

Summary
The nascent polypeptide-associated complex (NAC) is a conserved ribosome-associated protein biogenesis factor. Whether NAC exerts chaperone activity and whether this function is restricted to de novo protein synthesis is unknown. Here, we demonstrate that NAC directly exerts chaperone activity toward structurally diverse model substrates including polyglutamine (PolyQ) proteins, firefly luciferase, and Aβ40. Strikingly, we identified the positively charged ribosome-binding domain in the N terminus of the βNAC subunit (N-βNAC) as a major chaperone entity of NAC. N-βNAC by itself suppressed aggregation of PolyQ-expanded proteins in vitro, and the positive charge of this domain was critical for this activity. Moreover, we found that NAC also exerts a ribosome-independent chaperone function in vivo. Consistently, we found that a substantial fraction of NAC is non-ribosomal bound in higher eukaryotes. In sum, NAC is a potent suppressor of aggregation and proteotoxicity of mutant PolyQ-expanded proteins associated with human diseases like Huntington’s disease and spinocerebellar ataxias.
Keywords: proteostasis, chaperone, nascent polypeptide-associated complex, polyglutamine (PolyQ) proteins, Aβ40, protein aggregation, proteotoxicity, organismal fitness, age-related proteinopathies
Graphical Abstract
Highlights
-
•
The protein biogenesis factor NAC exhibits broad-spectrum chaperone activity
-
•
NAC exerts a ribosome-independent chaperone function
-
•
The positively charged N terminus of βNAC is a central chaperone entity of NAC
-
•
NAC suppresses aggregation and toxicity of disease-related polyglutamine proteins
NAC is a conserved protein biogenesis factor. Shen et al. demonstrate that NAC acts as a chaperone suppressing aggregation and toxicity of human disease-related polyglutamine-expanded proteins. They identify the positively charged domain of βNAC as the critical chaperone domain and show that NAC also acts independent of its ribosome association.
Introduction
A multifaceted chaperone network guards the integrity of the cellular proteome. This network comprises various conserved families of molecular chaperones operating in all cellular sub-compartments to promote the folding and function of their protein substrates and counteract proteotoxicity provoked by protein misfolding and aggregation (Kim et al., 2013). A subset of molecular chaperones is specialized for de novo protein folding, including the ribosome-associated complex (RAC) in eukaryotes or trigger factor in bacteria. These systems directly bind to translating ribosomes near the peptide exit tunnel to engage their substrates in a co-translational manner. Installed at the ribosome, they are enabled to assist protein folding at the earliest possible time when nascent chains are just reaching the cytoplasm. These chaperones thus lay the groundwork for the maintenance of protein homeostasis in the cell (Pechmann et al., 2013, Preissler and Deuerling, 2012).
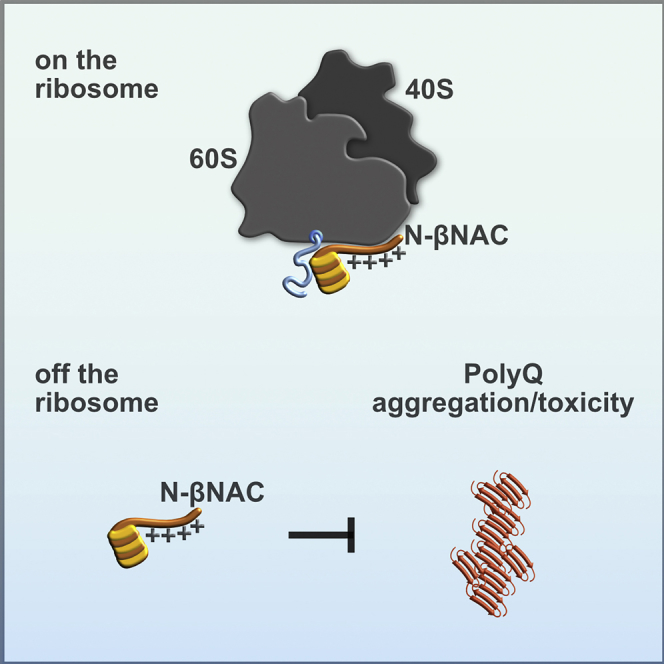
A major factor in eukaryotes that quantitatively associates with translating ribosomes near the peptide exit site is the ubiquitous nascent polypeptide-associated complex (NAC) (Wiedmann et al., 1994). It consists of two different subunits, αNAC and βNAC, that dimerize when their homologous NAC domains form a semi-β-barrel core (Liu et al., 2010, Wang et al., 2010). NAC is an abundant complex expressed at least equimolar relative to ribosomes; thus, most translating ribosomes likely associate with NAC (del Alamo et al., 2011, Raue et al., 2007). Essential for ribosome binding is an ∼40 aa domain found specifically in the N terminus of the βNAC subunit (herein N-βNAC). N-βNAC is highly conserved and exhibits a characteristic positive net charge. Deletion of either the first N-terminal 11 amino acids or mutation of a conserved positively charged central motif (RRKxxKK) abolishes ribosome binding in yeast, suggesting that this domain mediates the main ribosomal contact of NAC (Pech et al., 2010, Wegrzyn et al., 2006).
Because of its localization at the ribosomal tunnel exit, a proposed function of NAC is to act as a co-translational molecular chaperone similar to the ATP-independent trigger factor in bacteria. However, only indirect evidence supports this assumption, and mechanistic details of the proposed chaperone activity are entirely unknown (Duttler et al., 2013, Kirstein-Miles et al., 2013, Ott et al., 2015, Wang et al., 2013). Crosslinking data suggest that both NAC subunits can interact with protein clients, but the particular substrate binding site(s) of α- and βNAC and the substrate binding specificity are unknown (Martin et al., 2018, Wang et al., 1995). Further, whether NAC has a function aside from its co-translational ribosomal role is unknown.
Here, we conducted a series of in vitro and in vivo experiments to explore the potential chaperone function of NAC in greater detail. We found that NAC directly exerts chaperone activity as a holdase toward a set of structurally and physicochemically diverse model substrates. NAC effectively suppresses aggregation of disease-related polyglutamine-expanded (polyQ) proteins and amyloid-β 40 (Aβ40) peptides, as well as denatured firefly luciferase, independent from its ribosome association. Specifically, our data reveal that the ribosome-binding domain N-βNAC represents a central chaperone domain of NAC. Moreover, we found that NAC enhances organismal fitness of PolyQ-expressing C. elegans animals and prevents proteostasis collapse in neurons expressing PolyQ-expanded Huntingtin. These data suggest that NAC is a chaperone that acts as a potent modifier of age-related proteinopathies.
Results
NAC Suppresses Aggregation of Diverse PolyQ Proteins
NAC is a major ribosome-binding factor interacting broadly with nascent chains (del Alamo et al., 2011). However, its assumed chaperone function is poorly investigated. A previous study in C. elegans showed that depletion of NAC leads to increased aggregation of a model PolyQ protein (Kirstein-Miles et al., 2013). Although loss of NAC causes pleiotropic defects in C. elegans (Gamerdinger et al., 2015), this finding raises the possibility that NAC directly exerts a chaperone function on aggregation-prone proteins. In this case, overexpression of NAC should prevent aggregation of PolyQ proteins in vivo. Therefore, we used a C. elegans strain expressing 35 consecutive glutamine residues fused to YFP (PolyQ35::YFP) in body-wall muscle cells. This PolyQ length is close to the aggregation threshold in C. elegans muscle cells, leading to progressive, age-dependent aggregation starting at day 2 of adulthood (Morley et al., 2002). We generated transgenic animals that overexpress FLAG-tagged α- and βNAC under the control of the muscle-specific myo-3 promoter. PolyQ aggregation was assessed at day 3 of adulthood by fluorescence microscopy as well as semi-denaturing detergent agarose gel electrophoresis (SDD-AGE), which detects high-molecular weight oligomeric PolyQ species (Halfmann and Lindquist, 2008). The overexpression of NAC did not alter the overall morphology of C. elegans and expression levels of PolyQ35::YFP (Figure 1A, whole body images). However, we found that overexpression of NAC effectively suppressed PolyQ35::YFP aggregation in worms. This is evident by magnification of the head regions showing diffuse PolyQ35::YFP and less punctate PolyQ35::YFP structures when NAC was overexpressed (Figure 1A, head region images). Consistent with the fluorescence microscopy analysis, we found less insoluble high molecular weight aggregate species in NAC-overexpressing worms by SDD-AGE analysis (Figures 1B and S1A). These data indicate that NAC is a modifier of PolyQ aggregation. To directly test the potential chaperone function of NAC, we employed a peptide composed of 51 consecutive glutamines (PolyQ51) as a model substrate in a well-established in vitro aggregation assay. We initiated aggregation of PolyQ51 by cleaving a solubilizing GST tag with TEV protease and detected aggregates over time using a filter trap assay. Addition of purified human NAC strongly delayed the accumulation of PolyQ51 aggregates (Figures 1C and 1D), suggesting that NAC directly acts on the polyglutamine stretch and slows the rate of aggregation.
Figure 1.

NAC Suppresses Aggregation of Diverse PolyQ Proteins
(A) Fluorescence microscope images of PolyQ35::YFP C. elegans worms overexpressing NAC (FLAG-tagged α- and βNAC) in muscle cells. Images were taken at day 3 of adulthood. Scalebar, 500 μm in whole body images and 50 μm in images showing the head region. PolyQ35::YFP aggregates and cell nuclei are indicated by white and blue arrowheads, respectively. ev, empty vector.
(B) PolyQ35::YFP aggregation in animals as in (A) was further assessed by semi-denaturing agarose gel electrophoresis (SDD-AGE) immunoblot analysis. Total levels of SDS-soluble PolyQ35::YFP and FLAG-tagged NAC subunits were assessed by denaturing SDS-PAGE immunoblot analysis. Actin served as loading control.
(C) Filter trap aggregation assay of PolyQ51 peptide incubated with 5× molar excess of human NAC in vitro. Aggregation of GST-PolyQ51 was initiated by cleavage of the GST tag using the TEV protease. SDS-insoluble aggregates were detected with an S-tag antibody.
(D) Quantification of SDS-insoluble PolyQ51 aggregates obtained in filter trap from (C). Data are representative of at least 3 independent biological replicates.
(E) Filter trap aggregation assay of mutant Huntingtin (mHttQ51) incubated with 5× molar excess of human NAC or ovalbumin control (Ova) in vitro. Aggregation of GST-mHttQ51 was initiated by cleavage of the GST tag using the TEV protease. SDS-insoluble aggregates were detected with an S-tag antibody.
(F) Quantification of SDS-insoluble mHttQ51 aggregates obtained in filter trap from (E). Data are representative of at least 3 independent biological replicates.
See also Figure S1.
