

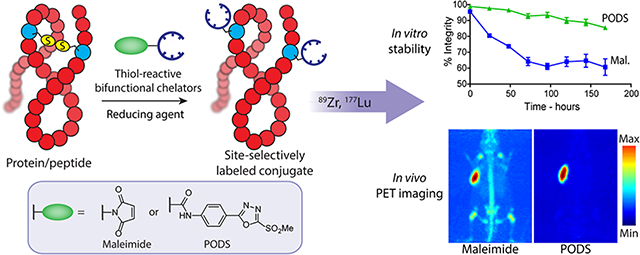
Abstract
Maleimide-bearing bifunctional chelators have been used extensively for the site-selective bioconjugation and radiolabeling of peptides and proteins. However, bioconjugates obtained using these constructs inevitably suffer from limited stability in vivo, a trait which translates into suboptimal activity concentrations in target tissue and higher uptake levels in healthy, non-target tissues. To circumvent this issue, phenyloxadiazolyl methylsulfones have previously been reported as alternatives to maleimides for thiol-based ligations, but these constructs have scarcely been used in the field of radiochemistry. In this report, we describe the synthesis of two thiol-reactive bifunctional chelators for 89Zr and 177Lu based on a new, easy-to-make phenyloxadiazolyl methylsulfone reagent: PODS. Radioimmunoconjugates created using these novel bifunctional chelators displayed higher in vitro stability than their maleimide-derived cousins. More importantly, PET imaging in murine models of cancer revealed that a 89Zr-labeled radioimmunoconjugate created using a PODS-bearing bifunctional chelator produced significantly lower uptake in non-target tissues than its analogous maleimide-based counterpart.
Keywords: site-specific bioconjugation, site-selective bioconjugation, maleimide, thiol, sulfhydryl, radioimmunoconjugate, positron emission tomography, PET, immunoPET
Graphical Abstract
INTRODUCTION
The ability of many biomolecules to selectively and specifically target cellular biomarkers has long been harnessed for both nuclear imaging and radiotherapy. This exploitation requires the attachment of a radioactive payload to the biomolecule. In the case of peptides and proteins, this is typically performed by reacting bifunctional probes with the amino acids within the biomolecule, most often lysines. While controlling the site of this conjugation is fairly easy with small peptides — which rarely possess more than one or two copies of each amino acid — this becomes a much bigger problem with larger biomolecules. For example, most antibodies contain dozens of lysines distributed throughout their macromolecule structure. The indiscriminate attachment of a bifunctional chelator to these lysines can lead to the formation of thousands of different regioisomers. Not surprisingly, this so-called “random” approach to bioconjugation has low reproducibility and produces highly heterogeneous conjugates. In addition, this strategy can result in the inadvertent grafting of payloads to bioactive sites in the biomolecule, rendering a portion of the immunoconjugates inoperative.
To circumvent these issues, researchers have turned their attention to strategies that allow for better control over the site of the ligation reaction, known as “site-specific” bioconjugations.1–5 Conjugations to cysteine — a thiol-bearing amino acid present in small numbers in proteins — with maleimide-bearing probes have become a staple of this field and have been used extensively over the last three decades.6 The prevalence of this strategy is rooted mainly in its simplicity and efficiency. Many proteins contain disulfide bridges that can be easily reduced to form reactive thiols, and the Michael addition between a maleimide and a sulfhydryl group can be performed at physiological pH and room temperature, reliably leading to the formation of a succinimidyl thioether linkage within an hour (Figure 1).
Figure 1.

Michael addition of a thiol-bearing biomolecule (green) and a radionuclide-bearing maleimide (yellow) to form a radiolabeled bioconjugate as well as the additional reactions that the radiolabeled construct can undergo in the presence of endogenous thiol-bearing molecules (pink).
While the ligation between thiols and maleimides represents an undeniable improvement over random conjugation methods, the reaction suffers from drawbacks as well. Indeed, maleimide-based conjugates display limited stability in physiological media because the succinimidyl thioether linkage can undergo a retro-Michael reaction that leads to the release of the payload or its exchange with other molecules containing free thiols (most often serum albumin, cysteine, and glutathione).7–11 Several studies focused on the development antibody-drug conjugates have reported on this phenomenon as well as the off-target uptake of payload that it creates.7,9–11 In response to this issue, these studies and others have explored the possibility of modifying the chemical environment of the succinimidyl moiety in order to accelerate its hydrolysis to a more stable thioether form that is immune to thiol exchange reactions. In the context of nuclear imaging and radiotherapy, this retro-Michael reaction can lead to the release of the radioactive payload and the subsequent in vivo radiolabeling of endogeneous biomolecules through thiol exchange reactions (Figure 1; top right). This “leakage” causes higher uptake in non-target tissues and lower uptake in target tissues, ultimately resulting in higher radiation doses to healthy tissues, reduced imaging contrast, and lower therapeutic ratios. Several alternative reagents which create more-stable linkages with thiols have been used for the bioconjugation and radiolabeling of antibodies, most notably tosylates, bromo- and iodo-acetyls, and vinyl sulfones.7,12–17 However, each of these options has drawbacks as well, including lower reaction rates with thiols as well as reactivity with other amino acids.18
An article published in 2013 holds particular promise in this area.19 In this work, the Barbas Laboratory described the creation of an oxadiazolyl methyl sulfone-based reagent that could selectively react with thiols at a rate comparable to maleimides and form more stable conjugates than those obtained with the latter (Scheme 1). This reagent was used to label different THIOMABs — antibodies engineered to bear free cysteine residues — with fluorescein, and these phenyloxadiazole-based conjugates displayed higher stability over time in serum than their maleimide-based cousins.20 Yet despite the benefits of this ligation over maleimide-thiol conjugations, this construct has scarcely been used since its publication. This is especially obvious in radiochemistry, in which only two reports describe the use of the reagent or its derivatives. In the first, Zhang et al. used this reagent along with other methylsulfone-oxadiazole derivatives to incorporate an azide moiety into an RGD peptide.21 In the second, a team in Switzerland designed an 18F-labeled prosthetic group based on this reagent which seemed to offer substantial advantages over traditional maleimide-based prosthetic groups for 18F-radiofluorinations.22
Scheme 1.

