

ABSTRACT
Molecular mechanisms that inform heterochronic adaptations of neurogenesis in Homo sapiens remain largely unknown. Here, we uncover a signature in the cell cycle that amplifies the proliferative capacity of human neural progenitors by input from microRNA4673 encoded in Notch-1. The miRNA instructs bimodal reprogramming of the cell cycle, leading to initial synchronization of neural precursors at the G0 phase of the cell cycle followed by accelerated progression through interphase. The key event in G0 synchronization is transient inhibition by miR4673 of cyclin-dependent kinase-18, a member of an ancient family of cyclins that license M–G1 transition. In parallel, autophagic degradation of p53/p21 and transcriptional silencing of XRCC3/BRCA2 relax G1/S cell cycle checkpoint and accelerate interphase by ≈2.8-fold. The resultant reprogrammed cell cycle amplifies the proliferative capacity and delays the differentiation of human neural progenitors.
KEYWORDS: MiRNA-4673, cell cycle, neurogenesis, differentiation
Introduction
Remarkable adaptations of human neurogenesis within the primate order despite minimal genomic innovation remain hard to explain. The proteomic homology of human and chimpanzee is estimated to be >99% [1] and the corresponding genomic homology is >95% [2]. Despite such remarkable genomic and proteomic similarities, the ontogeny of human brain diverges significantly from that of other primates. One aspect of brain development that shows characteristic interspecies differences is the timespan of neurogenesis [3]. Humans benefit [4,5] from the neuroplasticity and enhanced learning capacity provided by a protracted ontogeny [3]. Due to the prolonged development, interactions with external stimuli can enrich the postnatal wiring of the human brain as opposed to the predominance of intrinsic self-organization in other primates. It is likely that the protracted neurogenesis in human is induced by heterochronic reprogramming. Heterochrony refers to the evolution of complex morphological traits propelled by changes in the temporal dimension of ontogeny [6]. In axolotl, for example, the heterochronic reprogramming of ontogeny uncouples accelerated sexual maturity from metamorphosis [7]. The resultant heterochronic phenotype, such as retention of gills, enables axolotl to occupy deeper aquatic habitats. Heterochronic reprogramming of development occurs by rewiring the signaling cascades and requires minimal genomic innovation. Given the high genomic homology in the primate lineage, it was curious to know the molecular mechanisms that efficiently reprogram the temporal dimension of human neuro-ontogeny and amplify the proliferative capacity of the brain. Unraveling such a mechanism not only would shed light on aspects of the human evolutionary journey but may also provide clues regarding pathologies such as neurodegenerative disorders that affect the human nervous system.
While heterochronic regulators of neurogenesis in human remain equivocal, regulation of developmental clock in nematodes has been extensively studied. In nematodes, the developmental clock is effectively reprogrammed by a restricted group of small temporal microRNAs [8,9]. For example, the first larval stage (L1) of Caenorhabditis elegans hermaphrodite may be repeated or entirely skipped under instruction from lin-4 and lin-14/lin-28 microRNAs, respectively [10]. Modulation of the cell cycle by the latter microRNAs is key to the heterochronic regulation of larval cell fates. To induce heterochrony, activity of lin-14 and lin-28 at larval stage 1 delays entry into G1 phase of cell cycle and inhibition by lin-4 of lin-14/lin-28 reverses this effect by shortening G1 [10]. Regulation of cell cycle by microRNAs is not limited to nematodes. In Drosophila, the heterochronic bantam gene encodes a potent microRNA that stimulates cell proliferation and prevents apoptosis and hence promotes tissue growth [11]. Notably, reprogramming of the cell cycle is known to instruct evolutionary adaptations of the central nervous system of primates [12]. In particular, a short G1 phase is proposed to drive areal specialization during corticogenesis [13]. The abridged G1 signature suggests a potential involvement of upstream temporal microRNAs in heterochronic regulation of human neurogenesis, similar to lin-4 signaling in C. elegans. This scenario is potentially attractive as limited mutational changes are required to propel the evolution of mature microRNAs that induce major heterochronic adaptations. This is because microRNAs interact with various targets (≈200–300) leading to efficient rewiring of major developmental signaling cascades [6].
Herein we dissect the molecular blueprint of a key heterochronic cascade in human neurogenesis that is accessed by microRNA4673, encoded in the intron 4 of human Notch-1 [14]. The miRNA amplifies the neurogenic potential of notch-1 by reprogramming of the cell cycle of neural progenitors. The reprogrammed cell cycle enhances proliferative capacity and delays differentiation of human neural progenitors.
Materials and methods
All chemicals were purchased from Sigma–Aldrich Inc. unless stated otherwise. All primers were purchased from IDT DNA.
Immunohistochemistry
After blocking in incubation buffer containing 0.1 M phosphate-buffered saline, 1% BSA, 0.1% Tween-20, and 5% normal goat serum (for detection with rabbit Abs) or 5% normal rabbit serum (for detection with mouse Abs) for 40 min, sections were incubated with the primary antibodies overnight at 4°C and secondary antibodies for 1 h at room temperature. Specificity controls were carried out by incubating sections with rabbit or mouse IgG negative control antibodies.
Transmission electron microscopy
For transmission electron microscopy (TEM) analysis, cells were fixed in Karnovsky’s fixative for 4 h at room temperature followed by post-fixation in OsO4 for 1 h. Preparations were dehydrated in graded alcohols and embedded in low viscosity resin (TAAB Laboratory and Microscopy, United Kingdom). Ultrathin sections were mounted on Pioloform/formvar-coated slot grids, stained in uranyl acetate and lead citrate, and examined in a Phillips CM120 BioTWIN electron microscope.
Immuno-gold labeling
Cells were fixed with 4% paraformaldehyde (PFA)/0.1% glutaraldehyde in 0.1 M PBS for 2 h at room temperature. Immuno-gold labeling was performed using the Leica IGL processor (Leica Microsystems). After washing with PBS, sections were exposed to anti-rabbit IgG antibody conjugated with colloidal gold particles of 10-nm diameter (Biocell International, Medford, MA), diluted with PBS (1/40), for 2 h at room temperature. Specificity controls were carried out by incubating sections with rabbit IgG negative control antibody.
Cell culture
Human brain pericytes, with a demonstrated neural differentiation capacity [15], were purchased from ScienCell (Carlsbad, CA). DMEM/F12 supplemented with 10% fetal calf serum, recombinant human FGF-2 20 ng/mL (R&D Systems, 233-FB) and Antibiotic-Antimycotic (100X, Life Technologies) were used for culturing the neural progenitors for analysis of mitotic cycle. Cells were passaged every 24 h at 70–80% confluence stage. Experiments were performed at passages 8–10 [15].
Induction of neural differentiation
To differentiate neural progenitors, cells were cultured in differentiation medium containing Knockout™ DMEM (Gibco), GlutaMAX™ supplement, N2 supplement (Gibco), and brain-derived neurotrophic factor (10 ng/mL, Sigma) in the absence of FGF-2.
Amplification and inhibition of endogenous miRNA activity
Naked pre-miRNA4673 and antisense 2′-O-methyl inhibitor of the miRNA rendered RNAse-resistant by terminal N,N-diethyl-4-(4-nitronaphthalen-1-ylazo)-phenylamine (ZENTM, IDTDNA) were purchased from IDTDNA®. To amplify the effect of the miRNA and its cognate antisense inhibitor, pGeneClip® vector (Promega) was utilized. Pre-miRNA was cloned into pGeneClip® vector (Promega) using terminal adapters. Extraction and purification of the plasmids were achieved using EndoFree Plasmid Kit® (QIAGEN). A second plasmid was engineered to express miRNA inhibitor based on a “tough decoys” (TuDs) strategy. TuDs hybridize to and inhibit the activity of specific microRNAs via an imperfect structure due to an internal loop.
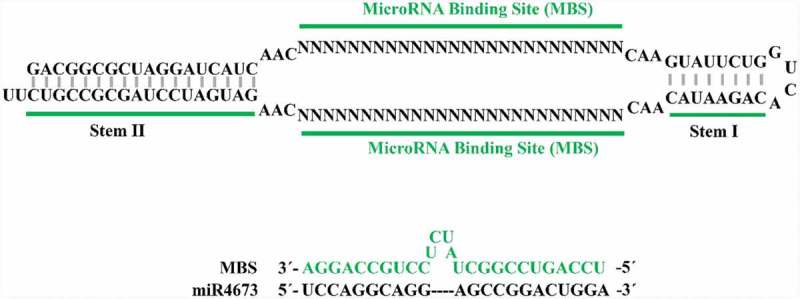
Figure legend. Structural representation of TuD inhibitor used for plasmid-driven suppression of miR4673. Two microRNA-binding sites (MBS) in TuD inhibitor bind to and inactivate the endogenous miRNA.
Electroporation
For electroporation with the miRNA, harvested cells were counted and resuspended in 400 μL of electroporation buffer (106 cells/400 μL). Electroporation buffer comprised 20 mM HEPES, 135 mM KCl, 2 mM MgCl2, 0.5% Ficoll 400, and 2 mM ATP/5 mM glutathione (pH 7.6). Naked pre-miRNA4673 was applied at a final concentration of 200 nM. Electroporation was carried out at 1700 V/cm, 700 μs, four pulses at 1-s intervals.
RNA extraction and reverse transcription
RNA was isolated using Trizol reagent (Invitrogen). After DNase treatment, reverse transcription of the extracted RNA was carried out using 1 μL of primers (250 nM final concentration), 4 μL total RNA, 1 μL dNTP Mix (10 mM each), 4 μL of 5x First-Strand Buffer, 2 μL of 0.1 M DTT, 1 μL of RNaseOUT (40 units/μL), 1 μL (200 units) of SuperScript-III reverse transcriptase, and 0.5 μL of T4 gene 32 protein (NEB).
Primer design
Gene sequence data and exon/intron boundaries were obtained from the GenBank database. In each of the primer sets (see supplementary Table 1), the common 3΄ or 5΄ primer spanned the adjacent exons to prevent amplification of genomic DNA.
Polymerase chain reaction
PCR reaction (40 cycles) comprised 4 μL of template cDNA, 250 nM forward/reverse primers (1 μL/primer), 12.5 μL of HotStarTaq Master Mix (Qiagen), and 6 μL of PCR-grade water. PCR algorithm was adopted from the manufacturer’s recommendations through 38 cycles of denaturation (94°C, 15 s), annealing (60°C, 30 s), and extension (72°C, 45 s) and a final single extension step (72°C, 7 min).
Real-time qPCR
Real-time quantitative PCR (38 cycles) was performed using SensiFAST™ SYBR® Lo-ROX reagents (BIOLINE®). Reaction mix comprised 2 μL of cDNA, 400 nM inner primers (1.5 μL/primer), 10 μL of 2x SensiFAST SYBR Lo-ROX Mix, and 5 μL of PCR-grade water on a Stratagene® Mx3000P real-time PCR instrument. Average efficiency of PCR amplification for each gene of interest was quantified based on a linear regression model using the LineRegPCR software. Relative expression of transcripts for each gene was plotted as the ratio between the average concentration of transcript in the test and control groups normalized to the average concentration of β-actin transcripts in both groups (above formula).
