

Abstract
Ataxia with oculomotor apraxia type 2 (AOA2) is a rare autosomal recessive cerebellar ataxia characterized by onset between 10–20 years of age and a range of neurological features that include progressive cerebellar atrophy, axonal sensorimotor neuropathy, oculomotor apraxia in a majority of patients, and elevated serum alpha-fetoprotein (AFP). AOA2 is caused by mutation of the SETX gene which encodes senataxin, a DNA/RNA helicase involved in transcription regulation, RNA processing, and DNA maintenance. Disruption of senataxin in rodents led to defective spermatogenesis and sterility in males uncovering a key role for senataxin in male germ cell survival. Here, we report the first clinical and cellular evidence of impaired spermatogenesis in AOA2 patients. We assessed sperm production in 3 AOA2 patients and testicular pathology in one patient and compared the findings to those of Setx knockout mice. Sperm production was impaired in all patients assessed (3/3, 100%). Analyses of testicular biopsies from an AOA2 patient recapitulate features of the histology seen in Setx knockout mice, strongly suggesting an underlying mechanism centering on DNA-damage-mediated germ cell apoptosis. These findings support a role for senataxin in human reproductive function and highlight a novel clinical feature of AOA2 that extends the extra-neurological roles of senataxin. This raises an important reproductive counselling issue for clinicians, and fertility specialists should be aware of SETX mutations as a possible diagnosis in young male patients presenting with oligospermia or azoospermia since infertility may presage the later onset of neurological manifestations in some individuals.
Keywords: Gait disorders/ataxia, Cerebellum, Genetics, Fertility, Sperm
Introduction
Ataxia oculomotor apraxia type 2 (AOA2, also known as SCAR1, MIM# 606002) is a member of the autosomal recessive spinocerebellar ataxias (SCAR, SCA recessive), a class of progressive neurological disorders that result from cerebellar atrophy 1. First described in 2000, AOA2 was subsequently mapped to chromosome 9q34 and the gene mutated in AOA2, SETX, was found to encode the protein senataxin, a DNA/RNA helicase involved in transcription regulation, RNA processing and in protecting the genome against oxidative damage 2–13. The major clinical features of AOA2 include onset in the second decade, progressive cerebellar ataxia with axonal sensorimotor peripheral neuropathy, diffuse cerebellar atrophy, frequent oculomotor apraxia, early loss of reflexes, loss of Purkinje cells in the cerebellar cortex, and elevated alpha-fetoprotein (AFP) serum level 14, 15. Disruption of the SETX gene in mice failed to recapitulate the neurological phenotype seen in human patients 16, however, it uncovered a new aspect of senataxin function in germ cell maturation by showing that Setx−/− male mice were infertile 16. Senataxin is essential for spermatogenesis since in its absence, germ cells failed to progress past the pachytene stage of meiosis prophase I 16. Furthermore, inefficient repair of Spo11-mediated DNA double strand breaks (DSB) was observed in Setx−/− spermatocytes resulting in the accumulation of Rad51 protein on autosomes at the pachytene stage and a failure to complete meiotic recombination and initiate crossovers 16. A defect in meiotic sex chromosome inactivation (MSCI) was also found in Setx−/− germ cells due to reduced recruitment of the chromatin remodeler CHD4 to the XY chromosomes, preventing the silencing of XY-linked genes at pachytene stage 17. Finally senataxin has been shown to attenuate the activity of RNA polymerase II at genes stimulated by viral infection to regulate the magnitude of the host response 16. Altogether, these animal studies revealed an important role for senataxin at the interface of transcription regulation, gene silencing, chromatin remodelling, RNA processing, and protection of the genome during germ cell maturation.
Several ataxic disorders have been associated with reproductive conditions.18 Gordon Holmes syndrome, for example, due to mutations in the PNPLA6, RNF216, or STUB1 genes, causes hypogonadotropic hypogonadism through effects on the endocrine system.18 Women with expansions in the FMR1 gene associated with Fragile X associated tremor ataxia syndrome may experience primary ovarian insufficiency.19 While there is currently no information on male fertility in AOA2, there are a few reports of problems in females. In two studies, two AOA2 females patients were reported to have entered menopause in their mid-twenties providing evidence of premature ovarian failure 15, 20. In addition, ovarian failure 21, 22, polycystic ovarian syndrome 23, and amenorrhoea secondary to hypogonadotrophic hypogonadism 14 have also been reported in patients with AOA2. These findings suggest a role for senataxin in reproductive function and perhaps, as shown in the Setx−/− mouse model, in germ cell maturation and survival. However, cellular gonadal investigation was not performed in these patients so it is unknown whether there was a primary ovarian defect. Moreover, the contrasting clinical features ranging from high ovarian reserve to suspected ovarian failure leave it unclear as to whether loss of senataxin compromises germ cell integrity.
