

Abstract
Lysosomal storage disorders (LSDs) comprise a clinically heterogeneous group of autosomal recessive or X-linked genetic syndromes caused by disruption of lysosomal biogenesis or function resulting in the accumulation of non-degraded substrates. Although LSDs are diagnosed predominantly in children, many show variable expressivity with clinical presentations possible later in life. Given the important role of lysosomes in neuronal homeostasis, neurologic manifestations, including movement disorders, can accompany many LSDs. Over the last decade, evidence from genetics, clinical epidemiology, cell biology, and biochemistry have converged to implicate links between LSDs and adult-onset movement disorders. The strongest evidence comes from mutations in Glucocerebrosidase, which cause Gaucher disease (GD), and are among the most common and potent risk factors for Parkinson’s disease (PD). However, recently many additional LSD genes have been similarly implicated, including SMPD1, ATP13A2, GALC, and others. Examination of these links can offer insight into pathogenesis of Parkinson’s disease and guide development of new therapeutic strategies. We systematically review the emerging genetic links between LSDs and PD.
Keywords: Genetics, Lysosomal Storage Disease, Movement Disorders, Parkinson’s Disease
The lysosome is a major site for the degradation of cellular proteins and organelles and is thus essential for cellular homeostasis. Neurons appear especially sensitive to lysosomal function as a wide range of neurological disorders have been attributed to defects in lysosome function. Mutations disrupting lysosomal biogenesis or function lead to the accumulation of nondegraded substrates and cause lysosomal storage disorders (LSDs). More than 50 LSDs have been identified, which show either autosomal recessive or X-linked inheritance. Although LSDs are predominantly pediatric disorders, there is substantial heterogeneity in both clinical presentations and manifestations, with infantile, juvenile, and adult forms recognized in many cases. Emerging evidence highlights links between LSDs in children and adult movement disorders, most notably, Parkinson’s disease (PD) (1) (Table 1). Examination of these links can inform our understanding of the different mechanisms of dysfunction that lead to onset of movement disorders and provide novel targets for therapeutic development.
Table 1.
Summary of Lysosomal Storage Disorder Associated with Parkinson’s disease
| Affected protein (Gene) |
Lysosomal storage disorder |
Parkinsonism in patients or carriers |
PD GWAS hit |
Pathogenic variant identified |
α-Synuclein pathology observed |
Relevant mouse modela |
|---|---|---|---|---|---|---|
| Glucocerebrosidase(GBA1) | Gaucher | X(118) | X(119) | X(12) | X(120) | X(121) |
| Acid Sphingomyelinase(SMPD1) | Niemann-Pick disease Type A,B |
X(36) | X(38) | X(35) | ||
| Niemann-Pick type C(NPC1) | Niemann-Pick type C | X(51) | X(48) | |||
| Galactosylceramidase(GALC) | Krabbe disease | X(70) | X(69) | X(69) | ||
| a-N-acetylglucosneuroaminidase(NAGLU) | Sanfilippo Syndrome B | X(79) | X(78) | X(122) | ||
| ATP13A2 (PARK9) | Neuronal Ceroid Lipofuscinosis Kufar-Rakeb syndrome |
X(88) | Xb,(87) | X(96) | X(123) | |
| Hexosamindase B (HEXB) | Sandhoff disease | X(109) | X(108) | X(107) |
Specifically animal mouse evidence of interaction with synuclein toxicity or PD mechanisms.
PD risk in PARK9 mutant carriers is controversial, but regardless can refer to PARK9 causing the related movement disorder, Kurfor-Rakeb syndrome, which includes parkinsonism.
The following review will examine the published links between lysosomal storage diseases in children and adult-onset PD. We have focused on overlaps which display a phenotypic or genetic association with PD and also display evidence of pathological association, namely alpha synuclein aggregation. Among the links described, only the link between GD and PD is definitively established. In most of the other cases we review additional investigation is required, including either genetic confirmation and/or mechanistic follow-up.
Glucocerebrosidase (GBA1)
Gaucher disease (GD) is a lysosomal storage disorder caused by autosomal recessive mutations in the GBA1 gene that encodes for the lysosomal enzyme glucocerebrosidase (GCase). GCase is a hydrolase which catalyzes the breakdown of glucosylceramide to glucose and ceramide (Figure 1). More than 300 mutations in GBA1 have been identified including point mutations, splice-site mutations, deletions, insertions, and mutations resulting from recombination events with the highly homologous pseudogene (2). These mutations lead to reduced lysosomal GCase activity due to either disrupted translation, misfolding, impaired trafficking, protein instability, or a combination of these defects. Very rarely, GD can also be caused by mutations in the PSAP gene which encode for Saposin C, and essential protein modulator for GCase (3).
Figure 1.
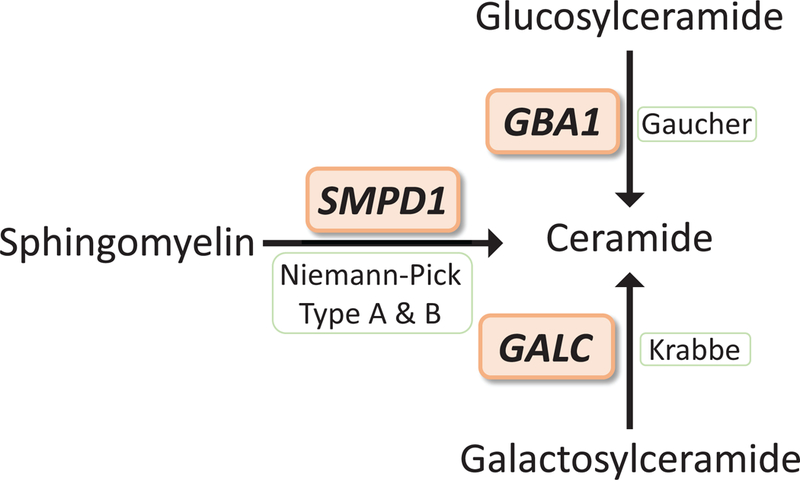
LSD genes important in ceramide metabolism are also implicated in PD. Mutations in GBA1, SMPD1, and GALC are responsible for Gaucher disease, Niemann-Pick disease type A and B, and Krabbe disease, respectively. GBA1 encodes for GCase which hydrolyzes glucosylceramide to glucose and ceramide, SMPD1 encodes for acid sphingomyelinase which hydrolyzes sphingomyelin to phosphocholine and ceramide, GALC encodes for galactosidase which hydrolyzes galactosyl moieties from lipids including the hydrolysis of galactosylceramide to galactose and ceramide.
Three clinical subtypes of GD are recognized based on age of onset and the presence of neurological manifestations. These subtypes are an attempt to categorize what is a phenotypic continuum with presentations ranging from perinatal lethality to nearly asymptomatic (4). Type 1 GD, is the mildest and most common form of disease (90% of cases in Europe and the USA), is the mildest form of the disease, typically presenting with hepatosplenomegaly and fatigue. The age at initial presentation of Type 1 GD is highly heterogeneous with onset reported in both juvenile and late-adult populations (5, 6). Type 2 GD is the most severe form of the disease with onset shortly after birth, including manifestation of severe neurological deficits, and rapidly progressive neurologic deterioration that is usually fatal by age 3. Type 3 GD includes the symptoms observed in type 1 GD along with more mild central nervous system involvement and variable age of onset (6).
As mentioned above, mutations in GBA1 lead to reduced GCase enzymatic activity. In general, greater decreases in enzymatic activity correlates with more severe phenotypes. For instance, mutations that result in complete or almost complete loss of enzymatic activity can be perinatal lethal and are more likely associated with the most severe, type 2 form of the disease (7, 8), while mutations with slightly higher residual activity display the least severe, type 1 phenotype. However, exceptions have also been observed (8). GBA1 loss-of-function mutations lead to accumulation of the GCase substrate glucosylceramide (GlcCer), particularly in macrophages, transforming them into classical Gaucher cells. Gaucher cells contain GlcCer aggregates in a characteristic twisted, fibrillar arrangement that are clearly distinguishable by electron microscopy. The monocyte lineage is especially vulnerable because they contain large amounts of glycosphingolipids. As GlcCer accumulates, it is converted to glucosylsphingosine by the enzyme acid ceramidase (9). GlcCer and Glucosylsphingosine spill over into the cytoplasm where they can be further metabolized by neutral GCase2 to ceramide and sphingosine respectively. Accumulation of glucosylsphingosine and sphingosine also contributes to toxicity in GD (10, 11).
