

Summary
High-grade gliomas (HGG) defined by histone 3 K27M driver mutations exhibit global loss of H3K27 trimethylation and reciprocal gain of H3K27 acetylation, respectively shaping repressive and active chromatin landscapes. We generated tumor-derived isogenic models bearing this mutation and show that it leads to pervasive H3K27ac deposition across the genome. In turn, active enhancers and promoters are not created de novo and instead reflect the epigenomic landscape of the cell of origin. H3K27ac is enriched at repeat elements, resulting in their increased expression, which in turn can be further amplified by DNA demethylation and histone deacetylase inhibitors providing an exquisite therapeutic vulnerability. These agents may therefore modulate anti-tumor immune responses as a therapeutic modality for this untreatable disease.
Keywords: Pediatric high-grade glioma, H3K27M, enhancer, epigenetic therapy, viral mimicry
Graphical Abstract
eTOC Blurb
Krug et al. reveal increased global H3K27ac deposition across the genome without creation of de novo active enhancers or promoters in high grade glioma (HGG) with H3K27M mutations. H3K27ac enrichment at repeat elements in H3K27M HGG increases their expression, conferring sensitivity to epigenetic therapies.
Introduction
High-grade gliomas (HGGs) are a leading cause of cancer-related death in children and young adults. These devastating primary brain tumors have less than 10% survival 2-years following diagnosis, with no targeted therapies currently available. Pediatric HGGs are characterized by epigenetic alterations directly or indirectly affecting the post-translational modification (PTM) of two major opposing chromatin marks, repressive H3K27me3 and active H3K36me3 (Fontebasso et al., 2014; Khuong-Quang et al., 2012; Schwartzentruber et al., 2012; Wu et al., 2012). The most frequent epigenetic modification in pediatric HGGs is a somatic heterozygous mutation in histone 3 (H3) variants leading to lysine-to-methionine substitutions at position 27 (H3K27M). This mutation characterizes more than 80% of midline gliomas, the most common HGGs in children, which include universally lethal diffuse intrinsic pontine gliomas (DIPG) (Khuong-Quang et al., 2012; Sturm et al., 2014; Wu et al., 2012). H3K27M leads to a global decrease in H3K27me3 levels, a PTM marking transcriptionally silent regions of the genome, shown in vitro to be due to a disruption of the catalytic activity of the polycomb repressive complex 2 (PRC2) (Bender et al., 2013; Lewis et al., 2013). H3K27M also leads to increased global H3K27 acetylation (H3K27ac) (Lewis et al., 2013), a PTM associated with active transcription (Creyghton et al., 2010). The role of residual H3K27me3 deposition in promoting oncogenesis in H3K27M is currently debated (Chan et al., 2013; Mohammad et al., 2017; Piunti et al., 2017). Increased H3K27ac was recently suggested to associate with aberrant deposition of heterotopic nucleosomes containing H3.3K27M-H3.3K27ac (Piunti et al., 2017). These aberrant nucleosomes are seemingly bound by bromodomain-containing proteins and suggested to act by excluding PRC2 from cellular differentiation genes regulated by Clusters of Regulatory Elements (COREs), stretch enhancers, or super enhancers (SEs) (Loven et al., 2013; Piunti et al., 2017; Whyte et al., 2013). This model, however, does not explain why K27M mutations in canonical H3.1 or H3.2, which have broader and distinct deposition patterns from noncanonical H3.3-containing nucleosomes, show exclusion of the PRC2 complex similar to H3.3. Our goal is thus to gain insight into the active cis-regulatory programs in H3.3K27M HGGs, delineate the effects of increased H3K27ac on active chromatin loci, their implications for gene expression, and uncover potential therapeutic vulnerabilities.
Results
Active chromatin landscape of pediatric high-grade glioma
We performed a comprehensive epigenomic characterization of a large panel of pediatric HGGs wild-type (WT, denoted H3K27WT) or carrying the H3.3K27M mutation. These included primary tumors, patient-derived xenografts (PDX), and cell lines, which were analyzed using quantitative histone mass spectrometry (n = 6), chromatin immunoprecipitation and sequencing (ChIP-seq) of H3K27ac (n = 38), ATAC-seq (n = 4) and RNA-seq (n = 41) (Figure S1A, Tables S1, S2). To quantify global alterations in histone modifications associated with H3.3K27M mutation we performed histone mass spectrometry of H3.3K27WT and H3.3K27M samples. We observed that H3.3K27M HGGs displayed a global loss of H3K27me3 and a global increase in H3K27ac, both on H3.3 and H3.1/H3.2 nucleosomes (Figure 1A). We asked whether this global increase in H3K27ac was associated with a distinct landscape of cis-regulatory elements, characterized by enhancers (peaks +/− 2.5 kb outside of transcription start sites, TSS) and promoters (at 2.5kb within TSS) across groups of pediatric HGG samples. Using unsupervised hierarchical clustering of the top 10,000 variant H3K27ac loci (Akhtar-Zaidi et al., 2012) identified by H3K27ac ChIP-seq, we found that patterns of H3K27ac separated H3.3K27M from H3K27WT models (including patient-derived primary cell lines and mouse xenografts) (Figure 1B, Tables S1, S2). Primary tumors that harbored the H3.3K27M mutation had distinct deposition of H3K27ac as compared to IDH1 mutated and H3K27WT samples (Figure 1C). Despite the global increase in H3K27ac, H3.3K27M and H3K27WT tumors had comparable numbers of active enhancers and promoters (Figure 1D; K27M: 43 333 and K27WT: 39 946 H3K27ac loci detected in at least 3 samples), indicating that increased H3K27ac in H3.3K27M results in a relatively modest difference in the total number of regulatory H3K27ac loci. We then defined the regions of gene transcription associated with differential H3K27ac deposition, by integrating RNA-seq with H3K27ac ChIP-seq patterns (Figure 1E, Table S3). H3K27ac associated gene expression changes specific to H3.3K27M HGGs included increased expression of LIN28B and PLAG1, two genes shown to be upregulated in H3K27M-mutant DIPGs (Funato et al., 2014) (Figure S1B), along with over-expression of additional genes including ADARB2. Conversely, H3K27WT HGGs were defined by distinct lineage markers expressed in different brain regions during development such as EN1, HOXD9, and HOXA10. Our findings reveal that H3.3K27M tumors harbor distinct H3K27ac deposition patterns when compared to H3K27WT HGGs and, unexpectedly, show minimal differences in absolute numbers of active enhancers and promoters despite the global increase in H3K27ac levels.
Figure 1. Active regulatory chromatin landscape of pediatric HGG.

(A) Quantification of H3K27me3 (top panel) levels and H3K27ac (bottom panel), abundance by mass spectrometry in H3.3-K27M HGG lines compared to WT lines (n=9; 3 technical replicates of 3 cell lines). Solid horizontal line indicates mean. Two-sided Wilcoxon-Rank Sum Test, ***p<0.001
(B) Unsupervised hierarchical clustering of pair-wise Spearman correlations performed between cell K27M and WT culture models based on read density within H3K27ac loci.
(C) t-SNE analysis on HGG cell lines, xenografts (denoted with the suffix -xn), and primary tissues using distinct H3K27ac loci between K27M and WT identified in cell lines.
(D) Number of H3K27ac positive enhancers and promoters detected in at least 3 samples out of either n=13 K27M or n=14 WT HGG samples shown in Figure 1C. Enhancers were defined by MACS1.4 peaks (p<1e-9) called greater than 2.5 kb upstream or downstream of the nearest TSS, and promoters defined by peaks within 2.5 kb.
(E) Identification of subgroup-specific genes with concordant changes in both expression and H3K27ac in patient-derived cell lines. X-axis: log2 fold-change (LFC) of gene expression between H3K27M (n=7 replicates) and H3K27WT (n=6 replicates) cell lines. Y-axis: LFC of H3K27ac at a promoter or enhancer associated with the gene. Genes with significantly differentially acetylated regulatory regions between H3K27M and H3K27WT lines are shown (p<0.05). Red: significantly upregulated in H3K27M. Blue: significantly upregulated in H3K27WT. Grey: no significant changes in expression. Filled circle: genes with the greatest magnitude of changes in both H3K27ac and expression. see also Figure s1 and Tables s1, s2, and s3.
Distinct core transcriptional regulatory circuitry programs define H3K27M and H3K27WT driven HGG
We next identified SEs within each group (Figures 2A–D, Table S4) using the Ranking of Super Enhancer (ROSE) algorithm (Loven et al., 2013; Whyte et al., 2013). Differential analysis identified SEs specific to H3.3K27M, including known (LIN28B and MYC) and additional (IRX2 and PAX3) developmental associated genes (Figure 2D). Notably, all H3.3K27M cell lines showed either MYC amplification (data not shown) or increased LIN28B, MYC, or MYCN expression (Figure S1C), possibly through potential SE association. H3K27WT specific SE genes included EN2, HOXA10, CDK6, LHX2, and EGFR (Figure 2D), consistent with known enrichment of EGFR and CDK6 amplifications in cortex HGGs (Sturm et al., 2014).
Figure 2. Core regulatory SE and TF programs delineate H3.3K27M and H3K27WT HGG.

(A-B) Super enhancers detected across all H3K27WT HGG samples (n=8) (A) and H3.3K27M HGG samples (n=8) (B) using the same samples listed in Figure 1B.
(C) Patterns of H3K27ac shown surrounding the EN1 locus (left panel) and LIN28B (right panel).
(D) Differential super enhancers restricted to H3K27WT and H3K27M samples as detected by DiffBind in DESeq2 mode.
(E) Core regulatory circuitry transcription factors shared or enriched in medulloblastoma, ependymoma, IDH1-glioma, H3K27WT (n=8) and H3K27M HGG (n=11) samples. Score indicates the number of core TFs detected within at least one core regulatory circuit in a given tumor type.
See also Table S4.
We then re-constructed the core transcription factor (TF) circuitry programs that regulate shared and specific H3.3K27M and H3K27WT SE networks, and compared their specificity to other known brain tumor entities (Saint-Andre et al., 2016). Using this approach, we identified core regulatory circuit TFs active in ependymoma, medulloblastoma, IDH1-glioma, HGG-H3.3K27M, or HGG-H3K27WT tumors (Lin et al., 2016; Mack et al., 2018) (Figure 2E). Several core TFs including SOX4, JUN, FOXO3, SP3, and NR4A2 were shared across this set of brain tumors while many others were disease specific, possibly acting as potential disease driver genes and/or lineage associated markers of cellular and developmental origins. Indeed, LHX1 and OTX2 have been shown to be drivers of medulloblastoma tumor formation, while RELA was identified as a core TF in the context of C11ORF95-RELA fusions in ependymoma. UNCX, highly expressed during embryonic development at day E14.5 in the mid-external granule layer (Machold et al., 2011), was in turn shown to be specific to medulloblastoma and to be a potential marker of tumor cellular origins. We identified two specific key core TFs in H3.3K27M HGGs, IRX2 and PAX3 (Figure 2E), in keeping with recent data from a study using single cell profiling of H3K27M HGGs, which demonstrated restricted expression of these TFs to H3K27M cells (Filbin et al., 2018). Interestingly, only a subset of H3K27M tumor derived cells expressed IRX2. These IRX2 positive cells had no specific association to the described astrocyte, oligodendrocyte, and cell cycle signatures (Filbin et al., 2018) (Figure S1D). In contrast, H3K27WT cells harbored other candidate lineage associated core TFs such as EN1, HOXA10, and HOXD8. EN1 expression in development is found within the mid-hindbrain junction coincident with midbrain dopaminergic neuronal development (Alves dos Santos and Smidt, 2011). Altogether, our results on typical enhancer, SE and core TF networks converge on lineage genes involved in brain development for both H3.3K27M mutant and H3K27WT HGGs, some of which may also represent tumor growth dependencies. In H3K27M tumors, these lineage-associated TF programs may intersect with other oncogenic programs, namely the LIN28B/MYC axis to promote tumorigenesis. Core TF regulation in pediatric HGGs resembles what is observed in other pediatric brain tumors such as ependymoma and medulloblastoma, leading us to hypothesize that they largely reflect cell state programs related to the cell identity and origin of these tumors (Lin et al., 2016; Mack et al., 2018). Thus, we conclude that these SEs and their core TF circuitries, which mark important brain developmental programs, are most likely an indication of cellular states associated with H3K27M tumor cells and may not necessarily be a direct consequence of H3K27M mutagenesis.