Stem-loop PCR for detection of endogenous miRNA
Small RNA from proliferating cells was isolated using mirVana™ miRNA isolation Kit (Ambion) according to the manufacturer’s protocol. Detection of the miRNA was accomplished using stem-loop RT-PCR. Specific primers were designed for reverse transcription and stem-loop RT-PCR amplification of the miRNA as shown in supplementary Table 2. Reverse transcription of the extracted small RNA was carried out using a mixture of 1 μL of 5 μM RT primer, 4 μL total RNA, 1 μL dNTP Mix (10 mM each), 4 μL of 5x First-Strand Buffer, 2 μL of 0.1 M DTT, 1 μL of RNaseOUT (40 units/μL), and 1 μL (200 units) of SuperScript-III reverse transcriptase. Reverse transcription was performed at 16°C for 30 min followed by 42°C for 30 min. PCR reaction (35 cycles) comprised 4 μL of template cDNA, 1 μL of 5 μM forward/reverse primers, 12.5 μL of HotStarTaq Master Mix (Qiagen), and 6 μL of PCR-grade water. PCR amplification was achieved through 40 cycles of denaturation (94°C, 15 s) and annealing (60°C, 45 s).
Cell cycle analysis by Premo™ Fucci sensor
The mihighΔpl (24-h post-electroporation) and control cells were transferred into 35 mm glass bottom dishes (MatTek®) and incubated for 12 h prior to experimentation. Transfection parameters were determined as follows:
Particles per cell: 40
Cell number per dish: ≈40,000–50,000
Volume of geminin-GFP or Cdt1-RFP used for each well = 20 μL
After adding the reagent based on the calculations above, cells were incubated overnight (16 h) and subsequently fixed and visualized using a fluorescence microscope.
Transcriptional imprint of endoplasmic reticulum stress
Unfolded protein response-mediated splicing of XBP-1 was used for transcriptional fingerprinting of the endoplasmic reticulum stress response based on the primers of supplementary Table 3.
Comet assay
Transfected cells were collected using a glass capillary micro-pipette (20 cells/group) and loaded onto pre-coated (1% agarose) microscopy-grade glass. In order to detect DNA double-stranded breaks, neutral lysis comet assay was performed. Slides were gently submerged in a dish containing neutral lysis solution (2% Sarkosyl, 0.5 M Na2EDTA, 0.5 mg/mL proteinase K, pH 8.0) and incubated at 37°C overnight. Slides were then washed three times at 30 min intervals using rinse/electrophoresis buffer (90 mM Tris buffer, 90 mM boric acid, 2 mM Na2EDTA, pH 8.5). Gel electrophoresis was conducted at 20 V and 7 mA for 25 min. Samples were then stained with 4′,6-diamidino-2-phenylindole (DAPI) and visualized under a fluorescence microscope.
Detection of telomerase activity by standard telomere repeat amplification (TRAP) assay
Lysis buffer comprised 10 mM Tris-HCl (pH: 7.5), 1 mM MgCl2, 1 mM EGTA, 0.1 mM PMSF, 5 mM β-mercaptoethanol (2-ME), 0.5% (v/v) CHAPS, 10% (v/v) glycerol, and DEPC water. After incubation of the cells with lysis buffer on ice for 30 min, lysates were centrifuged for 30 min at 40,000g and 4°C. A wax interface separated 0.1 μg of CX-primers (bottom phase, see supplementary Table 4) from the top phase comprising 20 mM Tris-HCl, pH 8.3, 1.5 mM MgCl2, 63 mM KCl, 0.005% (v/v) Tween-20, 1 mM EGTA, 50 μM dNTPs (Roche), 0.1 μg of TS-primer, 1 μg of T4g32 protein (NEB), 5 μg of BSA, 2 U of Taq DNA polymerase (Qiagen), DEPC-treated Milli-Q water, and cell lysates (0.5 μg). After incubation at room temperature for 30 min, the reaction was heated at 90° for 90 s and then subjected to 30 cycles of 94°C for 30 s, 50°C for 30 s, and 72°C for 45 s. The reaction products were then run on a 1.5% agarose gel at 5 V/cm, stained in SYBR Gold for 45 min, and de-stained in 1 × TAE buffer for 30 min before imaging.
UVC irradiation
For Ultraviolet-C (UVC) irradiation, transfected and control cells were trypsinized, collected, re-suspended in PBS, and transferred into an agar plate. A 30-W UVC generator at a distance of 40 cm from the agar plate was used as a source of ionizing radiation. The cells were exposed to six pulses of UVC (pulse duration: 10 s, intervals: 10 s). The cells were then incubated overnight and visualized after 24 and 48 h of incubation.
Live-imaging analyses and mathematical modeling
For live imaging analysis, cells were cultured in six-well plates overnight and transferred into the live-imaging platform (Leica DMI6000B live cell imaging microscope). Phase-contrast images were captured every 4 min for 36 h from multiple wells (10x magnification). To analyze mitotic activity, images were imported into FIJI (ImageJ) platform and cell divisions (mi: mitosis) were binned in 40-min periods (ti: periods) with M-phase defined as cell rounding until the end of cytokinesis. The cumulative division graphs (continuous lines of Figure 2b) for the data points (ti, mi) were generated as follows:
Figure 2.
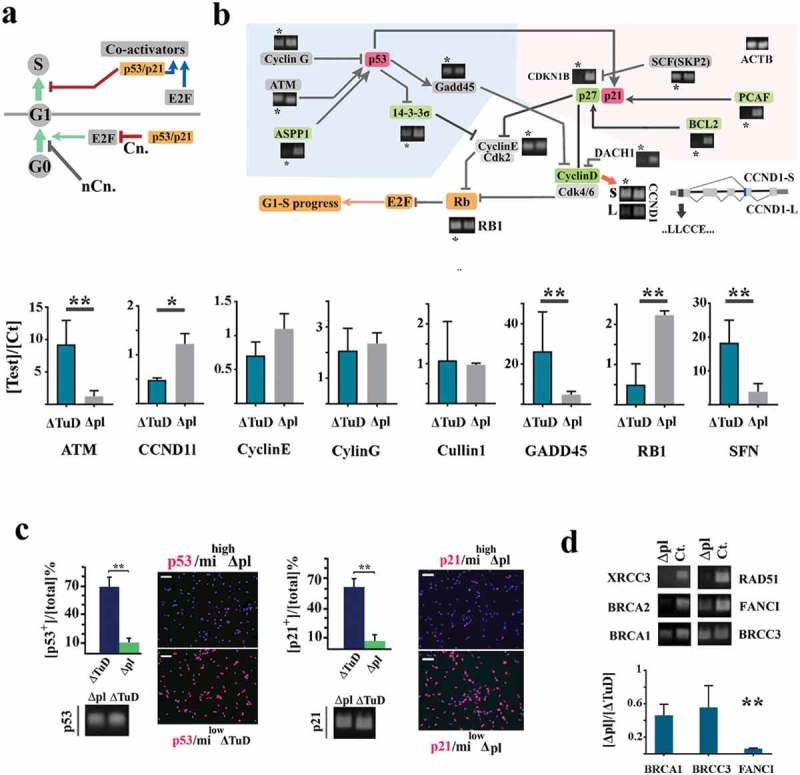
Amplification of miR4673 reprogrammed cell cycle by access to G1–S checkpoint. (a) In addition to canonical (Cn.) regulation of G0–G1 transition, p53/p21 signaling controls G1–S transition. Transient arrest at G0 may occur by alternative non-canonical (nCn.) pathways. (b) Transcriptional fingerprinting revealed transcriptional silencing of several genes (* indicates mihighΔpl cells and control cell are unlabeled) involved in regulation of cyclin-E and cyclin-D during early G1 (a representative sample of three biological replicates) (see supplementary Figure 8 for additional information). Bar graphs demonstrate real-time PCR analysis of targets that are not completely suppressed (**p < 0.01, *p < 0.05). (c) Amplification of miR4673 triggers degradation of p53 and p21 proteins despite active transcription (scale bar = 25 μm) for both mihighΔpl and ΔTuD treatments (n = 15 random fields from five samples/group; error bars: SD; ** p < 0.01). (d) The key components of M–G1 checkpoint machinery were transcriptionally downregulated in mihighΔpl cells. Bar graphs demonstrate real-time PCR analysis of targets that are not completely suppressed (values in mihighΔpl cells are normalized to corresponding values in milowΔTuD cells; **p < 0.01, *p < 0.05).
The cumulative linear mitotic rate (Lmr) defined the slope (α) of the least-squares regression line, where m and t correspond to the data points (ti, mi). The second parameter measures deviation (dmi) of the observed mitotic events (ti, mi) from those predicted by the linear regression model
Data were imported into MATLAB to build the regression model and calculate mitotic residuals using the curve fitting app of MATLAB.
Gyrification index
Histological sections (horizontal, sagittal, and coronal) of the studied species were obtained from Brain Biodiversity Bank of Michigan State University (https://msu.edu/~brains/index.html) and BrainMaps (http://brainmaps.org). Gyrification index (GI) in each plane was calculated according to the following equations:
SAgyr is a 2D representation of cerebral and cerebellar cortical surface area calculated as the average length of two lines drawn on external (green line) and internal (orange line) boundaries of the cortex. The line was drawn using a digital pen in ImageJ, and measurements were also done in the same platform. SAhull is the hull surface.
Neural organoids (generation and electroporation)
Neural organoids were generated using the hanging drop method. Briefly, the cultured neural progenitors were gently trypsinized, collected, and counted. The collected cells were resuspended in DMEM/F12 supplemented with 10% fetal calf serum, recombinant human FGF-2 (20 ng/mL), and Antibiotic-Antimycotic. A minimum of ≈104 cells were resuspended in 35 μL of the growth medium and loaded onto the cover of an agar plate. After incubation for 24 h, the aggregated organoids were gently transferred into a new agar plate and cultured in suspension for one more day. The organoids were then embedded in Matrigel and incubated in the growth medium for an additional 12 h. To electroporate the organoids, naked miR4673 or the cognate naked inhibitor was diluted in the electroporation buffer (final concentration 200 nM). The growth medium was replaced with the electroporation buffer, and Tweezertrodes (BTX, Harvard Apparatus) were used to electroporate the organoids (50 V, 25 ms, five pulses, interval: 1 s). Neural induction was initiated by substitution of the growth medium with neural induction medium and culturing for an additional 3 d in suspension. The organoids were then fixed, cryoprotected, and histologically processed for immunohistochemical staining.
Mathematical modeling of corticogenesis
To simulate the morphogenic signature of miR4673, we adopted the intermediate progenitor model of corticogenesis [16]. In the latter model, proliferation dynamics of the subventricular zone (SVZ) intermediate progenitor cells (IPC) will be mirrored in spatial micro- and macro-organization of the cortical plate. The cellular kinetics of this zone can be accurately described based on the universal morphogens of the Gray–Scott model of reaction-diffusion (R-D) [17]. It is noteworthy that the concept of morphogen in the R-D model is more universal than a simple graded morphogen in the biological sense such as secreted growth factors. The unit of the universal morphogen in the current R-D model is a neural progenitor cell that creates morphologies by the occupation of space. As such, our applied concept of a universal morphogen closely resembles that of a Turing morphogen [18].
