

Abstract
Acute intermittent porphyria (AIP) is an inborn error of heme biosynthesis due to the deficiency of hydroxymethylbilane synthase (HMBS) activity. Human AIP heterozygotes have episodic acute neurovisceral attacks that typically start after puberty, whereas patients with homozygous dominant AIP (HD-AIP) have early-onset chronic neurological impairment, including ataxia and psychomotor retardation. To investigate the dramatically different manifestations, knock-in mice with human HD-AIP missense mutations c.500G>A (p.Arg167Glu) or c.518_519GC>AG (p.Arg173Glu), designated R167Q or R173Q mice, respectively, were generated and compared with the previously established T1/T2 mice with ~30% residual HMBS activity and the heterozygous AIP phenotype. Homozygous R173Q mice were embryonic lethal, while R167Q homozygous mice (R167Q+/+) had ~5% of normal HMBS activity, constitutively elevated plasma and urinary 5-aminolevulinic acid (ALA) and porphobilinogen (PBG), profound early-onset ataxia, delayed motor development and markedly impaired rotarod performance. Central nervous system (CNS) histology was grossly intact, but CNS myelination was delayed and overall myelin volume was decreased. Heme concentrations in liver and brain were similar to those of T1/T2 mice. Notably, ALA and PBG concentrations in the cerebral spinal fluid and CNS regions were markedly elevated in R167Q+/+ mice compared with T1/T2 mice. When the T1/T2 mice were administered phenobarbital, ALA and PBG markedly accumulated in their liver and plasma, but not in the CNS, indicating that ALA and PBG do not readily cross the blood–brain barrier. Taken together, these studies suggest that the severe HD-AIP neurological phenotype results from decreased myelination and the accumulation of locally produced neurotoxic porphyrin precursors within the CNS.
Introduction
Acute intermittent porphyria (AIP) is an autosomal dominant disorder that is incompletely penetrant, resulting from half-normal activity of hydroxymethylbilane synthase (HMBS), the third enzyme in the heme biosynthetic pathway (1,2). Symptomatic AIP heterozygotes experience episodic life-threatening acute neurovisceral attacks that are characterized by autonomic, peripheral and central nervous system (CNS) symptoms, including excruciating abdominal pain, vomiting, hypertension, tachycardia, motor weakness, paralysis and transient psychosis. These attacks typically begin after puberty and are precipitated by factors such as fasting, certain porphyrinogenic drugs (particularly cytochrome P450-inducing drugs) and hormonal fluctuations, which induce the hepatic expression of the first and rate-limiting enzyme of the heme biosynthetic pathway, 5-aminolevulinic acid synthase 1 (ALAS1) (2,3). When hepatic ALAS1 is induced, the half-normal HMBS activity becomes rate limiting, leading to insufficient heme production and depletion of the hepatic ‘free’ heme pool. This results in derepression and further upregulation of hepatic ALAS1 expression, followed by the marked accumulation of the porphyrin precursors, 5-aminolevulinic acid (ALA) and porphobilinogen (PBG). Recent studies strongly support that the porphyrin precursors, ALA in particular, are neurotoxic and are the main mediators of the acute neurovisceral symptoms, rather than heme deficiency in the nervous tissues (4,5).
Homozygous dominant AIP (HD-AIP) is a rare condition due to biallelic HMBS loss-of-function mutations (5–9). In contrast to heterozygous AIP patients, HD-AIP patients have <4% residual HMBS activity and constitutively elevated levels of urinary ALA and PBG (5–9). Clinically, HD-AIP patients are characterized by infantile onset of severe and chronic neurological impairment, including early-onset ataxia, psychomotor retardation and nystagmus. They fail to thrive and typically die in early childhood.
Previously, a mouse model for AIP (designated T1/T2) that has ~30% of wild-type HMBS activity and slightly elevated baseline plasma and urinary ALA and PBG concentrations was gene-rated by homologous recombination (10). When the prototypic porphyrinogenic drug phenobarbital is administered, their hepatic ALAS1 activity is induced and plasma and urinary ALA and PBG concentrations are markedly elevated, thereby recapitulating the biochemical abnormality of human heterozygous AIP. Notably, the T1/T2 mice do not display overt neurologic manifestations during an induced attack like human AIP patients (10). Rather, T1/T2 mice develop late-onset neuromotor impairment, including a mild ataxia and poor motor coordination, regardless of whether they have been induced or not. (11–13). A progressive peripheral axonal degeneration that resembles the pattern seen in some human heterozygous AIP patients with severe or prolonged attacks becomes apparent by 6 months of age (11,14–18).
To investigate the pathogenic basis of the striking differences in the clinical presentation of human heterozygous AIP and HD-AIP, we generated knock-in mice that mimic the severe HD-AIP phenotype. The human HD-AIP mutations, c.500G>A (p.Arg167Glu) and c.518_519GC>AG (p.Arg173Glu), designated R167Q and R173Q, respectively (6,8), were introduced by homologous recombination. Mice homozygous for the R173Q mutation (R173Q+/+) were embryonic lethal, and mice heterozygous for both R167Q and R173Q mutations (R167+/−/R173+/−) all died before postnatal day (P)30. Homozygous R167Q mice (R167Q+/+) had a marked neurological phenotype closely resembling that of HD-AIP children, thereby facilitating investigation of disease pathogenesis. Here we describe the generation and clinical, biochemical and histopathologic characteristics of these mice. In particular, ALA, PBG and heme levels in CNS tissues were determined and compared with those in T1/T2 mice.
Results
R167Q+/+ mice are viable while R173Q+/+ mice are embryonic lethal
To select the HMBS mutations to be ‘knocked-in’ by homologous recombination, the human HD-AIP mutations, c.500G>A (p.Arg167Glu), c.499C>T (p.Arg167Trp) and c. 518_519GC > AG (p.Arg173Glu), were individually introduced into the murine Hmbs cDNA and expressed in Escherichia coli. These mutations were selected based on the fact that of the five reported HD-AIP patients, four had these mutations: one was homozygous for R167W (5), two were siblings and heterozygous for R167W and R167Q (8) and one was heterozygous for R167W and R173Q (6). The R167Q, R167W and R173Q mutant proteins had ~8%, 6.5% and 1% of wild-type activity, respectively (Supplementary Material, Table S1). The mutations with the highest (R167Q) and lowest (R173Q) HMBS activities were selected for construction of the knock-in targeting constructs (Fig. 1A), which were simultaneously electroporated into ES cells. PCR screening of ~200 G418-positive ES cell clones identified 42 (21.8%) targeted clones (data not shown), and selected clones were subsequently confirmed by southern blot analyses (Fig. 1B).
Figure 1.
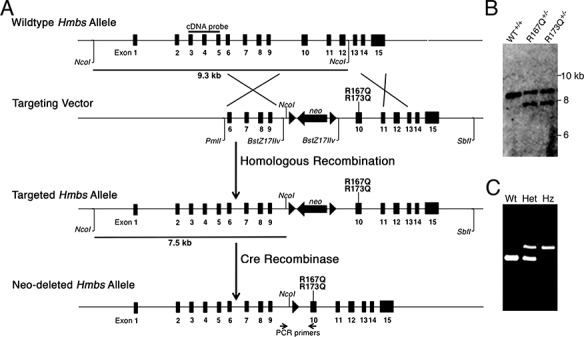
R167Q and R173Q knock-in mice were generated via gene targeting. (A) Schematic of gene targeting strategy to ‘knock-in’ human HD-AIP mutations c.500G>A (R167Q) and c.518_519GC>AG (R173Q) into the murine Hmbs locus. A targeting vector containing short (3.1 kb) and long (5.2 kb) arms of homology and a LoxP-flanked Neomycin (neo) cassette for positive selection was generated, and the R167Q and R173Q mutations were individually introduced into exon 10 by site-directed mutagenesis. Following homologous recombination, correctly targeted ES clones were identified by PCR and confirmed by southern blot analyses. Heterozygous founder mice for each mutation were identified and crossed with transgenic mice ubiquitously expressing Cre recombinase under the regulation of the EIIa-promoter to excise the LoxP-flanked Neomycin cassette. (B) Representative image of southern blot analysis following NcoI digestion. Using an ‘external’ cDNA probe spanning exons 3 to 5, a 9.3 kb band is detected for the wild-type allele, while a 7.5 kb band is detected for the targeted allele, due to an additional NcoI site provided by the neo cassette. (C) Genotyping of offspring was performed using a PCR primer pair in intron 9 and exon 10 that amplifies a product that is 105 base pairs (bp) larger in the targeted allele (521 bp) than in widltype allele (416 bp), due to the residual LoxP site. WT, wild-type; Het, heterozygous; Hz, homozygous.
