

Abstract
Viral and cellular double-stranded RNA (dsRNA) is recognized by cytosolic innate immune sensors, including RIG-I-like receptors. Some cytoplasmic dsRNA is commonly present in cells, and one source is mitochondrial dsRNA, which results from bidirectional transcription of mitochondrial DNA (mtDNA). Here we demonstrate that Trp53 mutant mouse embryonic fibroblasts contain immune-stimulating endogenous dsRNA of mitochondrial origin. We show that the immune response induced by this dsRNA is mediated via RIG-I-like receptors and leads to the expression of type I interferon and proinflammatory cytokine genes. The mitochondrial dsRNA is cleaved by RNase L, which cleaves all cellular RNA including mitochondrial mRNAs, increasing activation of RIG-I-like receptors. When mitochondrial transcription is interrupted there is a subsequent decrease in this immune-stimulatory dsRNA. Our results reveal that the role of p53 in innate immunity is even more versatile and complex than previously anticipated. Our study, therefore, sheds new light on the role of endogenous RNA in diseases featuring aberrant immune responses.
Keywords: mitochondrial dsRNA, p53, innate immunity, RNase L
INTRODUCTION
Type I interferon (IFN) secretion is a first line of defense against viral pathogens in most mammalian cells. IFN production and secretion is activated after host-specific pattern recognition receptors (PRRs) detect pathogen associated molecular patterns (PAMPs). A wide range of different types of molecules can serve as PAMPs, thus PRRs recognize specific types of ligands. MDA5 (melanoma differentiation-associated protein 5) and RIG-I (retinoic acid-inducible gene I) are two RIG-I-like receptors (RLRs), that detect double-stranded RNA (dsRNA), which is a replication intermediate for RNA viruses (Kang et al. 2002; Yoneyama et al. 2004). Nevertheless, those two receptors recognize different features of dsRNAs: RIG-I selectively binds ssRNA or short blunt-ended dsRNA (less than 1 kb) bearing uncapped 5′-di- or triphosphate whereas MDA-5 binds long dsRNA. Upon activation, both receptors interact via CARD domains with the mitochondrial antiviral signaling protein (MAVS). MAVS recruitment leads to nuclear translocation of the transcription factors IRF3 and NF-κB, and to the production of proinflammatory cytokines, chemokines and type I IFN and later to induction of hundreds of IFN-stimulated genes (ISGs) (Sato et al. 2000).
There are multiple sources of viral dsRNA; viral genomes (dsRNA viruses), viral replication (ssRNA viruses) or viral transcription (DNA viruses). Over the last 10–15 yr, many endogenous dsRNAs have been described, including pre-miRNA, rRNA stem–loops, inverted repeat Alu-elements (IR-Alu) (Chen et al. 2008; Sugimoto et al. 2015) and mitochondrial dsRNA, which is a result of bidirectional transcription of mitochondrial DNA (mtDNA) (Borowski et al. 2013; Dhir et al. 2018). In mice, mtDNA is a ∼16.5 kb long, circular DNA molecule, which is organized into mitochondrial nucleoids (Peralta et al. 2012). The mouse mitochondrial genome contains 37 genes coding for 13 proteins, 12S and 16S ribosomal RNAs (rRNAs), and 22 transfer RNAs (tRNAs) (Bibb et al. 1981; Bayona-Bafaluy et al. 2003). The mtDNA genes are arranged mostly on the Heavy (H) strand, which encompasses 12 of the 13 mRNAs, rRNAs and 14 of the 22 tRNAs, while the Light (L) strand codes only for one mRNA and eight tRNAs (Peralta et al. 2012). Almost the entire L strand transcript undergoes rapid decay by the RNA degradosome complex, which includes PNPase and hSuv3 helicase (Borowski et al. 2013). Those two enzymes are extremely important in restricting the levels of mitochondrial dsRNA, as the loss of either of them causes massive accumulation of mitochondrial dsRNA that escapes into the cytoplasm. This mitochondrial dsRNA triggers an MDA5-driven antiviral signaling pathway that results in a type I IFN response and therefore it is another cellular source of dsRNA (Dhir et al. 2018).
Another dsRNA sensing system is the oligoadenylate synthetase (OAS)/RNase L pathway (Li et al. 2017). OAS enzymes (OAS1, OAS2, OAS3, and OASL), are IFN-inducible, and upon sensing dsRNA they produce 2′–5′-oligoadenylates (2–5A) which activates the nuclease RNase L. In mice, there are five Oas genes, Oas1a and Oas1g, Oas2, Oas3 and Oasl2 that encode active enzymes and one, Oasl1 that encodes an enzymatically inactive protein (Kakuta et al. 2002; Kristiansen et al. 2011). Oasl1 was initially shown to inhibit the translation of IRF7 mRNA and to act as a negative regulator of type I IFN synthesis (Lee et al. 2013a), but new evidence shows that early in the antiviral response Oasl1 has an opposite role as it promotes RLR signaling by trapping viral RNA in stress bodies (Kang et al. 2018). Active RNase L cleaves all cellular RNAs predominantly in single-stranded regions at UpN dinucleotides (UA and UU > UG) (Silverman and Weiss 2014; Li et al. 2017). In the absence of the RNA editing enzyme, adenosine deaminase acting on RNA 1 (ADAR1), OAS can be activated by self-dsRNA, resulting in RNase L activity and cell death (Li et al. 2017). Recent studies show that RIG-I like receptors are activated by oligoadenylate synthetase-like protein 1 (OASL1). Loss of OASL1 expression reduced RIG-I signaling and enhanced virus replication in human cells. Conversely, OASL1 expression enhanced RIG-I-mediated IFN induction (Zhu et al. 2014).
P53 controls transcription and is a well-documented tumor suppressor. It is also an ISG, induced by IFN upon viral infection (Takaoka et al. 2003). It is thought that its role in innate immunity is to induce apoptosis, thus preventing the spread of viral infection. P53 that is posttranslationally modified is located in the cytoplasm and enhances the permeability of the mitochondrial outer membrane thus stimulating apoptosis (Green and Kroemer 2009). However, treating Trp53 mutant mouse embryonic fibroblasts (MEFs) with the DNA demethylating agent 5-aza-2′-deoxycytidine (5-aza-dC) (Leonova et al. 2013), was also reported to cause a huge increase in the level of transcripts encoding short interspersed nuclear elements (SINEs) and other species of noncoding RNAs that generated a strong type 1 IFN response. Thus it appears that another function of p53 in cells is to ensure the silencing of repeats that can accidentally induce an immune response.
Identifying endogenous, immune-stimulating dsRNA is especially important in relation to autoimmune diseases. Here we demonstrate that in the absence of transcription factor p53, an immunogenic, endogenous dsRNA is produced in cells. Surprisingly this endogenous dsRNA does not encode predominantly SINEs or other tandem repeats. Instead we show that this RNA is of mitochondrial origin and is processed by the OAS/RNase L system. Our study therefore sheds new light on the role of endogenous RNA in diseases with aberrant immune responses.
RESULTS
Endogenous dsRNA from Trp53 cells can induce immune responses when transfected into test cells
Our initial hypothesis was that dsRNA from Adar1 mutant cells is unedited and therefore these cells contain higher amounts of immunogenic dsRNAs. This would result in the activation of dsRNA-binding receptors and to subsequent activation of immune pathways leading to type I IFN production. However, MEFs generated from Adar1 mutant embryos where the entire gene is deleted are not viable and require an additional Trp53 mutation eliminating p53 protein expression for viability (Mannion et al. 2014). To validate our hypothesis and investigate if endogenous dsRNA can induce an immune response, total RNA was isolated from Adar1;Trp53 double mutant MEFs and from control wild-type and Trp53 mutant MEFs. DsRNA was isolated from these purified total RNA samples by in vitro immunoprecipitation with J2 antibody specific for dsRNA (Fig. 1A). The quality of the RNA was evaluated by microcapillary electrophoresis (Supplemental Fig. S1). To test the innate immune inducing potential of dsRNA from the different cell types the isolated dsRNA was then transfected back into cultured cells with lipofectamine and innate immune responses were measured; all of the three different MEF cell lines were tested as recipients.
FIGURE 1.