Here, we report the first clinical and cellular evidence of impaired spermatogenesis and the first cellular evidence of impaired gonadal function in AOA2 patients. Analyses of testicular biopsies from an AOA2 patient recapitulate features of the histology seen in Setx−/− mice. These biopsies revealed the presence of immature germ cells with high levels of DNA damage, an increased number of R-loop-containing germ cells, and elevated germ cell apoptosis. Together, this provides the first clinical and cellular evidence that an infertility phenotype exists in male AOA2 patients similar to that observed in Setx−/− male mice.
Patients and Methods
Patient recruitment
All patients were initially seen at a tertiary referral ataxia center. For enrollment in this study, patients were required to have positive SETX genetic testing and/or a clinical phenotype consistent with a diagnosis of AOA2. Patients were provided genetic counselling both before and after completion of the study. For semen analysis men were advised to abstain from ejaculation for 48 hours prior to testing, to provide the sample to the lab within 30–45 min of production, and to keep the sample warm. Written informed consent was obtained to collect biological samples for further analysis. All study methods were approved by the Institutional Review Board of the University of California, Los Angeles (UCLA).
Mouse tissue samples.
All animal work and experiments have been approved by The University of Queensland Animal Ethics Committee (HMRC/UQCCR/155/15/ARC). Testes from adult (35-day-old) mice were collected and fixed in PBS buffered 10% formalin, embedded in paraffin block and sectioned at 4 μm as previously described 16, 24.
Molecular analysis.
Testicular tissue was collected using standard procedures and immediately processed for paraffin embedding followed by histological sectioning and immunostaining. Briefly, testis biopsies were collected and fixed in PBS-buffered 10% formalin, embedded in paraffin block and sectioned at 4μm. Control testes sample was obtained from a tissue bank repository (Aquesta Pathology, QLD, Australia). Sections were stained with Hematoxylin & Eosin (H&E). Slides were examined under light microscope (Olympus CKX41 and objective CAchN 10X/0.25 PhP) and images captured by QIMAGING MicroPublisher 3.3 RTV camera and QCapture Pro 6.0 software.
Apoptosis (TUNEL) and Immunostaining
Paraffin sections from control, AOA2 patient, and Setx+/+ and Setx−/− testes were dewaxed and rehydrated with Shandon Varistain Gemini ES (Thermo Scientific, USA). Apoptotic cells were detected by Terminal deoxynucleotidyl transferase UTP Nick End Labelling (TUNEL) assay using the Fluorescence in situ Cell Death Detection Kit (Roche, Switzerland) following the manufacturer’s instructions. TUNEL is a method for detecting DNA fragmentation by labelling the terminal end of nucleic acids and a common method for detecting DNA fragmentation that results from apoptotic signalling cascades. DNA was stained with DAPI (1:10,000; Sigma-Aldrich) for 10 min at room temperature, and slides were mounted in Prolong Gold medium (Life Technologies, USA). Images were captured at room temperature using a digital camera (AxioCam 503 mono, Carl Zeiss Microimaging Inc., Germany) attached to a fluorescent microscope (AXIO Imager.M1, Carl Zeiss Microimaging Inc., Germany) and the Zen 2012 (Blue Edition) software (Carl Zeiss, Microimaging Inc. Germany). The objectives employed were Zeiss APOCHROMAT 10x/0,45 Ph1; Zeiss EC Neofluar 20x/0,5 and Zeiss EC Neofluar 40x/0,75 (Carl Zeiss, Germany). Images were subsequently assembled in Adobe Photoshop 10 (Adobe Systems Inc, USA), and contrast and brightness were adjusted on the whole image panel at the same time. For immunostaining, slides with tissue sections were dewaxed and enzymatic antigen retrieval was performed by incubating the sections with 1:10 Trypsin dilution in PBS for 20 min at 37ºC. Slides were washed 3 times for 5 min with PBS at room temperature for 5 min each. Tissues sections were blocked in (20% FCS, 2% BSA, 0.2% Triton X-100) for 1 h at room temperature. Slides were incubated with anti-R-loop (1:100, S9.6), anti-γH2AX (1:100, Y-P1016, Millipore) or anti-VASA (DDX4/MVH, 1:100, ab13840, Abcam) antibodies overnight at 4ºC in a humidified chamber. Slides were washed 5 times with 1xPBS containing 0.5% Triton X-100 for 5 min each at room temperature. Alexa-Dye488 or Alexa-Dye594-conjugated secondary antibody (Molecular Probes, Life technologies) was added for 1 h at 37ºC in a humidified chamber. Subsequently, slides were washed 3 times as before and DAPI (1:10,000; Sigma-Aldrich) was added for 10 min to staining nuclei. Slides were finally washed twice and glass coverslips were mounted in Prolong Gold (Life Technologies) medium for imaging. Images were subsequently assembled in Adobe Photoshop 10 (Adobe Systems, San Jose, CA, USA), and contrast and brightness were adjusted on the whole image panel at the same time. R-loop and apoptosis quantification was performed by examining a random field of view under the microscope and counting all the seminiferous tubules within it. The presence of one or more positive cells within a tubule defined that tubule as positive.
