

Abstract
A regioselective, nickel-catalyzed photoredox allylation of secondary, benzyl, and α-alkoxy radical precursors is disclosed. Through this manifold, a variety of linear allylic alcohols and allylated monosaccharides are accessible in high yields under mild reaction conditions. Quantum mechanical calculations [DFT and DLPNO-CCSD(T)] support the mechanistic hypothesis of a Ni(0) to Ni(II) oxidative addition pathway followed by radical addition and inner-sphere allylation.
Keywords: Photoredox; Nickel Catalysis; 1,4-Dihydropyridines; Allylation; Tsuji-Trost
Graphical Abstract
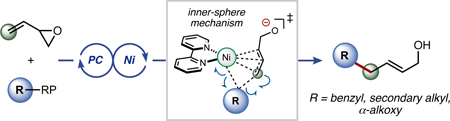
Shining New Light. Herein, we report a highly regioselective, intermolecular, nickel-catalyzed photoredox allylic substitution that expands both the radical and electrophile scope of dual photoredox/Ni-catalyzed reactions. Quantum mechanical calculations shed light on the disclosed mechanistic pathway, supporting a Ni(0) to Ni(II) oxidative addition followed by an inner-sphere radical addition.
Over the course of the past 30 years, the Tsuji-Trost allylation has been a workhorse reaction within the field of synthetic organic chemistry.1 A particularly striking advantage of the Tsuji-Trost reaction is the ability to forge C(sp3)–C(sp3) bonds under mild reaction conditions, typically using palladium as the transition metal in conjunction with an appropriate phosphine ligand.2 Although the breadth of electrophilic allyl partners has been quite thoroughly explored, nucleophilic partners have primarily been limited to two-electron carbon-or nitrogen-centered moieties (Scheme 1). 3 Specifically, for C–C bond formation, “soft” nucleophiles (e.g., malonates) are frequently employed, wherein addition occurs by an outer-sphere mechanism.1
Scheme 1.
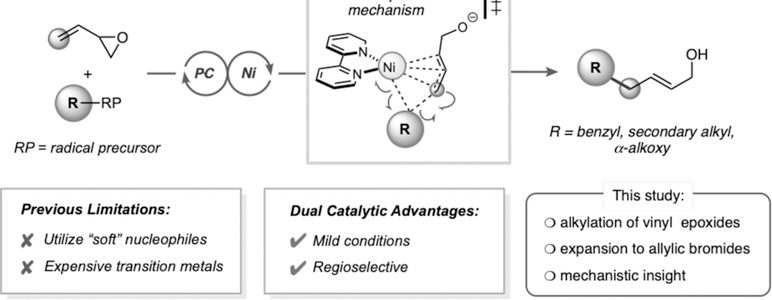
Exploring radical-based Tsuji-Trost reactivity under photoredox conditions.
