

Abstract
Humans are uniquely susceptible to typhoid fever caused by infection with Salmonella enterica serovar Typhi. Mathur et al. now report that mice lacking Toll-like receptor 11, which is absent in humans, can be lethally infected with S. Typhi, a breakthrough that promises to speed the development of better vaccines.
Typhoid fever is a global health problem estimated to cause more than 20,000,000 infections each year, resulting in more than 200,000 deaths (Crump et al., 2004). Although much has been learned about the pathogenesis of Salmonella infections in recent decades, studying S. Typhi has remained a challenge due to its exquisite specificity for the human host. In this issue, Mathur et al. (2012) report the breakthrough discovery that mice lacking the innate immunity receptor TLR11 can be lethally infected with S. Typhi via an oral route of infection. This promises to expand our understanding of typhoid pathogenesis and speed the development of better typhoid vaccines.
The clinical manifestations of typhoid are distinct from those of other Salmonella serovars such as S. Typhimurium, which typically cause self-limiting inflammatory diarrhea. In contrast, S. Typhi causes a debilitating systemic illness following a prolonged incubation period with absent or mild gastrointestinal symptoms (Parry et al., 2002). Immunity to nontyphoidal and typhoidal infections also appears to differ in fundamental respects, as individuals with chronic granulomatous disease, HIV infection, or other impairment of the interferon-γ/interleukin-12 (IFNγ/IL-12) signaling axis exhibit enhanced susceptibility to nontyphoidal salmonellosis, but not to typhoid fever (Gordon, 2008). The S. Typhi genome shows the presence of a number of putative and confirmed virulence determinants, such as the Vi capsular polysaccharide and typhoid toxin, that are not found in S. Typhimurium. Conversely, there is also evidence of considerable genomic decay in S. Typhi, with a large number of pseudogenes that have presumably undergone mutation following the evolutionary adaptation of S. Typhi to a narrow host range (Parkhill et al., 2001). It is generally assumed that genomic decay resulted in the loss of factors conferring resistance to host defense mechanisms that are absent in humans but present in other mammalian species. However, the identity of such host defense mechanisms has remained an enigma.
TLR11 is a Toll-like innate immune receptor that is expressed by mice, but not by humans. A previous study reported that TLR11 recognizes the parasitic protein profilin and regulates immune responses to Toxoplasma gondii (Yarovinsky et al., 2005). The new study shows that TLR11, like another Toll-like receptor called TLR5, can also recognize bacterial flagellar protein. TLR11 is highly expressed at mucosal surfaces, such as the bladder and intestinal epithelium, but is also expressed in viscera such as the liver and spleen and by macrophages, particularly in the absence of TLR5. TLR11 appears to control the expression of proinflammatory cytokines such as IL-6 and IL-12 (Figure 1). Mathur et al. (2012) show that TLR11-deficient mice are susceptible to S. Typhi infection by both oral and intraperitoneal routes of infection, suggesting that the presence of TLR11 may help to explain the intrinsic resistance of mice to S. Typhi infection. TLR11-deficient mice are also more susceptible to nontyphoidal S. Typhimurium. This contrasts with TLR5-deficient mice, which are more resistant to Salmonella infection. S. Typhi appears to be missing virulence factors that allow S. Typhimurium and other nontyphoidal Salmonella serovars to overcome the TLR11 barrier, although previous attempts to render S. Typhi virulent for mice by introducing S. Typhimurium DNA have not been successful.
Figure 1. TLR11-Mediated Innate Immunity to Salmonella Typhi in the Mouse Intestine.
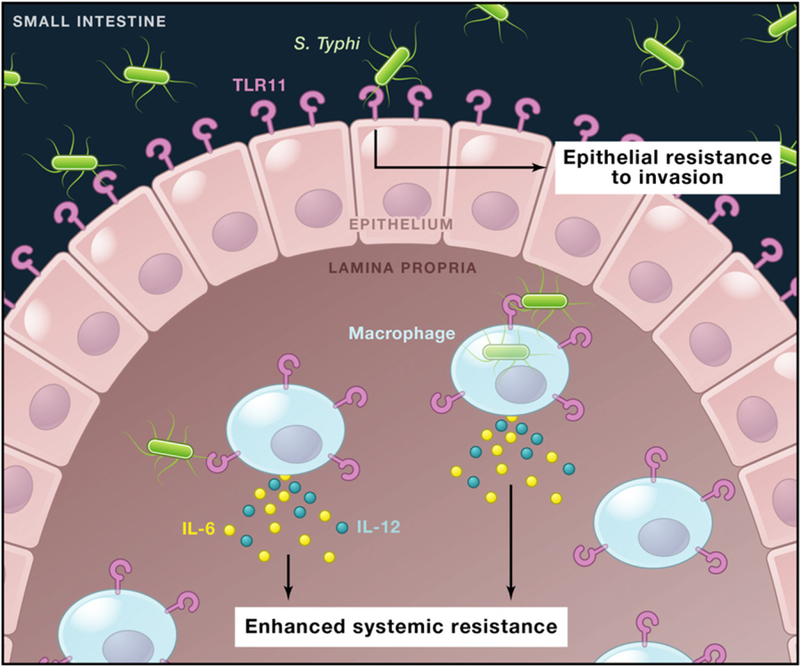
TLR11 is expressed by small intestinal epithelial cells and by mononuclear phagocytes in the lamina propria. Mathur et al. (2012) report that TLR11 recognition of the Salmonella Typhi flagellin protein enhances epithelial resistance to bacterial invasion and promotes cytokine production by mononuclear cells to enhance systemic resistance to bacterial dissemination. This may help to account for the specificity of S. Typhi for humans, who lack TLR11 expression.