Isogenic H3K27M models decouple lineage and oncogenic patterns associated with H3K27 acetylation
To address whether H3.3K27M directly impacts ‘active’ chromatin loci including enhancers, SEs, and associated TF programs, or regulates other uncharacterized genomic loci, we designed an isogenic tumor experimental system to interrogate solely the effect of the mutation. We introduced frameshift deletions/mutations in the H3F3A-K27M mutant allele in two tumor-derived primary cell lines, BT245 and DIPG-XIII, using CRISPR-Cas9 engineering (Figures 3A, S2A–B). H3.3K27M knockout abrogated mutant protein expression and was validated using mass spectrometry, which also confirmed that total H3.3 levels were similar in both edited and unedited clones (Figures 3A–C, S2B–D). CRISPR mediated editing decreased H3K27ac and increased H3K27me3 levels (Figures 1A, 3D–E, S2E–F).
Figure 3. Mapping of H3K27ac in isogenic models of H3K27M reveal minimal alterations of gene transcriptional programs.

(A) Immunoblot in parental and clonal H3K27M knockout lines of DIPG-XIII.
(B-E) Mass spectrometry analysis in DIPG-XIII for total (B), H3K27ac (C), K27M (D) and H3K27me3 (E) peptides among total H3, plot of mean of n=3 or 4 technical replicates. Solid horizontal lines indicate the mean.
(F-G) Pie graph (F) and heatmap (G) illustrating number and proportion of gained, lost, and retained H3K27ac sites consistent between BT245 and DIPG-XIII K27M expressing models as compared to KO lines.
(H) Gene plot illustrating patterns of H3K27ac and super enhancers between H3.3K27M cell models and isogenic KO clones.
See Figures S2 and S3 and Tables S5 and S6.
Surprisingly, when assessing H3K27ac changes common to both cell lines by ChIP-Rx (a quantitative ChIP-seq assay) (Orlando et al., 2014), the comparison of H3.3K27M mutant cell lines to their parental isogenic clones showed that only a very small proportion of H3K27ac sites (0.25%) were consistently lost in both lines while the vast majority of H3K27ac sites were unchanged (96.6%) upon H3.3K27M removal (Figures 3F–G). This genome-wide finding was also recapitulated when assessing changes occurring specifically in each of the two cell-lines compared to its isogenic H3K27WT clone (Figures S2G–H). One of the two H3.3K27M cell lines (DIPG-XIII) showed ~10% loss of H3K27ac loci when the K27M mutation was removed. However, except for rare genes including ADARB2, these H3K27ac changes did not significantly alter regulatory elements (enhancers or SEs) identified through H3K27ac profiling (Figures 3F–H, S2G–J, Table S5). Surprisingly, upon H3K27M knockout, we observed gains of H3K27ac at several loci in cells (Figure 3G). Interestingly, several genes associated with these H3K27ac gains have documented roles in neural differentiation and glial development and include DAAM1, NFIB, and EPHB2 (Table S6) (Kang et al., 2012; Zhu et al., 2017). These results support, at least in part, the hypothesis that distinct cell states associate with H3K27M or H3K27WT mutational status and the premise that H3K27M may lead to a blockade in differentiation which could be reversible upon removal of the mutation, similar to what we previously observed for another mutant histone H3K36M (Lu et al, Science 2016).
We further examined open chromatin distribution using ATAC-seq in H3.3K27M and H3K27WT HGGs, and in one isogenic HGG line carrying H3.3K27M mutation compared to H3.3K27M-KO. When overlaying open chromatin loci called by ATAC-seq with H3K27ac peaks, we observed that the overlap between both markers of open chromatin was significantly lower in H3.3K27M HGGs compared to H3K27WT lines (Figure S2K). Notably, removal of the mutation increased the overlap between ATAC-seq data and H3K27ac peaks from 53% to 73% (Figure S2K), a level typically observed in a standard ATAC-seq experiment. The lower overlap observed between ATAC-seq and H3K27ac loci in H3K27M HGG compared to H3K27WT HGG cell lines suggests regions of active chromatin outside well-defined H3K27ac peaks in H3.3K27M HGGs. Together with the unchanged number of canonical H3K27ac sites (Figures 1D, 3F–G), these findings indicate that the global increase in H3K27ac in H3K27M cells does not substantially alter or create new sites of focal H3K27ac deposition (including enhancers and promoters). Deposition of this active mark at these sites is largely independent of H3.3K27M, probably marking cell of origin state and lineage genes.
Based on their increased H3K27ac levels, H3.3K27M mutant cells have been suggested to exhibit cell growth dependencies on transcriptional activity mediated by bromodomain and extra-terminal domain (BET) proteins (Piunti et al., 2017; Nagaraja, et al., 2017). However, we did not observe differential sensitivity of H3.3K27M mutants to the BET inhibitors JQ-1 or I-BET762 in any of the H3.3K27M cell lines we tested compared to H3K27WT or isogenic H3K27M knockout cells (Figures S3A–C). Furthermore, when treating cells with inhibitors of the CREBBP/EP300 histone acetyltransferases (CBP-30 and C646) we observed a trend of H3K27WT line being more vulnerable to these agents (Figures S3D–I), indicative that the growth of H3.3K27M cells is not dependent on elevated histone acetyltransferase activity. Altogether, these findings support our observation that H3K27M SE landscapes reflect the cell state and/or lineage, and that H3K27ac gain does not create a sensitivity to BET or CREBBP/EP300 histone acetyltransferase inhibitors in K27M mutant cells.
H3.3K27M induces aberrant H3K27ac deposition at repetitive elements in human and mouse HGG
We investigated whether H3K27ac deposition occurs outside of traditional loci called with standard peak-calling algorithms. Indeed, both DIPG-XIII and BT245 H3.3K27M cell lines harbored, genome-wide, a greater number of domains with increased H3K27ac, including deposition of the mark in intergenic regions (Figures 4A–B, S4A–B). The human genome is mainly comprised (~70%) of repetitive elements broadly categorized to include LINEs, SINES, LTRs, DNA transposons and other repeat elements (de Koning et al., 2011). Genomic silencing of these repetitive sequences is important for the maintenance of genome integrity and tightly regulated using several layered mechanisms, which include combinatorial deposition of repressive epigenetic marks, namely H3K9me3 and H4K20me3, H3K27me3 or DNA methylation (Bestor and Bourc’his, 2004; Bourc’his and Bestor, 2004; Walsh et al., 1998). We examined whether this pervasive deposition of an active chromatin mark results in increased baseline expression of large regions of the genome that tend to be normally transcriptionally silent. Cell lines bearing H3.3K27M showed an increase of H3K27ac in unique reads mapping to these repetitive elements when compared to their isogenic H3K27M-KO primary HGG lines (Figures 4C, S4C); consistent with the genome-wide increase in H3K27ac observed by histone mass spectrometry.
Figure 4. H3K27ac deposits pervasively across intergenic genome and is enriched at repetitive elements.

(A) Quantitative H3K27ac ChIP-Rx scaled reads between DIPG-XIII K27M and KO cell cultures across chromosome 8.
(B) Enrichment of H3K27ac across the human genome in 1 MB windows comparing DIPG-XIII H3K27M versus KO clones using a Wilcoxon rank sum test, ***p<0.001. The horizontal line inside the box corresponds to the median, the lines above and below the box delimit the interquartile range (IQR), and the end of the whiskers delimit values up to 1.5 times the IQR.
(C) Enrichment of H3K27ac across repetitive regions (LINE, DNA, LTR, SINE) of the human genome annotated by the RepeatMasker database comparing DIPG-XIII K27M vs KO using a Wilcoxon rank sum test, ***p<0.001.
(D-E) Changes in deposition of H3K27ac (D) and transcription (E) of repeat elements in DIPG-XIII K27M relative to KO. Histograms show the distributions of the log2 fold-change (LFC) of genomic features in K27M relative to KO. Orange: H3K27ac deposition in repetitive elements (Repbase with ChIP-seq; DIPG-XIII n=1; DIPG-XIII-KO n=2). Green: transcription of repetitive elements (Repbase with RNA-seq; DIPG-XIII n=2; DIPG-XIII-KO n=2). Grey: H3K27ac (left) and transcription (right) of Ensembl genes (GRCh37). Elements with sufficiently high expression (baseMean > 100) and LFCs in [−3, 3] range shown here.
(F) Sensitive measurement of expression of a panel of ERV families by ddPCR in H3K27M lines compared to respective KO DIPG-XIII cells. Plot shows mean + SEM of 3 experimental replicates. Two-way ANOVA compares H3K27M to KO, with Bonferroni post-test significance shown by*** p<0.001. Y-axis is in log2 scale.
See also Figure S4.
We next refined the quantification at repetitive elements by alignment of ChIP-Rx reads to the human database of repetitive regions (Repbase), an approach that has been shown to provide robust quantification of transcriptional abundance at repeat elements (Solovyov et al., 2018). We observed in both cell lines increased H3K27ac deposition in H3.3K27M lines compared to isogenic H3K27M-KO controls for most repetitive elements present in the reference (Figures 4D, S4D). Notably, matching RNA-seq analyses showed upregulation of transcription of families of repeat elements in mutant lines (Figures 4E, S4E–F). At the transcriptional level, we observed increased baseline expression of specific families of repeat sequences, including endogenous retroviruses (ERVs) in H3.3K27M mutant cells when compared to their isogenic counterparts (data not shown). ERVs represent about 10% of the genome and have been associated with viral mimicry in cancer (Chiappinelli et al., 2015; Roulois et al., 2015). We selected ERV families previously implicated in viral mimicry (Chiappinelli et al., 2015; Roulois et al., 2015) and further validated this increase using droplet digital PCR (ddPCR), a highly sensitive technique to quantify changes in lowly expressed transcripts (Figures 4F, S4G).