The choice of nucleophile in these transformations is primarily limited by pKa constraints, because anions derived from pronucleophiles with pKa >25 are not appropriately tuned to react with the incipient π-allyl transition metal intermediate.4 Recent reports by Walsh and coworkers have pointed to the utility of diarylmethane nucleophiles (pKa up to 32) in Pd-catalyzed allylic substitution reactions, broadening the nucleophile window of Tsuji-Trost reactions.5 Alternatively, Zn,6 Sn, 7 B, 8 Mg, 9 Si, 10 and Li-based 11 enolates have been used successfully as nucleophilic partners in related transformations.2b, 12 However, a major limitation of enolate-based methods is the prerequisite for highly basic conditions. To overcome these limitations, pioneering work by Tunge shifted efforts toward radical-based intramolecular allylations via a palladium/photoredox cross-coupling, facilitating the decarboxylative allylation of α-amino carboxylic acids. Unfortunately, when Tunge and coworkers shifted to intermolecular allylation chemistry, only unsubstituted allyl groups were successfully coupled with alkyl carboxylic acid partners.13
Given the excellent precedent for merging photoredox and nickel catalysis in alkylation reactions established by our laboratory and others,14 we sought to expand this paradigm to radical-based Tsuji-Trost reactions (Scheme 1). In particular, numerous radical precursors (e.g., alkyltrifluoroborates, carboxylates, alkylsilicates, and 1,4-dihydropyridines) derived from diverse commodity chemicals have been developed.15 Although such reagents have been extensively utilized in conjunction with photoredox/Ni dual cross-coupling with aryl-16 and alkenyl 17 electrophiles, we have also begun to expand the scope of electrophilic partners for such transformations (e.g., using isocyanate, 18 acyl chloride, 19 carboxylic acid, 20 and acyl imide electrophiles21). In a continuation of that effort, the cost-effective nature of Ni-based catalysts and the slow α-hydride elimination of alkylnickel species relative to that of palladium was considered to investigate how this approach could be applied to the Tsuji-Trost reaction.22
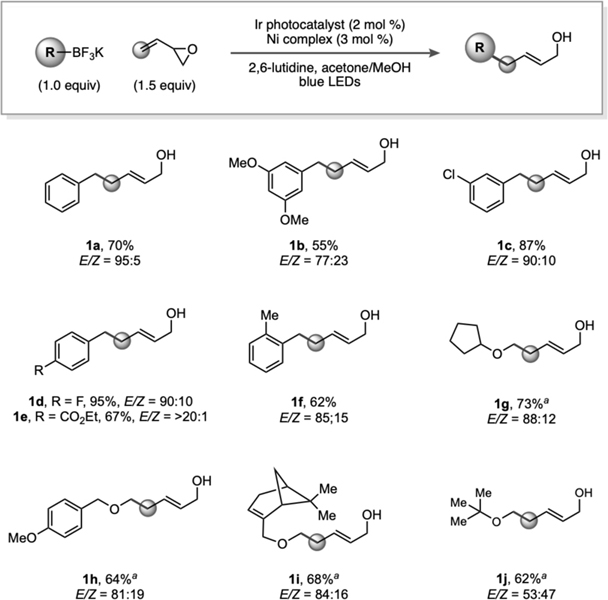
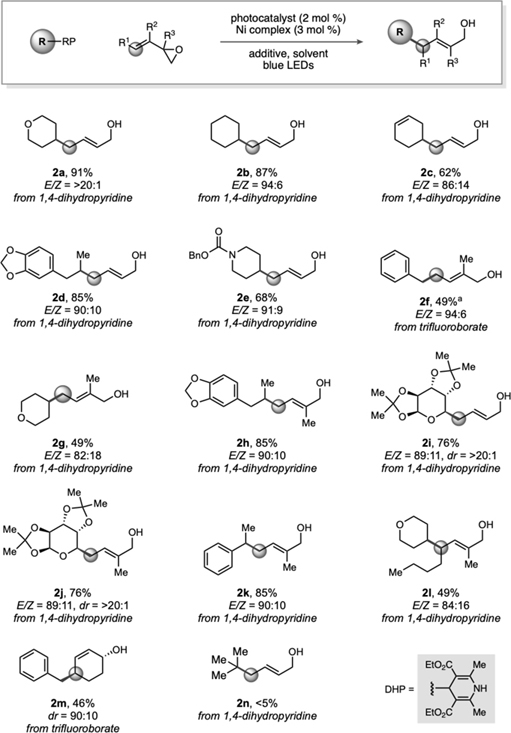
Vinyl epoxides have been employed previously in palladium-catalyzed allylations,23 but to the best of our knowledge, reports using a nickel-based catalyst — a non-precious, inexpensive catalyst — are scarce,24 with only a few examples being reported involving cycloaddition reactions. At the outset of substrate exploration, a variety of benzyltrifluoroborates were selected; early results demonstrated a diversity of electron-withdrawing and electron–donating groups were accommodated (Table 1). Additionally, modestly sterically hindered substrates could also be used. For example, ortho-substitution afforded 62% of desired product 1f. As the radical precursor search was expanded, α-alkoxytrifluoroborates were next targeted. Generally, E/Z selectivity was high, although the ratio was severely diminished to 53:47 when tert-butyl ether moieties were explored (1j).