Heterozygous founder mice for each mutation were identified and crossed with transgenic mice ubiquitously expressing Cre recombinase under the regulation of the EIIa-promoter to excise the LoxP-flanked Neomycin cassette. Genotyping was performed using PCR primers that amplified a product that was 105 base pairs (bp) larger in the targeted allele than in wild-type allele, due to the residual LoxP site and flanking sequence that was inserted as part of the neomycin cassette (Fig. 1C). Neomycin-deleted heterozygous R167Q or R173Q mice were then bred to homozygosity. Segregation analyses showed that the R173Q+/+ mice were embryonic lethal, while the R167Q+/+ mice were born at near expected Mendelian ratios (22%; Supplementary Material, Table S2), but only ~65% survived to adulthood. Notably, ~90% of the R167Q+/+ lethality occurred during P20–P30. Although compound heterozygous mice (R167Q+/−/R173Q+/−) had ~3% of mean wild-type hepatic HMBS activity and were also born at near expected ratios (23%; Supplementary Material, Table S2), they were severely runted, cachectic in appearance, and all died by P30. Thus, the R167Q+/+ mice were used for the following studies.
R167Q+/+ mice display clinical features of human HD-AIP
The R167Q+/+ mice were smaller than their wild-type littermates from infancy, and the gap in body weights widened with age (Fig. 2A). In addition to having shorter body lengths, the mice remained notably lean throughout life (Fig. 2B).
Figure 2.
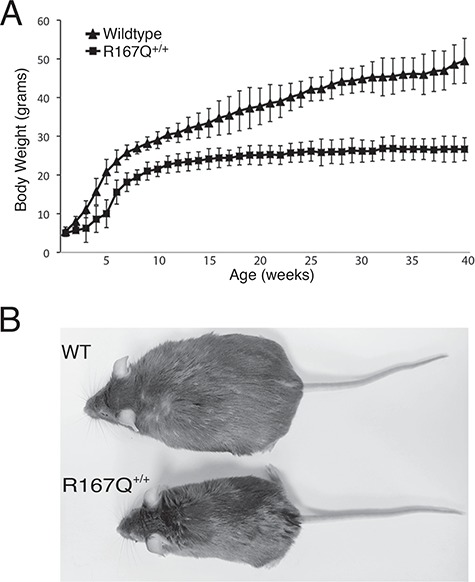
R167Q+/+ mice are runted. (A) Time course of body weight changes in R167Q+/+ male mice and sex-matched wild-type littermates. Data shown are means ± SDs (n = 7). P < 0.005 at all time points 2 weeks and beyond, Student’s t-test. (B) Representative image of a 1-year-old R167Q+/+ mouse (bottom) and wild-type littermate (top) shows that the R167Q+/+ mouse has a short body length and is also notably lean in comparison. WT, wild-type.
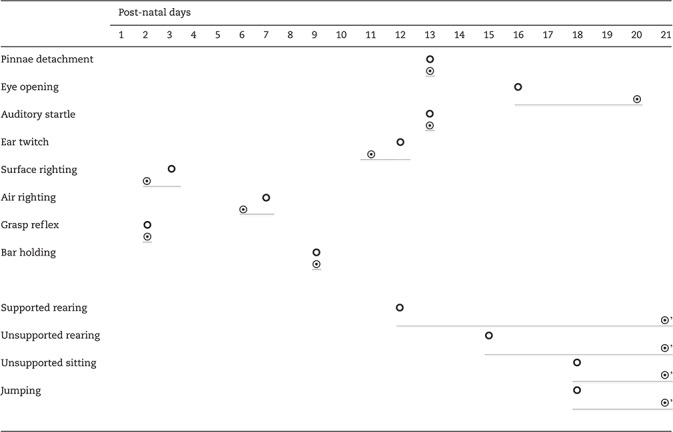
Eye opening, considered to be a measure of early neuro-development, was significantly delayed in the R167Q+/+ mice (Table 1). While the R167Q+/+ mice achieved sensory development (ear twitch and auditory startle) and acquisition of early reflexes (grasping, surface righting and air righting) at ages similar to their littermate controls (Table 1), they had markedly delayed and impaired performance for tasks requiring postural skills and complex motor coordination. By P7, the R167Q+/+ mice had jerky, ataxic gait and intension tremors. Extinguishing of pivoting was delayed, and they often propelled themselves using only their forelimbs, with their bellies on the ground. Their abilities to rear with or without support, sit without support and jump were delayed as well (Table 1). A 3 min open field test on days 9, 12, 15, 18 and 21 showed a significantly reduced number of boxes traveled compared with controls (Supplementary Material, Table S3). Testing was terminated prematurely in over 50% and 60% of wild-type mice at P18 and P21, respectively, due to the mice jumping out of the testing box. In contrast, none of the R167Q+/+ mice were able to jump out of the box. When hung by their tails, the R167Q+/+ mice displayed dystonic postures of their hind legs with self-clasping. Although the ataxia became less apparent and the mice were able to walk relatively normally after P30, rotarod performance was significantly impaired in 2-month-old R167Q+/+ mice and progressively worsened with age (Fig. 3).
Table 1.
Developmental reflexes and behavior studies in R167Q+/+ mice and wild-type littermates
|
 , wild-type mice;
, wild-type mice;  , R167Q+/+ mice;
, R167Q+/+ mice;
 , behavior not observed in more than half of the mice by P21. Data shown are means (n = 20).
, behavior not observed in more than half of the mice by P21. Data shown are means (n = 20).
Figure 3.
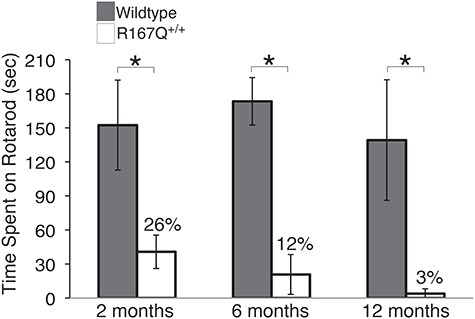
R167Q+/+ mice have progressive impairment of neuromotor function. Rotarod performance was assessed in R167Q+/+ and wild-type male mice at 2, 6, and 12 months, by placing them on a rotarod apparatus rotating at 16 rpm for a maximal time of 180 s. Data represent means ± SDs (n ≥ 6). *P < 5E-07, Student’s t-test.
Thus, the R167Q+/+ mice had clinical features that were strikingly similar to those observed in the HD-AIP patients, including developmental delay of motor skills, ataxia, dystonic postures and short stature (Table 2). The R167Q+/+ mice that survived beyond P30 were able to successfully mate with one another and had a normal lifespan.
Table 2.
Clinical and biochemical features of human HD-AIP and R167Q+/+ mice
| Human HD-AIP features | R167Q+/+ mice |
|---|---|
| Early onset of neurological impairment | + |
| Tremor | + |
| Ataxia | + |
| Dystonic posture | + |
| Early demisea | + |
| Short stature | + |
| Constitutively elevated urinary ALA and PBG | + |
| Porphyrinuria | + |
| Decreased CNS myelination | + |
+, Present in the R167Q+/+ mice.
aApproximately 35% of R167Q+/+ mice die before P30, but those that survive have a normal life span.
R167Q+/+ mice have biochemical features of human HD-AIP, including markedly decreased HMBS activity and constitutively elevated plasma and urinary ALA and PBG
HMBS activities were determined in erythrocytes and in various tissues, including liver, kidney, heart and brain of R167Q+/+ and wild-type mice (Table 3). The R167Q+/+ mice had ~5%–8% of wild-type HMBS activities in erythrocytes and all tissues examined, consistent with the results of the in vitro expression studies (Supplementary Material, Table S1). Despite the severe HMBS deficiency, the R167Q+/+ mice had normal complete blood count and were not anemic (data not shown). Human HD-AIP patients have been reported to be hematologically normal or to have a mild anemia (5,7).
Table 3.
Tissue and erythrocyte HMBS activities in R167Q+/+ and wild-type mice
| Tissue | HMBS activity (nmol/hr/mg protein) | ||
|---|---|---|---|
| Wild type | R167Q+/+ | Wild type, % | |
| Liver | 273 ± 47 | 13.5 ± 2.4 | 4.9 |
| Kidney | 216 ± 23 | 17.1 ± 9.8 | 7.9 |
| Heart | 222 ± 14 | 11.8 ± 2.5 | 5.3 |
| Brain | 90 ± 19 | 4.3 ± 0.4 | 4.8 |
| Erythrocytes | 509 ± 93 | 29.5 ± 2.8 | 5.8 |
HMBS activities are presented as means ± SDs (n = 3–5) and percentages of wild-type activity.
Plasma ALA and PBG were elevated approximately 7-fold and more than 134-fold, respectively, while urinary ALA and PBG were elevated approximately 5- and 60-fold, respectively, compared with mean wild-type levels (Table 4). Baseline urinary uroporphyrin I and III were markedly elevated by 15- and ~50-fold, respectively, while the coproporphyrin I and III isomers were only slightly elevated or normal (Table 4). Thus, the R167Q+/+ mice had constitutively elevated levels of plasma and urinary ALA and PBG and porphyrinuria without induction by a porphyrinogenic agent, similar to those of HD-AIP children (Table 2) (5–8).
Table 4.