Structure of the reagent reported by Barbas, et al. and scheme of its reactivity with thiols.19
The infrequent use of this reagent was somewhat surprising at first, especially since it is commercially available from Sigma-Aldrich. However, our experience in ordering it three separate times provides some hint as to this scarcity. In each instance, we received a complex mixture of degradation products containing less than 15% of the desired compound (Supplementary Figure S1). In light of these setbacks, we considered synthesizing the reagent according to the published procedure, but it appeared that this synthesis was far from trivial for radiochemical and molecular imaging laboratories that lack sophisticated organic chemistry equipment.
With this in mind, we have aimed to create an easily accessible phenyloxadiazole-based reagent for thiol conjugations that can be obtained through a robust route that could be carried out with basic organic chemistry equipment. Using this reagent — dubbed PODS — we have synthesized two bifunctional chelators for the bioconjugation and radiolabeling of thiol-bearing biomolecules with 89Zr and 177Lu. It is our hope that the efficacy of this PODS reagent as well as the simple and straightforward chemistry associated with its synthesis will encourage researchers from all fields to move from maleimide-based probes to a more reliable and stable alternative.
RESULTS AND DISCUSSION
Synthesis
The synthesis originally published by Toda, et al. involves extensive chromatographic purifications and overnight refluxes over its five steps.19 This approach requires specific equipment and skills, and while both are undoubtedly available in any organic chemistry laboratory, they are less common in laboratories focused on the synthesis of bioconjugates. Therefore, the first step of our investigation was designing an easily synthesized phenyloxadiazole-based reagent. Along these lines, our goal was to devise a synthetic route with minimal equipment, simple procedures, and no chromatographic purifications.
In the original synthesis, the formation of the oxadiazole ring is particularly tricky. Fortunately, several 5-phenyl-1,3,4-oxadiazole-2-thiol derivatives bearing substitutions at the phenyl para-position are commercially available, allowing us to circumvent this cumbersome synthesis. As amide linkages can be formed under milder conditions than the ether linkage created by Toda, et al., 5-(4-aminophenyl)-1,3,4-oxadiazole-2-thiol seemed like an ideal precursor for an easy-to-make reagent. Two other essential elements were included in the reagent as well: (i) a short PEG chain to make the construct more water-soluble and provide a spacer between the biomolecule and the payload and (ii) a terminal amine that can be readily coupled with amine-reactive bifunctional chelators and fluorophores.
Our phenyloxadiazolyl methyl sulfone derivative — dubbed ‘PODS’ — was obtained in four steps with a global yield of 48% (Scheme 2). All intermediates were obtained in >95% purity using minimal equipment and simple procedures without chromatographic purifications. The methyl thioether 1 was obtained quantitatively via the methylation of the thiol with methyl iodide; unfortunately, we were not able to replace the use of the toxic methyl iodide with any safer reagent. The coupling between 1 and the carboxylic acid-bearing PEG chain was performed using EDCI as a coupling agent. Overnight reaction at room temperature led to the complete consumption of the starting material but also the formation of degradation products. It was possible to separate 2 from the two unidentified impurities by precipitating it with a mixture of ethyl acetate and cyclohexane. The oxidation of 2 was performed using the peracid mCPBA. This lead to the formation of the methylsulfone 3 with a yield of 90%. Finally, PODS was obtained from 3 after the quantitative deprotection of the terminal amine using a solution of trifluoroacetic acid in dichloromethane. Solutions of PODS in DMSO were stable at room temperature and have been kept at room temperature for 8 months without any noticeable signs of degradation (Supplementary Figure S2).
Scheme 2.

Synthesis of PODS
Reactivity with model thiols
In order to study the reactivity of PODS with thiols, Boc-L-cysteine was used as a model thiol in conjunction with the mild reducing agent tris(2-carboxyethyl)phosphine (TCEP), whose role was the reduction of any latent cystine species in solution (Scheme 3). The reaction between PODS and Boc-L-cysteine was quantitative in less than 2 minutes at pH 7.5 (Supplementary Figure S3). In a move toward a model closer to proteins and peptides, a tripeptide containing a central cysteine — glutathione — was reacted with PODS as well. Again, the conjugation reaction reached completion before the first measurement could be acquired (<3 min at pH 7.5).
Scheme 3.

Coupling of PODS to thiol-bearing biomolecules.
Conjugation to a model antibody
Next, the utility of PODS for the site-selective modification of proteins was investigated. To this end, the well-characterized HER2-targeting humanized immunoglobulin trastuzumab was chosen as the model protein, and a fluorescent derivative of PODS (PODS-FL) was prepared via the reaction of PODS with fluorescein isothiocyanate. PODS-FL was conjugated with trastuzumab in the presence of TCEP, a mild reducing agent used to cleave the four inter-chain disulfide bonds within the immunoglobulin (Figure 2). The presence of several thiol moieties capable of reacting with PODS means that this approach to bioconjugation can best be characterized as “site-selective” rather than “site-specific”, as the latter suggests the formation of completely homogeneous immunoconjugates. Indeed, the grafting of cargoes onto any of the 8 available cysteine residues can lead to the formation of several regioisomers. Of course, “site-specificity” represents the ideal scenario. Yet still, site-selective conjugations easily outclass random modification methods due to the controlled location of the conjugation sites — i.e. in the hinge region, far from the CDR domains — as well as their limited number. The number of fluorescein moieties present on the final conjugate was assessed using UV-Vis spectrophotometry. As expected, the degree of labeling (DOL) increased with the number of equivalents of PODS-FL in the reaction mixture, reaching 2.04 ± 0.03 after 2 hours of incubation with 10 equivalents of PODS-FL. Interestingly, the DOL was only minimally affected by the temperature of the reaction: similar results were obtained at 25 °C and 37 °C. Even more importantly, the DOL of the immunoconjugates created via the incubation of 10 equivalents of PODS-FL with the antibody in the absence of TCEP or with the antibody following re-oxidization remained close to zero: 0.03 ± 0.02 and 0.1 ± 0.1, respectively. This last piece of data demonstrates the specificity of the reagent especially well.
Figure 2.