Results
Sperm abnormalities are observed in AOA2 patients.
To investigate possible abnormalities in spermatogenesis and fertility in AOA2 patients we assessed 3 subjects (Table I). The first patient (AOA2-P1), reported in more detail below, demonstrated a complete absence of sperm production. Another adult subject (AOA2-P2) was found to have oligospermia with 100% of their sperm non-motile. A third subject, AOA2-P3, also showed oligospermia with 73% non-motility. In total, sperm abnormalities were seen in all three subjects assessed (3/3, 100%) (Table I). We assessed hormonal levels in these 3 individuals as well as an additional 2 other male AOA2 patients and no clear correlation was observed (data not shown). Overall these findings are consistent with a reproductive defect in a subset of male patients with AOA2. In order to pursue these abnormalities in greater detail we obtained a testicular biopsy for analysis from patient AOA2-P1.
Table I.
Semen Testing in AOA2 Patients.
| Normal Values | AOA2-P1* | AOA2-P2 | AOA2-P3 | |
|---|---|---|---|---|
| Age at examination | Years | 34 | 35 | 17 |
| Sperm Concentration | > 15 million/mL | None | 2.0 | 3.5 |
| Sperm Morphology | > 4% normal | - | 0% | 7% |
| Sperm Progressive Motility | > 32% | - | 0% | 20% |
| Sperm Non-motility | < 60% | - | 100% | 73% |
, subject described in this report
Patient AOA2-P1 clinical presentation.
The patient is a 34 year old man of Eastern European descent with no other past medical history who developed slowly progressive ataxia starting at age 15. Neurological examination was notable for cerebellar ataxia characterized by abnormal eye movements (saccadic smooth pursuit, direction-changing nystagmus, overshoot and ocular dysmetria on tracking, but no oculomotor apraxia), moderately impaired limb coordination, severe gait and truncal ataxia, distal sensorimotor polyneuropathy with amyotrophy, and severe impairment of posterior column function. At the time of evaluation, his Scale for the Assessment and Rating of Ataxia (SARA-5) score was 22/40. Genetic testing demonstrated two novel compound heterozygous variations in the SETX gene; c.2747_2748insAT (p.Met917Leufs*2) and c.6689T>C(p.Met2230Thr). His sister is also affected with a similar clinical presentation and carries the identical mutations. The patient and his wife underwent genetic counselling and decided to proceed with childbearing. Unfortunately pregnancy did not occur so a fertility workup was initiated during which the patient was noted to have absence of sperm in the ejaculate (azoospermia) which prompted our further evaluation including a testicular biopsy.
Sterility in an AOA2 patient due to the absence of mature germ cells in seminiferous tubules.
We have previously shown that Setx−/− male mice have severe disruption of seminiferous tubules and lack mature germ cells due to a block at the pachytene stage of meiotic prophase I 16. To investigate the effect of senataxin deficiency on reproductive function in AOA2, we obtained a testicular biopsy from the infertile AOA2 man described above. Semen analysis performed on this patient revealed azoospermia. A small testicular biopsy was subsequently collected, fixed and embedded in paraffin to further characterize the histology of the germ cells or lack thereof defect in this patient. H&E staining of testicular sections showed the presence of immature germ cells ranging from spermatogonia to spermatocytes in the seminiferous tubules but an absence of mature sperm in the AOA2 patient compared to a control sample (Figure 1A). These findings are reminiscent of those obtained in adult Setx−/−mice which show a total absence of mature germ cells in contrast to wildtype littermates, which exhibit all stages of spermatogenic development (Figure 1B). Vacuolated seminiferous tubules in which both spermatozoa and mature spermatids are absent are observed in both AOA2 and Setx-/−. Taken together, this patient’s infertility is consistent with early maturation arrest. To confirm the presence of germ cells in the seminiferous tubules, sections were immunostained for the VASA (DDX4/MVH) protein (Figure 2), an RNA-dependent helicase essential for germ cell development that is expressed in fetal and adult gonadal germ cells in both males and females 25, 26. A normal cytoplasmic staining pattern was observed in both human (control and AOA2 patient) (Figure 2A) and in mouse seminiferous tubules (Figure 2B) as previously described 26.
Figure 1. Spermatogenesis is disrupted in an AOA2 patient as shown by the absence of mature germ cells in seminiferous tubules.