Porphyrin precursor and porphyrins in R167Q+/+ and T1/T2 mice
| Body Fluid | Porphyrin Precursor or Porphyrin |
Wild type
c
(Sv129/C57Bl6) |
R167Q
+/+
(Sv129/C57Bl6) |
Fold-change over Wild type |
Wild type
d
(C57Bl6) |
T1/T2
(C57Bl6) |
Fold change over Wild type |
|---|---|---|---|---|---|---|---|
| Plasmaa | ALA | 0.14 ± 0.05 | 0.98 ± 0.32 | 7 | 0.22 ± 0.03 | 0.53 ± 0.19 | 2.4 |
| PBG | UDL | 6.69 ± 2.76 | >134 | 0.08 ± 0.01 | 0.48 ± 0.21 | 6 | |
| Urineb | ALA | 11.53 ± 6.38 | 52.18 ± 28.21 | 4.5 | 14.8 ± 2.87 | 23.84 ± 6.57 | 1.6 |
| PBG | 2.10 ± 1.25 | 127 ± 21.47 | 60 | 0.91 ± 0.22 | 6.10 ± 1.52 | 6.7 | |
| URO I | 0.82 ± 0.83 | 12.15 ± 7.31 | 15 | 0.70 ± 0.32 | 10.99 ± 4.24 | 16 | |
| URO III | 0.32 ± 0.41 | 15.76 ± 11.47 | 49 | 0.18 ± 0.03 | 4.04 ± 1.97 | 22 | |
| 7-COOH I + III | 0.37 ± 0.21 | 1.09 ± 0.61 | 2.9 | 0.22 ± 0.06 | 0.93 ± 0.46 | 4.2 | |
| 6-COOH I + III | 1.18 ± 0.82 | 1.75 ± 0.68 | 1.5 | 0.92 ± 0.29 | 0.83 ± 1.21 | 0.9 | |
| 5-COOH I + III | 1.28 ± 0.90 | 1.91 ± 0.74 | 1.5 | 1.00 ± 0.31 | 2.01 ± 0.66 | 2.0 | |
| COPRO I | 0.86 ± 0.69 | 1.20 ± 0.32 | 1.4 | 0.59 ± 0.18 | 2.40 ± 1.05 | 4.1 | |
| COPRO III | 5.47 ± 3.92 | 6.07 ± 4.21 | 1.1 | 5.66 ± 1.83 | 8.97 ± 4.09 | 1.6 |
UDL, under detection limit of 0.05 μmol/L; URO, uroporphyrin; 7-COOH, monodecarboxylated URO; 6-COOH, didecarboxylated URO; 5-COOH, tridecarboxylated URO; COPRO, coproporphyrin.
aPlasma ALA and PBG are expressed in μmol/L.
bUrinary ALA and PBG are expressed in mmol/mol creatinine, while porphyrins are expressed in μmol/mol creatinine.
c,dR167Q+/+ and T1/T2 controls are Sv129/C57Bl6 and C57Bl6 wild-type mice, respectively.
Data shown are means ± SDs (n ≥ 4).
For comparison, the previously described T1/T2 mice with ~30% residual HMB-synthase activity had plasma and urinary ALA concentrations that were only increased by ~2-fold and a 6- to 7-fold increase in plasma and urinary PBG concentrations at baseline (Table 4). Urinary uroporphyrin I and III were increased by 16- and 22-fold, and coproporphyrin I and III were elevated by approximately 4- and 2-fold, respectively, compared with wild-type controls.
R167Q+/+ mice have normal hepatic Alas1 mRNA levels and are not readily inducible by porphyrinogenic stimuli
Baseline hepatic Alas1 mRNA levels in the R167Q+/+ mice were comparable to those of controls (Supplementary Material, Fig. S1). Hepatic Alas1 mRNA expression is known to be circadian regulated (19). Alas1 mRNA was relatively low at the beginning of the light cycle and increased toward the end of the light cycle, similar to controls, indicating that the circadian regulation of hepatic Alas1 mRNA was maintained in the R167Q+/+ mice (Supplementary Material, Fig. S1).
To evaluate whether hepatic Alas1 mRNA and the constitutively elevated plasma porphyrin precursor levels are further inducible, the R167Q+/+ mice were subjected to various porphyrinogenic stimuli, including fasting (48 h), pregnenolone-16α-carbonitrile (PCN), phenobarbital (PB) and a combination of PB and 3,5-diethoxycarbonyl-1,4-dihydro-2,4,6-trimethylpyridine (DDC). These stimuli were specifically selected, as they are known to induce hepatic ALAS1 expression through different transcription factors and/or nuclear receptors: fasting up-regulates peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1α) (20), while phenobarbital and PCN activate the constitutive androstane receptors and nuclear pregnane X receptors, respectively (21,22). DDC is an inhibitor of ferrochelatase (23), the last enzyme in the heme biosynthetic pathway, and therefore leads to hepatic heme depletion and marked upregulation of Alas1 when administered in combination with PB.
While fasting and PCN administration increased hepatic Alas1 mRNA expression by 5.3- and 5.5-fold, PB and PB/DDC administration only elevated hepatic Alas1 mRNA by 1.4- and 1.7-fold, respectively, in the R167Q+/+ mice (Fig. 4A). Plasma ALA and PBG remained at near baseline levels following all treatments, except for PB/DDC, which resulted in slight (approximately 2-fold) increases of ALA and PBG (Fig. 4B).
Figure 4.
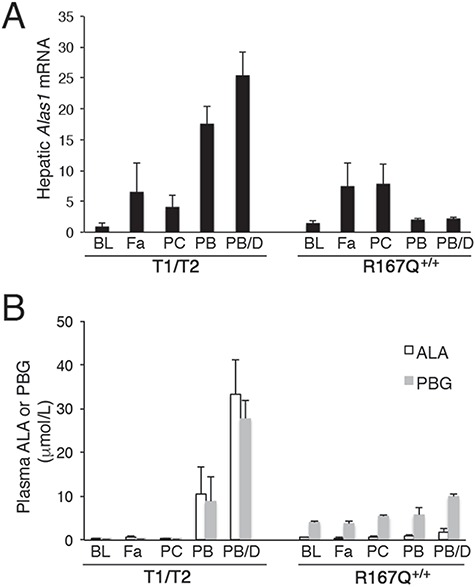
R167Q+/+ mice are not readily inducible by porphyrinognic factors. R167Q+/+ and T1/T2 male mice were subjected to various porphyrinogenic stimuli, including fasting (Fa), pregnenolone-16α-carbonitrile (PCN; PC), phenobarbital (PB) and a combination of PB and 3,5-diethoxycarbonyl-1,4-dihydro-2,4,6-trimethylpyridine (DDC) (PB/D), and (A) relative hepatic Alas1 mRNA and (B) plasma ALA and PBG concentrations were assessed. Baseline levels (BL) are shown for comparison. Data shown are means ± SDs (n ≥ 3).
The responses of the T1/T2 mice to fasting and PCN were similar to those of the R167Q+/+ mice, with hepatic Alas1 mRNA increased by approximately 6.5- and 4-fold, respectively, and no increases in plasma ALA and PBG levels (Fig. 4A and B). However, their responses to PB and PB/DDC differed dramatically from those of the R167Q+/+ mice, with hepatic Alas1 mRNA elevated by approximately 18- and 26-fold, ALA by 30 and 95-fold, and PBG by 45 and 150-fold (Fig. 4A and B), consistent with previous studies (24). Thus, unlike the T1/T2 mice, a biochemical acute attack was not induced by porphyrinogenic stimuli, including the potent and prototypic drug, phenobarbital, in the R167Q+/+ mice.
Of interest, relative basal Alas1 mRNA levels in the brains of the R167Q+/+ mice were elevated by approximately 1.9-fold compared with controls [1.87 ± 0.16 versus 1 ± 0.03 (mean ± SD; n = 4)] but were not further increased following treatment with PB/DDC (1.89 ± 0.1). Brains of T1/T2 mice had essentially wild-type levels of basal Alas1 mRNA (1.21 ± 0.14 versus 1 ± 0.25) and also were not increased following PB/DDC treatment (1 ± 0.1).
R167Q+/+ mice have marked accumulation of ALA and PBG in various tissues, including the CNS
In the liver, kidney, heart and spleen, the R167Q+/+ mice had ALA and PBG concentrations that were elevated by 1.5- to 2.5-fold and 73- to 390-fold over wild-type levels, respectively (Table 5). Erythrocyte porphyrin precursors were also elevated, with ALA and PBG increased by 1.5 and 38-fold compared with wild-type levels, respectively (Table 5).
Table 5.