Table 1.
Reactions were conducted on 0.50 mmol scale. Standard reaction conditions employed butadiene monoxide (1.0 equiv), alkyltrifluoroborate (1.5 equiv), Ir[dFCF3ppy]2(bpy)PF6 (2 mol %), [Ni(dtbbpy)(H2O)4]Cl2 (3 mol %), and 2,6-lutidine (3.5 equiv) in acetone/MeOH (9:1). aFor α-alkoxyalkyltrifluoroborates, a solvent mixture of acetonitrile/tert-butanol (10:1) was used.
 |
Moving forward, the breadth of carbon-centered radicals was expanded by targeting unstabilized secondary alkyl radical precursors. Under the standard and modified reaction conditions, secondary alkyltrifluoroborates formed complex mixtures (e.g., homocoupling, aldehydic side products).25 Therefore, DHPs (1,4-dihydropyridines) were investigated as alternative radical precursors 26–27 with 4CzIPN [2,4,5,6-tetra(9H-carbazol-9-yl)isophthalonitrile] organophotocatalyst.
During the exploration of various DHPs, heterocyclic (2a and 2e) and alkenyl moieties (2c) were successfully incorporated within the coupling partners under the optimized reaction conditions. A desire to integrate greater functional density within the radical precursor led to the exploration of monosaccharide moieties that were recently highlighted in publications from our group. 28 Pleasingly, pyranose DHP afforded the allylated products 2i-j in high E/Z ratios and diastereoselectivity.
Subsequently, substitution on the vinyl epoxide moiety was probed, starting with methyl substitution on the requisite epoxide (2f–2h). Notably, the stereochemical outcome of the photoredox/Ni dual catalyzed alkylation is complementary to that of the Pd-catalyzed transformations. The latter provide net retention via double inversion,26 whereas in the present case 1,2-epoxy-3,4-cyclohexene afforded primarily the trans isomer (2m), suggesting the nickel coordinates the alkene and adds to the opposite face of the epoxide, but then the resulting nickel complex engages the radical and undergoes reductive elimination by a stereoretentive, inner-sphere mechanism. This hypothesis was further supported by both DFT and DLPNO-CCSD(T) quantum mechanical calculations (vide infra).
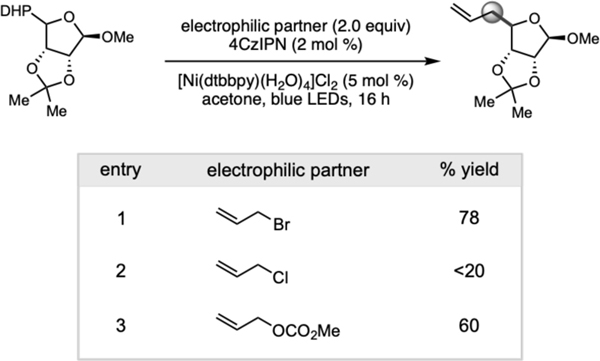
Finally, although the allylic alcohol products themselves are versatile moieties,29 we aimed to make the protocol more general by exploring electrophiles as π-allylnickel precursors. First, various leaving groups were surveyed (i.e., bromide, chloride, and carbonate), with allyl bromide affording the highest yield of product under the initial reaction conditions.