Endogenous dsRNA from cells lacking Trp53 induces an innate immune response in transfected cells. (A) Schematic representation of the experiment. Endogenous dsRNA was isolated from purified total RNA of wild-type, Trp53 mutant and Adar1;Trp53 double mutant MEFs by immunoprecipitation with J2 antibody and transfected back into reporter cells of the same lines. (B) Immunoblot showing the expression of MDA5 and RIG-I receptors after wild-type, Trp53 and Adar1;Trp53 reporter MEFs were transfected with dsRNA. Data are representative of two independent experiments. Black lines indicate where the image was cut to make the same alignment for all three immunoblot images; the original images are in Supplemental Figure S2. (C) ELISA showing mean levels of IL-6 in cell culture supernatants of Trp53 MEFs transfected with dsRNA. Results are normalized to cytokine expression in nontransfected cells. Data are mean ± SD of n = 3. (D) RT-qPCR analysis of transcripts encoding IFN-α, two dsRNA receptors, and three other ISG mRNAs in Trp53 MEFs transfected with dsRNA. Results are normalized to mRNA expression in nontransfected cells. Data are mean ± SD of three independent experiments. (*) P ≤ 0.05, (**) P ≤ 0.01, (***) P ≤ 0.001.
To investigate whether lack of ADAR1 editing in Adar1;Trp53 double mutant MEFs generated dsRNA that can induce an innate immune response after transfection into test cells, we first performed immunoblotting on the test cells after dsRNA transfection to measure the expression of two dsRNA-binding sensors; RIG-I and MDA5 (Fig. 1B; Supplemental Fig. S2). Unexpectedly, the immunoblots revealed that the dsRNA that induced the strongest response was the dsRNA isolated from Trp53 mutant MEFs. DsRNA isolated from Adar1;Trp53 MEFs also increased the expression of RIG-I and MDA5 receptors, but the effect was lower when compared to Trp53 mutant MEFs. This trend was observed for all the three cell lines transfected; WT MEFs, Trp53 MEFs and Adar1;Trp53 MEFs. Nonetheless, quantification relative to α-tubulin level (with ImageJ Software; data not shown), shows higher intensity for cells transfected with dsRNA from Trp53 cells. This experiment was repeated and the same results were observed. The results of immunoblots demonstrate that the lack of Trp53 in MEF cells generates dsRNA able to activate RIG-I and MDA5 receptors and this effect is independent of the genotype of the recipient cell.
To validate that dsRNA derived from Trp53 mutant MEFs activates an innate immune response in recipient transfected cells, an ELISA to detect the secretion of proinflammatory cytokine IL-6 was performed (Fig. 1C). The highest IL-6 secretion was observed for the cells transfected with dsRNA from Trp53 MEFs, lower for cells transfected with dsRNA from Adar1;Trp53 double mutant MEFs and lowest for dsRNA from WT MEFs. Those results confirmed that this RNA activates the whole pathway, as cytokine secretion is the last step.
Trp53 mutant MEFs were transfected with dsRNA derived from WT, Trp53 or Adar1;Trp53 MEFs and the expression level of mRNA transcripts was measured for Ifit3, Ifit1, Irf7 (three ISGs), IFN-α, Ifih1 (encoding MDA5) and Ddx58 (encoding RIG-I) (Fig. 1D). The results were normalized to mRNA expression in nontransfected cells. The expression of ISG transcripts was significantly up-regulated when cells were transfected with dsRNA from Trp53 MEFs, when compared to cells transfected with dsRNA from Adar1;Trp53 MEFs or WT MEFs. In general, changes in mRNA expression were more modest for Irf7 than for Ifit1 and Ifit3. In the case of IFN-α and Ifih1 transcripts the increase followed the same trend, with the highest increase for cells transfected with dsRNA from Trp53 MEFs. Transcripts encoding the two receptors, Ifih1(MDA5) and Ddx58 (RIG-I), had lower increases in expression in cells transfected with dsRNA from Trp53 MEFs (approximately fivefold) when compared to WT MEFs and Adar1;Trp53 MEFs (approximately twofold) dsRNA. The increase is especially low for Ddx58, which is probably due to its relatively high basal expression in nontransfected cells (as seen on immunoblot, Fig. 1B). Thus, results of immunoblots, ELISA and qPCR assays demonstrate that dsRNA from Trp53 MEFs can induce an immune response when transfected back into Trp53 MEFs. Next, we sought to determine which receptor recognizes endogenous dsRNA from Trp53 mutant cells. RNAi knockdown of transcripts encoding MDA5 and RIG-I receptors revealed that the immune response induced by this dsRNA is mediated mainly, but not exclusively, via MDA5 receptor (Fig. 2). Immunoblot results show an increase in the expression of MDA5 in cells with RIG-I knockdown. However, when MDA5 is knocked down, the increase in the expression of RIG-I receptor is minor. RIG-I receptor is therefore also involved in this immune response induced by endogenous dsRNA from Trp53 mutant cells, however to a lower extent (Fig. 2). There is variability in this experiment due to the batch difference in poly I:C, but this does not affect the results which is that after knockdown of MDA5 in Trp53 mutant cells the isolated dsRNA is less immune-stimulatory.
FIGURE 2.

The immune response induced by dsRNA from Trp53 MEFs is mediated primarily via the MDA5 receptor. (A) Immunoblot showing the expression of MDA5 and RIG-I receptors after transfection of Adar1;Trp53 MEFs with dsRNA from Trp53 MEFs. Prior to transfection, RIG-I was knocked down in the cells with esiRNA. Data are the mean ± SD of three independent experiments and quantified using Image J software. (B) Immunoblot showing the expression of MDA5 and RIG-I receptors after transfection of Adar1;Trp53 MEFs with dsRNA from Trp53 MEFs. Prior to transfection, MDA5 was knocked down in the cells with esiRNA. Data are mean ± SD of three independent experiments and quantified using Image J software. In cases where bands were not quantifiable for all biological replicates (e.g., MDA5 was not visible upon MDA5 knockdown), error bars were not included in the quantification diagram.
For isolation of dsRNA, we used the dsRNA-specific monoclonal antibody J2 that is widely used to detect viral dsRNA in animals and plants (Weber et al. 2006; Dhir et al. 2018). We verified the specificity of J2 for dsRNA in vitro by digesting immunoprecipitated dsRNA from Trp53 MEFs with RNase V1 prior to transfection into Adar1;Trp53 MEFs. RNase V1 is specific for double-stranded helical conformations in RNA (Lowman and Draper 1986; Nilsen 2013). In contrast to undigested and denatured dsRNA, dsRNA digested with RNase V1 was unable to induce immune response measured as expression of two dsRNA-binding sensors; RIG-I and MDA5 (Fig. 3A). This result confirms that the immune-stimulatory effect indeed is mediated by dsRNA. We then determined whether the response is caused by the cytoplasmic or nuclear fraction of dsRNA. For this we performed cellular fractionation with digitonin (Supplemental Fig. S4) first; total RNA was then isolated from cytoplasmic and nuclear fractions separately, followed by immunoprecipitation of dsRNA with the J2 antibody. Only dsRNA from the cytoplasmic fraction of Trp53 MEFs was able to induce the immune response (Fig. 3B).
FIGURE 3.

The immuno-stimulatory effect of dsRNA from Trp53 mutant cells depends mainly on the cytoplasmic fraction of dsRNA. Immunoblot showing levels of MDA5 and RIG-I receptors in Adar1;Trp53 MEFs after transfection with (A) dsRNA from Trp53 MEFs treated with RNase V1 or denatured dsRNA; (B) dsRNA isolated from total RNA of either cytoplasmic or nuclear fractions of Trp53 MEFs. Data are the mean ± SD of three independent experiments and quantified using Image J software.
RNA-seq analysis of dsRNA
We performed J2 immunoprecipitation-based dsRNA sequencing (dsRNA-seq) to identify the differences between dsRNA derived from wild-type MEF cells and the two mutants cell lines. For sequencing purposes, ribosomal RNA was first depleted from the dsRNA before library preparation; ribosomal RNA-depleted dsRNA was still immune-stimulatory (Supplemental Fig. S5). Chromosome-wise coverage analysis of dsRNA-seq revealed that the mitochondrial genome has the highest ratio of differentially expressed genes in dsRNA from Trp53 mutant versus wild-type cells and from Adar1;Trp53 versus wild-type cells (Fig. 4A). The results were normalized to the total number of protein-coding genes on each chromosome. The analysis of expression levels of individual, differentially expressed genes mapped to the mitochondrial chromosome shows higher fold increase in 12 out of 13 protein-coding genes in Trp53 than in Adar1;Trp53 when compared to wild-type (Fig. 4B). We do not observe an increase in sense and antisense mitochondrial RNA that could form intermolecular duplexes that would activate MDA5. This result is consistent with previous reports showing that MDA5 is not responding to longer stretches of paired sense and antisense strands but instead it recognizes dsRNA that was created as intramolecular duplexes (Runge et al. 2014).