Heterozygous mutations in the GBA1 gene are the most common genetic risk factor for PD with a predicted frequency of 7–10% in the PD population (12). This is an example of genetic pleiotropy, by which variation in a single gene (GBA1) can influence multiple, distinct clinical phenotypes (GD and PD), via autosomal recessive or dominant inheritance mechanisms, respectively. Overall GBA-PD exhibits a very similar symptomatic presentation to sporadic PD with a slightly earlier average age of onset. In addition, GBA-PD is characterized by a modest increased risk of cognitive impairment and dementia, and reduced bradykinesia and resting tremor relative to other PD patients (12).
Although the mechanism by which GBA1 mutations increase PD risk is not completely understood, GCase enzymatic activity appears to be a critical factor. Individuals with more severe GBA1 mutations, specifically mutations associated with lower enzymatic activity, have been shown to have increased risk for development of PD and an earlier age-of-onset (13). Specifically, carriers of more severe mutations, such as the c.84dupG or L444P mutation, which are often associated with neuronopathic GD, are more likely to develop PD than carriers of N370S of E326K mutations (13, 14). Moreover, stronger GBA1 loss-of-function alleles have also been implicated with more severe motor phenotypes and increased risk of non-motor manifestations, including hyposmia, cognitive impairment, and dementia (13–15). Interestingly, reduced GCase enzymatic activity has also been documented in sporadic PD (without known GBA1 mutations) (16, 17). Lastly, individuals with GD are also at risk of developing PD. The risk due to homozygous GBA1 loss-of-function alleles is significantly greater than that seen in heterozygous GBA1 mutations, although most individuals with GD will still not get PD (15, 18). Together, the accumulating evidence for genotype-phenotype relationships in GBA-PD strongly support a dose-dependent, loss-of-function risk model. There is also evidence that GCase unfolding and retention in the ER may also promote toxicity, perhaps via induction of ER stress. While this mechanism is unlikely to fully account for the GBA-PD link on its own, it could potentially be a relevant modifier contributing to disease heterogeneity (19). A similar modifier effect may also be driven by genes involved in regulation of GCase including SCARB2 and PSAP (3, 20, 21).
Work by our lab and others, has shown that reduced GCase activity is associated with lysosomal dysfunction and synuclein accumulation (22–24). This could be due to accumulation of the substrate, lysosomal glucosylceramide which was shown to increase alpha synuclein aggregation (25). Recent work has also shown that glucosylsphingosine, which is more abundant in the cytoplasm, might also contribute to alpha synuclein aggregation (11). More recently, we found that mitochondrial oxidant stress and increased oxidation of dopamine resulted in direct modification of GCase that led to its decreased activity and ultimately lysosomal dysfunction (24). This observation was made in dopaminergic neurons from patients with either idiopathic or familial PD indicating that this may represent a unifying mechanism linking GCase and PD (24). In support of this hypothesis, we found that mouse models exposed to increased oxidized dopamine displayed alpha synuclein accumulation, reduced GCase activity, and neurodegeneration in the substantia nigra (24). While alpha synuclein accumulation could inhibit GCase trafficking and lysosomal function in various cell types (23), our observation highlights one attractive mechanism for the increased vulnerability of midbrain dopaminergic neurons to loss of GCase activity (24).
Based on our current understanding, there are several strategies related to GCase ceramide metabolism that could be therapeutic for both GD and PD. While cellular regulation of ceramide is complex—involving de novo synthesis, recycling, and salvage pathways—several feasible strategies might target GCase specifically, including (i) increasing the stability or activity of the remaining GCase enzyme, (ii) GBA1 gene therapy, (iii) substrate removal therapy to reduce the accumulation of the glucosylceramide and glucosylsphingosine, or (iiii) restoring normal levels of ceramide. The first strategy involves the development of molecular chaperones to increase enzyme stability enabling increased trafficking of GCase to the lysosome and/or increasing protein half-life within the lysosome. Alternatively, the development of positive allosteric modulators could directly increase activity of GCase; although, this would only be effective if functional GCase is trafficked to the lysosome. The second strategy aims to increase functional GCase protein levels through gene therapy strategies. The third approach targets ceramide synthase in order to reduce accumulation of the GCase substrate, glucosylceramide. This strategy is already approved for use in GD patients and is currently in clinical trials for PD (26). Lastly, we have recently shown that a reduction in ceramide contributes to accumulation of alpha synuclein in GCase deficient cells (22). We found that inhibition of acid ceramidase, which catalyzes the hydrolysis of ceramide to sphingosine, could restore normal ceramide levels in the cell and rescue a GCase knockout phenotype (22).
Acid Sphingomyelinase (SMPD1)
Niemann-Pick disease (NPD) is a rare autosomal recessive disorder including neuropathic (Type A) and non-neuropathic (Type B) forms. NPD type A and B are caused by mutations in the gene SMPD1, which encodes for acid sphingomyelinase (ASM; Sphingomyelin phosphodiesterase). ASM catalyzes the hydrolysis of sphingomyelin to phosphocholine and ceramide in late endosomes and lysosomes (Figure 1). To date, over 180 mutations in SMPD1 have been identified, including point mutations (missense and frameshift), small deletions, and splice site mutations (27). These mutations appear to be equally distributed throughout the gene with no specific mutagenic regions identified.
Type A NPD is most common among individuals of Ashkenazi Jewish decent (66% of total type A), although it has also been reported in other populations (28). The most common SMPD1 mutations are R496L, L302P, and 990delC which account for 90% of type A alleles in the Ashkenazi Jewish population and the carrier frequency for these mutations within this population is 1:80 to 1:100, (29). NPD type B is considerably more pan-ethnic than type A with affected populations identified all around the world. Mutations in SMPD1 that lead to NPD type B display significantly reduced ASM activity, although it is thought that a small amount of residual activity remains, which accounts for the more mild phenotype in NPD type B patients relative to type A (30).
Type A NPD is characterized by hepatosplenomegaly with disease onset beginning months after birth. These individuals also display deteriorating psychomotor development and progressively worsening hypotonia (31). In contrast, patients with type B NPD display a significantly milder phenotype with age of onset ranging from infancy to late adulthood. These individuals have normal central nervous system development but exhibit hepatosplenomegaly, which can lead to liver failure. In addition, pulmonary function is often compromised and can lead to pulmonary infections, which is the most common cause of death in this population (31). Despite the classification of Type A and Type B, many intermediate cases have also been reported with mild to moderated neurological presentations (32). Several of these reported intermediate cases display compound heterozygous mutations, carrying one severe and one mild mutation. This observation highlights a genotype-phenotype association, although patients have also been identified as “intermediate” containing two mild mutations (32).
Mutations in SMPD1 lead to major loss of function of ASM. As a result, patients display accumulation of the ASM substrate sphingomyelin as well as bis(monoacylglycero)phosphate, lysosphingomyelin within the lysosome (33, 34). Under stress conditions, ASM is also translocated from the lysosome to the outer leaflet of the plasma membrane where sphingomyelin hydrolysis leads to reorganization of lipid microdomains (35). As a result, ASM knock-out animal models exhibit synaptic dysfunction (36, 37).
Carriers of SMPD1 mutations are at increased risk for PD (38). Specifically, heterozygous carriers of more severe mutations, which cause NPD type A, are implicated. The first mutation identified was L302P, which was found to substantially increase risk for PD (odds ratio estimate=9.4) (38). This was followed by identification of a less common mutation (R591C) in the Chinese population (39) as well as the fsP330 mutation (40). Interestingly, the R496L mutation, which is one of the most common point mutations in SMPD1, was not found to associate with increased PD (41).
Very little is known about the role of ASM and the pathology associated with SMPD1-linked PD, as this connection was only recently uncovered and is very rare in the general population. Patients with NPD type B display few neurological symptoms despite having only ~10% residual ASM activity (42). This creates uncertainties regarding the mechanism by which SMPD1 mutation carriers, anticipated to have at least 50% residual activity, would be at risk of PD. In addition, individuals with sporadic PD were found to have normal levels of ASM activity in a dried blood spot analysis (43). However, with the identification of SMPD1 mutations as risk factors for PD, it is likely that this connection will receive more attention.
