

Abstract
Antimicrobial resistance extracts high morbidity, mortality and economic costs yearly by rendering bacteria immune to antibiotics. Identifying and understanding antimicrobial resistance are imperative for clinical practice to treat resistant infections and for public health efforts to limit the spread of resistance. Technologies such as next-generation sequencing are expanding our abilities to detect and study antimicrobial resistance. This Review provides a detailed overview of antimicrobial resistance identification and characterization methods, from traditional antimicrobial susceptibility testing to recent deep-learning methods. We focus on sequencing-based resistance discovery and discuss tools and databases used in antimicrobial resistance studies.
Antimicrobials are small molecules that can inhibit or kill bacteria. These small molecules are commonly used as therapeutics for bacterial infections, but some bacteria can grow and survive despite antimicrobial pressures, a property known as antimicrobial resistance. In clinical settings, resistant bacterial infections decrease available treatment options and increase morbidity and mortality compared with those caused by susceptible bacteria1–5. Resistance is observed against nearly all antimicrobials (FIG. 1a,b), including so-called last-resort antimicrobials used in life-threatening, multidrug-resistant infections6–10. Bacteria resistant to first-line antimicrobials infect 2 million people in the USA yearly, and these infections exact a US$20 billion health-care cost11–13. This problem is not isolated to the USA. In the European Union, antimicrobial resistance has accounted for >30,000 deaths and nearly 900,000 disability-adjusted life-years14. In fact, multiple national and global public health organizations categorize antimicrobial resistance as an imminent danger and uniformly agree that tracking its emergence and prevalence is critical to minimize the threat to human health14–17. Antimicrobial susceptibility testing (AST) is the traditional method for assaying antimicrobial resistance in bacteria (Box 1). These culture-based tests determine how well bacteria can grow in the presence of antimicrobials. AST is widely used in hospital clinical microbiology laboratories because it provides actionable phenotypic resistance data to guide patient treatment decisions. Although culture-based resistance determination can provide critical information for patient management and resistance gene epidemiology, it has drawbacks in implementation and information content18. Conducting AST requires microbiology facilities and trained clinical microbiology personnel for accuracy. Additionally, AST is viable only for cultivable bacteria, precluding studies on the emergence and spread of antimicrobial resistance in diverse and complex microbial communities with large fractions of currently uncultured bacteria19.
Fig. 1 |. Antimicrobial targets and resistance mechanisms.
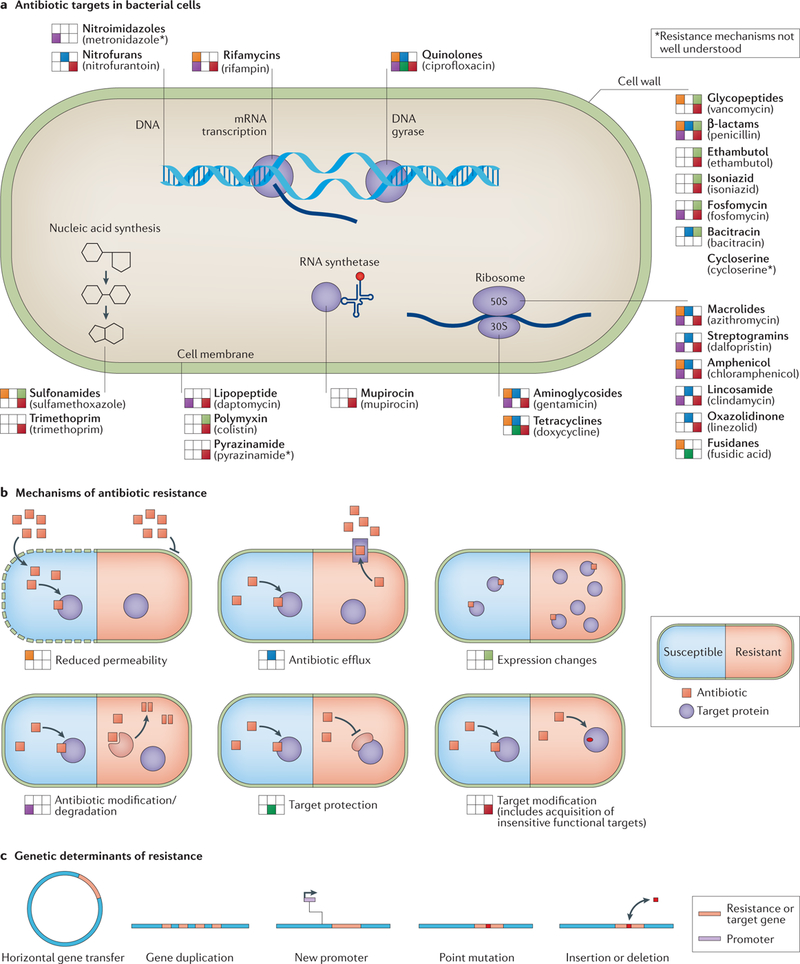
a | Antimicrobials are grouped by target site. Drug classes are in bold, and example drugs from that class are in parentheses below the drug classes. Antimicrobial resistance mechanisms that act on that class are depicted left of the antimicrobial class according to the layout shown in part b. b | Mechanisms of antimicrobial resistance are depicted with susceptible organisms represented on the left and resistant organisms represented on the right. To the left of each labelled mechanism is the legend annotation position used in part a. c | Genetic underpinnings of antimicrobial resistance are illustrated.
Box 1 |. Culture-based susceptibility testing.
Bacterial culture has a long history as an integral part of clinical microbiology115. researchers cultivate bacteria on agar plates or in liquid broth to probe bacterial phenotypes and to discover novel bacterial functions. Hospital clinical microbiology laboratories can use data gained in these assays to inform clinical treatment decisions.
Current techniques
For phenotypic testing, bacteria are isolated from patient or environmental samples by non-selective or selective agar plating for pure colonies. Isolated bacteria are then directly challenged with antimicrobials to test for antimicrobial resistance. In liquid media microbroth dilution antimicrobial susceptibility testing (ast), bacteria are challenged with decreasing antimicrobial concentrations to find the concentration at which they grow successfully. The lowest antibiotic concentration that inhibits the bacterial growth is called the minimum inhibitory concentration. solid media techniques use antimicrobial Kirby–Bauer disc diffusion116 or gradient diffusion strips117 to measure the clearance of the bacteria from the source of the antimicrobial. the area around the antimicrobial disc or strip is called the zone of inhibition (culture-based phenotypic testing is reviewed in detail elsewhere118). standards published by the Clinical and Laboratory Standards Institute (CLSI) or the European Committee on AST (EUCAST) are used to convert minimum inhibitory concentrations or zones of inhibition measurements to categorical resistance determinations.
New, sequence-independent methods of resistance determination are also being developed. They include matrix-assisted laser desorption/ionization time of flight mass spectrometry (MALDI–TOF-MS)119–124, fluorescence in situ hybridization (FISH)125–129 and microfluidics-based techniques, which reduced AST to ~30 min in one study130–134. This rapid testing is especially valuable for slow-growing organisms such as Mycobacterium tuberculosis, for which ast can take weeks135.
Challenges and remaining gaps
while ast is useful to provide phenotypic resistance results, it is low throughput. For each sample, a clinical microbiologist needs to culture the bacteria and to set up and read the AST results. This is limiting because not all health-care centres have trained clinical microbiologists on staff. For some bacteria (for example, M. tuberculosis), current laboratory diagnostic techniques feasible in low-resource areas have low sensitivity136. susceptibility standards are also not published for all combinations of antimicrobials and infection-causing, culturable bacteria. Additionally, the standards themselves are not uniform across different countries. Culture techniques can also fail in situations where multiple bacteria may cause symptomatic disease. In such cases, the cultured bacteria are assumed responsible, but several metagenomic studies from patient samples indicate that the bacteria detected in culture may not be responsible for disease symptoms137–140. Finally, these phenotypic methods have lower resolution in examining resistance determinants and provide less information about resistance gene epidemiology than whole-genome sequencing.
Innovations
Automation is rapidly being developed and implemented to conduct phenotypic assays, including ast141. Several systems, including VITEK142, ADAGIO143, accelerate Pheno144–147 and others, are already moving into clinical spaces. the automated platforms have several key advantages, including continuous monitoring systems, sensitive optical instruments for measuring susceptibility results148 and standardized internal clocks. these advantages can speed up susceptibility results for cultured bacteria118. they may also increase consistency of susceptibility results across different locations and reduce the burden of work for clinical microbiologists. Unfortunately, these automated platforms can be prohibitively expensive to set up.
Bacterial antimicrobial resistance is usually genetically encoded (FIG. 1c). Genetically encoded antimicrobial resistance can occur through a number of mechanisms, including overexpression or duplication of existing genes, point mutations or the acquisition of entirely new genes via horizontal gene transfer (HGT). Improvements in next-generation sequencing technologies and computational methods are facilitating rapid antimicrobial resistance gene identification and characterization in genomes and metagenomes. These developing technologies and methods complement traditional culture-based methods for clinical and surveillance applications and provide opportunities for quick and sensitive resistance determinations in cultivable and uncultivable bacteria. Large-scale and comparative studies of human, animal and environmental samples have provided unprecedented insights into the global distribution of antimicrobial resistance genes and the spread of multidrug-resistant bacteria20–24, resistance exchange networks25 and how different habitats and phylogeny affect the evolutionary dynamics of antimicrobial resistance worldwide26. Understanding and surveying genetic determinants of resistance using sequencing data pose unique challenges that are being addressed by improved computational algorithms that organize genomic data and predict antimicrobial resistance and by improving in vitro sequencing modalities.
In this Review, we discuss the strengths and weak-nesses of current and emerging methods for studying resistance, including computational strategies and resources for resistance gene identification in genomic and metagenomic samples. We also describe recent advancements to mitigate weaknesses in resistance detection methods, and we highlight areas requiring greater focus.
Sequencing-based resistance discovery
Advancements in sequencing technologies have increased bacterial sequence data availability, and continually decreasing costs have made sequencing a viable antimicrobial resistance surveillance tool. Several methods and tools have been published in recent years for detecting genetic determinants of antimicrobial resistance from whole-genome sequencing (WGS) and whole-metagenome sequencing (WMS) data (TABLE 1). Organizing sequencing data is an important pre-processing step before antimicrobial resistance gene analysis. Short reads, generated by technologies such as Illumina, can either be processed using assembly-based methods, whereby sequencing reads are first assembled into contiguous fragments (contigs) and then annotated by comparing with custom or public reference databases, or directly analysed using read-based methods, whereby resistance determinants are predicted by mapping reads directly to a reference database (FIG. 2).
