

ABSTRACT
TLR7 agonists are considered promising drugs for cancer therapy. The currently available compounds are not well tolerated when administered intravenously and therefore are restricted to disease settings amenable for topical application.
Here we present the preclinical characterization of SC1, a novel synthetic agonist with exquisite specificity for TLR7. We found that intravenously administered SC1 mediates systemic release of type I interferon, but not of proinflammatory cytokines such as TNFα and IL6, and results in activation of circulating immune cells. Tumors of SC1-treated mice have brisk immune cell infiltrates and are polarized towards a Th1 type signature. Intratumoral CD8+ T cells and CD11b+ conventional dendritic cells (cDCs) are significantly increased, plasmacytoid dendritic cells (pDCs) are strongly activated and macrophages are M1 phenotype polarized, whereas myeloid-derived suppressor cells (MDSCs) are decreased.
We further show that treatment of mice with SC1 profoundly inhibits the growth of established syngeneic tumors and results in significantly prolonged survival. Mice, which are tumor-free after SC1 treatment are protected from subsequent tumor rechallenge. The antitumor effect of SC1 depends on antigen-specific CD8+ T cells, which we found to be strongly enriched in the tumors of SC1-treated mice.
In conclusion, this study shows that systemically administered SC1 mobilizes innate and adaptive immunity and is highly potent as single agent in mice and thereby provides a rationale for clinical testing of this compound.
KEYWORDS: TLR7 ligand, CD8+ T cells, type I interferon, cancer immunotherapy
Introduction
Synthetic compounds mimicking pathogen–associated molecular patterns (PAMPSs) may overcome cancer-associated immune suppression and promote tumor cell killing by innate and adaptive immune mechanisms.1 Toll-like receptors (TLRs) are key sensors for such patterns. Engagement of TLRs induces recruitment of MyD88, interferon (IFN) regulatory factors and NF-κB, culminating in rapid induction of a type-1 IFN response together with the secretion of proinflammatory cytokines such as IL-1β, IL-6 and IL-12.2,3 The virus-sensing members of the TLR family are localized in specialized endosomal compartments, and detect pathogen genomes and replication intermediates. TLR7, the sensor for viral uridine- and guanosine/uridine-rich ssRNA, has moved into the spotlight as a target for novel immune-modulatory drugs. TLR7 is primarily expressed on plasmacytoid dendritic cells and on monocytes and B-cells.4,5 IFNα release is considered the critical mediator of anti-viral and anti-tumoral effects of TLR7-signaling.3,6-8
Various synthetic TLR7 agonists, including the imidazoquinolines imiquimod (R837), 852A, and resiquimod (R848), are being studied as immunotherapy drugs and have strong anti-tumor activity.9-11 So far only imiquimod is approved for human use. Repetitive intravenous (i.v.) administration of imiquimod at a systemically effective IFNα-inducing dose level is associated with dose-limiting adverse reactions.12,13 As a consequence, imiquimod is registered as a cream formulation for topical use in localized diseases such as basal cell carcinoma, genital warts, and bladder cancer.14 Similarly, repeat-dose i.v. regimens of other TLR7 agonists in development, e.g. R848 and 852A, are not well tolerated limiting the clinical use of these compounds to the treatment of localized disease and requiring their combination with other active drugs.15-17 We and others hypothesized that the unfavorable safety profile may be attributable to the release of a broader array of proinflammatory cytokines on top of the intended type 1 IFN induction.18,19
SC1, a novel TLR7-selective agonist, has been designed to mediate primarily IFNα-release as opposed to a broader array of cytokines. First indications of preclinical anti-tumor activity have been demonstrated in a pulmonary metastatic Renca model, in which i.v. SC1 prevented colonization of the lung with metastases.19 Moreover, subcutaneously administered SC1 in a treatment model of RMA-S lymphoma has been shown to revert NK cell anergy and to result in potent tumor-rejection.20
The objective of our study was to investigate immuno-pharmacodynamics and anti-tumor activity of systemic SC1 exposure in mouse tumor models in more detail with a particular focus on adaptive immunity. We demonstrate here that SC1 activates strong antitumor mechanisms including antigen-specific CD8+ T cells and long-term T-cell memory, which contribute to durable rejection of syngeneic tumors in mice. Our data extend the knowledge base on SC1 and inform drug development rationales for clinical testing of this unique compound.
Results
Intravenous administration of SC1 induces TLR7-dependent immune cell activation and IFNα–selective systemic cytokine release
We performed a series of experiments to determine a dose range, in which a single i.v. dose of SC1 would be well tolerated. At 10 mg/kg of SC1 no subclinical or clinical abnormalities as assessed per inspection related to SC1 were observed with regard to general health, body weight, activity, mobility, and behavior (Supplementary Figure1A right and data not shown). The tested parameters of a standard clinical chemistry panel in mice after a single dose of 15 mg/kg SC1 and of hematology in mice exposed to 10 mg/kg SC1 were largely within the normal reference range (Supplementary Figure 1A, B). Twenty-four hours after dosing 10 mg/kg SC1 lymphopenia was detected, which was transient and back to normal after 72 h. Liver enzymes were re-tested at a dose of 3 mg/kg SC1 and were found to be within the normal range (Supplementary Figure 1C).
Based on the finding that mice treated with SC1 at 3 mg/kg but not at 1 mg/kg and lower doses showed the intended IFNα release in combination with the lack of toxicity as described above, the dose range of 3–6 mg/kg was chosen for the pharmacodynamic studies.
A single dose of 3 mg/kg resulted in a steep and profound increase of IFNα, the key cytokine of TLR7 agonist activity, peaking 1 h post injection (Figure 1(a)) and returning to baseline within 24 h. IFNα release did not occur at doses of 1 mg/kg and lower. When 3 mg/kg SC1 was repeatedly administered every 5 d, IFNα release continued to be robustly induced by each dosing (Figure 1(b)).
Figure 1.
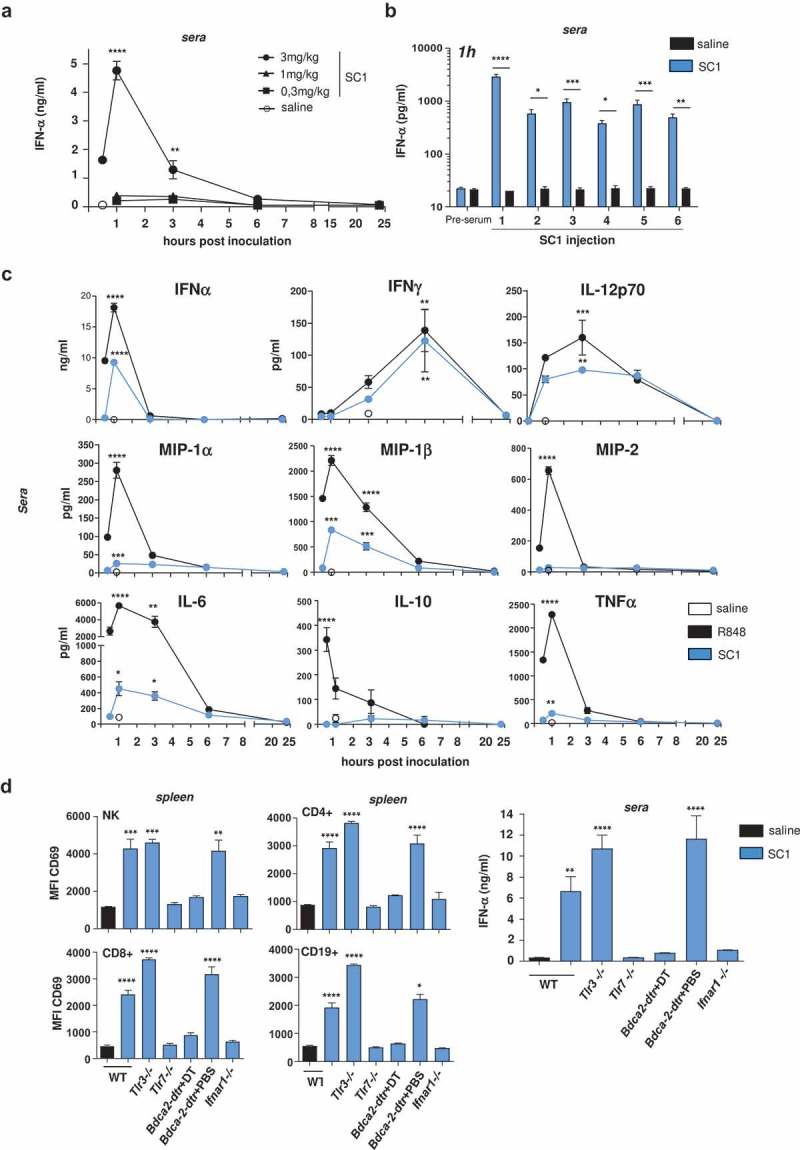
Intravenous administration of SC1 induces TLR7-dependent immune cell activation and IFNα-selective systemic cytokine release.