FIGURE 4.
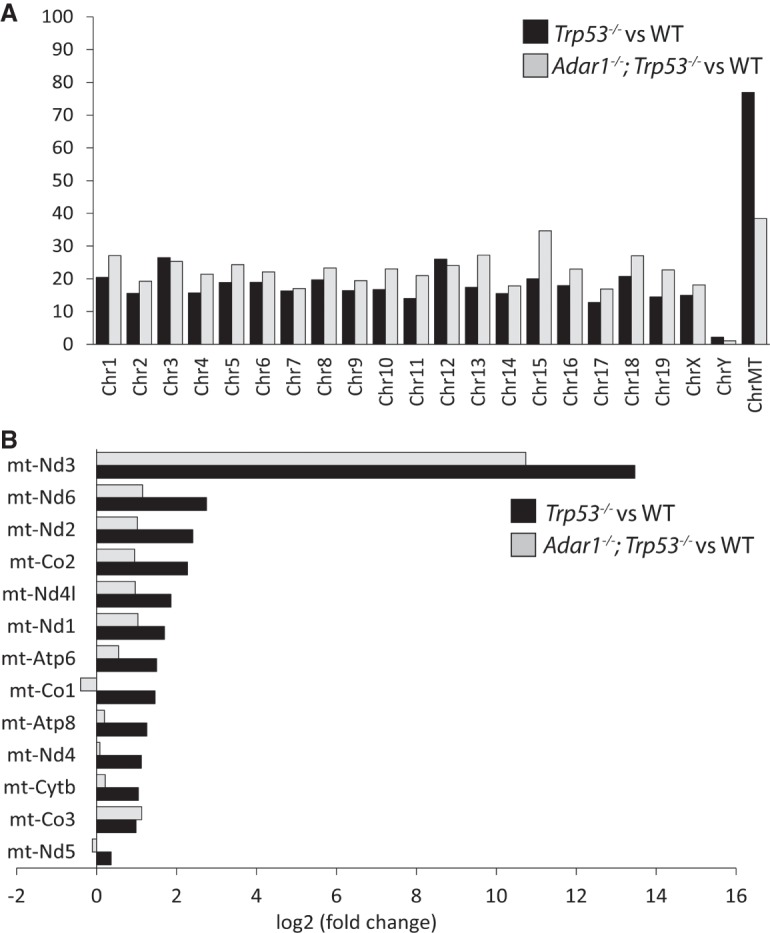
Sequences enriched in immunogenic dsRNA purified from untreated Trp53 versus wild-type and Adar1;Trp53 versus wild-type MEFs. (A) Chromosome-wise coverage showing the number of differentially expressed protein-coding genes, normalized to the total number of protein-coding genes on each chromosome. (B) Differential expression levels of differentially expressed genes mapped to the mitochondrial chromosome. All genes with an adjusted P-value <0.05 were considered as differentially expressed. Data are average of three independent experiments.
We also analyzed the repetitive sequence content of the J2-immunoprecipiated dsRNA material. For this analysis “RepBase repeat consensus” for mapping reads representing various types of repetitive elements in rodents was used (Supplemental Fig. S6; Supplemental Table S3). Immunoprecipitated dsRNA obtained from Adar1;Trp53 and from Trp53 MEFs did not show any significant enrichment in repetitive elements when compared to dsRNA from WT MEFs.
Adar1;Trp53 and Trp53 cells have an elevated immune response
To investigate whether there is an altered global transcriptional profile of genes involved in innate immune response in the Adar1;Trp53 and Trp53 cells from which the dsRNA was being purified, we performed next generation sequencing of total RNA isolated from these cells and wild-type controls. The resulting data revealed that transcripts of 1427 genes are up-regulated and 2153 down-regulated (at least 1.5 log2 fold and adjusted P-value <0.05) in Trp53 MEFs, while transcripts of 922 genes are up-regulated and 1440 down-regulated (at least log 1.5 fold) in Adar1;Trp53 MEFs (Supplemental Table S2; Supplemental Fig. S7).
Transcripts of some genes were up-regulated in both cell types, including classical proinflammatory and type I IFN-dependent genes encoding proteins of the Oas family (Oas3, Oasl1), IFN-induced protein with tetratricopeptide repeats 1 (Ifit1) and 3 (Ifit3), Irf7, Ilf3, Ifi205, and Isg15 (Fig. 5A). Two of the genes involved in immune responses, interleukin-1 receptor antagonist (Il1rn) and interleukin-4 receptor (Il4ra) were down-regulated in both cell lines. Interestingly, immunoglobulin-like domain-containing receptor 2 (Ildr2), which is a novel negative regulator for T cells involved in autoimmune response (Hecht et al. 2018), was highly down-regulated (over 10-fold) in Trp53 cells, but slightly up-regulated in Adar1;Trp53 cells. Sequencing results were confirmed by RT-qPCR comparing mRNA transcripts for Oas3, Ifit3 and Isg15 (Fig. 5B). Not all qPCR results correspond to sequencing results. For example, Oasl1 mRNA transcripts were higher in Adar1;Trp53 than in Trp53 MEFs (data not shown). Collectively, this finding suggests that, both Adar1;Trp53 and Trp53 MEFs when compared to wild-type cells, have altered transcriptional profiles of genes involved in innate immune responses, however transcripts of more genes are elevated in Trp53 MEFs. Additionally, Trp53 MEFs show a fivefold increase in Ifn-α mRNA transcript, compared to wild-type cells (Fig. 5B), whereas Adar1;Trp53 MEFs show an over fivefold increase in lfn-β mRNA transcript, compared to wild-type cells (Fig. 5B). Interestingly, chromosome-wise coverage analysis of total RNA-seq of differentially expressed genes again revealed that the mitochondrial genome has the highest ratio of differentially expressed genes in RNA from Trp53 versus wild-type cells and Adar1;Trp53 versus wild-type cells (Fig. 5C). The results were normalized to the total number of protein-coding genes on each chromosome. The effect is “diluted” compared to the dsRNA-seq results (Fig. 4A). This is not a surprise, as total RNA-seq does not enrich for the immune-stimulatory dsRNA.
FIGURE 5.

Aberrant immune response in Adar1;Trp53 double mutant MEFs and in Trp53 MEFs. (A) Differential expression of representative proinflammatory and type I IFN-dependent response genes in Trp53 versus wild-type and in Adar1;Trp53 versus wild-type MEFs. (B) RT-qPCR analysis of Oas3, Isg15, Ifit3, IFN-α, and IFN-β mRNA in wild-type, Trp53, and Adar1;Trp53 MEFs. Results are normalized to mRNA expression in wild-type cells. Data are the mean ± SD of n = 3–5. (C) Chromosome-wise coverage plot of differentially expressed protein-coding genes, normalized to the total number of protein-coding genes on each chromosome. All genes with an adjusted P-value <0.05 were considered as differentially expressed. Data are average of three independent experiments. (*) P ≤ 0.05, (**) P ≤ 0.01, (***) P ≤ 0.001.
Immunogenic dsRNA is a mitochondrial RNA cleaved by RNase L
RNA-seq of total RNA revealed that one of the most up-regulated genes in both Adar1;Trp53 and Trp53 MEFs, was Oas3 (in comparison to WT MEFs) which is a member of the OAS family. Oas3 upon binding to dsRNA, activates RNase L that cleaves cellular RNA into small fragments (Kakuta et al. 2002; Ibsen et al. 2014). Among all Oas proteins, Oas3 shows a dominant role in RNase L activation, with a higher affinity for dsRNA than either Oas1 or Oas2 (Li et al. 2016). We therefore hypothesized, that dsRNA from Trp53 MEFs that induces an innate immune response in transfected recipient cells, could be a product of RNase L cleavage. We chose to test this hypothesis using dsRNA from Trp53 MEFs as the mRNA encoding RNase L is slightly up-regulated in Trp53 MEFs whereas it is not up-regulated and remains at the same level in the WT cells as in Adar1;Trp53 MEFs (Fig. 6A). RNase L was knocked down in Trp53 MEF with esiRNAs (Supplemental Fig. S8), followed by total RNA extraction and immunoprecipitation of dsRNA (Fig. 6B). The J2 antibody only recognizes dsRNA that is greater than 40 perfectly paired bases so esiRNAs, which are 21 bp long, are not immunoprecipitated with this antibody. Isolated dsRNA was then transfected into Trp53 MEFs and the immune response was measured in the transfected cells. As a control, cells were transfected with dsRNA derived from Trp53 MEFs treated with a general, nonspecific siRNA.