Finally, we confirmed that this pattern is recapitulated in human tumors. Using ChIP-Rx in H3.3K27M and H3K27WT HGG tumors, we observed increased H3K27ac deposition in the H3.3K27M group genome-wide (Figures 5A) and at repetitive elements (Figures 5B–C). Notably, RNA-seq analysis of a large panel of H3.3K27M (n = 17) and H3K27WT (n = 15) HGGs showed increased transcription of these elements in the H3.3K27M group (Figure 5D). Furthermore, analysis of RNA-seq data generated in our recently published H3.3K27M-HGG mouse model (Pathania et al., 2017) provided cross-species validation for these findings. We observed a significant increase in the proportion of reads mapping to the murine repeat genome only in H3.3K27M HGG mouse tumors when compared to experimental controls (P<0.05, Figure 5E). In all, our convergent findings in primary tumors, in primary cell lines derived from patients and in our syngeneic H3.3K27M mouse model indicate that the global increase in H3K27ac induced by H3.3K27M leads to pervasive deposition of the mark throughout the genome, including at normally silent repeat elements, resulting in aberrant transcription in H3.3K27M tumors.
Figure 5. H3.3K27M tumors exhibit increased deposition of H3K27ac and transcription at repetitive elements.

(A) Enrichment of H3K27ac across the human genome in 1 MB windows comparing 2 H3.3K27M versus 3 H3K27WT tumors using a Wilcoxon rank sum test, ***p<0.001. The horizontal line inside the box corresponds to the median, the lines above and below the box delimit the interquartile range (IQR), and the end of the whiskers delimit values up to 1.5 times the IQR.
(B) Enrichment of H3K27ac across repetitive regions (LINE, DNA, LTR, SINE) of the human genome annotated by the RepeatMasker database comparing 2 H3.3K27M versus vs 3 H3K27WT tumors using a Wilcoxon rank sum test, ***p<0.001.
(C-D) Changes in H3K27ac (C) and transcription (D) of repeat elements in H3.3K27M tumors relative to H3K27WT tumors. Histograms show the distributions of the log2 fold-change (LFC) of genomic features in K27M relative to K27WT. Orange: H3K27ac deposition over repetitive elements (ChIP-seq; H3.3K27M n=2; H3K27WT n=3). Green: transcription of repetitive elements (RNA-seq; H3.3K27M n=17; H3K27WT n=15). Grey: H3K27ac (left) and transcription (right) of Ensembl genes (GRCh37). Elements with sufficiently high expression (baseMean > 100) and LFCs in the [−3,3] (C) and [−2,2] (D) ranges shown here.
(E) Proportion of RNA-seq reads that map to the repeat genome in K27M tumors relative to controls in a preclinical K27M mouse model. Black: neural progenitor cell (NPCs) controls (n=4). Teal: NPCs overexpressing H3.3WT (n=4). Red: K27M tumors (n=7). Preclinical K27M mouse model RNA-seq data taken from Pathania et al., 2017. Significance between conditions assessed using a two-sample t-test and is shown by **p<0.01; *p<0.05. Solid horizontal lines indicate the mean.
Epigenetic inhibitors exacerbate repeat element expression in a H3.3K27M HGGs
DNA methylation is important in the containment of ERVs, and manipulation of this epigenetic mark is the basis of several therapeutic strategies in cancer (Baylin and Jones, 2016; Chiappinelli et al., 2015; Roulois et al., 2015). Histone deacetylase inhibitors (HDAC inhibitors, HDACi), in turn, often used in combination with DNA demethylating agents, have a documented role in promoting ERV expression (Topper et al., 2017). We thus interrogated whether pharmacological modulation of baseline expression of repeat elements represents a specific cellular vulnerability in H3.3K27M versus H3K27WT HGGs. We tested the DNA demethylating agent 5-azacytidine and panobinostat, a pan-HDAC inhibitor currently in clinical trials for DIPG (https://clinicaltrials.gov/ct2/show/NCT02717455). We observed large transcriptomic changes, with the combination of 5-azacytidine and panobinostat inducing a greater increase in expression of repetitive elements in H3.3K27M lines compared to their H3.3K27M-KO isogenic counterparts (Figures 6A–B, S5A–C). This was further confirmed using ddPCR, which identified specific ERVs to be significantly induced by panobinostat or 5-azacytidine in H3.3K27M lines and showed that this induction was greatly attenuated upon H3.3K27M loss (Figures 6C–D, S5D–E). The pharmacologically-induced expression shift of repeat elements correlated with drug response, as H3.3K27M cell lines were more sensitive than H3K27WT HGG lines to DNA demethylation (Figure 6E). Consistent with a causal role of H3.3K27M, this sensitivity was decreased in H3.3K27M KO cells (Figure 6F, S6A).
Figure 6. Repeat element primed K27M cells are vulnerable to DNA methylation and histone deacetylase inhibitors.

(A-B) Induction of expression of repetitive element observed in DIPG-XIII (A) and KO lines (B) after combination treatment (panobinostat and 5-azacytidine). Histograms show the distributions of the log2 fold-change (LFC) of transcription of repetitive elements (green; Repbase) and Ensembl genes (grey; GRCh37) based on RNA-seq in treated compared to untreated cells (DIPG-XIII treated n=3, untreated n=3; DIPG-XIII-KO treated n=2, untreated n=2 biological replicates). Elements with sufficiently high expression (baseMean > 100) and LFCs in [−3, 3] range shown here.
(C-D) Measurement of ERV family expression by ddPCR upon 48 hr treatment with panobinostat (50 nM) or 5-azacytidine 5 μM), normalized to vehicle control, in DIPG-XIII (C) and KO lines (D). Plots show mean + SEM of 3 experimental replicates of the DIPG-XIII line. Two-way ANOVA compares vehicle to each drug, with Bonferroni post-test significance shown by ***p<0.001, **p<0.01, *p<0.05. Y-axis is in log2 scale.
(E) Dose-response curves of a panel of HGG lines to 5-azacytidine in a 7 day growth assay in H3K27M and WT lines. Plots show mean +/− SEM of 3 experimental replicates.
(F) The sensitivity of H3K27M and KO lines of BT245 and DIPG-XIII to 5-azacytidine in a 7-day growth assay is portrayed by IC50 values +/− 95% confidence intervals. ***p<0.001, *p<0.05.
(G) Western blot of total H3K27ac levels upon panobinostat treatment of DIPG-XIII at various doses (in nM) for 48 hr.
(H) Cell viability in 7 day growth assay under treatment of lines with sub-IC50 doses of panobinostat (15 nM), 5-azacytidine (1.5 μM) and their combination in DIPG-XIII and isogenic KO lines. Plots show mean + SEM of 3 experimental replicates. A two-tailed t-test compares combination treated lines, with **p<0.01.
(I) Mass spectrometry measurement of H3K27ac levels on histone H3 variants comparing treated and untreated DIPG-XIII cells with 50 nM panobinostat for 48 hr. Plot of mean of 3 experimental replicates. **p<0.01.
See also Figures S5 and S6.
HDACi have been reported to inhibit the growth of H3K27M DIPG cell lines despite the already high H3K27ac levels observed in this setting (Grasso et al., 2015). Treatment of H3K27M cell lines using panobinostat greatly elevated H3K27ac levels (Figure 6G) at doses that impaired cell viability. No differential cell growth impairment was observed between H3.3K27M and H3K27WT lines (and between isogenic H3.3K27M vs KO cell lines) when treating with panobinostat (Figures S6B–D). However, when used in combination, low doses of 5-azacytidine and panobinostat were additively toxic, resulting in a greater effect in H3.3K27M lines compared to their KO counterparts (Figures 6H, S6E) and to H3K27WT HGG lines (Figure S6F). Notably, mass spectrometry revealed the gain of acetylation induced by panobinostat mostly occurs in cells on non-canonical H3.3 (Figure 6I), which is known to be deposited in active regulatory elements as well as at repeat elements (Elsasser et al., 2015) and may thus lead to a further increase in repeat element expression.
Epigenetic inhibitors induce a cellular response to repetitive elements in primed H3.3K27M tumor cells
Elevated repeat element expression and formation of double-stranded RNA (dsRNA) stimulate innate immune responses mimicking a viral infectious state (i.e. viral mimicry), through induction of interferon type I signaling and expression of interferon stimulated genes (ISGs) (Chiappinelli et al., 2015; Roulois et al., 2015; Topper et al., 2017). H3K27M mutant HGGs can have a significant inflammatory infiltrate (Bechet et al., 2014) in keeping with induction of immunogenicity in these tumors. We found no consistent increase in expression of ISGs in H3.3K27M HGG compared to H3K27WT models or in K27M cell lines relative to KO (Figures 7A, S7A–B, Table S7). This is possibly due to the high MYC levels in H3.3K27M mutant cells (Figures 2D, S1C), as MYC expression has been shown to be a potent antagonist of interferon responses (Schlee et al., 2007) and has been shown to promote inhibition of apoptosis and immune evasion in other solid tumors (Topper et al., 2017).
Figure 7. Epigenetic therapies activate interferon response genes and dsRNA sensing pathways.

(A-B) Waterfall plots illustrating changes in the expression levels of ISGs in DIPG-XIII H3K27M cells relative to KO at baseline (A) and upon treatment with 5-azacytidine (B). Y-axis: log2 fold-change of expression in K27M relative to KO (baseline DIPG-XIII n=3, DIPG-XIII-KO n=2; treated DIPG-XIII n=3, DIPG-XIII-KO n=2 biological replicates). Red: upregulated genes in K27M. Blue: downregulated genes. Significantly deregulated genes highlighted (p<0.05; baseMean>100).
(C) The sensitivity of H3K27M and KO lines to 5-azacytidine upon expression of sh-NT (control) or pooled shRNAs targeting MAVS (sh-MAVS), is portrayed by IC50 values +/− 95% confidence intervals. ***p<0.001, **p<0.01. n=3
(D) Immunoblot of phosphorylated eIF2a (serine 51) induced by 48-hr treatment with panobinostat (50 nM) and 5-azacytidine (5 μM) or interferon gamma (IFN-γ, 1000 U/mL) in DIPG-XIII H3K27M cells and KO cells.
(E-G) Survival of mice bearing pcGBM2 (E), DIPGXIII (F), and BT245 (G) xenografts and treated with vehicle, 5-azacytidine, panobinostat, or combination of both drugs. Statistical significance measurements were determined using a Log-Rank test. n=9 animals per condition.
(H) Induction of interferon signature genes in patient-derived tumor xenografts treated. Mice were treated with vehicle, 5-azacytidine, panobinostat, or combination of both drugs, and tumors were isolated at end point and analyzed by qPCR. The fold change of expression compared to DMSO treated tumors is presented as mean + SD and statistical significance was determined using a two-way ANOVA test on 4 distinct biological replicates for each condition. ***p<0.001, **p<0.01 (what analysis was done (PCR?), what are the p values; what is the fold change relative to; how are the data presented (mean + SEM?)
