

Abstract
Age-associated changes to the mammalian DNA methylome are well documented and thought to promote diseases of aging, such as cancer. Recent studies have identified collections of individual methylation sites whose aggregate methylation status measures chronological age, referred to as the DNA methylation clock. DNA methylation may also have value as a biomarker of healthy versus unhealthy aging and disease risk; in other words, a biological clock. Here we consider the relationship between the chronological and biological clocks, their underlying mechanisms, potential consequences, and their utility as biomarkers and as targets for intervention to promote healthy aging and longevity.
DNA Methylation
Approximately 28 million CpG dinucleotides are unevenly distributed throughout the mammalian genome. Across most of the genome, these CpGs are relatively depleted but, elsewhere, are found in clusters that form so-called CpG islands, often at gene promoters (Schübeler, 2015). CpG dinucleotides are the target of the most abundant chemical modification of DNA in mammalian cells: methylation to generate 5-methylcytosine (5mC). Typically, the majority of CpG is in the methylated state, although graded levels at genes and regulatory sequences are a factor determining the level and integrity of gene expression. At gene bodies, methylation promotes expression and suppresses initiation of internal cryptic transcripts (Neri et al., 2017; Yang et al., 2014). Intermediate levels of DNA methylation are found at distal regulatory sequences, including enhancers (Elliott et al., 2015; Stadler et al., 2011), where methylation sometimes promotes gene silencing (Deaton and Bird, 2011; Hon et al., 2014; Lu et al., 2014). In normal cells, CpG islands are usually unmethylated regardless of their level of expression, but when they are methylated, this promotes gene silencing (Deaton and Bird, 2011). This mode of epigenetic silencing is a feature of some tumor suppressor genes in cancer cells (Greger et al., 1989; Herman et al., 1994). DNA methylation also contributes to X chromosome inactivation and allelic imprinting (Lessing et al., 2013; Li et al., 1993). DNA methylation can control gene expression by affecting binding of transcription factors and patterning of histone modifications that themselves influence gene expression (Reddington et al., 2013; Rose and Klose, 2014; Yin et al., 2017). Epigenome editing approaches have confirmed a causal role of DNA methylation in gene regulation (Gregory et al., 2013; Maeder et al., 2013; Saunderson et al., 2017). Accordingly, differences in methylation between cell types and tissues correlate with differences in gene expression (Davies et al., 2012; Deaton et al., 2011; Irizarry et al., 2009; Laurent et al., 2010; Nagae et al., 2011).
DNA methylation is not a static modification. 5mC is enzymatically deposited and removed by DNA methyl transferases (DNMTs) and demethylases of the ten-eleven translocation (TET) family, respectively. DNMT1 is responsible for copying DNA methylation onto the newly synthesized DNA strand after DNA replication (Schübeler, 2015). In contrast, DNMT3A and DNMT3B, together with DNMT3L, catalyze de novo methylation in non-proliferating cells. Mammals express three TET enzymes (TET1, TET2, and TET3) that convert 5mC to 5-hydroxymethylcytosine (5hmC), which, in turn, can be converted to unmethylated cytosine by TET-mediated conversion to 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC) and subsequent removal by base excision repair. 5mC and its derivatives can also be eliminated by passive demethylation during DNA replication because of inefficient copying of the hemi-modified strand (Wu and Zhang, 2017). Inactivation of TETs or DNMTs causes gains and losses of DNA methylation (Jeong et al., 2014; Rinaldi et al., 2016; Wu and Zhang, 2017), suggesting that the level of DNA methylation reflects a dynamic balance between methylation and demethylation. Recent studies have shown that, even in tissues comprised of largely slow or non-proliferating cells such as the liver, lung, and brain, the methylation status of a substantial proportion of CpGs oscillates with a circadian rhythm (Coulson et al., 2018; Lim et al., 2017; Oh et al., 2018). Methylation at enhancers is thought to be particularly dynamic (Feldmann et al., 2013; Hon et al., 2014; Lu et al., 2014). Feldmann et al. (2013) have proposed that active turnover of DNA methylation contributes to enhancer function, even in non-proliferating cells, where there are no replication-coupled dynamics. In stem cells exiting pluripotency, DNA methylation in single cells oscillates over a timescale of 2–3 hr, most markedly at enhancers, again pointing to a dynamic state independent of DNA replication (Rulands et al., 2018). In sum, DNA methylation is a dynamically controlled DNA modification that contributes to proper control of gene expression and genome function.
The Chronological Methylation Clock
In accord with DNA methylation being a dynamic modification, many reports have documented age-associated changes to DNA methylation (Fraga and Esteller, 2007). For example, DNA methylation tends to increase with age at some CpG islands (Ahuja et al., 1998; Issa et al., 1994, 1996, 2001; Kim et al., 2005a, 2005b; Ftakyan et al., 2010; So et al., 2006; Teschendorff et al., 2010; Waki et al., 2003; Yatabe et al., 2001), particularly at polycomb target genes (Rakyan et al., 2010; Teschendorff etal., 2010).
Based on age-associated DNA methylation changes that occur relatively consistently between individuals, several groups have identified so-called DNA methylation clocks that accurately measure the chronological age of the donor (Horvath and Raj, 2018; Nwanaji-Enwerem et al., 2018; Wagner, 2017; Zheng et al., 2016a). Although these DNA methylation clocks were first identified by groundbreaking studies in humans (Bocklandt et al., 2011; Garagnani et al., 2012; Hannum et al., 2013; Horvath, 2013; Weidner et al., 2014), they have since been identified in mice (Petkovich et al., 2017; Stubbs et al., 2017; Wang et al., 2017), dogs, wolves (Thompson et al., 2017), and humpback whales (Polanowski et al., 2014). Evidence suggests that the tick rate of epigenetic clocks may be related to species lifespan (Lowe et al., 2018). Each of the clocks is derived by a linear regression algorithm that trains against the chronological age of the sample donors and ultimately selects a set of CpG dinucleotides, from 3–353 CpGs in the human clocks (Bocklandt et al., 2011; Garagnani et al., 2012; Hannum et al., 2013; Horvath, 2013; Weidner et al., 2014) and 90–329 CpGs for the mouse clocks (Petkovich et al., 2017; Stubbs et al., 2017; Wang et al., 2017). The combined age-dependent methylation status of these CpGs yields an apparent DNA methylation age that correlates with chronological age (Figure 1; Box 1). The clock by Horvath (2013), built from data from many tissues, and the clock by Hannum et al. (2013), developed from whole-blood data, can measure age in many human tissues. The individual methylation changes that comprise the clock are variable in magnitude. For CpGs in the clock by Horvath (2013), the average difference in percent methylation is 3.2% per CpG, comparing individuals younger than 35 years and older than 55 years. However, in another study, a CpG in the ELOV2 gene was reported to range between 7% and 91% methylation over the lifespan (Garagnani et al., 2012). The clock-building algorithm weighs the contribution of each CpG to the clock according to the magnitude of its age-dependent change (Figure 1; Box 1). The accuracy of the clocks is measured by the correlation coefficient between actual chronological age and measured age and the average absolute difference between actual age and measured age. These clocks are remarkably accurate. For example, the two most accurate and most analyzed human clocks of Hannum et al. (2013) and Horvath (2013) have correlation coefficients of more than 0.9 and average errors of less than 5 years.
Figure 1. The DNA Methylation Clock: How It Works.

(A) The trajectory of change with age of methylation of eight CpGs, four with negative coefficients (left) and four with positive coefficients (right), colored by the rate of change with age. Darker colors indicate faster rates of change and, thus, stronger weights (numerically larger coefficients) in the epigenetic clock.
(B) Eight clock CpGs (columns) in three individuals of different chronological ages (rows) whose methylation values at each CpG are indicated by shading of the filled circle (fractional methylation, 0.0–1.0). The box color represents the coefficient of change over age (slope of line, top). In each colored box, the numerical product of the methylation value and the associated coefficient are shown. Summing across all boxes together with the intercept learned during clock building results in an epigenetic age that approximates chronological age.
Box 1. Building Epigenetic Clocks: Strategies and Limitations.
Epigenetic clocks have been reported for both humans (Hannum et al., 2013; Horvath, 2013) and mice (Petkovich et al., 2017; Stubbs et al., 2017; Wang et al., 2017). These clocks are represented as a linear model, where some models explicitly fit an intercept term and some do not (Equation 1). These clocks were predominantly built with ElasticNet regression (Zou and Hastie, 2005), a form of regularized regression. This algorithm automatically determines which CpG sites to use and the weights associated with each CpG. ElasticNet learns these parameters by minimizing the cost function, , shown in Equation 2. Regularization performs feature selection by scaling the least informative CpG with age to 0.
| (Equation 1) |
| (Equation 2) |
where λ2 = 0; λ1 = λ ~ Ridge Regression and λ1 = 0; λ2 = λ ~ LASSO.
The learned values in front of each CpG, β1, β2·····βN, indicate the amount that age changes in response to the change in methylation values. Therefore, negative coefficients indicate that the methylation value decreases as age increases, whereas positive coefficients indicate the opposite trend. Then the product of the methylation value at each CpG with the learned coefficient is summed, which yields the estimated age (Figure 1).
A strength of this approach is that it reduces overfitting because of the large number of CpGs that can be profiled in one experiment. ElasticNet automatically performs feature selection, keeping the most relevant features to predict age, and also scales these features appropriately. This allows linear models built using ElasticNet to perform well on new datasets. Indeed, many studies using the human epigenetic clock have demonstrated that this method is applicable across datasets (for example, see A Candidate Biological Methylation Clock).
However, there are some important limitations to consider. First, by formulating epigenetic clocks using linear regression, the model assumes that the amount each CpG contributes to age is additive and that the rate of methylation change is constant over the entire lifespan. However, the rate of methylation change at each CpG is not always linear and constant over the lifespan (Horvath, 2013; Petkovich et al., 2017). Strategies implemented during clock building work around this limitation by transforming age prior to clock building by using a non-linear function (Horvath, 2013; Petkovich et al., 2017; Stubbs et al., 2017; Wang et al., 2017) and/or learning a different non-linear function to convert calculated ages back into the desired time unit (Petkovich et al., 2017; Stubbs et al., 2017). Second, ElasticNet can be sensitive to differences in the underlying data used to formulate epigenetic clocks. Thus, many of the clocks reported may be equivalent in terms of their performance (e.g., errors in predicted age) but use completely different sets of CpG sites and are very likely to show tissue-dependent effects. This hinders our ability to assess whether the CpGs selected have any special or functional relationship to the aging process. Finally, when applying the model to new datasets, the accuracy of the age estimate is dependent on proper coverage of all CpG sites used in the model. For array-based methods, this is less of a concern because the standardized lllumina arrays can robustly measure methylation at the same CpG sites (notwithstanding differences between 27K, 450K, and infinium methylationEPIC arrays, of course), meaning that the age calculation can be readily applied to new datasets. However, for sequencing-based studies, this may not always be the case, which makes it difficult to automatically apply these clocks to other datasets, such as those generated in mice.