FIGURE 6.

Immunogenic dsRNA is a product of RNase L cleavage. (A) RNaseL mRNA expression in WT, Trp53, and Adar1;Trp53 MEFs. (B) Schematic representation of the experiment. Prior to isolation of endogenous dsRNA from Trp53 MEFs, RNaseL was knocked down in those cells with esiRNA. Isolated dsRNA was transfected back into Trp53 MEFs. (C) RT-qPCR analysis of Ifit3, Ifit1, Irf7, IFN-α, Ddx58, and Ifih1 mRNA in Trp53 MEFs transfected with dsRNA from Trp53, RNaseL knocked down MEFs and control dsRNA. Results are normalized to mRNA expression in nontransfected cells. (D) ELISA showing the mean levels of IL-6 in cell-culture supernatants of Trp53 MEFs transfected with dsRNA from Trp53, RNaseL knocked down MEFs and control dsRNA. Results are normalized to cytokine expression in nontransfected cells. Data are mean ± SD of three independent experiments. (*) P ≤ 0.05, (**) P ≤ 0.01, (***) P ≤ 0.001.
The expression of three chosen ISG mRNA transcripts, Ifit3, Ifit1, and Irf7, was significantly less induced when reporter cells were transfected with dsRNA derived from cells with RNase L knockdown, in comparison to cells transfected with dsRNA from Trp53 MEFs (Fig. 6C). Cells transfected with dsRNA obtained from Trp53 MEFs treated with negative control siRNAs show similar induction of ISGs to cells transfected with control dsRNA from Trp53 MEFs. This demonstrates that the ability of dsRNA to induce immune response relies on the presence of RNase L. The same reduced induction was observed for mRNA transcripts encoding IFN-α, Ifih1 and Ddx58, with significant decrease in expression in reporter cells transfected with dsRNA from cells with silenced RNase L (Fig. 6C).
To further confirm the effect of RNase L knockdown upon dsRNA immunogenicity, an ELISA assay was used to detect the secretion of proinflammatory cytokine IL-6 (Fig. 6D). The highest IL-6 secretion was observed from the cells transfected with dsRNA from Trp53 MEFs, lower from cells transfected with dsRNA from Trp53 MEFs with negative siRNAs and the lowest for dsRNA from Trp53 MEFs with RNase L knocked down. Taken together with qPCR results, this shows that the dsRNA from Trp53 MEFs that can induce an innate immune response may indeed be the product of RNase L cleavage, as the silencing of RNase L in those cells decreases the immunogenicity of dsRNA significantly.
Finally, to further support the idea that the dsRNA that triggers immune response is of mitochondrial origin, we inhibited mitochondrial transcription with low concentrations of ethidium bromide prior to dsRNA isolation (Fig. 7). Ethidium bromide causes decreased mitochondrial transcription without significantly affecting nuclear transcription (Hayakawa et al. 1998). In ethidium bromide–treated cells, analysis of transcripts by qPCR shows decreased mitochondrial transcription compared to untreated cells (Fig. 7B). DsRNA isolated from EtBr-treated cells was transfected into Adar1;Trp53 MEFs; immunoblots with RIG-I and MDA5 antibodies on proteins from transfected cells show that dsRNA isolated from ethidium bromide–treated cells triggers a weaker immune response (Fig. 7A). This result unambiguously demonstrates that in Trp53 MEFs there is an increase in mitochondrial dsRNA reaching the cytoplasm that can increase the hazard of triggering an innate immune response.
FIGURE 7.

Inhibition of mitochondrial transcription reduces the immune induction response to dsRNA from Trp53 mutant cells. (A) Immunoblots with RIG-I and MDA5 antibodies show that dsRNA isolated from ethidium bromide–treated cells triggers a reduced immune response in transfected Adar1;Trp53 MEFs. Data are the mean ± SD of three independent experiments and quantified using Image J software. (B) RT-qPCR analysis of mitochondrial transcripts in untreated or EtBr-treated Trp53 MEFs. Results are normalized to RNA expression in untreated cells. Gapdh was used as the housekeeping gene. (*) P ≤ 0.05, (**) P ≤ 0.01, (***) P ≤ 0.001.
DISCUSSION
The complex role of p53 in the immune system is increasingly appreciated (Muñoz-Fontela et al. 2016). Cells expressing p53 show p53-dependent apoptosis in response to viral infection (Turpin et al. 2005). As expected, mice deficient in p53 are prone to infection with viruses, including influenza A virus, probably due to the lack of an apoptotic response (Yan et al. 2015). Additionally, p53 directly activates transcription of a set of immune responsive genes, including Tlr3, and IFN regulatory factors 5 (Irf5) and 7 (Irf7) (Mori et al. 2002; Taura et al. 2008; Yan et al. 2015). In recent years, it has become evident that p53 is associated with the development of autoimmune diseases and suppresses the aberrant expression of proinflammatory factors (Takatori et al. 2014). Defective p53 is also now linked to the development of rheumatoid arthritis, SLE and dermatomyositis/polymyositis (Kovacs et al. 1997; Chauhan et al. 2004; Mimura et al. 2007). P53 directly inhibits the production of numerous cytokines by inhibiting signal transducer and activator of transcription 1 (STAT1) (Youlyouz-Marfak et al. 2008), and p53 deficiency in macrophages increases the production of proinflammatory cytokines, such as IL-1, IL-6, IL-12, and TNF-α (Komarova et al. 2005; Gudkov et al. 2011).
Here, we show that dsRNA from Trp53 mutant murine cell lines induces an aberrant innate immune response in transfected reporter cells. This effect is related to the Trp53 mutation, as dsRNA isolated from wild-type MEF cells is unable to induce this immune response. Interestingly, the innate immune inducing effect of the dsRNA appears to be increased by the presence of ADAR1 in those cells, and decreased when ADAR1 is absent. The response to transfected dsRNA is mediated mainly, but not exclusively, via the MDA5 receptor; the RIG-I receptor is also involved in this immune response, however to a lesser extent. Cellular sensing of this endogenous dsRNA through MDA5 and RIG-I leads to their interaction with MAVS signaling protein, translocation of transcription factor IRF7, and finally to the type I IFN and IL-6 secretion and up-regulation of ISGs.
We are confident that the observed effect relies on endogenous dsRNA, and not on dsDNA or other nucleic acids. Immunoprecipitation with an antibody specific for dsRNA was described in multiple publications (most recently in [Dhir et al. 2018]), which together with our control experiments prove the dsRNA specificity of the antibody. We can also control the dsRNA quality prior to transfection by analysis with microcapillary electrophoresis. In addition, the response is eliminated when we treat with a dsRNA-specific RNase.
The results presented here were surprising to us initially because we had expected that dsRNA not edited by ADAR1 would be more immune-stimulatory. One of the biological roles of ADAR1 is to negatively regulate the IFN response by editing endogenous dsRNA. This was observed in Adar1 deficient mice that die by embryonic day E12.5 with severe effects of massive interferon production, liver disintegration and widespread apoptosis (Hartner et al. 2004, 2009; Wang and Carmichael 2004; Mannion et al. 2014). Additionally, Adar1p150 mutant MEFs also show abnormal type I IFN response (Ward et al. 2011) and are not viable. The Adar1 null mutant we use here bears a deletion of exon 2 to 13, removing most of the open reading frame of the protein. Other Adar1 mutant alleles still contain dsRNA-binding domains and viable Adar1 MEFs can be generated from them (Wang et al. 2004). It is only after generating Adar1;Trp53 double mutant MEFs that we were able obtain viable cells. These cells were characterized by elevated immune responses, which were reduced after transfection of inosine containing dsRNA (Vitali and Scadden 2010; Mannion et al. 2014). These results demonstrated that the cell uses inosine to help discriminate between self and non-self dsRNA. If inosine is present in dsRNA it binds to RLRs and prevents activation of the innate immune response. However, if no inosine is present then the cell cannot discriminate “self” from “non-self” and treats the dsRNA as being of viral origin and activates a type I IFN response.