The aggregation suppression effect of NAC on the pure PolyQ substrate predicts that NAC may also inhibit aggregation of pathogenic proteins harboring an extended PolyQ tract. Thus, we investigated whether NAC prevents aggregation of mutant Huntingtin exon 1, the causative agent in the human neurodegenerative disorder Huntington’s disease (Labbadia and Morimoto, 2013). Using the same in vitro filter trap aggregation assay, we found that human NAC effectively suppressed aggregation of mutant Huntingtin exon 1 containing a pathogenic stretch of 51 glutamines (mHttQ51), whereas a molar equivalent ovalbumin control showed little effect (Figures 1E and 1F). Aggregation suppression of mHttQ51 by NAC was concentration dependent (Figure S1B) and was also observed, albeit to a lesser extent, with the C. elegans form of NAC (Figure S1C). Moreover, we found that NAC did not disaggregate preformed mHttQ51 aggregates in vitro (Figure S1D), suggesting that NAC exhibits a holdase function on early aggregation species to prevent further oligomerization, similar to the apical domain of the chaperonin TRiC (Tam et al., 2006).
A second pathogenic PolyQ substrate tested was full-length ataxin-3 harboring a stretch of 78 glutamines (AtxQ78), which causes spinocerebellar ataxia-3 in humans (Matos et al., 2011). Importantly, this protein shares no homology with Huntingtin exon 1 aside from the mutant expansion of a PolyQ repeat and aggregates via a separate kinetic mechanism (Saunders and Bottomley, 2009, Scarff et al., 2015). AtxQ78 exhibits lower aggregation propensity compared to PolyQ51 and mHttQ51, circumventing the requirement of a solubilizing tag. To assess whether NAC also affects AtxQ78 aggregation, we incubated the AtxQ78 substrate in the presence or absence of purified human NAC at 37°C and assessed aggregation by a filter trap assay. Similar to the effect on PolyQ51 and mHttQ51, addition of human NAC delayed the aggregation of AtxQ78 (Figures 2A and 2B). The C. elegans NAC homolog also prevented AtxQ78 aggregation but was slightly less effective than the human isoform (Figure S2A).
Figure 2.

NAC Suppresses Aggregation of Mutant Ataxin-3
(A) Filter trap aggregation assay of full-length Ataxin-3 containing a stretch of 78 glutamines (AtxQ78) incubated with 1× molar excess of NAC at 37°C for the indicated time. SDS-insoluble aggregates were detected with an anti-His antibody.
(B) Quantification of SDS-insoluble AtxQ78 aggregates obtained in filter trap from (A). Data are representative of at least 3 independent biological replicates.
(C) NAC and AtxQ78 were incubated in a 1:1 molar ratio and crosslinked using the homobifunctional crosslinker BS3. The schematic shows all intermolecular BS3 crosslinks identified by MS analysis of NAC-AtxQ78 complexes excised from the gel shown in Figure S2C.
Next, we mapped the PolyQ substrate binding site in NAC using a crosslinking and mass spectrometry (MS) approach. We used full-length AtxQ78 as our PolyQ model substrate for crosslinking to NAC for several reasons. First, AtxQ78 exhibits lower aggregation propensity than mHttQ51 or PolyQ51, allowing for crosslinking without concern for increasing the rate of the aggregation reaction. Second, the AtxQ78 construct does not require use of a solubilizing tag, avoiding the presence of additional factors during the crosslinking reaction. Third, while NAC may directly interact with the PolyQ region, glutamine does not contain any optimal functional groups for chemical crosslinking. However, AtxQ78 has many lysine residues, including several directly up-stream of the polyglutamine region, facilitating chemical crosslinking with amine-reactive crosslinkers (Figure S2B). Thus, crosslinks that occur within NAC to AtxQ78 regions adjacent to the polyglutamine tract may reflect the direct interaction between NAC and the PolyQ region, which we observed in vitro (Figures 1C and 1D). We incubated AtxQ78 with human NAC and used the amine-reactive homobifunctional crosslinker BS3 to trap transient chaperone interactions. Crosslinked NAC-AtxQ78 complexes visible on Coomassie-stained gels (Figure S2C) were excised, and crosslinked peptides were identified by LC-MS using StavroX (Götze et al., 2012). Strikingly, 11 out of 18 identified intermolecular crosslinks were to the C-terminal region of AtxQ78 close to the expanded PolyQ stretch (Figure 2C, Table S1). These data suggest that NAC acts by binding at, or close to, the PolyQ tract to suppress aggregation of AtxQ78, which agrees with the observed NAC effect on the pure PolyQ51 substrate (Figures 1C and 1D). Interestingly, NAC predominantly crosslinked to AtxQ78 via the N-terminal regions of αNAC and βNAC (Figure 2C, Table S1), suggesting that a crucial PolyQ binding site is located in these domains. However, single crosslinks were also identified to the NAC domains, the UBA domain of αNAC, and to the C-terminal domain of βNAC.
The Ribosome-Binding Domain of NAC Exerts Chaperone Activity
Our data show that NAC exhibits direct chaperone activity toward diverse PolyQ substrates. Previous crosslinking data (Martin et al., 2018, Wiedmann et al., 1994) and our AtxQ78-NAC crosslinking-MS analysis indicate that NAC may interact with protein clients via both subunits. However, the specific substrate-binding site(s) of NAC critical for its chaperone function are unknown. To answer this, we took a mutational approach based on the evolutionarily conserved regions of NAC including the NAC dimerization domains, the C-terminal UBA domain of αNAC, and the N-terminal ribosome-binding domain of βNAC harboring a conserved ribosome-binding motif (RRKxxKK) (Figure 3A). First, we asked which of these conserved domains may be crucial for preventing PolyQ aggregation in vivo. We generated three C. elegans strains overexpressing mutant NAC constructs under the control of the myo-3 promoter in the background of the PolyQ35::YFP strain. These included two deletion mutants lacking either the conserved αNAC UBA domain (ΔUBA-NAC) or the N-terminal βNAC domain (ΔNβ-NAC), as well as a mutation in the conserved ribosome-binding motif in the N terminus of βNAC (29RRK/AAA31-NAC) (Figure 3A), the latter of which abolished ribosome-binding of NAC in vivo (Figure S3A). All the NAC mutants expressed in C. elegans were stable and the expression levels were comparable to that of the overexpressed wild-type NAC complex (see FLAG immunoblot in Figures 3D and S3B). We assessed PolyQ35::YFP aggregation in these animals on day 3 of adulthood by fluorescence microscopy as well as SDD-AGE. We observed that both the ΔUBA-NAC and the RRK/AAA-NAC mutants suppressed PolyQ35::YFP aggregation similarly to WT-NAC, indicated by the increased diffuse PolyQ35::YFP signal in the head region of these animals in comparison to control worms (ev) in which the majority of the PolyQ35::YFP is aggregated in puncta (Figure 3C). This correlated well with the levels of aggregated PolyQ35::YFP detected by SDD-AGE (Figure 3D). Thus, neither the UBA domain nor ribosome-association of NAC is required to suppress PolyQ35::YFP aggregation. The latter finding suggests that NAC may serve additional chaperone functions off the ribosome in C. elegans. These data were intriguing because analysis of NAC distribution in yeast shows it is almost exclusively associated with ribosomes (Raue et al., 2007). However, polysome profile analysis of NAC distribution in both C. elegans and human cells showed that a large fraction of NAC was not associated with ribosomes (Figures 3B and S3C), suggesting that NAC may also exert a post-translational chaperone function in vivo. The finding of a large pool of non-ribosome associated NAC under steady-state conditions in C. elegans and human cells together with the observed aggregation suppression activity of a NAC variant that does not bind to ribosomes both support the idea of off-ribosomal chaperone functions for NAC in the cytosol.
Figure 3.

The Ribosome-Binding Domain of NAC Exerts Chaperone Activity
(A) Schematics showing the different heterodimeric NAC variants (α and β subunit) investigated in (C)–(E). Conserved domains (NAC and UBA) as well as the conserved ribosome-binding motif (RRKxxKK) in the β-subunit are highlighted.
(B) Sucrose density gradient analysis in wild-type N2 worms on day 2 of adulthood. Upper image shows polysome gradient profile (absorbance at 254 nm). Immunoblot images below show the distribution of NAC (α- and β-subunit) throughout the gradient. uL16 served as a ribosomal marker.
(C) Fluorescence microscope images of PolyQ35::YFP worms overexpressing WT-NAC or different mutant NAC versions shown in (A). Images were taken at day 3 of adulthood. Scalebar, 500 μm (upper row) and 50 μm (lower row). PolyQ35::YFP aggregates and cell nuclei are indicated by white and blue arrowheads, respectively. ev, empty vector.
(D) SDD-AGE immunoblot showing the PolyQ35::YFP aggregation in animals as in (C). Total levels of SDS-soluble PolyQ35::YFP and FLAG-tagged NAC variants were assessed by SDS-PAGE immunoblot analysis. Actin served as loading control.
(E) In vitro filter trap aggregation assay of mutant Huntingtin (mHttQ51) incubated with 5× molar excess of indicated NAC variants. Aggregation of GST-mHttQ51 was initiated by cleavage of the GST tag using the TEV protease. SDS-insoluble aggregates were detected with an S-tag antibody.
See also Figure S3.
Strikingly, deletion of the N-terminal domain of βNAC abrogated the ability of NAC to suppress PolyQ35::YFP aggregation (Figures 3C, 3D, and S3B), indicating that this region contains an essential interaction site for PolyQ tracts, which is consistent with our AtxQ78-NAC crosslinking-MS data (Figure 2C). To address this possibility directly, we tested the ability of the different NAC mutants to suppress mHttQ51 aggregation in vitro, using purified components. Consistent with the in vivo results, addition of purified ΔUBA- or RRK/AAA-NAC delayed mHttQ51 aggregation similar to WT-NAC, whereas ΔNβ-NAC lost the ability to suppress aggregation (Figure 3E). In sum, the crosslinking data combined with the in vitro and in vivo PolyQ aggregation analyses strongly suggest that the N-terminal domain of βNAC contains a crucial PolyQ substrate-binding site. Importantly, our data also imply that NAC suppression of pathogenic PolyQ aggregation via this domain can occur in a ribosome-independent manner.