Enzyme replacement therapies with ASM are currently in clinical trials for NPD patients. However, since these approaches do not reach the CNS, they are unlikely to be amenable for PD. Additional therapeutic strategies could be aimed at (i) increasing stability or activity of the residual enzyme, (ii) reducing the accumulation of the substrate, sphingomyelin, or (iii) increasing levels of products (phosphocholine and ceramide). Developing molecular chaperones could increase the stability and levels of protein in lysosomes, whereas positive allosteric modulators might enhance enzymatic activity. These efforts have been aided by the recent publication of the ASM crystal structure (44). Augmenting the activity of the similar enzyme, neutral sphingomyelinase, might be one approach to address the accumulation of sphingomyelin in NPD.
Niemann-Pick type C (NPC1)
Niemann-Pick type C (NPC) is an autosomal recessive lysosomal storage disorder primarily caused by mutations in the NPC1 gene (95% of cases). NPC1 is a large membrane protein that exists in late endosomes and lysosomes and participates in cholesterol efflux (45). More than 260 mutations have been identified in the NPC1 gene, most of which are missense mutations affecting the luminal domain of the protein, that are associated with onset the disease (46, 47). Similar to Niemann-Pick type A/B, NPC disease is characterized by hepatosplenomegaly and progressive neurodegeneration including ataxia, cognitive decline, and seizures. Disease onset usually begins between 2 and 4 years of age; however, similar to other LSDs, a milder form of the disease displays adult onset with slower progression (48, 49).
Several case reports have linked NPC1 to PD. Specifically, the discovery of phosphorylated synuclein pathology at autopsy in the brains of several patients with NPC, with classical lewy bodies described in the substantia nigra from two cases (50). In addition, several NPC patients and their family members have been reported to present with parkinsonism (51–53), consistent with a possible genetic link similar to GBA and SMPD1.
Loss of function of NPC1 leads to accumulation of cholesterol in the late endosome/lysosome of all cells (47, 54). Cholesterol has been shown to modulate alpha synuclein-membrane interactions (55), and thereby promoting aggregation and toxicity (56, 57). NPC1 mutant cells also accrue sphingolipids including glucosylceramide, glucosylsphingosine and sphingosine (58, 59). This is similar to GD and therefore potentially consistent with converging pathogenic mechanisms. The mechanism for accumulation of these additional lipids is unclear, but it could either reflect a direct role for NPC1 in lysosomal homeostasis or more indirectly, generalized lysosomal dysfunction following cholesterol accumulation. Further clinical and basic investigation is required to elucidate the potential association between NPC1 and PD.
One of the pathological hallmarks of NPC is the presence of neurofibrillary tangles comprised of the hyperphosphorylated tau isoform similarly observed in Alzheimer’s disease (60). Interestingly, the presence of the ApoE4 Alzheimer’s disease susceptibility allele in NPC1 patients may be associated with more rapid neurologic progression (61), and the coincident development of amyloid plaque pathology (50). Although confirmation of these findings in larger, independent cohorts is needed, the observations highlight both the potential role of genetic modifiers in modulating LSD phenotypes, and also the possibility of interactions between co-morbid brain pathologies. These factors likely contribute to the heterogeneity of disease phenotypes observed in many lysosomal storage disorders.
Galactosylceramidase (GALC)
Mutations in the gene GALC, which encodes for galactosylceramidase (GALC), are associated with onset of Globoid Cell Leukodystrophy, also known as Krabbe disease. GALC is a lysosomal enzyme that hydrolyses galactosyl moieties from lipids including galactosylcerebroside and galactosylsphingosine (Figure 1). More than 70 disease-causing mutations in the GALC gene have been identified, many of which are compound heterozygous (62, 63). A large number of these mutations are located outside the catalytic domain, yet still lead to substantially reduced enzymatic activity (~95%) (64). Therefore, these mutations are predicted to cause reduced RNA stability, protein stability, or impair protein trafficking (63). Similar to GD, age of onset and disease severity are often dictated by the degree to which enzymatic function is reduced (65). Enzymatic activity can be completely lost through splice site and nonsense mutations, while more mild, missense mutations show either normal or up to 22% reduced levels of function (64, 66). Considerable heterogeneity has been described in Krabbe disease, including among individuals with identical mutations, making it difficult to define genotype-phenotype relationship (66), and suggesting the possibility of other genetic or non-genetic modifiers.
Krabbe’s disease typically presents with infantile onset in the first 6 months of life. The disease commonly manifests as increased irritability, spasticity, and developmental delay along with unexplained fever, blindness and deafness (62). The disease course rapidly progresses to severe motor and mental deterioration (67). A small percentage of patients develop juvenile- or adult-onset forms of the disease. These forms are typically less severe, exhibit slower disease progression, and may not involve peripheral neuropathy (68).
Deficiency of lysosomal GALC leads to rapid accumulation of galactosylsphingosine in patients with Krabbe’s disease. Accumulation of galatosylsphingosine has been shown to be neurotoxic to oligodendrocytes and Schwann cells (69), and subsequent central and/or peripheral demyelination, respectively (70). In addition, on neuropathologic evaluation Krabbe’s disease may reveal cytoplasmic inclusions containing aggregated forms of alpha synuclein, which are thought to contribute to disease progression (71). Indeed, sphingolipid accumulation may directly promote and/or accelerate synuclein aggregation (11).
In a large GWAS meta-analysis, variants at the GALC locus were significantly associated with PD risk (72). The causal variant for the association of GALC locus with PD risk remains to be defined. Unlike the rare, loss-of-function mutations identified at GBA1 and SMPD1; the most strongly associated variant at GALC is a common polymorphism, rs8005172, located 12.6 kilobases proximal to the gene promoter, and located within an intron of an adjacent gene GPR65. Based on publicly available gene expression datasets (73) this variant is significantly associated with GALC gene expression across a large number of tissues, including in brain. Nevertheless, further studies are needed to definitively identify the causal variant(s), and confirm whether GALC is indeed responsible for the variant association with PD risk.
Further supporting a role of GALC in the onset of PD, a recent study revealed reduced GALC activity and associated sphingolipid accumulation in sporadic PD patients, when compared to healthy controls (74). Interestingly, GALC activity was also reduced in aging, which is the strongest risk factor for PD. However, similar to SMPD1-linked PD, the consideration of PD as part of the phenotypic spectrum of Krabbe’s disease is difficult to resolve. The disease phenotypes and symptomatic presentations are highly divergent, possibly suggestive of two separate disorders. It is possible that partial loss of GALC activity confers increased risk which, in combination with other factors, leads to the neurodegeneration observed in PD.
The discovery of a possible role of GALC in sporadic PD highlights the potential of this protein as a therapeutic target for idiopathic PD. While the current treatment of Krabbe’s disease is hematopoietic stem cell transplantation (75), recent research has identified molecular chaperones that effectively increase GALC protein and associated activity (63). These chaperones could potentially be therapeutically beneficial in PD as well. Alternatively, the development of allosteric modulators capable of enhancing GALC activity might have promising therapeutic potential in PD.
a-N-acetylglucosneuroaminidase (NAGLU)
Sanfilippo Syndrome B, also known as mucopolysaccharidosis III disease B (MPS-IIIB), is a lysosomal storage disorder caused by mutations in the NAGLU gene encoding α-N- acetylglucosminidase (NAG). NAG is a lysosomal hydrolase which degrades heparan sulfate glycosaminoglycans. Mutations in NAG lead to accumulation of heparan sulfate within affected individuals (76). Sanfilippo syndrome B is characterized by juvenile disease onset with progressive degeneration of the central nervous system. The rate of MPS-IIB progression varies considerably, which appears to correlate, in part, with the level of residual NAG enzymatic activity (77–79).
Common genetic variants at the NAGLU gene locus were initially discovered in association with PD risk on a candidate basis (80), and consistent results recently emerged from GWAS (81). In addition, examination of postmortem brain tissue from three MPS-IIIB cases revealed accumulations of phosphorylated alpha synuclein (82). Similar pathologic findings were also reported in the related LSD, MPS-IIIA. (80)
There are several possible mechanisms through which impaired NAG activity could promote synuclein accumulation. In-vitro studies have found that heparan sulfate dramatically increases the fibrillization of alpha synuclein (83). Synuclein aggregates can interact with heparan sulfates at the neuronal cell surface leading to cellular internalization (84). Thus, it is possible that increased heparan sulfates could accelerate this process enhancing the aggregation and subsequent propagation of alpha synuclein pathology. Additionally, glycosaminoglycans have been found to inhibit cathepsin D, a major lysosomal protease involved in synuclein degradation, leading to synuclein accumulation (85). Despite these promising observations, more investigation is needed to examine the potential overlap between MPS-IIIB and PD, including whether targeting NAG might have potential therapeutic benefit.