(A) The site-selective modification of trastuzumab with PODS-FL. The structure of trastuzumab-PODS-FL depicts only one regioisomer resulting from the conjugation reaction, though several are possible. (B) Plot of the degree of labeling (DOL) of the conjugates obtained after 2 h of reaction using different numbers of equivalent of PODS-FL under various conditions (10 equivalents of TCEP, unless specified otherwise).
In order to investigate the influence of antibody type on the efficacy of the conjugation reaction, several other antibodies — including human, humanized, chimeric, and murine constructs — were incubated with 10 equivalents of PODS-FL under the conditions described above (Table 1). Intriguingly, while the DOL of the fully human, humanized, and chimeric immunoconjugates all remained slightly above 2, the number of fluorophores per antibody dropped to ∼1.5 for the murine immunoglobulins. This difference may be due to dissimilarities in the regions surrounding the disulfide linkages in murine antibodies, but this hypothesis will require further investigation.
Table 1:
Degree of labeling of different antibodies following conjugation with PODS-FL
| Antibody | Type | Constant region | Ratio FL:Ab |
|---|---|---|---|
| Human plasma IgG | Human | Human IgG | 2.1±0.1 |
| Trastuzumab | Humanized | Human IgG1 | 2.0±0.1 |
| huA33 | Humanized | Human IgG1 | 2.1±0.1 |
| Cetuximab | Chimeric | Human IgG1 | 2.2±0.1 |
| AR 9.6 | Murine | Murine IgG1 | 1.4±0.1 |
| Mouse plasma IgG | Murine | Murine IgG | 1.5±0.1 |
Synthesis of bifunctional chelators
In light of these results, we moved forward with the synthesis of thiol-reactive bifunctional chelators based on PODS. Zirconium-89 and lutetium-177 were chosen as the proof-of-concept radionuclides for this study. Over the last decade, 89Zr has become an essential nuclide for immunoPET, and 177Lu is a β-emitting nuclide of growing interest for radioimmunotherapy.23–25 Accordingly, we chose to modify PODS with desferrioxamine (DFO) and CHX-A″-DTPA, the ‘gold standard’ chelators for 89Zr and 177Lu, respectively.26,27
PODS-CHX-A″-DTPA was obtained in 69% yield via the reaction of PODS with an isothiocyanate-bearing derivative of CHX-A″-DTPA chelator in the presence of base (Scheme 4). The synthesis PODS-DFO through a similarly straightforward approach turned out to be much trickier, as degradation products were commonly seen upon the analysis of the crude reaction mixture. Upon investigation, it became apparent that the hydroxylamines of the chelator react with PODS under basic conditions by substituting at the methylsulfonyl position, causing the formation of undesired side products (Supplemental Figure S5). To circumvent this, we ‘protected’ the hydroxylamine groups via the coordination of iron. The resulting p-SCN-Bn-DFO-Fe was conjugated with PODS in presence of base, leading to the formation of PODS-DFO-Fe in good yield (72%) (Scheme 4). We were able to obtain pure PODS-DFO by removing the iron with an aqueous solution of oxalic acid 1M. However — as could perhaps have been expected — free PODS-DFO underwent degradation in aqueous solution at pH > 4.5, almost certainly due to the hydroxylamine-mediated reactions discussed above. Given the low reaction rate between PODS and thiols at this pH (Supplemental Figure S3), unprotected PODS-DFO was deemed unsuitable for the modification of peptides and proteins.
Scheme 4.

Synthesis of the PODS-based bifunctional chelators for 89Zr and 177Lu.
Bioconjugation
The bifunctional chelators were then conjugated to a model antibody — trastuzumab — to produce DFO-PODS-trast and CHX-A″-DTPA-PODS-trast. A strategy similar to that developed by Verel et al. was used to obtain the former: PODS-DFO-Fe was conjugated to the antibody, and the iron was removed thereafter.28 The ideal conditions for this decomplexation procedure were investigated and optimized for PODS-DFO-Fe using UV-Vis spectrophotometry, and 30 min of incubation at pH 4.5 and 25 °C in presence of EDTA — 2.2 g/L — was found to lead to the removal of >95% of the iron from DFO (Supplementary Table S1). Informed by the experiments with PODS-FL, 10 equivalents of PODS-DFO-Fe and PODS-CHX-A″-DTPA were used with a goal of a final DOL of 2 chelators/mAb. Mass spectrometry confirmed that the immunoconjugates were indeed modified with PODS-DFO-Fe and PODS-CHX-A″-DTPA. However, the cleavage of the disulfide bonds resulted in the fragmentation of the antibody during MALDI-TOF which, in turn, rendered the accurate determination of the exact DOLs impossible. To facilitate in vitro comparisons, the maleimide-based counterparts of these two immunoconjugates — DFO-mal-trast and CHX-A″-DTPA-mal-trast — were also synthesized using identical reaction conditions.
Radiolabeling and in vitro stability
89Zr-DFO-PODS-trast and 89Zr-DFO-mal-trast were each obtained in good yield, high purity, and similar specific activity — 2.5 ± 0.2 and 2.4 ± 0.4 Ci/g, respectively — using standard 89Zr-radiolabeling protocols (Supplemental Table S2). Likewise, typical 177Lu-labeling protocols were used to produce 177Lu-CHX-A″-DTPA-PODS-trast and 177Lu-CHX-A″-DTPA-mal-trast in high yield, high purity, and identical specific activities: 1.3 ± 0.1 Ci/g.