NAC Exerts Broad-Spectrum Chaperone Activity
Next, we investigated whether the identified NAC aggregation suppression activity involving N-βNAC is PolyQ specific or reflects a broad-spectrum chaperone activity. As a non-PolyQ model substrate, we used chemically denatured firefly luciferase that is known to rely on molecular chaperones to become refolded and luminesce (Schröder et al., 1993). Indeed, we found that luciferase activity after denaturation with guanidine-HCl (GdmCl) was restored after addition of an ATP-driven Hsp70/Hsp40 folding chaperone system (Figure S4A, green curve). Interestingly, we found that purified human NAC alone had no effect on luciferase refolding (Figure S4A, red curve) and did not enhance luciferase refolding upon addition to the Hsp70/Hsp40 system (Figure S4A, gray curve). However, when NAC was added to chemically denatured luciferase first, subsequent refolding by the Hsp70/Hsp40 system was strongly enhanced over levels of just Hsp70/Hsp40 refolding (Figure 4A, red versus green curve). A molar equivalent control protein of similar size, GFP, showed no enhancing effect (Figure S4B, black curve). Similar results were obtained with C. elegans NAC (Figure S4C, pink curve). That NAC was only effective in the initial stages of luciferase refolding was strikingly similar to the necessity of NAC to be involved in the earlier stages to suppress aggregation of mHttQ51 (Figure S1C). Thus, NAC exerts a general holdase chaperone function and maintains unfolded luciferase or pathogenic aggregation substrates in a soluble and refolding competent state. Importantly, we found that the activity of ΔNβ-NAC was significantly reduced compared to WT-NAC (Figure 4A, yellow curve). Thus, the same domain crucial to prevent PolyQ aggregation is also necessary to hold luciferase in a refolding competent state. However, in contrast to the PolyQ substrates, ΔNβ-NAC still exerted residual chaperone activity toward denatured luciferase, suggesting that NAC contains additional unidentified chaperone domains that can bind luciferase, a protein with higher sequence complexity than PolyQ-expanded Htt or AtxQ78.
Figure 4.

NAC Exerts Broad-Spectrum Chaperone Activity
(A) In vitro chaperone refolding assays using guanidine-HCl (GdmCl)-denatured luciferase as substrate. Luciferase (0.02 μM) was preincubated with indicated NAC variants in a 1:1 molar ratio for 15 min at room temperature, and refolding was initiated by adding an Hsp70/Hsp40 chaperone system (3.2 μM/0.8 μM). Luciferase reactivation was analyzed by luminescence recording over 2 h at RT using luciferin as a substrate. Statistical significance was calculated by one-way ANOVA and Tukey post hoc test. a.u., arbitrary units. ∗p < 0.05 (n = 3).
(B) Fluorescence microscope images of C. elegans worms expressing a destabilized variant of firefly luciferase fused to EGFP (FlucDM-EGFP) and indicated NAC variants (FLAG-tagged α and βNAC) in muscle cells. Images were taken before heat shock (HS, 1 h at 33°C), directly after HS, and after 24 h recovery at 20°C. Scale bar, 20 μm. ev, empty vector.
(C) Kinetic aggregation assays of Aβ40 (32 μM, blue) incubated with an equimolar concentration of WT-NAC (red) or ΔNαβ-NAC (yellow) measured using ThioflavinT fluorescence. a.u., arbitrary units.
(D) Negative stain transmission electron micrographs of the reaction endpoint (at 20 h) for each sample shown in (C). Scale bar, 5 μm.
We further investigated NAC activity on luciferase refolding in vivo using a C. elegans strain expressing a structurally destabilized version of luciferase fused to EGFP (FlucDM-EGFP) in muscle cells (Gupta et al., 2011). This protein is soluble at moderate temperature but aggregates upon heat stress. After heat shock (1 h, 33°C) of worms, FlucDM-EGFP formed large punctate aggregates that were still evident in control animals after 24 h recovery at 20°C (Figure 4B, “ev”). Overexpression of wild-type NAC in these worms allowed FlucDM-EGFP aggregation to fully revert back to diffuse GFP signal within the 24 h recovery phase (Figure 4B, “WT-NAC”), showing that NAC promotes in vivo refolding of luciferase, consistent with the in vitro findings (Figure 4A). Moreover, overexpression of ΔNβ-NAC was inefficient to revert heat-shock-induced FlucDM-EGFP aggregates back to the diffuse, soluble form after the 24 h recovery period (Figure 4B, “ΔNβ-NAC,” and Figure S4D). Thus, efficient luciferase refolding depends on N-βNAC, in vitro and in vivo. Overall, these data corroborate that the N terminus of βNAC represents a central chaperone domain of NAC for luciferase as well as for PolyQ substrates.
Next, we investigated whether NAC suppresses aggregation of the Alzheimer’s disease-related Aβ40 peptide which, in contrast to PolyQ, has a more complex sequence, including regions more hydrophobic in character. Aβ40 aggregation was recorded over time in vitro using Thioflavin T (ThT) fluorescence as a readout for amyloid fibril formation. In the absence of NAC, we observed a rapid increase of ThT fluorescence, indicating Aβ40 aggregation (Figure 4C, “Buffer”), and electron microscope analysis confirmed formation of aggregates with fibrillar structure (Figure 4D). Remarkably, equimolar addition of purified human NAC completely suppressed Aβ40 aggregation and fibril formation (Figures 4C and 4D). In addition, ΔUBA- and RRK/AAA-NAC mutants also fully suppressed Aβ40 aggregation (Figure S4E). However, in contrast to the PolyQ substrates and luciferase, the N-terminal βNAC domain was dispensable for preventing aggregation of the highly hydrophobic Aβ40 substrate. Though crosslinking-MS analyses indicated an interaction of Aβ40 with the N-terminal domains of NAC similar to AtxQ78 (Figures S4F and S4G), NAC mutants lacking the βNAC N terminus (ΔNβ-NAC) or the N termini of both subunits (ΔNαβ-NAC) were fully active in suppressing Aβ40 aggregation comparably to WT-NAC (Figures 4C, 4D, and S4E). Thus, NAC likely contains other important substrate interaction sites that may specifically bind hydrophobic segments in substrates such as Aβ40 and luciferase.
In sum, these data show that NAC is able to chaperone diverse substrates with different structural and physicochemical properties, corroborating a broad-spectrum chaperone function of NAC. Moreover, our data reveal that the N-terminal βNAC domain confers substrate-specific chaperone function.
Functional Characterization of the N-Terminal βNAC Chaperone Domain
Our data show that the N-terminal domain of βNAC not only confers ribosome-binding but also has chaperone activity, preventing the aggregation of PolyQ proteins and promoting refolding of firefly luciferase. This small domain (∼40 aa) is characterized by a high positive net charge, in particular at its C-terminal half, which is predicted to be unstructured (Figure 5A). To gain more insight into the chaperone activity of this domain, we utilized synthetic peptides corresponding to the N-terminal region of βNAC from human and C. elegans in in vitro aggregation assays. We observed that these N-βNAC peptides alone were sufficient to suppress aggregation of mHttQ51 (Figure 5B). Similar aggregation suppression results were obtained when using the pure PolyQ51 substrate (Figure S5A), indicating a direct interaction of the N-βNAC peptides with the polyglutamine stretch.
Figure 5.

Functional Characterization of the N-Terminal βNAC Chaperone Domain
(A) (i) Peptide sequences of N-termini of βNAC from C. elegans and humans. Positively charged residues are highlighted in red. Both peptides exhibit a considerably high isoelectric point (pI). (ii) N-terminal peptide sequence of human αNAC exhibiting in contrast to βNAC peptides a low pI. Negatively charged residues are highlighted in blue.
(B) In vitro filter trap aggregation assay of mutant Huntingtin (mHttQ51) incubated with 5× or 10× molar excess of βNAC peptides shown in (A) or full-length NAC protein.
(C) (i) Chemical reaction scheme showing acylation of primary amines by Sulfo-NHS-acetate used to neutralize the positive charge in lysine residues of βNAC peptides shown in (A). (ii) In vitro filter trap aggregation assay of mutant Huntingtin (mHttQ51) incubated with 5× molar excess of peptides shown in (A) with and without Sulfo-NHS-acetate labeling.
(D) In vitro filter trap aggregation assay of mutant Huntingtin (mHttQ51) incubated with 5× molar excess of full-length NAC protein (NAC), full N-terminal βNAC peptide (Full N-term), or the N- and C-half of the peptide as indicated in the schematic below (N- and C-half highlighted in blue and red, respectively).
(E) In vitro filter trap aggregation assay of mutant Huntingtin (mHttQ51) incubated with 5× or 10× molar excess of human α- or βNAC peptides shown in (A).
See also Figure S5.
The most obvious characteristic of the N-βNAC peptides is the high positive net charge (Figure 5A). In addition to hydrophobic contacts, electrostatic interactions mediated via highly charged regions in chaperones are emerging to play an important role in client binding (He et al., 2016, Horowitz et al., 2016, Joachimiak et al., 2014). Thus, we asked whether the positively charged residues contribute to the ability of these peptides to suppress PolyQ aggregation. To address this, we acylated primary amines in lysine residues of the peptides using Sulfo-NHS-acetate in order to neutralize the positive charge (Figures 5Ci and S5B). Interestingly, we observed that upon labeling the lysine residues, the peptides significantly lost their ability to suppress mHttQ51 aggregation (Figure 5Cii). Thus, the positively charged residues in N-βNAC are critical for its chaperone activity. To investigate this in more detail, we split the human peptide into two halves, resulting in an N-terminal peptide containing a predicted conserved helical element with several hydrophobic residues and a C-terminal peptide encompassing most of the conserved positively charged residues (Figure 5D). Remarkably, the C-terminal half of N-βNAC was alone able to suppress mHttQ51 aggregation, whereas the N-terminal half showed no effect (Figure 5D). This finding corroborates that the primary PolyQ binding site is located in the positively charged stretch of the peptide. Although very potent, the C-terminal peptide was not as completely effective as the full-length peptide or the full-length NAC chaperone in suppressing mHttQ51 aggregation (Figure 5D). This suggests that additional regions in the N-βNAC peptide contribute to the chaperone activity. Of note, mixing the N-half peptide and the C-half peptides did not increase the aggregation suppression ability over the C-half peptide alone (Figure 5D). Thus, full function is only obtained by a cooperative activity that relies on the entire N-βNAC.
We also investigated the activity of the N terminus of the αNAC subunit, which is also highly charged, albeit with a net negative charge (Figure 5A). This domain is thought to be flexible, similar to the βNAC N-terminal region, and our AtxQ78-NAC crosslinking data indicated an interaction with the mutant PolyQ domain (Figure 2C). However, we found that N-αNAC peptides either added alone or in combination with the N-βNAC peptides had no effect on mHttQ51 aggregation (Figures 5E and S5C), even at 10× molar excess over the mHttQ51 substrate, underscoring the specificity of the aggregation suppression effect by the positively charged βNAC N-terminal domain.
NAC Suppresses Toxicity of PolyQ Proteins in Neuronal Cell Lines and Animals
Next, we investigated the relevance of NAC to mutant Huntingtin toxicity in neuronal cells. Here, we used a knockin mouse striatal cell line heterozygous for a 111-glutamine residue repeat mutation in the full-length Huntingtin protein (HttQ7/111), which is a well-established cell line to model Huntington’s disease pathology (Trettel et al., 2000). We compared the phenotype of this mutant Huntingtin cell line with that of a striatal mouse cell line homozygous for the wild-type Huntingtin allele (HttQ7/7). First, we assessed how knockdown of NAC would affect viability of mHtt-expressing cells. We depleted both NAC subunits by siRNA, and after 72 h determined the number of metabolically active cells by measuring ATP levels using the Cell Titer Glo assay. While knockdown of NAC decreased health of both cell lines, the mHtt-expressing HttQ7/111 cells were significantly more sensitive toward NAC depletion (Figure 6A). Knockdown efficiency of αNAC and βNAC was comparable in two Htt cell lines (Figures 6B and S6). Thus, while NAC generally promotes striatal neuron health, NAC is especially critical to maintain metabolic health of neurons expressing mutant, aggregation-prone Huntingtin. To address whether ribosome-binding of NAC was necessary for this protective function, we tried to overexpress different NAC variants in striatal cells. However, this was not possible, so it remains unclear whether suppression of Htt toxicity in neurons by NAC results from its co- and/or post-translational function.