ATP13A2 (PARK9)
ATP13A2 is a transmembrane P5-type ATPase that is involved in active transport of cations across the lysosomal membrane. Currently, only a single homozygous missense mutation in PARK9 (M810R) has been reported leading to onset of Neuronal Ceroid Lipofuscinosis (NCL) in humans (86); although a similar phenotype in canines has more commonly been reported following mutation in a PARK9 homolog (87). NCL’s are a heterogenous group of lysosomal storage disorders resulting from accumulation of lipopigments, predominantly in the brain and retina (88). PARK9-associated NCL initially presents with learning difficulties in childhood followed by progressive cognitive and motor decline.
Homozygous or compound heterozygous mutations in PARK9 are also linked to development of Kufor-Rakeb syndrome (KRS). To date, 14 mutations including deletions, duplications, missense, and splice site mutations in functionally important domains of the protein have been identified (89). KRS, which typically manifests in adolescence or young adulthood, is characterized by Parkinsonian motor signs (tremor, bradykinesia, rigidity) in association with spasticity, supranuclear gaze palsy, and dementia. Pathological features in KRS are unknown as autopsy studies have not been reported.
Heterozygous carriers of PARK9 mutations have been reported with increased PD risk in some (90–93), but not all (94, 95) studies. Interestingly, in one report, PARK9 variants were implicated to co-occur with either GBA1 variants or alleles of another PD gene, Leucine rich repeat kinase (LRRK2) (96). Given the rarity of PARK9 mutations, definitive confirmation of a potential association with PD risk will require sequencing of much larger sample sizes.
Mutations in ATP13A2 lead to reduced protein expression through nonsense-mediated mRNA decay, reduced protein stability, or impaired trafficking to the lysosome causing them to undergo ER-associated degradation (97). We and others have previously shown that loss of function in ATP13A2 leads to lysosomal and mitochondrial impairment (98, 99), possibly due to impaired intracellular Zn2+ homeostasis (100, 101). The role of ATP13A2 in PD is strengthened by the observation that ATP13A2 mRNA and protein levels are significantly increased in the dopaminergic neurons of patients with sporadic PD (102). This could indicate that dopaminergic neurons that express higher ATP13A2 protein levels are less susceptible to cell death. Potentially consistent with this, loss of ATP13A2 is associated with impairments in lysosomal function, exosome biogenesis/secretion, both of which may lead to accumulation of alpha synuclein (98, 103).
Hexosaminidase B (HEXB)
Mutations in HEXB, encoding β-hexosaminidase B, cause the recessive disorder, Sandhoff disease (GM2 gangliosidosis). HexB, which is also a lysosomal enzyme, is responsible for the metabolism of GM2 ganglioside. Over 20 mutations in HEXB, including both point mutations and deletions, have been described (104). A juvenile-onset form of Sandhoff disease is caused by strong mutations that nearly abolish enzymatic function (<2% residual activity) (104). These patients present with reduced attention, weakness, and hypotonia, followed by progressive psychomotor impairment (105). HEXB mutations in which at least slightly higher enzymatic activity is maintained result in a milder, adult-onset form (106), including muscle weakness, ataxia and other movement related symptoms (107).
While no direct genetic connection has been reported, studies of animal models, human postmortem tissue, along with clinical case reports support a possible link between Sandhoff disease and PD. Notable, HEXB knockout mice manifest alpha synuclein pathology in the substantia nigra, along with lysosomal dysfunction (108). Brain autopsy studies in Sandhoff disease have similarly revealed accumulation of GM2 ganglioside along with alpha synuclein deposits (109, 110). Lastly, several case reports describe the manifestation of parkinsonian motor symptoms/signs in patients with either childhood- or adult-onset Sandhoff (110–112).
The exact driver of synuclein accumulation in HexB deficient cells remains unclear. It is possible that accumulation of GM2 ganglioside may directly modulate alpha synuclein aggregation, or alternatively, this may represent a more indirect consequence of lysosomal dysfunction. Thus, further studies are needed to determine if modulation of GM2 ganglioside metabolism might be beneficial in PD.
Discussion
The accumulating evidence for genetic connections between LSD and PD, has led to the intriguing hypothesis that beyond the pleiotropic effects identified for a handful of genes/variants, a deeper mechanistic link may exist between LSDs and PD (113, 114). By extension, this would potentially implicate as many as 60 other genes known to cause LSDs. As introduced above, a wealth of evidence, including studies of human brain tissue and experiments in numerous animal models highlight a clear role for lysosomal dysfunction in PD. Given the rarity of most LSD-causing variants and allowing for the possibility of incomplete- and age-dependent penetrance, definitive assessment of each gene for an association with PD risk will require very large case/control cohorts with sequencing. Based on recent studies in Alzheimer’s disease (115), it now seems likely that sample sizes including more than 10,000 subjects will be necessary. While studies of this scope may be possible in the near future, rather than focusing on individual genes where statistical power is more limited, we meanwhile have pursued a complementary approach examining aggregate genetic risk among 54 LSD genes. Indeed, among ~3,000 PD cases and controls with exome sequencing data, we discovered a significant “burden” of putative damaging variants among this LSD gene set (116). This result was robust to the exclusion of GBA1 and was consistent in 2 independent datasets, suggesting that other LSD genes besides GBA1 likely contribute to PD risk. Importantly, more than half of our PD cohort (56%) had more than 1 LSD gene variant, and 21% carried 2 or more potential risk alleles in multiple LSD genes (116). These results therefore support the possibility of an oligogenic risk model for PD, in which multiple genetic hits act in combination to degrade cellular metabolism, enhancing disease susceptibility (Figure 2). Evidence for the involvement of additional LSD genes in PD risk, also comes from a recently-completed GWAS, including more than 50,000 cases and 1 million control subjects. This largest-ever study identifies newly significant variant associations at NAGLU, GUSB, NEU1, and GRN (81). Although additional work will be required to confirm whether these genes are in fact responsible for the variant associations, the results of both rare and common variant genetic association studies are converging to implicate significant overlap in susceptibility loci between PD and heterogeneous LSDs.
Figure 2.

A hypothetical model for oligogenic risk in PD. Genetic mutations associated with LSDs cause severe impairment in lysosomal function resulting in significant lipid accumulations and leading to the severe phenotypes observed in juvenile onset LSDs. In cases where mutations result in modest impairments that are associated with PD, lysosomal function is maintained to an extent where lipid metabolism can be relatively normal. However, mild impairments in overall lysosomal protein degradation efficiency caused by one or more LSD genetic variants in combination other genetic and/or non-genetic risk factors as well as aging may further disrupt cellular proteostasis, leading to accumulation of alpha synuclein and ultimately, PD.
Despite the genetic, neuropathological, and mechanistic studies implicating lysosomal dysfunction in PD pathogenesis, the involvement of this pathway in disease is not unique to PD. There is abundant evidence of lysosomal dysfunction in several other neurodegenerative disorders including Alzheimer’s (117) and Huntington’s disease (118), suggesting that dysfunction of the lysosomal system is an important contributor to pathogenesis across many neurodegenerative disorders. As a result, lysosomal dysfunction alone cannot explain the preferential neuronal vulnerability in PD and it is important to consider other responsible factors. In PD, as discussed above, dopamine oxidization may exacerbate lysosomal dysfunction and provide at least a partial explanation for the selective vulnerability of dopaminergic neurons. Increased cytoplasmic oxidation of dopamine may result from synaptic deficits that lead to impaired packaging of dopamine or mitochondrial stress, which increases oxidative potential (24, 119). In addition, elevation of alpha synuclein levels that occurs during lysosomal dysfunction may also explain neuronal vulnerability and could further contribute to lysosomal, mitochondrial, and synaptic dysfunction. Nevertheless, these genetic forms of PD that are linked to lysosomal genes provide a foundation for development of targeted therapies—e.g. activation of GCase in GBA-PD. Despite the potential for complicated interactions and distributed failure of lysosomal, mitochondrial, and synaptic mechanisms in the PD brain, there is promise that targeted approaches, such as activating GCase to enhance lysosomal function may halt the vicious cycle, enhancing mitophagy, promoting degradation of alpha synuclein, and blocking accumulation of potentially harmful dopamine oxidation products.
