1B mutations found in diseased liver samples would be predicted to reduce transformation risk.
Another example is PKD1, which is involved in autosomal dominant polycystic kidney and liver disease. Despite having whole-body haploinsufficiency, PKD1 patients are not at higher risk for HCC, even though patients can develop massive cysts throughout the liver. These patients also do not suffer from liver dysfunction (Hogan et al., 2015). Likewise, PKD1 mutations are rarely observed in HCC or cholangiocarcinoma, and there is evidence that PKD2 overexpression is oncogenic in HCC (Zhu et al., 2016). We showed that Pkd1 heterozygosity resulted in reduced hepatocyte death and fibrosis after injury, raising the possibility that these mutations could make the microenvironment less cancer-prone. Although one might expect that a fitness promoting mutation should represent a step closer to transformation, we have shown that regenerative expansion of hepatocytes is distinct from the clonal expansion of cancer cells. We challenge the simplicity of the concept that cells accumulate mutations on a linear path toward malignancy, since some mutations could protect or even suppress cancer.
A consistent finding from this and other normal tissue sequencing studies is the highly frequent accumulation of mutant clones that do not seem destined for cancer. Besides the specific genetic mechanisms involving PPARGC1B and PKD1, what other mechanisms might prevent mutant clones from transforming? One possibility for the liver is tissue architecture: lobule boundaries likely prevent unfettered expansion. Bridging fibrosis could further serve to restrain expanding nodules, especially in the context of mutations that drive hepatocyte proliferation. Our study shows that mutation burden is tightly correlated with fibrosis stage. Instead of mutations driving tissue injury and fibrosis, it is possible that fibrosis could also be a mechanical “fence” to constrain transformation. Another possibility is that the immune system culls mutant clones before they can expand. Indeed, we found that mutations which give rise to computationally predicted neoantigens have lower VAFs than other non-immunogenic mutations (Figure S7A,B). This suggested that somatic mutations in damaged livers can already stimulate immunosurveillance, as is observed in cancers. This mechanism is consistent with prior studies demonstrating immunosurveillance of hepatocytes expressing mutant cancer genes (Kang et al., 2011). Our study shows that mutations in cirrhosis are common, functional, and in some cases adaptive. Besides defining mutations and their impact on normal tissues, the understanding of mutation burden in liver disease also has clinical implications for cirrhosis staging and early cancer detection using deep sequencing approaches.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources should be directed to and will be provided by the Lead Contact, Hao Zhu (hao.zhu@utsouthwestern.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All mice were handled in accordance with the guidelines of the Institutional Animal Care and Use Committee at UT Southwestern. Fah KO mice were obtained from Yecuris. In Aridla floxed mice, induced deletion between the two loxP sites produces cells lacking exon 8 of Aridla (X. Gao et al., 2008). Aridla floxed mice were crossed with Albumin-Cre mice to generate Arid1afl/+; Alb-Cre mice. Kmt2d floxed mice were obtained from Kai Ge’s lab (JE. Lee et al, 2013). Pkd1 floxed mice were obtained from the UT Southwestern George M. O’Brien Kidney Research Core (NIH P30DK079328). All experiments were done in 6 to 10 week old mice. Both male and female mice were used in this study. In each experiment, mice were age and sex matched.
Human samples
All patients provided informed consent under IRB #STU 062013–063 for liver tissues and #STU 092013–010 for blood samples. A total of 129 pieces of non-malignant liver tissues from 61 UT Southwestern patients were selected for genomic studies. 59 patients underwent liver resections and had different stages of liver fibrosis. One was a healthy liver transplant donor. Another was a patient with colon cancer metastases in the liver. The mean age of the patients was 60 years old. 48 patients were male and 13 were female. Genomic DNA was extracted from 4–28 mg tissue fragments using the Qiagen Allprep DNA/RNA miniprep kit. Pathological review and fibrosis staging were confirmed by a board-certified pathologist specializing in gastrointestinal oncology and liver cancer (P.G.). The samples had different stages of liver fibrosis but none contained dysplastic, potentially malignant (adenoma), or malignant (HCC or cholangiocarcinoma) nodules. Only regenerative or cirrhotic nodules and no dysplastic nodules were identified in the UT Southwestern samples based on WHO criteria (Supplemental Table 1). We used histologic scoring systems developed to grade inflammation and stage fibrosis (Guido et al., 2011; Lefkowitch, 2007). The determinants of inflammatory activity were lymphocytic piecemeal necrosis, lobular necroinflammation, and portal inflammation, which were graded 0 to 4. A patient was considered to have cirrhosis if the fibrosis stage was 4. All clinical information including disease stage, gender, and age are in Supplemental Table 1.
METHOD DETAILS
Histology, immunohistochemistry, and immunofluorescence
Tissue samples were fixed in 4% paraformaldehyde (PFA) and embedded in paraffin. Primary antibodies against Ki-67 (Abcam, ab15580), PTEN (Cell Signaling, CST#9559S), and FAH (Yecuris, 20–0034) were used. Detection was performed with the Elite ABC Kit and DAB Substrate (Vector Laboratories), followed by hematoxylin counterstaining (Sigma).
