

Summary
Hyperactivity and disturbances of attention are common behavioral disorders whose underlying cellular and neural circuit causes are not understood. We report the discovery that striatal astrocytes drive such phenotypes through a hitherto unknown synaptic mechanism. We found that striatal medium spiny neurons (MSNs) triggered astrocyte signaling via GABAB receptors. Selective chemogenetic activation of this pathway in striatal astrocytes in vivo resulted in acute behavioral hyperactivity and disrupted attention. Such responses also resulted in up-regulation of the synaptogenic cue thrombospondin-1 (TSP1) in astrocytes, increased excitatory synapses, enhanced corticostriatal synaptic transmission, and increased MSN action potential firing in vivo. All of these changes were reversed by blocking TSP1 effects. Our data identify a form of bidirectional neuron-astrocyte communication, and demonstrate that acute reactivation of a single, latent astrocyte synaptogenic cue alters striatal circuits controlling behavior, revealing astrocytes and the TSP1 pathway as therapeutic targets in hyperactivity, attention deficit and related psychiatric disorders.
Keywords: hyperactivity, attention deficit, astrocyte, behavior, striatum, microcircuit, thrombospondin, gabapentin
Graphical Abstract
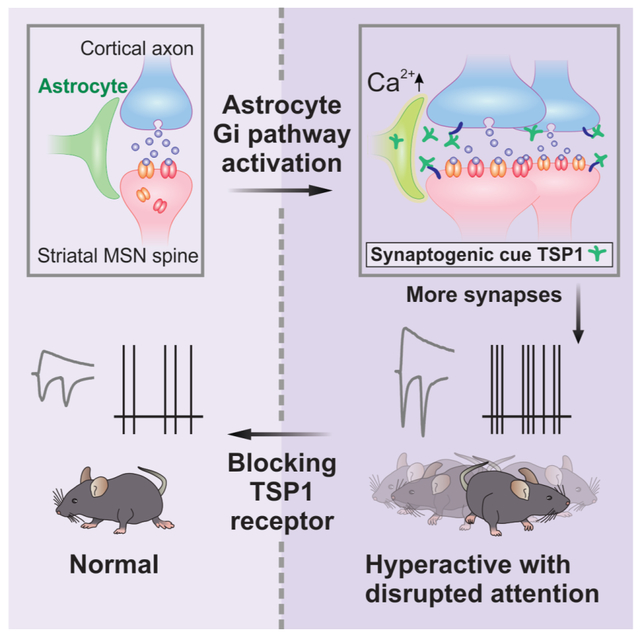
In Brief
Bi-directional communication between striatal neurons and astrocytes drive acute behavioral hyperactivity and disrupted attention
Introduction
Hyperactivity and disturbances of attention are common behavioral disorders (DSM-5, 2013; Fayyad et al., 2007; Polanczyk et al., 2007) whose underlying causes are unknown and which lack adequate treatment (Curatolo et al., 2010; de la Peña et al., 2018). Such disorders involve dysfunctions in the striatum based on imaging studies in humans (Cubillo et al., 2012; Riva et al., 2018). The striatum is the largest nucleus of the basal ganglia, a group of interconnected subcortical nuclei involved in movement, repetitive behaviors, obsessions, habits, tics and diverse neuropsychiatric conditions (Graybiel, 2008). In the current study, we report the unexpected discovery that latent synaptogenic cues derived from striatal astrocytes drive behavioral hyperactivity with disrupted attention in adult mice.
Initially documented over a century ago, astrocytes represent about 40% of all brain cells. They are the most numerous type of glia and tile the entire central nervous system (Barres, 2008). During development, astrocytes provide important cues to regulate synapse formation and removal (Allen and Lyons, 2018), whereas in adults the finest astrocyte processes from these “bushy” cells continue to contact neurons, synapses, blood vessels and other glial cells. In these locations, astrocytes mediate multiple active and homeostatic functions (Attwell et al., 2010; Khakh and Sofroniew, 2015; Volterra et al., 2014). Astrocytes also display CNS area specific properties and functions (Chai et al., 2017; Haim and Rowitch, 2017; Molofsky et al., 2014). Despite these advances, the mechanisms of astrocyte-neuron signaling, its effects on the functions of intact neural circuits, their behavioral outputs and their contributions to brain diseases remain to be fully elucidated.
Replete with molecularly defined astrocytes, the striatum represents an important circuit to explore astrocyte biology within adult mice (Chai et al., 2017; Kelley et al., 2018). As the major input nucleus of the basal ganglia, the striatum integrates converging excitatory and inhibitory signals from numerous parts of the brain and is involved in action selection and motor function (Graybiel, 2008). We used several recently developed striatal astrocyte selective genetic, transcriptomic, imaging, behavioral and electrophysiology approaches (Bakhurin et al., 2016; Chai et al., 2017; Srinivasan et al., 2016; Yu et al., 2018) to interrogate the roles of bidirectional neuron-astrocyte interactions in the function of striatal microcircuits in vivo. We discovered an unexpected mechanism for astrocyte-neuron mediated synaptic plasticity, a hitherto unknown role for astrocytes in hyperactivity and disrupted attention phenotypes and potential therapeutic strategies within astrocytes to treat such psychiatric diseases.
Results
The results of statistical comparisons, n numbers and P values are shown in the figure panels or figure legends with the relevant average data. When the average data are reported in the text, the statistics are also reported there. However, all statistical tests are reported in Supplementary Table 1 for every experiment.
Striatal MSN-to-astrocyte signaling via GABA
We expressed the genetically encoded Ca2+ indicator GCaMP6f (Chen et al., 2013) in striatal astrocytes (Srinivasan et al., 2016) and depolarized medium spiny neurons (MSNs) to physiological upstate-like membrane potential transitions (Wilson and Kawaguchi, 1996) via whole-cell patch-clamp (Figure 1A-C). MSN depolarization by ~20–30 mV resulted in action potential (AP) firing and significantly increased the frequency of Ca2+ signals in nearby astrocytes (<50 μm away from MSN somata or dendrites) from 1.4 ± 0.2 to 2.4 ± 0.3 min−1 (Figure 1B,C; n = 24 astrocytes, 6 mice, P < 0.001). The amplitude of the Ca2+ signals was unaltered (0.3 ± 0.04 to 0.3 ± 0.04 dF/F; P > 0.05, n = 24 astrocytes, 6 mice), but their duration increased (2.8 ± 0.04 to 4.0 ± 0.4 s; P < 0.01, n = 24 astrocytes, 6 mice) likely reflecting merged events. No change in Ca2+ signals was observed by current injection via an open pipette, indicating that the astrocyte responses were not due to mechanical effects (Figure 1B-C; n = 20 astrocytes, 5 mice). Furthermore, astrocytes responded similarly when either D1 or D2 MSNs were depolarized (Figure S1A-C), likely reflecting developmental maturity in adult mice (Martín et al., 2015) and consistent with anatomical data (Octeau et al., 2018). MSN depolarization-evoked astrocyte Ca2+ signals were resistant to tetrodotoxin (TTX; 300 nM; Figure 1C, n = 21 astrocytes, 5 mice), which blocked all APs (Figure S1F). However, astrocyte Ca2+ signals were abolished by Cd2+ (50 μM, n = 20 astrocytes, 5 mice) and nimodipine (20 μM, n = 23 astrocytes, 4 mice), which both block MSN L-type Ca2+ channels (Bargas et al., 1994; Carter and Sabatini, 2004) (Figure 1C). The depolarization-evoked astrocyte Ca2+ signals were also blocked by MSN dialysis with the Ca2+ chelator BAPTA (10 mM, n = 21 astrocytes, 4 mice) or with the light-chain of tetanus toxin (LC-TeNT; 1 μM; Figure 1C; n = 24 astrocytes, 5 mice), which blocks vesicular release. Together with imaging of MSN activity during upstate-like heightened excitability (Figure S1D-G), these data show that MSN membrane potential transitions open high-voltage activated Ca2+ channels and cause Ca2+-dependent vesicular release of a substance from MSNs that communicates to nearby astrocytes to cause intracellular Ca2+ elevations.
Figure 1: MSN GABA release activated striatal astrocyte Ca2+ signaling in situ and in vivo.

(A) Whole-cell recording from MSNs (filled with Alexa 568) and imaging from nearby cytosolic GCaMP6f expressing astrocytes.
(B) MSN depolarization to upstate like levels for 5 s (117 ± 11 APs evoked) increased the frequency of astrocyte Ca2+ signals (blue traces, 5 representative cells). This did not occur without patching (gray traces, 5 representative cells).
(C) Graph of astrocyte Ca2+ signal frequency before and after MSN depolarization in control and various experimental configurations (n = 18-24 astrocytes from 4-6 mice per condition).
(D-E) Striatal astrocyte specific qPCR (D, n = 4 mice) and Western blotting (E, n = 6 experiments from 20 mice) revealed GABAB receptor enrichment in astrocytes.
(F) Left, Baclofen bath application increased the frequency of astrocyte Ca2+ signals (3 representative cells). The right summary graph shows astrocyte Ca2+ signal frequency before and after drug application or MSN depolarization in various experimental configurations (n = 15-30 astrocytes from 4-5 mice).
(G) Left, astrocyte Ca2+ signals (3 representative cells) from the striatum in which Gabbr1 was deleted. The right summary graphs show astrocyte Ca2+ signal frequency before and after baclofen application or MSN depolarization in Gabbr1 f/f mice with or without Cre (n = 18-25 astrocytes from 4 mice).
(H) Cartoon illustrating AAV microinjection into the dorsal striatum to delete Gabbr1 in astrocytes by delivering AAV2/5 GfaABC1D-Cre and the method used to activate neurons in vivo with optical stimulation following expression of ChR2(H134R). tdTomato was expressed in order to visualize astrocytes.
(I) ChR2-based striatal neuron excitation in vivo resulted in c-Fos expression in astrocytes, which was attenuated by Gabbr1 deletion in astrocytes (n = 4 mice).
Paired t-test or Wilcoxon signed ranks test between before (basal) and after stimulation (C, F, G). Paired t-test (D, E). Two-way ANOVA test followed by Tukey’s post-hoc test (I). Scale bars, 20 μm (A, I). Data are shown as mean ± s.e.m. Full details of n numbers, precise P values and statistical tests are reported in Supplementary Table 1. * indicates P < 0.05, ** indicates P < 0.01, *** indicates P < 0.001, **** indicates P < 0.0001, NS indicates not significantly different. See also Fig S1-3.
Since MSNs are GABAergic, we explored roles for GABA in MSN-to-astrocyte signaling. A role for GABA was supported by RNA-seq (Chai et al., 2017) and qPCR data showing enrichment of GABAB receptor Gabbr1 and Gabbr2 mRNAs in striatal astrocytes (Figure 1D; 4 mice). GABAB receptor type-1 (GB1R) proteins (gene: Gabbr1) were abundant in striatal astrocytes isolated by FACS (Chai et al., 2017) from Aldh1l1-eGFP mice (Figure 1E; n = 6; 20 mice). Furthermore, consistent with functional expression of GABA receptors in astrocytes, bath application of GABA (300 μM, n = 24 astrocytes, 5 mice) and the GABAB receptor agonist baclofen (50 μM, n = 20 astrocytes, 5 mice) increased astrocyte Ca2+ signals (Figure 1F, P < 0.01). The effect of baclofen was blocked by the GABAB receptor antagonist CGP55845 (10 μM, n = 15 astrocytes, 4 mice; Figure 1F), which also blocked the MSN-depolarization evoked astrocyte Ca2+ signals (Figure 1F, n = 18 astrocytes, 4 mice). Astrocyte Ca2+ responses evoked by baclofen and by MSN-depolarization were abolished (Figure 1G) in mice in which GB1Rs were deleted from the striatum (Figure 1G & S2A-B).
We explored if MSN depolarization stimulated astrocytes via GABA in vivo. We expressed ChR2(H134R) in MSNs and assessed immediate early gene (c-Fos) expression in astrocytes following optical stimulation. We detected GB1R-dependent c-Fos expression in astrocytes following MSN ChR2(H134R) stimulation: the optically stimulated increase in c-Fos expression within astrocytes was significantly reduced when GB1R was deleted (Figure 1H-I; 4 mice). Ca2+ signals represent a readout of diverse astrocyte G-protein coupled receptors (GPCRs) (Porter and McCarthy, 1997). GB1Rs couple to Gi proteins, which in astrocytes (Haustein et al., 2014) leads to Ca2+ elevations by activation of phospholipase C, which we confirmed for the GABAB receptor responses (Figure S2C,D). Furthermore, MSNs intermingle extensively with astrocytes (Chai et al., 2017; Octeau et al., 2018) and their dendrites were closely juxtaposed with astrocyte somata and processes (Figure S2E,F; n = 26 images, 4 mice), providing the proximity for MSN released GABA to stimulate astrocyte GABA receptors. We hypothesize that MSNs release GABA from their dendrites to mediate astrocyte responses: dendritic release of neurotransmitters including GABA is known (Waters et al., 2005). It has also been suggested that hippocampal astrocytes respond to glutamate, ATP and/or endocannabinoid release from dendrites (Bernardinelli et al., 2011; Navarrete and Araque, 2008) via release mechanisms that are not yet delineated. Taken together, our data provide strong evidence for MSN-to-astrocyte signaling mediated by neuronal GABA release acting on astrocyte GABAB receptors (Figures 1, S1-S2).
We comment on our use of mice carrying a floxed (f/f) Gabbr1 allele and the use of AAVs. In the preceding sections, we deleted GB1R from astrocytes using striatal AAV2/5 GfaABC1D-Cre microinjections. We could identify astrocytes based on their bushy morphologies as well as by marker expression (Figure S2A,B), and we could therefore easily monitor the consequences of deleting GB1Rs in single cell evaluations (Figure 1G,I). However, as reported in Fig S3A-D and the associated legend, we could not use Gabbr1 f/f mice for astrocyte-selective evaluations of more complex phenomena such as animal behavior. To explore the consequences of GB1R Gi-pathway activation within astrocytes, in the following section we used chemogenetic approaches that have been fully validated for astrocyte selectivity (Adamsky et al., 2018; Chai et al., 2017).
Striatal astrocyte Gi pathway activation in vivo
GABAB receptors exist in multiple brain cells including neurons, therefore GABAB receptor agonists cannot be used in vivo to interrogate astrocyte GABAB receptor mediated physiology. Furthermore, currently available genetic strategies cannot selectively delete GABAB receptors only from striatal astrocytes in the adult brain (Figure S3A-D). Hence, to specifically explore the consequences of striatal astrocyte GABAB Gi pathway activation in vivo, which is necessary to interpret behavioral effects, we expressed hM4Di Designer Receptors Exclusively Activated by Designer Drugs (DREADDs) (Roth, 2016) using established methods that result in selective expression within 84 ± 3% of striatal astrocytes (Chai et al., 2017; Yu et al., 2018) using AAV (Figure S3E-G; 4 mice). hM4Di and GCaMP6f were also co-expressed so that the consequences of hMD4i activation could be imaged (Figure 2A, n = 34 mice). We confirmed that intrastriatal microinjection of AAV2/5-delivered cargo was astrocyte selective and restricted to the striatum, although there was a little expression proximal to the needle tract in astrocytes of the cortex and sometimes of the corpus callosum (Figure S3E). We suspect such expression occurred in all past studies employing viruses, as it is impossible to reach subcortical brain structures without advancing the needle through the overlying tissue: all studies employing microinjections (including ours) need to be interpreted with this anatomical caveat in mind.
Figure 2: Astrocyte-specific Gi pathway activation by a Gi-DREADD hM4Di.

(A) Cartoon illustrating AAVs used for expressing GCaMP6f with and without mCherry-fused hM4Di in astrocytes in the dorsal striatum. The lower images show GCaMP6f and hM4Di-mCherry expressing astrocytes in striatal slices were colocalised (Chai et al., 2017).
(B-C) Kymographs and ΔF/F traces of astrocyte Ca2+ responses evoked by bath application of 1 μM CNO in control AAV injected and hM4Di injected mice. The bar graph shows the CNO-evoked integrated area of astrocyte Ca2+ signals in the hM4Di group, and in the controls (n ≥ 11 cells from ≥ 3 mice).
(D) Schematic illustrating 1 mg/kg CNO was administrated i.p. in vivo 2 hr prior to harvesting brains for imaging.
(E) Kymographs and ΔF/F traces of astrocyte Ca2+ responses in control and hM4Di groups. The bar graphs summarize the integrated areas of the spontaneous Ca2+ signals in hM4Di and control mice that received CNO i.p. 2 hr prior (n ≥ 21 astrocytes from ≥ 3 mice). These data show that a single in vivo dose of CNO evoked a long lasting increase in astrocyte Ca2+ signaling.
(F) hM4Di activation with in vivo CNO administration increased c-Fos expression in striatal S100β positive astrocytes (4 mice).
Scale bars, 20 μm (A, F). Data are shown as mean ± s.e.m. Full details of n numbers, precise P values and statistical tests are reported in Supplementary Table 1. * indicates P < 0.05, **** indicates P < 0.0001. See also Fig S3.