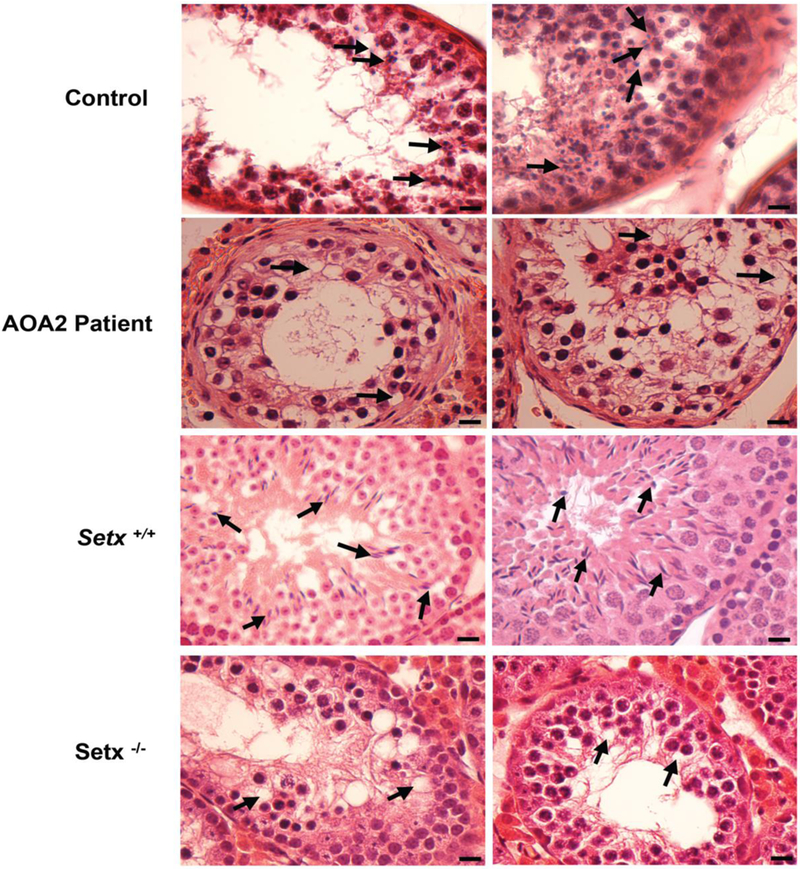
Hematoxylin and eosin (H&E)-stained sections of testis from A. control and AOA2 patient sample, and B. adult Setx+/+ and Setx−/− mice. Black arrows indicate mature germ cells in the control and Setx+/+ while vacuolated seminiferous tubules in which both spermatozoa and mature spermatids are absent are observed in both AOA2 patient and Setx−/− seminiferous tubules. Scale bar, 20 μm.
Figure 2. Presence of germs cells in control, AOA2, and Setx mice seminiferous tubules.
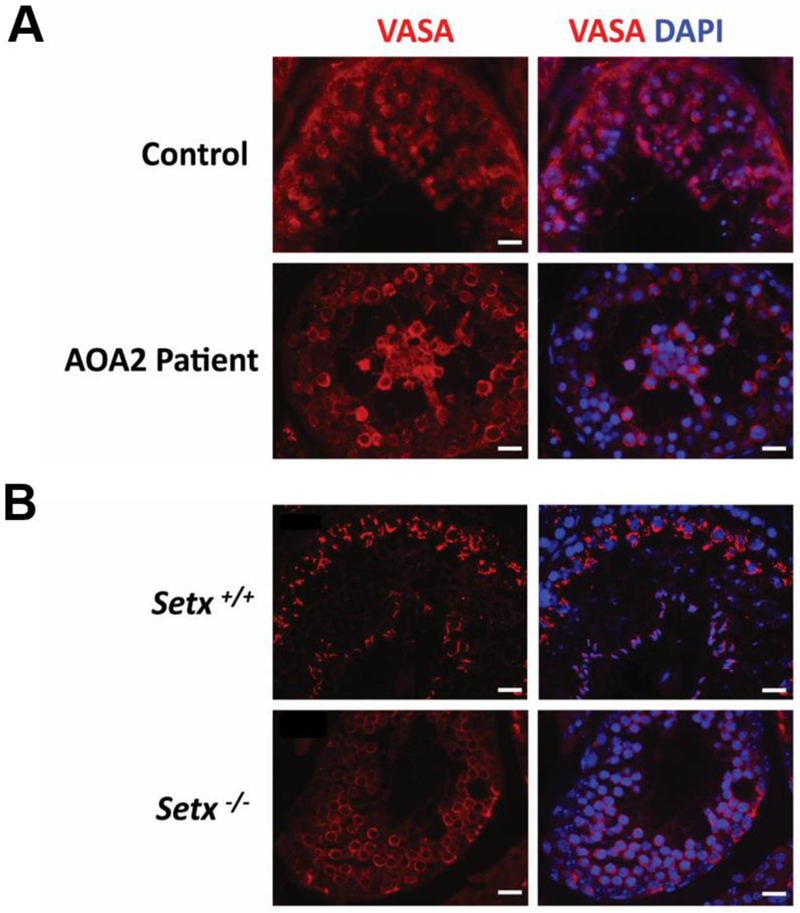
A. Immunostaining of germ cells in human AOA2 and control testis biopsy sections, and B. mouse Setx+/+ and Setx−/− seminiferous tubules. Germ cells were stained for VASA (DDX4/MVH), an RNA binding protein with an RNA-dependent helicase that is essential for germ cell development25. Nuclei were counterstained with DAPI. Scale bar, 20 μm.