Whole exome sequencing, processing, and mutation calling
Exome sequencing was performed by Admera Health. Exome capture was performed using the xGen Exome Research Panel (Integrated DNA Technologies) according to the manufacturer’s protocols. Captured nucleotides were subjected to 150bp paired-end sequencing on an Illumina Hiseq platform at Admera Health. We used a mutation calling pipeline for somatic mutation calling developed by the Quantitative Biomedical Research Center (QBRC) at UT Southwestern. Exome-seq data quality was examined by fastqc (https://www.bioinformatics.babraham.ac.uk/proiects/fastqc/). Exome-seq reads were aligned to the GRCh38 genome by BWA-MEM (Li and Durbin, 2009). Picard was used to add read group information and sambamba was used to mark PCR duplicates. The calculation of read coverage was performed after duplicate removal. The GATK toolkit (DePristo et al., 2011; McKenna et al., 2010; Van der Auwera et al., 2013) was used to perform base quality score recalibration and local realignment around Indels. MuTect (Cibulskis et al., 2013), VarScan (Koboldt et al., 2012), Shimmer (Hansen et al., 2013), SpeedSeq (Chiang et al., 2015), Manta (Chen et al., 2016), and Strelka2 (Saunders et al., 2012) were used to call SNPs and Indels. A mutation called by ≥ any 3 of these algorithms was retained. Annovar was used to annotate SNPs and Indels (Wang et al., 2010). All SNPs and Indels were combined and kept if there were > 7 total (wild-type and variant) reads in the blood sample and > 3 variant reads in the liver sample. Somatic mutations and germline mutations were annotated according to the VAFs in the liver and normal blood samples. Mutations were filtered out if they appear with >1% population frequencies in any of the ESP6500, ExAC, and 1000 Genome cohorts. As we were working with a small cohort of unrelated patients, a mutation was further excluded if it appeared in more than 25% of all patients. CNVkit was used to call copy number variation (Talevich et al., 2016).
Ultra-deep targeted sequencing, processing, and mutation calling
To detect mutations with low VAFs in highly heterogeneous tissues, we performed ultra-deep target resequencing with Agilent SureSelectXT HS with molecular barcodes to exclude false positive mutations derived from sequence or amplification errors. 136 genes from 129 liver samples obtained from 61 patients were sequenced. Blood DNA from the same patient was used as a control. 100ng of input genomic DNA was used for library construction. DNA was sheared to 150–200bp using the Covaris ME220 system. PCR cycling conditions were chosen according to the Agilent library preparation protocol. Targeted libraries were submitted to Admera Health for sequencing at a depth of 2000x using the Illumina HiSeq platform. Sequences were aligned and processed using the same QBRC pipeline described above, except that we also used the Agilent Genomics NextGen Toolkit to process the molecular barcode data and curate the consensus read sequences between the alignment and recalibration. We also added LoFreq-star (Wilm et al., 2012) to the mutation caller pipeline.
Mutant clone calculations and nodule volume measurements
Mutant clone volumes were calculated as VAF × 2 × m/ρ for each mutation. m refers to the mass of the cirrhotic sample (Supplemental Table 4) and ρ is an estimate of liver tissue density, previously measured to be 1.051g/ml (Overmoyer et al., 1987). For nodule volume measurements, H&E stained slides were scanned with a Hamamatsu Nanozoomer 2.0HT scanner (UT Southwestern Whole Brain Microscopy Facility). The diameters of each nodule (long and short) were measured using NDP.view2 software. When images obtained with the scanner were not clear enough, we used a microscope to take pictures with a 4x objective and analyzed the data with ImageJ. Nodule volumes were calculated using the formula for ellipsoid volume: Volume = (4/3) *π*a*b*c where a = L/2,b = S/2,c = (a + b)/2, where L = long diameter and S = short diameter (Figure 3A). For each piece of tissue, 1–3 nodules were measured. Please also see Supplemental Table 4 for measurement data.
Neoantigen prediction pipeline
We used a QBRC pipeline for neoantigen calling. We started neoantigen analysis with somatic mutations called by the QBRC mutation calling pipeline. We used only frameshift, non-frameshift, missense, and stoploss mutations that were predicted to lead to protein coding changes. We kept only somatic mutations whose VAFs were < 0.02 (2%) in the blood sample and > 0.05 (5%) in the liver samples. For class I HLA proteins (A, B, C), we predicted the neoantigens of 8–11 amino acid in length, and for class II HLA proteins (DRB1 and DQB1/DQA1), we predicted the neoantigens of 15 amino acid in length. Class I and II HLA subtypes were predicted by the ATHLATES tool (Liu et al., 2013). Putative neoantigens with amino acid sequences exactly matching known human protein sequences were filtered out. For class I bindings, IEDB recommended mode (http://tools.iedb.org/main/) was used for prediction of binding affinities, while for class II binding, NetMHCIIpan embedded in the IEDB toolkit was used for prediction of binding affinities. Neoantigens were kept only if the predicted ranks of binding affinities were <2% when compared to an atlas of wild-type peptides. Liver RNA-seq data were aligned to the hg38 reference genome using the STAR aligner (Dobin et al., 2013). FeatureCounts was used to summarize gene expression levels (Liao et al., 2014). Neoantigens whose corresponding mutations were in genes with expression level <1 RPKM in either the specific exon or the whole transcript were filtered out.