In brain slices from control mice, the hM4Di agonist clozapine-N-oxide (CNO at 1 μM) had no effect on astrocyte Ca2+ signals (Figure 2B,C; Movie S1; n = 14 astrocytes, 4 mice). However, in brain slices from mice expressing hM4Di in striatal astrocytes, CNO evoked significant astrocyte Ca2+ elevations (Figure 2B,C, Movie S2; n = 11 astrocytes, 4 mice, P < 0.0001). These were similar to those mediated by GABAB receptors (Figure 1F) and other endogenous GPCRs (the CNO-evoked response area was 52.1 ± 8.4 dF/F.sec, whereas that for phenylephrine (Srinivasan et al., 2016) acting on α1 receptors was 62.5 ± 8.8 dF/F.sec; n = 11 & 12 astrocytes, n = 4 & 3 mice). Furthermore, 2 hrs after acute in vivo administration of CNO (Alexander et al., 2009), striatal astrocytes in brain slices displayed significantly elevated spontaneous Ca2+ signals (Figure 2E,F; n = 21 & 28 astrocytes, n = 3 & 4 mice). in vivo hM4Di activation by CNO increased c-Fos expression in striatal astrocytes (Figure 2F; n = 4 mice). Thus, CNO stimulated hM4Di-expressing striatal astrocytes to a level similar to that mediated by endogenous GPCRs (Porter and McCarthy, 1997; Shigetomi et al., 2016) recalling data with exogenous and endogenous GABA (Figure 1).
Hyperactivity and disrupted attention following striatal astrocyte Gi pathway stimulation
We prepared mice with bilateral expression of hM4Di in striatal astrocytes and assessed behavior 2 hrs after intraperitoneal (i.p.) CNO (Alexander et al., 2009) (Figure 3). Since CNO can have off-target effects (Gomez et al., 2017), we performed three controls for every behavior experiment. We prepared mice with a control AAV (tdTomato) and administered either vehicle or CNO (i.e. “AAV + Veh” and “AAV + CNO” groups in Figure 3A,B). We also prepared hM4Di expressing mice, which received either vehicle or CNO (“hM4Di + Veh” and “hM4Di + CNO” groups in Figure 3A,B). hM4Di + CNO mice showed heightened ambulation in the open-field when compared to the AAV + CNO control, or to the hM4Di + Veh and AAV + Veh groups (Figure 3B, C). There were no differences between groups on the accelerating rotarod that we could ascribe to altered motor function (Figure S4A). Interestingly, the hM4Di + CNO mice showed heightened rearing behavior on this task (Figure S4B) increasing their tendency to fall off the rotarod on Day 1. However, after multiple days of testing there were no significant differences between the groups (Figure S4A). There were also no differences in the footprint assay, showing that the mice had intact motor co-ordination (Figure S4C). Consistent with an overall hyperlocomotion phenotype disturbing the bedding, hM4Di + CNO mice buried significantly more marbles (Figure S4D).
Figure 3: Astrocyte-specific Gi pathway activation in vivo induced hyperactivity and disrupted attention.

(A) Cartoon illustrating the AAV2/5 reagents and approaches for selectively expressing hM4Di-mCherry or tdTomato (as a control AAV) bilaterally in striatal astrocytes. Once such mice were prepared, behavior was assessed 3 weeks later and 2 hrs after intraperitoneal administration of 1 mg/kg CNO or vehicle.
(B) The representative open-field activity tracks show the 4 experimental groups used in behavioral analyses to control for potential off target effects of CNO and to control for AAV microinjections.
(C) Distance travelled by the mice over 20 min in an open field chamber, divided into 5 min epochs and also pooled over 20 mins for the 4 experimental groups.
(D) Cartoon of the modified open field test with a light stimulus.
(E) Distance travelled in the modified open field chamber before, during and after light stimulation (in 1 min epochs). Notably, the hM4Di + CNO group showed no significant increase in ambulation in response to light stimulation, whereas all the other groups did so.
(F) Behavioral layout of the novel object recognition task for the 4 experimental groups. No significant difference was found between hM4Di + Veh and AAV + Veh groups across all the behavioral tests, but there were clear differences between the AAV + CNO and the hM4Di + CNO groups (B-F).
Data: mean ± s.e.m. Full details of n numbers, precise P values and statistical tests are reported in Supplementary Table 1. * indicates P < 0.05, ** indicates P < 0.01, *** indicates P < 0.001, NS indicates not significantly different. See also Fig S3-4.
In humans, hyperactivity is often associated with a lack of attention to environmental stimuli (e.g. Attention Deficit Hyperactivity Disorder, ADHD) (DSM-5, 2013). To explore this association in CNO-treated hM4Di mice we used a well-characterized modified open field task (Godsil et al., 2005a; Godsil and Fanselow, 2004; Godsil et al., 2005b) (Figure 3D). In this initially dark open field, onset of a localized visual stimulus drives investigatory activity. hM4Di+CNO mice were initially hyperactive in the dark relative to AAV+CNO controls (Figure 3E upper panel). However, unlike controls, which showed pronounced investigatory response to the light, hM4Di+CNO mice appeared oblivious to stimulus onset. Again, the hM4Di+CNO mice did not react to light termination while control mice decreased their activity to pre light levels. hM4Di+Veh mice were indistinguishable from controls (Figure 3E lower panel). Furthermore, in the novel object recognition task, hM4Di + CNO mice spent significantly less time with the novel object as compared to controls (Figure 3F, 6–8 mice). Overall, activation of an astrocyte-specific Gi pathway produced inattentive hyperactivity in mice reminiscent of human ADHD.
Astrocyte Gi pathway augmented MSN excitatory synapses and increased AP firing
The striatum is involved in common hyperactivity disorders in humans (Cubillo et al., 2012; Riva et al., 2018), which lack mechanistic understanding, but must involve synaptic and circuit dysfunctions. To explore such possibilities, we evaluated synaptic mechanisms accompanying hyperactivity phenotypes following acute striatal astrocyte Gi-pathway activation in vivo (Figure 3). In sagittal brain slices, we stimulated glutamatergic corticostriatal axons to assess fast excitatory postsynaptic currents (EPSCs) onto MSNs in the four experimental groups (Figure 4A,B). We recorded AMPA receptor-mediated evoked EPSCs, paired-pulse responses (at −70 mV) and NMDA receptor-mediated evoked EPSCs (at +40 mV; Figure 4A). We found no significant changes in any of these metrics for the three control groups (AAV + Veh, AAV + CNO, hM4Di + Veh). However, in the hM4Di + CNO group, we detected significantly potentiated AMPA and NMDA EPSCs (Figure 4A,B), with no change in paired-pulse or AMPA/NMDA ratios (Figure 4A,B), arguing against altered neurotransmitter release probability or D-serine levels. For the experiments shown in Figure 4AB, we used the same stimulation for each slice, but we also examined evoked AMPA EPSCs at multiple stimulation intensities (Figure 4C and C’; n = 12–13 MSNs from 4 mice) and found the same results. To our knowledge, the boosting of both AMPA and NMDA receptor-mediated EPSCs is a previously unreported synaptic phenotype mediated by astrocytes, prompting us to explore the underlying mechanisms.
Figure 4: Increased corticostriatal excitatory synaptic transmission and elevated MSN firing in vivo by acute astrocyte Gi pathway activation.

(A) Cartoon illustrating whole-cell patch-clamp recording from an MSN (filled with Biocytin) surrounded by hM4Di-expressing astrocytes. The lower traces show representative traces for evoked AMPA.R EPSCs due to paired stimuli at membrane potentials of −70 mV (i) and for NMDA.R EPSCs due to single stimuli at +40 mV (ii) from the indicated 4 experimental groups.
(B) Summary of multiple experiments such as those illustrated with representative traces in panel A (n = 12-13 MSNs from 4 mice). Notably, the AMPA.R and the NMDA.R EPSC amplitude in the hM4Di + CNO group was greater compared to other control groups, but there was no significant change in PPR and AMPA/NMDA ratios.
(C) The graphs plot the AMPA EPSC amplitudes with varying stimulation intensities delivered to the cortico-striatal pathway in brain slices from the indicated 4 experimental groups. Plots in light colors show individual data from each MSN and those in dark colors and thicker lines indicate averaged data (n = 12-13 MSNs from 4 mice). The subpanel C’ shows average plots from indicated 4 experimental groups.
(D) Illustration and scanning electron microscope image of the silicon microprobes used to record neuronal activity in vivo. The probes were coated with DiD fluorescent dye, which was deposited at the implantation site, allowing reconstruction of their position post hoc. The subpanel D’ shows that the microprobes (indicated in white by the dye) were positioned near hM4Di-expressing astrocytes (indicated in red owing to mCherry).
(E) Representative extracellularly recorded MSN AP.
(F) The graphs plot the MSN firing rate before and following i.p. CNO administration to AAV control mice and to hM4Di mice. The scatter graphs on the right summarize such experiments. Notably, MSN firing rate was significantly increased 120 min after CNO i.p. administration in hM4Di mice, but not in control mice (n = 7 mice).
Scale bars, 20 μm in panel (A), 1 mm in the large image of panel D, 10 μm in the small image of the microprobes in D, and 200 μm in panel D’. Data are shown as mean ± s.e.m. Full details of n numbers, precise P values and statistical tests are reported in Supplementary Table 1. * indicates P < 0.05, ** indicates P < 0.01, **** indicates P < 0.0001, NS indicates not significantly different. See also Fig S4.
To determine how astrocyte Gi-pathway activation affected striatal microcircuits in vivo, we used silicon probes (Figure 4D) to record from MSNs in awake head-fixed mice before, during and after acute i.p. CNO administration (Bakhurin et al., 2016). We recorded from probes inserted near hM4Di-expressing astrocytes (Figure 4D’). Extracellular APs were detected and those from MSNs identified by their characteristic waveform duration and baseline firing properties (Bakhurin et al., 2016) (Figure 4E). We recorded 300 units from the AAV + CNO control group and 492 units from the hM4Di + CNO group (7 mice each). Consistent with previously reported striatal neuron type composition, the majority of these units were putative MSNs (~70% of recorded units) with smaller proportions of tonically active neurons (TANs), fast spiking interneurons (FSIs) and unclassified neurons (Figure S4E). Within 30 min of CNO, MSN firing in the hM4Di + CNO group increased, but not in the AAV + CNO control group (Figure 4F). The increase in MSN firing measured from hM4Di + CNO mice stabilized at ~2 hrs post CNO (Figure 4F; P <0.0001), corresponding to the time point when mouse behavior was assessed (Figure 3). Consistent with behavioral observations in Figure 3, CNO significantly increased locomotor activity of the mice on the treadmill in the hM4Di + CNO group (Figure S4G). We detected no change in FSI firing rate (Figure S4F). We explored whether an increase in MSN firing in vivo may reflect intrinsic MSN excitability following astrocyte Gi pathway activation. However, we found no evidence for this from whole-cell current-clamp measurements of MSN excitability in brain slices (Figure S4H-K). Our data show that acute striatal astrocyte Gi-pathway activation in vivo leads to rapid augmentation of synaptic excitation (Figure 4A-C), and elevated MSN firing (Figure 4D-F) that accompany behavioral hyperactivity (Figure 3).
Activation of a synaptogenic cue (TSP1) by the astrocyte Gi pathway
There are no data to indicate the mechanism(s) by which Gi pathway activation in striatal astrocytes may lead to hyperlocomotion with disrupted attention, potentiated fast excitatory synaptic transmission and elevated firing of MSNs. We therefore performed RNA-seq to explore agnostically the mechanisms that underlie Gi-pathway mediated changes in striatal synapses, circuits and behavior (Figures 1–4). We used recently reported RiboTag AAVs (Yu et al., 2018) to deliver the ribosomal subunit Rpl22-HA to astrocytes that received hM4Di in the dorsal striatum (Figure 5A). In this approach (Chai et al., 2017; Yu et al., 2018), we immunoprecipitated astrocyte specific RNA from cells following acute in vivo Gi pathway activation with CNO for 2 hrs and from vehicle controls (Figure 5A; Figure S5A; 4 mice; Supplementary Table 2). We analyzed the RNA-seq data by FPKM (>5) and with a false discovery rate (FDR) of < 0.05 to identify differentially expressed genes (DEGs) induced by Gi-pathway activation and identified ~2300 DEGs in the IP samples (Figure 5B). With a >2-fold change cut-off, we identified ~250 DEGs between control and hM4Di + CNO groups (Figure 5B). Fig 5C reports the top 50 altered genes between hM4Di + Veh and hM4Di + CNO groups, along with their proposed functions. Among the top 10, Thbs1 was notable because its gene product, thrombospondin 1 (TSP1), has roles in developmental synapse formation (Christopherson et al., 2005; Crawford et al., 2012; Eroglu et al., 2009). Figure 5D compares the fold-change in Thbs1 and 13 other astrocyte genes implicated in synapse formation or loss. Of these, only Thbs1 was significantly upregulated in the hM4Di + CNO groups by ~ 40-fold (Figure 5D; n = 4 mice). To further explore this, we performed RNAscope mRNA analysis in single cells and found Thbs1 was expressed at low levels within adult astrocytes in all three-control groups, but its expression was increased significantly in the hM4Di + CNO group (Figure 5E; n = 3 mice, P < 0.0001). Thus, Gi-pathway stimulation activates TSP1, a latent synaptic synaptogenic cue in adult mice (Christopherson et al., 2005).
Figure 5: Astrocyte transcriptomes following astrocyte Gi pathway activation revealed Thbs1 upregulation.

(A) Cartoon illustrating AAVs for selectively expressing Rpl22-HA and hM4Di-mCherry in astrocytes in the dorsal striatum via intracranial microinjections for RNA-seq 2 hrs after intraperitoneal administration of 1 mg/kg CNO or vehicle.
(B) The number of differentially expressed genes (DEGs) in RNA-seq, with no fold-change cutoff and with a > 2-fold change cut off.
(C) Heatmaps of FPKM for the top 50 DEGs. Log2(FPKM) ranged from −4 (blue, relatively low expression) to 8 (red, relatively high expression). The proposed functions of the gene based on gene ontology analyses are also shown.
(D) Fold-change of genes implicated in astrocyte-dependent synapse formation and removal.
(E) RNAscope based assessment of Thbs1 mRNA expression in the dorsal striatum of the four experimental groups (n = 20-21 astrocytes from 4 mice per group). Significant upregulation of Thbs1 mRNA was observed in the hM4Di + CNO group.
Scale bars, 2 μm in panel (E). Data are shown as mean ± s.e.m. Full details of n numbers, precise P values and statistical tests are reported in Supplementary Table 1. **** indicates P < 0.0001. See also Fig S5.
Rescue of astrocyte Gi-pathway mediated cellular, circuit and hyperactivity phenotypes
Gabapentin (GBP) is an antagonist of TSP1 receptor α2δ−1 on neurons and was used to selectively block TSP1 actions in vivo in adult wild-type mice (Crawford et al., 2012; Eroglu et al., 2009), because TSP1 and α2δ−1 deletion mice have quite severe baseline dysfunctions (Crawford et al., 1998; Risher et al., 2018) that vitiate behavioral and synaptic assessments in our experimental design. Using GBP and in support of a causal role for TSP1 in our observations, we measured significantly increased density of dendritic spines in the hM4Di + CNO group relative to the AAV + CNO control group (Figure 6A, P < 0.0001, 39–46 dendritic segments, 4 mice in each group). We also determined how persistent these synaptic effects were. We found that the increased density of dendritic spines in the hM4Di + CNO group was reversible 48 hrs after CNO administration in vivo (Figure S6A). In accord, the boosted AMPA and NMDA EPSCs and the behavioral hyperactivity were also reversible at 48 hrs (Figure S6B-F). Furthermore, this increase in dendritic spines in the hM4Di + CNO group was abolished by pretreatment with GBP (100 mg/kg) for 1 hr before the administration of CNO (Figure 6A). The increased numbers of dendritic spines were associated with vGlut1 positive presynaptic puncta, indicative of increased corticostriatal synapse formation (Figure S7A,B). Consistent with this, we recorded significantly increased frequency and amplitude of mEPSCs in the hM4Di + CNO group, and this was rescued by GBP pretreatment (Figure 6B, n = 1518 MSNs, n = 5 – 6 mice).
Figure 6: Gabapentin (GBP) rescued astrocyte Gi pathway-induced morphological, electrophysiological, and behavioral phenotypes.

(A) Increase in spine density and spine head size of MSNs in hM4Di + CNO mice compared to AAV + CNO mice, which was rescued by GBP i.p. administration (n = 4 mice per group).
(B-C) Increased mEPSC frequency and evoked EPSC amplitude in hM4Di + CNO mice was rescued by GBP i.p. administration (n = 15-18 MSNs from 5-6 mice per group for B and 12-19 MSNs from 4 mice per group for C).
(D) CNO i.p. administration did not change MSN firing rate after in vivo administration of GBP (n = 7 mice for AAV + GBP, n = 5 mice for hM4Di + GBP).