Table 1 |.
sequencing-based tools for antimicrobial resistance detection
| name | Description | Accessibility | Year | Link | status |
|---|---|---|---|---|---|
| Assembly-based tools | |||||
| Resfinder72 | Tool for detecting acquired AR genes from sequenced or partially sequenced bacterial isolates | Web and/or standalone | 2012 | https://cge.cbs.dtu.dk/services/ResFinder/ | Active |
| ARG-ANNOT66 | Tool for pairwise comparison of query sequence with ARG-ANNOT database | Standalone | 2014 | Not available | Archived |
| RGI67 | • Pairwise comparison of query sequence with the CARD • Uses curated AR detection models to predict intrinsic resistance genes, dedicated resistance genes and acquired resistance from mutations in drug targets |
Web and/or standalone | 2015 | https://card.mcmaster.ca/analyze/rgi | Active |
| ARGs-OAP (v2)74 | • Online analysis pipeline for AR genes • Detection from metagenomic data using an integrated structured database of AR sequences |
Web and/or standalone | 2016 | https://galaxyproject.org/use/args-oap/ | Active |
| ARIBA52 | Tool for rapid AR genotyping directly from sequencing reads using curated public databases | Standalone | 2017 | https://github.com/sanger-pathogens/ariba | Active |
| PointFinder73 | Web tool for WGS-based detection of AR associated with chromosomal point mutations in bacterial pathogens | Web and/or standalone | 2018 | https://cge.cbs.dtu.dk/services/ResFinder/ | Active |
| NCBI-AMRFinder | Tool for identification of acquired resistance genes using NCBI’s curated AR database and curated collection of HMMs | Standalone | 2018 | https://www.ncbi.nlm.nih.gov/pathogens/antimicrobial-resistance/AMRFinder/ | Active |
| Read-based tools | |||||
| SRST2 (REF.50) | Tool for direct mapping of reads to curated AR databases | Standalone | 2014 | https://github.com/katholt/srst2 | Active |
| SEAR54 | Cloud-compatible pipeline and web interface for rapidly detecting AR genes directly from sequencing reads | Web and/or standalone (archived) | 2015 | https://github.com/will-rowe/SEAR | Archived |
| ShortBRED61 | Tool to profile protein families in the metagenomic data using short peptide marker sequences | Standalone | 2015 | http://huttenhower.sph.harvard.edu/shortbred | Active |
| PATRIC176 | A unique resource for studying AR | Web | 2016 | www.patricbrc.org | Active |
| SSTAR177 | Tool to identify known, putative new alleles and truncated versions of existing AR genes from WGS data | Standalone | 2016 | https://github.com/tomdeman-bio/Sequence-Search-Tool-for-Antimicrobial-Resistance-SSTAR- | Active |
| KmerResistance51 | Program that compares the co-occurrence of k-mers from raw reads with k-mers from multiple databases | Web | 2016 | https://cge.cbs.dtu.dk/services/KmerResistance/ | Active |
| GROOT56 | Software that enables resistome profiling by mapping metagenomic reads to graph representation of reference gene sets | Standalone | 2018 | https://github.com/will-rowe/groot | Active |
| DeepArgs110 | A deep-learning approach for predicting AR genes from metagenomic data | Web | 2018 | https://bench.cs.vt.edu/deeparg | Active |
AR, antimicrobial resistance; ARG-ANNOT, Antibiotic Resistance Gene Annotation; CARD, Comprehensive Antibiotic Resistance Database; GROOT, Graphing Resistance Out of Metagenomes; HMM, hidden Markov model; NCBI, National Center for Biotechnology Information; PATRIC, Pathosystems Resource Integration Center; RGI, Resistance Gene Identifier; SEAR, Search Engine for Antimicrobial Resistance; ShortBRED, Short, Better Representative Extract Dataset; WGS, whole-genome sequencing.
Fig. 2 |. Assembly versus read mapping.
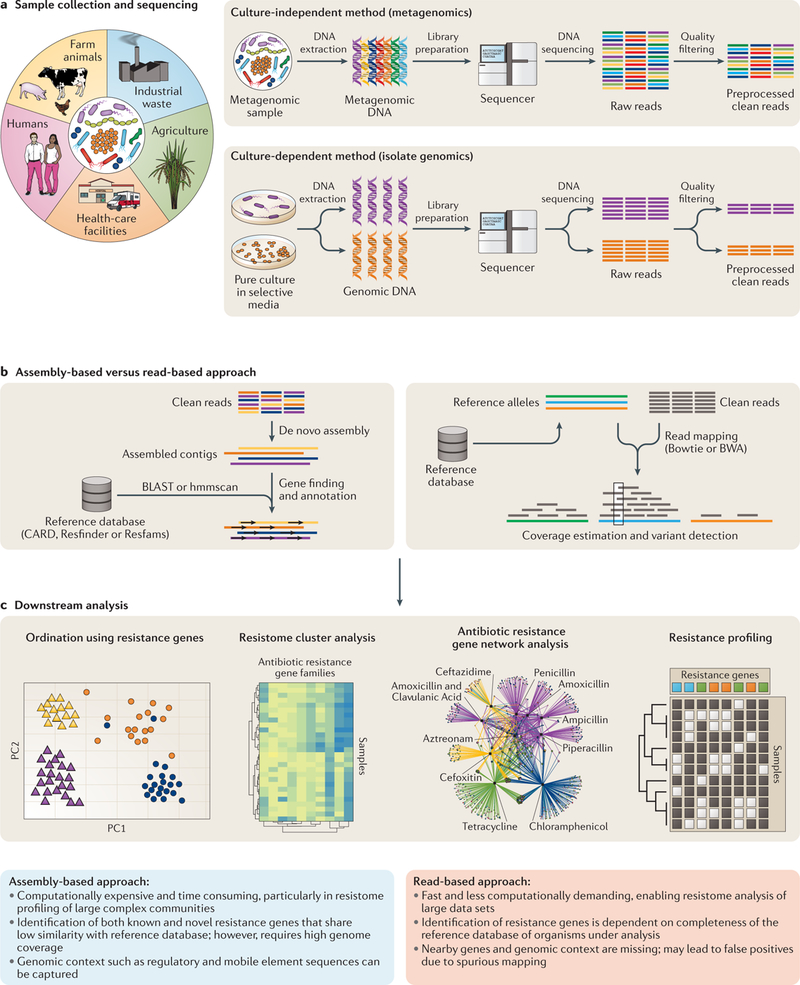
a | The process for sequencing data generation for metagenomic and genomic samples. b | Steps for the read-based and the assembly-based methods of in silico resistance gene identification are contrasted. c | Examples of analysis that can be conducted on samples after resistance gene identification. CARD, Comprehensive Antibiotic Resistance Database.
Assembly-based methods.
The de novo assembly of WGS of bacterial genomes from short-read data is generally performed by De Bruijn graph (DBG)-based assemblers such as SPAdes27, Velvet28, ABySS29 and SOAPdenovo30. In this approach, sequencing reads are divided into shorter overlapping sub-sequences (called k-mers) of length k (where k is less than the read length) and are used to form a network graph. The assemblers then reconstruct the genome sequence by finding an optimum path (euler’s path) through the graph that visits each edge once (see reF.31 for more information on DBG-based assembly). Although the DBG approach is computationally efficient in handling high-volume sequencing data, it is greatly affected by errors introduced during sequencing32. Errors in sequencing data introduce false k-mers in the graph, resulting in fragmented assemblies. Several assemblers (for example, SPAdes and Velvet) heuristically eliminate these errors before finding a Euler’s path in the graph31,33. Assembling WMS data is more complicated than single-isolate assembly (FIG. 2a), as the algorithms need to account for unknown abundances of different organisms with unknown phylogenetic relationships32. In single-genome assembly, uniform sequencing coverage across the genome is used by assemblers to correct sequencing errors and to identify repetitive sequences and plasmids — several assemblers exploit the higher coverage of plasmids owing to copy number to distinguish between chromosome and plasmid sequences in isolate genomes34–37 — but uneven coverage of different organisms in WMS data makes detecting repeats difficult. Long stretches of identical sequences in unrelated species further complicate assembly by making it difficult to assign reads to a particular species. Thus, algorithms developed for single-genome assembly cannot be directly applied to assemble metagenomes. Several metagenome-specific assemblers have been developed to overcome these challenges, either by partitioning or optimizing the graph for uneven sequencing depths32. Some notable metagenomic assemblers are IDBA-UD38, MEGAHIT39, MetaSPAdes40 and MetaVelvet41 (extensions of SPAdes and Velvet for metagenomes). The CAMI project42, now starting its second iteration43, seeks to benchmark these assemblers on highly complex and close to real data sets for users. However, currently, there is no single assembler that stands out as the best one that would accurately reconstruct known genomes and capture the majority of the taxonomic diversity in real data sets. Both biological factors (such as sample source and microbial community structure) and technical factors (such as library preparation method, sequencing depth and sequencing platform choice) affect the ability of an assembler to generate accurate and larger contigs. Thus, it is recommended to apply multiple assemblers on a subset of samples to determine the best fit for a given data set.
Following assembly, genomic or metagenomic contigs are annotated for resistance determinants by predicting protein-coding regions on contigs and then comparing them against antimicrobial resistance reference databases using similarity-based search tools (for example, BLAST44, USEARCH45 or DIAMOND46). Although pairwise alignment between the query and antimicrobial resistance reference sequences is the most commonly applied approach for characterizing the resistome from contigs, an inherent bias of databases towards human-associated organisms is reflected in prediction outputs, so choosing the appropriate databases to compare assembled contigs with reference sequences is imperative47.
Given sufficient coverage, assembly-based methods can construct whole genomes or large contigs with protein-coding genes, regulatory sequence information and the complete surrounding genomic context. This information can be used to study co-associated genes and biological pathways that are involved in resistance determination. Assembly and annotation of WMS data can identify antimicrobial resistance genes that are more divergent from and lack homology to known sequences in the reference databases. However, the process of de novo assembly and annotation is computationally expensive, time consuming and requires higher genome coverage than reference-based assembly or read mapping-based methods, which can be difficult to achieve for all samples, specifically when dealing with metagenomic samples with high microbial diversity and uneven taxonomic composition.
Read-based methods.
Antimicrobial resistance genes in a sample can be detected without genome assembly either by aligning reads to the reference databases using pairwise alignment tools such as Bowtie2 (REF.48) or BWA49, or by splitting reads into k-mers and mapping them to the reference databases.
SRST2 (REF.50) is one widely used tool that aligns reads to a custom reference database using Bowtie2 to predict antimicrobial resistance genes in the sample. Alternatively, KmerResistance51 splits reads into k-mers, maps them and counts the co-occurrence of k-mers between reads and a reference database to predict resistance genes and associated species. Both methods can identify antimicrobial resistance genes even in the presence of contaminants (for example, background noise in the raw reads owing to the presence of laboratory or host contamination) and in samples for which insufficient reads are available for de novo assembly, but they cannot predict antimicrobial resistance conferred by single-nucleotide polymorphisms (SNPs). By contrast, ARIBA52 uses a hybrid approach where reference sequences in the database are first clustered using CD-HIT53 before sequences from each cluster are assembled independently. Resulting contigs are then compared with the closest reference to identify allelic variants. Additionally, ARIBA provides information on whether genes are complete or fragmented and reports sequence variants along with their potential effects (for example, missense, nonsense or frameshift mutations and small insertions and deletions (indels)). Clustering reference sequences and using a representative sequence from the cluster to map reads considerably reduce ambiguous alignments54, but using a single linear representative locus masks subtle yet important variation between subtypes and subfamilies of genes within clusters54,55. To account for this information loss, Graphing Resistance Out of Metagenomes (GROOT)56, a newly established tool for resistome profiling of metagenomes, builds a variation graph for reference gene sets and aligns sequence reads to these graphs. Variation graphs are bidirectional acyclic sequence graphs that represent overall sequence variation within a given population. The alignment of reads against variation graphs effectively removes reference bias and facilitates accurate annotation of antimicrobial resistance genes. Before aligning sequences against a variation graph, traversals within the graphs are indexed by either Burrows–Wheeler transform or hash-map (minHash), indexing algorithms that considerably improve the mapping rate of large-scale sequencing reads to the graphs48,57. The read-based approach is generally fast and less computationally demanding because it bypasses de novo assembly, protein-coding gene prediction and pairwise alignment to public databases. For this reason, read-based methods have gained traction in recent years, especially in clinical diagnostics where conducting real-time sequencing-based resistance prediction is crucial.