Elevated levels of DNA damage, R-loops and apoptosis in AOA2 germ cells consistent with infertility secondary to maturation arrest.
We have previously shown that the persistence of unrepaired DNA damage in Setx−/− mouse spermatocytes prevented cross-over formation and triggered the mid-pachytene checkpoint resulting in the elimination of DNA-damage-containing cells 16, 24. To assess whether DNA repair is also compromised in AOA2 patient germ cells, we carried out immunostaining for pSer139 of histone H2AX (γH2AX), a well-characterized maker of DNA double strand breaks 27. AOA2 spermatocytes exhibited increased levels of γH2AX staining compared to the spermatocytes from a healthy control patient (Figure 3) suggesting that defective senataxin function hinders meiotic DSB repair and leads to the accumulation of unrepaired DNA breaks that can threaten genomic integrity in these cells. These findings mirror those of Setx−/− mice (Figure 3) confirming that senataxin is essential for the effective repair of programmed meiotic DSBs 16.
Figure 3. Elevated levels of DNA damage in AOA2 patient germ cells compared to control.
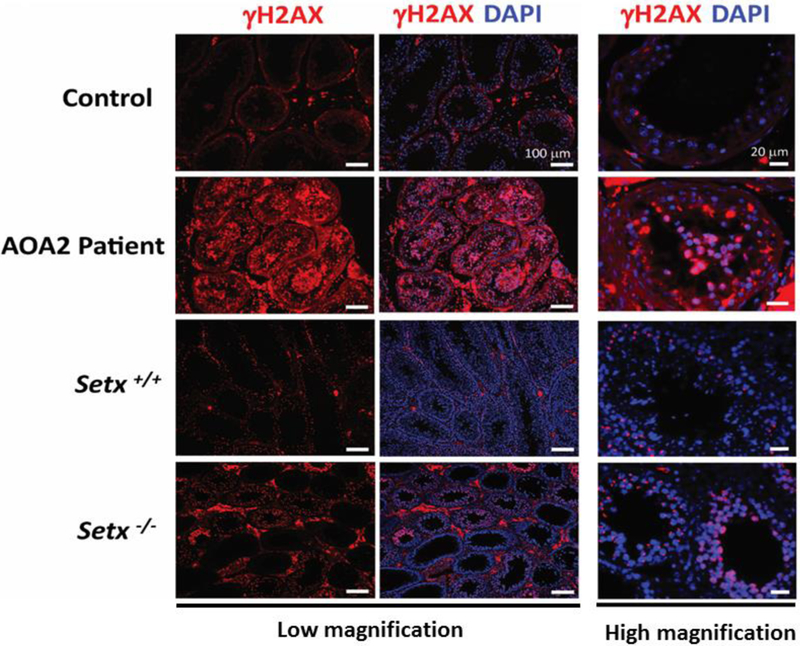
Increased levels of DNA breaks are shown by immunostaining with anti-γH2AX (pSer139 H2AX) antibody were detected in AOA2 patient germ cells and Setx−/− seminiferous tubules as compared to Control and Setx+/+. Both low and high magnification images are shown with scale bars of 100μm and 20μm, respectively. Nuclei were stained with DAPI.
Recent studies have demonstrated that senataxin, through its DNA/RNA helicase activity, resolves DNA:RNA hybrid structures known as R-loops that form during normal DNA transcription when the RNA transcription machinery encounters a GC rich region and stalls, allowing the nascent RNA strand to pair with the DNA template 12. Both in vitro and in vivo studies have provided compelling evidence for the formation and/or accumulation of these structures in Setx-defective proliferating cells 4, 14, 16, 24. Indeed, increased levels of R-loops were detected in Setx−/− spermatocytes compared to their wild type counterpart, and this was not only limited to germ cells but was also found in other tissues such as the spleen and intestine of Setx−/− mice, two tissues characterized by a high proportion of proliferating cells 24. To determine whether this would also be the case in AOA2 male patient germ cells, we looked for the presence of R-loops by immunostaining using the well-characterized R-loop-specific (S9.6) antibody 28, 29. A greater proportion of tubules containing R-loop positive cells (57%) were detected in the AOA2 patient testis compared to that of the control (8%) in agreement with findings in the Setx−/− mouse model 16, 24 (Figure 4A). However, the number of R-loop positive cells per tubule in the AOA2 patient was less than that observed in Setx−/− mouse testis (Figure 4B and 4C). For each positive seminiferous tubule, only 1 to 2 R-loop positive cells were detected in the AOA2 patient, while multiple R-loop positive cells (4 to 40; mean 18±9) were detected in Setx−/− mouse tubules (Figure 4B and 4C). This difference in the number of R-loop positive germ cells per tubule may possibly reflect differences in phenotypic expressivity in patients, disease variance across tissues, a milder phenotype of the patient’s AOA2 mutation(s) as compared to the complete ablation of the Setx gene in the mouse, or an inherent difference between humans and rodents.