All four radioimmunoconjugates were incubated in human serum at 37 °C for one week, during which the integrity of the constructs was assessed using instant thin layer chromatography (ITLC) (Supplemental Figure S6). This trial clearly revealed that the PODS-based conjugates were more stable than their maleimide-based cousins. 89Zr-DFO-PODS-trast was 81 ± 5% intact after seven days of incubation, whereas the integrity of 89Zr-DFO-mal-trast fell well below 70% by the same point: 63 ± 12%. A similar difference — though admittedly of lesser magnitude — was obtained with the 177Lu-labeled immunoconjugates: 177Lu-CHX-A″-DTPA-PODS-trast and 177Lu-CHX-A″-DTPA-mal-trast were 82 ± 1% and 76 ± 2 % intact, respectively, after 7 days. These difference in the stability of the maleimide- and PODS-based immunoconjugates are consistent with previously obtained data for antibody-fluorophore conjugates.20
In vivo behavior
We chose a different model system for a comparison of the in vivo behavior of 89Zr-DFO-PODS- and 89Zr-DFO-mal-based radioimmunoconjugates: huA33, a humanized antibody that targets the A33 antigen, a transmembrane glycoprotein expressed in more than 95% of colorectal cancers.29–31 There are two reasons for this switch. First, we wanted to interrogate the modularity of our PODS-based approach by validating our bioconjugation, stability, and in vitro results with a different antibody. Second, many HER2-expressing cell lines (e.g. BT474, SKOV3, etc.) exhibit irregular growth patterns in mice, resulting in heterogeneous tumors. In contrast, the two most often used A33 antigen-expressing cell lines — SW1222 and LS174T — grow extremely predictably in athymic nude mice, reliably forming xenografts with similar size and shape. Given that the goal of this phase of this investigation was to make comparisons between the in vivo behavior of PODS- and maleimide-based radioimmunoconjugates, we decided to make the homogeneity of our xenografts a top priority.
Using the procedures we have described above, DFO-PODS-huA33 and DFO-mal-huA33 were successfully synthesized and radiolabeled with 89Zr to produce 89Zr-DFO-PODS-huA33 and 89Zr-DFO-mal-huA33 in high yield, purity, and specific activity (2.8 ± 0.4 and 2.8 ± 0.2 Ci/g, respectively). The incubation of these radioimmunoconjugates in human serum for seven days at 37 °C yielded stability profiles similar to those obtained for the trastuzumab-based radioimmunoconjugates: after a week, 89Zr-DFO-PODS-huA33 remained 86 ± 1 % intact, while its maleimide-based cousin was only 61 ± 5 % intact (Supplementary Figure S6). The in vitro immunoreactivity of the two radioimmunoconjugates was determined via saturation binding assay with A33 antigen-expressing SW1222 human colorectal cancer cells. The immunoreactive fractions were similar for both constructs: 0.95 ± 0.04 and 0.92 ± 0.06 for 89Zr-DFO-PODS-huA33 and 89Zr-DFO-mal-huA33, respectively.
89Zr-DFO-PODS-huA33 and 89Zr-DFO-mal-huA33 were injected into athymic mice bearing subcutaneous A33 antigen-expressing SW1222 human colorectal cancer xenografts. PET imaging revealed striking differences between the in vivo behavior of the two constructs (Figure 3). These disparities are particularly evident in the maximum intensity projections. 89Zr-DFO-PODS-huA33 displayed better tumor-to-background activity concentration ratios than the 89Zr-DFO-mal-huA33 at 24, 48, and 120 h post-injection. For 89Zr-DFO-mal-huA33, uptake in the bone was clearly visible as early as 24 h after administration and increased over time, and significant uptake in the kidneys and liver could also be observed. 89Zr-DFO-PODS-huA33, in contrast, produced much lower activity concentrations in each of these three tissues. The two constructs produced similar activity concentrations in the tumor: around 50–60 %ID/g for each.
Figure 3.

Planar (left) and maximum intensity projection (right) PET images of athymic nude mice bearing A33 antigen-expressing SW1222 colorectal cancer xenografts (white arrow) following the injection of 89Zr-DFO-PODS-huA33 and 89Zr-DFO-mal-huA33 (140 μCi, 60–65 μg). The coronal slices intersect the center of the tumors.
The biodistribution data tells a similar story (Figure 4 and Supplemental Table S3). Most notably, 89Zr-DFO-mal-huA33 produces higher activity concentrations in the kidneys and bone than 89Zr-DFO-PODS-huA33. Similar tumoral activity concentrations were observed for the two radioimmunoconjugates; the decreases observed for both constructs between 48 h and 120 h post-injection were likely due to the growth of the tumors between the two time points (Supplemental Figure S7). The efficient clearance of 89Zr-DFO-PODS-huA33 from healthy organs over the course of the study led to it producing lower uptake values in all non-target tissues — except the large intestine — at 120 h post-injection compared to 89Zr-DFO-malhuA33. In contrast, 89Zr-DFO-mal-huA33 produced increasing activity concentrations in the bone over the course of the study and did not display any appreciable clearance from the liver and the spleen over 5 days. The tumor-to-organ activity concentration ratios obtained with 89Zr-DFO-PODS-huA33 are generally superior to those created by 89Zr-DFO-mal-huA33 at 120 h post-injection (Supplemental Table S4). More specifically, the tumor-to-organ activity concentration ratios for the four organs with the highest uptake — liver, spleen, kidney and bone — are nearly double for 89Zr-DFO-PODS-huA33 compared to 89Zr-DFO-mal-huA33: 24.4 ± 8.5 to 13.6 ± 2.4 (tumor-to-liver), 23.2 ± 13.4 and 14.9 ± 1.6 (tumor-to-spleen), 19.4 ± 5.2 and 12.0 ± 2.3 (tumor-to-kidney), and 6.5 ± 1.9 and 3.0 ± 0.7 (tumor-to-bone). Indeed, the only organ for which the activity concentration ratio is lower for 89Zr-DFO-PODS-huA33 compared to 89Zr-DFO-mal-huA33 is the large intestine: 67 ± 18 and 132 ± 30, respectively.
Figure 4.

Biodistribution data after the administration of 89Zr-DFO-PODS-huA33 and 89Zr-DFO-mal-huA33 (30 μCi, 15–18 μg) to athymic nude mice bearing A33 antigen-expressing subcutaneous SW1222 human colorectal cancer xenografts. The values for the stomach, small intestine, and large intestine include contents.