Sanger sequencing
Target regions of genomic DNA were PCR amplified (see primers from Supplemental Table 6) and products were TA cloned into pCR™4Blunt-TOPO vector using Zero Blunt TOPO Cloning Kit (Invitrogen). Single clones were picked and submitted to Genewiz for Sanger sequencing using the M13 reverse primer.
mRNA sequencing
18 liver samples underwent RNA-sequencing. Total RNA was extracted from 15–25 mg of tissue with the Invitrogen PureLink RNA mini kit. RNA was sent to Admera Health for RNA library preparation and sequencing. Truseq stranded library preparation with Ribozero rRNA depletion was performed. 100 million paired-end 150bp reads were sequenced for each sample on the Novo-seq platform.
In vivo CRISPR screening
We custom designed the sgRNAs for 147 mutated genes identified in our sequencing studies. The library consisted of 882 sgRNAs, which included 6 guides targeting each of the 147 genes and 15 non-targeting control guides (Supplemental Table 5). sgRNA sequences were extracted from the mouse Gecko v2 library (Shalem et al., 2014), which were designed against coding exons. The sgRNAs were synthesized and subcloned into our transposon vector with good representation (>99%) and high uniformity across sgRNAs (Figure 5A). 5μg of plasmid and 1μg of SB100 transposase plasmid were hydrodynamically injected into Fah KO mice. NTBC water was withdrawn immediately after injection. After one month, the liver was collected and DNA was extracted from the whole liver. The sgRNA was amplified and sequenced on a NextSeq500. sgRNA representation was analyzed by the MAGeCK algorithm.
Liver injury experiments
To induce acute liver injury, CCl4 (Sigma) was diluted 1:10 in corn oil and injected once intraperitoneal (IP) at a dose of 0.5ml/kg (Beer et al., 2008). Blood was collected at baseline prior to and 24 hours after injection. 48 hours after CCl4 injection, mice were euthanized and the livers were analyzed by histology. To induce liver fibrosis, mice were injected with CCl4 twice per week for a total of 12 weeks. The T3 hormone diet was made by adding 4 PPM (0.0004%) T3 (Sigma) to standard rodent chow (TestDiet). Mice were put on T3 diet for 1 month before hepatocyte proliferation was assessed using Ki-67. DDC diet was made by adding 0.1% DDC (Santa Cruz) to standard rodent chow (TestDiet). Mice were put on DDC diet to induce biliary injury. Liver function was monitored at baseline prior to and 2 weeks after DDC diet was provided.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis
The data in most figure panels reflect multiple experiments performed on different days using mice derived from different litters. Variation is indicated using standard error of the mean (SEM) and presented as mean ± SEM. Unless otherwise stated in the figure legends, two-tailed Student’s t-tests (two-sample equal variance) were used to test the significance of differences between two groups. Statistical significance is displayed as * (P < 0.05), ** (P < 0.01), *** (P < 0.001), ****(P < 0.0001).
DATA AND SOFTWARE AVAILABILITY
Data availability
The whole exome sequencing data, ultra-deep targeted sequencing data, and RNA sequencing data reported by this paper were deposited in the European Genome-phenome Archive (EGA) database: https://www.ebi.ac.uk/ega/home. The identifier is EGAS00001003496.
Code availability
The QBRC somatic mutation and neoantigen calling pipelines are available on GitHub: https://github.com/Somatic-pipeline/Somatic-pipeline and https://github.com/Neoantigen-pipeline/Neoantigen-pipeline.
Supplementary Material
Supplemental Figure S1. Data related to Figure 1.
A. Clinical information for 60 UT Southwestern patients.
B. H&E histology of samples with F0 and F4 fibrosis. Scale bar is 200pm for 4x pictures and 100μm for 10x pictures.
C. Waterfall plot for mutations called in 319 TCGA HCC samples.
Supplemental Figure S6. AAV-TBG-GFP and AAV-TBG-Cre do not exert independent biological effects. Data related to Figures 6 and 7.
A. Wild-type CD1 mice were either uninjected or injected with AAV-TBG-GFP or AAV-TBG-Cre virus at a dose of 5×1010 viral particles per mouse. 11 days after virus injection, all three groups of mice were injected with one dose of 10% CCl4 intraperitoneally to induce liver injury. Left panel: H&E staining showing hepatic necrosis 48 hours after CCl4 injury. Scale bar: 200μm. Right panel: Necrotic area was quantified (n = 8,8,9).
B. Pkd1+/+ and Pkd1fl/+ mice were injected with AAV-TBG-GFP or AAV-TBG-Cre virus. 11 days after virus injection, mice were injected with CCl4. Left panel: H&E staining showing hepatic necrosis 48 hours after CCl4 injury. Scale bar: 200μm. Right panel: Necrotic area was quantified (n = 10, 10, 9, 10).
Supplemental Figure S7. Neoantigen prediction and analysis. Data related to Figure 1.
A. Number of predicted neoantigens in 17 patients.
B. VAF of clones that have mutations predicted to result in neoantigen presentation. The average VAFs of mutations that generate neoantigens, divided by the average VAF of all mutations. Then a logarithmic transformation is performed. Mutations with negative VAF differences are selected against by T cells, suggestive of a disadvantaged survival fitness of immunogenic mutations.
Supplemental Table 1: Clinical and pathological data about the patients sequenced in the study. This is related to Figures 1–4 and STAR Methods.
Supplemental Table 2: Whole exome sequencing data for 82 patients in the study. This is related to Figure 1.
