

SUMMARY
Our bodies are equipped with powerful immune surveillance to clear cancerous cells as they emerge. How tumor-initiating stem cells (tSCs) that form and propagate cancers equip themselves to overcome this barrier remains poorly understood. To tackle this problem, we designed a skin cancer model for squamous cell carcinoma (SCC) that can be effectively challenged by adoptive cytotoxic T cell transfer (ACT)-based immunotherapy. Using single cell-RNA-seq and lineage-tracing, we found that TGFβ-responding tSCs are superior at resisting ACT and form the root of tumor relapse. Probing mechanism, we discovered that during malignancy, tSCs selectively acquire CD80, a surface ligand previously identified on immune cells. Moreover, upon engaging cytotoxic T lymphocyte antigen-4 (CTLA4), CD80-expressing tSCs directly dampen cytotoxic T cell activity. Conversely, upon CTLA4 or TGFβ-blocking immunotherapies, or Cd80- ablation, tSCs become vulnerable, diminishing tumor relapse after ACT treatment. Our findings place tSCs at the crux of how immune checkpoint pathways are activated.
Keywords: Tumor stem cell, squamous cell carcinoma, immune-stem cell interactions, single cell RNA-sequencing, lineage-tracing, immune evasion, adoptive T cell transfer therapy
Graphical Abstract
In Brief
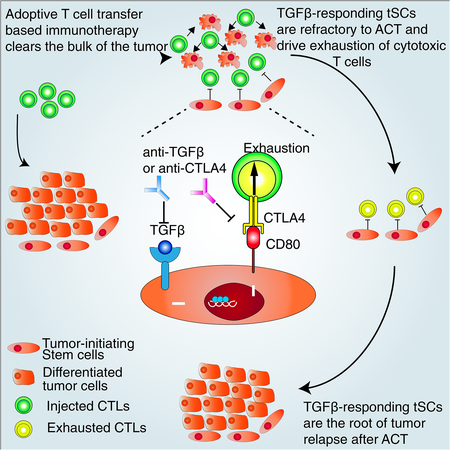
A subset of TGFβ-responsive squamous cell carcinoma stem cells is refractory to immunotherapy via expression of CD80 leading to dampening of cytotoxic T cell responses, and these tumor stem cells are competent to drive tumor relapse.
INTRODUCTION
Increasing evidence suggests that cancers are fueled by specialized cell populations that resemble normal adult tissue stem cells in their ability to maintain and propagate homeostasis and regeneration (Batlle and Clevers, 2017; Kreso and Dick, 2014). While their molecular properties are often strikingly different from their tissue stem cell counterparts, tumor-initiating stem cells (tSCs) possess a potent capacity for selfrenewal and differentiation, and therefore, are often able at low cell number to fuel and sustain tumor growth. To achieve these feats, tSCs must first overcome the powerful immune surveillance barrier which otherwise clears cancerous cells as they emerge. The mechanisms that enable tSCs to escape anti-tumor immunity are largely unknown.
This problem has become increasingly important with the emergence of immunotherapies, revolutionary treatments for cancers that are based upon their ability to harness the power of the immune system to fight cancer cells (Mellman et al., 2011). A major current approach is Adoptive T cell Transfer (ACT), which includes increasingly used ‘CAR-T therapy’. In this procedure, cytotoxic T cells from cancer patients are genetically engineered in vitro to recognize an antigen specific to the cancer, such that when they are activated and transferred back to the patients, they can selectively attack the cancer (Restifo et al., 2012). Despite promising clinical outcomes, patients receiving ACT therapy often experience tumor relapse (Landsberg et al., 2012; Mackensen et al., 2006). Although direct experimental evidence is lacking, the ability of a regressed tumor to grow back implies that at least some tSCs of the cancer have acquired ability to resist enhanced anti-tumor immunity. A priori, surviving tumor cells may simply lose the targeted antigen, thus bypassing ACT therapy (DuPage et al., 2012; Khong and Restifo, 2002). Indeed, studies on normal tissue stem cells (SCs) have led to speculation that tSCs may maintain only limited antigen presentation levels, and hence are invisible to the immune system (Agudo et al., 2018). However, it is also possible that tSCs possess antigen-editing, independent molecular features that may endow them with the capacity to evade immune attack.
Squamous cell carcinomas (SCCs) provide an excellent tumor model to address whether tSCs are uniquely equipped to resist cancer immunotherapy. As a cohort, these cancers are the 6th most deadly world-wide, affecting a variety of epithelial tissues including skin, head and neck, esophagus, lung and cervix. In mice, skin SCCs originate from HRAS-driven mutations in epidermal and/or hair follicle SCs (Lapouge et al., 2011; Li et al., 2018; White et al., 2011). They go through a benign, papilloma state prior to progressing to malignant SCCs, where they can then metastasize to the lymph nodes and lung. The tSCs of these cancers have been identified and characterized. In limit-dilution orthotopic transplantation assays in host recipient mice, they initiate SCCs efficiently (Lapouge et al., 2012; Schober and Fuchs, 2011).
Skin SCC-tSCs mainly reside at the tumor-stroma interface, and are typified by their robust expression of Integrin α6, CD34, CD44, SOX2 and SOX9 (Schober and Fuchs, 2011) (Boumahdi et al., 2014; Oshimori et al., 2015). A subset of SCC-tSCs that reside near the tumor vasculature possess particularly potent tumorigenic activity (Beck et al., 2011; Oshimori et al., 2015). Responding to enriched TGFβ in the perivasculature, these tSCs become slower-cycling and invasive. In limited dilution transplantation assays, they are also more tumorigenic than tSCs more distant from blood vessels and which are not receiving a TGFβ stimulus (David et al., 2016; Oshimori et al., 2015). Moreover, these TGFβ-responding tSCs are also the primary tumor cells that survive chemotherapy and are responsible for tumor relapse following treatment (Brown et al., 2017; Oshimori et al., 2015). Notably, external signaling ligands such as TGFβ are known to activate transcriptional cascades that can dramatically change a SC’s behavior (David et al., 2016) by adjusting their metabolism, stress responses and cellular behavior, as well as their interactions with the microenvironment to better endure any damage brought by chemotherapies (Oshimori et al., 2015).
Here, we show that TGFβ-responding tSCs are also able to evade immune attack, and we dissect the underlying mechanisms involved. To do so, we devised a novel mouse skin papilloma/SCC model in which neoantigen expression is coupled to the oncogene activation, so that we could unearth novel resistance mechanisms to ACT immunotherapy that go beyond mere loss of the targeted antigen. Using lineage tracing, we track the fate of the ACT survivors and show that they are enriched for TGFβ-responding tSCs. Employing single cell analysis of molecular signatures, we show that these tSCs and their human counterparts express CD80, a well-known immune cell surface ligand that is capable of interacting with T lymphocytes. Employing genetics and in vitro co-culture analyses, we show that tSC-specific CD80 is responsible for tumor relapse following ACT and that it acts directly on cytotoxic T cells to affect their ability to successfully mount a tumor attack.
RESULTS
A Model for ACT Immunotherapy and Tumor Cell Resistance.
To uncover possible immune evasion mechanisms that go beyond antigen editing, we began by designing a model in which at the onset of oncogenic HRASG12V activation, the transformed epidermal SCs are activated to concomitantly present a peptide (SIINFEKL) derived from ovalbumin antigen (OVA). Importantly, we used the same regulatory mechanism to drive both OVA (Rosenblum et al., 2011) and HRASG12V (Chin et al., 1999), making it difficult for the tumor to lose the neoantigen by gene silencing (Figure 1A).
Figure 1. A Murine Skin Tumor Model for Adoptive T Cell Transfer-Based Immunotherapy.

See also Figure S1
(A) Schematic. Lentivirus harboring rtTA3 and mCherry, both under the control of a constitutive phosphoglycerate kinase (PGK) promoter. Sparse, selective transduction of surface ectoderm was achieved by in utero ultrasound-guided microinjection of low titer LV into the amniOT-Ic sacs of living E9.5 CD45.2 TRE-HrasG12V TRE-OVA embryos. Doxy-induction of HRasG12V concomitantly initiates tumorigenesis and OVA expression in these transduced clonal patches of skin epithelium. Cytotoxic T cells for ACT were prepared from the spleen of CD45.1 OT-I mice and then activated in vitro as shown. Activated CTLs were then injected into tail veins of mice bearing OVA+ tumors. Mice were given cyclophosphamide 6 hrs prior to ACT to clear their endogenous immune repertoire.
(B) Mice, H&E staining, and immunofluorescence (IMF) of skin tissue sections of a representative tumor induced as in (A). Integrin α6 demarcates tumor-stroma interface; keratin 5 (K5) marks progenitors and K10, K6 and loricrin confirm the well-differentiated nature of these early skin tumors.
(C) Flow cytometry analysis of OT-I CD8+ T cell infiltration into OVA− control or OVA+ tumors at one week post-ACT. CD45.1 distinguished donor from host immune cells.
(D) IMF images of CD8+ T cells within tumors sampled before (naive) and one week after ACT.
(E) Changes in tumor size following ACT treatment of mice harboring OVA− control or OVA+ tumors. Tumor volumes were normalized to their original size. n=12; data are mean ± SEM.
(F) Flow cytometry analysis of OVA antigen presentation on the surface of lineage-negative tumor cells ± ACT.
(G) Flow cytometry analyses of proliferation maker (Ki67), anti-tumor cytokine production (granzyme) and T cell exhaustion markers (CTLA4, PD1, Tim3 and LAG3) in OT-I T cells following ACT. Naïve CD8+ T cells from donor spleen were used as negative control.
(H) Decrease in tumor-infiltrating OT-I T cells following ACT treatment; data are mean ± SEM.
(I, J) After one (I) and even two (J) ACT treatments, both active CTLs and tSCs still persist, since tumors regrow immediately following CTL depletion. Tumor volumes were normalized to their original size; the curves from different tumors are distinguished by different colors. data are mean ± SEM. All scale bars=50 μm.
We used our powerful and highly selective in utero lentiviral delivery method at low titer to stably transduce a few skin keratinocytes specifically and exclusively within the single layer of epidermal progenitors that covers the surface of E9.5 living C57BL/6 mouse embryos (Beronja et al., 2013; Beronja et al., 2010). The lentivirus (LV) harbored PGK-rtTA3-P2A-mCherry, encoding a Doxycline (Doxy)-inducible transactivator that can bind and activate TRE and a fluorescent reporter. By administering Doxy, we could then co-induce oncogenic HRASG12V and OVA specifically in these clonal, transduced mCherry+ patches of skin epidermal cells (Figure 1A).
Upon activating oncogene expression at birth, mCherry+ tumors arose within one month (Figure 1B). Reflective of the methodology, (Beronja et al., 2010; Oshimori et al., 2015) skin tumors were exclusively epithelial in origin. Initial tumors were non-invasive and still well-differentiated, replete with eosinophilic keratin pearls displaying both epidermal and tumor markers (Figure 1B). Judged by immune landscape profiling, these early stage naïve tumors exhibited only limited infiltration of endogenous antitumor immune cells, e.g. CD19+ B lymphocytes, CD8+ cytotoxic T lymphocytes (CTLs), and NK1.1+ natural killer (NK) cells, but were enriched for suppressive immune cells, including CD4+ Foxp3+ regulatory T (Treg) cells, CD11b+Ly6G+Ly6Cl° polymorphonuclear (PMN) or CD11b+Ly6G−Ly6Chi monocytic (M)-myeloid-derived suppressor cells (MDSCs) and CD11b+CD64+ tumor associated macrophages (TAMs) (Figures S1A–D).
For adoptive T cell transfer therapy, we first isolated cytotoxic T lymphocytes from the spleen of OT-I mice, whose CD8+ T cells express genetically engineered T cell receptors that specifically recognize OVA-derived peptide presented by class I MHC molecules (Figure 1A). Adapting a recently described ACT strategy to efficiently target established spontaneous human solid tumors (Tran et al., 2016), we next pre-treated our tumor-bearing mice with a low dose of cyclophosphamide to keep endogenous immune cells low/absent in the tumors (Figure S1B–D), and then injected the tail vein with OVA-recognizing CD8+ CTLs, following activation in vitro (Figure 1A). Paralleling human CAR-T therapy, this method provided several weeks for the injected T cells to attack the tumor in the absence of counteracting endogenous immune cells, especially Treg cells, MDSCs and TAMs (Figure S1B–D).
As judged by flow cytometry and immunofluorescence, the injected OT-I CD8+ T cells infiltrated robustly into OVA+ tumors, when compared to their OVA− littermates (Figure 1C and 1D). During the first week post-injection, OT-I T cells exhibited robust activated/effector phenotypes (CD25, CD69 and CD44 upregulation, CD62L downregulation) (Figure S1E). Correspondingly, within four weeks, while control (OVA−) tumors had nearly doubled in size, most OVA+ tumors shrank to less than ~10% of their original size, with some macroscopically invisible (Figure 1E). However, as has been observed in human patients who receive ACT treatments, despite injecting ~107 cytotoxic T cells, ~70% of these regressed tumors began to relapse after several months.
As strategized, surviving tumor cells still displayed OVA-derived peptide bound to class I MHC molecules, indicating that the relapse was not a consequence of cells losing the neoantigen (Figure 1F). Instead, even though OT-I T cells were proliferative and granzyme-producing at the start of ACT, within two weeks, they began to dampen their antitumor activities and upregulate inhibitory receptors (CTLA4, PD1, Tim3 and LAG3) and by four weeks, T cell numbers had plummeted (Figures 1G, H).
We performed two experiments to determine if tumor equilibrium and regrowth merely reflected OT-I T cell exhaustion (Wherry and Kurachi, 2015). First, we administered CD8 antibody (Ab) to deplete any remaining OT-I T cells, should they exist (Figure 1I). Second, we administered a second ACT treatment (107 OT-I T cells plus cyclophosphamide), and after 4 weeks we again treated with CD8 Ab (Figure 1J). Interestingly, although the second ACT enhanced the efficacy of tumor elimination, many tumor cells that survived the first ACT treatment still persisted through the second ACT. Moreover, whether mice received one or two ACT treatments, the majority of these treated tumors regrew following OT-I T cell ablation. These results suggested that some tSCs might be refractory to ACT.