Figure 6.

NAC Suppresses Toxicity of PolyQ Proteins
(A) Viability of mouse striatal neurons either homozygous for wild-type Huntingtin (Q7/7) or heterozygous for mutant Huntingtin (Q7/111). Metabolically active cells were quantified by measuring ATP levels using the Cell Titer Glo assay after 3-day treatment with siRNAs targeting both NAC subunits. Scrambled (Scrm) siRNA sequences were used as control. Data are represented as mean ± SEM. Statistical significance was determined by two-way ANOVA. Data are representative of at least 5 independent biological replicates. a.u., arbitrary units.
(B) Quantification of α/βNAC knockdown by qPCR in the mouse striatal cell lines Q7/7 and Q7/111. Actin was used as a housekeeping gene control. Knockdown of α/βNAC was compared to the scrambled control condition.
(C) (i) Homozygous wild-type (Q7/7) and heterozygous mutant (Q7/111) Huntingtin cells were treated with NAC siRNA or a scrambled control (Scrm) for 3 days. Fluorescence microscope images show cells stained with Proteostat fluorescent dye to assess protein aggregation. Cells treated with the proteasome inhibitor MG132 (5 μM, 6 h) were used as positive aggregation control. Scale bar, 15 μm. Hoechst was used to label nuclei of cells. (ii) Diagram shows the ratio of Proteostat fluorescence to Hoechst fluorescence in cells shown as in (i). Data are represented as mean ± SEM (n = 3).
(D) Diagram shows the percentage of non-paralyzed PolyQ35::YFP C. elegans worms overexpressing either wild-type NAC (WT-NAC), ribosome-binding deficient NAC (RRK/AAA), or NAC lacking the N-terminal region of βNAC (ΔNβ-NAC) between days 6 and 10 of adulthood. Data are represented as mean ± SEM (n = 3). Statistical significance was calculated by Student’s t test. ∗∗/∗∗∗p < 0.01/0.001 versus WT-NAC. EV, empty vector.
See also Figure S6.
To understand the specific need for NAC in maintaining metabolic function of mHtt-expressing striatal neurons, we asked whether NAC was especially crucial for neuronal protein homeostasis in the presence of mutant Huntingtin. Therefore, we analyzed the protein aggregation burden in both wild-type (HttQ7/7) and mutant Htt-expressing cells (HttQ7/111) upon silencing of NAC. Protein aggregation was assessed using the Proteostat dye, which recognizes aggregates from a broad range of protein substrates (Shen et al., 2011). We observed that in wild-type cells, knockdown of NAC had a negligible effect on protein aggregation, whereas protein aggregates strongly accumulated in the mHtt-expressing cells (Figure 6C). Thus, NAC is essential to counteract the increased burden on the protein homeostasis machinery provoked by mHtt expression, demonstrating the essential, physiologically protective role NAC plays in maintaining protein homeostasis.
Finally, because protein aggregation has been tightly linked to age-associated organismal degeneration (Sala et al., 2017), we asked whether NAC is also essential for organismal fitness and healthy aging. We overexpressed wild-type NAC and mutant ΔNβ-NAC in C. elegans expressing the aggregation-prone PolyQ35::YFP and measured age-associated paralysis (Cohen et al., 2006, Morley et al., 2002). We found that overexpressing wild-type NAC significantly improves motility of PolyQ35::YFP-expressing worms during aging (Figure 6D), suggesting that overexpression of NAC alone can improve protein homeostasis and organismal health. In addition, we found that this improvement in motility strictly depends on the presence of the N terminus of βNAC (Figure 6D), highlighting the importance of this domain in maintaining protein homeostasis in aging. Importantly, the protective activity of N-βNAC is mostly independent from its ribosome-binding role as overexpression of the ribosome-binding deficient RRK/AAA-NAC variant still improved motility during aging, albeit less effective than WT-NAC (Figure 6D). Thus, ribosome-binding of NAC contributes but is not essential under these conditions per se for improving protein homeostasis and organismal health in aging. Overall, these data show the tight linkage between cellular and organismal proteostatic health and the chaperone role of NAC as mediated by the N terminus of the βNAC subunit.
Discussion
Our data show that the same domain of NAC critical for ribosome-binding also exerts chaperone activity. Thus, N-βNAC has a dual role and may serve chaperone functions on and off the ribosome. NAC may contact nascent substrates via N-βNAC to promote co-translational folding, and, likewise, this domain binds misfolded cytosolic proteins post-translationally to prevent aggregation. Indeed, NAC exists in an equilibrium between a ribosome-bound and unbound state under steady-state conditions with a large non-ribosomal population. Our data also clearly show that the chaperone function of NAC is not restricted to co-translational de novo protein synthesis as revealed by the ribosome-independent aggregation suppression effect of NAC on PolyQ proteins in vivo and in vitro. We suggest a model in which the canonical activity of NAC in co-translational protein transport and folding (del Alamo et al., 2011, Gamerdinger et al., 2015) is complemented by its off-ribosomal chaperone activity to prevent aggregation of misfolded cytosolic protein species. Whether ribosome-associated NAC also actively dissociates from the ribosome during protein stress to chaperone aggregation-prone substrates is an attractive hypothesis and remains to be further explored. Previous data suggested an elegant mechanism for how NAC binding to misfolded protein species might be coupled to ribosome detachment under high proteotoxic stress (Kirstein-Miles et al., 2013). This stress-induced ribosome dissociation of NAC could be mediated by other chaperone co-factors activated by the presence of protein aggregation or by bulk association upon accumulation of protein aggregates.
NAC has broad chaperone activity toward distinct types of substrates, including PolyQ, Aβ40, and luciferase. The type of misfolded or unfolded domain that NAC recognizes remains unclear. We find that NAC is most effective in the early stages of misfolding of aggregation-prone pathogenic proteins (Figure S1D). This suggests that NAC may act as a holdase chaperone that recognizes a misfolded intermediate appearing early in the aggregation reaction. NAC may then prepare these early-aggregation species substrates for further manipulation by other chaperones as demonstrated for luciferase in this study (Figure 4A) or sequester aggregation-prone domains critical in the early stages of oligomerization. It is tempting to envision a similar activity for NAC when acting on nascent polypeptides. The affinity of NAC for different misfolded protein domains remains to be determined, and affinity may well depend both on the sequence and conformational properties of the client, as is observed for other ATP-independent chaperones (Saio et al., 2014, Stull et al., 2016). Our in vitro experiments demonstrate a concentration dependence of NAC suppression of PolyQ aggregation (Figure S1B), with higher molar excess of NAC over the client resulting in greater aggregation suppression. Such an observation is consistent with findings on other ATP-independent chaperones that bind their substrates weakly, such that an excess of chaperone is required to enable chaperone binding to compete effectively with aggregation. Importantly, NAC is an abundant protein in vivo and is at least stoichiometric with the ribosome (Raue et al., 2007), which would enable effective chaperoning even for weakly binding clients that are highly aggregation-prone. Thus, even if only a small percentage of NAC associates with misfolded Htt substrates at any given time, rapid binding and release, combined with potential remodeling of the protein client in the bound state, could also enhance folding and decrease the probability of aggregation. This would enable these proteins to remain soluble so that they can fold spontaneously or be bound by other chaperones that complete folding or target misfolded proteins to degradation pathways (Balchin et al., 2016, Saibil, 2013).
The discovery that just the positively charged N terminus of βNAC is sufficient to potently suppress mutant PolyQ aggregation in vitro, as well as necessary to delay age-associated paralysis in C. elegans, has important implications for a general binding mechanism of chaperones to mutant PolyQ substrates. While the PolyQ itself is uncharged, its polarity can still take part in weak electrostatic interactions with a charged surface like the βNAC N terminus. Indeed, hydrophilic regions have been associated with chaperone-substrate recognition patterns in addition to the more canonical hydrophobic regions. For example, the TRiC chaperonin, a potent suppressor of mHtt aggregation and toxicity, contains bipartite hydrophilic and hydrophobic substrate recognition sites (Joachimiak et al., 2014). It has been proposed that both of these domains may recognize either hydrophobic or hydrophilic regions of mHtt to mediate suppression of aggregation (Joachimiak et al., 2014, Tam et al., 2006, Tam et al., 2009). DNAJB6 suppresses aggregation of expanded PolyQ tracts through a serine/threonine-rich domain, which disrupts formation of stabilizing hydrogen bonding among the PolyQ residues (Kakkar et al., 2016). It is intriguing that there seems to be specificity with respect to charge: while the N-terminal region of the αNAC subunit is also highly charged, it is net negatively charged and has little to no effect on Htt aggregation (Figure 5E). Thus, it seems that an overall net positive charge is specific for suppression of PolyQ aggregation, but reasons why are still unclear.