(E-G) Increased ambulation in an open field (E), blunted responses to bright light stimuli (F) and novel objects (G) observed in hM4Di + CNO mice relative to controls were rescued by GBP i.p. administration (n = 14-16 mice per group for E, 12-13 mice per group for F and 7-8 mice per group for G). Scale bars, 20 μm in the left image and 2 μm in the right image (A). Full details of n numbers, precise P values and statistical tests are reported in Supplementary Table 1. * indicates P <0.05, ** indicates P < 0.01, **** indicates P <0.0001, NS indicates not significantly different. See also Fig S6-7.
We next explored if the synaptic, circuit and behavioral phenotypes reported in Figures 3–4 were downstream of TSP1 given that its expression was elevated and resulted in increased excitatory synapses assessed with neuroanatomical or physiological measurements (Figure 6C-G). First, the larger AMPA and NMDA receptor mediated evoked EPSCs observed in the hM4Di + CNO group were rescued by GBP (Figure 6C; n = 12 – 19 MSNs, 4 mice). Second, following treatment with GBP, CNO failed to evoke increased MSN firing in vivo in hMD4i mice (Figure 6D; 7 & 5 mice, P > 0.05). Third, the increase in open-field activity (i.e. hyperlocomotion) caused by CNO in hM4Di mice was rescued by GBP (Figure 6E; 14 – 16 mice). Fourth, the diminished responses to bright light stimulation in the open-field for hM4Di + CNO mice (i.e. disturbed attention) were rescued by GBP (Figure 6F; 12 – 13 mice). Fifth, the diminished exploration of a novel object observed in the hM4Di + CNO group was rescued by GBP (Figure 6G; 7–8 mice). Moreover, although GBP reversed the synaptic, circuit and behavioral responses triggered by CNO in hM4Di mice, it did not affect any of these parameters in control mice (Figure 6), indicating the selectivity of TSP1 inhibition in mediating the observed phenotypes. We performed a set of experiments to determine if GBP affected CNO responses either when GBP was acutely applied to brain slices before and during CNO (Figure S7C), or following in vivo administration (Figure S7D). We detected no significant effect of GBP on CNO-evoked responses in either condition (Figure S7C,D; n = 22–36 cells from 4 mice in each condition). Taken together, our data provide strong molecular evidence for the activation of an astrocyte latent synaptogenic cue (TSP1) in adult mice following Gi-pathway activation with clear hyperactivity and disrupted attention related phenotypic readouts at cellular, circuit and in vivo levels.
Discussion
We report a bidirectional astrocyte-neuron signaling mechanism that boosts fast excitatory synaptic transmission with clear neural circuit and behavioural effects in fully developed mice. As far as we know, this is the first example that an acute, selective and physiologically relevant manipulation of astrocytes can lead to psychiatric phenotypes of hyperactivity with disturbed attention in adult mice. The key findings are summarized in Figure 7. These data advance the concept that reactivation of latent astrocyte synaptogenic cues can reversibly drive psychiatric phenotypes in adults, portending the exploitation of such mechanisms for therapeutics.
Figure 7: Summary and model for Gi GPCR-mediated MSN-astrocyte bidirectional interactions.

When MSNs were depolarized to levels associated with upstates, they released GABA (step i) which activated Gi-protein coupled GABAB GPCRs on striatal astrocytes, leading to increase in intracellular Ca2+ signals (step ii). Selectively stimulating the Gi-pathway with DREADDS and CNO evoked Ca2+ signals in striatal astrocytes (step iii), upregulated the astrocyte synaptogenic molecule TSP-1, boosted excitatory synapse formation, boosted fast excitatory synaptic transmission (step iv) and increased firing of MSNs (step v), which together resulted in hyperactivity with disrupted attention phenotypes in mice (step vi). The synaptic, circuit and behavioral effects resulting from Gi-pathway activation in vivo (steps iv-vi) were all reversed by blocking TSP-1 actions on neuronal α2δ–1 receptors with gabapentin.
We propose a model in which MSNs communicate with astrocytes via the release of GABA during heightened activity such as upstate transitions. Such functional interactions are supported by the proximity of ~12 MSNs and ~51,000 excitatory synapses within the territory of single striatal astrocytes (Chai et al., 2017). Furthermore, astrocyte processes make extensive contacts with excitatory synapses onto MSNs, with 82% of such synapses being located within 100 nm of an astrocyte process (Octeau et al., 2018). Our data are most consistent with the hypothesis that GABA is released from MSN dendrites (Waters et al., 2005), which has been suggested for other neuroactive substances (Bernardinelli et al., 2011; Navarrete and Araque, 2008). Once released from MSNs, GABA activates Gi-coupled GABAB GPCRs, which are highly expressed in rodent and human astrocytes (Chai et al., 2017; Srinivasan et al., 2016; Zhang et al., 2014). GABAB receptor activation in turn results in the elevation of intracellular Ca2+ levels via release from stores (Jiang et al., 2016; Yu et al., 2018). In these regards, intracellular Ca2+ imaging represents a precise, quantifiable readout of GABAB receptor activation in living tissue, because multiple astrocyte GPCRs, including Gi-coupled GPCRs, result in Ca2+ elevations (Chai et al., 2017; Kang et al., 1998; Porter and McCarthy, 1997; Shigetomi et al., 2016). Selective activation of the Gi-pathway in astrocytes resulted in elevated Thbs1 gene expression, and the resultant TSP-1 actions increased excitatory synapse formation, synaptic function, increased MSN firing in vivo and for the behavioural hyperactivity and disturbed attention phenotypes triggered by the striatal astrocyte Gi-pathway. The effects on Thbs1 were selective in relation to a variety of other molecules implicated in synapse formation and removal (Allen and Lyons, 2018).
Advances with in vivo imaging and with mouse models of disease have fueled new hypotheses and have necessitated the need to understand how astrocytes regulate neural circuits and behavior (Bazargani and Attwell, 2016; Nimmerjahn and Bergles, 2015). Indeed, it has long been suggested that astrocytes and neurons may functionally interact to regulate circuits (Barres, 2008; Kuffler, 1967; Smith, 1994) and ultimately behavior, but the mechanisms and consequences have been difficult to identify and study (Halassa and Haydon, 2010). In one proposed mechanism, astrocytes regulate neurons via GPCR mediated signaling that has been documented in vitro and in vivo in multiple species, including in human astrocytes (Shigetomi et al., 2016). There has been important progress in exploring hippocampal astrocytes and the release of d-serine gliotransmitter (Adamsky et al., 2018; Henneberger et al., 2010), but the biology of the Gi pathway that is preferentially enriched within striatal astrocytes relative to hippocampal astrocytes has been unknown (Chai et al., 2017). We found that this pathway is physiologically engaged in striatal astrocytes and we made the discovery that its activation regulates behavior associated with hyperactivity and disturbed attention phenotypes in mice. Our findings show that activation of a single astrocyte-derived synaptogenic cue (TSP1) in adult mice via Gi-pathway signaling causes acute behavioral hyperactivity with disrupted attention via a synaptic mechanism. The consequences of Gi-pathway activation in vivo were reversible. The finding that astrocyte signaling can reactivate latent synaptogenic cues provides an unappreciated mechanism by which astrocytes regulate synapses and circuits. This mechanism may operate in parallel with or separately from gliotransmission (Araque et al., 2014), but is the underlying cause of the responses reported herein.
We speculate on additional settings under which astrocyte Gi-pathway activation mediated by MSN GABA would occur. Our data suggest that astrocyte Ca2+ signaling would accompany heightened activity of MSNs such as during upstates. Upstates occur during convergent glutamatergic excitation from multiple synaptic inputs into the striatum, which overrides the influence of MSN inward rectifier K+ channels resulting in depolarized membrane voltages from which APs emerge (Gerfen and Surmeier, 2011). In addition, heightened MSN activity occurs during dopamine release from the nigro-striatal pathway: the consensus is that such modulation increases D1 MSN excitability (Gerfen and Surmeier, 2011). Dopamine plays known roles in the control of motor skills, higher cognitive functions and the appetitive and consummatory aspects of reward (Tritsch and Sabatini, 2012). Elevated and altered MSN activity is also observed in a variety of neurological and psychiatric conditions such as Huntington’s disease (HD), Parkinson’s disease, OCD and addiction (Gerfen and Surmeier, 2011; Kreitzer and Malenka, 2008), although the nature and magnitude of the change almost certainly varies with disease progression in some or all of these cases. With the exception of HD (Khakh et al., 2017), the contributions of astrocytes to these conditions are essentially unexplored. Our data raise the possibility that the TSP1-dependent bidirectional astrocyte-neuron signaling mechanism might contribute to the phenotypes associated with the aforementioned physiologies and pathologies, either causally and/or correlatively.
Overall, our findings show not only that physiological activity of neurons triggers astrocyte signaling, but that signaling from astrocytes to neurons is also sufficient per se to alter synapses, circuits and behavior in adults, which has broad relevance to brain plasticity and neural network dynamics on time scales beyond fast neuronal activity alone (Cui et al., 2018). More conceptually, therefore, the findings suggest that behavioral phenotypes accompanying diverse brain disorders that currently lack mechanistic understanding in adults may have an astrocytic component and that identifying and exploiting astrocyte based neuromodulation affords new therapeutic opportunities for ADHD-like and possibly other psychiatric diseases.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Baljit S. Khakh (bkhakh@mednet.ucla.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All animal experiments were conducted in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals and were approved by the Chancellor’s Animal Research Committee at the University of California, Los Angeles. All mice were housed with food and water available ad libitum in a 12 hr light/dark environment. All animals were healthy with no obvious behavioral phenotype, were not involved in previous studies, and were sacrificed during the light cycle. Data for experiments were collected from adult mice (8–14 weeks old). For behavior tests and in vivo electrophysiology, only male wild-type C57BL/6NJ mice purchased from Jackson Laboratories were used. For other experiments, both male and female C57BL/6NTac mice were used. Mice were generated from in house breeding colonies or purchased from Taconic Biosciences.
Mouse models
Dldla-tdTomato transgenic mice were kindly provided from Michael Levine’s laboratory at UCLA. To selectively express GCaMP6f in astrocytes, Ai95 mice were crossed with Aldh1l1-CreERT2 BAC mice (B6N.FVB-Tg(Aldh1l1-cre/ERT2)1Khakh/J, JAX Stock # 029655) and injected with 75 mg/kg tamoxifen dissolved in corn oil for 5 days at 6 weeks of age. Floxed Gabbr1 mice were kindly provided by Dr. Henriette van Praag at the NIH and maintained in the Balb/c genetic background at UCLA.
METHODS DETAILS
Stereotaxic microinjections of adeno-associated viruses (AAV)
All surgical procedures were conducted under general anesthesia using continuous isoflurane (induction at 5%, maintenance at 1–2.5% vol/vol). Depth of anesthesia was monitored continuously and adjusted when necessary. Following induction of anesthesia, the mice were fitted into a stereotaxic frame with their heads secured by blunt ear bars and their noses placed into a veterinary grade anesthesia and ventilation system (David Kopf Instruments). Mice were administered 0.1 mg/kg of buprenorphine (Buprenex, 0.1 mg/ml) subcutaneously before surgery. The surgical incision site was then cleaned three times with 10% povidone iodine and 70% ethanol (vol/vol). Skin incisions were made, followed by craniotomies of 2–3 mm in diameter above the left frontal or parietal cortex using a small steel burr (Fine Science Tools) powered by a high-speed drill (K.1070, Foredom). Saline (0.9%) was applied onto the skull to reduce heating caused by drilling. Unilateral viral injections were carried out by using a stereotaxic apparatus (David Kopf Instruments) to guide the placement of beveled glass pipettes (1B100–4, World Precision Instruments). For the left striatum: the coordinates were 0.8 mm anterior to bregma, 2 mm lateral to midline, and 2.4 mm from the pial surface. Adeno-associated virus (AAV) was injected by using a syringe pump (Pump11 PicoPlus Elite, Harvard Apparatus). Following AAV microinjections, glass pipettes were left in place for at least 10 min prior to slow withdrawal. Surgical wounds were closed with external 5–0 nylon sutures. Following surgery, animals were allowed to recover overnight in cages placed partially on a low-voltage heating pad. Buprenorphine was administered two times per day for up to 2 days after surgery. In addition, trimethoprim sulfamethoxazole was provided in food to the mice for 1 week. Virus injected mice were used for experiments at least two weeks post surgery. Viruses used were: 0.5 μl of AAV2/5 GfaABC1D-cyto-GCaMP6f virus (2.3 × 1013 genome copies); 0.8 μl of AAV2/5 GfaABC1D-hM4Di-mCherry virus (1.1 × 1013 genome copies/ml); 0.4 μl of AAV2/5 GfaABC1D-tdTomato virus (1.0 × 1014 genome copies/ml); 0.7 μl of AAV2/5 GfaABC1D-PI-CRE virus (1.3 × 1013 genome copies/ml); 0.8 μl of AAV1 hSyn-hChR2(H134R)-EYFP virus (2.9 × 1013 genome copies); 0.7 μl of AAV2/5 GfaABC1D-Rpl22-HA virus (2.1 × 1013 genome copies/ml).
In vivo activation of hM4Di
Two to three weeks after appropriate microinjection of AAV2/5 hM4Di-mCherry into the striatum, CNO was administered to animals by intraperitoneal injection (1 mg/kg; dissolved in saline). Two hours after CNO administration, animals were used for behavior tests, or sacrificed for brain slice experiments or immunohistochemistry. For in vivo electrophysiology, CNO was intraperitoneally injected 30 min after baseline recording (please see In vivo electrophysiology section for details).
Optical stimulation of neurons in vivo
Construction of the fiber optic cannula:
we constructed a fiber optic cannula from the DIY-cannula kit (Prizmatix). Briefly, the fiber optic was cleaved into the desired length using a metal scribe. A small droplet of epoxy resin was applied on the flat opening of the cannula. The cleaved fiber optic was inserted through the epoxy into the cannula until it protruded approximately 1 mm from the opposite end of the cannula. Epoxy was then cured using a heat gun to secure the cannula in place. The fiber optic cannula was allowed to cool down for 2 hours and then made transparent using polishing paper of increasing grits (from 4500–60000 grits) on the convex end of cannula. The cannulated fibers were connected to the external LED source (Prizmatix) using a patch cord and tested for their integrity and maximum light output.
Fiber optic implantation surgery:
we implanted the cannula into the brain of anesthetized mice just after AAV microinjections. After the microinjection needle was removed, the cannula was slowly lowered into the striatum and secured in place using vetbondTM and a thin layer of dental cement. Thin and uniform layers of dental cement were applied around the cannula. The mouse was allowed to recover from the surgery for 3 weeks. Once the mouse had recovered and the virus had expressed, the mouse was connected to the optical stimulation system. The optical cannula was connected to the patch cord through the mating sleeve. An optical stimulation (3–4 mW) paradigm consisting of 2.5 s light-on and 27.5 s light-off (mimicking MSN up-state like excitability) for a period of 60 min was administered to each mouse. The mice were perfused at 8 hr post stimulation for IHC.
Immunohistochemistry (IHC)
Frozen sections:
For transcardial perfusion, mice were euthanized with pentobarbitol (i.p.) and perfused with 10% buffered formalin (Fisher #SF100-20). Once all reflexes subsided, the abdominal cavity was opened and heparin (50 units) was injected into the heart to prevent blood clotting. The animal was perfused with 20 ml ice cold 0.1 M phosphate buffered saline (PBS) followed by 60 ml 10% buffered formalin. After gentle removal from the skull, the brain was postfixed in 10% buffered formalin overnight at 4°C. The tissue was cryoprotected in 30% sucrose PBS solution the following day for at least 48 hours at 4°C until use. 40 μm coronal sections were prepared using a cryostat microtome (Leica) at −20°C and processed for immunohistochemistry. Sections were washed 3 times in 0.1 M PBS for 10 min each, and then incubated in a blocking solution containing 10% NGS in 0.1 M PBS with 0.5% Triton-X 100 for 1 hr at room temperature with agitation. Sections were then incubated with agitation in primary antibodies diluted in 0.1 M PBS with 0.5% Triton-X 100 overnight at 4°C. The following primary antibodies were used: chicken anti-GFP (1:1000; Abcam ab13970), mouse anti-NeuN (1:500; Millipore MAB377), rabbit anti-S100b (1:1000; Abcam ab41548), rabbit anti-c-Fos (1:1000; Millipore ABE457), rabbit anti-RFP (1:1,000; Rockland 600–401-379). The next day the sections were washed 3 times in 0.1 M PBS for 10 min each before incubation at room temperature for 2 hr with secondary antibodies diluted in 0.1 M PBS. Alexa conjugated (Molecular Probes) secondary antibodies were used at 1:1000 dilution except streptavidin conjugated Alexa 647 at 1:250 dilution. The sections were rinsed 3 times in 0.1 M PBS for 10 min each before being mounted on microscope slides in fluoromount-G. Fluorescent images were taken using UplanSApo 20X 0.85 NA, UplanFL 40X 1.30 NA oil immersion or PlanApo N 60X 1.45 NA oil immersion objective lens on a confocal laser-scanning microscope (FV10-ASW; Olympus). Laser settings were kept the same within each experiment. Images represent maximum intensity projections of optical sections with a step size of 1.0 μm. Images were processed with ImageJ. Cell counting was done on maximum intensity projections using the Cell Counter plugin; only cells with soma completely within the region of interest (ROI) were counted. For astrocyte-dendrite proximity analysis (Figure S2E,F), image was taken with a step size of 0.33 μm and maximum intensity projections of 3 slices (1 μm stack) was obtained. A line ROI was made across the dendrite that is apposed to astrocyte processes. The peak of obtained from each profile was defined as the center of dendrite or astrocyte process and the distance between them was measured. For Figure 1I, c-Fos expression in astrocytes were analyzed within 800 μm from the end of fiber optics.