Choosing the right approach.
Presently, there is no consensus on which sequence analysis approach is better, and the choice of analysis mainly depends on the type of sequencing (WGS versus WMS), availability of computational resources and the study objective. Both approaches have trade-offs, as assembly causes information loss compared with direct read analysis58 but enables identification of protein-coding genes and for investigation of upstream and downstream regulatory elements, whereas direct read analysis lacks the positional information required to analyse upstream and downstream factors of identified resistance genes. New sequencing technologies, such as long-read sequencing and chromosome conformation capture-derived assays, are helping to alleviate this information loss by improving assembly fidelity (BOX 2).
Box 2 |. Sequencing-based innovations.
Chromosome conformation capture assays
Chromosome conformation capture uses crosslinking, ligation and short-read sequencing to understand the spatial relationships of genetic material within cells149,150. these methods have been applied effectively in bacterial isolates and in metagenomic samples151–155. Using spatial information from chromosome conformation capture, researchers can increase their understanding of antimicrobial resistance gene regulation and begin to untangle more complicated resistance mechanisms. For example, these data may help resolve epistatic antimicrobial resistance gene relationships because gene spatial colocalization may indicate coordinated function. Chromosome conformation capture is also useful for identifying plasmid genes from chromosomal genes. this is important for antimicrobial resistance in particular because of the role of plasmids in horizontal gene transfer (HGt). Perhaps most interesting is the applicability of chromosome conformation capture to metagenomic samples. In addition to the points mentioned above, crosslinking information in metagenomic samples greatly reduces the complexity of metagenomic assembly because it enables researchers to group sequences by cell. thus, antimicrobial resistance annotation methods that rely on assembled contigs are more viable if chromosome conformation capture information is also available.
Long-read sequencing
Long-read sequencing by single-molecule real-time sequencing (SMRT-seq) or nanopore sequencing can yield reads from 10 kb to over 100 kb (REFS156,157). these long reads have several advantages over short reads for studying antimicrobial resistance in bacterial isolates and metagenomic samples. Long-read sequencing can greatly reduce the complexity of assembly for both bacterial isolates and metagenomic samples and, in some cases, can provide finished bacterial genomes (higher error rates than short reads means that sometimes a combination of short-read and long-read sequencing is needed to achieve these results)158–162. Better assemblies enable stronger interpretation of genomic context, providing similar benefits as with chromosome capture. Also similar to chromosome capture, long-read sequencing provides easier resolution of plasmid sequences, enabling more in-depth studies to understand antimicrobial resistance HGt. additionally, both approaches give information about DNa methylation, which can inform genome assembly in metagenomic samples162–164. Finally, long-read sequencing run times tend to be faster than short-read sequencing. this is useful for potential clinical deployments where speed may affect patient outcomes. Nanopore sequencing in particular excels in this area owing to its real-time data; these real-time data have already been used to provide rapid (within minutes) antimicrobial resistance gene-agnostic antimicrobial susceptibility testing (AST) predictions in Streptococcus pneumoniae165.
Transcriptomics
Transcriptomics techniques such as RNA sequencing (RNA-seq) facilitate analysis of bacterial gene expression, and these expression data have the potential to fill antimicrobial resistance knowledge gaps166–169. One major area where transcriptional data can contribute to antimicrobial resistance understanding is in connecting genotypic antimicrobial resistance data to phenotypic antimicrobial resistance results170. In cases where antimicrobial resistance genes are present but no resistance phenotype is found or in cases where no obvious antimicrobial resistance genes are present but antimicrobial resistance is confirmed, transcriptomic data may bridge the knowledge gap and point to novel antimicrobial resistance genes169,171–173. transcriptomics also has the potential to identify combinatorial gene effects resulting in antimicrobial resistance174. Finally, transcriptomics can successfully identify cases in which non-coding regulatory RNAs result in resistant bacterial phenotypes168,175.
The read-based approaches scale well with ever-increasing query sequences and antimicrobial resistance reference data. More importantly, they enable identification of antimicrobial resistance genes from low-abundance organisms present in complex communities, which may be missed by assembly-based methods owing to incomplete or poor assemblies. However, mapping reads directly to large data sets can inflate false-positive predictions, as reads derived from protein-coding sequences may spuriously align to other genes as a result of local sequence homology59. Thus, it is important that the reference databases are comprehensive and contain all variants of the reference genes. Database choice is especially critical when identifying antimicrobial resistance genes from large and complex communities such as soil and ocean, as novel or distant homologues of antimicrobial resistance genes present in understudied, less characterized environmental communities may be missed.
Well-studied sample types, such as the human gut, are now extensively characterized, even for low-abundance microorganisms and, thus, read-based approaches can be more confidently applied60. However, analysis of diverse samples is confounded by the lack of reference sequences, so the antimicrobial resistance genes in these environments are likely underestimated. To address this problem, a marker-based method, Short, Better Representative Extract Dataset (ShortBRED)61, was developed that enables fast and accurate profiling of the resistome in metagenomic data sets. ShortBRED first identifies marker sequences (short peptide sequences) representative of antimicrobial resistance protein families from the reference database and then maps reads to these markers to quantify the relative abundance of the associated antimicrobial resistance protein families. Several studies have applied this method to quantify the abundance of resistance genes in large and complex metagenomic data sets, including human25,62, animal63 and environmental data sets64. Downstream analysis of resistomes from metagenomic samples can be performed similarly to taxonomic and functional profiling. A comprehensive discussion on processing and analysing metagenomic samples has been previously published60.
Antimicrobial resistance databases
Both assembly-based and read-based approaches for the computational prediction of antimicrobial resistance in pathogens and environmental bacteria depend largely on curated antimicrobial resistance gene databases that link known genetic determinants of resistance to the antimicrobials they confer phenotypic resistance against (TABLE 2). These databases usually represent information accumulated from multiple studies that include AST of bacteria harbouring specific antimicrobial resistance genes.
Table 2 |.
summary of antimicrobial resistance reference databases
| Database | Description | Link | status |
|---|---|---|---|
| General databases | |||
| CARD67 | • Ontology-based database that provides comprehensive information of AR genes and their resistance mechanisms • Currently contains >2,200 protein homologues and includes a curated set of resistance-conferring chromosomal mutations in protein-coding genes |
https://card.mcmaster.ca/ | Active; launched in 2013; updated monthly |
| Resfinder72 | Collation of AR genes involved in HGT events | https://cge.cbs.dtu.dk//services/ResFinder/ | Active; started in 2012; update regularly; last update in February 2019 |
| ResfinderFG84 | Collection of resistance gene variants identified in multiple functional metagenomics studies | https://cge.cbs.dtu.dk/services/ResFinderFG/ | Active; last update in November 2016 |
| Resfams26 | A profile HMM-based curated database confirmed for AR function | http://www.dantaslab.org/resfams/ | Active; last update in January 2015 |
| ARDB65 | • First centralized resource of AR gene information • Manually curated; contains >4,500 AR sequences |
https://ardb.cbcb.umd.edu/ | Archived; last updated in 2009 |
| MEGARes178 | • Collation of multiple databases (CARD, ARG-ANNOT and ResFinder) to avoid redundancy between entries • For high-throughput screening and statistical analysis |
https://megares.meglab.org/ | Active; last update in December 2016 |
| NDARO | • Collated and curated data from multiple databases (CARD, Lahey, Pasteur Institute β-Lactamases and ResFinder) • Contains 4,500 AR sequences |
https://www.ncbi.nlm.nih.gov/bioproject/PRJNA313047 | Active; started in 2016 |
| ARG-ANNOT66 | • Repository of >1,800 AR sequences collated from scientific literature and online resources • Also includes point mutation data for select AR-associated chromosomal genes |
Not available | Archived; last update in May 2018 |
| Mustard85 | Resource containing 6,095 AR determinants from 20 families, including curated sets of AR genes identified in functional metagenomics studies | http://mgps.eu/Mustard/ | Active; last update in November 2018 |
| FARME database83 | Curated set of microbial sequences functionally screened to confer resistance in various functional metagenomics studies of different habitats | http://staff.washington.edu/jwallace/farme/ | Active; last update in 2017 |
| SARG (v2)74 | • Hierarchical structured database derived from ARDB, CARD and NCBI-NR database • Contains >12,000 AR genes; also includes profile HMMs for 189 AR genes subtypes |
http://smile.hku.hk/SARGs | Active |
| Lahey list of β-lactamases70 | First initiative to compile known β-lactamases and assign nomenclature to novel ones | http://www.lahey.org/Studies/ | Archived; last update in 2015 |
| BLDB179 | Manually curated database for AR enzymes classified by class, family and subfamily | http://bldb.eu/ | Active; last update in November 2018 |
| LacED68,69 | Curated database of TEM and SHV β-lactamases, including a curated set of known TEM and SHV variants | http://www.laced.uni-stuttgart.de/ | TEMLacED active: last update in 2017; SHVED archived: last update in April 2010 |
| CBMAR71 | Database that identifies and characterizes novel β-lactamases on the basis of Ambler classification |
http://proteininformatics.org/mkumar/lactamasedb/ | Last update in September 2014 |
| Species-specific databases | |||
| MUBII-TB-DB76 | Database of mutations associated with AR in Mycobacterium tuberculosis | https://umr5558-bibiserv.univ-lyon1.fr/mubii/mubii-select.cgi | Last update in December 2013 |
| u-CARE180 | User-friendly, comprehensive AR repository for Escherichia coli | http://www.e-bioinformatics.net/ucare | Last update in 2016 |
AR, antimicrobial resistance; ARDB, Antibiotic Resistance Genes Database; ARG-ANNOT, Antibiotic Resistance Gene Annotation; BLDB, β-Lactamase Database; CARD, Comprehensive Antibiotic Resistance Database; CBMAR, Comprehensive β-Lactamase Molecular Annotation Resource; FARME, Functional Antibiotic Resistance Metagenomic Element; HGT, horizontal gene transfer; HMM, hidden Markov model; LacED, Lactamase Engineering Database; NDARO, National Center for Biotechnology Information (NCBI) Bacterial AR Reference Gene Database/National Database of Antibiotic Resistant Organisms.
Generalized versus specialized databases.
