

Abstract
Chimeric antigen receptor (CAR) T cell therapy is a promising novel therapeutic approach for cancer but also for chronic infection. We have developed a fully human, second-generation CAR directed against the envelope protein of hepatitis B virus on the surface of infected cells (S-CAR). The S-CAR contains a human B cell-derived single-chain antibody fragment and human immunoglobulin G (IgG) spacer, CD28- and CD3-signaling domains that may be immunogenic in mice. Because immunosuppression will worsen the clinical course of chronic hepatitis B, we aimed at developing a preclinical mouse model that is immunocompetent and mimics chronic hepatitis B but nevertheless allows evaluating efficacy and safety of a fully human CAR. The S-CAR grafted on T cells triggered antibody responses in immunocompetent animals, and a co-expressed human-derived safeguard, the truncated epidermal growth factor receptor (EGFRt), even induced B and T cell responses, both limiting the survival of S-CAR-grafted T cells. Total body irradiation and transfer of T cells expressing an analogous, signaling-deficient S-CAR decoy and the safeguard induced immune tolerance toward the human-derived structures. S-CAR T cells transferred after immune recovery persisted and showed long-lasting antiviral effector function. The approach we describe herein will enable preclinical studies of efficacy and safety of fully human CARs in the context of a functional immune system.
Keywords: T cell therapy, chimeric antigen receptor, CAR, HBV, AAV, rejection, scFv expression, immunocompetent mice, immunotherapy
Graphical Abstract
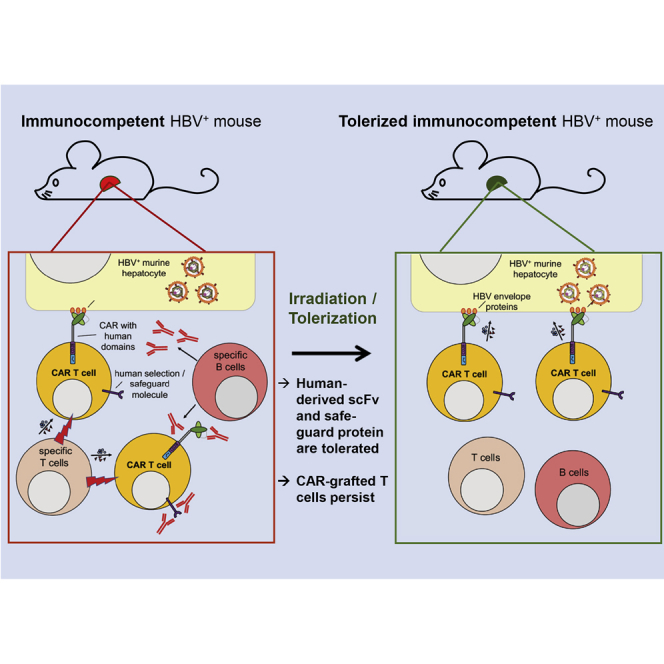
Protzer, Wisskirchen, and colleagues provide a preclinical mouse model that allows studying novel immune and cell therapies in the context of a fully functional immune system. After irradiation and subsequent tolerization, therapeutic efficacy of cells expressing human-derived proteins can be studied without being attacked by the murine immune system.
Introduction
T cell therapies utilizing chimeric antigen receptors (CARs) have emerged as a revolutionary approach to treat cancers and infections with a high specificity during the last two decades.1 Anti-CD19 CAR T cell therapy has a significant benefit for patients with B cell malignancies not responding to first-line chemo- and immunotherapies, and recently two CAR T cell products have been approved for clinical use.2, 3 CAR T cells for other cancer types, including solid tumors, have also been developed and are currently being evaluated in clinical trials. However, it became obvious that CAR T cell therapy of solid tumors is a more complex scenario.4 Targets for CAR T cell therapies include tumor-associated antigens but also viral antigens displayed on the surface of malignant or infected cells. CARs that exploit binders recognizing viral envelope proteins have been developed for chronic infections with hepatitis B virus (HBV),5 human cytomegalovirus,6 hepatitis C virus,7 and HIV.8, 9
A CAR is composed of a single-chain variable fragment (scFv) that determines the target specificity, an extracellular spacer linking the scFv to the signaling domains, a transmembrane domain, and intracellular signaling domains. In clinical application, ideally a fully human CAR should be utilized to prevent rejection of CAR T cells by the patient’s immune system. Furthermore, accessory molecules co-expressed to purify CAR T cells or as a safeguard to be able to deplete T cells if needed could also be recognized as foreign if they contain non-human-derived domains. In fact, CAR T cells carrying a murine scFv were rejected, and their use led to decreased response rates in patients.10
Before advancing to clinical application, CAR constructs have to be studied in preclinical models. In particular, immunocompetent preclinical models are urgently needed to study the efficacy but also potential side effects of a CAR T cell therapy, because these can be largely influenced by bystander effects of other immune cells or mediators. Immune competence of the animals, however, can limit the preclinical investigation of a CAR with human domains, since allogenic immune rejection could limit CAR T cell persistence in these models. To prevent such an immune response by the endogenous murine immune system, most preclinical studies are performed in immunodeficient mouse models.11 In the case of anti-CD19 CAR T cell transfer for hematological malignancies, patients are preconditioned with chemotherapeutic lymphodepleting regimens, and, hence, using immunodeficient mice mimics this particular clinical situation.12 However, immunosuppressive or lymphodepleting regimens will most likely not be applied in the clinics for CAR T cell approaches targeting certain solid tumors or fighting viral diseases. We therefore aimed at generating an experimental, preclinical system in which the recipient is immunocompetent and which at the same time allows the study of CARs with human-derived sequences.
The target of the CAR T cell approach presented here is HBV. Chronic hepatitis B and HBV-associated hepatocellular carcinoma (HCC) are a major health concern with >250 million humans affected and 887,000 deaths per year due to HBV-associated liver diseases.13 Current treatment regimens suppress viral replication but are curative only in rare cases. HBV still is the major cause of HCC development worldwide, and, mainly due to a lack of therapeutic options, HCC became the number two cause of cancer-related death.14 CAR T cell therapy is a promising approach to address this medical need.15
We have generated a fully human CAR that is specific for the small envelope protein “S” of HBV and targets the S domain (S-CAR) of all HBV envelope proteins,5 which are found on the surface of HBV virions and subviral particles, therefore called hepatitis B surface antigen (HBsAg), but are also located on the surface of HBV-infected hepatocytes and HBV-induced hepatoma cells.16 Previous results from our laboratory indicated that the S-CAR can redirect T cells against HBV-infected hepatocytes in vitro and eliminate HBV from infected cell cultures5 but that the therapeutic effect of adoptively transferred murine S-CAR T cells into HBV-transgenic immunocompetent mice was limited. After an initial expansion and a very good antiviral effect, S-CAR T cells vanished and viral parameters rose again.17
In the study presented here, we show that an immune response against the human domains of the S-CAR limited CAR T cell persistence in immunocompetent preclinical mouse models, but not in immunocompromised animals. We were able to overcome the problem of S-CAR T cell rejection by specifically tolerizing immunocompetent mice against the allogenic CAR domains. In this setting, S-CAR T cells persisted at high numbers and induced a sustained antiviral effect.
Results
A Repeated Transfer of S-CAR T Cells into Immunocompetent Mice Does Not Lead to Quantitative or Functional Reconstitution of S-CAR T Cells
A loss of S-CAR T cell function that has been observed after transfer into HBV-transgenic mice17 could be due to T cell exhaustion, activation-induced cell death, or an immune response against the transferred cells. To address this question, we investigated whether a second transfer of S-CAR T cells would maintain the antiviral effect. We engineered murine CD45.1+ CD8+ T cells to express the S-CAR (schematically depicted in Figure S1), and we transferred them on day 0 and again on day 20 into CD45.1-negative HBV-transgenic mice. A second group of mice received the T cell product only on day 20 (Figure 1A). The congenic marker CD45.1 allowed us to differentiate transferred cells from endogenous CD45.2+ cells. We detected comparable numbers of total CD45.1+ transferred cells on day 25, i.e., 5 days after first or second transfer, that dropped until day 33, i.e., 2 weeks after transfer, in both groups (Figure 1B). In contrast to total transferred cells, S-CAR-expressing T cells were only detected after the first, but not after the second transfer (Figure 1C).
Figure 1.