The superior stability of the PODS-thiol linkage can explain these results. Indeed, the in vivo radiolabeling of endogenous thiol-bearing molecules and proteins caused by the retro-Michael reaction leads to off-target uptake of the radiometal and slower clearance from the non-target organs. Further, the metabolism of these radiolabeled endogeneous biomolecules likely causes the release of 89Zr into the blood and translates into higher activity concentrations in the bone.32–34
CONCLUSION
In this investigation, we have described the synthesis of a new thiol-reactive phenyloxadiazolyl methylsulfone reagent — dubbed PODS — for the site-selective modification of peptides and proteins. This reagent was obtained in four steps with a global yield of 48% using a simple and straightforward synthetic route. PODS displayed rapid and selective reactivity with thiols and was successfully used to site-selectively modify a model antibody (trastuzumab) with a fluorophore. We then synthesized two bifunctional chelators based on PODS: PODS-DFO (for 89Zr) and PODS-CHX-A″-DTPA (for 177Lu). Both of these constructs were used successfully for the site-selective modification and radiolabeling of trastuzumab, and 89Zr- and 177Lu-labeled radioimmunoconjugates based on PODS proved more stable in vitro than their maleimide-based analogs. Finally, the in vivo behavior of 89Zr-DFO-PODS-huA33 was compared to that of 89Zr-DFO-mal-huA33 in a murine model of human colorectal cancer. The performance of the former was clearly superior, especially at later time points (>48 h p.i.). In light of the clinical preference for imaging time points ranging from 72 – 144 h, the use of PODS-based radioimmunoconjugates in lieu of their maleimide-derived cousins could substantially improve the quality of clinical 89Zr-immunoPET scans. Moreover, the improved stability of 177Lu-DOTA-PODS-labeled antibodies compared to maleimide-based constructs could be beneficial in radioimmunotherapy as well, providing radioimmunoconjugates with lower uptake in healthy tissues and thus higher therapeutic indices.
While this study has focused on the modification of IgGs, PODS-based reagents hold equal promise for the site-selective modification of cysteine-bearing antibody fragments, peptides, and proteins as well. In addition, bifunctional chelators similar to the ones described in this report could be created to accommodate virtually any radiometal, as amine-reactive derivatives of a wide range of chelators are currently commercially available. Ultimately, we sincerely hope that this work — and especially the simple and straightforward chemistry that we have used here — will help promote the use of phenyloxadiazolyl reagents for thiol-based conjugations across a wide variety of disciplines and encourage researchers to move from maleimides to more reliable and more stable alternatives.
EXPERIMENTAL PROCEDURES
Synthesis of 4-(5-(methylthio)-1,3,4-oxadiazol-2-yl)aniline (1)
In a glass vessel protected from light with aluminium foil, 5-(4-aminophenyl)-1,3,4-oxadiazole-2-thiol (100 mg; 0.517 mmol; 1 eq.) was dissolved in 3 mL of methanol, and diisopropylethylamine (DIPEA; 360 μL; 2.07 mmol; 4 eq.) was added to the solution. The mixture was stirred at room temperature for 10 minutes before the slow addition of iodomethane (32 μL; 0.517 mmol; 1 eq.). After 45 minutes, the solvent was removed under reduced pressure. The white solid was dissolved in 3 mL of ethyl acetate and washed with a 0.1 M solution of sodium carbonate and then washed with water until reaching pH 7. The organic phase was dried over MgSO4 before the evaporation of the volatiles under reduced pressure, ultimately affording white needles (107 mg; yield: 100%). Because of its slight light-sensitiveness, this compound was kept in foil-covered glass vials. TLC (Ethyl acetate:triethylamine, 9:1): Rf 0.65. 1H NMR (500 MHz, CDCl3) 7.79 (2H, d, J = 8.5 Hz), 6.72 (2H, d, J = 8.5 Hz), 4.04 (2H, br s), 2.75 (3H, s). 13C NMR (125 MHz, CDCl3)166.3, 163.7, 149.7, 128.5, 114.8, 113.5, 14.8. HRMS-ESI m/z Calcd for [C9H9N3OS+H]+: 208.0539; found: 208.0539; Δ: −0.80 ppm.
Synthesis of tert-Butyl (18-((4-(5-(methylthio)-1,3,4-oxadiazol-2-yl)phenyl)amino)-15,18-dioxo-4,7,10-trioxa-14-azaoctadecyl)carbamate (2)
To a solution of N-Boc-N’-succinyl-4,7,10-trioxa-1,13-tridecanediamine (386.5 mg; 0.919 mmol; 1 eq.) in dichloromethane (3 mL) in a glass vessel protected from light with aluminum foil was added diisopropylethylamine (480 μL; 3 eq.) and N-ethyl-N’-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDCI; MW = 191.7 g/mol; 1.379 mmol; 1.5 eq.). To this solution was added 200 mg of 1 (0.965 mmol; 1.05 eq.). The reaction mixture was stirred at room temperature overnight. The mixture was then washed three times with 5 mL of an aqueous solution of 1 M hydrochloric acid. The organic phase was washed twice with 5 mL of an aqueous solution of 1 M Na2CO3 and then with water until pH neutral. The organic phase was dried on MgSO4 and evaporated. The off-white solid residue was dissolved in 10 mL of ethyl acetate, and the target compound was precipitated by slow addition of 30 mL of cyclohexane. After filtration, the final product was obtained as a white powder (310 mg; yield: 55%). Because of its slight light-sensitiveness, this compound was kept in foil-covered glass vials. TLC (acetonitrile:water, 3:1): Rf 0.73. 1H NMR (500 MHz, CDCl3) 9.68 (1H, s), 7.91 (2H, d, J= 9.0 Hz), 7.71 (2H, d, J= 8.5 Hz), 6.82 (1H, s), 4.99 (1H, s), 3.70–3.45 (12H, m), 3.41 (2H, q, J= 6.0 Hz), 3.20 (2H, q, J= 6.5 Hz), 2.76 (3H, s), 2.71 (2H, m), 2.63 (2H, m), 1.80–1.70 (4H, m), 1.42 (9H, s). 13C NMR (125 MHz, CDCl3) 172.6, 171.3, 165.8, 164.6, 156.2, 141.8, 127.7, 119.6, 118.6, 79.2, 70.6, 70.5, 70.3, 70.1, 69.6, 38.8, 38.5, 33.5, 31.6, 29.9, 28.6, 14.8. HRMS-ESI m/z Calcd for [C28H43N5O8S+Na]+: 632.2725; found: 632.2722; Δ: 0.35 ppm.