In the model, universal morphogen U is added to the reaction at a fixed rate f. Biologically, symmetric division of IPCs in the SVZs determines the feed rate or f. Subsequently, morphogen U is converted into B. From a biological perspective, differentiation of IPCs into neuro-glial elements corresponds to such a conversion. Eventually, V is depleted at a fixed kill rate (k). In our model, migration to the cortical plate of differentiated cells provides the biological correlate of the abstract parameter k. Therefore, our model simulated cellular kinetics that occur in a narrow transitional slice between SVZ and cortical plate based on the following equations:
Simulation of the Gray–Scott system was achieved using the open-source software READY. To simulate the R-D reaction on a spherical surface, we utilized a method originally developed by Turk [19]. A spherical mesh (polygonal mesh, 32,256 cells, and 16,130 nodes) was generated using Paraview platform and imported into READY. The simulation was initialized with U = 1 and V = 0, and a small area was fed with V = 1 to begin the R-D reaction. To simulate Macaque brain morphogenesis (in the absence of bimodal regulation of cell cycle), we applied the values f = 0.035, k = 0.057, Du = 0.05, and Dv = 0.041. These values resulted in the most accurate simulation of the Macaque brain. To alter the R-D parameters based on bimodal regulation of cell cycle, we utilized findings from the collective locomotory landscape of neural progenitors after amplification (mihighΔmi) and following inhibition of the endogenous miRNA4673 (milowΔAs). The feed (f) and the diffusion rates of U (Du) were scaled up by ≈ 74% (the difference in locomotion mihighΔmi and milowΔAs cells). As such, the human brain, based on bimodal alteration of the cell cycle, was modeled with the values f = 0.06, k = 0.061, Du = 0.087, and Dv = 0.041. In the simulated pattern, areas with a high concentration of U correspond to domains of active proliferation that generate tangential spatial expansion of the cortex (gyri). Areas with a high concentration of V, on the other hand, represent cellular differentiation zones that are non-proliferative and hence generate the brain sulci. The resultant spherical map of morphogenesis was subsequently imported into Paraview to generate a 3D interpretation based on the logic explained above. In Paraview, cell attributes were transformed into point attributes by applying CellDatatoPointData filter. Subsequently, surface normals were generated and warped by applying the warpbyScale filter of Paraview (scale factor = −10).
Quantification and statistical analysis
SPSS statistical software (SPSS v.16, Chicago, IL, USA) was used for the statistical analysis of data. The relative expression levels of genes of interest were compared using univariate ANOVA and non-parametric Mann–Whitney U test. In the present study, a p-value of <0.01 was considered statistically significant.
Results
MiRNA4673 instructs bimodal reprogramming of cell cycle
We used human neural progenitors [15] with endogenous (miend ct.) miR4673 activity (Figure 1a) to investigate the dynamics of miRNA signaling. Amplification of the endogenous miRNA was achieved by application of the pre-miRNA cloned into pGeneClip-U1 vector (mihighΔpl, 100 nM/2 × 106 cells) or the naked pre-miRNA (mihighΔmi, 200 nM/2 × 106 cells). The mihighΔmi cells mimic transient physiological outbursts of the endogenous miRNA. The mihighΔpl cells provided additional mechanistic insight into the miRNA activity by sustained expression of supra-physiological levels of the miRNA (supplementary Figure 1). Inhibition of the endogenous miRNA was achieved by utilizing 2′-O-methyl antisense RNA inhibitors (milowΔAs, 200 nM/2 × 106 cells) rendered RNAse-resistant by terminal N,N-diethyl-4-(4-nitronaphthalen-1-ylazo)-phenylamine (ZENTM, IDTDNA) modification or the tough decoy strategy [20] in pGeneClip-U1 plasmid (milowΔTuD, 100 nM/2 × 106 cells). The logic for inhibition of the endogenous miRNA was similar to that explained for mihighΔmi and mihighΔpl.
Figure 1.
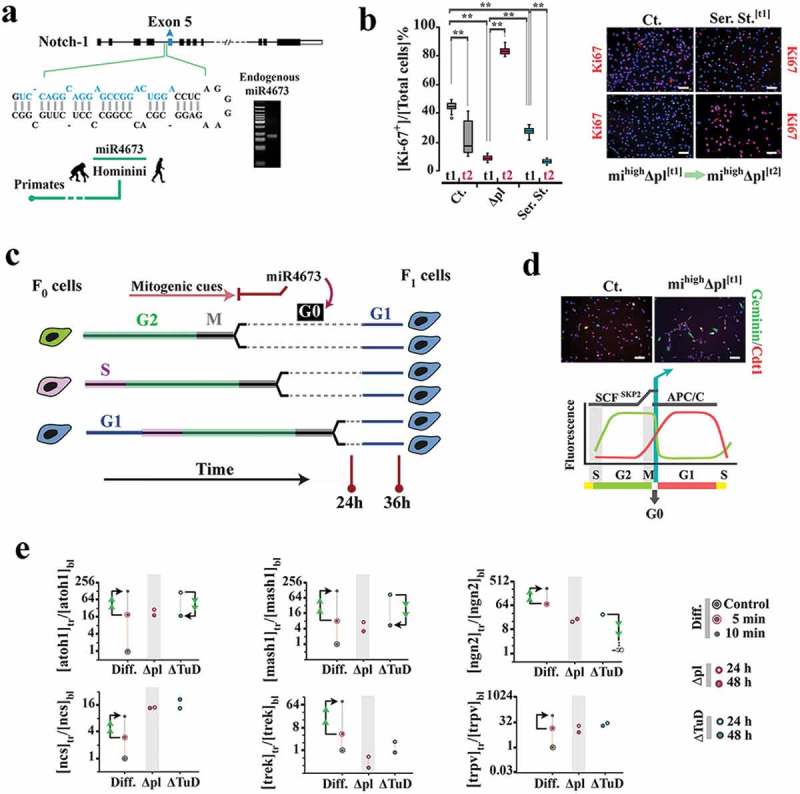
Amplification of miR4673 reprograms G0–G1 transition.(a) MiR4673 is expressed endogenously in cycling human neural progenitors. (b) Ki-67 profile of untreated control (ct.), serum-starved (ser. st.) control, and mihighΔpl populations after 24 h (t1) and 36 h (t2) (blue: nuclear DAPI) (n = 20 random fields from five samples/group; error bars: SD; ** p < 0.01) (scale bars = top left: 30 μm, top right: 50 μm, bottom left: 50 μm, bottom right: 30 μm). (c) The amplification of miR4673 transiently arrests the cycling mihighΔpl cells at G0 followed by synchronized entry into G1. (d) The mihighΔpl cells dwell longer in G0 where Geminin (green probe) is degraded by Anaphase-promoting complex (APC/C) and Cdt-1 (red probe) is degraded by SCF (Skp2) E3 ligase (hence the colorless phase as shown in the bottom schematic diagram) (scale bars = 30 μm). (e) To measure the differentiation propensity of synchronized cells, the transcriptional profile of control cells cultured in neural differentiation (Diff.) medium was compared to that instructed by the amplification (Δpl) and inhibition (ΔTuD) of miR4673 in the growth medium (tr: post-transfection, bl: baseline control, arrowheads indicate temporal directionality of change in gene expression). All values are normalized to β-actin and referenced to the control cells cultured in growth medium. Subsequent to induction of neurogenesis, marked upregulation of pro-neural transcription factors atoh1, mash1, and neurogenin-2 was noted in the control progenitor cells. Upregulation of the pro-neural transcription factors in Δpl cells (in growth medium) mainly reflects the reduction of population heterogeneity (≈5-fold from Ki-67+ profile shown in Figure 1b) by synchronization. True upregulation of atoh1 and ngn2 following the inhibition of miR4673 (ΔTuD, 24 h), to levels comparable to that of differentiating cells and despite the absence of differentiation cues, highlighted anti-differentiation signature of the miRNA. The late differentiation markers, potassium channel trek and the cation channel trpv1, were not upregulated in Δpl and ΔTuD cells.
We used Ki-67 immunoreactivity to disclose the population of cycling cells following the amplification of miRNA4673. Within 24 h of amplifying the endogenous miRNA, mihighΔpl cells became ki-67− despite the presence of mitogenic signals such as FGF-2 (Figure 1b). Subsequently, an abrupt population-level inversion of Ki-67− to Ki-67+ mihighΔpl cells was noted from 24 to 36 h post-transfection (Figure 1b). The inversion suggested a transient synchronization of the cycling cells at Ki-67− G0 phase prior to coordinated progression through Ki-67+ interphase (Figure 1c). Application of Fucci cell cycle reporter confirmed transient arrest at G0 of mihighΔpl cells (Figure 1d). While the Geminin− (DNA replication inhibitor) profile of mihighΔpl cells indicated a license to enter G1, the Cdt-1− (DNA replication factor) fingerprint was consistent with transient arrest at G0 prior to entry into G1 (Figure 1d). We measured the transcriptional level of pro-neural transcription factors (atoh1, mash1, and ngn2) to probe the differentiation propensity of synchronized cells. Compared to differentiating control cells, the level of these transcription factors did not change significantly (Figure 1e). Hence, re-entry of Ki-67− mihighΔpl cells into the cell cycle was enforced by the miRNA-mediated repression of the differentiation propensity in the G0-synchronized population (Figure 1e). Having established that miR4673 can access and reprogram the G0 phase, we addressed the mechanistic basis for such reprogramming.
Induction of G0 commonly occurs by inhibitory input from two parallel cascades that converge to p53 and p21/p27 mediators [21] (Figure 2a). We, therefore, fingerprinted the two cascades in mihighΔpl cells. We noted transcriptional suppression of ASPP1 (PPP1R13B), a trans-activating partner of p53 [22], in mihighΔpl cells (Figure 2b). In these cells, p27 and the associated co-activators PCAF/BCL2 [23,24] were also silenced (Figure 2b). Simultaneously, p53 and p21 proteins were eliminated by post-transcriptional degradation in mihighΔpl cells (Figure 2c, supplementary Figure 2). We also detected an alternative splicing switch from Cyclin D1 short isoform (CCND1-s) to the long isoform (CCND1-s) following amplification of endogenous miRNA in mihighΔpl cells (Figure 2b). The short isoform of Cyclin D1 lacks the cyclin box required for cdk4 activation and release of E2F proteins from inhibitory Rb1 [25]. Therefore, inhibition of short isoform of Cyclin D1 would propel entry into G1. The complete suppression of the canonical inducers of G0 suggested that miRNA4673 functions by accessing a non-canonical G0-induction cascade. This would also explain the suppression of the canonical cascade genes that lack the seed region required for direct targeting by miR4673 (supplementary Data 1). The suppression may occur in response to the non-canonical induction of G0 cascade in order to initiate and energize progression into G1 (Figure 2a). We, therefore, anticipated accelerated progression of G0-synchronized cells subsequent to entry into G1.