Although the algorithms used to generate the clocks select only relatively few CpG sites, the methylation changes that form the basis of the clock are many and widely distributed. Mammalian genomes appear to contain tens of thousands of candidate clock CpGs whose methylation status tracks chronological age (Hannum et al., 2013; Wang et al., 2017). Human clock sites that are syntenic in the mouse measured mouse age comparably to a random selection of sites (Stubbs et al., 2017), both with a mean absolute error of only about 11 weeks. This supports the idea that epigenetic clocks are not based on a small number of loci that are conserved between human and mouse; instead, potential clock CpGs appear to be abundant in both species. These numerous clock CpGs do not appear to localize exclusively to any particular genomic feature, and they are not obviously excluded from regulatory regions. In some cases, CpGs whose methylation increases with age are overrepresented near polycomb target genes (Horvath, 2013; Thompson etal., 2017; Weidner etal., 2014), in line with previous observations (Rakyan et al., 2010; Teschendorff et al., 2010). In the mouse, many clock CpGs localize to genes and promoters (Figure 2; Petkovich et al., 2017; Stubbs et al., 2017; Wang et al., 2017), and some studies also implicate hypomethylation of enhancers as a component of the mouse clock (Cole et al., 2017; Petkovich et al., 2017; Wang et al., 2017). A previous study also showed age-associated hypomethylation of enhancers in mouse pancreatic β cells (Avrahami et al., 2015).
Figure 2. Distribution of Mouse Clock CpGs across the Genome.
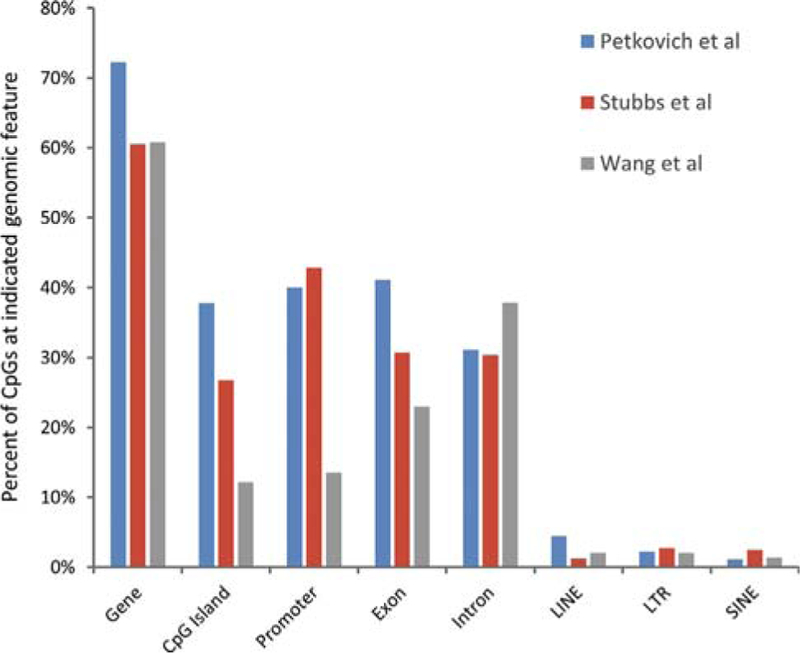
The mouse clock CpGs described by Petkovich et al. (2017), Stubbs et al. (20 7), and Wang etal. (2017) were assigned to the indicated genome features. Some CpGs map to more than one feature. Hence, the sum of percentages for any one mouse clock is greater than 100.
A closer comparison of the three reported mouse clocks showed that there are no CpGs in common between the clock developed by Petkovich et al. (2017) from mouse blood and the clock developed from mouse liver by Wang et al. (2017) and from liver, heart, brain, and lung by Stubbs et al. (2017). There are only two overlapping CpGs in the clocks of Wang et al. (2017) and Stubbs et al. (2017). Consistent with this low overlap, there are also differences in the distribution of CpGs from the 3 mouse clocks across different genome features (Figure 2). This low overlap likely largely reflects the fact that the algorithms used to build the clocks select only a small subset of the available clock CpGs (see above). Tissue differences may partly account for the low overlap between the mouse clocks. Previous studies showed that, although some age-associated methylation changes are shared across tissues, others are tissue-specific (Christensen et al., 2009; Day et al., 2013; Maegawa et al., 2010; Thompson et al., 2010). Although Stubbs et al. (2017) primarily generated a pan-tissue mouse clock, they also presented evidence that distinct tissue-specific clocks can also be formulated. Thus, the limited overlap between the clocks of Stubbs et al. (2017) and Wang et al. (2017) may reflect the shared liver component used to generate these clocks. However, the low overlap and differences in genome distribution between the mouse clocks is probably also due to variations in the datasets used to train the clocks. Of note, the mouse clocks were all generated largely from reduced representation bisulfite sequencing (RRBS) data, whose quality and genome representation can vary between sources, more so than lllumina arrays used to develop the human clocks. We have attempted to cross-validate the three mouse clocks with variable success, perhaps again reflecting variations in the training data and tissue specificity of some of the clocks (Box 1). An array-based method for profiling the methylation status of relevant CpGs would help to move the mouse field forward.
To conclude this section, our earlier fragmented view of the aging msthylome has progressed to reveal a methylation landscape that incurs many widespread age-linked alterations of variable magnitude. These can be computationally distilled down to much smaller subsets of CpGs whose methylation status across tissues calculates an apparent DNA methylation age. Remarkably, across a population, this DNA methylation age correlates strongly with chronological age and, in many human individuals, it measures chronological age to within a few years.
A Candidate Biological Methylation Clock
By definition, a chronological age clock measures time elapsed since birth. Hence, individuals born on the same day share the same chronological age throughout life, regardless of lifestyle, health, or disease (Figure 3A). In contrast, individuals of identical chronological age may have different biological ages. Biological age, although not well-defined, is intended to measure tissue and organismal functional decline, risk of and actual age-associated disease, morbidity, and mortality (Jackson et al., 2003). Individuals of the same chronological age can, of course, differ greatly in their burden of age-associated dysfunction, disease, and risk of mortality and so can, correspondingly, be regarded as of different biological age (Figure 3A). This concept of biological age is important because it can potentially provide a means to predict disease and assess disease risk that is more accurate than chronological age alone. However, although panels of functional and physiological markers reflecting physical capability, musculoskeletal function, cognitive function, cardiovascular and lung function, glucose metabolism, endocrine function, inflammation, and other molecular markers (e.g., telomere length and features of cell senescence) have been proposed, there is currently no consensus as to how to best measure biological age (Mitnitski et al., 2015). This likely reflects the complex, multi-dimensional nature of aging and the qualitative, not just quantitative, inter-individual variations in the aging process.
Figure 3. Chronological versus Biological Age and Their Assessment by Methylation Clocks.

(A) Two people, blue and red, born at the same time, will always share the same chronological age (gray arrow timeline measured in years). However, because of genetic, epigenetic, and environmental factors and lifestyle choices, they may progress through the functional decline that characterizes biological aging at different rates. Shown here, red ages biologically more quickly than blue, likely associated with earlier onset of lethal disease. As illustrated, in early life, red and blue are assumed to have the same biological age.
(B) By definition, a perfect chronological clock deft), whether based on DNA methylation or any other molecular parameter, measures time elapsed since birth. Therefore, it cannot distinguish between individuals that biologically age fast (red) or slow (blue). In contrast, a biological clock (center) can distinguish between unhealthy (red) versus healthy (green) aging but is a less accurate chronological clock. A hybrid clock (right) tracks closely with chronological age, but deviation from the position of the 45° perfect chronological clock is a reflection of biological age. However, the hybrid clock is likely a less accurate predictor of age and disease than a bona fide biological clock. The human clocks calibrated against chronological age are likely hybrid clocks (Hannum et al., 2013; Horvath, 2013; Weidner et al., 2014). The colors of the filled circles indicate the donor, red or blue, from (A).
Despite these complexities, recent studies have indicated that the difference between apparent DNA methylation age and true chronological age (referred to as ∆age) reflects biological age, at least to some extent. In other words, if an individual has a chronological age of 55 years but a calculated methylation age of 62 years, then he or she can be considered to be biologically older than the chronological age would suggest. Several lines of evidence support this idea. HIV infection is associated with methylation age advancement, concordant with phenotypic and functional data indicative of premature biological aging in HIV-infected individuals (Gross et al., 2016; Horvath and Levine, 2015; Levine et al., 2016a). Some human genetic syndromes thought to accelerate biological aging, such as Werner’s syndrome (Maierhofer et al., 2017) and Down syndrome (Horvath et al., 2015a), also exhibit an accelerated methylation clock. Conversely, super-centenarian humans and their offspring have been reported to have a decelerated methylation clock in the blood (Horvath et al., 2015b). Many other cross-sectional studies support the notion that advanced biological age is associated with an accelerated DNA methylation age. Human conditions shown to be associated with advanced DNA methylation age include decreased mental and physical fitness (Marioni et al., 2015b), menopause and bilateral oophorectomy (Levine et al., 2016b), obesity, BMI and metabolic syndrome (Horvath et al., 2014; Nevalainen et al., 2017; Quach et al., 2017) (although BMI not in all studies; Hannum et al., 2013), non-alcoholic steatohepatitis (NASH) (Loomba et al., 2018), Parkinson’s disease (Horvath and Ritz, 2015), Alzheimer’s disease (Levine et al., 2015b), Huntington’s disease (Horvath et al., 2016), and other early indications of brain-degenerative disorders (Raina et al., 2017). Insomnia and extreme lifestyle stresses are also associated with accelerated methylation aging (Carroll et al., 2017; Jovanovic et al., 2017; Zannas et al., 2015).
In light of these studies, there has been considerable excitement regarding the use of methylation age as a measure of biological age and, hence, as a predictive biomarker of disease and mortality. Baker and Sprott (1988) defined a biomarker as a molecular, physiological, or functional readout that predicts future disease better than chronological age. Indeed, in some cases, an accelerated methylation clock not only correlates cross-sectionally with apparent biological age, but is predictive of future phenotypes of aging, such as cancer, cardiovascular disease, and all-cause mortality (Ambatipudi et al., 2017; Chen et al., 2016; Christiansen et al., 2016; Durso et al., 2017; Levine et al., 2015a; Marioni et al., 2015a; Pema et al., 2016; Zheng et al., 2016b). A 5-year elevation of DNA methylation age compared with chronological age is associated with a 16% higher mortality risk, adjusting forage, sex, and other health and social parameters (Marioni et al., 2015a). A measure that also considers age-associated change in blood cell composition (so-called extrinsic epigenetic age-acceleration [EEAA]) performed better (Chen et al., 2016).