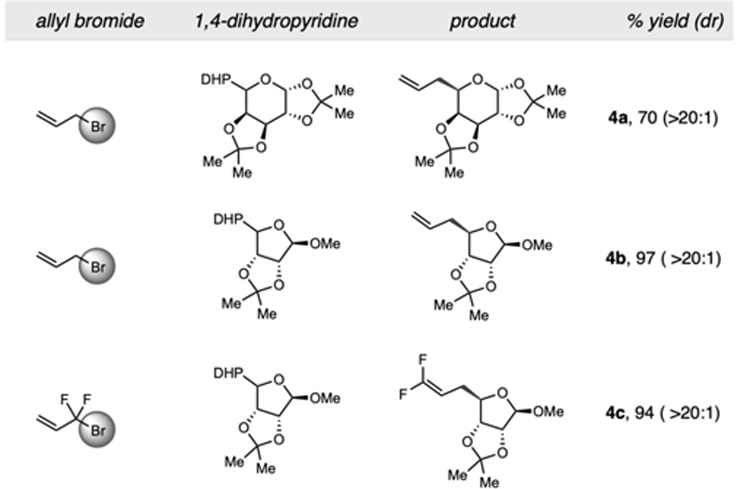
Motivated by the advantageous biological properties demon-strated by molecules containing alkyl-substituted carbohydrate substructures, 30 DHP monosaccharides were successfully allylated under the dual catalytic conditions with modest to high diastereoselectivities (Table 4). Pyranose and furanose backbones with benzyl-, methoxy-, and dioxolane protecting groups were conserved. Further studies on substituent effects were conducted using difluoroallyl bromide. Under the established reaction conditions, 4c was isolated as a single regioisomer. Presumably, oxidative addition to allyl bromide followed by bromide expulsion generates the key π-allylnickel complex which, via an inner-sphere mechanism (vide infra), intercepts the α-alkoxy radical to undergo diastereoselective reductive elimination, forming the allylated products.
Table 4.
All reactions were performed using 0.75 mmol of allyl bromide, 0.25 mmol of 1,4-dihydropyridine, 4CzIPN (2 mol %), [Ni(dtbbpy)(H2O)4]Cl2 (3 mol %) in acetone with blue LED irradiation for 24 h.
 |
Intrigued by the stereoselectivity of these processes and questions regarding the order of reaction events, quantum mechanical calculations were undertaken to gain insight into the underlying mechanism of this transformation.31 As shown in Figure 1 (squares), complexation of Ni(0) to vinyl epoxide (A’’ is energetically favored over that of complexation to benzyl radical (A’; presumably formed from the photocatalytic cycle, see Supporting Information for energetics) by 8.9 kcal/mol. Subsequently, this vinyl epoxide-Ni(0) (A’’) complex can quickly undergo an SN2-like ring-opening (via A’’-TS; barrier is only 3.1 kcal/mol from A’’) to form π-allylnickel(II) intermediate B’’. As anticipated by previous calculations,32 the nickel(II) intermediate B” can quickly engage the benzyl radical, generated by SET oxidation of BnBF3K by the photocatalyst (see Supporting Information for energetics), to form Ni(III) intermediate C (via B’’-C-TS). Overall, radical addition is facile (the barrier is ca. 1–3 kcal/mol, dependent on the method). Finally, inner-sphere C(sp2)-C(sp3) bond formation via C-TSL (the barrier is 8.9 kcal/mol from C) leads to formation of the linear product (PL), 50.3 kcal/mol downhill in energy.
Figure 1.

Competing Ni(0)/Ni(I)/Ni(III) (circles), Ni(0)/Ni(II)/Ni(III) (squares), Ni(0)/Ni(II) (squares-triangles), pathways leading to C(sp3)-C(sp3) bond formation.