A Population of ACT Survivors with Stem Cell Characteristics.
To probe the molecular features of these enduring tumor cells, we waited until two weeks post-ACT when tumors had shrunk, and then used Fluorescence Activated Cell Sorting (FACS) to exclude immune cells, melanocytes, fibroblasts, other non-epithelial lineages and dead cells, and enrich for mCherry+ tumor cells (Figure S2A). We then transcriptionally profiled (Smartseq2) individual ACT-surviving epithelial tumor cells (Picelli et al., 2014) (Figure 2A).
Figure 2. Single Cell Transcriptome Analysis of ACT-Surviving Tumor Cells.

See also Figure S2.
(A) Experimental scheme to characterize tumors 2 wk post-ACT by single cell RNA-seq (SmartSeq2). Principle component analyses (right) reveals four major clusters.
(B) C1 shows classical features of tSCs (top row) and stem cell-specific, super-enhancer regulated genes (bottom row). Note: C3 and C4 display signatures of suprabasal, differentiating tumor cells, while C2 likely represent early suprabasal tumor cells (intga6loCd34neg) (Figure S2).
(C) Gene ontology analyses of C1 transcripts changed by ≥2X compared to other tumor cell clusters (p<0.05).
(D) (Top) Schematic of the LV construct for marking and monitoring TGFβ-responding tSCs. (Bottom left) Representative image of a naïve skin tumor immunolabeled for TGFβ-reporter (red) and (top) integrin α6 (green) or (bottom) pSMAD2(green). (Bottom right) Flow cytometry quantification of CD34+CD44+ tSCs within the integrin α6hiTGFβ-reporter+ cells. Scale bar, 50 μm
(E) Single cell transcriptome of TGFβ-reporter+ tSCs from skin tumors (described later) overlaps with C1 ACT survivors.
Principle component (PC) analysis, t-distributed stochastic neighbor embedding (t-SNE), and unsupervised hierarchical clustering revealed four distinct populations (Figure 2A, S2B). Clusters 3 and 4 were enriched for genes encoding differentiation markers, and their low integrin expression further suggested that they were unlikely to contribute to tumor regrowth (Oshimori et al., 2015) (Figure S2C). Cluster 2 was the smallest population, and although C2 cells expressed integrins and a few tSC markers, they showed reduced features of bona fide tSCs, such as CD34.
By contrast, cluster 1 (C1) was enriched for Itga6, Cd34 and Cd44, which from prior limit-dilution orthotopic transplantation studies were known to have potent tumor-initiating capacity (Lapouge et al., 2012; Oshimori et al., 2015; Schober and Fuchs, 2011). Representing ~25% of post-ACT mCherry+ tumor cells, this cohort also expressed SCC-tSC genes Sox9 and Nrp2 (Beck et al., 2011; Lapouge et al., 2012; Lapouge et al., 2011; Oshimori et al., 2015; Schober and Fuchs, 2011) and Plau, Hmga2 and Pthlh, known to be regulated by SCC-tSC super-enhancers (Yang et al., 2015) (Figure 2B).
TGFβ-Responding Stem Cells At the Root of Tumor Relapse
Gene ontology (GO) analyses of our single cell RNA-seq data indicated that compared to the other clusters, C1 cells displayed signs of TGFβ-signaling, a hallmark of invasive SCC-tSCs (Figure 2C, Tables S1–S3) (Oshimori et al., 2015). Moreover, the pSMAD2/3-SMAD4 transcriptional program triggered by TGFβ-signaling has been shown to confer tSC resistance to chemotherapy (Brown et al., 2017; Oshimori et al., 2015).
To further test whether the C1 cells surviving ACT are indeed TGFβ-responding tSCs, we adapted our PGK-rtTA3 lentivirus by adding a pSMAD2/3 Binding Enhancer element (SBE) to drive mCherry and then initiated tumorigenesis as before (Figure 2D). As reported previously (Oshimori et al., 2015), TGFβ-reporter activity faithfully mirrored nuclear pSMAD2 and was mainly found in a subpopulation of integrin α6+CD34+CD44+ tSCs basal cells (Figure 2D). Not detected under homeostatic conditions, reporter activity arose in HRASG12V-induced, integrin-rich cells within developing skin tumors.
When we transcriptionally profiled FACS-purified TGF-β reporter+ tSCs, and basal or suprabasal non-SCs within the tumor, it was clear that the α6hi TGF-β reporterhi tSC signature overlapped with the C1 cluster which survived ACT (Figures 2E and S2D–S2G). Intriguingly, C1 cells also expressed lower levels of cytokines and chemokines that keratinocytes normally employ to stimulate immune responses, as well as reduced expression of genes associated with apoptosis (Figure 2C and Tables S1–S3).
To test the hypothesis that TGF-β-responding tSCs constitute the major population that survives ACT, we infused activated OT-1 CD8+ T cells into our tumor-bearing mice, and then monitored the temporal death of tumor cells. Interestingly, whereas most tumor cells underwent apoptosis within a week of ACT treatment, enzymatically active Caspase3 was mutually exclusive with TGFβ reporter+ tSCs (Figure 3A). Moreover, as quantified by flow cytometry, the percentage of integrin α6hi TGFβ reporter+ tSCs increased >10X, from only ~1–3% of total naïve tumor cells to >20% of post-ACT survivors (Figure 3B). We further confirmed by Ki67 immunofluorescence and EdU incorporation that these TGFβ reporter+ tSCs were not actively proliferating (Figures S3A, B). Therefore, the enrichment of these TGFβ reporter+ tSCs did not appear to result from SC expansion, but rather from a superior capacity to survive ACT.
Figure 3. TGFβ-Responding tSCs Persist and Form the Roots of Tumor Relapse Following ACT Treatment.

See also Figure S3.
(A) TGFβ-responding tSCs are persistent after ACT. Representative image and quantification of immunolabeling for active Caspase3 (green) and TGFβ-reporter (red) in naïve tumor or tumor at 1 week post-ACT. Three tumors and >150 cells from each of two sagittal sections per tumor were analyzed for each time point.
(B) Flow cytometry quantification of integrin α6hi TGFβ-reporter+ tSCs at 1 week post-ACT. Normal back skin cells labeled with the same Ab cocktail were used as fluorescence minus one (FMO) negative control to set the gate for mCherry.
(C) Lineage-tracing of TGFβ-responding tSCs following ACT therapy. Experimental scheme (top), quantification (middle), and representative images (bottom) from lineage-traced tumor regrowth following ACT. Three tumors and >150 cells from each of two sagittal sections per tumor were analyzed for each time point. All scale bars=50 μm.
To address whether these ACT-surviving cells are responsible for tumor relapse, we turned to lineage-tracing. For this purpose, we generated Rosa26-Loxp-Stop-Loxp-tdTomato;TRE-OVA;TFIE-HrasG12V mice, and transduced skin patches at E9.5 in utero with a lentivirus in which mCherry was swapped for tamoxifen-inducible CreER, so that Cre recombinase activity would be restricted to TGFβ-responding tSCs (Figure 3C). We then induced tumor formation in postnatally and subjected the OVA-expressing tumors to OT-I CD8+ T cell treatment as before.
When the tumor size shrank following OVA-targeting T cell treatment, we injected tamoxifen to activate Cre recombinase activity selectively in TGFβ-responding cells (see Figure 3C schematic). Within several days, >80% of dtTomato+ cells were also integrin CD34+CD44+, establishing their identity as tSCs (Figure S3C). Moreover, upon tumor regrowth, >90% of relapsed tumor cells were tdTomato+ (Figure 3C).
Recent studies indicate that some differentiated cells can de-differentiate to become tSCs (Landsberg et al., 2012; Schwitalla et al., 2013). To determine whether in SCCs, tSCs that are present at the end of ACT arose from tSCs that had survived ACT treatment or rather tSC progeny that had dedifferentiated, we modified our lentiviral vector to drive both mCherry and CreER from the TGFβ-sensing enhancer, and transduced at low titer the skin of Rosa26-Loxp-Stop-Loxp-YFP,TRE-OVA;TRE-HrasG12V mice (Figure S3D). This enabled us to selectively label TGFβ-responding mCherry+ tSCs with YFP prior to ACT, and subsequently check for mCherry+ tSCs that might arise from dedifferentiation of the YFPneg tSC progeny that were present in the tumor at the time we injected CTLs. As shown in Figure S3D, >98% of α6Hi CD34+CD44+mCherry+ cells after ACT were YFP+. These results suggested strongly that the TGFβ-responding tSCs were not generated de novo, but rather had survived ACT immunotherapy.
Most immune cells, including Tregs, were depleted by our cyclophosphamide-ACT regimen, but since some MDSCs and TAMs lingered (Figure S1C, D), we wondered if they might have created an immune privileged niche to protect the TGFβ-responding tSCs from the injected CTLs. However, even when we enhanced MDSC and TAM depletion with Gr1 and CSF1R blocking Abs (Figure S1C, D), TGFβ-responding tSCs still survived ACT (Figure S3E). Thus, we turned to the possibility that TGFβ-responding tSCs may be uniquely equipped with special molecular features that enable them to modulate T cell activity for self-protection.
CD80 as a Surprising Surface Protein Expressed by tSCs
tSCs still presented OVA antigen, ruling out antigen loss or defective antigen presentation as their means of ACT survival (Figure 4A). Therefore, we searched for a feature that might account for the ability of tSCs to be spotted by T cells, but resist their attack. PDL1 and CD47 are well-known immune modulatory factors that have been implicated in the ability of tumors to evade immune responses. However, PDL1 (Cd274) was not strongly expressed in our tumor model, and Cd47 was similarly distributed across all the tumor cells (Figure S3F). Turning to an unbiased approach that could provide insights into the superior ability of TGF-β responding tSCs to resist ACT treatment and regrow the tumor, we then analyzed single cell transcriptome of naïve skin tumors.
Figure 4. TGF-β-Responding tSCs Express CD80.

See also Figure S4.
(A) Flow cytometry analysis of OVA antigen presentation on the surface of Integrin α6hi TGFβ-reporter+ tSCs before and after ACT treatment.
(B) Heatmap of single cell transcriptome profiles of naïve tumors reveals genes differentially expressed between TGFβ-responding tSCs and other tumor cells (Integrin α6hi or low). Note that Cd80 is among the most highly up-regulated transcripts in naïve tSCs.
(C) t-SNE plots showing that the high Cd80 expression is restricted to C1 cluster that represents the TGFβ-reporter+ tSCs of the naïve tumor.
(D) t-SNE plots showing high Cd80 expression only in the C1 cluster of ACT survivors that represent the tSCs (Integrin α6hi).
(E) CD80 expression in skin tumors. (Upper) Flow cytometry analysis shows that integrin α6hi TGFβ-reporter+ tSCs are the non-immune (CD45neg cell) source of surface CD80. (Lower left) qRT-PCR analysis of Cd80 in naïve tumor cells. Data are mean ± SEM. (Lower Right) Flow cytometry analysis of CD80 protein different FACS-purified tumor populations.
(F-H) IMF and quantifications of CD80 and integrin α6hi in TGFβ-reporter+ tSCs of mouse (F,G) and human (H) skin SCCs. Data are from 3 tumors and 2 sagittal sections (>150 cells each) for each tumor and time point. Scale bars, 50 μm.
(I) Flow cytometry shows that CD80 on Integrin a6+CD34+CD44+ tSCs was significantly reduced when TGFβ blocking Abs were administered to tumor bearing mice for one month.
When CD34+CD44+α6+TGFβ-reporter+ tSCs were compared to all the other epithelial cells within the naive tumors, Cd80 emerged as a top distinguishing feature, enriched by >6X in naive tSCs as well as tSCs that survived ACT (Figures 4B–4D and Table S4). Although CD80 (or B7–1) is known to be expressed by some immune cells (Carreno and Collins, 2002), we had gated out these cells in our FACS-purified tumor cells, and those genes (Cd45, Cd11b, Cd11c, Cd207) were not expressed in any of the profiled cells (Figures S2A, S2D and S2G). Rather, although not every itgaβHi cell expressed Cd80, those that did also expressed other tSC markers, such as Cd44, Cd34 and Sox9 (Figures 4C, 4D and S2F).
Flow cytometry and rtPCR confirmed the presence of surface CD80 on CD45-negative, non-immune tumor cells, >95% of which were integrin α6hi and TGFβ reporter+ (Figure 4E). Immunofluorescence further confirmed the specificity of surface CD80 protein in TGFβ reporter+ tSCs near or at the tumor-stromal interface, in both naive tumors as well as those that had relapsed after ACT (Figures 4F and S4A). Consistent with our single cell analyses of ACT survivors, ACT-refractory CD80+ tumor cells showed no signs of apoptosis (Figure 4G). CD80 was also detected in Keratin14+ pSMAD2/3+ progenitors from 7,12-Dimethylbenz[a]anthracene (DMBA) and 12-O-tetradecanoylphorbol-13-acetate (TPA) induced skin tumors (Figure S4B), >20% of which bear oncogenic mutations in Kras, Trp53, Hras1, Myh9 and/or Notch1 (Li et al., 2018; Nassar et al., 2015; Nicolas et al., 2003; Schramek et al., 2014) Moreover, the specific acquisition of CD80 appeared to be independent of immune selection, as it was seen even in progenitors from skin tumors that were generated by DMBA/TPA treatment of Rag2-null mice, which lack an adaptive immune system (Figure S4B).
Importantly, our findings also appeared to be relevant to human cancers, as CD80 was expressed in pSMAD2/3+Keratin14+ primary skin SCC progenitors directly collected from human patients (Figure 4H). We also detected CD80 both in xenograft-derived human skin SCCs grown on Severe Combined Immunodeficient (SCID) mice and also in skin SCCs that had metastasized to lung (Figure S4C). Taken together, these data suggest that TGFβ-responding tSCs are a significant source of CD80 across a broad cohort of mouse and human tumors, including those relapsed from ACT.