Sequential Transfers of S-CAR T Cells into HBV-Transgenic Mice
(A) Scheme of the experimental procedure. CD45.2+ HBV-transgenic mice were injected once (day 20, gray symbols) or twice (day 0 and day 20, black symbols) with 4 × 106 CD45.1+ S-CAR+ T cells each (n = 7 per group). Transferred, CD45.1+ cells in peripheral blood and serum parameters were monitored over time. (B) Numbers of CD45.1+ T cells per microliter peripheral blood, (C) numbers of S-CAR+ T cells in peripheral blood, and (D) ALT activity in sera at the indicated time points. (E) Lymphocytes were isolated from liver and spleen on day 33 and cultured on HBsAg- or anti-CD3 and anti-CD28- or PBS-coated control plates overnight. Activation of CD45.1+ T cells was determined by intracellular staining or IFN-γ and TNF-α followed by flow cytometry analysis. (B–D) Data points represent individual animals and mean values ± SD are indicated. (E) Data are given as mean values ± SD. ns, not significant; *p < 0.05, **p < 0.01, ***p < 0.001 (Mann-Whitney test).
Concomitantly, liver damage indicated by a rise in serum alanine amino transferase (ALT) levels 5–7 days after transfer was exclusively detected after the first, but not after the second injection of S-CAR T cells (Figure 1D). On day 33, lymphocytes from liver and spleen were isolated and stimulated on plate-bound HBsAg or anti-CD3 and anti-CD28 antibodies as a positive control. PBS-treated plates served as a negative control. Intracellular cytokine staining (ICS) did not reveal HBsAg-specific activation of lymphocytes (Figures 1E and S2), although a comparable S-CAR signal was still detected by qPCR in both groups (Figure S3). The fact that neither S-CAR T cells nor liver cytotoxicity were detected after the second adoptive transfer suggested an immune response against the transferred cells rather than activation-induced cell death, or a lack of antigenic stimulation or T cell exhaustion, both of which would not lead to reduced cell numbers in a short term.
Adaptive Immunity Limits S-CAR T Cell Persistence
To find out if the murine immune system would react to the human-derived domains on S-CAR T cells, we first transferred T cells that expressed the S-CAR or a non-functional S-decoy (Δ)-CAR, both in combination with a truncated human epidermal growth factor receptor (EGFRt), into HBV-naive C57BL/6J mice. The SΔ-CAR construct17 contains the same extracellular domains as the S-CAR, but intracellular T cell signaling domains have been exchanged to the cytoplasmic domain of the nerve growth factor receptor, rendering the SΔ-CAR incapable of activating T cells (Figure S1). EGFRt can be targeted by depleting antibodies, and it serves as a potential safeguard when in vivo toxicity is observed, but also as an additional transduction and selection marker.18, 19 Both CARs contained a mutated, human immunoglobulin G (IgG)1 spacer with decreased Fc receptor-binding capacity (Figure S1).20
The T cell products had transduction rates of 85% (S-CAR+/EGFRt+) and 74% (SΔ-CAR+/EGFRt+), as determined by flow cytometry (Figure 2A). Expansion and persistence of S-CAR and SΔ-CAR T cells were limited in immunocompetent animals compared to that of mock T cells without transgene expression, although all cell products were detected at comparable numbers on day 3 (Figure 2B). When we transferred S-CAR or SΔ-CAR T cells into B cell- and T cell-deficient Rag2−/− mice, cells expanded and persisted at least as well as mock T cells (Figure 2B). Hence, S-CAR and SΔ-CAR T cells vanished irrespectively of the presence of antigen or ability of CAR T cells to be activated, but only in immunocompetent mice and not in immunodeficient mice. This indicated that neither a lack of antigenic stimulation nor tonic signaling is the leading cause of S-CAR T cell depletion but that an immune response caused the fate of S-CAR T cells.
Figure 2.

S-CAR T Cell Engraftment in Immunocompetent and Immunodeficient Mice
2.7 × 106 CD45.1+ S-CAR+/EGFRt+, SΔ-CAR+/EGFRt+, or mock T cells were transferred into HBV-naive CD45.2+ wild-type C57BL/6J (n = 5 per group) or Rag2−/− mice (n = 3 per group). (A) CAR and EGFRt expression on CD8+ T cells determined by flow cytometry at day of transfer. (B) Numbers of transferred CD45.1+ cells in peripheral blood were determined over time by flow cytometry. (C–F) AAV-HBV-infected CD45.2+ Rag2−/−/IL-2Rγ−/− mice received 1 × 106 S-CAR+/EGFRt+ T cells each (n = 5, gray triangles) or remained untreated (n = 3, open triangles). CD45.1+ cells in peripheral blood and serum parameters were monitored over time. (C) Numbers of S-CAR+ or EGFRt+ cells per microliter blood, (D) serum ALT activity, (E) HBsAg level, and (F) HBeAg level were determined. All data are given as mean values ± SD. ns, not significant; *p < 0.05 (Mann-Whitney test).
To investigate the antiviral potential of S-CAR T cells in the absence of anti-CAR immunity, we adoptively transferred the cells into adeno-associated virus (AAV)-HBV-infected Rag2−/−/interleukin-2 (IL-2)Rγ−/− mice. AAV-mediated HBV genome transfer to the mouse liver allows persistence of the HBV genome over months,21 generating a preclinical model that a priori allows the study not only of HBV persistence but also of HBV cure. A cure can be achieved in AAV-HBV-infected mice because only a proportion of hepatocytes is infected and the HBV genome remains episomal, allowing the elimination of infected hepatocytes. In AAV-HBV-infected Rag2−/−/IL-2Rγ−/− mice, S-CAR T cells expanded and were detected for >30 days after transfer (Figure 2C). S-CAR T cell therapy induced moderate liver damage, indicated by a transient increase of serum ALT to 70–190 U/L (Figure 2D). To assess the antiviral activity of S-CAR T cells, we determined viral HBsAg (Figure 2E) and e antigen (HBeAg) (Figure 2F) in serum. Notably, HBsAg significantly decreased by about 2 log10 until day 13 and then remained detectable at a low level. HBeAg decreased more slowly by 60% until day 38. These results indicated that S-CAR T cells expand and exhibit a continuous antiviral effect if they are not targeted by an adaptive immune response.
Immunocompetent Mice Mount CD8+ T Cell and Antibody Responses against S-CAR T Cells
As shown in Figure 2B, S-CAR T cells persisted in Rag2−/− mice that harbor functional natural killer (NK) cells but neither B nor T cells. Hence, both B and T cells could be responsible for the reduced survival of S-CAR T cells. When we analyzed the expression of either S-CAR or EGFRt by flow cytometry after T cell transfer, both markers disappeared in immunocompetent, but not in immunodeficient mice (Figures 3A and S4), confirming the loss of S-CAR T cells. Next, we determined if T cell responses contributed to S-CAR T cell rejection in the immunocompetent animals. Thus, we co-cultured splenocytes from wild-type recipient mice that had received 2.7 × 106 T cells grafted with both the S-CAR and the EGFRt overnight with CD8+ T cells as target cells expressing either the S-CAR or the EGFRt. ICS revealed that endogenous CD45.2+ CD8+ T cells from the treated mice became activated and expressed interferon (IFN)-γ if co-cultured with EGFRt-expressing, but not with S-CAR-expressing target cells (Figure 3B). This indicated a CD8+ T cell response against the human-derived EGFRt. In contrast, we did not detect a CD8+ T cell response against the S-CAR, although it also contains human-derived domains, namely, the extracellular scFv C8, a human IgG1 spacer, a transmembrane domain from human CD28, and intracellular signaling domains of human CD28 and CD3ζ.
Figure 3.