BALB/c and C57BL/6 wild-type (WT) mice and knock-out variants were injected i.v. with a single or repetitive dose of SC1 (3 mg/kg or as otherwise indicated) or vehicle (saline) every 5 d. (a) Kinetics of IFNα levels in sera of BALB/c WT mice were measured after a single injection of SC1. Data representative of two independent experiments. (b) BALB/c WT mice (n = 3) were given 6 serial i.v. injections of 3 mg/kg SC1 or vehicle (saline) every 5 d. Serum IFNα levels were measured 1 h after each injection. Data representative of two independent experiments. (c) Kinetics of cytokine levels in sera of BALB/c WT mice were measured after a single injection of SC1 or equimolar dose of R848 was measured by Multiplex. (d) CD69 surface expression on different lymphocyte subtypes in the spleen of C57BL/6 WT and knock-out mice 24 h after a single injection of SC1 was measured by flow cytometry (left); IFNα levels in sera 1 h after injection of SC1 was measured by ELISA (right). Data shown as mean ± s.e.m of (a) n = 4–15, (B-D) n = 3–5 mice per group, p*<0.05, p**<0.01, p***<0.001, p****<0.0001 using one-way ANOVA-test using saline as reference group, (c) at the peak of the expression.
Next, we studied serum levels of cytokines commonly associated with pharmacodynamics of TLR7 agonist exposure in mice treated intravenously with a single 3 mg/kg SC1 dose as compared to an equimolar dose of R848 (resiquimod). IFNα, IFNγ, and IL-12p70 were induced at similar levels by both TLR7 agonists (Figure 1(c)). However, whereas R848 treatment resulted in high levels of cytokines considered as strongly pyrogenic such as MIP-1α (CCL3), IL-6, MIP-2 (CXCL2), as well as in robust induction of TNFα, and MIP-1β (CCL4), and of IL-10, these cytokines were barely or moderately induced by SC1 (Figure 1(c)). This data confirmed and further extended findings of a previous study by Wiedemann et al. in the C57BL/6 mice strain using a different injection route,20 with the exception that SC1-induced IL6 response had been reported to be closer to those levels induced by R848. We showed that SC1-induced IL6 response in BALB/c mice does not depend on the injection route (Supplementary Figure 2), however, cannot exclude that other mouse strains react differently.
Furthermore, in spleens of mice treated with SC1 we found CD4+ and CD8+ T cells, NK cells, and B cells, to be strongly activated as indicated by CD69 upregulation (Figure 1(d) left and middle).
SC1-mediated IFNα release (Figure 1(d) right) as well as immune cell activation (Figure 1(d) left, middle) were strongly impaired in mice deficient for TLR7 (Tlr7−/-) and for the IFNα receptor (Ifnar1−/-). SC1 effects were also abrogated in Bdca2-DTR mice, which were depleted of plasmacytoid dendritic cells (pDCs) by diphtheria toxin (DT) injection (Figure1(d) right).
In summary, these data indicate that SC1 treatment induces TLR7-dependent immune cell activation and IFNα–selective systemic cytokine release in mice.
SC1 treatment of mice with established tumors mediates efficient pDC and IFNα signaling-dependent tumor rejection and survival benefit
The therapeutic effect of treatment with repeated doses of SC1 was assessed in three syngeneic mouse tumor models.
First, in a pulmonary metastasis model initiated by orthotopic injection of 4T1 breast cancer cells, SC1 was shown to effectively prevent the formation of spontaneous lung metastases, whereas vehicle-treated mice (saline) as control developed a massive pulmonary tumor load (Figure 2(a)).
Figure 2.
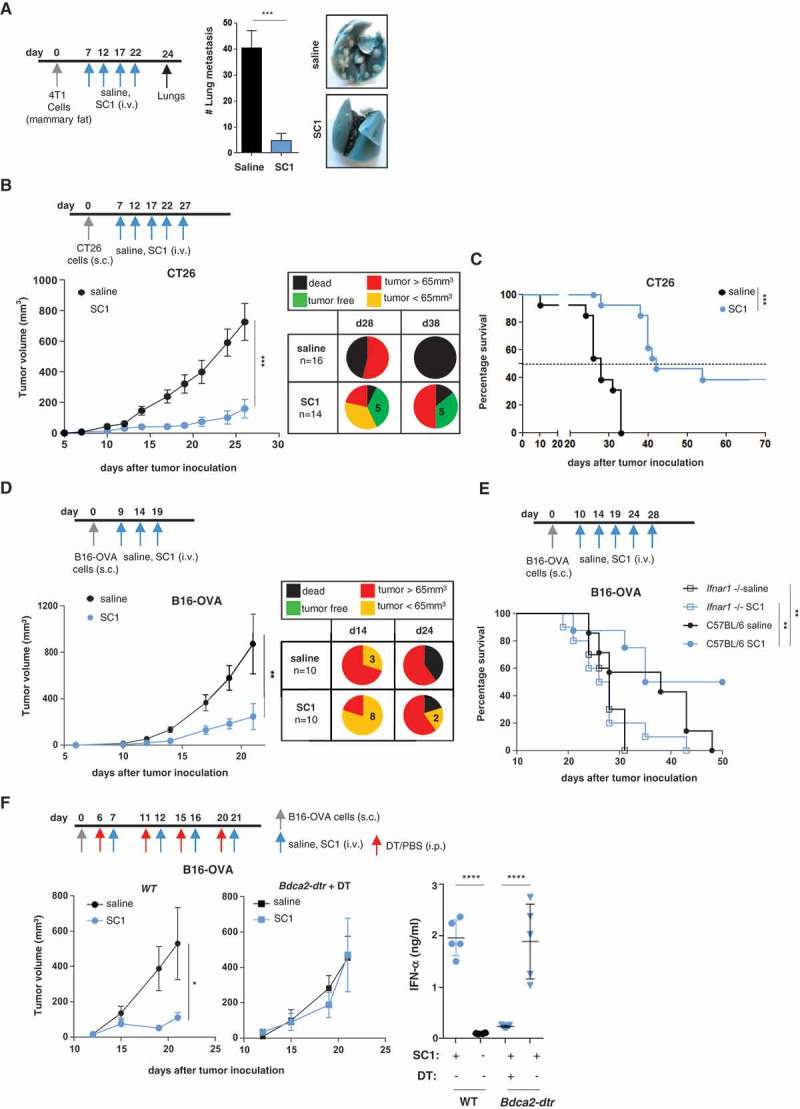
SC1 treatment of mice with established tumors mediates efficient pDC- and IFNα-dependent tumor rejection and survival benefit.