Synthesis of tert-Butyl (18-((4-(5-(methylsulfonyl)-1,3,4-oxadiazol-2-yl)phenyl)amino)-15,18-dioxo-4,7,10-trioxa-14-azaoctadecyl)carbamate (3)
In a glass vessel protected from light with aluminium foil, 30 mg of 2 (0.049 mmol, 1 eq.) was dissolved in 4 mL of dichloromethane before the slow addition of 48.5 mg of m-chloroperbenzoic acid (70%, 0.197 mmol, 4 eq.). The reaction mixture was stirred at room temperature overnight. The resulting yellow mixture was washed three times with 8 mL of an aqueous 0.1 M solution of NaOH and then with water until the pH of the solution reached 7. The organic phase was dried over MgSO4 and evaporated under reduced pressure to give a pale solid (28.5 mg, yield: 90%). TLC (acetonitrile): Rf 0.51. 1H NMR (500 MHz, CDCl3) 9.99 (1H, s), 7.98 (2H, d, J= 9.0 Hz), 7.75 (2H, d, J= 8.5 Hz), 6.88 (1H, s), 4.99 (1H, s), 3.66–3.50 (15H, m), 3.41 (2H, q, J= 6.0 Hz), 3.20 (2H, q, J= 6.5 Hz), 2.71 (2H, m), 2.65 (2H, m), 1.80–1.70 (4H, m), 1.43 (9H, s). 13C NMR (125 MHz, CDCl3) 172.6, 171.5, 166.5, 161.6, 156.1, 143.4, 128.7, 119.6, 116.4, 79.1, 70.5, 70.4, 70.2, 70.0, 69.4, 43.0, 38.8, 38.4, 33.2, 31.3, 29.7, 28.4. HRMS-ESI m/z Calcd for [C28H43N5O10S+H]+: 642.2803; found: 642.2797; Δ: 1.06 ppm.
Synthesis of N1-(3-(2-(2-(3-Aminopropoxy)ethoxy)ethoxy)propyl)-N4-(4-(5-(methylsulfonyl)-1,3,4 oxadiazol-2-yl)phenyl)succinamide (PODS)
To a solution of 3 in dichloromethane (30.0 mg, 46.8 μmol; in 1.6 mL) was added trifluoroacetic acid (400 μL). The reaction mixture was stirred at room temperature for 3 hours, and the volatiles were then removed by evaporation under reduced pressure. The oily residue was dissolved in 7 mL of water and 4 mL of ethyl acetate. The aqueous phase was then washed twice with 4 mL of ethyl acetate. The aqueous layer was lyophilized to afford the final product as a white powder (25.0 mg, yield: 98%). TLC (acetonitrile:water, 3:1): Rf 0.38.1H NMR (500 MHz, D2O) 7.85 (2H, d, J = 9.0 Hz), 7.55 (2H, d, J = 8.5 Hz), 3.60–3.45 (15H, m), 3.45 (2H, t, J= 6.5 Hz), 3.20 (2H, t, J= 6.5 Hz), 3.04 (2H, t, J= 7.0 Hz), 2.67 (2H, t, J= 6.5 Hz), 2.54 (2H, t, J= 6.5 Hz), 1.87 (2H, qt, J= 6.5 Hz), 1.70 (2H, qt, J= 6.5 Hz).13C NMR (125 MHz, D2O) 174.5, 173.2, 166.8, 161.4, 142.2, 128.6, 120.3, 116.6, 69.4, 69.4, 69.3, 69.2, 68.2, 68.2, 42.5, 37.6, 36.2, 31.9, 30.7, 28.2, 26.4. HRMS-ESI m/z Calcd. for [C23H35N5O8S+H]+: 542.2279; found: 542.2281; Δ: −0.36 ppm.
Synthesis of PODS-FL
To a solution of 6.0 mg of PODS in 600 uL of DMF (11.1 μmol, 1 eq.) were added 3.9 μL of DIPEA (22.2 μmol, 2 eq.) and 122 μL of a 0.1 M solution of fluorescein-isothiocyanate in DMSO (12.2 μmol, 1.1 eq.). The mixture was protected from light and let to react overnight at room temperature. The product was then purified by HPLC (gradient MeCN/H2O + 0.1% TFA, 0% MeCN to 100% in 30 min, Rt = 22.5 min) to afford 3.8 mg of an orange powder (yield: 37%). HRMS-ESI m/z Calcd. for [C44H46N6O13S2+H]+: 931.2637; found: 931.2634; Δ: 0.27 ppm.
Synthesis of PODS-CHX-A″-DTPA
To a solution of PODS (2.1 mg, 3.84 μmol, 1 eq.) in 200 μL of DMSO was added DIPEA (6.7 μL, 10 eq.). To this solution was added 3.0 mg of p-SCN-Bn-CHX-A″-DTPA•3HCl (4.25 μmol, 1.1 eq.; Macrocyclics, Inc.), and the mixture was stirred at room temperature overnight. The product was then purified by HPLC (gradient MeCN/H2O + 0.1% TFA, 0% MeCN to 100% in 25 min, Rt = 19.5 min) to afford 3.0 mg of a white powder (yield: 69%).HRMS-ESI m/z Calcd. for [C49H69N9O18S2+H]+: 1136.4275; found: 1136.4273; Δ: 0.18 ppm.
Synthesis of PODS-DFO-Fe
To a solution of p-SCN-Bn-DFO (5.0 mg, 6.65 μmol, 1.2 eq.; Macrocyclics, Inc.) in 100 μL of DMSO was added 1.8 mg of FeCl3 hexahydrate (6.65 μmol, 1.2 eq.). The resulting dark red solution was added to a solution of PODS (3.0 mg, 5.54 μmol, 1 eq.) in 100 μL of DMSO with DIPEA (4.83 μL, 5 eq.). The mixture was stirred at room temperature overnight. The product was then purified by HPLC (gradient MeCN/H2O +0.1% TFA, 30% MeCN to 100% in 30 min, Rt = 16.5 min) to afford 5.2 mg of a dark red solid (yield: 72%).HRMS-ESI m/z Calcd. for [C56H84FeN13O16S3+H]+: 1347.4745; found: 1347.4735; Δ: 0.74 ppm.
Preparation of mAb-PODS-FL conjugates
To a suspension of 200 μg of antibody in PBS pH 7.4 (1 mg/mL) was added 1.33 μL of a fresh TCEP solution (10 mM in water, 10 eq.) and the appropriate volume of a solution of PODS-FL (1 mM in DMSO). The reaction mixture was stirred on a thermomixer (25°C or 37°C) for 30 min, 2 h, or 24 h. The conjugate was then purified on a size exclusion column (Sephadex G-25 M, PD-10 column, GE Healthcare; dead volume = 2.5 mL, eluted with 2 mL of PBS, pH 7.4) and concentrated using centrifugal filtration units with a 50,000 Da molecular weight cut off (Amicon™ Ultra 4 Centrifugal Filtration Units, Millipore Corp. Billerica, MA).