B and T Cell Responses against S-CAR and EGFRt after T Cell Transfer
(A–E) 2.7 × 106 CD45.1+ S-CAR+/EGFRt+ CD8+ T cells were transferred into CD45.2+ HBV-naive wild-type (WT, mock, open circles; S-CAR, black boxes; n = 5 per group) or Rag2−/− mice (S-CAR, gray boxes; n = 3) (see also Figure 2B). (A) Surface expression levels of S-CAR and EGFRt were determined by median fluorescence intensity (MFI) on CD45.1+ CD8+ T cells in peripheral blood at the indicated time points after transfer. (B) Splenocytes were isolated from treated WT mice on day 18 post-transfer and co-cultured overnight with mock, S-CAR-, or EGFRt-expressing CD8+ T cells. IFN-γ expression by endogenous CD45.2+ CD8+ T cells was determined via ICS. (C) Detection of S-CAR- and EGFRt-specific antibodies in serum of mice on day 18 post-transfer. Binding of antibodies to S-CAR- or EGFRt-expressing target cells was determined via bound fluorochrome-labeled secondary anti-mouse IgG antibody by flow cytometry. (D) Detection of anti-hIgG1 or (E) anti-scFv C8 antibodies in serial dilutions of mouse sera from day 3 or day 18 post-transfer by ELISA. (F and G) 2 × 106 CD45.1+ T cells expressing an S-CAR with either human (n = 3, black boxes) or murine IgG1 spacer domains (n = 4, gray triangles) were transferred into CD45.2+ HBV-transgenic mice. (F) Numbers of transferred, CD45.1+ cells per microliter peripheral blood. (G) Detection of anti-hIgG1 or anti-scFv C8 antibodies in 1:200 diluted mouse sera from day 26 by ELISA. (A, D, and E) Data are given as mean values ± SD. (B, C, F, and G) Data points represent individual animals and mean values are indicated (in B, C, and F, ±SD). ns, not significant; **p < 0.01 (Mann-Whitney test).
This finding, together with the time kinetics of vanishing S-CAR or EGFRt stainings on day 5 or 7, respectively (Figure 3A), indicated an additional immunological mechanism playing a role in S-CAR T cell rejection. We therefore decided to also test for anti-S-CAR and anti-EGFRt antibodies; we incubated our target cells expressing either the S-CAR or EGFRt with mouse sera of recipient mice and stained for bound murine IgG antibodies. Flow cytometry analysis revealed antibody production against both the S-CAR and EGFRt molecules (Figure 3C). To confirm anti-S-CAR antibodies and to investigate which domains of the S-CAR were targeted by the antibodies, an ELISA was established. This ELISA confirmed the presence of antibodies against both extracellular domains of the S-CAR, namely, the human IgG1 spacer (Figure 3D) and the scFv C8 (Figure 3E).
As an attempt to prevent an antibody response against the S-CAR, the number of immunogenic extracellular epitopes was reduced by exchanging the spacer to a murine IgG1 domain (Figure S1) and excluding the EGFRt. The intracellular signaling domains were left unaltered as no T cell response against the S-CAR had been detected. Thus, in this murine IgG1 S-CAR construct, only the human-derived scFv C8 remained as a potentially immunogenic extracellular epitope. To study the effect of an antibody response against the scFv C8, we transferred murine IgG1 S-CAR T cells into HBV-transgenic mice. In murine IgG1 S-CAR T cell-treated mice, the numbers of transferred cells declined with kinetics comparable to those in mice treated with S-CAR T cells containing a human IgG1 (Figure 3F). While no anti-human IgG1 (hIgG1) antibodies were detected anymore, we still detected antibodies directed against the human-derived scFv C8 domain (Figure 3G).
Taken together, the EGFRt elicited B and T cell responses in immunocompetent mice, while the S-CAR elicited only antibody responses. Antibody responses were directed against the scFv C8 binder, which is of human origin, as well as the human IgG1 spacer domain within the S-CAR. Since the human scFv binder was still targeted by antibodies, a reduction of immunogenic epitopes did not prevent rejection of S-CAR T cells by the endogenous immune system. Exchanging the scFv, however, does not allow preclinical evaluation of a CAR anymore. Thus, alternative models are required for preclinical evaluation of a human CAR.
Irradiation Allows Long-Term Persistence of S-CAR T Cells
Sublethal total body irradiation is an option to prevent the rejection of cells expressing alloantigens.22 To establish tolerance against S-CAR- and EGFRt-expressing T cells in AAV-HBV-infected immunocompetent mice, recipients were irradiated 1 day before T cell transfer (Figure 4A). Under this condition, S-CAR T cells expanded and persisted until day 140 in peripheral blood (Figures 4B and 4C; gating strategy depicted in Figure S5A). Importantly, even when B cells and CD8+ T cells (Figure 4D) as well as CD4+ T cells and NK cells (Figure S6) had reached physiological concentrations again 80 days after irradiation, the concentration of S-CAR T cells remained stable.
Figure 4.

S-CAR T Cell Engraftment in Irradiated Mice
(A) Scheme of the experimental procedure. AAV-HBV-infected CD45.2+ wild-type mice were injected with 1 × 106 CD45.1+ S-CAR+/EGFRt+ or mock T cells per animal 1 day after sublethal total body irradiation (S-CAR, black boxes; mock, open circles) or without prior irradiation (S-CAR, gray triangles) (n = 4 per group). (B) Exemplary flow cytometry plot of (transferred) CD45.1+ and (endogenous) CD45.1− CD8+ T cells in peripheral blood on day 28. (C) The amount of S-CAR+ or EGFRt+ T cells per microliter peripheral blood was determined by flow cytometry at the indicated time points. (D) Amount of (endogenous) CD45.1− CD19+ B cells (left) or CD8+ T cells (right) in peripheral blood. Arrows mark the time point of irradiation. (E) Count of S-CAR+ or EGFRt+ cells in liver and spleen on day 140. (F) Phenotype of CD8+ T cell subsets of transferred CD45.1+ T cells in irradiated mice determined by flow cytometry after staining for CD62L and CD127: effector (CD62L− CD127−), effector memory (CD62L− CD127+), intermediate (CD62L+ CD127−), and naive or central memory (CD62L+ CD127+). (G) Expression of exhaustion markers (PD-1, CTLA-4, and Tim-3) on CD45.1+ lymphocytes isolated from liver and spleen of irradiated mice on day 140. (H) Ex vivo functionality of S-CAR T cells of irradiated mice determined by overnight culture on plate-bound HBsAg or PBS as control. ICS is shown for IFN-γ and TNF-α. (I) Sera from days −8, 14, and 140 were analyzed by ELISA for anti-hIgG1 and anti-scFv C8 antibodies. (C, D, F, and H) Data are given as mean values ± SD. (E, G, and I) Data points represent individual animals and mean values ± SD are indicated. Dotted lines represent the background determined in mock T cell-treated mice. *p < 0.05 (Mann-Whitney test).
At day 140 after transfer, S-CAR T cells were still detected at high numbers in liver and spleen (Figure 4E) and allowed characterizing their phenotype. The majority of S-CAR T cells (60%–70%) in both organs exhibited an effector phenotype (CD62L− CD127−) (Figures 4F and S7A). Mock-transduced CD8+ T cells, in contrast, showed a phenotype of naive or central memory T cells (CD62L+ CD127+, 60%–70% in liver, 89%–94% in spleen). When exhaustion markers were analyzed, a high percentage of S-CAR T cells were positive for PD-1, but only about 25% expressed Tim-3 and CTLA-4 was barely detected at all (Figures 4G and S7B). To investigate the functionality of S-CAR T cells after in vivo circulation for more than months, cells from liver and spleen isolated on day 140 after transfer were re-stimulated ex vivo on plate-bound HBsAg. ICS revealed that the transduced S-CAR T cells could still be activated and expressed the proinflammatory cytokines IFN-γ and, to a lower extent, tumor necrosis factor alpha (TNF-α) upon antigen encounter (Figure 4H). Furthermore, irradiation prevented the development of anti-hIgG1 and anti-scFv C8 antibodies upon S-CAR T cell transfer (Figure 4I). In summary, irradiation of immunocompetent mice prior to T cell transfer allowed the expansion and long-term persistence of S-CAR T cells, which developed an effector phenotype and to a considerable proportion remained functional.
S-CAR T Cells Have Long-Term Antiviral Function in Irradiated Immunocompetent Mice
Having shown that transferred S-CAR T cells could expand and survive in AAV-HBV-infected and irradiated wild-type mice, we next determined their antiviral effect in this model. Mice that had received S-CAR T cells, both with and without prior irradiation, displayed 2- to 4-fold elevated serum ALT levels on day 7 (Figure 5A). Around day 40, irradiated mice treated with S-CAR T cells, but not the other groups, showed a moderate ALT elevation again. Serum HBsAg levels dropped by 1 log10 until day 30 in both S-CAR T cell-treated groups independent of prior irradiation. However, HBsAg subsequently rebounded in mice without prior irradiation, reaching pretreatment levels again around day 80 (Figure 5B). One mouse (not irradiated) developed spontaneous anti-HBV immunity >80 days after S-CAR T cell treatment and seroconverted, with a drop in HBsAg of >3 log10 and anti-HBs antibodies detectable on day 140 (Figure 5C).
Figure 5.