ALA and PBG concentrations in various non-CNS and CNS tissues
| Tissue |
Wild type
c
(Sv129/C57Bl6) ALA, PBG |
R167Q
+/+
(Sv129/C57Bl6) ALA, PBG |
Fold change over wild type |
Wild type
d
(C57Bl6) ALA, PBG |
T1/T2
(C57Bl6) ALA, PBG |
Fold change over wild type |
|---|---|---|---|---|---|---|
| Non-CNS tissues and cells | ||||||
| Liver | 10.1 ± 2.6, 2.4 ± 0.9 |
19.0 ± 2.9, 934 ± 225 |
1.9 390 |
6 ± 2.5, UDL |
7.1 ± 0.6, 2.6 ± 0.6 |
1.2 N/A |
| Kidney | 22.6 ± 12.5, 8.0 ± 4.5 |
38.3 ± 4.6, 582 ± 245 |
1.7 73 |
ND, ND |
ND, ND |
N/A N/A |
| Heart | 34.6 ± 10.0, UDL |
87.6 ± 13.4, 1,006 ± 20.8 |
2.5 N/A |
ND, ND |
ND, ND |
N/A N/A |
| Spleen | 36.2 ± 12.3, 3.4 ± 0.5 |
53.8 ± 5.3, 376 ± 176 |
1.5 110 |
ND, ND |
ND, ND |
N/A N/A |
| Erythrocytes | 1.8 ± 0.6, 1.2 ± 0.2 |
2.7 ± 0.8, 45.3 ± 11.9 |
1.5 38 |
ND, ND |
ND, ND |
N/A N/A |
| CNS tissues and fluids | ||||||
| Brain (whole) | 6.2 ± 5.6 UDL |
33.7 ± 4.4, 886 ± 118 |
5.4 N/A |
5.6 ± 1.8, UDL |
6.4 ± 1.7, 23.6 ± 5.1 |
1.1 N/A |
| Cerebrum | 6.5 ± 2.5 UDL |
29.1 ± 10.7, 856 ± 403 |
4.5 N/A |
ND, ND |
ND, ND |
N/A N/A |
| Cerebellum | 7.9 ± 4.0 UDL |
30.3 ± 6.8, 687 ± 96.6 |
3.8 N/A |
ND, ND |
ND ND |
N/A N/A |
| Upper brain stema | 10.9 ± 5.7 UDL |
43.2 ± 19.2, 1,162 ± 211 |
4 N/A |
ND, ND |
ND, ND |
N/A N/A |
| Lower brain stemb | 16.5 ± 10.5 UDL |
35.7 ± 7.9, 1,124 ± 32 |
2.2 N/A |
ND, ND |
ND, ND |
N/A N/A |
| Spinal cord | 7.3 ± 0.3, UDL |
68 ± 5.4, 1,759 ± 391 |
9.3 N/A |
9.8 ± 0.3, UDL |
25.6 ± 4.8, 306 ± 57 |
2.6 N/A |
| CSF | 0.07 ± 0.01, 0.12 ± 0.07 |
0.15 ± 0.04, 8.9 ± 5.1 |
2.1 74 |
0.07 ± 0.01 0.12 ± 0.07 |
0.09 ± 0.01, 1.73 ± 0.25 |
1.3 14 |
UDL, under detection limit of 0.05 μmol/L; ND, not determined; N/A, not applicable
a,bUpper and lower brain stems include midbrain—pons and the medulla, respectively.
c,dR167Q+/+ and T1/T2 controls are Sv129/C57Bl6 and C57Bl6 wild-type mice, respectively.
Data presented are means ± SDs (n ≥ 4) expressed as pmol/mg protein in tissues and μmol/L in CSF.
ALA and PBG levels were also markedly elevated in the CNS tissues of the R167Q+/+ mice. In the whole brain, ALA and PBG concentrations were 33.7 ± 4.4 and 886 ± 118 pmol/mg protein, respectively, whereas they were 6.2 ± 5.6 pmol/mg protein and undetectable in wild-type (Table 5). Analysis of various CNS regions, including cerebrum, cerebellum, upper brain stem (midbrain—pons) and lower brain stem (medulla), showed that ALA and PBG were globally elevated within the brain and brain stem (Table 5). In the spinal cord, ALA and PBG were increased to 68 ± 5.4 and 1,759 ± 391 pmol/mg protein, respectively, whereas they were 7.3 ± 0.3 pmol/mg protein and undetectable in wild-type. In the cerebral spinal fluid (CSF), ALA and PBG were elevated to 0.15 ± 0.04 and 8.9 ± 5.1 μmol/L, while in wild type they were 0.07 ± 0.01 and 0.12 ± 0.07 μmol/L, respectively (Table 5). These levels were markedly higher compared with those in the T1/T2 mice, which had ALA and PBG levels of 6.4 ± 1.7 and 23.6 ± 5.1 pmol/mg protein in whole brain, 25.6 ± 4.8 and 306 ± 57 pmol/mg protein in spinal cord and 0.09 ± 0.01 and 1.73 ± 0.25 μmol/L in CSF, respectively (Table 5).
To investigate whether the elevated ALA and PBG in the CNS were derived from local production or via the blood, T1/T2 mice were induced with three daily doses of PB to markedly increase their plasma ALA and PBG concentrations, and CNS tissues were analyzed for porphyrin precursor concentrations. Following induction by PB, ALA and PBG concentrations were markedly elevated in the livers of the T1/T2 mice, by approximately 18- and 1225-fold over baseline levels, respectively (Fig. 5A). Similarly, plasma ALA and PBG concentrations were elevated approximately 34- and approximately 56-fold over baseline levels, respectively (Fig. 5B). Despite the fact that these levels were much higher compared with baseline levels in the R167Q+/+ mice (Fig. 5A and B), ALA and PBG concentrations in the brain and spinal cord tissues were only increased by approximately 1.3- to 1.5-fold following PB administration (Fig. 5C and D). Taken together, these findings indicate that the blood–brain barrier is not, or is only weakly permeable to ALA and PBG, and that the elevated porphyrin precursor concentrations in the CNS of the R167Q+/+ mice are predominantly derived from local production within the CNS tissues.
Figure 5.
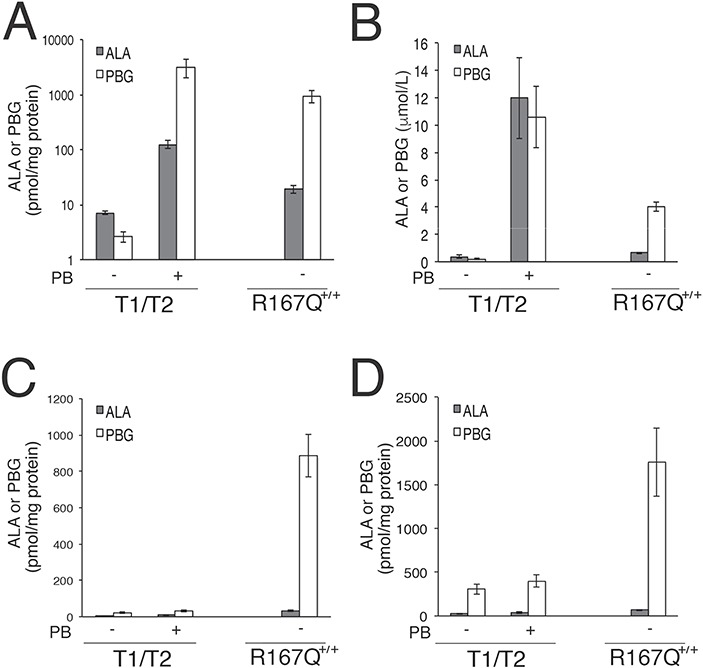
ALA and PBG do not readily cross the BBB. T1/T2 male mice were administered phenobarbital to markedly increase ALA and PBG concentrations in their livers and plasma to assess whether porphryin precursor concentrations are increased in CNS tissues. Means ± SDs (n ≥ 3) of ALA and PBG concentrations in (A) livers, (B) plasma, (C) whole brains and (D) spinal cords are shown. Basal levels in the R167Q+/+ mice are shown for comparison. Note that panel (A) shows data on a logarithmic scale.
Of note, porphyrins, predominantly the uroporphyrin I isomer, were elevated in erythrocytes and various tissues of the R167Q+/+ mice, including liver, kidney, and brain (Supplementary Material, Table S4). These findings are consistent with the marked elevation of PBG, which, when in excess, is converted non-enzymatically to the non-physiologic metabolite, uroporphyrin I.
CNS myelination is delayed and defective in the R167Q+/+ mice
Cerebrums were isolated from the R167Q+/+ mice and wild-type littermates at P10, 13, 14 and P15, and myelin basic protein (MBP) levels were assessed by western blot analyses to evaluate myelination status. In wild-type mice, MBP protein was not detectable, or barely detectable, at P10 (data not shown) and P13 but highly expressed from P14 onward (Fig. 6A). The R167Q+/+ mice, on the other hand, had barely detectable MBP levels at P14 and did not express relatively high levels of MBP until P15 (Fig. 6A). Thus, CNS myelination was slightly delayed in the R167Q+/+ mice.
Figure 6.
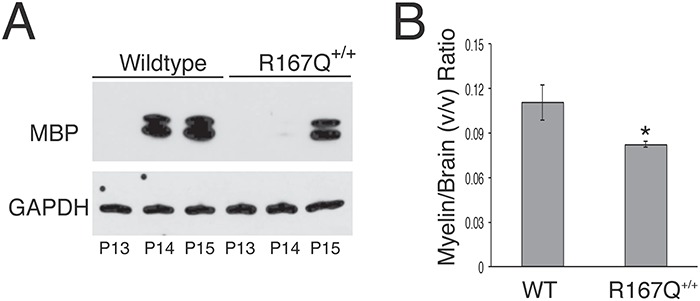
CNS myelination is delayed and decreased in R167Q+/+ mice. (A) Representative western blot assessing MBP levels in cerebrums of R167Q+/+ males and sex-matched wild-type littermates at postnatal days (P)13, 14 and 15. (B) Micro-MRI evaluation of brain myelin volumes. Shown are the means ± SDs (n = 3) of brain myelin volumes normalized by brain volumes in 3-month-old R167Q+/+ males and wild-type (WT) male littermates. *P < 0.01, Student t-test.