Finally, the ability of the βNAC N-terminal peptide to suppress aggregation makes this an important sequence for possible therapeutic development. How NAC is able to recognize different misfolded substrates is an intriguing question for further exploration. A strong overexpression of full-length NAC is poorly tolerated on the organismal level (Gamerdinger et al., 2015), potentially due to its binding mode at the ribosomal exit site that is competitive with other essential protein biogenesis factors. This restricts a potential therapeutic intervention strategy that aims to increase NAC expression levels to combat PolyQ diseases. However, delivery or overexpression of just the N-terminal peptide, which is sufficient to suppress PolyQ aggregation, may be much better tolerated in an organism and thus an effective an anti-aggregation therapeutic strategy. In sum, this study provides a detailed understanding of a chaperone activity of NAC off the ribosome and highlights a substrate recognition mechanism that is based on positive charges which provides a possible avenue for a peptide-based therapeutics approach in Huntington’s disease and related PolyQ disorders.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| GFP | Covance | RRID:AB_10063778 |
| FLAG | Sigma-Aldrich | RRID:AB_262044 |
| Actin | Santa Cruz | RRID:AB_2714189 |
| Beta-NAC (BTF3) | Abcam | RRID:AB_1141066 |
| HRP-anti-mouse IgG | Jackson | RRID:AB_2340771 |
| HRP-anti-rabbit IgG | Jackson | RRID:AB_ 2340585 |
| Alpha-NAC/Beta-NAC (C. elegans) | Kirstein-Miles et al., 2013 | N/A |
| uL24 (C. elegans) | Deuerling lab | N/A |
| uL16 (RPL10L) | Abgent | Cat#AP17603a |
| Goat Anti-S tag | Abcam | RRID:AB_777789 |
| Anti-6x HIS tag | Abcam | RRID:AB_2732046 |
| RPLP0 | Immunovision | Cat#HPO-0100 |
| Tubulin | Gift from Thomas Mayer, University of Konstanz | N/A |
| Amyloid-beta (clone 6E10) | Absolute Antibody | Cat#ABA-AB00714-1.7 |
| Bacterial and Virus Strains | ||
| DH5α | Thermo-Fisher | Cat#18265017 |
| OP50 | CGC | Strain OP50 |
| BL21 (DE3) Rosetta | Merck | Cat#70954 |
| Rosetta 2 (DE3) pLysS | EMD Millipore | Cat#71401-4 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Complete EDTA-free protease inhibitor cocktail | Roche | Cat#5056489001 |
| Cycloheximide | Sigma-Aldrich | Cat#C7698 |
| DNase I | Sigma-Aldrich | Cat#DN25 |
| Ni-IDA matrix, Protino | Roth | Cat#CN08.3 |
| Floxuridine | LKT Laboratories | Cat#50-91-9 |
| Levamisole | LKT Laboratories | Cat#16595-80-5 |
| T4 DNA Ligase | New England Biolabs | Cat#M0202S |
| Phusion DNA Polymerase | Deuerling lab | N/A |
| C. elegans betaNAC N-term peptide | Genscript | Custom order, C-terminal amidation |
| Human betaNAC N-term peptide | Genscript | Custom order, C-terminal amidation |
| Human betaNAC N-term peptide (N-half) | Genscript | Custom order, C-terminal amidation |
| Human betaNAC N-term peptide (C-half) | Genscript | Custom order, C-terminal amidation |
| Human alphaNAC N-term peptide | Genscript | Custom order, C-terminal amidation |
| Sulfo-NHS Acetate | Thermo-Fisher | Cat#26777 |
| GST-mHttQ51 recombinant protein | This paper | N/A |
| GST-Q51 recombinant protein | This paper | N/A |
| Ataxin-3 recombinant protein | This paper | N/A |
| NAC recombinant variants (C. elegans + human) | This paper | N/A |
| Ovalbumin | Sigma-Aldrich | Cat#A5503 |
| acTEV protease | Thermo-Fisher | Cat#12575015 |
| TNBS | G Biosciences | Cat#BC86 |
| Glutathione Sepharose 4B | GE Healthcare | Cat#17-0756-05 |
| Cellulose acetate membrane OE66 | GE Healthcare | Cat#10404180 |
| 7K MWCO Zeba Spin desalting columns | Thermo Fisher | Cat#89882 |
| Strataclear Resin | Agilent | Cat#400714 |
| BS3-d0/BS3-d4 (used for Ataxin-3-NAC xlinking) | Thermo-Fisher | Cat#10066323 |
| Q Sepharose Fast Flow | GE Healthcare | Cat#17051001 |
| GeneRuler 1 kb | Thermo-Fisher | Cat#SM0312 |
| BS3-H12/BS3-d12 (used for Abeta40-NAC xlinking) | Creative Molecules | Cat#001SS |
| Ulp-1 | Deuerling lab | N/A |
| Hsp-1 (Hsp-70) | This paper | N/A |
| Dnj-13 (Hsp40) | This paper | N/A |
| Firefly Luciferase | This paper | N/A |
| Protein G Agarose | Thermo-Fisher | Cat#20398 |
| Critical Commercial Assays | ||
| Proteostat Aggresome Detection Kit | Enzo Life Sciences | Cat#ENZ-51035-K100 |
| CellTiter-Glo Luminescent Cell Viability Assay | Promega | Cat#G7572 |
| Experimental Models: Cell Lines | ||
| ST HDH Q7/7 | Coriell Cell Repositories | Cat#CH00096 |
| ST HDH Q7/111 | Coriell Cell Repositories | Cat#CH00097 |
| HEK293T | ATCC | Cat# CRL-3216 |
| Experimental Models: Organisms/Strains | ||
| N2 | CGC | WormBase ID: N2 |
| AM140 rmIs132 [unc-54p::Q35::YFP] | CGC | AM140 |
| FlucDM-EGFP marIs135 [unc-54p::FlucDM::EGFP] | Ulrich Hartl, Gupta et al., 2011 | N/A |
| Oligonucleotides | ||
| alphaNAC siRNA | Dharmacon | Cat#L-041821-01-0005 |
| betaNAC siRNA | Dharmacon | Cat#L-052370-01-0005 |
| Scrambled siRNA | Dharmacon | Cat#D-001206-14-05 |
| Recombinant DNA | ||
| pET11a-ataxin3-78Q | Generated in Radford lab. Orginal construct from Sandra Macedo-Ribeiro (Instituto de Biologia Molecular e Celular and Instituto de Investigação e Inovação em Saúde, University of Porto) | N/A |
| pETSac-mAβ40 | Provided by Dr. Sara Linse (Lund University, Sweden) and Prof. Dominic Walsh (Harvard Institute of Medicine, USA). | N/A |
| pCFJ90 (myo-2p::mCherry) | Addgene, Frøkjaer-Jensen et al., 2008 | RRID:Addgene_19327 |
| pPD61_125 | Addgene, gift from Andrew Fire | RRID:Addgene_1508 |
| pPD61_125_myo-3p::3xFLAG-alphaNAC::unc-53-3′utr | This paper | N/A |
| pPD61_125_myo-3p::3xFLAG-betaNAC::unc-53-3′utr | This paper | N/A |
| pPD61_125_myo-3p::3xFLAG-deltaUBA-alphaNAC::unc-53-3′utr | This paper | N/A |
| pPD61_125_myo-3p::3xFLAG-RRK/AAA-betaNAC::unc-53-3′utr | This paper | N/A |
| pPD61_125_myo-3p::3xFLAG-deltaN-betaNAC::unc-53-3′utr | This paper | N/A |
| p6xHis-SUMO-Hsp-1 (Hsp70 expression vector) | This paper | N/A |
| p6xHis-SUMO-Dnj-13 (Hsp40 expression vector) | This paper | N/A |
| pDS56-6xHis-Luciferase | This paper | N/A |
| p6xHis-SUMO-alphaNAC/betaNAC (C. elegans) | Kirstein-Miles et al., 2013 | N/A |
| p6xHis-SUMO-alphaNAC/betaNAC (human) | This paper | N/A |
| p6xHis-SUMO-alphaNAC/RRK/AAA-betaNAC (C. elegans) | This paper | N/A |
| p6xHis-SUMO-alphaNAC/deltaN-betaNAC (C. elegans) | This paper | N/A |
| p6xHis-SUMO-deltaUBA-alphaNAC/betaNAC (C. elegans) | This paper | N/A |
| p6xHis-SUMO-deltaN-alphaNAC/deltaN-betaNAC (human) | This paper | N/A |
| Software and Algorithms | ||
| Fiji Image software | Schindelin et al., 2012 | https://fiji.sc/ |
| Prism 7 | Graphpad | https://www.graphpad.com/scientific-software/prism/ |
| MassLynx 4.1 | Waters | http://www.waters.com/waters/en_US/MassLynx-MS-Software |
| Peaks 7/8 | Bioinformatics Solutions | http://www.bioinfor.com/peaks-studio/ |
| StavroX | Götze et al., 2012 | https://www.stavrox.com |
| Image Studio Lite | Li-Cor | https://www.licor.com/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| WinDaq | DataQ Instruments | https://www.dataq.com |
Contact for Reagent and Resource Sharing
Further information and requests for reagents should be directed to Lead Contact Elke Deuerling (elke.deuerling@uni-konstanz.de).
Experimental Model and Subject Details
Cell lines
Cell lines (ST HDH Q7/7 and ST HDH Q7/111) were purchased from the Coriell Cell Repository and not authenticated for this study. Cells were cultured in DMEM under 5% CO2.
C. elegans strains
C. elegans strain AM140 (rmIs132 [unc-54p::Q35::YFP]) was obtained from Caenorhabditis Genetics Center (CGC). Strain FlucDM-EGFP (marIs135 [unc-54p::FlucDM::EGFP] (Gupta et al., 2011) was obtained from Ulrich Hartl, Max-Planck-Institute of Biochemistry, Martinried, Germany. Worms were cultured according to standard techniques with E. coli OP50 as food source (Brenner, 1974).
Method details
Protein purification
PolyQ51 and mHttQ51 (Exon 1 of HTT with 51 glutamine repeat) plasmids were constructed as previously described (Tam et al., 2006). Proteins were expressed in Rosetta 2(DE3) pLysS competent cells (Agilent Technologies) in LB media supplemented with carbenicilin and chloramphenicol. Cultures were induced with 1 mM IPTG for 2.5h at 16°C. For purification, pellets were resuspended in 50 mM sodium phosphate, pH 8.0; 150 mM NaCl; 1 mM EDTA and lysed using an Emulsiflex (Avestin). Lysate was incubated with GSH-Sepharose resin (GE Healthcare) and washed with 0.1% (v/v) Triton, 500 mM NaCl, and 5 mM Mg-ATP before eluting protein with 15 mM glutathione. Protein was concentrated and buffer exchanged with 50 mM Tris-HCl, pH 8.0; 100 mM NaCl; 5% (v/v) glycerol. Concentrated protein was 0.2 μm filtered before storage at −80°C.
Wild-type NAC and NAC mutants from both human (αNAC = NACA; βNAC = BTF3) and C. elegans (αNAC = icd-2; βNAC = icd-1) were recombinantly expressed in Rosetta (DE3) cells as His-SUMO fusion constructs. Cultures were induced with 0.5 mM IPTG over night at 20°C. Cell pellets were resuspended in lysis buffer (20 mM sodium phosphate pH 7.5, 300 mM NaCl, 6 mM MgCl2, 2 mM β-mercaptoethanol, 2 mM PMSF, 10 μg/mL DNase I, 10% (v/v) glycerol) and lysed by French Press. Proteins were captured using Ni-IDA matrix (Protino; Macherey-Nagel) and eluted with lysis buffer containing 250 mM imidazole. Elution fractions were dialyzed overnight in the presence of 8 μg Ulp-1 per mg protein for proteolytic cleavage of the His-SUMO tag. Ion exchange chromatography using Resource Q column (GE Healthcare) was used for further purification. Elution fractions containing αNAC and βNAC in a 1:1 ratio were pooled, frozen in liquid nitrogen and stored at −80°C.
His-tagged AtxQ78 (ATXN3) was purified as described in Scarff et al. (Scarff et al., 2015) using nickel affinity chromatography and size-exclusion chromatography.
