

Abstract
Much evidence has accumulated in the literature over the last fifteen years that indicates vitamin A has a role in metabolic disease prevention and causation. This literature proposes that vitamin A can affect obesity development and the development of obesity-related diseases including insulin resistance, type 2 diabetes, hepatic steatosis and steatohepatitis, and cardiovascular disease. Retinoic acid, the transcriptionally active form of vitamin A, accounts for many of the reported associations. However, a number of proteins involved in vitamin A metabolism, including retinol-binding protein 4 (RBP4) and aldehyde dehydrogenase 1A1 (ALDH1A1, alternatively known as retinaldehyde dehydrogenase 1 or RALDH1), have also been identified as being associated with metabolic disease. Some of the reported effects of these vitamin A-related proteins are proposed to be independent of their roles in assuring normal retinoic acid homeostasis. This review will consider both human observational data as well as published data from molecular studies undertaken in rodent models and in cells in culture. The primary focus of the review will be on the effects that vitamin A per se and proteins involved in vitamin A metabolism have on adipocytes, adipose tissue biology, and adipose-related disease, as well as on early stage liver disease, including non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH).
Keywords: Retinoid, Retinoic Acid, Adipocyte, Obesity, Insulin Resistance, Type 2 Diabetes, Non-Alcoholic Fatty Liver Disease (NAFLD)
I. Introduction.
By definition vitamin A is all-trans-retinol. However, in normal scientific usage, vitamin A is taken to signify not only this compound but also the natural metabolites of all-trans-retinol. The natural metabolites of vitamin A include retinaldehyde (retinal), retinoic acid, retinyl esters, oxidized forms of retinol and retinoic acid, and conjugates of retinol and retinoic acid. The chemical structures for some of these are provided in Figure 1. Throughout this review, vitamin A will be used to refer collectively to retinol and all of its natural metabolites.
Figure 1. Chemical structures for vitamin A metabolites mentioned in the text.
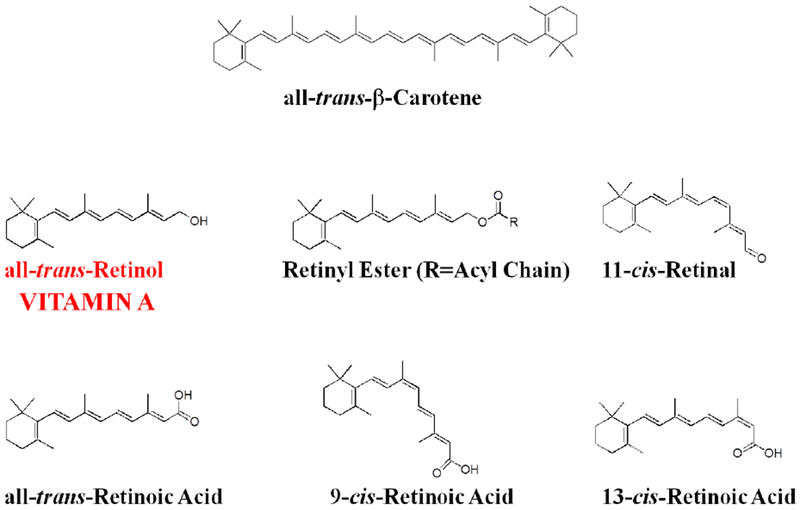
By definition, vitamin A is all-trans-retinol. There are a number of different retinyl esters found within the body, all of these possess long chain acyl groups. The most abundant retinyl esters in the body are retinyl palmitate, retinyl sterate, retinyl oleate, and retinyl linoleate, accounting for greater than 95% of the body’s total retinyl ester pool. Short chain retinyl esters like retinyl acetate do not occur in nature and are found only in food supplements. All-trans-β-carotene is the prototypic provitamin A carotenoid that can be enzymatically converted to vitamin A.
The preponderance of the vitamin A present in the body is in the all-trans-configuration. However, 11-cis-retinaldehyde is the chromophore for the visual pigment rhodopsin (Wald 1968; Palczewski 2012) and 9-cis-retinoic acid and its’ oxidized metabolites have been identified to be transcriptional regulators (Chambon, 1996; Gudas, 2011; Al Tanoury et al., 2013). If the isomeric configuration of a vitamin A metabolite is not specifically identified in the text, this should be taken to refer to the all-trans-isomer. Thus, retinyl ester refers to all-trans-retinyl ester.
In the late 1970s, Sporn coined the term retinoid (Sporn et al., 1976). A retinoid is any chemical compound that bears a structural resemblance to all-trans-retinol, with or without the biological activity of vitamin A. Thus, the term retinoid refers to both natural metabolites of retinol as well as its synthetic analogs. Many synthetic retinoids, produced by academic and pharmaceutical chemists, are now available and many have found use in the clinic. The reader should note that the terms vitamin A and retinoid are used interchangeably. Since this review will focus predominantly on natural metabolites of vitamin A, the term retinoid will only be used in the text to refer to synthetic retinoids and not natural forms of vitamin A.
There has been a longstanding interest in understanding the actions of vitamin A in preventing and treating disease. Until about fifteen years ago, most of this research interest was focused on proliferative disorders, primarily on cancers and dermatologic disease (Hong & Lotan, 1993; Dollé & Neiderreither, 2015). However, research interest in vitamin A and its relationship to disease processes have migrated so as to have now a central focus on metabolic disorders. This review will consider primarily liver disease and how this may be associated with vitamin A homeostasis, and adipose tissue vitamin A physiology and its associations with obesity and insulin resistance. There is a great deal of current research interest in understanding vitamin A’s role in endocrine pancreas physiology and disease. This topic will not be covered in this review and the reader is referred to Brun et al. (2016) for a review on this topic. There also presently exists an extensive literature on vitamin A and its actions in the cardiovascular system but this topic too will only be briefly touched upon by this review. The reader is referred to (Rhee et al., 2012; Pan et al., 2014; D’Aniello & Waxman, 2015) for excellent reviews on vitamin A and cardiovascular physiology and disease.
II. Overview of Vitamin A Biology and Signaling.
In order to consider the role of vitamin A in metabolic disease, it is first necessary to have a general understanding of vitamin A biology and vitamin A actions within tissues and cells. In large part, this is because the metabolism and actions of vitamin A involve a number of proteins and processes that are unique to vitamin A. Moreover, a number of these specialized proteins and processes are proposed to have key roles in metabolic disease causation. Consequently, a general overview of these topics is provided immediately below.
It is important to keep in mind that vitamin A and its metabolites are lipids and, hence are insoluble within the aqueous environment of the body. Consequently, within cells and in the extracellular space, vitamin A is found either bound to one of a number of different, but specific, vitamin A-binding proteins or present within intracellular lipid droplets. Binding proteins include ones that bind retinoic acid, retinol and/or retinaldehyde. They function either intracellularly or extracellularly but usually not in both compartments. Some of these are listed in Table 1. The longstanding understanding of the intracellular vitamin A-binding proteins has been that they transport an insoluble vitamin A form within the cell to allow for its metabolism and/or storage. They are usually proposed to be involved in metabolically channeling a vitamin A form from one protein/location to another (Napoli, 2017). The extracellular binding proteins are required for transporting vitamin A from one cell type to another, or from one tissue to another. Several of these vitamin A-binding proteins have been implicated as having important roles in obesity, diabetes and liver disease.
TABLE 1.
| Original Nomenclature | Genetic Nomenclature | Ligands | Tissue Distribution |
|---|---|---|---|
| Retinol-binding protein (RBP) | RBP4 | retinol | serum/plasma |
| Cellular retinol-binding protein I (CRBPI) | RBP1 | retinol & retinal | most tissues |
| Cellular retinol-binding protein II (CRBPII) | RBP2 | retinol & retinal | small intestine |
| Cellular retinol-Binding protein III (CRBPIII) | RBP7 | retinol & retinal | adipose and muscle |
| Interphotoreceptor retinol-binding protein (IRBP) | RBP3 | retinol & retinal & other lipids | eye, retina retinal pigmented epithelium |
| Cellular retinoic acid-binding protein I (CRABPI) | CRABP1 | retinoic acid & metabolites | many tissues |
| Cellular retinoic acid-binding protein II (CRABPII) | CRABP2 | retinoic acid | skin, adipose, others |
| Fatty acid-binding (FABP5) protein | FABP5 | fatty acids & retinoic acid | adipose, liver, others |
Table 1 does not provide an inclusive listing of all vitamin A-binding proteins that have been identified in the literature. Rather the listing is limited primarily to those considered in the text.
RBP1, RBP2, RBP7, CRABP1, CRABP2, and FABP5 are all members of the fatty acid-binding protein family of proteins. RBP4 is a member of the lipocalin protein family.
A. The Physiological Actions of Vitamin A and the Molecular Basis for These.
Vitamin A is required for maintaining normal growth and development (by regulating cell proliferation and differentiation, and cell death), immunity, barrier integrity, male and female reproduction, and vision (Dollé & Niederreither, 2015). Vitamin A actions to maintain normal physiology can be attributed primarily to one of three mechanisms. Retinoic acid and some of its metabolites are very potent transcriptional regulators acting through members of the nuclear hormone superfamily of ligand-dependent transcription factors (Chambon, 1996; Gudas, 2011; Al Tanoury et al., 2013). There also is growing evidence that retinoic acid can act outside of the nucleus affecting cellular signal transduction pathways (Al Tanoury et al., 2013; Rochette-Egly, 2015). Since the early work of Wald and others in the 1930s, it has been known that 11-cis-retinaldehyde acts as the visual chromophore in rhodopsin, initiating the visual process (Wald, 1968; Palczewski, 2012).
1. Transcriptional activation.
At the molecular level, most of the physiological actions of vitamin A are thought to be mediated by nuclear hormone receptors (ligand-dependent transcription factors) that bind retinoic acid or one of its metabolites (Chambon, 1996; Gudas, 2011; Al Tanoury et al., 2013). Canonically, the three retinoic acid receptors (RARα, RARβ and RARγ) along with the three retinoid X receptors (RARα, RARβ and RARγ) are thought to mediate the great majority of the transcriptional regulatory activity of vitamin A. In excess of 500 diverse genes are known to be transcriptionally responsive to vitamin A (Balmer & Blomhoff, 2001). The RARs and RXRs have been extensively studied at both the gene and protein levels and the molecular details of how these transcription factors activate/repress transcription are known in exquisite detail. These topics will not be considered below. The reader is referred to in depth recent reviews on these topics (Bourguet & Moras, 2015; Urban et al., 2015; Mendoza-Parra et al., 2015).
All-trans-retinoic acid can bind to each of the RARs with high affinity and is universally considered to be the physiological ligand for these nuclear receptors (Chambon, 1996; Gudas, 2011; Al Tanoury et al., 2013). The early literature regarding the properties of the RXRs proposed that 9-cis-retinoic acid is the physiological ligand for the RXRs (Heyman et al., 1992; Allenby et al., 1993). However, there is presently no consensus as to whether 9-cis-retinoic acid is truly a physiologically relevant ligand for the RXRs. This lack of consensus primarily centers around the analytical methods used to assess tissue and cellular 9-cis-retinoic acid concentrations. Some authors have failed to detect significant (measurable) concentrations of 9-cis-retinoic acid within tissues obtained from male mice and female rats raising a question as to the physiological relevance of 9-cis-retinoic acid for facilitating RXR-dependent transcription activity (Kane et al., 2008b). Whereas other authors have reported low but measureable concentrations of 9-cis-retinoic acid as well as other low abundance retinoic acid metabolites in human serum obtained from males (Arnold et al., 2012). Both of these studies involved the use of very sensitive and specific liquid chromatography tandem mass spectrometry-based analytical procedures and it is not readily possible to reconcile the differences. However, it should be noted that measurement of tissue all-trans- and 9-cis-retinoic acid is fraught with difficulties involving strong tissue matrix effects. There is a clear need for further investigations of 9-cis-retinoic acid tissue concentrations and whether these are sufficient to confirm its physiological role in regulating RXR actions. Owing to the lack of clear consensus on this issue, Rochette-Egly and colleagues (Al Tanoury et al., 2013) have conservatively summarized this situation as “Indeed RXRs cannot bind all-trans-retinoic acid, and although its 9-cis isomer was initially considered as a bona fide RXR ligand, it is now controversial due to the inability to detect this compound in vivo.” Never-the-less, 9-cis-retinoic acid is a potent agonist for bringing about activation of RXR-mediated gene expression, albeit one that is not present at readily detectable concentrations using state-of-the-are analytical approaches in most tissues (Heyman et al., 1992; Allenby et al., 1993; Kane et al., 2008b)).
A number of naturally occurring molecules, most of them are unrelated to vitamin A, including phytanic acid and β-apo 13-carotenone, are reported to be able to bind to RXRs with relatively high affinity and able to influence gene transcription in vitro (Hiebl et al., 2018). Presently though, there is no general agreement as to whether any of these is a physiologically significant natural RXR ligand. Recently, 9-cis-13,14-dihydroretinoic acid, a natural metabolite of 9-cis-retinoic acid that is reported to be abundant in cells and tissues, has been proposed to be a natural endogenous RXR ligand (RÜhl et al., 2015). At present, it remains to be confirmed whether this vitamin A metabolite is truly responsible in vivo for regulating RXR transactivation.
The literature also proposes that retinoic acid is able to bind and modulate the transcriptional activities of other members of the nuclear hormone receptor superfamily, including peroxisome proliferator-activator receptor β/δ (PPARβ/δ) (Shaw et al., 2003; Noy, 2016a), RORβ (Stehlin-Gaon et al., 2003), COUP-TFII (Kruse et al., 2008) and TR4 (Zhou et al., 2011). The most well studied of these interactions involves all-trans-retinoic acid interactions with PPARβ/δ and how this modulates expression of PPAR β/δ-responsive genes within cells. The PPARs regulate genes as heterodimers with RXRs (Chambon, 1996; Gudas, 2011; Al Tanoury et al., 2013) and are recognized to be lipid sensors that can be activated by fatty acids and their metabolites, as well as by other lipids (Palomer et al., 2018). The binding of retinoic acid to PPARβ/δ is proposed to regulate energy homeostasis and insulin responses (Palomer et al., 2018). Noy and colleagues (Shaw et al., 2003; Noy, 2016a) have reported studies showing that all-trans-retinoic acid binds with nanomolar affinity, modulates the conformation of the receptor, promotes interaction with the transcriptional coactivator SRC-1, and efficiently activates PPARβ/δ-mediated transcription in transactivation assays. This effect of all-trans-retinoic acid was not observed for PPARα or PPARγ. Subsequent studies from the Noy laboratory established a role for cellular retinoic acid-binding protein, type 2 (CRABP2) in mediating retinoic acid-dependent PPARβ/δ-transcription (Berry et al., 2010). Noy has further proposed a role for fatty acid-binding protein 5 (FABP5) in controlling all-trans-retinoic acid-dependent PPARβ/δ transcription. Specifically, all-trans-retinoic acid will only efficiently activate PPARβ/δ when the FABP5/CRABP2 ratio within cells is high (Noy, 2016a).
Although the proposed actions of vitamin A on PPARβ/δ-regulated gene expression are intriguing, this hypothesis is not yet universally accepted. A number of studies specifically focused on this hypothesis have reached the conclusion that all-trans-retinoic acid does not act as a ligand for PPARβ/δ (Pieck et al., 2008; Borland et al., 2008; Borland et al. 2011). Studies by Pieck et al. (2008), using several different mouse and human cell lines, were unable to establish a role for all-trans-retinoic acid as a PPARβ/δ activator in a number of different reporter assays. In addition, these authors were unable to establish all-trans-retinoic acid responsive transcriptional activity for bona fide PPARβ/δ target genes (ADRP and AN-GPTL4) but were able to demonstrate all-trans-retinoic acid-mediated transcriptional effects on known RAR-responsive genes. Borland et al. (2008, 2011) reported findings from studies employing cultured human HaCaT keratinocytes. Data reported by these authors support the conclusion that all-trans-retinoic acid acts in these cells in a manner that is independent of PPARβ/δ transcriptional regulation. In addition, Borland et al. (2011), studying HaCaT keratinocytes that expressed different levels and consequentially different ratios of FABP5 and CRABP2, concluded that FABP5 does not transport all-trans-retinoic acid to the nucleus to modulate PPARβ/δ transcription. Finally, a recent report from the Gudas lab (Laursen et al. 2018), involving the combinatorial knockout of all three RARs in murine embryonic stem cells, reached the conclusion, that within the context of their cell system, the RARs accounted for all of the transcriptional actions of all-trans-retinoic acid.
2. Direct effects of retinoic acid on cellular signal transduction pathways.
Until recently, it had been thought that most or all of retinoic acid actions in the body could be explained by its actions in the nucleus (Chambon, 1996; Gudas, 2011; Al Tanoury et al., 2013). However, recent work has suggested a role for retinoic acid outside of the nucleus, where retinoic acid acts in a non-genomic manner (Al Tanoury et al., 2013; Rochette-Egly, 2015; Park et al., 2018). These non-genomic actions are proposed to involve a number of different processes including ones involving RARs located outside of the nucleus and cellular retinoic acid-binding protein, type 1 (CRABP1). The non-genomic actions of retinoic acid are considered in greater detail in reviews by Rochette-Egly and colleagues (Al Tanoury et al., 2013; Rochette-Egly, 2015).
It is now recognized that retinoic acid treatment of cells can give rise to non-genomic activation of kinase-dependent signaling cascades that do not directly involve genomic signaling. Studies aimed at comparing responses to retinoic acid treatments of normal and malignant prostate cancer cells established a rapid, non-genomic effect of retinoic acid on cytoplasmic signaling pathways (Pasquali et al., 2005). The authors were able to demonstrate that this effect specifically involved extracellular signal-regulated kinase 1/2 (Erk 1/2) activation. Studies by others established that retinoic acid treatment of testicular Sertoli cells decreased cell viability and promoted apoptotic cell death (Zanotto-Filho et al., 2008). Apoptosis was reported to be dependent on rapid and non-classical stimulation of the Erk 1/2 signaling pathway. For retinoic acid-treated cells, Erk 1/2 was found to induce caspase-3 activation. Other published work from several laboratories has established that activation of cellular signal transduction pathways, including Erk 1/2 signaling, involves a pool of RARs that are localized outside of the nucleus in membrane lipid rafts (Masiá et al., 2007; Piskunov & Rochette-Egly, 2012). To date, a number of different signaling cascades, including the Erk 1/2, p38 mitogen-activated protein kinase (p38MAPK), phosphoinositide 3-kinase, and Src kinase pathways, have been identified as being responsive to the non-genomic actions of retinoic acid (Rochette-Egly, 2015). However, based on the presently available information, it appears that the specific effects of retinoic acid on intracellular signal transduction pathways may be cell type dependent.
Wei and colleagues had recently identified a role for CRABP1 that is independent of RAR signaling in modulating cell proliferation and learning in male mice (Lin et al., 2017). These investigators showed that before retinoic acid enters the nucleus, it binds to CRABP1 to effect signaling involving the ERK1/2 pathway, modulating cell cycle control of proliferation. Subsequent studies from this group, ones carried out using male Crabp1-null mice, established that CRABP1 directly inhibits calcium-calmodulin protein kinase II (CaMKII) by competing with calmodulin for interaction with CaMKII (Park et al., 2018).
Presently, there is considerable research interest focused on the non-genomic actions of retinoic acid. As far as the author is aware, there now is little published evidence to indicate that these non-genomic pathways are important for the development of metabolic disease. However, it would not be too surprising if such information were to become available in the future.
B. Vitamin A Metabolism and Storage.
The processing of dietary vitamin A in the intestine and its postprandial uptake into the body, as a component of chylomicrons, shares many common features with other neutral lipids, especially triglycerides and cholesterol. However, once absorbed into the body, most aspects of vitamin A metabolism and storage are unique to vitamin A. The author has published several extensive reviews covering vitamin A uptake in the intestine, transport in the blood, storage in tissues and metabolism and the reader is referred to these for more details (D’Ambrosio et al., 2011; Blaner & Li, 2015; Blaner et al., 2016).
1. Metabolism.
A generalized metabolic scheme for the metabolism of vitamin A within cells is provided in Figure 2. This scheme may not hold for all cell types but it summarizes the major metabolic steps that are important for assuring vitamin A homeostasis in the body. Briefly, vitamin A comes to cells in the blood as retinol bound to retinol-binding protein 4 (RBP4). Some cells, especially those of the retinal pigmented epithelium in the eye and adipocytes, possess a cell surface receptor for RBP4 known as stimulated by retinoic acid 6 (STRA6), but most cells in the body do not express STRA6 (Berry et al. 2013). How STRA6 takes up retinol from RBP4 has been described at the molecular level in published crystallographic studies (Chen et al., 2016). The retinol internalized by cells can either be stored as retinyl ester or undergo two oxidative steps, the first forming retinaldehyde and the second retinoic acid. Retinyl esters are a storage form of vitamin A and are found primarily in hepatic stellate cells (HSCs) within the liver, retinal pigment epithelial cells within the eye, adipocytes, and some other cells throughout the body (O’Byrne & Blaner, 2013). Retinoic acid, formed through the two-step oxidation of retinol, is needed by most cells in the body to maintain normal vitamin A-dependent gene expression and signaling (see above).
Figure 2. Generalized metabolic scheme for the major steps important to vitamin A metabolism.
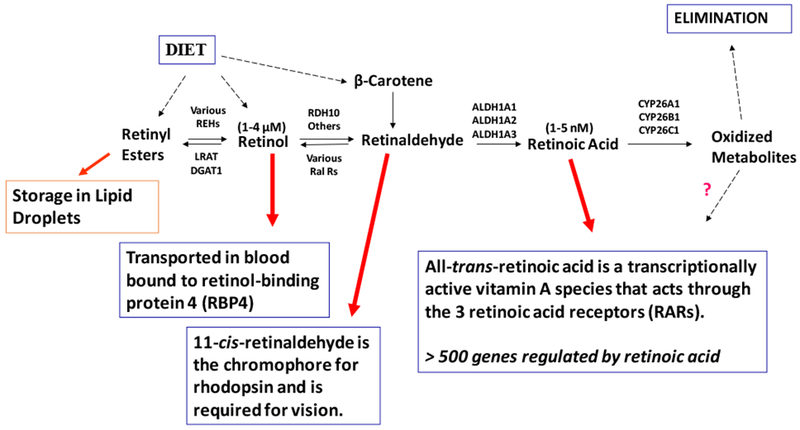
All vitamin A is derived from the diet either as preformed vitamin A (predominantly retinyl esters and retinol) or as provitamin A carotenoids such as β-carotene. Once internalized, vitamin A (retinol) can be converted to its retinyl ester storage forms primarily through the actions of lecithin:retinol acyltransferase (LRAT), but also in some instances by diacylglycerol acyltransferase 1 (DGAT1). The stored retinyl esters undergo hydrolysis through the actions of a retinyl ester hydrolase (REH). The molecular identities of physiologically important REHs remain to be elucidated. Retinol can be oxidized to retinaldehyde primarily through the actions of retinol dehydrogenase 10 (RDH10), although other enzymes have also been proposed to catalyze this oxidation in vivo. A number of enzymes are able to catalyze the reverse reaction, the reduction of retinaldehyde to retinol. These enzymes are referred to as retinaldehyde reductases (RalRs). Retinol can also be secreted from cells/tissues into the circulation bound to retinol-binding protein 4 (RBP4). The concentration of retinol that is present within the blood is in the low μM range. Retinaldehyde undergoes oxidation to retinoic acid, the vitamin A form needed for transcriptional regulation. Retinoic acid formation is catalyzed by one of three aldehyde dehydrogenase, ALDH1A1, ALDH1A2 or ALDH1A3. The concentration of retinoic acid present in cells and tissues is in the low nM range. Retinoic acid can undergo further oxidative metabolism catalyzed by CYP26A1, CYP26B1 or CYP26C1 forming a number of oxidized products. Although some of these oxidized metabolites have been proposed to have transcriptional modulatory activity, these metabolites are destined predominantly for elimination from the body.
Some cells, especially ones in tissues that accumulate relatively high levels of retinyl esters, like liver, retinal pigment epithelium, and adipose tissue, are able to synthesize and secrete RBP4 (Kanai et al., 1968; Tsutsumi et al., 1992; Soprano & Blaner, 1994). This allows for the mobilization of vitamin A bound to RBP4 in times of insufficient dietary vitamin A intake from these stores. Since the discovery and first description of RBP4 in the late 1960s, the sole function of RBP4 in the body had been thought to be the mobilization of retinol from vitamin A stores and deliver it to target tissues requiring vitamin A actions (Kanai et al., 1968; Soprano & Blaner, 1994). However, Kahn and colleagues reported studies that allowed them to suggest that RBP4 synthesized in adipose tissue acts as a signal that lessens insulin responsiveness in the body (Yang et al., 2005; Graham et al., 2006). This proposal remains the focus of much research and some controversy. The role of RBP4 in the development of obesity, insulin resistance, and liver disease will be considered in much more detail below.
The two oxidative steps needed for the conversion of retinol to retinoic acid are catalyzed sequentially by a number of retinol dehydrogenases (RDHs) and retinal dehydrogenases (RALDHs). More than a dozen RDHs have been described in the literature (Kedishvili, 2013). However, outside of the eye, only a few of these are thought to have a physiologically relevant role in the generation of retinoic acid (Kedishvili, 2013). Some of these enzymes have physiologic roles in catalyzing the reverse reaction of retinaldehyde reduction to retinol (acting as retinaldehyde reductases). There is now a general consensus that RDH10 is the major enzyme involved in the synthesis of retinoic acid within the body (Kedishvili, 2013). RDH1 and Dhrs9 too are proposed by some investigators to have a physiologic role in this metabolic process (Kedishvili, 2013; Yang et al., 2017; Yang et al., 2018). The RDHs, ones that are now understood to be important for catalyzing retinol oxidation as the first enzymatic step needed for retinoic acid formation, are members of the short chain dehydrogenase/reductase protein family. The older literature had proposed that proteins from the medium chain dehydrogenase protein family, specifically a number of alcohol dehydrogenases (ADHs), act physiologically in catalyzing this oxidation. This older notion is no longer considered to be valid. There is also general consensus that only three retinaldehyde dehydrogenases (sometimes referred to in the literature as RALDH1, RALDH2, and RALDH3) are physiologically important for catalyzing retinaldehyde oxidation to retinoic acid (Kedishvili, 2013). The three retinaldehyde dehydrogenases are all members of the aldehyde dehydrogenase (ALDH) protein family (RALDH1 = ALDH1A1, RALDH2 = ALDH1A2, and RALDH3 = ALDH1A3). Given the lack of an absolute substrate specificity of these enzymes for retinaldehyde, the enzymes will be referred to throughout the review as ALDHs.