Since CD80 occurred mainly on TGF-β responding tSCs, we wondered whether the two events might be linked. Notably, treatment with TGF-β blocking Ab significantly decreased CD80 surface expression on Integrin α6HiCD34+CD44+ tSCs (Figure 4I). This result builds upon increasing evidence that TGF-β signaling can elicit immune suppression (Travis and Sheppard, 2014).
CD80 is Necessary for tSCs to Endure an Immune Attack
In ACT therapy, the endogenous immune system is suppressed. To interrogate the importance of tumor CD80 in withstanding a full-scale immune attack, we turned to an engrafted SCC model, where we could carefully control for the numbers of tumor-initiating cells, the rates of tumor growth and the differentiated status of developing tumors. We retained the oncogenic driver from our initial ACT experiments by choosing a well-characterized HRasG12V SCC line (PDVC57) derived from DMBA-treated skin keratinocytes isolated from C57/BL6 mice (Bremner and Balmain, 1990). Importantly, this line could initiate SCCs even in adult, wild-type C57/BL6 mice, and although PCVC57-derived tumors were robustly infiltrated with anti-tumor immune cells, particularly cytotoxic T cells, SCCs grew and were lethal to mice.
With this line, we could address whether CD80 is an essential feature of tumor growth. To do so, we employed CRISPR/Cas gene editing to target Cd80 ablation in otherwise isogenic PDVC57 tumor cells (Figure 5A). In comparison to their unedited PDVC57 counterparts, Cd80-null PDVC57 tumor cells displayed blunted tumor growth (Figure 5B).
Figure 5. CD80 Protects Tumor Stem Cells from Cytotoxic T Cells.

See also Figure S5.
(A) Schematic of CRISPR/Cas9-mediated Cd80 ablation in the skin SCC line PDVC57. Immunoblots confirmed the status of CD80 in CD80(+) and CD80(−) isogenic PDVC57 SCC cells.
(B) Effect of CD80 on SCC growth in C57BL/6 mice (left: tumor volume; right: tumor size at week 5). Note that CD80 deficiency diminishes tumorigenesis on these immunocompetent mice. n=15; data are mean ± SEM.
(C) Effect of CD80 on SCC growth in C57BL/6 mice lacking all B and T lymphocytes (left: tumor volume; right: tumor size at week 5). Note that CD80 deficiency does not diminish tumorigenesis when mice lack an adaptive immune system. n=15, data are mean ± SEM.
(D) Effect of CD80 on SCC growth in C57BL/6 mice immunodepleted for their CD8+ CTLs (left: tumor volume; right: tumor size at week 5). Note that CD80 deficiency does not diminish tumorigenesis when CD8 T cells are not present. n=15; data are mean ± SEM.
(E,F) Effect of CD80 on (E) CD8+ T cell infiltration in tumors (Left, percentage of total CD45+ cells; Right, exact T cell number per 0.1 mg tumor) and (F) CD8+ T cell production of Granzyme, IFNγ and TNFα (percentage of total CD8+ T cells). Quantifications are by flow cytometry.
If the impaired tumor growth arises from an inability of Cd80-null PDVC57 tumor cells to efficiently withstand immune attack, then the difference in growth of Cd80(+) (WT) and CD80(−) tumors should be erased on the background of immunocompromised mice. Indeed, parental and Cd80-null PDVC57 cells grew SCCs at comparable rates in C57/BL6 Rag2-null mice, which lack an adaptive immune system (Figure 5C).
Since in ACT, CD8+ cytotoxic T cells are injected to combat the cancer, we next asked whether tumor-CD80 can specifically suppress the functions of these CD8+ T cells. Indeed, Ab depletion of CD8+ cells erased the diminished tumor progression caused by CD80 deficiency, indicating that cytotoxic T cells were involved in CD80’s actions (Figure 5D). Moreover, CD8+ T cell infiltration, proliferation and anti-tumor activity (as judged by the production of Granzyme B, IFN-γ and TNF-α) became significantly enhanced when Cd80 was ablated (Figures 5E, 5F and S5A). These findings were specific for Cd80 ablation, as similar effects were observed when shRNAs were used to knockdown Cd80 in the same cell line (Figure S5 B–D).
Tumor Cell CD80 Suppresses Cytotoxic T Cell Activity in a CTLA4-Dependent Manner.
Since the majority of endogenous immune suppressive cells were eliminated prior to ACT with cytotoxic T cells, we surmised that tSCs have the capacity to directly suppress cytotoxic CD8 T cell functions, independent of possible priming of other suppressive immune cells, such as regulatory T cells. Thus, we concentrated our efforts on how CD80 on tSCs might attenuate cytotoxic T cell activities. In this regard, we were intrigued by studies on innate immune cells, whose CD80 can directly engage with CTLA4 on the surface of activated CTLs and silence their activity (Walker and Sansom, 2011).
To explore whether this immune effector might act analogously when present on tSCs, we designed an in vitro co-culture assay. We first purified CD8+ T cells from spleen, then prestimulated them with immobilized CD3/CD28-Abs, and finally exposed them to either cultured CD80(+) or CD80(−) PDVC57 SCC cells. Interestingly, CD80(+) SCC cells potently inhibited CTL proliferation and production of granzyme (Figures 6A, B). These suppressive effects were markedly attenuated with CD80(−) SCC cells, underscoring the importance of tumor CD80.
Figure 6. tSC-CD80 Attenuates Cytotoxic T Cell Activities Both by Direct Engagement and Indirectly by Elevating Treg Numbers.

See also Figure S6.
(A) Primary CD8+ T cells were activated by incubation with CD3/CD28 Abs and labeled with CFSE, a dye that is only diluted with subsequent cell divisions. These T cells were then co-cultured to measure the effects of CD80(+) and CD80(−) SCC cells on their dye dilution as a proxy for T cell activity. Note that CD80(+) SCC cells robustly impaired T cell activity, and that this effect was abrogated by CTLA4 blocking Abs. This combined effect was comparable to that of SCC cells lacking CD80. n=15; data are mean ± SEM.
(B) Primary CD8+ T cells, activated as in (A), were exposed to IL2 and IL12 for 3d and then cocultured to measure the effects of CD80(+) or CD80(−) SCC cells on their granzyme production as a proxy for CTL activity. Note that CD80(+) SCC cells robustly impaired T cell cytokine production, and that this effect was abrogated by silencing CD80 or CTLA4 blocking Abs. Data are mean ± SEM.
(C) CD80(+) or CD80(−) PDVC57 SCC cells were transplanted into C57/BL6 mice treated with either isotype control or CTLA4 blocking Abs, and tumor progression was analyzed at times indicated. Note that the effects of CD80 loss are as potent as CTLA4 Abs. n=15; data are mean ± SEM.
(D) Flow cytometry quantifications of the CD4+CD25HiFoxp3+ Treg cells that infiltrated CD80(+) and CD80(−) tumors (Left, percentage of total CD4 T cells; Right, exact Treg number per 0.1 mg tumor).
(E) CD80(+) or CD80(−) SCC tumors grown on Foxp3DTR mice treated with vehicle or with diphtheria toxin to eliminate Foxp3+ Treg cells. n=20; data are mean ± SEM.
To further test the possibility that tSC CD80 acts through CTLA4 receptors on activated CTLs, we used CTLA4 blocking Abs and repeated the co-culture experiment. Indeed, this treatment significantly reinvigorated T cell activities in the presence of CD80(+) SCCs (Figures 6A, B). To test for specificity, we also stimulated naïve T cells by incubating OT-I splenocytes with OVA (257–264) peptide, and then used negative selection to remove all other immune cells from the mixture (see methods). CD80(+) or CD80(−) SCC cells were then added to the purified, OVA-stimulated CTLs (Figure S6A). In this system, CTLA4 blocking Abs still diminished the suppressive effects of CD80(+) SCC cells on activated CTLs. Notably, however, CTLA4 Abs showed no added reinvigoration to T cell activity in CD80(−) SCC co-cultures, consistent with the notion that CTLA4 and CD80 interact in a common pathway (Figure S6B).
CTL receptor CD28 can also bind to innate immune cell CD80, where its effects on CTLs are stimulatory (Alegre et al., 2001). As expected, since activated OT-I CTLs are CTLA4Hi|, we were not surprised to find that CD28 stimulatory Abs did not alter the suppressive effects of SCCs on OT-I CTL proliferation (Figure S6C). Interestingly, however, while CD28 blocking Abs (Papotto et al., 2017) on their own showed no effect on activated OT-I CTL proliferation, they did reverse the rescue effects that CTLA4 blocking Abs had on CD80(+) SCC-mediated suppression of CTL proliferation (Figure S6C). While modest, these effects were significant across six independent replicates. Placed in the framework of prior knowledge on innate immune cell CD80, our data suggest that tSC CD80 preferentially engages with CTLA4 on activated CTLs, but if this interaction is blocked, CD80 binds to CD28 and becomes stimulatory.
Finally, PDL1 is closely related to CD80, and in T cells and some cancer cells, it interacts with CTL PD1 receptor to activate an immune checkpoint and dampen lymphocyte targeting potency. Recently, it was shown that immune cell PDL1 can bind in Cis to CD80, where it can impair T cell proliferation (Chaudhri et al., 2018). While not expressed in the tSCs of our HRasG12V skin SCC tumor model, PDL1 (Cd274) was expressed by PDVC57 SCC cells. However in these SCC cells, PDL1 acted parallel rather than linear to CD80, and the loss of CD80 restored CTL proliferation to a greater extent than PDL1 blocking Abs (Figure S6D). Although the focus of our study here was CD80, PDL1 blocking Abs did give a modest boost in combination to loss of CD80 alone in these assays.
Given the promise of our in vitro findings, we turned to addressing whether CTLA4 immunotherapy would be effective at counteracting in vivo tumor progression of PDVC57-derived SCCs, and whether eliminating CD80 from the cancer cells would dampen CTLA4 immunotherapy’s potency. Indeed, while CTLA4 blocking Abs were effective in checking the growth of the CD80(+) parental SCCs, CD80-deficient SCCs were refractory (Figures 6C and S7A).
Direct Effects of tSC CD80 on Cytotoxic T Cells and tSC Survival In Vivo
Since Tregs are known to be depleted upon CTLA4 Ab treatment (Peggs et al., 2009) and since mice engrafted with PDVC57-SCC cells had their full immune regalia, we also examined the effects of tumor CD80 on Tregs. Interestingly, in untreated mice, the frequency and absolute numbers of Tregs were reduced in CD80(−) compared to CD80(+) SCCs (Figure 6D). This was consistent with the reduction in tumor growth upon Cd80 ablation, which was as potent as CTLA4 blocking Abs. However, when we transplanted CD80(+) and CD80(−) SCC cells into Foxp3DTR mice whose Tregs had been efficiently depleted by diphtheria toxin (DT), we found that CD80(+) tumors still grew faster compared to CD80(−) tumors (Figure 6E). Thus even when Tregs were absent, tSC CD80 still suppressed CTL functions.
Since Tregs are mostly depleted during the cyclophosphamide and tumor clearance phase of ACT (Figure S1B), and since tSC CD80 exerts an effect independent of Tregs, we concentrated our efforts on whether and how tumor CD80 promotes stem cell survival in ACT. To directly test this possibility in vivo, we silenced Cd80 selectively in the tSCs (Figure 7A). We achieved this goal by crossing our earlier ACT tumor model (TRE-OVA X TRE-HrasG12V) to Rosa26-LSL-Cas9-P2A-GFP mice, and then utilized in utero lentiviral delivery to generate clonal patches of skin epithelia transduced with three elements: a) PGK-rtTA to induce tumor formation and OVA upon Doxy; b) a pSMAD2/3 enhancer driving both mCherry and CreER; and c) CRISPR small guide (sg) RNAs targeting either a scramble control sequence (Scr) or Cd80. Prior to ACT, we administered tamoxifen to activate CAS9 and delete Cd80 specifically in the TGFβ-responding tSCs of naïve tumors (Figure 7A).
Figure 7. CD80 is Critical for TGFβ-responding tSCs to Dampen Immune Attack and Survive ACT.

See also Figure S7.
(A) (Left) Strategy to achieve TGFβ-responding tSC-specific silencing of Cd80 in a spontaneous HRasG12V-driven skin tumor. (Right) qPCR and representative IMF reveal efficient CD80 targeting and CD80 Ab efficacy.
(B) Flow cytometry analysis of proliferation (Ki67), cytokine production (granzyme) and T cell exhaustion markers (CTLA4, PD1, Tim3 and LAG3) in OT-I T cells that infiltrated Control (Scr-sg) or Cd80-null (Cd80-sg) tumors at 2 week post-ACT. The naïve CD8+ T cells from donor spleen was used as negative control.
(C) Flow cytometry analysis of T cell frequency in the control tumors or CD80 sgRNA transduced tumors at 2 week post-ACT.
(D) IMF and quantifications of active Caspase3 (green) and TGFβ-reporter (red) in OVA+ Scr sgRNA (left) or Cd80 sgRNA (right) transduced tumors at 1 week post-ACT. Five tumors with two slides from each tumor were analyzed; >150 cells were counted from each slide.
(E) Flow cytometry quantifications of integrin α6hi TGFβ-reporter+ tSCs in Scr sgRNA or Cd80 sgRNA transduced tumors at 1 week post-ACT showing that the enrichment of tSCs is dramatically reduced when Cd80 is silenced in tSCs.
(F) Flow cytometry quantifications of integrin α6hi TGFβ-reporter+ tSCs in mice treated with control or CTLA4 blocking Ab at 1 week post-ACT.
(G) Flow cytometry quantifications of integrin α6hi TGFβ-reporter+ tSCs in mice treated with control or TGFβ blocking Ab without ACT treatment or at 1 week post-ACT.
(H) Tracking Scr sgRNA or Cd80 sgRNA transduced tumors after ACT. n=15 for control; n=21 for tSC-specific targeting of Cd80. All Scale bars=50 μm.