The abolished activity of canonical G0 inducers could also accelerate progression in G1 by another parallel mechanism. The canonical G0 inducers contribute to G1–S checkpoint machinery after the progression of cells into G1 (Figure 2a), and suppression of these proteins facilities G1–S transition. Further fingerprinting of mihighΔpl cells revealed transcriptional silencing of several other G1–S checkpoint components (rad51, xrcc3, and brca2) (Figure 2d) that are also trans-activated by p53 [26] pathway. Notably, reduced activity of G1–S checkpoint is the key signature that sustains the proliferative capacity of embryonic stem cells by instructing an abridged G1 [27]. We concluded that non-canonical induction of G0 not only synchronizes the cycling cells but would also expedite progression in interphase. Confirmation of this bimodal signature required the application of naked miRNA4673 that was accomplished in subsequent stoichiometric experiments. Prior to that, we attempted to identify the non-canonical cascade targeted by miRNA4673 to induce G0 synchronization. There is one more cascade that is known to be a potent inducer of G0 arrest in yeast, that is Pho80-Pho85 signaling cascade [28]. This cascade induces G0 arrest upon sensing environmental stressors [29].
Inhibition of cdk-18 by miR4673 underpins reprogramming of cell cycle
MiR4673 inhibits cyclin-dependent kinase-18 (cdk-18) (Figure 3a), the closest mammalian homolog of yeast Pho85 cyclin (supplementary Data 2). In yeast, Pho85 licenses G0–G1 transition [30]. Severe environmental stressors mediate inhibition of Pho85 [29]. In consequence, autophagy is activated [31] leading to cell cycle reprogramming [32] by lengthening G0 and shortening G1 [33]. The resultant Pho85− phenotype improves the fitness of stressed yeast cells. As such, inhibition of cdk-18 by miR4673 simulated a faux stress response in neural progenitors activating endoplasmic reticulum stress [34] and autophagy [14] (Figure 3a, b). Subsequent to the induction of autophagy [14], the cells were arrested at G0 (Figure 1b). The enhanced autophagic flux also bolstered the abridged G1 signature of mihighΔpl cells by accelerating degradation of cell cycle inhibitors p53 [35] and p21 (Figure 2c, supplementary Figure 2) and intracellular Notch-1 [36] (Figure 2c, supplementary Figure 3). On the other hand, the cell cycle propellant Catenin-β1 [37] evaded proteasomal degradation and accumulated in the cytoplasm (Figure 2d).
Figure 3.
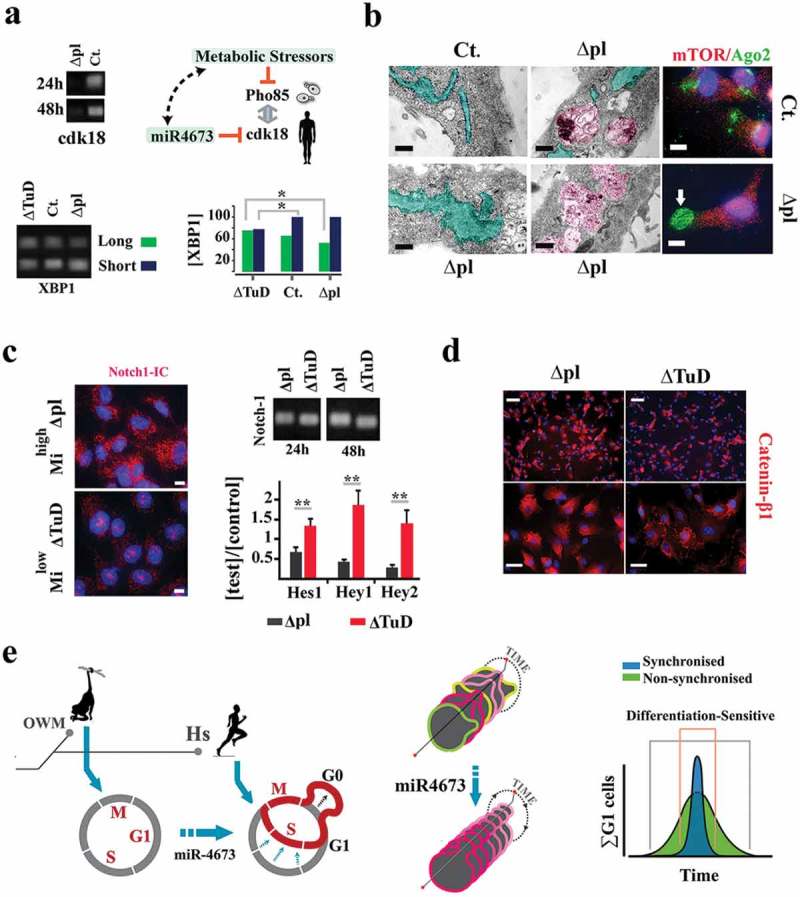
Cdk-18 mediates coupling of miR4673 to cell cycle.(a) MiR4673 silences cdk-18 by hybridization to two potential flanking seed regions in the 3ˊ-UTR of the transcript (SI Appendix, Fig S4). MiR4673 mimics a faux-stressor by inhibiting cdk-18, similar to the suppression of Pho85 by metabolic stressors in yeast. Enhanced endoplasmic reticulum stress in mihighΔpl cells was confirmed by alternative splicing switch to the short isoform of xbp-1 (bottom left). Bar plot (bottom right) demonstrates the concentrations of short and long isoforms of xbp-1 in transfected cells (tr) normalized to the contraction of the long isoform of xbp-1 in control (ct.) cells (*p < 0.05). (b) Dilated endoplasmic reticulum (turquoise) and autophagosome formation (purple) in mihighΔpl cells (scale bars = 1.5 μm). Enhanced Ago-2 activity suggests activation of stress-mediated RNA silencing in mihighΔpl cells (left scale bar = 1.5 μm, right scale bar = 5 μm). (c) Minimal level of activated Notch-1 in the nucleus after amplification of miR4673 (left) despite active transcription (top right). Notch-1 downstream mediators were downregulated in mihighΔpl cells consistent with the post-translational degradation of the protein (scale bar = 5 μm). n = 9; error bars: SD; **p < 0.01. (d) Enhanced cytoplasmic catenin-β1 following amplification of miR4673 (top scale bar = 50 μm; bottom scale bar = 15 μm). (e) Reprogrammed cell cycle (G0 expansion/G1 contraction) of Homo sapiens (Hs) by miR4673 is contrasted with the putative ancestral cell cycle in old-world monkeys (OWM) that lack the miRNA. Synchronization of the cycling population by miR4673 minimizes the impact of differentiation cues.
Reduced activity of Notch-1 [38] and enhanced signaling from Wnt/Catenin-β1 [37] promote the proliferative capacity of neural stem cells. Hence, autophagic degradation of p53, p21, and Notch-1 complemented the transcriptional signature of miR4673 in shortening G1 phase. Shortening of G1 phase can also diminish differentiation likelihood of neural progenitors following entry into interphase [39,40]. G0 synchronization, on the other hand, restricts differentiation-sensitive G1 phases of cycling cells to a narrow temporal window compared to dispersion of G1 phases in an asynchronous population (Figure 2e). By this mechanism, proliferative capacity may be indirectly enhanced due to the minimized impact of differentiation cues on a cycling population (Figure 2e). Having established the molecular signature of miR4673, we measured the stoichiometric impact of miR4673 signaling on synchronization and proliferation rate of cycling human neural progenitors.
Activity of miR4673 reduces population-level entropy of cell cycle
We used high-resolution single-cell tracking by live imaging to establish the rate and relative synchronicity of mitotic events. Manipulations in these experiments were achieved by application of the naked miRNA (mihighΔmi) and the naked inhibitor (milowΔAs) to precisely titrate these moieties against the endogenous miRNA. The observed mitotic events (mi) were binned in 40-min intervals (ti, i = 50 time points), and cumulative graphs for the mitotic events (x = ti, y = mi) were generated (Figure 4a) as follows:
Figure 4.
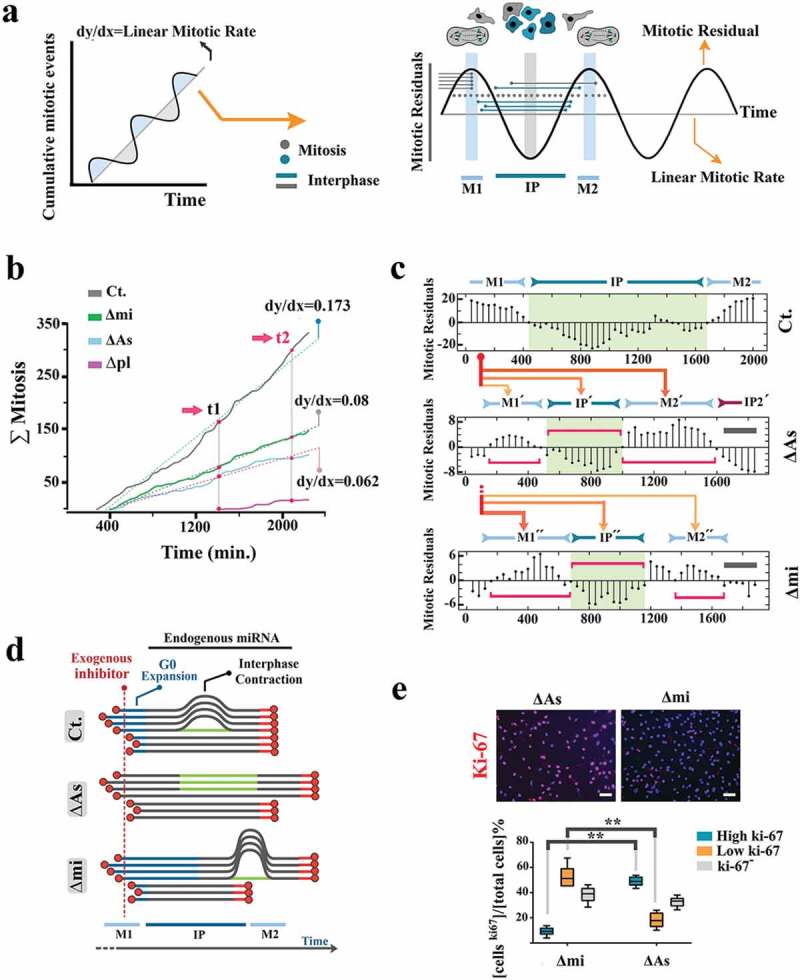
Endogenous miR4673 enhances the proliferative capacity of neural precursors. (a) Linearity of cumulative mitotic rate indicates asynchronous mitotic entry (individual cells represented by gray circles in graphical depiction on right). Positive deviation from linearity ensues from synchronous mitotic events (M1, M2: mitosis-rich period). Negative deviation from linearity occurs when synchronized neural progenitors are collectively in the interphase (IP: interphase-rich period). (b) Amplitude of population-level mitotic oscillations reflects relative synchronization of the cycling cells. Acute modulation of miR4673 alters cumulative mitotic rate of cycling cells. (c) Bar plots demonstrate oscillatory deviations of the observed cumulative mitotic events from those predicted by a linear regression model as demonstrated in Figure 3a (left) (detailed in SI Appendix, Methods). The horizontal red brackets highlight periodicity of synchronized oscillations. The gray lines show the terminal amplitude of oscillations. The x-axis shows time points (in min). The orange lines demonstrate redistribution of the M1 wave subsequent to suppression/amplification of endogenous miRNA. Expansion of early G1 and shortening of the longer cell cycle improve the temporal alignment of cycling cells. (d) Demonstration of population-level cycle dynamics following transient suppression and amplification of endogenous miR4673. De-synchronization in the milowΔAs cells follows the expansion of the contracted interphase and ablation of G0 expansion. De-synchronization of the mihighΔmi cells results from further expansion of G0. (e) Expression of Ki-67 diminishes from G1 towards M phase and eventually becomes undetectable at G0. The strong expression of ki-67 in milowΔAs cells is consistent with a prolonged G1 as opposed to the weak expression in mihighΔmi cell with a contracted G1 phase (n = 20 random fields from five samples/group; error bars: SD; ** indicates p < 0.01) (scale bar = 30 μm).