However, using DNA methylation age as a measure of biological age in this way is not straightforward. First, despite the fact that, in humans, advancing chronological age universally leads to a decline in biological function, a perfect chronological clock, by definition, contains no information on inter-individual variation in biological age; i.e., if a methylation clock accurately judges a group of 50-year-olds to all be 50 years of age, then it cannot also report on variations in biological age between them (Figure 3B, left). The aforementioned methylation clocks, including Hannum et al. (2013) and Horvath (2013), were trained on chronological age. Given their accuracy at measuring chronological age, they are likely not ideal for measuring biological age (Figure 3B, compare center and right). Second, different tissues may age at different relative rates in different people, accounting for why some 70-year-olds are afflicted with Alzheimer’s disease and others with cardiovascular disease. Thus, a pan-tissue clock measured in peripheral white blood cells will not likely accurately predict future tissue-specific diseases, as required by current clinical practice. Third, the current human methylation clocks may not sample the most biologically informative CpGs. In part, this may be because the most informative biological clock CpGs may have been discarded by the modeling algorithm because their methylation is not the best correlate of chronological age. It may also be because the lllumina 450K arrays, used to generate the DNA methylation data, do not probe some of the most biologically informative CpGs.
Multiple studies have attempted to build human DNA methylation biological clocks, distinct from chronological clocks (Figure 3B, center). In light of previously reported patterns of age-associated DNA methylation and their relationship to cancer (Ahuja et al., 1998; Issa et al., 1994,1996,2001; Kim et al., 2005a, 2005b; Ohm et al., 2007; Rakyan et al., 2010; Schlesinger et al., 2007; So et al., 2006; Teschendorff et al., 2010; Waki et al., 2003; Widschwendter et al., 2007; Yatabe et al., 2001), Yang et al. (2016) rationally built the so-called EpiTOC from a set of promoter CpGs that is marked by the PRC2 complex in human embryonic stem cells (ESCs), constitutively unmethylated in many different fetal tissue types, whose methylation tends to increase with age. Although derived from blood DNA methylation data, the EpiTOC appears to be a biological clock that reflects cumulative cell divisions and is accelerated in premalignant lesions in other tissue types and buccal tissue of smokers. Zhang et al. (2017) profiled DNA methylation in blood using lllumina 450K arrays, and searched not for correlates of chronological age but for DNA methylation signatures linked to risk of mortality, one plausible but imperfect measure of biological age. Ultimately, 10 selected CpGs were used to calculate a mortality risk score that was validated in another cohort. There was no apparent overlap between these mortality-related CpGs and chronological clock CpGs (Hannum et al., 2013; Horvath, 2013). This might be because each set of CpGs is only a small subset of a much larger combined total, or it may be because biological and chronological clock CpGs are largely distinct loci, reflecting different underlying processes. Most recently, Levine et al. (2018) trained human DNA methylation data from whole blood against a multi-system measure of biological age, or phenotypic age, to generate a DNA methylation clock called DNAm PhenoAge, comprised of 513 CpG sites. The measures of phenotypic age included 9 markers of tissue function, immune function, and also chronological age.
At least in some cases, these candidate biological clocks perform better than the original chronological DNA methylation clocks in predicting disease and mortality. For example, DNAm PhenoAge is a better predictor of 10- and 20-year survival than either the Hannum et al. (2013) or Horvath (2013) chronological clocks (Levine et al., 2018). As well as all-cause mortality, the risk score of Zhang et al. (2017) also specifically identifies individuals with substantially increased risk of death by cancer and cardiovascular disease. It is exciting to speculate that these early biological clocks of Yang et al. (2016), Zhang et al. (2017), and Levine et al. (2018) might presage effective predictors of future disease; for example, future malignant disease in the case of the EpiTOC (Yang et al., 2016). However, there are also limitations to these candidate biological clocks and their application. As noted above, DNA methylation in blood is likely of limited relevance to disease in other tissues and, if the clock is trained against mortality, there are other determinants of mortality aside from biological age; for example, risky lifestyle choices.
Given the obvious challenges of access to normal healthy human tissues for testing the predictive power of candidate, potentially tissue-specific, biological clocks, mouse models will be important to test the predictive power of DNA methylation clocks. Like the human clock, the mouse DNA methylation clocks, although calibrated against chronological age, also appear to partly reflect biological age. Physical insults considered detrimental to health or lifespan (namely, ovariectomy and a high-fat diet) accelerate methylation age (Stubbs et al., 2017). Conversely, the mouse methylation clocks are retarded by interventions that slow biological aging, such as calorie restriction and rapamycin treatment (Petkovich et al., 2017; Stubbs et al., 2017; Wang et al., 2017). Some studies in mice have suggested the existence of a candidate biological clock at intragenic enhancers of highly expressed genes (Cole et al., 2017; Petkovich et al., 2017; Wang et al., 2017). In the mouse liver, age-associated hypomethylation is enriched at enhancers that are over-represented in the mouse clock (Wang et al., 2017), and hypomethylation at these sites is suppressed by pro-longevity interventions that presumably delay biological aging (Cole et al., 2017; Petkovich et al., 2017; Wang et al., 2017). In mice, it is feasible to test the ability of a candidate DNA methylation biological clock, assessed in a prospectively biopsied aged tissue, to predict the subsequent response of that tissue to an insult.
In conclusion, preliminary analyses indicate that age-associated DNA methylation changes may, in part, reflect biological age and comprise a biological age clock. However, by definition, chronological and biological clocks are not the same, and a biological clock may be best defined by comparing DNA methylation directly with one or more candidate functional or physiological measures of biological age, perhaps also considering chronological age. This approach is complicated by the fact that there is no consensus definition of biological age, and, unlike chronological age, it may not be possible to express biological age as a single quantitative value derived from a single tissue. Despite these complexities, there is reason to believe that bona fide biological clocks can be powerful predictors of future disease and aging. Additional studies are required in humans and animal models to select the best CpG sites and approach to training of the clock, to appreciate the importance of tissue and disease specificity, and to test the predictive value of candidate biological clocks.
What Is the Mechanism Underlying the Clocks?
The chronological clock is not tightly linked to cell division, proliferation, or onset of cell senescence (Horvath, 2013; Lowe et al., 2016; Lu et al., 2018). Nor is the chronological clock wholly a reflection of age-associated changes in tissue composition (Gross et al., 2016; Horvath and Ritz, 2015). Although the algorithms use only a relatively small number of clock CpGs, potential chronological clock-like CpGs appear to be numerous and widely distributed across the epigenome (Hannum et al., 2013; Stubbs et al., 2017; Wang et al., 2017). In line with this, the ticking of the clock appears to reflect a general progression of high- and low-methylated CpGs to an intermediate level nearer 50% methylation. This suggests a smoothening with age of the epigenetic landscape and a chronological clock driven by an increase in entropy (Hannum et al., 2013; Weidner et al., 2014).
In contrast, other observations are intuitively more consistent with the chronological clock reflecting a programmed process, perhaps selected for through evolution. First, as already noted, the methylation clock provides a remarkably accurate measure of chronological age. Second, there are apparent links between the chronological clock and cell and tissue development. Methylation changes underlying the clock are apparent during early growth and development (Horvath, 2013; Petkovich et al., 2017), prior to sexual maturation, and before the time when the cell and tissue decline associated with aging is conventionally thought to begin (Kirkwood, 2005). In diverse species, genes most closely linked to clock CpGs (and/or other clock-like CpGs whose methylation changes with age) are enriched for genes linked to development and differentiation (Horvath, 2013; Maegawa et al., 2017; Petkovich et al., 2017; Stubbs et al., 2017; Thompson et al., 2017), including polycomb targets (Horvath, 2013; Thompson et al., 2017). Adult breast, a tissue that passes through an enhanced development phase during puberty, was reported by Horvath (2013) to show an accelerated clock (Sehl et al., 2017) (although this was not noted by Hannum et al., 2013). In girls, faster ticking of the clock is associated with earlier puberty (Binder et al., 2018). Although these observations might initially suggest that the chronological clock and, by extension, aging itself are programmed and, in some way, a continuation of development, as has been proposed previously (Blagosklonny, 2013), these observations are also consistent with the entropic model. The apparent continuity between epigenetic changes associated with development and aging may simply reflect dynamic control of DNA methylation across a large proportion of the genome in both processes, linked to cell lineage specification and maturation, either during tissue growth and development or during tissue maintenance and repair (Bock et al., 2012; Ji et al., 2010). The consistency between individuals of a species (i.e., the precision of the chronological clock) can be embedded within the initial set point of the system and the dynamic regulatory mechanisms that evolved to control and maintain epigenome function over the reproductive lifespan of that species (Lowe et al., 2018). As is the case for other age-associated changes and damage accumulation (Kirkwood, 2005), there is no need to invoke a specific evolved program of aging to account for the DNA methylation clock, and there are strong evolutionary arguments against such an evolved program (Kirkwood, 2005). Based on these considerations, we propose that ticking of the chronological clock primarily represents widespread limited entropic decay, or smoothening, of the epigenetic landscape.
In renewable tissues underpinned by division of stem and progenitor cells, a component of entropic decay may be due to lack of fidelity of methylation patterns coupled to DNA replication (Issa, 2014). However, this is likely not solely responsible (Horvath, 2013; Lowe et al., 2016; Lu et al., 2018), and entropic decay of the DNA methylome in non-proliferating cells may also result from errors in its dynamic maintenance by cycles of TET and DNMT activity, perhaps especially at regions of more open and dynamic chromatin. This view fits with the observation that CpGs whose methylation changes with age partly overlap features where methylation is thought to be particularly dynamic; namely, enhancers and CpGs subject to circadian oscillation (Feldmann et al., 2013; Hon et al., 2014; Lu et al., 2014; Oh et al., 2018; Petkovich et al., 2017; Rulands et al., 2018; Wang et al., 2017). Age-associated decay because of maintenance errors may be exacerbated by age-altered expression of TETs and DNMTs, changes in their targeting, and/or altered levels of essential substrates, such as S-adenosylmethionine (SAM) for DNMTs or α-ketoglutarate (α-KG) for TETs (Berger and Sassone-Corsi, 2016). Expression of DNMT3B decreases with age in at least some tissues, perhaps promoting age-associated hypomethylation of some clock CpGs (Armstrong et al., 2014; Ciccarone et al., 2016). Levels of SAM have been reported to decrease with age (Geller et al., 1988; Hoffman et al., 1979; Stramentinoli et al., 1977; Trolin et al., 1994). Mitochondrial function, important for production of α-KG, also declines with age (Sun et al., 2016). DNA damage and repair (O’Hagan et al., 2008, 2011), an ongoing process even in healthy unstressed cells, is also linked to altered targeting of the DNA methylation machinery.