BALB/c or C57BL/6 mice were injected with tumor cells. When tumors reached sizes of 20–50 mm3 mice were treated i.v. with SC1 or with vehicle (saline) every 5 d. (a) Macroscopic spontaneous lung metastasis count in BALB/c mice (n = 7 per group) on d 24 after orthotopic 4T1 cell injection. Data representative of two independent experiments. (b) Tumor growth in BALB/c mice (n = 16/group) injected s.c. with CT26 cells. Pie charts show the proportion of mice displaying the indicated tumor stage at d 28 and 38 after CT26 inoculation. Tumor-free (TF), dead mice and mice bearing different size of tumors are indicated. (c) Survival of CT26-tumor-bearing mice. Data representative of more than three independent experiments. (d) Tumor growth in C57BL/6 mice (n = 10) injected s.c. with B16-OVA cells. Pie charts show the proportion of mice displaying the indicated tumor stage at d 14 and 24 after tumor inoculation. (e) Survival of C57BL/6 and Ifnar1−/- mice (n = 7–10) injected with B16-OVA cells. (f) Tumor growth in C57BL/6 WT (n = 8, left) and Bdca2-dtr mice (n = 6, center) injected s.c. with B16-OVA cells and i.p. with DT. IFNα levels in the sera of mice 1 h after the first i.v. injection with SC1 or vehicle (saline) was measured by ELISA (right). Data shown as mean ± s.e.m.of the indicated experimental numbers, p*<0.05, p**<0.01, p***<0.001, p****<0.0001 using Mann–Whitney test (A, B, D, and F, left) or one-way ANOVA-test (F, right) or Log-rank (Mantel-Cox) test (c,e).
Next, we studied SC1 treatment in two models with established subcutaneous tumors, one being CT26 colon carcinoma in BALB/c mice and the other B16-OVA melanoma in C57BL/6 mice (Figure 2(b-f)). In the CT26 tumor model treatment with SC1 as compared to control resulted in tumor growth inhibition, sustained tumor rejection in about one-third of mice (Figure 2(b)) and significantly improved median overall survival (42 versus 28 d) (Figure 2(c)). In the B16-OVA melanoma model, the capacity of SC1 to inhibit the growth of established tumors and to convey survival benefit was confirmed (Figure 2(d,e)).
These treatment experiments were performed with tumors in the size range of 20–50 mm3 reached 7–13 d after inoculation which are well palpable, well-engrafted and have an established tumor microenvironment and commonly accepted models for assessing single-agent activity of new compounds, particularly TLR agonists.10,20-22 We further challenged the potency of SC1 by treating mice at a more advanced stage with large-sized fast progressing tumors (average size at treatment start 110–160 mm3) and found single agent SC1 not to be efficacious at this stage (Supplementary Figure 3).
SC1-mediated tumor growth inhibition and survival in these mouse models were found to depend critically on pDCs and on IFNα signaling, as they were abrogated in mice, which were Ifnar1−/- (Figure 2(e)), pDC-depleted (Figure 2(f)) or treated with a blocking IFNAR1 monoclonal antibody (Supplementary Figure 4).
Next, we benchmarked SC1 activity in the CT26 model to that of equimolar amounts of other imidazoquinolines. SC1 significantly outperformed 852A with regard to both IFNα induction and prolongation of median overall survival (Supplementary Fig. 5A). As repeated i.v. administration of R848 was not tolerated at the respective dose level, we injected both R848 and SC1 intratumorally for the purpose of comparison and again found SC1 to be significantly superior in the induction of IFNα, however comparable effect for tumor survival (Supplementary Fig. 5B).
Intravenous administered SC1 is retained in the tumor environment and associated with activation of immune cells and TH1 polarization
A series of experiments investigated SC1 effects at the tumor site. First, we studied the biodistribution of intravenously administered SC1 in mice bearing established subcutaneous CT26 tumors. One hour after dosing comparably high levels of SC1 were detectable in well-vascularized organs such as liver and spleen, but also in the tumor tissue. Whereas clearance from hepatic and splenic tissue was rapid, SC1 retention in tumor tissue was markedly prolonged and still robustly detectable after 24 h (Figure 3(a)).
Figure 3.
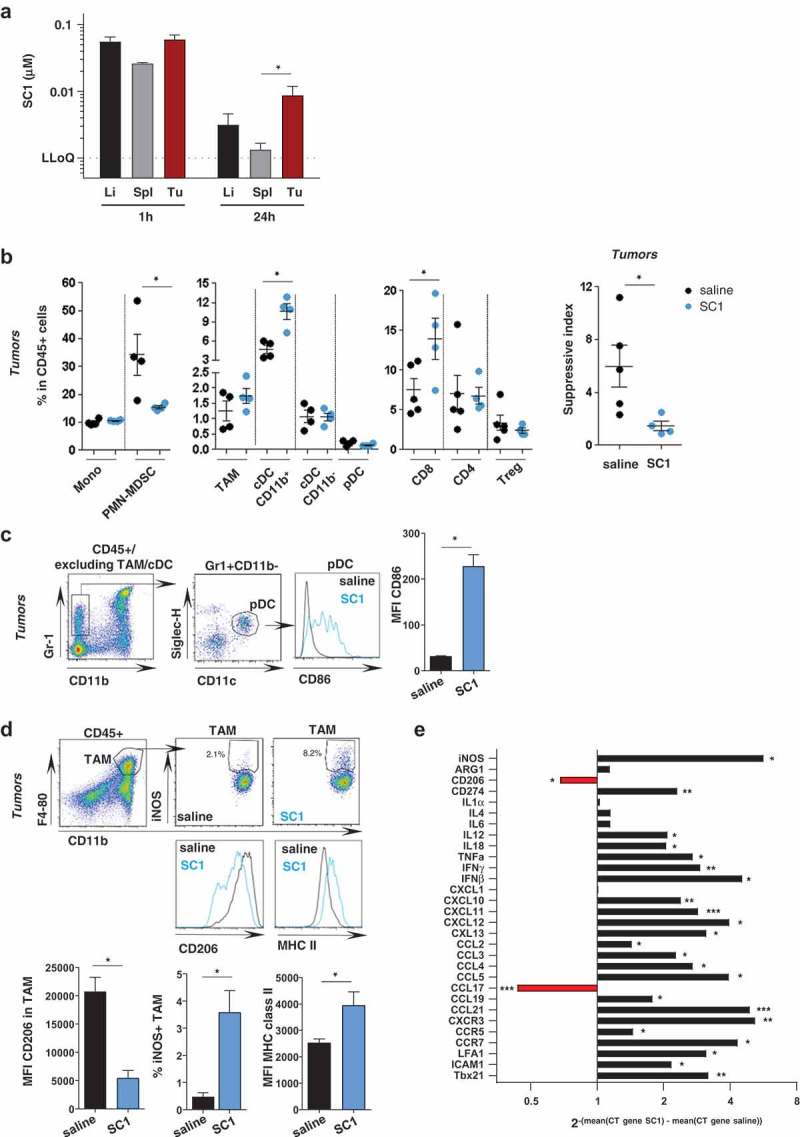
Intravenous administered SC1 is retained in the tumor environment and associated with activation of immune cells and Th1 polarization.