Public databases vary considerably in the scope of the resistance mechanisms19 that they cover and in the type of information they provide for annotations. Generalized antimicrobial resistance databases, such as the now archived Antibiotic Resistance Genes Database (ARDB)65 or the active Antibiotic Resistance Gene Annotation (ARG-ANNOT)66 and Comprehensive Antibiotic Resistance Database (CARD)67, cover broad spectrums of antimicrobial resistance genes and mechanism information, whereas specialized antimicrobial resistance databases provide comprehensive information for specific gene families or species (TABLE 2). For example, targeted databases such as Lactamase Engineering Database (LacED)68,69, the Lahey database of β-lactamases70, National Center for Biotechnology Information (NCBI) β-Lactamase Alleles Initiative, and the Comprehensive β-Lactamase Molecular Annotation Resource (CBMAR)71 focus on β-lactamases, a family of antimicrobial resistance enzymes that facilitate hydrolysation of the key β-lactam rings in β-lactam antimicrobials, thus protecting the bacteria from the antimicrobial activity. Resfinder72 is a web-based and standalone tool for detecting acquired antimicrobial resistance genes from sequenced or partially sequenced bacterial isolates (TABLE 1). Unlike other databases that require contigs as an input, Resfinder72 also accepts short reads as an input for comparison against known acquired resistance genes in bacterial genomes. In 2017, Resfinder72 updated its web-based service to enable identification of chromosomal mutations using PointFinder73. However, the identification of antimicrobial resistance-conferring chromosomal mutations is available for only a limited set of pathogenic microorganisms (Campylobacter, Escherichia coli, Mycobacterium tuberculosis, Neisseria gonorrhoeae, Plasmodium falciparum and Salmonella). Similar to Resfinder72, CARD67 offers its own tool, known as Resistance Gene Identifier (RGI), which uses curated antimicrobial resistance detection models to predict intrinsic antimicrobial resistance genes, dedicated resistance genes and acquired resistance from mutations in drug targets. RGI uses two antimicrobial resistance detection models: Protein Homologue Model for detecting functional homologues of antimicrobial resistance proteins and Protein Variant Model for the detection of mutations conferring antimicrobial resistance in otherwise sensitive targets. ARGs-OAP (v2)74 uses a custom database constructed from ARDB65 and CARD67, called SARG, with a hybrid UBLAST and BLASTX algorithm, reflecting the critical need for a comprehensive database combined with lower identity matching for antimicrobial resistance gene annotation of metagenomic sequence data.
Species-specific databases exist for pathogenic or model bacteria such as M. tuberculosis (for example, Tuberculosis Drug Resistance Database75 or MUBII-TB-DB76) and E. coli (TABLE 2). These species-specific databases are invaluable for understanding resistance in these specific organisms but also highlight the importance of considering antimicrobial resistance genes in their phylogenetic context, especially as some bacteria can have intrinsic resistance to some antimicrobials (reviewed previously77). Species-centric databases enable rapid and effective curation of new antimicrobial resistance genes and chromosomal mutations and can offer quick preliminary screening for characterization. Such screening has proved highly effective for pathogens such as M. tuberculosis in which HGT events are rare and drug resistance originates mainly from chromosomal mutations78. The CRyPTIC Consortium and 100,000 Genomes Project demonstrated this effectiveness in M. tuberculosis with resistance predictions with over 90% sensitivity and specificity for all four first-line anti-tuberculosis drugs79.
While these tools are all steps in the right direction, a continuously updating and comprehensive database with extensive gene metadata and the ability to find both point mutation matches and remote homologues is needed.
Hidden Markov model-based databases.
One major limitation of these databases is that the antimicrobial resistance genes they contain are heavily biased towards human pathogens and easily cultivable model organisms, making it difficult to identify remote homologues or novel resistance sequences present in fastidious or uncultured bacteria80. This bias complicates antimicrobial resistance gene identification across less commonly studied bacteria, a difficulty that is magnified by the diverse and complicated mechanisms that cause resistance81. One potential solution to overcome this bias is to use hidden Markov model (HMM) databases. Derived from the multiple sequence alignment of known sequences, an HMM can find sequences with similar function but low sequence identity82. Resfams26 is an HMM database of antimicrobial resistance proteins derived from multiple sequence alignments of manually curated sets of representative antimicrobial resistance protein sequences obtained from the generalized CARD and the specialized LacED69 and Lahey database70 (TABLE 2). The authors of the Resfams26 database showed that it can identify a substantially greater number of novel antimicrobial resistance genes and remote homologues of known antimicrobial resistance genes than other databases such as ARDB and CARD that rely on BLAST-based methods for gene identification. A direct comparison of manually curated antimicrobial resistance gene sets showed that Resfams26 identified 64% more antimicrobial resistance genes in both soil and human gut microbiota than the BLAST-based search of CARD and ARDB. This increased sensitivity demonstrates the versatility of the HMM in annotating sequences from non-clinical samples with sparser representation in publicly available resistance gene databases. However, HMM-based approaches may have poor specificity (yield higher number of false-positive hits) and may not be able to distinguish between protein families with closely related functions. This could occur owing to the higher probability of selecting sequences from other subfamilies on the basis of domains common to the family. To mitigate the lack of specificity, Resfams26 (like the Pfam database) uses curated thresholds (for example, a gathering threshold) for each profile HMM. These profile-specific gathering threshold values set an inclusion or exclusion bit score cut-off by comparing it with test data sets containing negative sequences. Currently, Resfams26 contains 166 profile HMMs that represent major antimicrobial resistance gene families. HMM-based antimicrobial resistance databases could be valuable in identifying large and diverse arrays of resistance determinants in understudied environmental samples compared with BLAST-based databases. However, current HMM-based databases do not identify resistance arising from chromosomal mutations. To further facilitate the detection of antimicrobial resistance genes in large complex environments, the Functional Antibiotic Resistance Metagenomic Element (FARME)83 database comprises a curated set of microbial sequences excluded from current databases but functionally screened to confer resistance in various functional metagenomics studies of different habitats. Apart from predicted protein-coding antimicrobial resistance sequences, the FARME database also includes regulatory elements, mobile genetic elements and predicted proteins flanking antimicrobial resistance genes.
A similar database, the functional resistance database (ResfinderFG)84, was built by aggregating data from four functional metagenomics studies selected against 23 antimicrobials. When comparing this database with the Resfinder72 database, the authors noted that they found different results by total antimicrobial use; this observation may represent a difference in how resistance is conferred when putative resistance determinants are cloned into E. coli as compared with when they are expressed in their native bacterial host.
The Mustard85 antimicrobial resistance determinants database uses an innovative approach of incorporating 3D protein structure to help predict resistance genes. When this approach was applied to predicted proteins from metagenomic samples, it predicted >6,000 resistance genes compared with 67 genes identified by BLASTP and 50 by Resfinder72, suggesting higher sensitivity.
Remaining challenges.
Considerable developments in biocuration of antimicrobial resistance sequences have enabled the identification and characterization of antimicrobial resistance genes from genomes and metagenomes, but several limitations still preclude cost-effective and rapid antimicrobial resistance surveillance. One major bottleneck is the lack of effective curation strategies. With few exceptions, antimicrobial resistance databases lack efficient and sustainable curation pipelines, so they tend to receive active maintenance for a few years before becoming outdated.
Many antimicrobial resistance genes can be assigned names on the basis of nucleotide sequences and protein sequences, leading to conflicting naming schemes. Conflicting gene names and synonyms create redundancy across databases and confuse users (for example, dihydrofolate reductase is referred to as dhfr in some databases and dfrA in others)86. This problem is exacerbated by assigning gene names by sequence identity. A plethora of different sequence identity-based systems exists for assigning nomenclature to a new resistance gene. These systems offer different cut-offs and are not in consensus with the reference87.
Antimicrobial resistance genomic data are an ever expanding data source. HGT events and selection pressures that proliferate new antimicrobial resistance mutations require active biocuration strategies whereby entries can be curated as they are recognized. The propagation of new colistin resistance mechanisms such as mcr-1, which was first described in 2016 from Chinese bacterial isolates9 and then subsequently identified worldwide in newly collected and previously stored isolates88–96, demonstrates the need for frequent database updating and curation. When properly implemented, they facilitate rapid collection of epidemiological data for recently discovered resistant determinants97. Indeed, antimicrobial resistance annotation should be a continuous effort, as all downstream analyses depend on the accuracy of reference databases. Establishing best practices for biocuration, systematically assigning annotations to newly discovered genes and preventing misinterpretations will pay dividends for public health and basic science.
Another important limitation of current antimicrobial resistance databases is their focus on the identification and characterization of protein-coding resistance genes; they ignore other potential antimicrobial resistance mechanisms such as genomic changes or de novo mutations in ribosomal RNA (rRNA) genes and regulatory elements and drug target mutations. Recent efforts by CARD67 and Resfinder72 have tried to address this issue.
Functional metagenomics
In addition to sequence-based metagenomics, functional metagenomics is a powerful, culture-independent, sequence-unbiased approach for characterizing resistomes98,99. In this method, a metagenomic library is generated by cloning the total community DNA extracted from a sample into an expression vector. This library is transformed into a susceptible indicator host strain and is assayed for antimicrobial resistance by plating on selective media that are lethal to the wild-type host. The selected inserts from the surviving recombinant, antimicrobial-resistant host cells are then sequenced, and resulting sequences are subsequently assembled and annotated (FIG. 3). Parallel Annotation and Reassembly of Functional Metagenomic Selections (PARFuMS)100 is a custom computational pipeline that assembles reads from functional metagenomic selections into contigs using the Velvet28 and Phrap101 assemblers and annotates the assemblies for antimicrobial resistance genes using MetaGeneMark102 and Resfams26. This approach enables high-throughput analysis of large genomic content (up to 50 Gbp of unique metagenomic DNA interrogated per library), and antimicrobial resistance phenotypes can be associated directly with causative genes, obviating the need to culture individual antimicrobial resistance gene carriers.
Fig. 3 |. Functional metagenomics to interrogate acquired resistance genes in different environments and human pathogens.
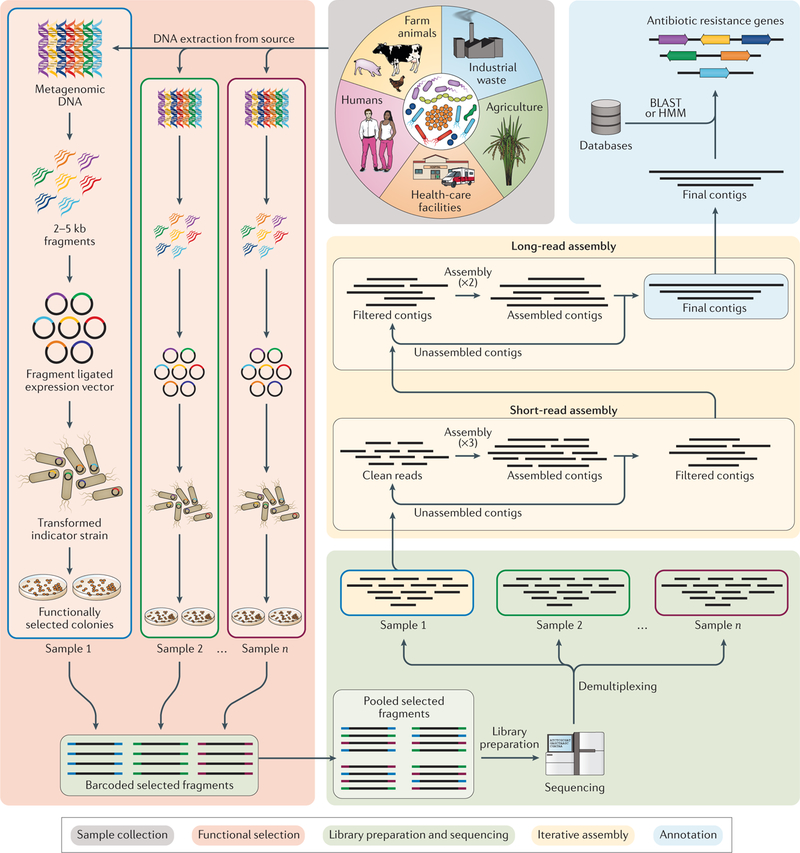
A summary of experimental and computational steps involved in functional metagenomics. First, sample collection and extraction occur. Metagenomic DNA is isolated from the sample (for example, soil or faeces). Second, functional selection using an expression vector and the host system is performed. The metagenomic DNA is sheared to a target size of 2–5 kb, and the fragments are then cloned into an expression vector and transformed into a host system (for example, Escherichia coli). The transformants are then selected using antimicrobials at concentrations that are inhibitory to the wild-type host system. Third, barcoded sequencing of pooled DNA fragments is performed. The resistance-conferring fragments are PCR amplified, barcoded and pooled together for sequencing. The sequencing reads are computationally demultiplexed using barcode assembly and quality trimmed to obtain high-quality clean reads. Fourth, iterative assembly of sequencing reads by sample is performed. The clean reads are assembled with computational pipeline Parallel Annotation and Reassembly of Functional Metagenomic Selections (PARFuMS), in which ensemble-based assembly is performed using multiple rounds of a short-read assembler (Velvet), and intermediate contigs are then used in a long-read assembler (Phrap) to give full-length contigs. Finally, resistance gene annotation of assembled reads is performed. The annotation of contigs is accomplished using BLAST-based and hidden Markov model (HMM)-based databases.