Both RDHs and ALDHs have been implicated in the literature are being causally associated with metabolic disease development. Although much of the individual data are convincing, the entirety of the data is not definitive. The weakness of this literature is that RDHs and ALDHs respectively, do not show absolute substrate specificities for retinol and retinaldehyde. For instance, both human and rat RDH1 act as 3α-hydroxysteroid dehydrogenases, efficiently catalyzing the conversion of 3α-adiol to dihydrotestosterone (Biswas & Russell, 1997). Similarly, ALDH1A1, which is widely studied as a retinaldehyde dehydrogenase for its possible involvement in metabolic disease causation, has been shown to play an important role in the detoxification of lipid-derived aldehydes. These aldehydes include 4-hydroxy-2-nonenal and acrolein, compounds that mediate oxidative stress-related disease (Makia et al., 2011). These issues complicate interpretation of the vitamin A-related literature. The possible roles of RDHs and ALDHs in metabolic disease will be discussed in detail below.
Retinoic acid levels within tissues are tightly controlled. There is a clear need for this regulation given the potency of retinoic acid as a transcriptional regulator. The control of cell and tissue retinoic acid concentrations involves the breakdown of retinoic acid by cytochrome P450 enzymes (Thatcher and Isoherranen, 2009; Ross & Zolfaghari, 2011; Kedishvili, 2013). The importance of this regulation is underscored by the identification of a patient with a deletion on a portion of a chromosome that includes the genes CYP26A1 and CYP26C1 (Nilsson et al., 2016). This patient was found to possess markedly elevated plasma concentrations of total retinoic acid and 13-cis-retinoic acid. The elevation in retinoic acid concentrations was associated with accelerated skeletal and dental development, retinal scarring, and autism-spectrum disorder. The CYP26 family, which includes CYP26A1, CYP26B1 and CYP26C1, is both transcriptionally regulated by, and is active towards all-trans-retinoic acid (Thatcher and Isoherranen, 2009; Ross & Zolfaghari, 2011). Consequently, when intracellular retinoic acid levels become too great, the CYP26 enzymes will be induced and catalyze the breakdown of the excessive retinoic acid.
The CYP26s catalyze the oxidative metabolism of retinoic acid to more polar compounds including 4-hydroxy-retinoic acid and 4-oxo-retinoic acid (Thatcher and Isoherranen, 2009; Ross & Zolfaghari, 2011). These oxidized products, as well as retinoic acid itself, can undergo enzymatic glucuronidation to form glucuronide conjugates that will be eliminated from the body in urine and feces (Ross & Zolfaghari, 2011; Barua et al., 1989). Thus, the oxidative degradation of retinoic acid is a key metabolic step for regulating vitamin A metabolism and actions.
2. Storage.
Based on measures of tissue vitamin A levels, Blomhoff and coauthors (Blomhoff et al., 1991) concluded that for a nutritionally vitamin A-sufficient rat greater than 90% of the total vitamin A present in the body is found in the liver. These authors went on to conclude that this percentage will be linked to vitamin A nutritional status, with a substantially lower percentage present in the livers of animals experiencing insufficient dietary vitamin A intake. A more recent study by Kane et al. (2008a), one systematically exploring total vitamin A levels in tissues of 2- to 4-month old male sv129 mice fed the AIN-93M diet containing 4 IU vitamin A/g diet from the time of weaning, provides data which are consistent with the conclusion that approximately 90% of whole body vitamin A was present in the livers of these mice (Kane et al. 2008a). This estimate for mice agrees well with the estimate by Blomhoff and coauthors for the male rat (Blomhoff et al., 1991).
Within the liver, two distinct hepatic cell types play important roles in the storage and metabolism of vitamin A, hepatocytes and the non-parenchymal HSCs (Blaner et al., 1985; Blomhoff et al., 1985). There is presently no evidence that the other non-parenchymal cell types present in the liver have a significant role in hepatic vitamin A physiology (Blaner et al., 1985; Blomhoff et al., 1985). High performance liquid chromatography measures involving sensitive UV-Vis detection of cellular total retinol (retinol + retinyl ester) concentrations present in well characterized freshly isolated hepatocytes, HSC, hepatic endothelial cell and Kupffer cell preparations from male rats leave no doubt that hepatocytes and HSCs are the cell types within the liver that account for greater than 99% of the total retinol present in the liver. This conclusion is further substantiated by sensitive radioimmunoassay measures of RBP4 and RBP1 as well as activity measures of retinyl ester hydrolase and esterifying activities establishing that the preponderance of these measured parameters within the intact liver are accounted for by hepatocytes and HSCs.
a. The Hepatocyte.
After traversing the lymphatic system and entering the general circulation, vitamin A-containing chylomicrons undergo a process of remodeling that involves the hydrolysis of triglycerides by lipoprotein lipase (LpL) and the acquisition of new apolipoprotein components, especially apolipoprotein E (Cooper, 1992; Abumrad & Davidson, 2012). This results in the formation of much smaller and relatively triglyceride-poor chylomicron remnants. The hepatocyte is the cellular site of chylomicron remnant clearance in the liver, and consequently the cellular uptake site of dietary vitamin A (Blomhoff et al., 1982; Blomhoff et al., 1984; Cooper, 1992; Abumrad & Davidson, 2012; Blaner & Li, 2015).
Hepatocytes constitute approximately two-thirds of all cells present in the liver and approximately 90% of total hepatic protein (Blaner et al., 1985; Geerts, 2001; Friedman, 2008). The hepatocyte is also the sole cellular site of RBP4 synthesis in the liver (Blaner et al., 2009). Thus, hepatocytes are involved in both the uptake of dietary vitamin A by the liver and its mobilization from the liver. But hepatocytes account for only a relatively small proportion of the total vitamin A present within the liver. For vitamin A-sufficient rats, estimates of the hepatic total vitamin A that is present in hepatocytes range from 10-20% (Blaner et al., 2009). The remainder, 80-90%, is found in HSCs.
The process of RBP4 secretion from hepatocytes is similar to that of other secretory proteins. RBP4 is synthesized as a pre-protein with a signal peptide that is cleaved prior to secretion (Soprano et al., 1981). Retinol is loaded into newly synthesized apo-RBP4 in the endoplasmic reticulum, although how loading takes place has not been established (Suhara et al., 1990; Soprano & Blaner, 1994). When retinol is unavailable, for instance, in times of dietary vitamin A-insufficiency, apo-RBP4 is not secreted and accumulates in the endoplasmic reticulum to levels that are 3- to 10-fold higher than those of a vitamin A-sufficient liver (Soprano & Blaner, 1994). There is evidence that newly synthesized RBP4 also binds transthyretin (TTR) within the hepatocyte (Melhus et al., 1991). However, RBP4 binding to TTR does not appear to be required for secretion to take place since RBP4 is secreted from Ttr-deficient mouse hepatocytes (Wei et al., 1995).
b. The Hepatic Stellate Cell (HSC).
HSCs, along with Kupffer and hepatic endothelial cells, constitute the non-parenchymal cells of the liver (Wake, 1980; Geerts, 2001; Friedman, 2008). They account for approximately 8% of the total cells present in the liver and about 1% of hepatic protein (Blaner et al., 1985; Geerts, 2001; Friedman, 2008). Yet approximately 80-90% of the total vitamin A in the liver of a healthy well-nourished human or animal is present in HSCs (Blaner et al., 2009). Since the liver accounts for approximately 90% of the total vitamin A present in the body, the great majority of the vitamin A present in the body is localized to this small and relatively unabundant hepatic cell type. Within the HSC, vitamin A is stored as retinyl ester within prominent HSC cytoplasmic lipid droplets, which occupy most of the cytoplasm. HSC lipid droplets have an unusual lipid composition with a relatively high vitamin A content, which is unlike the lipid droplets present in adipocytes or hepatocytes that consist almost entirely of triglyceride (Blaner et al., 2009). Moriwaki et al., (1988) reported that lipid droplets purified from HSCs isolated from male rats fed a purified diet containing 8 IU vitamin A/g diet had an average lipid composition that consisted of 39.5% retinyl ester, 31.7% triglyceride, 15.4% cholesteryl ester, 4.7% cholesterol, 6.3% phospholipids and 2.4% free fatty acids (Moriwaki et al., 1988). These authors further reported that the HSC lipid droplet lipid composition was very responsive to changes in dietary vitamin A intake but not to changes in dietary fat intake (Moriwaki et al., 1988). Based on their retinyl ester content and their responses to dietary vitamin A intake, it would appear that HSC lipid droplets are highly specialized for vitamin A storage. The HSC lipid droplets are both functionally and biochemically very similar to lipid droplets isolated from the bovine retinal pigmented epithelium, which are reported to have a lipid composition of 42.2% retinyl esters, 13.1% retinol, 12.2% triglyceride, 10.4% cholesteryl ester, 6.5% cholesterol, 2.5% phospholipids, and 12.7% free fatty acids (Orban et al., 2011). But the lipid droplets present in HSCs and retinal pigmented epithelium cells are unlike lipid droplets found in hepatocytes or adipocytes that consist primarily of triglyceride and contain relatively little retinyl ester or retinol.
Since the hepatocyte is the cellular site of dietary vitamin A uptake by the liver and the HSC is the cellular site of vitamin A storage within the liver, this raises a question as to how newly absorbed dietary vitamin A is transferred from the hepatocyte to the HSC. A considerable amount of in vivo and in vitro work carried out in the 1980s and 1990s had suggested that this required the actions of RBP4 (Blomhoff et al., 1988; Blomhoff et al., 1991). Although when it was first proposed this hypothesis was generally thought to be valid, subsequent investigations involving mice which totally lacked expression of Rbp4 in all tissues, proved that this was indeed incorrect (Quadro et al., 2004). Quadro et al. observed no differences in the cellular distribution of hepatic total vitamin A between matched male wild type, Rbp4-deficient, or Rbp4-deficient mice expressing human RBP4 in skeletal muscle, and therefore concluded that RBP4 is not required for intrahepatic transport and storage of vitamin A in HSCs (Quadro et al., 2004). It also had been suggested early on that retinol-binding protein 1 (RBP1 or CRBP1, see Table 1), which is highly expressed in both hepatocytes and HSCs (Blaner et al. 1985; Blomhoff et al., 1985), may play a role in this process (Eriksson et al., 1984). But this too now seems unlikely, since totally Rbp1-deficient mice accumulate vitamin A in HSCs (Ghyselinck et al., 1999). Thus, at present, it remains to be established through what process(es) dietary vitamin A is transferred from hepatocytes to HSCs for storage or from HSCs to hepatocytes for secretion bound to RBP4.
It is important to note that HSCs, which play the central role in the storage of vitamin A in the body, are also a central cell type involved in the development of hepatic disease (Tsuchida & Friedman, 2017). Hepatic injury activates HSCs resulting in the acquisition of a proliferative myofibroblast-like phenotype. HSC activation is accompanied by a complete loss of the vitamin A-containing HSC lipid droplets and greatly increased HSC production of extracellular matrix. Thus, HSCs not only store the majority of vitamin A that is present in the body but they also are centrally involved in the development of hepatic fibrosis, cirrhosis and hepatocellular carcinoma.
c. The Adipocyte.
Although the hepatocyte is the cellular site where most postprandial vitamin A is cleared from the circulation (Cooper, 1992; Abumrad & Davidson, 2012), during chylomicron remodeling in the circulation, some chylomicron retinyl ester is hydrolyzed by LpL. This results in the product retinol being taken up by extrahepatic tissues where LpL is expressed, especially adipose tissue (Blaner et al., 1994; van Bennekum et al., 1999). This LpL-dependent process confers to adipose tissue, a key organ contributing to metabolic disease development, a role in vitamin A storage and metabolism.
Adipose tissue accumulates both retinol and retinyl esters (Tsutsumi et al., 1992). Tsutsumi et al. reported that the inguinal, dorsal, mesenteric, epididymal, perinephric, and brown adipose depots of chow fed male rats each contains approximately 6-7 μg total vitamin A (retinol + retinyl ester) per gram tissue (Tsutsumi et al., 1992). Based on these levels and estimates of the total adipose tissue present in a 300-450 g rat, these authors speculated that depending on nutritional status, adipose tissue may contains as much as 15-20% of the total vitamin A found in the liver (Tsutsumi et al., 1992). However, based on adipose tissue total vitamin A levels reported by Kane et al. (Kane et al., 2008a) for mice fed a purified diet and by O’Byrne et al. (O’Byrne et al., 2005) for mouse fed a chow diet, it would appear that the mouse accumulates slightly less vitamin A in adipose tissue than the rat, approximately 2-4% of liver levels. Studies of lecithin:retinol acyltransferase-deficient male mice (an animal model where retinyl esters are found only in adipose tissue) fed a totally vitamin A-deficient diet have established that adipose tissue vitamin A is indeed mobilized to defend circulating vitamin A levels (Liu and Gudas 2005; O’Byrne et al., 2005; Liu et al., 2008).
Tsutsumi et al. further reported that, of the cell types present in male rat adipose tissue, only adipocytes contain detectable amounts of vitamin A (as retinol and retinyl ester), at levels ranging from 0.60 to 0.85 μg vitamin A/106 adipocytes (Tsutsumi et al., 1992). By comparison, rat hepatocytes are reported to contain between 0.10 and 0.34 μg total vitamin A/106 cells and HSCs 10.9 μg total vitamin A/106 cells (Blaner et al., 1985). The adipocyte is also the cellular site of RBP4 synthesis in and secretion from adipose tissue. mRNA levels of RBP4 in adipose tissue of chow-fed rats are approximately 15-20% those of the liver (Tsutsumi et al., 1992). Investigations of BFC-1β preadipocyte differentiation to adipocytes established that RBP4 mRNA expression is indeed differentiation-dependent, showing a temporal pattern of expression that resembles those of the adipocyte-specific genes encoding LpL and fatty acid-binding protein 4 (Zovich et al., 1992).
Bhat and colleagues (Sima et al., 2011) explored possible differences in vitamin A levels and enzymes and binding proteins involved in vitamin A metabolism in visceral (epididymal and mesenteric) adipose versus subcutaneous adipose (inguinal) tissue from male mice. These authors report that visceral adipose tissue of mice expresses higher levels of Rbp4 mRNA compared to subcutaneous fat. Also more highly expressed in visceral fat were Rbp1, Rdh10, Cyp26a1, and Cyp26b1.
Primary human adipocytes isolated from mammary subcutaneous adipose tissue obtained from healthy women undergoing reductive breast surgery are reported to synthesize and secrete RBP4 (Janke et al., 2006). Other studies of RBP4 expression carried out using human adipose tissue explants showed that adipose RBP4 mRNA levels are higher in women than in men and that RBP4 protein secretion from this ex vivo model is stimulated by leptin but not by insulin (Kos et al., 2011). Other investigations involving 26 non-obese and 33 obese women established that RBP4 mRNA expression levels are approximately 4-fold higher in subcutaneous adipose tissue compared to visceral adipose tissue (Baljzová et al., 2008). No differences in RBP4 mRNA levels were reported between the non-obese and obese groups.
III. Vitamin A and Metabolic Disease.
As discussed earlier in the text, vitamin A is required for mediating many physiologically important processes in the body, involving multiple genes and signal transduction pathways. Moreover, some of the processes for metabolism and storage of vitamin A overlap with lipid-related parameters (related to cholesterol and triglyceride metabolism) that when dysregulated contribute to disease progression. Consequently, it is not too surprising that the literature has implicated vitamin A and/or vitamin A-related parameters (vitamin A-binding proteins and enzymes involved in vitamin A metabolism) to have a role, or actually multiple different roles, in metabolic disease development. However, at present, there is little consensus in this literature regarding how vitamin A causes or prevents metabolic disease development. In fact, much of this literature is controversial and/or contradictory.
The remainder of this review will focus on the role of vitamin A and vitamin A-related parameters in early stage liver disease, obesity, and some aspects of insulin resistance. These diseases are interrelated so it is not possible to consider one disease without referring to others. The literature on these topics has focused primarily on the actions of vitamin A signaling per se, RBP4, ALDH1A1, and other vitamin A-related proteins in disease causation with a smaller focus on other vitamin A-related parameters. The text below reflects this focus.
It should be noted that it is often assumed by investigators and readers that the actions of many of the vitamin A-related proteins proposed to be involved in metabolic disease, like RBP4 and ALDH1A1, involves effects of the protein on vitamin A levels and signaling. Thus, for instance, changes in RBP4 levels that would affect vitamin A delivery to tissues are often assumed to affect vitamin A actions in these tissues. But most studies relating RBP4 levels to metabolic disease do not report levels of retinol, retinyl esters, or retinoic acid. There needs to be some caution in how data obtained from investigations of vitamin A-related proteins and their relationship to metabolic disease are interpreted. When interpreting data, one needs to be open minded to the possibility that these proteins may have actions in metabolic disease that are independent of their roles in vitamin A physiology.
Finally, it should be noted that each investigatory approach has its inherent limitations that qualifies and limits data interpretation. Retrospective human studies are generally considered to be less compelling than prospective ones. Animal studies involving different diets and/or genetically manipulated mice can only be interpreted in the context of the diets and/or animals studied. There may be gender-based differences in findings that may be missed when comparing results from different studies. Cell culture studies provide mechanistic insights into molecular processes but are less informative regarding whole body physiology. The biochemical approaches employed to measure gene expression levels are also fraught with potential issues relevant for interpreting data. Most frequently, mRNA levels are reported as a measure of gene expression while protein expression may be ignored. With regards to vitamin A related gene expression, it is well established that RBP4 mRNA and protein levels are well correlated but mRNA levels often do not correlate well with LRAT protein levels. It is also the experience of this reviewer that hepatic Cyp26a1 and Cyp26b1 mRNA levels do not always correlate well with corresponding protein levels. Thus, one must be cautious in how one evaluates the strength of a particular study and its significance. It is best when multiple different investigatory approaches are brought to bear on a research question, since only then can a full understanding of how metabolic disease may be related to vitamin A and vitamin A-related parameters.
A. Vitamin A, Vitamin A-Related Proteins and Liver disease.
The literature is convincing that impairments in vitamin A signaling and metabolism can contribute to nonalcoholic fatty liver disease (NAFLD) and steatohepatitis (NASH), as well as later stage liver disease (fibrosis, cirrhosis, and hepatocellular carcinoma (HCC)). But the underlying mechanisms responsible for this remain to be completely established.
NAFLD is an umbrella term that describes a variety of conditions characterized by an excessive accumulation of fat within hepatocytes and is a major cause of later stage liver disease (Silverman et al., 1990). NAFLD is present in approximately 90% of adults who are morbidly obese where the disease can range from mild hepatic steatosis to the more severe NASH. If not resolved, these early disease stages can progressively worsen to fibrosis, cirrhosis and HCC (Sliverman et al., 1990). Because, unlike later stage liver disease, NAFLD and NASH are reversible, there is now considerable research activity focused on understanding the pathobiology of NAFLD and NASH and how these conditions can be prevented and reversed. Many patients with NAFLD display other metabolic complications including insulin resistance, muscle atrophy, and cardiovascular disease that can contribute to the morbidity and mortality of liver disease (Fotbolcu & Zorlu, 2016).
Since both hepatocytes and HSCs play key roles in hepatic vitamin A metabolism and storage, it should be noted that the early stages of liver disease usually involve insults to hepatocytes (Tsuchida & Friedman, 2017). This is true for both NASH and viral infection. Mediators released by injured hepatocytes promote HSC activation facilitating later stage hepatic disease.
1. Vitamin A per se.
Perhaps the most convincing work demonstrating a direct role for vitamin A in hepatic disease was provided by Shiota and colleagues (Yanagitani et al., 2004). These investigators systematically investigated the importance of retinoic acid-mediated transcription in the liver using a mouse model that expresses a well characterized dominant negative RARα transgene specifically in hepatocytes. This dominant-negative transgene is known to diminish but not completely ablate all RAR-mediated retinoic acid signaling in tissues (inhibiting RARα, -β, and -γ signaling). By 4-months-of-age, chow diet fed male transgenic mice displayed NASH that was not seen in matched wild type controls. This was accompanied by decreased expression of genes involved in mitochondrial β-oxidation, suggesting a role for retinoic acid in regulating hepatic mitochondrial fatty acid β-oxidation. Feeding of high levels of retinoic acid significantly decreased steatosis in the transgenic animals, further confirming a role of retinoic acid in the disease process. By 12-months-of-age, 50% of the transgenic mice had developed HCC compared to none of the matched wild type mice. These data convincingly establish that dysregulated retinoic acid-mediated transcriptional activation contributes to the development of progressively worsening liver disease.
Subsequent published reports have agreed with and extended the findings of Yanagitani et al. (Yanagitani et al., 2004) regarding a role for vitamin A and RARs in modulating mitochondrial β-oxidation in hepatocytes. Studies carried out in human HepG2 hepatocytes showed that treatment of the cells with all-trans-retinoic acid resulted in an upregulation of expression of genes encoding carnitine palmitoyl transferase-1, a key protein involved in mitochondrial β-oxidation (Amengual et al., 2012). Other investigators using both cultured HepG2 cells and male mice treated with adenoviral expression vectors encoding either fibroblast growth factor 21 (FGF21) or RARβ, were able to demonstrate that the Fgf21 gene is a target gene for RARβ. Upregulation of Fgf21 expression by all-trans-retinoic acid mediated by RARβ was found to enhance hepatic fatty acid oxidation and ketogenesis and to increase whole body energy expenditure in the male mice (Li et al., 2013). Trasino et al. (2016a), studying both high fat diet-induced obesity in male mice and genetically obese ob/ob and db/db mice, found that oral treatment of the animals with synthetic retinoid RARβ2 agonists reduced hepatic fat accumulation and increased mRNA levels for genes involved in β-oxidation and lessened expression levels for genes encoding proteins important for de novo lipogenesis. Other published studies by these same investigators showed that oral treatment of wild type male mice fed a high fat diet for 4 months with a highly selective synthetic RARβ2 agonist markedly reduced expression of a marker for HSC activation, α-smooth muscle actin (Tarasino et al., 2016b). Collectively, these published data argue for roles for retinoic acid and RARs, especially RARβ, in regulating expression of genes important for hepatic fat metabolism. All of these authors suggested that vitamin A or synthetic retinoids might prove to be useful therapeutic agents for application in liver disease.
Trasino et al. (2015) found that hepatic retinol, retinyl palmitate, and RARβ2 mRNA levels in human patients with NAFLD show a strong inverse correlation with the severity of the disease. Similar findings were reported from study of obese male mice generated by feeding a high fat diet for 12 weeks. The authors took this to indicate that overnutrition and obesity can give rise to very diminished tissue vitamin A levels that may have unappreciated effects on human health in obese individuals (Trasino et al., 2015a). This finding certainly merits confirmation and further investigation.
ALDH1A1 actions in hepatic steatosis have been studied by Kiefer et al. (2012a) in chow fed weight-matched female Aaldh1a1-deficient and wild type mice, and in primary hepatocytes isolated from the mice. These authors report that Aldh1a1-deficiency results in downregulated hepatic glucose production and repressed hepatic triglyceride production, where serum triglyceride levels are reduced by 28% compared to wild type mice. Treatment of primary hepatocytes isolated from Aldh1a1-deficient mice with all-trans-retinoic acid, but not treatment with all-trans-retinaldehyde, resulted in an induction of the gluconeogenic enzymes phosphoenolpyruvate carboxykinase and glucose 6-phosphatase. The authors’ data point to the conclusion that ALDH1A1 plays an important role in regulating hepatic glucose and triglyceride production. However, this conclusion, at face value, appears to contradict the findings of in vivo studies discussed above. Since ALDH1A1 absence would downregulate or diminish retinoic acid synthesis and the Aldh1a1-deficient mice show a better metabolic phenotype than that of matched wild type mice, this is in apparent contradiction of the studies concluding that retinoic acid availability blocks hepatic disease development. Further studies are needed to establish whether these apparently contradictory findings can be reconciled.
2. RBP4 and liver disease.
There is a considerable literature exploring possible relationships between circulating RBP4 levels and adipose tissue RBP4 expression and the development of NAFLD. This research interest has grown out of the proposal that elevated adipose tissue expression of RBP4 and the resultant elevation in serum RBP4 levels is associated with insulin resistance and other components of the metabolic syndrome, including NAFLD (Yang et al., 2005; Graham et al., 2006). This literature includes both observational studies in humans and mechanistic studies carried out in mouse models and cultured cells.
a. Studies in Humans.
Human observational studies exploring relationships between serum/plasma RBP4 and NAFLD occurrence are largely in agreement with the conclusion that serum/plasma RBP4 levels are elevated in patients with NAFLD. A representative sample of, but not all correlative studies, include ones involving patients with type II diabetes (Wu et al., 2008), obese children (Romanowska et al., 2011), healthy 6- to 12-year-old children (Huang & Yang, 2013), subjects diagnosed with NAFLD but not type II diabetes (Yan et al., 2013), obese and lean patients diagnosed with NAFLD (Zwolak et al., 2016), postmenopausal women diagnosed with NALFD (Cai et al., 2018) and a community-based cross-sectional study involving 2938 participants 40-70-year-olds (Chen et al., 2017). Each of these studies reported significantly elevated serum/plasma RBP4 concentrations for patients with NAFLD but provided little insight into possible cause/effect relationships.
Contrary to the positive associations reported for the human studies considered above, a meta-analysis carried out to determine whether NAFLD or NASH patients have altered blood RBP4 levels was unable to identify such a relationship (Zhou et al., 2017). The meta-analysis involved published data from 12 studies comprising 4247 participants including 2271 NAFLD patients and 1976 controls. The authors concluded that circulating RBP4 levels may not be associated with NAFLD.
b. Molecular studies.
The molecular basis for how RBP4 contributes to liver disease development has been investigated in both cells in culture and in mouse models. Tan et al. (2011) reported the generation of a RNA oligonucleotide (oligo) that was able to knockdown expression of mouse Rbp4 in 3T3-L1 adipocytes. This anti-Rbp4 oligo was then used in male mice fed a high fat diet fed for 10 weeks to establish the effects of RBP4 knockdown on metabolic disease development. After 10 weeks of feeding of the diet, mice were separated into 4 groups that received either oral doses of the PPARγ agonist rosiglitazone, a scrambled control oligo, or the anti-RBP4 oligo, and followed for an additional 4 weeks with high fat diet feeding. The anti-Rbp4 oligo treated group was found to have RBP4 protein levels in liver that were approximately 60% of controls, accompanied by visceral adipose tissue levels that were approximately 33% of controls. Rosiglitazone diminished Rbp4 expression in visceral adipose tissue by approximately 30% but had no effect on hepatic RBP4 protein levels. Mice receiving either rosiglitazone or anti-Rbp4 oligo were found to have significantly improved responses to intraperitoneal (IP) glucose and insulin tolerance tests. Both the grade of hepatic steatosis and hepatic triglyceride levels were significantly reduced in the anti-Rbp4 treatment group, whereas rosiglitazone had no effect on hepatic steatosis. Thus, the combined knockdown of RBP4 expression in liver and visceral adipose lessened high fat diet induced hepatic triglyceride accumulation. This provides direct evidence for a role of elevated RBP4 levels/expression in early stage liver disease.