Compared to control sgRNA tumors, OT-I T cells within tumors whose tSCs lacked CD80 were more proliferative, produced more anti-tumor granzyme, and displayed diminished expression of inhibitory receptors (Figure 7B). This delay in CTL exhaustion was also reflected by an increase in OT-I CTL frequency within tumors whose tSCs lacked CD80 (Figure 7C). Moreover, these tumor-suppressive effects of CD80 tSCs appeared to be mediated through CTLA4, since the consequences to ACT of ablating Ctla4 in OT-I CTLs were similar to those of Cd80 ablation in tSCs (Figures S7B, C).
It was noteworthy that tSCs lacking CD80 were also more vulnerable to this influx of CTLs. This was evidenced by the marked increase in apoptotic tSCs and decline in tSC survival through ACT therapy (Figures 7D, E). CTLA4-deficient OT-I CTLs and CTLA4 blocking Abs administered during ACT had similar deleterious effects on tSC survival in established tumors (Figures 7F and S7D) and led to enhanced tumor clearance. Of additional note, lineage tracing again revealed that even when TGFβ-responding tSCs became susceptible to ACT, dedifferentiation did not appear to function as a means to replenish the depleted tSCs (Figure S7E).
Finally, we surveyed the impact of our findings on SCC therapy. First, although TGFβ blocking Abs alone caused a significant expansion of CD34+CD44+ tSCs, injecting OT-I CTLs into TGFβ blocking Ab-treated mice abolished this survival advantage (Figure 7G). Second, by ablating tSC-specific CD80, not only was the exhaustion program of injected OT-I CTLs delayed, but in addition, tumor relapse was diminished (Figure 7H). Third, in contrast to the immediate rescue of CD80(+) tumor growth upon depleting OT-I CTLs after ACT, many tumors failed to regrow in the absence of CD80 (Figure S7F). Even in the few Cd80-null tumors that eventually relapsed, tumor regrowth was significantly delayed (Figure S7G), underscoring the deleterious effects of losing CD80 on tSC survival. Of note, relapsed tumors arising after ACT therapy on tSC-Cd80 targeted tumors harbored CD80+ cells (Figure S7H), leaving open the possibility that some tSCs may not have been responding to TGFβ at the time ACT was initiated but became TGFβ-responsive and hence activated CD80 during tumor relapse.
Taken together, our data uncovered and highlighted a key role for tSC CD80 in orchestrating the exhaustion program of injected CTLs during ACT, thereby enabling survival of TGFβ-responding tSCs throughout treatment and allowing for subsequent tumor relapse.
DISCUSSION
A Surprising New Player in Evading Targeted Cytotoxic T Cell Therapy and Driving Tumor Relapse.
It has been increasingly recognized that tSCs are endowed with unique molecular features to drive tumorigenesis, as well as to promote resistance to cancer therapies. By establishing an effective mouse ACT model in which we could track skin SCC-tSCs and directly test and compare their susceptibility to antigen-specific anti-tumor immune responses, we unearthed compelling evidence that TGFβ-responding tSCs show pronounced resistance to enhanced cytotoxic T cell responses. For SCCs, we further discovered that these TGFβ-responding tSCs are responsible for tumor recurrence following ACT therapy. To our surprise, we also found that within the tumor, these stem cells express an immune cell ligand, CD80, which when silenced, blunts their resistance to immune surveillance and dampens tumor relapse.
Both CTLA4 and CD28 are expressed by CD8+ cytotoxic T cells. CD80 is mostly considered to be stimulatory, as when expressed on immune cells, it can work through CD28 to activate T cells (Alegre et al., 2001). However, CD80’s affinity is higher for CTLA4 than for CD28, and its interaction with CTLA4 can have an inhibitory effect (van der Merwe et al., 1997). Importantly, when cytotoxic T cells receive their activating stimulus in peripheral lymph nodes and then infiltrate a solid tumor, they display markedly elevated CTLA4 (Alegre et al., 2001). Thus, within the tumor, the CD80 on stem cells is more likely to engage CTLA4 than CD28, thereby dampening the effectiveness of cytotoxic T cells at attacking the tumor. Supporting this hypothesis, when CTLA4-mediated interactions with SCC cells were blocked in vitro, tumor-CD80 engaged instead with CD28 on activated CTLs (Figure S5).
The biochemical details of these various immune engagements with tumor-CD80 will be interesting to explore in the future. For the purposes here, we’ve concentrated on the discovery and physiological relevance of tSC-specific CD80. Overall, our findings, initially with ACT therapy, then interrogated through in vitro co-culture and with tumor transplantations on Treg-depleted mice and finally challenged in vivo with CD80-deficient SCC cells and CTLA4-deficient CTLs, all highlight that a direct interaction between CD80(+) tSCs and CTLA4(+) cytotoxic T cells acts as an important, hitherto unappreciated contributor to resistance to immune surveillance and tumor recurrence in cancer.
A Possible Role For tSC-CD80 in Priming Tregs
Although the major focus of the current manuscript was to investigate how tSCs persist through adaptive T cell transfer therapies involving cytotoxic T cells, our discovery of CD80 as an immune evasive ligand unique to tSCs prompted us to interrogate the effects of tSC-CD80 on Tregs, which like cytotoxic T cells express CTLA4. Indeed, tSC-derived CD80 also impacted Tregs, whose numbers declined upon tumor-specific Cd80 ablation. Since regulatory T cells are themselves potent suppressors of cytotoxic T cells, this provides an additional means by which CD80 endows tumor stem cells with an ability to evade an immune attack, even when it is unleashed to its fullest extent. Although probing deeper into the mechanistic details of tSC-CD80 on Treg-mediated suppression of cytotoxic T cells is outside the scope of the current investigation, it is possible that, instead of engaging with CTLA4 on Tregs, tSC-CD80 might bind to CD28, which has been reported to be critical for the development of optimal suppressive Treg functions (Zhang et al., 2013).
Irrespective of the underlying mechanisms involved, an indirect suppression of cytotoxic T cells through tSC-CD80-mediated priming of Tregs might come into play when the host immune system begins to recover from ACT. Recovering MDSCs and TAMs may also provide sources of CD80 to further prime the function of Tregs at these later stages following ACT and curb an endogenous cytotoxic T cell attack. Thus, although we’ve focused on how a direct interaction between CD80(+) tSCs and CTLA4hi cytotoxic T cells enables tSCs to survive ACT-mediated immunotherapy, indirect immune suppressive effects of tSCCD80, as well as the CD80 from other suppressive immune cells are likely to enter into play during the recovery phase of ACT or in natural tumor progression.
tSCs At the Core of Orchestrating a Tumor Immune Evasion Program
Our study revealed that tSCs are specifically equipped with surface CD80, endowing tumor stem cells with the powers to both actively orchestrate cytotoxic T cell exhaustion but also fuel their own tumor growth. Although future research will be needed to dissect the relative contributions of stem cell-derived CD80 on establishing immune suppression in various spontaneous tumor models, our studies on SCCs place so-called “cancer stem cells” at the crux of how immune evasion is established.
Interestingly, CD80 was only activated in the subset of tSCs that actively responded to TGFβ and its expression could be reduced by blocking TGFβ signaling. Since in SCCs, TGFβ arises primarily from the perivasculature, and the vasculature of a growing tumor is dynamic (Oshimori et al., 2015), this heterogeneity in the tumor microenvironment implies that the ability of SCC-tSCs to resist immunotherapy may change over time. The importance of TGFβ in protecting cancer cells from anti-tumor immunity has also been reported for bladder and colon cancer, in this case with conventional PDL1 immunotherapy (Ganesh and Massague, 2018; Mariathasan et al., 2018; Tauriello et al., 2018)
The depth of the arsenal of immune-evasive mechanisms endowed to tSCs is still in its infancy, as is the question of how TGFβ-signaling might activate CD80 and confer these tumor-evasive mechanisms to stem cells. We did nOT-Ice that the Cd80 expression was not completely abolished upon blocking TGFβ signals, suggesting that additional signaling might function in tSC Cd80 activation. Given the newfound importance of tSC CD80 in immune evasion, it will be important to uncover these pathways in the future.
In closing, our studies shed new light on the basis for immune resistance and tumor relapse from immunotherapy. We’ve not only unveiled a new player in the game of tumor immune resistance, but also raised the possibility that TGFβ-signaling may be at the nexus of immune evasion and malignancy of tSCs in other cancers as it is for SCCs. Clinically, our findings also help to shape the underlying biological basis for the effectiveness of current immune checkpoint blockade drugs that target CTLA4. Moreover, given that TGFβ-responding tSCs are also the major population of cancer cells that escape chemotherapies (Oshimori et al., 2015), our finding that they are also at the roots of immune evasion suggest that a combination of cisplatin, CTLA4 blocking Abs, adaptive T cell therapy and short-term TGFβ inhibitors might be particularly effective in future SCC therapeutics that target the tSCs at the core of immuno- and chemo- resistance.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Elaine Fuchs (fuchslb@rockefeller.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
TRE-OVA (Rosenblum et al., 2011) and TRE-Hras1G12V (Chin et al., 1999) mice have been described before. The original TRE-Hras1G12V mice were back bred 10 generations to C57BL/6J and then bred to TRE-OVA (B6, CD45.2) mice to generate the ACT tumor model. Wild type C57BL/6J, B6;129S6-Gt(ROSA>26Sortm9(CAG-tdTomato)Hze/J (R26-LSL-tdTomato), B6; 129-Gt(ROSA>26Sortm1(CAG-cas9*,-EGFP>Fezh/J (R26-LSL-Cas9) (Platt et al., 2014), B6(Cg)-Rag2tm1.1Cgn/J (Rag2−/−), C57BL/6-Tg(TcraTcrb)1100Mjb/J (OT-I), B6.SJL-Ptprca Pepcb/BoyJ (CD45.1), and B6.129(Cg)-Foxp3tm3(DTR/GFP)Ayr/J (Foxp3-DTR) mice were obtained from The Jackson Laboratories. Animals were maintained in an Association for Assessment and Accreditation of Laboratory Animal Care (AALAC)-accredited animal facility, and procedures were performed with Institutional Animal Care and Use Committee (IACUC)-approved protocols. The animals were maintained and bred under specific-pathogen free conditions at Comparative Bioscience Center at Rockefeller University and all the procedures are in accordance with the Guide for the Care and Use of Laboratory Animals. For ACT treatment, both female and male mice were used and the mean age of mice at enrollment was 60 ± 10 days. For tumor transplantation, only 8 weeks old female mice were used.
Cell lines
Mouse skin SCC line PDVC57 was cultured in E low calcium medium (contains 50mM calcium), made in house. Human skin SCC line A431 for tumor grafting and HEK293T cells for packaging lentivirus were cultured in Dulbecco’s modified eagle medium (DMEM) in 10% FBS, 100 units/mL streptomycin and 100 mg/mL penicillin with 2mM glutamine. Sex of squamous cell carcinoma cell lines is not available.
Human tumor samples
Human skin SCC tumors originating from different body regions (e.g. leg, scalp, and forehead) were removed in the course of normal surgical procedures from cancer patients at Weill Cornell Medical College, in compliance with federal and state laws, and National Institute of Health guidelines. Small tissue samples normally discarded, were analyzed for CD80 and pSMAD2/3 expression.
METHOD DETAILS
Tumor formation and drug treatment
To induce tumor formation and OVA antigen expression, the TRE-HrasG12V mice with or without TRE-OVA allele were fed with Doxy (2mg/kg) chow (Bioserv) to activate rtTA3 at postnatal day 1. To activate Cre in TGFβ-responding tSCs, 3 or 5×100 μg of tamoxifen (Sigma) was introduced by intraperitoneal (i.p) injection. For tumor transplantation, 1×105 PDVC57 mice SCC cells were mixed with high concentration Matrigel membrane matrix (Corning) and injected subcutaneously in WT, Rag2−/− mice or Foxp3DTR mice. To deplete CD8 T cells or blocking CTLA4 during tumor grafting, three dose of 0.5 mg CD8 Ab (YTS 169.4) or 0.1 mg CTLA4 Ab (UC10–4F10–11) were injected intraperitonelly (i.p.) prior to the tumor transplantation. To block TGFβ signaling in the tumor-bearing mice, 0.1 mg TGFβ blocking ab (1D11.16.8) were injected i.p. every other day for 4 weeks. To deplete MDSCs or TAMs before ACT, 0.2 mg Gr1 (RB6–8C5) or CSF1R (AFS98) Ab were injected i.p. every other day for one week, prior ACT, then continue injection every other day after ACT for another week. Tregs were depleted in Foxp3DTR mice using modified protocol. Briefly, diphtheria toxin (DT, Sigma-Aldrich) was diluted to 2.5 μg/ml, and 200 μl (0.5 μg) was injected intraperitonelly (i.p.) into WT or Foxp3 DTR mice that had received PDVC57 SCC cells previously at Day 10, 12, 14 post tumor-transplantation and maintain by one dose of DT treatment (0.5 μg) every four days.
Adoptive T cell transfer
Mice were enrolled for ACT treatment when the tumor size on their back skin exceeded 10 mm. The mean age of mice at enrollment was 60 ± 10 days. 6 days prior ACT, the splenocytes were isolated from CD45.1 OT-I mice, and pulsed with OVA 257–264 (SIINFEKL) peptide (2 μg/ml) (InvivoGen) for 6 hours followed by washing and culturing in T cell medium containing β-mercaptoethanol (50 μM). Animals received one low dose (180 mg/kg) of cyclophosphamide at enrollment, and followed 6 hours later by intravenous infusion of 1 × 107 in vitro activated CD45.1 OT-I T cells. The ACT treated mice were then receiving recombinant hIL2 (2×104 IU, i.p.) (PeproTech) every other day for 8 days, in order to promote survival and expansion of donor OT-I T cells.