Supplemental Table 3: Targeted ultra-deep sequencing data for 61 patients in the study. This is related to Figures 2–4.
Supplemental Table 4: Mutant clone and nodule volume measurements. This is related to Figure 3.
Supplemental Table 5: sgRNAs used in the CRISPR screen are in the first sheet and the screen results are in the second sheet. This is related to Figure 5.
Supplemental Table 6: Primers for Sanger sequencing. This is related to the STAR Methods.
Supplemental Figure S2. Characterization of ultra-deep targeted sequencing results. Data related to Figure 2.
A. Mutation counts in each of the 61 patients and each of the 129 samples. An additional patient without liver disease was added to this ultra-deep sequencing group.
B. Types of mutations. Among 214 mutations, 131 are missense, 23 are frameshift, 8 are in-frame deletions, 6 are nonsense, and 49 are synonymous.
C. SNV classes for 131 missense mutations are shown here.
D. Correlation between mutation count and fibrosis stage. The number of patients with each fibrosis score is listed under the graph. The p-value is calculated based on the one-way Jonckheere trend test.
E. VAF distribution for ultra-deep targeted sequencing data.
F. Mutations identified in ARID1A, KMT2D, and PKD1 are shown with respect to protein domains.
Supplemental Figure S3. Sanger sequencing confirms select recurrent mutations. Data related to Figure 2.
Supplemental Figure S4. Chromosome level CNVs are observed but not frequently identified in diseased liver tissues. Data related to Figure 1.
A. Mean CNVs of all 317 HCC samples from TCGA called by the CNVkit algorithm.
B Correlation between mRNA expression and copy number. More genes are positively correlated with their CNVs than negatively correlated (Y-axis). This trend is clearer when visualizing genes that are more actively going through CNV changes (larger median absolute deviation of copy number) (X-axis), which could have a stronger impact on transcription. We analyzed all 17 patients who underwent both whole exome and RNA sequencing.
C. Regions with copy number loss have fewer mutations detected.
D. CNVs from a representative sample (HS122) with few alterations.
E. CNVs for two samples with chromosome 1 and 8 gains (HS37, HS10).
F. Mean CNVs from all 82 liver samples.
Supplemental Figure S5. The tyrosine metabolism pathway is defective in hereditary tyrosinemia, a disease caused by deleterious mutations in Fah. Related to Figure 5.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Ki-67 | Abcam | Cat# ab15580; RRID: AB_44320 |
| Anti-PTEN (138G6) | Cell Signaling | Cat# 9559; RRID: AB_390810 |
| Anti-FAH | Yecuris | Cat# 20–0034; RRID: N/A |
| Bacterial and Virus Strains | ||
| AAV8.TBG.PI.eGFP.WPRE.bGH | Addgene | Cat# 105535-AAV8 |
| AAV8.TBG.PI.Cre.rBG | Addgene | Cat# 107787-AAV8 |
| Biological Samples | ||
| Human liver samples | UT Southwestern and Parkland Memorial Hospital | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Carbon tetrachloride | Sigma-Aldrich | Cat# 289116 |
| The Triiodothyronine (T3) hormone | Sigma-Aldrich | Cat# T6397–250MG |
| 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC) | Santa-Cruz Biotechnology | Cat# SC-239721 |
| T3 diet (4 PPM, 0.0004%) | TestDiet | 1816922–204 |
| 0.1% DDC diet | TestDiet | 1816695–204 |
| Critical Commercial Assays | ||
| SureSelect XT HS Low Input | Agilent | Cat# G9707B |
| Deposited Data | ||
| Whole exome sequencing | This paper | EGAS00001003496 |
| Ultra-deep targeted sequencing | This paper | EGAS00001003496 |
| RNA sequencing | This paper | EGAS00001003496 |
| Experimental Models: Cell Lines | ||
| None | ||
| Experimental Models: Organisms/Strains | ||
| Mouse: Arid1a exon 8 floxed | Laboratory of Zhong Wang (Michigan) | X. Gao et al., 2008 |
| Mouse: Alb-Cre | The Jackson Lab | https://www.jax.org/strain/003574 |
| Mouse: Kmt2dfl/fl | Laboratory of Kai Ge | JE. Lee et al., 2013 |
| Mouse: Pkd1fl/fl | UTSW George M. O’Brien Kidney Research Core | N/A |
| Mouse: CD1 | Charles River | Strain #022 |
| Mouse: Fah KO | Yecuris | N/A |
| Oligonucleotides | ||
| sgPten: CACCGACTTGTCCTCCCGCCGCGT | Reference: Inducible in vivo genome editing with CRISPR/Cas9 | Dow et al., 2015 |
| Guides for CRISPR screening, see Supplemental Table 5 | the mouse Gecko v2 library | Shalem et al., 2014 |
| Primers for Sanger sequencing, see Supplemental Table 6 | IDT | N/A |
| Recombinant DNA | ||
| pT-U6-sgRNA-Fah-P2A-Cas9 | This paper | N/A |
| Software and Algorithms | ||
| QBRC mutation calling pipeline | Tao Wang Lab, Quantitative Biomedical Research Center, UT Southwestern | https://qbrc.swmed.edu/labs/wanglab/software.php |
| QBRC neoantigen calling pipeline | Tao Wang Lab, Quantitative Biomedical Research Center, UT Southwestern | https://qbrc.swmed.edu/labs/wanglab/software.php |
| GATK toolkit (version 3.5) | DePristo et al., 2011; McKenna et al., 2010; Van der Auwera et al., 2013 | https://software.broadinstitute.org/gatk/ |
| Mutect (version 1.1.7) | Cibulskis et al., 2013 | https://software.broadinstitute.org/cancer/cga/mutect |