Antiviral Effect of S-CAR T Cells in Irradiated Mice
Identical experimental setup as in Figure 4. (A) ALT activity and (B) HBsAg levels in serum over time. (C) Inverse correlation of HBsAg and anti-HBs antibody concentrations in a single mouse that underwent spontaneous seroconversion. This animal was excluded from (B). (D) HBeAg levels in serum over time. (E) Intrahepatic AAV- and HBV-DNA copies per cell were determined by qPCR and normalized to cell numbers using the single-copy gene PRNP. Individual animals are indicated relative to the mean value determined in mock-treated animals (set to 100%). (A, B, and D) Data are given as mean values ± SD. (E) Data points represent individual animals and mean values ± SD are indicated. ns, not significant; *p < 0.05 (Mann-Whitney test).
In all irradiated and S-CAR T cell-treated mice, HBsAg continued to decrease to <1% of pretreatment values until day 140 (Figure 5B) and HBeAg continuously dropped (Figure 5D). This was not observed in non-irradiated or mock T cell-treated animals. The antiviral effect was confirmed by qPCR analysis of liver DNA. AAV as well as HBV DNA copies in the liver were significantly reduced in irradiated and S-CAR T cell-treated mice compared to the other groups (Figure 5E). Thus, when the initiation of immune responses against the human-derived S-CAR was prevented by irradiation, S-CAR T cells expanded, persisted long-term, and elicited a significant antiviral effect in AAV-HBV-infected mice. Whether low-level persistence of HBsAg and HBeAg was due to the fact that the observation period was limited to 140 days or to the fact that the largely reduced antigen levels were not sufficient to stimulate the S-CAR anymore cannot be clarified.
S-CAR-Specific Tolerization of Mice Allows T Cell Persistence and Antiviral Efficacy
To further improve the model and allow S-CAR T cell transfer into fully immunocompetent animals, we aimed at inducing antigen-specific tolerance to the human-derived CAR domains and EGFRt before S-CAR T cell transfer. To this end, non-functional SΔ-CAR T cells co-expressing EGFRt were transferred into AAV-HBV-infected mice 1 day after irradiation (Figure 6A). SΔ-CAR T cells should neither proliferate nor show any effector function in AAV-HBV-infected mice. We hypothesized that the presence of the human alloantigens from SΔ-CAR and EGFRt during recovery of the endogenous immune system would allow us to induce specific immune tolerance.
Figure 6.

S-CAR T Cell Persistence and Antiviral Effect after Tolerization of Immunocompetent Mice
(A) Scheme of the experimental procedure. AAV-HBV-infected CD45.2+ wild-type mice underwent total body irradiation 1 day prior to the transfer of 5 × 106 non-functional, CD45.1+ SΔ-CAR+/EGFRt+ T cells per animal (n = 5 per group). Three months later, mice were injected with 3 × 106 functional CD45.1+/CD45.2+ mock (open circles) or S-CAR+ and EGFRt+ T cells per animal (day 0). Mice that received S-CAR+/EGFRt+ T cells were either only irradiated (irradiation, black squares) or were irradiated and received SΔ-CAR+/EGFRt+ T cells (tolerization, gray triangles). Mice that received mock T cells were not pretreated. Mice were sacrificed 110 days after transfer of functional S-CAR T cells. (B) Sera from day 27 were analyzed by ELISA for anti-hIgG1 and anti-scFv C8 antibodies. (C) Splenocytes isolated at the end of the experiment were co-cultured with EGFRt-expressing target cells before ICS. IFN-γ expression of endogenous CD45.1− CD8+ T cells upon antigen encounter is shown. (D) Numbers of CD45.1+/CD45.2+ S-CAR+ or EGFRt+ T cells per microliter peripheral blood determined at the indicated time points. Dotted line represents the background determined in mock T cell-treated mice. (E) ALT activity, (F) HBsAg level, and (G) HBeAg level in serum measured over time. (H) Intrahepatic AAV- and HBV-DNA copies per cell determined by qPCR normalized to the cellular single-copy gene PRNP. Values are shown relative to the mean value determined in mock-treated mice. (D–G) Data are given as mean values ± SD. (B, C, and H) Data points represent individual animals and mean values ± SD are indicated. ns, not significant; *p < 0.05, **p < 0.01 (Mann-Whitney test).
As observed before, irradiation of mice induced depletions of endogenous B and T cell populations that were restored in numbers after 2 months (Figures S8A–S8C). SΔ-CAR T cells injected at the time of irradiation persisted at a low concentration for more than 3 months (Figure S8D). After 3 months, functional S-CAR T cells that were additionally grafted with EGFRt were injected. Mice that had been irradiated and had received SΔ-CAR T cells neither mounted an antibody response against the human IgG1 or scFv C8 domains of the S-CAR (Figure 6B) nor developed a CD8+ T cell response against the EGFRt (Figure 6C). In contrast, mice only irradiated, but not tolerized using SΔ-CAR T cells, mounted B and T cell responses against the human alloantigens. Functional S-CAR T cells proliferated well and persisted in tolerized mice until the end of the study, i.e., day 110 after S-CAR T cell transfer (Figure 6D), but they rapidly vanished from peripheral blood in mice that had not been tolerized. This shows that functionality of the endogenous immune system against the foreign antigens S-CAR and EGFRt had been re-established after irradiation at the time point when functional S-CAR T cells were transferred. On the other hand, it proves that antigen-specific immune tolerance induced by the SΔ-CAR T cell transfer after irradiation was sufficient to allow the persistence of S-CAR T cells.
In the tolerized animals, in which S-CAR T cells expanded and survived, we then determined the antiviral effect of S-CAR T cells. Serum ALT levels remained slightly elevated after S-CAR T cell treatment, although statistically significant only on day 110 (Figure 6E). Viral HBsAg decreased by 2 log10 in tolerized animals, but it remained unaltered in the other groups (Figure 6F). Both, serum HBeAg levels (Figure 6G) as well as AAV-DNA and HBV-DNA copies in the liver (Figure 6H) decreased about 60% in comparison to control groups. Again, despite being quite efficient, S-CAR T cell therapy did not fully cure mice from HBV infection and a proportion of HBV-positive hepatocytes persisted.
Discussion
Appropriate mouse models are needed for thorough preclinical investigations of CAR T cell products. While a CAR ideally consists of only human-derived domains when applied to a patient, the same construct may be recognized as foreign in immunocompetent mice. Hence, therapeutic efficacy and safety profiles might be altered due to limited CAR T cell persistence. Here we show that the induction of adaptive immunity is indeed an issue when investigating CAR T cells harboring human domains in an immunocompetent mouse model.
After an initial expansion, HBV-specific S-CAR T cell numbers rapidly declined and a second S-CAR T cell transfer was unable to induce an antiviral effect anymore. Cells that expressed either the CAR or EGFRt as a safeguard could be used to detect antibodies in the serum of treated mice and CD8+ T cell responses via flow cytometry-based assays. These experiments showed that EGFRt was targeted by both humoral and cellular immune responses. While we could only detect humoral immune responses against the S-CAR, these were, unfortunately, at least partially directed against the scFv binder, as confirmed by a specifically developed ELISA. The scFv C8 binder as the only essential domain of human origin was still sufficient to induce antibody responses and a loss of S-CAR T cells. The rapid decrease of CAR T cells also occurred in HBV-negative mice or when CAR T cells lacked signaling domains or harbored a spacer with reduced Fc receptor-binding capacity (Figure S1).20 Hence, we concluded that neither T cell exhaustion nor activation-induced cell death, due to overactivation by antigen or by binding of Fc receptors to the CAR, could have played a role in reduced S-CAR T cell persistence.
When mice were irradiated directly before T cell transfer, we were able to induce long-term tolerance to the S-CAR and EGFRt alloantigens, with S-CAR T cells being detected at high numbers even 140 days after transfer in peripheral blood, spleen, and liver. Since S-CAR T cells were transferred only 1 day after irradiation, when the immune system was strongly depressed, one could argue that the mice were not fully immunocompetent. Therefore, we investigated if we could induce a specific tolerance to the alloantigens by an immediate transfer of non-functional SΔ-CAR T cells. Although these cells persisted only at low numbers due to a lack of an activation signal, their numbers were sufficient to induce specific tolerance against the human-derived domains, and functional S-CAR T cells were able to persist for more than 3 months in constantly high numbers and to elicit an antiviral function, even when encountering a fully reconstituted immune system.