Functional metagenomics has enabled the discovery of several new antimicrobial resistance mechanisms and their related genes103. One such example is the recently discovered tetracycline resistance mechanism by tetracycline destructases104, whereby soil functional metagenomics led to the discovery of nine genes that confer tetracycline resistance through enzymatic inactivation. Further analysis and biochemical characterization revealed that these enzymes catalyse tetracycline oxidation in an FAD-dependent manner, thereby inactivating tetracycline104.
While the preceding study shows the strength and usefulness of functional metagenomics, this approach has certain limitations. For example, a gene has to be functional outside its native microbial host to be identified by functional metagenomic selections. Many times, differences between a recombinant expression host such as E. coli and the original host (for example, some Gram-positive organisms) do not confer the same phenotype for the same gene. This problem was highlighted by studies showing effects of different hosts on the same metagenomic libraries97,105. Thus, there is a need to include a phylogenetically diverse group of hosts that can be used for functional metagenomic selections. In addition, genes outside their genomic context, such as syntenic regulatory elements, may have different phenotypes in the recombinant expression host from those in the original host106. Thus, it is important that novel antimicrobial resistance genes identified by functional metagenomics screens be characterized microbiologically and biochemically. Extension of the current functional metagenomics approach and development of new techniques to discover novel resistance genes are deserving research directions.
Machine learning for resistance prediction
Numerous studies have explored machine learning algorithms for studying antimicrobial resistance, highlighting its role in predicting resistance phenotype directly from genotype. Machine learning approaches can be implemented as supervised learning or unsupervised learning approaches. In supervised learning, the training data set with outcome of interest can be utilized to build a prediction model that can be further applied to query sequences to predict their outcome. Several studies have used gene presence or absence and AST outcomes as features to create the training set for models. In one study, a logistic regression approach was used to develop a model based on 14 gene parameters and 3 molecular typing markers that can differentiate between vancomycin-susceptible and vancomycin-intermediate Staphylococcus aureus using publicly available genomic data and patient isolates107. The model performance was tested by a leave-one-out validation method, and it showed 84% classification accuracy. Although this accuracy level does not meet clinical standards, the approach provides an important proof of concept that motivates the development of more sophisticated models for identifying antimicrobial resistance. Another study evaluated a rules-based and a machine learning-based approach (that is, logistic regression) for predicting antimicrobial resistance profiles and showed that the machine learning-based approach had higher accuracy with novel variants in known antimicrobial resistance genes than the rules-based approach24.
Recent studies and tools use k-mers derived from whole genomes of antimicrobial-resistant and antimicrobial-susceptible species along with their AST outcomes to develop prediction models. Mykrobe predictor108, a fast k-mer screening tool, is used to identify antimicrobial resistance genes and SNPs in S. aureus and M. tuberculosis. It utilizes the curated genetic information of resistant and susceptible alleles of the same species to build reference graphs (DBG) of these two categories and to map k-mers derived from sequencing reads to these graphs. Mykrobe predictor showed 99.1% and 82.6% sensitivity and 99.6% and 98.5% specificity for S. aureus and M. tuberculosis, respectively, on an independent validation set and provided important insights on potential antimicrobial resistance elements.
By contrast, Rapid Annotation using Subsystem Technology (RAST)109 is a k-mer-based tool that uses a machine learning classifier (AdaBoost) based on the Pathosystems Resource Integration Center (PATRIC) database to identify target-specific antimicrobial resistance genes in a specific collection of pathogens. RAST is trained on k-mer data derived from the contigs of each genome. These k-mer counts were converted to a binary matrix of 1s and 0s to depict whether a particular k-mer is present in that genome or not. The binary matrix along with AST outcome is then used to form a classifier model as well as to identify putative k-mers associated with resistance. The RAST classifier could identify carbapenem resistance in Acinetobacter baumannii, methicillin resistance in S. aureus and β-lactam and co-trimoxazole resistance in Streptococcus pneumoniae with accuracies of 88–99%109.
One major shortcoming of any machine learning classifier is its dependency on the training data or existing knowledge base. To apply machine learning classifiers in clinical diagnostics, a large data set of curated antimicrobial resistance genes that contains accurate genotypic data linked to curated AST data (Box 1) will be required to build an effective and robust machine learning-based classifier for antimicrobial-resistant organisms. In addition to differentiating between an antimicrobial-resistant and antimicrobial-susceptible organism, machine learning approaches are currently being applied to predict antimicrobial resistance genes in metagenomic data. DeepArgs110 is a newly established tool that applies deep learning111 to identify antimicrobial resistance genes. On the basis of curated data sets of CARD and ARDB combined with Uniprot protein data, DeepArgs built a dissimilarity matrix between antimicrobial resistance proteins and non-antimicrobial resistance proteins and used it to train two deep-learning models: DeepArg-LS for assembled genes and DeepArg-SS for short reads. These models can be used to predict antimicrobial resistance genes in new test data.
Although the application of machine learning to antimicrobial resistance prediction and classification is promising, these techniques have a long way to go before they can be used for rapid diagnostic purposes and replace traditional culture techniques and AST, which can take days or weeks to yield results.
Conclusions and future perspectives
Antimicrobial resistance is a major public health threat. Monitoring and understanding the prevalence, mechanisms and spread of antimicrobial resistance are priorities for both individual patient care and global infection control strategies. Despite stellar advancements, hurdles for antimicrobial resistance detection and understanding persist. Costs are decreasing for sequencing and for automated antimicrobial resistance detection instruments, but start-up and operation costs still outstrip many health-care budgets. Further cost reductions for these technologies will be important for widespread adoption.
The accurate identification of resistance determinants and the correlation of antimicrobial resistance gene profiles to antimicrobial treatment outcomes will facilitate personalized approaches to developing treatment regimens. The success of this approach depends heavily on the comprehensiveness and quality of public antimicrobial resistance gene databases, which have major roles in the development of biological assays and computational tools that expand our ability to detect resistance genes in single isolates and in microbial communities. While progress has been made in building comprehensive antimicrobial resistance gene databases, lack of standardization across databases and long update intervals hold back their potential. Moreover, complex resistance mechanisms (FIG. 1b) are difficult to capture in antimicrobial resistance databases. For example, resistance can arise from epistatic relationships between multiple genes such as in the case of carbapenem resistance, which can arise from the combination of extended-spectrum β-lactamases and efflux pumps or porin impermeability112. Resistance can even occur via overexpression of normal genes such as those encoding efflux pumps, and detection of these resistance mechanisms requires transcriptional measurements113,114 (FIG. 1b,c). These complex resistance mechanisms, coupled with the fact that known antimicrobial resistance genes may not always be expressed, contribute to the difficulty in accurately predicting phenotypic antimicrobial resistance from genotypic antimicrobial resistance data. Machine learning algorithms have made headway in using isolate genomic sequence data and antimicrobial resistance gene databases to predict phenotypic resistance, but these techniques tend to be specialized to specific bacteria or are not accurate and consistent enough for general clinical deployment. To realize the goal of making phenotypic predictions from genotypic data, we need more comprehensive databases that link specific antimicrobial resistance genes to specific AST results. Importantly, these databases should include a broad diversity of bacteria with full sequence and antimicrobial resistance gene prediction metadata and report AST results with exact zone sizes or minimum inhibitory concentrations (BOX 1) rather than categorical guideline interpretations. Parallel improvements in AST and sequence-based antimicrobial resistance gene prediction will augment efforts to mitigate the clinical impact of antimicrobial resistance.
Although techniques for novel antimicrobial resistance gene discovery exist, such as functional metagenomics, these techniques still have major caveats in the types of antimicrobial resistance genes that they can detect. Innovative methods to determine other antimicrobial resistance gene mechanisms are sorely needed. Moreover, robust models to predict which resistance genes will spread both on the local level within a health-care setting and on the global scale between countries are needed. These models will likely need to incorporate not only the antimicrobial resistance gene sequence and mechanism but also the genomic context, host bacterial species and geographic location.
Rapid and accurate identification of resistance genes in isolate and metagenomic samples would augment the ability of clinicians to make treatment plans for bacterial infections, facilitating a future where sequence-based personized medicine is routine. It would also ease antimicrobial resistance surveillance efforts and enable low-resource areas to benefit more fully from rapidly decreasing sequencing costs.
Antimicrobial resistance
Bacterial ability to survive or grow in otherwise lethal or inhibitory antimicrobial concentrations.
Antimicrobial susceptibility testing
(AST). Challenge of bacteria with antimicrobials to determine whether they have phenotypic antimicrobial resistance.
Horizontal gene transfer
(HGT). Passage of resistance genes from one bacterium to another when neither bacteria is the parent or daughter cell. This process usually occurs through transduction, conjugation or transformation.
Metagenomes
Collections of genes from all organisms of a given habitat or sample.
Resistance exchange networks
Interconnected groups of environments or bacteria that transfer resistance genes with each other.
Phylogeny
The evolutionary ancestral relationships between organisms.
Contigs
Contiguous sequences assembled from sequencing reads.
De Bruijn graph
(DBG). Directional graphing algorithm commonly used for short-read assembly.
Euler’s path
A walk through a directed graph that crosses each edge in the graph only once. euler’s path is used to reconstruct genome sequences from De Bruijn graphs.
Isolate assembly
Gathering of sequencing reads from a bacterial isolate into longer contiguous sequences representative of their state within the bacterium.
Resistome
All antimicrobial resistance genes within a given sample of bacteria.
Annotation
Identification and labelling of genes within a genome.
Burrows–Wheeler transform
A reversible data transformation algorithm to organize text with repeated sequences for efficient compression. This algorithm is implemented in bioinformatics software owing to frequent repeated sequences in biological data.
Metagenomic assembly
Deconvolution and assembly of sequencing reads from a metagenomic sample.
Hidden Markov model
(HMM). A probabilistic model of antimicrobial resistance process where hidden states emit observable outputs. These models are commonly used for sequence annotation.
Microbiota
A community of microorganisms from a given habitat or sample.
Functional metagenomics
A biological assay in which a metagenomic library of DNA is expressed in a naive host and then the host is exposed to a selection pressure to select for DNA that confers a fitness advantage against the selection pressure.
Biocuration
The collection and organization of biological data in a data structure useful for future analysis.
Carbapenem resistance
Resistance against the broad-spectrum carbapenem class of β-lactam antimicrobials, which are often used as drugs of last resort.
Methicillin resistance
Resistance against methicillin, a narrow-spectrum penicillin derivative. Methicillin resistance is often seen in the context of methicillin-resistant Staphylococcus aureus (MRSA), a common human pathogen. This resistance is commonly gained by horizontal transfer of a modified target protein (see Fig. 1b and 1c).