Aβ40 peptide was expressed and purified as described previously (Stewart et al., 2017, Walsh et al., 2009). In short, BL21 (DE3) cells were transformed with pETSac-mAβ40. Expression of Aβ40 was induced by the addition of IPTG to a final concentration of 1 mM. The cells were allowed to grow for an additional three hours before collection. Inclusion bodies were extracted from the cells by means of sonication followed by centrifugation. Aβ40 was purified from the inclusion body lysate by Q-Sepharose purification followed by two rounds of SEC. The purified peptides were lyophilized and stored at −20°C. The purity of the peptides was confirmed by SDS-PAGE and LC-MS.
In vitro aggregation assays
Mutant Huntingtin and PolyQ51 aggregation reactions were performed at concentration 3 μM of mHttQ51, 0.044 Units/μl acTEV protease (Invitrogen, Carlsbad, CA, USA), and respective concentrations of Ovalbumin (Sigma) or purified NAC chaperone variants. Aggregation was conducted in TEV reaction buffer (Invitrogen) and incubated at 30°C. AtxQ78 was buffer exchanged into TEV reaction buffer using 7K MWCO Zeba Spin Desalting Columns (Thermo Fisher) to initiate aggregation. AtxQ78 aggregation reactions were performed at 30 μM of Ataxin-3 in reaction buffer (20 mM sodium phosphate pH 7.5, 25 mM NaCl, 6 mM MgCl2, 2 mM DTT, 5% (v/v) glycerol) and incubated at 37°C. Samples at varied time-points were then taken and combined in a 1:1 ratio with a 4% (w/v) SDS, 100 mM DTT solution, boiled for 5 min at 95°C, and stored at −20°C. Samples were then filtered through a 0.22 μm cellulose acetate membrane (Whatman) and washed with 0.1% (w/v) SDS. Membrane was probed using an S-tag antibody (Abcam) for mHttQ51 and PolyQ51, and with a His-tag antibody (Abcam) for AtxQ78.
Thioflavin T fluorescence assay
Lyophilized Aβ40 was resuspended at 320 μM in 20 mM sodium phosphate pH 7.4, 0.2 mM EDTA, 0.01% (w/v) sodium azide and stored on ice. The NAC proteins were diluted to 100 μM in storage buffer (20 mM sodium phosphate pH 7.5, 25 mM NaCl, 6 mM MgCl2, 2 mM β-mercaptoethanol, 5% (v/v) glycerol) and buffer exchanged into 20 mM sodium phosphate pH 7.4, 0.2 mM EDTA, 0.01% (w/v) sodium azide, 1x complete™ mini protease inhibitor, EDTA free (Roche) by means of ZebaSpin 7 kDa MWCO spin columns (Thermo Scientific). Samples were prepared that contained equimolar concentrations of Aβ40 and NAC variants. Thioflavin T was added to a final concentration of 10 μM. The samples were transferred to a 96 well half-area clear bottom microplate (Corning GmbH, Wiesbaden, Germany), with 95 μL of sample in each well. The fluorescence (excitation: 440 nm, emission: 480 nm) was measured using a BMG Omega plate reader (BMG Labtech) incubating samples at 37°C, quiescently.
Transmission electron microscopy
After 20 h, samples were taken from the Thioflavin T plate and fixed on carbon coated copper grids, made in house. The samples were negative stained with 2% (w/v) uranyl acetate. The samples were imaged on a JEOL 1400 TEM at the Astbury structural biology laboratory, University of Leeds.
Cell viability assays and real-time PCR
siRNA knockdown was completed using the DharmaFECT reverse transfection protocol. Striatal knock-in cell lines (homozygous wild-type HttQ7/7 and heterozygous mutant HttQ7/111) were plated in 96-well plates (1.25 × 10ˆ4 cells/well) in complete medium (DMEM with high glucose, 10% FBS) at 32°C. Experiments were plated to have four technical replicates per siRNA treatment for each experiment, with each independent experiment repeated at least three times. 72 hours post transfection, cells were incubated with Cell Titer Glo reagent (Promega) for at least 10 min before recording luminescence signal. For real-time PCR (RT-PCR), RNA was harvested from cells using the Zymo Quick-RNA kit, cDNA was synthesized using the iScript kit. RT-PCR was completed using the SYBR Green Master Mix from Biorad and fold-knockdown was calculated using the 2–ΔΔCt method.
Fluorescence microscopy of cells
Cells were imaged on a Zeiss LSM 700 confocal microscope (Carl Zeiss). Cells were prepared similarly as above but plated on a poly-lysine coated coverslip in 24-well plate. Post transfection, the cells were stained with a 1:2000 Proteostat solution (Enzo Life Sciences, Farmingdale, NY, USA) for 1 hour, followed by a 0.67 μg/mL Hoechst stain for 5 min, prior to imaging. For proteasome inhibition, cells were treated with 5 μM MG132 for 6 hours prior to imaging.
Polysome analysis in human cells
Prior to harvesting, HEK293T cells were treated with 100 μg/mL cycloheximide (CHX) for 5 min at 37C. Cells were then washed twice in 100 μg/mL CHX in PBS and harvested in the same buffer on ice. Pelleted cells were then resuspended in lysis buffer (10 mM HEPES pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, 1% Triton X-100, 100 μg/mL CHX) and lysed by trituration through a 26G needle for 10 passes. The sample was centrifuged at 1500 x g for 5 min at 4°C. RNA concentration of the supernatant was then measured by Nanodrop. 200 μg (in RNA) of lysate was loaded onto a 12 mL 10%–50% (w/v) linear sucrose gradient (Gradient Mate, Biocomp Instruments) prepared in gradient buffer (100 mM KCl, 20 mM HEPES pH 7.6, 5 mM MgCl2, 100 μg/mL CHX, 1U/μl RNase inhibitor) and centrifuged for 2 h at 36000 rpm in a swinging bucket rotor (SW-41, Beckmann). Gradients were then fractionated from top to bottom with a density gradient fractionator (Brandel), and A260 was monitored to detect cytosolic fractions, ribosomal subunits, monosomes, and polysomes. Data were recorded and processed with WinDaq (Dataq Instruments). For one gradient, 15 fractions with 1 mL each were collected. For subsequent western analysis, 500 μl of gradient buffer and 10 μl of Strataclear resin slurry (Agilent Technologies) was added to each fraction and incubated on rotation at 4°C. Fractions were centrifuged twice at 3000 x g at 4°C for 5 min to remove supernatant. The remaining resin was then resuspended in 20 μl 2X Laemmli Buffer, centrifuged at 3000 x g for 5 min, and the residual supernatant was loaded into an SDS-PAGE gel and then transferred onto a nitrocellulose membrane. Different dilutions of primary antibodies were applied (1:400 anti-βNAC, 1:3000 anti-RPLP0).
Sulfo-NHS peptide labeling
NAC N-terminal peptides were ordered from Genscript. Peptides were dissolved to 1 mM in 0.1 M sodium carbonate buffer, pH 8.5. Peptide solutions were added to a 25-fold molar excess of Sulfo-NHS Acetate (Thermo Scientific, Waltham, MA, USA) to amine groups in the sample and incubated for 1 hour at room temperature. Reaction was quenched using a 1 M Tris-HCl, pH 7.5 solution. To quantify the efficiency of this labeling, a lysine standard curve was established; samples were assayed for free primary amines by adding 0.01% (w/v) TNBS in 0.1 M sodium bicarbonate, pH 8.5 solution (G Biosciences) to each sample and standard, and incubated at 37°C for 2 hours. 10% SDS and 1N HCl was then added to stop the reaction. Absorbance was measured at 335 nm.
Chemical crosslinking and mass spectrometry
For crosslinking AtxQ78 was buffer exchanged into 10 mM sodium phosphate (pH 7.2) and added to NAC at a 1:1 ratio (20 μM NAC + 20 μM AtxQ78). A 1:1 mixture of BS3-d0 and BS3-d4 was added to the proteins at 20x and 50x molar excess and the reaction allowed to proceed at room temperature for 1 hour before quenching with the addition of 50 mM Tris-Cl (pH 7.5). Samples were diluted with 2x loading buffer and separated on Tris-tricine gels followed by staining with InstantBlue (Expedeon).
Aβ40 (18 μM) was incubated with purified NAC in a 1:1 molar ratio in Aβ aggregation buffer (20 mM sodium phosphate pH 7.5, 0.2 mM EDTA, 0.01% (w/v) NaN3) at 37°C for 2 h. BS3-h12d12 was added to the proteins at 20x molar excess and incubated for 30 min at 37°C before quenching with 50 mM NH4HCO3. Crosslinked samples were gel-filtrated using a Superdex 75 column (GE Healthcare) and fractions containing crosslinked Aβ40-NAC complexes subjected to co-immunoprecipitation using Aβ antibody 6E10 (Biozol). Captured proteins were denatured with 1x SDS sample buffer under non-reducing conditions to avoid splitting of the antibody in heavy and light chains.
Gel pieces containing the crosslinked complexes were washed with 25 mM ammonium bicarbonate (pH 7.8) for 1 h with shaking. The solution was removed and the pieces destained three times with 25 mM ammonium bicarbonate in 60% acetonitrile. Gel pieces were dehydrated with 100% acetonitrile for 10 min and left to air-dry in a laminar flowhood for 1 h. Rehydration of the gel pieces was achieved by adding 0.1 mg/mL trypsin solution and incubating the samples on ice for 30 min. Excess trypsin was removed and the samples were incubated at 37°C and 1000 rpm overnight. Peptides were extracted from the gel using 3 washes with 60% acetonitrile/5% formic acid. The extracts were pooled and concentrated using a SpeedVac before being analyzed using a nanoACQUITY LC-system coupled to a Synapt HDMS G2Si mass spectrometer. Peptides were injected onto a C18 column equilibrated with 0.1% formic acid in water and eluted using an increasing gradient of 0.1% formic acid in acetonitrile over 60 min at a flow rate of 0.3 μl/min. The Synapt HDMS G2Si was operated in positive mode using a capillary voltage of 3.0 kV, cone voltage of 40 V, backing pressure of 3.6 mbar and a trap bias of 2.0 V. The source temperature was 80°C and the trap pressure was 8.70 × 10−3 mBar. Glu-fibrinogen and Leucine Enkephalin were infused as lock mass calibrants. Data acquisition was achieved using Data Dependent Analysis (DDA) with a one second MS scan over an m/z range of 250–3000 being followed by three 1 s MS/MS scans taken from the five most intense ions in the MS spectrum over an m/z range of 50–2000. Data processing was performed using the MassLynx v4.1 suite of software supplied with the mass spectrometer and PEAKS 7/8 (Bioinformatics Solutions). Crosslinks were identified using StavroX software (Götze et al., 2012) and verified manually.