We found that endogenous dsRNA can indeed induce an immune response and this effect was associated to the p53 deficiency. To examine this dsRNA in more detail, we performed dsRNA-seq. The results revealed the mitochondrial chromosome as the chromosome with the highest proportion of genes differentially expressed between Trp53 and wild-type cells, and between Adar1;Trp53 and wild-type cells. The analysis of expression levels of individual, differentially expressed genes mapped to the mitochondrial chromosome, shows higher fold increase in 12 out of 13 protein-coding genes, in Trp53 compared to wild-type than in Adar;Trp53 compared to wild-type, with the only exception being the mitochondrially encoded cytochrome C oxidase III (MT-CO3). In contrast to recently published results (Borowski et al. 2013; Dhir et al. 2018), most of the detectable cellular dsRNA is not encoded by the mitochondrial genome; however, our results still indicate that the ability of endogenous dsRNA to induce immune response relies mostly on the mitochondrial fraction and not on dsRNA in general. We also find that in whole transcriptomes of both Trp53 MEF and Adar1;Trp53 MEF cell lines the highest percentage of differentially expressed genes was identified in the mitochondrial genome. In addition, when mitochondrial transcription was decreased by growing the cells in low concentrations of ethidium bromide, the dsRNA isolated from these cells was less immune-stimulatory when transfected into Adar1;Trp53 MEFs. This demonstrates that the endogenous dsRNA that we isolated with dsRNA antibodies is of mitochondrial origin and is not encoding SINEs or other RNAs with tandem repeats as was found when the DNA-demethylating agent 5-aza-dC was used to treat Trp53MEFs (Leonova et al. 2013).
In human cells, mitochondrial RNA can induce immune response to a similar extent as bacterial RNA (Dhir et al. 2018). Mitochondrial dsRNA is particularly dangerous as it can lead to the activation of potent innate immune defense mechanisms that have evolved to protect vertebrates against microbial and viral attack. This may result in autoimmune disorders. Under the normal circumstances, this RNA is strictly controlled by the degradosome components, mitochondrial RNA helicase SUV3 and polynucleotide phosphorylase (PNPase). Loss of either of those enzymes results in massive accumulation of mitochondrial dsRNA that escapes into the cytoplasm and drives type I IFN response (Dhir et al. 2018). Another recent publication has demonstrated that Protein kinase RNA-activated (PKR) also binds mitochondrial dsRNA which can regulate its phosphorylation and signaling (Kim et al. 2018).
We found that Trp53 mutant MEFs, both with and without concurrent knockout of Adar1, have elevated levels of multiple genes involved in the immune response. This was observed in cells not stimulated with dsRNA. Since Trp53 cells have fivefold higher IFN-α secretion than wild-type and Adar1;Trp53 cells, it was then crucial to identify the main transcripts differing between the Trp53 mutant and the other two lines. Among the most up-regulated genes in both Trp53 and Adar1;Trp53 cells, when compared to wild-type, were members of the Oas family, Oasl1 and Oas3. Oas family enzymes catalyzes the synthesis of oligoadenylates of the general structure ppp(A2′p)nA (2′–5′), which upon binding, activate the endoribonuclease RNase L. When activated, RNase L catalyzes the degradation of both viral and cellular RNAs (Hovanessian and Justesen 2007). Oas1-3 upon binding to dsRNA, activate RNase L, with Oas3 having the dominant role in this process (Ibsen et al. 2014; Li et al. 2016). The role of mouse Oasl1 however is still puzzling. Mouse Oasl1 gene is the orthologue of the human OASL gene and is enzymatically inactive (Kristiansen et al. 2011). Until recently, the only reported role of Oasl1 was the negative regulation of IFN response (Kristiansen et al. 2011; Lee et al. 2013b; Oh et al. 2016). However, a recent report demonstrates that Oasl1 plays context-dependent roles in the antiviral response (Kang et al. 2018). In early stages of viral infection, Oasl1 forms stress granules trapping viral RNAs and promoting efficient RLR signaling. Stress granule formation is dependent on the RNA-binding activity of Oasl1. However, in the late stages of infection, Oasl1 inhibits translation resulting in down regulation of IFN production (Kang et al. 2018). These results demonstrate that Oasl1 has a more complicated role in response to viral dsRNA, than previously appreciated.
The level of RNaseL transcript in unstimulated cells is higher in Trp53 cells than in wild-type or Adar1;Trp53. Therefore, we studied the role of the OAS/RNase L system in the production of immunogenic dsRNA in this cell line. Nevertheless, RNase L exists in the cell in inactive form and is activated by dimerization, which occurs upon 2-5A binding. Knocking down RNase L in Trp53 MEFs results in the loss of the immune-stimulating ability of the endogenous dsRNA. This can be seen on multiple levels of the type I IFN response in transfected reporter cells, starting with the lowered expression of MDA5 and RIG-I receptors, through smaller production of IFN-α and IL-6, and finally, much lower expression of ISGs. Active RNase L cleaves all cellular RNAs predominantly in single-stranded regions at UpN dinucleotides (UA and UU > UG), however the cleaved RNA may have single- and double-stranded regions (Wreschner et al. 1981). The J2 antibody used for immunoprecipitation recognizes continuous duplex structures of at least 40 bp in length (Bonin et al. 2000).
The OAS system is localized in multiple cellular compartments. Whereas RNase L can be either cytoplasmic or mitochondrial (Le Roy et al. 2007; Kjær et al. 2014), OAS can be localized in mitochondria, cytoplasm, but additionally also in the ER and nucleus (Lin et al. 2009). The cellular localization of the OAS/RNase L system may also depend on the stage of cell life. Activation of RNase L results in the degradation of all RNA within the cell, viral and endogenous, which leads to apoptosis of mammalian cells in a caspase-dependent manner. At the beginning of apoptosis, RNase L and OAS are localized in the mitochondria and cytosol fractions, while at the onset of apoptosis both enzymes are largely in mitochondria (Domingo-Gil and Esteban 2006). Based on the obtained results it is not possible to confirm with full certainty whether isolated mitochondrial dsRNA was cleaved by mitochondrial or cytoplasmic RNase L. Both possibilities seem plausible. Nevertheless, we are confident that the immunoprecipitated RNA, and therefore RNA that can induce immune response, is mitochondrial and that it was processed by the OAS/RNase L system.
Overall, our results demonstrate a role of p53—OAS axis in mitochondrial RNA processing and preventing self-nucleic acid such as dsRNA from aberrantly activating innate immune responses. The lack of p53 transcription factor causes activation of the OAS/RNase L system in the absence of a danger signal such as viral infection.
MATERIALS AND METHODS
Cell culture
Mouse embryonic fibroblasts (MEF) were derived from mouse embryos of the same genetic background (C57BL/6J) as previously described (Mannion et al. 2014). Cells were grown in DMEM high glucose medium (BioSera) in the presence of 10% fetal bovine serum (FBS). To silence RNase L, MEF Trp53 cells were transfected with 30 nM of 21 bp esiRNA, targeting mouse RNase L (MISSION, Sigma), with Lipofectamine 3000 for 48 h. The silencing effectiveness was measured by qPCR, with primers specific for RNase L (see Supplemental Table S1). The silencing specificity of siRNA was assessed by parallel transfection with 30 nM of siRNA Universal Negative Control (MISSION, Sigma). To silence RIG-I and MDA5 receptors, Adar1;Trp53 MEFs were transfected with 30 nM of esiRNA targeting mouse Ifih1 or 30 nM of esiRNA targeting mouse Ddx58 (MISSION, Sigma), with Lipofectamine 3000 for 72 h.
RNA extraction
Total RNA was obtained from cells by phenol:chloroform extraction with an overnight isopropanol precipitation. RNA samples were treated with 1 µL DNase I (Qiagen) per 20 µg of total RNA and then precipitated in 70% isopropanol with 150 mM sodium acetate. A 2 µg aliquot of total RNA was reverse transcribed into cDNA with HyperScript Reverse Transcriptase with 100 µM Oligo(dT)15 primer (GeneAll). Samples were incubated for 1 h at 55°C and the reaction terminated by heating for 5 min at 70°C.
Quantitative RT-PCR
For all qPCR experiments, the QuantStudio 12K Flex Real-Time PCR System (Thermo Scientific) was used. PCR was performed with FastStart Universal SYBR Green Master (Rox) (Sigma) with standard two-step cycling protocol: one cycle at 95°C for 60 sec, then 40 cycles of 95°C for 15 sec and 60°C for 45 sec. PCR amplification was performed with gene-specific primers (see Supplemental Table S1). Results were normalized to the mRNA expression of β-actin or Gapdh. To analyze relative quantification of genes, the comparative CT Method (ΔΔCT) was used. Samples were analyzed in technical duplicates and biological triplicates.