BALB/c mice were injected s.c. with CT26 cells. (a) After tumors reached sizes of 70–100 mm3, mice (n = 3–5/group) were i.v injected with single dose of SC1 or with vehicle (saline). Kinetics of SC1 quantification detected in liver (Li), spleen (Sp) and tumor (Tu) at 1 h and 24 h after injection were performed by LC-MS/MS. LLOQ: lower limit of quantification. Data representative of two independent experiments. B-E) After tumors reached sizes of 20–50 mm3, mice were treated i.v. with SC1 or with vehicle (saline) every 5 d. 24 h after treatment with fourth dose mice were sacrificed and tumors collected for flow cytometric analysis (B-D, data representative of two independent experiments) and for differential gene expression analysis (E). (b) Indicated leukocyte populations identified in CT26 tumors as a percentage of total CD45+ infiltrated cells (left). Suppressive index was calculated as the ratio of the frequencies of PMN-MDSC and CD8+ T cells (right). (c) Representative flow cytometry plots of the intratumoral pDC and CD86 expression. (d) Representative flow cytometry plots of TAM and level of expression of iNOS, CD206 and MHC class II. (e) mRNA expression levels of immuno-modulators in tumor samples from SC1 and vehicle (saline) treated mice. Data shown as mean ± s.e.m. of the indicated numbers of mice, p*<0.05, p**<0.01 using Mann–Whitney test (A-D) and unpaired t-test. (E).
Next, we analyzed the tumor environment in samples resected from SC1- and vehicle-treated mice. Phenotyping of immune cell infiltrates by flow cytometry (Supplementary Fig. 6) showed that SC1 treatment significantly increases the frequency of CD8+ T cells among CD45+ cells, while decreasing polymorphonuclear myeloid-derived suppressor cells (PMN-MDSC) (Figure 3(b)), resulting in a significantly lower suppressive index within the tumor environment (Figure 3(b) right). Frequencies of tumor-infiltrating CD4+ and regulatory T cells were not affected by SC1 treatment (Figure 3(b)). Furthermore, CD11b+ cDCs were significantly increased in frequency (Figure 3(b)). pDCs were not changed in number by SC1-treatment, but exhibited an activated phenotype with increased surface expression of CD86 (Figure 3(c)). Also, tumor-associated macrophages (TAMs) did not significantly change numerically in SC1-treated mice but were strongly skewed towards an activated M1-like phenotype, with downregulation of CD206 and upregulation of iNOS and MHC class II (Figure 3(d)).
Comprehensive gene expression analysis confirmed these findings and revealed further substantial differences between the tumors of SC1- and vehicle (saline) treated mice as control. Markers for Th1 (Il12, Il18, Tnfα, Ifnγ, Cxcl10, Cxcl11, Cxcr3, and Ccr7), but not for Th2 orientation (Il4, Il6) were strongly upregulated. Proinflammatory markers (Ifnγ, Tnfα, Ccl2, Ccl3), markers of T-cell activation (Ccl5, Ccr5), T-cell attraction and extravasation (Cxcr1, Icam-1, Lfa-1, Ccl21, Ccl19) were all significantly upregulated. Markers for type 2 macrophages (CD206) were downregulated, whilst molecules associated with type 1 macrophage were upregulated (CD274, iNos, Ccr7, Cxcl9, Cxcl10, Cxcl11, Cxcl13) (Figure 3(e)).
Altogether, these data indicate that systemic treatment with SC1 has a strong local effect on the tumor environment and polarizes towards an inflammatory, Th1 cytotoxic immune contexture.
Antigen-specific CD8+ T cells are critical for durable SC1-mediated tumor control and protection from tumor rechallenge
Next, we investigated the effect of SC1 on antigen-specific immune responses.
T-cells directed against gp70, the immune dominant antigen of CT26 cells, were assessed by tetramer staining in CT26-tumor-bearing mice after treatment with SC1 or saline as control. In the peripheral CD8+ T-cell population of SC1-treated mice, the frequencies of gp70-specific CD8+ T cells were markedly increased (Figure 4(a)). In contrast, we did not detect circulating gp70-specific CD8+ T cells in mice treated i.v. with equimolar doses of 852A in line with the lack of beneficial effect on survival (Supplementary Fig. 5A right). R848 was equally potent as SC1 at inducing gp70-specific CD8+ T cells in blood when both these compounds were injected intratumorally (Supplementary Fig. 5B right).
Figure 4.
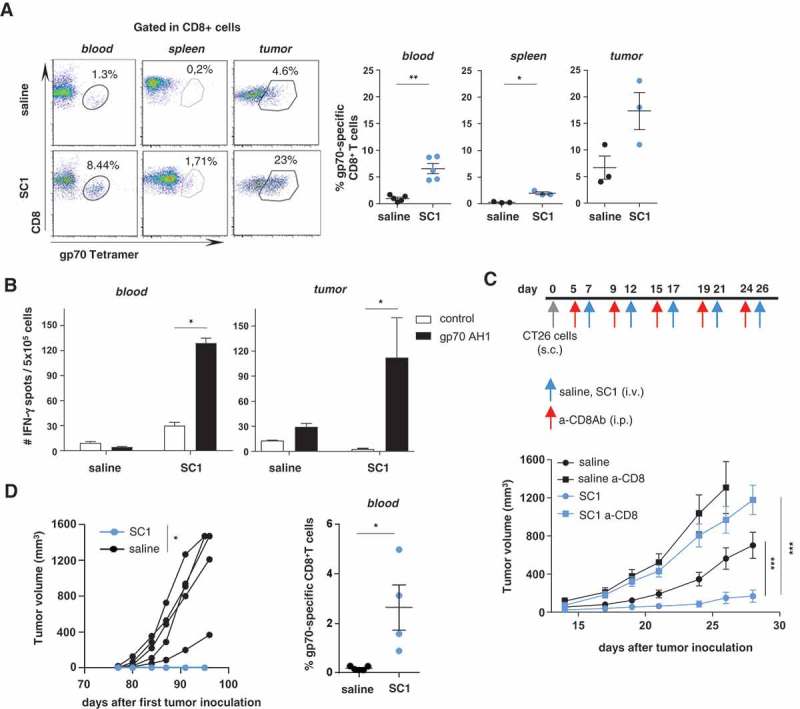
Antigen-specific CD8+ T cells are critical for SC1-mediated tumor control, and protection from tumor rechallenge.
(a-d) BALB/c mice were engrafted with CT26 cells and treated with four doses of SC1 or vehicle (saline) every 5 d. (a) The percentage of gp70-specific CD8+ T cells in blood (at d 21 after tumor inoculation, three independent experiments), spleens and tumors (on d 26, 2 independent experiments) was measured by tetramer staining by flow cytometry. (b) Lymphocytes were isolated from blood at d 21 (n = 5) (left) and from tumors at d 26 (n = 3) (right) and the number of IFNγ-producing cells was measured by ELISpot. (c) BALB/c mice (n = 10) were injected s.c. with CT26 cells, then treated with anti-CD8 antibody prior to each i.v. dose of SC1 or vehicle (saline). Tumor growth over time is shown. (d) Mice were treated as described in Figure 2(b), and all SC1-treated, tumor-free mice (n = 4) were rechallenged s.c. with CT26 cells on d 77 after the first tumor injection. Control mice (n = 5). Tumor growth in mice is shown (left). The percentage of gp70-specific CD8+ T cells in the blood at d 90 after first CT26 tumor inoculation (d 13 after the CT26 tumor rechallenge) was measured by tetramer staining (right). Data shown as mean ± s.e.m, of the indicated numbers of mice. p*<0.05, p**<0.01, p***<0.001, using Mann–Whitney test (A, B, and D), one-way ANOVA-test (C).