A series of reductive elimination transition states leading to formation of branched product (e.g., C-TSB) were also located, but these were much higher in energy (>5 kcal/mol) than C-TSL, presumably because of unfavorable electronic repulsion and steric interactions between the charged oxygen and the benzyl group. Moreover, C(sp2)-C(sp3) bond formation via an outer sphere pathway (squares-triangles) was also found higher in energy. Formation of Ni(III) complex C from a Ni(0)-Ni(I)-Ni(III) pathway (Figure 1, circle) is also overall higher in energy than the Ni(0)-Ni(II)-Ni(III) pathway (squares). Albeit low, the barrier for radical addition (A-TS) from Ni(0) is still 7.3 kcal/mol higher in energy than oxidative addition to vinyl epoxide (via A’’-TS). However, it is likely that formation of C is highly dependent on the local concentration of benzyl radical and vinyl epoxide. Overall, DFT and DLPNO-CCSD(T) calculations support a Ni(0)-Ni(II)-Ni(III) reaction pathway and an inner sphere reductive elimination as the product-determining step, leading to formation of the linear product as the major product.33 This model is in accord with the stereochemical results observed with cyclohexadiene monoxide (Table 2, 2m). Further, although computational studies favored the linear product, the branched pathway is only 5 kcal/mol higher in energy, suggesting branched products may be observed in some cases (likely dependent on the nature of the substrate, ligand, etc.). To test this hypothesis, we subjected sterically hindered aryl-substituted vinyl epoxides to the standard reaction conditions and indeed found a mixture of linear and branched products, albeit in low yields (Scheme 2).
Table 2.
Reactions were conducted on 0.50 mmol scale. Standard reaction conditions employed butadiene monoxide (1.0 equiv), 1,4-dihydropyridine (1.5 equiv), 4CzIPN (2 mol %), and [Ni(dtbbpy)(H2O)4]Cl2 (3 mol %) in acetone. For trifluoroborates, standard reaction conditions employed butadiene monoxide (1.0 equiv), alkyltrifluoroborate (1.5 equiv), Ir[dFCF3ppy]2(bpy)PF6 (2 mol %), [Ni(dtbbpy)(H2O)4]Cl2 (3 mol %), and 2,6-lutidine (3.5 equiv) in acetone/MeOH (9:1). For α-alkoxyalkyltrifluoroborates, a solvent mixture of acetonitrile/tert-butanol (10:1) was used.
 |
Scheme 2.
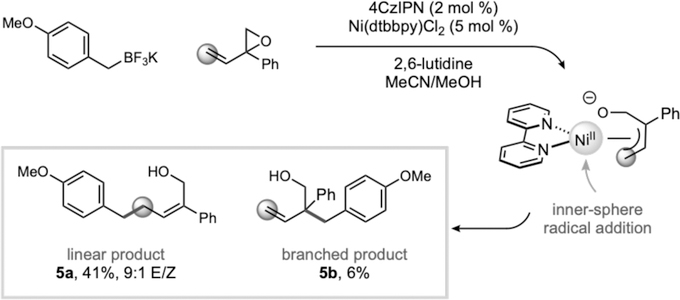
Observation of linear and branched mixtures.
In summary, a dual catalytic allylation reaction is reported that demonstrates complementarity to traditional Tsuji-Trost reactions. In juxtaposition with classical reactivity in the Pd-catalyzed transformations (stabilized anionic carbon nucleophiles), carbon-centered radicals were employed, enabling the allylation of secondary, benzyl, and α-alkoxy radical precursors. Additionally, net inversion of stereochemistry is observed in the photoredox/Ni dual catalyzed systems, as opposed to the net retention via double inversion observed in the traditional Pd-catalyzed processes.
To gain insight into the mechanistic pathway, studies incorporating quantum mechanical calculations [using dispersion corrected (U)DFT and DLPNO-CCSD(T) methods] were conducted to determine the energetic favorability of the oxidative addition step. Here, a Ni(0) to Ni(II) oxidative addition step was calculated to occur before radical addition to the nickel center; this is a deviation from previous mechanistic studies of photoredox/Ni dual cross-coupling pathways where Ni(0) to Ni(I) has been involved in the putative pathway.32 Given the mild reaction conditions and availability of numerous radical precursors, the disclosed method provides a useful complement to the traditional, palladium-catalyzed Tsuji-Trost reaction.
Supplementary Material
Table 3.