Studies carried out in human HepG2 hepatocytes and in male wild type and peroxisome proliferator-activated receptor-γ coactivator 1β (Pgc-1β)-deficient mice led to the proposal that RBP4-induced hepatic lipid accumulation involves RBP4 effects mediated by sterol regulatory element-binding protein (SREBP-1) (Xia et al., 2013). When cultured HepG2 cells or primary hepatocytes isolated from wild type mice were treated with high doses of recombinant human RBP4 this increased hepatocyte triglyceride levels and was associated with increased de novo lipogenesis. This was also accompanied by an increase in SREBP-1 activation, as well as increased expression of SREBP-1 target genes. When recombinant RBP4 was infused directly into the circulations of wild type mice, an induction of hepatic SREBP-1c mRNA as well as hepatic and serum triglyceride levels was observed. To explain their data, Xia et al. (2013) proposed that elevated levels of circulating RBP4 induce SREBP1 upregulating triglyceride synthesis and accumulation within hepatocytes. A limitation for understanding this study is that the doses of RBP4 used in these in vitro and in vivo studies were very large, well above those which would ever be found in the circulations of obese NAFLD patients. This presents a caveat for understanding the report from Xia et al. (2013) and renders the interpretation of the findings somewhat equivocal.
Using a newly generated transgenic mouse model that expresses low levels of human RBP4 in visceral, sub-cutaneous and brown adipose tissue, Lee et al. (2016) reported that an inflammatory response within adipose tissue is largely responsible for the elevated triglyceride accumulation observed in the livers of male transgenic mice. Although circulating levels of total (mouse + human) RBP4 were only elevated by approximately 10% over those of matched control mice, transgenic mice fed a chow diet displayed significantly higher levels of hepatic triglycerides as well as adipose tissue inflammation compared to controls. This elevation was associated with increased lipolysis and mobilization of fatty acids from adipose tissue and increased circulating unesterified fatty acid levels. Lee et al. (2016) also presented evidence for increased uptake of circulating unesterified fatty acids by the liver. These authors however were not able to obtain evidence for decreased fatty acid oxidation, fatty acid export or de novo lipogenesis within the livers of the transgenic mice. Importantly, there was no evidence for differences in serum, hepatic or adipose tissue levels of retinol, retinyl esters or all-trans-retinoic acid between matched chow fed control and transgenic mice. These data were taken to indicate that inflammatory events occurring within adipose tissue, resulting in increased triglyceride hydrolysis and nonesterified fatty acid release from adipocytes, account for the hepatic phenotype of the mice rather than some RBP4-dependent process occurring directly within the liver.
A number of proteins proposed to have direct roles in catalyzing hepatic vitamin A metabolism have been proposed to be associated with NAFLD. One of these, patatin-like phospholipase domain-containing 3 (PNPLA3), shows a strong linkage to NAFLD (Dongiovanni et al., 2011; Anstee & Day, 2015). PNPLA3 is expressed in the liver, especially in HSCs, where it is thought to be involved in lipid metabolism. The I148M protein variant of PNPLA3 has been identified to be a major determinant of liver fat content. This form of PNPLA3 is proposed to be a modifier of disease outcome across the full spectrum of liver disease, from NAFLD to advanced fibrosis and HCC. PNPLA3 has been reported in the literature to be a retinyl ester hydrolase responsible for the hydrolysis HSC retinyl ester stores (Pirazzi et al., 2014). Patients expressing the I148M form of PNPLA3 are reported to display lower circulating levels of both RBP4 and retinol. (Mondual et al., 2015). Although it is very provocative to propose PNPLA3 involvement in vitamin A-mediated liver disease, it has not yet been convincingly established that PNPLA3 is a physiologically relevant enzyme for catalyzing retinyl ester hydrolysis and retinol mobilization from the liver. Aside from Pirazzi et al. (2014) and Mondual et al. (2015) there is little published information on the retinyl ester hydrolase activity of PNPLA3. Moreover, there are at least 4 other candidate enzymes that have repeatedly been proposed to be physiologically significant hepatic retinyl ester hydrolases (Blaner & Li, 2015). The role of PNPLA3 in hepatic vitamin A metabolism needs to be definitively established before this proposed linkage between PNPLA3, vitamin A, and NAFLD can be accepted.
B. Vitamin A, Vitamin A-Related Proteins and Adipose Biology and Adipose-Related Disease.
Both white adipose tissue (WAT) and brown adipose tissue (BAT) play central roles in whole body energy expenditure (Villarroya et al., 2017a). The predominate cell type present in WAT, the white adipocyte, contains a single triglyceride-rich lipid droplet that occupies most of the cell volume and is responsible for storing fat for use when food is unavailable. BAT, in contrast, consists of brown adipocytes that contain multiple lipid droplets and are very enriched in mitochondrial content. The brown adipocytes oxidize metabolic substrates to produce heat and are consequently important for non-shivering thermogenesis. This activity is accomplished by an uncoupling of the respiratory chain and oxidative phosphorylation in brown adipocyte mitochondria arising from the actions of uncoupling protein-1 (UCP1). Vitamin A and retinoic acid are proposed to be important for maintaining normal WAT and BAT physiology. Moreover, there is growing evidence that retinoic acid may be able to contribute to the “browning” of WAT resulting in a beneficial effect for preventing excessive triglyceride accrual. These and other adipose-related topics will be considered in more depth below.
1. Retinoic acid effects on adipocyte differentiation.
It has long been known that all-trans-retinoic acid treatment of cells that are able to be differentiated in culture from preadiocytes to adipocytes blocks differentiation. Studying 3T3-F442A preadipocytes cultured on plastic plates, Kuri-Harcuch (1982) demonstrate that treatment of the cells with either 1.0 or 10 μM all-trans-retinoic acid blocked formation of adipocytes, but had little effect on proliferation of the preadipocytes. Subsequent studies by Kuri-Harcuch and colleagues of 3T3-F442A cell differentiation (Castro-Muñozledo et al., 1987) led these investigators to propose that retinoic acid was acting to affect lipogenic enzyme expression in a non-specific manner that involves changes in cytoskeletal structure.
Stone & Bernlohr (1990), studying the conversion of 3T3-L1 cells to adipocytes, reported that treatment of these cells with 1.0 μM all-trans-retinoic acid failed to block proliferation but markedly inhibited the early adipocyte differentiation program. Inhibition of adipogenesis was associated with marked alterations in expression levels of c-Jun and Jun-B mRNAs. These authors concluded that retinoic acid must be acting in some way that affects regulation of gene expression by transcription factors. Other studies exploring the effect of retinoic acid in blocking differentiation of 3T3-L1 cells led Kamei et al. (1994) to propose that RARα signaling was involved (Kamei et al., 1994). Thus, based on this early understanding, retinoic acid acting through RARα was proposed to block adipogenesis in its early stages, before the differentiating cells can accumulate substantial amounts of fat.
Studies of 3T3-L1 cells by Lazar and colleagues established that the treatment of these cells with all-trans-retinoic acid at the time the cells are stimulated to undergo adipogenesis completely blocks adipocyte differentiation, blocking induction of peroxisome proliferator-activated receptor γ (PPARγ), a key transcription factor needed for fat accrual in adipocytes (Schwarz et al., 1997). These investigators showed the retinoic acid liganded RARs block the transcription of both CCAAT/enhancer-binding protein-α (C/EBPα) and CCAAT/enhance-binding protein-β (C/EBPβ). Their data suggested that retinoic acid-induced blockage of adipocyte differentiation likely involves inhibition of C/EBPα-dependent gene transcription. The downregulation of C/EBPα-mediated transcription, which is required for adipogenesis, and the accompanying downregulation of PPARγ are now generally accepted as the primary molecular events underlying the inhibitory effects of retinoic acid treatment on adipogenesis.
The literature considering the molecular actions of retinoic acid in blocking 3T3-L1 adipocyte differentiation is quite extensive and will be summarized briefly here. A number of intracellular signaling pathways have been implicated to have roles in mediating retinoic acid blockage of adipogenesis. Action by the transcription factor Smad3 is reported to be needed for the inhibition of adipogenesis by retinoic acid (Marchildon et al., 2010). These authors show that retinoic acid acts to specifically stimulate the expression of Smad3, its nuclear accumulation, and its transcriptional activity. Activated Smad3 is proposed to interact with C/EBPα interfering with the binding of C/EBPα to DNA affecting adipogenesis. Retinoic acid treatment of 3T3-L1 cells also has been shown to enhance the transcriptional activity of β-catenin, as well as Wnt gene expression, during adipocyte differentiation (Kim et al., 2013). This effect of retinoic acid is also proposed to help facilitate the blockage of adipocyte differentiation. Another report established that all-trans-retinoic acid treatment of differentiating 3T3-L1 cells results in suppressed upregulation of the amino acid transporter ASCT2, in a manner that is dependent on the dose of retinoic acid (Takahashi et al., 2015). These authors also showed that inhibition of ASCT2 with other unrelated inhibitors resulted in inhibition of adipogenesis. Takahashi et al. (2015) suggested retinoic acid actions to block ASCT2 are important for mediating its effects on adipogenesis. There is also recent evidence obtained from both studies of cells in culture and male mice that retinoic acid blocks adipogenesis through inhibition of expression of the transcription factor zinc finger protein 423 (Zfp423), a transcription factor needed for maintain white adipose identity, by blocking DNA demethylation in the promoter of the Zfp423 gene (Wang et al., 2017a). The significance of each of these proposed actions of retinoic acid in blocking adipocyte differentiation requires further investigation and confirmation.
Others have confirmed that all-trans-retinoic acid suppression of 3T3-L1 adipocyte differentiation involves a mechanism the reduces C/EBPα, PPARγ, and PPARγ target gene expression (Wang et al., 2014). However, these authors extend understanding of how this takes place by showing that PPARγ transcriptional regulation is dampened by the interaction of RARγ, and not RARα, with C-Fos protein to specifically inhibit transcriptional regulation by PPARγ2, leading to inhibition of adipocyte differentiation.
Recent literature suggests retinoic acid actions on 3T3-L1 preadipocyte differentiation may be different when the cells are cultured in low versus high glucose containing medium (Eldaim et al., 2017). These investigators propose that retinoic acid effects in blocking 3T3-L1 adipose conversion depend on the medium glucose concentration. When cells were cultured in 5.5 mM glucose and treated with 1 μM all-trans-retinoic acid, a blockage in adipogenesis was observed. However, when cultured in 25 mM glucose and treated with 1 μM all-trans-retinoic acid, the treatment was reported to increase triglyceride accumulate in the cells. This was attributed to a glucose concentration-dependent upregulation in SREBP-1 expression. Although intriguing, this finding needs to be confirmed and assessed in more depth.
All-trans-retinoic acid treatment is reported also to affect gene expression in mature 3T3-L1 adipocytes (8 days after induction of differentiation) giving rise to diminished triglyceride accumulation in the cells (Mercader et al., 2007). This was accompanied by statistically significant changes in mRNA levels for a number of genes including those encoding RXRα and C/EBPα, but not PPARγ. The authors concluded that all-trans-retinoic acid treatment of 3T3-L1 adipocytes promotes a remodeling of mature adipocytes towards increased capacity for oxidative metabolism and reduced capacity for lipogenesis.
Noy and colleagues, in a series of publications, proposed that all-trans-retinoic acid acting transcriptionally through PPARβ/δ modulates adipocyte differentiation and responses and whole body adiposity (Berry and Noy, 2009; Berry et al., 2010; Berry et al., 2013; Noy, 2013; Noy, 2016a). These authors proposed an explanation for why all-trans-retinoic acid can block preadipocyte differentiation into adipocytes but yet be needed for mediating retinoic acid-dependent transcriptional signaling in mature adipocytes. Using cultured mature 3T3-L1 adipocytes and male mice fed a high fat- and high carbohydrate-containing, Berry and Noy (2009) were able to show that adipogenesis was accompanied by a downregulation of RAR signaling and an upregulation of PPARβ/δ signaling. Noy and colleagues proposed that all-trans-retinoic acid is able to signal through both RARs and PPARβ/δ to induce expression of multiple genes involved in regulation of energy homeostasis and insulin responses. Administration of all-trans-retinoic acid to obese mice from subcutaneously implanted slow release pellets was found to lead to a loss of fat mass and improved insulin tolerance. These effects could be traced to activation of both RAR- and PPARβ/δ-mediated gene expression. Studies involving 3T3-L1 cells and human preadipocytes established that a downregulation of Crabp2, which is highly expressed in preadipocytes, at the onset of adipocyte differentiation is critical for allowing adipocyte differentiation (Berry et al., 2010). Importantly, onset of adipocyte differentiation was accompanied by a marked upregulation of Fabp5. Diminished ability of retinoic acid to activate RAR signaling following induction of differentiation was proposed to arise from the downregulation of Crabp2 expression. The consequent increase in intracellular FABP5/CRABP2 ratios was proposed to shift retinoic acid signaling to the PPARβ/δ pathway. Thus, CRABP2 sensitizes preadiocytes to retinoic acid-induced inhibition of differentiation. The downregulation of CRABP2 sets the stage for proper retinoic acid-signaling in mature adipocytes and the failure of all-trans-retinoic acid to block differentiation at later stages of adipogenesis (Berry et al., 2010). Subsequent studies confirmed that administration of all-trans-retinoic acid from subcutaneously implanted pellets in male mice fed a high fat high carbohydrate diet markedly reduced body weight gain and obesity development and upregulated a number of PPARβ/δ-responsive genes (Berry et al., 2012a). Noy and colleagues further identified changes in expression of a number of genes that they proposed were important for mediating the retinoic acid-induced block in adipogenesis. Finally, employing heterozygous Crabp2-deficient mice, these investigators were able to establish that decreased retinoic acid signaling through the CRABP2/RAR pathway promotes diet-induced adipogenesis and obesity in vivo (Berry et al., 2012a). When considered collectively, these data provide strong evidence to support the proposed involvement of CRABP2 and PPARβ/δ in adipocyte differentiation.
Similar findings to those reported regarding retinoic acid actions in murine adipocyte differentiation have been obtained for human preadipocytes. Investigations of human adipocyte stem cells establish that all-trans-retinoic acid also inhibits human adipocyte differentiation at early stages but not at later stages of the differentiation program (Takeda et al., 2016). This effect of retinoic acid was especially pronounced for differentiation of visceral adipocytes. Importantly, Takeda et al. (2016) showed that many genes involved in vitamin A and retinoic acid metabolism were differentially expressed in adipose stem cells obtained from subcutaneous and visceral fat. Endogenous levels of all-trans-retinoic acid were higher in visceral adipose stem cells and this was associated with an upregulation of genes involved in retinoic acid synthesis.
Other studies exploring retinoic acid effects on human adipose-derived stem cells increasing the frequency of Sub-G1 cells and decreased it is G1, establishing where in the cell cycle retinoic acid acts (de Carvalho Schweich et al., 2017). Gene expression data obtained for treated cells suggested that retinoic acid treatment leads to mitochondrial membrane permeabilization and the consequent release of proapoptotic factors including BAK and BAX. The authors took these data to suggest that retinoic acid not only interferes with adipocyte differentiation during its early stages but also facilitates apoptosis of stem cells, preadipocytes and adipocytes.
The older literature reports that some proteins can become post-translationally modified through the covalent addition of a retinoic acid molecule to the protein, see [(Brun et al., 2013)] for review. This process is referred to as protein retinoylation. This action of retinoic acid has been proposed by Dave et al. (2014) to effect adipocyte differentiation. These authors report that when 3T3-L1 adipocytes are treated with either 0.1 or 1.0 μM all-trans-retinoic acid they are able to detect retinoylation of the protein exportin, a protein that regulates nuclear export to the cytoplasm. This modification was associated with disrupted export of mitogen-activated protein kinase 1 from the nucleus resulting in a sequestration of PPARγ and a blockage of the adipocyte differentiation program. The authors further report that retinoic acid binding to RARs will not account for their findings (Dave et al., 2014).
There is also literature regarding the actions of retinoic acid in mediating the commitment of mouse embryonic stem (ES) cells into the adipocyte lineage. Retinoic acid is reported to be necessary if ES cells are to become committed to the adipocyte lineage (Bost et al., 2002). These authors report that retinoic acid treatment of mouse ES cells results in a prolonged activation of the ERK 1/2 pathway but not the c-Jun N-terminal kinase (JNK), p38 mitogen-activated protein kinase (p38MAPK), or phosphoinositide 3-kinase pathways. Based on their data, Bost et al. (2002) maintain that retinoic acid acting through the ERK 1/2 pathway is required at this very early stage of preadipocyte formation. Thus, somewhat paradoxically, retinoic acid is needed both to ensure ES commitment to the adipocyte lineage but also to block preadipocyte differentiation to adipocytes.
Recent studies reported by Yang et al. (2018) are consistent with this early literature (Bost et al., 2002) regarding the effects of retinoic acid on mouse ES cell commitment to the adipocyte lineage. Yang et al. (2018) report that modest decreases in endogenous all-trans-retinoic acid concentrations in male and female mice heterozygous for the Rdh10 gene were associated with metabolic abnormalities including effects on glucose and insulin sensitivity and adipogenesis. In vitro investigations involving the use of mouse embryonic fibroblasts (MEFs) identified all-trans-retinoic acid levels to be diminished by approximately 50% in cells heterozygous for the Rdh10-null allele. Adult heterozygous Rdh10-null mice were found to be more sensitive and to display a more severe phenotype of both diet-induced obesity and insulin resistance than wild type mice. Male heterozygotes Rdh10-null mice were reported to be more sensitive to developing high fat diet induced hepatic steatosis. These data from Yang et al. are fully consistent with the hypothesis that Rdh10-derived all-trans-retinoic acid is needed to maintain a normal state of adipocyte differentiation and adipose tissue accrual and that this is important for maintaining a healthy metabolic phenotype.
Interestingly, Liu et al. (2016) reported an association between circulating retinoic acid levels and the development of metabolic syndrome for measurements using sera from 1042 nondiabetic 50-70-year-old adult participants in the population-based Nutrition and Health of Aging Population in South China study. The authors report that serum retinoic acid levels are inversely associated with the development of diseases that compose the metabolic syndrome, including central obesity, dyslipidemia, hyperglycemia, insulin resistance and hypertension. These novel data were taken to suggest that retinoic acid is needed to prevent or slow the development of the metabolic syndrome. However, there are some technical ambiguities with this study. Retinoic acid levels in serum were measured using a commercial ELISA kit, a method that is not often used in published studies. Is not clear whether the ELISA measures only all-trans-retinoic acid or total retinoic acid levels which may include 13-cis-retinoic acid and oxidized retinoic acid metabolites. It has been persuasively argued that only liquid chromatography tandem mass spectrometry-based protocols are valid for measuring retinoic acid concentrations in tissues (Kane et al., 2008b). This is a widely accepted view.
2. Retinoic acid and uncoupling protein-1 (UCP1).
Although it has long been understood that BAT plays an important role in thermogenesis and energy utilization, until recently, it was thought that BAT is not present in adult humans. However, this view markedly changed in 2009 with the simultaneous publication of two reports showing that BAT is indeed present at significant levels in healthy human adults and that BAT activities are reduced in overweight and obese humans (Cypress et al., 2009; van Marken Lichtenbelt et al., 2009). These reports immediately implicated BAT actions as being key for regulating human body weight. There is now very substantial research interest in BAT activity focusing primarily on how the loss or acquisition of BAT activities affects the development of obesity and related metabolic disease. One component of this research is focused on UCP1 expression and how this may be manipulated to benefit metabolic disease prevention.
UCP1 is the key protein needed for uncoupling mitochondrial respiration resulting in lessened energy production and fat accumulation in WAT. UCP1 ablation in mice induces obesity and alters the thermogenic capacity of the animals (Feldmann et al., 2009). The transcriptional regulation of UCP1 expression is complex. The literature indicates that this can involve a myriad of transcription factor actions including those of C/EBPα and –β, PPARβ and –γ, RARs, RXRs, the thyroid hormone receptor, the cAMP response element-binding protein and zinc finger protein-516 (Villarroya et al., 2017b). Also importantly involved in UCP1 transcriptional regulation is the transcriptional coregulator PGC-1α. Published evidence supporting a role for retinoic acid and RAR-mediated expression of UCP1 in BAT will be considered below.
Early work by Alvarez et al. (1995) showed that treatment of differentiating mouse brown adipocytes in culture with all-trans-retinoic acid increased Ucpl mRNA levels by several-fold. The effect of retinoic acid was reported to display both dose- and time-dependence. Through the use of electrophoretic mobility shift assays, a retinoic acid response element was identified in the −2357/−2330 upstream region of the rat Ucpl gene (Alvarez et al., 1995) Another early report by Puigserver et al. (1996) demonstrated that treatment of primary mouse BAT precursor cells or H1B 1B brown adipocyte cells with either all-trans- or 9-cis-retinoic acid for 7 days resulted in fewer cells that had a more fibroblast-like appearance than control non-treated cells. This observation appears to be consistent with the notion that retinoic acid blocks to some extent brown adipocyte differentiation. However, treated brown adipocytes were found to express higher levels of Ucp1 mRNA and protein. The effectiveness of retinoic acid as a Ucp1 inducer was reported to be dependent on the stage of brown adipocyte differentiation, being maximal in confluent primary brown adipocytes. Treatment of mice with all-trans-retinoic acid for 4 days also was reported to result in increased BAT UCP1 content in vivo.
Treatment of rats with all-trans-retinoic acid has been reported to increase BAT Ucp1 mRNA expression within 5 hours after administration of retinoic acid (Kumar & Scarpase, 1998). In contrast, WAT leptin mRNA levels were reported to be significantly reduced by the same treatment. Follow-up studies by these investigators involving feeding of a vitamin A-supplemented diet, one supplemented with approximately 10-times the amount of vitamin A present in the control diet to male rats for 8 weeks, resulted in a statistically significant 31% increase in BAT Ucp1 mRNA levels (Kumar et al., 1999). Although the dietary level of vitamin A being studied was relatively high, this was probably not a toxic level given the 8-week duration of the study. This finding was taken to suggest that dietary vitamin A consumption can be effective for inducing Ucp1 in BAT.
The involvement of RARs and RXRs in Ucp1 induction was studied by Alvarez et al. (2000). Using receptor-type specific agonists and primary cultures of mature murine brown adipocytes, these authors were able to show that treatment of the brown adipocytes increased Ucp1 mRNA levels by up to approximately 8-fold. Moreover, using transactivation assays, Alvarez et al. (2000) demonstrated that these actions involved upstream promoter elements present in the Ucp1 gene. A somewhat contrary finding was reported by Ribot et al. (2004), who reported that all-trans-retinoic acid (100 mg/kg body weight for 4 days) treatment of mice maintained on a vitamin A-deficient diet resulted in a reduction in RARα expression in BAT and that this was accompanied by an induction of Ucp1 expression. Another report reaching a contrary conclusion involved the study of fetal brown adipocytes obtained from 20-day-old rat fetuses treated with either the PPARγ agonist rosiglitazone, or all-trans-retinoic acid, or 9-cis-retinoic acid has implicated p38MAPK and not direct RAR-mediated transcriptional activation as having a role in retinoic acid-induced Ucp1 expression (Teruel et al., 2003). Collectively, these published reports all support the conclusion that retinoic acid treatment of brown adipocytes results in an induction of Ucp1 expression. However, diverse mechanisms are proposed to account for this.
When MEFs prepared from 14-day-old mouse embryos were differentiated into white adipocytes using standard protocols and then exposed to all-trans-retinoic acid, the treated cells were found to express Ucp1 mRNA and protein (Mercader et al., 2010). The induction of Ucp1 expression levels showed a direct dose-dependence on all-trans-retinoic acid concentration. Induction of Ucp1 expression could be reproduced upon treatment with either RAR agonists or retinaldehyde and was reported to require p38MAPK activity. The authors proposed that their data underscore the potential use of vitamin A to induce Ucp1 gene expression in adipocytes.
Later studies, however, raise a question as to whether the inductive effect of retinoic acid on Ucp1 expression may be limited to rodents (Murholm et al., 2013). Carrying out studies in hMADS cells, a human brown adipocyte line, in primary human adipocytes, and in a number of different mouse cells including C3H10T1/2 mesenchymal stem cells, 3T3-L1 cells, and day 14 MEFs, these authors reported that all-trans-retinoic acid treatment as well as treatments with a number of synthetic RAR agonists and antagonists failed to induce UCP1 expression in human brown adipocytes. These same treatments were effective in inducing Ucp1 expression in murine cells. Data from experiments employing RAR agonists and antagonists showed that the effect of retinoic acid on Ucp1 gene expression is mediated by RARs. Murholm et al. (2013) were unable to identify a role for PPARβ/δ in regulating Ucp1 gene expression, nor were they able to establish a role for PGC-1α.
A potential complicating observation for understanding the actions of retinoic acid on UCP1 activity comes from a report that all-trans-retinoic acid can bind with high affinity to purified hamster UCP1 protein (Tomas et al., 2002). If binding between UCP1 protein and retinoic acid can occur in vivo, and if this holds for UCP1 protein species present in other species, this may have an impact on how we understand retinoic acid actions in BAT. As far as the author is aware, no other evidence exists to support this finding regarding direct retinoic acid binding to UCP1 or its physiological significance.
3. Retinoic acid and the browning of white adipose tissue.
As suggested above, there is now considerable research interest in the conversion of white adipocytes into ones with brown adipocyte-like character (Villarroya et al., 2017a). Brown adipocyte-like cells, referred to as beige or brite adipocytes, appear in WAT depots in the mammalian body under conditions of thermogenic activity. This process is commonly referred to as browning (or beiging) of WAT and involves induction of expression of UCP1 in beige adipose tissue. There are obvious therapeutic benefits that may be gained from the browning of WAT.