Importantly, 5-azacytidine (5-azaC) treatment led to a significantly stronger induction of a large number of ISGs in both H3.3K27M mutant cell lines compared to their isogenic wild-type clones (Figure 7B, Table S7), reaching more than 5-fold higher induction for some genes. Notably, relative to KO cells lacking the K27M mutation, 31/61 of the ISGs were significantly overexpressed in K27M-mutant cells in response to 5-azaC treatment, in line with a stronger ISG response in the presence of the mutation, which seemingly amplifies the ability of the cancer cells to induce an IFN response when exposed to DNA demethylases. Interestingly, a minor increase in ISGs compared to baseline was seen when we used the HDACi panobinostat (Figures S7C–D, Table S7). In all, our findings argue that H3.3K27M cells are primed for viral mimicry in a H3K27M-dependent manner, as evidenced by the baseline ERV expression in mutant cells relative to their WT or KO counterparts and their increased expression following further de-repression of silent repeat elements by epigenetic modulators, with preferential induction of ISGs in K27M-mutant cells following the use of DNA demethylating agents.
We then characterized the functional role of dsRNA sensing pathways in the mechanism of epigenetic therapy action in H3.3K27M cell lines. Mitochondrial antiviral signaling protein (MAVS) is the signaling adaptor common to two known cytoplasmic viral receptors, retinoic acid inducible gene 1 (RIG-1) and melanoma differentiation associated protein (MDA5), which recognize dsRNA, and is responsible for activating interferon responses in virally infected cells (Belgnaoui et al., 2011). These proteins have also been implicated in viral mimicry responses to cancer cells treated with DNA demethylating agents. Short hairpin RNA interference mediated knockdown of MAVS attenuated the sensitivity of H3.3K27M but not H3K27M-KO HGG lines to 5-azacytidine (Figures 7C, S7E–I), suggesting the transduction of signals from dsRNA receptors to the interferon pathway is active in the presence of H3.3K27M but not in cells where this mutation was removed. Furthermore, phosphorylation of eukaryotic initiation factor alpha (eIF2α), a key target of the dsRNA sensor protein kinase R (PKR), was induced by panobinostat and 5-azacytidine in H3.3K27M lines but not in H3K27M-KO lines (Figures 7D, S7J). These two responses to dsRNA sensing and adaptation were specific to H3.3K27M HGG cells, further suggesting that H3.3K27M tumors are primed to innate immune responses through global alterations of histone modifications.
Last, we assessed tumor growth following systemic administration of 5-azacytidine, panobinostat or a combination of these drugs in an orthotopic NOD SCID-IL2R gamma-chain deficient mice (NSG) mouse model. Isogenic lines lacking the H3.3K27M mutation have defective growth in vivo. We thus used parental H3.3K27M DIPG-XIII and BT245 and, as control, an H3.3K27M wild-type line, pcGBM2. Each group of mice was subjected to one of the four arms: 5-azacytidine, panobinostat as previously described (Borodovsky et al., 2013; Grasso et al., 2015; Yamashita et al., 2018), a combination of both drugs, or control vehicle. Compared to vehicle-treated control mice, we observed extended survival following administration of panobinostat in K27M-mutant BT245 and DIPG-XIII derived tumors as previously described (Grasso et al., 2015), as well as in K27M-wild-type tumors derived from the pcGBM2 cell line (Figure 7E–G). Notably, administration of 5-azacytidine resulted in improved survival in mice carrying H3.3K27M-mutant cell lines compared to panobinostat or the vehicle alone, while no significant effect of the drug was observed in mice injected with pcGBM2 (Figure 7E–G, S7K). Moreover, in mice injected with H3.3K27M cell lines, combined administration of 5-azacytidine and panobinostat resulted in further improved survival compared to either drug delivered alone (Figure 7E–G, S7K). Evaluation of a set of ISGs in the tumors extracted at end-point from the mouse brain showed their significant induction in tumors carrying the H3.3K27M mutation following the use of 5-azacytidine or the combination therapy (Figure 7H). Limited ISGs induction was observed in tumors wild-type for H3K27, while panobinostat had modest effect on the expression of these genes both in K27M-mutant and in K27M wild-type tumors (Figure 7H). These results confirm our in vitro data and further indicate that the use of DNA demethylating agents and this drug combination may be effective in K27M-mutant HGGs.
Discussion
Our integrative epigenetic analysis of pediatric HGG, including tumors with H3K27M mutations, provides a comprehensive analysis of the specific transcriptional programs associated with H3.3K27M and H3K27WT HGGs, while revealing factors important in defining their respective cellular identity. Specifically, we show that H3.3K27M HGGs have a specific promoter, SE and core TF circuitry program which is indicative of cell lineage and identity, and seemingly independent of the H3K27M mutation. Importantly, analyses of our isogenic models show that the increased H3K27ac in H3K27M HGGs is mainly attributed to pervasive deposition of the H3K27ac mark across the genome following H3K27me3 loss and does not directly activate oncogenic SE programs. This pervasive acetylation induces increased expression of repetitive elements including ERVs in H3.3K27M cells, which can prime cells for innate immune responses (Figure 8). We show that use of DNA demethylating agents and HDAC inhibitors further increase this baseline expression of ERVs and potentially help overcome possible MYC-driven immune evasion, allowing robust induction of ISGs following DNA demethylating therapies, while significantly impairing cell growth in vitro and in vivo in H3.3K27M mutant HGGs. These findings uncover an exquisite vulnerability to available epigenetic therapies in deadly H3.3K27M HGGs.
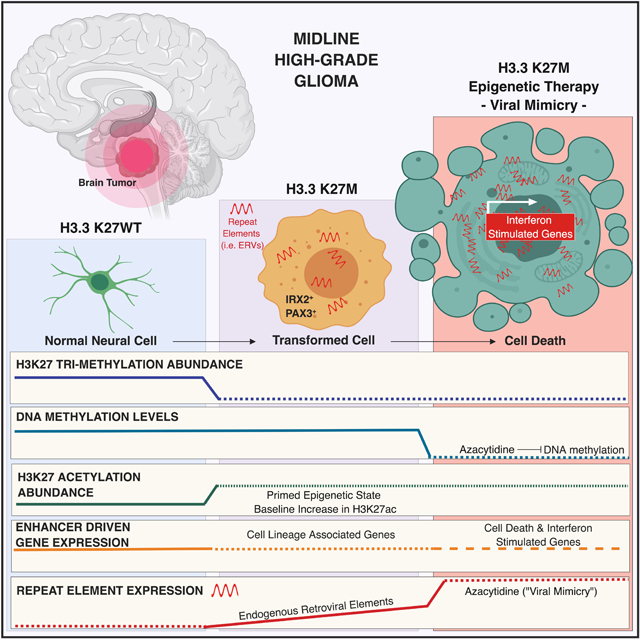
Figure 8. Schematic summary of findings resulting from this study.

The global loss of H3K27me3 by H3.3K27M leads to pervasive gain of H3K27ac across broad regions of chromatin, including repeat elements and endogenous retroviruses. This state primes them for activation by DNA demethylation and HDAC inhibition, a therapeutic vulnerability caused by this mutation.
Our refined mapping of the cis-regulatory landscape of H3.3K27M and H3K27WT tumors reveal potentially important core regulatory circuitry TFs that will be relevant to developing animal models to study this disease as applied to other pediatric brain tumors(Lin et al., 2016; Mack et al., 2018). In addition to epigenetic mapping by H3K27ac ChIP-seq, ATAC-seq and RNA-seq, our isogenic models are further validated by histone mass spectrometry to being faithful representations of the disease. While several studies have proposed epigenetic therapies to target H3K27M tumors (Grasso et al., 2015; Mohammad et al., 2017; Piunti et al., 2017) our comparison of isogenic models expressing H3.3K27M is of value to identify those with direct effect on K27M mutant cells while determining their mechanism of action. Thus, in support of our model that H3.3K27M does not create significant de novo oncogenic SE features, disruption of transcriptional activity through BET or HAT inhibition is not a specific vulnerability of H3.3K27M mutant lines. This does not exclude the importance of these compounds as agents which may be effective across pediatric HGG, however they may not be specific to K27M mutagenesis as initially postulated and their use in the context of priming the immune system may be counterproductive.
Repetitive elements are silenced by epigenetic marks including DNA methylation and H3K9 and H3K27 trimethylation. We show that loss of intergenic H3K27me3 results in pervasive H3K27ac deposition, which induces baseline expression of these normally silenced elements including ERVs. This effect can be further enhanced when manipulating the epigenome using DNA demethylating agents and using of HDAC inhibitors. Notably, the effects of HDACi in increasing H3K27ac were prominent on H3.3, which is the main H3 variant deposited in nucleosomes on repeat elements (Elsasser et al., 2015), potentially accounting for the further increased ERV expression using these inhibitors. We also show that, despite the high MYC expression resulting from SE regulation in H3.3K27M, DNA demethylating agents could induce a strong ISG response in mutant cells in vitro and in vivo. Importantly, the use of DNA demethylating agents in combination to HDACi resulted in significant growth inhibition in H3.3K27M mutant cells compared to their isogenic counterpart in vitro or to wild-type cells in in vivo orthotopic mouse models. We thus propose a model where pervasive H3K27ac leads to expression of RNA transcripts from repetitive regions and de-repression of ERVs. Repeat element and ERV de-repression may be potentiated by specific epigenetic therapies, representing a unique vulnerability of H3.3K27M tumors. In line with other solid tumors, modulating the epigenome (with agents such as 5-azacytidine and HDACi) at repeat elements can trigger viral mimicry and induce cellular interferon responses, thus promoting tumor cell death and immune cell activation (Jones et al., 2019). This effect is likely not specific to H3.3K27M HGGs and could be of relevance in a significant number of other cancers exhibiting loss of PRC2 activity and altered H3K27me3 levels including intractable posterior fossa ependymoma group A and malignant peripheral neural sheath tumors (Lee et al., 2014). In line with our findings of a primed immune state in H3.3K27M mutants, several groups have shown that DIPGs have a level of immune infiltrate of unknown origin and significance including in recent data from single cell transcriptomic of H3K27M mutant DIPGs (Bechet et al., 2014; Filbin et al., 2018) suggesting that further modulation of interferon signaling could achieve productive anti-tumor responses in vivo.
It is also important to consider that treatment of K27M HGG cells with panobinostat or 5-azacytidine have relevant cell intrinsic effects. We provide support that the release of dsRNA and ERVs normally silenced in midline high-grade glioma cells activates a potential viral mimicry response. This occurs through recognition of dsRNAs by intracellular DNA pattern receptors (i.e. RIG-1 and MDA5), activation of MAVS, and induction of interferon response genes (Jones et al., 2019; Roulois et al., 2015). We show that MAVS expression is important for mediating response to 5-azacytidine preferentially in K27M cells. In a K27M specific context, we show that eIF2α is phosphorylated at the serine 51 position when cells are treated with panobinostat or 5-azacytidine. As a result, eIF2α phosphorylation inhibits translation initiation and therefore protein synthesis, representing one possible mechanism of cell growth inhibition. Another important consideration is that a small subset of genes (such as the p16 tumor suppressor) retain epigenetic silencing in H3K27M cells despite global loss of H3K27me3 and DNA methylation (Cordero et al., 2017). P16 may be re-expressed upon treatment with epigenetic inhibitors and may also negatively impact tumor cell growth.
