

Abstract
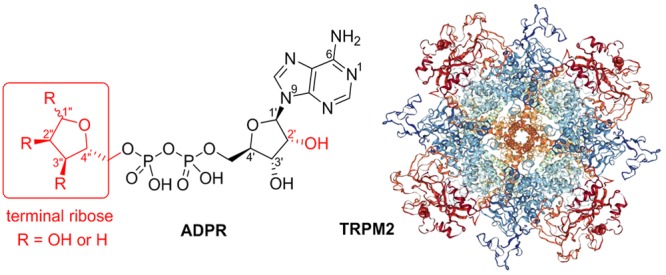
TRPM2 (transient receptor potential cation channel, subfamily M, member 2) is a nonselective cation channel involved in the response to oxidative stress and in inflammation. Its role in autoimmune and neurodegenerative diseases makes it an attractive pharmacological target. Binding of the nucleotide adenosine 5′-diphosphate ribose (ADPR) to the cytosolic NUDT9 homology (NUDT9H) domain activates the channel. A detailed understanding of how ADPR interacts with the TRPM2 ligand binding domain is lacking, hampering the rational design of modulators, but the terminal ribose of ADPR is known to be essential for activation. To study its role in more detail, we designed synthetic routes to novel analogues of ADPR and 2′-deoxy-ADPR that were modified only by removal of a single hydroxyl group from the terminal ribose. The ADPR analogues were obtained by coupling nucleoside phosphorimidazolides to deoxysugar phosphates. The corresponding C2″-based analogues proved to be unstable. The C1″- and C3″-ADPR analogues were evaluated electrophysiologically by patch-clamp in TRPM2-expressing HEK293 cells. In addition, a compound with all hydroxyl groups of the terminal ribose blocked as its 1″-β-O-methyl-2″,3″-O-isopropylidene derivative was evaluated. Removal of either C1″ or C3″ hydroxyl groups from ADPR resulted in loss of agonist activity. Both these modifications and blocking all three hydroxyl groups resulted in TRPM2 antagonists. Our results demonstrate the critical role of these hydroxyl groups in channel activation.
Introduction
The nonselective cation channel TRPM2 (transient receptor potential cation channel, subfamily M, member 2) is activated in a Ca2+-dependent manner after binding of the nucleotide ADP-ribose (ADPR) to its cytosolic C-terminal NUDT9H domain that shares homology with a mitochondrial nucleotide pyrophosphatase NUDT9.1 While earlier studies indicated that the NUDT9H domain also has a low pyrophosphatase activity, hydrolyzing ADPR to AMP and ribose 5-phosphate (R5P),2 a recent study showed that the Nudix box motif in TRPM2 does not support catalysis and the production of AMP might have been due to spontaneous hydrolysis of ADPR at alkaline pH.3
Reactive oxygen species (ROS) and genotoxic stress can result in release of ADPR from the nucleus due to the activation of the poly(ADP-ribose) polymerase-1 (PARP-1) and poly(ADP-ribose) glycohydrolase pathways.4,5 The ADPR so generated can then activate TRPM2, resulting in prolonged Ca2+-entry, mitochondrial Ca2+-overload, and apoptosis,6 thereby contributing to cell damage in post ischemic reperfusion injury during myocardial infarction7 and stroke.8
Besides this role in cell death, TRPM2 also participates in physiological processes like inflammation.9 In neutrophil granulocytes and dendritic cells, TRPM2 contributes to chemotaxis.10−12 The chemotaxis of murine neutrophils in response to fMLP is independent of PARP-1 but can be inhibited by 8-Br-ADPR, a compound that inhibits activation of TRPM2 by ADPR, and by knock-out of CD38, a glycohydrolase that produces ADPR from NAD.10 In macrophages and monocytes, TRPM2 is involved in secretion of chemokines and cytokines in response to ROS and pro-inflammatory cytokines,13,14 whereas in effector T cells, it plays a role in proliferation and secretion of pro-inflammatory cytokines.15 Inhibition of TRPM2 has been shown to reduce tissue damage after stroke by preventing invasion of neutrophils,16 and knock-out of TRPM2 ameliorates the symptoms of experimentally induced autoimmune encephalomyelitis, a model for multiple sclerosis.15 TRPM2 is thus an attractive pharmacological target for the treatment of neurodegenerative and autoimmune diseases.17,18
Further investigations into the role of TRPM2 in physiology and pathophysiology require specific modulators of channel function, and their rational design will benefit from a better understanding of its structure–activity relationship (SAR). Recently, TRPM2 structures from three different species were elucidated by cryo-EM. The structure of RPM2 from the sea anemone Nematostella vectensis nvTRPM2 lacks the NUDT9H domain, probably because of the flexibility of this part of the molecule.19 TRPM2 from zebrafish (Danio rerio) drTRPM2 has been solved in the apo state and in an ADPR-bound state.20 While ADPR could not be localized in the NUDT9H domain, electron density corresponding to ADPR was found in the N-terminal MHR1/2 domain. Comparison of the conformations of the apo and the ADPR-bound state and mutational analysis indicate that in drTRPM2, gating of the channel occurs via binding of ADPR to the MHR1/2 domain instead of NUDT9H.20 Structures of human TRPM2 (hTRPM2) have been resolved for the apo state, an ADPR bound (primed) state and an open conformation bound to ADPR and Ca2+.21 Mutagenesis of the MHR1/2 domain and removal of the NUDT9H domain show that in the human TRPM2 channel, gating occurs after binding of ADPR to the NUDT9H domain, but again poor resolution prevents the placement of ADPR in the presumed binding pocket. Structure-based drug design therefore still awaits a high-resolution structure of either the full-length channel or the isolated NUDT9H domain of TRPM2.
Recently, we synthesized ADPR analogues to explore the role of the adenosine, pyrophosphate, and terminal ribose motifs in activation of TRPM2.22,23 The pyrophosphate-forming couplings used employed morpholidate- or CDI-mediated methodologies. Yields were generally low and reaction times were generally long and improvements are warranted. To our surprise, the majority of structural modifications of ADPR led to compounds that do not activate TRPM2, indicating that all three moieties are required for channel opening.23 These studies also revealed that hydroxyl group removal at C2′ resulted, in the case of ADPR, in a compound that is a TRPM2 agonist with significantly higher efficacy than ADPR itself23 and also improved the antagonist 8-Ph-ADPR (IC50 11 μmol/L compared to 8-Ph-2′-deoxy-ADPR, IC50 3 μmol/L).22 We also highlighted the importance of the terminal ribose for TRPM2 activation by evaluating a series of modified ADPR analogues.29 While simple ADP neither activated nor antagonized the channel, introduction of substituents at the β-phosphate that increasingly resembled the terminal ribose (Figure 1) returned antagonist properties, but none of the analogues exhibited agonist activity. The fact that β-(tetrahydrofuran-2-yl)methyl-ADP, an analogue with the ribofuranose backbone, but lacking the hydroxyl groups, did not activate the channel but instead antagonized TRPM2 indicated that one or more of the hydroxyl groups might be important for the gating of TRPM2. Previous studies have shown hydroxyl group deletion to be a valuable tool in SAR elucidation for complex bioactive molecules.24−26 While such endeavors may be synthetically protracted in order to effect a desired precision edit to the parent molecule, they can divulge key mechanistic information.27,28
Figure 1.