In utero lentiviral transduction
The tetracycline element-regulated oncogene and OVA antigen expression was induced on the mice skin via the ultrasound-guided in utero injection of the lentivirus carrying rtTA3. Briefly, female mice were anesthetized with isoflurane (Hospira) at day 9.5 of gestation, and low titer lentivirus was injected into the amniOT-Ic sacs, in order to selectively transduce a small number of progenitors within the surface ectoderm. At E9.5, the embryo surface consists of a single layer of progenitors that will give rise to the skin epithelium (Beronja et al., 2010). With this method, lentivirus only infects this single layered epithelium, which is homogenous and consists of keratinocytes at this stage. Importantly, the injection procedure does not alter embryonic development or prompt an immune reaction, and once integrated (~24 hr post-infection), the DNA harbored within the lentivirus is stably propagated to the epithelial progenitor progeny (Beronja et al., 2010). Postnatally, the skin contains clonal patches of transduced cells that can then induce tumorigenesis upon doxycycline administration.
Cell sorting and flow cytometry
To sort target tumor populations, tumors were dissected from mice skin at designated time points, minced and digested with 0.25% collagenase (Sigma) in HBSS (Gibco) for 20 minutes followed by Trypsin treatment for 10 minutes at 37°C. Single cells sus pension was prepared and stained with a cocktail Ab at predetermined concentration in staining buffer (PBS with 5% FBS and 1% HEPES): CD49f/integrin α6-PerCP Cy5.5 (1:250, BD); CD34-FITC (1:100, eBiosciene); CD44-PE-Cy7 (1:100, BD); CD45-APC (1:500, Biolegend); CD31-APC (1:100, Biolegend); CD117-APC (1:100, Biolegend); CD140a-APC (1:100, Biolegend). DAPI was used to remove dead cells. The sortings were performed on FACSAria. To profile immune population and measure tumor-infiltrating T cell activity, the tumor was digested in Liberase (25 mg/ml) (Roche) for 60 minutes at 37°C with adapted protocol. After preparation of single cell suspension, lymphocytes were enriched via density gradient centrifugation in the Lymphoprep™ medium. The enriched lymphocytes were then either treated with Leukocyte Activation Cocktail with GolgiPlug™ (BD Bioscience) for 4 hour at 37°C or directly stained with LIVE/DEAD Fixable Dead Cell Stain (1:200 Thermo Fisher) for 30 minutes on ice, and then with a cocktail of Abs for surface antigens at pre-determined concentrations (CD45-AF700 1:200, Biolegend; TCRβ-PerCP Cy5.5 1:200 Biolegend; CD4 PE-Cy7 1:200, Biolegend; CD8b APC-Cy7 1:200 Biolegend; CD25-BV421 1:200, Biolegend; CD19-BV605 1:200, Biolegend; NK1.1-PE-Cy5.5 1:200, Biolegend). The cells were then fixed and permeabilized with Cytofix/Cytoperm™ (BD Bioscience) or Foxp3 staining buffer set (eBioscience) and stained with Ab recognizing intracellular antigens (Granzyme-PE 1:100, Thermo Fisher; IFNγ-PE 1:200, Biolegend; TNFα-PE 1:100, Biolegend; Foxp3-FITC 1:100, Thermo Fisher). The stained cells were analyzed with LSR II Analyzer (BD Biosciences) and with FlowJo program.
Single cell cDNA synthesis and sequencing library preparation
Single cell RNA-seq was performed using Smart-seq2 protocol with modifications (Picelli et al., 2014) Briefly, single cells from target tumor population were sorted into 2μl lysis buffer (0.2% Triton X-100, 20 U/μl SUPERase In RNAse inhibitor (Thermo Fisher), 0.25 μM oligo-dT30VN primer, and 1:8,000,000 ERCC spike-in RNAs control (Ambion)). The 96-well plates containing sorted cells were then heated at 72°C for 3 min to lyse the cells followed by reverse transcription with 4U/ml Maxima H-transcriptase (Thermo Fisher), template switching reaction and PCR pre-amplification (15 cycles) according to the protocol (Picelli et al., 2014). PCR products were then cleaned and purified with 0.8 × AMPure XP beads and the quality of cDNA libraries was evaluated by qPCR amplifying housekeeping gene Ppib2 to exclude any empty wells or low quality libraries. 50–100 pg of cDNA library was then used to generating Illumina sequencing library using the Nextera XT DNA library preparation kits (Illumina). The pooled sequencing libraries were then sequenced with Illumina Nextseq 500 instrument using 75 bp paired-end-reads setting.
SCC tumor/T cell co-culture
CD8+ T cells were purified from the spleen of the WT C57/BL6 mice, through the use of a Mojo CD8 T cell Negative Selection kit. Naïve T cells were then labeled with 5 μM Alexa Fluor 488 conjugated Carboxyfluorescein succinimidyl ester (CFSE, Invitrogen) dye for 10 minutes at 37°C. The purified CD8 T cells were then activated by plate-coated or Dynabeads-coated CD3/CD28 Ab. In the meantime, CD80 (+) or CD80(−) PDVC57 SCC cells were pre-treated with mitomycin c (4 μg/ml) for 2 hours to arrest their proliferation, and then added on Day 2 post T cell activation and added to T cells at 1:1 ratio in T cell medium.
T cell proliferation, a sign of successful activation, was measured by flow cytometry quantification of CFSE retention in T cells after 3 days co-culture. For tumor/OT-I T cell co-cultures, splenocytes from OT-I mice were activated with OVA 257–264 (SIINFEKL) peptide (2 μg/ml) (InvivoGen) for 2 days, and then the CD8+ T cells were purified, labeled with CFSE, and added to CD80 (+) or CD80 (−) SCC cells at 1:1 ratio, together with 10 μg/ml CD28 agonist Ab (clone 37.51 or D665), 10 μg/ml CD28 antagonist Ab (mPEG PV1-Fab’)(Papotto et al., 2017), 25 μg/ml CTLA4 blocking Ab (clone UC10–4F10–11), or 25 μg/ml PDL1 blocking Ab (clone 10F.9G2) and co-culture in T cell medium for 3 days
Immunofluorescence and imaging
The dissected tumor were fixed with 4% PFA in PBS for 1 hour at 4°C, washed then incubated in 30% sucrose in PBS O/N at 4°C, then embedded in OCT (Tissue Tek). Cryosections were permeabilized, blocked and stained with the following primary Ab: Integrin a6 (rat,1:2,000, BD), RFP/mCherry (Guinea Pigs, 1:5,000, Fuchs Lab), K5 (Guinea Pigs, 1:2,000, Fuchs Lab), K10 (Rabbit, 1:2,000, Fuchs Lab), K14 (Chicken, 1:1000, Covance), mCD80 (Goat, 1:50, R&D), hCD80 (Goat, 1:50, R&D), pSMAD2 (rabbit, 1:1000, Cell Signaling), cleaved Caspase-3 (rabbit, 1:500, Cell Signaling), CD8 (Rat, 1:100, Abcam). For pSMAD2 immuno-staining, the Tyramide SuperBoost™ kit (Thermo Fisher) kit were used to enhance the signal. The slides were then stained with secondary Abs conjugated with Alexa 488, 546, or 647 (Life Technologies) and imaged on Zeiss Axio Observer Z1 equipped with ApoTome.2. The images were collected using Zeiss ZEN software and analyzed using ImageJ Software.
RNA purification and qRT-PCR
The FACS-purified target tumor populations were sorted directly into TRI Reagent (Sigma) and the total RNA was purified using Direct-zol RNA MiniPrep Kit (Zymo Research) in accordance to manufacture’s instructions. For real-time qRT-PCR analysis of any target genes, equal amount of RNA were reverse transcribed using SuperScript VILO cDNA synthesis kit (Life Technologies). cDNAs were then mixed with specific gene primes and SYBE green qPCT Master Mix (Sigma) and the qRT-PCR was performed on an Applied Biosystems 7900HT Fast Real-Time PCR system.
CRISPR mediated CD80 knockout
The Alt-R® CRISPR-Cas9 System (IDT) was used to generate the Cd80-null PDVC57 line. Briefly, a ribonucleoprotein (RNP) formed by an efficient sgRNA targeting Exon 3 of mouse CD80 (CATCAATACGACAATTTCCC, synthesized by IDT), an ATTO™ 550 fluorescent dye conjugated transactivating crRNA (IDT), and a recombinant Cas9 endonuclease (IDT) was transfected into PDVC57 line using RNAiMax (Thermo Fisher). The transfected cells were FACS purified, and then each single cell was sorted into an individual well of a 96 well plate. Single cell colonies were then expanded, and clones deficient for CD80 were selected for experiments.
shRNA for Cd80 knockdown
Hairpin from Broad Institute’s Mission TRC mouse library targeting mouse Cd80 (CCGAGTATAAGAACCGGACTT) were cloned into pLKO-H2B-GFP vector and transduced into PDVC57 cells in order to complement our CRISPR-mediated Cd80 knockout, and thus ensure that any phenotype from the CRISPR-mediated procedure was not a result of non-specific targeting of the sgRNA.
OT-I-specific Ctla4 silencing
Hairpin targeting mouse Ctla4 (GGAGCATGAACAGAGAGCT) was cloned into a modified pMKO.1 vector (pMKO.1-Thy1.1). OT-I CTLs were isolated and stimulated (1 μg/ml CD3 antibody, 0.5 μg/ml CD28 antibody, and 100U/ml recombinant IL2) for 24 hours. The OT-I T cells were then spin-infected with Thy1.1-Ct/a4 or Thy1.1-Scramble Control shRNA retrovirus (1000g 90 min) in the retronectin (20 μg/ml) coated plate. 48 hours later, the tranduced OT-I T cells were purified by positive selection using biotinylated Thy1.1 antibody (Biolegend) with magnetic nanobeads (R&D). The Thy1.1+ OT-I T cells were then mixed with irradiated splenocytes pulsed with OVA 257–264 (SIINFEKL) peptide (2 μg/ml). 5 days later, the expanded Thy1.1+ OT-I T cells were injected into OVA+ skin SCC for ACC.
QUANTIFICATION AND STATISTICAL ANALYSIS
Single-cell RNA-seq analysis
To analyze the sequencing data, the reads were first aligned to the mouse reference genome (Version mm10 from UCSC), together with the reads for ERCC spike-ins using Bowtie2 (version 2.2.9). Using RSEM (v 1.2.30), the expression level for each gene were quantified as TPM (Li and Dewey, 2011) which was then transformed to log2(TPM + 1). Any cells with less than 4000 genes detected or any genes expressed in less than 5 cells were removed. Genes with level of variation above the technical variation and FDR value less than 0.1 were considered as highly variable. The list of highly variable genes were analyzed with principle component analysis (PCA), in order to identify cell clusters. The significant PCs were applied to unsupervised hierarchical clustering using ward’s method and Euclidean distance. The results of PCA were then used to perform t-distributed stochastic neighbor embedding (t-SNE), to present the high dimensional data in two-dimensional space, and the expression level of any interested genes was represented in the t-SNE plots. Additionally, to identify the gene signatures of each cluster, we normalized raw counts which were then applied to DEseq package (Anders and Huber, 2010) using R. Any genes with fold change between clusters >2 and FDR <0.1 or 0.3 were considered differentially expressed. Signature genes of each cluster were defined as genes that are >2× differentially expressed, and unique for each cluster. Gene Ontology (GO) analysis was performed with differentially expressed genes between clusters using DAVID v6.721,22. (Huang da et al., 2009) Pathways enriched with FDR<0.25 were considered to be significant.
Statistics
Statistics analysis (unpaired two-tailed student’s t test, with a 95% confidence interval under the untested assumption of normality) was performed in Prism 6 (GraphPad). Data are presented as mean ± SEM or mean ± 95% confidence interval. Group size was determined by power analysis on the basis of preliminary experiment results. Experiments were not performed in a blinded manner, Significant difference between two groups were noted by asterisks (* p<0.01; ** p<0.005: *** p<0.001).
DATA AND SOFTWARE AVAILABILITY
Raw and analyzed data is available at NCBI and the accession number for the single cell RNA sequencing data reported in this paper are NCBI GEO: GSE108679
Supplementary Material
(A) Flow cytometry quantifications of tumor infiltrating CD8+ CTLs, CD19+ B cells and NK1.1+ NK cells in a naïve HRASG12V skin tumor before ACT treatment.
(B) Flow cytometry and numerical analysis of tumor infiltrating CD4+ Foxp3+ Regulatory T (Treg) cells in a naïve HRASG12V skin tumor, HRASG12V skin tumor receiving one dose of cyclophosphamide (Cyclo), or cyclophosphamide plus ACT treatment.
(C) Flow cytometry and numerical analysis of tumor infiltrating CD11b+Ly6G+Ly6Cl° polymorphonuclear (PMN) or CD11b+Ly6G−Ly6Chi monocytic (M)-myeloid-derived suppressor cells (MDSCs) in a naïve HRASG12V skin tumor, HRASG12V skin tumor receiving one dose of cyclophosphamide (Cyclo), or cyclophosphamide plus ACT treatment ± Gr1 blocking Ab to deplete MDSCs.
(D) Flow cytometry and numerical analysis of infiltrating CD11b+MHCII+CD64+ tumor associated macrophages (TAMs) in a naïve HRASG12V skin tumor, HRASG12V skin tumor receiving one dose of cyclophosphamide (Cyclo), or cyclophosphamide plus ACT treatment ± CSF1R blocking Ab to deplete TAMs.
(E) Flow cytometry analysis of T cell activation markers (CD25, CD69, CD44 and CD62L) in the OT-I T cells at the first and second weeks after ACT. The naïve CD8+ T cells from donor spleen were used as negative control.
Table S1. Differentially expressed genes in C1 (CSC) cluster compared to other tumor cell clusters (C2, C3, C4). Related to Figure 2
Table S2. Gene ontology analyses of C1 transcripts up-regulated by ≥2X compared to other tumor cell clusters (C2, C3, C4) (p<0.05). Related to Figure 2
Table S3. Gene ontology analyses of C1 transcripts down-regulated by ≥2X compared to other tumor cell clusters (C2, C3, C4) (p<0.05). Related to Figure 2
Table S4. Differentially expressed genes in FACS sorted Integrin a6Hi TGFβ reporter+ (CSC) C1 cluster compared to other tumor cell populations (C2: Integrin a6Hi TGFβ reporter− basal cells; C3: Integrin a6− TGFβ reporter− suprabasal cells). Related to Figure 4.