Luciferase refolding assay
Firefly Luciferase refolding activity was measured as previously described (Sun et al., 2012). Recombinant luciferase (2.5 μM) was chemically denatured for 45 min at room temperature in denaturing buffer (25 mM HEPES/KOH, pH 7.4, 50 mM KCl, 15 mM MgCl2, 1 mM ATP, 10 mM DTT, 0.05 mg/mL BSA, 5 M GdmCl). To test for refolding activity, 0.02 μM denatured luciferase was preincubated in the presence or absence of 0.02 μM NAC variants for 15 min at room temperature. Luciferase refolding was induced by addition of 3.2 μM CeHsc70 (HSP-1), 0.8 μM CeHsp40 (DNJ-13) and luminescence buffer (75 mM HEPES/KOH, pH 7.4, 50 mM KCl, 15 mM MgCl2, 1 mM ATP, 2 mM DTT, 0.05 mg/mL BSA, 240 μM Coenzyme A, 0.1 mM luciferin, 10 mM PEP, 50 μg/mL pyruvate kinase). Luminescence was measured in 96-well LIA-plates (Greiner) over 2 hours at room temperature in a microplate reader (BertholdTech TriStar2S).
C. elegans transformation
Transgenic strains were generated using standard microinjection protocols (Mello and Fire, 1995). Constructs for overexpression of NAC in body wall muscles were generated by cloning the coding sequences of icd-1 (βNAC) and icd-2 (αNAC) into pPD61_125 vector containing the myo-3 promoter and the unc-54 3′ untranslated region (UTR). The NAC genes were N-terminally tagged with 3x FLAG. Mutant NAC constructs were generated by standard mutagenesis protocols. AM140 (rmIs132 [unc-54p::Q35::YFP]) and FlucDM-EGFP (marIs135 [unc-54p::FlucDM::EGFP]) worms were injected with 25 ng/μl of each NAC plasmid together with CFJ90 myo-2p::mCherry (2.5 ng/μl) (Frøkjaer-Jensen et al., 2008) and DNA ladder (100 ng/μl, GeneRuler 1 kb, Thermo Scientific). Control strains were obtained by injecting 50 ng/μl empty vector, 2.5 ng/μl myo-2p::mCherry and 100 ng/μl DNA ladder. For each transformation, at least two independent transgenic lines carrying extrachromosomal arrays were obtained showing similar results. Detailed strain information is available in Table S3.
Synchronization of C. elegans
Synchronization of worms for microscopic studies was carried out by a timed egg-lay for 5 h. Large age-synchronized C. elegans cultures for SDD-AGE analyses were obtained by collecting embryos from gravid adult worms using a 20% alkaline hypochlorite bleaching for 5 min. Embryos were allowed to hatch overnight in M9 buffer to get arrested L1s. Transgenic L1 larvae were sorted based on the myo-2p::mCherry marker using a COPAS FlowPilot system (Union Biometrica). The synchronized, transgenic L1s were transferred to OP50 seeded plates and incubated at 20°C. After two days the young adult worms were transferred to new plates containing 150 μM 5-fluorodeoxyuridine (LKT Laboratories) to prevent the culture from reproducing.
Fluorescence microscopy of worms
Worms were immobilized on 3% agarose pads and anaesthetized using 25 mM levamisole (LKT Laboratories). Images were taken with a confocal laser-scanning microscope TCS SP8 (Leica) with 5x (whole body images) and 63x objectives (head region). Images were adjusted as necessary in Fiji (ImageJ) (Schindelin et al., 2012) using cropping, brightness and contrast tools.
SDD-AGE
SDD-AGE was carried out as previously described (Halfmann and Lindquist, 2008). For sample preparation, worms were extracted in lysis buffer (100 mM Tris-Cl pH 7.5, 50 mM NaCl, 10 mM β-mercaptoethenol, 1x complete protease inhibitor) by sonication (four times, 10 pulses, duty cycle = 40, output control = 2; Branson sonifier). Lysed worms were centrifuged for 1 min at 500 g to remove debris and the supernatant was transferred to a new tube. ¼ volume of 4x SDD-AGE sample buffer (2x TAE, 20% glycerol, 8% SDS, 0.05% bromphenol blue) was added to the lysates and incubated for 15 min at RT. Samples were loaded onto a 1.2% agarose gel in 1x TAE buffer (40 mM Tris-Cl, pH 7.6, 20 mM acetic acid, 1 mM EDTA) containing 0.1% SDS and proteins were blotted on nitrocellulose membranes by capillary transfer in 1x Tris buffer (150 mM NaCl, 50 mM Tris-Cl pH 7.5) over night at room temperature. The membrane was analyzed using an anti-GFP antibody (Covance).
Polysome analysis of worms
C. elegans N2 worms were cultivated in liquid culture at 20°C in presence of E. coli OP50 as food source. Day 2 post-L4 worms were harvested on ice with 0.1 M NaCl and seperated from bacteria via sucrose floatation. After an additional washing step the nematodes were flash frozen in liquid nitrogen. Worm pellets were cryo-genic grinded using a cryo-mill (Retsch) for 30 s at 22 Hz. Frozen worm powder was resuspended in lysis buffer (30 mM HEPES/KOH pH 7.4, 50 mM KoAc, 5 mM MgCl2, 5% (w/v) mannitol, 100 μg/mL cycloheximide, 2 mM β-mercaptoethanol, 1 x complete protease inhibitor) and centrifuged at 18,000 g for 15 min at 4°C. The supernatant was adjusted to 20 A260 U/mL and 500 μl were loaded on a sucrose gradient (15%–45% in lysis buffer). Ribosomal species were separated by ultracentrifugation (TH-641 rotor) at 39,000 rpm for 2.5 hours (4°C). Gradients were fractionated using a density gradient fractionator (Teledyne Isco, Inc.) monitoring the A254 and fractions were directly analyzed by immunoblotting.
Paralysis assay
To analyze the percentage of paralyzed worms, 100 semi-synchronized (timed egg lay for 5 h) young adult worms of each strain were placed on a plate containing 150 μM 5-fluorodeoxyuridine. Screening of paralyzed worms was started at day 6 of adulthood. Worms were scored as paralyzed when they only moved their heads but failed to undergo a full body wave propagation upon repeated prodding with a platinum wire worm picker.
Immunoblot analysis in C. elegans
Protein samples were applied to SDS–PAGE and electroblotted onto nitrocellulose membranes according to standard protocols. Commercial antibodies used throughout this study were GFP (Covance, MMS-118P), FLAG (Sigma, F1804), Actin (Santa Cruz, sc-47778), and uL16 (Abgent, AP176039). Polyclonal antibody against C. elegans NAC (αNAC + βNAC) was described previously (Kirstein-Miles et al., 2013). Antibody against uL24 was raised in rabbits immunized with recombinant full-length C. elegans RPL-26 protein. Tubulin antibodies were a kind gift from Thomas Mayer, University of Konstanz. Blots were probed with secondary antibodies coupled to HRP (Jackson, anti-mouse 715-035-151; anti-rabbit 711-005-152).
Quantification and Statistical Analysis
Experimental procedure
Each experiment was conducted at least three times to generate three biological repeats. For western blots, dot blots, and fluorescence microscope images the most representative experiment is shown.
Statistics
Bar graphs for quantifications show the average of at least three independent experiments ± SEM. Statistical parameters, including the exact value of n and statistical significance are reported in the Figure Legends. Data are judged to be statistically significant when p < 0.05 by the comparison test indicated in the Figure legends. In figures, asterisks denote statistical significance (∗, p < 0.05, ∗∗, p < 0.01, ∗∗∗, p < 0.001) as compared to appropriate controls.
Data and Software Availability
Raw data have been deposited to Mendeley Data and are available at https://doi.org/10.17632/r6dgtfjt2y.1.
Acknowledgments
We thank Christina Schlatterer for preparing the graphical abstract. We thank the members of our research groups for valuable support and discussion of the manuscript, Sandra Macedo-Ribeiro for collaborations on Atx-3, and Alison Ashcroft and James Ault for collaborations on biological MS. This work was supported by research grants from the German Science Foundation (DFG; SFB969; A01 and A07) to E.D. and M.G., from Human Frontier in Science (HFSP) to S.E.R., E.D., and J.F., and by a guest-professorship for J.F. by the State Baden-Württemberg. E.M.M. was supported by HFSP (RGP0025) and the Wellcome Trust Institutional Strategic Fund (RGP0025/2012). S.E.R. acknowledges the Wellcome Trust (09154/Z/15/Z) and the European Research Council (ERC) under European Union’s Seventh Framework Programme (FP7/2007–2013) ERC grant agreement no. 322408 for funding. We also thank the BBSRC for funding A.N.C., P.D.K., and K.L.S. (BB/P000037/1, BB/J014443/1 and BB/K01451X/1, respectively) and the Wellcome Trust and BBSRC for funding the Astbury Centre’s mass spectrometry equipment (BB/M012573/11 and 208385/Z/17/Z). We are thankful for NIH grants GM056433 and NS092525 to J.F.
Author Contributions
Conceptualization, M.G., K.S., S.E.R., J.F., and E.D.; Investigation, K.G., R.C., E.M.M., N.S., P.D.K., R.S., A.N.C., K.L.S., L.L., A.B., K.S., and M.G.; Writing – Original Draft, M.G. and K.S.; Writing – Review & Editing, S.E.R., J.F., and E.D.; Funding Acquisition, M.G., S.E.R., J.F., and E.D.
Declaration of Interests
The authors declare no competing interests.
Published: April 11, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.molcel.2019.03.012.
Contributor Information
Sheena E. Radford, Email: s.e.radford@leeds.ac.uk.
Judith Frydman, Email: jfrydman@stanford.edu.
Elke Deuerling, Email: elke.deuerling@uni-konstanz.de.