Age-associated DNA methylation changes may also be secondary to other changes to chromatin and/or chromatin modifiers (Booth and Brunet, 2016). Histone modifications affect DNA methylation (Rose and Klose, 2014). For example, DNA methylation is excluded from gene promoters by H3K4me3 (histone H3, lysine 4 trimethylation) but recruited to gene bodies and heterochromatin by H3K36me3 (histone H3, lysine 36 trimethylation) and H3K9me3 (histone H3, lysine 9 trimethylation), respectively (Rose and Klose, 2014). Polycomb complexes have also been reported to recruit DNMTs and DNA methylation (Mohammad et al., 2009; Viré et al., 2006). Conversely, DNA hypomethylation causes a redistribution of polycomb and H3K27me3 (histone H3, lysine 27 trimethylation; Reddington et al., 2013). By this view, the DNA methylation clock can be depicted as an “epigenetic network clock,” and methylation changes may be secondary to other epigenetic or even linked metabolic changes (Berger and Sassone-Corsi, 2016), that comprise this network.
Although the chronological clock reflects methylation changes that are shared between individuals, by definition, a biological methylation clock parallels inter-individual variations in tissue dysfunction and disease or disease risk and so ought to reflect epigenetic divergence between individuals. Such age-associated epigenetic divergence of individuals has been referred to as “epigenetic drift” (Fernández et al., 2015; Fraga et al., 2005; Issa, 2014; Talens et al., 2012). Accordingly, it is tempting to equate the biological clock to epigenetic drift (Fraga et al., 2005; Issa, 2014), at least in part. Concordant with this view, drift can be suppressed by pro-longevity interventions (Maegawa et al., 2017), which presumably also slow biological aging (Cole et al., 2017; Hahn et al., 2017; Wang et al., 2017). The mechanism of epigenetic drift is unclear. Given the diversity of cell and tissue stresses, it is likely that there is not a single cause of drift and presumptive biological clocks. For example, ticking of the EpiTOC, which is accelerated in pre-cancerous lesions and cancer and in normal epithelial cells exposed to a carcinogen, reflects methylation of polycomb targets through mitotic divisions (Yang et al., 2016), perhaps because of errors in DNA replication-coupled epigenetic transmission (Issa, 2014). This may be potentiated by expression of telomerase (Lu et al., 2018); for example, in stem cells. The all-cause mortality clock of Zhang et al. (2017) in part reflects smoking exposure. Smoking has been repeatedly reported to affect DNA methylation (Lee and Pausova, 2013), and recent studies showed that chronic smoke exposure causes widespread epigenetic changes, including increased sequential recruitment of the polycomb protein complex and DNA methylation to promoters of candidate tumor suppressor genes, dysregulation of signaling and growth control genes, and predisposition to transformation (Vaz et al., 2017). Conceivably, smoking may also exert its effects on DNA methylation via DNA damage and/or reactive oxygen species (O’Hagan et al., 2008, 2011). Although the drivers of the EpiTOC and the all-cause mortality clock are likely not identical, these biological clocks underscore the complex but very evident relationship between DNA methylation and polycomb, whose dysregulation is a recurring feature in the relationship between aging, cancer, the epigenome and, more recently, epigenetic clocks. The presumptive biological clock manifest in hypomethylation of genic enhancers of highly expressed genes might reflect epigenetic dynamics associated with high gene and enhancer activity (Cole et al., 2017; Feldmann et al., 2013; Wang et al., 2017), perhaps linked to circadian or other cycles of net TET and DNMT activity (Coulson et al., 2018; Lim et al., 2017; Oh et al., 2018; Rulands et al., 2018).
In sum, the mechanisms underlying DNA methylation chronological and biological clocks are likely different but, perhaps, also overlapping. The chronological clock likely reflects entropic decay of the methylation landscape linked to biochemical activity. The biological clock may overlap with the phenomenon previously referred to as epigenetic drift. Both methylation clocks may be part of wider epigenetic clocks, encompassing both DNA methylation and chromatin changes. Tools for integration of diverse datasets to build better epigenetic networks are being developed (Cabezas-Wallscheid et al., 2014; Chen and Li, 2016). Viewing age-associated epigenetic changes as an interconnected network of DNA and histone modifications, and even histone variants, nucleosome density, and chromatin binding proteins, can have important implications for understanding the cause, consequence, and targets for intervention. For example, drug interventions that primarily target histone methylation might have secondary effects on the entire epigenetic network, including the DNA methylation clock.
What is the Consequence of the Clocks, if Any?
Although some human disease syndromes caused by mutations in the DNA methylation machinery, such as immunodeficiency-centromeric instability-facial (ICF) anomalies syndrome and DNMT3A overgrowth syndrome (Ehrlich et al., 2006; Tatton-Brown et al., 2014), are not obviously indicative of premature aging, in other cases, altered DNA methylation can be causative of some diseases of aging, including cancer and degenerative diseases. Methylation at polycomb targets has previously been suggested to promote cancer (Ohm et al., 2007; Schlesinger et al., 2007; Widschwendter et al., 2007). In support of this, epigenome editing to direct methylation of the CDKN2A locus, a polycomb target encoding expression of the tumor suppressors p16 and p14ARF, prevents onset of senescence, a potent tumor suppressor mechanism (Saunderson et al., 2017). DNMT3B is an oncogene, notably in colon cancer (Lin et al., 2006; Linhart et al., 2007). DNMT3A is a tumor suppressor in hematologic malignancies (Mayle et al., 2015). TET DNA demethylases are also recurrently mutated in diverse cancers (Rasmussen and Helin, 2016). At least in hematologic malignancies, mutation of DNMT3A and TET2 is thought to be an early event that initiates disease progression through altered DNA methylation, acting as a precursor to the onset of these diseases in the aged (Jan et al., 2017). DNMT1 mutations cause two related adult-onset and age-dependent neurodegenerative diseases, hereditary sensory and autonomic neuropathy type 1 (HSAN1E) and autosomal dominant cerebellar ataxia, deafness, and narcolepsy (ADCA-DN) (Baets et al., 2015; Klein et al., 2011,2013; Winkel-mann et al., 2012). Based on these documented links between altered DNA methylation and disease, it is reasonable to consider whether age-associated DNA methylation changes, reflected in the chronological and biological clocks, might also promote dysfunction and disease.
The chronological clock encompasses methylation changes at regulatory regions of the genome (Horvath, 2013; Petkovich et al., 2017; Thompson et al., 2017; Wang et al., 2017; Weidner et al., 2014), and Hannum et al. (2013) noted that, at least in blood, genes that show age-associated changes in expression are close to CpGs that show changes in methylation. Thus, ticking of the chronological clock may contribute to age-associated phenotypes that, more or less, track chronological age in most individuals; for example, hair greying, skin wrinkling, cognitive decline, sarcopenia, and many others. Although these phenotypes progress at different rates in different individuals, a shared component may be driven by a chronological clock. If, as proposed above, ticking of the chronological clock reflects increased entropy and a decay of the epigenetic landscape that underpins epigenome function, then ticking might result in individual epigenetic features (genes, promoters, enhancers) being less well resolved from each other. Although the effect of decay at any one locus might be very small, the consequences for the functionality of the whole genome and cell can be substantial, perhaps in part because of many long-range and synergistic regulatory interactions and a large cumulative effect of many, many small individual changes across the genome. This could lead to the characteristic decline in cell and tissue function with age.
In contrast to the chronological clock, ticking of a biological clock is more likely to contribute to the characteristic inter-individual variability in age-associated functional decline and disease. The all-cause mortality clock of Zhang et al. (2017) is seemingly enriched for genes previously implicated in age-associated diseases, such as cancer, type 2 diabetes, and cardiovascular disease, suggesting that methylation might affect disease progression by control of these genes. Ticking of the EpiTOC reflects methylation of polycomb target genes (Yang et al., 2016); as noted above, this process has previously been suggested to promote cancer (Ohm et al., 2007; Schlesinger et al., 2007; Widschwendter et al., 2007). Enhancer hypomethylation, a candidate biological clock, for example, in the liver (Cole et al., 2017; Wang et al., 2017), might affect enhancer activity and, therefore, basal or induced expression of genes, change enhancer-gene interactions, or promote dysfunctional cryptic transcription from within genic enhancers (Neri et al., 2017). In the liver, such dysregulation might contribute to steatosis, steatohepatitis, type 2 diabetes, and metabolic syndrome (Michelotti et al., 2013).
If ticking of the clocks contributes to age-associated cell dysfunction, it may also be that the cell harbors mechanisms to restrain those epigenetic changes to promote phenotypic and functional stability over the lifespan. We have previously proposed the existence of mechanisms of chromatin homeostasis—or “chromostasis” —to maintain a chromatin steady state and, hence, a stable cell phenotype over the life course and promote healthy aging (Figure 4; Rai et al., 2014). If so, more efficient chromostasis may equate to reduced epigenetic drift and slowed ticking of a biological clock
Figure 4. Chromostasis, a Presumptive Process that Prevents Age-Associated Epigenetic and Phenotypic Change, Promotes Healthy Aging and Longevity.