Our results show that a specific immune tolerance has been induced by transfer of the non-functional SΔ-CAR T cells, preventing rejection of S-CAR T cells after immune reconstitution. Full reconstitution of the immune system at the time point of S-CAR T cell transfer was indicated, since mice that had only been irradiated but did not receive an early transfer of SΔ-CAR T cells rejected the S-CAR T cells. Immune tolerance can be achieved by two distinct means, namely, central and peripheral tolerance. We propose that central tolerance is the mode of action of tolerance induction to transferred S-CAR T cells.
Central tolerance is induced in the thymus when, during T cell development and after T cell receptor gene rearrangement, T cells are assessed for their specificity.23 Only T cells with a non-self T cell receptor specificity can leave the thymus and become part of the pool of mature peripheral T cells. Since autoreactive T cells are excluded this way, the T cell pool usually does not target self-tissue and auto-immune diseases remain a rare event.23 If autoreactive T cells escape negative selection in the thymus or an antigen is only encountered later in life, peripheral tolerance comes into play.24 Tissue damage is prevented by conversion of T cells to regulatory T cells (Tregs), induction of T cell apoptosis, T cell exhaustion, or anergy by, e.g., metabolic alteration. B cells experience similar selection mechanisms.25 In their case, central tolerance is achieved during maturation in the bone marrow. If autoreactive B cells escape negative selection, absent CD4+ T cell help in the periphery prevents B cell activation and antibody production.
In our setting, the alloantigens expressed on transferred T cells (namely extracellular domains of the S-CAR and the EGFRt) were present during replenishment of the immune cell pool after irradiation. It was previously reported that intrathymic antigen inoculation after total body irradiation can induce selective non-responsiveness to bovine gamma-globulin as an alloantigen in rats.22 Similarly, intrathymic transplantation of pancreatic islet allografts after lymphodepletion led to the acceptance of islet grafts both inside and outside the thymus.26 Hereby, clonal deletion induced by the recognition of alloantigens was identified as the mode of action for selective non-responsiveness.27 For our case, we propose the following concept of non-responsiveness to the human-derived antigens: adoptively transferred SΔ-CAR T cells are distributed throughout the body and will migrate to the thymus. Here, cross-presentation of peptides by thymic dendritic cells28 induces negative selection for both CD4+ and CD8+ T cells with specificities for S-CAR or EGFRt epitopes. This would directly prevent CD8+ T cell responses and indirectly B cell responses because of a lacking CD4+ T cell help. Our data indicate that the presence of the alloantigens during recovery from total body irradiation deluded the immune system in a way that the human-derived domains are considered self-antigens and must not be targeted.
The situation may be comparable to the clinical setting. Murine components of CAR T cell products have been reported to be immunogenic in humans. Stronger lymphodepleting preconditioning with fludarabine instead of only cyclophosphamide-based regimens leads to improved and sustained engraftment of CAR T cells. However, the mechanism by which fludarabine increased CAR T cell survival and whether immune responses against murine domains were delayed or prevented remain unclear.10 It may well be the same mechanism we describe in our mouse model.
In our setting, S-CAR T cell therapy had a sustained antiviral effect without inducing apparent therapy-limiting side effects; however, it was not yet able to cure the AAV-HBV infection during the 110 days of treatment. One possible explanation would be an insufficient antigenic stimulation, i.e., that the affinity of the scFv C8 is not high enough to detect low amounts of S protein on the cellular membrane. This has been described for an anti-CD20 CAR.29 Alternatively, S protein may not be present on the membrane in a proportion of hepatocytes. T cell cytokines can downregulate HBV protein expression30, 31 and can even deplete the HBV persistence form32, 33 without killing infected cells. This will largely reduce the antigen expression level in HBV-positive cells and prevent elimination by T cells. In a clinical setting, this limitation may be overcome if patients were selected for high and homogeneous expression of viral proteins on the cell surface of HBV-infected cells or HBV-induced HCC tissue in liver biopsies.
A second explanation could be inefficient endogenous bystander immunity targeting HBV in the irradiated mice. Since HBV antigens are also continuously present in high amounts when the immune system recovers from irradiation, clonal deletion resulting in selective non-responsiveness27 to HBV is possible. A third explanation could be the liver microenvironment where the anti-HBV immunity needs to become effective. To preserve integrity of the liver as an essential organ, its microenvironment is particularly prone to allowing foreign antigens to escape immunity. This is, e.g., exploited by pathogens like HBV, hepatitis C virus, or malaria sporozoites that persist in the liver and by this frequently escape immune clearance, but it also explains the extraordinary tolerance of orthotopic liver transplants.34, 35 This may be overcome if the S-CAR T cell-induced antiviral immune response paved the way for endogenous immune cells to fight the infection.
For reliable preclinical assessment of T cell therapies, it seems important that the chosen preclinical model closely reflects the anticipated clinical scenario. In particular, if chronic viral infections or particular solid cancers shall be treated, there will be a need to apply CAR T cells without prior immunosuppression, although lymphodepletion seems to support CAR T cell efficacy. As a potential reason for the beneficial effect of lymphodepletion, competition for cytokines with endogenous immune cells but also alteration of the tumor microenvironment, or simply a lack of space for the transferred cells to expand have been discussed.36 To fully understand the benefit of lymphodepletion prior to T cell transfer, orthotopic and immunocompetent preclinical models are required and will help to improve clinical efficacy of CAR T cell therapy in settings other than hematological malignancies.37
For adoptive T cell therapy of chronic HBV infection and HBV-associated HCC, it is essential to rely on preclinical models using immunocompetent animals as this reflects the clinical situation. For both diseases, lymphodepleting regimens are contraindicated. Several studies have reported that chemotherapy in chronic HBV carriers leads to virus reactivation.38, 39, 40 Especially lymphodepletion via cyclophosphamide, which is commonly used before T cell transfer, leads to reactivation of HBV in up to one-third of patients.41 Already the depletion of B cells using anti-CD20 antibodies results in life-threatening HBV reactivation.42 Therefore, preclinical evaluation of S-CAR T cell therapy in an immunocompetent rather than an immunocompromised mouse model is needed, since it is likely to provide an efficacy and safety profile that has relevance for clinical application.
Importantly, the model described here is transferable to other CAR T cell approaches, e.g., the treatment of solid tumors, that shall be evaluated in immunocompetent preclinical models and utilize human scFv. In comparison to models using immunodeficient mice, the tolerized animals offer the advantage that they have a fully functional immune system at the time of CAR T cell transfer, allowing investigation of interactions with and activation of the endogenous immune system and how this influences efficacy and safety of the therapy. Tumor infiltration by bystander immune cells will certainly contribute to an anti-tumor response but potentially also to a cytokine storm.37 To evaluate all consequences of a cytokine storm, cytokine receptors matching between transferred T cells and host tissue are required, as is the case in our model. Furthermore, in this model, a combination therapy with checkpoint inhibitors targeting CAR T cells but also endogenous immune cells can be evaluated.
The model described here is the only model that allows studying a CAR with human-derived domains in the context of an intact immune system besides humanized mouse models harboring human immune cells. In contrast to humanized mouse models, our model allows transfer of syngeneic murine T cells, which is of special importance for long-term studies of cell-cell interactions and to avoid misinterpretation of anti-tumor efficacy by graft-versus-host reactions.43 Compared to humanized mouse models, our model is less laborious, cheaper, and more physiological.
Taken together, by irradiation and subsequent tolerization with a signaling-deficient CAR, we were able to induce long-lasting, specific tolerance to human-derived CAR domains, and we could thereby study the engraftment, proliferation, long-term persistence, and antiviral effector function of S-CAR T cells in fully immunocompetent mice. We believe that this model can be transferred to other CAR T cell approaches in case they require preclinical evaluation in the context of a fully functional immune system. It will allow for the study of interactions with the different arms of the endogenous immune system, bystander immune cell activation, and combination therapies with checkpoint inhibitors. Thus, it will help to bring better characterized, more efficient, and safer cell products into the clinics.
Materials and Methods
Animal Models
HBVtg HBV1.3xfs mice (HBV genotype D, serotype ayw), Rag2−/− mice, Rag2−/−/IL-2Rγ−/− mice, and CD45.1+ C57BL/6 donor mice were bred in-house in specific pathogen-free animal facilities. AAV serotype 2 containing the 1.2 overlength genome of HBV genotype D (AAV-HBV) was packed with an AAV serotype 8 capsid, as previously described.21 Viral vector was produced by Plateforme de Thérapie Génique (Nantes, France). For the AAV-HBV model, 8-week-old male wild-type C57BL/6J mice were purchased from Janvier (Le Genest-Sain-Isle, France) and infected with 2 × 1010 viral particles 3–4 weeks before T cell transfer. The study was conducted according to the German Law for the Protection of Animals and was approved by the local authorities.