Immunoprecipitation of dsRNA
Forty micrograms of total RNA was isolated as described above and incubated with the J2 anti-dsRNA IgG2a monoclonal antibody (Scicons) in the presence of IP buffer (50 mM Tris-HCl pH 8, 150 mM NaCl, 1% Triton X-100, and 1 mM EDTA) at 4°C for 16 h. Next, 70 µL of Protein A-Sepharose 4B Fast Flow beads (Sigma) was added to the RNA-J2 mix and incubated for another 4 h at 4°C. The beads coupled with J2-RNA complexes were then washed gently three times at 4°C with IP buffer. DsRNA was isolated from the beads with the standard phenol–chloroform RNA extraction protocol. The concentration and quality of the isolated RNA was measured by TapeStation (Agilent 2200) according to the manufacturer's instructions.
Transfection with dsRNA
In the experiments, three cell lines, WT MEF, Trp53 MEF, and Adar1;Trp53 MEF, were transfected with dsRNA (also derived from WT MEF, Trp53 MEF, and Adar1;Trp53 MEF). Always, the same amount of immunoprecipitated RNA was transfected into cells with Lipofectamine 3000. This was 2 µg transfected into ∼1.6 × 105 of MEFs in 2.3 mL of media.
ELISA
Cell culture media was stored at −80°C and thawed at room temperature prior to ELISA. The amount of IL-6 secreted by MEF cells (confluent 4 cm2 in 1 mL of medium) was measured with Mouse IL-6 ELISA Set (BD OptEIA) according to the manufacturer instructions. Samples were analyzed in technical duplicates and biological triplicates.
Immunoblotting
Cells (1–1.2 × 106) were lysed in 50 mM Tris-HCl pH 8, 150 mM NaCl, 1% Triton X-100, and 1 mM EDTA with the addition of protease inhibitors (cOmplete, Roche). Protein samples were separated at 100 V by 10% SDS-PAGE electrophoresis and blotted on a nitrocellulose membrane. Membranes were incubated with specific antibodies: mouse anti-α-tubulin (1:8000, Sigma-Aldrich), rabbit anti-MDA5 (1:800, Cell Signaling), rabbit anti-RIGI (1:800, Cell Signaling), rabbit anti-fibrillarin (1:10,000, Abcam). HRP-conjugated anti-rabbit (1:80,000) and anti-mouse (1:5000) antibodies were purchased from Sigma-Aldrich. Proteins were revealed with Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific). All immunoblots blots were performed in triplicate and the quantification of the protein bands from all three experiments is shown in each figure.
Subcellular fractionation
Fractionation protocol was adapted from Holden and Horton (2009) and Liu and Fagotto (2011). Trp53 MEFs cells were grown on 15 cm dishes until they reached ∼85% confluency. Cells on the dish were washed with cold 1× PBS and permeabilized with 6 mL of digitonin solution (1× NEH Buffer containing 45 µg/mL digitonin, 10 mM DTT, and 10 mM MgCl2) by gentle rocking at 4°C for 10 min. (10× NEH Buffer: 1500 mM NaCl, 2 mM EDTA, 200 mM HEPES-NaOH pH 7.4). The released cytoplasmic fraction was collected and cleared by centrifugation at 500g, 4°C for 10 min. Cytoplasmic RNA was isolated from clear supernatant with TriPure Reagent (Sigma-Aldrich) as per manufacturer's instructions. Cell residues on the dish were washed with cold 1× PBS and collected with 3 mL of Buffer 2 (150 mM NaCl, 50 mM HEPES-NaOH pH 7.4, 1% NP40) by scraping, followed by 30 min incubation on ice. Nuclei were pelleted by centrifugation at 7000g, 4°C for 15 min. The supernatant containing other organelles was discarded and the pelleted nuclei were washed with cold 1× PBS. The nuclear pellet from one 15 cm dish was disrupted by incubating for 30 min at 37°C with 10 U of TURBO DNase (Thermo Fisher) in 10× Reaction Buffer with occasional pipetting using 100 µL tips. Afterward, nuclear RNA was isolated with TriPure Reagent as per manufacturer's instructions. A small part of both cytoplasmic and nuclear fraction was saved for immunoblotting to verify the fractions’ purity.
Ethidium bromide treatment of MEFs to block mitochondrial transcription
Trp53 MEFs were seeded in 10 cm plates and grown until they reached ∼80% confluency. Then, ethidium bromide (AppliChem) was added to the cell culture medium at a final concentration of 0.05 µg/mL (Hayakawa et al. 1998; Surovtseva et al. 2011). Cells were grown in ethidium bromide-supplemented medium for 24 h. Cells were washed 3× with PBS, then total RNA was isolated and treated with DNase as described above. Expression of mitochondrial genes was analyzed by quantitative RT-PCR as described above with RNA from untreated Trp53 MEFs as a control. Random hexamers were used for reverse transcription instead of oligo(dT)15. Cycling conditions for qPCR: one cycle of 95°C for 10 min, then 40 cycles of 95°C for 10 sec, 55°C for 20 sec, and 72°C for 8 sec. Total RNA from ethidium bromide–treated cells was also used for dsRNA immunoprecipitation using the same protocol as described above.
NGS library preparation
Total RNA was isolated from cells using the phenol–chloroform extraction method and treated with DNase I (Ambion). RNA was depleted of rRNA with the RiboCop rRNA Depletion Kit (Lexogen) and used for library preparation using the SENSE Total RNA-seq Library Prep Kit (Lexogen). For the sequencing of the dsRNA, immunoprecipitation was performed with J2 (dsRNA-specific antibody), then 1 µg per sample was depleted of rRNA with RiboCop rRNA Depletion Kit (Lexogen). The rRNA depleted dsRNA was used for library preparation with SENSE Total RNA-seq Library Prep Kit (Lexogen). The libraries were prepared with TruSeq Ilumina adapters. Sequencing was performed on the NextSeq 500/550 sequencer.
Differential gene expression
The raw data generated during sequencing were quality checked using FastQC and preprocessed with Trimmomatic. Adapter sequences and low-quality ends were trimmed (Phred score <3; both 5′ and 3′ ends). Alignment was performed by STAR to the mouse reference genome–GRCm38 primary assembly; Ensembl release 86 (NGS of total RNA) and release 91 (NGS of dsRNA). Raw gene counts were counted only from uniquely mapped reads by featureCounts (NGS of total RNA). Estimated raw gene counts were counted from both uniquely and multimapped reads by RSEM (NGS of total RNA). Strandedness of the sequencing was considered during the counting. Differential gene expression analysis was performed by DESeq2 Bioconductor package and the raw P-values were adjusted for multiple testing error using Benjamini–Hochberg method. Samples were analyzed in biological triplicates.
Analysis of dsRNA sequences immunoprecipitated with J2 antibody
Raw alignment coverage was calculated using DeepTools and normalized to counts-per-million (CPM). Signals of each of the strands were calculated separately. All positions that did not have coverage of at least 0.5 CPM were filtered out. Only signals with a continuous length of at least 150 bp (S9 A) or 500 bp (S9 B) were kept. The regions were selected only if they were present in two out of three replicates for each condition (WT, Trp53 and Adar1;Trp53). Visualization of the strands was done in ggplot2 R package.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
Core Facility Bioinformatics of CEITEC Masaryk University is gratefully acknowledged for obtaining the scientific data presented in this paper. We acknowledge the CF Genomics CEITEC MU supported by the NCMG research infrastructure (LM2015091 funded by MEYS CR) for their support with obtaining scientific data presented in this paper. Computational resources were provided by the CESNET LM2015042 and the CERIT Scientific Cloud LM2015085, provided under the program “Projects of Large Research, Development, and Innovations Infrastructures.” We would like to thank the reviewers for their insightful comments regarding this manuscript. This work was supported by the European Union's Seventh Framework Programme for research, technological development and demonstration under grant agreement no. 621368 to M.A.O.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.069625.118.