Deep learning
An extension of representational machine learning methods where the algorithm uses multiple transformation layers between raw data and output rather than one layer. This often improves results for more complex machine learning tasks.
Acknowledgements
The authors thank K. Sukhum and M. Pandey for reading through a draft of this paper. This work was supported in part by awards to G.D. through the National Institute of Allergy and Infectious Diseases (NIAID), the Eunice Kennedy Shriver National Institute of Child Health & Human Development and the National Center for Complementary and Integrative Health of the US National Institutes of Health (NIH) under award numbers R01AI123394, R01HD092414 and R01AT009741, respectively. A.W.D. received support from the Institutional Program Unifying Population and Laboratory-Based Sciences Burroughs Wellcome Fund Grant to Washington University. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
Footnotes
Competing interests
The authors declare no competing interests.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Reviewer information
Nature Reviews Genetics thanks J. Parkhill, E. Ruppé and other anonymous reviewer(s) for their contribution to the peer review of this work.
References
- 1.Cosgrove SE & Carmeli Y The impact of antimicrobial resistance on health and economic outcomes. Clin. Infect. Dis 36, 1433–1437 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Hawkey PM The growing burden of antimicrobial resistance. J. Antimicrob. Chemother 62 (Suppl. 1), i1–i9 (2008). [DOI] [PubMed] [Google Scholar]
- 3.Acar JF Consequences of bacterial resistance to antibiotics in medical practice. Clin. Infect. Dis 24 (Suppl. 1), 17–18 (1997). [DOI] [PubMed] [Google Scholar]
- 4.Cosgrove SE The relationship between antimicrobial resistance and patient outcomes: mortality, length of hospital stay, and health care costs. Clin. Infect. Dis 42 (Suppl. 2), 82–89 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Tillotson GS & Zinner SH Burden of antimicrobial resistance in an era of decreasing susceptibility. Expert Rev. Anti. Infect. Ther 15, 663–676 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Poirel L & Nordmann P Carbapenem resistance in Acinetobacter baumannii: mechanisms and epidemiology. Clin. Microbiol. Infect 12, 826–836 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Johnson AP & Woodford N Global spread of antibiotic resistance: the example of New Delhi metallo-beta-lactamase (NDM)-mediated carbapenem resistance. J. Med. Microbiol 62, 499–513 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Gupta N, Limbago BM, Patel JB & Kallen AJ Carbapenem-resistant enterobacteriaceae: epidemiology and prevention. Clin. Infect. Dis 53, 60–67 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Liu YY et al. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: a microbiological and molecular biological study. Lancet Infect. Dis 16, 161–168 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Ventola CL The antibiotic resistance crisis: part 1: causes and threats. P T 40, 277–283 (2015). [PMC free article] [PubMed] [Google Scholar]
- 11.Centers for Disease Control and Prevention. Antibiotic/antimicrobial resistance (AR/AMR): biggest threats and data CDC; http://www.cdc.gov/drugresistance/threat-report-2013/ (updated 26 November 2018). [Google Scholar]
- 12.Smith R & Coast J The true cost of antimicrobial resistance. BMJ 346, f1493 (2013). [DOI] [PubMed] [Google Scholar]
- 13.O’Neill J Tackling Drug-Resistant Infections Globally: Final Report and Recommendations (Review on Antimicrobial Resistance, 2016).
- 14.Cassini A et al. Attributable deaths and disability-adjusted life-years caused by infections with antibiotic-resistant bacteria in the EU and the European Economic Area in 2015: a population-level modelling analysis. Lancet. Infect. Dis 19, 56–66 (2019).This paper demonstrates the impact of antimicrobial resistance on the health-care system and identifies major priorities for future mitigation efforts.
- 15.World Health Organization. Global Action Plan on Antimicrobial Resistance 2015 (Report No. 9789241509763) (WHO, 2015). [DOI] [PubMed] [Google Scholar]
- 16.Tacconelli E et al. Surveillance for control of antimicrobial resistance. Lancet. Infect. Dis 18, e99–e106 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Wernli D et al. Mapping global policy discourse on antimicrobial resistance. BMJ Global Health 2, e000378 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Didelot X, Bowden R, Wilson DJ, Peto TE & Crook DW Transforming clinical microbiology with bacterial genome sequencing. Nat. Rev. Genet 13, 601–612 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.D’Costa VM, McGrann KM, Hughes DW & Wright GD Sampling the antibiotic resistome. Science 311, 374–377 (2006).This article shows that soil bacteria are a reservoir for resistance determinants.
- 20.Wang R et al. The global distribution and spread of the mobilized colistin resistance gene mcr-1. Nat. Commun 9, 1179 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nordmann P, Naas T & Poirel L Global spread of carbapenemase-producing Enterobacteriaceae. Emerg. Infect. Dis 17, 1791–1798 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Canton R et al. Rapid evolution and spread of carbapenemases among Enterobacteriaceae in Europe. Clin. Microbiol. Infect 18, 413–431 (2012). [DOI] [PubMed] [Google Scholar]
- 23.Potter RF, D’Souza AW & Dantas G The rapid spread of carbapenem-resistant Enterobacteriaceae. Drug Resist. Updat 29, 30–46 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pesesky MW et al. KPC and NDM-1 genes in related Enterobacteriaceae strains and plasmids from Pakistan and the United States. Emerg. Infect. Dis 21, 1034–1037 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pehrsson EC et al. Interconnected microbiomes and resistomes in low-income human habitats. Nature 533, 212–216 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gibson MK, Forsberg KJ & Dantas G Improved annotation of antibiotic resistance determinants reveals microbial resistomes cluster by ecology. ISME J 9, 207–216 (2015).This paper describes the creation of a profile HMM-based resistance database and presents an application of this database showing that environmental-based and human-based samples have different resistance profiles.
- 27.Bankevich A et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol 19, 455–477 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zerbino DR & Birney E Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18, 821–829 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Simpson JT et al. ABySS: a parallel assembler for short read sequence data. Genome Res 19, 1117–1123 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luo R et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1, 18 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Compeau PE, Pevzner PA & Tesler G How to apply de Bruijn graphs to genome assembly. Nat. Biotechnol 29, 987–991 (2011).This short paper explains how DBGs are used in genome assembly.
- 32.Ghurye JS, Cepeda-Espinoza V & Pop M Metagenomic assembly: overview, challenges and applications. Yale J. Biol. Med 89, 353–362 (2016). [PMC free article] [PubMed] [Google Scholar]
- 33.Miller JR, Koren S & Sutton G Assembly algorithms for next-generation sequencing data. Genomics 95, 315–327 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Antipov D et al. plasmidSPAdes: assembling plasmids from whole genome sequencing data. Bioinformatics 32, 3380–3387 (2016). [DOI] [PubMed] [Google Scholar]
- 35.Rozov R et al. Recycler: an algorithm for detecting plasmids from de novo assembly graphs. Bioinformatics 33, 475–482 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roosaare M, Puustusmaa M, Mols M, Vaher M & Remm M PlasmidSeeker: identification of known plasmids from bacterial whole genome sequencing reads. PeerJ 6, e4588 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lanza VF et al. Plasmid flux in Escherichia coli ST131 sublineages, analyzed by plasmid constellation network (PLACNET), a new method for plasmid reconstruction from whole genome sequences. PLOS Genet 10, e1004766 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peng Y, Leung HC, Yiu SM & Chin FY IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28, 1420–1428 (2012). [DOI] [PubMed] [Google Scholar]
- 39.Li D, Liu CM, Luo R, Sadakane K & Lam TW MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Nurk S, Meleshko D, Korobeynikov A & Pevzner PA metaSPAdes: a new versatile metagenomic assembler. Genome Res 27, 824–834 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Namiki T, Hachiya T, Tanaka H & Sakakibara Y MetaVelvet: an extension of Velvet assembler to de novo metagenome assembly from short sequence reads. Nucleic Acids Res 40, e155 (2012).Peng et al. (2012), Li et al. (2015), Nurk et al. (2017) and Namiki et al. (2012) are method papers of metagenomic assemblers developed to assemble complex metagenomics data sets with uneven sequencing depths.
- 42.Sczyrba A et al. Critical assessment of metagenome interpretation-a benchmark of metagenomics software. Nat. Methods 14, 1063–1071 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bremges A & McHardy AC Critical assessment of metagenome interpretation enters the second round. mSystems 3, e00103–18 (2018).Together with Sczyrba et al., this paper describes the CAMI project designed to evaluate the differences between different metagenomics tools for metagenomic assembly, taxonomic classification and assembled contig binning.
- 44.Altschul SF, Gish W, Miller W, Myers EW & Lipman DJ Basic local alignment search tool. J. Mol. Biol 215, 403–410 (1990). [DOI] [PubMed] [Google Scholar]
- 45.Edgar RC Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461 (2010). [DOI] [PubMed] [Google Scholar]
- 46.Buchfink B, Xie C & Huson DH Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60 (2015). [DOI] [PubMed] [Google Scholar]
- 47.Werner JJ et al. Impact of training sets on classification of high-throughput bacterial 16s rRNA gene surveys. ISME J 6, 94–103 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li H & Durbin R Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Inouye M et al. SRST2: rapid genomic surveillance for public health and hospital microbiology labs. Genome Med 6, 90 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Clausen PT, Zankari E, Aarestrup FM & Lund O Benchmarking of methods for identification of antimicrobial resistance genes in bacterial whole genome data. J. Antimicrob. Chemother 71, 2484–2488 (2016). [DOI] [PubMed] [Google Scholar]
- 52.Hunt M et al. ARIBA: rapid antimicrobial resistance genotyping directly from sequencing reads. Microb. Genom 3, e000131 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fu L, Niu B, Zhu Z, Wu S & Li W CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rowe W et al. Search engine for antimicrobial resistance: a cloud compatible pipeline and web interface for rapidly detecting antimicrobial resistance genes directly from sequence data. PLOS ONE 10, e0133492 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Munk P et al. A sampling and metagenomic sequencing-based methodology for monitoring antimicrobial resistance in swine herds. J. Antimicrob. Chemother 72, 385–392 (2017). [DOI] [PubMed] [Google Scholar]
- 56.Rowe WPM & Winn MD Indexed variation graphs for efficient and accurate resistome profiling. Bioinformatics 34, 3601–3608 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Langmead B, Trapnell C, Pop M & Salzberg SL Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Henson J, Tischler G & Ning Z Next-generation sequencing and large genome assemblies. Pharmacogenomics 13, 901–915 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Carr R & Borenstein E Comparative analysis of functional metagenomic annotation and the mappability of short reads. PLOS ONE 9, e105776 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Quince C, Walker AW, Simpson JT, Loman NJ & Segata N Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol 35, 833–844 (2017).This detailed review discusses the best strategies used in shotgun metagenomics studies.
- 61.Kaminski J et al. High-specificity targeted functional profiling in microbial communities with ShortBRED. PLOS Comput. Biol 11, e1004557 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gibson MK et al. Developmental dynamics of the preterm infant gut microbiota and antibiotic resistome. Nat. Microbiol 1, 16024 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tsukayama P et al. Characterization of wild and captive baboon gut microbiota and their antibiotic resistomes. mSystems 3, e00016–18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hsu T et al. Urban transit system microbial communities differ by surface type and interaction with humans and the environment. mSystems 1, e00018–16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu B & Pop M ARDB—antibiotic resistance genes database. Nucleic Acids Res 37, D443–D447 (2009).ARDB was one of the first general antimicrobial resistance gene databases, and this paper spawned several other efforts to compile resistance gene information across drug classes and bacterial species.