Micro-MRI evaluation performed at 3 months of age revealed that overall myelin volume in the R167Q+/+ mouse brains was reduce by ~30% compared with those of wild-type littermates (mean ± SD; 30.2 ± 2.3 versus 43.4 ± 4.8 mm3), while total brain volume was ~9% smaller in the R167Q+/+ mice relative to wild-type littermates (367 ± 20.1 versus 392 ± 7.6 mm3). When normalized for brain volume, brain myelin volume was decreased by ~25% in the R167Q+/+ mice compared with wild-type mice (Fig. 6B).
The severe neurological impairment in the R167Q+/+ mice is not due to CNS or hepatic heme deficiency
Total heme content in the livers and brains of the R167Q+/+ mice were ~93% and ~80% of their wild-type controls, respectively (Fig. 7A). Interestingly, these levels were slightly higher compared with those in the T1/T2 mice, which had ~57% in the liver and ~70% in the brain tissues compared with their controls.
Figure 7.
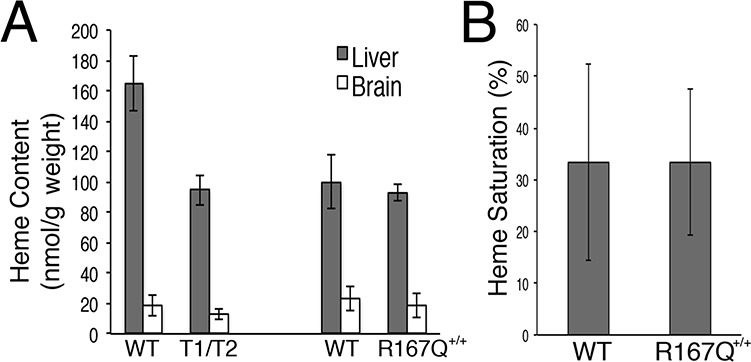
Heme content in the R167Q+/+ mice is similar to that of the T1/T2 mice. (A) Total heme content in liver and brain tissues from wild-type (WT), R167Q+/+ and T1/T2 male mice and (B) heme saturation of tryptophan dioxygenase in wild-type and R167Q+/+ livers are presented as means ± SDs (n ≥ 3).
Further, hepatic heme saturation levels of tryptophan 2,3-dioxygenase (TDO) were comparable between the R167Q+/+ and wild-type mice, at 33 ± 14% and 33 ± 19%, respectively (Fig. 7B). TDO is a heme-dependent liver enzyme involved in tryptophan metabolism that has been previously shown to be sensitive to decreased heme availability (24–26). Taken together, these data indicate that generalized heme deficiency in the liver or brain is not responsible for the marked neurological impairment of the R167Q+/+ mice.
R167Q+/+ mice exhibit normal CNS and peripheral motor nerve histology
Hematoxylin and eosin (H&E) and Nissl staining revealed grossly normal brain and spinal cord structure in 12-month-old R167Q+/+ mice (data not shown). Femoral nerves isolated from 6-month-old R167Q+/+ mice were also grossly normal, whereas those from the age-matched T1/T2 mice had marked motor axonal degeneration, as previously described (data not shown) (11). Thus, the severe neurological defects of the R167Q+/+ mice are not attributable to structural changes in central and peripheral nervous tissues.
Discussion
In humans, HD-AIP is manifested by early onset, progressive neurological dysfunction and is distinct from the autosomal dominant form, which is characterized by episodic acute neurovisceral attacks that are triggered by precipitating factors and occur after puberty. While the pathogenesis of AIP has been long debated, recent studies strongly indicate that ALA and/or PBG are the main mediators of the acute attacks, rather than neuronal heme deficiency (4,5). ALA in particular has been suggested to be neurotoxic, as it is elevated in all four acute hepatic porphyrias (i.e. PBG is not elevated in ALA-dehydratase deficient porphyria) and has been shown to modulate GABAergic activity in both in vitro and in vivo models (27–29). ALA has also been shown to have pro-oxidant properties in vitro (30–33). The toxicity of PBG is much less studied, although it has been reported that it triggered seizures with the same potency as ALA when directly injected into rat brains (34).
In the present study, a mouse model of HD-AIP was generated and compared with the previously established T1/T2 AIP mouse model to investigate the pathogenesis of HD-AIP. The R167Q+/+ mice recapitulated many of the clinical and biochemical abnormalities observed in HD-AIP patients, including early-onset neuromotor retardation, ataxia, constitutively elevated plasma and urinary ALA and PBG and porphyrinuria (Table 2). Despite their striking phenotype, the R167Q+/+ mice had grossly normal CNS histopathology and their hepatic and brain heme contents were comparable or slightly higher than those of the milder T1/T2 mice (Fig. 7A). Further, 6-month-old R167Q+/+ mice did not develop peripheral axonal degeneration, which was previously suggested to occur as a result of chronic neuronal heme deficiency in the T1/T2 mice (11). Thus, the severe neurological impairments in the R167Q+/+ mice are not attributable to structural changes or heme deficiency in nervous tissues, although the mild heme deficiency (~80% of normal) in the CNS may also contribute.
Notably, the R167Q+/+ mice had markedly greater elevations of ALA and PBG in their CNS tissues compared with the T1/T2 mice (Table 5). Most or all of the accumulated ALA and PBG in the CNS were presumably derived from local production, as the blood–brain barrier was not, or was only slightly permeable to ALA and PBG (Figs. 5A-D). That most of the accumulated porphyrin precursors in the CSF and CNS tissues was PBG rather than ALA suggests that ALA-dehydratase rapidly converts the ALA to PBG in the CNS. Interestingly, CSF samples taken from a 2.5-year-old HD-AIP child with severe neurological impairment had an essentially normal ALA level while PBG was elevated over 250-fold (Yasuda et al., unpublished data). These findings suggest that either ALA is very potent and has pronounced neurotoxic effects even at slight elevations, or PBG, too, is neurotoxic. Additionally, the R167Q+/+ mice had delayed and impaired CNS myelin formation (Fig. 6A and B), which was also demonstrated in a HD-AIP child (5). It is likely that the deficient myelin formation also contributes to the marked neuromotor impairment in human HD-AIP, as many early-onset dysmelinating and demyelinating disorders present with symptoms similar to those of HD-AIP, including ataxia, nystagmus and psychomotor retardation. Notably, ALA has been shown to impair peripheral myelin formation in vitro through oxidative damage (31).
Recently, Homedan et al. demonstrated that the activities of TCA cycle and/or downstream respiratory chain complexes were markedly decreased in the liver, brain and muscle tissues of the T1/T2 mice following administration of PB, suggesting that mitochondrial energetic defects may also play a role in AIP pathogenesis (35,36). The mitochondrial defects in the liver were presumably due to a large amount of succinyl-CoA being shuttled away from the TCA cycle to be used as substrate for the markedly increased ALAS1 enzyme (35). Mitochondrial impairments in the brain and muscle were postulated to be due to the neurotoxic effects of ALA overproduced by the liver, although in the brain, the activity of heme-dependent respiratory enzymes were decreased, suggesting that heme deficiency may also contribute (36).
Given that the R167Q+/+ mice have essentially normal hepatic Alas1 mRNA levels (Supplementary Material, Fig. S1), succinyl CoA-cataplerosis and hepatic mitochondrial defects associated with it are not expected to occur in these mice. Unlike the PB-induced T1/T2 mice, in which the marked production of ALA and PBG are essentially restricted to the liver (37), the R167Q+/+ mice have constitutively elevated porphyrin precursor levels in various tissues, including the liver, kidney, heart and brain (Table 5), although hepatic ALA and PBG levels are considerably lower compared with that of the PB-induced T1/T2 mice (Fig. 5). Thus, it is possible that mitochondrial energetic defects occur in the R167Q+/+ mice as well, especially in the brain, where ALA and PBG levels were markedly higher compared with that of the T1/T2 mice (Table 5).
Of particular interest was the finding that the R167Q+/+ mice were not ‘inducible’ by porphyrinogenic stimuli, especially PB, which markedly increased hepatic Alas1 expression and plasma ALA and PBG in the T1/T2 mice (Fig. 4A and B). While this difference in response may be due to mouse strain differences (i.e. R167Q+/+ mice are Sv129/C57Bl6, whereas T1/T2 mice are pure C57Bl6), it is also possible that it is a unique feature of HD-AIP. The aforementioned 2.5-year-old HD-AIP patient had uncontrollable seizures and was subjected to porphyrinogenic stimuli, including a brief ketogenic diet (i.e. low carbohydrate diet) and cannabidiol, which is metabolized by induction of the cytochrome P450 isozymes 2C19 and 3A4 (38). However, the patient did not develop acute attack symptoms or further elevations of urinary ALA and PBG (Yasuda et al., unpublished data). To the best of our knowledge, thus far, there have been no reports of acute attack episodes in HD-AIP patients.