REFERENCES
- Bayona-Bafaluy MP, Acin-Perez R, Mullikin JC, Park JS, Moreno-Loshuertos R, Hu P, Perez-Martos A, Fernandez-Silva P, Bai Y, Enriquez JA. 2003. Revisiting the mouse mitochondrial DNA sequence. Nucleic Acids Res 31: 5349–5355. 10.1093/nar/gkg739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibb MJ, Van Etten RA, Wright CT, Walberg MW, Clayton DA. 1981. Sequence and gene organization of mouse mitochondrial DNA. Cell 26: 167–180. 10.1016/0092-8674(81)90300-7 [DOI] [PubMed] [Google Scholar]
- Bonin M, Oberstrass J, Lukacs N, Ewert K, Oesterschulze E, Kassing R, Nellen W. 2000. Determination of preferential binding sites for anti-dsRNA antibodies on double-stranded RNA by scanning force microscopy. RNA 6: 563–570. 10.1017/S1355838200992318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borowski LS, Dziembowski A, Hejnowicz MS, Stepien PP, Szczesny RJ. 2013. Human mitochondrial RNA decay mediated by PNPase-hSuv3 complex takes place in distinct foci. Nucleic Acids Res 41: 1223–1240. 10.1093/nar/gks1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan R, Handa R, Das TP, Pati U. 2004. Over-expression of TATA binding protein (TBP) and p53 and autoantibodies to these antigens are features of systemic sclerosis, systemic lupus erythematosus and overlap syndromes. Clin Exp Immunol 136: 574–584. 10.1111/j.1365-2249.2004.02463.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LL, Decerbo JN, Carmichael GG. 2008. Alu element-mediated gene silencing. EMBO J 27: 1694–1705. 10.1038/emboj.2008.94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhir A, Dhir S, Borowski LS, Jimenez L, Teitell M, Rötig A, Crow YJ, Rice GI, Duffy D, Tamby C, et al. 2018. Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature 560: 238–242. 10.1038/s41586-018-0363-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingo-Gil E, Esteban M. 2006. Role of mitochondria in apoptosis induced by the 2-5A system and mechanisms involved. Apoptosis 11: 725–738. 10.1007/s10495-006-5541-0 [DOI] [PubMed] [Google Scholar]
- Green DR, Kroemer G. 2009. Cytoplasmic functions of the tumour suppressor p53. Nature 458: 1127–1130. 10.1038/nature07986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudkov AV, Gurova KV, Komarova EA. 2011. Inflammation and p53: a tale of two stresses. Genes Cancer 2: 503–516. 10.1177/1947601911409747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartner JC, Schmittwolf C, Kispert A, Müller AM, Higuchi M, Seeburg PH. 2004. Liver disintegration in the mouse embryo caused by deficiency in the RNA-editing enzyme ADAR1. J Biol Chem 279: 4894–4902. 10.1074/jbc.M311347200 [DOI] [PubMed] [Google Scholar]
- Hartner JC, Walkley CR, Lu J, Orkin SH. 2009. ADAR1 is essential for the maintenance of hematopoiesis and suppression of interferon signaling. Nat Immunol 10: 109–115. 10.1038/ni.1680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa T, Noda M, Yasuda K, Yorifuji H, Taniguchi S, Miwa I, Sakura H, Terauchi Y, Hayashi J, Sharp GW, et al. 1998. Ethidium bromide-induced inhibition of mitochondrial gene transcription suppresses glucose-stimulated insulin release in the mouse pancreatic β-cell line βHC9. J Biol Chem 273: 20300–20307. 10.1074/jbc.273.32.20300 [DOI] [PubMed] [Google Scholar]
- Hecht I, Toporik A, Podojil JR, Vaknin I, Cojocaru G, Oren A, Aizman E, Liang SC, Leung L, Dicken Y, et al. 2018. ILDR2 is a novel B7-like protein that negatively regulates T cell responses. J Immunol 200: 2025–2037. 10.4049/jimmunol.1700325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holden P, Horton WA. 2009. Crude subcellular fractionation of cultured mammalian cell lines. BMC Res Notes 2: 243 10.1186/1756-0500-2-243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovanessian AG, Justesen J. 2007. The human 2′–5′oligoadenylate synthetase family: unique interferon-inducible enzymes catalyzing 2′–5′ instead of 3′–5′ phosphodiester bond formation. Biochimie 89: 779–788. 10.1016/j.biochi.2007.02.003 [DOI] [PubMed] [Google Scholar]
- Ibsen MS, Gad HH, Thavachelvam K, Boesen T, Desprès P, Hartmann R. 2014. The 2′–5′ oligoadenylate synthetase 3 (OAS3) enzyme potently synthesizes the 2′–5′ oligoadenylates required for RNase L activation. J Virol 88: 14222–14231. 10.1128/JVI.01763-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakuta S, Shibata S, Iwakura Y. 2002. Genomic structure of the mouse 2′,5′-oligoadenylate synthetase gene family. J Interferon Cytokine Res 22: 981–993. 10.1089/10799900260286696 [DOI] [PubMed] [Google Scholar]
- Kang DC, Gopalkrishnan RV, Wu Q, Jankowsky E, Pyle AM, Fisher PB. 2002. mda-5: an interferon-inducible putative RNA helicase with double-stranded RNA-dependent ATPase activity and melanoma growth-suppressive properties. Proc Natl Acad Sci 99: 637–642. 10.1073/pnas.022637199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JS, Hwang YS, Kim LK, Lee S, Lee WB, Kim-Ha J, Kim YJ. 2018. OASL1 traps viral RNAs in stress granules to promote antiviral responses. Mol Cells 41: 214–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Park J, Kim S, Kim M, Kang MG, Kwak C, Kang M, Kim B, Rhee HW, Kim VN. 2018. PKR senses nuclear and mitochondrial signals by interacting with endogenous double-stranded RNAs. Mol Cell 71: 1051–1063 e1056. 10.1016/j.molcel.2018.07.029 [DOI] [PubMed] [Google Scholar]
- Kjær KH, Pahus J, Hansen MF, Poulsen JB, Christensen EI, Justesen J, Martensen PM. 2014. Mitochondrial localization of the OAS1 p46 isoform associated with a common single nucleotide polymorphism. BMC Cell Biol 15: 33 10.1186/1471-2121-15-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komarova EA, Krivokrysenko V, Wang K, Neznanov N, Chernov MV, Komarov PG, Brennan ML, Golovkina TV, Rokhlin OW, Kuprash DV, et al. 2005. p53 is a suppressor of inflammatory response in mice. FASEB J 19: 1030–1032. 10.1096/fj.04-3213fje [DOI] [PubMed] [Google Scholar]
- Kovacs B, Patel A, Hershey JN, Dennis GJ, Kirschfink M, Tsokos GC. 1997. Antibodies against p53 in sera from patients with systemic lupus erythematosus and other rheumatic diseases. Arthritis Rheum 40: 980–982. 10.1002/art.1780400531 [DOI] [PubMed] [Google Scholar]
- Kristiansen H, Gad HH, Eskildsen-Larsen S, Despres P, Hartmann R. 2011. The oligoadenylate synthetase family: an ancient protein family with multiple antiviral activities. J Interferon Cytokine Res 31: 41–47. 10.1089/jir.2010.0107 [DOI] [PubMed] [Google Scholar]
- Le Roy F, Silhol M, Salehzada T, Bisbal C. 2007. Regulation of mitochondrial mRNA stability by RNase L is translation-dependent and controls IFNα-induced apoptosis. Cell Death Differ 14: 1406–1413. 10.1038/sj.cdd.4402130 [DOI] [PubMed] [Google Scholar]
- Lee MS, Kim B, Oh GT, Kim YJ. 2013a. OASL1 inhibits translation of the type I interferon-regulating transcription factor IRF7. Nat Immunol 14: 346–355. 10.1038/ni.2535 [DOI] [PubMed] [Google Scholar]
- Lee MS, Park CH, Jeong YH, Kim Y-J, Ha S-J. 2013b. Negative regulation of type I IFN expression by OASL1 permits chronic viral infection and CD8+ T-cell exhaustion. PLoS Pathog 9: e1003478 10.1371/journal.ppat.1003478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonova KI, Brodsky L, Lipchick B, Pal M, Novototskaya L, Chenchik AA, Sen GC, Komarova EA, Gudkov AV. 2013. p53 cooperates with DNA methylation and a suicidal interferon response to maintain epigenetic silencing of repeats and noncoding RNAs. Proc Natl Acad Sci 110: E89–98. 10.1073/pnas.1216922110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Banerjee S, Wang Y, Goldstein SA, Dong B, Gaughan C, Silverman RH, Weiss SR. 2016. Activation of RNase L is dependent on OAS3 expression during infection with diverse human viruses. Proc Natl Acad Sci 113: 2241–2246. 10.1073/pnas.1519657113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Banerjee S, Goldstein SA, Dong B, Gaughan C, Rath S, Donovan J, Korennykh A, Silverman RH, Weiss SR. 2017. Ribonuclease L mediates the cell-lethal phenotype of double-stranded RNA editing enzyme ADAR1 deficiency in a human cell line. eLife 6: e25687 10.7554/eLife.25687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin RJ, Yu HP, Chang BL, Tang WC, Liao CL, Lin YL. 2009. Distinct antiviral roles for human 2′,5′-oligoadenylate synthetase family members against dengue virus infection. J Immunol 183: 8035–8043. 10.4049/jimmunol.0902728 [DOI] [PubMed] [Google Scholar]