Two parameters shaped the mitotic landscape (Figure 4a, supplementary Figure 4). The cumulative linear mitotic rate (Lmr) represents the slope of the least-squares regression line (dotted lines in Figure 4b) that optimally describes the data points (ti, mi). The second parameter measures deviation (dmi) of the observed mitotic events (ti, mi) from those predicted by the linear regression model (mitotic residuals in Figure 4a, c). The oscillations (periodicity and amplitude) of deviations (ti, dmi) inform the temporal behavior of the cycling cells based on synchronized mitotic entry (Figure 4a, supplementary Figure 4).
Transient inhibition of the endogenous miRNA by the naked antisense inhibitor (milowΔAs) reduced the linear mitotic rate by ≈2.8-fold (Figure 4b). Inhibition of miR4673 also restored inherent temporal entropy of population cell cycle length that is otherwise masked by the endogenous miRNA-mediated reprogramming of cell cycle (Figure 1b–d). This is evidenced by ≈5-fold repression of the highly synchronized M1 wave and temporal expansion into M1´+IP´+M2´ waves in milowΔAs cells (Figure 4c). The reduced linear mitotic rate and de-synchronized cycling of milowΔAs cells are aligned to the enhanced activity of G1–S checkpoint machinery that lengthens G1 phase, reduces mitotic rate, and increases entropy of population-level cell cycle (Figure 4d). This finding is also aligned to the higher expression level of miR4673 in cycling cells compared to quiescent cells (supplementary Figure 5). Overexpression of the miRNA (mihighΔmi) also decreased the mitotic rate (≈2.1-fold) by lengthening the G0 delay (Figure 4b–d). The G1–specific strong expression of Ki-67 in mihighΔAs cells (long G1) compared to the weak expression in mihighΔmi cells (long G0, short G1) corroborated our interpretation of reprogramming by miR4673 of the mitotic landscape (Figure 4e). We next investigated the impact of reprogrammed cell cycle on the migratory behavior of neural progenitors.
Locomotory landscape of neural progenitors adapts to the reprogrammed cell cycle
The mihighΔpl cells, synchronized at expanded G0, expressed chemo-repulsive Slit1 [41] as opposed to the lack of expression in control cells (Figure 5a). Expression of Slit-1 communicates positive migratory signals to Robo-2+ neighboring cells [42]. On the other hand, slit-1+ mihighΔpl cells did not express EphB2 (Figure 5a) that stimulates migration by activating RhoA [43]. Synchronization at G0 establishes EphB2− stationary cells (Figure 5b, supplementary Movie S1) that present Slit-1+ repulsive cues to the adjacent cells and are therefore predicted to indirectly enhance population-level locomotion (Figure 5c). The locomotory landscape was constructed by measuring the average velocity of cells (Vav) in a field of view:
where N represents the number of mobile cells tracked (N = 120) and binned in 60-min intervals (i = 30 frames). As expected, the mihighΔmi cells exhibited amplified collective locomotion by an average of ≈30% (Figure 5d, supplementary Movies S2, 3). The enhanced locomotion is aligned to the expansion of G0-synchronized Slit-1+ cells acting as chemo-repulsive nodes. In contrast, prolonged dwelling in late G1 by milowΔAs cells decelerated the collective locomotion due to reduction of repulsive interactions (Figure 5d, supplementary Movies S2, 4). Given the impact of miR4673 on proliferative capacity and collective locomotory behavior of neural progenitors, we sought to overlay these diverse outcomes into a morphogenic signature.
Figure 5.
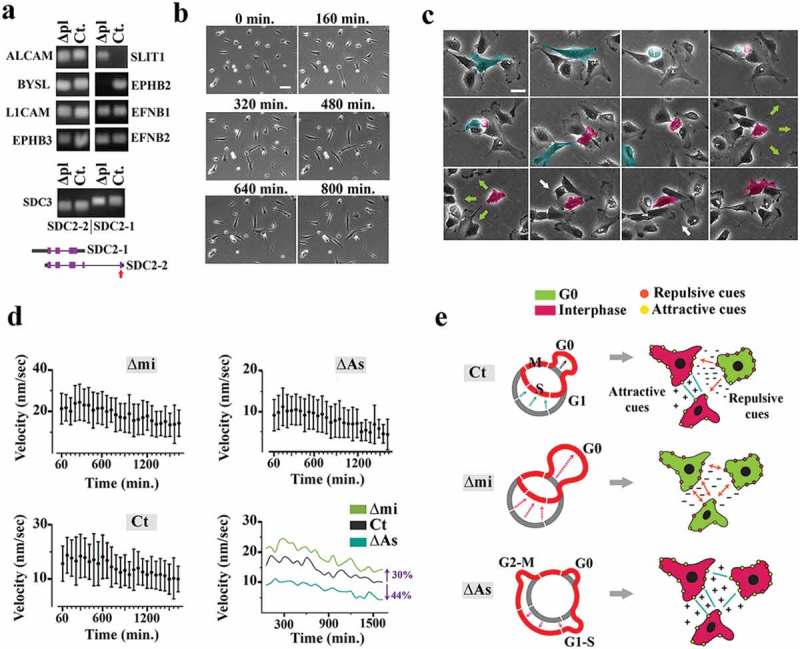
Population-level synchronization of cell cycle reshapes the locomotory landscape of neural progenitors.(a) Profiling of genes coding for attractive and repulsive guidance cues in the mihighΔpl and control cells revealed transcriptional inhibition of ephB2 and simultaneous activation of slit1 (short-range repulsive cue) in the mihighΔmi cells synchronized to G0 (a representative sample of three biological replicates). Also, the longer isoform of sdc-3 (syndecan-3, sdc-1), a heparan sulfate proteoglycan required for the activity of Slit1, was upregulated in mihighΔpl cells (bottom). Only the longer isoform of sdc-3 contains the secretory signal sequence (red arrow). (b) Live imaging confirmed the static nature of mihighΔpl cells (scale bar = 100 μm). (c) Live imaging of control untreated progenitors demonstrates transient arrest of a daughter cell at G0 after asymmetric division (pseudo-colored red). Note the stationary nature of the arrested cell and repulsion of the neighboring cells. Entry into the cycle establishes the attractive cues. The green arrows show directionality of repulsive interactions, and the white arrows point to the attractive interactions (scale bar = 15 μm). (d) Graphs show the population-level average linear velocity of the mihighΔmi, milowΔAs, and control cells. Vertical lines demonstrate standard deviation of velocity in each interval (60 min) and circles represent the mean values. (e) Schematic demonstration of the miRNA-mediated alteration of the locomotory landscape by resetting population-level cell cycle.
Bimodal reprogramming of cell cycle informs a higher morphogenic dimension of neuro-ontogeny
A morphogenic signature evolves from regulation of proliferation rate and differentiation outcome that informs spatial micro-organization and, eventually, macro-anatomical patterns. Having addressed the proliferation rate, we focused on the differentiation outcome. The differentiation tendency and sublineage fate choice were investigated separately.
Modulation of the differentiation probability was calculated from the altered signature of the cell cycle. In the reprogrammed progenitor, synchronization is instructed by agonistic activity of G0 expansion/G1 contraction. The enhanced mitotic rate is, however, positively impacted by G1 contraction and negatively influenced by G0 expansion. Based on the latter logic, we calculated the miR-mediated reprograming parameters of cell cycle based on:
By solving the latter equations, we calculated the impact of miR4673 on cell cycle reprogramming as ≈3.7-fold contraction of G1 and ≈1.3-fold expansion of G0. The reprogrammed cycle is estimated to reduce differentiation likelihood (Figure 3e) of individual cycling cells by ≈3.7-fold (by G1 contraction) and that of the population by ≈5-fold (by synchronization) despite an enhanced proliferation rate (2.8-fold). Corroborating our findings, we noted suppression of Hippo signaling pathway (Hippo°ff) evidenced by nuclear translocation of Yap1 and Taz as well as enhanced telomerase activity in the mihighΔmi cells (Figure 6a, supplementary Figure 6). Parallel to downregulation of Hippo cascade post-amplification of miR4673, we noted a resistance to differentiation in neural progenitors (Figure 6b). Downregulation of Hippo cascade is known to delay differentiation of neural precursors and enlarge brain size in Drosophila melanogaster [44].
Figure 6.
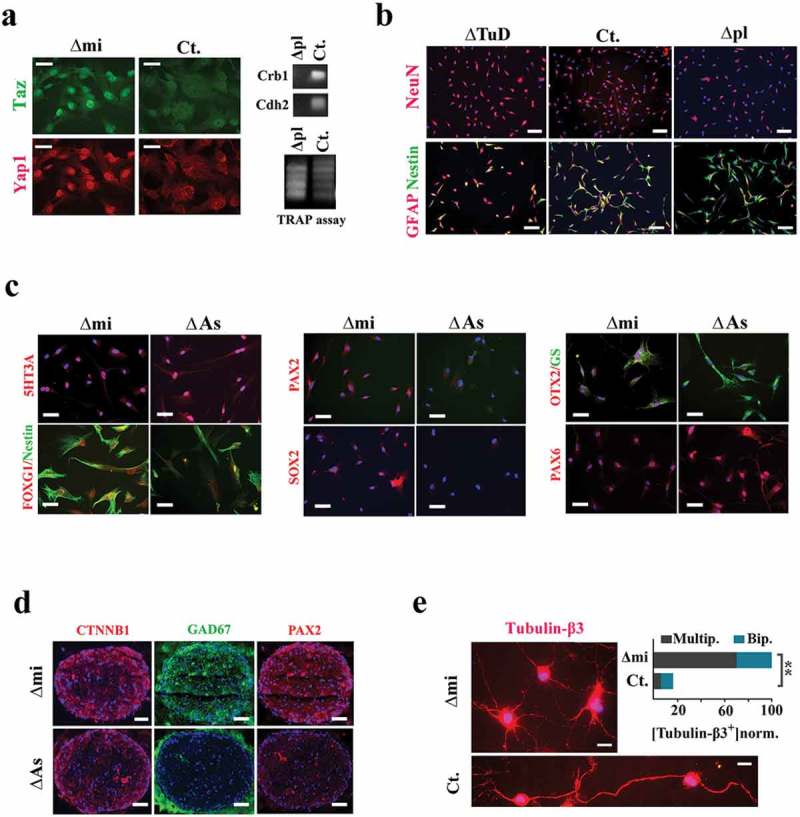
Phenotypic impact of the bimodal reprogramming of cell cycle.(a) Immunohistochemical localization of Hippo cascade mediators shows upregulation of Yap1 and Taz after application of the exogenous miRNA in growth medium (scale bars = 10 μm). The Hippo cascade antagonists, crb-1 (36) and cdh-2 (37) (N-cadherin), were silenced in the mihighΔpl cells (top). TRAP assay demonstrates higher activity of telomerase in mihighΔpl cells compared to control cells (bottom). (b) In neural differentiation medium, NeuN−/nestin+ fingerprint of differentiation-resistant mihighΔpl cells contrasts sharply with NeuN+/nestin− fingerprint of differentiating milowΔTuD cells (scale bars = 40 μm). (c) Molecular fingerprint of differentiating human neural progenitors with suppressed (milow∆As) and amplified (mihigh∆mi) miR4673 activity. Neural induction was initiated immediately after electroporation and fingerprinting was done 24 h after the induction of differentiation. The bias towards the assumption of an interneuronal fate under instruction from miR4673 (mihigh∆mi) was evident based on upregulation of FOXG1/OTX2/PAX2/SOX2 transcription factors in differentiating mihigh∆mi cells (scale bars = 50 μm). (d) Differentiation of neural organoids in Matrigel resulted in a neuron-rich structure (demonstrated by reactivity for CTNNBI, GAD67, and PAX2) under instruction from amplified miR4673 (scale bar = 150 μm). (e) The mihighΔpl cells mainly differentiated into multipolar neurons (top) as opposed to a few scattered bipolar neurons in the control group (top scale bar = 10 μm, bottom scale bar = 8 μm). Abundance of neurons in the control group is normalized to mihighΔmi group and is presented as percentage (**p < 0.01).