Remarkably, CT26 tumors of SC1 – but not vehicle-treated mice were heavily infiltrated with gp70-specific CD8+ T cells constituting about 20% of the intratumoral CD8+ T cell population (Figure 4(a)). The tumor-infiltrating as well as circulating gp70-specific CD8+ T cells in SC1-treated mice were functional and capable of secreting IFNγ upon antigen recognition in vitro (Figure 4(b)).
The antitumor effect of SC1 treatment in CT26-bearing mice was completely abolished by depletion of CD8+ T cells, indicating the critical role of antigen-specific cytotoxic T lymphocytes as effectors for SC1 activity (Figure 4(c)). Splenectomy of mice prior to treatment with SC1, however, did neither compromise the antitumor effect of this compound nor the induction of circulating antigen-specific T cells suggesting that SC1 activity does not depend on this immune compartment (Supplementary Fig. 7).
SC1-treated mice, which experienced full tumor rejection remained tumor-free and on rechallenge with CT26 tumor cells spontaneously rejected these (Figure 4(d) left). In these mice circulating gp70-specific CD8+ T cells were detectable about two weeks after the CT26 tumor rechallenge (Figure 4(d) right). In contrast, naïve control mice that were grafted with CT26 cells for the first time experienced fast tumor growth and died within 27 d.
These data indicate that in mice SC1 efficiently induces antigen-specific CD8+ T cells, which are potent mediators of durable tumor clearance and protective memory.
Discussion
The scope of this study was to assess the antitumor activity of the TLR7 ligand SC1 with a focus on adaptive immunity in preclinical tumor models and investigate its mode-of-action. SC1 was developed under the educated assumption that the dose-limiting side effects observed with i.v. administration of other TLR7 agonists may be attributable to the systemic release of a broader array of proinflammatory and pyrogenic cytokines to which a cross-reactivity to TLR8 may contribute.13,18,19
As shown by benchmarking to 852A and resiquimod (R848), SC1 has distinctive features in this regard. For the one, SC1 dosing releases considerably higher levels of the therapeutically relevant IFNα. Second, whereas other TLR7 agonists have been reported to render the IFN response refractory and thus induce strong tachyphylaxis,22-24 we showed that each dosing of a 5-daily repeat-drug regimen with SC1 is still capable of inducing a substantial IFNα serum peak. Third, further extending previous reports,20 we showed that distinctively SC1 was not associated with release of pyrogenic cytokines, e.g., IL6, TNFα, MIP1β. These observations indicate that in terms of its activity and its tolerability profile SC1 may differ from other TLR agonists.
Whereas systemic cytokine release by SC1 appears to be type 1 IFN dominated, locally at the tumor site we found activation of a broader array of cytokines resulting in a strong proinflammatory immune signature and reversion of the suppressive milieu. In this regard, it is noteworthy, that in the tumors of mice treated with SC1 intravenously, we found high concentrations and prolonged retention of SC1.
Another key finding is the strong activity of SC1 as a single agent at systemically tolerated doses, which we demonstrated in melanoma and colorectal cancer tumor models. SC1 prevents spontaneous lung metastasis, retards tumor growth, results in tumor rejection, prolongs survival and, most importantly, protects from tumor rechallenge.
We showed that innate mechanisms such as TLR7, IFNα signaling, and pDCs, which are most likely the major source of IFNα, are required to actualize in vivo activity of SC1. Of note, tumors of SC1 treated mice were found to be strongly enriched for M1 polarized macrophages and for activated pDCs and their downstream effects, e.g., IL18, IFNβ upregulation.8
Our data revealed that tumors of SC1 treated mice display a signature, which is associated with promoting local recruitment and activation of effector T cells. CXCL10, CXCL11, CXCL13 CCL2, CXCR3 and integrins LFA-1, ICAM-1, which are known to promote the mobilization and transmigration of antigen-specific CD8+ T cells into the tumor bed, are upregulated. These factors have been associated with local immune stimulation and regression of tumors upon therapeutic treatments.10,25 We also found the CCR7/CCL19-CCL21 axis, which in melanoma patients and mouse models is involved in the recruitment of memory T cells and dendritic cells,26 to be upregulated by SC1.
In line with these findings, we demonstrated that SC1 executes its activity through antigen-specific CD8+ T cells as mediators of adaptive immunity. We detected antigen-specific CD8 + T cells not only in the circulation but also as significant fraction of the tumor-infiltrating CD8 + T cells. That the in vivo antitumor effect of SC1 treatment is not abrogated in splenectomized mice implies that the draining lymph node may be the place for efficient priming of these T cells.
We showed that the induced antigen-specific T cells were functional and capable of releasing IFNγ, were durable and protective against rechallenge.
With this, we demonstrate for the first time that the primary pharmacodynamic effects as well as the antitumor activity of SC1 depend not only on innate but also on adaptive immune mechanisms.
In summary, our findings provide evidence that SC1 at systemically tolerated doses activates innate and adaptive immunity and is an attractive candidate for further exploration in the clinical setting.
Material and methods
Cell lines
All cell lines used in this study were free of Mycoplasma and maintained as specified by the vendor. The 4T1 breast and CT26 colon cancer cell lines were purchased from ATCC. B16-OVA, a murine B16F10 melanoma cell line expressing the chicken ovalbumin gene (OVA) containing the H2-Kb-restricted OVA257-264 epitope (SIINFEKL) was a gift from U. Hartwig.
Mice and ethical clearance
The following strains of age-matched female 8–10 weeks old mice were used in experiments: BALB/c mice from Janvier Labs, C57BL/6 mice from Envigo RMS GmbH (former Harlan Laboratories), Bdca2-dtr8 and Tlr3-/-27 mice from Jackson Laboratory. Tlr7−/− mice on a C57BL/6 background were a kind gift from H. J. Schild, C57BL/6 Ifnar1−/− mice,28 derived from 129Sv Ifnar−/- mice29 via backcrossing, were a gift from J. Kirberg (Paul-Ehrlich-Institute). Mice were kept under standard conditions at the Animal Research Center of the Johannes Gutenberg – University of Mainz. Animal studies were performed in compliance with the German animal welfare act and regulations of the state of Rhineland-Palatinate. All animal studies were carried out in strict accordance with national and European guidelines for the care and use of laboratory animals (European regulations; 2010/63/EU). Animal studies were approved by either the Regierungsbehörde of Rhineland-Palatinate, Koblenz (authorization number G14-8–025) or of Oberbayern, München (authorization number 55.2–1-54–2532.2–9-11). All procedures were in compliance with the German Animal Welfare Act and German regulations (TierSchG/TierSchVersV).