| Shimmer (version 0.1.1) | Hansen et al., 2013 | https://github.com/nhansen/Shimmer/blob/master/ |
| SpeedSeq (version 0.1.2) | Chiang et al., 2015 | https://github.com/hall-lab/speedseq/blob/master/bin/speedseq |
| Manta (version 1.3.1) | Chen et al., 2016 | https://github.com/Illumina/manta |
| Strelka(version 2.8.3) | Saunders et al., 2012 | https://github.com/Illumina/strelka |
| Varscan(version 2.4.2) | Koboldt et al., 2012 | http://varscan.sourceforge.net |
| CNVkit(version 0.9.0) | Talevich et al., 2016 | https://cnvkit.readthedocs.io/en/stable/ |
| lofreq_star(version 2.1.3.1) | Wilm et al., 2012 | http://csb5.github.io/lofreq/ |
| ATHLATES tool | Liu et al., 2013 | https://www.broadinstitute.org/viral-genomics/athlates |
| NetMHCIIpan (version 3.2) | N/A | http://www.cbs.dtu.dk/services/NetMHCIIpan/ |
| STAR aligner (version 2.5.2b) | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| FeatureCounts(version 1.6) | Liao et al., 2014 | http://subread.sourceforge.net |
Highlights.
Deep seq reveals an accumulation of mutations in chronic liver disease tissues.
PKD1, PPARGC1B, KMT2D, and ARID1A are recurrently mutated
In vivo CRISPR screens validate functional relevance of Pkd1, Kmt2d, and Aridla
Mutations seen in liver tissues but not in cancer promote hepatocyte fitness
Acknowledgments
We would like to thank Helen Hobbs and Teresa Eversole for contributing human samples, Sean Morrison, Joshua Mendell, Branden Tarlow, and Jian Xu for constructive comments on the manuscript, Cheryl Lewis and John Shelton for histopathology, and the CRI Sequencing Core (Jian Xu, Xin Liu) and Admera Health (Yun Zhao) for genomics. T.W. is supported by a R03ES026397-01. T.W. and X.L. were supported by CPRIT (RP150596). H.Z. was supported by the Pollack Foundation, an NIH/NIDDK R01 grant (DK111588), a Burroughs Wellcome Career Award for Medical Scientists, a CPRIT Scholar Award (R1209), a Stand Up To Cancer Innovative Research Grant (SU2C-AACR-IRG 10-16). Stand Up To Cancer is a program of the Entertainment Industry Foundation and its research grants are administered by the American Association for Cancer Research, the scientific partner of SU2C.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aihara T, Noguchi S, Sasaki Y, Nakano H, and Imaoka S (1994). Clonal analysis of regenerative nodules in hepatitis C virus-induced liver cirrhosis. Gastroenterology 107, 1805–1811. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. Electronic address: wheeler@bcm.edu, and Cancer Genome Atlas Research Network (2017). Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell 169, 1327–1341.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Kallberg M, Cox AJ, Kruglyak S, and Saunders CT (2016). Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32, 1220–1222. [DOI] [PubMed] [Google Scholar]
- Chiang C, Layer RM, Faust GG, Lindberg MR, Rose DB, Garrison EP, Marth GT, Quinlan AR, and Hall IM (2015). SpeedSeq: ultra-fast personal genome analysis and interpretation. Nat. Methods 12, 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, and Getz G (2013). Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol 31, 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet 43, 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan AW, Taylor MH, Hickey RD, Hanlon Newell AE, Lenzi ML, Olson SB, Finegold MJ, and Grompe M (2010). The ploidy conveyor of mature hepatocytes as a source of genetic variation. Nature 467, 707–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan AW, Hanlon Newell AE, Bi W, Finegold MJ, Olson SB, Beaudet AL, and Grompe M (2012a). Aneuploidy as a mechanism for stress-induced liver adaptation. J. Clin. Invest 122, 3307–3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan AW, Hanlon Newell AE, Smith L, Wilson EM, Olson SB, Thayer MJ, Strom SC, and Grompe M (2012b). Frequent aneuploidy among normal human hepatocytes. Gastroenterology 142, 25–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanti M, Singh S, Ledda-Columbano GM, Columbano A, and Monga SP (2014). Triiodothyronine induces hepatocyte proliferation by protein kinase A-dependent β-catenin activation in rodents. Hepatology 59, 2309–2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellous TG, Islam S, Tadrous PJ, Elia G, Kocher HM, Bhattacharya S, Mears L, Turnbull DM, Taylor RW, Greaves LC, et al. (2009). Locating the stem cell niche and tracing hepatocyte lineages in human liver. Hepatology 49, 1655–1663. [DOI] [PubMed] [Google Scholar]
- Font-Burgada J, Shalapour S, Ramaswamy S, Hsueh B, Rossell D, Umemura A, Taniguchi K, Nakagawa H, Valasek MA, Ye L, et al. (2015). Hybrid Periportal Hepatocytes Regenerate the Injured Liver without Giving Rise to Cancer. Cell 162, 766–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese G, Kähler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, Chambert K, Mick E, Neale BM, Fromer M, et al. (2014). Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med 371, 2477–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong L, Li Y-H, Su Q, Chu X, and Zhang W (2010). Clonality of nodular lesions in liver cirrhosis and chromosomal abnormalities in monoclonal nodules of altered hepatocytes. Histopathology 56, 589–599. [DOI] [PubMed] [Google Scholar]