Acknowledgments
Financial Disclosures: D.Y. was supported by NIH grant T32NS041234, J.M.S. was supported by Huffington Foundation, Jan and Dan Duncan Neurological Research Institute at Texas Children’s Hospital, and a Career Award for Medical Scientists from the Burroughs Welcome Fund. D.K. was supported by NIH grants R01 NS076054, R37 NS096241, R01 NS096240, U01 NS 094148 and grants from the Michael J. Foundation for Parkinson’s Research.
Funding Sources: D.Y. was supported by NIH grant T32NS041234, J.M.S. was supported by Huffington Foundation, Jan and Dan Duncan Neurological Research Institute at Texas Children’s Hospital, and a Career Award for Medical Scientists from the Burroughs Welcome Fund. D.K. was supported by NIH grants R01 NS076054, R37 NS096241, R01 NS096240.
Footnotes
Relevant conflicts of interest/financial disclosures: The authors declare no conflicts of interest
References
- 1.Shachar T, Lo Bianco C, Recchia A, Wiessner C, Raas-Rothschild A, Futerman AH. Lysosomal storage disorders and Parkinson’s disease: Gaucher disease and beyond. Mov Disord. 2011;26(9):1593–604. [DOI] [PubMed] [Google Scholar]
- 2.Hruska KS, LaMarca ME, Scott CR, Sidransky E. Gaucher disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum Mutat. 2008;29(5):567–83. [DOI] [PubMed] [Google Scholar]
- 3.Vaccaro AM, Motta M, Tatti M, Scarpa S, Masuelli L, Bhat M, et al. Saposin C mutations in Gaucher disease patients resulting in lysosomal lipid accumulation, saposin C deficiency, but normal prosaposin processing and sorting. Hum Mol Genet. 2010;19(15):2987–97. [DOI] [PubMed] [Google Scholar]
- 4.Sidransky E Gaucher disease: complexity in a “simple” disorder. Mol Genet Metab. 2004;83(1–2):6–15. [DOI] [PubMed] [Google Scholar]
- 5.Kaplan P, Andersson HC, Kacena KA, Yee JD . The clinical and demographic characteristics of nonneuronopathic Gaucher disease in 887 children at diagnosis. Arch Pediatr Adolesc Med. 2006;160(6):603–8. [DOI] [PubMed] [Google Scholar]
- 6.Grabowski GA, Zimran A, Ida H. Gaucher disease types 1 and 3: Phenotypic characterization of large populations from the ICGG Gaucher Registry. Am J Hematol. 2015;90 Suppl 1:S12–8. [DOI] [PubMed] [Google Scholar]
- 7.Mignot C, Gelot A, Bessieres B, Daffos F, Voyer M, Menez F, et al. Perinatal-lethal Gaucher disease. Am J Med Genet A. 2003;120A(3):338–44. [DOI] [PubMed] [Google Scholar]
- 8.Stone DL, Tayebi N, Orvisky E, Stubblefield B, Madike V, Sidransky E. Glucocerebrosidase gene mutations in patients with type 2 Gaucher disease. Hum Mutat. 2000;15(2):181–8. [DOI] [PubMed] [Google Scholar]
- 9.Ferraz MJ, Marques AR, Appelman MD, Verhoek M, Strijland A, Mirzaian M, et al. Lysosomal glycosphingolipid catabolism by acid ceramidase: formation of glycosphingoid bases during deficiency of glycosidases. FEBS Lett. 2016;590(6):716–25. [DOI] [PubMed] [Google Scholar]
- 10.Mistry PK, Liu J, Sun L, Chuang WL, Yuen T, Yang R, et al. Glucocerebrosidase 2 gene deletion rescues type 1 Gaucher disease. Proc Natl Acad Sci U S A. 2014;111(13):4934–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taguchi YV, Liu J, Ruan J, Pacheco J, Zhang X, Abbasi J, et al. Glucosylsphingosine Promotes alpha-Synuclein Pathology in Mutant GBA-Associated Parkinson’s Disease. J Neurosci. 2017;37(40):9617–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med. 2009;361(17):1651–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gan-Or Z, Giladi N, Rozovski U, Shifrin C, Rosner S, Gurevich T, et al. Genotype-phenotype correlations between GBA mutations and Parkinson disease risk and onset. Neurology. 2008;70(24):2277–83. [DOI] [PubMed] [Google Scholar]
- 14.Liu G, Boot B, Locascio JJ, Jansen IE, Winder-Rhodes S, Eberly S, et al. Specifically neuropathic Gaucher’s mutations accelerate cognitive decline in Parkinson’s. Ann Neurol. 2016;80(5):674–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thaler A, Gurevich T, Bar Shira A, Gana Weisz M, Ash E, Shiner T, et al. A “dose” effect of mutations in the GBA gene on Parkinson’s disease phenotype. Parkinsonism Relat Disord. 2017;36:47–51. [DOI] [PubMed] [Google Scholar]
- 16.Rocha EM, Smith GA, Park E, Cao H, Brown E, Hallett P, et al. Progressive decline of glucocerebrosidase in aging and Parkinson’s disease. Ann Clin Transl Neurol. 2015;2(4):433–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chiasserini D, Paciotti S, Eusebi P, Persichetti E, Tasegian A, Kurzawa-Akanbi M, et al. Selective loss of glucocerebrosidase activity in sporadic Parkinson’s disease and dementia with Lewy bodies. Mol Neurodegener. 2015;10:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bultron G, Kacena K, Pearson D, Boxer M, Yang R, Sathe S, et al. The risk of Parkinson’s disease in type 1 Gaucher disease. J Inherit Metab Dis. 2010;33(2):167–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ron I, Horowitz M. ER retention and degradation as the molecular basis underlying Gaucher disease heterogeneity. Hum Mol Genet. 2005;14(16):2387–98. [DOI] [PubMed] [Google Scholar]
- 20.Rothaug M, Zunke F, Mazzulli JR, Schweizer M, Altmeppen H, Lullmann-Rauch R, et al. LIMP-2 expression is critical for beta-glucocerebrosidase activity and alpha-synuclein clearance. Proc Natl Acad Sci U S A. 2014;111(43):15573–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Velayati A, DePaolo J, Gupta N, Choi JH, Moaven N, Westbroek W, et al. A mutation in SCARB2 is a modifier in Gaucher disease. Hum Mutat. 2011;32(11):1232–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim MJ, Jeon S, Burbulla LF, Krainc D. Acid ceramidase inhibition ameliorates alpha-synuclein accumulation upon loss of GBA1 function. Hum Mol Genet. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mazzulli JR, Zunke F, Isacson O, Studer L, Krainc D. alpha-Synuclein-induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proc Natl Acad Sci U S A. 2016;113(7):1931–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burbulla LF, Song P, Mazzulli JR, Zampese E, Wong YC, Jeon S, et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science. 2017;357(6357):1255–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mazzulli JR, Zunke F, Tsunemi T, Toker NJ, Jeon S, Burbulla LF, et al. Activation of beta-Glucocerebrosidase Reduces Pathological alpha-Synuclein and Restores Lysosomal Function in Parkinson’s Patient Midbrain Neurons. J Neurosci. 2016;36(29):7693–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sardi SP, Cedarbaum JM, Brundin P. Targeted Therapies for Parkinson’s Disease: From Genetics to the Clinic. Mov Disord. 2018;33(5):684–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zampieri S, Filocamo M, Pianta A, Lualdi S, Gort L, Coll MJ, et al. SMPD1 Mutation Update: Database and Comprehensive Analysis of Published and Novel Variants. Hum Mutat. 2016;37(2):139–47. [DOI] [PubMed] [Google Scholar]
- 28.Zhang H, Wang Y, Gong Z, Li X, Qiu W, Han L, et al. Identification of a distinct mutation spectrum in the SMPD1 gene of Chinese patients with acid sphingomyelinase-deficient Niemann-Pick disease. Orphanet J Rare Dis. 2013;8:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Genetics ACo. ACOG Committee Opinion No. 442: Preconception and prenatal carrier screening for genetic diseases in individuals of Eastern European Jewish descent. Obstet Gynecol. 2009;114(4):950–3. [DOI] [PubMed] [Google Scholar]