Tissue preparation
Blood for ELISA, multiplex cytokine analysis and clinical chemistry parameters was collected from the retro-orbital sinus in heparin-coated serum tubes (BD Microtainer). Spleen single-cell suspensions were prepared in PBS by passing the tissue through a 70-μm cell strainer (BD Falcon) using the plunger of a 3 ml syringe. Erythrocytes were removed by hypotonic lysis (154 mM NH4CL, 10 mM KHCO3, 127 µM EDTA). Tumor samples were cut into small pieces and incubated for 20 min in digestion buffer (1 mg/ml collagenase type IV, 40 μg/ml DNase I and 500 U/mg hyaluronidase). After passing through a cell strainer, the erythrocytes were removed by hypotonic lysis.
Enzyme-linked immunosorbent assay
Serum levels of IFNα were measured by sandwich ELISA (Pbl Assay Systems) according to manufacturer's instructions.
Cytokine multiplex analysis
Serum cytokine levels were measured by Multiplex analysis (Procartaplex, ProcartaPlex Analyst 1.0, ThermoFisher Scientific) according to manufacturer's instructions.
Clinical chemistry parameters and blood counts
Peripheral cell populations after SC1 administration was measured using VetScan HM5 Hematology Analyzer, and LDH, ALT, AST, and bilirubin levels were analyzed with the IndikoTM clinical and specialty chemistry analyzer, Thermo Fisher using kits purchased from ThermoScientific (clinical chemistry diagnostics) according to manufacturer's instructions.
Tumor models and treatments
BALB/c or C57BL/6 mice were inoculated s.c. with 2.5 × 105 CT26-WT or 1 × 105 B16-OVA cells into the flank, respectively. The 4T1 cell line (1x105 cells) was injected orthotopically in the mammary fat pad. Once tumors reached palpable sizes, mice were randomly divided into treatment and control groups. Tumor volume was measured unblinded with a caliper and calculated using the formula (a × b2)/2 (a being the largest and b being the smallest diameter of the tumor). Tumor growth was documented as mean tumor size with standard error disregarding single distant outliers. Treatment was initiated after 7–10 d with tumors having reached a volume of 20–50 mm3. 3 mg/kg (75 µg/mouse; BALB/c mice) or 6 mg/kg (150 µg/mouse; C57BL/6 mice) SC1 (4SC Discovery GmbH, Martinsried, Germany) was administered by i.v. injection to the retro-orbital venous plexus. Control groups received an equal volume of 100 µl saline vehicle (0.1% 1 M HCl, 0.1% 1 M NaOH, 99.8% isotonic NaCl solution pH 5.63). For depletion of pDC, Bdca2-dtr mice were injected with diphtheria toxin (DT) from corynebacterium diphtheria (Sigma D0564) (4,5 ng/g mouse, i.p.) 24 h previously each SC1 injection. 852A and R848 (BioNTech Small Molecules GmbH) were injected in equimolar dose to SC1. Due to the known toxicity induced by systemic delivery of R848, we performed a comparative study using intra-tumoral delivery to mimic local treatment. A maximum of eight treatments was administered every 4–5 d during the course of tumor growth, alternating the injection site. When tumor volume reached 1500 mm3, mice were sacrificed. Spontaneous lung metastases in the 4T1 tumor model were assessed on d 28 shortly before mice exhibited impaired breathing. Mice were killed and tumor burden was quantified unblinded after intratracheal ink (85 ml PBS, 15 ml ink) injection, and fixation with Fekete’s solution (5 ml 70% ethanol, 0.5 ml formalin, and 0.25 ml glacial acetic acid).30 After 2–6 h, tumor lesions were bleached whereas normal lung tissue remained stained. BALB/c mice injected s.c. with CT26 cells were treated i.p. with 250 µg/mouse anti-CD8 depleting antibody (clone YTS169.4, BioXcell) or 100 µg/mouse anti-IFNAR blocking antibody (MAR1-5A3, BioXcell) 18-24 h prior to each i.v. dose of SC1 or vehicle (saline) as control. SC1-treated mice showing rejection of CT26 tumors and long-term survival were rechallenged on d 80 with the same number of tumor as at initial challenge, but not treated with SC1.
Splenectomy
Mice were anesthetized using 120 mg/kg Ketamine and 16 mg/kg Xylazine i.p. and placed on a warming mat. Before and after operation mice received 500 µl PBS s.c. Mice were shaved on the left flank, and after disinfection the abdominal cavity was opened carefully with scissors. The spleen was removed by cauterization of supplying blood vessels. After ensurance for absence of bleeding, abdominal cavity and skin were sutured closed. Mice were kept under a warming lamp while recovering from anesthesia to maintain body temperature. Sham control mice are equivalently treated but omitting the resection of the spleen. To alleviate pain, mice were dosed with two drops Metamizol orally before surgery as well as with 5 mg/kg Carprofen s.c. 3 h after the operation and once daily for 2–3 consecutive d.
SC1 tissue quantification
Two replicates per organ (80–120 mg) were transferred into tubes (Precellys®24-Dual, 2 mL ceramic mix bead CKM 1.4/2.8 mm) with PBS buffer pH (7.4) and homogenized using the Precellys®24-Dual (Bertin Technologies) at 6500 rpm for 20 sec twice). Acetonitrile (ACN) was added, followed by homogenization (6000 rpm; 1 × 30 sec). All steps were performed on ice with pre-chilled buffers. After centrifugation (6000 x g; 15 min; 4°C) 400 µl supernatant was transferred to HPLC Micronics tubes and either analyzed by LC-MS/MS or stored at −80°C.
Flowcytometry on tumor-infiltrating leukocytes
Cells from blood or tissues were washed with PBS after erythrocyte lysis. Live/dead staining was performed using LiveDead Blue (eBioscience). Cells were washed once with PBS containing 2% FBS (FACS buffer). Single-cell suspensions were pre-incubated with Fc Blocker (anti-mouse CD16/CD32) for 10 min on ice followed by incubation with conjugated antibodies at 4°C for 30 min in the dark. Cells were washed twice with FACS buffer prior to acquisition. For intracellular staining (for Foxp3, CD206, and iNOS staining) extracellularly labeled cells were washed and incubated with FACS intracellular buffers (BD Pharm fix and perm). FACS acquisition was performed using Fortessa (BD) flow cytometer. Data were analyzed using Flowjo version 7.6.5 (Treestar). Monoclonal antibodies for extracellular staining are listed in Table 1. CD8+ T cells recognizing gp70 AH1-A1 were detected with H-2Ld/AH1423-431 (SPSYVYHQF) tetramer kindly provided by the NIH tetramer core facility (Emory University Vaccine Center).
Table 1.