Expanding the allylic handle.
 |
Acknowledgements
The authors are grateful for the financial support provided by NIGMS (R01 GM 113878 to G.M.) and NSF (CAREER, 1751568 to O.G.). J.K.M. is supported by the Bristol-Myers Squibb Graduate Fellowship for Synthetic Organic Chemistry. O.G. is grateful to the University of Maryland College Park for start-up funds and computational resources from UMD Deepthought2 and MARCC/BlueCrab HPC clusters and XSEDE (CHE160082 and CHE160053). We thank Dr. Charles W. Ross, III (University of Pennsylvania) for assistance in obtaining HRMS data. Additionally, Dr. Jun Gu (University of Pennsylvania) and Matt Bunner (University of Pennsylvania) assisted in the acquisition and analysis of NMR data. The alkyltrifluoroborates and the iridium (for photocatalyst synthesis) were generously donated by Frontier and Johnson-Matthey, respectively.
Abbreviations
- DHP
1,4-dihydropyridine
- SET
Single Electron Transfer
- 4CzIPN
2,4,5,6-tetra(9H-carbazol-9-yl)isophthalonitrile
Footnotes
Conflict of interest
The author declare no conflict of interest.
Contributor Information
Jennifer K. Matsui, Department of Chemistry, University of Pennsylvania, Roy and Diana Vagelos Laboratories, Philadelphia, Pennsylvania 19104-6323. United States of America
Álvaro Gutiérrez-Bonet, Department of Chemistry, University of Pennsylvania, Roy and Diana Vagelos Laboratories, Philadelphia, Pennsylvania 19104-6323. United States of America.
Madeline Rotella, Department of Chemistry and Biochemistry, University of Maryland, College Park, Maryland, 20742, United States of America.
Rauful Alam, Department of Chemistry, University of Pennsylvania, Roy and Diana Vagelos Laboratories, Philadelphia, Pennsylvania 19104-6323. United States of America.
Osvaldo Gutierrez, Department of Chemistry and Biochemistry, University of Maryland, College Park, Maryland, 20742, United States of America.
Gary A. Molander, Department of Chemistry, University of Pennsylvania, Roy and Diana Vagelos Laboratories, Philadelphia, Pennsylvania 19104-6323. United States of America
References
- 1.Trost BM, Acc. Chem. Res 1980, 13, 385–393. [Google Scholar]
- 2.(a) Tsuji J, Organic Synthesis with Palladium Compounds, Springer: New York, 1980. [Google Scholar]; (b) Trost BM, Van Vranken DL, 1996, 96, 395–422. [DOI] [PubMed] [Google Scholar]
- 3.(a) Consiglio G, Waymouth RM, Chem. Rev 1989, 89, 257–276. [Google Scholar]; (b) Heumann A, Reglier M, Tetradedron 1995, 51, 975–1015. [Google Scholar]
- 4.Trost BM, Machacek MR, Aponick A, Acc. Chem. Res 2006, 39, 747. [DOI] [PubMed] [Google Scholar]
- 5.Sha S-C, Zhang J, Carroll PJ, Walsh PJ, J. Am. Chem. Soc 2013, 135, 17602–17609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Son S, Fu GC, J. Am. Chem. Soc 2008, 130, 2756–2757. [DOI] [PubMed] [Google Scholar]
- 7.(a) Trost BM, Self CR, J. Org. Chem 1984, 49, 468–473. [Google Scholar]; (b) Trost BM, Keinan E, Tetrahedron Lett 1980, 21, 2591–2594. [Google Scholar]
- 8.Negishi E, Matsushita H, Chatterjee S, John RA, J. Org. Chem 1982, 47, 3188–3190. [Google Scholar]
- 9.Braun M, Meier T, Angew. Chem. Int. Ed 2006, 45, 6952–6955. [DOI] [PubMed] [Google Scholar]
- 10.Graening T, Hartwig JF, J. Am. Chem. Soc 2005, 127, 17192–17193. [DOI] [PubMed] [Google Scholar]
- 11.Trost BM, Schroeder GM, J. Am. Chem. Soc 1999, 121, 6759–6760. [Google Scholar]