In summary, our findings identify a potential therapeutic vulnerability in H3.3K27M mutant HGGs. The mutation primes the cells for viral mimicry, a state we show can be further enhanced by specific epigenetic drugs which may act by making the tumor “hot” for the immune system. While further work using immune competent models is warranted, several of these epigenetic therapies are FDA approved and HDACi and immune checkpoint inhibitors are currently in clinical trials for DIPGs as single agents, as are DNA demethylating agents which are used in combination with cytotoxic agents in several solid tumors including HGGs. It is important to note that while epigenetic therapies such as nucleoside analogs (5-azacytidine) may incorporate and target actively dividing cells, at higher doses, such agents exhibit high levels of cytotoxicity particularly within the bone marrow (Jones et al., 2019). Low-dose combinations of these agents with immunomodulatory compounds could be rapidly tested at the bedside and may represent an effective therapeutic strategy against primed H3.3K27M HGGs, providing a needed alternative in an untreatable deadly disease.
STAR Methods
Contact for reagent and resource sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Nada Jabado (nada.jabado@mcgill.ca).
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Sample isolation and processing
The derivation of tumor cell lines was approved by and procedures conform to the standards of the Institutional Review Boards of McGill University, the University of Toronto, Stanford University, the Dana-Farber Cancer Center and the Hospital San Juan de Deu. Informed consent was obtained from all cases.
Cell lines
Tumor-derived cell lines were maintained in Neucult NS-A proliferation media (StemCell Technologies) supplemented with bFGF (10ng/mL) (StemCell Technologies), rhEGF (20 ng/mL) (StemCell Technologies) and heparin (0.0002%) (StemCell Technologies) on plates coated in poly-L-ornithine (0.01%) (Sigma) and laminin (0.01 mg/mL) (Sigma) in an incubator at 37 degrees Celsius and 5% carbon dioxide. All lines tested negative for mycoplasma contamination, checked monthly using the MycoAlert Mycoplasma Detection Kit (Lonza). Tumor-derived cell lines were confirmed to match original tumors by STR fingerprinting, where tumors were available. The sex of all cell lines are reported in Table S1.
Animals used in PDOX studies
All in vivo procedures in mice were approved by and conform to the standards of the Research Animal Care and user committee at McGill University, the Canadian Council of Animal Care, the Animal Welfare and Ethical Review body and institutional and UK Home Office guidelines (Project license 70/8240, 70/7428 and 80/2325). Housing, breeding and procedures performed were in keeping with the US National Institute of Health guide for the care and use of laboratory animals. Both genders were equally used in all experiments. PDOX of two patient-derived H3.3K27M mutant cell lines, DIPGXIII, BT245, and a wild-type high-grade glioma cell line, pcGBM2, were generated as previously described (Grasso et al., 2015). All mice used were NOD SCID-IL2R gamma-chain deficient mice (NSG, Jackson Laboratory) aged 4–6 weeks.
METHOD DETAILS
Identification and quantification of histone modifications with nLC-MS
The complete workflow for histone extraction, LC/MS, and data analysis was recently described in detail (Sidoli et al., 2016). Briefly, cell pellets (approx. 1×106 cells) were lysed on ice in nuclear isolation buffer supplemented with 0.3% NP-40 alternative. Isolated nuclei were incubated with 0.4 N H2SO4 for 3 hr at 4°C with agitation. 100% trichloroacetic acid (w/v) was added to the acid extract to a final concentration of 20% and samples were incubated on ice overnight to precipitate histones. The resulting histone pellets were rinsed with ice cold acetone + 0.1% HCl and then with ice cold acetone before resuspension in water and protein estimation by Bradford assay. Approximately 20 μg of histone extract was then resuspended in 100 mM ammonium bicarbonate and derivatized with propionic anhydride. 1 μg of trypsin was added, and samples were incubated overnight at 37°C. After tryptic digestion, a cocktail of isotopically-labeled synthetic histone peptides was spiked in at a final concentration of 250 fmol/μg and propionic anhydride derivatization was performed a second time. The resulting histone peptides were desalted using C18 Stage Tips, dried using a centrifugal evaporator, and reconstituted using 0.1% formic acid in preparation for nanoLC-MS analysis. nanoLC was performed using a Thermo Scientific™ Easy nLC™ 1000 equipped with a 75 μm × 20 cm in-house packed column using Reprosil-Pur C18-AQ (3 μm; Dr. Maisch GmbH, Germany). Buffer A was 0.1% trifluoroacetic acid and Buffer B was 0.1% trifluoroacetic acid in 80% acetonitrile. Peptides were resolved using a two-step linear gradient from 5% to 33% B over 45 min, then from 33% B to 90% B over 10 min at a flow rate of 300 nL/min. The HPLC was coupled online to an Orbitrap Elite mass spectrometer operating in the positive mode using a Nanospray Flex™ Ion Source (Thermo Scientific) at 2.3 kV. Two full MS scans (m/z 300–1100) were acquired in the orbitrap mass analyzer with a resolution of 120,000 (at 200 m/z) every 8 DIA MS/MS events using isolation windows of 50 m/z each (e.g. 300–350, 350–400…650–700). MS/MS spectra were acquired in the ion trap operating in normal mode. Fragmentation was performed using collision-induced dissociation (CID) in the ion trap mass analyzer with a normalized collision energy of 35. AGC target and maximum injection time were 10e6 and 50 ms for the full MS scan, and 10e4 and 150 ms for the MS/MS can, respectively. Raw files were analyzed using EpiProfile 2.0.
CRISPR/Cas9 genome editing
pSpCas9(BB)-2A-GFP (PX458v2) was a gift from Feng Zhang (Addgene plasmid # 48138). CRISPR-Cas9 editing was carried out as described in Ran et al (Ran et al., 2013). Constructs were transfected with lipofectamine 2000 (Thermo Fischer Scientific) according the manufacturer’s protocol. Flow cytometry sorted single GFP+ cells in 96 well plates, 72 hr post-transfection. Clones were expanded, and the target locus sequenced by Sanger sequencing. Select clones were screened by Illumina MiSeq system for the target exon to confirm complete mutation of the K27M allele. Mass spectrometry confirmed the absence of K27M mutant peptide in these clones (Figure 3C, S2D). The guide RNA sequence and the tracks of MiSeq data demonstrating editing of the K27M allele are shown in Figures S2A–B.
Cell viability assay
Cells were plated at a density of 5000 cells per well in 96 well plate, and 24 hr following plating were treated with agents for a 7-day period, with media replaced every 4 days. All agents were dissolved in DMSO (Sigma-Aldrich) which was the vehicle serving as control to normalize cell indices. We used agents CBP-30, JQ-1, Panobinostat from the Structural Genomics Consortium (Toronto) and 5-azacytidine (Sigma-Aldrich). Cell index was measured using Alamar Blue Cell Viability Reagent (ThermoFisher Scientific) according to manufacturer’s protocol. Absorbance at 570nm and 600nm was determined using i-Control microplate reader software by Tecan. Normalized cell index was calculated by (raw cell index – blank media reading) / (vehicle control index). Cell index response curves were plotted using Graphpad Prism using nonlinear regression of variable slope (four parameters) using least squares (ordinary) fitting method. Differential agent sensitivity was determined by comparing LogIC50 values between cell lines, using the Extra sum-of-squares F test of this parameter. The p value of this comparison is stated in plots. Graphs of IC50 values carry error bars of 95% confidence intervals for pairwise comparisons. Plots of normalized cell index portray mean and SEM of three experimental replicates for each cell line.
MAVS Knockdown
MAVS Human shRNA lentiviral particles (TL307181V) were purchased from OriGene. A pool of four shRNAs targeting MAVS were transduced into cell lines at a concentration of 2 TU/cell, alongside a scramble control shRNA particle (sh-NT). Transduced cells were selected with 2 μg/mL puromycin 48 hr post-transduction and puromycin selection was maintained for the duration of the experiment. Loss of MAVS protein was confirmed by western blot at the time of assay for 5-azacytidine sensitivity (Figure S7E).
Droplet Digital PCR
RNA was extracted from cells using the Aurum Total RNA Mini Kit (Bio-Rad) and concentration was quantified on the BioDrop uLite (Montreal Biotech). cDNA was generated using iScript Reverse Transcription Supermix (Bio-Rad). Target concentration was determined using the QX200 ddPCR EvaGreen Supermix assay (Bio-Rad) using 20 uL per reaction containing 10 ng of cDNA, using manufacturer’s protocol cycling conditions with a 58 degrees annealing temperature and 40 cycles. Droplets were assayed using the QX200 Droplet Reader (Bio-Rad) and manually scored for positive signal using QuantaSoft Software (Bio-Rad). The concentration of positive droplets per target was normalized to the concentration of GAPDH. The relative target abundance is shown as the average of three biological replicates (distinct passages of each cell line) determined by a single technical replicate. Primer sequences are found in Table S8.
Western blotting
Cells were lysed using RIPA buffer with added protease inhibitors (Roche) for non-histone proteins. Histone lysates were extracted using the Histone Extraction Kit (Abcam). Lysate protein concentration was determined with the Bradford assay reagent (Bio-Rad). Ten micrograms of non-histone protein, or 3 micrograms of histone, was separated on NuPAGE Bis-Tris 10% gels (ThermoFischer Scientific) and wet-transferred to a PVDF membrane (GE Healthcare). Membrane blocking was performed with 5% skim milk in tris buffered saline (50 mM Tris, 150 mM NaCl, 0.1% Tween 20, pH 7.4) (TBST) for 1 hr. Membranes were incubated overnight with primary antibody solutions in 1% skim milk in TBST. Membranes were washed 3 times in TBST, and the ECL anti-rabbit IgG Horseradish Perixidase linked whole antibody (GE Healthcare) was applied for 1 hr, at 1:1000 dilution in 1% skim milk in TBST. Membranes were washed 3 times and the signal was resolved with Amersham ECL Prime Western Blotting Detection Reagent (GE Healthcare) and imaged on a ChemiDoc MP Imaging System (Bio-Rad).
Chromatin Immunoprecipitation and sequencing
Cells were fixed with 1% formaldehyde (Sigma). Fixed cell preparations were washed, pelleted and stored at −80°C. Sonication of lysed nuclei (lysed in a buffer containing 1% SDS) was performed on a BioRuptor UCD-300 for 60 cycles, 10 s on 20 s off, centrifuged every 15 cycles, chilled by 4°C water cooler. Samples were checked for sonication efficiency using the criteria of 150–500 bp by gel electrophoresis. After the sonication, the chromatin was diluted to reduce SDS level to 0.1% and before ChIP reaction ~5% of sonicated drosophila S2 cell chromatin was spiked-in the samples for quantification of total levels of histone mark after the sequencing (see below). ChIP reaction for histone modifications was performed on a Diagenode SX-8G IP-Star Compact using Diagenode automated Ideal ChIP-seq Kit. 25ul Protein A beads were washed and then incubated with 6 μg of H3K27ac antibody (Diagenode, C15410196), and 2 million cells of sonicated cell lysate combined with protease inhibitors for 10 hr, followed by 20 min wash cycle with provided wash buffers. Reverse cross linking took place on a heat block at 65°C for 4 hr. ChIP samples were then treated with 2ul RNase Cocktail (Life Technologies) at 65°C for 30 min followed by 2 μl Proteinase K (Thermo Fisher Scientific) at 65°C for 30 min. Samples were then purified with QIAGEN MiniElute PCR purification kit as per manufacturers’ protocol. In parallel, input samples (chromatin from about 50,000 cells) were reverse crosslinked and DNA was isolated following the same protocol.