Treatments of male mice with relatively large doses of all-trans-retinoic acid (10, 50 and 100 mg/kg body weight), given by subcutaneous injection over 4 successive days, were found to give rise to a marked upregulation of Ucp1, Ppara, and Pgc1a mRNA expression in epididymal and retroperitoneal WAT depots but had little or no effect on expression in inguinal WAT (Mercader et al., 2006). However, UCP1 protein levels were found to be upregulated by approximately 2-fold in inguinal WAT when the mice were maintained for one week at thermoneutrality (30 °C) and treated with all-trans-retinoic acid (50 mg/kg body weight) for the final 4 days prior to sacrifice. Treatment was also reported to increase the number of multilocular adipocytes present in the inguinal WAT for mice maintained at thermoneutrality. In a subsequent report, these investigators showed that the all-trans-retinoic acid significantly reduced RBP4 mRNA and protein expression in epididymal, inguinal, and retroperiotoneal WAT and in BAT but not in liver from treated mice (Mercader et al., 2008).
All-trans-retinoic acid treatment of male mice, administered at a dose of 50 mg/kg body weight by subcutaneous injection over 4 days immediately prior to sacrifice, is reported to increase the number of mitochondria in WAT (Tourniaire et al., 2015). This increase was accompanied by increased expression of genes associated with mitochondrial oxidative metabolism as well as increased expression of genes that are markers for beige/brite adipocytes. Other studies have been reported that involve administration of a smaller dose of all-trans-retinoic acid (5 mg/kg body weight), by gavage for 4 days immediately prior to sacrifice, to male mice that on the day of sacrifice received an IP injection of E. coli lipopolysaccharide (LPS) to induce inflammation (Karkeni et al., 2017). Retinoic acid treatment was found to significantly reduce inflammatory cytokine and chemokine expression levels in the LPS-mediated inflammation present in epididymal adipose tissue. Karkeni et al. (2017) showed that this effect arises due to diminished NF-κB activation and was associated with decreased macrophage infiltration of the epididymal fat. Since increased macrophage infiltration of adipose tissue plays a critical role in metabolic disease development (Ferrante, 2013; Mathis, 2013), the authors proposed that retinoic acid supplementation might represent a useful nutritional strategy for preventing obesity and its complications.
Retinoic acid has been shown to induce the browning of WAT by increasing adipose vascularity and inducing beige adipogenesis of PDGFRα+ adipose progenitors (Wang et al., 2017b). Retinoic acid was found to upregulate expression of vascular endothelial growth factor (VEGF) signaling and increase the number of blood vesicles and PDGFRα+adipose precursors present in WAT. This was evidenced by both increased vascularization and the presence of PDGFRα+ progenitors in inguinal WAT. Using PDGFRα tracking mice, Wang et al. (2017b) were able to establish that treatment of the mice with all-trans-retinoic acid (10 mg/kg body weight administered by daily IP injection) promotes the differentiation of PDGFRα+ positive cells into beige adipocytes in vivo. This was accomplished by showing increased expression of Ucp1 and other marker genes. VEGF signaling was reported to be required in order to achieve retinoic acid-induced WAT browning. The data reported by Wang et al. (2017b) are strong and provide support for the notion that retinoic acid can induce browning of WAT.
4. ALDH1A1 actions in adipocytes and obesity.
ALDH1A1 (RALDH1) is one of 3 enzymes in the body that catalyzes the final oxidative step needed for the synthesis of retinoic acid from retinol; the oxidation of all-trans-retinaldehyde to all-trans-retinoic acid (Kedishvili, 2013). There has been much research interest focused on ALDH1A1 and its actions as a retinaldehyde dehydrogenase. It is now convincingly established that ALDH1A1 expression is required for mediating normal WAT accrual and white adipocyte differentiation.
The importance of ALDH1A1 in WAT was first shown by Plutzky and colleagues, who reported that female Aldh1a1-deficient mice are resistant to high fat diet-induced obesity and insulin resistance (Ziouzenkova et al., 2007). The null mice were reported to show increased energy dissipation and this was proposed to account partially for the observed resistance to diet-induced obesity. Consistent with a role for ALDH1A1 in retinoic acid formation, these investigators were able to show that WAT retinaldehyde levels were significantly elevated by approximately 2-fold over those of matched wild type mice. Ziouzenkova et al. (2007) also showed that retinaldehyde levels were significantly reduced in WAT obtained from high fat diet fed female wild type mice compared to matched control diet fed mice. This suggested that retinaldehyde might have a role in regulating adipose tissue responses. To confirm this possibility, genetically obese ob/ob mice that were treated with retinaldehyde for 3 weeks were found to have less visceral fat accumulation than vehicle-treated mice. In vitro studies established a role for retinaldehyde in inhibiting the transactivational responses of PPARγ and RXR (Ziouzenkova et al. 2007). Based on their findings, Ziouzenkova et al. (2007) proposed that all-trans-retinaldehyde acts as a transcriptional regulator of metabolic responses to a high fat diet. This was the first report that retinaldehyde per se is important for directly regulating physiologic responses outside of the eye, acting as a transcriptional regulator.
Other studies by this same group employing 3T3-L1 adipocytes and MEFs obtained from female wild type and Aldh1a1-deficient mouse embryos, were able to establish that Aldh1a1 expression is required for adipogenesis to take place normally (Reichert et al., 2011). Aldh1a1-deficiency was reported to impair both retinoic acid production and adipogenesis in cultured cells. The Aldh1a1-dependent impairments to adipocyte differentiation were associated with markedly reduced expression of PPARγ that was reported to arise through the regulatory actions of the transcription factor Zfp423. Treatment of Aldh1a1-deficienct cells with retinoic acid or overexpression of any of the three Aldh genes partially restored adipogenesis.
Following-up these earlier reports, Kiefer et al. (2012b) proposed that Aldh1a1 and its substrate retinaldehyde are determinants of adipocyte plasticity and adaptive thermogenesis. This work identified that whole body Aldh1a1-deficiency in mice activates a BAT-like transcriptional program in WAT that drives uncoupled respiration and adaptive thermogenesis. Ucp1 mRNA and protein were found to be significantly induced in perigonadal WAT of chow fed female Aldh1a1-deficient mice. Citrate synthase activity, the rate limiting step of the tricarboxylic acid cycle, was reported to be significantly increased in perigonadal WAT but not in BAT of chow fed Aldh1a1-deficient mice. The authors reported that when C3H10T1/2 cells were induced to undergo adipocyte differentiation, Aldh1a1 expression was also induced. When differentiating C3H10T1/2 cells were treated with the ALDH inhibitor diethyl aminobenzaldehyde a marked elevation compared to vehicle treated cells of Ucp1 gene expression was observed, suggesting a normal role for ALDH1A1 in blocking a BAT-like differentiation of WAT.
Later studies involving many of these same investigators established that female Aldh1a1−/− mice, but not males, were resistant to high fat diet induced visceral WAT formation (Yasmeen et al., 2013). However, subcutaneous WAT formation was reduced similarly in both male and female Aldh1a1−/− mice. This sexual dimorphism in visceral fat was attributed to elevated adipose triglyceride lipase protein expression localized in clusters of multilocular UCP1 positive cells in female but not male mice. The authors went on to show an effect of estrogen on obesity development and proposed that this may account for the susceptibility of women to visceral obesity. They further reported that their results obtained in mice were paralleled by increased expression of ALDH1A1 in obese women.
The finding that normal expression of ALDH1A1 is required for maintaining normal WAT adipogenesis has been confirmed by other laboratories (Haenish et al., 2017; Landrier et al., 2017). When male wild type mice were fed a high fat diet for 8 weeks followed by an additional 9 weeks of feeding the high fat diet with or without supplementation of the specific ALDH1A1 inhibitor WIN 18,446, Haenish et al., (2017) found that the WIN 18,446-treated group gained less weight and had decreased adipose tissue weights and adipocyte size compared to untreated mice. Macrophage infiltration of adipose tissue was reported to be diminished and Ucp1 expression elevated in WIN 18,446 treated mice.
Investigations by others, established that feeding of a high fat diet to mice results in a statistically significant upregulation of Aldh1a1 expression in WAT (Landrier et al., 2017). Elevated ALDH1A1 expression was also observed in human adipose biopsies obtained from obese patients compared to those from controls. Surprisingly, for wild type mice fed the high fat diet, WAT levels of all-trans-retinoic acid were reported to be lower than in mice fed a control diet. This was the case even though Aldh1a1 expression was elevated (Landrier et al., 2017). Experiments carried out using 3T3-L1 adipocytes treated with 1.0 μM all-trans-retinoic acid resulted in significantly increased Aldh1a1 expression. These authors agreed that ALDH1A1 has a role in WAT physiology but did not address a role for retinaldehyde.
The notion that all-trans-retinaldehyde acts transcriptionally to modulate adipose tissue and other tissue metabolic responses, one that was first proposed more than 10 years ago (Ziouzenkova et al., 2007), has not yet found general acceptance. This has hindered understanding of how ALDH1A1 acts in metabolic disease causation and prevention. A recent publication from Yang et (2018) reports very rigorous investigations that led these authors to conclude that ALDH1A1 functions within adipose tissue to modulate adipogenesis and adipocyte differentiation through mechanisms that are independent of both retinaldehyde and retinoic acid. These investigations involved the use of both male and female Aldh1a1-deficient mice fed a high fat diet and MEFs isolated from the mice, and employed well validated LC/MS/MS measurements of WAT retinaldehyde and retinoic acid concentrations. Male epididymal WAT, female parametrial WAT, and femoral WAT from both male and female mice fed a high fat diet were not found to display diet-dependent differences in either tissue retinaldehyde or retinoic acid levels. Contrary to the findings of Yasmeen et al. (2013), both female and male Aldh1a1-deficient mice were reported to be resistant to high fat diet-induced weight gain. Loss of Aldh1a1 expression was not found to affect retinoic acid synthesis when the MEFs were treated with retinol. Yang et al. (2018) conclude that actions of ALDH1A1 not related to its role in vitamin A metabolism likely account for the effects of this enzyme is causing resistance to weight gain. The authors further noted that ALDH1A1 catalyzes other important oxidizations within cells including those of 4-hydroxy-2-nonenal and acrolein, two toxic aldehyde products formed through lipid peroxidation (Makia et al., 2011).
Interestingly, a signaling role for 4-hydroxy-2-nonenal in regulating mitochondrial uncoupling has been proposed (Echtay et al., 2003). Thus, both retinaldehyde and 4-hydroxy-2-nonenal, two distinct substrates for ALDH1A1, are reported to be able to induce mitochondrial uncoupling. This raises a question as to whether accumulation of only one or possibly both substrates may be contributing to the phenotypes observed for Aldh1a1-deficient mice.
5. Roles of other vitamin A-related proteins in obesity.
Studies of mice lacking Rbp1 in all tissues have led to the proposal that RBP1 has a role in blocking adipogenesis (Zizola et al., 2010). In adipose tissue, RBP1 is expressed solely in the stromal-vascular cell fraction and is not expressed in adipocytes (Tsutsumi et al., 1991). When Rbp1-deficient mice were fed a high fat diet, the mice showed increased fat pad mass compared to matched wild type controls (Zizola et al., 2010). This was accompanied by statistically significant increases in mRNA for PPARγ, as well as for a number of genes important for mediating fatty acid uptake by adipocytes. The high fat diet fed Rbp1-deficient mice remained more glucose and insulin tolerant than wild type mice. Knockdown of Rbp1 expression in 3T3-L1 preadipocytes increased adipocyte differentiation and triglyceride accumulation. Based on this study, Zizola et al. (2010) proposed that RBP1 present in preadipocytes regulates adipocyte differentiation through effects on PPARγ actions.
Possibly related to the findings reported by Zizola et al (2010) regarding RBP1 actions in adipocyte differentiation, Rbp1-deficient mice were found to lack any 9-cis-13,14-dihydroretinoic acid in serum, liver and brain, although adipose tissue levels for 9-cis-13,14-dihydroretinoic acid were not reported (RÜhl et al., 2015). As noted earlier, this retinoic acid metabolite is proposed to be a natural endogenous ligand for modulating the transcriptional activities of the 3 RXRs (RÜhl et al., 2015).
Retinol saturase (RetSat), which catalyzes the conversion of all-trans-retinol to all-trans-13,14-dihydroretinol, saturating the 13-14 double bond of all-trans-retinol, has also been implicated as having a role in adipogenesis (Moise et al., 2004). RetSat is expressed in many tissues and all-trans-13,14-dihydroretinol is found in these tissue (Moise et al., 2004). Interestingly, RÜhl et al. (2015) have suggested that RetSat may have a role in generating 13,14-dihydroretinols that are precursors needed for 9-cis-13,14-dihydroretinoic acid synthesis.
A number of published studies establish a role for RetSat in promoting adipogenesis (Schupp et al., 2009; Moise et al., 2010; Pang et al., 2017). RetSat was reported to be expressed in adipocytes and is required for 3T3-L1 adipocyte differentiation (Schupp et al. 2009). These investigators report that RetSat expression is induced during adipocyte differentiation through transcriptional activation by PPARγ. Ablation of RetSat markedly inhibited adipogenesis, but addition of all-trans-13,14-dihydroretinol to the culture medium did not overcome the blockage. Surprisingly, RetSat expression was reported to be downregulated in adipose tissue obtained from male genetically obese ob/ob mice or from male mice who had become obese mice upon feeding of a high fat diet. It was suggested that this downregulation of RetSat expression in obesity may involve the actions of factors produced by macrophages that have infiltrated the adipose tissue (Schupp et al., 2009).
RetSat-null mice, both males and females, which display normal levels of retinol and retinyl palmitate in liver, serum and adipose tissue, are deficient of all-trans-13,14-dihydroretinol in tissues (Moise et al., 2010). Despite accumulating more fat, RetSat-null mice maintained on either a low fat or a high fat diet did not consume more food or gain more weight than matched wild type controls. The increased adiposity of the null mice was associated with elevated PPARγ expression. From these initial studies of mixed genetic background (C57BL6 X 129sv) RetSat-null mice, Moise et al. (2010) proposed that dihydrovitamin A metabolites produced by RetSat control physiological processes that influence PPARγ activity. In follow-up studies carried out by Moise and colleagues, where the RetSat-null mice had been crossed into the C57BL/6N genetic background (Pang et al., 2017), the RetSot-null mice showed a much more pronounced weight gain phenotype when maintained on either a chow or high fat diet compared to matched wild type mice that were accompanied by statistically significant elevations in hepatic triglyceride levels. Pang et al. (2017) also reported data suggesting that RetSat is a potent modulator of cellular responses to oxidative stress and the generation of reactive oxygen species (Pang et al., 2017). This led Pang et al. (2017) to suggest that RetSat actions in adipose accrual is independent of its role in the formation of all-trans-13,14-dihydroretinol but rather dependent upon other pathophysiological processes related to lipid metabolism and oxidative stress. Studies of RetSat actions in both human liver and male mice led Heidenreich et al. (2017) to conclude that the enzyme coordinates liver metabolism by regulating carbohydrate response element binding protein, a cellular hexose-phosphate sensor and inducer of lipogenesis. Based on their data, Heidenreich et al. (2017) concluded that RetSat, by functioning as an upstream regulator of hepatic carbohydrate response element binding protein, directly affecting human and mouse circulating and hepatic triglyceride concentrations. Thus, although the data are clear that RetSat affects both adipogenesis and hepatic triglyceride metabolism and is affected by obesity, it remains to be established whether this involves vitamin A-dependent processes or possibly other as yet to be determined pathways.
Homozygous disruption of the mouse Rdh1 gene, gives rise to mice that show increased overall size and adiposity when compared with matched controls (Zhang et al., 2007). Although Rdh1 ablation does not impair embryonic development, unlike disruption of Rdh10 (Yang et al. 2018), Rdh1-null mice at 33-weeks-of-age were found to have significantly heavier mesenteric, femoral, epididymal retroperitoneal, brown and inguinal adipose depots than controls. Since the Rdh1-null mice are larger than matched controls, Zhang et al. (2007) also normalized fat pad size to body weight. When normalized to the body weight, the mesenteric, femoral and inguinal fat pads were still significantly larger than those of controls. Although no differences in endogenous all-trans-retinoic acid levels for the Rdh1-null versus control mice were found, the authors identified significant differences between the genotypes in both hepatic retinol and retinyl ester concentrations. Based on these data, it would appear that RDH1 and/or its products act in a manner that affects adipose tissue accrual.
6. Proposed clinical use of retinoic acid to prevent or treat obesity.
The well-established findings regarding retinoic acid effects in blocking adipocyte differentiation in cultured cells, blocking weight gain when fed in high doses in high fat diet fed rodents, and inducing Ucp1 expression have often led to the proposal by investigators that dietary vitamin A supplementation or pharmacological retinoic acid administration may be used therapeutically to prevent development of obesity. However, these proposals are premature for a number of reasons. It still has not been convincingly established whether the cell culture and animal model data can be translated to human therapies. In addition, there are important questions arising out of rodent studies that still need to be answered.
The doses of all-trans-retinoic acid used in rodent model studies that showed efficacy for blocking or reversing adipose accumulation range from 5 to 100 mg/kg body weight. When scaled up on the basis of body weight this would reflect 300 to 6000 mg/60 kg human being. These are very large doses. High dose all-trans-retinoic acid treatment (45 mg/m2/d or approximately 80 mg per dose) is a standard therapy for patients with acute promyelocytic leukemia (APL) (Agadir & Chomienne, 1999; Fenaux et al., 1999). This dose of all-trans-retinoic acid used in treating critically ill APL patients is much lower than those that have shown efficacy for blocking adipose accrual in mice. Moreover, the administration of this dose of all-trans-retinoic acid to APL patients is not without serious side effects. Some APL patients receiving this effective therapeutic dose develop a potentially fatal syndrome referred to as “Retinoic Acid Syndrome” or more recently as “Differentiation Syndrome” (Montesinos & Sanz, 2011). Amongst the many adverse symptoms associated with this dose of all-trans-retinoic acid are elevated blood cholesterol and/or triglyceride levels (up to 60% of patients) and weight gain (23% of patients) ((Agadir & Chomienne, 1999; Fenaux et al., 1999). Moreover, excessive weight gain is considered to be one of the symptoms that might be indicative of onset of Differentiation Syndrome (Fenaux et al., 1999; Montesinos & Sanz, 2011). Similarly, the usage of 13-cis-retinoic acid (a naturally occurring form of vitamin A (Kane et al., 2008b)) in clinical dermatology has established that administration of large doses of this naturally occurring retinoic acid form can result in a high risk of hyperlipidemia and other components of the metabolic syndrome in treated patients, including increased fat accumulation in adipose tissue (Rodondi et al., 2002). The mechanisms responsible for the hyperlipidemia observed in patients receiving 13-cis-retinoic acid are not fully understood. Early studies proposed that this may involve a transcriptional effect resulting in the upregulation of apolipoprotein C-III expression, a known regulator of plasma triglyceride metabolism (Vu-Dac et al., 1998). Taken together, the clinical experience in chronically using retinoic acid to treat human disease does not suggest that retinoic acid is a good candidate drug for limiting or reversing obesity. This reservation needs to be addressed before one can seriously consider the use of retinoic acid, either pharmacologically or as a dietary supplement, to limit or reverse obesity development in humans.
Early studies by Jeyakumar et al. (2006) established that the feeding of a control diet supplement with an approximate 50-fold excess of vitamin A to 7-month-old obese male WIN/Ob rats resulted in decreased adiposity and increased BAT Ucp1 expression. Based on these data, Jeyakumar et al. (2006) proposed that dietary supplementation with vitamin A may be useful for controlling obesity. However, in a subsequent study, where 30-week-old obese male WNIN/GR-Ob rats were fed the same vitamin A supplemented diet, a different outcome was obtained. Obese WNIN/GR-Ob rats fed the vitamin A supplemented diet were found to display significantly increased visceral adiposity and a two-fold increase in weight gain compared to male rats receiving the control diet (Jeyakumar et al., 2015). Feeding of the same diets to lean WIN/GR-Ob rats resulted in a nine-fold increase in weight gain for the rats fed the vitamin A-supplemented diet compared to controls. The only difference between the two studies was the genetic background of the rats used in each study. Collectively, these studies suggest that there are genetic factors that can modulate the actions of vitamin A in obesity (Jeyakumar et al., 2015). This needs to be taken into account in future rodent model studies.
In summary, although there is much enthusiasm amongst investigators for using retinoic acid as a treatment to limit obesity and associated metabolic disease, a number of significant research questions still need to be addressed before this enthusiasm can achieve reality.
C. RBP4, Adipose Tissue and Adipose-Related Metabolic Disorders.
Until 2005, the sole known function for RBP4 was to mobilize retinol from tissue stores and deliver it to vitamin A-responsive cells where it can be converted to retinoic acid for use in regulating vitamin A-dependent transcription and functions. In 2005, Kahn and colleagues reported that circulating RBP4 levels affect glucose clearance, with high RBP4 levels inducing insulin resistance (Yang et al., 2005; Graham et al., 2006). Specifically, Kahn and colleagues proposed that adipocyte-derived RBP4 is a signal that contributes to the pathogenesis of type 2 diabetes, linking obesity with type 2 diabetes, as well as other obesity-related metabolic diseases.
Working with mice that fail to express the glucose transporter GLUT4 in adipose tissue, Kahn and colleagues showed that that Rbp4 expression was elevated in adipocyte-specific male Glut4-null mice (Yang et al., 2005). These investigators showed further that serum RBP4 levels were elevated in the insulin-resistant mice but this could be normalized upon treatment with rosiglitazone, an insulin-sensitizing drug. Both male transgenic mice overexpressing RBP4 and injection of RBP4 into the circulations of normal mice were found to be associated with insulin resistance. Moreover, increased serum RBP4 levels were found to induce hepatic expression of the gluconeogenic enzyme phosphoenolpyruvate carboxykinase and to impair insulin signaling in muscle. Male Rbp4-null mice showed better responsiveness to challenges with either glucose or insulin. Based on these data, Kahn and colleagues proposed that RBP4 is an adipocyte-derived signal that may contribute to impaired glucose and insulin responsiveness and the pathogenesis of type 2 diabetes (Yang et al., 2005).
The mouse model studies were followed-up by studies of RBP4 levels in lean, obese and diabetic human subjects (Graham et al., 2006). Studies involving 3 distinct cohorts, ones from San Diego California, Leipzig Germany, and Goteborg Sweden, allowed Kahn and colleagues to reach the identical conclusion from their human studies as they did for the mouse studies. Human RBP4 was found to be an adipocyte-secreted molecule that is elevated in human serum before the development of frank diabetes. The authors proposed that elevated serum RBP4 levels identified insulin resistance and associated cardiovascular risk factors in subjects with varied clinical presentations.
These findings have given rise to considerable research interest in adipose-derived RBP4 and its role in obesity and obesity-related metabolic disease. This research involves both human-based cohort studies as well as mechanism-based studies involving mouse models and cells in culture.
1. RBP4, adipose tissue and adipose-related metabolic disorders---human studies.
A large number of published reports have reported a linkage between RBP4 synthesized by adipocytes and obesity and insulin signaling. Although the majority of this literature supports the view that circulating levels of RBP4 and/or adipose tissue levels of RBP4 are associated with the insulin resistance and other metabolic disorders associated with obesity, there is a significant literature that disagrees with this conclusion. At present, it is still not possible to draw unequivocal conclusions from this literature. A listing of some of the gaps in the literature focused on RBP4 and metabolic disease that are in need of resolution are provided in Table 2. These are discussed in more detail below.
TABLE 2.
QUESTIONS REGARDING RBP4 ACTIONS IN METABOLIC DISEASE IN NEED OF RESOLUTION
| 1. | Are the actions of RBP4 dependent upon or independent of its ligand retinol? |
| 2. | What roles do circulating holo-RBP4 versus apo-RBP4 have in the disease process? |
| 3. | Do the observed effects involve circulating RBP4, or local RBP4 actions in adipose tissue, or both? |
| 4. | To what extent do methodological differences in how circulating RBP4 levels are measured account for apparent discrepancies in findings obtained from human cohorts? |
| 5. | To what extent do genetic factors influence outcomes of human studies as well as studies carried out in animal models? To what extent do genetic differences account for some of the apparent disagreement in the literature. |
| 6. | To what extent does local inflammation in adipose tissue account for observed metabolic phenotypes? Does this explain all RBP4-induced metabolic disease. |
| 7. | To what extent does RBP4 binding to its cell surface receptor STRA6 account for observed metabolic phenotypes? Does this explain all RBP4-induced metabolic disease. |
a. Studies supporting a relationship between circulating RBP4 levels and metabolic disease.
The literature reporting an association between circulating RBP4 levels and metabolic disease is relatively large. Only a representative sampling of this literature will be considered below to illustrate the scope of the literature.
A cross-sectional study of 3289 50- to 70-year-olds living in Beijing and Shanghai found that elevated circulating RBP4 levels were strongly and independently associated with body mass index (BMI), waist circumference, insulin resistance, and hyperlipidemia (Qi et al., 2007). Another study reported similar findings of a direct association between fasting RBP4 levels and insulin resistance in type 2 diabetic patients (Cheng & Zhang, 2009). A retrospective cohort study involving 51 overweight, post-pubertal non-Hispanic black teenage participants carried out by Goodman et al. (2009) provided evidence that increased RBP4 levels were associated with significantly higher odds of worsening insulin resistance and hypertriglyceridemia in this population. Another study involving elderly patients (mean age of 75.9 ± 4.8 years) found a significant positive correlation between RBP4 levels and fasting insulin levels, insulin resistance and triglyceride levels (Lee et al., 2009). Obese Scottish subjects were reported to have significantly higher serum RBP4 levels in comparison to normal and overweight subjects (Hoggard et al., 2012). However, for this study adipose tissue RBP4 protein levels were found to be similar for the normal, overweight and obese subjects (Hoggard et al., 2012). Results from a two-year prospective study of middle-aged Asian Indian men with impaired glucose tolerance where 71 incident type 2 diabetes and 76 non-diabetic cases were studied, found that baseline RBP4 levels were independently associated with incident diabetes (Ram et al. 2015). Studies involving measures of both circulating RBP4 and TTR levels in age-matched individuals with normal glucose tolerance (n = 90), impaired glucose tolerance (n = 70) and type 2 diabetes (n = 90) identified that circulating levels of both RBP4 and TTR showed significant associations with glucose intolerance, obesity, and type 2 diabetes (Pandey et al. 2015). Additionally, RBP4 but not TTR levels were found to be positively associated with insulin resistance (Pandey et al., 2015). Serum RBP4 levels are reported to be elevated in in 267 newly diagnosed Chinese hypertensive patients compared to controls (Deng et al., 2014). However, no association between serum RBP4 levels and obesity was observed.