- Liu X, Fagotto F. 2011. A method to separate nuclear, cytosolic, and membrane-associated signaling molecules in cultured cells. Sci Signal 4: pl2 10.1126/scisignal.2002165 [DOI] [PubMed] [Google Scholar]
- Lowman HB, Draper DE. 1986. On the recognition of helical RNA by cobra venom V1 nuclease. J Biol Chem 261: 5396–5403. [PubMed] [Google Scholar]
- Mannion NM, Greenwood SM, Young R, Cox S, Brindle J, Read D, Nellåker C, Vesely C, Ponting CP, McLaughlin PJ, et al. 2014. The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep 9: 1482–1494. 10.1016/j.celrep.2014.10.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimura Y, Yazawa N, Tamaki Z, Ashida R, Jinnin M, Asano Y, Tada Y, Kubo M, Ihn H, Tamaki K. 2007. Anti-p53 antibodies in patients with dermatomyositis/polymyositis. Clin Rheumatol 26: 1328–1331. 10.1007/s10067-006-0473-1 [DOI] [PubMed] [Google Scholar]
- Mori T, Anazawa Y, Iiizumi M, Fukuda S, Nakamura Y, Arakawa H. 2002. Identification of the interferon regulatory factor 5 gene (IRF-5) as a direct target for p53. Oncogene 21: 2914–2918. 10.1038/sj.onc.1205459 [DOI] [PubMed] [Google Scholar]
- Muñoz-Fontela C, Mandinova A, Aaronson SA, Lee SW. 2016. Emerging roles of p53 and other tumour-suppressor genes in immune regulation. Nat Rev Immunol 16: 741–750. 10.1038/nri.2016.99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen TW. 2013. RNA structure determination using nuclease digestion. Cold Spring Harb Protoc 2013: 379–382. 10.1101/pdb.prot072330 [DOI] [PubMed] [Google Scholar]
- Oh JE, Lee MS, Kim YJ, Lee HK. 2016. OASL1 deficiency promotes antiviral protection against genital herpes simplex virus type 2 infection by enhancing type I interferon production. Sci Rep 6: 19089 10.1038/srep19089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peralta S, Wang X, Moraes CT. 2012. Mitochondrial transcription: lessons from mouse models. Biochim Biophys Acta 1819: 961–969. 10.1016/j.bbagrm.2011.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runge S, Sparrer KM, Lässig C, Hembach K, Baum A, Garcia-Sastre A, Söding J, Conzelmann KK, Hopfner KP. 2014. In vivo ligands of MDA5 and RIG-I in measles virus-infected cells. PLoS Pathog 10: e1004081 10.1371/journal.ppat.1004081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, Nakaya T, Katsuki M, Noguchi S, Tanaka N, et al. 2000. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-α/β gene induction. Immunity 13: 539–548. 10.1016/S1074-7613(00)00053-4 [DOI] [PubMed] [Google Scholar]
- Silverman RH, Weiss SR. 2014. Viral phosphodiesterases that antagonize double-stranded RNA signaling to RNase L by degrading 2-5A. J Interferon Cytokine Res 34: 455–463. 10.1089/jir.2014.0007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto Y, Vigilante A, Darbo E, Zirra A, Militti C, D'Ambrogio A, Luscombe NM, Ule J. 2015. hiCLIP reveals the in vivo atlas of mRNA secondary structures recognized by Staufen 1. Nature 519: 491–494. 10.1038/nature14280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surovtseva YV, Shutt TE, Cotney J, Cimen H, Chen SY, Koc EC, Shadel GS. 2011. Mitochondrial ribosomal protein L12 selectively associates with human mitochondrial RNA polymerase to activate transcription. Proc Natl Acad Sci 108: 17921–17926. 10.1073/pnas.1108852108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaoka A, Hayakawa S, Yanai H, Stoiber D, Negishi H, Kikuchi H, Sasaki S, Imai K, Shibue T, Honda K, et al. 2003. Integration of interferon-α/β signalling to p53 responses in tumour suppression and antiviral defence. Nature 424: 516–523. 10.1038/nature01850 [DOI] [PubMed] [Google Scholar]
- Takatori H, Kawashima H, Suzuki K, Nakajima H. 2014. Role of p53 in systemic autoimmune diseases. Crit Rev Immunol 34: 509–516. 10.1615/CritRevImmunol.2014012193 [DOI] [PubMed] [Google Scholar]
- Taura M, Eguma A, Suico MA, Shuto T, Koga T, Komatsu K, Komune T, Sato T, Saya H, Li JD, et al. 2008. p53 regulates Toll-like receptor 3 expression and function in human epithelial cell lines. Mol Cell Biol 28: 6557–6567. 10.1128/MCB.01202-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turpin E, Luke K, Jones J, Tumpey T, Konan K, Schultz-Cherry S. 2005. Influenza virus infection increases p53 activity: role of p53 in cell death and viral replication. J Virol 79: 8802–8811. 10.1128/JVI.79.14.8802-8811.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitali P, Scadden AD. 2010. Double-stranded RNAs containing multiple IU pairs are sufficient to suppress interferon induction and apoptosis. Nat Struct Mol Biol 17: 1043–1050. 10.1038/nsmb.1864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Carmichael GG. 2004. Effects of length and location on the cellular response to double-stranded RNA. Microbiol Mol Biol Rev 68: 432–452. 10.1128/MMBR.68.3.432-452.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Miyakoda M, Yang W, Khillan J, Stachura DL, Weiss MJ, Nishikura K. 2004. Stress-induced apoptosis associated with null mutation of ADAR1 RNA editing deaminase gene. J Biol Chem 279: 4952–4961. 10.1074/jbc.M310162200 [DOI] [PubMed] [Google Scholar]
- Ward SV, George CX, Welch MJ, Liou LY, Hahm B, Lewicki H, de la Torre JC, Samuel CE, Oldstone MB. 2011. RNA editing enzyme adenosine deaminase is a restriction factor for controlling measles virus replication that also is required for embryogenesis. Proc Natl Acad Sci 108: 331–336. 10.1073/pnas.1017241108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber F, Wagner V, Rasmussen SB, Hartmann R, Paludan SR. 2006. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J Virol 80: 5059–5064. 10.1128/JVI.80.10.5059-5064.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wreschner DH, McCauley JW, Skehel JJ, Kerr IM. 1981. Interferon action–sequence specificity of the ppp(A2′p)nA-dependent ribonuclease. Nature 289: 414–417. 10.1038/289414a0 [DOI] [PubMed] [Google Scholar]
- Yan W, Wei J, Deng X, Shi Z, Zhu Z, Shao D, Li B, Wang S, Tong G, Ma Z. 2015. Transcriptional analysis of immune-related gene expression in p53-deficient mice with increased susceptibility to influenza A virus infection. BMC Med Genomics 8: 52 10.1186/s12920-015-0127-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. 2004. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol 5: 730–737. 10.1038/ni1087 [DOI] [PubMed] [Google Scholar]
- Youlyouz-Marfak I, Gachard N, Le Clorennec C, Najjar I, Baran-Marszak F, Reminieras L, May E, Bornkamm GW, Fagard R, Feuillard J. 2008. Identification of a novel p53-dependent activation pathway of STAT1 by antitumour genotoxic agents. Cell Death Differ 15: 376–385. 10.1038/sj.cdd.4402270 [DOI] [PubMed] [Google Scholar]
- Zhu J, Zhang Y, Ghosh A, Cuevas RA, Forero A, Dhar J, Ibsen MS, Schmid-Burgk JL, Schmidt T, Ganapathiraju MK, et al. 2014. Antiviral activity of human oligoadenylate synthetases-like (OASL) is mediated by enhancing retinoic acid-inducible gene I (RIG-I) signaling. Immunity 40: 936–948. 10.1016/j.immuni.2014.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.