Library preparation was carried out using Kapa HTP Illumina library preparation reagents. Briefly, 25 μl of ChIP sample was incubated with 45 μl end repair mix at 20°C for 30 min followed by Ampure XP bead purification. A tailing: bead bound sample was incubated with 50ul buffer enzyme mix for 30°C 30 min, followed by PEG/NaCl purification. Adaptor ligation: bead bound sample was incubated with 45 μl buffer enzyme mix and 5 μl of different TruSeq DNA adapters (Illumina) for each sample, for 20°C 15 min, followed by PEG/NaCl purification (twice). Library enrichment: 12 cycles of PCR amplification. Size selection was performed after PCR using a 0.6x/0.8x ratio of Ampure XP beads (double size selection) set to collect 250–450bp fragments. ChIP libraries were sequenced using Illumina HiSeq 2000, 2500 or 4000 at 50 bp SE reads, and NovaSeq 6000 at 50 bp PE reads.
RNA-seq library preparation and sequencing
Total RNA was extracted from cell pellets using the RNeasy mini kit (Qiagen) according to instructions from the manufacturer. Library preparation was performed with ribosomal RNA (rRNA) depletion according to instructions from the manufacturer (Epicentre) to achieve greater coverage of mRNA and other long non-coding transcripts. Paired-end sequencing was performed on the Illumina HiSeq 2000, 2500 and 4000 platforms.
ATAC-seq library preparation and sequencing
Adherent cell cultures were dissociated and 100 000 cells were washed twice in cold PBS at 4°C. Cells were resuspended in 100 μL Hypotonic Cell Lysis Buffer (0.1% Sodium Citrate Tribasic Dihydrate, 0.1% Triton X-100) and titurated until cells were dissolved. Samples were incubated on ice 30 minutes, centrifuged at 2000 g for 5 min at 4°C, and pellet resuspended in 100 μL Normal Cell Lysis Buffer (10 mM Tris-HCl, pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% IGEPAL CA-630), titurated, incubated on ice 30 min, centrifuged at 2000 g for 5 min at 4°C, and supernatant removed. The transposase reaction was carried out by titurating in 25 μL per sample in TD Buffer (10 mM Tris-HCl, pH 8.00, 5 mM Magnesium Chloride) with 5 μL Transposase (Illumina Nextera Kit), and incubated at 37°C for 30 min, and 8.5 uL of 100 mM EDTA was added, samples transferred to ice, and DNA recovered using MinElute PCR Purification columns (Qiagen). Libraries were generated by PCR in 50 μL reaction (25 μL sample, 10 μL 5x Phusion HF buffer, 0.5 μL Phusion Polymerase, 1 μL 10 mM dNTPs, 0.5 μL of each custom Illumina primers at 12.5 μM). The PCR reaction followed 98°C for 30 s, followed by 12 cycles of 98°C for 10 s, 63°C for 30 s, 72°C for 1 min, followed by 72°C for 5 min. DNA was recovered using GeneRead Purification columns (Qiagen). The libraries were sequenced to 50 million reads per sample on Illumina HiSeq 2500 using Nextera Sequencing Primers.
PDOX generation and treatment
In single cell suspension, 100,000 cells were stereotactically injected in the fourth ventricle/pons of. Tumor cells were allowed to engraft for 7 days. Mice were then randomly assigned to a control group where only the vehicle control was administered using intra-peritoneal (i.p) injections, or to one of the three therapeutic groups: 5-azacytidine (Sigma Aldrich, MO; 3 mg/kg diluted in sterile water) daily i.p. injections for five days followed by a two-day rest period every 4 weeks as previously described (Borodovsky et al., 2013; Yamashita et al., 2018); panobinostat (Selleck Chemicals, 10mg/kg diluted in DMSO) five days a week alternating with 5 days rest for 4 weeks as previously described (Grasso et al., 2015); or a combination of both drugs. For each of the three cell lines used to generate PDOX in mice, nine animals were used for each experimental condition. Mice were then observed until they became moribund, at which point they were sacrificed, and the presence of intracranial tumors was confirmed.
Quantitative PCR for Interferon Signature Genes.
Quantitative PCR (qPCR) was performed to assess for ISGs induction in tumors removed following euthanasia of terminally morbid animals. Total RNA was extracted from tumors derived from H3.3K27M BT245 and DIPG-XIII or pcGBM2 using the miRNeasy mini kit (Qiagen) according to manufacturer’s instructions with purity and integrity assessed utilizing Nanodrop (Thermo-Fisher) and Experion (Biorad) methodologies. 100ng of RNA was used for reverse-transcription using the iScript RT Supermix (BioRad) following manufacturer’s instructions. Real-time PCR for OAS1, STAT1, IL15, IRF7 and SCOS1 was performed on cDNA extracted from 4 biological replicates for each experimental condition (vehicle, 5-azacytidine, panobinostat, combination) using primers described in (Roulois et al., 2015). Samples were run on a Lightcycler 96 (Roche) with the SsoFast Evagreen SuperMix kit (BioRad). Cycling conditions were: 95°C for 30 sec followed by 40 cycles 95°C for 5sec/60 °C for 20 sec. Fold change values were calculated utilizing the 2−ΔΔCt method with ACTB expression and vehicle treated cells used as the calibrator, comparing each treatment group to the vehicle control group.
QUANTIFICATION AND STATISTICAL ANALYSIS
The description of statistical details for each experiment can be found in figure legends. Significance was assigned by tests deriving a p value less than 0.05. The largest sample size of tumors and cell lines that were capable of collection were used and no available data was excluded. For functional experiments, two independent cell lines were modeled by CRISPR-Cas9 editing of H3.3K27M KO, and two independent clones per line were profiled, and followed the same trend in all analyses.
Analysis of ChIP-seq data.
Data processing, peak calling and annotations.
Sequence reads were aligned to the human genome (HG19) using Bowtie2 (v2.1.0) (Langmead and Salzberg, 2012) under default settings. PCR duplicates were removed using PICARD tools generating.BAM files (Li et al., 2009). Read processing and alignment for analysis of repeat elements was performed separately (see below for details). Significant peaks were identified using Model-Based Analysis for ChIP-seq (MACS 1.4) (Zhang et al., 2008)with a p value cutoff of 1e−9. Peaks were annotated using HOMER (v3.12) (Heinz et al., 2010) with promoter regions classified as any peak within +/− 2.5 kb of a transcriptional start site (TSS), and enhancer region greater than 2.5 kb from a TSS. Peaks were also annotated using ChIP-atlas annotating distal enhancers to genes based upon public CHIA-PET datasets. Super enhancers were identified using the ROSE algorithm with exclusion of peaks within +/− 2.5 kb of a TSS and a stitch distance of 12.5 kb. For visualization of H3K27 acetylation profiles, BAM alignment files were normalized to RPKM values using DeepTools (Ramirez et al., 2014) and visualized in Integrated Genome Viewer (v2.3.40).
Clustering and visualization of samples based on H3K27ac.
For unsupervised clustering of H3K27ac patterns, a matrix of the normalized H3K27ac density was generated in DiffBind based upon the consensus H3K27ac peaks identified in at least 3 samples. The top 10,000 most variable loci (defined by the variation in H3K27ac signal across all samples used for clustering using Median Absolute Deviation as a distance metric) were selected as features. Unsupervised hierarchical clustering using spearman correlation of samples was then performed on these 10,000 most variant peaks, using Spearman correlation as a distance metric. t-stochastic neighbor embedding (t-SNE) (van der Maaten and Hinton, 2008) was also used on consensus H3K27ac peaks to visualize samples based on H3K27ac.
Differential H3K27ac enrichment analysis.
Utilizing the read-depth normalized matrix of H3K27ac signal for all consensus H3K27ac peaks, differential H3K27ac loci between H3K27WT and H3K27M tumors was determined using DiffBind, employing the DESeq2 method. Significant regions were further filtered to events with an FDR < 0.05. The same approach was used to detect differential super enhancer loci between H3K27WT and H3K27M samples.
Quantitative ChIP-Rx, sequencing, and analysis.
The spiked-in drosophila chromatin was used for quantification of histone mark, as described previously (Orlando et al., 2014). After sequencing, the reads were aligned to human (hg19) and drosophila (dm3) genomes, then the number of reads mapping to each genome (from H3K27ac and input samples) were used to compute the relative levels of histone mark using the following formula for each sample:
where iphg is the number of reads in the target sample mapping to the human genome; ipdm, to the drosophila genome; inputhg, the number of reads in the input sample mapping to the human genome; and inputhg, to the drosophila genome.
Regulatory networks for all SE associated transcription factors.
We used core regulatory circuitry (CRC) analysis with default parameters to quantify the interaction network of transcription factor (TF) regulation at super enhancers. Briefly, for all promoters within 100 kb, the most acetylated promoter was assigned as the target of the SE (excluding promoters that overlap SEs, which are automatically assigned the target. If there were no active promoters within 100kb, the SE was assigned to the nearest active promoter. All SE-associated promoters annotated to regulate a TF were considered as the node-list for network construction. For any given TF (TFi) The IN degree was defined as the number of TFs with an enriched binding motif at the proximal SE or promoter of TFi. The OUT degree was defined as the number of TF associated SEs containing an enriched binding site for TFi. Within any given SE, enriched TF binding sites were determined at putative nucleosome free regions (valleys) flanked by high levels of H3K27ac. Valleys were calculated using an algorithm adapted from Ramsey et al., 2010 (Ramsey et al., 2010). In these regions, we searched for enriched TF binding sites using the FIMO59 algorithm with TF position weight matrices defined in the TRANSFAC database (Matys et al., 2006). An FDR cutoff of 0.01 was used to identify enriched TF binding sites. Group specific core TFs were defined as TFs identified within CRCs present in at least one sample, and statistically assessed between groups using a Wilcoxon Rank Sum test.
Analysis of RNA-seq data.
Data processing.
Adaptor sequences and the first four nucleotides of each read were removed from the read sets using Trimmomatic (Bolger et al., 2014) (v0.32). Reads were scanned from start to end and truncated if and when the average quality of a 4-nucleotide sliding window fell too low (phred33<30). Short reads (<30 bp) were subsequently discarded. Multiple quality control metrics were obtained using FASTQC (v0.11.2), samtools (Li et al., 2009) (v0.1.19), BEDtools (Quinlan and Hall, 2010) (v2.17.0) and custom scripts.
Gene expression analysis.
The remaining clean set of reads were then aligned to the reference genome build hg19 (GRCh37) with STAR (Dobin et al., 2013) (v2.3.0e) using the default parameters. Only primary alignments of reads mapping to 9 or fewer locations in the genome were retained. Analysis of the repeat genome was performed separately (see below for details). Gene expression levels were estimated by quantifying primary alignments of reads mapping to 2 or fewer locations in the genome (MAPQ>3) falling into exonic regions defined by the ensGene annotation set from Ensembl (GRCh37; N=60234 genes) using featureCounts (Liao et al., 2014) (v1.4.4). Normalization (mean-of-ratios), variance-stabilized transformations of the data, as well as differential expression analysis, were performed using DESeq2 (Love et al., 2014). Unless otherwise stated, all reported p values have been adjusted for multiple testing using the Benjamini-Hochberg procedure.