- 12.Braun M, Meier T, Angew. Chem. Int. Ed 2006, 45, 6952–6955. [DOI] [PubMed] [Google Scholar]
- 13.Lang SB, O’Nele KM, Tunge JA, J. Am. Chem. Soc 2014, 136, 13606–13609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Tellis JC, Primer DN, Molander GA, Science 2014, 345, 433–436. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zuo Z, Ahneman DT, Chu L, Terett JA, Doyle AG, MacMillan DWC, Science 2014, 345, 437–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Corce V, Chamoreau L–M, Derat E, Goddard J–P, Ollivier C, Fensterbank L, Angew. Chem. Int. Ed 2015, 54, 11414–11418. [DOI] [PubMed] [Google Scholar]; (b) Jouffroy M, Primer DN, Molander GA, J. Am. Chem. Soc 2016, 138, 475–478. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Nakajima K, Nojima S, Nishibayashi Y, Angew. Chem. Int. Ed 2016, 55, 14106–14316. [DOI] [PubMed] [Google Scholar]; (d) Gutierrez-Bonet A, Tellis JC, Matsui JK, Vara BA, Molander GA, ACS Catal 2016, 6, 8004–8008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Photoredox catalysis reviews: Tellis JC, Kelly CB, Primer DN, Jouffroy M, Patel NR, Molander GA, Acc. Chem. Res 2016, 49, 1429–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Matsui JK, Lang SB, Heitz DR, Molander GA, ACS Catal 2017, 7, 2563–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Prier CK, Rankic DA, MacMillan DWC, Chem. Rev 2013, 113, 5322–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Hopkinson MN, Sahoo B, Li J–L, Glorius F, Chem. Eur. J 2014, 20, 3874–3886. [DOI] [PubMed] [Google Scholar]
- 17.Patel NR, Kelly CB, Jouffroy M, Molander GA, Org. Lett 2016, 18, 764–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zheng S, Primer DN, Molander GA, ACS Catal 2017, 7, 7957–7961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.(a) Amani J, Sodagar E, Molander GA, Org. Lett 2016, 18, 732–735. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Amani J, Molander GA, Org. Lett 2017, 82, 1856–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Amani J, Molander GA, Org. Lett 2017, 19, 3612–3615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Amani J, Alam R, Badir S, Molander GA, Org. Lett 2017, 19, 2426–2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tasker SZ, Standley EA, Jamison TF, Nature 2014, 509, 299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Trost BM, Molander GA, J. Am. Chem. Soc 1981, 103, 5969–5972. For an extensive review article, see: He J., Ling J., Chiu P., Chem. Rev. 2014, 114, 8037–8128. [Google Scholar]
- 24.Crotti S, Bertolini F, Macchia F, Pineschi M, Org. Lett 2009, 11, 3762 During the course of our studies, Gong and coworkers published a method describing a reductive allylation of tertiary alkyl halides using allylic carbonates: Chen H., Jia X., Yu Y., Qian Q., Gong H., Angew. Chem. Int. Ed. 2017, 56, 13103–13106. Their report includes deuterium studies supporting the formation of a π-allylnickel intermediate. [DOI] [PubMed] [Google Scholar]