- 30.Levran O, Desnick RJ, Schuchman EH. Identification and expression of a common missense mutation (L302P) in the acid sphingomyelinase gene of Ashkenazi Jewish type A Niemann-Pick disease patients. Blood. 1992;80(8):2081–7. [PubMed] [Google Scholar]
- 31.Schuchman EH. The pathogenesis and treatment of acid sphingomyelinase-deficient Niemann-Pick disease. J Inherit Metab Dis. 2007;30(5):654–63. [DOI] [PubMed] [Google Scholar]
- 32.Pavlu-Pereira H, Asfaw B, Poupctova H, Ledvinova J, Sikora J, Vanier MT, et al. Acid sphingomyelinase deficiency. Phenotype variability with prevalence of intermediate phenotype in a series of twenty-five Czech and Slovak patients. A multi-approach study. J Inherit Metab Dis. 2005;28(2):203–27. [DOI] [PubMed] [Google Scholar]
- 33.Chuang WL, Pacheco J, Cooper S, McGovern MM, Cox GF, Keutzer J, et al. Lyso-sphingomyelin is elevated in dried blood spots of Niemann-Pick B patients. Mol Genet Metab. 2014;111(2):209–11. [DOI] [PubMed] [Google Scholar]
- 34.Kirkegaard T, Roth AG, Petersen NH, Mahalka AK, Olsen OD, Moilanen I, et al. Hsp70 stabilizes lysosomes and reverts Niemann-Pick disease-associated lysosomal pathology. Nature. 2010;463(7280):549–53. [DOI] [PubMed] [Google Scholar]
- 35.Schuchman EH. Acid sphingomyelinase, cell membranes and human disease: lessons from Niemann-Pick disease. FEBS Lett. 2010;584(9):1895–900. [DOI] [PubMed] [Google Scholar]
- 36.Galvan C, Camoletto PG, Cristofani F, Van Veldhoven PP, Ledesma MD. Anomalous surface distribution of glycosyl phosphatidyl inositol-anchored proteins in neurons lacking acid sphingomyelinase. Mol Biol Cell. 2008;19(2):509–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Buccinna B, Piccinini M, Prinetti A, Scandroglio F, Prioni S, Valsecchi M, et al. Alterations of myelin-specific proteins and sphingolipids characterize the brains of acid sphingomyelinase-deficient mice, an animal model of Niemann-Pick disease type A. J Neurochem. 2009;109(1):105–15. [DOI] [PubMed] [Google Scholar]
- 38.Gan-Or Z, Ozelius LJ, Bar-Shira A, Saunders-Pullman R, Mirelman A, Kornreich R, et al. The p.L302P mutation in the lysosomal enzyme gene SMPD1 is a risk factor for Parkinson disease. Neurology. 2013;80(17):1606–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Foo JN, Liany H, Bei JX, Yu XQ, Liu J, Au WL, et al. Rare lysosomal enzyme gene SMPD1 variant (p.R591C) associates with Parkinson’s disease. Neurobiol Aging. 2013;34(12):2890 e13–5. [DOI] [PubMed] [Google Scholar]
- 40.Dagan E, Schlesinger I, Ayoub M, Mory A, Nassar M, Kurolap A, et al. The contribution of Niemann-Pick SMPD1 mutations to Parkinson disease in Ashkenazi Jews. Parkinsonism Relat Disord. 2015;21(9):1067–71. [DOI] [PubMed] [Google Scholar]
- 41.Gan-Or Z, Orr-Urtreger A, Alcalay RN, Bressman S, Giladi N, Rouleau GA. The emerging role of SMPD1 mutations in Parkinson’s disease: Implications for future studies. Parkinsonism Relat Disord. 2015;21(10):1294–5. [DOI] [PubMed] [Google Scholar]
- 42.Viana MB, Giugliani R, Leite VH, Barth ML, Lekhwani C, Slade CM, et al. Very low levels of high density lipoprotein cholesterol in four sibs of a family with non-neuropathic Niemann-Pick disease and sea-blue histiocytosis. J Med Genet. 1990;27(8):499–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Alcalay RN, Wolf P, Levy OA, Kang UJ, Waters C, Fahn S, et al. Alpha galactosidase A activity in Parkinson’s disease. Neurobiol Dis. 2018;112:85–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gorelik A, Illes K, Heinz LX, Superti-Furga G, Nagar B. Crystal structure of mammalian acid sphingomyelinase. Nat Commun. 2016;7:12196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kwon HJ, Abi-Mosleh L, Wang ML, Deisenhofer J, Goldstein JL, Brown MS, et al. Structure of N-terminal domain of NPC1 reveals distinct subdomains for binding and transfer of cholesterol. Cell. 2009;137(7):1213–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Runz H, Dolle D, Schlitter AM, Zschocke J. NPC-db, a Niemann-Pick type C disease gene variation database. Hum Mutat. 2008;29(3):345–50. [DOI] [PubMed] [Google Scholar]
- 47.Karten B, Peake KB, Vance JE. Mechanisms and consequences of impaired lipid trafficking in Niemann-Pick type C1-deficient mammalian cells. Biochim Biophys Acta. 2009;1791(7):659–70. [DOI] [PubMed] [Google Scholar]
- 48.Crocker AC. The cerebral defect in Tay-Sachs disease and Niemann-Pick disease. J Neurochem. 1961;7:69–80. [DOI] [PubMed] [Google Scholar]
- 49.Shulman LM, David NJ, Weiner WJ. Psychosis as the initial manifestation of adult-onset Niemann-Pick disease type C. Neurology. 1995;45(9):1739–43. [DOI] [PubMed] [Google Scholar]
- 50.Saito Y, Suzuki K, Hulette CM, Murayama S. Aberrant phosphorylation of alpha-synuclein in human Niemann-Pick type C1 disease. J Neuropathol Exp Neurol. 2004;63(4):323–8. [DOI] [PubMed] [Google Scholar]
- 51.Josephs KA, Matsumoto JY, Lindor NM. Heterozygous Niemann-Pick disease type C presenting with tremor. Neurology. 2004;63(11):2189–90. [DOI] [PubMed] [Google Scholar]
- 52.Coleman RJ, Robb SA, Lake BD, Brett EM, Harding AE. The diverse neurological features of Niemann-Pick disease type C: a report of two cases. Mov Disord. 1988;3(4):295–9. [DOI] [PubMed] [Google Scholar]
- 53.Kluenemann HH, Nutt JG, Davis MY, Bird TD. Parkinsonism syndrome in heterozygotes for Niemann-Pick C1. J Neurol Sci. 2013;335(1–2):219–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zervas M, Dobrenis K, Walkley SU. Neurons in Niemann-Pick disease type C accumulate gangliosides as well as unesterified cholesterol and undergo dendritic and axonal alterations. J Neuropathol Exp Neurol. 2001;60(1):49–64. [DOI] [PubMed] [Google Scholar]
- 55.Fantini J, Yahi N. The driving force of alpha-synuclein insertion and amyloid channel formation in the plasma membrane of neural cells: key role of ganglioside- and cholesterol-binding domains. Adv Exp Med Biol. 2013;991:15–26. [DOI] [PubMed] [Google Scholar]
- 56.Ysselstein D, Joshi M, Mishra V, Griggs AM, Asiago JM, McCabe GP, et al. Effects of impaired membrane interactions on alpha-synuclein aggregation and neurotoxicity. Neurobiol Dis. 2015;79:150–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ysselstein D, Dehay B, Costantino IM, McCabe GP, Frosch MP, George JM, et al. Endosulfine-alpha inhibits membrane-induced alpha-synuclein aggregation and protects against alpha-synuclein neurotoxicity. Acta Neuropathol Commun. 2017;5(1):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lloyd-Evans E, Morgan AJ, He X, Smith DA, Elliot-Smith E, Sillence DJ, et al. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat Med. 2008;14(11):1247–55. [DOI] [PubMed] [Google Scholar]
- 59.te Vruchte D, Lloyd-Evans E, Veldman RJ, Neville DC, Dwek RA, Platt FM, et al. Accumulation of glycosphingolipids in Niemann-Pick C disease disrupts endosomal transport. J Biol Chem. 2004;279(25):26167–75. [DOI] [PubMed] [Google Scholar]
- 60.Auer IA, Schmidt ML, Lee VM, Curry B, Suzuki K, Shin RW, et al. Paired helical filament tau (PHFtau) in Niemann-Pick type C disease is similar to PHFtau in Alzheimer’s disease. Acta Neuropathol. 1995;90(6):547–51. [DOI] [PubMed] [Google Scholar]