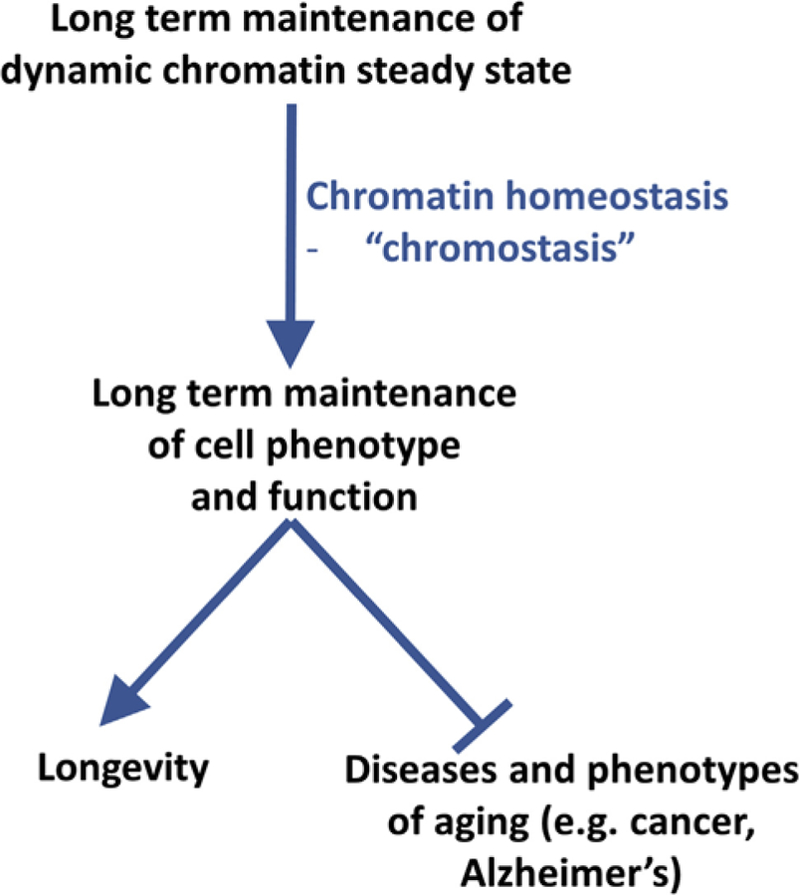
Healthy aging and longevity depend upon maintenance of cell phenotype. Because chromatin, in part, determines cell phenotype, this preservation of phenotype depends on a level of chromatin stability. Because chromatin is a dynamic structure, this, in turn, depends on chromatin homeostasis (chromostasis). Presumptive, but largely unknown, mechanisms of chromostasis may retard the ticking of DNAmethylation and epigenetic clocks and, perhaps, be a target for interventions to promote healthy aging and longevity.
Although it is tempting to attribute consequences to the clocks, it is important to note that one or both clocks might be passive, without consequence. Alternatively, they might reflect protective mechanisms to counter other detrimental changes in the aged cell. Indeed, Horvath (2013) has proposed that the DNA methylation clock reflects the activity of an epigenetic maintenance system (EMS) designed to maintain epigenetic stability.
It is important to know the consequences, if any, of the clocks, because this will dictate the outcome if the rate of ticking of the clocks can be manipulated. In mice, epigenetic drift can also be slowed by calorie restriction (CR) (Maegawa et al., 2017). Prolongevity interventions, including reduced growth hormone (GH) and insulin-like growth factor (IGF) signaling, CR, and rapamycin, also slow down ticking of the presumptive biological clock (Petkovich et al., 2017; Wang et al., 2017). However, whether slowing of the clock contributes to lifespan extension or vice versa is not clear. Models where apparent rejuvenation has been achieved primarily via targeting the epigenome (Ocampo et al., 2016; Wahlestedt et al., 2013) offer credence to the idea that intervening at the level of a biological epigenetic clock can be a route to healthy aging.
To conclude, although it is possible to speculate regarding the functional consequences of both chronological and biological DNA methylation clocks, the predominantly correlative nature of the studies to date makes it very difficult to distinguish between the clocks as causes, consequences, or passive bystanders of aging. A better understanding of the drivers of the chronological and biological clocks, as well as sophisticated epigenome-editing approaches in mice, will facilitate experiments to test their functional consequence, if any. If the clocks contribute to pathologies of aging, then it may ultimately be possible to intervene to promote healthy aging. If, on the other hand, the clocks are consequences or bystanders of aging, then they can still be valuable biomarkers of aging.
Summary and Outstanding Questions
Many years of research have documented age-associated changes to DNA methylation. However, only recently have computational analyses identified collections of individual CpG sites whose aggregate methylation status provides an accurate measure of chronological age, the DNA methylation clock. Ticking of this clock most likely reflects age-dependent genome-wide entropic decay of the DNA methylation landscape, linked to physiological dynamic turnover of DNA methylation. Although the DNA methylation clock has initially been developed as a molecular biomarker of chronological age, evidence suggests that DNA methylation may also have value as a biomarker of healthy versus unhealthy aging and disease risk; in other words, a biological clock.
There are a number of outstanding questions and areas for further investigation (Figure 5). First, DNA methylation clocks to date have been generated using methylation data that reflect the average in populations of cells. However, at any one CpG, this population average methylation value obscures heterogeneity between cells (Angermueller et al., 2016; Gravina et al., 2016; Smallwood et al., 2014). When considering the causes and potential consequences of methylation clocks, it will be important to consider this cell-to-cell heterogeneity. Second, although the evidence for a DNA methylation-based biological clock or clocks is provocative, future studies should attempt to build dedicated, perhaps tissue-specific, biological clocks by correlating DNA methylation directly with aging phenotypes and dysfunctions of interest. Ultimately, this may provide predictive biomarkers for aging and disease. Third, the molecular mechanisms underlying the chronological clock and potential biological clocks are very poorly understood. Of special interest is the distinction between the chronological and biological clocks at the molecular level. Fourth, the consequences, if any, of either the chronological clock or biological clocks are unknown. Potentially, although a chronological clock might contribute to features of age-associated functional decline that are shared between individuals, tissue-specific biological clocks might contribute to the inter-individual differences in dysfunction and disease that characterize the aging process. A better understanding of molecular mechanisms underlying the clocks, together with sophisticated epigenome editing approaches, can help to establish whether the clocks have functions or consequences. In turn, this knowledge will inform whether the clocks can be targets for intervention to promote disease prevention and healthy aging.
Figure 5. Some Outstanding Questions.

(A) In the mouse liver, hypomethylation of enhancers is linked to chronological age and contributes to the clock, but the rate of hypomethylation is decreased by interventions thought to slow biological aging (calorie restriction (CR), rapamycin (rapa), and Ames dwarfism [not shown]), suggesting that enhancers are a hybrid chronological and biological clock (but potentially separable) (Cole et al„ 2017; Wang et al., 2017).
(B-F) Many questions remain, although the answers to these questions likely differ for different clock CpGs (e.g., at enhancers versus promoters).
(B) To date, methylation clocks have been generated from data obtained from populations of cells, is there cell-to-cell variation in ticking of the clock?
(C) Does enhancer hypomethylation result from downregulation of DNMTs or their methyl donor substrate (SAM), increased TET demethylase activity, increased TET cofactor α-KG, increased passive demethylation, or another mechanism?
(D) At enhancers, does hypomethylation affect gene expression, expression of cryptic transcripts that are normally silenced by DNA methylation, enhancer-gene interactions, or all or none of these?
(E) Can a biological clock, probably tissue-specific, predict disease with sufficient sensitivity and specificity to be clinically useful?
(F) How do pro-longevity interventions slow ticking of the clock, and does this contribute to their pro-longevity and/or pro-health aging benefits?
ACKNOWLEDGMENTS
Work in the lab of P.D.A. is funded by the NIA grant P0l AG031862. Work in the lab of T.l. is funded by the NIH grant R01ES014811 and the CIRM Center of Excellence for Stem Cell Genomics.
REFERENCES
- Ahuja N, Li Q, Mohan AL, Baylin SB, and Issa JP (1998). Aging and DNA methylation in colorectal mucosa and cancer. Cancer Res 58, 5489–5494. [PubMed] [Google Scholar]
- Ambatipudi S, Horvath S, Perrier F, Cuenin C, Hernandez-Vargas H, Le Calvez-Kelm F, Durand G, Byrnes G, Ferrari P, Bouaoun L, et al. (2017). DNA methylome analysis identifies accelerated epigenetic ageing associated with postmenopausal breast cancer susceptibility. Eur. J. Cancer 75,299–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angermueller C, Clark SJ, Lee HJ, Macaulay IC, Teng MJ, Hu TX, Krueger F, Smallwood S, Ponting CP, Voet T, et al. (2016). Parallel single-cell sequencing links transcriptional and epigenetic heterogeneity. Nat. Methods 13, 229–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong VL, Rakoczy S, Rojanathammanee L, and Brown-Borg HM (2014). Expression of DNA methyltransferases is influenced by growth hormone in the long-living Ames dwarf mouse in vivo and in vitro. J. Gerontol. A Biol. Sci. Med. Sci 69, 923–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avrahami D, Li C, Zhang J, Schug J, Avrahami R, Rao S, Stadler MB, Burger L, Schübeler D, Glaser B, and Kaestner KH (2015). Aging-dependent demethylation of regulatory elements correlates with chromatin state and improved p cell function. Cell Metab 22, 619–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baets J, Duan X, Wu Y, Smith G, Seeley WW, Mademan I, McGrath NM, Beadell NC, Khoury J, Botuyan MV, et al. (2015). Defects of mutant DNMT1 are linked to a spectrum of neurological disorders. Brain 138, 845–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker GT 3rd, and Sprott RL (1988). Biomarkers of aging. Exp. Gerontol 23, 223–239. [DOI] [PubMed] [Google Scholar]
- Berger SL, and Sassone-Corsi P (2016). Metabolic signaling to chromatin. Cold Spring Harb. Perspect. Biol 8, a019463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder AM, Corvalan C, Mericq V, Pereira A, Santos JL, Horvath S, Shepherd J, and Michels KB (2018). Faster ticking rate of the epigenetic clock is associated with faster pubertal development in girls. Epigenetics 13, 85–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blagosklonny MV (2013). Aging is not programmed: genetic pseudo-program is a shadow of developmental growth. Cell Cycle 12, 3736–3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock C, Beerman I, Lien WH, Smith ZD, Gu H, Boyle P, Gnirke A, Fuchs E, Rossi DJ, and Meissner A (2012). DNA methylation dynamics during in vivo differentiation of blood and skin stem cells. Mol. Cell 47, 633–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocklandt S, Lin W, Sehl ME, Sánchez FJ, Sinsheimer JS, Horvath S, and Vilain E (2011). Epigenetic predictor of age. PLoS ONE 6, e14821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth LN, and Brunet A (2016). The aging epigenome. Mol. Cell 62, 728–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabezas-Wallscheid N, Klimmeck D, Hansson J, Lipka DB, Reyes A, Wang Q, Weichenhan D, Lier A, von Paleske L, Renders S, et al. (2014). Identification of regulatory networks in HSCs and their immediate progeny via integrated proteome, transcriptome, and DNA methylome analysis. Cell Stem Cell 15, 507–522. [DOI] [PubMed] [Google Scholar]