- Grompe M, Lindstedt S, al-Dhalimy M, Kennaway NG, Papaconstantinou J, Torres-Ramos CA, Ou CN, and Finegold M (1995). Pharmacological correction of neonatal lethal hepatic dysfunction in a murine model of hereditary tyrosinaemia type I. Nat. Genet 10, 453–460. [DOI] [PubMed] [Google Scholar]
- Guido M, Mangia A, Faa G, Gruppo Italiano Patologi Apparato Digerente (GIPAD), and Società Italiana di Anatomia Patologica e Citopatologia Diagnostica/International Academy of Pathology, Italian division (SIAPEC/IAP) (2011). Chronic viral hepatitis: the histology report. Dig. Liver Dis 43 Suppl 4, S331–43. [DOI] [PubMed] [Google Scholar]
- Hansen NF, Gartner JJ, Mei L, Samuels Y, and Mullikin JC (2013). Shimmer: detection of genetic alterations in tumors using next-generation sequence data. Bioinformatics 29, 1498–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan MC, Abebe K, Torres VE, Chapman AB, Bae KT, Tao C, Sun H, Perrone RD, Steinman TI, Braun W, et al. (2015). Liver involvement in early autosomal-dominant polycystic kidney disease. Clin. Gastroenterol. Hepatol 13, 155–64.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshida Y, Villanueva A, Kobayashi M, Peix J, Chiang DY, Camargo A, Gupta S, Moore J, Wrobel MJ, Lerner J, et al. (2008). Gene expression in fixed tissues and outcome in hepatocellular carcinoma. N. Engl. J. Med 359, 1995–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda A, Shimizu T, Matsumoto Y, Fujii Y, Eso Y, Inuzuka T, Mizuguchi A, Shimizu K, Hatano E, Uemoto S, et al. (2014). Leptin receptor somatic mutations are frequent in HCV-infected cirrhotic liver and associated with hepatocellular carcinoma. Gastroenterology 146, 222–32.e35. [DOI] [PubMed] [Google Scholar]
- Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, et al. (2014). Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med 371, 2488–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang T-W, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, Hohmeyer A, Gereke M, Rudalska R, Potapova A, et al. (2011). Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 479, 547–551. [DOI] [PubMed] [Google Scholar]
- Kim D-H, Rhee JC, Yeo S, Shen R, Lee S-K, Lee JW, and Lee S (2015). Crucial roles of mixed-lineage leukemia 3 and 4 as epigenetic switches of the hepatic circadian clock controlling bile acid homeostasis in mice. Hepatology 61, 1012–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D-H, Kim J, Kwon J-S, Sandhu J, Tontonoz P, Lee S-K, Lee S, and Lee JW (2016). Critical roles of the histone methyltransferase MLL4/KMT2D in murine hepatic steatosis directed by ABL1 and pparY2. Cell Rep. 17, 1671–1682. [DOI] [PubMed] [Google Scholar]
- Knouse KA, Wu J, Whittaker CA, and Amon A (2014). Single cell sequencing reveals low levels of aneuploidy across mammalian tissues. Proc Natl Acad Sci USA 111, 13409–13414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, and Wilson RK (2012). VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulik L, and El-Serag HB (2019). Epidemiology and management of hepatocellular carcinoma. Gastroenterology 156, 477–491.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee-Six H, Ellis P, Osborne RJ, Sanders MA, Moore L, Georgakopoulos N, Torrente F, Noorani A, Goddard M, Robinson P, et al. (2018a). The landscape of somatic mutation in normal colorectal epithelial cells. BioRxiv. [DOI] [PubMed]
- Lee-Six H, ∅bro NF, Shepherd MS, Grossmann S, Dawson K, Belmonte M, Osborne RJ, Huntly, Martincorena I, Anderson E, et al. (2018b). Population dynamics of normal human blood inferred from somatic mutations. Nature 561, 473–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefkowitch JH (2007). Liver biopsy assessment in chronic hepatitis. Arch. Med. Res 38, 634–643. [DOI] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, and Liu XS (2014). MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 15, 554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Lin S, Nascimento EM, Gajera CR, Chen L, Neuhofer P, Garbuzov A, Wang S, and Artandi SE (2018). Distributed hepatocytes expressing telomerase repopulate the liver in homeostasis and injury. Nature 556, 244–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W-R, Lim S-N, McDonald SAC, Graham T, Wright VL, Peplow CL, Humphries A, Kocher HM, Wright NA, Dhillon AP, et al. (2010). The histogenesis of regenerative nodules in human liver cirrhosis. Hepatology 51, 1017–1026. [DOI] [PubMed] [Google Scholar]