- 66.Gupta SK et al. ARG-ANNOT, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob. Agents Chemother 58, 212–220 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jia B et al. CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res 45, D566–D573 (2017).This paper describes recent updates to the CARD and tools that are associated with the database.
- 68.Thai QK & Pleiss J SHV lactamase engineering database: a reconciliation tool for SHV beta-lactamases in public databases. BMC Genomics 11, 563 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Thai QK, Bos F & Pleiss J The lactamase engineering database: a critical survey of TEM sequences in public databases. BMC Genomics 10, 390 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bush K & Jacoby GA Updated functional classification of β-lactamases. Antimicrob. Agents Chemother 54, 969–976 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Srivastava A, Singhal N, Goel M, Virdi JS & Kumar M CBMAR: a comprehensive beta-lactamase molecular annotation resource. Database (Oxford) 2014, bau111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zankari E et al. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother 67, 2640–2644 (2012).This article describes Resfinder, a widely used tool for the identification of acquired antimicrobial resistance genes in whole-genome data.
- 73.Zankari E et al. PointFinder: a novel web tool for WGS-based detection of antimicrobial resistance associated with chromosomal point mutations in bacterial pathogens. J. Antimicrob. Chemother 72, 2764–2768 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yin X et al. ARGs-OAP v2.0 with an expanded SARG database and hidden Markov models for enhancement characterization and quantification of antibiotic resistance genes in environmental metagenomes. Bioinformatics 34, 2263–2270 (2018). [DOI] [PubMed] [Google Scholar]
- 75.Sandgren A et al. Tuberculosis drug resistance mutation database. PLOS Med 6, e2 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Flandrois JP, Lina G & Dumitrescu O MUBII-TB-DB: a database of mutations associated with antibiotic resistance in Mycobacterium tuberculosis. BMC Bioinformatics 15, 107 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cox G & Wright GD Intrinsic antibiotic resistance: mechanisms, origins, challenges and solutions. Int. J. Med. Microbiol 303, 287–292 (2013). [DOI] [PubMed] [Google Scholar]
- 78.Gygli SM, Borrell S, Trauner A & Gagneux S Antimicrobial resistance in Mycobacterium tuberculosis: mechanistic and evolutionary perspectives. FEMS Microbiol. Rev 41, 354–373 (2017). [DOI] [PubMed] [Google Scholar]
- 79.Allix-Beguec C et al. Prediction of susceptibility to first-line tuberculosis drugs by DNA sequencing. N. Engl. J. Med 379, 1403–1415 (2018).This paper shows the effectiveness of a sequencing approach to phenotypic antimicrobial resistance predictions in M. tuberculosis.
- 80.McArthur AG & Tsang KK Antimicrobial resistance surveillance in the genomic age. Ann. NY Acad. Sci 1388, 78–91 (2017). [DOI] [PubMed] [Google Scholar]
- 81.Yelin I & Kishony R Antibiotic resistance. Cell 172, 1136–1136 (2018). [DOI] [PubMed] [Google Scholar]
- 82.Eddy SR Profile hidden Markov models. Bioinformatics 14, 755–763 (1998). [DOI] [PubMed] [Google Scholar]
- 83.Wallace JC, Port JA, Smith MN & Faustman EM FARME DB: a functional antibiotic resistance element database. Database (Oxford) 2017, baw165 (2017).This paper compiles putative resistance determinants from functional antimicrobial selections in public databases to identify resistance determinants that are not well represented in databases built primarily from clinical bacterial isolates.
- 84.Munk P et al. Abundance and diversity of the faecal resistome in slaughter pigs and broilers in nine European countries. Nat. Microbiol 3, 898–908 (2018).This article presents a new technique for identifying antimicrobial resistance determinants by including 3D information.
- 85.Ruppe E et al. Prediction of the intestinal resistome by a three-dimensional structure-based method. Nat. Microbiol 4, 112–123 (2019). [DOI] [PubMed] [Google Scholar]
- 86.Xavier BB et al. Consolidating and exploring antibiotic resistance gene data resources. J. Clin. Microbiol 54, 851–859 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hall RM & Schwarz S Resistance gene naming and numbering: is it a new gene or not? J. Antimicrob. Chemother 71, 569–571 (2016). [DOI] [PubMed] [Google Scholar]
- 88.Carnevali C et al. Occurrence of mcr-1 in colistin-resistant salmonella enterica isolates recovered from humans and animals in Italy, 2012 to 2015. Antimicrob. Agents Chemother 60, 7532–7534 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ortega-Paredes D, Barba P & Zurita J Colistin-resistant Escherichia coli clinical isolate harbouring the mcr-1 gene in Ecuador. Epidemiol. Infect 144, 2967–2970 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Teo JQ et al. mcr-1 in multidrug-resistant blaKPC-2-producing clinical enterobacteriaceae isolates in Singapore. Antimicrob. Agents Chemother 60, 6435–6437 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fernandes MR et al. First report of the globally disseminated IncX4 plasmid carrying the mcr-1 gene in a colistin-resistant Escherichia coli sequence type 101 isolate from a human infection in Brazil. Antimicrob. Agents Chemother 60, 6415–6417 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Delgado-Blas JF, Ovejero CM, Abadia-Patino L & Gonzalez-Zorn B Coexistence of mcr-1 and blaNDM-1 in Escherichia coli from Venezuela. Antimicrob. Agents Chemother 60, 6356–6358 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kline KE et al. Investigation of first identified mcr-1 gene in an isolate from a U. S. patient - Pennsylvania, 2016. MMWR Morb. Mortal. Wkly. Rep 65, 977–978 (2016). [DOI] [PubMed] [Google Scholar]
- 94.Wong SC et al. Colistin-resistant enterobacteriaceae carrying the mcr-1 gene among patients in Hong Kong. Emerg. Infect. Dis 22, 1667–1669 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Brauer A et al. Plasmid with colistin resistance gene mcr-1 in extended-spectrum-beta-lactamase-producing Escherichia coli strains isolated from pig slurry in Estonia. Antimicrob. Agents Chemother 60, 6933–6936 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.von Wintersdorff CJ et al. Detection of the plasmid-mediated colistin-resistance gene mcr-1 in faecal metagenomes of Dutch travellers. J. Antimicrob. Chemother 71, 3416–3419 (2016). [DOI] [PubMed] [Google Scholar]
- 97.Crofts TS, Gasparrini AJ & Dantas G Next-generation approaches to understand and combat the antibiotic resistome. Nat. Rev. Microbiol 15, 422–434 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Riesenfeld CS et al. Uncultured soil bacteria are a reservoir of new antibiotic resistance genes. Environ. Microbiol 6, 981–989 (2015).This is one of the initial studies to demonstrate the application of functional metagenomics selections for discovering novel antibiotic resistance genes.
- 99.Pehrsson EC, Forsberg KJ, Gibson MK, Ahmadi S & Dantas G Novel resistance functions uncovered using functional metagenomic investigations of resistance reservoirs. Front. Microbiol 4, 145 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Forsberg KJ et al. The shared antibiotic resistome of soil bacteria and human pathogens. Science 337, 1107–1111 (2012).This paper applies a functional metagenomics approach and assembly pipeline to show evidence of resistance gene exchange between human pathogens and soil bacteria.
- 101.de la Bastide M & McCombie WR Assembling genomic DNA sequences with PHRAP. Curr. Protoc. Bioinformatics 17, 11.4.1–11.4.15 (2007). [DOI] [PubMed] [Google Scholar]
- 102.Zhu W, Lomsadze A & Borodovsky M Ab initio gene identification in metagenomic sequences. Nucleic Acids Res 38, e132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Torres-Cortes G et al. Characterization of novel antibiotic resistance genes identified by functional metagenomics on soil samples. Environ. Microbiol 13, 1101–1114 (2011). [DOI] [PubMed] [Google Scholar]
- 104.Forsberg KJ, Patel S, Wencewicz TA & Dantas G The tetracycline destructases: a novel family of tetracycline-inactivating enzymes. Chem. Biol 22, 888–897 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Martinez A et al. Genetically modified bacterial strains and novel bacterial artificial chromosome shuttle vectors for constructing environmental libraries and detecting heterologous natural products in multiple expression hosts. Appl. Environ. Microbiol 70, 2452–2463 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Dantas G & Sommer MO Context matters - the complex interplay between resistome genotypes and resistance phenotypes. Curr. Opin. Microbiol 15, 577–582 (2012).This article covers the importance of genomic context in understanding how genotypic resistance determinants result in varied phenotypic antimicrobial susceptibility profiles.
- 107.Rishishwar L, Petit RA 3rd, Kraft CS & Jordan IK Genome sequence-based discriminator for vancomycin-intermediate Staphylococcus aureus. J. Bacteriol 196, 940–948 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bradley P et al. Rapid antibiotic-resistance predictions from genome sequence data for Staphylococcus aureus and Mycobacterium tuberculosis. Nat. Commun 6, 10063 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Davis JJ et al. Antimicrobial resistance prediction in PATRIC and RAST. Sci. Rep 6, 27930 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Arango-Argoty G et al. DeepARG: a deep learning approach for predicting antibiotic resistance genes from metagenomic data. Microbiome 6, 23 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.LeCun Y, Bengio Y & Hinton G Deep learning. Nature 521, 436–444 (2015). [DOI] [PubMed] [Google Scholar]
- 112.Baroud M et al. Underlying mechanisms of carbapenem resistance in extended-spectrum beta-lactamase-producing Klebsiella pneumoniae and Escherichia coli isolates at a tertiary care centre in Lebanon: role of OXA-48 and NDM-1 carbapenemases. Int. J. Antimicrob. Agents 41, 75–79 (2013). [DOI] [PubMed] [Google Scholar]
- 113.Shigemura K et al. Association of overexpression of efflux pump genes with antibiotic resistance in Pseudomonas aeruginosa strains clinically isolated from urinary tract infection patients. J. Antibiot 68, 568–572 (2015). [DOI] [PubMed] [Google Scholar]
- 114.Depardieu F, Podglajen I, Leclercq R, Collatz E & Courvalin P Modes and modulations of antibiotic resistance gene expression. Clin. Microbiol. Rev 20, 79–114 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Isenberg HD Clinical microbiology: past, present, and future. J. Clin. Microbiol 41, 917–918 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bauer AW, Kirby WM, Sherris JC & Turck M Antibiotic susceptibility testing by a standardized single disk method. Am. J. Clin. Pathol 45, 493–496 (1966). [PubMed] [Google Scholar]
- 117.Brown DF & Brown L Evaluation of the E test, a novel method of quantifying antimicrobial activity. J. Antimicrob. Chemother 27, 185–190 (1991). [DOI] [PubMed] [Google Scholar]
- 118.Jorgensen JH & Ferraro MJ Antimicrobial susceptibility testing: a review of general principles and contemporary practices. Clin. Infect. Dis 49, 1749–1755 (2009).This is a review of traditional microbiology techniques and of several automation innovations, including disc diffusion, microbroth dilution and a Vitek system.