Preparation of mAb-PODS-DFO conjugates
To a suspension of 1 mg of antibody in PBS pH 7.4 (1 mg/mL) was added 6.7 μL of a fresh TCEP solution (10 mM in water, 10 eq.) and 33 μL of a solution of PODS-DFO-Fe (2 mM in DMSO). The reaction mixture was stirred on a thermomixer at 2 5°C for 2 hours. To this yellow solution was then added 100 μL of EDTA (tetrasodic salt, 25 g/L in water), and the pH was adjusted to 4.5 with 0.25 M H2SO4. The mixture was stirred at 25 °C for 30 min to yield a colorless solution, indicating the transchelation of the Fe(III) from the DFO. The conjugate was then purified on a size exclusion column (Sephadex G-25 M, PD-10 column, GE Healthcare; dead volume = 2.5 mL, eluted with 2 mL of PBS, pH 7.4) and concentrated using centrifugal filtration units with a 50,000 Da molecular weight cut off (Amicon™ Ultra 4 Centrifugal Filtration Units, Millipore Corp. Billerica, MA).
Supplementary Material
ACKNOWLEDGEMENTS
The authors are grateful for the generous financial support of the National Institutes of Health (4R00CA178205–02 and R01CA204167), the TeamConnor Childhood Cancer Foundation, and the National Institute on Minority Health and Health Disparities (G12MD007599). Services provided by the MSKCC Small-Animal Imaging Core Facility were supported in part by NIH grants R24 CA83084 and P30 CA08748.
ABBREVIATIONS
- DOL
Degree of labeling
- FL
fluorescein
- IgG
immunoglobulin G
- DFO
desferrioxamine
- CHX-A″-DTPA
2,2’-((2-(((1S,2S)-2-(bis(carboxymethyl)amino)cyclohexyl) (carboxymethyl)amino)ethyl)azanediyl)diacetic acid
- PET
positron emission tomography
Footnotes
SUPPORTING INFORMATION DESCRIPTION
General information; synthetic protocols; chemical characterization data; PODS-thiol reactivity assay; preparation of mAb-PODS-FL conjugates; preparation of mAb-PODS-DFO and mAb-PODS-CHX-A″-DTPA conjugates; radiolabeling protocols; in vitro stability assays; PET imaging; biodistribution assay; supplementary data.
REFERENCES
- 1.Dozier JK & Distefano MD (2015) Site-specific PEGylation of therapeutic proteins. Int. J. Mol. Sci 16, 25831–25864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Freidel C, Kaloyanova S & Peneva K (2016) Chemical tags for site-specific fluorescent labeling of biomolecules. Amino Acids 48, 1357–1372 [DOI] [PubMed] [Google Scholar]
- 3.Behrens CR & Liu B (2014) Methods for site-specific drug conjugation to antibodies. mAbs 6, 46–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Agarwal P & Bertozzi CR (2015) Site-specific antibody–drug conjugates: The nexus of bioorthogonal chemistry, Protein Engineering, and Drug Development. Bioconjug. Chem 26, 176–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Adumeau P, Sharma SK, Brent C & Zeglis BM (2016) Site-specifically labeled immunoconjugates for molecular imaging—Part 1: Cysteine residues and glycans. Mol. Imaging Biol 18, 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aslam M & Dent A (1998) Bioconjugation: protein coupling techniques for the biomedical sciences (London: Macmillan Reference, 1998), Nature Publishing Group. [Google Scholar]
- 7.Alley SC, Benjamin DR, Jeffrey SC, Okeley NM, Meyer DL, Sanderson RJ & Senter PD (2008) Contribution of linker stability to the activities of anticancer immunoconjugates. Bioconjug. Chem 19, 759–765 [DOI] [PubMed] [Google Scholar]
- 8.Baldwin AD & Kiick KL (2011) Tunable degradation of maleimide–thiol adducts in reducing environments. Bioconjug. Chem 22, 1946–1953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shen B-Q, Xu K, Liu L, Raab H, Bhakta S, Kenrick M, Parsons-Reponte KL, Tien J, Yu S-F, Mai E, et al. (2012) Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat. Biotechnol 30, 184–189 [DOI] [PubMed] [Google Scholar]
- 10.Jackson D, Atkinson J, Guevara CI, Zhang C, Kery V, Moon S-J, Virata C, Yang P, Lowe C, Pinkstaff J, et al. (2014) In vitro and in vivo evaluation of cysteine and site specific conjugated herceptin antibody-drug conjugates. PLOS ONE 9, e83865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ponte JF, Sun X, Yoder NC, Fishkin N, Laleau R, Coccia J, Lanieri L, Bogalhas M, Wang L, Wilhelm S, et al. (2016) Understanding how the stability of the thiolmaleimide linkage impacts the pharmacokinetics of lysine-linked antibody–maytansinoid conjugates. Bioconjug. Chem. 27, 1588–1598 [DOI] [PubMed] [Google Scholar]
- 12.Stimmel JB, Merrill BM, Kuyper LF, Moxham CP, Hutchins JT, Fling ME & Kull FC (2000) Site-specific conjugation on serine → cysteine variant monoclonal antibodies. J. Biol. Chem 275, 30445–30450 [DOI] [PubMed] [Google Scholar]
- 13.Li L, Olafsen T, Anderson A-L, Wu A, Raubitschek AA & Shively JE (2002) Reduction of kidney uptake in radiometal labeled peptide linkers conjugated to recombinant antibody fragments. site-specific conjugation of DOTA-peptides to a Cys-diabody. Bioconjug. Chem. 13, 985–995 [DOI] [PubMed] [Google Scholar]
- 14.Li J, Wang X, Wang X & Chen Z (2006) Site-specific conjugation of bifunctional chelator BAT to mouse IgG1 Fab1 fragment. Acta Pharmacol. Sin 27, 237–241 [DOI] [PubMed] [Google Scholar]
- 15.Tinianow JN, Gill HS, Ogasawara A, Flores JE, Vanderbilt AN, Luis E, Vandlen R, Darwish M, Junutula JR, Williams, et al. (2010) Site-specifically 89Zr-labeled monoclonal antibodies for ImmunoPET. Nucl. Med. Biol 37, 289–297 [DOI] [PubMed] [Google Scholar]