Clustering and visualization.
Global changes in expression levels were evaluated by hierarchical clustering of samples and principal component analysis (PCA) using normalized expression data coupled with variance-stabilized transformation. For hierarchical clustering, Euclidean distance was used as the distance metric, and complete linkage as the agglomeration method. For visualization, normalized Bigwig tracks were generated using BEDtools (Quinlan and Hall, 2010) and UCSC tools. Integrative Genomic Viewer (Thorvaldsdottir et al., 2013) was used for data visualization.
Analysis of ATAC-Seq data.
The ATAC-seq libraries were sequenced with 125 bp paired-end reads and trimmed for Nextera sequencing adaptors using trimgalore with default settings. The trimmed reads were then mapped to hg19. Reads were filtered to remove duplicates, unmapped or poor quality (phred33<30) reads, mitochondrial reads, and those overlapping the ENCODE blacklist (Carroll et al., 2014). Following alignment, accessible chromatin regions/peaks were called using MACS2. Default parameters were used, except for the following: --keep-dup all -B --nomodel --SPMR -q 0.05 --slocal 6250 -- llocal 6250. The signal intensity was calculated as the fold enrichment of the signal per million reads in a sample over a modelled local background using the bdgcmp function in MACS2 (Zhang et al., 2008). Custom scripts along with the bedtools suite were used to identify chromatin accessibility peaks unique to specific groups of samples and to identify overlaps with H3K27ac peaks. ENCODE’s Genome Structure Correction tool (Bickel et al., 2011) was used to calculate the significance of the change in agreement between the H3K27ac and ATAC-seq signal.
Analysis of repeat elements
Repeat element H3K27ac analysis.
Quantifying reads in repetitive regions of the genome is challenging due to the mapping uncertainty induced by sequence similarity and the high number of occurrences in the genome. To obtain a robust quantification, we used several conservative and complementary strategies, by including or excluding multimapping reads, and using either the repeat genome or the complete human genome as reference. Only results consistent across approaches are reported.
First, we started with the more conservative approach, as previously described (Goke et al., 2015), where ChIP-rx of H3K27ac experiments were aligned to the human genome (hg19) using Bowtie2 (v2.1.0) (Langmead and Salzberg, 2012). We counted the number of reads over each of the repetitive elements annotated in RepeatMasker using HOMER (v3.12), considering each occurrence of a repetitive elements in the genome to be a distinct entity. In the process, we discarded duplicate reads as well as multimapping reads, and retained only primary alignments with the highest alignment scores. This approach underestimates the true signal since only uniquely mapped reads are considered. Finally, ChIP-Rx spike-in scaling factors were used to scale read counts. Repetitive elements were subsequently grouped into families (i.e. SINE, LINE, LTR, DNA) using annotations provided by RepeatMasker (Smit A et al., 2013–2015).
In a second approach, reads were processed as described for RNAseq, and then aligned using the human repeat genome as reference, as previously described (Solovyov et al., Cell Reports 2018). Repbase reference genome was used (Bao et al., 2015) (v23.03) (http://www.girinst.org/repbase). Since Repbase largely consists of family consensus sequences, reads are assigned to these sequences regardless of where and how many times they are present in the genome, mitigating mapping uncertainty. We combined humrep.ref and humsub.ref into a single reference of repeat sequences for the human genome, covering a total of 1132 consensus elements, and counted the number of reads mapping into each of these elements using featureCounts (MAPQ≥3). Differential enrichment of H3K27ac at the consensus sequences were then calculated using DESeq2 (Love et al., 2014). The inverse of ChIP-Rx spike-in scaling factors (i.e. 1/rx) were supplied to DESeq2 as normalization factors (i.e. size factors).
We repeated the two approaches with and without discarding multimapping reads for quantification of repeats. Only results robust to inclusion/exclusion or multimapping reads are reported in this work. Results obtained with uniquely mapped reads (i.e. the most conservative approach) are reported in figures and tables.
Repeat element expression analysis.
The clean set of rRNA-depleted RNA-seq reads were aligned to the human repeat genome (Repbase) using STAR. Normalization (library size) factors derived from canonical genes (Ensembl ensGene annotation) using mean-of-ratios (as described for regular RNA-seq gene expression analysis) were used to normalize the expression of repeat elements. To ensure that the results were robust with respect to the treatment of multimapping reads, we repeated the analysis considering (a) only uniquely mapped reads (MAPQ=255), (b) primary alignments of reads mapping to 2 or fewer locations in the genome (MAPQ≥3), and (c) primary and secondary alignments of reads mapping up to 1000 locations in the repeat genome (MAPQ≥0; STAR with “--outFilterMultimapNmax=1000”). Next, we confirmed the findings using a second aligner, BWA (v.7.15), considering only MAPQ≥3 mapped reads. While all four approaches produced the same results, we report the findings obtained with STAR using approach (b) for the sake of brevity.
Repeat element expression analysis in K27M mouse model.
The rRNA-depleted RNA-seq data was taken from (Pathania et al., 2017) and processed using the same approach as for the human samples. The reference repeat sequences rodrep.ref and rodsub.ref were downloaded from Repbase (Bao et al., 2015) (v23.03) (http://www.girinst.org/repbase) and combined into a single reference for the murine repeat genome. We calculated the proportion of reads mapping to the repeat genome using the following formula:.
Statistical analysis of PDOX studies.
Sample size calculations to detect a 50% difference in tumor growth between two groups with an alpha of 0.05 and power 0.8 indicated a minimum group size of three animals. A log-rank (Mantel-Cox) test was used to compare survival between each pair of treatment groups. A two-way ANOVA was used to compared fold change in expression of ISGs in tumors by qPCR, comparing fold change in pcGBM2 (WT line) to both BT245 and DIPG-XIII (K27M lines).
DATA AND SOFTWARE AVAILABILITY
The raw human data have been deposited in Gene Expression Omnibus (GEO) under ID code GSE128745 and in European Genome-Phenome Archive (EGA) under ID code EGAS00001003572.
Supplementary Material
Table S1. Related to Figure 1. Sample characteristics.
Table S2. Related to Figure 1. Sample sequencing characteristics and quality control.
Table S3. Related to Figure 1. Subgroup-specific genes with concordant changes in both expression and H3K27ac enrichment at promoter or enhancer regions.
Table S4. Related to Figure 2. Differential super enhancers between H3K27M and H3K27WT samples as detected by DiffBind.
Table S5. Related to Figure 3. H3K27ac peaks detected in BT245 and DIPG-XIII in a K27M specific, KO, or shared manner.
Table S6. Related to Figure 3. Pathway enrichment analysis of H3K27ac peaks gained and lost in BT245 and DIPG-XIII relative to KO.
Table S7. Related to Figure 7. Differential expression of H3.3K27M versus K27WT and KO samples.
Table S8. Related to STAR Methods.
Significance.
Elevated levels of H3K27ac are linked to oncogenesis in H3K27M-mutant midline HGGs through altering enhancer landscapes. We find that that increased H3K27ac leads to a therapeutic vulnerability in H3K27M tumors. By removing the confounding effects due to the epigenetic landscape of the cell of origin, we show that increased H3K27ac is pervasively distributed across the genome in H3K27M HGGs. This renders repeat elements susceptible to activation by epigenetic therapies, including DNA demethylating agents. Insight into the induction of endogenous retroviruses and activation of innate immune pathways in response to epigenetic therapies in H3K27M tumors suggests that patients with these intractable tumors may benefit from combining these agents with immune modulating therapies.
Highlights.
H3K27M-mutant gliomas exhibit an enhancer landscape reflecting lineage of origin
Elevated H3K27 acetylation is pervasively distributed across the epigenome
Repeat element expression is de-repressed by H3K27M mutation
H3K27M cells are more vulnerable to DNA demethylation triggering viral mimicry
Acknowledgements
We thank Jacek Majewski for invaluable guidance in experimental design and critical reading of manuscript. Funding: This work was supported by funding from: US National Institutes of Health (NIH grant P01-CA196539 to NJ, PWL, BAG; T32GM008275 and TL1TR001880 to BAG), the Canadian Institutes for Health Research (CIHR grant MOP-286756 and FDN-154307 to NJ, EP1–120608 to TP and PJT-156086 to CLK), the Fonds de Recherche du Québec en Santé (FRQS) salary award to CLK and fellowships to NDJ and ASH. NJ is a member of the Penny Cole Laboratory and the recipient of a Chercheur Boursier, Chaire de Recherche Award from the FRQS. This work was performed within the context of the International CHildhood Astrocytoma INtegrated Genomic and Epigenomic (ICHANGE) consortium with funding from Genome Canada and Genome Quebec. Computational infrastructure was provided by Compute Canada and Calcul Québec. MKM is funded by a CIHR Banting postdoctoral fellowship. PWL is a Pew Scholar in the Biomedical Sciences. We are especially grateful for the generous philanthropic donations of Kat D DIPG, Poppies for Irene and We Love You Connie Foundations. SCM is supported by Cancer Prevention Research Institute of Texas (CPRIT) scholar award (RR170023), Alex’s Lemonade Stand Foundation (ALSF) A award and Young investigator award, RALLY research grant, BEAR Necessities Pediatric Cancer Foundation Grant, Children’s Cancer Research Fund award, Children’s Brain Tumor Foundation Award, and Baylor College of Medicine Junior Faculty Award.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare no competing interests.