Inhibition of Hippo cascade also modulates sublineage fate choice by neural progenitors; the Hippo°ff state can reduce astrocytic differentiation with no obvious impact on the differentiation of neurons [45]. We employed neural organoids as well as 2D differentiation assays to investigate the impact on sublineage fate determination of miR4673. The activity of endogenous miRNA was amplified in 2D cultures and neural organoids embedded in Matrigel prior to final induction of neural differentiation (see Methods). Post-differentiation, mihighΔmi cells demonstrated remarkable upregulation of FOXG1/PAX2/PAX6/OTX2 transcription factors (Figure 6c). These transcription factors regulate differentiation of the neocortical interneurons [46–49]. Likewise, the mihighΔmi organoids after 4 d exhibited high activity of Wnt/cateninβ1 and were mainly composed of GAD67+/PAX2+ interneurons (Figure 6d). Hence, miR4673 signaling enhances differentiation into inhibitory GABAergic interneurons at the cost of a reduced glial population. The relaxation of cell cycle checkpoint machinery by miR4673 signaling may facilitate differentiation into neurons as DNA double-stranded breaks remove the transcription blocks from the promoters of neuronal early response genes such as NPAS4 [50]. After final neuronal maturation (2 wk in the induction medium), the mature mihighΔmi/tubulin-β3+ interneurons were mainly multipolar (Figure 6e), consistent with the amplified activity of Wnt/Cateninβ1 cascade in these cells [51]. Therefore, the delayed differentiation anticipates an inter-neuronal fate bias in response to the amplification of miR4673.
We finally attempted to gain insight into the morphogenic signature that may result from the bimodal modulation of the cell cycle. It is noteworthy that such a signature does not exclusively result from the activity of miR4673; the activity of this microRNA only foreshadows a common morphogenic platform that may be accessed and reprogrammed by other similar molecular entities to instruct heterochronic specialization of the central nervous system. We adopted a mathematical model (see Methods) with demonstrated success in prediction of biological pattern formation [52] including brain morphogenesis [17]. Based on reaction–diffusion dynamics of the Gray–Scott model, we suppose that brain morphogenesis is driven by the universal morphogens (forces) U and V [18]. The model evolves based on Turing-style instabilities at the interface of proliferation and differentiation zones (Figure 7a). Proliferating cells provide static repulsive cues (U, the inhibitor) and differentiating cells, generated from U, constitute the mobile diffusible phase (V, the activator).
Figure 7.
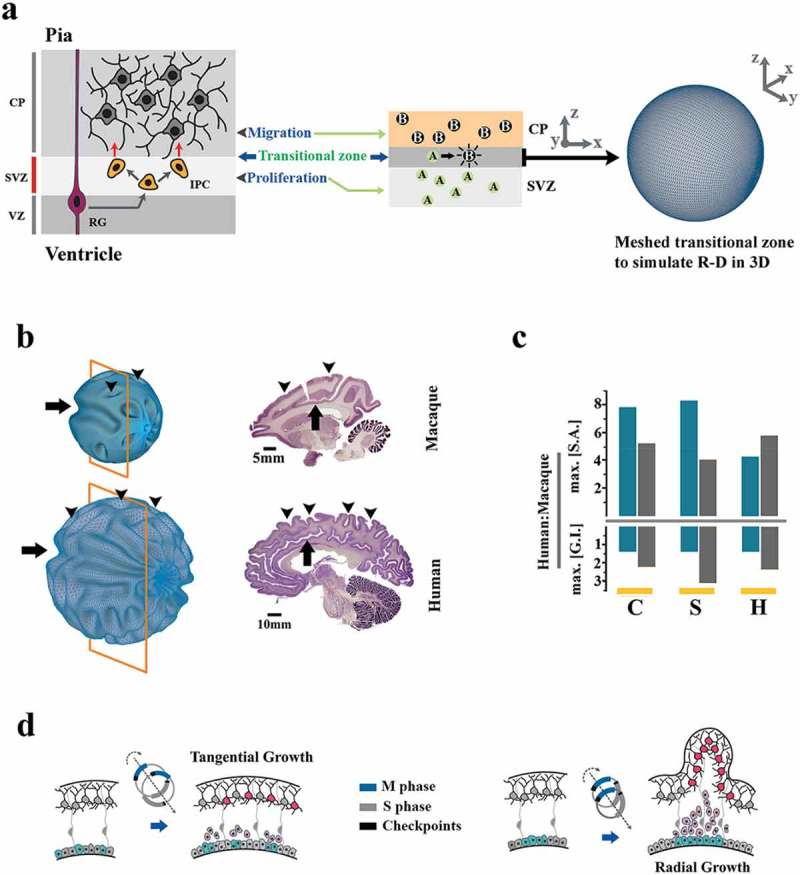
Mathematical modeling of the morphogenic signature of miR4673. (a) Schematic demonstration of modeling brain morphogenesis based on Reaction-Diffusion system. In the intermediate progenitor model (top left), the transitional zone between the subventricular zone (SVZ) and the cortical plate (CP) is occupied by proliferative intermediate progenitor cells (IPCs labeled as A) that generate an expansive morphogenic force (U). Subsequent to differentiation into neuro-glia (labeled as B), the second morphogenic component (V) is generated. Local instabilities due to the interactions between repulsive A and migratory B instructs morphogenic features of a developing brain. This zone was modeled on a spherical meshed surface and the local instabilities were projected onto the spherical mesh (right). (b) Simulation of macaque and human brain growth based on a reaction–diffusion model and input values that correspond to the bimodal alteration of the cell cycle (see Figure 3) by miR4673 (human) and unaltered cell cycle in the absence of miR4673 (macaque). Note conservation of the paracingulate sulcus (arrow) and amplification of secondary sulci (arrowheads) subsequent to altered cell cycle anatomy mediated by miR4673. Images were obtained from Brain Biodiversity Bank and BrainMaps databases. (c) Bar plots demonstrate the surface area (SA) and gyrification index (GI) of human cerebrum/cerebellum normalized to that of macaque (C: coronal, S: sagittal, H: horizontal). Note that the growth of gyrification index from macaque (an old-world monkey that lacks miR4673) to human is ≈1.5-fold for cerebrum (blue columns) and ≈2–3-fold for cerebellum (gray columns). These values are closely aligned to the prediction of GI growth based on the miR4673 signature. (d) The MiR-mediated bimodal cell cycle reprogramming favors radial growth and hence gyrification of the brain by synchronized division of neural progenitors.
The dynamic behavior of such a system is overall described by the following equations:
where F is the feed rate (replenishment rate) of U and k represents the kill rate (removal rate) of V. We modeled the growth of a macaque brain with the values F = 0.035, k = 0.057, Du = 0.05, and Dv = 0.041 (Figure 7b, Supplementary Figure 7). The human brain, based on bimodal alteration of the cell cycle, was modeled with the values F = 0.06, k = 0.061, Du = 0.087, and Dv = 0.041 (Figure 7b, Supplementary Figure 7). The feed (F) and the diffusion rates of the inhibitor (Du) were enhanced by ≈74% in modeling growth of the human brain based on the population-level impact on the entropy of miR4673 (from Figure 5d, see also Methods). The modeling outcome based on the bimodal modulation of cell cycle regenerated the surface topology of the human and macaque brains (Figure 7b, Supplementary Figure 7). The numerical estimates based on the growth of GI from macaque to human (Figure 7c) confirmed the results of topological modeling. It seems plausible to postulate that bimodal alteration of the cell cycle may be a universal platform to induce species-specific specializations of brain regions in primates.
The predictions of the reaction–diffusion model are also intuitively accessible. Enhanced gyrification is an inevitable accompaniment to the bimodal reprogramming that alters the growth pattern from tangential to radial due to the synchronized cell division that exceeds the rate of spatial expansion (Figure 7d). It is, therefore, not surprising that a nonsense mutation in Cdk5 (a paralog of Cdk-18/Pho85), that also regulates M–G1 transition, manifests as autosomal recessive lissencephaly with cerebellar hypoplasia [53].
Discussion
Data presented herein provide primary evidence regarding a heterochronic signature in the human neurogenic landscape underpinned by the activity of miR4673 that modulates temporal anatomy of the cell cycle. To induce a heterochronic signature, the miRNA capitalizes on the plasticity of cell cycle anatomy. The signature effectively combines a transient arrest at G0 with an accelerated progression at G1 that leads to enhanced proliferative capacity and synchronized cycling of neural progenitors. Non-canonical arrest at G0 is triggered by miRNA-mediated suppression of cdk-18 that licenses G0–G1 transition. Downregulation of canonical inducers of G0 and G1 arrest, in response to non-canonical induction of G0, generates a momentum that accelerates the progression of synchronized cells in interphase subsequent to progression into G1 (Figure 8). Delayed differentiation of neural progenitors and eventual assumption of a neuronal fate bias are further consequences of the altered cell cycle anatomy (Figure 8).
Figure 8.