Supplemental Information
References
- Balchin D., Hayer-Hartl M., Hartl F.U. In vivo aspects of protein folding and quality control. Science. 2016;353:aac4354. doi: 10.1126/science.aac4354. [DOI] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen E., Bieschke J., Perciavalle R.M., Kelly J.W., Dillin A. Opposing activities protect against age-onset proteotoxicity. Science. 2006;313:1604–1610. doi: 10.1126/science.1124646. [DOI] [PubMed] [Google Scholar]
- del Alamo M., Hogan D.J., Pechmann S., Albanese V., Brown P.O., Frydman J. Defining the specificity of cotranslationally acting chaperones by systematic analysis of mRNAs associated with ribosome-nascent chain complexes. PLoS Biol. 2011;9:e1001100. doi: 10.1371/journal.pbio.1001100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duttler S., Pechmann S., Frydman J. Principles of cotranslational ubiquitination and quality control at the ribosome. Mol. Cell. 2013;50:379–393. doi: 10.1016/j.molcel.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frøkjaer-Jensen C., Davis M.W., Hopkins C.E., Newman B.J., Thummel J.M., Olesen S.P., Grunnet M., Jorgensen E.M. Single-copy insertion of transgenes in Caenorhabditis elegans. Nat. Genet. 2008;40:1375–1383. doi: 10.1038/ng.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamerdinger M., Hanebuth M.A., Frickey T., Deuerling E. The principle of antagonism ensures protein targeting specificity at the endoplasmic reticulum. Science. 2015;348:201–207. doi: 10.1126/science.aaa5335. [DOI] [PubMed] [Google Scholar]
- Götze M., Pettelkau J., Schaks S., Bosse K., Ihling C.H., Krauth F., Fritzsche R., Kühn U., Sinz A. StavroX--a software for analyzing crosslinked products in protein interaction studies. J. Am. Soc. Mass Spectrom. 2012;23:76–87. doi: 10.1007/s13361-011-0261-2. [DOI] [PubMed] [Google Scholar]
- Gupta R., Kasturi P., Bracher A., Loew C., Zheng M., Villella A., Garza D., Hartl F.U., Raychaudhuri S. Firefly luciferase mutants as sensors of proteome stress. Nat. Methods. 2011;8:879–884. doi: 10.1038/nmeth.1697. [DOI] [PubMed] [Google Scholar]
- Halfmann R., Lindquist S. Screening for amyloid aggregation by Semi-Denaturing Detergent-Agarose Gel Electrophoresis. J. Vis. Exp. 2008;17:838. doi: 10.3791/838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L., Sharpe T., Mazur A., Hiller S. A molecular mechanism of chaperone-client recognition. Sci. Adv. 2016;2:e1601625. doi: 10.1126/sciadv.1601625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horowitz S., Salmon L., Koldewey P., Ahlstrom L.S., Martin R., Quan S., Afonine P.V., van den Bedem H., Wang L., Xu Q. Visualizing chaperone-assisted protein folding. Nat. Struct. Mol. Biol. 2016;23:691–697. doi: 10.1038/nsmb.3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joachimiak L.A., Walzthoeni T., Liu C.W., Aebersold R., Frydman J. The structural basis of substrate recognition by the eukaryotic chaperonin TRiC/CCT. Cell. 2014;159:1042–1055. doi: 10.1016/j.cell.2014.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakkar V., Månsson C., de Mattos E.P., Bergink S., van der Zwaag M., van Waarde M.A.W.H., Kloosterhuis N.J., Melki R., van Cruchten R.T.P., Al-Karadaghi S. The S/T-rich motif in the DNAJB6 chaperone delays polyglutamine aggregation and the onset of disease in a mouse model. Mol. Cell. 2016;62:272–283. doi: 10.1016/j.molcel.2016.03.017. [DOI] [PubMed] [Google Scholar]
- Kim Y.E., Hipp M.S., Bracher A., Hayer-Hartl M., Hartl F.U. Molecular chaperone functions in protein folding and proteostasis. Annu. Rev. Biochem. 2013;82:323–355. doi: 10.1146/annurev-biochem-060208-092442. [DOI] [PubMed] [Google Scholar]
- Kirstein-Miles J., Scior A., Deuerling E., Morimoto R.I. The nascent polypeptide-associated complex is a key regulator of proteostasis. EMBO J. 2013;32:1451–1468. doi: 10.1038/emboj.2013.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbadia J., Morimoto R.I. Huntington’s disease: underlying molecular mechanisms and emerging concepts. Trends Biochem. Sci. 2013;38:378–385. doi: 10.1016/j.tibs.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Hu Y., Li X., Niu L., Teng M. The crystal structure of the human nascent polypeptide-associated complex domain reveals a nucleic acid-binding region on the NACA subunit. Biochemistry. 2010;49:2890–2896. doi: 10.1021/bi902050p. [DOI] [PubMed] [Google Scholar]
- Martin E.M., Jackson M.P., Gamerdinger M., Gense K., Karamonos T.K., Humes J.R., Deuerling E., Ashcroft A.E., Radford S.E. Conformational flexibility within the nascent polypeptide-associated complex enables its interactions with structurally diverse client proteins. J. Biol. Chem. 2018;293:8554–8568. doi: 10.1074/jbc.RA117.001568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matos C.A., de Macedo-Ribeiro S., Carvalho A.L. Polyglutamine diseases: the special case of ataxin-3 and Machado-Joseph disease. Prog. Neurobiol. 2011;95:26–48. doi: 10.1016/j.pneurobio.2011.06.007. [DOI] [PubMed] [Google Scholar]
- Mello C., Fire A. DNA transformation. Methods Cell Biol. 1995;48:451–482. [PubMed] [Google Scholar]
- Morley J.F., Brignull H.R., Weyers J.J., Morimoto R.I. The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA. 2002;99:10417–10422. doi: 10.1073/pnas.152161099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott A.K., Locher L., Koch M., Deuerling E. Functional dissection of the nascent polypeptide-associated complex in saccharomyces cerevisiae. PLoS ONE. 2015;10:e0143457. doi: 10.1371/journal.pone.0143457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pech M., Spreter T., Beckmann R., Beatrix B. Dual binding mode of the nascent polypeptide-associated complex reveals a novel universal adapter site on the ribosome. J. Biol. Chem. 2010;285:19679–19687. doi: 10.1074/jbc.M109.092536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pechmann S., Willmund F., Frydman J. The ribosome as a hub for protein quality control. Mol. Cell. 2013;49:411–421. doi: 10.1016/j.molcel.2013.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preissler S., Deuerling E. Ribosome-associated chaperones as key players in proteostasis. Trends Biochem. Sci. 2012;37:274–283. doi: 10.1016/j.tibs.2012.03.002. [DOI] [PubMed] [Google Scholar]
- Raue U., Oellerer S., Rospert S. Association of protein biogenesis factors at the yeast ribosomal tunnel exit is affected by the translational status and nascent polypeptide sequence. J. Biol. Chem. 2007;282:7809–7816. doi: 10.1074/jbc.M611436200. [DOI] [PubMed] [Google Scholar]
- Saibil H. Chaperone machines for protein folding, unfolding and disaggregation. Nat. Rev. Mol. Cell Biol. 2013;14:630–642. doi: 10.1038/nrm3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saio T., Guan X., Rossi P., Economou A., Kalodimos C.G. Structural basis for protein antiaggregation activity of the trigger factor chaperone. Science. 2014;344:1250494. doi: 10.1126/science.1250494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala A.J., Bott L.C., Morimoto R.I. Shaping proteostasis at the cellular, tissue, and organismal level. J. Cell Biol. 2017;216:1231–1241. doi: 10.1083/jcb.201612111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders H.M., Bottomley S.P. Multi-domain misfolding: understanding the aggregation pathway of polyglutamine proteins. Protein Eng. Des. Sel. 2009;22:447–451. doi: 10.1093/protein/gzp033. [DOI] [PubMed] [Google Scholar]
- Scarff C.A., Almeida B., Fraga J., Macedo-Ribeiro S., Radford S.E., Ashcroft A.E. Examination of Ataxin-3 (atx-3) aggregation by structural mass spectrometry techniques: a rationale for expedited aggregation upon polyglutamine (polyQ) expansion. Mol. Cell. Proteomics. 2015;14:1241–1253. doi: 10.1074/mcp.M114.044610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder H., Langer T., Hartl F.U., Bukau B. DnaK, DnaJ and GrpE form a cellular chaperone machinery capable of repairing heat-induced protein damage. EMBO J. 1993;12:4137–4144. doi: 10.1002/j.1460-2075.1993.tb06097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen D., Coleman J., Chan E., Nicholson T.P., Dai L., Sheppard P.W., Patton W.F. Novel cell- and tissue-based assays for detecting misfolded and aggregated protein accumulation within aggresomes and inclusion bodies. Cell Biochem. Biophys. 2011;60:173–185. doi: 10.1007/s12013-010-9138-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart K.L., Hughes E., Yates E.A., Middleton D.A., Radford S.E. Molecular Origins of the Compatibility between Glycosaminoglycans and Aβ40 Amyloid Fibrils. J. Mol. Biol. 2017;429:2449–2462. doi: 10.1016/j.jmb.2017.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stull F., Koldewey P., Humes J.R., Radford S.E., Bardwell J.C.A. Substrate protein folds while it is bound to the ATP-independent chaperone Spy. Nat. Struct. Mol. Biol. 2016;23:53–58. doi: 10.1038/nsmb.3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L., Edelmann F.T., Kaiser C.J., Papsdorf K., Gaiser A.M., Richter K. The lid domain of Caenorhabditis elegans Hsc70 influences ATP turnover, cofactor binding and protein folding activity. PLoS ONE. 2012;7:e33980. doi: 10.1371/journal.pone.0033980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam S., Geller R., Spiess C., Frydman J. The chaperonin TRiC controls polyglutamine aggregation and toxicity through subunit-specific interactions. Nat. Cell Biol. 2006;8:1155–1162. doi: 10.1038/ncb1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam S., Spiess C., Auyeung W., Joachimiak L., Chen B., Poirier M.A., Frydman J. The chaperonin TRiC blocks a huntingtin sequence element that promotes the conformational switch to aggregation. Nat. Struct. Mol. Biol. 2009;16:1279–1285. doi: 10.1038/nsmb.1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trettel F., Rigamonti D., Hilditch-Maguire P., Wheeler V.C., Sharp A.H., Persichetti F., Cattaneo E., MacDonald M.E. Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum. Mol. Genet. 2000;9:2799–2809. doi: 10.1093/hmg/9.19.2799. [DOI] [PubMed] [Google Scholar]
- Walsh D.M., Thulin E., Minogue A.M., Gustavsson N., Pang E., Teplow D.B., Linse S. A facile method for expression and purification of the Alzheimer’s disease-associated amyloid beta-peptide. FEBS J. 2009;276:1266–1281. doi: 10.1111/j.1742-4658.2008.06862.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S., Sakai H., Wiedmann M. NAC covers ribosome-associated nascent chains thereby forming a protective environment for regions of nascent chains just emerging from the peptidyl transferase center. J. Cell Biol. 1995;130:519–528. doi: 10.1083/jcb.130.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Zhang W., Wang L., Zhang X.C., Li X., Rao Z. Crystal structures of NAC domains of human nascent polypeptide-associated complex (NAC) and its αNAC subunit. Protein Cell. 2010;1:406–416. doi: 10.1007/s13238-010-0049-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F., Durfee L.A., Huibregtse J.M. A cotranslational ubiquitination pathway for quality control of misfolded proteins. Mol. Cell. 2013;50:368–378. doi: 10.1016/j.molcel.2013.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegrzyn R.D., Hofmann D., Merz F., Nikolay R., Rauch T., Graf C., Deuerling E. A conserved motif is prerequisite for the interaction of NAC with ribosomal protein L23 and nascent chains. J. Biol. Chem. 2006;281:2847–2857. doi: 10.1074/jbc.M511420200. [DOI] [PubMed] [Google Scholar]
- Wiedmann B., Sakai H., Davis T.A., Wiedmann M. A protein complex required for signal-sequence-specific sorting and translocation. Nature. 1994;370:434–440. doi: 10.1038/370434a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data have been deposited to Mendeley Data and are available at https://doi.org/10.17632/r6dgtfjt2y.1.