- Carroll JE, Irwin MR, Levine M, Seeman TE, Absher D, Assimes T, and Horvath S (2017). Epigenetic aging and immune senescence in women with insomnia symptoms: findings from the Women’s Health Initiative Study. Biol. Psychiatry 81,136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BS, and Li CW (2016). Constructing an integrated genetic and epigenetic cellular network for whole cellular mechanism using high-throughput next-generation sequencing data. BMC Syst. Biol 10,18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BH, Marioni RE, Colicino E, Peters MJ, Ward-Caviness CK, Tsai PC, Roetker NS, Just AC, Demerath EW, Guan W, et al. (2016). DNA methylation-based measures of biological age: meta-analysis predicting time to death. Aging (Albany NY) 8,1844–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen BC, Houseman EA, Marsit CJ, Zheng S, Wrensch MR, Wiemels JL, Nelson HH, Karagas MR, Padbury JF, Bueno R, et al. (2009). Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet 5, e1000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiansen L, Lenart A, Tan Q, Vaupel JW, Aviv A, McGue M, and Christensen K (2016). DNA methylation age is associated with mortality in a longitudinal Danish twin study. Aging Cell 15,149–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccarone F, Malavolta M, Calabrese R, Guastafierro T, Bacalini MG, Reale A, Franceschi C, Capri M, Hervonen A, Hurme M, et al. (2016). Age-dependent expression of DNMT1 and DNMT3B in PBMCs from a large European population enrolled in the MARK-AGE study. Aging Cell 15, 755–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole JJ, Robertson NA, Rather MI, Thomson JP, McBryan T, Sproul D, Wang T, Brock C, Clark W, Ideker T, et al. (2017). Diverse interventions that extend mouse lifespan suppress shared age-associated epigenetic changes at critical gene regulatory regions. Genome Biol 18, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulson RL, Yasui DH, Dunaway KW, Laufer BI, Vogel Ciernia A, Zhu Y, Mordaunt CE, Totah TS, and LaSalle JM (2018). Snordl 16-dependent diurnal rhythm of DNA methylation in mouse cortex. Nat. Commun 9,1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies MN, Volta M, Pidsley R, Lunnon K, Dixit A, Lovestone S, Coarfa C, Harris RA, Milosavljevic A, Troakes C, et al. (2012). Functional annotation of the human brain methylome identifies tissue-specific epigenetic variation across brain and blood. Genome Biol 13, R43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day K, Waite LL, Thalacker-Mercer A, West A, Bamman MM, Brooks JD, Myers RM, and Absher D (2013). Differential DNA methylation with age displays both common and dynamic features across human tissues that are influenced by CpG landscape. Genome Biol 14, R102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deaton AM, and Bird A (2011). CpG islands and the regulation of transcription. Genes Dev 25,1010–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deaton AM, Webb S, Kerr AR, Illingworth RS, Guy J, Andrews R, and Bird A (2011). Cell type-specific DNA methylation at intragenic CpG islands in the immune system. Genome Res 21,1074–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durso DF, Bacalini MG, Sala C, Pirazzini C, Marasco E, Bonafé M, do Valle IF, Gentilini D, Castellani G, Faria AMC, et al. (2017). Acceleration of leukocytes’ epigenetic age as an early tumor and sex-specific marker of breast and colorectal cancer. Oncotarget 8, 23237–23245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich M, Jackson K, and Weemaes C (2006). Immunodeficiency, centromeric region instability, facial anomalies syndrome (ICF). Orphanet J. Rare Dis 1, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott G, Hong C, Xing X, Zhou X, Li D, Coarfa C, Bell RJ, Maire CL, Ligon KL, Sigaroudinia M, et al. (2015). Intermediate DNA methylation is a conserved signature of genome regulation. Nat. Commun 6, 6363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmann A, Ivanek R, Murr R, Gaidatzis D, Burger L, and Schübeler D (2013). Transcription factor occupancy can mediate active turnover of DNA methylation at regulatory regions. PLoS Genet 9, e 003994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández AF, Bayón GF, Urdinguio RG, Toraño EG, García MG, Carella A, Petrus-Reurer S, Ferrero C, Martinez-Camblor P, Cubillo I, et al. (2015). H3K4me1 marks DNA regions hypomethylated during aging in human stem and differentiated cells. Genome Res 25, 27–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraga MF, and Esteller M (2007). Epigenetics and aging: the targets and the marks. Trends Genet 23, 413–418. [DOI] [PubMed] [Google Scholar]
- Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, Heine-Suñer D, Cigudosa JC, Urioste M, Benitez J, et al. (2005). Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. USA 102,10604–10609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garagnani P, Bacalini MG, Pirazzini C, Gori D, Giuliani C, Mari D, Di Blasio AM, Gentilini D, Vitale G, Collino S, et al. (2012). Methylation of ELOVL2 gene as a new epigenetic marker of age. Aging Cell 11,1132–1134. [DOI] [PubMed] [Google Scholar]
- Geller AM, Kotb MY, Jernigan HM Jr., and Kredich NM (1988). Methionine adenosyltransferase and S-adenosylmethionine in the developing rat lens. Exp. Eye Res 47,197–204. [DOI] [PubMed] [Google Scholar]
- Gravina S, Dong X, Yu B, and Vijg J (2016). Single-cell genome-wide bisulfite sequencing uncovers extensive heterogeneity in the mouse liver methylome. Genome Biol 17,150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greger V, Passarge E, Höpping W, Messmer E, and Horsthemke B (1989). Epigenetic changes may contribute to the formation and spontaneous regression of retinoblastoma. Hum. Genet 83,155–158. [DOI] [PubMed] [Google Scholar]
- Gregory DJ, Zhang Y, Kobzik L, and Fedulov AV (2013). Specific transcriptional enhancement of inducible nitric oxide synthase by targeted promoter demethylation. Epigenetics 8,1205–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross AM, Jaeger PA, Kreisberg JF, Licon K, Jepsen KL, Khosroheidari M, Morsey BM, Swindells S, Shen H, Ng CT, et al. (2016). Methylome-wide analysis of chronic HIV infection reveals five-year increase in biological age and epigenetic targeting of HLA. Mol. Cell 62,157–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn O, Grönke S, Stubbs TM, Ficz G, Hendrich O, Krueger F, Andrews S, Zhang Q, Wakelam MJ, Beyer A, et al. (2017). Dietary restriction protects from age-associated DNA methylation and induces epigenetic reprogramming of lipid metabolism. Genome Biol 78, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, Klotzle B, Bibikova M, Fan JB, Gao Y, et al. (2013). Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 49,359–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman JG, Latif F, Weng Y, Lerman MI, Zbar B, Liu S, Samid D, Duan DS, Gnarra JR, Linehan WM, et al. (1994). Silencing of the VHL tumor-suppressor gene by DNA methylation in renal carcinoma. Proc. Natl. Acad. Sci. USA 91, 9700–9704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman DR, Cornatzer WE, and Duerre JA (1979). Relationship between tissue levels of S-adenosylmethionine, S-adenylhomocysteine, and transmethylation reactions. Can. J. Biochem 57, 56–65. [DOI] [PubMed] [Google Scholar]
- Hon GC, Song CX, Du T, Jin F, Selvaraj S, Lee AY, Yen CA, Ye Z, Mao SQ, Wang BA, et al. (2014). 5mC oxidation by Tet2 modulates enhancer activity and timing of transcriptome reprogramming during differentiation. Mol. Cell 56, 286–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S (2013). DNA methylation age of human tissues and cell types. Genome Biol 14, R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S, and Levine AJ (2015). HIV-1 infection accelerates age according to the epigenetic clock. J. Infect. Dis 212,1563–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S, and Raj K (2018). DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet 19, 371–384. [DOI] [PubMed] [Google Scholar]
- Horvath S, and Ritz BR (2015). Increased epigenetic age and granulocyte counts in the blood of Parkinson’s disease patients. Aging (Albany NY) 7, 1130–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S, Erhart W, Brosch M, Ammerpohl O, von Schönfels W, Ahrens M, Heits N, Bell JT, Tsai PC,Spector TD, et al. (2014). Obesity accelerates epigenetic aging of human liver. Proc. Natl. Acad. Sci. USA 111, 15538–15543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S, Garagnani P, Bacalini MG, Pirazzini C, Salvioli S, Gentilini D, Di Blasio AM, Giuliani C, Tung S, Vinters HV, and Franceschi C (2015a). Accelerated epigenetic aging in Down syndrome. Aging Cell 14, 491–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S, Pirazzini C, Bacalini MG, Gentilini D, Di Blasio AM, Delle-donne M, Mari D, Arosio B, Monti D, Passarino G, et al. (2015b). Decreased epigenetic age of PBMCs from Italian semi-supercentenarians and their offspring. Aging (Albany NY) 7,1159–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S, Langfelder P, Kwak S, Aaronson J, Rosinski J, Vogt TF, Eszes M, Faull RL, Curtis MA, Waldvogel HJ, et al. (2016). Huntington’s disease accelerates epigenetic aging of human brain and disrupts DNA methylation levels. Aging (Albany NY) 8,1485–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, et al. (2009). The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet 41,178–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issa JP (2014). Aging and epigenetic drift: a vicious cycle. J. Clin. Invest 124, 24–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issa JP, Ottaviano YL, Celano P, Hamilton SR, Davidson NE, and Baylin SB (1994). Methylation of the oestrogen receptor CpG island links ageing and neoplasia in human colon. Nat. Genet 7, 536–540. [DOI] [PubMed] [Google Scholar]
- Issa JP, Vertino PM, Boehm CD, Newsham IF, and Baylin SB (1996). Switch from monoallelic to biallelic human IGF2 promoter methylation during aging and carcinogenesis. Proc. Natl. Acad. Sci. USA 93,11757–11762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issa JP, Ahuja N, Toyota M, Bronner MP, and Brentnall TA (2001). Accelerated age-related CpG island methylation in ulcerative colitis. Cancer Res 61, 3573–3577. [PubMed] [Google Scholar]
- Jackson SH, Weale MR, and Weale RA (2003). Biological age-what is it and can it be measured? Arch. Gerontol. Geriatr 36,103–115. [DOI] [PubMed] [Google Scholar]
- Jan M, Ebert BL, and Jaiswal S (2017). Clonal hematopoiesis. Semin. Hematol 54, 43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong M, Sun D, Luo M, Huang Y, Challen GA, Rodriguez B, Zhang X, Chavez L, Wang H, Hannah R, et al. (2014). Large conserved domains of low DNA methylation maintained by Dnmt3a. Nat. Genet 46, 17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji H, Ehrlich LI, Seita J, Murakami P, Doi A, Lindau P, Lee H, Aryee MJ, Irizarry RA, Kim K, et al. (2010). Comprehensive methylome map of lineage commitment from haematopoietic progenitors. Nature 467, 338–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic T, Vance LA, Cross D, Knight AK, Kilaru V, Michopoulos V, Klengel T, and Smith AK (2017). Exposure to violence accelerates epigenetic aging in children. Sci. Rep 7, 8962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Siegmund KD, Tavaré S, and Shibata D (2005a). Age-related human small intestine methylation: evidence for stem cell niches. BMC Med 3,10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Tavaré S, and Shibata D (2005b). Counting human somatic cell replications: methylation mirrors endometrial stem cell divisions. Proc. Natl. Acad. Sci. USA 102,17739–17744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkwood TB (2005). Understanding the odd science of aging. Cell 120, 437–447. [DOI] [PubMed] [Google Scholar]