Acute sections:
300 μm fresh brain slices were placed into 10% buffered formalin overnight at 4°C and processed as follows for IHC. Sections were washed 3 times in 0.1 M PBS with 2% Triton-X 100 for 5 min each, and then incubated in a blocking solution containing 10% NGS in 0.1 M PBS with 1% Triton-X 100 for 1 hr at room temperature with agitation. Sections were then incubated with agitation in primary antibodies diluted in 0.1 M PBS with 0.4% Triton-X 100 for 3 days at 4°C. The primary antibody was guinea pig anti-vGlut1 (1:2000; Synaptic Systems 135302). Sections were washed 3 times in 0.1 M PBS with 0.4% Triton-X 100 for 10 min each before incubation 3 days at 4°C with streptavidin conjugated Alexa 647 (1:250) diluted in 0.1 M PBS with 0.4% Triton-X 100. The sections were rinsed 3 times in 0.1 M PBS for 10 min each before being mounted on microscope slides in fluoromount-G. Images were obtained in the same way as IHC for frozen sections except a step size of 0.33 μm. For quantification of spine density, we only analyzed spines on dendritic shafts that are parallel to the imaging plane to minimize the possibility of rotational artifacts. Spine density was calculated by dividing the number of spines by the length of the dendritic segment. For quantification of spine head size, a line ROI across the maximum diameter of the spine was made and a profile that has a single peak and is closer to a Gaussian curve was obtained. Full-Width Half-Maximum of that was defined as a spine head size to avoid the point spread function. For counting the number of vGlut1-positive synapse, only spines that are off from optical plane were analyzed. As shown in Figure S7A, a line ROI was made over MSN spine and vGlut-1 puncta that is closest to the spine. FWHM of each profile was measured. A MSN spine was recognized as forming vGlut-1-positive synapse when each FWHM is overlapped (see Figure S7A), while recognized as not forming vGlut-1-positive synapse when there is a gap between each FWHM (see Figure S7A). As a result, the ratio of vGlut-1-positive synapse number to total number of MSN spines was 47 ± 6%, which is reasonably matched with previous synapses analysis of MSN using EM (Doig et al., 2010).
Dual in situ hybridization (ISH) with RNAscope and IHC
Cryosections were prepared as described above and stored at −80°C. ISH was performed using Multiplex RNAscope (ACDBio 320851). Sections were washed at least for 15 min with 0.1 M PBS, and then incubated in 1X Target Retrieval Reagents (ACDBio 322000) for 5 min at 99–100°C. After washing with ddH2O twice for 1 min each, they were dehydrated with 100% ethanol for 2 min and dried at RT. Then, the sections were incubated with Protease Pretreat-4 solution (ACDBio 322340) for 30 min at 40°C. The slides were washed with ddH2O twice for 1 min each and then incubated with probe(s) for 2 hours at 40°C. The following probes were used: Mm-Gabbr1-C2 (ACDBio 425181-C2), Mm-Aldh1l1-C3 (ACDBio 405891-C3) and Mm-Thbs1-C3 (ACDBio 457891-C3). The sections were incubated in AMP 1-FL for 30 min, AMP2-FL for 15 min, AMP3-FL for 30 min and AMP4-FL for 15 min at 40°C with washing in 1X Wash Buffer (ACDBio 310091) twice for 2 min each prior to the first incubation and in between incubations. All the incubations at 40°C were performed in the HybEZ™ Hybridization System (ACDBio 310010). Slices were washed in 0.1 M PBS three times for 10 min each, followed by IHC that was performed as described above except with antibody dilutions. Following primary antibodies were used: chicken anti-GFP (1:250; Abcam ab13970) to stain GCaMP and rabbit anti-RFP (1:250; Rockland 600–401-379) to stain tdTomato or mCherry. Images were obtained in the same way as IHC described above except a step size of 0.8 μm. Images were processed with ImageJ (NIH). Astrocyte somata were demarcated based on GFP or RFP signal, and number of puncta and intensity of probe signals within somata were measured.
Acute brain slice preparation for imaging and electrophysiology
Sagittal striatal slices were prepared from 8–11 week old C57 WT mice, or C57 WT mice with AAV injection plus CNO or vehicle I.P. injection for EPSCs recording. For other experiments, coronal striatal slices were prepared from 8–11 week old C57 WT mice with AAV injection or Aldh1l1-cre/ERT2 × Ai95 mice. Briefly, animals were deeply anesthetized with isoflurane and decapitated with sharp shears. The brains were placed and sliced in ice-cold modified artificial CSF (aCSF) containing the following (in mM): 194 sucrose, 30 NaCl, 4.5 KCl, 1 MgCl2, 26 NaHCO3, 1.2 NaH2PO4, and 10 D-glucose, saturated with 95% O2 and 5% CO2. A vibratome (DSK-Zero1) was used to cut 300 μm brain sections. The slices were allowed to equilibrate for 30 min at 32–34°C in normal aCSF containing (in mM); 124 NaCl, 4.5 KCl, 2 CaCl2, 1 MgCl2, 26 NaHCO3, 1.2 NaH2PO4, and 10 D-glucose continuously bubbled with 95% O2 and 5% CO2. Slices were then stored at 21–23°C in the same buffer until use. All slices were used within 4–6 hours of slicing.
Electrophysiological recordings in the striatal slices
Electrophysiological recordings were performed using standard methods as described belows. Slices were placed in the recording chamber and continuously perfused with 95% O2 and 5% CO2 bubbled normal aCSF. pCLAMP10.4 software and a Multi-Clamp 700B amplifier was used for electrophysiology (Molecular Devices). Whole-cell patch-clamp recordings were made from medium spiny neurons (MSNs) in the dorsolateral striatum using patch pipettes with a typical resistance of 56 MΩ. MSNs were morphologically and electrophysiologically identified. In some experiments, D1- and D2-MSNs were selected based on tdTomato fluorescence. The intracellular solution for MSN EPSCs recordings comprised the following (in mM) : 120 CsMeSO3, 15 CsCl, 8 NaCl, 10 HEPES, 0.2 EGTA, 0.3 Na-GTP, 2 Mg-ATP, 10 TEA-Cl, with pH adjusted to 7.3 with CsOH. The intracellular solution for other experiments comprised the following (in mM): 135 potassium gluconate, 5 KCl, 0.5 CaCl2, 5 HEPES, 5 EGTA, 2 Mg-ATP and 0.3 Na-GTP, pH 7.3 adjusted with KOH. To assess evoked EPSCs, electrical field stimulation (EFS) was achieved using a bipolar matrix electrode (FHC) that was placed on the dorsolateral corpus callosum to evoke glutamate release from the cortico-striatal pathway. The MSNs to be assessed were typically located ~300-400 μm away from the stimulation site to avoid the EFS-evoked astrocyte calcium increase that occurs nearby the stimulating electrode. In order to construct stimulus-response curves for each striatal slice, electrical stimulation current intensity was varied from 0 to 800 μA in 50 μA steps for each cell. We determined full stimulation-response curves for the AMPA EPSCs at −70 mV: cells in which the stimulation evoked antidromic spiking at current levels below 800 μA were recorded only over the current range that evoked EPSCs, i.e. none of the recordings shown in the study were contaminated with antidromic spiking. We could not determine full stimulation-response curves for NMDA EPSCs, because clamping the cells at +40 mV for the prolonged periods needed for such assessments decreased the quality of whole-cell recording. However, we evaluated AMPA and NMDA EPSCs equivalently when stimulation intensities were set to 250 μA that approximately evoke responses at 50% maximal amplitude of the AMPA EPSCs. To isolate the AMPAR- and NMDAR-mediated evoked EPSCs, MSNs were voltage-clamped at −70 mV or +40 mV in the presence of 10 μM bicuculline. Paired pulses were delivered at 50 ms inter-pulse intervals. The AMPAR-mediated EPSC was measured at the peak amplitude of the EPSC at −70 mV, while the amplitude of the EPSC 50 ms after stimulation at +40 mV was used to estimate the NMDAR-mediated component. To isolate mEPSCs, MSNs were voltage-clamped at −70 mV and pre-incubated with 10 μM bicuculline and 300 nM TTX for 5 min before recording. In some cases, 1 mg/ml biocytin (Tocris, 3349) was added to the intracellular solution to subsequently visualize patched neuron. All recordings were performed at room temperature, using pCLAMP10 (Axon Instruments, Molecular Devices) and a MultiClamp 700B amplifier (Axon Instruments, Molecular Devices). Cells with Ra that exceeded 20 MΩ were excluded from analysis. Analysis was performed using ClampFit 10.4 software.
Astrocyte intracellular Ca2+ imaging
Imaging:
Slice preparation was performed as described above. Cells for all the experiments were imaged using a confocal microscope (Fluoview 1200; Olympus) with a 40X water-immersion objective lens with a numerical aperture (NA) of 0.8 and at a digital zoom of two to three. We used the 488 nm line of an Argon laser, with the intensity adjusted to 9% of the maximum output of 10 mW. Astrocytes were typically ~20 to ~30 μm below the slice surface and scanned at 1 frame per second for imaging sessions. When imaging was performed along with MSN depolarization, astrocytes were located nearby (< 50 μm) from MSN somata or dendrites that were visualized by MSN dialysis with Alexa Fluor 568 hydrazide (Thermo Fisher Scientific, A10441).
Drug applications:
the following agonists were applied in the bath: Phenylephrine (Tocris Bioscience 2838), γ-Aminobutyric acid (Sigma Aldrich A2129), R-Baclofen (Tocris Bioscience 0796), Clozapine N-oxide (CNO, Tocris Bioscience 4936). Inhibitors and antagonists were applied in the bath at least 5 min prior to recording to allow adequate equilibration. The following inhibitors and antagonists were used: Tetrodotoxin (Cayman Chemical 14964), CdCl2 (Sigma Aldrich 439800), Nimodipine (Tocris Bioscience CAS 0600), CGP55845 (Tocris Bioscience 1248), Bicuculline (Abcam ab120110 or Tocris Bioscience 0131) and NNC711 (Tocris Bioscience 1779). PLC inhibitor U73122 (Tocris Bioscience 1268) or its control analog U73433 (Tocris Bioscience 4133) was applied in bath for 40 min prior to recording. 1,2-Bis(2-Aminophenoxy)ethane-N,N,N’,N’-tetraacetic acid (BAPTA from Sigma Aldrich A4926) or Recombinant Light Chain from Tetanus Toxin (List Biological Laboratories 650A) was dialyzed in MSN through patch pipette prior to MSN depolarization at least for 20 min or 10 min, respectively. A constant flow of fresh buffer perfused the imaging chamber at all times.
Neuron intracellular Ca2+ imaging
Slice preparation was performed as described above. MSNs were dialyzed via the whole-cell patch pipette with 100 μM Fluo-4 (Thermo Fisher Scientific F14200) at least 5 min before imaging. A line ROI was made on MSN somata and line scan imaging was performed at 500 Hz with a 40X water-immersion objective lens. Imaging and analyses were conducted using FV10-ASW from Olympus.
qPCR experiments
Amplified cDNA from RNA samples (RiboTag IP and FACS) was generated using Ovation PicoSL WTA System V2 (Nugen). The cDNA was then purified with a QIAquick PCR Purification Kit (Qiagen) and quantified with a Nanodrop 2000. qPCR was performed in a LightCycler 96 Real-Time PCR System (Roche). Amplified cDNA from eGFP-positive cell populations from three separate sorts was used. Ten nanograms of cDNA were loaded per well and the expression of Gabbr1, Gabbr2, and Arbp was analyzed using the primers shown below:
| Gene | Sequence | Amplicon (bp) |
|---|---|---|
| Gabbr1 | Forward 5’ ACAGACCAAATCTACCGGGC 3’ Reverse 5’ GTGCTGTCGTAGTAGCCGAT 3’ |
152 |
| Gabbr2 | Forward 5’ AAGCTCAAGGGGAACGACG 3’ Reverse 5’ ACTTGCTGCCAAACATGCTC 3’ |
115 |
| Arbp | Forward 5’ TCCAGGCTTTGGGCATCA 3’ Reverse 5’ AGTCTTTATCAGCTGCACATCAC 3’ |
76 |
Arbp was used as an internal control to normalize RNA content. To calculate the expression of gene of interest, the following formula was used: 2-ΔCt (Gene of interest-Arbp).
Western blot analyses
Standard SDS-PAGE was performed. Each lane contained protein extracted from one fluorescence-activated cell sorting (FACS) experiment. Aldh1l1-eGFP mice were used to purify astrocytes by FACS. Whole striata from heterozygous P30 mice were dissociated. Briefly, the striata from four mice (two male and two females) were dissected and digested together for 45 min at 36°C in a 35 mm Petri dish with 2.5 ml of papain solution (1x EBSS, 0.46% D-glucose, 26 mM NaHCO3, 50 mM EDTA, 75 U/ml DNase1, 200 units of papain for hippocampal and 300 units of papain for striatal tissue, and 2 mM L-cysteine) while bubbling with 5% CO2 and 95% O2. After digestion, the tissue was washed four times with ovomucoid solution (1x EBSS, 0.46% D-glucose, 26 mM NaHCO3, 1 mg/ml ovomucoid, 1 mg/ml BSA, and 60 U/ml DNase1) and mechanically dissociated with two fire-polished borosilicate glass pipettes with different bore sizes. A bottom layer of concentrated ovomucoid solution (1× EBSS, 0.46% D-glucose, 26 mM NaHCO3, 5. mg/ml ovomucoid, 5.5 mg/ml BSA, and 25 U/ml DNase1) was added to the cell suspension. The tubes were centrifuged at room temperature at 300 g for 10 min and the resultant pellet was re-suspended in D-PBS with 0.02% BSA and 13 U/ml of DNase1, and filtered with a 20 μm mesh. FACS was performed in a FACSAria II (BD Bioscience) with a 70 μm nozzle using standard methods at the University of California, Los Angeles (UCLA) Cell Sorting Core. For protein extraction, cells were collected in D-PBS and, right after FACS, cells were incubated with lysis buffer (150 mM NaCl, 1% Triton X-100, 12 mM Na+-Deoxycholate, 3.5 mM sodium dodecyl sulfate, 50 mM Tris pH8, 1:100 Halt Protease Inhibitor cocktail (Thermo Scientific)) at 4°C for 40 mins. The extracted protein was subsequently precipitated with trifluoroacetic acid and acetone. Protein pellet was dried and resuspended in a proteomics compatible buffer (0.5% Na-Deoxycholate, 12 mM N-Lauroylsarcosine sodium salt, 50 mM triethylammonium bicarbonate), boiled at 95 °C for 10 min and stored at −80°C. A total of six separate cell sorts from 26 mice were included in the analysis. We probed for GABAB1R and β-actin using guinea pig anti-GABAB1R (Millipore AB2256) and rabbit antî-actin (Abcam ab8227) primary antibodies at 1:1000 dilution. The secondary antibodies IRDye 680RD anti-guinea pig (Li-Cor 925–68077) and IRDye 800CW anti-rabbit (Li-Cor 827–08365) were added to visualize the proteins using a Li-Cor Odyssey imager. Signal intensities were quantified with Image J (NIH) and normalized to β-actin.
Behavioral tests
Behavioral tests were performed during the light cycle. Only male mice were used in behavioral tests because of gender-dependent differences known for striatal physiology. All the experimental mice were transferred to the behavior testing room at least 30 min before the tests to acclimatize to the environment and to reduce stress. Temperature and humidity of the experimental rooms were kept at 23 ± 2°C and 55 ± 5%, respectively. The brightness of the experimental room was kept < 15 fc unless otherwise stated. Background noise (60–65 dB) was generated by Air Purifier 50150-N from Honeywell Enviracaire. 1 mg/kg CNO or vehicle (0.86% DMSO) was intraperitoneally injected to mice 2 hours before the initiation of test. 100 mg/kg Gabapentin (Tocris Bioscience 0806) or saline was intraperitoneally injected to mice 3 hours before the initiation of test.
Open field test:
For Figure 3, the open field chamber consisted of a square arena (28 × 28 cm) enclosed by walls made of Plexiglass (19 cm tall). Locomotor activity was then recorded for 20 min using an infrared camera located underneath the open field chamber. Recording camera was connected to a computer operating an automated video tracking software Noldus Ethovision. Parameters analyzed included distance traveled with 5 min and 20 min time bins. For Figure 6, the open field chamber consisted of a square arena (26.7 × 26.7 cm) enclosed by walls made of translucent polyethylene (20 cm tall). Locomotor activity was then recorded for 30min using an infrared camera located above the open field chamber. Recording camera was connected to a computer operating an automated video tracking software Topscan from CleverSys. Parameters analyzed included distance traveled with 5 and 30 min time bins.