Retroviral Transduction and Adoptive T Cell Transfer
Splenocytes were isolated from donor mice and enriched for CD8+ T cells using CD8a MACS beads (Miltenyi Biotec, Bergisch Gladbach, Germany). A total of 1.5 × 106 CD8+ T cells/well were stimulated for 24 h with 5 ng/mL interleukin-12 (IL-12) (kindly provided by E. Schmitt, University of Mainz) on tissue-treated 12-well plates that were precoated with anti-CD3 and anti-CD28 antibodies (kindly provided by R. Feederle, Helmholtz Zentrum München) for 2 h at 37°C (10 μg/mL in PBS). The next day, cells were transferred to uncoated 12-well plates and retrovirally transduced 2 days in a row. Retroviral supernatants were obtained from Platinum-E packaging cells transfected with MP71 retroviral plasmids containing CAR-coding sequences. Activated CD8+ T cells and retroviral supernatants were supplemented with 2 μg/mL protamine sulfate (Leo Pharma, Neu-Isenburg, Germany) and spinoculated (850 × g, 32°C, 2 h). The day after the second transduction, cells were analyzed by flow cytometry and the transduction rate determined as described below. Cells were washed twice with PBS, resuspended in PBS, and transferred intraperitoneally in 200 μL into mice that were grouped by HBsAg and HBeAg levels. If applicable, mice were irradiated with 5 Gy 1 day before T cell transfer.
Isolation of Splenocytes, Liver-Associated Lymphocytes, and Peripheral Blood Mononuclear Cells
Spleens were mashed through a 100-μm cell strainer and erythrocytes lysed using ammonium-chloride-potassium (ACK) lysis buffer (8 g NH4Cl, 1 g KHCO3, and 37 mg Na2EDTA, added to 1 L H2O [pH 7.2–7.4]) for 2 min at room temperature (RT). Livers were perfused with PBS to eliminate circulating lymphocytes in blood and mashed through a 100-μm cell strainer. Mashed liver tissue was digested with 4,500 U collagenase type 4 (Worthington Biochemical, Lakewood, NJ, USA) (20 min, 37°C), and leukocytes were purified in an 80%/40% Percoll (GE Healthcare, Solingen, Germany) gradient (1,400 × g, 20 min, RT, without brake). For peripheral blood mononuclear cell isolation, peripheral blood was collected into Microvette 500 LH-Gel tubes (Sarstedt, Nümbrecht, Germany), and 15 μL blood was incubated with 250 μL ACK lysis buffer for 2 min at RT and then resuspended in fluorescence-activated cell sorting (FACS) buffer (0.1% BSA in PBS).
Flow Cytometry
Staining of cells was performed for 30 min in the dark on ice in FACS buffer (0.1% BSA in PBS). The following antibodies were used and purchased from different suppliers: CD4, CD8, CD19, CD45.1, IFN-γ, and TNF-α (BD Biosciences, Heidelberg, Germany); CD3, CD45.2, CD62L, CD127, NK1.1, and PD-1 (Thermo Fisher Scientific, Germering, Germany); and CTLA-4 and Tim-3 (BioLegend, Koblenz, Germany). Viable cells were determined with LIVE/DEAD cell marker (Thermo Fisher Scientific). For ICS, cells were permeabilized using Cytofix/Cytoperm (BD Biosciences) prior to incubation with antibodies, following the manufacturer’s instructions. Total cell numbers were determined by the addition of CountBright Absolute Counting Beads (Thermo Fisher Scientific). Cells were analyzed on a FACS Canto II (BD Biosciences) or CytoFLEX S (Beckman Coulter, Munich, Germany). If a CAR, EGFRt and other surface markers were analyzed: first, the CAR was stained with an anti-human-IgG (Abcam, Cambridge, UK) or anti-murine-IgG (BD Biosciences) antibody, followed by the primary staining of EGFRt with biotin-labeled cetuximab (Merck, Darmstadt, Germany). In a last step, bound cetuximab was stained with fluorochrome-labeled streptavidin (Thermo Fisher Scientific) together with additional antibodies against surface markers.
Cultivation of Murine Cells
Primary murine cells were cultured in murine T cell medium (RPMI Dutch modified, 10% fetal calf serum (FCS), 1% glutamine, 1% penicillin and strepomycin, 1% sodium pyruvate, and 50 μM β-mercaptoethanol; Thermo Fisher Scientific).
Ex Vivo T Cell Stimulation
The functionality of S-CAR T cells was determined by culturing 5 × 105 splenocytes or liver-associated lymphocytes per well on tissue-treated 96-well plates precoated with HBsAg (2.5 μg/mL in PBS, overnight, 4°C; Roche Diagnostics, Mannheim, Germany) or anti-CD3 and anti-CD28 antibodies (10 μg/mL in PBS, overnight, 4°C). To determine an immune response against the S-CAR and EGFRt, 1 × 106 splenocytes were cultured with 1 × 105 S-CAR+ or EGFRt+ CD8+ T cells. After 1 h of culture, 1 μg/mL brefeldin A (Sigma-Aldrich, Munich, Germany) was added. Cytokine expression was determined the following day via an ICS and flow cytometry analysis.
Cell-Based Anti-S-CAR and Anti-EGFRt Antibody Detection
Platinum-E cells were transfected with MP71 plasmids encoding the S-CAR or EGFRt. After 48 h, cells were harvested and a flow cytometry staining was performed. Cells were stained with serum diluted 1:200 in FACS buffer, and, in a subsequent staining step, they were incubated with phycoerythrin (PE)-labeled anti-mouse-IgG antibody (12-4010-82; Thermo Fisher Scientific). Median fluorescence intensity was determined on a CytoFLEX S (Beckman Coulter).
ELISA-Based Anti-C8 and Anti-IgG1 Antibody Detection
ELISA plates were precoated overnight with recombinant scFv C8 (1 μg/mL in PBS) or IgG1 cetuximab antibody (1 μg/mL in PBS; Merck) at 4°C. The next day, plates were blocked with assay diluent (1% BSA in PBS) for 1 h at RT. Diluted serum of treated mice was incubated on wells for 2 h, and bound antibodies were detected with a horseradish peroxidase (HRP)-labeled anti-mouse-IgG antibody (1:1,000, Sigma-Aldrich). 3,3',5,5'-tetramethylbenzidine (TMB) substrate (Thermo Fisher Scientific) conversion (OD450 nm − OD560 nm) was measured on an infinite F200 photometer (Tecan, Männedorf, Switzerland), and the signal of serum on uncoated wells was subtracted.
Intrahepatic AAV- and HBV-DNA Copies
DNA was extracted from approximately 20 mg liver tissue using the Nucleo Spin Tissue Kit (Macherey-Nagel, Berlin, Germany), following the manufacturer’s instructions. qPCR was performed with SyBrGreen (Roche Diagnostics) on a LightCycler 480 II (Roche Diagnostics) using the following primers: AAV forward, 5′-AACCCGCCATGCTACTTATCTACGT-3′; AAV reverse, 5′-CACACAGTCTTTGAAGTAGGCC-3′; HBV forward, 5′-GCCTCATCTTCTTGTTGGTTC-3′; and HBV reverse, 5′-GAAAGCCCTACGAACCACTGAAC-3′. Results were normalized to cell numbers using the single-copy gene PRNP (PRNP forward, 5′-TGCTGGGAAGTGCCATGAG-3′; and PRNP reverse, 5′-CGGTGCATGTTTTCACGATAGTA-3′).
Serological Analyses
Peripheral blood was collected into Microvette 500 LH-Gel tubes (Sarstedt) and centrifuged to separate serum (10 min, 5,000 × g, RT). ALT activity was determined 1:4 diluted with PBS using the Reflotron ALT test (Roche Diagnostics). Serum HBsAg, HBeAg and anti-HBs antibodies were quantified in different dilutions with PBS on an Architect platform using the quantitative HBsAg test (6C36-44; cut-off, 0.25 IU/mL), the HBeAg Reagent Kit (6C32-27) with HBeAg Quantitative Calibrators (7P24-01; cut-off, 0.20 PEI U/mL), and the anti-HBs test (7C18-27; cut-off, 12.5 mIU/mL) (Abbott Laboratories, Wiesbaden, Germany).