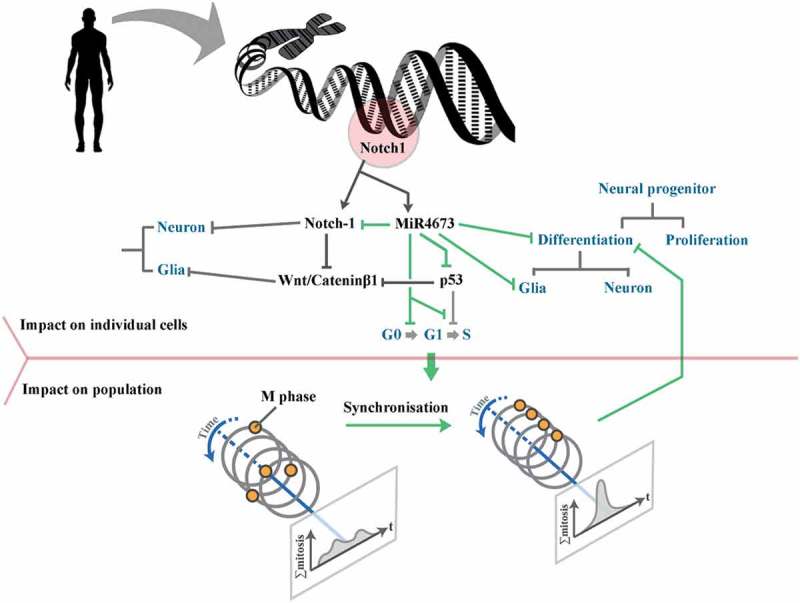
Schematic demonstration of the altered morphogenic signature of human Notch-1 subsequent to the heterochronic regulation by co-expressed miR4673. In order to instruct the morphogenic signature, the miRNA operates at the individual cell level and at the population level. At the individual cell level, miR4673 antagonizes Notch-1 and p53 and empowers Catenin-β1. This profile alters the growth arrest/gliogenic bias that characterizes Notch-1 activity to the proliferation/neurogenic bias that is coerced by the co-expressed miRNA-4673. At the population level, synchronization of the cell cycle by miR4673 minimizes the impact of differentiation cues and hence improves the proliferative capacity of the progenitor cells.
Reprogramming of the cell cycle is not an underutilized facet of ontogeny. Indeed, the cell cycle length of neural precursors shows significant inter-species variability [54]. This heterogeneity is mainly attributable to G1/S checkpoint that can be accessed and reprogrammed by endogenous species-specific mechanisms [55]. Such programmability is a powerful driving force in generating major heterochronic adaptations, particularly in the central nervous system. For example, the increased proliferation rate of neural precursors by shortening G1 phase underpins higher numbers of neurons in the striate cortex of primates [12]. Conversely, lengthening of the G1 phase prevents expansion of neuronal precursors within the dentate gyrus of rodents [56]. The heterochronic signature of miR4673, however, reveals a novel programmable dimension of the cell cycle, the plasticity of G0. To access G0, the miRNA simulates a faux stress response by targeting cdk-18, an ancient metabolic stress sensor. Non-canonical induction of G0 combined with the shortening of G1 underpins synchronized and accelerated cycling of human neural progenitors with morphogenic consequences. To this end, it is known that coordinated synchronization and desynchronization of population cell cycle dynamics during ontogeny constitutes a developmental switch for spatial patterning of mitosis and hence morphogenic patterning during neurulation [57]. The predicted morphogenic signature that arises from the heterochronic activity of miR4673 is compatible with aspects of human brain anatomy.
Regulation of cell cycle by microRNAs commonly occurs in development [58]. One major group of miRNAs promote differentiation. These miRNAs target Cdk2/4/6 complex and induce G1 arrest [59]. The let-7 family members operate in this mode to induce G1 synchronization. The second group of miRNAs enhance the proliferative capacity of cycling cells. The mouse miR-290 cluster, for example, downregulates major inhibitors of cell cycle, including RB, RBL1, RBL2, p21, and LATS2, and thus accelerates transition into S phase [60]. Suppression of RB proteins occurs downstream to signaling by various other embryonic miRNAs and facilitates the G1/S transition of cycling cells [61]. Reprogramming of cell cycle by miR4673 occurs by a unique mode. It targets cdk-18 (a non-canonical inducer of G0) and synchronizes the cycling cells while anticipating an accelerated progression through interphase due to suppression of G1–S checkpoint components. Therefore, the cycling pace and the synchronicity of proliferating cells increase simultaneously with morphogenic consequences that are explained by mathematical modeling. In a broader context, the morphogenic activity of miR4673 reveals an underlying universal platform to induce heterochronic adaptations of the central nervous system via bimodal regulation of cell cycle. Subtle reprogramming of the latter platform may trigger significant adaptive changes that enable survival of species at times of environmental crisis.
The current paper focuses mainly upon the neurogenic signature of miR4673. However, signaling by Notch-1 is central to the proliferation and differentiation of many other cell types [62,63]. As such, it would be interesting to study the heterochronic impact of miR4673 in development of other organ systems. Also, experimental proof regarding the morphogenic impact of miR4673 (e.g. on gyrification of the human brain) would strengthen the predictions of the current study. Introduction into the mouse genome of a synthetic miR4673 homolog (or paralog) that targets cdk18 (with partial selectivity against some other important targets of the miRNA) could provide experimental evidence for the predicted heterochronic regulatory activity of the miRNA.
These findings add a novel mechanistic twist to the evolutionary journey of Homo sapiens as a selected heterochronic mutant of the primate order. The proposed perspective may also have broader implications for mechanistic re-interpretation and targeted therapy of human syndromes.
Funding Statement
This work was supported by the National Institute of Dental and Craniofacial Research [R01 DE015272].
Acknowledgments
This study was supported by NIDCR grant R01 DE015272 and Australian National Health. R.M.F. conceived, designed, and performed experiments and mathematical modeling, analyzed and interpreted data, and wrote the manuscript. S.RL. assisted R.M.F. with experiments and analysis. M.S. performed electron microscopy. N.H. conceived experiments, contributed to interpretation of the data, and contributed to writing the manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Suppplemental data for this article can be accessed here.
References
- [1].King MC, Wilson AC.. Evolution at 2 levels in humans and chimpanzees. Science. 1975;188(4184):107–116.PubMed PMID: WOS:A1975W051500004; English. [DOI] [PubMed] [Google Scholar]
- [2].Britten RJ. Divergence between samples of chimpanzee and human DNA sequences is 5%, counting indels. Proc Natl Acad Sci U S A. 2002. October 15;99(21):13633–13635. PubMed PMID: WOS:000178635700054; English. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Somel M, Franz H, Yan Z, et al. Transcriptional neoteny in the human brain. Proc Natl Acad Sci U S A. 2009. April 7;106(14):5743–5748. PubMed PMID: 19307592; PubMed Central PMCID: PMCPMC2659716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Johnson MH. Functional brain development in humans. Nat Rev Neurosci. 2001. July;2(7):475–483.PubMed PMID: 11433372. [DOI] [PubMed] [Google Scholar]
- [5].Miller DJ, Duka T, Stimpson CD, et al. Prolonged myelination in human neocortical evolution. Proc Natl Acad Sci U S A. 2012. October 9;109(41):16480–16485. PubMed PMID: 23012402; PubMed Central PMCID: PMCPMC3478650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Moss EG. Heterochronic genes and the nature of developmental time. Curr Biol. 2007. June 5;17(11):R425–34. PubMed PMID: 17550772. [DOI] [PubMed] [Google Scholar]
- [7].Denoel M, Joly P. Neoteny and progenesis as two heterochronic processes involved in paedomorphosis in Triturus alpestris (Amphibia: Caudata). Proc R Soc B Biol Sci. 2000. July 22;267(1451):1481–1485. PubMed PMID: WOS:000088677300016; English. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Banerjee D, Slack F. Control of developmental timing by small temporal RNAs: a paradigm for RNA-mediated regulation of gene expression. Bioessays. 2002. February;24(2):119–129.PubMed PMID: 11835276. [DOI] [PubMed] [Google Scholar]
- [9].Ambros V, Horvitz HR. Heterochronic mutants of the nematode Caenorhabditis elegans. Science. 1984. October 26;226(4673):409–416. PubMed PMID: 6494891. [DOI] [PubMed] [Google Scholar]
- [10].Euling S, Ambros V. Heterochronic genes control cell cycle progress and developmental competence of C. elegansvulva precursor cells. Cell. 1996. March 8;84(5):667–676. PubMed PMID: 8625405. [DOI] [PubMed] [Google Scholar]
- [11].Brennecke J, Hipfner DR, Stark A, et al. bantam encodes a developmentally regulated microRNA that controls cell proliferation and regulates the proapoptotic gene hid in Drosophila. Cell. 2003. April 4;113(1):25–36. PubMed PMID: 12679032. [DOI] [PubMed] [Google Scholar]
- [12].Dehay C, Giroud P, Berland M, et al. Modulation of the cell cycle contributes to the parcellation of the primate visual cortex. Nature. 1993. December 2;366(6454):464–466. PubMed PMID: 8247154. [DOI] [PubMed] [Google Scholar]
- [13].Lukaszewicz A, Savatier P, Cortay V, et al. G1 phase regulation, area-specific cell cycle control, and cytoarchitectonics in the primate cortex. Neuron. 2005. August 4;47(3):353–364. PubMed PMID: 16055060; PubMed Central PMCID: PMCPMC1890568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Dökümcü K, Simonian M, Farahani RM. miR4673 improves fitness profile of neoplastic cells by induction of autophagy. Cell Death Dis. 2018;9(11):1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Farahani R, Rezaei-Lotfi S, Simonian M, et al. Neural microvascular pericytes contribute to human adult neurogenesis. J Comp Neurol. 2018. In Press. DOI: 10.1002/cne.24565. [DOI] [PubMed] [Google Scholar]
- [16].Noctor SC, Martinez-Cerdeno V, Ivic L, et al. Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat Neurosci. 2004. February;7(2):136–144. PubMed PMID: WOS:000188468500011; English. [DOI] [PubMed] [Google Scholar]
- [17].Lefevre J, Mangin JF, A reaction-diffusion model of human brain development. PLoS Comput Biol. 2010. April 22;6(4):e1000749 PubMed PMID: 20421989; PubMed Central PMCID: PMCPMC2858670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Harrison LG, Wehner S, Holloway DM, Complex morphogenesis of surfaces: theory and experiment on coupling of reaction-diffusion patterning to growth. Faraday Discuss. 2001;120:277–294. discussion 325–51. PubMed PMID: 11901681. [DOI] [PubMed] [Google Scholar]
- [19].Turk G, Generating textures on arbitrary surfaces using reaction-diffusion. Comp Graph. 1991;25:289–298. PubMed PMID: WOS:A1991BU58Y00033; English. [Google Scholar]