- Liu C, Yang X, Duffy B, Mohanakumar T, Mitra RD, Zody MC, and Pfeifer JD (2013). ATHLATES: accurate typing of human leukocyte antigen through exome sequencing. Nucleic Acids Res. 41, e142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martincorena I, Roshan A, Gerstung M, Ellis P, Van Loo P, McLaren S, Wedge DC, Fullam A, Alexandrov LB, Tubio JM, et al. (2015). Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 348, 880–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martincorena I, Fowler JC, Wabik A, Lawson ARJ, Abascal F, Hall MWJ, Cagan A, Murai K, Mahbubani K, Stratton MR, et al. (2018). Somatic mutant clones colonize the human esophagus with age. Science 362, 911–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nault JC, Mallet M, Pilati C, Calderaro J, Bioulac-Sage P, Laurent C, Laurent A, Cherqui D, Balabaud C, and Zucman-Rossi J (2013). High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat. Commun 4, 2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nault JC, Calderaro J, Di Tommaso L, Balabaud C, Zafrani ES, Bioulac-Sage P, Roncalli M, and Zucman-Rossi J (2014). Telomerase reverse transcriptase promoter mutation is an early somatic genetic alteration in the transformation of premalignant nodules in hepatocellular carcinoma on cirrhosis. Hepatology 60, 1983–1992. [DOI] [PubMed] [Google Scholar]
- Overmoyer BA, McLaren CE, and Brittenham GM (1987). Uniformity of liver density and nonheme (storage) iron distribution. Arch. Pathol. Lab. Med 111, 549–554. [PubMed] [Google Scholar]
- Overturf K, Al-Dhalimy M, Tanguay R, Brantly M, Ou CN, Finegold M, and Grompe M (1996). Hepatocytes corrected by gene therapy are selected in vivo in a murine model of hereditary tyrosinaemia type I. Nat. Genet 12, 266–273. [DOI] [PubMed] [Google Scholar]
- Paradis V, Laurendeau I, Vidaud M, and Bedossa P (1998). Clonal analysis of macronodules in cirrhosis. Hepatology 28, 953–958. [DOI] [PubMed] [Google Scholar]
- Piccinin E, Peres C, Bellafante E, Ducheix S, Pinto C, Villani G, and Moschetta A (2018). Hepatic peroxisome proliferator-activated receptor γ coactivator 1β drives mitochondrial and anabolic signatures that contribute to hepatocellular carcinoma progression in mice. Hepatology 67, 884–898. [DOI] [PubMed] [Google Scholar]
- Piontek KB, Huso DL, Grinberg A, Liu L, Bedja D, Zhao H, Gabrielson K, Qian F, Mei C, Westphal H, et al. (2004). A functional floxed allele of Pkd1 that can be conditionally inactivated in vivo. J. Am. Soc. Nephrol 15, 3035–3043. [DOI] [PubMed] [Google Scholar]
- Qiu W, Federico L, Naples M, Avramoglu RK, Meshkani R, Zhang J, Tsai J, Hussain M, Dai K, Iqbal J, et al. (2008). Phosphatase and tensin homolog (PTEN) regulates hepatic lipogenesis, microsomal triglyceride transfer protein, and the secretion of apolipoprotein B-containing lipoproteins. Hepatology 48, 1799–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders CT, Wong WSW, Swamy S, Becq J, Murray LJ, and Cheetham RK (2012). Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 28, 1811–1817. [DOI] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, et al. (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singal AG, and El-Serag HB (2015). Hepatocellular Carcinoma From Epidemiology to Prevention: Translating Knowledge into Practice. Clin. Gastroenterol. Hepatol 13, 2140–2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Chuang J-C, Kanchwala M, Wu L, Celen C, Li L, Liang H, Zhang S, Maples T, Nguyen LH, et al. (2016). Suppression of the SWI/SNF component arid1a promotes mammalian regeneration. Cell Stem Cell 18, 456–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahvanainen E, Tahvanainen P, Kaariainen H, and Hockerstedt K (2005). Polycystic liver and kidney diseases. Ann. Med 37, 546–555. [DOI] [PubMed] [Google Scholar]
- Talevich E, Shain AH, Botton T, and Bastian BC (2016). CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput. Biol 12, e1004873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, et al. (2013). From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinformatics 11, 11.10.1–11.10.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Zhao L, Fish M, Logan CY, and Nusse R (2015). Self-renewing diploid Axin2(+) cells fuel homeostatic renewal of the liver. Nature 524, 180–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, and Hakonarson H (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White DL, Kanwal F, and El-Serag HB (2012). Association between nonalcoholic fatty liver disease and risk for hepatocellular cancer, based on systematic review. Clin. Gastroenterol. Hepatol 10, 1342–1359.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilm A, Aw PPK, Bertrand D, Yeo GHT, Ong SH, Wong CH, Khor CC, Petric R, Hibberd ML, and Nagarajan N (2012). LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 40, 11189–11201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama A, Kakiuchi N, Yoshizato T, Nannya Y, Suzuki H, Takeuchi Y, Shiozawa Y, Sato Y, Aoki K, Kim SK, et al. (2019). Age-related remodelling of oesophageal epithelia by mutated cancer drivers. Nature 565, 312–317. [DOI] [PubMed] [Google Scholar]
- Zare F, Dow M, Monteleone N, Hosny A, and Nabavi S (2017). An evaluation of copy number variation detection tools for cancer using whole exome sequencing data. BMC Bioinformatics 18, 286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Cheng Y, Guo Y, Chen J, Chen F, Luo R, and Li A (2016). Protein kinase D2 contributes to TNF-α-induced epithelial mesenchymal transition and invasion via the PI3K/GSK-3β/β-catenin pathway in hepatocellular carcinoma. Oncotarget 7, 5327–5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure S1. Data related to Figure 1.