Production of Recombinant scFv C8
E. coli XL1-blue were transformed with scFv C8-encoding pHOG21 plasmid and a single-clone colony inoculated overnight in 5 mL lysogeny broth (LB) media (10 g Tryptone, 5 g yeast extract, and 10 g NaCl, added to 1 L H2O). The next day, 3 L 2x yeast extract tryptone (YT) (17 g Tryptone, 10 g yeast extract, and 5 g NaCl, added to 1 L H2O) was inoculated 1:1,000 and grown for approximately 10 h until the optical density (OD)600 reached 0.6. Induction was performed overnight at 18°C with 0.1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) (Carl Roth, Karlsruhe, Germany). Large-scale protein purification was performed by fast protein liquid chromatography under native conditions on an ÄKTA avant (GE Healthcare). Bacterial cells of the overnight induction culture were harvested (15 min, 5,000 × g, RT) and resuspended in 10 mL ÄKTA binding buffer (20 mM disodium phosphate, 500 mM NaCl, and 20 mM imidazole [pH 7.4]) per 1 g bacterial pellet. 3 U/mL benzonase (Merck) and 0.2 mg/mL lysozyme (Thermo Fisher Scientific) were added, followed by incubation for 20 min on ice. The samples were then submitted to five cycles of sonication (1 min each) and centrifuged (30 min, 5,000 × g, 4°C). Samples were continuously kept on ice.
A HisTrap FF crude 5 mL column (GE Healthcare) was connected to the ÄKTA avant (GE Healthcare) and loaded with lysate. Samples were eluted by gradually increasing the proportion of elution buffer (20 mM disodium phosphate, 500 mM NaCl, and 500 mM imidazole [pH 7.4]) with a flow rate of 5 mL/min collecting 1-mL fractions. The protein content of the eluent was measured by UV monitoring at 280 nm. According to the chromatographic peaks, the respective fractions were analyzed by SDS-PAGE and Coomassie staining in order to confirm protein presence. Positive fractions were pooled and dialyzed to storage buffer (25 mM Tris-HCl, 100 mM KCl, 1 mM EDTA, 1 mM MgCl2, and 10% Glycerol [pH 7.4]) overnight at 4°C. The final sample was filtered and protein concentration was measured via a Nanodrop One (Thermo Fisher Scientific).
Author Contributions
M.M.F., J.F., T.A., and K.W. conducted the experiments. S.S. and J.S. produced critical reagents and helped to perform experiments. S.P.F. helped with experimental setup and performed irradiation. D.H.B. provided essential infrastructure and technical support. M.M.F., K.W., M.A.M.-H., and U.P. designed the experiments. M.M.F., K.W., and U.P. wrote the paper.
Conflicts of Interest
U.P. and K.W. are co-founders of and M.M.F. was employed part-time by SCG Cell Therapy, Singapore. K.W. is consulting for LION TCR/SCG Cell Therapy, Singapore. U.P. serves as ad-hoc scientific advisor for Roche, GILEAD, J&J, Abbvie, Merck, Arbutus, and VIR Biotechnology.
Acknowledgments
We are grateful to Michael Jensen for providing the EGFRt construct, Hinrich Abken for providing CAR constructs, and Marie-Louise Michel for providing the AAV-HBV1.2 construct. We thank Philipp Hagen, Natalie Röder, and Romina Bester for excellent technical assistance and Claudia Dembek for critical reading of the manuscript. The work was funded by the German Research Foundation (DFG) via TRR36 and the German Center for Infection Research (DZIF).
Footnotes
Supplemental Information includes eight figures and can be found with this article online at https://doi.org/10.1016/j.ymthe.2019.02.001.
Contributor Information
Karin Wisskirchen, Email: karin.wisskirchen@helmholtz-muenchen.de.
Ulrike Protzer, Email: protzer@tum.de.
Supplemental Information
References
- 1.Gill S., Maus M.V., Porter D.L. Chimeric antigen receptor T cell therapy: 25years in the making. Blood Rev. 2016;30:157–167. doi: 10.1016/j.blre.2015.10.003. [DOI] [PubMed] [Google Scholar]
- 2.Brower V. First Chimeric Antigen Receptor T-Cell Therapy Approved. J. Natl. Cancer Inst. 2017;109:djx251. doi: 10.1093/jnci/djx251. [DOI] [PubMed] [Google Scholar]
- 3.Mullard A. Second anticancer CAR T therapy receives FDA approval. Nat. Rev. Drug Discov. 2017;16:818. doi: 10.1038/nrd.2017.249. [DOI] [PubMed] [Google Scholar]
- 4.Hartmann J., Schüßler-Lenz M., Bondanza A., Buchholz C.J. Clinical development of CAR T cells-challenges and opportunities in translating innovative treatment concepts. EMBO Mol. Med. 2017;9:1183–1197. doi: 10.15252/emmm.201607485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bohne F., Chmielewski M., Ebert G., Wiegmann K., Kürschner T., Schulze A., Urban S., Krönke M., Abken H., Protzer U. T cells redirected against hepatitis B virus surface proteins eliminate infected hepatocytes. Gastroenterology. 2008;134:239–247. doi: 10.1053/j.gastro.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 6.Full F., Lehner M., Thonn V., Goetz G., Scholz B., Kaufmann K.B., Mach M., Abken H., Holter W., Ensser A. T cells engineered with a cytomegalovirus-specific chimeric immunoreceptor. J. Virol. 2010;84:4083–4088. doi: 10.1128/JVI.02117-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sautto G.A., Wisskirchen K., Clementi N., Castelli M., Diotti R.A., Graf J., Clementi M., Burioni R., Protzer U., Mancini N. Chimeric antigen receptor (CAR)-engineered T cells redirected against hepatitis C virus (HCV) E2 glycoprotein. Gut. 2016;65:512–523. doi: 10.1136/gutjnl-2014-308316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hale M., Mesojednik T., Romano Ibarra G.S., Sahni J., Bernard A., Sommer K., Scharenberg A.M., Rawlings D.J., Wagner T.A. Engineering HIV-Resistant, Anti-HIV Chimeric Antigen Receptor T Cells. Mol. Ther. 2017;25:570–579. doi: 10.1016/j.ymthe.2016.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leibman R.S., Richardson M.W., Ellebrecht C.T., Maldini C.R., Glover J.A., Secreto A.J., Kulikovskaya I., Lacey S.F., Akkina S.R., Yi Y. Supraphysiologic control over HIV-1 replication mediated by CD8 T cells expressing a re-engineered CD4-based chimeric antigen receptor. PLoS Pathog. 2017;13:e1006613. doi: 10.1371/journal.ppat.1006613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turtle C.J., Hanafi L.A., Berger C., Gooley T.A., Cherian S., Hudecek M., Sommermeyer D., Melville K., Pender B., Budiarto T.M. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Invest. 2016;126:2123–2138. doi: 10.1172/JCI85309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Siegler E.L., Wang P. Preclinical Models in Chimeric Antigen Receptor-Engineered T-Cell Therapy. Hum. Gene Ther. 2018;29:534–546. doi: 10.1089/hum.2017.243. [DOI] [PubMed] [Google Scholar]
- 12.Hay K.A., Turtle C.J. Chimeric Antigen Receptor (CAR) T Cells: Lessons Learned from Targeting of CD19 in B-Cell Malignancies. Drugs. 2017;77:237–245. doi: 10.1007/s40265-017-0690-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.WHO (2018). Hepatitis B, fact sheet. https://www.who.int/en/news-room/fact-sheets/detail/hepatitis-b.