- [20].Bak RO, Hollensen AK, Primo MN, et al. Potent microRNA suppression by RNA Pol II-transcribed ‘Tough Decoy’ inhibitors. Rna. 2013. February;19(2):280–293. PubMed PMID: 23249752; PubMed Central PMCID: PMCPMC3543086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Liu Y, Elf SE, Miyata Y, et al. p53 regulates hematopoietic stem cell quiescence. Cell Stem Cell. 2009. January 9;4(1):37–48. PubMed PMID: 19128791; PubMed Central PMCID: PMCPMC2839936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Bergamaschi D, Samuels Y, Jin B, et al. ASPP1 and ASPP2: common activators of p53 family members. Mol Cell Biol. 2004. February;24(3):1341–1350. PubMed PMID: 14729977; PubMed Central PMCID: PMCPMC321425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Love IM, Sekaric P, Shi D, et al. The histone acetyltransferase PCAF regulates p21 transcription through stress-induced acetylation of histone H3. Cell Cycle. 2012. July 1;11(13):2458–2466. PubMed PMID: 22713239; PubMed Central PMCID: PMCPMC3404877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Vairo G, Soos TJ, Upton TM, et al. Bcl-2 retards cell cycle entry through p27(Kip1), pRB relative p130, and altered E2F regulation. Mol Cell Biol. 2000. July;20(13):4745–4753. PubMed PMID: 10848600; PubMed Central PMCID: PMCPMC85901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Narasimha AM, Kaulich M, Shapiro GS, et al. Cyclin D activates the Rb tumor suppressor by mono-phosphorylation [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t]. eLife. 2014;3 PubMed PMID: 24876129; PubMed Central PMCID: PMC4076869. eng DOI: 10.7554/eLife.02872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Arias-Lopez C, Lazaro-Trueba I, Kerr P, et al. p53 modulates homologous recombination by transcriptional regulation of the RAD51 gene. EMBO Rep. 2006. February;7(2):219–224. PubMed PMID: 16322760; PubMed Central PMCID: PMCPMC1369244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ruiz S, Panopoulos AD, Herrerias A, et al. A high proliferation rate is required for cell reprogramming and maintenance of human embryonic stem cell identity. Curr Biol. 2011. January 11;21(1):45–52. PubMed PMID: 21167714; PubMed Central PMCID: PMCPMC3034649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wanke V, Pedruzzi I, Cameroni E, et al. Regulation of G0 entry by the Pho80-Pho85 cyclin-CDK complex. Embo J. 2005. December 21;24(24):4271–4278. PubMed PMID: 16308562; PubMed Central PMCID: PMCPMC1356330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Moffat J, Huang D, Andrews B, Functions of Pho85 cyclin-dependent kinases in budding yeast. Prog Cell Cycle Res. 2000;4:97–106. PubMed PMID: 10740818. [DOI] [PubMed] [Google Scholar]
- [30].Aerne BL, Johnson AL, Toyn JH, et al. Swi5 controls a novel wave of cyclin synthesis in late mitosis. Mol Biol Cell. 1998. April;9(4):945–956. PubMed PMID: 9529390; PubMed Central PMCID: PMCPMC25320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Yang Z, Geng J, Yen WL, et al. Positive or negative roles of different cyclin-dependent kinase Pho85-cyclin complexes orchestrate induction of autophagy in Saccharomyces cerevisiae. Mol Cell. 2010. April 23;38(2):250–264. PubMed PMID: 20417603; PubMed Central PMCID: PMCPMC2861662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Song Q, Kumar A, An overview of autophagy and yeast pseudohyphal growth: integration of signaling pathways during nitrogen stress. Cells. 2012. July 4;1(3):263–283. PubMed PMID: 24710476; PubMed Central PMCID: PMCPMC3901118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kron SJ, Styles CA, Fink GR. Symmetric cell division in pseudohyphae of the yeast Saccharomyces cerevisiae. Mol Biol Cell. 1994. September;5(9):1003–1022. PubMed PMID: 7841518; PubMed Central PMCID: PMCPMC301123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Palmer KJ, Konkel JE, Stephens DJ. PCTAIRE protein kinases interact directly with the COPII complex and modulate secretory cargo transport [Research Support, Non-U.S. Gov’t]. J Cell Sci. 2005. September 1;118(Pt 17):3839–3847. PubMed PMID: 16091426; eng. [DOI] [PubMed] [Google Scholar]
- [35].Pluquet O, Qu LK, Baltzis D, et al. Endoplasmic reticulum stress accelerates p53 degradation by the cooperative actions of Hdm2 and glycogen synthase kinase 3beta [Research Support, Non-U.S. Gov’t]. Mol Cell Biol. 2005. November;25(21):9392–9405. PubMed PMID: 16227590; PubMed Central PMCID: PMC1265800. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wu X, Fleming A, Ricketts T, et al. Autophagy regulates Notch degradation and modulates stem cell development and neurogenesis. Nat Commun. 2016. February;3(7):10533 PubMed PMID: 26837467; PubMed Central PMCID: PMCPMC4742842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Shtutman M, Zhurinsky J, Simcha I, et al. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci U S A. 1999. May 11;96(10):5522–5527. PubMed PMID: 10318916; PubMed Central PMCID: PMCPMC21892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Guentchev M, McKay RD. Notch controls proliferation and differentiation of stem cells in a dose-dependent manner. Eur J Neurosci. 2006. May;23(9):2289–2296.PubMed PMID: 16706837. [DOI] [PubMed] [Google Scholar]
- [39].Calegari F, Haubensak W, Haffner C, et al. Selective lengthening of the cell cycle in the neurogenic subpopulation of neural progenitor cells during mouse brain development. J Neurosci. 2005. July 13;25(28):6533–6538. PubMed PMID: 16014714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Coronado D, Godet M, Bourillot PY, et al. A short G1 phase is an intrinsic determinant of naive embryonic stem cell pluripotency. Stem Cell Res. 2013. January;10(1):118–131. PubMed PMID: 23178806. [DOI] [PubMed] [Google Scholar]
- [41].Zhu Y, Li H, Zhou L, et al. Cellular and molecular guidance of GABAergic neuronal migration from an extracortical origin to the neocortex. Neuron. 1999. July;23(3):473–485. PubMed PMID: 10433260. [DOI] [PubMed] [Google Scholar]
- [42].Halperin-Barlev O, Kalcheim C. Sclerotome-derived Slit1 drives directional migration and differentiation of Robo2-expressing pioneer myoblasts. Development. 2011. July;138(14):2935–2945.PubMed PMID: 21653616. [DOI] [PubMed] [Google Scholar]
- [43].Wang SD, Rath P, Lal B, et al. EphB2 receptor controls proliferation/migration dichotomy of glioblastoma by interacting with focal adhesion kinase. Oncogene. 2012. December 13;31(50):5132–5143. PubMed PMID: 22310282; PubMed Central PMCID: PMCPMC3349801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Poon CL, Mitchell KA, Kondo S, et al. The hippo pathway regulates neuroblasts and brain size in Drosophila melanogaster. Curr Biol. 2016. April 25;26(8):1034–1042. PubMed PMID: 26996505. [DOI] [PubMed] [Google Scholar]
- [45].Huang Z, Hu J, Pan J, et al. YAP stabilizes SMAD1 and promotes BMP2-induced neocortical astrocytic differentiation. Development. 2016. July 1;143(13):2398–2409. PubMed PMID: 27381227; PubMed Central PMCID: PMCPMC4958318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Bernard C, Prochiantz A. Otx2-PNN Interaction to regulate cortical plasticity. Neural Plast. 2016;2016:7931693.PubMed PMID: 26881132; PubMed Central PMCID: PMCPMC4736602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Burrill JD, Moran L, Goulding MD, et al. PAX2 is expressed in multiple spinal cord interneurons, including a population of EN1(+) interneurons that require PAX6 for their development. Development. 1997. November;124(22):4493–4503. PubMed PMID: WOS:000071111800008; English. [DOI] [PubMed] [Google Scholar]
- [48].Maricich SM, Herrup K. Pax- 2 expression defines a subset of GABAergic interneurons and their precursors in the developing murine cerebellum. J Neurobiol. 1999. November 5;41(2):281–294. PubMed PMID: 10512984. [DOI] [PubMed] [Google Scholar]
- [49].Shen W, Ba R, Su Y, et al. Foxg1 regulates the postnatal development of cortical interneurons. Cereb Cortex. 2018. April 18 DOI: 10.1093/cercor/bhy051 PubMed PMID: 29912324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Madabhushi R, Gao F, Pfenning AR, et al. Activity-induced DNA breaks govern the expression of neuronal early-response genes. Cell. 2015. June 18;161(7):1592–1605. PubMed PMID: 26052046; PubMed Central PMCID: PMCPMC4886855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Boitard M, Bocchi R, Egervari K, et al. Wnt signaling regulates multipolar-to-bipolar transition of migrating neurons in the cerebral cortex. Cell Rep. 2015. March 3;10(8):1349–1361. PubMed PMID: 25732825. [DOI] [PubMed] [Google Scholar]
- [52].Kondo S, Miura T. Reaction-diffusion model as a framework for understanding biological pattern formation. Science. 2010. September 24;329(5999):1616–1620. PubMed PMID: 20929839. [DOI] [PubMed] [Google Scholar]
- [53].Magen D, Ofir A, Berger L, et al. Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with a loss-of-function mutation in CDK5. Hum Genet. 2015. March;134(3):305–314. PubMed PMID: 25560765. [DOI] [PubMed] [Google Scholar]
- [54].Kornack DR, Rakic P. Changes in cell-cycle kinetics during the development and evolution of primate neocortex. Proc Natl Acad Sci U S A. 1998. February 3;95(3):1242–1246. PubMed PMID: 9448316; PubMed Central PMCID: PMCPMC18732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Fluckiger AC, Marcy G, Marchand M, et al. Cell cycle features of primate embryonic stem cells. Stem Cells. 2006. March;24(3):547–556. PubMed PMID: 16239321; PubMed Central PMCID: PMCPMC1934406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Beukelaers P, Vandenbosch R, Caron N, et al. Cdk6-dependent regulation of G(1) length controls adult neurogenesis. Stem Cells. 2011. April;29(4):713–724. PubMed PMID: 21319271. [DOI] [PubMed] [Google Scholar]
- [57].Ogura Y, Sasakura Y. Developmental control of cell-cycle compensation provides a switch for patterned mitosis at the onset of chordate neurulation. Dev Cell. 2016. April 18;37(2):148–161. PubMed PMID: 27093084. [DOI] [PubMed] [Google Scholar]
- [58].Chivukula RR, Mendell JT. Circular reasoning: microRNAs and cell-cycle control. Trends Biochem Sci. 2008. October;33(10):474–481.PubMed PMID: 18774719; PubMed Central PMCID: PMCPMC2824243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Bueno MJ, Malumbres M. MicroRNAs and the cell cycle. Biochim Biophys Acta. 2011. May;1812(5):592–601.PubMed PMID: 21315819. [DOI] [PubMed] [Google Scholar]
- [60].Yuan K, Ai WB, Wan LY, et al. The miR-290-295 cluster as multi-faceted players in mouse embryonic stem cells. Cell Biosci. 2017. August 7;7 DOI: 10.1186/s13578-017-0166-2 PubMed PMID: WOS:000407998100001; English. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Hao J, Duan FF, Wang Y, MicroRNAs and RNA binding protein regulators of microRNAs in the control of pluripotency and reprogramming. Curr Opin Genet Dev. 2017. October;46:95–103. PubMed PMID: 28753462. [DOI] [PubMed] [Google Scholar]
- [62].Canalis E, Notch signaling in osteoblasts. Sci Signal. 2008. April 29;1(17):pe17 PubMed PMID: 18445833. [DOI] [PubMed] [Google Scholar]
- [63].Chau MD, Tuft R, Fogarty K, et al. Notch signaling plays a key role in cardiac cell differentiation. Mech Dev. 2006. August;123(8):626–640. PubMed PMID: 16843648; PubMed Central PMCID: PMCPMC1567976. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.