The current standard treatment for the acute attacks of AIP is intravenous infusion of hemin, which exerts its therapeutic effect by providing exogenous heme for the negative feedback inhibition of hepatic ALAS1. Recent preclinical and phase I/II clinical trials have indicated that a liver-directed RNAi therapeutic targeting ALAS1 mRNA effectively and safely prevents and treats acute porphyric attacks, therefore emerging as a promising alternative therapy (24). That baseline hepatic Alas1 levels were essentially normal in the HD-AIP mice and were not induced upon exposure to porphyrinogenic stimuli (Fig. 4A and B; Supplementary Material, Fig. S1) indicates that therapies that aim to reduce hepatic ALAS1 levels, including hemin and RNAi therapy, are not expected to be beneficial for these patients. Indeed, three daily intravenous administrations of hematin (4 mg/kg) to the R167Q+/+ mice (n = 5) resulted in only slight decreases of plasma ALA (0.67 ± 0.06 versus 0.58 ± 0.25 μmol/L) and PBG (4.27 ± 0.57 versus 3.16 ± 0.63 μmol/L). Restoration of liver HMBS activity via liver transplantation or liver-targeted gene therapy is unlikely to be beneficial as well, as these therapeutic approaches do not address the markedly elevated porphyrin precursor levels or myelination defects in the CNS.
In conclusion, a mouse model that closely mimics the clinical and biochemical features of HD-AIP was generated. By comparing these mice with the milder inducible T1/T2 model, these studies suggest that the severe neurological phenotype of HD-AIP is due to increased accumulation of ALA and PBG in the CNS and decreased myelination. Thus, these mice have provided new insights into HD-AIP disease pathogenesis and permit future investigations aimed to develop novel therapeutic approaches for HD-AIP.
Materials and Methods
Prokaryotic expression of Hmbs missense mutations
A ~1.4 kb wild-type murine Hmbs cDNA (RefSeq NM_013551.2) was subcloned into the pET16b vector (Novagen, Madison, WI, USA) and designated pET-WT. Mutant constructs carrying the c.500G>A (p.Arg167Glu), c.499C>T (p.Arg167Try) or c.518_519GC>AG (p.Arg173Glu) substitutions, designated pET-R167Q, pET-R167W and pET-R173Q, respectively, were generated using the Qiagen Quickchange site-directed mutagenesis kit (Valencia, CA, USA) and primer pairs provided in the Supplementary Material, Table S5. The constructs were confirmed by sequencing and transformed into E. coli BL21 cells (Millipore Sigma, St. Louis, MO, USA). Following growth in LB medium, induction with 1mM Isopropyl-β-D-1-thiogalactopyranoside (IPTG) and inoculation for 4 h, the cells were lysed and the supernatants were used to assay HMBS activities as previously described (39).
Construction of targeting vectors and generation of HD-AIP mice
A 129/SvEv murine BAC clone containing the entire genomic Hmbs gene was obtained (clone RPCI22.HYB; Thermo Fisher Scientific, Carlsbad, CA, USA) and gene-targeting constructs were generated as depicted in Figure 1A. In brief, an 8.3 kb genomic fragment spanning introns 5 (PmlI) to 15 (SbfI) of the Hmbs gene was isolated and subcloned into the pKO Scrambler vector (Stratagene, La Jolla, CA, USA), designated pKO-Hmbs. The c.500G>A (p.R167Q) and c.518_519GC>AG (p.R173Q) mutations were individually introduced into exon 10 using Qiagen Quickchange site-directed mutagenesis kit. Additional subcloning steps were performed to ensure that additional unexpected mutations were not introduced. A LoxP-flanked Neomycin cassette carrying a novel NcoI site was inserted into intron 9 for positive selection with G418. The two targeting constructs were sequence-confirmed, linearized and simultaneously electroporated into male Sv129/C57Bl6 hybrid ES cells. Correct targeting of ES cells was confirmed by PCR screening and subsequent Southern blot analysis, using an external cDNA probe spanning exons 3–5 (Fig. 1B). The‘knocked-in’ mutations were verified by sequence analysis and targeted ES cells were microinjected into C57/Bl6 blastocysts using standard procedures.
Animal studies
Animal procedures were reviewed and approved by the Icahn School of Medicine Institutional Animal Care and Use Committee. Early development, including reflexes (surface and air righting, grasping), physiologic milestones (eye opening, pinnae detachment), sensory function (ear twitch and auditory startle) and strength (bar holding), was evaluated every day from postnatal day 2, according to previously described methods (40). Open-field testing (one 3 min trial) was performed on postnatal days 9, 12, 15, 18 and 21, as previously described (41), to evaluate motor development and coordination, including the ability to rear, sit and jump. For rotarod analysis, the mice were trained for 3 days (two trials per day, 60 s maximum per trial) and tested on the fourth day, at a rotation speed of 16 rpm (two trials per day, 180 s maximum) at 2, 6 and 12 months (13,24). For all other studies, 1- to 3-month-old male mice were used, unless specified otherwise. The R167Q+/+ mice are on Sv129/C57Bl6 mixed background, while the T1/T2 mice are congenic C57Bl6. Thus, Sv129/C57Bl6 and C57Bl6 wild-type males were used as R167Q+/+ and T1/T2 controls, respectively. Of note, when the R167Q line was backcrossed onto the C57Bl6 background, homozygous R167Q+/+ mice were no longer obtained.
Phenobarbital induction was performed as previously described (10,24), by administering 110, 120 and 130 mg/kg per day intraperitoneally (i.p.), or at reduced doses (80, 90 and 100 mg/kg per day, i.p.) when given in combination with DDC (20 mg/kg per day, oral gavage). PCN (100 mg/kg per day) was administered for three consecutive days via i.p. injection. Urines (24 h) were collected in metabolic cages, while plasma samples were collected via the facial vein technique. For CSF collection, mice were deeply anesthetized with ketamine/xylazine and their dura mater were exposed. 75 mm × 1.2 mm capillary tubes that were modified to have pointed ends were carefully inserted into the cisterna magna and CSF was collected by capillary action. Mice were deeply anesthetized and euthanized at the indicated times by cardiac perfusion with phosphate-buffered saline, unless specified otherwise. Tissues were harvested, snap frozen in liquid nitrogen and stored in the dark at −80°C until use.
HMBS enzyme activity and porphyrin/porphyrin precursor assays
Tissues were homogenized on ice in three volumes/tissue weight of chilled reporter lysis buffer (Promega, Valencia, WI, USA), and HMBS enzyme activities were determined as previously described (39). Plasma, urinary and tissue ALA and PBG concentrations were determined by LC–MS/MS (42), while porphyrins were separated and quantified by UPLC as previously described (43). Urinary creatinine was measured using a colorimetric assay based on the picric acid method (44).
RNA extraction and real-time PCR for hepatic Alas1 mRNA quantification
RNA isolation, reverse transcription and real-time PCR were performed as previously described (24). In brief, total RNA (1 μg) was isolated from tissues using TRIzol reagent and reverse transcribed with SuperscriptII (both from Thermo Fisher Scientific, Carlsbad, CA, USA). Real-time PCR was performed using predesigned Taqman assays for Alas1 (Thermo Fisher Scientific, Assay ID: Mm01235914_m1) and β-actin (Mm02619580_g1), and transcript levels were quantitated with an ABI Prism 7900 sequence detection system. Relative Alas1 mRNA levels were calculated by the comparative Ct method using β-actin as an internal control. Experiments were performed in triplicates.
Quantification of total heme content in brain and liver tissues
CNS and hepatic tissues (~100 mg) collected from the saline-perfused mice were mixed with 40 μL of 50 mM potassium phosphate (KPi) buffer, pH 7.4 and ground up in a Potter-Elvehjem glass-teflon homogenizer with 10 pairs of up and down strokes. Protein concentrations of the homogenates were determined using bicinchoninic acid reagent and adjusted to 10 mg/mL with KPi buffer. Extraction solvent (EA) was prepared by mixing four volumes of ethyl acetate to one volume of glacial acetic acid. Homogenates (50 μL) were mixed with freshly prepared EA (200 μL), vortexed vigorously for 60 s and then centrifuged at 16 000g for 30 s. The resulting supernatant, ~90% of the original volume, was collected into a new tube. A total of 10 μL of the extract was injected into a Waters Acquity UPLC system, which included a binary solvent manager, sample manager, photodiode array detector (PDA), fluorescence detector (FLR), column heater and an Acquity UPLC BEH C18, 1.7 μM, 2.1 × 100 mm column. The PDA was set to measure hemin absorbance at 398 nm while the FLR was set to measure fluorescence of protoporphyrin IX (PPIX) at 404 nm excitation and 630 nm emission. Flow rate was 0.4 ml/min. Solvent A was 0.2% aqueous formic acid, while solvent B was 0.2% formic acid in methanol. Each sample injection was run at 60°C for 7 min. The following were the successive linear gradient settings for run time in min versus solvent A: 0.0, 80%; 2.5, 1%; 4.5, 1%; 5.0, 80%. For standards, extract solutions of known concentrations of authentic hemin and PPIX initially dissolved in 1% aqueous trimethylamine, and then diluted with buffer, were used. Heme content was expressed as nmol/g tissue weight.