A. Clinical information for 60 UT Southwestern patients.
B. H&E histology of samples with F0 and F4 fibrosis. Scale bar is 200pm for 4x pictures and 100μm for 10x pictures.
C. Waterfall plot for mutations called in 319 TCGA HCC samples.
Supplemental Figure S6. AAV-TBG-GFP and AAV-TBG-Cre do not exert independent biological effects. Data related to Figures 6 and 7.
A. Wild-type CD1 mice were either uninjected or injected with AAV-TBG-GFP or AAV-TBG-Cre virus at a dose of 5×1010 viral particles per mouse. 11 days after virus injection, all three groups of mice were injected with one dose of 10% CCl4 intraperitoneally to induce liver injury. Left panel: H&E staining showing hepatic necrosis 48 hours after CCl4 injury. Scale bar: 200μm. Right panel: Necrotic area was quantified (n = 8,8,9).
B. Pkd1+/+ and Pkd1fl/+ mice were injected with AAV-TBG-GFP or AAV-TBG-Cre virus. 11 days after virus injection, mice were injected with CCl4. Left panel: H&E staining showing hepatic necrosis 48 hours after CCl4 injury. Scale bar: 200μm. Right panel: Necrotic area was quantified (n = 10, 10, 9, 10).
Supplemental Figure S7. Neoantigen prediction and analysis. Data related to Figure 1.
A. Number of predicted neoantigens in 17 patients.
B. VAF of clones that have mutations predicted to result in neoantigen presentation. The average VAFs of mutations that generate neoantigens, divided by the average VAF of all mutations. Then a logarithmic transformation is performed. Mutations with negative VAF differences are selected against by T cells, suggestive of a disadvantaged survival fitness of immunogenic mutations.
Supplemental Table 1: Clinical and pathological data about the patients sequenced in the study. This is related to Figures 1–4 and STAR Methods.
Supplemental Table 2: Whole exome sequencing data for 82 patients in the study. This is related to Figure 1.
Supplemental Table 3: Targeted ultra-deep sequencing data for 61 patients in the study. This is related to Figures 2–4.
Supplemental Table 4: Mutant clone and nodule volume measurements. This is related to Figure 3.
Supplemental Table 5: sgRNAs used in the CRISPR screen are in the first sheet and the screen results are in the second sheet. This is related to Figure 5.
Supplemental Table 6: Primers for Sanger sequencing. This is related to the STAR Methods.
Supplemental Figure S2. Characterization of ultra-deep targeted sequencing results. Data related to Figure 2.
A. Mutation counts in each of the 61 patients and each of the 129 samples. An additional patient without liver disease was added to this ultra-deep sequencing group.
B. Types of mutations. Among 214 mutations, 131 are missense, 23 are frameshift, 8 are in-frame deletions, 6 are nonsense, and 49 are synonymous.
C. SNV classes for 131 missense mutations are shown here.
D. Correlation between mutation count and fibrosis stage. The number of patients with each fibrosis score is listed under the graph. The p-value is calculated based on the one-way Jonckheere trend test.
E. VAF distribution for ultra-deep targeted sequencing data.
F. Mutations identified in ARID1A, KMT2D, and PKD1 are shown with respect to protein domains.
Supplemental Figure S3. Sanger sequencing confirms select recurrent mutations. Data related to Figure 2.
Supplemental Figure S4. Chromosome level CNVs are observed but not frequently identified in diseased liver tissues. Data related to Figure 1.
A. Mean CNVs of all 317 HCC samples from TCGA called by the CNVkit algorithm.
B Correlation between mRNA expression and copy number. More genes are positively correlated with their CNVs than negatively correlated (Y-axis). This trend is clearer when visualizing genes that are more actively going through CNV changes (larger median absolute deviation of copy number) (X-axis), which could have a stronger impact on transcription. We analyzed all 17 patients who underwent both whole exome and RNA sequencing.
C. Regions with copy number loss have fewer mutations detected.
D. CNVs from a representative sample (HS122) with few alterations.
E. CNVs for two samples with chromosome 1 and 8 gains (HS37, HS10).
F. Mean CNVs from all 82 liver samples.
Supplemental Figure S5. The tyrosine metabolism pathway is defective in hereditary tyrosinemia, a disease caused by deleterious mutations in Fah. Related to Figure 5.
Data Availability Statement
Data availability
The whole exome sequencing data, ultra-deep targeted sequencing data, and RNA sequencing data reported by this paper were deposited in the European Genome-phenome Archive (EGA) database: https://www.ebi.ac.uk/ega/home. The identifier is EGAS00001003496.
Code availability
The QBRC somatic mutation and neoantigen calling pipelines are available on GitHub: https://github.com/Somatic-pipeline/Somatic-pipeline and https://github.com/Neoantigen-pipeline/Neoantigen-pipeline.
The whole exome sequencing data, ultra-deep targeted sequencing data, and RNA sequencing data reported by this paper were deposited in the European Genome-phenome Archive (EGA) database: https://www.ebi.ac.uk/ega/home. The identifier is EGAS00001003496.