- Klein CJ, Botuyan MV, Wu Y, Ward CJ, Nicholson GA, Hammans S, Hojo K, Yamanishi H, Karpf AR, Wallace DC, et al. (2011). Mutations in DNMT1 cause hereditary sensory neuropathy with dementia and hearing loss. Nat. Genet 43, 595–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein CJ, Bird T, Ertekin-Taner N, Lincoln S, Hjorth R, Wu Y, Kwok J, Mer G, Dyck PJ, and Nicholson GA (2013). DNMT1 mutation hot spot causes varied phenotypes of HSAN1 with dementia and hearing loss. Neurology 80, 824–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent L, Wong E, Li G, Huynh T, Tsirigos A, Ong CT, Low HM, Kin Sung KW, Rigoutsos I, Loring J, and Wei CL (2010). Dynamic changes in the human methylome during differentiation. Genome Res 20, 320–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KW, and Pausova Z (2013). Cigarette smoking and DNA methylation. Front. Genet 4,132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessing D, Anguera MC, and Lee JT (2013). X chromosome inactivation and epigenetic responses to cellular reprogramming. Annu. Rev. Genomics Hum. Genet 14, 85–110. [DOI] [PubMed] [Google Scholar]
- Levine ME, Hosgood HD, Chen B, Absher D, Assimes T, and Horvath S (2015. a). DNA methylation age of blood predicts future onset of lung cancer in the women’s health initiative. Aging (Albany NY) 7, 690–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine ME, Lu AT, Bennett DA, and Horvath S (2015b). Epigenetic age of the pre-frontal cortex is associated with neuritic plaques, amyloid load, and Alzheimer’s disease related cognitive functioning. Aging (Albany NY) 7, 1198–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine AJ, Quach A, Moore DJ, Achim CL, Soontornniyomkij V, Masliah E, Singer EJ, Gelman B, Nemanim N, and Horvath S (2016a). Accelerated epigenetic aging in brain is associated with pre-mortem HIV-associated neurocognitive disorders. J. Neurovirol 22, 366–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine ME, Lu AT, Chen BH, Hernandez DG, Singleton AB, Ferrucci L, Bandinelli S, Salfati E, Manson JE, Quach A, et al. (2016b). Menopause accelerates biological aging. Proc. Natl. Acad. Sci. USA 113, 9327–9332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine ME, Lu AT, Quach A, Chen BH, Assimes TL, Bandinelli S, Hou L, Baccarelli AA, Stewart JD, Li Y, et al. (2018). An epigenetic biomarker of aging for lifespan and healthspan. Aging (Albany NY) 10, 573–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li E, Beard C, and Jaenisch R (1993). Role for DNA methylation in genomic imprinting. Nature 366, 362–365. [DOI] [PubMed] [Google Scholar]
- Lim AS, Klein HU, Yu L, Chibnik LB, Ali S, Xu J, Bennett DA, and De Jager PL (2017). Diurnal and seasonal molecular rhythms in human neocortex and their relation to Alzheimer’s disease. Nat. Commun 8,14931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H, Yamada Y, Nguyen S, Linhart H, Jackson-Grusby L, Meissner A, Meletis K, Lo G, and Jaenisch R (2006). Suppression of intestinal neoplasia by deletion of Dnmt3b. Mol. Cell. Biol 26, 2976–2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linhart HG, Lin H, Yamada Y, Moran E, Steine EJ, Gokhale S, Lo G, Cantu E, Ehrich M, He T, et al. (2007). Dnmt3b promotes tumorigenesis in vivo by gene-specific de novo methylation and transcriptional silencing. Genes Dev 21, 3110–3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomba R, Gindin Y, Jiang Z, Lawitz E, Caldwell S, Djedjos CS, Xu R, Chung C, Myers RP, Subramanian GM, et al. (2018). DNA methylation signatures reflect aging in patients with nonalcoholic steatohepatitis. JCI Insight 3, 96685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe D, Horvath S, and Raj K (2016). Epigenetic clock analyses of cellular senescence and ageing. Oncotarget 7, 8524–8531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe R, Barton C, Jenkins CA, Ernst C, Forman O, Fernández-Twinn DS, Bock C, Rossiter SJ, Faulkes CG, Ozanne SE, et al. (2018). Ageing-associated DNA methylation dynamics are a molecular readout of lifespan variation among mammalian species. Genome Biol 79, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu F, Liu Y, Jiang L, Yamaguchi S, and Zhang Y (2014). Role of Tet proteins in enhancer activity and telomere elongation. Genes Dev 28,2103–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu AT, Xue L, Salfati EL, Chen BH, Ferrucci L, Levy D, Joehanes R, Murabito JM, Kiel DP, Tsai PC, et al. (2018). GWAS of epigenetic aging rates in blood reveals a critical role for TERT. Nat. Commun 9, 387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeder ML, Angstman JF, Richardson ME, Linder SJ, Cascio VM, Tsai SQ, Ho QH, Sander JD, Reyon D, Bernstein BE, et al. (2013). Targeted DNA demethylation and activation of endogenous genes using programmable TALE-TET1 fusion proteins. Nat. Biotechnol 31,1137–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maegawa S, Hinkal G, Kim HS, Shen L, Zhang L, Zhang J, Zhang N, Liang S, Donehower LA., and Issa, J.P. (2010). Widespread and tissue specific age-related DNA methylation changes in mice. Genome Res 20, 332–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maegawa S, Lu Y, Tahara T, Lee JT, Madzo J, Liang S, Jelinek J, Colman RJ, and Issa JJ (2017). Caloric restriction delays age-related methylation drift. Nat. Commun 8, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maierhofer A, Flunked J, Oshima J, Martin GM, Haaf T, and Horvath S (2017). Accelerated epigenetic aging in Werner syndrome. Aging (Albany NY) 9,1143–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marioni RE, Shah S, McRae AF, Chen BH, Colicino E, Harris SE, Gibson J, Henders AK, Redmond P, Cox SR, et al. (2015a). DNA methylation age of blood predicts all-cause mortality in later life. Genome Biol 16,25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marioni RE, Shah S, McRae AF, Ritchie SJ, Muniz-Terrera G, Harris SE, Gibson J, Redmond P, Cox SR, Pattie A, et al. (2015b). The epigenetic clock is correlated with physical and cognitive fitness in the Lothian Birth Cohort 1936. Int. J. Epidemiol 44,1388–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayle A, Yang L, Rodriguez B, Zhou T, Chang E, Curry CV, Challen GA, Li W, Wheeler D, Rebel VI, and Goodell MA (2015). Dnmt3a loss predisposes murine hematopoietic stem cells to malignant transformation. Blood 125, 629–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelotti GA, Machado MV, and Diehl AM (2013). NAFLD, NASH and liver cancer. Nat. Rev. Gastroenterol. Hepatol 10, 656–665. [DOI] [PubMed] [Google Scholar]
- Mitnitski A, Collerton J, Martin-Ruiz C, Jagger C, von Zglinicki T, Rock-wood K, and Kirkwood TB (2015). Age-related frailty and its association with biological markers of ageing. BMC Med 13, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammad HP, Cai Y, McGarvey KM, Easwaran H, Van Neste L, Ohm JE, O’Hagan HM, and Baylin SB (2009). Polycomb CBX7 promotes initiation of heritable repression of genes frequently silenced with cancer-specific DNA hypenmethylation. Cancer Res 69, 6322–6330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagae G, Isagawa T, Shiraki N, Fujita T, Yamamoto S, Tsutsumi S, Nonaka A, Yoshiba S, Matsusaka K, Midorikawa Y, et al. (2011). Tissue-specific demethylation in CpG-poor promoters during cellular differentiation. Hum. Mol. Genet 20, 2710–2721. [DOI] [PubMed] [Google Scholar]
- Neri F, Rapelli S, Krepelova A, Incarnato D, Parlato C, Basile G, Maldotti M, Anselmi F, and Oliviero S (2017). Intragenic DNA methylation prevents spurious transcription initiation. Nature 543, 72–77. [DOI] [PubMed] [Google Scholar]
- Nevalainen T, Kananen L, Marttila S, Jylhävä J, Mononen N, Kähönen M, Raitakari OT, Hervonen A, Jylhä M, Lehtimäki T, and Hurme M (2017). Obesity accelerates epigenetic aging in middle-aged but not in elderly individuals. Clin. Epigenetics 9, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nwanaji-Enwerem JC, Weisskopf MG, and Baccarelli AA (2018). Multi-tissue DNA methylation age: Molecular relationships and perspectives for advancing biomarker utility. Ageing Res. Rev 45,15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hagan HM, Mohammad HP, and Baylin SB (2008). Double strand breaks can initiate gene silencing and SIRT1 -dependent onset of DNA methylation in an exogenous promoter CpG island. PLoS Genet 4, e1000155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hagan HM, Wang W, Sen S, Destefano Shields C, Lee SS, Zhang YW, Clements EG, Cai Y, Van Neste L, Easwaran H, et al. (2011). Oxidative damage targets complexes containing DNA methyltransferases, SIRT1, and polycomb members to promoter CpG Islands. Cancer Cell 20, 606–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocampo A, Reddy P, Martinez-Redondo P, Platero-Luengo A, Hatanaka F, Hishida T, Li M, Lam D, Kurita M, Beyret E, et al. (2016). In vivo amelioration of age-associated hallmarks by partial reprogramming. Cell 167,1719–1733.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh G, Ebrahimi S, Carlucci M, Zhang A, Nair A, Groot DE, Labrie V, Jia P, Oh ES, Jeremian RH, et al. (2018). Cytosine modifications exhibit circadian oscillations that are involved in epigenetic diversity and aging. Nat. Commun 9, 644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohm JE, McGarvey KM, Yu X, Cheng L, Schuebel KE, Cope L, Mohammad HP, Chen W, Daniel VC, Yu W, et al. (2007). A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat. Genet 39, 237–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perna L, Zhang Y, Mons U, Holleczek B, Saum KU, and Brenner H (2016). Epigenetic age acceleration predicts cancer, cardiovascular, and all-cause mortality in a German case cohort. Clin. Epigenetics 8, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkovich DA, Podolskiy DI, Lobanov AV, Lee SG, Miller RA, and Gladyshev VN (2017). Using DNA methylation profiling to evaluate biological age and longevity interventions. Cell Metab 25, 954–960.