(A) Sorting strategy for purifying live, non-epithelial lineage negative, mCherry+ epithelial cells from tumors remaining after ACT treatment.
(B) Heatmap of RNA-seq analyses of individual ACT-surviving tumor cells from (A), revealing transcriptome similarities between single cells as measured by Pearson’s correlation coefficient matrix.
(C) t-SNE plots showing expression of various epithelial-specific keratin markers in ACT survivors. Krt18 is expressed specifically by SCC basal cells and is seen in the C1 cluster. Krt1, Krt10, Krt6, and Krt16 are markers of suprabasal cells of well-differentiated SCCs and are seen in C2, C3, C4 clusters.
(D) Sorting strategy for purifying integrin α6hi TGFβ-reporter+ tSCs (green), as well as non-TGFβ-responding basal progenitors (red) and suprabasal cells (black) from naïve HRasG12V skin tumors.
(E) Single cell RNA-seq of individual tumor cells. Shown are unbiased clustering of transcriptomes of FACs purified individual integrin α6hi TGFβ reporter+ tSCs (C1), non-TGFβ-responding basal progenitors (C2), and suprabasal cells (C3). Each cell is represented as a dot, colored by clustering algorithm and plotted on the t-SNE graph.
(F) t-SNE plots showing enriched expression of stem cell signature genes in the C1 cluster representing the integrin α6hi TGFβ-reporter+ tSCs.
(G) t-SNE plot showing the absence of immune cell specific markers, including Cd45 for pan immune cells, Cd11b for myeloid cells, Cd11c for dendritic cells and Cd207for Langerhan cells.
(A) Representative IMF and quantification of Ki67 (green), TGFβ-reporter (red) and Integrin α6 (blue) in tumor at 1 week post-ACT. Three tumors and >150 cells from each of two sagittal sections per tumor were analyzed. Scale bars, 50 μm.
(B) Representative IMF and quantification of EdU (green), TGFβ-reporter (red) and Integrin α6 (blue) in tumor at 1 week post-ACT after two pulses of EdU at 24 hrs and 12 hrs before collection. Three tumors and >150 cells from each of two sagittal sections per tumor were analyzed. Scale bars, 50 μm.
(C) (Top left) Experimental scheme for tSC lineage tracing. (Top right), quantifications of tSC markers expressed by Tomato+ cells following short-term tamoxifen treatment. (Bottom)
Representative IMF images confirming that the TGFβ-reporter drives CreER activity specifically in stem cells, which are positive for CD44 and CD34. Note that most tSCs (CD44Hi and CD34Hi) are marked by Tomato. >150 cells from each of two sagittal sections per tumor were analyzed. Scale bars, 50 μm.
(D) Lineage-tracing of TGFβ-responding tSCs following ACT therapy. Construct design (Top), experimental scheme (middle), and flow cytometry based quantification (bottom) from lineage-traced tumor at 1 week post-ACT.
(E) Flow cytometry quantifications of integrin α6hi TGFβ-reporter+ tSCs in tumors that are treated with control or blocking Abs targeting Gr1 or CSF1R for one week, then followed by ACT plus the same Ab treatment for another week.
(F) t-SNE plots showing the low expression of PDL1 (Cd274), and broad distribution of Cd47 across different tumor populations.
(A) Immunofluorescence and quantifications of CD80 (green) and pSMAD2+ (blue) cells in Tomato+ tumor cells that relapsed following ACT treatment (red). Data are from 3 tumors and two sagittal sections (>150 cells each) for each tumor analyzed at each time point. All scale bars=50 μm.
(B) Immunofluorescence co-labeling of CD80 (green), pSMAD2/3 (red) and Keratin 14 (blue) in the skin epithelium of a DMBA/TPA induced tumor generated on wild-type C57/BL6 mice (left) or Rag2 −/− mice (right), which lack a functional adaptive immune system.
(C) Immunofluorescence labeling of CD80 (red) and Keratin 14 (green) epithelial cancer basal cells in (left) xenografts of the human skin SCC line A431 on SCID mice or (right) cells that have undergone metastasis to the lung following injection of A431 skin SCC cells into the tail vein of SCID mice.
(A) Flow cytometry quantification of proliferation marker (Ki67) in cytotoxic T cells that infiltrated CD80(+) or CD80(−) PDVC57 C57/Bl6 SCCs generated from engraftment into C57Bl/6 immune competent mice.
(B) Isogenically matched, PDVC57 SCC cells were transduced with LV expressing Scrambled shRNA or Cd80 shRNA and engrafted onto WT C57/Bl6 mice. Note that Cd80 silencing diminished tumorigenesis, analogous to what we observed with CRISPR/CAS-mediated ablation of Cd80. Data are mean ± SEM. ***P < 0.001, unpaired Student’s t test, n=6.
(C) Cells as in (A) were engrafted onto Rag2−/− mice, which lack an adaptive immune system. Note that loss of adaptive immunity eliminates the tumorigenesis defects from CD80 silencing. n=15; data are mean ± SEM.
(D) Cells as in (A) were engrafted onto wild-type mice that had been immunodepleted for CD8+ T cells. Note that loss of cytotoxic T cells eliminated the tumorigenic defects resulting from Cd80 silencing in the SCC cells. n=15; data are mean ± SEM.
(E) Flow cytometry quantifications of the CD8+ T cells infiltrating Scrambled shRNA or Cd80 shRNA transduced tumors (percentage of total CD45+ cells).
(A) Scheme showing the experimental setup for SCC cell co-culture with activated OT-I cytotoxic T cells to dissect the relative contributions of putative CTL receptors for the surface CD80 on SCC tSCs.
(B) Measurement of OT-I T cell proliferation as monitored by flow cytometry quantification of CFSE dye dilution in activated primary OT-I T cells when co-cultured with CD80(+) or CD80(−) PDVC57 SCC cells, with or without CTLA4 blocking Abs. Data are mean ± SEM. **P < 0.005, unpaired Student’s t test, n=6.
(C) Measurement of OT-I T cell proliferation as monitored by flow cytometry quantifications of CFSE dye dilution in activated primary OT-I T cells when co-cultured with CD80(+) or CD80(−) PDVC57 SCC cells, with or without CTLA4 blocking Abs, CD28 agonist Abs or CD28 antagonist Abs. Data are mean ± SEM. **P < 0.005, unpaired Student’s t test, n=6.
(D) Measurement of OT-I T cell proliferation as monitored by flow cytometry quantifications of CFSE dye dilution in activated primary OT-I CD8+ T cells when co-cultured with CD80(+) or CD80(−) PDVC57 SCC cells, with or without PDL1 blocking Abs, which eliminate possible interactions between PDL1 on the surface of PDVC57 tumor cells and either PD1 on the surface of CD8+ T cells or tumor CD80 (CIS interactions). See text for further details. Data are mean ± SEM. **P <0.005, unpaired Student’s t test, n=6.
(A) CD80(+) or CD80(−) PDVC57 SCC cells were transplanted into C57/BL6 mice treated with either isotype control or CTLA4 blocking Abs, and tumor weight was analyzed at 4 weeks post-grafting. Data are mean ± SEM.
(B) Scheme showing the experimental setup for generating OT-I T cell-specific CTLA4 silencing for use in ACT.
(C) Flow cytometry analysis of T cell exhaustion markers (CTLA4, PD1, Tim3 and LAG3) and proliferation marker (Ki67) in the injected OT-I T cells that had been transduced with control or Ctla4 shRNA and then collected at 2 week post-ACT. The naïve CD8+ T cells from donor spleen were used as negative control.
(D) Flow cytometry quantifications of integrin α6hi TGFβ-reporter+ tSCs in mice treated with control or Ctla4 shRNA transduced OT-I T cells at 1 week post-ACT.
(E) Flow cytometry based quantification of lineage-traced (GFP+), TGFβ-responding (mCherry+) Cd80-null tSCs in Cd80-null tumors following 1 week post-ACT. Note that CreER was selectively activated in naive tSCs at a time when all differentiated tumor cells were GFP(−). Thus, had the tSCs present in ACT-treated tumors been derived from de-differentiation of differentiated tumor cells, they would be mCherry+ but many would be GFP(−).
(F) Changes in tumor size following depletion of injected OT-I T cells after ACT treating the TGFβ-responding tSC specific CD80 KO tumors. Tumor volumes are normalized to their original size. Data are mean ± SEM.
(G) Volume changes of the relapsed Scr sgRNA or Cd80 sgRNA transduced tumors following ACT. n=12 for control; n=7 for tSC-specific targeting of Cd80. Data are mean ± SEM.
(H) Immunofluorescence for CD80 (green), integrin α6hi (blue) and TGFβ-reporter (red) to examine tumor cells that relapsed following ACT treatment. Prior to treatment, Cd80 was depleted specifically in TGFβ-responding tSCs within the tumors of these mice. Note that, whereas majority of a6+TGFβ-reporter+ tSCs in these relapsed tumor are CD80 negative (arrowhead), a few TGFβ-reporter+ tSCs are CD80 positive (arrow). All scale bars=50 μm.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mCherry/RFP, guinea pig polyclonal | Fuchs lab | N/A |
| Anti-K6, rabbit polyclonal | Fuchs lab | N/A |
| Anti-K5, guinea pig polyclonal | Fuchs lab | N/A |
| Anti-Loricrin, Rabbit polyclonal | Fuchs lab | N/A |
| Anti-K10, rabbit polyclonal | Covance | PRB-159P |
| Anti-K14, chicken polyclonal | Covance | SIG-3476 |
| Biotin conjugated anti-CD45, rat monoclonal (Clone 30-F11) | Biolegend | Cat#103104 |
| Biotin conjugated anti-CD31, rat monoclonal (Clone MEC13.3) | Biolegend | Cat#102504 |
| Biotin conjugated anti-CD117, rat monoclonal (Clone 2B8) | Biolegend | Cat#105804 |
| Biotin conjugated anti-CD140a, rat monoclonal (Clone APA5) | Biolegend | Cat#135910 |
| Biotin conjugated anti-Thy1.1, mouse monoclonal (Clone OX7) | Biolegend | Cat#202510 |
| PE/Cy7 anti-mouse/human CD44, rat monoclonal (Clone IM7) | Biolegend | Cat#103030 |
| PerCP/Cy5.5 anti-mouse CD49f, rat monoclonal (Clone GoH3) | Biolegend | Cat# 313617 |
| Alexa Fluor® 700 anti-mouse CD45 rat monoclonal (Clone 30-F11) | Biolegend | Cat# 103128 |
| PE/Cy7 anti-mouse CD4 rat monoclonal (Clone GK1.5) | Biolegend | Cat# 100421 |
| APC/Cy7 anti-mouse CD8b rat monoclonal (Clone YTS156.7) | Biolegend | Cat# 126619 |
| Brilliant Violet 605™ anti-mouse CD19 rat monoclonal (Clone 6D5) | Biolegend | Cat# 115539 |
| Brilliant Violet 421™ anti-mouse CD25 rat monoclonal (Clone PC61) | Biolegend | Cat# 102033 |
| PE/Cy5 anti-mouse NK-1.1 mouse monoclonal (Clone PK136) | Biolegend | Cat# 108715 |
| PerCP/Cy5.5 anti-TCR β Hamster monoclonal (Clone H57–597) | Biolegend | Cat# 109227 |
| FITC anti-mouse CD80 Hamster monoclonal (Clone 16–10A1) | Biolegend | Cat# 104705 |
| FITC anti-mouse Foxp3 Rat monoclonal (Clone FJK-16s) | eBioscience | Cat# 11-5773-82 |
| Anti-Integrin-a6/CD49f, rat monoclonal (Clone GoH3) | eBioscience | Cat# 14-0495 |
| PE anti-TNF alpha rat monoclonal (Clone MP6-XT22) | eBioscience | Cat# 12-7321-41 |
| FITC Anti-Mouse CD34 rat monoclonal (Clone RAM34) | eBioscience | Cat# 11-0341-82 |
| APC Anti-Mouse OVA257–264 (SIINFEKL)/MHCI Mouse monoclonal (Clone eBio25-D1.16) | eBioscience | Cat# 17-5743 |
| PE anti- IFNγ rat monoclonal (Clone XMG1.2) | eBioscience | Cat# 12-7311-41 |
| PE anti- Granzyme B rat monoclonal (Clone NGZB) | eBioscience | Cat# 12-8898-80 |
| Anti-CD3e Hamster monoclonal (Clone 145–2C11) | eBioscience | Cat# 16-0031-82 |
| Anti- CD28 monoclonal (Clone 37.51) | eBioscience | Cat# 16-0281-82 |
| Anti-CD8 alpha rat monoclonal (Clone YTS 169.4) | BioXCell | Cat# BE0117 |
| Anti-CTLA-4 (CD.152) Hamster monoclonal (Clone UC10–4F10–11) | BioXCell | Cat# BE0032 |
| Anti-TGFβ monoclonal (Clone 1D11.16.8) | BioXCell | Cat# BE0057 |
| Anti-Ly6G/Ly6C(Gr1) monoclonal (Clone RB6–8C5) | BioXCell | Cat# BE0075 |
| Anti-CSF1 R(CD115) monoclonal (Clone AFS98) | BioXCell | Cat# BE0213 |
| Anti- Mouse B7–1/CD80 Goat polyclonal | R&D | Cat# AF740 |
| Anti- Human B7–1/CD80 Goat polyclonal | R&D | Cat# AF140 |
| Anti-Anti-CD80 Rabbit polyclonal | Abcam | Cat# ab64116 |
| Anti-CD8 rat monoclonal (Clone YTS169.4) | Abcam | Cat# ab22378 |
| Anti-Cleaved Caspase-3(Asp175) Rabbit monoclonal(Clone 5A1E) | Cell Signaling | Cat# 9664 |
| Anti-Phospho-Smad2 (Ser465/467) Rabbit monoclonal(Clone 138D4) | Cell Signaling | Cat# 3108 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Tamoxifen | Sigma | Cat#T5648 |
| Diptheria toxin | Sigma | Cat# D0564 |
| Cyclophosphamide | Sigma | Cat# PHR1404 |
| 7,12-Dimethylbenz[a]anthracene | Sigma | Cat# D3254 |
| 12-O-T etradecanoylphorbol 13-acetate | Sigma | Cat# P1585 |
| Matrigel Matrix (High Concentration) | Corning | Cat# 354262 |
| OVA 257–264 | InvivoGen | Cat# vac-sin |
| Recombinant Mouse IL-2 | R&D | Cat# 402-ML-020 |
| Recombinant Human IL-2 | PeproTech | Cat# 200–02 |
| Critical Commercial Assays | ||
| CellTrace™ CFSE Cell Proliferation Kit, for flow cytometry | Thermo | Cat# C34554 |
| Leukocyte Activation Cocktail with GolgiPlug TM | BD | Cat# 550583 |
| Nextera DNA Sample Preparation Kit | Illumina | Cat# FC-121-1030 |
| Alexa Fluor™ 647 Tyramide SuperBoost™ Kit | Thermo | Cat# B40926 |
| Agencourt AMPure XP | Beckman Coulter | Cat# A63880 |
| Foxp3 / Transcription Factor Staining Buffer Set | eBioscience | Cat# 00-5523-00 |
| MagCollect Streptavidin Ferrofluid | R&D | Cat# MAG999 |
| MojoSort™ Mouse CD8 T Cell Isolation Kit | Biolegend | Cat# 480035 |
| Deposited Data | ||
| Single cell RNaseq data | This Study | GEO: GSE108679 |
| Experimental Models: Cell Lines | ||
| Mouse SCC PDVC57 | (Bremner and Balmain, 1990) | N/A |
| Human SCC A431 | (Yang et al., 2015) | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: TRE-OVA | (Rosenblum et al., 2011) | N/A |
| Mouse: TRE-HrasG12V | (Chin et al., 1999) | N/A |
| Mouse: C57BL/6J | The Jackson Laboratory | 000664 |
| Mouse: B6;129S6-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J | The Jackson Laboratory | 007905 |