- 25.Secondary alkyltrifluoroborates have significantly high reduction potentials at +1.50 V vs SCE (see Primer DN, Karakaya K, Tellis JC, Molander GA, J. Am. Chem. Soc 2015, 137, 2195–2198). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.(a) Tewari N, Dwivedi N, Tripathi R, Tetrahedron Lett 2004, 45, 9011–9014. [Google Scholar]; (b) Adibi H, Samimi HA, Beygzadeh M, Catal. Commun 2007, 8, 2119–2124. [Google Scholar]; (c) Heydari A, Khaksar S, Tajbakhsh M, Bijanzadeh HR, J. Fluorine Chem 2009, 130, 609–614. [Google Scholar]; (d) Zhang D, Wu L-Z, Zhou L, Han X, Yang Q-Z, Zhang L-P, Tung C-H, J. Am. Chem. Soc 2004, 126, 3440–3441. [DOI] [PubMed] [Google Scholar]; (e) Wei X, Wang L, Jia W, Du S, Wu L, Liu Q, Chin. J. Chem 2014, 32, 1245–1250. [Google Scholar]; (f) Fukuzumi S, Suenobu T, Patz M, Hirasaka T, Itoh S, Fujitsuka M, Ito O, J. Am. Chem. Soc 1998, 120, 8060–8068. [Google Scholar]; (g) Wang D, Liu Q, Chen B, Zhang L, Tung C, Wu L, Chin. Sci. Bull 2010, 55, 2855–2858. [Google Scholar]
- 27.(a) Nakajima K, Nojima S, Sakata K, Nishibayashi Y, ChemCatChem 2016, 8, 1028–1032. [Google Scholar]; (b) Chen W, Liu Z, Tian J, Li J, Ma J, Cheng X, Li G, J. Am. Chem. Soc 2016, 138, 12312–12315. [DOI] [PubMed] [Google Scholar]; (c) Buzzetti L, Prieto A, Roy SR, Melchiorre Angew P. Chem. Int. Ed 2017, 56, 15039–15043. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Verrier C, Alandini N, Pezzetta C, Moliterno M, Buzzetti L, Hepburn HB, Vega-Penaloza A, Silvi M, Melchiorre P, ACS Catal 2018, 8, 1062–1066. For a recent review, see: Huang W., Cheng X., Synlett 2017, 28, 148–158. [Google Scholar]
- 28.(a) Badir SO, Dumoulin A, Matsui JK, Molander GA, Angew. Chem. Int. Ed 2018, 57, 6610–6613. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dumoulin A, Matsui JK, Gutierrez-Bonet A, Molander GA, Angew. Chem. Int. Ed 2018, 57, 6614–6618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Butt NA, Zhang W, Chem. Soc. Rev 2015, 44, 7929–7967. [DOI] [PubMed] [Google Scholar]
- 30.Khalil ZG, Salim AA, Vuong D, Crombie A, Lacey E, Blumental A, Capon RJ, J. Antibiot 2017, 70, 1097–1103. [DOI] [PubMed] [Google Scholar]; (b) Bayliss M; Donaldson MI, Nepogodiev SA, Pergolizzi G, Scott AE, Harmer NJ, Field RA, Prior JL, Carbohydr. Res 2017, 452, 17–24. Synthetic efforts: [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhao S, Mankad NP, Angew. Chem. Int. Ed 2018, 130, 5969–5972. [Google Scholar]; (d) Szekreyi A, Garrabou X, Parella T, Joglar J, Bujons J, Clapes P, Nat. Chem 2015, 7, 724–729. [DOI] [PubMed] [Google Scholar]
- 31.For all of the calculations, dispersion-corrected, broken-spin (U)DFT functionals (UM06/6–311+G(d,p)-SMD(water)//UB3LYP/6–31G(d) and UB3LYP-D3/6–311+G(d,p)-SMD(water)//UB3LYP/6–31G(d)) were used. Further, the energy profile using dynamic correlation, open-shell domain-based local pair natural orbital coupled-cluster calculations (DLPNO-CCSD(T)/def2-TZVPP-SMD(water)//UB3LYP/6–31G(d)) were compared, which are known to provide accurate energies (within 3 kJ mol−1) with the computational cost comparable to DFT calculations. Overall, all methods provided similar conclusions (See Supporting Information for further details). For simplicity, only UM06/6–311+G(d,p)-SMD(water)//UB3LYP/6–31G(d) energies will be discussed in the manuscript.
- 32.Gutierrez O, Tellis JC, Primer DN, Molander GA, Kozlowski MC, J. Am. Chem. Soc 2015, 137, 4 896–4899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.We also considered the protonation of B’’ followed by: (i) outer-sphere C(sp2)–C(sp3) bond formation and (ii) radical addition, with subsequent inner sphere C(sp2)–C(sp3) bond formation. However, the barriers for both protonated inner sphere and outer sphere pathways were higher in energy than the anionic pathway (see Supporting Information).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.