Anti-mouse antibodies for flow cytometry.
| Target | Fluorochrome | Clone | Company | Reference |
|---|---|---|---|---|
| CD45 | V-500 | 30-F11 | BD | 561487 |
| CD11b | PerCPCy5.5 | M1-70 | BD | 550993 |
| CD11c | APC | N418 | Miltenyi | 30–091-844 |
| CD11c | PE-Cy7 | HL3 | BD | 558079 |
| CD16/32 (Fc-block) | purified | 24G.2 | BD | 553142 |
| CD19 | PE | 1D3 | BD | 553786 |
| CD206 | A488 | C068C2 | BioLegend | 141712 |
| iNOS | PE-efl610 | CXNFT | eBioscience | 61–5920-82 |
| CD25 | PE-Cy7 | PC61.5 | eBioscience | 25–0251-82 |
| CD4 | PerCPCy5.5 | RM4-5 | BD | 553052 |
| CD8 | APC-Cy7 | 53–6.7 | BD | 557654 |
| CD49b | FITC | DX5 | BD | 553857 |
| F4-80 | BV421 | BM8 | BioLegend | 123131 |
| Foxp3 | A647 | FJK-16s | eBioscience | 51–5773 |
| Gr1 | APC-Cy7 | RB6-8C5 | BD | 557661 |
| Singlec-H | A647 | ebio440c | eBioscience | 51–0333-82 |
| CD69 | PE | H1.2F3 | BD | 553237 |
| CD86 | PE | GL-1 | BD | 553692 |
| Tetramer AH1 | APC | NIH | ||
| CD40 | PE | MR1 | BD | 553658 |
| LIVE/DEAD® Fixable Dead Cell Stain | Blue, Em 450nm | Invitrogen | L23105 |
Enzyme-linked immunospot (ELISpot)
ELISpot assay was carried out as previously described.31 In brief, responder cells were cultured overnight at 37°C in Multiscreen 96-well plates (Millipore) coated with anti-IFNγ antibody (10 µg/mL, clone AN18, Mabtech) and cytokine secretion was detected with an anti–IFNγ antibody (1 µg/mL, clone R4-6A2, Mabtech) after T-cell stimulation with 2 µg/mL gp70 AH1 or VSV peptide. For analysis of antigen specificity of tumor-infiltrating lymphocytes tumors were digested as described above. Dead cells were removed via density gradient centrifugation (Ficoll-Paque PREMIUM 1,084, GE Healthcare) followed by removal of CT26 tumor cells via plastic adhesion (2–3 h at 37°C) and of dead cells via magnetic beads (Miltenyi). Counted leukocytes were finally re-stimulated with syngeneic peptide-pulsed BMDC. For analysis of T-cell responses in the blood, PBMC were isolated via density gradient centrifugation, counted and re-stimulated by addition of peptide. All samples were tested in duplicates or triplicates and spots counted using Cellular Technology Ltd. Immunospot Reader.
Synthetic peptides
Peptides derived virus H2 – from vesicular-stomatitis virus nucleoprotein (H2-Kb-restricted VSV-NP52-59), and the murine Leukemia Ld restricted gp70423-431 SPSYAYHQF (AH1) peptide were obtained from Jerini Peptide Technologies.
Quantitative reverse transcriptase-PCR
RNA was extracted using the RNeasy Mini Spin kit (QIAGEN) according to the manufacturer’s manual. cDNA was generated with PrimeScript™ RT Reagent Kit with gDNA Eraser (Takara Bio Inc.). qRT-PCR was performed using the BioMark™ HD system (Fluidigm®). Samples and assays were prepared and analyzed according to the “Fast Gene Expression Analysis” using TaqMan® Gene Expression Assays on the BioMark™ or “BioMark™ HD System Fluidigm® Advanced Development Protocol 28”. The 96 × 96 Gene Expression Dynamic Array IFCs were loaded using the IFC Controller HX. Expression values were generated using ΔΔ-Ct calculation method relative to house-keeping gene HPRT1.
Statistics
Biological replicates were used in all experiments unless stated otherwise. Mann–Whitney t-test was used for comparison of two groups. One-way analysis of variance (ANOVA) test was performed when more than two groups were compared with Tukey’s post-hoc test. Survival benefit was determined with the log-rank (Mantel-cox) test. p* ≤ 0.05 was considered significant. All statistical analyses were performed with GraphPad PRISM 7.0. In all experiments, representative images, dot plots, and histograms are shown. No statistical methods were used to pre-determine the sample size for animal or other experiments.
Funding Statement
This work was supported by the technology innovation program of the Rhineland Palatinate government, the ImmunoTransporter 13N13340 (BMBF), the CI3 Cutting Edge Cluster Funding of the German Ministry of Technology (BMBF).
Acknowledgments
The authors thank R. Roth, U. Schmitt, A. König, C. Worm, N. Krimmel, I. Beulshausen, I. Eichelbrönner, M. Suchan, B. Schrörs, M. Löwer, S. Clausen and U. Neupert for technical support; K. Chu for critical reading; NIH Tetramer Core Facility for providing gp70 MHC class I tetramer.
Disclosure of potential conflicts of interests
U.Sahin, O.Türeci, K. Walzer. M.Vormehr, S.Strobl, and R.Rösemann, are employees at BioNTech Corporation (Mainz, Germany). M.Diken and S.Kreiter are working as consultants for BioNTech Corporation (Mainz, Germany). Aforementioned authors may therefore have a financial interest in the development of novel TLR7/8 agonists for cancer therapies. All other authors have no potential conflict of interest.
Author contribution
U.Sahin was responsible for conception and experimental strategy of the study.
Design analysis of the experiments an interpretation of the data was done by F.Vascotto, J.Petschenka., M.Diken, S.Kreiter, and S. Strobl.
F.Vascotto, J.Petschenka, K.Walzer, M.Brkic, M. Vormehr, and R. Rösemann performed the experiments and acquired the data.
F.Vascotto, J.Petschenka, M.Diken, S.Kreiter, Ö.Türeci, and U.Sahin drafted the manuscript.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Adams JL, Smothers J, Srinivasan R, Hoos A.. Big opportunities for small molecules in immuno-oncology. Nat Rev Drug Discov. 2015;14(9):603–622. eng. doi: 10.1038/nrd4596. [DOI] [PubMed] [Google Scholar]
- 2.Swiecki M, Colonna M.. The multifaceted biology of plasmacytoid dendritic cells. Nat Rev Immunol. 2015;15(8):471–485. eng. doi: 10.1038/nri3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G. Type I interferons in anticancer immunity. Nat Rev Immunol. 2015;15(7):405–414. eng. doi: 10.1038/nri3845. [DOI] [PubMed] [Google Scholar]
- 4.Barchet W, Wimmenauer V, Schlee M, Hartmann G. Accessing the therapeutic potential of immunostimulatory nucleic acids. Curr Opin Immunol. 2008;20(4):389–395. eng. doi: 10.1016/j.coi.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 5.Kobold S, Wiedemann G, Rothenfußer S, Endres S. Modes of action of TLR7 agonists in cancer therapy. Immunotherapy. 2014;6(10):1085–1095. eng. doi: 10.2217/imt.14.75. [DOI] [PubMed] [Google Scholar]
- 6.Drobits B, Holcmann M, Amberg N, Swiecki M, Grundtner R, Hammer M, Colonna M, Sibilia M. Imiquimod clears tumors in mice independent of adaptive immunity by converting pDCs into tumor-killing effector cells. J Clin Invest. 2012;122(2):575–585. eng. doi: 10.1172/JCI61034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Le Mercier I, Poujol D, Sanlaville A, Sisirak V, Gobert M, Durand I, Dubois B, Treilleux I, Marvel J, Vlach J, et al. Tumor promotion by intratumoral plasmacytoid dendritic cells is reversed by TLR7 ligand treatment. Cancer Res. 2013;73(15):4629–4640. eng. doi: 10.1158/0008-5472.CAN-12-3058. [DOI] [PubMed] [Google Scholar]