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polanowski AM, Robbins J, Chandler D, and Jarman SN (2014). Epigenetic estimation of age in humpback whales. Mol. Ecol. Resour 14,976–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quach A, Levine ME, Tanaka T, Lu AT, Chen BH, Ferrucci L, Ritz B, Bandinelli S, Neuhouser ML, Beasley JM, et al. (2017). Epigenetic clock analysis of diet, exercise, education, and lifestyle factors. Aging (Albany NY) 9, 419–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai TS, Cole JJ, Nelson DM, Dikovskaya D, Faller WJ, Vizioli MG, Hewitt RN, Anannya O, McBryan T, Manoharan I, et al. (2014). HIRA orchestrates a dynamic chromatin landscape in senescence and is required for suppression of neoplasia. Genes Dev 28, 2712–2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raina A, Zhao X, Grove ML, Brassier J, Gottesman RF, Guan W, Pankow JS, Boerwinkle E, Mosley TH, and Fornage M (2017). Cerebral white matter hyperintensities on MRI and acceleration of epigenetic aging: the atherosclerosis risk in communities study. Clin. Epigenetics 9, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakyan VK, Down TA, Maslau S, Andrew T, Yang TP, Beyan H, Whittaker P, McCann OT, Finer S, Valdes AM, et al. (2010). Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res 20, 434–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen KD, and Helin K (2016). Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev 30, 733–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddington JP, Perricone SM, Nestor CE, Reichmann J, Youngson NA, Suzuki M, Reinhardt D, Dunican DS, Prandergast JG, Mjoseng H, et al. (2013). Redistribution of H3K27me3 upon DNA hypomethylation results in de-repression of Polycomb target genes. Genome Biol 14, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaldi L, Datta D, Serrat J, Morey L, Solanas G, Avgustinova A, Blanco E, Pons JI, Matallanas D, Von Kriegsheim A, et al. (2016). Dnmt3a and Dnmt3b associate with enhancers to regulate human epidermal stem cell homeostasis. Cell Stem Cell 19, 491–501. [DOI] [PubMed] [Google Scholar]
- Rose NR, and Klose RJ (2014). Understanding the relationship between DNA methylation and histone lysine methylation. Biochim. Biophys. Acta 1839,1362–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rulands S, Lee HJ, Clark SJ, Angermueller C, Smallwood SA, Krueger F, Mohammed H, Dean W, Nichols J, Rugg-Gunn P, et al. (2018). Genome-scale oscillations in DNA methylation during exit from pluripotency. Cell Syst 7, 63–76.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunderson EA, Stepper P, Gomm JJ, Hoa L, Morgan A, Allen MD, Jones JL, Gribben JG, Jurkowski TP, and Ficz G (2017). Hit-and-run epigenetic editing prevents senescence entry in primary breast cells from healthy donors. Nat. Commun 8,1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlesinger Y, Straussman R, Keshet I, Farkash S, Hecht M, Zimmerman J, Eden E, Yakhini Z, Ben-Shushan E, Reubinoff BE, et al. (2007). Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat. Genet 39,232–236. [DOI] [PubMed] [Google Scholar]
- Schübeler D (2015). Function and information content of DNA methylation. Nature 517, 321–326. [DOI] [PubMed] [Google Scholar]
- Sehl ME, Henry JE, Storniolo AM, Ganz PA, and Horvath S (2017). DNA methylation age is elevated in breast tissue of healthy women. Breast Cancer Res. Treat 164, 209–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smallwood SA, Lee HJ, Angenmueller C, Krueger F, Saadeh H, Peat J, Andrews SR, Stegle O, Reik W, and Kelsey G (2014). Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat. Methods 11, 817–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- So K, Tamura G, Honda T, Homma N, Waki T, Togawa N, Nishizuka S, and Motoyama T (2006). Multiple tumor suppressor genes are increasingly methylated with age in non-neoplastic gastric epithelia. Cancer Sci 97, 1155–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler MB, Murr R, Burger L, Ivanek R, Lienert F, Schöler A, van Nim-wegen E, Wirbelauer C, Oakeley EJ, Gaidatzis D, et al. (2011). DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 480, 490–495. [DOI] [PubMed] [Google Scholar]
- Stramentinoli G, Gualano M, Catto E, and Algeri S (1977). Tissue levels of S-adenosylmethionine in aging rats. J. Gerontol 32, 392–394. [DOI] [PubMed] [Google Scholar]
- Stubbs TM, Bonder MJ, Stark AK, Krueger F, von Meyenn F, Stegle O, and Reik W; Bl Ageing Clock Team (2017). Multi-tissue DNA methylation age predictor in mouse. Genome Biol 18, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun N, Youle RJ, and Finkel T (2016). The mitochondrial basis of aging. Mol. Cell 61, 654–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talens RP, Christensen K, Putter H, Willemsen G, Christiansen L, Kremer D, Suchiman HE, Slagboom PE, Boomsma DI, and Heijmans BT (2012). Epigenetic variation during the adult lifespan: cross-sectional and longitudinal data on monozygotic twin pairs. Aging Cell 11, 694–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatton-Brown K, Seal S, Ruark E, Harmer J, Ramsay E, Del Vecchio Duarte S, Zachariou A, Hanks S, O’Brien E, Aksglaede L, et al. ; Childhood Overgrowth Consortium (2014). Mutations in the DNA methyltransferase gene DNMT3A cause an overgrowth syndrome with intellectual disability. Nat. Genet 46, 385–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teschendorff AE, Menon U, Gentry-Maharaj A, Ramus SJ, Weisen-berger DJ, Shen H, Campan M, Noushmehr H, Bell CG, Maxwell AP, et al. (2010). Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res 20, 440–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RF, Atzmon G, Gheorghe C, Liang HQ, Lowes C, Greally JM, and Barzilai N (2010). Tissue-specific dysregulation of DNA methylation in aging. Aging Cell 9, 506–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson MJ, vonHoldt B, Horvath S, and Pellegrini M (2017). An epigenetic aging clock for dogs and wolves. Aging (Albany NY) 9, 1055–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trolin CG, Lofberg C,Trolin G, andOreland L (1994). Brain ATP:L-methionine S-adenosyltransferase (MAT), S-adenosylmethionine (SAM) and S-adenosylhomocysteine (SAH): regional distribution and age-related changes. Eur. Neuropsychopharmacol 4, 469–477. [DOI] [PubMed] [Google Scholar]
- Vaz M, Hwang SY, Kagiampakis I, Phallen J, Patil A, O’Hagan HM, Murphy L, Zahnow CA, Gabrielson E, Velculescu VE, et al. (2017). Chronic cigarette smoke-induced epigenomic changes precede sensitization of bronchial epithelial cells to single-step transformation by KRAS mutations. Cancer Cell 32, 360–376.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viré E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, Morey L, Van Eynde A, Bernard D, Vanderwinden JM, et al. (2006). The Polycomb group protein EZH2 directly controls DNA methylation. Nature 439, 871–874. [DOI] [PubMed] [Google Scholar]
- Wagner W (2017). Epigenetic aging clocks in mice and men. Genome Biol 18,107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlestedt M, Norddahl GL, Sten G, Ugale A, Frisk MA, Mattsson R, Deierborg T, Sigvardsson M, and Bryder D (2013). An epigenetic component of hematopoietic stem cell aging amenable to reprogramming into a young state. Blood 121, 4257–4264. [DOI] [PubMed] [Google Scholar]
- Waki T, Tamura G, Sato M, and Motoyama T (2003). Age-related methylation of tumor suppressor and tumor-related genes: an analysis of autopsy samples. Oncogene 22, 4128–4133. [DOI] [PubMed] [Google Scholar]
- Wang T, Tsui B, Kreisberg JF, Robertson NA, Gross AM, Yu MK, Carter H, Brown-Borg HM, Adams PD, and Ideker T (2017). Epigenetic aging signatures in mice livers are slowed by dwarfism, calorie restriction and rapamycin treatment. Genome Biol 18, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidner CI, Lin Q, Koch CM, Eisele L, Beier F, Ziegler P, Bauerschlag DO, Jöckel KH, Erbel R, Mühleisen TW, et al. (2014). Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol 15, R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widschwendter M, Fiegl H, Egle D, Mueller-Holzner E, Spizzo G, Marth C, Weisenberger DJ, Campan M, Young J, Jacobs I, and Laird PW (2007). Epigenetic stem cell signature in cancer. Nat. Genet 39,157–158. [DOI] [PubMed] [Google Scholar]
- Winkelmann J, Lin L, Schormair B, Kornum BR, Faraco J, Plazzi G, Melberg A, Cornelio F, Urban AE, Pizza F, et al. (2012). Mutations in DNMT1 cause autosomal dominant cerebellar ataxia, deafness and narcolepsy. Hum. Mol. Genet 21, 2205–2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, and Zhang Y (2017). TET-mediated active DNAdemethylation: mechanism, function and beyond. Nat. Rev. Genet 18, 517–534. [DOI] [PubMed] [Google Scholar]
- Yang X, Han H, De Carvalho DD, Lay FD, Jones PA, and Liang G (2014). Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell 26, 577–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Wong A, Kuh D, Paul DS, Rakyan VK, Leslie RD, Zheng SC, Widschwendter M, Beck S, and Teschendorff AE (2016). Correlation of an epigenetic mitotic clock with cancer risk. Genome Biol 17, 205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatabe Y, Tavaré S, and Shibata D (2001). Investigating stem cells in human colon by using methylation patterns. Proc. Natl. Acad. Sci. USA 98, 10839–10844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, Morgunova E, Jolma A, Kaasinen E, Sahu B, Khund-Sayeed S, Das PK, Kivioja T, Dave K, Zhong F, et al. (2017). Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science 356, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zannas AS, Arloth J, Carrillo-Roa T, lurato S, Roh S, Ressler KJ, Nemeroff CB, Smith AK, Bradley B, Heim C, et al. (2015). Lifetime stress accelerates epigenetic aging in an urban, African American cohort: relevance of glucocorticoid signaling. Genome Biol 16, 266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Wilson R, Heiss J, Breitling LP, Saum KU, Schöttker B, Holleczek B, Waldenberger M, Peters A, and Brenner H (2017). DNA methylation signatures in peripheral blood strongly predict all-cause mortality. Nat. Commun 8,14617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SC, Widschwendter M, and Teschendorff AE (2016a). Epigenetic drift, epigenetic clocks and cancer risk. Epigenomics 8, 705–719. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Joyce BT, Colicino E, Liu L, Zhang W, Dai Q, Shrubsole MJ, Kibbe WA, Gao T, Zhang Z, et al. (2016b). Blood epigenetic age may predict cancer incidence and mortality. EBioMedicine 5, 68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou H, and Hastie T (2005). Regularization and variable selection via the elastic net. J. R. Stat. Soc. Series B Stat. Methodol 67, 301–320. [Google Scholar]