- 61.Fu R, Yanjanin NM, Elrick MJ, Ware C, Lieberman AP, Porter FD. Apolipoprotein E genotype and neurological disease onset in Niemann-Pick disease, type C1. Am J Med Genet A. 2012;158A(11):2775–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wenger DA, Rafi MA, Luzi P, Datto J, Costantino-Ceccarini E. Krabbe disease: genetic aspects and progress toward therapy. Mol Genet Metab. 2000;70(1):1–9. [DOI] [PubMed] [Google Scholar]
- 63.Lee WC, Kang D, Causevic E, Herdt AR, Eckman EA, Eckman CB. Molecular characterization of mutations that cause globoid cell leukodystrophy and pharmacological rescue using small molecule chemical chaperones. J Neurosci. 2010;30(16):5489–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fiumara A, Barone R, Arena A, Filocamo M, Lissens W, Pavone L, et al. Krabbe leukodystrophy in a selected population with high rate of late onset forms: longer survival linked to c.121G>A (p.Gly41Ser) mutation. Clin Genet. 2011;80(5):452–8. [DOI] [PubMed] [Google Scholar]
- 65.Harzer K, Knoblich R, Rolfs A, Bauer P, Eggers J. Residual galactosylsphingosine (psychosine) beta-galactosidase activities and associated GALC mutations in late and very late onset Krabbe disease. Clin Chim Acta. 2002;317(1–2):77–84. [DOI] [PubMed] [Google Scholar]
- 66.Tappino B, Biancheri R, Mort M, Regis S, Corsolini F, Rossi A, et al. Identification and characterization of 15 novel GALC gene mutations causing Krabbe disease. Hum Mutat. 2010;31(12):E1894–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wenger DA, Rafi MA, Luzi P. Krabbe disease: One Hundred years from the bedside to the bench to the bedside. J Neurosci Res. 2016;94(11):982–9. [DOI] [PubMed] [Google Scholar]
- 68.Kolodny EH, Raghavan S, Krivit W. Late-onset Krabbe disease (globoid cell leukodystrophy): clinical and biochemical features of 15 cases. Dev Neurosci. 1991;13(4–5):232–9. [DOI] [PubMed] [Google Scholar]
- 69.Cantuti Castelvetri L, Givogri MI, Hebert A, Smith B, Song Y, Kaminska A, et al. The sphingolipid psychosine inhibits fast axonal transport in Krabbe disease by activation of GSK3beta and deregulation of molecular motors. J Neurosci. 2013;33(24):10048–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Suzuki K, Suzuki K. Myelin pathology in the twitcher mouse. Ann N Y Acad Sci. 1990;605:313–24. [DOI] [PubMed] [Google Scholar]
- 71.Smith BR, Santos MB, Marshall MS, Cantuti-Castelvetri L, Lopez-Rosas A, Li G, et al. Neuronal inclusions of alpha-synuclein contribute to the pathogenesis of Krabbe disease. J Pathol. 2014;232(5):509–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chang D, Nalls MA, Hallgrimsdottir IB, Hunkapiller J, van der Brug M, Cai F, et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat Genet. 2017;49(10):1511–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Consortium GT, Laboratory DA, Coordinating Center -Analysis Working G, Statistical Methods groups-Analysis Working G, Enhancing Gg, Fund NIHC, et al. Genetic effects on gene expression across human tissues. Nature. 2017;550(7675):204–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Marshall MS, Jakubauskas B, Bogue W, Stoskute M, Hauck Z, Rue E, et al. Analysis of age-related changes in psychosine metabolism in the human brain. PLoS One. 2018;13(2):e0193438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang RY, Bodamer OA, Watson MS, Wilcox WR, Diseases AWGoDCoLS. Lysosomal storage diseases: diagnostic confirmation and management of presymptomatic individuals. Genet Med. 2011;13(5):457–84. [DOI] [PubMed] [Google Scholar]
- 76.Valstar MJ, Ruijter GJ, van Diggelen OP, Poorthuis BJ, Wijburg FA. Sanfilippo syndrome: a mini-review. J Inherit Metab Dis. 2008;31(2):240–52. [DOI] [PubMed] [Google Scholar]
- 77.Yogalingam G, Weber B, Meehan J, Rogers J, Hopwood JJ. Mucopolysaccharidosis type IIIB: characterisation and expression of wild-type and mutant recombinant alpha-N-acetylglucosaminidase and relationship with sanfilippo phenotype in an attenuated patient. Biochim Biophys Acta. 2000;1502(3):415–25. [DOI] [PubMed] [Google Scholar]
- 78.Chinen Y, Tohma T, Izumikawa Y, Uehara H, Ohta T. Sanfilippo type B syndrome: five patients with an R565P homozygous mutation in the alpha-N-acetylglucosaminidase gene from the Okinawa islands in Japan. J Hum Genet. 2005;50(7):357–9. [DOI] [PubMed] [Google Scholar]
- 79.Valstar MJ, Neijs S, Bruggenwirth HT, Olmer R, Ruijter GJ, Wevers RA, et al. Mucopolysaccharidosis type IIIA: clinical spectrum and genotype-phenotype correlations. Ann Neurol. 2010;68(6):876–87. [DOI] [PubMed] [Google Scholar]
- 80.Winder-Rhodes SE, Garcia-Reitbock P, Ban M, Evans JR, Jacques TS, Kemppinen A, et al. Genetic and pathological links between Parkinson’s disease and the lysosomal disorder Sanfilippo syndrome. Mov Disord. 2012;27(2):312–5. [DOI] [PubMed] [Google Scholar]
- 81.Nalls MA, Blauwendraat C, Vallerga C, Heilbron K, Bandres-Ciga S, Chang D, et al. Parkinson’s disease genetics: identifying novel risk loci, providing causal insights and improving estimates of heritable rist. bioRxiv; 2018. [Google Scholar]
- 82.Hamano K, Hayashi M, Shioda K, Fukatsu R, Mizutani S. Mechanisms of neurodegeneration in mucopolysaccharidoses II and IIIB: analysis of human brain tissue. Acta Neuropathol. 2008;115(5):547–59. [DOI] [PubMed] [Google Scholar]
- 83.Cohlberg JA, Li J, Uversky VN, Fink AL. Heparin and other glycosaminoglycans stimulate the formation of amyloid fibrils from alpha-synuclein in vitro. Biochemistry. 2002;41(5):1502–11. [DOI] [PubMed] [Google Scholar]
- 84.Ihse E, Yamakado H, van Wijk XM, Lawrence R, Esko JD, Masliah E. Cellular internalization of alpha-synuclein aggregates by cell surface heparan sulfate depends on aggregate conformation and cell type. Sci Rep. 2017;7(1):9008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lehri-Boufala S, Ouidja MO, Barbier-Chassefiere V, Henault E, Raisman-Vozari R, Garrigue-Antar L, et al. New roles of glycosaminoglycans in alpha-synuclein aggregation in a cellular model of Parkinson disease. PLoS One. 2015;10(1):e0116641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bras J, Verloes A, Schneider SA, Mole SE, Guerreiro RJ. Mutation of the parkinsonism gene ATP13A2 causes neuronal ceroid-lipofuscinosis. Hum Mol Genet. 2012;21(12):2646–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Farias FH, Zeng R, Johnson GS, Wininger FA, Taylor JF, Schnabel RD, et al. A truncating mutation in ATP13A2 is responsible for adult-onset neuronal ceroid lipofuscinosis in Tibetan terriers. Neurobiol Dis. 2011;42(3):468–74. [DOI] [PubMed] [Google Scholar]
- 88.Bennett MJ, Rakheja D. The neuronal ceroid-lipofuscinoses. Dev Disabil Res Rev. 2013;17(3):254–9. [DOI] [PubMed] [Google Scholar]
- 89.Park JS, Blair NF, Sue CM. The role of ATP13A2 in Parkinson’s disease: Clinical phenotypes and molecular mechanisms. Mov Disord. 2015;30(6):770–9. [DOI] [PubMed] [Google Scholar]
- 90.Di Fonzo A, Chien HF, Socal M, Giraudo S, Tassorelli C, Iliceto G, et al. ATP13A2 missense mutations in juvenile parkinsonism and young onset Parkinson disease. Neurology. 2007;68(19):1557–62. [DOI] [PubMed] [Google Scholar]
- 91.Lin CH, Tan EK, Chen ML, Tan LC, Lim HQ, Chen GS, et al. Novel ATP13A2 variant associated with Parkinson disease in Taiwan and Singapore. Neurology. 2008;71(21):1727–32. [DOI] [PubMed] [Google Scholar]
- 92.Chen CM, Lin CH, Juan HF, Hu FJ, Hsiao YC, Chang HY, et al. ATP13A2 variability in Taiwanese Parkinson’s disease. Am J Med Genet B Neuropsychiatr Genet. 2011;156B(6):720–9. [DOI] [PubMed] [Google Scholar]