Western blot analyses to evaluate MBP levels in brain tissues
Cerebrums were homogenized in chilled lysis buffer [62.5 mM Tris–HCl, 2.5% SDS, 0.002% Bromophenol Blue, 5% β-mercapto-ethanol, 10% glycerol, pH 6.8] and total protein lysates (~20 μg) were denatured and separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and then transferred to a polyvinylidene difluoride (PVDF) membrane. The membranes were blocked with 5% non-fat dry milk in buffer (50 mM Tris, 150 mM NaCl, pH 7.6 and 0.05% Tween-20) for 1 h at room temperature, incubated with anti-human MBP antibody (Santa Cruz Biotechnology, cat# sc-376995, Dallas, TX, USA) overnight at 4°C (1:2000 dilution), and then incubated with HRP-conjugated anti-mouse IgG antibodies (1:20 000 dilution) for 45 min at room temperature. Signals were detected using the Chemiluminescence HRP substrate (Millipore Sigma, Burlington, MA, USA). The membranes were stripped and re-probed with anti-GAPDH antibodies (Abcam, cat# ab125247, Cambridge, MA, USA), and subsequently, HRP-conjugated anti-mouse IgG antibodies, to normalize loading amounts.
Histological studies of CNS and femoral motor nerves
Quadriceps nerves were fixed in 3% glutaraldehyde for a minimum of 24 h, EPON embedded and sectioned to 1 μm, then stained with toluidine blue staining using standard procedures. Brain and spinal cord tissues were collected from mice that were intracardially perfused with saline and fixed overnight in 4% formalin. Specimens were sectioned to 15 μm and H&E and Nissl staining were performed using standard procedures.
Ex vivo micro-MRI analyses to quantify brain and myelin volume
10- to 12-week-old mice were euthanized and brains were post-fixed with a mixture of formaldehyde and 0.5% gadolinium-based contrast agent, Prohance (Bracco Diagnostics Inc., Monroe Township, NJ, USA), for 48–72 h. Ex vivo 3D MR images were acquired using a 15.2 T Biospec horizontal bore scanner (Bruker BioSpin, Ettlingen, Germany) with BFG6S-100 actively shielded gradient system (1 T/m maximum gradient strength). A four-channel receive coil (Bruker BioSpin) was used. Fast low-angle shot (FLASH) 3D sequence was used with TR/TE 150/5.6 ms, 1.6 × 1.6 × 0.8 cm3 field of view, 40 × 50 × 50 μm3 spatial resolution, 6 averages. Total imaging time was 13 h.
All images were first bias-corrected using SPM 12 software. Each 3D image was manually segmented using Amira 6.1 (Visualization Science Group, Burlington, MA, USA). Paxinos’ mouse brain atlas (45) was used as a reference. Total brain volume was calculated based on manual segmentation, while total white matter (myelin) volume was delineated based on automated segmentation and signal intensity threshold. All volumetric measurements were done in Amira 6.1, and final volumes are given in mm3.
Statistical analyses
Statistical analyses were performed by unpaired one-tailed Student’s t-test. Differences of P ≤ 0.05 were considered statistically significant.
Supplementary Material
Acknowledgements
We thank Zhikai Zhang (Albert Einstein College of Medicine), Hector Bergonia (University of Utah School of Medicine) and Sonia Clavero (Icahn School of Medicine at Mount Sinai) for their excellent technical assistance in ES cell electroporation/ES cell culture, quantifying tissue heme concentrations and determining tissue porphyrin concentrations, respectively, and Dr Rita De Gasperi (Icahn School of Medicine at Mount Sinai) for her help in analyzing nervous tissue histopathology. The Porphyrias Consortium is a part of the National Institutes of Health (NIH) Rare Diseases Research Network (RDCRN) and supported through collaboration between the NIH Office of Rare Diseases Research (ORDR) at the National Center for Advancing Translational Science (NCATS) and National Institutes of Diabetes and Digestive and Kidney Diseases (NIDDK). The CIHD is an NIDDK Cooperative Center of Excellence in Hematology. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. All authors confirm independence from the sponsors and that the content of the article has not been influenced by the sponsors.
MRI analyses were performed by the Preclinical Imaging Facility at Vienna Biocenter Core Facilities (VBCF), member of the Vienna Biocenter (VBC), Austria.
Conflict of Interest statement. M.Y. and R.J.D. are inventors on intellectual property licensed to Alnylam Pharmaceuticals. R.J.D. is a consultant for Recordati Rare Diseases and Alnylam Pharmaceuticals. None of the other authors have competing interests.
Funding
National Institutes of Health (K01 DK087971 to M.Y., U54 DK083909 to R.J.D., U54 DK110858 to J.D.P. and R01 CA076329 to W.E).
References
- 1. Anderson K.E., Sassa S., Bishop D.F. and Desnick R.J. (2001) Disorders of heme biosynthesis: X-linked sideroblastic anemia and the porphyrias In Scriver C., Beaudet A., Sly W. and Valle D. (eds), The Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill, New York, NY, pp. 2961–3062. [Google Scholar]
- 2. Puy H., Gouya L. and Deybach J.C. (2010) Porphyrias. Lancet, 375, 924–937. [DOI] [PubMed] [Google Scholar]
- 3. Bissell D.M., Anderson K.E. and Bonkovsky H.L. (2017) Porphyria. N. Engl. J. Med., 377, 862–872. [DOI] [PubMed] [Google Scholar]
- 4. Dowman J.K., Gunson B.K., Bramhall S., Badminton M.N. and Newsome P.N. (2011) Liver transplantation from donors with acute intermittent porphyria. Ann. Intern. Med., 154, 571–572. [DOI] [PubMed] [Google Scholar]
- 5. Solis C., Martinez-Bermejo A., Naidich T.P., Kaufmann W.E., Astrin K.H., Bishop D.F. and Desnick R.J. (2004) Acute intermittent porphyria: studies of the severe homozygous dominant disease provides insights into the neurologic attacks in acute porphyrias. Arch. Neurol., 61, 1764–1770. [DOI] [PubMed] [Google Scholar]
- 6. Beukeveld G.J., Wolthers B.G., Nordmann Y., Deybach J.C., Grandchamp B. and Wadman S.K. (1990) A retrospective study of a patient with homozygous form of acute intermittent porphyria. J. Inherit. Metab. Dis., 13, 673–683. [DOI] [PubMed] [Google Scholar]
- 7. Hessels J., Voortman G., Wagen A., Elzen C., Scheffer H. and Zuijderhoudt F.M. (2004) Homozygous acute intermittent porphyria in a 7-year-old boy with massive excretions of porphyrins and porphyrin precursors. J. Inherit. Metab. Dis., 27, 19–27. [DOI] [PubMed] [Google Scholar]
- 8. Llewellyn D.H., Smyth S.J., Elder G.H., Hutchesson A.C., Rattenbury J.M. and Smith M.F. (1992) Homozygous acute intermittent porphyria: compound heterozygosity for adjacent base transitions in the same codon of the porphobilinogen deaminase gene. Hum. Genet., 89, 97–98. [DOI] [PubMed] [Google Scholar]
- 9. Picat C., Delfau M.H., Rooij F.W., Beukeveld G.J., Wolthers B.G., Wadman S.K., Nordmann Y. and Grandchamp B. (1990) Identification of the mutations in the parents of a patient with a putative compound heterozygosity for acute intermittent porphyria. J. Inherit. Metab. Dis., 13, 684–686. [DOI] [PubMed] [Google Scholar]
- 10. Lindberg R.L., Porcher C., Grandchamp B., Ledermann B., Burki K., Brandner S., Aguzzi A. and Meyer U.A. (1996) Porphobilinogen deaminase deficiency in mice causes a neuropathy resembling that of human hepatic porphyria. Nat. Genet., 12, 195–199. [DOI] [PubMed] [Google Scholar]
- 11. Lindberg R.L., Martini R., Baumgartner M., Erne B., Borg J., Zielasek J., Ricker K., Steck A., Toyka K.V. and Meyer U.A. (1999) Motor neuropathy in porphobilinogen deaminase-deficient mice imitates the peripheral neuropathy of human acute porphyria. J. Clin. Invest., 103, 1127–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Unzu C., Sampedro A., Mauleon I., Alegre M., Beattie S.G., Salamanca R.E., Snapper J., Twisk J., Petry H., Gonzalez-Aseguinolaza G. et al. (2011) Sustained enzymatic correction by rAAV-mediated liver gene therapy protects against induced motor neuropathy in acute porphyria mice. Mol. Ther., 19, 243–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yasuda M., Bishop D.F., Fowkes M., Cheng S.H., Gan L. and Desnick R.J. (2010) AAV8-mediated gene therapy prevents induced biochemical attacks of acute intermittent porphyria and improves neuromotor function. Mol. Ther., 18, 17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cavanagh J.B. and Mellick R.S. (1965) On the nature of the peripheral nerve lesions associated with acute intermittent porphyria. J. Neurol. Neurosurg. Psychiatry, 28, 320–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Defanti C.A., Sghirlanzoni A., Bottacchi E. and Peluchetti D. (1985) Porphyric neuropathy: a clinical, neurophysiological and morphological study. Ital. J. Neurol. Sci., 6, 521–526. [DOI] [PubMed] [Google Scholar]