The relationship between blood RBP4 and insulin sensitivity was explored through use of hyperinsulinemic-euglycemic clamps studying both young subjects (20-50 years-of-age) and elderly subjects (60-83-years-of-age) (Gavi et al., 2008). Significant associations between RBP4 levels and insulin sensitivity, percent trunk fat, plasma triglycerides and low density lipoprotein levels were identified for the young subjects. However, none of these associations were present for the elderly group. The authors concluded that the associations between RBP4 levels with insulin sensitivity and the other parameters measured were influenced by age.
There has been substantial research interest in whether polymorphisms in the RBP4 gene may be associated with obesity, insulin resistance and other related metabolic diseases. Kovacs et al. (2007) sequenced the RBP4 gene in DNA samples obtained from 48 nonrelated Caucasian subjects and identified five novel and three previously known SNPs. These investigators found that a haplotype of six common SNPs was significantly increased in 934 subjects with type 2 diabetes compared to 537 healthy controls (Kovacs et al., 2007). Two single SNPs were reported to be associated with BMI, waist-to-hip ratio, and fasting plasma insulin. Kovacs et al. also found that subjects carrying one of the SNPs had significantly higher RBP4 mRNA levels in visceral adipose tissues for a subgroup of nondiabetic subjects. Based on their findings, Kovacs et al. (2007) concluded that there is a role for RBP4 genetic variation in susceptibility to type 2 diabetes and insulin resistance.
Munkhtulga et al. (2007) reported the identification of 9 signal nucleotide polymorphisms (SNPs) that are present in the promoter region of the RBP4 gene. A case-control study involving 511 control and 281 diabetic Mongolian subjects identified that for four of these SNPs, the rare alleles were associated with increased risk of diabetes. One of these SNPs (SNP −803G>A) was found to increase the efficiency of RBP4 transcription in cultured hepatocytes. This RBP4 SNP was also found to be associated with increased serum RBP4 levels in diabetic patients. Follow-up studies where carried out using adipose tissue biopsies obtained from Mongolian and Japanese subjects (Munkhtulga et al., 2010). These studies established that the minor allele carriers of the regulatory RBP4 SNP −803 G>A display significantly higher BMIs in Japanese men and women and in Mongolian women. Relative quantitation of RBP4 transcripts in SNP −803 G>A heterozygotes showed that the minor allele-linked haplotype-derived mRNA was significantly more abundant than the transcript from the major allele. Based on these and other cell culture data, Munkhtulga et al. (2010) reported that the minor allele of this RBP4 regulatory SNP enhanced its expression in adipocytes and may be associated with adipogenesis.
Other published studies have also provided evidence that RBP4 gene variants are associated with insulin resistance. Hu et al. (2008) identified ten SNPs present in the RBP4 gene for a population of 32 Chinese subjects. A combination of non-coding SNPs was reported to be associated with circulating RBP4 concentrations, as well as with phenotypes related to glucose metabolism (Hu et al., 2008). In a prospective study, homozygous carriers of the RBP4 SNP −803 G>A were found to be at increased risk of type 2 diabetes (van Hoek et al., 2008). The increased risk for diabetes could not be explained by dietary retinol intake or plasma retinol levels. A study involving 3,210 Chinese subjects found that RBP4 polymorphisms were significantly associated with plasma RBP4 levels and risk of hypertriglyceridemia (Wu et al., 2009). Another study, one involving 2002 patients living in South India, explored the effects of SNPs in both the RBP4 gene and the gene for STRA6 (Nair et al., 2010). These investigators reported that they were unable to find significant associations between RBP4 SNPs and type 2 diabetes but they identified three significant associations between SNPs in the STRA6 gene and type 2 diabetes (Nair et al., 2010). An association between RBP4 genetic variants with childhood obesity and metabolic parameters associated with cardiovascular risk factors has been reported for a study of 97 obese and 82 normal weight Spanish Caucasian children (Codoner-French et al., 2015). The authors concluded that childhood obesity may be associated with RBP4 gene variants and that the presence of some SNPs in the RBP4 gene may account for obesity-related metabolic complications (Codoñer-French et al., 2015). A prospective study of 100 women with gestational diabetes and 100 gestationally normal women identified 2 SNPs in the RBP4 gene that were associated with insulin resistance and insulin levels in the women with gestational diabetes (Saucdo et al., 2014). These investigators did not assess circulating RBP4 levels in their studies (Saucdo et al., 2014)
Nearly all investigators exploring the relationship between circulating RBP4 levels and disease have measured circulating levels of total RBP4 (apo- + holo-RBP4). However, in the circulation both apo- and holo-RBP4 are present, with holo-RBP4 accounting for approximately 85% of the total RBP4 present in the circulations of healthy individuals (the remaining 15% is present as apo-RBP4) (Soprano & Blaner, 1994). Two published reports have provided measures of both apo- and holo-RBP4 levels in patients. One study of normal and obese men and women, reported that total serum RBP4 was strongly associated with both BMI and measures of insulin resistance (Mills et al., 2008). For this study, serum apo-RBP4 levels were approximately two-fold higher in obese patients compared to controls and the retinol:RBP4 ratio, a measure of relative apo- and holo-RBP4 levels, was reported to be significantly lower in obese patients (Mills et al., 2008). Other investigators have reached the same conclusion that excess RBP4 relative to retinol (increased apo-RBP4) is more indicative of type 2 diabetes than total RBP4 levels (Erikstrup et al., 2009). These investigators also reported a positive correlation between adipose tissue RBP4 mRNA levels and plasma tumor necrosis factor-α (TNF-α) levels. This later finding raises a question regarding the possible role of adipose inflammation in RBP4-related disease (Erikstrup et al., 2009). Collectively, these two reports raise a question as to whether both apo- and holo-RBP4 levels should be routinely measured for assessing linkages between circulating RBP4 levels and disease incidence. Importantly, they also raise a caveat for interpreting reports were only total RBP4 was reported.
Circulating RBP4 levels have also been associated with risk of coronary heart disease (Sun et al., 2013). The authors assessed plasma RBP4 levels for 468 women who developed coronary heart disease and for 472 matched controls in the Nurses’ Health Study cohort during 16 years of follow-up. Higher levels of circulating RBP4 were associated with increased risk of coronary heart disease in a time–dependent manner. In another study, a significant correlation was identified between serum RBP4 levels and various established risk factors for cardiovascular disease in women (Alkharfy et al., 2012). This study which involved 139 male and 145 female patients was unable to establish this relationship in men (Alkharfy et al., 2012). Feng et al. (2015) reported that blood RBP4 levels were positively correlate with carotid atherosclerosis in type 2 diabetes patients for a study involving 1,076 patients.
b. Studies that fail to support a relationship between circulating RBP4 levels and metabolic disease.
Investigations of human adipose tissue obtained from healthy pre- and postmenopausal women, as well as primary human adipocytes isolated from the adipose tissue, led Janke et al. (2006) to conclude that adipocyte-derived-RBP4 does not play an important role in signaling insulin-responsiveness in humans. Although these authors established that primary human adipocytes are able to synthesize and secrete RBP4, they observed a downregulation of RBP4 mRNA levels in subcutaneous adipose tissue obtained from obese women. Circulating RBP4 concentrations were not different for normal weight, overweight, or obese women. A 5% body weight loss was found to be associated with decreased adipose RBP4 expression, but weight loss did not affect circulating RBP4 concentrations. Thus, although Janke et al. (2006) confirmed that RBP4 is an adipokine, they failed to establish that the regulation of RBP4 expression in human adipose tissue or the regulation of circulating RBP4 concentrations resembles that reported for mice.
Similar findings were reported by Kos et al. (2011). These investigators explored RBP4 expression in adipose tissue obtained from lean and obese human subjects. RBP4 protein levels were found to be higher in subcutaneous adipose tissue compared to visceral fat. Women were found to have higher RBP4 levels than men for both adipose depots. However, these differences were not reflected in circulating RBP4 levels which failed to show gender-based differences. Leptin treatments of visceral adipose tissue explants were found to markedly increase RBP4 levels. The authors’ data provided no support for the contention that circulating RBP4 is associated with insulin resistance in humans, nor for the notion that insulin may alter adipose tissue derived RBP4 expression.
A study of 72 adult Mexican-Americans (16 lean subjects with normal glucose tolerance, 17 obese subjects with normal glucose tolerance, and 39 subjects with impaired fasting glucose or impaired glucose tolerance or type 2 diabetes) led the investigators to conclude that plasma RBP4 levels are elevated in type 2 diabetes and associated with impaired glucose tolerance but not associated with obesity or insulin resistance or impaired insulin secretion (Chavez et al., 2009). The research involved the use of a number of different clinical tests including oral glucose tolerance tests and euglycemic-hyperinsulinemic clamp studies, as well as measurement of hepatic glucose production rates and tissue RBP4 levels. Although plasma RBP4 concentrations were reported to be elevated in type 2 diabetic patients, no correlations with insulin resistance or insulin secretion were found. The authors concluded that this indicates that in Mexican Americans elevated plasma RBP4 does not play a role in the development of insulin resistance and type 2 diabetes (Chavez et al., 2009).
Bajzová et al. (2008), who explored RBP4 mRNA levels in biopsies obtained from both obese and non-obese adult women, reported that they were unable to confirm a direct relationship between adipose tissue RBP4 expression and plasma RBP4 levels. Moreover, these investigators were unable to demonstrate a relationship between RBP4 plasma levels and either adiposity or insulin resistance.
RBP4 levels have been reported to be associated with dyslipidemia but not with insulin resistance (von Eynatten et al., 2007). Serum RBP4 levels were measured in 365 men (126 with type 2 diabetes, 143 with coronary artery disease, and 96 controls). RBP4 levels were not found to be associated with insulin resistance in males with diabetes or coronary artery disease. However significant associations between serum RBP4 levels and pro-atherogenic lipoproteins and enzymes involved in lipoprotein metabolism were identified for patients both with and without the metabolic syndrome. These authors concluded that RBP4 may have a role in affecting plasma lipid metabolism but not in insulin resistance (von Eynatten et al., 2007).
A number of less extensive observational studies have been reported that also fail to establish a relationship between RBP4 levels in blood and insulin resistance. An investigation involving 98 obese children studied in a tertiary care children’s hospital failed to establish a significant positive association between blood RBP4 levels and insulin resistance (Thiruvengadam et al., 2015). A longitudinal study of 206 overweight subjects who were followed for 36 months failed to show any changes in plasma RBP4 levels for the subjects who developed insulin resistance. This led to the authors to conclude that RBP4 is not a marker of insulin resistance in overweight humans (Lewis et al., 2008). Another published report found a relationship between markers for adipose tissue inflammation and RBP4 mRNA expression for both human visceral and subcutaneous adipose tissue but not between RBP4 mRNA levels and insulin resistance (Yao-Borengasser et al., 2007)
In contrast to the many positive studies reported above that were able to establish associations between RBP4 SNPs, elevated circulating RBP4 levels and insulin resistance, Friebe et al. (2011) were unable to find an association between the −803 G>A RBP4 promoter SNP with BMI, parameters of glucose and lipid metabolism or blood pressure. This study, which involved 304 lean and 283 obese Caucasian children, reports a finding that directly contradicts the findings that were reported for Japanese and Mongolian populations (Munkhtula et al., 2007; Munkhtulga et al., 2010). Possibly other genetic factors may account for this discrepancy.
It is clear from the above text that a significant number of studies have failed to identify a relationship in patients between circulating or tissue RBP4 levels and metabolic disease. A number of investigators have noted a potential cofounding problem for studies involving only measurement of circulating RBP4 levels. Studies of diabetic patients and controls reported by Schweigert and colleagues (Raila et al., 2007) led these authors to propose that blood RBP4 levels in type 2 diabetic patients are affected by incipient nephropathy. Owing to this, Schweigert and colleagues suggested that studies evaluating RBP4 as a regulator of systemic insulin resistance and type 2 diabetes need to take into consideration renal function. Subsequent studies by this same group, involving a larger cohort of type 2 diabetics and controls, provided further support for this proposal and led the authors to conclude that elevated circulating RBP4 levels observed in type 2 diabetic patients is the result of moderate renal insufficiency rather than due to linkages between RBP4 and obesity or type 2 diabetes (Henze et al., 2008). A similar conclusion was reached by Murata et al. (2009) from a study of both type 2 diabetic patients and controls. These authors concluded that alterations in patient blood RBP4 levels do not associate with either insulin resistance and microvascular disease but rather with renal impairments. This same conclusion was also reached by Akbay et al. (2010) who also assessed circulating RBP4 levels in type 2 diabetes patients. Jing et al. (2016) reported from a study of 21 healthy patients, 45 patients with chronic kidney disease, and 10 patients on renal dialysis that both plasma RBP4 and retinol are elevated in patients with chronic kidney disease or on renal dialysis. This and other data led Jing et al. (2016) to suggest that chronic kidney disease is associated with altered vitamin A homeostasis, including altered retinoic acid homeostasis. Thus, it would appear that renal complications may account in some studies for observed elevations in blood RBP4 levels. This certainly confounds interpretations of data from studies where possible renal complications were not directly considered.
2. RBP4, adipose tissue and adipose-related metabolic disorders---molecular studies.
The literature involving mouse model and cell culture studies is generally consistent with the notion that elevated adipose tissue expression of RBP4 is associated with the adverse metabolic phenotypes that were first reported by Kahn and colleagues to result from elevated RBP4 levels (Yang et al., 2005). However, the literature on this topic is not uniformly supportive of this conclusion. Moreover, the molecular mechanisms underlying disease causation have not been convincingly established and remain controversial. Kahn and colleagues provided evidence that RBP4 acts to increase adipose tissue inflammation and that this ultimately is responsible for RBP4-induced disease. A diagram of how these authors believe RBP4 acts to induce adipose tissue inflammation is provided in Figure 3. Noy and colleagues proposed that RBP4 binding to its cell surface receptor STRA6 is responsible for disease causation. A summary of how STRA6 is proposed to act in mediating RBP4-induced metabolic disease is provided in Figure 4. In addition, as recounted below, other investigators have provided data suggesting still other possibilities.
Figure 3. Depiction of RBP4 immunomodulatory actions locally within and how this gives rise to insulin resistance.
RBP4 synthesized by adipocytes (A) (or possibly arriving from the circulation) activates resident adipose tissue macrophages (M) and dendritic/antigen presenting cells (APC). Through a JNK-dependent pathway, this induces proinflammatory cytokine secretion (TNFα and IL-1β) from macrophages and expression of major histocompatibility complex class II (MHCII) molecules as well as costimulatory molecules. The proinflammatory molecules and cytokines directly contribute to adipose tissue inflammation and insulin resistance. The activated antigen presenting cells induce CD4 T cell proliferation and Th1 polarization increasing levels of TNFα and IFN-γ which further stimulate adipose tissue macrophages bringing about increased local inflammation and systemic insulin resistance. Adapted from Moraes-Vieira et al. (2014).
Figure 4. Diagramatic providing a summary of how STRA6 acts to mediated RBP4-induced metabolic disease.
The process is depicted as involving six steps. The mechanism links vitamin A uptake by STRA6-expressing cells with a signaling cascade that regulates expression of multiple STAT target genes. Step 1 commences with the binding of holo-RBP4 to its binding site on the extracellular surface of STRA6. STRA6 is primed with apo-RBP1 bound to its intracellular side. In Step 2 retinol is released by RBP4 and traverses through a pore in STRA6. The movement of retinol is proposed to trigger the phosphorylation of JAK2. Phospho-JAK2 initiates Step 3 which results in the phosphorylation of STRA6 and the frees RBP1 to be released from STRA6. This is followed by Step 4 where holo-RBP1 is released from STRA6 and STAT3 or STAT5 are recruited and undergo activation through phosphorylation by phospho-JAK2. In Step 5, holo-RBP1 delivers retinol to LRAT which converts the retinol to retinyl ester releasing apo-RBP1. This occurs concurrently with the movement of activated phospho-STAT3/5 to the nucleus where it induces expression of STAT target genes including suppressor of cytokine signaling 3 (SOCS3). This results in an inhibition of insulin signaling and PPARg activation. Step 6 involves the binding of apo-RBP1 to STRA6 and the dephosphorylation of JAK2 and STRA6, priming STRA6 for another round of binding with holo-RBP4. Figure 4 was adapted from [(Noy 2016b)].
The possibility of immune cell involvement in RBP4-induced metabolic disease was first suggested by the work of Norseen et al. (2012). These studies showed that incubation of recombinant hRBP4 with isolated mouse or human macrophages induced expression and secretion of a number of proinflammatory cytokines, including TNF-α, IL-6 and monocyte chemoattractant protein-1. These authors went on to show that this could directly inhibit insulin signaling in co-cultures of macrophages and adipocytes. The basis for this was reported to involve activation of c-Jun-N-terminal protein kinase (JNK) and Toll-like receptor 4 (TLR4) pathways and was reported to be independent of STRA6. Interestingly, apo-hRBP4 was found to be as potent as retinol-bound hRBP4 in inducing proinflammatory cytokines in cultured macrophages.
Other published findings from this laboratory involved the use of male transgenic mice that overexpress hRBP4 in skeletal muscle (Moraes-Vieira et al., 2014). This investigation led to the conclusion that RBP4 activates adipose tissue antigen-presenting cells that induce CD4 T cell Th1 polarization and adipose tissue inflammation (Moraes-Vieira et al., 2014). The RBP4 overexpressing mice were found to be insulin resistant and glucose intolerant and to have increased adipose tissue macrophage and CD4 T cell infiltration. This effect was proposed to result from direct activation of adipose tissue antigen-presenting cells by RBP4 and was found to be sufficient to cause insulin resistance. The authors reached this conclusion based on data showing that transfer of RBP4-activated antigen-presenting cells into normal mice induced adipose tissue inflammation as well as impaired glucose tolerance and insulin sensitivity (Moraes-Vieira et al., 2014). In subsequent studies, these authors established that male Rbp4-null mice fed a high fat diet to induce obesity are more insulin sensitive and display less adipose tissue inflammation than matched wild type mice (Moraes-Vieira et al., 2016). This later finding agrees well with the data obtained from the RBP4 overexpressing mice.
Further studies of the male RBP4 overexpressing mice explored the molecular process that are responsible for RBP4-induced antigen-presenting cell activation (Moraes-Vieira et al., 2016). These investigations led to the conclusion that antigen presentation by macrophages activated by RBP4 was dependent on the p38MAPK and NF-κB signaling pathways. Adipose tissue macrophages obtained from the overexpressing mice displayed enhanced JNK, ERK 1/2 and p38MAPK phosphorylation. Inhibition of these pathways and of NF-κB was found to reduce RBP4-induced activation of macrophages and CD4 T cells. It also was found that MyD88, an adaptor protein involved in proinflammatory signaling acting upstream of p38MAPK and NF-κB, is importantly involved in the process since RBP4 failed to induce adipose tissue inflammation in MyD88-deficient mice.
Collectively, these published data from the Kahn laboratory provide support for the conclusion that adipose tissue inflammation, induced by RBP4, accounts for the adverse effects of RBP4 on glucose and insulin metabolism. As mentioned earlier, adipose tissue inflammation induced upon RBP4 transgene expression in adipocytes is reported to be responsible for the hepatic steatosis phenotype associated with elevated adipocyte RBP4 expression (Lee et al., 2016). Interestingly, and possibly relevant for understanding these observations, there is a growing literature establishing that resident macrophages and especially dendritic cells within tissues, including adipose tissue, are sites of retinoic acid synthesis (Dalmas et al., 2017; Bazewicz et al., 2018; de Mendonca Oliveira et al., 2018).
Published studies by Thompson et al. (2017) have added a complication for understanding RBP4-mediated induction of adipose inflammation. These authors report that mouse adipocytes do not synthesis or secrete significant levels of RBP4 into the circulation even in the setting of diet-induced insulin resistance. Thompson et al. (2017) generated a hepatocyte-specific knockout for Rbp4 in mice. When fed a high fat high sucrose diet, male mutant mice gained significantly more body weight than matched control mice but only developed similar degrees of insulin resistance and glucose intolerance. Unexpectedly, RBP4 protein could not be detected in either the general or portal circulations of the hepatocyte-specific Rbp4-knockout mice. This was despite the finding that perigonadal WAT explants taken from hepatocyte-specific knockout mice and from matched control mice were found to secrete equal amounts of RBP4 into the culture medium. This suggests that RBP4 is being synthesized and secreted from adipocytes but little of this RBP4 finds its way to the general circulation. Based on their data, Thompson et al. (2017) concluded that adipocyte RBP4 is not a significant source of circulating RBP4, even in the setting of insulin resistance, and went on to propose that adipocyte RBP4 is an important autocrine/paracrine factor whose actions are confined to the adipose tissue compartment (Thompson et al., 2017).
Through the use of antisense oligonucleotides able to knockdown hepatic expression of Ttr in male mice, Zemany et al. (2015) were able to demonstrate that treatments aimed at lowering circulating RBP4 levels were effective in improving insulin sensitivity and lowering hepatic glucose production. Since TTR is required for preventing renal filtration of RBP4 and its loss in the urine (Soprano & Blaner 1994), knockdown of TTR levels in ob/ob mice was found to effectively lower plasma RBP4 levels and to improve insulin sensitivity without altering body composition (Zemany et al., 2015). The treatment also improved glucose and insulin homeostasis in high fat diet-induced obese mice, including improved performance during hyperinsulinemic-euglycemic clamp studies. The findings from these studies appear to be in conflict with the studies discussed immediately above that support the notion that local actions of RBP4 in adipose tissue are the underlying cause of insulin resistance and glucose intolerance. The data from Zemany et al. (2015) suggest a role for circulating RBP4 in disease causation. Possibly this involves some action of RBP4 in skeletal muscle that were not investigated in the previously mentioned studies.
Noy and colleagues reported that binding of retinol-RBP4, but not apo-RBP4, to STRA6 triggers phosphorylation of a tyrosine residue in the cytosolic domain of STRA6, resulting in recruitment and activation of the Janus kinase 2 (JAK2) and the transcription factors STAT3 and STAT5 (Berry et al., 2011). Using HepG2 cells transfected to express STRA6, Berry et al. (2011) were able to establish that the holo-RBP4/STRA6/JAK2/STAT3/5 signaling cascade induces expression of STAT target genes including suppressor of cytokine signaling 3 (SOCS3), which is known to inhibit insulin signaling and PPARy activation. Since STRA6 is expressed in both WAT and skeletal muscle, Berry et al. (2011) undertook further studies in mice. These studies identified the relevance of this signaling cascade in vivo. Injection of recombinant RBP4 into the circulations of mice was shown to induce expression of STAT target genes and to increase phosphorylation of STRA6, JAK2, and STAT3/5 in adipose tissue and skeletal muscle. These findings led the authors to propose a role for STRA6 in mediating RBP4-induced metabolic disease (Berry et al., 2011).
Other studies by Noy and colleagues aimed at confirming STRA6 involvement in metabolic disease development established that cellular retinol uptake mediated by STRA6 from the retinol-RBP4 complex is required for STRA6 activation of JAK/STAT signaling (Berry et al., 2012b). This action by STRA6 is reported to involve the recruitment of RBP1 to STRA6 to accept the retinol that has been removed from RBP4 by STRA6. Overexpression of RBP1 in HepG2 cells was found to enhance the phosphorylation of STRA6 as well as its downstream effectors JAK2 and STAT5. Berry et al. (2012b) proposed that STRA6 functions both like a classical cytokine receptor to recruit the kinase JAK2 to activate the transcription factor STAT5 as well as a sensor of plasma holo-RBP4 concentrations coupling vitamin A homeostasis and metabolism to the activation of a signaling cascade that regulates transcriptional signaling.
This was followed-up by a study involving whole body male Stra6-null mice (Berry et al., 2013) which extended understanding of STRA6 actions as a dual modulator of vitamin A homeostasis and transcriptional signaling. Stra6-deficiency was found to have only a modest effect on the uptake of vitamin A into cells and tissues except for the eye. However, ablation of Stra6 was reported to have a striking effect for protecting mice from RBP4-induced suppression of insulin signaling (Berry et al., 2013). Infusion of holo-RBP4 into wild type mice induced JAK2 phosphorylation and increased expression of target genes but this effect was not observed for Stra6-null mice. Glucose tolerance tests established that high fat diet-induced obese Stra6-null mice were significantly more glucose tolerant than their wild type counterparts. Accordingly, hyperinsulinemic-euglycemic clamp studies revealed that the glucose infusion rate needed to maintain euglycemia was significantly higher in obese Stra6-null mice than obese wild type mice. This was taken to indicate that ablation of Stra6 protects animals from high fat diet induced insulin resistance (Berry et al., 2013). More recent studies from the Noy laboratory involving both male and female mice have shown that adipose tissue STRA6 undergoes circadian patterning that is driven in part by the nuclear transcription factor REV-ERBα (Gliniak et al., 2017). STRA6 was found to be necessary for diurnal rhythmicity of insulin signaling and JAK/STAT signaling in adipose tissue. Collectively, the data provided by Noy and colleagues supports the contention that STRA6 acts in adipose tissue and skeletal muscle to mediate RBP4 effects on glucose and insulin tolerance (Berry et al., 2011; Berry et al., 2012b; Berry et al., 2013; Gliniak et al., 2018).
Two human observational studies have maintained that apo-RBP4 and not holo-RBP4 is predictive of metabolic disease development (Mills et al., 2008; Erikstrup et al., 2009), suggesting that increased apo-RBP4 concentrations may contribute to disease development. However, there is no general consensus in the human literature on this point. Cell culture studies by Kahn and colleagues suggest that apo-hRBP4 is as potent as holo-RBP4 in modulating insulin responsiveness in adipocytes (Norseen et al., 2012). But other cell culture studies by Noy and colleagues show that retinol-bound RBP4, but not apo-RBP4, upon binding to STRA6 activates a signaling cascade that culminates in induction of STAT target genes (Berry et al., 2011; Berry et al., 2012b). These conflicting data suggest that it may not be possible to reconcile directly the findings of Kahn and colleagues with those of Noy and colleagues.