References
- Akhtar-Zaidi B, Cowper-Sal-lari R, Corradin O, Saiakhova A, Bartels CF, Balasubramanian D, Myeroff L, Lutterbaugh J, Jarrar A, Kalady MF, et al. (2012). Epigenomic enhancer profiling defines a signature of colon cancer. Science 336, 736–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alves dos Santos MT, and Smidt MP (2011). En1 and Wnt signaling in midbrain dopaminergic neuronal development. Neural development 6, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao W, Kojima KK, and Kohany O (2015). Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile DNA 6, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baylin SB, and Jones PA (2016). Epigenetic Determinants of Cancer. Cold Spring Harb Perspect Biol 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechet D, Gielen GG, Korshunov A, Pfister SM, Rousso C, Faury D, Fiset PO, Benlimane N, Lewis PW, Lu C, et al. (2014). Specific detection of methionine 27 mutation in histone 3 variants (H3K27M) in fixed tissue from high-grade astrocytomas. Acta neuropathologica 128, 733–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belgnaoui SM, Paz S, and Hiscott J (2011). Orchestrating the interferon antiviral response through the mitochondrial antiviral signaling (MAVS) adapter. Curr Opin Immunol 23, 564–572. [DOI] [PubMed] [Google Scholar]
- Bender S, Tang Y, Lindroth AM, Hovestadt V, Jones DT, Kool M, Zapatka M, Northcott PA, Sturm D, Wang W, et al. (2013). Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer cell 24, 660–672. [DOI] [PubMed] [Google Scholar]
- Bestor TH, and Bourc’his D (2004). Transposon silencing and imprint establishment in mammalian germ cells. Cold Spring Harb Symp Quant Biol 69, 381–387. [DOI] [PubMed] [Google Scholar]
- Bickel P, Boley N, Brown J, Huang H, and Zhang N (2011). Subsampling Methods for Genomic Inference, Vol 4). [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics (Oxford, England) 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borodovsky A, Salmasi V, Turcan S, Fabius AW, Baia GS, Eberhart CG, Weingart JD, Gallia GL, Baylin SB, Chan TA, and Riggins GJ (2013). 5-azacytidine reduces methylation, promotes differentiation and induces tumor regression in a patient-derived IDH1 mutant glioma xenograft. Oncotarget 4, 1737–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourc’his D, and Bestor TH (2004). Meiotic catastrophe and retrotransposon reactivation in male germ cells lacking Dnmt3L. Nature 431, 96–99. [DOI] [PubMed] [Google Scholar]
- Carroll TS, Liang Z, Salama R, Stark R, and de Santiago I (2014). Impact of artifact removal on ChIP quality metrics in ChIP-seq and ChIP-exo data. Frontiers in genetics 5, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan KM, Fang D, Gan H, Hashizume R, Yu C, Schroeder M, Gupta N, Mueller S, James CD, Jenkins R, et al. (2013). The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev 27, 985–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, Hein A, Rote NS, Cope LM, Snyder A, et al. (2015). Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 162, 974–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordero FJ, Huang Z, Grenier C, He X, Hu G, McLendon RE, Murphy SK, Hashizume R, and Becher OJ (2017). Histone H3.3K27M Represses p16 to Accelerate Gliomagenesis in a Murine Model of DIPG. Molecular cancer research : MCR 15, 1243–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. (2010). Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A 107, 21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Koning AP, Gu W, Castoe TA, Batzer MA, and Pollock DD (2011). Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet 7, e1002384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics (Oxford, England) 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsasser SJ, Noh KM, Diaz N, Allis CD, and Banaszynski LA (2015). Histone H3.3 is required for endogenous retroviral element silencing in embryonic stem cells. Nature 522, 240–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filbin MG, Tirosh I, Hovestadt V, Shaw ML, Escalante LE, Mathewson ND, Neftel C, Frank N, Pelton K, Hebert CM, et al. (2018). Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science 360, 331–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontebasso AM, Papillon-Cavanagh S, Schwartzentruber J, Nikbakht H, Gerges N, Fiset PO, Bechet D, Faury D, De Jay N, Ramkissoon LA, et al. (2014). Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nature genetics 46, 462–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funato K, Major T, Lewis PW, Allis CD, and Tabar V (2014). Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science 346, 1529–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goke J, Lu X, Chan YS, Ng HH, Ly LH, Sachs F, and Szczerbinska I (2015). Dynamic transcription of distinct classes of endogenous retroviral elements marks specific populations of early human embryonic cells. Cell stem cell 16, 135–141. [DOI] [PubMed] [Google Scholar]
- Grasso CS, Tang Y, Truffaux N, Berlow NE, Liu L, Debily MA, Quist MJ, Davis LE, Huang EC, Woo PJ, et al. (2015). Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nature medicine 21, 555–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Molecular cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PA, Ohtani H, Chakravarthy A, and De Carvalho DD (2019). Epigenetic therapy in immune-oncology. Nature reviews Cancer. [DOI] [PubMed] [Google Scholar]
- Kang P, Lee HK, Glasgow SM, Finley M, Donti T, Gaber ZB, Graham BH, Foster AE, Novitch BG, Gronostajski RM, and Deneen B (2012). Sox9 and NFIA coordinate a transcriptional regulatory cascade during the initiation of gliogenesis. Neuron 74, 79–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khuong-Quang DA, Buczkowicz P, Rakopoulos P, Liu XY, Fontebasso AM, Bouffet E, Bartels U, Albrecht S, Schwartzentruber J, Letourneau L, et al. (2012). K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta neuropathologica 124, 439–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nature methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W, Teckie S, Wiesner T, Ran L, Prieto Granada CN, Lin M, Zhu S, Cao Z, Liang Y, Sboner A, et al. (2014). PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nature genetics 46, 1227–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis PW, Muller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, Garcia BA, Muir TW, Becher OJ, and Allis CD (2013). Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 340, 857–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics (Oxford, England) 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics (Oxford, England) 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Lin CY, Erkek S, Tong Y, Yin L, Federation AJ, Zapatka M, Haldipur P, Kawauchi D, Risch T, Warnatz HJ, et al. (2016). Active medulloblastoma enhancers reveal subgroup-specific cellular origins. Nature 530, 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, and Young RA (2013). Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153, 320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machold R, Klein C, and Fishell G (2011). Genes expressed in Atoh1 neuronal lineages arising from the r1/isthmus rhombic lip. Gene expression patterns : GEP 11, 349–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack SC, Pajtler KW, Chavez L, Okonechnikov K, Bertrand KC, Wang X, Erkek S, Federation A, Song A, Lee C, et al. (2018). Therapeutic targeting of ependymoma as informed by oncogenic enhancer profiling. Nature 553, 101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matys V, Kel-Margoulis OV, Fricke E, Liebich I, Land S, Barre-Dirrie A, Reuter I, Chekmenev D, Krull M, Hornischer K, et al. (2006). TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic acids research 34, D108–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammad F, Weissmann S, Leblanc B, Pandey DP, Hojfeldt JW, Comet I, Zheng C, Johansen JV, Rapin N, Porse BT, et al. (2017). EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nature medicine 23, 483–492. [DOI] [PubMed] [Google Scholar]
- Orlando DA, Chen MW, Brown VE, Solanki S, Choi YJ, Olson ER, Fritz CC, Bradner JE, and Guenther MG (2014). Quantitative ChIP-Seq normalization reveals global modulation of the epigenome. Cell Rep 9, 1163–1170. [DOI] [PubMed] [Google Scholar]
- Pathania M, De Jay N, Maestro N, Harutyunyan AS, Nitarska J, Pahlavan P, Henderson S, Mikael LG, Richard-Londt A, Zhang Y, et al. (2017). H3.3(K27M) Cooperates with Trp53 Loss and PDGFRA Gain in Mouse Embryonic Neural Progenitor Cells to Induce Invasive High-Grade Gliomas. Cancer cell 32, 684–700 e689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piunti A, Hashizume R, Morgan MA, Bartom ET, Horbinski CM, Marshall SA, Rendleman EJ, Ma Q, Takahashi YH, Woodfin AR, et al. (2017). Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. Nature medicine 23, 493–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics (Oxford, England) 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F, Dundar F, Diehl S, Gruning BA, and Manke T (2014). deepTools: a flexible platform for exploring deep-sequencing data. Nucleic acids research 42, W187–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey SA, Knijnenburg TA, Kennedy KA, Zak DE, Gilchrist M, Gold ES, Johnson CD, Lampano AE, Litvak V, Navarro G, et al. (2010). Genome-wide histone acetylation data improve prediction of mammalian transcription factor binding sites. Bioinformatics (Oxford, England) 26, 2071–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nature protocols 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roulois D, Loo Yau H, Singhania R, Wang Y, Danesh A, Shen SY, Han H, Liang G, Jones PA, Pugh TJ, et al. (2015). DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell 162, 961–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saint-Andre V, Federation AJ, Lin CY, Abraham BJ, Reddy J, Lee TI, Bradner JE, and Young RA (2016). Models of human core transcriptional regulatory circuitries. Genome research 26, 385–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlee M, Schuhmacher M, Holzel M, Laux G, and Bornkamm GW (2007). c-MYC impairs immunogenicity of human B cells. Adv Cancer Res 97, 167–188. [DOI] [PubMed] [Google Scholar]
- Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DA, Tonjes M, et al. (2012). Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482, 226–231. [DOI] [PubMed] [Google Scholar]
- Sidoli S, Bhanu NV, Karch KR, Wang X, and Garcia BA (2016). Complete Workflow for Analysis of Histone Post-translational Modifications Using Bottom-up Mass Spectrometry: From Histone Extraction to Data Analysis. J Vis Exp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit A, Hubley R, and P. G (2013–2015). RepeatMasker Open-4.0 http://www.repeatmasker.org. [Google Scholar]
- Solovyov A, Vabret N, Arora KS, Snyder A, Funt SA, Bajorin DF, Rosenberg JE, Bhardwaj N, Ting DT, and Greenbaum BD (2018). Global Cancer Transcriptome Quantifies Repeat Element Polarization between Immunotherapy Responsive and T Cell Suppressive Classes. Cell Rep 23, 512–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturm D, Bender S, Jones DT, Lichter P, Grill J, Becher O, Hawkins C, Majewski J, Jones C, Costello JF, et al. (2014). Paediatric and adult glioblastoma: multiform (epi)genomic culprits emerge. Nature reviews Cancer 14, 92–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdottir H, Robinson JT, and Mesirov JP (2013). Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Briefings in bioinformatics 14, 178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topper MJ, Vaz M, Chiappinelli KB, DeStefano Shields CE, Niknafs N, Yen RC, Wenzel A, Hicks J, Ballew M, Stone M, et al. (2017). Epigenetic Therapy Ties MYC Depletion to Reversing Immune Evasion and Treating Lung Cancer. Cell 171, 1284–1300 e1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Maaten L, and Hinton G (2008). Visualizing Data using t-SNE. J Mach Learn Res 9, 2579–2605. [Google Scholar]
- Walsh CP, Chaillet JR, and Bestor TH (1998). Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nature genetics 20, 116–117. [DOI] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, and Young RA (2013). Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, Qu C, Ding L, Huether R, Parker M, et al. (2012). Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nature genetics 44, 251–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita AS, da Costa Rosa M, Borodovsky A, Festuccia WT, Chan T, and Riggins GJ (2018). Demethylation and epigenetic modification with 5-Azacytidine reduces IDH1 mutant glioma growth in combination with Temozolomide. Neuro Oncol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome biology 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Krishna S, Garcia C, Lin CJ, Mitchell BD, Scott KL, Mohila CA, Creighton CJ, Yoo SH, Lee HK, and Deneen B (2017). Daam2 driven degradation of VHL promotes gliomagenesis. eLife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Related to Figure 1. Sample characteristics.
Table S2. Related to Figure 1. Sample sequencing characteristics and quality control.
Table S3. Related to Figure 1. Subgroup-specific genes with concordant changes in both expression and H3K27ac enrichment at promoter or enhancer regions.
Table S4. Related to Figure 2. Differential super enhancers between H3K27M and H3K27WT samples as detected by DiffBind.
Table S5. Related to Figure 3. H3K27ac peaks detected in BT245 and DIPG-XIII in a K27M specific, KO, or shared manner.
Table S6. Related to Figure 3. Pathway enrichment analysis of H3K27ac peaks gained and lost in BT245 and DIPG-XIII relative to KO.
Table S7. Related to Figure 7. Differential expression of H3.3K27M versus K27WT and KO samples.
Table S8. Related to STAR Methods.
Data Availability Statement
The raw human data have been deposited in Gene Expression Omnibus (GEO) under ID code GSE128745 and in European Genome-Phenome Archive (EGA) under ID code EGAS00001003572.