- 93.Djarmati A, Hagenah J, Reetz K, Winkler S, Behrens MI, Pawlack H, et al. ATP13A2 variants in early-onset Parkinson’s disease patients and controls. Mov Disord. 2009;24(14):2104–11. [DOI] [PubMed] [Google Scholar]
- 94.Rakovic A, Stiller B, Djarmati A, Flaquer A, Freudenberg J, Toliat MR, et al. Genetic association study of the P-type ATPase ATP13A2 in late-onset Parkinson’s disease. Mov Disord. 2009;24(3):429–33. [DOI] [PubMed] [Google Scholar]
- 95.Fei QZ, Cao L, Xiao Q, Zhang T, Zheng L, Wang XJ, et al. Lack of association between ATP13A2 Ala746Thr variant and Parkinson’s disease in Han population of mainland China. Neurosci Lett. 2010;475(2):61–3. [DOI] [PubMed] [Google Scholar]
- 96.Lubbe SJ, Escott-Price V, Gibbs JR, Nalls MA, Bras J, Price TR, et al. Additional rare variant analysis in Parkinson’s disease cases with and without known pathogenic mutations: evidence for oligogenic inheritance. Hum Mol Genet. 2016;25(24):5483–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yang X, Xu Y. Mutations in the ATP13A2 gene and Parkinsonism: a preliminary review. Biomed Res Int. 2014;2014:371256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Usenovic M, Tresse E, Mazzulli JR, Taylor JP, Krainc D. Deficiency of ATP13A2 leads to lysosomal dysfunction, alpha-synuclein accumulation, and neurotoxicity. J Neurosci. 2012;32(12):4240–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Grunewald A, Arns B, Seibler P, Rakovic A, Munchau A, Ramirez A, et al. ATP13A2 mutations impair mitochondrial function in fibroblasts from patients with Kufor-Rakeb syndrome. Neurobiol Aging. 2012;33(8):1843 e1–7. [DOI] [PubMed] [Google Scholar]
- 100.Tan J, Zhang T, Jiang L, Chi J, Hu D, Pan Q, et al. Regulation of intracellular manganese homeostasis by Kufor-Rakeb syndrome-associated ATP13A2 protein. J Biol Chem. 2011;286(34):29654–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tsunemi T, Krainc D. Zn(2)(+) dyshomeostasis caused by loss of ATP13A2/PARK9 leads to lysosomal dysfunction and alpha-synuclein accumulation. Hum Mol Genet. 2014;23(11):2791–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ramirez A, Heimbach A, Grundemann J, Stiller B, Hampshire D, Cid LP, et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet. 2006;38(10):1184–91. [DOI] [PubMed] [Google Scholar]
- 103.Tsunemi T, Hamada K, Krainc D. ATP13A2/PARK9 regulates secretion of exosomes and alpha-synuclein. J Neurosci. 2014;34(46):15281–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mahuran DJ. Biochemical consequences of mutations causing the GM2 gangliosidoses. Biochim Biophys Acta. 1999;1455(2–3):105–38. [DOI] [PubMed] [Google Scholar]
- 105.Hendriksz CJ, Corry PC, Wraith JE, Besley GT, Cooper A, Ferrie CD. Juvenile Sandhoff disease--nine new cases and a review of the literature. J Inherit Metab Dis. 2004;27(2):241–9. [DOI] [PubMed] [Google Scholar]
- 106.Kang SY, Song SK, Lee JS, Choi JC, Kang JH. Adult Sandhoff disease with 2 mutations in the HEXB gene presenting as brachial amyotrophic diplegia. J Clin Neuromuscul Dis. 2013;15(2):47–51. [DOI] [PubMed] [Google Scholar]
- 107.Delnooz CC, Lefeber DJ, Langemeijer SM, Hoffjan S, Dekomien G, Zwarts MJ, et al. New cases of adult-onset Sandhoff disease with a cerebellar or lower motor neuron phenotype. J Neurol Neurosurg Psychiatry. 2010;81(9):968–72. [DOI] [PubMed] [Google Scholar]
- 108.Suzuki K, Iseki E, Katsuse O, Yamaguchi A, Katsuyama K, Aoki I, et al. Neuronal accumulation of alpha- and beta-synucleins in the brain of a GM2 gangliosidosis mouse model. Neuroreport. 2003;14(4):551–4. [DOI] [PubMed] [Google Scholar]
- 109.Keilani S, Lun Y, Stevens AC, Williams HN, Sjoberg ER, Khanna R, et al. Lysosomal dysfunction in a mouse model of Sandhoff disease leads to accumulation of ganglioside-bound amyloid-beta peptide. J Neurosci. 2012;32(15):5223–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Suzuki K, Iseki E, Togo T, Yamaguchi A, Katsuse O, Katsuyama K, et al. Neuronal and glial accumulation of alpha- and beta-synucleins in human lipidoses. Acta Neuropathol. 2007;114(5):481–9. [DOI] [PubMed] [Google Scholar]
- 111.Inzelberg R, Korczyn AD. Parkinsonism in adult-onset GM2 gangliosidosis. Mov Disord. 1994;9(3):375–7. [DOI] [PubMed] [Google Scholar]
- 112.Jellinger K, Anzil AP, Seemann D, Bernheimer H. Adult GM2 gangliosidosis masquerading as slowly progressive muscular atrophy: motor neuron disease phenotype. Clin Neuropathol. 1982;1(1):31–44. [PubMed] [Google Scholar]
- 113.Klein AD, Mazzulli JR. Is Parkinson’s disease a lysosomal disorder? Brain. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Futerman AH, Hardy J. Perspective: Finding common ground. Nature. 2016;537(7621):S160–1. [DOI] [PubMed] [Google Scholar]
- 115.Bis JC, Jian X, Kunkle BW, Chen Y, Hamilton-Nelson KL, Bush WS, et al. Whole exome sequencing study identifies novel rare and common Alzheimer’s-Associated variants involved in immune response and transcriptional regulation. Mol Psychiatry. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Robak LA, Jansen IE, van Rooij J, Uitterlinden AG, Kraaij R, Jankovic J, et al. Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain. 2017;140(12):3191–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Orr ME, Oddo S. Autophagic/lysosomal dysfunction in Alzheimer’s disease. Alzheimers Res Ther. 2013;5(5):53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Cortes CJ, La Spada AR. The many faces of autophagy dysfunction in Huntington’s disease: from mechanism to therapy. Drug Discov Today. 2014;19(7):963–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Nguyen M, Krainc D. LRRK2 phosphorylation of auxilin mediates synaptic defects in dopaminergic neurons from patients with Parkinson’s disease. Proc Natl Acad Sci U S A. 2018;115(21):5576–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lwin A, Orvisky E, Goker-Alpan O, LaMarca ME, Sidransky E. Glucocerebrosidase mutations in subjects with parkinsonism. Mol Genet Metab. 2004;81(1):70–3. [DOI] [PubMed] [Google Scholar]
- 121.Pankratz N, Beecham GW, DeStefano AL, Dawson TM, Doheny KF, Factor SA, et al. Meta-analysis of Parkinson’s disease: identification of a novel locus, RIT2. Ann Neurol. 2012;71(3):370–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Saunders-Pullman R, Hagenah J, Dhawan V, Stanley K, Pastores G, Sathe S, et al. Gaucher disease ascertained through a Parkinson’s center: imaging and clinical characterization. Mov Disord. 2010;25(10):1364–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Xu YH, Sun Y, Ran H, Quinn B, Witte D, Grabowski GA. Accumulation and distribution of alpha-synuclein and ubiquitin in the CNS of Gaucher disease mouse models. Mol Genet Metab. 2011;102(4):436–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Beard H, Hassiotis S, Gai WP, Parkinson-Lawrence E, Hopwood JJ, Hemsley KM. Axonal dystrophy in the brain of mice with Sanfilippo syndrome. Exp Neurol. 2017;295:243–55. [DOI] [PubMed] [Google Scholar]
- 125.Schultheis PJ, Fleming SM, Clippinger AK, Lewis J, Tsunemi T, Giasson B, et al. Atp13a2-deficient mice exhibit neuronal ceroid lipofuscinosis, limited alpha-synuclein accumulation and age-dependent sensorimotor deficits. Hum Mol Genet. 2013;22(10):2067–82. [DOI] [PMC free article] [PubMed] [Google Scholar]