Open field test with bright light stimulus:
As previously described (Godsil and Fanselow, 2004), the modified open-field arena was a white, translucent polyethylene box (Model CB-80, Iris USA, Pleasant Prairie, WI) with internal dimensions of 69 cm long × 34 cm wide × 30 cm high (Godsil et al., 2005a; Godsil and Fanselow, 2004; Godsil et al., 2005b). Three lamps containing single 100 W white light bulbs were positioned at one end of the table (see Figure 3D). One lamp was positioned 14 cm from the center of a short wall of the rectangular arena; one lamp flanked each long side of the arena, 14 cm from the long walls and 15 cm from the original short wall. All three lamps were situated 18 cm above the base of the arena. The test is divided into three phases. Illumination levels were measured with a light meter (Model 403125, Extech Instruments, Waltham, MA). Cameras suspended from the ceiling or floor monitored and captured activity of animals. Locomotive activity was measured using the software Topscan from CleverSys. Parameters analyzed included distance traveled with 1 min and 4 min time bins. Phase 1 minutes 1-4 is the dark phase where the lights are turned off (< 0.5 fc). Phase 2 is the light phase minutes 5-8 where the lights will be turned on and create an illumination gradient across the arena (~100 fc at one end of the open field with light). Phase 3 minutes 9-12 the lights will be turned off (< 0.5 fc). The change in locomotion was calculated as follows: distance travelled in Phase 1 divided by the distance travelled in Phase 2.
Rearing behavior:
Mice were placed individually into plastic cylinders (15 cm in diameter and 35 cm tall) and allowed to habituate for 20 min. Rearing behavior was recorded for 10 min. A timer was used to assess the cumulative time spent in rearing behavior, in which mice support their weight freely on its hind legs without using its tail or forepaws.
Rotarod test:
Mice were held by the tails and placed on a single lane rotarod apparatus (ENV-577M, Med Associates Inc.), facing away from the direction of rotation. Mice were habituated on the rod for 1 min just before the trial. The rotarod was set with a start speed of 4 rpm. Acceleration started 10 seconds later and was set to 20 rpm per minute with a maximum speed 40 rpm. Each mouse received 5 trials at least 5 min apart per day for two consecutive days and the latency to fall was recorded for each trial.
Footprint test:
A one-meter long runway (8 cm wide) was lined with paper. Each mouse with hind paws painted with non-toxic ink was placed at an open end of the runway and allowed to walk to the other end with a darkened box. For the gait analysis, stride length and width were measured and averaged for both left and right hindlimbs over 5 steps.
Marble burying test:
a fresh, unscented soft wood chip bedding was added to polycarbonate cages (21 cm × 43 cm × 20.5 cm) to a depth of 5 cm. Sanitized 15 glass marbles were gently placed on the surface of the bedding in 5 rows of 3 marbles. Mice were allowed to remain in the cage undisturbed for 30 min. A marble was scored as buried if two-thirds of its surface area was covered by bedding.
Novel Object Recognition test:
At day 1 and day 2, mice were placed in an empty open chamber (26.7 × 26.7 cm) for 10 minutes for habituation. At day 3 (training day), mice were placed in the same open chamber containing two identical objects evenly spaced apart; trial was video recorded for 10 minutes. At day 4 (testing day), 24 hours after training, mice were placed in the same open chamber, but one of the two objects has been replaced with a novel object; trial is video recorded for 10 minutes. Time exploring around the objects was measured. Recognition index was calculated as follows: (time exploring the novel object – time exploring the familiar object) / (time exploring both objects) – 50.
In vivo electrophysiology
Surgeries and habituation:
All animals underwent surgical procedures under aseptic conditions and isoflurane anesthesia on a stereotaxic apparatus. We attached rectangular head fixation bars on each side of the skull (9 × 7 × 0.76 mm dimensions, 0.6 g weight, laser cut from stainless steel at Fab2Order). Animals were allowed to recover for 2 weeks before beginning habituation. Carprofen (5 mg/kg, s.c.) was administered daily for the first three days post-operatively and Analgesics (ibuprofen) and antibiotics (amoxicillin) were administered in the drinking water for the first week post-operatively. Animals were habituated to the head fixation apparatus and the circular treadmill (a freely-rotating spherical styrofoam ball) in the dark for 4 days at least for 30 min per each day. To habituate to I.P. injection, saline was administered intraperitoneally on halfway through the habituation. No stimuli were present during habituation. On the recording day, animals underwent a brief craniotomy surgery above the striatum and cerebellum under isoflurane anesthesia. The dura was removed. During a recovery period, the craniotomies were sealed with a silicone elastomer compound (Kwik-Cast, World Precision Instruments).
Recording:
Neural recordings were carried out either with a 128 or 256-electrode silicon microprobe. The 128-electrode device consisted of 4 prongs spaced by 330 μm, 32 electrodes per prong in a staggered array pattern spanning a depth of 990 μm. The 256-electrode device consisted of 4 prongs spaced by 200 μm, 64 electrodes per prong in a honeycomb array pattern spanning a depth of 1.05 mm). Prior to their first use electrodes were gold plated with constant pulses (−2.5 V relative to a Pt wire reference, 1-5 s) until their impedence reached below 0.5 MΩ to improve signal-to-noise ratio. Subsequently, awake animals were head restrained, a silver/silver-chloride electrical reference wire was placed in contact with CSF above the cerebellum, and the microprobe was inserted into the striatum under the control of a motorized micromanipulator. The target coordinates of the most lateral silicon prong were 0.8 mm anterior, 2.5 mm lateral, 4.0 mm ventral to bregma. Mineral oil was placed on the craniotomy to prevent drying. Data acquisition commenced 30 min after device insertion, using custom-built hardware at a sampling rate of 25 kHz per electrode. Treadmill velocity was sampled at a rate of 10 kHz and animal speed was calculated as the mean rotational velocity. The distance the mice traveled was calculated as the integrated area of the speed. No stimuli were present during recording. The microprobe was cleaned after each recording session in a trypsin solution and deionized water and ethanol, and reused in subsequent experiments.
RNA-Seq analysis of striatal astrocyte transcriptomes
AAV2/5 GfaABC1D-hM4Di-mCherry virus and AAV2/5 GfaABC1D-Rpl22-HA virus were microinjected into the dorsal striatum of adult (P44–45) male C57Bl/6NJ mice. 18-19 days later, RNA was collected from striata of those mice (at P63). Briefly, freshly dissected tissues were collected from four animals and individually homogenized. RNA was extracted from 10-20% of cleared lysate as input. The remaining lysate was incubated with mouse anti-HA antibody (1:250; Covance #MMS-101R) with rocking for 4 hours at 4°C followed by addition of magnetic beads (Invitrogen Dynabeads #110.04D) and overnight incubation with rocking at 4°C. The beads were washed three times in high salt solution. RNA was purified from the IP and corresponding input samples (Qiagen Rneasy Plus Micro #74034). RNA concentration and quality were assessed with nanodrop and Agilent 2100 Bioanalyzer. RNA samples were used for the Ribo-Zero rRNA reduction prep. Sequencing was performed on Illumina HiSeq 4000 using paired-end 75 bp reads. Data quality check was done on Illumina SAV. Demultiplexing was performed with Illumina Bcl2fastq2 v 2.17 program. Reads (60 to 84M per sample) were aligned to the latest mouse mm10 reference genome using the STAR spliced read aligner. Between 79 and 91% of the reads mapped uniquely to the mouse genome and were used for subsequent analyses. Differential gene expression analysis was performed with genes with FPKM > 5 at least 4 samples per condition and Log2FC > 1 or < −1, using Bioconductor packages edgeR and limmaVoom with false discovery rate (FDR) threshold set at < 0.1 or 0.05 (http://www.bioconductor.org) and Htseq-count were used (Anders et al., 2015; Law et al., 2014). RNAseq data has been deposited within the Gene Expression Omnibus (GEO) repository (www.ncbi.nlm.nih.gov/geo), accession ID # of GSE119058.
QUANTIFICATION AND STATISTICAL ANALYSIS
In vivo electrophysiology data analysis:
Spike sorting and all neural activity analyses were carried out with custom MATLAB scripts. Striatal units were classified as putative medium spiny neurons (MSNs), fast spiking interneurons (FSIs), or tonically active neurons (TANs), based on spike waveform peak-to-trough width, and coefficient of variation of the baseline firing rate (Bakhurin et al., 2016). FSIs were characterized by a narrow spike waveform (maximum width = 0.475 ms). MSNs and TANs both have wider waveforms (minimum width = 0.55 ms, maximum width = 1.25 ms). TANs were separated from MSNs by the regularity of their baseline firing (maximum coefficient of variation = 1.5).
Imaging data analysis:
Analyses of time-lapse image series were performed using ImageJ (NIH). XY drift was corrected using ImageJ. The data were analyzed essentially as previously reported (Chai et al., 2017; Jiang et al., 2016; Octeau et al., 2018; Tong et al., 2014; Yu et al., 2018). Time traces of fluorescence intensity were extracted from the ROIs and converted to dF/F values. For analyzing spontaneous Ca2+ signaling, regions of interest (ROIs) were defined in normal aCSF (control). Using Origin 2016 (Synaptosoft), Ca2+ events were manually marked. Event amplitudes, half width, event frequency per ROI per min, the integrated area-under-the-curve (AUC) of dF/F traces were measured. Events were identified based on amplitudes that were at least 2-fold above the baseline noise of the dF/F trace.
Statistical tests were run in OriginPro 2016. Summary data are presented as mean ± s.e.m. Sample sizes were not determined a priori and were based on past studies that are cited at the relevant sections of the manuscript and methods. Statistical tests were chosen as described below. For each set of data to be compared, we determined within OriginPro whether the data were normally distributed or not. If they were normally distributed, we used parametric tests. If the data were not normally distributed, we used non-parametric tests. Paired or unpaired Student’s t-test, Wilcoxon signed-rank test or Mann–Whitney tests was used for statistical analyses with two samples (as appropriate). One-way or two-way ANOVA tests followed by Tukey’s post-hoc test were used for statistical analyses with more than three samples. Significant difference was declared at P < 0.05. In the figures, P values were stated by asterisk(s): *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P <0.0001. Exact P values, sample numbers (n numbers for replicates and the numbers of mice for every experiment) as well as the details of statistical analyses in each case are provided fully in Supplementary Table 1 for every experiment. All the experiments were replicated and the details of these replications are provided in Supplementary Table 1. All mice were assigned to particular experimental groups at random. Blinding could not be performed, because the experimenter had to know which AAVs to inject for the various mice that are reported. No data points were excluded for any of the experiments and all of the raw data used to generate the graphs shown in this study are provided at www.data.mendeley.com (ID jprg767fk6.1).
DATA AND SOFTWARE AVAILABILITY
All of the raw and normalized RNA-Seq data have been deposited in the Gene Expression Omnibus repository (http://www.ncbi.nlm.nih.gov/geo) with accession number GSE119058. All of the FPKM RNA-Seq values are also provided in Supplementary Excel file 2. All of the raw data used to generate the graphs shown in the figures are available at www.data.mendeley.com (ID jprg767fk6.1).
Supplementary Material
Supplementary Movie 1 (related to Figure 2): Representative movie for intracellular Ca2+ signals in hM4Di-negative astrocytes before, during and after CNO application in brain slices. Confocal microscopy imaging of GCaMP6f-expressing striatal astrocytes showing that bath application of 1 μM CNO did not change astrocyte Ca2+ signals in absence of hM4Di expression.
Supplementary Movie 2 (related to Figure 2): Representative movie for intracellular Ca2+ signals in hM4Di-expressing astrocytes before, during and after CNO application in brain slices. Confocal microscopy imaging of GCaMP6f- and hM4Di-expressing striatal astrocytes showing that bath application of 1 μM CNO strongly evoked astrocyte Ca2+ signals.
Supplementary Table 1 (related to all the Figures): reports all the statistical analyses, n numbers and relevant parameters for all figure panels in the main and supplemental data. This table has been submitted as an Excel file.
Supplementary Table 2 (related to Figure 5): reports the RNA-seq data in full for all samples and has been submitted as an Excel file. The raw data have also been deposited at the Gene Expression Omnibus (GEO) with accessions ID # of GSE119058. Three tables are included in this single Excel file for RNA-seq.
Figure S1 (related to Figure 1): MSN depolarization, which induced Ca2+ influx into MSNs, activated astrocyte Ca2+ signaling irrespective of MSN subtypes. (A) Representative images showing tdTomato-positive (i, D1) and tdTomato-negative (ii, D2) MSNs from Drd1a-tdTomato mice before and after whole-cell patching (dialyzed with Alexa 568). (B) Depolarization of D1 or D2 MSNs to upstate like levels (96 ± 21 action potentials in i and 130 ± 6 action potentials in ii) increased the frequency of astrocyte Ca2+ signals (3 representative cells for each). (C) Astrocyte Ca2+ signal frequency before and after D1 or D2 MSN depolarization (n = 4 mice per group). ≥ 20% increase in Ca2+ signal frequency was observed in 7 out of 9 astrocytes (i) and 7 out of 10 astrocytes (ii). (D-E) Simultaneous electrophysiological recording and “fast” line scan intracellular Ca2+ imaging from MSNs filled with Fluo-4 via the patch pipette. (F) Representative traces from a MSN during depolarizing current injections (400 pA). MSNs displayed no action potentials (AP) in the presence of 300 nM TTX (with or without Cd2+) in the bath. However, intracellular BAPTA dialysis did not block MSN APs. (G) Representative line scan data of MSN intracellular Ca2+ levels before, during and after somatic depolarization (400 pA current injections). (H) The left graph shows representative traces for MSN intracellular Ca2+ line scan imaging data under the various conditions shown. The right bar graphs summarize average data from such experiments for basal and peak Fluo-4 intensity (n = 5-6 MSNs from 3-5 mice). Overall, in these experiments MSN-depolarization evoked intracellular Ca2+ elevations in MSNs were not abolished by TTX, but were abolished by bath application of Cd2+ and by BAPTA dialysis. Paired t-test between before (basal) and after MSN depol (C). One-way ANOVA test (F). Scale bars, 20 μm (A) and 40 μm in (E). Data are shown as mean ± s.e.m. Full details of n numbers, precise P values and statistical tests are reported in Supplementary Table 1. * indicates P < 0.05, ** indicates P < 0.01, **** indicates P < 0.0001.
Figure S2 (related to Figure 1): Gabbr1 deletion in striatal astrocytes, intracellular mechanism and proximity of striatal astrocytes to MSNs. (A) To delete Gabbr1 from striatal astrocytes, floxed Gabbr1 mice received an AAV2/5 GfaABC1D for selectively expressing Cre in astrocytes (AAV Cre+) along with GCaMP6f. Control mice were floxed Gabbr1 mice that received only astrocyte selective AAV2/5 GCaMP6f (AAV Cre−). Representative images for in situ hybridization (RNAscope) for Gabbr1 and Aldh1l1 followed by IHC using anti-GFP antibodies to visualize GCaMP6f expressing astrocytes (outlined by dotted lines). (B) Average data of fluorescence intensity and puncta number of Gabbr1 revealed with RNAscope showing its reduction in striatal astrocytes. n = 32-33 astrocytes from 3 mice. Unpaired t-test (B). Scale bars, 20 μm (A) and 10 μm (A’). Data are shown as mean ± s.e.m. Full details of n numbers, precise P values and statistical tests are reported in Supplementary Table 1. * indicates P < 0.05, **** indicates P < 0.0001. (C, D) PLC-dependent astrocyte Ca2+ signaling evoked by PE and baclofen. (C) 10 μM U73122, a PLC inhibitor, blocked PE and baclofen-evoked increases in striatal astrocyte Ca2+ signals (D), while 10 μM U73433, a control analogue for U73122, did not (C). The traces shown are from 3 representative cells in each case and the scatter graphs are average data. n = 8-20 astrocytes from 3-4 mice. Wilcoxon signed ranks test (A, B). Data are shown as mean ± s.e.m. Full details of n numbers, precise P values and statistical tests are reported in full in Table S1. ** indicates P < 0.01, NS indicates not significantly different. (E ,F) MSN dendrites were juxtaposed with astrocyte somata and processes. (E) Representative image of a tdTomato-expressing astrocyte and YFP-expressing neuronal soma and dendrites in the striatum showing close apposition of astrocytes and MSN dendrites (arrows). (F) Average line-profile data showing the distance between the center of the dendrite and the center of the astrocyte somata and/or processes. The scatter graph reports average data from analyses such as those in b (n = 26 images, 4 mice). Scale bar, 20 μm (A). Data are shown as mean ± s.e.m.