| Mouse: B6;129-Gt(ROSA)26Sortm1(CAG-cas9*,-EGFP)Fezh/J | The Jackson Laboratory | 024857 |
| Mouse: B6(Cg)-Rag2tm1.1Cgn/J | The Jackson Laboratory | 008449 |
| Mouse: C57BL/6-Tg(TcraTcrb)1100Mjb/J | The Jackson Laboratory | 003831 |
| Mouse: B6.SJL-Ptprca Pepcb/BoyJ (CD45.1) | The Jackson Laboratory | 002014 |
| Mouse: B6.129(Cg)-Foxp3tm3(DTR/GFP)Ayr/J | The Jackson Laboratory | 016958 |
| OligonucleOT-Ides | ||
| Mouse Cd80 sgRNA1: CATCAATACGACAATTTCCC | This Study | N/A |
| Mouse Cd80 sgRNA2: CGTGTCAGAGGACTTCACCT | This Study | N/A |
| Mouse Cd80 shRNA1: CCGAGTATAAGAACCGGACTT | This Study | N/A |
| Mouse Ctla4 shRNA1: GGAGCATGAACAGAGAGCT | This Study | N/A |
| Recombinant DNA | ||
| pLKO-rtTA-P2A-mCherry | This Study | N/A |
| pLKO-PGK-rtTA-SBE-mCherry | This Study | N/A |
| pLKO- PGK-rtTA-SBE-CreER | This Study | N/A |
| pLKO-PGK-rtTA-SBE-mCherry-P2A-CreER-U6-Scr Control sgRNA | This Study | N/A |
| pLKO-PGK-rtTA-SBE-mCherry-P2A-CreER-U6-Cd80 sgRNA | This Study | N/A |
| pMKO.1-Thy1.1-Scr Control shRNA | This Study | N/A |
| pMKO.1-Thy1.1-Ctla4 shRNA | This Study | N/A |
| Software and Algorithms | ||
| Fiju (ImageJ) | https://fiji.sc/ | N/A |
| Bowtie2 | (Langmead and Salzberg, 2012) | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| RSEM | (Li and Dewey, 2011) | http://deweylab.github.io/RSEM/ |
| DESeq | (Anders and Huber, 2010) | http://bioconductor.org/packages/release/bioc/html/DESeq.html |
| Gene Set Enrichment Analysis | (Subramani an et al., 2005) | http://software.broadinstitute.org/gsea/index.jsp |
| Mclust (R package) | (Fraley and Raftery, 2002) | https://cran.r-project.org/ |
| Picard | Broad Institute | https://github.com/broadinstitute/picard/releases/tag/2.7.1 |
| FlowJo | https://www.flowjo.com | N/A |
| Other | ||
| BD FACSAria II Cell Sorter | BD Biosciences | N/A |
| BD LSR-Fortessa analyzer | BD Biosciences | N/A |
| Illumina Nexseq 500 instrument | Illumina | N/A |
Highlights:
Tumor-initiating stem cells have enhanced resistance to T-cell immunotherapy
TGFβ-responding stem cells form the root of relapse after adoptive cell transfer
Tumor-initiating stem cells can acquire CD80 to modulate cytotoxic T cell attack
CTLA4 or TGFβ-blocking antibodies can render stem cell susceptibility to treatment
ACKNOWLEDGMENTS
We thank M. Nikolova, E. Wong and J. Racelis for technical assistance; N. Oshimori, S. Naik, S. Larsen, S. Liu for discussions; H.Johnson (Weill Cornell Medical College) for human SCC samples; Semova, S. Han and S. Tadesse (Rockefeller University Flow Cytometry Core) for conducting FACS sorting; C. Lai (Rockefeller University Genomics Core) for single cell RNA-sequencing and raw data analyses. We thank A. Balmain (University of California San Francisco) for PDVC57 cells and B. Vanhove (Ash Clinical Research Training Institute) for CD28 blocking Ab. E.F. is a Howard Hughes Medical Investigator. Y.M. is a Jane Coffin Childs Postdoctoral Fellow. This study was supported by grants to E.F. from the National Institutes of Health (R01-AR050452, R37-AR27883) and New York State (C32585GG).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF FINANCIAL INTERESTS.
The authors declare no competing financial interests.
REFERENCES
- Agudo J, Park ES, Rose SA, Alibo E, Sweeney R, Dhainaut M, Kobayashi KS, Sachidanandam R, Baccarini A, Merad M, et al. (2018). Quiescent Tissue Stem Cells Evade Immune Surveillance. Immunity 48, 271–285 e275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alegre ML, Frauwirth KA, and Thompson CB (2001). T-cell regulation by CD28 and CTLA-4. Nat Rev Immunol 1, 220–228. [DOI] [PubMed] [Google Scholar]
- Anders S, and Huber W (2010). Differential expression analysis for sequence count data. Genome Biol 11, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batlle E, and Clevers H (2017). Cancer stem cells revisited. Nat Med 23, 1124–1134. [DOI] [PubMed] [Google Scholar]
- Beck B, Driessens G, Goossens S, Youssef KK, Kuchnio A, Caauwe A, Sotiropoulou PA, Loges S, Lapouge G, Candi A, et al. (2011). A vascular niche and a VEGF-Nrp1 loop regulate the initiation and stemness of skin tumours. Nature 478, 399–403. [DOI] [PubMed] [Google Scholar]
- Beronja S, Janki P, Heller E, Lien WH, Keyes BE, Oshimori N, and Fuchs E (2013). RNAi screens in mice identify physiological regulators of oncogenic growth. Nature 501, 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beronja S, Livshits G, Williams S, and Fuchs E (2010). Rapid functional dissection of genetic networks via tissue-specific transduction and RNAi in mouse embryos. Nat Med 16, 821–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boumahdi S, Driessens G, Lapouge G, Rorive S, Nassar D, Le Mercier M, Delatte B, Caauwe A, Lenglez S, Nkusi E, et al. (2014). SOX2 controls tumour initiation and cancer stem-cell functions in squamous-cell carcinoma. Nature 511, 246–250. [DOI] [PubMed] [Google Scholar]
- Bremner R, and Balmain A (1990). Genetic changes in skin tumor progression: correlation between presence of a mutant ras gene and loss of heterozygosity on mouse chromosome 7.Cell 61, 407–417. [DOI] [PubMed] [Google Scholar]
- Brown JA, Yonekubo Y, Hanson N, Sastre-Perona A, Basin A, Rytlewski JA, Dolgalev I, Meehan S, Tsirigos A, Beronja S, et al. (2017). TGF-beta-Induced Quiescence Mediates Chemoresistance of Tumor-Propagating Cells in Squamous Cell Carcinoma. Cell Stem Cell 21, 650–664 e658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreno BM, and Collins M (2002). The B7 family of ligands and its receptors: new pathways for costimulation and inhibition of immune responses. Annu Rev Immunol 20, 29–53. [DOI] [PubMed] [Google Scholar]
- Chaudhri A, Xiao Y, Klee AN, Wang X, Zhu B, and Freeman GJ (2018). PD-L1 binds to B7–1 only in cis on the same cell surface. Cancer Immunol Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin L, Tam A, Pomerantz J, Wong M, Holash J, Bardeesy N, Shen Q, O’Hagan R, Pantginis J, Zhou H, et al. (1999). Essential role for oncogenic Ras in tumour maintenance. Nature 400, 468–472. [DOI] [PubMed] [Google Scholar]
- David CJ, Huang YH, Chen M, Su J, Zou Y, Bardeesy N, Iacobuzio-Donahue CA, and Massague J (2016). TGF-beta Tumor Suppression through a Lethal EMT. Cell 164, 1015–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuPage M, Mazumdar C, Schmidt LM, Cheung AF, and Jacks T (2012). Expression of tumour-specific antigens underlies cancer immunoediting. Nature 482, 405–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraley C, and Raftery AE (2002). Model-based clustering, discriminant analysis, and density estimation. J Am Stat Assoc 97, 611–631. [Google Scholar]
- Ganesh K, and Massague J (2018). TGF-beta Inhibition and Immunotherapy: Checkmate. Immunity 48, 626–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, and Lempicki RA (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Khong HT, and Restifo NP (2002). Natural selection of tumor variants in the generation of “tumor escape” phenotypes. Nat Immunol 3, 999–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreso A, and Dick JE (2014). Evolution of the cancer stem cell model. Cell Stem Cell 14, 275–291. [DOI] [PubMed] [Google Scholar]
- Landsberg J, Kohlmeyer J, Renn M, Bald T, Rogava M, Cron M, Fatho M, Lennerz V, Wolfel T, Holzel M, et al. (2012). Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation. Nature 490, 412–416. [DOI] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapouge G, Beck B, Nassar D, Dubois C, Dekoninck S, and Blanpain C (2012). Skin squamous cell carcinoma propagating cells increase with tumour progression and invasiveness. EMBO J 31, 4563–4575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapouge G, Youssef KK, Vokaer B, Achouri Y, Michaux C, Sotiropoulou PA, and Blanpain C (2011). Identifying the cellular origin of squamous skin tumors. Proc Natl Acad Sci U S A 108, 7431–7436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Balmain A, and Counter CM (2018). A model for RAS mutation patterns in cancers: finding the sweet spot. Nat Rev Cancer 18, 767–777. [DOI] [PubMed] [Google Scholar]
- Mackensen A, Meidenbauer N, Vogl S, Laumer M, Berger J, and Andreesen R (2006). Phase I study of adoptive T-cell therapy using antigen-specific CD8+ T cells for the treatment of patients with metastatic melanoma. J Clin Oncol 24, 5060–5069. [DOI] [PubMed] [Google Scholar]
- Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, Kadel EE III, Koeppen H, Astarita JL, Cubas R, et al. (2018). TGFbeta attenuates tumour response to PDL1 blockade by contributing to exclusion of T cells. Nature 554, 544–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellman I, Coukos G, and Dranoff G (2011). Cancer immunotherapy comes of age. Nature 480, 480–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassar D, Latil M, Boeckx B, Lambrechts D, and Blanpain C (2015). Genomic landscape of carcinogen-induced and genetically induced mouse skin squamous cell carcinoma. Nat Med 21, 946–954. [DOI] [PubMed] [Google Scholar]
- Nicolas M, Wolfer A, Raj K, Kummer JA, Mill P, van Noort M, Hui CC, Clevers H, Dotto GP, and Radtke F (2003). Notch1 functions as a tumor suppressor in mouse skin. Nat Genet 33, 416–421. [DOI] [PubMed] [Google Scholar]
- Oshimori N, Oristian D, and Fuchs E (2015). TGF-beta promotes heterogeneity and drug resistance in squamous cell carcinoma. Cell 160, 963–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papotto PH, Marengo EB, Sardinha LR, Carvalho KI, de Carvalho AE, Castillo-Mendez S, Jank CC, Vanhove B, Goldberg AC, and Rizzo LV (2017). Novel CD28 antagonist mPEG PV1-Fab’ mitigates experimental autoimmune uveitis by suppressing CD4+ T lymphocyte activation and IFN-gamma production. PLoS One 12, e0171822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peggs KS, Quezada SA, Chambers CA, Korman AJ, and Allison JP (2009). Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J Exp Med 206, 1717–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picelli S, Faridani OR, Bjorklund AK, Winberg G, Sagasser S, and Sandberg R (2014). Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc 9, 171–181. [DOI] [PubMed] [Google Scholar]
- Platt RJ, Chen S, Zhou Y, Yim MJ, Swiech L, Kempton HR, Dahlman JE, Parnas O, Eisenhaure TM, Jovanovic M, et al. (2014). CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 159, 440–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Restifo NP, Dudley ME, and Rosenberg SA (2012). Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol 12, 269–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblum MD, Gratz IK, Paw JS, Lee K, Marshak-Rothstein A, and Abbas AK (2011). Response to self antigen imprints regulatory memory in tissues. Nature 480, 538–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schober M, and Fuchs E (2011). Tumor-initiating stem cells of squamous cell carcinomas and their control by TGF-beta and integrin/focal adhesion kinase (FAK) signaling. Proc Natl Acad Sci U S A 108, 10544–10549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramek D, Sendoel A, Segal JP, Beronja S, Heller E, Oristian D, Reva B, and Fuchs E (2014). Direct in vivo RNAi screen unveils myosin IIa as a tumor suppressor of squamous cell carcinomas. Science 343, 309–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwitalla S, Fingerle AA, Cammareri P, Nebelsiek T, Goktuna SI, Ziegler PK, Canli O, Heijmans J, Huels DJ, Moreaux G, et al. (2013). Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell 152, 25–38. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, Sevillano M, Ibiza S, Canellas A, Hernando-Momblona X, et al. (2018). TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 554, 538–543. [DOI] [PubMed] [Google Scholar]
- Tran E, Robbins PF, Lu YC, Prickett TD, Gartner JJ, Jia L, Pasetto A, Zheng Z, Ray S, Groh EM, et al. (2016). T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N Engl J Med 375, 2255–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travis MA, and Sheppard D (2014). TGF-beta activation and function in immunity. Annu Rev Immunol 32, 51–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Merwe PA, Bodian DL, Daenke S, Linsley P, and Davis SJ (1997). CD80 (B7–1) binds both CD28 and CTLA-4 with a low affinity and very fast kinetics. J Exp Med 185, 393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker LS, and Sansom DM (2011). The emerging role of CTLA4 as a cell-extrinsic regulator of T cell responses. Nat Rev Immunol 11, 852–863. [DOI] [PubMed] [Google Scholar]
- Wherry EJ, and Kurachi M (2015). Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 15, 486–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White AC, Tran K, Khuu J, Dang C, Cui Y, Binder SW, and Lowry WE (2011). Defining the origins of Ras/p53-mediated squamous cell carcinoma. Proc Natl Acad Sci U S A 108, 7425–7430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Schramek D, Adam RC, Keyes BE, Wang P, Zheng D, and Fuchs E (2015). ETS family transcriptional regulators drive chromatin dynamics and malignancy in squamous cell carcinomas. Elife 4, e10870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Huynh A, Whitcher G, Chang J, Maltzman JS, and Turka LA (2013). An obligate cell-intrinsic function for CD28 in Tregs. J Clin Invest 123, 580–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Flow cytometry quantifications of tumor infiltrating CD8+ CTLs, CD19+ B cells and NK1.1+ NK cells in a naïve HRASG12V skin tumor before ACT treatment.