A few caveats should be mentioned. If TLR11 activation by S. Typhi flagellin is indeed responsible for the resistance of mice to typhoid, then the abrogation of either TLR11 or flagellin expression would be predicted to render S. Typhi virulent in the mouse model. TLR11 deficiency in the host does in fact have the predicted effect, but a bacterial flagellar mutant does not. This suggests the possibility that other bacterial ligands in addition to flagellin might be able to stimulate TLR11. An alternative possibility is that flagella are essential for S. Typhi virulence due to a TLR11-independent mechanism. This would not have been anticipated, given the dispensibility of flagella for S. Typhimurium virulence in the mouse model (Lockman and Curtiss, 1990), but must be seriously considered in view of observations by Mathur et al. (2012) and others that aflagellate S. Typhi mutants exhibit reduced growth in human macrophages (Sabbagh et al., 2012).
Another open question is whether the TLR11-deficient mouse faithfully recapitulates the immunological and pathological features of human typhoid. Following high-dose oral S. Typhi administration, TLR11-deficient mice develop intense destruction of the intestinal mucosa and bloody diarrhea. Although gastrointestinal bleeding and even intestinal perforation have been described as late complications (Parry et al., 2002), severe intestinal destruction is not typically characteristic of human typhoid. The production of IL-12 is also striking and somewhat unexpected, given the lack of apparent involvement of IFNγ-IL-12 signaling in human typhoid. Protection against an S. Typhi challenge is conferred by adoptive transfer of serum, implying the involvement of specific antibody. However, although a robust antibody response to the Vi capsular antigen is protective against typhoid, considerable evidence argues for the importance of cellular responses in typhoid immunity (Sztein, 2007). Further pathological and immunological characterization of experimental S. Typhi infection in TLR11-deficient mice will be of considerable interest. This will be especially important when evaluating the suitability of the TLR11-deficient mouse as a model for the evaluation of novel typhoid vaccine candidates.
Humanized mice engrafted with human hematopoietic cells have recently been shown to be susceptible to S. Typhi infection and appear to exhibit some of the pathological and immunological characteristics of human typhoid (Libby et al., 2010). However, TLR11-deficient mice have the potential to provide a less costly alternative that does not require the transplantation of human tissue and exhibits less animal-to-animal variability. The TLR11-deficient mouse may also be useful for the study of other human enteric pathogens that are unable to cause infections in mice. The development of multiple small animal models for typhoid is an exciting development that is expected to yield important new insights into the pathogenesis and immunity of this enigmatic and neglected disease.
ACKNOWLEDGMENTS
The authors have received support from the National Institutes of Health (AI91966 and AI90882 to F.C.F. and AI44170 and AI76246 to A.J.B.).
REFERENCES
- Crump JA, Luby SP, and Mintz ED (2004). Bull. World Health Organ 82, 346–353. [PMC free article] [PubMed] [Google Scholar]
- Gordon MA (2008). J. Infect 56, 413–422. [DOI] [PubMed] [Google Scholar]
- Libby SJ, Brehm MA, Greiner DL, Shultz LD, McClelland M, Smith KD, Cookson BT, Karlinsey JE, Kinkel TL, Porwollik S, et al. (2010). Proc. Natl. Acad. Sci. USA 107, 15589–15594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockman HA, and Curtiss R III. (1990). Infect. Immun 58, 137–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathur R, Oh H, Zhang D, Park SG, Seo J, Koblansky A, Hayden MS, and Ghosh S (2012). Cell 151, this issue, 590–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhill J, Dougan G, James KD, Thomson NR, Pickard D, Wain J, Churcher C, Mungall KL, Bentley SD, Holden MT, et al. (2001). Nature 413, 848–852. [DOI] [PubMed] [Google Scholar]
- Parry CM, Hien TT, Dougan G, White NJ, and Farrar JJ (2002). N. Engl. J. Med 347, 1770–1782. [DOI] [PubMed] [Google Scholar]
- Sabbagh SC, Lepage C, McClelland M, and Daigle F (2012). PLoS ONE 7, e36643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sztein MB (2007). Clin. Infect. Dis 45(Suppl 1), S15–S19. [DOI] [PubMed] [Google Scholar]
- Yarovinsky F, Zhang D, Andersen JF, Bannenberg GL, Serhan CN, Hayden MS, Hieny S, Sutterwala FS, Flavell RA, Ghosh S, and Sher A (2005). Science 308, 1626–1629. [DOI] [PubMed] [Google Scholar]