- 16. Wikberg A., Andersson C. and Lithner F. (2000) Signs of neuropathy in the lower legs and feet of patients with acute intermittent porphyria. J. Intern. Med., 248, 27–32. [DOI] [PubMed] [Google Scholar]
- 17. Wochnik-Dyjas D., Niewiadomska M. and Kostrzewska E. (1978) Porphyric polyneuropathy and its pathogenesis in the light of electrophysiological investigations. J. Neurol. Sci., 35, 243–256. [DOI] [PubMed] [Google Scholar]
- 18. Yamada M., Kondo M., Tanaka M., Okeda R., Hatakeyama S., Fukui T. and Tsukagoshi H. (1984) An autopsy case of acute porphyria with a decrease of both uroporphyrinogen I synthetase and ferrochelatase activities. Acta Neuropathol., 64, 6–11. [DOI] [PubMed] [Google Scholar]
- 19. Kaasik K. and Lee C.C. (2004) Reciprocal regulation of haem biosynthesis and the circadian clock in mammals. Nature, 430, 467–471. [DOI] [PubMed] [Google Scholar]
- 20. Handschin C., Lin J., Rhee J., Peyer A.K., Chin S., Wu P.H., Meyer U.A. and Spiegelman B.M. (2005) Nutritional regulation of hepatic heme biosynthesis and porphyria through PGC-1alpha. Cell, 122, 505–515. [DOI] [PubMed] [Google Scholar]
- 21. Honkakoski P., Zelko I., Sueyoshi T. and Negishi M. (1998) The nuclear orphan receptor CAR-retinoid X receptor heterodimer activates the phenobarbital-responsive enhancer module of the CYP2B gene. Mol. Cell. Biol., 18, 5652–5658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kliewer S.A., Moore J.T., Wade L., Staudinger J.L., Watson M.A., Jones S.A., McKee D.D., Oliver B.B., Willson T.M., Zetterstrom R.H. et al. (1998) An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway. Cell, 92, 73–82. [DOI] [PubMed] [Google Scholar]
- 23. Marks G.S., Allen D.T., Johnston C.T., Sutherland E.P., Nakatsu K. and Whitney R.A. (1985) Suicidal destruction of cytochrome P-450 and reduction of ferrochelatase activity by 3,5-diethoxycarbonyl-1,4-dihydro-2,4,6-trimethylpyridine and its analogues in chick embryo liver cells. Mol. Pharmacol., 27, 459–465. [PubMed] [Google Scholar]
- 24. Yasuda M., Gan L., Chen B., Kadirvel S., Yu C., Phillips J.D., New M.I., Liebow A., Fitzgerald K., Querbes W. et al. (2014) RNAi-mediated silencing of hepatic Alas1 effectively prevents and treats the induced acute attacks in acute intermittent porphyria mice. Proc. Natl. Acad. Sci. U. S. A., 111, 7777–7782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Badawy A.A. (1978) Tryptophan pyrrolase, the regulatory free haem and hepatic porphyrias. Early depletion of haem by clinical and experimental exacerbators of porphyria. Biochem. J., 172, 487–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cebrian M.E., Albores A., Connelly J.C. and Bridges J.W. (1988) Assessment of arsenic effects on cytosolic heme status using tryptophan pyrrolase as an index. J. Biochem. Toxicol., 3, 77–86. [DOI] [PubMed] [Google Scholar]
- 27. Becker D.M., Cayanis E. and Kramer S. (1980) The effect of delta-aminolevulinic acid on the synthesis and metabolism of GABA in rabbit brain homogenates. S. Afr. Med. J., 57, 458–460. [PubMed] [Google Scholar]
- 28. Brennan M.J. and Cantrill R.C. (1979) The effect of delta-aminolaevulinic acid on the uptake and efflux of [3H]GABA in rat brain synaptosomes. J. Neurochem., 32, 1781–1786. [DOI] [PubMed] [Google Scholar]
- 29. Puy H., Deybach J.C., Bogdan A., Callebert J., Baumgartner M., Voisin P., Nordmann Y. and Touitou Y. (1996) Increased delta aminolevulinic acid and decreased pineal melatonin production. A common event in acute porphyria studies in the rat. J. Clin. Invest., 97, 104–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Demasi M., Penatti C.A., DeLucia R. and Bechara E.J. (1996) The prooxidant effect of 5-aminolevulinic acid in the brain tissue of rats: implications in neuropsychiatric manifestations in porphyrias. Free Radic. Biol. Med., 20, 291–299. [DOI] [PubMed] [Google Scholar]
- 31. Felitsyn N., McLeod C., Shroads A.L., Stacpoole P.W. and Notterpek L. (2008) The heme precursor delta-aminolevulinate blocks peripheral myelin formation. J. Neurochem., 106, 2068–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Neal R., Yang P., Fiechtl J., Yildiz D., Gurer H. and Ercal N. (1997) Pro-oxidant effects of delta-aminolevulinic acid (delta-ALA) on Chinese hamster ovary (CHO) cells. Toxicol. Lett., 91, 169–178. [DOI] [PubMed] [Google Scholar]
- 33. Onuki J., Chen Y., Teixeira P.C., Schumacher R.I., Medeiros M.H., Van Houten B. and Di Mascio P. (2004) Mitochondrial and nuclear DNA damage induced by 5-aminolevulinic acid. Arch. Biochem. Biophys., 432, 178–187. [DOI] [PubMed] [Google Scholar]
- 34. Pierach C.A. and Edwards P.S. (1978) Neurotoxicity of delta-aminolevulinic acid and porphobilinogen. Exp. Neurol., 62, 810–814. [DOI] [PubMed] [Google Scholar]
- 35. Homedan C., Laafi J., Schmitt C., Gueguen N., Lefebvre T., Karim Z., Desquiret-Dumas V., Wetterwald C., Deybach J.C., Gouya L. et al. (2014) Acute intermittent porphyria causes hepatic mitochondrial energetic failure in a mouse model. Int. J. Biochem. Cell Biol., 51, 93–101. [DOI] [PubMed] [Google Scholar]
- 36. Homedan C., Schmitt C., Laafi J., Gueguen N., Desquiret-Dumas V., Lenglet H., Karim Z., Gouya L., Deybach J.C., Simard G. et al. (2015) Mitochondrial energetic defects in muscle and brain of a Hmbs−/− mouse model of acute intermittent porphyria. Hum. Mol. Genet., 24, 5015–5023. [DOI] [PubMed] [Google Scholar]
- 37. Johansson A., Moller C., Fogh J. and Harper P. (2003) Biochemical characterization of porphobilinogen deaminase-deficient mice during phenobarbital induction of heme synthesis and the effect of enzyme replacement. Mol. Med., 9, 193–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jiang R., Yamaori S., Takeda S., Yamamoto I. and Watanabe K. (2011) Identification of cytochrome P450 enzymes responsible for metabolism of cannabidiol by human liver microsomes. Life Sci., 89, 165–170. [DOI] [PubMed] [Google Scholar]
- 39. Chen C.H., Astrin K.H., Lee G., Anderson K.E. and Desnick R.J. (1994) Acute intermittent porphyria: identification and expression of exonic mutations in the hydroxymethylbilane synthase gene. An initiation codon missense mutation in the housekeeping transcript causes ``variant acute intermittent porphyria” with normal expression of the erythroid-specific enzyme. J. Clin. Invest., 94, 1927–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hill J.M., Lim M.A. and Stone M.M. (2008) Developmental milestones in the newborn mouse In Gonzes I. (ed), Neuropeptide Techniques. Humana Press, Inc., Totow, NJ, Vol. 39, pp. 131–149. [Google Scholar]
- 41. Glynn D., Sizemore R.J. and Morton A.J. (2007) Early motor development is abnormal in complexin 1 knockout mice. Neurobiol. Dis., 25, 483–495. [DOI] [PubMed] [Google Scholar]
- 42. Zhang J., Yasuda M., Desnick R.J., Balwani M., Bishop D. and Yu C. (2011) A LC-MS/MS method for the specific, sensitive, and simultaneous quantification of 5-aminolevulinic acid and porphobilinogen. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci., 879, 2389–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wakabayashi A., Ulirsch J.C., Ludwig L.S., Fiorini C., Yasuda M., Choudhuri A., McDonel P., Zon L.I. and Sankaran V.G. (2016) Insight into GATA1 transcriptional activity through interrogation of cis elements disrupted in human erythroid disorders. Proc. Natl. Acad. Sci. U. S. A., 113, 4434–4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lamb E., Newman D.J. and Price C.P. (2006) Kidney funtion tests In Burtis C.A., Ashwood E.R. and Bruns D.E. (eds), Tietz Tetbook of Clinical Chemistry. Saunders, Philadelphia, PA, pp. 798–801. [Google Scholar]
- 45. Paxinos G. and Franklin K. (2012) The Mouse Brain in Stereotaxic Coordinates. Elsevier Acdemic Press, San Diego, CA. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.