(B) Flow cytometry and numerical analysis of tumor infiltrating CD4+ Foxp3+ Regulatory T (Treg) cells in a naïve HRASG12V skin tumor, HRASG12V skin tumor receiving one dose of cyclophosphamide (Cyclo), or cyclophosphamide plus ACT treatment.
(C) Flow cytometry and numerical analysis of tumor infiltrating CD11b+Ly6G+Ly6Cl° polymorphonuclear (PMN) or CD11b+Ly6G−Ly6Chi monocytic (M)-myeloid-derived suppressor cells (MDSCs) in a naïve HRASG12V skin tumor, HRASG12V skin tumor receiving one dose of cyclophosphamide (Cyclo), or cyclophosphamide plus ACT treatment ± Gr1 blocking Ab to deplete MDSCs.
(D) Flow cytometry and numerical analysis of infiltrating CD11b+MHCII+CD64+ tumor associated macrophages (TAMs) in a naïve HRASG12V skin tumor, HRASG12V skin tumor receiving one dose of cyclophosphamide (Cyclo), or cyclophosphamide plus ACT treatment ± CSF1R blocking Ab to deplete TAMs.
(E) Flow cytometry analysis of T cell activation markers (CD25, CD69, CD44 and CD62L) in the OT-I T cells at the first and second weeks after ACT. The naïve CD8+ T cells from donor spleen were used as negative control.
Table S1. Differentially expressed genes in C1 (CSC) cluster compared to other tumor cell clusters (C2, C3, C4). Related to Figure 2
Table S2. Gene ontology analyses of C1 transcripts up-regulated by ≥2X compared to other tumor cell clusters (C2, C3, C4) (p<0.05). Related to Figure 2
Table S3. Gene ontology analyses of C1 transcripts down-regulated by ≥2X compared to other tumor cell clusters (C2, C3, C4) (p<0.05). Related to Figure 2
Table S4. Differentially expressed genes in FACS sorted Integrin a6Hi TGFβ reporter+ (CSC) C1 cluster compared to other tumor cell populations (C2: Integrin a6Hi TGFβ reporter− basal cells; C3: Integrin a6− TGFβ reporter− suprabasal cells). Related to Figure 4.
(A) Sorting strategy for purifying live, non-epithelial lineage negative, mCherry+ epithelial cells from tumors remaining after ACT treatment.
(B) Heatmap of RNA-seq analyses of individual ACT-surviving tumor cells from (A), revealing transcriptome similarities between single cells as measured by Pearson’s correlation coefficient matrix.
(C) t-SNE plots showing expression of various epithelial-specific keratin markers in ACT survivors. Krt18 is expressed specifically by SCC basal cells and is seen in the C1 cluster. Krt1, Krt10, Krt6, and Krt16 are markers of suprabasal cells of well-differentiated SCCs and are seen in C2, C3, C4 clusters.
(D) Sorting strategy for purifying integrin α6hi TGFβ-reporter+ tSCs (green), as well as non-TGFβ-responding basal progenitors (red) and suprabasal cells (black) from naïve HRasG12V skin tumors.
(E) Single cell RNA-seq of individual tumor cells. Shown are unbiased clustering of transcriptomes of FACs purified individual integrin α6hi TGFβ reporter+ tSCs (C1), non-TGFβ-responding basal progenitors (C2), and suprabasal cells (C3). Each cell is represented as a dot, colored by clustering algorithm and plotted on the t-SNE graph.
(F) t-SNE plots showing enriched expression of stem cell signature genes in the C1 cluster representing the integrin α6hi TGFβ-reporter+ tSCs.
(G) t-SNE plot showing the absence of immune cell specific markers, including Cd45 for pan immune cells, Cd11b for myeloid cells, Cd11c for dendritic cells and Cd207for Langerhan cells.
(A) Representative IMF and quantification of Ki67 (green), TGFβ-reporter (red) and Integrin α6 (blue) in tumor at 1 week post-ACT. Three tumors and >150 cells from each of two sagittal sections per tumor were analyzed. Scale bars, 50 μm.
(B) Representative IMF and quantification of EdU (green), TGFβ-reporter (red) and Integrin α6 (blue) in tumor at 1 week post-ACT after two pulses of EdU at 24 hrs and 12 hrs before collection. Three tumors and >150 cells from each of two sagittal sections per tumor were analyzed. Scale bars, 50 μm.
(C) (Top left) Experimental scheme for tSC lineage tracing. (Top right), quantifications of tSC markers expressed by Tomato+ cells following short-term tamoxifen treatment. (Bottom)
Representative IMF images confirming that the TGFβ-reporter drives CreER activity specifically in stem cells, which are positive for CD44 and CD34. Note that most tSCs (CD44Hi and CD34Hi) are marked by Tomato. >150 cells from each of two sagittal sections per tumor were analyzed. Scale bars, 50 μm.
(D) Lineage-tracing of TGFβ-responding tSCs following ACT therapy. Construct design (Top), experimental scheme (middle), and flow cytometry based quantification (bottom) from lineage-traced tumor at 1 week post-ACT.
(E) Flow cytometry quantifications of integrin α6hi TGFβ-reporter+ tSCs in tumors that are treated with control or blocking Abs targeting Gr1 or CSF1R for one week, then followed by ACT plus the same Ab treatment for another week.
(F) t-SNE plots showing the low expression of PDL1 (Cd274), and broad distribution of Cd47 across different tumor populations.
(A) Immunofluorescence and quantifications of CD80 (green) and pSMAD2+ (blue) cells in Tomato+ tumor cells that relapsed following ACT treatment (red). Data are from 3 tumors and two sagittal sections (>150 cells each) for each tumor analyzed at each time point. All scale bars=50 μm.
(B) Immunofluorescence co-labeling of CD80 (green), pSMAD2/3 (red) and Keratin 14 (blue) in the skin epithelium of a DMBA/TPA induced tumor generated on wild-type C57/BL6 mice (left) or Rag2 −/− mice (right), which lack a functional adaptive immune system.
(C) Immunofluorescence labeling of CD80 (red) and Keratin 14 (green) epithelial cancer basal cells in (left) xenografts of the human skin SCC line A431 on SCID mice or (right) cells that have undergone metastasis to the lung following injection of A431 skin SCC cells into the tail vein of SCID mice.
(A) Flow cytometry quantification of proliferation marker (Ki67) in cytotoxic T cells that infiltrated CD80(+) or CD80(−) PDVC57 C57/Bl6 SCCs generated from engraftment into C57Bl/6 immune competent mice.
(B) Isogenically matched, PDVC57 SCC cells were transduced with LV expressing Scrambled shRNA or Cd80 shRNA and engrafted onto WT C57/Bl6 mice. Note that Cd80 silencing diminished tumorigenesis, analogous to what we observed with CRISPR/CAS-mediated ablation of Cd80. Data are mean ± SEM. ***P < 0.001, unpaired Student’s t test, n=6.
(C) Cells as in (A) were engrafted onto Rag2−/− mice, which lack an adaptive immune system. Note that loss of adaptive immunity eliminates the tumorigenesis defects from CD80 silencing. n=15; data are mean ± SEM.
(D) Cells as in (A) were engrafted onto wild-type mice that had been immunodepleted for CD8+ T cells. Note that loss of cytotoxic T cells eliminated the tumorigenic defects resulting from Cd80 silencing in the SCC cells. n=15; data are mean ± SEM.
(E) Flow cytometry quantifications of the CD8+ T cells infiltrating Scrambled shRNA or Cd80 shRNA transduced tumors (percentage of total CD45+ cells).
(A) Scheme showing the experimental setup for SCC cell co-culture with activated OT-I cytotoxic T cells to dissect the relative contributions of putative CTL receptors for the surface CD80 on SCC tSCs.
(B) Measurement of OT-I T cell proliferation as monitored by flow cytometry quantification of CFSE dye dilution in activated primary OT-I T cells when co-cultured with CD80(+) or CD80(−) PDVC57 SCC cells, with or without CTLA4 blocking Abs. Data are mean ± SEM. **P < 0.005, unpaired Student’s t test, n=6.
(C) Measurement of OT-I T cell proliferation as monitored by flow cytometry quantifications of CFSE dye dilution in activated primary OT-I T cells when co-cultured with CD80(+) or CD80(−) PDVC57 SCC cells, with or without CTLA4 blocking Abs, CD28 agonist Abs or CD28 antagonist Abs. Data are mean ± SEM. **P < 0.005, unpaired Student’s t test, n=6.
(D) Measurement of OT-I T cell proliferation as monitored by flow cytometry quantifications of CFSE dye dilution in activated primary OT-I CD8+ T cells when co-cultured with CD80(+) or CD80(−) PDVC57 SCC cells, with or without PDL1 blocking Abs, which eliminate possible interactions between PDL1 on the surface of PDVC57 tumor cells and either PD1 on the surface of CD8+ T cells or tumor CD80 (CIS interactions). See text for further details. Data are mean ± SEM. **P <0.005, unpaired Student’s t test, n=6.
(A) CD80(+) or CD80(−) PDVC57 SCC cells were transplanted into C57/BL6 mice treated with either isotype control or CTLA4 blocking Abs, and tumor weight was analyzed at 4 weeks post-grafting. Data are mean ± SEM.
(B) Scheme showing the experimental setup for generating OT-I T cell-specific CTLA4 silencing for use in ACT.
(C) Flow cytometry analysis of T cell exhaustion markers (CTLA4, PD1, Tim3 and LAG3) and proliferation marker (Ki67) in the injected OT-I T cells that had been transduced with control or Ctla4 shRNA and then collected at 2 week post-ACT. The naïve CD8+ T cells from donor spleen were used as negative control.
(D) Flow cytometry quantifications of integrin α6hi TGFβ-reporter+ tSCs in mice treated with control or Ctla4 shRNA transduced OT-I T cells at 1 week post-ACT.
(E) Flow cytometry based quantification of lineage-traced (GFP+), TGFβ-responding (mCherry+) Cd80-null tSCs in Cd80-null tumors following 1 week post-ACT. Note that CreER was selectively activated in naive tSCs at a time when all differentiated tumor cells were GFP(−). Thus, had the tSCs present in ACT-treated tumors been derived from de-differentiation of differentiated tumor cells, they would be mCherry+ but many would be GFP(−).
(F) Changes in tumor size following depletion of injected OT-I T cells after ACT treating the TGFβ-responding tSC specific CD80 KO tumors. Tumor volumes are normalized to their original size. Data are mean ± SEM.
(G) Volume changes of the relapsed Scr sgRNA or Cd80 sgRNA transduced tumors following ACT. n=12 for control; n=7 for tSC-specific targeting of Cd80. Data are mean ± SEM.
(H) Immunofluorescence for CD80 (green), integrin α6hi (blue) and TGFβ-reporter (red) to examine tumor cells that relapsed following ACT treatment. Prior to treatment, Cd80 was depleted specifically in TGFβ-responding tSCs within the tumors of these mice. Note that, whereas majority of a6+TGFβ-reporter+ tSCs in these relapsed tumor are CD80 negative (arrowhead), a few TGFβ-reporter+ tSCs are CD80 positive (arrow). All scale bars=50 μm.
Data Availability Statement
Raw and analyzed data is available at NCBI and the accession number for the single cell RNA sequencing data reported in this paper are NCBI GEO: GSE108679