- 119.Seng P et al. Ongoing revolution in bacteriology: routine identification of bacteria by matrix-assisted laser desorption ionization time-of-flight mass spectrometry. Clin. Infect. Dis 49, 543–551 (2009). [DOI] [PubMed] [Google Scholar]
- 120.Nagy E, Maier T, Urban E, Terhes G & Kostrzewa M Species identification of clinical isolates of Bacteroides by matrix-assisted laser-desorption/ionization time-of-flight mass spectrometry. Clin. Microbiol. Infect 15, 796–802 (2009). [DOI] [PubMed] [Google Scholar]
- 121.Eigner U et al. Performance of a matrix-assisted laser desorption ionization-time-of-flight mass spectrometry system for the identification of bacterial isolates in the clinical routine laboratory. Clin. Lab 55, 289–296 (2009). [PubMed] [Google Scholar]
- 122.Vrioni G et al. MALDI-TOF mass spectrometry technology for detecting biomarkers of antimicrobial resistance: current achievements and future perspectives. Ann. Transl Med 6, 240 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kostrzewa M, Sparbier K, Maier T & Schubert S MALDI-TOF MS: an upcoming tool for rapid detection of antibiotic resistance in microorganisms. Proteomics Clin. Appl 7, 767–778 (2013). [DOI] [PubMed] [Google Scholar]
- 124.Sparbier K, Schubert S & Kostrzewa M MBT-ASTRA: a suitable tool for fast antibiotic susceptibility testing? Methods 104, 48–54 (2016). [DOI] [PubMed] [Google Scholar]
- 125.Yilmaz O & Demiray E Clinical role and importance of fluorescence in situ hybridization method in diagnosis of H pylori infection and determination of clarithromycin resistance in H pylori eradication therapy. World J. Gastroenterol 13, 671–675 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Moter A & Gobel UB Fluorescence in situ hybridization (FISH) for direct visualization of microorganisms. J. Microbiol. Methods 41, 85–112 (2000). [DOI] [PubMed] [Google Scholar]
- 127.Juttner S et al. Reliable detection of macrolide-resistant Helicobacter pylori via fluorescence in situ hybridization in formalin-fixed tissue. Mod. Pathol 17, 684–689 (2004). [DOI] [PubMed] [Google Scholar]
- 128.Haas M, Essig A, Bartelt E & Poppert S Detection of resistance to macrolides in thermotolerant campylobacter species by fluorescence in situ hybridization. J. Clin. Microbiol 46, 3842–3844 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Werner G et al. Detection of mutations conferring resistance to linezolid in Enterococcus spp. by fluorescence in situ hybridization. J. Clin. Microbiol 45, 3421–3423 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Choi J et al. A rapid antimicrobial susceptibility test based on single-cell morphological analysis. Sci. Transl Med 6, 267ra174 (2014). [DOI] [PubMed] [Google Scholar]
- 131.Kalashnikov M et al. Rapid phenotypic stress-based microfluidic antibiotic susceptibility testing of Gram-negative clinical isolates. Sci. Rep 7, 8031 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mohan R et al. A multiplexed microfluidic platform for rapid antibiotic susceptibility testing. Biosens. Bioelectron 49, 118–125 (2013). [DOI] [PubMed] [Google Scholar]
- 133.Hou HW, Bhattacharyya RP, Hung DT & Han J Direct detection and drug-resistance profiling of bacteremias using inertial microfluidics. Lab. Chip 15, 2297–2307 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Baltekin O, Boucharin A, Tano E, Andersson DI & Elf J Antibiotic susceptibility testing in less than 30 min using direct single-cell imaging. Proc. Natl Acad. Sci. USA 114, 9170–9175 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Choi J et al. Rapid drug susceptibility test of Mycobacterium tuberculosis using microscopic time-lapse imaging in an agarose matrix. Appl. Microbiol. Biotechnol 100, 2355–2365 (2016). [DOI] [PubMed] [Google Scholar]
- 136.Seki M, Kim CK, Hayakawa S & Mitarai S Recent advances in tuberculosis diagnostics in resource-limited settings. Eur. J. Clin. Microbiol. Infect. Dis 37, 1405–1410 (2018). [DOI] [PubMed] [Google Scholar]
- 137.Wolfe AJ et al. Evidence of uncultivated bacteria in the adult female bladder. J. Clin. Microbiol 50, 1376–1383 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Rudkjobing VB et al. Comparing culture and molecular methods for the identification of microorganisms involved in necrotizing soft tissue infections. BMC Infect. Dis 16, 652 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Fok CS et al. Urinary symptoms are associated with certain urinary microbes in urogynecologic surgical patients. Int. Urogynecol. J 29, 1765–1771 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Mowat A Commentary on: urinary symptoms are associated with certain urinary microbes in urogynecologic surgical patients. Int. Urogynecol. J 29, 1773 (2018). [DOI] [PubMed] [Google Scholar]
- 141.Patel JB, Tenover FC, Turnidge JD & Jorgensen JH in Manual of Clinical Microbiology 10th edn (eds Versalovic J et al.) (American Society for Microbiology, 2011). [Google Scholar]
- 142.Shetty N, Hill G & Ridgway GL The Vitek analyser for routine bacterial identification and susceptibility testing: protocols, problems, and pitfalls. J. Clin. Pathol 51, 316–323 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Idelevich EA et al. Evaluation of an automated system for reading and interpreting disk diffusion antimicrobial susceptibility testing of fastidious bacteria. PLOS ONE 11, e0159183 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lutgring JD et al. Evaluation of the accelerate pheno system: results from two academic medical centers. J. Clin. Microbiol 56, e01672–17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Marschal M et al. Evaluation of the accelerate pheno system for fast identification and antimicrobial susceptibility testing from positive blood cultures in bloodstream infections caused by gram-negative pathogens. J. Clin. Microbiol 55, 2116–2126 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Florio W, Morici P, Ghelardi E, Barnini S & Lupetti A Recent advances in the microbiological diagnosis of bloodstream infections. Crit. Rev. Microbiol 44, 351–370 (2018). [DOI] [PubMed] [Google Scholar]
- 147.Peker N, Couto N, Sinha B & Rossen JW Diagnosis of bloodstream infections from positive blood cultures and directly from blood samples: recent developments in molecular approaches. Clin. Microbiol. Infect 24, 944–955 (2018). [DOI] [PubMed] [Google Scholar]
- 148.Fredborg M et al. Rapid antimicrobial susceptibility testing of clinical isolates by digital time-lapse microscopy. Eur. J. Clin. Microbiol. Infect. Dis 34, 2385–2394 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Dekker J, Rippe K, Dekker M & Kleckner N Capturing chromosome conformation. Science 295, 1306–1311 (2002). [DOI] [PubMed] [Google Scholar]
- 150.Hakim O & Misteli T SnapShot: chromosome confirmation capture. Cell 148, 1068–1068.e2 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Trussart M et al. Defined chromosome structure in the genome-reduced bacterium Mycoplasma pneumoniae. Nat. Commun 8, 14665 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Yildirim A & Feig M High-resolution 3D models of Caulobacter crescentus chromosome reveal genome structural variability and organization. Nucleic Acids Res 46, 3937–3952 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Marbouty M, Baudry L, Cournac A & Koszul R Scaffolding bacterial genomes and probing host-virus interactions in gut microbiome by proximity ligation (chromosome capture) assay. Sci. Adv 3, e1602105 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Stewart RD et al. Assembly of 913 microbial genomes from metagenomic sequencing of the cow rumen. Nat. Commun 9, 870 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Press MO et al. Hi-C deconvolution of a human gut microbiome yields high-quality draft genomes and reveals plasmid-genome interactions. Preprint at bioRxiv 10.1101/198713 (2017). [DOI] [Google Scholar]
- 156.Eid J et al. Real-time DNA sequencing from single polymerase molecules. Science 323, 133–138 (2009). [DOI] [PubMed] [Google Scholar]
- 157.Clarke J et al. Continuous base identification for single-molecule nanopore DNA sequencing. Nat. Nanotechnol 4, 265–270 (2009). [DOI] [PubMed] [Google Scholar]
- 158.Wick RR, Judd LM, Gorrie CL & Holt KE Completing bacterial genome assemblies with multiplex MinION sequencing. Microb. Genom 3, e000132 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Liao YC, Lin SH & Lin HH Completing bacterial genome assemblies: strategy and performance comparisons. Sci. Rep 5, 8747 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Wick RR, Judd LM, Gorrie CL & Holt KE Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLOS Comput. Biol 13, e1005595 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Frank JA et al. Improved metagenome assemblies and taxonomic binning using long-read circular consensus sequence data. Sci. Rep 6, 25373 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Driscoll CB, Otten TG, Brown NM & Dreher TW Towards long-read metagenomics: complete assembly of three novel genomes from bacteria dependent on a diazotrophic cyanobacterium in a freshwater lake co-culture. Stand. Genomic. Sci 12, 9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Beaulaurier J et al. Metagenomic binning and association of plasmids with bacterial host genomes using DNA methylation. Nat. Biotechnol 36, 61–69 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Bertrand D et al. Nanopore sequencing enables high-resolution analysis of resistance determinants and mobile elements in the human gut microbiome. Preprint at bioRxiv 10.1101/456905 (2018). [DOI] [Google Scholar]
- 165.Břinda K et al. Lineage calling can identify antibiotic resistant clones within minutes. Preprint at bioRxiv 10.1101/403204 (2018). [DOI] [Google Scholar]
- 166.Croucher NJ & Thomson NR Studying bacterial transcriptomes using RNA-seq. Curr. Opin. Microbiol 13, 619–624 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Wang Z, Gerstein M & Snyder M RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet 10, 57–63 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Dersch P, Khan MA, Muhlen S & Gorke B Roles of regulatory RNAs for antibiotic resistance in bacteria and their potential value as novel drug targets. Front. Microbiol 8, 803 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Khaledi A et al. Transcriptome profiling of antimicrobial resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother 60, 4722–4733 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Suzuki S, Horinouchi T & Furusawa C Prediction of antibiotic resistance by gene expression profiles. Nat. Commun 5, 5792 (2014).This paper uses expression profiles to help predict phenotypic resistance from genotypic data, showing the power of combining multiple omics techniques.
- 171.Qin H et al. Comparative transcriptomics of multidrug-resistant Acinetobacter baumannii in response to antibiotic treatments. Sci. Rep 8, 3515 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Low YM et al. Elucidating the survival and response of carbapenem resistant Klebsiella pneumoniae after exposure to imipenem at sub-lethal concentrations. Pathog. Glob. Health 112, 378–386 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Cho H & Kim KS Escherichia coli OxyS RNA triggers cephalothin resistance by modulating the expression of CRP-associated genes. Biochem. Biophys. Res. Commun 506, 66–72 (2018). [DOI] [PubMed] [Google Scholar]
- 174.Schniederjans M, Koska M & Häussler S Transcriptional and mutational profiling of an aminoglycoside-resistant Pseudomonas aeruginosa small-colony variant. Antimicrob. Agents Chemother 61, e01178–17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Felden B & Cattoir V Bacterial adaptation to antibiotics through regulatory RNAs. Antimicrob. Agents Chemother 62, e02503–17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Antonopoulos DA et al. PATRIC as a unique resource for studying antimicrobial resistance. Brief. Bioinform 10.1093/bib/bbx083 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.de Man TJ & Limbago BM SSTAR, a stand-alone easy-to-use antimicrobial resistance gene predictor. mSphere 1, e00050–15 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Lakin SM et al. MEGARes: an antimicrobial resistance database for high throughput sequencing. Nucleic Acids Res 45, D574–D580 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Naas T et al. Beta-lactamase database (BLDB) - structure and function. J. Enzyme Inhib. Med. Chem 32, 917–919 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Saha SB, Uttam V & Verma V u-CARE: user-friendly Comprehensive Antibiotic resistance Repository of Escherichia coli. J. Clin. Pathol 68, 648–651 (2015). [DOI] [PubMed] [Google Scholar]