Figure 4. Detection of R-loops (DNA:RNA hybrid structures) in AOA2, control, Setx+/+ and Setx−/− testis.
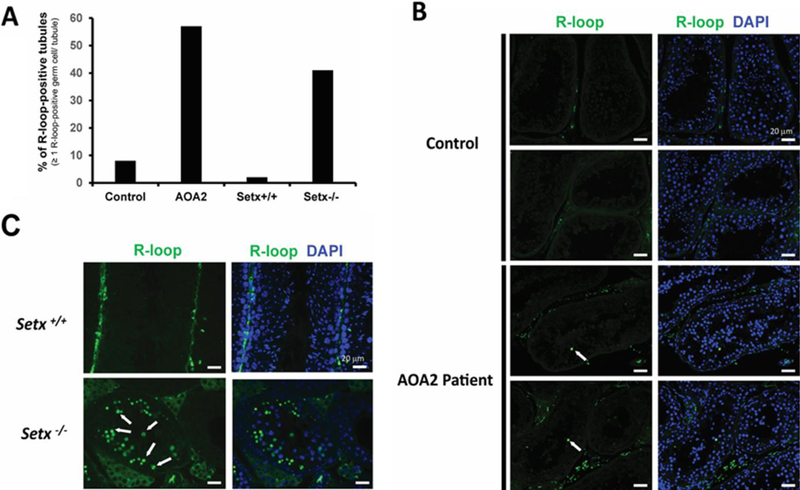
A. Percentage of R-loop-positive tubules in Control, AOA2, Setx+/+ and Setx−/− samples. Tubules containing ≥ 1 R-loop-positive germ cell were scored as R-loop-positive tubule. B. Example of R-loop staining in control and AOA2 patient testis biopsy sections. C. R-loop staining in Setx+/+ and Setx−/− mouse testis seminiferous tubules. R-loops were detected in germ cells as shown by the white arrow. While many R-loop-positive germs cells were detected per tubule in Setx−/− mice as shown in (C), only one to two R-loop-positive germ cells were detected per tubule in the AOA2 patient sample (B).
The accumulation of DNA breaks, the increased number of R-loop-containing cells, and the absence of mature germ cells in the AOA2 patient testis prompted us to next investigate the level of germ cell apoptosis. As expected, elevated levels of apoptosis were detected in the AOA2 patient (29%) compared to the control (5%) using TUNEL assay (Figure 5A, Figure 5B, upper panel) and these levels were comparable to those observed in Setx−/− (34%) and Setx+/+ (6%) mice (Figure 5B, lower panel). Overall, these data suggest that in humans, as in mice, persistent DNA breaks secondary to defective senataxin function leads to the induction of a stage IV mid-pachytene checkpoint that results in male germ-cell depletion and severe infertility.
Figure 5. Elevated levels of germ cells apoptosis in an AOA2 patient.

A. TUNEL-stained testis sections from control, AOA2 patient and adult Setx+/+ and Setx−/− mice. White arrow indicates TUNEL-positive germ cells. DAPI was used to stain nuclei. Scale bar, 20 μm. B. Quantitation of the number of apoptotic tubules (tubules containing ≥ 1 TUNEL positive cell). More than 200 tubules were scored for each sample.
Discussion
Our earlier work identified a severe male infertility phenotype in transgenic mice lacking the Setx gene and characterised its underlying mechanism. In this report, we describe the observation of a similar phenotype characterised by defective spermatogenesis and lack of mature germ cells in male AOA2 patients, confirming the essential role of senataxin in male reproductive function. The similarities between the mouse model and the AOA2 patient phenotype are striking and strongly suggest that similar molecular mechanisms apply to both.