- 8.Swiecki M, Gilfillan S, Vermi W, Wang Y, Colonna M. Plasmacytoid dendritic cell ablation impacts early interferon responses and antiviral NK and CD8(+) T cell accrual. Immunity. 2010;33(6):955–966. eng. doi: 10.1016/j.immuni.2010.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheadle EJ, Lipowska-Bhalla G, Dovedi SJ, Fagnano E, Klein C, Honeychurch J, Illidge TM. A TLR7 agonist enhances the antitumor efficacy of obinutuzumab in murine lymphoma models via NK cells and CD4 T cells. Leukemia. 2017;31(10):2278 eng. doi: 10.1038/leu.2017.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Doorduijn EM, Sluijter M, Salvatori DC, Silvestri S, Maas S, Arens R, Ossendorp F, van der Burg SH, van Hall T. CD4+ T cell and NK cell interplay key to regression of MHC class ilow tumors upon TLR7/8 agonist therapy. Cancer Immunol Res. 2017;5(8):642–653. eng. doi: 10.1158/2326-6066.CIR-16-0334. [DOI] [PubMed] [Google Scholar]
- 11.Inglefield JR, Dumitru CD, Alkan SS, Gibson SJ, Lipson KE, Tomai MA, Larson CJ, Vasilakos JP. TLR7 agonist 852A inhibition of tumor cell proliferation is dependent on plasmacytoid dendritic cells and type I IFN. J Interferon Cytokine Res. 2008;28(4):253–263. eng. doi: 10.1089/jir.2007.0097. [DOI] [PubMed] [Google Scholar]
- 12.Witt PL, Ritch PS, Reding D, McAuliffe TL, Westrick L, Grossberg SE, Borden EC. Phase I trial of an oral immunomodulator and interferon inducer in cancer patients. Cancer Res. 1993;53(21):5176–5180. eng. [PubMed] [Google Scholar]
- 13.Holldack J. Toll-like receptors as therapeutic targets for cancer. Drug Discov Today. 2014;19(4):379–382. eng. doi: 10.1016/j.drudis.2013.08.020. [DOI] [PubMed] [Google Scholar]
- 14.Geisse J, Caro I, Lindholm J, Golitz L, Stampone P, Owens M. Imiquimod 5% cream for the treatment of superficial basal cell carcinoma: results from two phase III, randomized, vehicle-controlled studies. J Am Acad Dermatol. 2004;50(5):722–733. eng. doi: 10.1016/j.jaad.2003.11.066. [DOI] [PubMed] [Google Scholar]
- 15.Geller MA, Cooley S, Argenta PA, Downs LS, Carson LF, Judson PL, Ghebre R, Weigel B, Panoskaltsis-Mortari A, Curtsinger J, et al. Toll-like receptor-7 agonist administered subcutaneously in a prolonged dosing schedule in heavily pretreated recurrent breast, ovarian, and cervix cancers. Cancer Immunol Immunother. 2010;59(12):1877–1884. eng. doi: 10.1007/s00262-010-0914-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weigel BJ, Cooley S, DeFor T, Weisdorf DJ, Panoskaltsis-Mortari A, Chen W, Blazar BR, Miller JS. Prolonged subcutaneous administration of 852A, a novel systemic toll-like receptor 7 agonist, to activate innate immune responses in patients with advanced hematologic malignancies. Am J Hematol. 2012;87(10):953–956. eng. doi: 10.1002/ajh.23280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Christian LM, Porter K, Karlsson E, Schultz-Cherry S. Proinflammatory cytokine responses correspond with subjective side effects after influenza virus vaccination. Vaccine. 2015;33(29):3360–3366. eng. doi: 10.1016/j.vaccine.2015.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gorden KB, Gorski KS, Gibson SJ, Kedl RM, Kieper WC, Qiu X, Tomai MA, Alkan SS, Vasilakos JP. Synthetic TLR agonists reveal functional differences between human TLR7 and TLR8. J Immunol. 2005;174(3):1259–1268. eng. [DOI] [PubMed] [Google Scholar]
- 19.Hamm S, Rath S, Michel S, Baumgartner R. Cancer immunotherapeutic potential of novel small molecule TLR7 and TLR8 agonists. J Immunotoxicol. 2009;6(4):257–265. eng. doi: 10.3109/15476910903286733. [DOI] [PubMed] [Google Scholar]
- 20.Wiedemann GM, Jacobi SJ, Chaloupka M, Krächan A, Hamm S, Strobl S, Baumgartner R, Rothenfusser S, Duewell P, Endres S, et al. A novel TLR7 agonist reverses NK cell anergy and cures RMA-S lymphoma-bearing mice. Oncoimmunology. 2016;5(7):e1189051 eng. doi: 10.1080/2162402X.2016.1189051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yin P, Liu X, Mansfield AS, Harrington SM, Li Y, Yan Y, Dong H. CpG-induced antitumor immunity requires IL-12 in expansion of effector cells and down-regulation of PD-1. Oncotarget. 2016;7(43):70223–70231. eng. doi: 10.18632/oncotarget.11833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bourquin C, Hotz C, Noerenberg D, Voelkl A, Heidegger S, Roetzer LC, Storch B, Sandholzer N, Wurzenberger C, Anz D, et al. Systemic cancer therapy with a small molecule agonist of toll-like receptor 7 can be improved by circumventing TLR tolerance. Cancer Res. 2011;71(15):5123–5133. eng. doi: 10.1158/0008-5472.CAN-10-3903. [DOI] [PubMed] [Google Scholar]
- 23.Koga-Yamakawa E, Murata M, Dovedi SJ, Wilkinson RW, Ota Y, Umehara H, Sugaru E, Hirose Y, Harada H, Jewsbury PJ, et al. TLR7 tolerance is independent of the type I IFN pathway and leads to loss of anti-tumor efficacy in mice. Cancer Immunol Immunother. 2015;64(10):1229–1239. eng. doi: 10.1007/s00262-015-1730-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsitoura D, Ambery C, Price M, Powley W, Garthside S, Biggadike K, Quint D. Early clinical evaluation of the intranasal TLR7 agonist GSK2245035: use of translational biomarkers to guide dosing and confirm target engagement. Clin Pharmacol Ther. 2015;98(4):369–380. eng. doi: 10.1002/cpt.157. [DOI] [PubMed] [Google Scholar]
- 25.Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, Vitale I, Goubar A, Baracco EE, Remédios C, et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med. 2014;20(11):1301–1309. eng. doi: 10.1038/nm.3708. [DOI] [PubMed] [Google Scholar]
- 26.Fankhauser M, Broggi MAS, Potin L, Bordry N, Jeanbart L, Lund AW, Da Costa E, Hauert S, Rincon-Restrepo M, Tremblay C, et al. Tumor lymphangiogenesis promotes T cell infiltration and potentiates immunotherapy in melanoma. Sci Transl Med. 2017;9(407). eng. doi: 10.1126/scitranslmed.aal4712. [DOI] [PubMed] [Google Scholar]
- 27.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by toll-like receptor 3. Nature. 2001;413(6857):732–738. eng. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 28.Kamphuis E, Junt T, Waibler Z, Forster R, Kalinke U. Type I interferons directly regulate lymphocyte recirculation and cause transient blood lymphopenia. Blood. 2006;108(10):3253–3261. eng. doi: 10.1182/blood-2006-06-027599. [DOI] [PubMed] [Google Scholar]
- 29.Müller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264(5167):1918–1921. eng. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- 30.Wexler H. Accurate identification of xperimental pulmonary metastases. J Natl Cancer Inst. 1966;36(4):641–645. eng. doi: 10.1093/jnci/36.4.641. [DOI] [PubMed] [Google Scholar]
- 31.Kreiter S, Selmi A, Diken M, Koslowski M, Britten CM, Huber C, Türeci O, Sahin U. Intranodal vaccination with naked antigen-encoding RNA elicits potent prophylactic and therapeutic antitumoral immunity. Cancer Res. 2010;70(22):9031–9040. eng. doi: 10.1158/0008-5472.CAN-10-0699. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.