Figure S3 (related to Figure 1 and 2): Gene expression using GfaABC1D AAV2/5 in the dorsal striatum. (A) Cartoon illustrating AAV2/5 microinjection into the dorsal striatum to express GCaMP6f by delivering AAV2/5 GfaABC1D-Cre into Ai95 mice that have a floxed-STOP cassette preventing expression of GCaMP6f. AAV2/5 GfaABC1D-tdTomato was co-injected. (B) Images showing tdTomato and GCaMP6f were expressed in the dorsal striatum (dSt) which is underneath the corpus callosum (CC) in the AAVs-injected side, but not in the contralateral side. (C) Representative image of S100β positive astrocytes that express tdTomato and are GCaMP positive (arrows). Bar graphs show that 86% of the S100β positive astrocytes were tdTomato positive (red bar graph) and that 91% of the S100β positive astrocytes were GCaMP6f positive due to Cre (green bar graph). This indicates both AAVs were delivered into most of astrocytes in dSt. n = 4 mice. (D) Representative image showing that NeuN positive neurons did not express tdTomato which was driven under the GfaABC1D promoter. However, although Cre expression was driven under the same promoter, some NeuN positive neurons were GCaMP6f positive (arrows). Open arrows indicate GCaMP6f negative neurons. Bar graphs show that none of the NeuN positive neurons were tdTomato positive (left bar graph), but that 61% of the NeuN positive neurons were GCaMP6f positive (green bar graph). n = 4 mice. (E) Image of the brain distribution of hM4Di-mCherry, which was restricted to the striatum when AAV2/5 GfaABC1D hM4Di-mCherry was microinjected in the dorsal striatum. (F) Representative image of hM4Di-mCherry positive striatal astrocytes (arrows) that were S100β positive and NeuN negative. (G) Bar graphs show that 98% of the hM4Di-mCherry positive astrocytes were S100β positive (red bar graph). Furthermore, ~84% of the S100β positive astrocytes in the dorsal striatum expressed hM4Di-mCherry (green bar graph) following AAV2/5 microinjections. However, an insignificant number of the hM4Di-mCherry positive cells were NeuN positive (~2%; right bar graph). Taken together, these data indicate that AAV2/5 GfaABC1D mediated delivery of hM4Di-mCherry to the dorsal striatum was astrocyte selective and targeted most of the striatal astrocytes. n = 4 mice. Scale bars, 200 μm in panel B, and 20 μm in panel C,D and F, 2 mm in E. Data are shown as mean ± s.e.m.
Additional note on Gabbr1 f/f mice and AAV2/5 GfaABC1D-Cre in relation to panels A-D: In data reported in the main text, we deleted GB1R from astrocytes using striatal AAV2/5 GfaABC1D-Cre microinjections. We could identify astrocytes based on their bushy morphologies as well as by marker expression (Figure S2A,B), and we could therefore easily monitor the consequences of deleting GB1Rs in single cell evaluations. However, we could not use Gabbr1 f/f mice for astrocyte-selective evaluations of more complex phenomena such as animal behavior. There are two reasons. First, there is no Cre mouse line that targets only striatal astrocytes, and thus crossing Gabbr1 f/f mice with even the best available Aldh1l1-Cre/ERT2 mice would delete GB1Rs from all astrocytes in the CNS, and from all Aldh1l1-expressing cells in the periphery (Chai et al., 2017; Srinivasan et al., 2016). Such an approach would not allow meaningful assessment of astrocyte GB1Rs functions for striatum-dependent behaviors. Second, we could not use AAV2/5 GfaABC1D-Cre microinjections, because as reported previously these viruses lead to low-level Cre expression within striatal neurons, which was sufficient to cause excision of floxed genomic sequences (Srinivasan et al., 2016). It is important to note, however, that the AAV2/5 GfaABC1D constructs do not lead to expression of other cargo such as reporters, channels and receptors in neurons (e.g. GFP, tdTomato, Kir4.1, DREADDS and GCaMP). For these molecules, the AAVs are demonstrably astrocyte selective (Adamsky et al., 2018; Anderson et al., 2016; Bonder and McCarthy, 2014; Cui et al., 2018; Haustein et al., 2014; Jiang et al., 2016; Jiang et al., 2014; Octeau et al., 2018; Rungta et al., 2016; Shigetomi et al., 2013; Srinivasan et al., 2015; Srinivasan et al., 2016; Stobart et al., 2018; Tong et al., 2014; Yu et al., 2018). The explanation is that for such cargo to be measurably functional requires quite high expression, which the tropism of AAV2/5 and the GfaABC1D promoter favors for astrocytes, but prevents for neurons. However, in the case of Cre expression, even a very low number of recombinase molecules are sufficient to cause excision of floxed genomic sequences, resulting in observable effects in both neurons and astrocytes, as previously shown (Srinivasan et al., 2016). We report a specific set of experiments in panels A-D to document that the AAV constructs were astrocyte selective for reporter expression, but not for Cre-mediated excision of genomic sequences. Reporter expression was observed in 86 ± 4% of astrocytes and no neurons (0%), but Cre-mediated excision was observed in 91 ± 3% of astrocytes and 61 ± 3% of striatal neurons (C-D, n = 4 mice for each). For these reasons, we could not use the Gabbr1 f/f mice to evaluate reliably astrocyte GB1R contributions to mouse behavior. We emphasize that these limitations of AAV2/5 GfaABC1D-Cre apply to the striatum and hippocampus (Srinivasan et al., 2016). In the future, it may be possible to develop intersectional genetic approaches to target striatal astrocytes in the adult brain for selective gene deletion experiments.
Figure S4 (related to Figure 3 and 4): Additional assessments of the consequences of astrocyte Gi signaling in vivo. (A) Time spent on the accelerating Rotarod for individual trials completed at days 1 and 2 (5 trials per day). Note that there was a trend for the hM4Di + CNO mice to fall off the Rotarod sooner than the controls on Day 1, but this did not reach statistical significance at Day 2. We suspect this trend occurred because the mice appeared to rear more on the Rotarod on Day 1 and during this inattentive period they fell off. Consistent with this, panel (B) shows significantly increased rearing in the hM4Di + CNO group relative to controls. (C) Representative raw data for footprint tracks of mice walking on paper with their rear paws painted with black paint. The bar graphs show average data for footprint length and width, which is a measure of gate. (D) Marble burying test following activation of astrocyte Gi signaling. The number of buried marbles was significantly greater in the hM4Di + CNO group compared to AAV + CNO group. This is because the hM4Di + CNO group displayed hyperlocomotion (see main text), which resulted in greater disturbance of the bedding in the cage and thus more buried marbles n = 7-8 mice per group. (E) The pie charts show the types of neurons that were recorded with in vivo silicon probes in AAV control and hM4Di mice. TAN = tonically active neurons, FSI = fast spiking interneurons, MSN = medium spiny neurons, uncl = unclassified neurons. (F) Graphs show that the FSI firing rate was not altered after CNO i.p. administration at t = 0 min in hM4Di mice (7 mice) and control mice (5 mice). The inset shows a mean spike waveform of a representative FSI. (G) The graphs plot the distance that head-restrained mice travelled on a spherical treadmill before and following i.p. CNO administration. The distance was binned in 30 min blocks and normalized to the distance travelled in the 30 min before CNO i.p. injection (basal). Data from each mouse is shown in light traces, whereas the average data are shown in dark traces. The bar graph on the right summarizes the experiments and shows that hM4Di-expressing mice displayed significantly increased ambulation compared to control AAV-expressing mice. (H-K) Whole-cell current-clamp recordings from MSNs near hM4Di-expressing astrocytes following in vivo i.p. CNO administration revealed that the resting membrane potential (H), membrane resistance (I) and rheobase (J, K) of MSNs were not altered by activation of astrocyte Gi signaling under conditions that did later behavior as reported in the main manuscript. The right panels show representative traces from whole-cell current-clamp MSN recordings (current injections: −100, 100, 200, 300 and 400 pA). n = 15-16 MSNs from 4 mice. Two-way ANOVA test (A). Unpaired t-test (B, D, G). Paired t-test (F). Data are shown as mean ± s.e.m. Full details of n numbers, precise P values and statistical tests are reported in Supplementary Table 1. * indicates P < 0.05, ** indicates P < 0.01, NS indicates not significantly different.
Figure S5 (related to Figure 5): Astrocyte transcriptomes from adult mouse striatum following astrocyte Gi signaling activation in vivo for 2 hrs. (A) Cartoon illustrating the experimental design employing AAV2/5 GfaABC1D Rpl22-HA and the outline of the protocol for RNA-seq (Yu et al., 2018). (B) Heat map showing relative enrichment (red) or depletion (blue) of the top 200 adult striatal astrocyte markers. Results from eight RNA-seq samples for both input and IP from four mice that received astrocyte specific hM4Di in the dorsolateral striatum are shown. The row Z scores were calculated using the FPKM values. These data show that the RNA-seq data were replete with known astrocyte markers, which served to validate the approach. (C) Representative Western blot showing that Rpl22-HA was preserved in the IP sample, whereas β-actin was depleted in the IP sample (in relation to input). In contrast, Rpl22HA was not evident in the supernatant, whereas β-action was. n = three biological replicates. (D) Gene expression levels of cell-specific markers for astrocytes, neurons, oligodendrocytes, and microglia in IP samples from the hM4Di+Veh group. n = 4 mice per group. Data are shown as mean ± s.e.m All the RNA-seq data are provided the Supplementary Table 2.
Figure S6 (related to Figure 3, 4 and 6): Astrocyte Gi signaling-mediated changes returned to baseline at 48 hrs after in vivo CNO administration. (A) Images on the left show spines from biocytin filled MSNs. Right two graphs show no significant difference in spine density and spine head size of MSNs in the indicated 2 experimental groups at 48 hrs after CNO (n = 4 mice per group). (B) Representative traces for evoked AMPA EPSCs due to paired stimuli at membrane potentials of −70 mV (i) and for NMDA EPSCs due to single stimuli at +40 mV (ii) from the two indicated experimental groups. (C) Summary of multiple experiments such as those illustrated with representative traces in panel B (n = 20 MSNs from 4 mice). There was no significant change in EPSC amplitudes. (D) The two graphs on the left plot the AMPA EPSC amplitudes with multiple stimulation intensities to the cortico-striatal pathway in brain slices from the two indicated experimental groups. Plots in light colors show individual data from each MSN and those in dark colors indicate averaged data. The right graph show averaged plots from the two indicated experimental groups. (E) Cartoon illustrating the AAV2/5 reagents and approaches for selectively expressing hM4Di-mCherry bilaterally in striatal astrocytes. Once such mice were prepared, open field test (OFT) was performed twice: (i) 2 weeks after AAV surgery (before injection) and (ii) 2 hrs after intraperitoneal (i.p.) injection of 1 mg/kg CNO or vehicle. Bar graphs show that distance travelled by the mice over 30 min in an open field chamber for the 2 experimental groups. The distance travelled was not different between groups before i.p. injection. At 2 hrs after i.p. injection, the CNO injected group showed hyperactivity compared to the vehicle injected group. n = 8 mice per group. (F) Cartoon illustrating the AAV2/5 reagents and approaches for selectively expressing hM4Di-mCherry bilaterally in striatal astrocytes. OFT was performed twice: (i) 2 weeks after AAV surgery and (ii) 48 hrs after i.p. injection of 1 mg/kg CNO or vehicle. Bar graphs show that distance travelled by the mice over 30 min in an open field chamber. The distance travelled was not different between groups before and after i.p. injection, n = 8 mice per group. Thus, the statistically significant effects observed at 2 hrs after CNO abated by 48 hrs. Mann-Whitney test or unpaired Student’s t-test as appropriate (A,C,E,F). Two-way ANOVA repeated measure followed by Tukey’s post-hoc test (D). Scale bar, 2 μm (A). Data are shown as mean ± s.e.m. Full details of n numbers, precise P values and statistical tests are reported in Supplementary Table 1. * indicates P <0.05, NS indicates not significantly different.
Figure S7 (related to Figure 6): Activation of astrocyte Gi signaling increased vGlut1-positive synapses onto MSNs, and Gabapentin did not alter astrocyte Ca2+ signals mediated by Gi-signaling. (A) Representative image of a dendritic shaft and dendritic spines of a MSN filled with biocytin via intracellular dialysis during whole-cell recordings, in relation to vGlut1 + puncta. Such images were used to assess synapses. Putative synapses for the open squares 1 and 2 are shown in A1 and A2, respectively. A1 and A2 illustrate how we defined vGlut1-positive or negative synapses (see methods for detail). Briefly, vGlut1 positive synapses were defined as such if the vGlut1 and dendritic spine fluorescence signals overlapped at the full-width half maxima of the cognate optical signals from line profile analyses. The outcome of this analysis showed that 47 ± 6% of MSN dendritic spines formed vGlut1 positive synapses in control mice (n = 138-177 spines from 6 MSNs from 3 mice), which is similar to previous work on MSNs (Doig et al., 2010). (B) Summary graph shows that astrocyte Gi signaling activation resulted in increased vGlut1-positive synapses onto MSNs relative to the control group (6 MSNs, mice). (C) Kymographs and ΔF/F traces of astrocyte Ca2+ responses before and during bath application of 1 μM CNO in the absence or presence of 50 μM Gabapentin in the bath. The bar graph shows the CNO-evoked astrocyte Ca2+ signals (integrated area) with and without Gabapentin in bath (n ≥ 25 cells from 4 mice). (D) Kymographs and ΔF/F traces of astrocyte Ca2+ responses. The bar graphs summarize the integrated areas of the spontaneous astrocyte Ca2+ signals in hM4Di and control mice that received either Gabapentin or saline i.p. 3 hr prior, and CNO i.p. 2 hr prior to dissection (n ≥ 22 astrocytes from 4 mice). These data show that a single in vivo dose of CNO evoked a long lasting increase in astrocyte Ca2+ signaling and in vivo Gabapentin administration did not alter the CNO response. Unpaired t-test (B). Mann-Whitney test (C). Two-way ANOVA followed by Tukey’s post-hoc test (D). Scale bar, 2 μm (A). Data are shown as mean ± s.e.m. Full details of n numbers, precise P values and statistical tests are reported in Supplementary Table 1. * indicates P < 0.05, ** indicates P < 0.01, NS indicates not significantly different.
Highlights.