The absence of mature germ cells in the AOA2 patient suggests a blockage in meiosis and perhaps the elimination of aberrant germ cells at the mid-pachytene checkpoint as previously observed in the Setx−/− mouse model 16. This observation identifies a protein with a significant role in protecting the brain as also playing a role in spermatogenesis. This is consistent with the association of gonadal dysfunction with other genes involved in protecting the genome, such as premature ovarian failure seen with mutation of ATM in ataxia-telangiectasia,30 another ataxia disorder, or NBN in Nijmegen breakage syndrome.31 Our observation is also reminiscent of how the generation of a mouse model of pantothenate kinase deficiency revealed the presence of azoospermia, which was not previously appreciated in the human disorder,32 emphasizing the importance of such models in identifying associated phenotypes or pathologies in rare neurological disorders such as cerebellar ataxia. Furthermore, this is one of the few examples of male infertility in humans in which the cellular defect underpinning spermatogenic failure has been elucidated. These findings are the first evidence of male infertility in male patients with AOA2 and support essential extra-neurological roles for senataxin in germ cell maturation and survival in humans. As indicated in Table I, the phenotype may be variable among male AOA2 patients so future studies will be necessary to examine the role of senataxin in normal human spermatogenesis and connections to this and potentially other forms of azoospermia. Interestingly, during this study, we identified one male AOA2 patient who had successfully fathered a child at age 30, suggesting either variability to the phenotype, similar to what has been reported with female fertility issues 15, 20–23, or a successful pregnancy in the setting of oligospermia. This raises an important reproductive counselling issue for clinicians to address in their male patients with AOA2.
These findings have major implications for the investigation and management of male infertility due to spermatogenic failure. Genetic causes of severe spermatogenic defects have typically been linked with sex chromosome abnormalities such as Y chromosome micro-deletions or sex chromosome aneuploidies such as Klinefelter syndrome (47, XXY males). Our data uncover a novel cause of spermatogenic impairment in humans that can be ascribed to an autosomal mutation. It is possible that other SETX variants that do not incur significant neurological sequelae could affect male infertility and future study will be needed to address this.
Conclusion
In this study we report the first clinical and cellular evidence of impaired spermatogenesis in AOA2 patients, a finding not previously observed in this patient population, and consistent with the phenotype seen in Setx knockout mice, both clinically and histologically. This supports a novel role for senataxin in human germ cell development and reproductive function and extends its extra-neurological roles. Fertility specialists should be made aware of SETX mutations as a possible diagnosis in young male patients who present with severe oligozoospermia or azoospermia since infertility may potentially presage the later onset of neurological manifestations in some individuals.
Acknowledgements
The authors thank the patients and their families for their participation in this project. We thank John Luff and HMRC animal house staff for the maintenance of the mice and Dr. Abrey Yeo for assistance with the preparation of the figures.
Funding: This work was supported by the Australian Research Council [ARC, DP 130100389 to M.F.L] and the National Institute for Neurological Disorders and Stroke [R01NS082094 to B.L.F]. BLF acknowledges support through donations to the University of California by the DeMint Family and the Ruehl Family.
Footnotes
Conflict of interest: All the authors declare that there is no conflict of interest.
References
- 1.Palau F, Espinos C. Autosomal recessive cerebellar ataxias. Orphanet J Rare Dis 2006;1:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nemeth AH, Bochukova E, Dunne E, et al. Autosomal recessive cerebellar ataxia with oculomotor apraxia (ataxia-telangiectasia-like syndrome) is linked to chromosome 9q34. Am J Hum Genet 2000;67:1320–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moreira MC, Klur S, Watanabe M, et al. Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2. Nat Genet 2004;36:225–227. [DOI] [PubMed] [Google Scholar]
- 4.Yuce O, West SC. Senataxin, defective in the neurodegenerative disorder ataxia with oculomotor apraxia 2, lies at the interface of transcription and the DNA damage response. Mol Cell Biol 2013;33:406–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Suraweera A, Becherel OJ, Chen P, et al. Senataxin, defective in ataxia oculomotor apraxia type 2, is involved in the defense against oxidative DNA damage. J Cell Biol 2007;177:969–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Suraweera A, Lim Y, Woods R, et al. Functional role for senataxin, defective in ataxia oculomotor apraxia type 2, in transcriptional regulation. Hum Mol Genet 2009;18:3384–3396. [DOI] [PubMed] [Google Scholar]