Schupp and colleagues (Muenzner et al., 2013) have identified another role for both RBP4 and STRA6 in modulating adipogenesis. Studying 3T3-L1 and C3H10T1/2 cells along with mouse adipose tissue and cells obtained from wild type male mice fed a high fat diet, these authors were able to show that STRA6 is expressed in adipocyte precursor cells and that when the precursor cells are exposed to retinol-RBP4 (holo-RBP4), this blocked adipocyte differentiation via STRA6-dependent activation of RARα. Interestingly, apo-RBP4 treatment of cells was found to promote adipocyte differentiation in a STRA6-dependent manner. Apo-RBP4 also was reported to trigger retinol efflux from the cells and this reduced cellular vitamin A levels, RARα activity and target gene expression, and enhanced adipogenesis synergistically with ectopic STRA6 expression. These authors further report that STRA6 depletion from adipocyte precursors impairs adipocyte differentiation in a manner that was dependent on the presence of holo-RBP4. Muenzner et al. (2013) proposed from their studies that STRA6 presence in adipocyte precursor cells links nuclear RARα activity to the circulating RBP4 isoforms (apo- versus holo-), whose ratio in the blood can limit the adipogenic potential of precursor cells. Although the findings of Muenzner et al. (2013) have not yet been confirmed by others, they do raise provocative questions regarding the possible effects of circulating RBP4 levels and STRA6 on adipocyte differentiation and adipose tissue accrual.
The effects of RBP4 on primary human adipocytes isolated from subcutaneous adipose tissue from patients undergoing elective surgery was studied by Öst et al. (2018). These investigators reported that overnight incubation of primary human adipocytes with physiologically-relevant levels of commercially obtained human RBP4 (50 μg/ml) mimics the situation seen in adipocytes obtained from type 2 diabetic patients. RBP4-treated adipocytes exhibited the same molecular defects observed in insulin signaling, affecting insulin receptor substrate 1 (IRS1) and p38MAPK, as observed for type 2 diabetes patients. Without affecting autophosphorylation of the insulin receptor, RBP4 blocked the insulin–stimulated phosphorylation of IRS1 at serine 307 and concomitantly increased by 4-fold the EC50 for insulin stimulation of IRS1 phosphorylation. The authors further showed a decreased sensitivity towards insulin-stimulated phosphorylation downstream of ERK1/2, that also resembles that observed in adipocytes isolated from type 2 diabetic patients. Contrary to the findings of Yang et al, (2005), Öst et al. (2018) reported that endogenous adipocyte levels of RBP4 were markedly reduced in adipocytes from obese or type 2 diabetic patients, whereas expression levels of RBP4 mRNA were unaffected. This finding led Öst et al. (2017) to propose that RBP4 may be released from diabetic adipocytes and act locally to inhibit phosphorylation of IRS1 at serine 307, a phosphorylation site that they propose may integrate nutrient sensing with insulin signaling.
Bhat and colleagues (Manolescu et al., 2009) report that administration for up to 16 days of an oral dose of all-trans-retinoic acid (100 μg per day) to female ob/ob mice fed a standard chow diet resulted in significantly diminished circulating levels of RBP4, by approximately 20%. This was accompanied by improved glucose clearance and insulin sensitivity, as well as diminished basal serum glucose levels. Manolescu et al. (2009) suggested that retinoic acid might be an effective antidiabetic agent. Other authors have also reported that treatment of male mice with all-trans-retinoic acid results in increased insulin sensitive and leads to a reduction of Rbp4 mRNA and protein levels in adipose tissue (Mercader et al., 2008).
Many investigators who have been unable to establish an association between circulating RBP4 levels and insulin resistance in humans have concluded that findings obtained from studies in mice regarding RBP4 and metabolic disease may not be valid for humans. However, there are mouse studies that do not support the proposed role for elevated circulating RBP4 levels in insulin resistance. Motani et al. (2009) identified a compound, A1120, that is not a retinoid but is able to bind to RBP4 and affect RBP4 interactions with TTR. A1120 binding induces RBP4 loss in the urine, since TTR binding prevents renal filtration or RBP4 (Soprano & Blaner, 1994). As expected, administration of A1120 to male mice lowered circulating RBP4 by up to 80%. This reduction in RBP4 levels was accompanied by reduced circulating retinol concentrations. Yet, this did not improve insulin sensitivity nor did it protect against high fat diet-induced insulin resistance. Motani et al. (2009), based on their studies in mice concluded that the development of drug therapies aimed at lowering RBP4 concentrations in the circulation may not be an effective strategy for treating diabetes.
Two very recent studies (Perduca et al., 2018a; Perduca et al., 2018b) have identified human plasma RBP4 as a fatty acid-binding protein. These authors report high resolution three-dimensional structures of human RBP4 naturally lacking bound retinol that was purified from plasma, urine and amniotic fluid. All three crystal structures were found to contain a fatty acid molecule bound in the RBP4 hydrophobic ligand-binding site, a result that was confirmed by mass spectrometry measurements. The fatty acids present in the RBP4 binding pocket were identified as being palmitate and laurate. The authors’ data raise the possibility that RBP4 may have some previously unsuspected role in lipid metabolism and this could provide a linkage for better understanding RBP4 and its relationship to metabolic disease development.
3. RBP4 and ocular disease.
A study exploring serum levels of RBP4 in patients with type 2 diabetes with and without diabetic retinopathy identified an association between elevated serum RBP4 levels and proliferative diabetic retinopathy (Li et al., 2010). Serum RBP4 levels were assessed in 92 patients that were divided into three subgroups: those without diabetic retinopathy (n = 40), those with simple diabetic retinopathy (n = 37), and those with proliferative diabetic retinopathy (n = 15). A significant positive correlation between serum RBP4 levels was identified for the proliferative diabetic retinopathy group compared the groups without diabetic retinopathy or with simple diabetic retinopathy. The authors took their data to suggest a relationship between elevated circulating RBP4 levels and retinal disease.
The molecular basis for the relationship between elevated RBP4 levels and retinal disease has been studied in cells in culture and in mouse models. Investigations of the effects of bacterially generated recombinant human RBP4 on primary cultures of human retinal capillary endothelial cells and human umbilical vein endothelial cells were reported by Farjo et al. (2012). The authors report that both apo-RBP4 and retinol-bound RBP4 induce production of inflammatory molecules including VCAM-1, ICAM-1, E-selectin, MCP-1, and IL-6 in both macro- and microvascular human endothelial cells. This effect of RBP4 occurred in part through activation of NADPH oxidase and NF-κB. Subsequent studies of cultured human retinal capillary endothelial cells treated with RBP4 led these investigators to conclude that RBP4-induced inflammation is largely mediated by TLR4 (Du et al., 2017a). In vivo studies involving the use of both male and female transgenic mice that overexpress human RBP4 in skeletal muscle giving rise to very high levels of circulating RBP4 also have been reported by these investigators (Du et al., 2015). This transgenic mouse model was found to progressively develop retinal dysfunction and degeneration characterized by photoreceptor ribbon synapse deficiency followed by bipolar cell loss. Retinal degeneration was associated with increased levels of pro-interleukin-18 and activated interlukin-18. The eyes of the transgenic mice were found to possess vitamin A levels that were not different from those of matched wild type mice. Treatment of the RBP4 overexpressing transgenic mice with the drug A1120, which disrupts RBP4-TTR interactions resulting in lowered circulating RBP4 levels (Motani et al., 2009), did not prevent retinal dysfunction in either male or female transgenic mice (Du et al. 2017b). Thus, it would appear that elevated circulating levels of RBP4 are not directly responsible for the retinal phenotype.
IV. Summary.
The literature concerning the actions of retinoic acid in metabolic disease is strong and points to the conclusion that normal retinoic acid signaling is required for preventing disease. This is especially true for preclinical studies of NAFLD where it is clear from the literature that dysregulation of retinoic acid signaling mediated by RARs contributes to hepatic disease development and progression. The actions of retinoic acid in white adipocyte differentiation are also clearly defined but it remains unclear whether this involves only RAR signaling and/or PPARβ/δ signaling as proposed by some authors. This literature also convincingly suggests a role for retinoic acid in the browning of WAT and in maintaining normal BAT functions. These actions of retinoic acid will undoubtedly receive further research attention in the future.
Although the literature proposes a number of distinct actions for RBP4 in obesity, insulin resistance, type 2 diabetes, NAFLD and other related metabolic diseases, definitive understanding of how RBP4 contributes to these disease states remains elusive. A majority of the human literature finds associations between circulating RBP4 levels or adipose tissue RBP4 expression and metabolic disease. However, a large number of human studies have failed to establish associations between RBP4 levels and disease. It has been suggested that methodological differences in how RBP4 concentrations are measured may account for some of this apparent discrepancy (Graham et al., 2007). RBP4 protein levels are routinely measured using immunoassays purchased from commercial suppliers. There is considerable variability in the quality of these commercial products. There is a clear need for standardization of methods used to assess tissue RBP4 levels. Yang et al. (2012) have suggested that mass spectrometry be used in the measurement of intact and C-terminal proteolyzed RBP4. Although this approach would allow for truly reproducible quantitative measures of RBP4 to be made, one wonders whether this method is a practical one for use in routine clinical studies.
The molecular or mechanistic literature regarding RBP4 actions in cells and animal models generally supports the notion that RBP4 has a role in disease causation, although not all published studies support this conclusion. It is the view of this author that RBP4-induced inflammatory responses within adipose tissues are probably responsible for most of these effects, especially RBP4-induced NAFLD and retinal disease. However, the possibility that RBP4 binding to STRA6 may account for some of disease causation cannot be dismissed and this merits further study. A very important question that needs to be addressed is whether circulating RBP4 contributes to disease causation or whether only RBP4 synthesized by adipocytes provokes disease.
ALDH1A1 (RALDH1) is reported to have a role in a number of key metabolic processes involved in WAT physiology, including the browning of WAT. These actions were originally proposed to involve the actions of retinaldehyde as a transcriptional regulator. Later studies suggested that retinoic acid also contributes to the ALDH1A1-mediated effects in WAT. However, a recently published report proposes that neither retinaldehyde nor retinoic acid is involved in mediating ALDH1A1 actions in WAT, suggesting rather that other ALDH1A1 substrate/product combinations may account for the observed effects of ALDH1A1 in WAT. More research will be required if we are to understand whether ALDH1A1 actions in WAT are related to the role of this enzyme in vitamin A metabolism.
A number of other proteins involved in vitamin A metabolism and transport, including RetSat, RBP1, RDH1, and CRAPB2, have been proposed to have significant roles in adipose tissue accrual and adipose-related disease. However, the literature regarding the actions of these and other vitamin A-related proteins in metabolic disease is still relatively limited and in its early stages. Clearly more research in this area will be needed if we are truly to understand the actions of these and other vitamin A-related proteins in metabolic disease.
In summary, the literature is convincing that vitamin A and related proteins have roles in the development and prevention of obesity and diseases related to obesity. But the literature is not sufficiently mature to allow for definitive conclusions to be drawn regarding whether these effects involve a common mechanistic pathway involving retinoic acid signaling or multiple different pathways. This is the key question that needs to be addressed in future research if we are to understand how vitamin A and related proteins act in metabolic disease development and prevention.
ACKNOWLEDGEMENTS.
The author receives support from National Institutes of Health Grants R01 DK068437 and R01 DK101251. This grant support was responsible for cited research that was carried out in the author’s laboratory and partially allowed for the writing of this review.
ABBREVIATIONS.
- ALDH1A1
Aldehyde dehydrogenase 1A1 (Retinaldehyde dehydrogenase 1 (RALDH1))
- APL
Acute promyelocytic leukemia
- BAT
Brown adipose tissue
- BMI
Body mass index
- CaMKII
Calcium-calmodulin protein kinase II
- C/EBPα
CCAAT/enhancer-binding protein-α
- CRABP1
Cellular retinoic acid-binding protein type 1
- CRABP2
Cellular retinoic acid-binding protein type 2
- Erk 1/2
Extracellular signal-regulated kinase ½
- ES
Embryonic stem
- FABP5
Fatty acid-binding protein 5
- FGF21
Fibroblast growth factor 21
- HCC
Hepatocellular carcinoma
- HSC
Hepatic stellate cell
- IP
Intraperitoneal
- JAK2
Janus kinase 2
- JNK
c-Jun-N-terminal
- LpL
Lipoprotein lipase
- MEF
Mouse embryonic fibroblast
- NAFLD
Nonalcoholic fatty liver disease
- NASH
Nonalcoholic steatohepatitis
- p38MAPK
p38 Mitogen-activated protein kinase
- PGC-1α
Peroxisome proliferator-activator receptor activator 1-α
- PNPLA3
Patatin-like phospholipase domain-containing 3
- PPARα
Peroxisome proliferator-activated receptor α
- PPAR β/δ
Peroxisome proliferator-activator receptor β/δ
- PPAR γ
Peroxisome proliferator-activator receptor γ
- RARα
Retinoic acid receptor α
- RARβ
Retinoic acid receptor β
- RARγ
Retinoic acid receptor γ
- RBP1
Retinol-binding protein 1 (or cellular retinol-binding protein, type 1)
- RBP4
Retinol-binding protein 4 (or serum retinol-binding protein (RBP))
- RetSat
Retinol saturase
- RXRα
Retinoid X receptor α
- RXRβ
Retinoid X receptor β
- RXRγ
Retinoid X receptor γ
- SOCS3
Suppressor of cytokine signaling 3
- SNP
Single nucleotide polymorphism
- STRA6
Stimulated by retinoic acid 6
- TLR4
Toll-like receptor 4
- TNFα
Tumor necrosis factor-α
- UCP1
Uncoupling protein-1
- WAT
White adipose tissue
- Zfp423
Zinc-finger protein 423
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST STATEMENT
The author declares that there are no conflicts of interest.
REFERENCE LIST
- Abumrad NA, & Davidson NO (2012) Role of the gut in lipid homeostasis. Physiol. Rev 92, 1061–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agadir A, & Chomienne C (1999). Retinoids and diffentiation of normal and malignant hematopoietic cells In Nau H & Blaner WS (Eds), Retinoids: The Biochemical and Molecular Basis of Vitamin A and Retinoid Actions (pp. 277–300). Berlin Heidelberg: Springer-Verlag. [Google Scholar]
- Akbay E, Muslu N, Nayir E, Ozhen O, & Kiykim A (2010) Serum retinol binding protein 4 level is related with renal function in Type 2 diabetes. J. Endocrinol. Invest 33, 725–729. [DOI] [PubMed] [Google Scholar]
- Alkharfy KM, Al-Daghri NM, Vanhoutte PM, Krishnaswarmy S, Xu A. 2012. Serum retinol-binding protein 4 as a marker for cardiovascular disease in women. PLoS One 7(10):e48612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allenby G, Bocquel M-T, Saunders M, Kazmer S, Speck J, Bosenberger M, Lovey A, Kastner P, Grippo JF, Chambon P, & Levin AA (1993) Retinoic acid receptors and retinoid X receptors: Interactions with endogenous retinoic acids. Proc. Natl. Acad. Sci. USA 90, 30–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Tanoury Z, Piskunov A, and Rochette-Egly C (2013) Vitamin A and retinoid signaling: genomic and nongenomic effects. J. Lipid Res 54, 761–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez R, de Andrés J, Yubero P, Viñas O, Manpel T, Iglesias R, Giralt M, & Villarroya F (1995) A novel regulatory pathway of brown fat thermogenesis. J. Biol. Chem 270, 5666–5673. [DOI] [PubMed] [Google Scholar]
- Alvarez R Checa ML, Brun S, Viňas O, Mampel T, Iglesias R, Giralt M, & Villarroya F (2000) Both retinoic-acid-receptor- and retinoid-X-receptor-dependent signaling pathways mediate the induction of the brown-adipose-tissue-uncoupling-protein-1 gene by retinoids. Biochem. J 345, 91–97. [PMC free article] [PubMed] [Google Scholar]
- Amengual J, Petrov P, Bonet ML, Ribot J & Palou A (2012) Induction of carnitine palmitoyl transferase 1 and fatty acid oxidation by retinoic acid in HepG2 cells. Internal. J. Bochem. Cell Biol 44, 2019–2017. [DOI] [PubMed] [Google Scholar]
- Anstee QM, & Day CP (2015) The genetics of nonalcoholic fatty liver disease: Spotlight on PNPLA3 and TM6SF2. Semin. Liver Dis 35, 270–290. [DOI] [PubMed] [Google Scholar]
- Arnold SLM, Amory JK, Walsh TJ, & Isoherranen N (2012) A sensitive and specific method for measurement of multiple retinoids in human serum with UHPLC-MS/MS. J. Lipid Res 53, 587–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajzová M, Kováčiková M, Víitková M, Klimčková E, Polák J, Kováčiková Z, Vigurie N, Vedral T, Mikulášek L, Šrámkova P, Srp A, Hejnová J, Langin D, & Stich V (2008) Retinol-binding protein 4 expression in visceral and subcutaneous fat in human obesity. Physiol. Res 57, 927–934. [DOI] [PubMed] [Google Scholar]
- Balmer JE and Blomhoff R (2001) Gene expression regulation by retinoic acid. J. Lipid Res 43, 773–1808. [DOI] [PubMed] [Google Scholar]
- Barua AB, Batres RO, & Olson JA (1980) Characterization of retinyl beta-glucuronide in human blood. Am. J. Clin. Nutr 50, 370–374. [DOI] [PubMed] [Google Scholar]
- Bazewicz CG, Dinavahi SS, Schell TD, & Robertson GP (2018) Aldehyde dehydrogenase in regulatory T-cell development, immunity and cancer. Immunology 156:47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry DC & Noy N (2009) All-trans-retinoic acid represses obesity and insulin resistance by activating both peroxisome proliferation-activated receptor β/δ and retinoic acid receptor. Mol. Cell. Biol 29, 3286–3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry DC, Soltanian H, & Noy N (2010) Repression of cellular retinoic acid-binding protein II during adipocyte differentiation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry DC, Jin H, Majumdar A, & Noy N (2011) Signaling by vitamin A and retinol-binding protein regulates gene expression to inhibit insulin responses. Proc. Natl. Acad. Sci. USA 108, 4340–4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry DC, DeSantis D, Soltanian H, Croniger CM, & Noy N (2012a) Retinoic acid upregulates preadipocyte genes to block adipogenesis and suppress diet-induced obesity. Diabetes 61, 1112–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry DC, O’Byrne SM, Vreeland AC, Blaner WS, & Noy N (2012b) Cross talk between signaling and vitamin A transport by the retinol-binding protein receptor STRA6. Mol. Cell. Biol 32, 3164–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry DC, Jacobs H, Marwarha G, Gely-Pernot A, O’Byrne SM, DeSantis D, Klopfenstein M, Feret B, Dennefeld C, Blaner WS, Croniger CM, Mark M, Noy N, & Ghyselinck NB (2013) The Stra6 receptor is essential for retinol-binding protein-induced insulin resistance but not for maintaining vitamin A homeostasis in tissues other than the eye. J. Biol. Chem 288, 24528–24539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas MG, & Russell DW (1997) Expression cloning and characterization of oxidative 17β- and 3α-hydroxysteroid dehydrogenases from rat and human prostate. J. Biol. Chem 272, 15959–15966. [DOI] [PubMed] [Google Scholar]
- Blaner WS, Henriks HF, Brouwer A, de Leeuw AM, Knook DL, & Goodman DS (1985) retinoids, retinoid-binding proteins, and retinyl palmitate hydrolase distributions in different types of rat liver cells. J. Lipid Res 26, 1241–1251. [PubMed] [Google Scholar]
- Blaner WS, Obunike JC, Kurlandsky SB, al-Haideri M, Piantedosi R, Deckelbaum RJ, & Goldbert IJ. (1994) Lipoprotein lipase hydrolysis of retinyl ester. Possible implications for retinoid uptake by cells. J. Biol. Chem 269, 16559–16565. [PubMed] [Google Scholar]
- Blaner WS, O’Byrne SM, Wongsiriroj N, Kluwe J, D’Ambrosio DD, Jiang H, Schwabe RF, Piantedosi R, Hillman ECM & Libien J (2009) Hepatic stellate cell lipid droplets: A specialized lipid droplet for retinoid storage. Biochem. Biophys. Acta 1791, 467–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaner WS, & Li Y (2015). Vitamin A metabolism, storage and tissue delivery mechanisms In Dollé P & Niederreither K (Eds.), The Retinoids; Biology, Biochemistry, and Disease (pp. 3–34). Hoboken, New Jersey: WILEY Blackwell. [Google Scholar]
- Blaner WS, Li Y, Brun PJ, Yuen JJ Lee SA, & Clugston RD (2016) Vitamin A absorption, storage and metabolism. Subcell. Biochem 81, 95–125. [DOI] [PubMed] [Google Scholar]
- Blomhoff R, Helgerud P, Rasmussen M, Berg T, & Norum KR (1982). In vivo uptake of chylomicron [3H]retinyl ester by rat liver: evidence for retinol transfer from parenchymal to nonparenchymal cells. Proc. Natl. Acad. Sci. USA 79, 7326–7330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomhoff R, Holte K, Naess L, & Berg T (1984) Newly administered [3H]retinol is transferred from hepatocytes to stellate cells in liver for storage. Exp. Cell Res 150, 186–193. [DOI] [PubMed] [Google Scholar]
- Blomhoff R, Rasmussen M, Nilsson A, Norum KR, Berg T, Blaner WS, Kato M, Mertz JR, Goodman DS, Eriksson U, & Peterson PA (1985) Hepatic retinol metabolism. Distribution of retinoids, enzymes, and binding proteins in isolated rat liver cells. J. Biol. Chem 260, 13560–13565. [PubMed] [Google Scholar]
- Blomhoff R, Berg T, & Norum KR (1988) Transfer of retinol from parenchymal to stellate cells in liver is mediated by retinol-binding protein. Proc. Natl. Acad. Sci. USA 85, 3455–3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomhoff R, Green MH, Green JB, Berg T, & Norum KR (1991) Vitamin A metabolism: new perspectives on absorption, transport, and storage. Physiol. Rev 71, 951–990. [DOI] [PubMed] [Google Scholar]
- Borland MG, Foreman JE, Girroir EE, Zolaghari R, Sharma AK, Amin S, Gonzalez FJ, Ross AC, & Peters JM (2008) Ligand activation of peroxisome proliferator-activated receptor-β/δ inhibits cell proliferation in human HaCaT keratinocytes. Mol. Pharmol 74, 1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borland MG, Khozoie C, Albrect PP, Zhu B, Lee C, Lahoti TS, Gonzalez FJ, & Peters JM (2011) Stable over-expression of PPARβ/δ and PPARγ to examine receptor signaling in human HaCaT keratinocytes. Cellular Signalling 23, 2039–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bost F, Caron L, Marchetti I, Dani C, & Le Marchand-Brustel Y (2002) Retinoic acid activation of the ERK pathway is required for embryonic stem cell commitment into the adipocyte lineage. Biochem J. 361, 621–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourguet W, & Moras E (2015). Retinoid receptors: Protein structure, DNA recognition and structure-function relationships In Dollé P & Niederreither K (Eds.), The Retinoids; Biology, Biochemistry, and Disease (pp. 131–150). Hoboken, New Jersey: WILEY Blackwell. [Google Scholar]
- Brun P-J, Yang KJZ, Lee S-A, Yuen JJ & Blaner WS (2013) Retinoids: Potent regulators of metabolism. BioFactors 39, 151–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brun P-J, Wongsiriroj N, & Blaner WS (2016) Retinoids in the pancreas. Hepatobiliary Surg. Nutr 5, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Muñozledo F, Marsch-Moreno M, Beltràn-Langarica A, & Kuri-Harcuch W (1987) Commitment of adipocyte differentiation in 3T3 cells is inhibited by retinoic acid, and the expression of lipogenic enzymes is modulated through cytoskeleton stabilization. Differentiation 36, 211–219. [DOI] [PubMed] [Google Scholar]
- Chambon P (1996) A decade of molecular biology of retinoic acid receptors. FASEB J. 10, 940–954. [PubMed] [Google Scholar]
- Chavez AO, Coletta DK, Kamath S, Cormack DT, Monroy A, Folli F, DeFronzo RA, & Tripathy D (2009) Retinol-binding protein 4 is associated with impaired glucose tolerance but not with whole body or hepatic insulin resistance in Mexican Americans. Am. J. Physiol. Endocrinol. Metab 296, E758–E764. [DOI] [PubMed] [Google Scholar]