Striatal MSNs release GABA to activate astrocyte Gi-coupled GABAB receptors
Astrocyte Gi pathway activation resulted in hyperactivity with disrupted attention
Astrocyte Gi pathway activation increased fast synaptic excitation and MSN firing
Behavioral and synaptic effects were due to reactivation of TSP1 within astrocytes
Acknowledgments
Supported by NIH grants NS060677 and MH104069 (BSK). JN was partly supported by a JSPS Overseas Research Fellowship (H28-729) and the Uehara Memorial Foundation Overseas Postdoctoral Research Fellowship (201730082). We acknowledge the NINDS Informatics Center for Neurogenetics and Neurogenomics (P30 NS062691 to GC) and the Genetics, Genomics and Informatics Core of the Semel Institute of Neuroscience at UCLA (U54HD087101-01 from the Eunice Kennedy Shriver National Institute of Child Health and Human Development). AKR and MSF were supported by RO1 MH062122, NARSAD 26612 (AKR) and the Staglin Center for Brain and Behavioral Health. Behavioral experiments utilized the UCLA Behavioral Testing Core. AH and SM were supported NIH grants NS100050, DA042739, and NSF NeuroNex Technology Hub Award 1707408. We thank the UCLA Neuroscience Genomics Core (www.semel.ucla.edu/ungc) for assistance with sequencing, and Fuying Gao for helping with RNA-seq data analysis. Thanks to Michael V. Sofroniew and Christopher J. Evans for sharing equipment. Many thanks to Drs. Yu, Beaumont, Flint and Sofroniew for valuable comments.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare no commercial interests
References
- Adamsky A, Kol A, Kreisel T, Doron A, Ozeri-Engelhard N, Melcer T, Refaeli R, Horn H, Regev L, Groysman M, et al. (2018). Astrocytic Activation Generates De Novo Neuronal Potentiation and Memory Enhancement. Cell 174, 59–71. [DOI] [PubMed] [Google Scholar]
- Alexander GM, Rogan SC, Abbas AI, Armbruster BN, Pei Y, Allen JA, Nonneman RJ, Hartmann J, Moy SS, Nicolelis MA, et al. (2009). Remote control of neuronal activity in transgenic mice expressing evolved G protein-coupled receptors. Neuron 63, 27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen NJ, and Lyons DA (2018). Glia as architects of central nervous system formation and function. Science 362, 181–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson MA, Burda JE, Ren Y, Ao Y, O’Shea TM, Kawaguchi R, Coppola G, Khakh BS, Deming TJ, and Sofroniew MV (2016). Astrocyte scar formation aids central nervous system axon regeneration. Nature 532, 195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araque A, Carmignoto G, Haydon PG, Oliet SH, Robitaille R, and Volterra A (2014). Gliotransmitters travel in time and space. Neuron 81, 728–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwell D, Buchan AM, Charpak S, Lauritzen M, Macvicar BA, and Newman EA (2010). Glial and neuronal control of brain blood flow. Nature 468, 232–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhurin KI, Mac V, Golshani P, and Masmanidis SC (2016). Temporal correlations among functionally specialized striatal neural ensembles in reward-conditioned mice. J Neurophysiol 115, 1521–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargas J, Howe A, Eberwine J, Cao Y, and Surmeier DJ (1994). Cellular and molecular characterization of Ca2+ currents in acutely isolated, adult rat neostriatal neurons. J Neurosci 14, 6667–6686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barres BA (2008). The mystery and magic of glia: a perspective on their roles in health and disease. Neuron 60, 430–440. [DOI] [PubMed] [Google Scholar]
- Bazargani N, and Attwell D (2016). Astrocyte calcium signalling: the third wave. Nat Neurosci 19, 182–189. [DOI] [PubMed] [Google Scholar]
- Bernardinelli Y, Salmon C, Jones EV, Farmer WT, Stellwagen D, and Murai KK (2011). Astrocytes display complex and localized calcium responses to single-neuron stimulation in the hippocampus. J Neurosci 31, 8905–8919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonder DE, and McCarthy KD (2014). Astrocytic Gq-GPCR-linked IP3R-dependent Ca2+ signaling does not mediate neurovascular coupling in mouse visual cortex in vivo. J Neurosci 34, 13139–13150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter AG, and Sabatini BL (2004). State-dependent calcium signaling in dendritic spines of striatal medium spiny neurons. Neuron 44, 483–493. [DOI] [PubMed] [Google Scholar]
- Chai H, Diaz-Castro B, Shigetomi E, Monte E, Octeau JC, Yu X, Cohn W, Rajendran PS, Vondriska TM, Whittelegge JP, et al. (2017). Neural circuit-specialized astrocytes: transcriptomic, proteomic, morphological and functional evidence. Neuron 95, 531–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TW, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, Schreiter ER, Kerr RA, Orger MB, Jayaraman V, et al. (2013). Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499, 295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P, and Barres BA (2005). Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell 120, 421–433. [DOI] [PubMed] [Google Scholar]
- Crawford DC, Jiang X, Taylor A, and Mennerick S (2012). Astrocyte-derived thrombospondins mediate the development of hippocampal presynaptic plasticity in vitro. J Neurosci 32, 13100–13110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford SE, Stellmach V, Murphy-Ullrich JE, Ribeiro SM, Lawler J, Hynes RO, Boivin GP, and Bouck N (1998). Thrombospondin-1 is a major activator of TGF-beta1 in vivo. Cell 93, 1159–1170. [DOI] [PubMed] [Google Scholar]
- Cubillo A, Halari R, Smith A, Taylor E, and Rubia K (2012). A review of fronto-striatal and fronto-cortical brain abnormalities in children and adults with Attention Deficit Hyperactivity Disorder (ADHD) and new evidence for dysfunction in adults with ADHD during motivation and attention. Cortex 48, 194–215. [DOI] [PubMed] [Google Scholar]
- Cui Y, Yang Y, Ni Z, Dong Y, Cai G, Foncelle A, Ma S, Sang K, Tang S, Li Y, et al. (2018). Astroglial Kir4.1 in the lateral habenula drives neuronal bursts in depression. Nature 554, 323–327. [DOI] [PubMed] [Google Scholar]
- Curatolo P, D’Agati E, and Moavero R (2010). The neurobiological basis of ADHD. Ital J Pediatr December 22;36(1):79. doi: 10.1186/1824-7288-36-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Peña JB, Dela Peña IJ, Custodio RJ, Botanas CJ, Kim HJ, and Cheong JH (2018). Exploring the Validity of Proposed Transgenic Animal Models of Attention-Deficit Hyperactivity Disorder (ADHD). Mol Neurobiol 55, 3739–3754. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doig NM, Moss J, and Bolam JP (2010). Cortical and thalamic innervation of direct and indirect pathway medium-sized spiny neurons in mouse striatum. J Neurosci 30, 14610–14618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DSM-5 (2013). American Psychiatric Association: Diagnostic and Statistical Manual of Mental Disorders (DSM-5). 5th edition. [Google Scholar]
- Eroglu C, Allen NJ, Susman MW, O’Rourke NA, Park CY, Ozkan E, Chakraborty C, Mulinyawe SB, Annis DS, Huberman AD, et al. (2009). Gabapentin receptor alpha2delta-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell 139, 380–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fayyad J, De Graaf R, Kessler R, Alonso J, Angermeyer M, Demyttenaere K, De Girolamo G, Haro JM, Karam EG, Lara C, et al. (2007). Cross-national prevalence and correlates of adult attention-deficit hyperactivity disorder. Br J Psychiatry 190, 402–409. [DOI] [PubMed] [Google Scholar]
- Gerfen CR, and Surmeier DJ (2011). Modulation of striatal projection systems by dopamine. Annu Rev Neurosci 34, 441–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godsil BP, Blackmore MA, and Fanselow MS (2005a). Modulation of an activity response with associative and nonassociative fear in the rat: a lighting differential influences the form of defensive behavior evoked after fear conditioning. Learn Behav 33, 454–463. [DOI] [PubMed] [Google Scholar]
- Godsil BP, and Fanselow MS (2004). Light stimulus change evokes an activity response in the rat. Learn Behav 32, 299–310. [DOI] [PubMed] [Google Scholar]
- Godsil BP, Stefanacci L, and Fanselow MS (2005b). Bright light suppresses hyperactivity induced by excitotoxic dorsal hippocampus lesions in the rat. Behav Neurosci 119, 1339–1352. [DOI] [PubMed] [Google Scholar]
- Gomez JL, Bonaventura J, Lesniak W, Mathews WB, Sysa-Shah P, Rodriguez LA, Ellis RJ, Richie CT, Harvey BK, Dannals RF, et al. (2017). Chemogenetics revealed: DREADD occupancy and activation via converted clozapine. Science 357, 503–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graybiel AM (2008). Habits, rituals, and the evaluative brain. Annu Rev Neurosci 31, 359–387. [DOI] [PubMed] [Google Scholar]
- Haim LB, and Rowitch DH (2017). Functional diversity of astrocytes in neural circuit regulation. Nat Rev Neurosci 18, 31–41. [DOI] [PubMed] [Google Scholar]
- Halassa MM, and Haydon PG (2010). Integrated brain circuits: astrocytic networks modulate neuronal activity and behavior. Annu Rev Physiol 72, 335–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haller C, Casanova E, Muller M, Vacher CM, Vigot R, Doll T, Barbieri S, Gassmann M, and Bettler B (2004). Floxed allele for conditional inactivation of the GABAB(1) gene. Genesis 40, 125–130. [DOI] [PubMed] [Google Scholar]
- Haustein MD, Kracun S, Lu XH, Shih T, Jackson-Weaver O, Tong X, Xu J, Yang XW, O’Dell TJ, Marvin JS, et al. (2014). Conditions and constraints for astrocyte calcium signaling in the hippocampal mossy fiber pathway. Neuron 82, 413–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henneberger C, Papouin T, Oliet SH, and Rusakov DA (2010). Long-term potentiation depends on release of D-serine from astrocytes. Nature 463, 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang R, Diaz-Castro B, Tong X, Looger LL, and Khakh BS (2016). Dysfunctional calcium and glutamate signaling in striatal astrocytes from Huntington’s disease model mice. J Neurosci 36, 3453–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang R, Haustein MD, Sofroniew MV, and Khakh BS (2014). Imaging intracellular Ca2+ signals in striatal astrocytes from adult mice using genetically-encoded calcium indicators. J Vis Exp November 19;(93):e51972. doi: 10.3791/51972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J, Jiang L, Goldman SA, and Nedergaard M (1998). Astrocyte-mediated potentiation of inhibitory synaptic transmission. Nat Neurosci December;1(8):683–92. [DOI] [PubMed] [Google Scholar]
- Kelley KW, Nakao-Inoue H, Molofsky AV, and Oldham MC (2018). Variation among intact tissue samples reveals the core transcriptional features of human CNS cell classes. Nat Neurosci 21, 1171–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khakh BS, Beaumont V, Cachope R, Munoz-Sanjuan I, Goldman SA, and Grantyn R (2017). Unravelling and Exploiting Astrocyte Dysfunction in Huntington’s Disease. Trends Neurosci 40, 422–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khakh BS, and Sofroniew MV (2015). Diversity of astrocyte functions and phenotypes in neural circuits. Nat Neurosci 18, 942–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, and Malenka RC (2008). Striatal plasticity and basal ganglia circuit function. Neuron 60, 543–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuffler SW (1967). Neuroglial cells: physiological properties and a potassium mediated effect of neuronal activity on the glial membrane potential. Proc R Soc Lond B Biol Sci 168, 1–21. [DOI] [PubMed] [Google Scholar]
- Law CW, Chen Y, Shi W, and Smyth GK (2014). voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biology 15, R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín R, Bajo-Grañeras R, Moratalla R, Perea G, and Araque A (2015). Circuit-specific signaling in astrocyte-neuron networks in basal ganglia pathways. Science 349, 730–734. [DOI] [PubMed] [Google Scholar]
- Molofsky AV, Kelley KW, Tsai HH, Redmond SA, Chang SM, Madireddy L, Chan JR, Baranzini SE, Ullian EM, and Rowitch DH (2014). Astrocyte-encoded positional cues maintain sensorimotor circuit integrity. Nature 509, 189–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarrete M, and Araque A (2008). Endocannabinoids mediate neuron-astrocyte communication. Neuron 57, 883–893. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, and Bergles DE (2015). Large-scale recording of astrocyte activity. Curr Opin Neurobiol 32, 95–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Octeau JC, Chai H, Jiang R, Bonanno SL, Martin KC, and Khakh BS (2018). An Optical Neuron-Astrocyte Proximity Assay at Synaptic Distance Scales. Neuron 98, 49–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polanczyk G, de Lima MS, Horta BL, Biederman J, and Rohde LA (2007). The worldwide prevalence of ADHD: a systematic review and metaregression analysis. Am J Psychiatry 164, 942–948. [DOI] [PubMed] [Google Scholar]
- Porter JT, and McCarthy KD (1997). Astrocytic neurotransmitter receptors in situ and in vivo. Prog Neurobiol 51, 439–455. [DOI] [PubMed] [Google Scholar]
- Risher WC, Kim N, Koh S, Choi JE, Mitev P, Spence EF, Pilaz LJ, Wang D, Feng G, Silver DL, et al. (2018). Thrombospondin receptor α2δ−1 promotes synaptogenesis and spinogenesis via postsynaptic Rac1. J Cell Biol 217, 3747–3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riva D, Taddei M, and Bulgheroni S (2018). The neuropsychology of basal ganglia. Eur J Paediatr Neurol 22, 321–326. [DOI] [PubMed] [Google Scholar]
- Roth BL (2016). DREADDs for Neuroscientists. Neuron 89, 683–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rungta R, Bernier L-P, Dissing-Olesen L, Groten C, LeDue J, Drissler S, and MacVicar B (2016). Ca2+ transients in astrocyte fine processes occur via Ca2+ influx in adult mouse hippocampus. Glia 64, 2093–2103. [DOI] [PubMed] [Google Scholar]
- Sanz E, Yang L, Su T, Morris DR, McKnight GS, and Amieux PS (2009). Cell-type-specific isolation of ribosome-associated mRNA from complex tissues. Proc Natl Acad Sci U S A 106, 13939–13944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigetomi E, Bushong EA, Haustein MD, Tong X, Jackson-Weaver O, Kracun S, Xu J, Sofroniew MV, Ellisman MH, and Khakh BS (2013). Imaging calcium microdomains within entire astrocyte territories and endfeet with GCaMPs expressed using adeno-associated viruses. J Gen Physiol 141, 633–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigetomi E, Patel S, and Khakh BS (2016). Probing the Complexities of Astrocyte Calcium Signaling. Trends Cell Biol 26, 300–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith S (1994). Neural signalling. Neuromodulatory astrocytes. Curr Biol 4, 807–810. [DOI] [PubMed] [Google Scholar]
- Srinivasan R, Huang BS, Venugopal S, Johnston AD, Chai H, Zeng H, Golshani P, and Khakh BS (2015). Ca(2+) signaling in astrocytes from Ip3r2(−/−) mice in brain slices and during startle responses in vivo. Nat Neurosci 18, 708–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan R, Lu T-Y, Chai H, Xu J, Huang BS, Golshani P, Coppola G, and Khakh BS (2016). New Transgenic Mouse Lines for Selectively Targeting Astrocytes and Studying Calcium Signals in Astrocyte Processes In Situ and In Vivo. Neuron 92, 1181–1195: PMID: 27939582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stobart JL, Ferrari KD, Barrett MJP, Gluck C, Stobart MJ, Zuend M, and Weber B (2018). Cortical Circuit Activity Evokes Rapid Astrocyte Calcium Signals on a Similar Timescale to Neurons. Neuron 98, 726–735. [DOI] [PubMed] [Google Scholar]
- Tong X, Ao Y, Faas GC, Nwaobi SE, Xu J, Haustein MD, Anderson MA, Mody I, Olsen ML, Sofroniew MV, et al. (2014). Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington’s disease model mice. Nat Neurosci 17, 694–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tritsch NX, and Sabatini BL (2012). Dopaminergic modulation of synaptic transmission in cortex and striatum. Neuron 76, 33–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volterra A, Liaudet N, and Savtchouk I (2014). Astrocyte Ca(2)(+) signalling: an unexpected complexity. Nat Rev Neurosci 15, 327–335. [DOI] [PubMed] [Google Scholar]
- Waters J, Schaefer A, and Sakmann B (2005). Backpropagating action potentials in neurones: measurement, mechanisms and potential functions. Prog Biophys Mol Biol, 145–170. [DOI] [PubMed] [Google Scholar]
- Wilson CJ, and Kawaguchi Y (1996). The origins of two-state spontaneous membrane potential fluctuations of neostriatal spiny neurons. J Neurosci 16, 2397–2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Taylor AMW, Nagai J, Golshani P, Evans CJ, Coppola G, and Khakh BS (2018). Reducing astrocyte calcium signaling in vivo alters striatal microcircuits and causes repetitive behavior. Neuron 99, 1170–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. (2014). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34, 11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Movie 1 (related to Figure 2): Representative movie for intracellular Ca2+ signals in hM4Di-negative astrocytes before, during and after CNO application in brain slices. Confocal microscopy imaging of GCaMP6f-expressing striatal astrocytes showing that bath application of 1 μM CNO did not change astrocyte Ca2+ signals in absence of hM4Di expression.
Supplementary Movie 2 (related to Figure 2): Representative movie for intracellular Ca2+ signals in hM4Di-expressing astrocytes before, during and after CNO application in brain slices. Confocal microscopy imaging of GCaMP6f- and hM4Di-expressing striatal astrocytes showing that bath application of 1 μM CNO strongly evoked astrocyte Ca2+ signals.
Supplementary Table 1 (related to all the Figures): reports all the statistical analyses, n numbers and relevant parameters for all figure panels in the main and supplemental data. This table has been submitted as an Excel file.
Supplementary Table 2 (related to Figure 5): reports the RNA-seq data in full for all samples and has been submitted as an Excel file. The raw data have also been deposited at the Gene Expression Omnibus (GEO) with accessions ID # of GSE119058. Three tables are included in this single Excel file for RNA-seq.
Figure S1 (related to Figure 1): MSN depolarization, which induced Ca2+ influx into MSNs, activated astrocyte Ca2+ signaling irrespective of MSN subtypes. (A) Representative images showing tdTomato-positive (i, D1) and tdTomato-negative (ii, D2) MSNs from Drd1a-tdTomato mice before and after whole-cell patching (dialyzed with Alexa 568). (B) Depolarization of D1 or D2 MSNs to upstate like levels (96 ± 21 action potentials in i and 130 ± 6 action potentials in ii) increased the frequency of astrocyte Ca2+ signals (3 representative cells for each). (C) Astrocyte Ca2+ signal frequency before and after D1 or D2 MSN depolarization (n = 4 mice per group). ≥ 20% increase in Ca2+ signal frequency was observed in 7 out of 9 astrocytes (i) and 7 out of 10 astrocytes (ii). (D-E) Simultaneous electrophysiological recording and “fast” line scan intracellular Ca2+ imaging from MSNs filled with Fluo-4 via the patch pipette. (F) Representative traces from a MSN during depolarizing current injections (400 pA). MSNs displayed no action potentials (AP) in the presence of 300 nM TTX (with or without Cd2+) in the bath. However, intracellular BAPTA dialysis did not block MSN APs. (G) Representative line scan data of MSN intracellular Ca2+ levels before, during and after somatic depolarization (400 pA current injections). (H) The left graph shows representative traces for MSN intracellular Ca2+ line scan imaging data under the various conditions shown. The right bar graphs summarize average data from such experiments for basal and peak Fluo-4 intensity (n = 5-6 MSNs from 3-5 mice). Overall, in these experiments MSN-depolarization evoked intracellular Ca2+ elevations in MSNs were not abolished by TTX, but were abolished by bath application of Cd2+ and by BAPTA dialysis. Paired t-test between before (basal) and after MSN depol (C). One-way ANOVA test (F). Scale bars, 20 μm (A) and 40 μm in (E). Data are shown as mean ± s.e.m. Full details of n numbers, precise P values and statistical tests are reported in Supplementary Table 1. * indicates P < 0.05, ** indicates P < 0.01, **** indicates P < 0.0001.