- 14.Sartorius K., Sartorius B., Aldous C., Govender P.S., Madiba T.E. Global and country underestimation of hepatocellular carcinoma (HCC) in 2012 and its implications. Cancer Epidemiol. 2015;39:284–290. doi: 10.1016/j.canep.2015.04.006. [DOI] [PubMed] [Google Scholar]
- 15.Gehring A.J., Protzer U. Targeting Innate and Adaptive Immune Responses to Cure Chronic HBV Infection. Gastroenterology. 2019;156:325–337. doi: 10.1053/j.gastro.2018.10.032. [DOI] [PubMed] [Google Scholar]
- 16.Safaie P., Poongkunran M., Kuang P.P., Javaid A., Jacobs C., Pohlmann R., Nasser I., Lau D.T. Intrahepatic distribution of hepatitis B virus antigens in patients with and without hepatocellular carcinoma. World J. Gastroenterol. 2016;22:3404–3411. doi: 10.3748/wjg.v22.i12.3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krebs K., Böttinger N., Huang L.R., Chmielewski M., Arzberger S., Gasteiger G., Jäger C., Schmitt E., Bohne F., Aichler M. T cells expressing a chimeric antigen receptor that binds hepatitis B virus envelope proteins control virus replication in mice. Gastroenterology. 2013;145:456–465. doi: 10.1053/j.gastro.2013.04.047. [DOI] [PubMed] [Google Scholar]
- 18.Wang X., Chang W.C., Wong C.W., Colcher D., Sherman M., Ostberg J.R., Forman S.J., Riddell S.R., Jensen M.C. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood. 2011;118:1255–1263. doi: 10.1182/blood-2011-02-337360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paszkiewicz P.J., Fräßle S.P., Srivastava S., Sommermeyer D., Hudecek M., Drexler I., Sadelain M., Liu L., Jensen M.C., Riddell S.R., Busch D.H. Targeted antibody-mediated depletion of murine CD19 CAR T cells permanently reverses B cell aplasia. J. Clin. Invest. 2016;126:4262–4272. doi: 10.1172/JCI84813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hombach A., Hombach A.A., Abken H. Adoptive immunotherapy with genetically engineered T cells: modification of the IgG1 Fc ‘spacer’ domain in the extracellular moiety of chimeric antigen receptors avoids ‘off-target’ activation and unintended initiation of an innate immune response. Gene Ther. 2010;17:1206–1213. doi: 10.1038/gt.2010.91. [DOI] [PubMed] [Google Scholar]
- 21.Dion S., Bourgine M., Godon O., Levillayer F., Michel M.L. Adeno-associated virus-mediated gene transfer leads to persistent hepatitis B virus replication in mice expressing HLA-A2 and HLA-DR1 molecules. J. Virol. 2013;87:5554–5563. doi: 10.1128/JVI.03134-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Staples P.J., Gery I., Waksman B.H. Role of the thymus in tolerance. 3. Tolerance to bovine gamma globulin after direct injection of antigen into the shielded thymus of irradiated rats. J. Exp. Med. 1966;124:127–139. doi: 10.1084/jem.124.2.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Starr T.K., Jameson S.C., Hogquist K.A. Positive and negative selection of T cells. Annu. Rev. Immunol. 2003;21:139–176. doi: 10.1146/annurev.immunol.21.120601.141107. [DOI] [PubMed] [Google Scholar]
- 24.Mueller D.L. Mechanisms maintaining peripheral tolerance. Nat. Immunol. 2010;11:21–27. doi: 10.1038/ni.1817. [DOI] [PubMed] [Google Scholar]
- 25.Nemazee D. Mechanisms of central tolerance for B cells. Nat. Rev. Immunol. 2017;17:281–294. doi: 10.1038/nri.2017.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Posselt A.M., Barker C.F., Tomaszewski J.E., Markmann J.F., Choti M.A., Naji A. Induction of donor-specific unresponsiveness by intrathymic islet transplantation. Science. 1990;249:1293–1295. doi: 10.1126/science.2119056. [DOI] [PubMed] [Google Scholar]
- 27.Turvey S.E., Hara M., Morris P.J., Wood K.J. Mechanisms of tolerance induction after intrathymic islet injection: determination of the fate of alloreactive thymocytes. Transplantation. 1999;68:30–39. doi: 10.1097/00007890-199907150-00007. [DOI] [PubMed] [Google Scholar]
- 28.Proietto A.I., Lahoud M.H., Wu L. Distinct functional capacities of mouse thymic and splenic dendritic cell populations. Immunol. Cell Biol. 2008;86:700–708. doi: 10.1038/icb.2008.63. [DOI] [PubMed] [Google Scholar]
- 29.Watanabe K., Terakura S., Martens A.C., van Meerten T., Uchiyama S., Imai M., Sakemura R., Goto T., Hanajiri R., Imahashi N. Target antigen density governs the efficacy of anti-CD20-CD28-CD3 ζ chimeric antigen receptor-modified effector CD8+ T cells. J. Immunol. 2015;194:911–920. doi: 10.4049/jimmunol.1402346. [DOI] [PubMed] [Google Scholar]
- 30.Guidotti L.G., Ishikawa T., Hobbs M.V., Matzke B., Schreiber R., Chisari F.V. Intracellular inactivation of the hepatitis B virus by cytotoxic T lymphocytes. Immunity. 1996;4:25–36. doi: 10.1016/s1074-7613(00)80295-2. [DOI] [PubMed] [Google Scholar]
- 31.Guidotti L.G., Rochford R., Chung J., Shapiro M., Purcell R., Chisari F.V. Viral clearance without destruction of infected cells during acute HBV infection. Science. 1999;284:825–829. doi: 10.1126/science.284.5415.825. [DOI] [PubMed] [Google Scholar]
- 32.Lucifora J., Xia Y., Reisinger F., Zhang K., Stadler D., Cheng X., Sprinzl M.F., Koppensteiner H., Makowska Z., Volz T. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science. 2014;343:1221–1228. doi: 10.1126/science.1243462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xia Y., Stadler D., Lucifora J., Reisinger F., Webb D., Hösel M., Michler T., Wisskirchen K., Cheng X., Zhang K. Interferon-γ and Tumor Necrosis Factor-α Produced by T Cells Reduce the HBV Persistence Form, cccDNA, Without Cytolysis. Gastroenterology. 2016;150:194–205. doi: 10.1053/j.gastro.2015.09.026. [DOI] [PubMed] [Google Scholar]
- 34.Protzer U., Maini M.K., Knolle P.A. Living in the liver: hepatic infections. Nat. Rev. Immunol. 2012;12:201–213. doi: 10.1038/nri3169. [DOI] [PubMed] [Google Scholar]
- 35.Knolle P.A., Thimme R. Hepatic immune regulation and its involvement in viral hepatitis infection. Gastroenterology. 2014;146:1193–1207. doi: 10.1053/j.gastro.2013.12.036. [DOI] [PubMed] [Google Scholar]
- 36.Berger C., Turtle C.J., Jensen M.C., Riddell S.R. Adoptive transfer of virus-specific and tumor-specific T cell immunity. Curr. Opin. Immunol. 2009;21:224–232. doi: 10.1016/j.coi.2009.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Srivastava S., Riddell S.R. Chimeric Antigen Receptor T Cell Therapy: Challenges to Bench-to-Bedside Efficacy. J. Immunol. 2018;200:459–468. doi: 10.4049/jimmunol.1701155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cheng J.C., Liu M.C., Tsai S.Y., Fang W.T., Jer-Min Jian J., Sung J.L. Unexpectedly frequent hepatitis B reactivation by chemoradiation in postgastrectomy patients. Cancer. 2004;101:2126–2133. doi: 10.1002/cncr.20591. [DOI] [PubMed] [Google Scholar]
- 39.Lok A.S., Liang R.H., Chiu E.K., Wong K.L., Chan T.K., Todd D. Reactivation of hepatitis B virus replication in patients receiving cytotoxic therapy. Report of a prospective study. Gastroenterology. 1991;100:182–188. doi: 10.1016/0016-5085(91)90599-g. [DOI] [PubMed] [Google Scholar]
- 40.Nakamura Y., Motokura T., Fujita A., Yamashita T., Ogata E. Severe hepatitis related to chemotherapy in hepatitis B virus carriers with hematologic malignancies. Survey in Japan, 1987-1991. Cancer. 1996;78:2210–2215. doi: 10.1002/(sici)1097-0142(19961115)78:10<2210::aid-cncr24>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 41.Ma B., Yeo W., Hui P., Ho W.M., Johnson P.J. Acute toxicity of adjuvant doxorubicin and cyclophosphamide for early breast cancer -- a retrospective review of Chinese patients and comparison with an historic Western series. Radiother. Oncol. 2002;62:185–189. doi: 10.1016/s0167-8140(02)00003-8. [DOI] [PubMed] [Google Scholar]
- 42.Perceau G., Diris N., Estines O., Derancourt C., Lévy S., Bernard P. Late lethal hepatitis B virus reactivation after rituximab treatment of low-grade cutaneous B-cell lymphoma. Br. J. Dermatol. 2006;155:1053–1056. doi: 10.1111/j.1365-2133.2006.07451.x. [DOI] [PubMed] [Google Scholar]
- 43.Shultz L.D., Brehm M.A., Garcia-Martinez J.V., Greiner D.L. Humanized mice for immune system investigation: progress, promise and challenges. Nat. Rev. Immunol. 2012;12:786–798. doi: 10.1038/nri3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.