- Chen Y, Clarke OB, Kim J, Stowe S, Kim YK, Assur Z, Cavalier M, Godoy-Ruiz R, von Alpen DC, Manzini C, Blaner WS, Frank J, Quadro L, Weber DJ, Shapiro L, Hendrickson WA, & Mancia F (2016) Structure of the STRA6 receptor for retinol uptake. Science 353(6302). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Shen T, Li Q, Chen X, Li Y, Li D, Chen G, Ling W, & Chen YM (2017) Retinol binding protein 4 levels and non-alcoholic fatty liver disease: A community-based cross sectional study. Sci. Rep 7:45100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X & Zhang H (2009) Serum retinal-binding protein 4 is positively related to insulin resistance in Chinese subjects with type 2 diabetes. Diabetes Res. Clin. Pract 84, 58–60. [DOI] [PubMed] [Google Scholar]
- Cia H, Lu S, Chen Y, Das S, Niu Z, Zhuo G, Lai L, & Zhang Z Serum retinol binding protein 4 and galectin-3 binding protein as novel markers for postmenopausal nonalcoholic fatty liver disease. Clin. Biochem 2018, (in press). [DOI] [PubMed] [Google Scholar]
- Cooper AD (1992) Hepatic clearance of plasma chylomicron remnants. Semin. Liver Dis 12, 386–396. [DOI] [PubMed] [Google Scholar]
- Codoñer-French P, Carrasco-Luna J, Allepuz P, Codoñer-Alejos A, & Guillem V (2016) Association of RBP4 genetic variants with childhood obesity and cardiovascular risk factors. Pediatric Diabetes 17, 576–583. [DOI] [PubMed] [Google Scholar]
- Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB, Kuo FC, Palmer EL, Tseng YH, Doria A, Kolodny GM, & Kahn CR (2009) Identification and importance of brown adipose tissue in humans. N. Engl. J. Med 360, 1509–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalmas E, Lehmann FM, Dror E, Wueest S, Thienot C, Borsigova M, Stawiski M, Traunecker E, Lucchini FC, Dapito DH, Kaliart SM, Guigas B, Pattou F, Kerr-Conte J, Maechier P, Girard J-P, Konrad D, Wolfrum C, Böni-Schnetzler M, Finke D, & Donath MY (2017) lnterleukin-33-activated islet-resistant innate lymphoid cells promote insulin secretion through myeloid cell retinoic acid production. Immunity 47:928–942. [DOI] [PubMed] [Google Scholar]
- D’Ambrosio DN, Clugston RD, and Blaner WS (2011) Vitamin A metabolism: An update. Nutrients 3, 63–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Aniello ED, & Waxman JS (2015) Input overload: Contributions of retinoic acid signaling feedback mechanisms to heart development and teratogenesis. Developmental Dynamics 244, 513–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dave S, Nanduri R, Dkhar HK, Bhagyaraj E, Rao A, & Gupta P (2012) Nuclear MEKI1 sequesters PPARγ and bisects MEK1/ERK signaling: A non-canonical pathway of retinoic acid inhibition of adipocyte differentiation. PLoS ONE 9:el00862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweich L. de Carvalho, de Oliveira E.J. Tores, Pesarini JR, Hermento L. Corrêa, Camassola M, Nardi N. Beyer, Brochado T.M. Milan, Antoniolli-Silva A.C. Milan Brochado, & Oliverira RJ (2017) All-trans retinoic acid induces mitochondrial-mediated apoptosis of human adipose-derived stem cells and affects the balance of adipogenic differentiation. Biomed. Pharmacotherapy 96, 1267–1274. [DOI] [PubMed] [Google Scholar]
- Olivria L. de Mendoca, Teixeira FME, & Sato M. Natoni (2018) Impact of retinoic acid on immune cells and inflammatory disease. Mediators Inflamm. 2018:3067126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W, Zhang Y, Zheng Y, Jiang Y, Wu Q, Liang Z, Yang G, & Chen B (2014) Serum retinol-binding protein 4 levels are elevated but do not contribute to insulin resistance in newly diagnosed Chinese hypertensive patients. Diabetol. Metab. Syndr 6:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dollé P, & Niederreither K (2015). The Retinoids: Biology, biochemistry, and disease. New York: Wiley Blackwell. [Google Scholar]
- Dongiovanni P, Donati B, Fares R, Lombardi R, Mancina RM, Romeo S, & Valenti L (2013) PNPLA3 I148M polymorphism and progressive liver disease. World J. Gastroenterol 19, 6969–6978, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du M, Otalora L, Martin AA, Moiseyev G, Vanlandingham P, Wang Q, Farjo R, Yangeneh A, Quiambao A, and Farjo KM (2015) Transgenic mice overexpressing serum retinol-binding protein develop progressive retinal degeneration through a retinoid-independent mechanism. Mol. Cell. Biol 35, 2771–2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du M, Martin A, Hays F, Johnson J, Farjo RA, & Farjo KM (2017a) Serum retinol-binding protein-induced endothelial inflammation is mediated through the activation of toll-like receptor 4. Mol. Vis 23, 185–197. [PMC free article] [PubMed] [Google Scholar]
- Du M, Phelps E, Balangue MJ, Dockins A, Moiseyev G, Shin Y, Kane S, Otalora L, Ma JX, Farjo R, & Farjo KM (2017b) Transgenic mice over-expressing RBP4 have RBP4-dependent and light-independent retinal degeneration. Invest. Ophthalmol. Vis. Sci 58, 4375–4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echtay KS, Esteves TC, Pakay JL, Jekabsons MB, Lambert AJ, Portero-Otin M, Pamplona R, Vidal-Puig AJ, Wang S, Roebuck SJ & Brand MD (2002) A signaling role for 4-hydroxy-2-nonenal in regulation of mitochondrial uncoupling. EMBO J. 22, 4103–4110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldaim AAA, Matsuoka S, Okamatsu-Ogura Y, Kamikawa A, Ahmed MM, Terao A, Nakajima K-I, & Kimura K (2017) Retinoic acid modulates lipid accumulation glucose concentration dependently through inverse regulation of SREBP-1 expression in 3T3L1 adipocytes. Genes to Cells 22, 568–582. [DOI] [PubMed] [Google Scholar]
- Eriksson U, Das K, Busch C, Nordlinder H, Rask L, Sundelin J, Sallstrom J, & Peterson PA (1984) Cellu7lar retinol-binding protein. Quantitation and distribution. J. Biol. Chem 259, 13464–13470. [PubMed] [Google Scholar]
- Erkstrup C, Mortensen OH, Nielsen AR, Fischer CP, Plomgaard P, Petersen AM, Krough-Madsen R, Lindegaard B, Erhardt JG, Ullum H, Benn CS, & Pedersen BK (2009) RBP-to-retinol ratio, but not total RBP, is elevated in patients with type 2 diabetes. Diabetes Obes. Metabol 11, 204–209. [DOI] [PubMed] [Google Scholar]
- Farjo KM, Farjo RA, Halsey S, Moiseyev G, & Ma JX (2012) Retinol-binding protein 4 induces inflammation in human endothelial cells by an NADPH oxidase- and nuclear factor kappa B-dependent and retinol-independent mechanism. Mol. Cell. Biol 32, 5103–5115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman HM, Golozoubova V, Cannon B, & Nedergaard J (2009) UCP1 ablation induces obesity and abolishes diet-induced thermogenesis in mice exempt from thermal stress by living at thermoneutrality. Cell Metabolism 9, 203–209. [DOI] [PubMed] [Google Scholar]
- Fenaux P, Chastang C, Chevret S, Sanz M, Dombert H, Archimbaud E, Fey M, Rayon C, Huguet F, Sotto J-J, Gardin C, Makhoul PC, Travade P, Solary E, Fegueux N, Bordessoule D, San Miguel J, Link H, Desablens B, Stamatoullas A, Deconinck E, Maloisel F, Castaigne S, Preudhomme C, & Degos L for the European APL Group. (1999) A randomized comparison of all transretinoic acid (ATRA) followed by chemotherapy and ATRA plus chemotherapy and the role of maintenance therapy in newly diagnosed acute promyleocytic leukemia. Blood 94, 1192–1200. [PubMed] [Google Scholar]
- Feng S, Zhu Y, Yan C, Wang Y, & Zhang Z (2015) Retinol binding protein 4 correlates with and is an early predictor of carotid atherosclerosis in type 2 diabetes mellitus patients. J. Biomed. Res 3, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante AW Jr (2013) Macrophages, fat, and the emergence of immunometabolism. J. Clin. Invest 123, 4992–4993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fotbolcu H & Zorlu E (2016) Nonalcoholic fatty liver disease is a multi-systemic disease. World J. Gastroenterol 22, 4079–4090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friebe D, Kovacs P, Neff M, Bldher S, Schleinitz D, Kiess W, & Komer A (2011) The promoter variant −803 G>A in the RBP4 gene is not associated with BMI, metabolic parameters or blood pressure in Caucasian children. Exp. Clin. Endocrinol. Diabetes 119, 628–632. [DOI] [PubMed] [Google Scholar]
- Friedman S,L. (2008) Hepatic Stellate Cells: Protean, Multifunctional, and Enigmatic Cells of the Liver. Physiological Rev. 88, 125–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavi S, Qurashi S, Stuart LM, Lau R, Melendez MM, Mynarcik DC, McNurlan MA, & Gelato MC (2008) Influence of age on the assocaition of retinol-binding protein 4 with metabolic syndrome. Obesity 16, 893–895. [DOI] [PubMed] [Google Scholar]
- Geerts A (2001) History, heterogeneity, developmental biology, and functions of quiescent hepatic stellate cells. Semin. Liver Dis 21, 311–335. [DOI] [PubMed] [Google Scholar]
- Ghyselinck NB, Bavik C, Sapin V, Mark M, Bonnier D, Hindelang C, Dierich A, Nilsson CB, Hakansson H, Sauvant P, Azai’s-Braesco V, Frasson M, Picaud S, & Chambon P (1999) Cellular retinol-binding protein 1 is essential for vitamin A homeostasis. EMBO J. 18, 4903–4914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gliniak CM, Brown JM, & Noy N (2017) The retinol binding-protein receptor STRA6 regulates diurnal insulin responses. J. Biol. Chem 292, 15080–15093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman E, Graham TE, Dolan LM, Daniels SR, Goodman ER, & Kahn BB (2009) The relationship of retinol binding protein 4 to changes in insulin resistance and cardiometabolic risk in overweight black adolescents. J. Pediatr 154, 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham TE, Yang Q, BlCiher M, Hammarstedt A, Ciaraldi TP, Henry RR, Wason CJ, Oberbach A, Jansson P-A, Smith U, & Kahn BB (2006) Retinol-Binding Protein 4 and Insulin Resistance in Lean, Obese, and Diabetic Subjects. New England J. Med 354, 2552–2563. [DOI] [PubMed] [Google Scholar]
- Graham TE, Wasson CJ, Bldher M, & Kahn BB (2007) Shortcomings in methodology complicate measurement of serum retinol binding protein (RBP4) in insulin-resistant human subjects. Diabetologia 50, 814–823. [DOI] [PubMed] [Google Scholar]
- Gudas LJ (2011) Retinoids, retinoic acid receptors, and cancer. Annu. Rev. Pathol. Mech. Dis 6, 345–364. [DOI] [PubMed] [Google Scholar]
- Haenisch M, Treuting PM, Brabb T, Goldstein AS, Berkseth K, Amory JK, & Paik J (2018) Pharmacological inhibition of ALDH1A enzymes suppresses weight gain in a mouse model of diet-induced obesity. Obes. Res. Clin. Pract 12, 93–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidenreich S, Witte N, Weber P, Goehring I, Tolkachov A, von Loeffelholz C, Docke S, Bauer M, Stockmann M, Pfeiffer AFH, Birkenfeld AL, Pietzke M, Kempa S, Muenzner M, & Schupp M (2017) Retinol saturase coordinates liver metabolism by regulating ChREBP activity. Nature Communications 8, 384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henze A, Frey SK, Raila J, Tepel M, Schotze A, Pfeiffer AFH, Weikert MO, Spranger J, &Schweigert FJ (2008) Diabetes 57, 3323–3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyman RA, Mangelsdorf DJ, Dyck JA, Stein RB, Eichele G, Evans RM, & Thaller C (1992) 9-Cis retinoic acid is a high affinity lignd for the retinoid X receptor. Cell 68, 397–406. [DOI] [PubMed] [Google Scholar]
- Hiebl V, Landurner A, Latkolik S, & Dirsch VM (2018) Natural products as modulators of the nuclear receptors and metabolic sensors LXR, FXR and RXR. Biotechnology Advances (in press). [DOI] [PubMed] [Google Scholar]
- Hoggard N, Agouni A, Mody N, & Delibegovic M (2012) Serum levels of RBP4 and adipose tissue levels of PTP-1B are increased in obese men resident in northeast Scotland without associated changes in ER stress response genes. Int. J. Gen. Med 5, 403–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong WK, & Lotan R (1993) Retinoids in Oncology, (pp. 1–338). New York: Marcel Dekker, Inc. [Google Scholar]
- Hu C, Jia W, Zhang R, Wang C, Lu J, Wu H, Fang Q, Ma X & Xiang K (2008) Effect of RBP4 gene variants on circulating RBP4 concentration and type 2 diabetes in a Chinese population. Diabet. Med 25, 11–18. [DOI] [PubMed] [Google Scholar]
- Huang SC, & Yang YJ (2013) Serum retinol-binding protein 4 is independently associated with pediatric NAFLD and fasting triglyceride level. J. Pediatr. Gastroenterol. Nutr 56, 145–150. [DOI] [PubMed] [Google Scholar]
- Janke J, Engele S, Boschmann M, Adams F, Bohnke J, Luft FC, Sharma AM, & Jordan J (2006) Retinol-binding protein 4 in human obesity. Diabetes 55, 2805–2810. [DOI] [PubMed] [Google Scholar]
- Jeyakumar SM, Vajreswari A, & Giridharan NV (2006) Chronic dietary vitamin A supplementation regulates obesity in an obese mutant WNIN/ob rat model. Obesity 13, 52–59. [DOI] [PubMed] [Google Scholar]
- Jeyakumar SM, Sheril A, & Vajreswari A (2015) Chronic vitamin A-enriched diet feeding induces body weight gain and adiposity in lean and glucose-intolerant obese rats of WNIN/GR-Ob strain. Exp. Physiol 10011, 1352–1361. [DOI] [PubMed] [Google Scholar]
- Jing J, Isoherranen N, Robinson-Cohen C, Petrie I, Kestenbaum BR, & Yeung CK (2016) Chronic kidney disease alters vitamin A homeostasis via effects on hepatic RBP4 protein expression and metabolic enzymes. Clin. Transl. Sci 9, 207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamei Y, Kawada T, Mizukami J, & Sugimoto E (1994) The prevention of adipose differentiation of 3T3-L1 cells caused by retinoic acid is elicited through retinoic acid receptor alpha. Lif Sci. 55, PL307–312. [DOI] [PubMed] [Google Scholar]
- Kanai M, Raz A, & Goodman DS (1968) Retinol-binding protein: The transport protein for vitamin A in human plasma. J. Clin Invest 47, 2025–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane MA, Folias AE, & Napoli JL (2008a) HPLC/UV quantitation of retinal, retinol, and retinyl esters in serum and tissues. Anal. Blochem 378, 71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane MA, Folias AE, Wang C, & Napoli JL (2008b) Quantitative profiling of endogenous retinoic acid in vivo and in vitro by tandem mass spectrometry. Anal. Chem 80, 1702–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karkeni E, Bonnet L, Astier J, Couturier C, Dalifard J, Tourniaire F, & Landrier J-F (2017) All-trans-retinoic acid represses chemokine expression in adipocytes and adipose tissue by inhibiting NF-κB signaling. J. Nutr. Biochem 42, 101–107. [DOI] [PubMed] [Google Scholar]
- Kedishvili NY (2013) Enzymology of retinoic acid biosynthesis and degradation. J. Lipid Res 54, 1744–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiefer FW, Orasanu G, Nallamshetty S, Brown JD, Wang H, Luger P, Qi NR, Burant CF, Duester G, & Plutzky J (2012a) Retinaldehyde dehydrogenase 1 coordinates hepatic gluconeogenesis and lipid metabolism. Endocrinology 153, 3089–3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiefer FW, Vernochet C, O’Brein P, Spoerl S, Brown JD, Nallamshetty S, Zeyda M, Stulnig TM, Cohen DE, Kahn CR & Plutzky J (2012b) Retinaldehyde dehydrogenase 1 regulates a thermogenic program in white adipose tissue. Nature Medicine 18, 918–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DM, Choi H-R, Park A, Shin S-M, Bae K-H, Lee SC Kim 1-C, & Kim WK (2013) Retinoic acid inhibits adipogenesis via activation of Wnt signaling pathway in 3T3-L1 preadipocytes. Biochem. Biophys. Res. Commun 434, 455–459. [DOI] [PubMed] [Google Scholar]
- Kos K, Wong S, Tan BT, Kerrigan D, Randeva HS, Pinkney JH, & Wilding JPH (2011) Human RBP4 adipose tissue expression is gender specific and influenced by leptin. Clin. Endrocrinol 74, 197–205. [DOI] [PubMed] [Google Scholar]
- Kovacs P, Geyer M, Berndt J, Kloting N, Graham TE, Bottcher Y, Enigk B, Tonjes A, Schleinitz D, Schon MR, Kahn BB, BlOher M, & Stumvoll M (2007) Effects of genetic variation in human retinol binding protein-4 (RBP4) on insulin resistance and fat depot-specific mRNA expression. Diabetes 56, 3095–3100. [DOI] [PubMed] [Google Scholar]
- Kruse SW, Suino-Powell K, Zhou R, Kretschman JE, Reynolds R, Vonrhein C, Xu Y, Wang I, Tsai SY, Tsai MJ, & Xu HE (2008) Identification of COUP-TFII orphan nuclear receptor as a retinoic acid-activated receptor PLoS Biol. 6:e221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar MV & Scarpase PJ (1998) Differential effects of retinoic acid on uncoupling protein-1 and leptin gene expression. J. Endocrinology 157, 237–243. [DOI] [PubMed] [Google Scholar]
- Kumar MV, Sunvold GD, & Scarpase PJ (1999) Dietary vitamin A supplementation in rats: Suppression of leptin and induction of UCP1 mRNA. J. Lipid Res 40, 824–829. [PubMed] [Google Scholar]
- Kuri-Harcuch W (1982) Differentiation of 3T3-F442A cells into adipocytes is inhibited by retinoic acid. Differentiation 23, 164–169. [DOI] [PubMed] [Google Scholar]
- Landrier JF, Kasiri E, Karkeni E, Mihaly J, Beke G, Weiss K, Lucas R, Aydemir G, Salles J, Waltrand S, de Lera AR, & ROhl R (2017) Reduced adiponectin expression after high-fat diet is associated with selective up-regulation of ALDH1A1 and further retinoic acid receptor signaling in adipose tissue. FASEB J. 31, 203–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laursen KB & Gudas LJ (2018) Combinatorial knockout of RARα, RARβ, and RARγ completely abrogates transcriptional responses to retinoic acid in murine embryonic stem cells. J. Biol. Chem 293, 11891–11900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JW, Im JA, Park KD, Lee HR, Shim JY, & Lee DC (2009) Retinol binding protein 4 and insulin resistance in apparently healthy elderly subjects. Clin. Chim. Acta 400, 30–32. [DOI] [PubMed] [Google Scholar]
- Lee SA, Yuen JJ, Jiang H, Kahn BB, & Blaner WS (2016) Adipocyte-specific overexpression of retinol-binding protein 4 causes hepatic steatosis in mice. Hepatology 64, 1534–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis JG, Shand BI, Frampton CM, Elder PA, & Scott RS (2008) Plasma retinol-binding protein is not a marker of insulin resistance in overweight subjects: a three year longitudinal study. Clin. Biochem 41, 1034–1038. [DOI] [PubMed] [Google Scholar]
- Li Y, Wong K, Walsh K, Gao B, & Zang M (2013) Retinoic acid receptor β stimulates hepatic induction of fibroblast growth factor 21 to promote fatty acid oxidation and control whole-body energy homeostasis in mice. J. Biol. Chem 288, 10490–10504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z-Z, Lu Z-Z, Liu J-B, & Chen L (2010) Serum retinol-binding protein levels in patients with diabetic retinopathy. J. Internat. Med. Res 38, 95–99. [DOI] [PubMed] [Google Scholar]
- Lin Y-L, Persaud SD, Nhieu J, & Wei L-N (2017) Cellular retinoic acid-binding protein 1 modulates stem cell proliferation to affect learning and memory in male mice. Endocrinology 158, 3004–3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, & Gudas LJ (2005) Disruption of the lecithin:retinol acyltransferase gene makes mice more susceptible to vitamin A deficiency. J. Biol. Chem 280, 40226–40234. [DOI] [PubMed] [Google Scholar]
- Liu L, Tang XH, & Gudas LJ (2008) Homeostasis of retinol in lecithin:retinol acyltransferase gene knockout mice fed a high retinol diet. Biochem. Pharmacol 75, 2316–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Chen H, Mu D, Fan J, Song J, Zhong Y, Li D, & Xia M (2016) Circulating retinoic acid levels in the development of metabolic syndrome. J. Clin. Endocrinol. Metab 104, 1686–1692. [DOI] [PubMed] [Google Scholar]
- Makia N, Bojang P, Falkner KC, Conklin DJ, & Prough RA (2011) Murine hepatic aldehyde dehydrogenase lal is a major contributor to oxidation of aldehydes formed by lipid peroxidation. Chemico-Biological Interactions 191, 278–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolescu D-C, Sima A, & Bhat P (2009) All-trans retinoic acid lowers serum retinol-binding protein 4 concentrations and increases insulin sensitivity in diabetic mice. J. Nutr 140, 311–316. [DOI] [PubMed] [Google Scholar]
- Marchildon F, St. Louis C Akter R, Roodman V, & Wiper-Bergeron NL (2010) Transcription factor Smad3 is required for the inhibition of adipogenesis by retinoic acid. J. Biol. Chem 285, 13274–13284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masia M, Alvarez S, de Lera AR, & Barettino D (2007) Rapid, nongenomic actions of retinoic acid on phosphatidylinositol-3-kinase signaling pathway mediated by the retinoic acid receptor. Molecular Endocrinology 21, 2391–2402. [DOI] [PubMed] [Google Scholar]
- Mathis D (2013) Immunological goings-on in visceral adipose tissue. Cell Metabol. 17, 851–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melhus H, Milsson T, Peterson PA, & Rask L (1991) Retinol-binding protein and transthyretin expressed in HeLa cells form a complex in the endoplasmic reticulum in both the absence and the presence of retinol. Exp. Cell Res 197, 119–124. [DOI] [PubMed] [Google Scholar]
- Mendoza-Parra M-A, Bourguet W, de Lera AR, & Gronemeyer H (2015). Retinoid receptor-selective modulators: Chemistry, 3D structures and systems biology In Dollé P & Niederreither K (Eds.), The Retinoids; Biology, Biochemistry, and Disease (pp. 165–192). Hoboken, New Jersey: WILEY Blackwell. [Google Scholar]
- Mercader J, Ribot J, Murano I, Felipe F, Cinti S, Bonet ML, & Palou A (2006) Remodeling of white adipose tissue after retinoic acid administration in mice. Endocrinology. 147, 5325–5332. [DOI] [PubMed] [Google Scholar]
- Mercader J, Madsen L, Felipe F, Palou A, Kristiansen K, & Bonet ML (2007) All-trans retinoic acid increases oxidative metabolism in mature adipocytes. Cell. Physiol. Biochem 20, 1061–1072. [DOI] [PubMed] [Google Scholar]
- Mercader J, Granados N, Bonet ML, & Palou A (2008) All-irons retinoic acid decreases murine adipose retinol binding protein 4 production. Cell. Physiol. Biochem 22, 363–372. [DOI] [PubMed] [Google Scholar]
- Mercader J, Palou A, & Bonet ML (2010) Induction of uncoupling protein-1 in mouse embryonic fibroblast-derived adipocytes by retinoic acid. Obesity, 18, 655–662. [DOI] [PubMed] [Google Scholar]
- Mills JP, Furr HC, & Tanumihardjo SA (2008) Retinol to retinol-binding protein (RBP) is low in obese adults due to elevated apo-RBP. Exp. Biol. Med (Moywood) 233, 1255–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moise AR, Kuksa V, Imanishi Y, and Palczewski K (2004) Identification of all-trans-retinol:all-trans-13,14-dihydroretinol saturase. J. Biol. Chem 279, 50230–50242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moise AR, Lobo GP, Erokwu B, Wilson DL, Peck D, Alvarez S, Dominguez M, Alvarez R Flask CA, de Lera AR von Lintig J, & Palczewski K (2010) Increased adiposity in the retinol saturase-knockout mouse. FASEB J. 24, 1261–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondul A, Mancina RM, Merlo A, Dongiovanni P, Rametta R, Montalcini T, Valenti L, Albanes D, & Romeo S (2015) PNPLA3 I148M variant influences circulating retinol in adults with nonalcoholic fatty liver disease or obesity. J. Nutr 145, 1687–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montesinos P, & Sanz MA (2011) The Differentiation Syndrome in patients with acute promyelocytic leukemia: Experience of the Pethema Group and review of the literature. Mediterr. J. Hemotol. Infect. Dis 3:e2011059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moraes-Vieira PM, Yore MM, Dwyer PM, Syed I, Aryal P, & Kahn BB (2014) RBP4 activates antigen-presenting cells, leading to adipose tissue inflammation and systemic insulin resistance. Cell Metobol. 19, 512–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moraes-Vieira PM, Castoldi A, Aryal P, Wellenstein K, Peroni OD, & Kahn BB (2016) Antigen presentation and T-cell activation are critical for RBP4-induced insulin resistance. Diabetes 65, 1317–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriwaki H, Blaner WS, Piantedosi R, & Goodman DS (1988) Effects of dietary retinoid and triglyceride on the lipid composition of rat liver stellate cells and stellate cell lipid droplets. J. Lipid Res 29, 1523–1534. [PubMed] [Google Scholar]
- Motani A, Wang Z, Conn M, Siegler K, Zhang Y, Liu Q, Johnstone S, Xu H, Thibault S, Wang Y, Fan P, Connors R, Le H, Xue G, Walker N, Xhan B, & Coward P (2009) Identification and characterization of a non-retinoid ligand for retinol-binding protein 4 which lowers serum retinol-binding protein 4 levels in vivo. J. Biol. Chem 284, 7673–7680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muenzner M, Tuvia N, Deutschmann C, Witte N, Tolkachov A, Valai A, Henze A, Sander LE, Raila J, & Schupp M (2013) Retinol-binding protein 4 and its membrane receptor STRA6 control adipogenesis by regulating cellular retinoid homeostasis and retinoic acid receptor α activity. Mol. Cell. Biol 33, 4068–4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munkhtulga L, Nakayama K, Utsumi N, Yanagisawa Y, Gotoh T, Omi T, Kumada M, Erdenebulgan B, Zolzaya K, Lkhagvasuren T, & Iwamoto S (2007) Identification of a regulatory SNP in the retinol binding protein 4 gene associated with type 2 diabetes in Mongolia. Hum. Genet 120, 879–888. [DOI] [PubMed] [Google Scholar]
- Munkhtulga L, Nagashima S, Nakayama K, Utsumi N, Yanagisawa Y, Gotoh T, Omi T, Kumada M, Zolzaya K, Lkhagvasuren T, Kagawa Y, Fujiwara H, Hosoya Y, Hyodo M, Horie H, Kojima M, Ishibashi S, & Iwamoto S (2010) Regulatory SNP in the RBP4 gene modified the expression in adipocytes and associated with BMI. Obesity (Silver Spring) 18, 1006–1014. [DOI] [PubMed] [Google Scholar]