- 16.Li L, Crow D, Turatti F, Bading JR, Anderson A-L, Poku E, Yazaki PJ, Carmichael J, Leong D, Wheatcroft MP, et al. (2011) Site-specific conjugation of monodispersed DOTA-PEGn to a thiolated diabody reveals the effect of increasing PEG size on kidney clearance and tumor uptake with improved 64-Copper PET imaging. Bioconjug. Chem 22, 709–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khalili H, Godwin A, Choi J, Lever R & Brocchini S (2012) Comparative binding of disulfide-bridged PEG-Fabs. Bioconjug. Chem 23, 2262–2277 [DOI] [PubMed] [Google Scholar]
- 18.Koniev O & Wagner A (2015) Developments and recent advancements in the field of endogenous amino acid selective bond forming reactions for bioconjugation. Chem. Soc. Rev 44, 5495–5551 [DOI] [PubMed] [Google Scholar]
- 19.Toda N, Asano S & Barbas CF (2013) Rapid, stable, chemoselective labeling of thiols with Julia–Kocieński-like reagents: A serum-stable alternative to maleimide-based protein conjugation. Angew. Chem. Int. Ed 52, 12592–12596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patterson JT, Asano S, Li X, Rader C & Barbas CF (2014) Improving the serum stability of site-specific antibody conjugates with sulfone linkers. Bioconjug. Chem 25, 1402–1407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Q, Dall’Angelo S, Fleming IN, Schweiger LF, Zanda M & O’Hagan D (2016) Last-step enzymatic [18F]-fluorination of cysteine-tethered RGD peptides using modified Barbas linkers. Chem. – Eur. J 22, 10998–11004 [DOI] [PubMed] [Google Scholar]
- 22.Chiotellis A, Sladojevich F, Mu L, Herde AM, Valverde IE, Tolmachev V, Schibli R, Ametamey SM & Mindt TL (2016) Novel chemoselective 18F-radiolabeling of thiol-containing biomolecules under mild aqueous conditions. Chem. Commun 52, 6083–6086 [DOI] [PubMed] [Google Scholar]
- 23.Deri MA, Zeglis BM, Francesconi LC & Lewis JS (2013) PET imaging with 89Zr: from radiochemistry to the clinic. Nucl. Med. Biol 40, 3–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jauw YWS, M. der H. van Oordt WC, Hoekstra OS, Hendrikse NH, Vugts DJ, Zijlstra JM, Huisman MC, van Dongen GAMS (2016) Immuno-positron emission tomography with Zirconium-89-labeled monoclonal antibodies in oncology: What can we learn from initial clinical trials? Front. Pharmacol 7:131, 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Banerjee S, Pillai MRA & Knapp FF (Russ). (2015) Lutetium-177 therapeutic radiopharmaceuticals: linking chemistry, radiochemistry, and practical applications. Chem. Rev 115, 2934–2974 [DOI] [PubMed] [Google Scholar]
- 26.Fischer G, Seibold U, Schirrmacher R, Wängler B & Wängler C (2013) 89Zr, a radiometal nuclide with high potential for molecular imaging with PET: chemistry, applications and remaining challenges. Molecules 18, 6469–6490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parus JL, Pawlak D & Duatti RM and A. (2015) Chemistry and bifunctional chelating agents for binding 177Lu. Curr. Radiopharm 8, 86–94 [DOI] [PubMed] [Google Scholar]
- 28.Verel I, Visser GWM, Boellaard R, Walsum MS, Snow GB & Dongen GAMS van. (2003) 89Zr Immuno-PET: comprehensive procedures for the production of 89Zr-labeled monoclonal antibodies. J. Nucl. Med. 44, 1271–1281 [PubMed] [Google Scholar]
- 29.King DJ, Antoniw P, Owens RJ, Adair JR, Haines AM, Farnsworth AP, Finney H, Lawson AD, Lyons A & Baker TS (1995) Preparation and preclinical evaluation of humanised A33 immunoconjugates for radioimmunotherapy. Br. J. Cancer 72, 1364–1372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sakamoto J, Kojima H, Kato J, Hamashima H & Suzuki H (2000) Organ-specific expression of the intestinal epithelium-related antigen A33, a cell surface target for antibody-based imaging and treatment in gastrointestinal cancer. Cancer Chemother. Pharmacol 46, S27–S32 [DOI] [PubMed] [Google Scholar]
- 31.Sakamoto J, Oriuchi N, Mochiki E, Asao T, Scott AM, Hoffman EW, Jungbluth AA, Matsui T, Lee FT, Papenfuss A, et al. (2006) A phase I radioimmunolocalization trial of humanized monoclonal antibody huA33 in patients with gastric carcinoma. Cancer Sci. 97, 1248–1254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perk LR, Visser GWM, Vosjan MJWD, Walsum MS, Tijink BM, Leemans CR & Dongen G. A. M. S. van. (2005) 89Zr as a PET surrogate radioisotope for scouting biodistribution of the therapeutic radiometals 90Y and 177Lu in tumor-bearing nude mice after coupling to the internalizing antibody cetuximab. J. Nucl. Med 46, 1898–1906 [PubMed] [Google Scholar]
- 33.Heskamp S, Laarhoven H. W. M. van, Molkenboer-Kuenen JDM, Franssen GM, Versleijen-Jonkers YMH, Oyen WJG, Graaf W. T. A. van der & Boerman OC (2010) ImmunoSPECT and immunoPET of IGF-1R expression with the radiolabeled Antibody R1507 in a triple-negative breast cancer model. J. Nucl. Med. 51, 1565–1572 [DOI] [PubMed] [Google Scholar]
- 34.Holland JP, Divilov V, Bander NH, Smith-Jones PM, Larson SM & Lewis JS (2010) 89Zr-DFO-J591 for immunoPET of prostate-specific membrane antigen expression in vivo. J. Nucl. Med 51, 1293–1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.