Figure S2 (related to Figure 1): Gabbr1 deletion in striatal astrocytes, intracellular mechanism and proximity of striatal astrocytes to MSNs. (A) To delete Gabbr1 from striatal astrocytes, floxed Gabbr1 mice received an AAV2/5 GfaABC1D for selectively expressing Cre in astrocytes (AAV Cre+) along with GCaMP6f. Control mice were floxed Gabbr1 mice that received only astrocyte selective AAV2/5 GCaMP6f (AAV Cre−). Representative images for in situ hybridization (RNAscope) for Gabbr1 and Aldh1l1 followed by IHC using anti-GFP antibodies to visualize GCaMP6f expressing astrocytes (outlined by dotted lines). (B) Average data of fluorescence intensity and puncta number of Gabbr1 revealed with RNAscope showing its reduction in striatal astrocytes. n = 32-33 astrocytes from 3 mice. Unpaired t-test (B). Scale bars, 20 μm (A) and 10 μm (A’). Data are shown as mean ± s.e.m. Full details of n numbers, precise P values and statistical tests are reported in Supplementary Table 1. * indicates P < 0.05, **** indicates P < 0.0001. (C, D) PLC-dependent astrocyte Ca2+ signaling evoked by PE and baclofen. (C) 10 μM U73122, a PLC inhibitor, blocked PE and baclofen-evoked increases in striatal astrocyte Ca2+ signals (D), while 10 μM U73433, a control analogue for U73122, did not (C). The traces shown are from 3 representative cells in each case and the scatter graphs are average data. n = 8-20 astrocytes from 3-4 mice. Wilcoxon signed ranks test (A, B). Data are shown as mean ± s.e.m. Full details of n numbers, precise P values and statistical tests are reported in full in Table S1. ** indicates P < 0.01, NS indicates not significantly different. (E ,F) MSN dendrites were juxtaposed with astrocyte somata and processes. (E) Representative image of a tdTomato-expressing astrocyte and YFP-expressing neuronal soma and dendrites in the striatum showing close apposition of astrocytes and MSN dendrites (arrows). (F) Average line-profile data showing the distance between the center of the dendrite and the center of the astrocyte somata and/or processes. The scatter graph reports average data from analyses such as those in b (n = 26 images, 4 mice). Scale bar, 20 μm (A). Data are shown as mean ± s.e.m.
Figure S3 (related to Figure 1 and 2): Gene expression using GfaABC1D AAV2/5 in the dorsal striatum. (A) Cartoon illustrating AAV2/5 microinjection into the dorsal striatum to express GCaMP6f by delivering AAV2/5 GfaABC1D-Cre into Ai95 mice that have a floxed-STOP cassette preventing expression of GCaMP6f. AAV2/5 GfaABC1D-tdTomato was co-injected. (B) Images showing tdTomato and GCaMP6f were expressed in the dorsal striatum (dSt) which is underneath the corpus callosum (CC) in the AAVs-injected side, but not in the contralateral side. (C) Representative image of S100β positive astrocytes that express tdTomato and are GCaMP positive (arrows). Bar graphs show that 86% of the S100β positive astrocytes were tdTomato positive (red bar graph) and that 91% of the S100β positive astrocytes were GCaMP6f positive due to Cre (green bar graph). This indicates both AAVs were delivered into most of astrocytes in dSt. n = 4 mice. (D) Representative image showing that NeuN positive neurons did not express tdTomato which was driven under the GfaABC1D promoter. However, although Cre expression was driven under the same promoter, some NeuN positive neurons were GCaMP6f positive (arrows). Open arrows indicate GCaMP6f negative neurons. Bar graphs show that none of the NeuN positive neurons were tdTomato positive (left bar graph), but that 61% of the NeuN positive neurons were GCaMP6f positive (green bar graph). n = 4 mice. (E) Image of the brain distribution of hM4Di-mCherry, which was restricted to the striatum when AAV2/5 GfaABC1D hM4Di-mCherry was microinjected in the dorsal striatum. (F) Representative image of hM4Di-mCherry positive striatal astrocytes (arrows) that were S100β positive and NeuN negative. (G) Bar graphs show that 98% of the hM4Di-mCherry positive astrocytes were S100β positive (red bar graph). Furthermore, ~84% of the S100β positive astrocytes in the dorsal striatum expressed hM4Di-mCherry (green bar graph) following AAV2/5 microinjections. However, an insignificant number of the hM4Di-mCherry positive cells were NeuN positive (~2%; right bar graph). Taken together, these data indicate that AAV2/5 GfaABC1D mediated delivery of hM4Di-mCherry to the dorsal striatum was astrocyte selective and targeted most of the striatal astrocytes. n = 4 mice. Scale bars, 200 μm in panel B, and 20 μm in panel C,D and F, 2 mm in E. Data are shown as mean ± s.e.m.
Additional note on Gabbr1 f/f mice and AAV2/5 GfaABC1D-Cre in relation to panels A-D: In data reported in the main text, we deleted GB1R from astrocytes using striatal AAV2/5 GfaABC1D-Cre microinjections. We could identify astrocytes based on their bushy morphologies as well as by marker expression (Figure S2A,B), and we could therefore easily monitor the consequences of deleting GB1Rs in single cell evaluations. However, we could not use Gabbr1 f/f mice for astrocyte-selective evaluations of more complex phenomena such as animal behavior. There are two reasons. First, there is no Cre mouse line that targets only striatal astrocytes, and thus crossing Gabbr1 f/f mice with even the best available Aldh1l1-Cre/ERT2 mice would delete GB1Rs from all astrocytes in the CNS, and from all Aldh1l1-expressing cells in the periphery (Chai et al., 2017; Srinivasan et al., 2016). Such an approach would not allow meaningful assessment of astrocyte GB1Rs functions for striatum-dependent behaviors. Second, we could not use AAV2/5 GfaABC1D-Cre microinjections, because as reported previously these viruses lead to low-level Cre expression within striatal neurons, which was sufficient to cause excision of floxed genomic sequences (Srinivasan et al., 2016). It is important to note, however, that the AAV2/5 GfaABC1D constructs do not lead to expression of other cargo such as reporters, channels and receptors in neurons (e.g. GFP, tdTomato, Kir4.1, DREADDS and GCaMP). For these molecules, the AAVs are demonstrably astrocyte selective (Adamsky et al., 2018; Anderson et al., 2016; Bonder and McCarthy, 2014; Cui et al., 2018; Haustein et al., 2014; Jiang et al., 2016; Jiang et al., 2014; Octeau et al., 2018; Rungta et al., 2016; Shigetomi et al., 2013; Srinivasan et al., 2015; Srinivasan et al., 2016; Stobart et al., 2018; Tong et al., 2014; Yu et al., 2018). The explanation is that for such cargo to be measurably functional requires quite high expression, which the tropism of AAV2/5 and the GfaABC1D promoter favors for astrocytes, but prevents for neurons. However, in the case of Cre expression, even a very low number of recombinase molecules are sufficient to cause excision of floxed genomic sequences, resulting in observable effects in both neurons and astrocytes, as previously shown (Srinivasan et al., 2016). We report a specific set of experiments in panels A-D to document that the AAV constructs were astrocyte selective for reporter expression, but not for Cre-mediated excision of genomic sequences. Reporter expression was observed in 86 ± 4% of astrocytes and no neurons (0%), but Cre-mediated excision was observed in 91 ± 3% of astrocytes and 61 ± 3% of striatal neurons (C-D, n = 4 mice for each). For these reasons, we could not use the Gabbr1 f/f mice to evaluate reliably astrocyte GB1R contributions to mouse behavior. We emphasize that these limitations of AAV2/5 GfaABC1D-Cre apply to the striatum and hippocampus (Srinivasan et al., 2016). In the future, it may be possible to develop intersectional genetic approaches to target striatal astrocytes in the adult brain for selective gene deletion experiments.
Figure S4 (related to Figure 3 and 4): Additional assessments of the consequences of astrocyte Gi signaling in vivo. (A) Time spent on the accelerating Rotarod for individual trials completed at days 1 and 2 (5 trials per day). Note that there was a trend for the hM4Di + CNO mice to fall off the Rotarod sooner than the controls on Day 1, but this did not reach statistical significance at Day 2. We suspect this trend occurred because the mice appeared to rear more on the Rotarod on Day 1 and during this inattentive period they fell off. Consistent with this, panel (B) shows significantly increased rearing in the hM4Di + CNO group relative to controls. (C) Representative raw data for footprint tracks of mice walking on paper with their rear paws painted with black paint. The bar graphs show average data for footprint length and width, which is a measure of gate. (D) Marble burying test following activation of astrocyte Gi signaling. The number of buried marbles was significantly greater in the hM4Di + CNO group compared to AAV + CNO group. This is because the hM4Di + CNO group displayed hyperlocomotion (see main text), which resulted in greater disturbance of the bedding in the cage and thus more buried marbles n = 7-8 mice per group. (E) The pie charts show the types of neurons that were recorded with in vivo silicon probes in AAV control and hM4Di mice. TAN = tonically active neurons, FSI = fast spiking interneurons, MSN = medium spiny neurons, uncl = unclassified neurons. (F) Graphs show that the FSI firing rate was not altered after CNO i.p. administration at t = 0 min in hM4Di mice (7 mice) and control mice (5 mice). The inset shows a mean spike waveform of a representative FSI. (G) The graphs plot the distance that head-restrained mice travelled on a spherical treadmill before and following i.p. CNO administration. The distance was binned in 30 min blocks and normalized to the distance travelled in the 30 min before CNO i.p. injection (basal). Data from each mouse is shown in light traces, whereas the average data are shown in dark traces. The bar graph on the right summarizes the experiments and shows that hM4Di-expressing mice displayed significantly increased ambulation compared to control AAV-expressing mice. (H-K) Whole-cell current-clamp recordings from MSNs near hM4Di-expressing astrocytes following in vivo i.p. CNO administration revealed that the resting membrane potential (H), membrane resistance (I) and rheobase (J, K) of MSNs were not altered by activation of astrocyte Gi signaling under conditions that did later behavior as reported in the main manuscript. The right panels show representative traces from whole-cell current-clamp MSN recordings (current injections: −100, 100, 200, 300 and 400 pA). n = 15-16 MSNs from 4 mice. Two-way ANOVA test (A). Unpaired t-test (B, D, G). Paired t-test (F). Data are shown as mean ± s.e.m. Full details of n numbers, precise P values and statistical tests are reported in Supplementary Table 1. * indicates P < 0.05, ** indicates P < 0.01, NS indicates not significantly different.
Figure S5 (related to Figure 5): Astrocyte transcriptomes from adult mouse striatum following astrocyte Gi signaling activation in vivo for 2 hrs. (A) Cartoon illustrating the experimental design employing AAV2/5 GfaABC1D Rpl22-HA and the outline of the protocol for RNA-seq (Yu et al., 2018). (B) Heat map showing relative enrichment (red) or depletion (blue) of the top 200 adult striatal astrocyte markers. Results from eight RNA-seq samples for both input and IP from four mice that received astrocyte specific hM4Di in the dorsolateral striatum are shown. The row Z scores were calculated using the FPKM values. These data show that the RNA-seq data were replete with known astrocyte markers, which served to validate the approach. (C) Representative Western blot showing that Rpl22-HA was preserved in the IP sample, whereas β-actin was depleted in the IP sample (in relation to input). In contrast, Rpl22HA was not evident in the supernatant, whereas β-action was. n = three biological replicates. (D) Gene expression levels of cell-specific markers for astrocytes, neurons, oligodendrocytes, and microglia in IP samples from the hM4Di+Veh group. n = 4 mice per group. Data are shown as mean ± s.e.m All the RNA-seq data are provided the Supplementary Table 2.
Figure S6 (related to Figure 3, 4 and 6): Astrocyte Gi signaling-mediated changes returned to baseline at 48 hrs after in vivo CNO administration. (A) Images on the left show spines from biocytin filled MSNs. Right two graphs show no significant difference in spine density and spine head size of MSNs in the indicated 2 experimental groups at 48 hrs after CNO (n = 4 mice per group). (B) Representative traces for evoked AMPA EPSCs due to paired stimuli at membrane potentials of −70 mV (i) and for NMDA EPSCs due to single stimuli at +40 mV (ii) from the two indicated experimental groups. (C) Summary of multiple experiments such as those illustrated with representative traces in panel B (n = 20 MSNs from 4 mice). There was no significant change in EPSC amplitudes. (D) The two graphs on the left plot the AMPA EPSC amplitudes with multiple stimulation intensities to the cortico-striatal pathway in brain slices from the two indicated experimental groups. Plots in light colors show individual data from each MSN and those in dark colors indicate averaged data. The right graph show averaged plots from the two indicated experimental groups. (E) Cartoon illustrating the AAV2/5 reagents and approaches for selectively expressing hM4Di-mCherry bilaterally in striatal astrocytes. Once such mice were prepared, open field test (OFT) was performed twice: (i) 2 weeks after AAV surgery (before injection) and (ii) 2 hrs after intraperitoneal (i.p.) injection of 1 mg/kg CNO or vehicle. Bar graphs show that distance travelled by the mice over 30 min in an open field chamber for the 2 experimental groups. The distance travelled was not different between groups before i.p. injection. At 2 hrs after i.p. injection, the CNO injected group showed hyperactivity compared to the vehicle injected group. n = 8 mice per group. (F) Cartoon illustrating the AAV2/5 reagents and approaches for selectively expressing hM4Di-mCherry bilaterally in striatal astrocytes. OFT was performed twice: (i) 2 weeks after AAV surgery and (ii) 48 hrs after i.p. injection of 1 mg/kg CNO or vehicle. Bar graphs show that distance travelled by the mice over 30 min in an open field chamber. The distance travelled was not different between groups before and after i.p. injection, n = 8 mice per group. Thus, the statistically significant effects observed at 2 hrs after CNO abated by 48 hrs. Mann-Whitney test or unpaired Student’s t-test as appropriate (A,C,E,F). Two-way ANOVA repeated measure followed by Tukey’s post-hoc test (D). Scale bar, 2 μm (A). Data are shown as mean ± s.e.m. Full details of n numbers, precise P values and statistical tests are reported in Supplementary Table 1. * indicates P <0.05, NS indicates not significantly different.
Figure S7 (related to Figure 6): Activation of astrocyte Gi signaling increased vGlut1-positive synapses onto MSNs, and Gabapentin did not alter astrocyte Ca2+ signals mediated by Gi-signaling. (A) Representative image of a dendritic shaft and dendritic spines of a MSN filled with biocytin via intracellular dialysis during whole-cell recordings, in relation to vGlut1 + puncta. Such images were used to assess synapses. Putative synapses for the open squares 1 and 2 are shown in A1 and A2, respectively. A1 and A2 illustrate how we defined vGlut1-positive or negative synapses (see methods for detail). Briefly, vGlut1 positive synapses were defined as such if the vGlut1 and dendritic spine fluorescence signals overlapped at the full-width half maxima of the cognate optical signals from line profile analyses. The outcome of this analysis showed that 47 ± 6% of MSN dendritic spines formed vGlut1 positive synapses in control mice (n = 138-177 spines from 6 MSNs from 3 mice), which is similar to previous work on MSNs (Doig et al., 2010). (B) Summary graph shows that astrocyte Gi signaling activation resulted in increased vGlut1-positive synapses onto MSNs relative to the control group (6 MSNs, mice). (C) Kymographs and ΔF/F traces of astrocyte Ca2+ responses before and during bath application of 1 μM CNO in the absence or presence of 50 μM Gabapentin in the bath. The bar graph shows the CNO-evoked astrocyte Ca2+ signals (integrated area) with and without Gabapentin in bath (n ≥ 25 cells from 4 mice). (D) Kymographs and ΔF/F traces of astrocyte Ca2+ responses. The bar graphs summarize the integrated areas of the spontaneous astrocyte Ca2+ signals in hM4Di and control mice that received either Gabapentin or saline i.p. 3 hr prior, and CNO i.p. 2 hr prior to dissection (n ≥ 22 astrocytes from 4 mice). These data show that a single in vivo dose of CNO evoked a long lasting increase in astrocyte Ca2+ signaling and in vivo Gabapentin administration did not alter the CNO response. Unpaired t-test (B). Mann-Whitney test (C). Two-way ANOVA followed by Tukey’s post-hoc test (D). Scale bar, 2 μm (A). Data are shown as mean ± s.e.m. Full details of n numbers, precise P values and statistical tests are reported in Supplementary Table 1. * indicates P < 0.05, ** indicates P < 0.01, NS indicates not significantly different.
Data Availability Statement
All of the raw and normalized RNA-Seq data have been deposited in the Gene Expression Omnibus repository (http://www.ncbi.nlm.nih.gov/geo) with accession number GSE119058. All of the FPKM RNA-Seq values are also provided in Supplementary Excel file 2. All of the raw data used to generate the graphs shown in the figures are available at www.data.mendeley.com (ID jprg767fk6.1).