- Murata M, Saito T, Otani T, Sasaki M, Ikoma A, Toyoshima H, Kawakami M, & Ishikawa S-e. (2009) An increase in serum retinol-binding protein 4 in Type 2 diabetic subjects with nephropathy. Endocrine J. 56, 287–294. [DOI] [PubMed] [Google Scholar]
- Murholm M, Isidor MS, Basse AL, Winther S, Sørensen C, Skovgaard-Petersen J, Nielsen MM, Hansen AS, Quistorff B, & Hansen JB (2013) Retinoic acid has different effects on UCP1 expression in mouse and human adipocytes. BMC Cell Biology 14:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair AK, Sugunan D, Kumar H, & Anikumar G (2010) Case-control analysis of SNPs in GLUT4, RBP4, and STRA6: association of SNPs in STRA6 with type 2 diabetes in a South Indian population. PLoS ONE 5:ell444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napoli JL (2017) Cellular retinoid-binding proteins, CRBP, CRABP, FABP5: Effects on retinoid metabolism, function and related diseases. Pharmocology & Therapeutics 173, 19–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson 0, Isoherranen N, Guo MH, Lui JC Jee YH, Guttmann-Bauman I, Acerini C, Lee W, Allikmets R, Yan ovski JA, Dauber A, & Baron J (2016) Accelerated skeletal maturation in disorders of retinoic acid metabolism: A case report and Focused review of the literature. Norm. Metab. Res 48, 737–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norseen J, Hosooka T, Hammarstedt A, Yore MM, Kant S, Aryal P, Kierman UA, Phillips DA, Maruyama H, Kraus BJ Usheva A, Davis RJ, Smith U, & Kahn BB (2012) retinol-binding protein 4 inhibits insulin signaling in adipocytes by inducing proinflammatory cytokines in macrophages through a c-Jun N-terminal kinase- and toll-like receptor 4-dependent and retinol-independent mechanism. Mol. Cell. Biol 32, 2010–2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noy N (2013) The onetow punch. Retinoic acid suppressess obesity both by promiting energy expenditure and by inhibiting adipogenesis. Adipocyte 2, 184–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noy N (2016a) Non-classical transcriptional activity of retinoic acid. Subcell. Biochem 81, 179–199. [DOI] [PubMed] [Google Scholar]
- Noy N (2016b) Vitamin A in regulation of insulin responsiveness: mini review. Proc. Nutr. Soc 75, 212–215. [DOI] [PubMed] [Google Scholar]
- O’Byrne SM, Wongsiriroj N, Libien J, Vogel S, Goldberg IJ, Baehr W, Palczewski K, & Blaner WS (2005) Retinoid absorption and storage is impaired in mice lacking lecithin:retinol acyltransferase (LRAT). J. Biol. Chem 280, 35647–35657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Byrne SM, & Blaner WS (2013) Retinol and retinyl esters: Biochemistry and physiology. J. Lipid Res 54, 1731–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orban T, Palczewska G, & Palczewski K (2011) Retinyl ester storage particles (retinosomes) from the retinal pigmented epithelium resemble lipid droplets in other tissues. J. Biol. Chem 286, 17248–17258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Öst A, Danielsson A, Lidén M, Ericksson U, Nystrom FH, & Stråalfors P (2018) Retinol-binding protein-4 attenuates insulin-induced phosphorylation of IRS1 and ERK1/2 in primary human adipocytes. FASEBJ. 21, 3696–3704. [DOI] [PubMed] [Google Scholar]
- Palczewski K (2012). Chemistry and Biology of Vision. J. Biol. Chem 287, 1612–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palomer X, Barroso E, Pizarro-Delgado J, Peña L, Botteri G, Zarei M, Aguilar D, Montori-Grau M, & Vázquez-Carrera M (2018) PPARβ/δ: A key therapeutic target in metabolic disorders. Int. J. Mol. Sci 19, 913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J, Guleria RS, Zhu S, & Baker KM (2014) Molecular mechanisms of retinoid receptors in diabetes-induced cardiac remodeling. J. Clin. Med 3, 566–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey GK, Balasubaramanyam J, Balakumar M, Deepa M, Anjana RM, Abhijit S, Kaviya A, Velmurugan K, Miranda P, Balsubramanyam M, Mohan V, & Gokulakrishnan K (2015) Altered circulating levels of retinol binding protein 4 and transthyretin in relation to insulin resistance, obesity, and glucose intolerance in Asian Indians. Endocr. Pract 21, 861–869. [DOI] [PubMed] [Google Scholar]
- Pang X-Y, Wang S, Jurczak MJ, Shulman GI, & Moise AR (2017) Retinol saturase modulates lipid metabolism and the production of reactive oxygen species. Arch Biochem. Biophys 633, 93–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SW, Persaud SD, Ogokeh S, Meyers TA, Townsend DeW., & Wei L-N (2018) CRABP1 protects the heart from isoproterenol-induced acute and chronic remodeling. J. Endocrinology 236, 151–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquali D, Chieffi P, Deery WJ, Nicoletti G, Bellastella A, & Sinisi AA (2005) Differential effects of all-trans-retinoic acid (RA) on Erkl/2 phosphorylation and cAMP accumulation in normal and malignant human prostate epithelial cells: Erkl/2 inhibition restores RA-induced decrease of cell growth in malignant prostate cell. Eur. J. Endocrinology 152, 663–669. [DOI] [PubMed] [Google Scholar]
- Perduca M, Nicolis S, Mannucci B, Balliano M, & Monaco HL (2018a) Human plasma retinol-binding protein (RBP4) is also a fatty acid-binding protein. Biophys. Biophys. Acta 1863, 458–466, [DOI] [PubMed] [Google Scholar]
- Perduca M, Nicolis S, Mannucci B, Galliano M, & Monaco HL (2018b). High resolution crystal structure data of human plasma retinol-binding protein (RBP4) bound to retinol and fatty acids. Data in Brief 18, 1073–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piskunov A & Rochette-Egly C (2012) A retinoic acid receptor RARα pool present in membrane lipid rafts forms complexes with G protein αQ to activate p38MAPK. Oncogene 31, 3333–3345. [DOI] [PubMed] [Google Scholar]
- Pirazzi C, Valenti L, Motta BM, Pingitore P, Hedfalk K, Mancina RM, Burza MA, Indiveri C, Ferro Y, Montalcini T, Maglio C, Dongiovanni P, Fargion S, Rametta R, Pujia A, Andersson L, Ghosai S, Levin M, Wiklund O, lacovino M, Borén J, & Romeo S (2014) PNPLA3 has retinyl-palmitate lipase activity in human hepatic stellate cell. Hum. Mol. Genet 23, 4077–4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puigserver P, Vazquez F, Bonet ML, Pico C, Palou A (1996) In vitro and in vivo induction of brown adipocyte uncoupling protein (thermogenin) by retinoic acid. Biochem. J 317, 827–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Q, Yu Z, Ye X, Zhao F, Huang P, Hu FB, Franco OH, Wang J, Li H, Liu Y, & Lin X (2007) Elevated retinol-binding protein levels are associated with metabolic syndrome in Chinese people. J. Clin. Endocrinol. Metab 92, 4827–4834. [DOI] [PubMed] [Google Scholar]
- Quadro L, Blaner WS, Hamberger L, Novikoff PM, Vogel S, Piantedosi R, Gottesman ME, & Colantuoni V (2004) The role of extrahepatic retinol binding protein in the mobilization of retinoid stores. J. Lipid Res 45, 1975–1982. [DOI] [PubMed] [Google Scholar]
- Raila J, Henze A, Spranger J, Möhlig M, Pfeiffer AFH, & Schweigert FJ (2007) Microalbuminuria is a major determinant of elevated plasma retinol-binding protein 4 in type 2 diabetic subjects. Kidney International 72, 505–511. [DOI] [PubMed] [Google Scholar]
- Ram J, Snehalatha C, Selvam S, Sanditha A, Shetty AS, Godsland IF, Johnston DG, & Ramachandran A (2015) Retinol binding protein-4 predicts incident diabetes in Asian Indian men with prediabetes. Biofactors 41, 160–165. [DOI] [PubMed] [Google Scholar]
- Reichert B, Yasmeen R, Jeyakumar SM, Yang F, Thomou T, Alder H, Duster G, Maiseyeu A, Mihai G, Harrison EH, Rajagopalan S, Kirkland JL, & Ziouzenkova O (2011) Concerted action of aldehyde dehydrogenases influence depot-specific fat formation. Mol. Endocrinol 25, 799–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieck M, Meissner W, Ries S, Müller-Brüsselbach S, & Müller R (2008) Ligand-mediated regulation of peroxisome proliferator-activated receptor (PPAR) β/δ: A comparative analysis of PPAR-selective agonists and all-irons retinoic acid. Mol. Pharmacol 74, 1269–1277. [DOI] [PubMed] [Google Scholar]
- Rhee E-J, Nallamshetty S, & Plutzky J (2012) Retinoid metabolism and its effects on the vascular. Biochim. Biophys. Acta 1821, 230–240. [DOI] [PubMed] [Google Scholar]
- Ribot J, Felipe F, Bonet ML, & Palou A (2004) Retinoic acid administration and vitamin A status modulate retinoid X receptor and retinoic acid receptor alpha levels in brown adipose tissue. Mol. Cell. Biochem 266, 25–30. [DOI] [PubMed] [Google Scholar]
- Rochette-Egly C (2015) Retinoic acid signaling and mouse embryonic stem cell differentiation: Cross talk between genomic and non-genomic effects of RA. Biochim. Biophys. Acta 1851, 66–75. [DOI] [PubMed] [Google Scholar]
- Romanowska A, Lebensztejn DM, Skia E, Trasów E, & Kaczmarski M (2011) retinol binding protein-4 as a serum biomarker of intrahepatic lipid content in obese children—preliminary report. Acta Biochim. Pol 58, 35–38. [PubMed] [Google Scholar]
- Rodondi N, Darioli R, Ramelet A-A, Hohl D, Lenain V, Perdrix J, Wietlisbch V Riesen WF, Walther T, Medinger L, Nicod P, Desvergne B, & Mooser V (2002) High risk of hyperlipidemia and the metabolic syndrome after an episode of hypertriglyceridemia during 13-cis retinoic acid therapy for acne: A pharmacogenetic study. Ann. Intern. Ned 136, 582–589. [DOI] [PubMed] [Google Scholar]
- Ross AC, & Zolfaghari R (2011) Cytochrome P450s in the regulation of cellular retinoic acid metabolism. Anna. Rev. Nutr 31, 65–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rühl R, Krżyzosiak A, Niewiadomska-Cimicka A, Rochel N, Szeles L, Vaz B, Wietrzych-Schindler M, Ȧlvarez S, Szklenar M, Nagy L, de Lera AR, & Krężel W (2015) 9-cis-13,14-Dihydroretinoic acid is an endogenous retinoid acting as RXR ligand in mice. PLoS Genet ll:e1005213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saucedo R, Zarate A, Basurto L, Hernandez M, Puello E, Mendoza-Lorenzo P, & Ostrosky-Wegman P (2014) RBP4 gene variants are associated with insulin resistance in women with previous gestational diabetes. Dis. Markers 2014:269208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schupp M, Lefteova MI, Janke J, Leitner K, Cristancho AG, Mullican SE, Qatanani M, Szwergold N, Steger DJ, Curtin JC, Kim RJ, Shu M-J, Albert MR, Engeli S, Gudas LJ, & Lazar MA (2009) Retinol saturase promotes adipogenesis and is downregulated in obesity. Proc. Natl. Acad. Sci. USA 106, 1105–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz EJ, Reginato MJ, Shao D, Krakow SL, & Lazar MA (1997) Retinoic acid blocks adipogenesis by inhibiting C/EBP-mediated transcription. Mol. Cell. Biol 17, 1552–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw N, Elholm M, & Noy N (2003) Retinoic acid is a high affinity selective ligand for the peroxisome proliferator-activated receptor β/δ. J. Biol. Chem 278, 41589–41592. [DOI] [PubMed] [Google Scholar]
- Sima A, Manolescu D-C, & Bhat P (2011) Retinoids and retinoid-metabolic gene expression in mouse adipose tissue. Biochem. Cell Biol 89, 578–584. [DOI] [PubMed] [Google Scholar]
- Sliverman JF, O’Brein KF, Long S, Leggett N, Khazanie PG, Pories WJ, Norris HT, & Caro JF (1990) Liver pathology in morbidly obese patients with and without diabetes. Am. J. Gastroenterol 85, 1349–1355. [PubMed] [Google Scholar]
- Soprano DR, Pickett CB, Smith JE, & Goodman DS (1981) Biosynthesis of plasma retinol-binding protein in liver as a larger molecular weight precursor. J. Biol. Chem 256, 8256–8258. [PubMed] [Google Scholar]
- Soprano DR, & Blaner WS (1994) Plasma retinol-binding protein, In: Sporn MB, Roberts AB, & Goodman DS (Eds.), The Retinoids, Biology, Chemistry, and Medicine, 2nd Edition, (pp. 257–282). New York: Raven Press. [Google Scholar]
- Sporn MB, Dunlop NM, Newton DL, & Smith JM (1976) Prevention of chemical carcinogenesis by vitamin A and its synthetic analogs (retinoids). Fed Proc 35, 1332–1328. [PubMed] [Google Scholar]
- Stehlin-Gaon C, Willmann D, Zeyer D, Sanglier S, van Dorsselaer A, Renauld JP, Moras D, & Schule R (2003) All-trans-retinoic acid is a ligand for the orphan nuclear receptor ROR beta. Nat. Struct. Biol 10 820–825. [DOI] [PubMed] [Google Scholar]
- Stone RL & Bernlohr DA (1990) The molecular basis for inhibition of adipose conversion of murine 3T3-L1 cells by retinoic acid. Differentiation 45, 119–127. [DOI] [PubMed] [Google Scholar]
- Suhara A, Kato M, & Kanai M (1990) Ultrastructural localization of plasma retinol-binding protein in rat liver. J. Lipid Res 31, 1669–1681. [PubMed] [Google Scholar]
- Sun Q, Kierman UA, Shi L, Phillips DA, Kahn BB, Hu FB, Manson JE, Albert CM, & Rexrode KM (2013) Plasma retinol-binding protein 4 (RBP4) levels and risk of coronary heart disease. Circulation 127, 1938–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Uchida N, Kitanaka C, Sagara C, Imai M, & Takahashi N (2015) Inhibition of ASCT2 is essential in all-trans retinoic acid-induced reduction of adipogenesis in 3T3-L1 cells. FEBS Open Bio. 5, 571–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K, Sriram S, Chan XHD, Ong WK, Yeo CR, Tan B, Lee S-A, Kong KV, Hoon S, Jiang H, Yuen JJ, Perumal J, Agraway M, Vas C, So J, Shabbir A, Blaner WS, Olivo M, Han W, Tanavde V, Toh S-A, & Sugii S (2016) Retinoic acid mediates visceral specific adipogenic defects of human adipose-derived stem cells. Diabetes 65, 1164–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Y, Sun L-Q, Kamal MA, Wang X, Seale JP, & Qu X (2011) Suppression of retinol-binding protein 4 with RNA oligonucleotide prevents high-fat diet-induced metabolic syndrome and non-alcoholic fatty liver disease in mice. Biochim. Biophys. Acta 1811, 1045–1053. [DOI] [PubMed] [Google Scholar]
- Teruel T, Hernandez R, Benito M, & Lorenzo M (2003) Rosiglitazone and retinoic acid induce uncoupling protein-1 (UCP-1) in a p38 mitogen-activated protein kinase-dependent manner in fetal primary brown adipocytes. J. Biol. Chem 278, 263–269. [DOI] [PubMed] [Google Scholar]
- Thatcher JE, & Isoherranen N (2009) The role of CYP26 enzymes in retinoic acid clearance. Expert Opin. Drug Metab. Toxicol 5, 875–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiruvengadam V, Amperayanui S, Babu RP, & Uppulura R (2015) Correlation of childhood obesity and related insulin resistance with leptin and retinol binding protein 4. Indian J. Pediatr 82, 799–804. [DOI] [PubMed] [Google Scholar]
- Thompson SJ, Sargsyan A, Lee S-A, Yuen JJ, Cai J, Smalling R, Ghyselinck N, Mark M, Blaner WS, & Graham TE (2017) Hepatocytes are the principal source of circulating RBP4 in mice. Diabetes, 66, 58–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomas P, Ledesma A, & Rial E (2002) Photoaffinity labeling of the uncoupling protein UCP1 with retinoic acid: Ubiquinone favors binding. FEBS Letters 526, 63–65. [DOI] [PubMed] [Google Scholar]
- Tourniaire F, Musinovic H, Gouranton E, Astier J, Marcotorchino J, Arreguin A, Bernot D, Palou A, Bonet LM, Ribot J, & Landrier J-F (2015) All-trans retinoic acid induces oxidative phosphorylation and mitochondria biogenesis in adipocytes. J. Lipid Res 56, 1100–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trasino SE, Tang X-H, Jessurun J, & Gudas LJ (2015) Obesity leads to tissue, but not serum vitamin A deficiency. Sci. Reports 5:15893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trasino SE, Tang X-H, Jessurun J, & Gudas LJ (2016a) Retinoic acid receptor β2 agonist restore glycaemic control in diabetes and reduce steatosis. Diabetes, Obesity & Metabolism 18, 142–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trasino SE, Tang X-H, Jessurun J, & Gudas LJ (2016b) A retinoic acid receptor β2 agonist reduces hepatic stellate cell activation in nonalcoholic fatty liver disease. J. Mol. Med 94 1143–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchida T & Friedman SL (2017) Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol 14, 397–411. [DOI] [PubMed] [Google Scholar]
- Tsutsumi C, Okuno M, Tannous L, Piantedosi R, Allan M, Goodman DS &, Blaner WS (1992) Retinoids and retinoid-binding protein expression in rat adipocytes. J. Biol. Chem 267, 1805–1810. [PubMed] [Google Scholar]
- Urban S, Ye T, & Davidson I (2015). How the RAR-RXR heterodimer recognizes the genome In Dollé P & Niederreither K (Eds.), The Retinoids; Biology, Biochemistry, and Disease (pp. 151–164). Hoboken, New Jersey: WILEY Blackwell. [Google Scholar]
- van Bennekum AM, Kako Y, Weinstock PH, Harrison EH, Deckelbaum RJ, Goldberg IJ, & Blaner WS (1999) Lipoprotein lipase expression levels influences tissue clearance of chylomicron retinyl ester. J. Lipid Res 40, 565–574. [PubMed] [Google Scholar]
- van Hoek M, Dehghan A, Zillikens MC, Hofman A, Witteman JC, & Sijbrands EJ (2008) An RBP4 polymorphism increases risk of type 2 diabetes. Diabetologia 51, 1323–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lchtenbelt M.D. van Marken, Vanhommerig JW, Smulders NM, Drossaerts JMAFL, Kemerink GJ, Bouvy ND, Schrauwen P, & Teule G.J. Jaap (2009) Cold-activated brown adipose tissue in healthy men. N. Engl. J. Med 360, 1500–1508. [DOI] [PubMed] [Google Scholar]
- Villarroya F, Gavaldà-Navarro A, Peyrou M, Villarroya J, & Giralt M (2017a) The lives and times of brown adipokines. Trends Endocrinol. Metabol 28, 855–866. [DOI] [PubMed] [Google Scholar]
- Villaroya F, Peyrou M, & Giralt M (2017b) Transcriptional regulation of the uncoupling protein-1 gene. Biochemie 134, 86–92. [DOI] [PubMed] [Google Scholar]
- von Eynatten M, Lepper PM, Liu D, Lang K, Baumann M, Naworth PP, Bierhaus A, Dugi KA, Heemann U, Allolio B, & Humpert PM (2007) Retinol-binding protein 4 is associated with components of the metabolic syndrome, but not with insulin resistance, in men with type 2 diabetes or coronary artery disease. Diabetologia 50, 1930–1937. [DOI] [PubMed] [Google Scholar]
- Vu-Dac N, Gervois P, Torra I. Pineda, Fruchart J-C, Kosykh V, Kooistra T, Princen HMG, Daillongeville J, & Staels B (1998) Retinoids increase human apo C-lll expression at the transcriptional level via the retinoid X receptor. J. Clin. Invest 162, 625–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wake K (1980) Perisinusoidal stellate cells (fat-storing cells, interstitial cells, lipocytes), their related structure in and around the liver sinusoids, and vitamin A-storing cells in extrahepatic organs. Int. Rev. Cytol 66, 303–353. [DOI] [PubMed] [Google Scholar]
- Wald G (1968) Molecular Basis of Visual Excitation. Science 162, 230–239. [DOI] [PubMed] [Google Scholar]
- Wang X, Yang P, Liu J, Wu H, Yu W, Zhang T, Fu H, Liu Y, & Hai C (2014) RARγ-C-Fos-PPARγ2 signaling rather than ROS generation is critical for all-trans retinoic acid-inhibited adipocyte differentiation. Biochemie 106, 121–130. [DOI] [PubMed] [Google Scholar]
- Wang B, Fu X, Zhu M-J, & Du M (2017a) Retinoic acid inhibits white adipogenesis by disrupting GADD45A-mediated Zfp423 DNA demethylation. J. Mol. Cell. Biol 9, 338–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Fu X, Liang X, Deavila JM, Wang Z, Zhao L, Tian Q, Zhao J, Gomez NA, Trombetta SC, Zhu M-J, & Du M (2017b) Retinoic acid induces white adipose tissue browning by increasing adipose vascularity and inducing beige adipogenesis of PDGFRα+ adipose progenitors. Cell Discovery 3, 17036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei S, Episkopou V, Piantedosi R Maeda S, Shimada K, Gottesman ME, & Blaner WS (1995) Studies on the metabolism of retinol and retinol-binding protein in transthyretin-deficient mice produced by homologous recombination. J. Biol. Chem 270, 886–870. [DOI] [PubMed] [Google Scholar]
- Wu H, Jia W, Bao Y, Lu J, Zhu J, Wang R, Chen Y, & Xiang K (2008) Serum retinol binding protein 4 and nonalcoholic fatty liver disease in patients with type 2 diabetes mellitus. Diabetes Res. Clin. Pract 79, 185–190. [DOI] [PubMed] [Google Scholar]
- Wu Y, Li H, Loos RJ, Qi Q, Hu FB, Liu Y, & Lin X (2009) RBP4 variants are significantly associated with plasma RBP4 levels and hypertriglyceridemia risk in Chinese Han. J. Lipid Res 50, 1479–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia M, Liu Y, Guo H, Wang D, & Ling W (2013) Retinol binding protein 4 stimulates hepatic sterol regulatory element-binding protein 1 and increases lipogenesis through the peroxisome proliferator-activated receptor-β coactivator 1β-dependent pathway. Hepatology 58, 564–575. [DOI] [PubMed] [Google Scholar]
- Yan H, Chang X, Xia M, Bian H, Zhang L, Lin H, Chen G, Zheng M, & Gao X (2013) Serum retinol binding protein 4 is negatively related to beta cell function in Chinese women with non-alcoholic fatty liver disease: a cross-sectional study. Lipids Health Dis. 12:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagitani A, Yamada S, Yasui S, Shimomura T, Murai R, Murarwaki Y, Hashiguchi K, Kanbe T, Saeki T, Ichiba M, Tanabe Y, Yoshida Y, Morino S-l., Kurimasa A, Usuda N, Yamazaki H, Kunisada T, Ito H, Murawaki Y, & Shiota G (2004) Retinoic acid receptor a dominant negative form causes steatohepatitis and liver tumors in transgenic mice. Hepatology 40, 366–375. [DOI] [PubMed] [Google Scholar]
- Yang D, Krois CR, Huang P, Wang J, Min J, Yoo HS, Deng Y, & Napoli JL (2017) Raldh1 promotes adiposity during adolescence independently of retinal signaling. PLoS One 12(ll):e0187669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, Vuckovic MG, Smullin CP, Kim M, Lo CP-S, Devericks E, Yoo HS, Tintcheva M, Deng Y, & Napoli JL (2018) Modest decreases in endogenous all-trans-retinoic acid produced by mouse Rdh10 heterzygote provoke major abnormalities in adipogenesis and lipid metabolism. Diabetes 67, 662–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q, Graham TE, Mody N, Preitner F, Peroni OD, Zabolotny JM, Kotani K, Quadro L, & Kahn BB (2005) Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature 436, 356–362. [DOI] [PubMed] [Google Scholar]
- Yang Q, Eskurza I, Kieman UA, Phillips DA, B1ϋher M, Graham TE, & Kahn BB (2012) Quantitative measurement of full-length and C-terminal proteolyzed RBP4 in serum of normal and insulin-resistant humans using a novel mass spectrometry immunoassay. Endocrinology 153, 1519–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao-Borengasser A, Varma V, Bodies AM, Rasouli N, Phanavanh B, Lee MJ, Starks T, Kern LM, Spencer HJ 3rd., Rashidi AA, McGehee RE Jr., Fried SK, & Kern PA (2007) retinol binding protein 4 expression in humans: relationship to insulin resistance inflammation and response to pioglitazone. J. Clin Endocrinol. Metabol 92, 2590–2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasmeen R, Reichert B, Deiuliis J, Yang F, Lynch A, Meyers J, Sharlach M, Shin S, Volz KS, Green KB, Lee K, Alder H, Duester G, Zechner R, Rajagopalan S, & Ziouzenkova O (2013) Autocrine function of aldehyde dehydrogenase 1 as a determinant of diet- and sex-specific differences in visceral adiposity. Diabetes 62, 124–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanotto-Filho A, Cammarota M, Gelain DP Oliveria RB, Delgado-Cañedo A, Dalmolin RJS, Pasquali MAB, & Moreira JCF (2008) Toxicology in vitro 22, 1205–1212. [DOI] [PubMed] [Google Scholar]
- Zemany L, Bhanot S, Peroni OD, Murray SF, Moraes-Vieira PM, Castoldi A, Manchem P, Guo S, Monia BP, & Kahn BB (2015) Transthyretin antisense oligonucleotides lower circulating RBP4 levels and improve insulin sensitivity in obese mice. Diabetes 64, 1603–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Hu P, Krois CR, Kane MA, & Napoli JL (2007) Altered vitamin A homeostasis and increased size and adiposity in the rdh1-null mouse. FASEB J. 21, 2886–2896. [DOI] [PubMed] [Google Scholar]
- Zhou XE, Suino-Powell KM, Xu Y, Chan CW, Tanabe O, Kruse SW, Reynolds R, Engle JD, & Xu HF (2011) The orphan nuclear receptor TR4 is a vitamin A-activated nuclear receptor. J. Biol. Chem 286, 2877–2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Chen H, Ju H, & Sun M (2017) Circulating retinol binding protein 4 levels in nonalcoholic fatty acid disease: a systematic review and meta-analysis. Lipids Health Dis. 20, 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zizola CF, Frey SK, Jitngarmkusol S, Kadereit B, Yan N, & Vogel S (2010) Cellular retinol-binding protein type I (CRBP-1) regulates adipogenesis. Mol. Cell. Biol 30, 3412–3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziouzenkova O, Orasanu G, Sharlach M, Akiyama TE, Berger JP, Viereck J, Hamilton JA, Tang G, Dolnikowski GG, Vogel S, Duester G, & Plutsky J (2007) Retinaldehyde represses adipogenesis and diet-induced obesity. Nature Med. 13, 696–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zovich DC, Orologa A, Okuno M, Kong LW, Talmage DA, Piantedosi R, Goodman DS, & Blaner WS (1992) Differentiation-dependent expression of retinoid-binding proteins in BFC-1 beta adipocytes. J. Biol. Chem 267, 13884–13889. [PubMed] [Google Scholar]
- Zwolak A, Szuster-Ciesielska A, Daniluk J, Semeniuk J, & Dandefer-Szerszen M (2016) Chemerin, retinol binding protein-4, cytokeratin-18 and transgelin-2 presence in sera of patients with non-alcoholic liver fatty disease. Ann. Hepatol 15, 862–869. [DOI] [PubMed] [Google Scholar]