- 7.Roda RH, Rinaldi C, Singh R, Schindler AB, Blackstone C. Ataxia with oculomotor apraxia type 2 fibroblasts exhibit increased susceptibility to oxidative DNA damage. J Clin Neurosci 2014;21:1627–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Becherel OJ, Sun J, Yeo AJ, et al. A new model to study neurodegeneration in ataxia oculomotor apraxia type 2. Hum Mol Genet 2015;24:5759–5774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fogel BL, Cho E, Wahnich A, et al. Mutation of senataxin alters disease-specific transcriptional networks in patients with ataxia with oculomotor apraxia type 2. Hum Mol Genet 2014;23:4758–4769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miller MS, Rialdi A, Ho JS, et al. Senataxin suppresses the antiviral transcriptional response and controls viral biogenesis. Nature immunology 2015;16:485–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Amicis A, Piane M, Ferrari F, Fanciulli M, Delia D, Chessa L. Role of senataxin in DNA damage and telomeric stability. DNA Repair (Amst) 2011;10:199–209. [DOI] [PubMed] [Google Scholar]
- 12.Skourti-Stathaki K, Proudfoot NJ, Gromak N. Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol Cell 2011;42:794–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Richard P, Feng S, Manley JL. A SUMO-dependent interaction between Senataxin and the exosome, disrupted in the neurodegenerative disease AOA2, targets the exosome to sites of transcription-induced DNA damage. Genes Dev 2013;27:2227–2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anheim M, Monga B, Fleury M, et al. Ataxia with oculomotor apraxia type 2: clinical, biological and genotype/phenotype correlation study of a cohort of 90 patients. Brain 2009;132:2688–2698. [DOI] [PubMed] [Google Scholar]
- 15.Criscuolo C, Chessa L, Di Giandomenico S, et al. Ataxia with oculomotor apraxia type 2: a clinical, pathologic, and genetic study. Neurology 2006;66:1207–1210. [DOI] [PubMed] [Google Scholar]
- 16.Becherel OJ, Yeo AJ, Stellati A, et al. Senataxin plays an essential role with DNA damage response proteins in meiotic recombination and gene silencing. PLoS Genet 2013;9:e1003435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yeo AJ, Becherel OJ, Luff JE, Graham ME, Richard D, Lavin MF. Senataxin controls meiotic silencing through ATR activation and chromatin remodeling. Cell Discov 2015;1:15025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manto M, Hampe CS. Endocrine disorders and the cerebellum: from neurodevelopmental injury to late-onset ataxia. Handb Clin Neurol 2018;155:353–368. [DOI] [PubMed] [Google Scholar]
- 19.Fink DA, Nelson LM, Pyeritz R, et al. Fragile X Associated Primary Ovarian Insufficiency (FXPOI): Case Report and Literature Review. Front Genet 2018;9:529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Le Ber I, Bouslam N, Rivaud-Pechoux S, et al. Frequency and phenotypic spectrum of ataxia with oculomotor apraxia 2: a clinical and genetic study in 18 patients. Brain 2004;127:759–767. [DOI] [PubMed] [Google Scholar]
- 21.Lynch DR, Braastad CD, Nagan N. Ovarian failure in ataxia with oculomotor apraxia type 2. Am J Med Genet A 2007;143A:1775–1777. [DOI] [PubMed] [Google Scholar]
- 22.Gazulla J, Benavente I, Lopez-Fraile IP, Modrego P, Koenig M. Sensorimotor neuronopathy in ataxia with oculomotor apraxia type 2. Muscle Nerve 2009;40:481–485. [DOI] [PubMed] [Google Scholar]
- 23.Fogel BL, Lee JY, Perlman S. Aberrant splicing of the senataxin gene in a patient with ataxia with oculomotor apraxia type 2. Cerebellum 2009;8:448–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yeo AJ, Becherel OJ, Luff JE, et al. R-loops in proliferating cells but not in the brain: implications for AOA2 and other autosomal recessive ataxias. PLoS ONE 2014;9:e90219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Castrillon DH, Quade BJ, Wang TY, Quigley C, Crum CP. The human VASA gene is specifically expressed in the germ cell lineage. Proc Natl Acad Sci U S A 2000;97:9585–9590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Noce T, Okamoto-Ito S, Tsunekawa N. Vasa homolog genes in mammalian germ cell development. Cell Struct Funct 2001;26:131–136. [DOI] [PubMed] [Google Scholar]
- 27.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem 1998;273:5858–5868. [DOI] [PubMed] [Google Scholar]
- 28.Phillips DD, Garboczi DN, Singh K, Hu Z, Leppla SH, Leysath CE. The sub-nanomolar binding of DNA-RNA hybrids by the single-chain Fv fragment of antibody S9.6. J Mol Recognit 2013;26:376–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boguslawski SJ, Smith DE, Michalak MA, et al. Characterization of monoclonal antibody to DNA.RNA and its application to immunodetection of hybrids. J Immunol Methods 1986;89:123–130. [DOI] [PubMed] [Google Scholar]
- 30.Gatti R, Perlman S. Ataxia-Telangiectasia In: Adam MP, Ardinger HH, Pagon RA, et al. , eds. GeneReviews((R)) Seattle (WA)2016. [Google Scholar]
- 31.Varon R, Demuth I, Chrzanowska KH. Nijmegen Breakage Syndrome In: Adam MP, Ardinger HH, Pagon RA, et al. , eds. GeneReviews((R)) Seattle (WA)2017. [Google Scholar]
- 32.Kuo YM, Duncan JL, Westaway SK, et al. Deficiency of pantothenate kinase 2 (Pank2) in mice leads to retinal degeneration and azoospermia. Hum Mol Genet 2005;14:49–57. [DOI] [PMC free article] [PubMed] [Google Scholar]