Structure of ADPR and known terminal ribose analogues.29
We report here the synthesis and electrophysiological evaluation of novel terminal-ribose-modified analogues of the TRPM2 agonists ADPR and 2′-deoxy-ADPR to study the role of the individual hydroxyl groups of the terminal ribose in TRPM2 activation. The critical step of analogue formation was achieved by combining a sugar phosphate with a P-activated nucleotide to form a single molecule linked by a pyrophosphate bond. A recently reported improved procedure for the preparation of NDP sugars from nucleoside phosphorimidazolides36 uses 2–4 equiv of magnesium chloride to achieve high reaction yields and short reactions times for nucleoside 5-phosphorimidazolide coupling to sugar phosphates.
Results and Discussion
The terminal ribose of ADPR is essential for the ligand-driven activation of the cation channel TRPM2.29 To investigate the SAR of this part of ADPR in more detail, we focused upon the synthesis of all three possible 1″-deoxy, 2″-deoxy, and 3″-deoxy terminal ribose ADPR derivatives 2, 3, and 4 by selective deletion of the appropriate hydroxyl group (Figure 2). The similar reactivity of the ribose hydroxyl groups and potential for ring opening of the cyclic hemi-acetal required development of multistep synthetic routes with selective masking and deprotection. The modified ribose building blocks were then coupled to either AMP or 2′-deoxy-AMP via pyrophosphate coupling reaction (Figure 2). We also synthesized a corresponding analogue 1 with all hydroxyl groups present, but blocked with small alkyl groups to interrogate the H-bond donating capability of the terminal ribose (Figure 2). The chemically stable analogues were evaluated regarding their agonist and antagonist activity by patch-clamp experiments in whole cell configuration using HEK cells with stable expression of human TRPM2.
Figure 2.

Pyrophosphate coupling reaction and ADPR analogues prepared in this study.
At first, routes to the required modified terminal ribose 5-phosphates were designed. It was envisaged that such phosphates would be ideal coupling partners for activated nucleotide imidazolides. Protected ribose monophosphate 9 was chosen to exploit the ligand space around the terminal ribose, H-bond donating capability, and its good susceptibility for coupling with activated nucleosides. The precursor of 9, compound 8, was obtained from protected ribose307 by phosphitylation with dibenzyl N,N-diisopropylphosphoramidite and subsequent oxidation using hydrogen peroxide31 to afford 8 (79%) (Scheme 1A). Compound 8 was then hydrogenated using Pd/C to afford 9 (82%). Ribose-5-monophosphate 9 was converted to its tributylammonium salt and freeze-dried. Synthesis of 1-deoxyribose-5-phosphate 10, to explore the role of the anomeric OH group of the terminal ribose, started from 2,3-O-isopropylidene ribose 13 that was protected at position 5 by a trityl group, to afford 14 (65%) (Scheme 1B). Reduction of 14 by sodium borohydride led to compound 15 (85%). Reaction of 15 with tosyl chloride in pyridine gave no significant result at room temperature, but heating the reaction mixture up to 60 °C helped to afford the protected 1-deoxyribose 16 (80%). However, deprotection of 16 with acetic acid failed after multiple attempts, as both the 5-O-trityl and 2,3-O-isopropylidene protecting groups were cleaved, leading to the undesired fully deprotected 1-deoxyribose or to complex mixtures. Using the alternative deprotection of 16 with formic acid in diethylether32 afforded compound 17 (32%).33 Phosphitylation and subsequent oxidation of 17 with di-tert-butyl N,N-diisopropylphosphoramidite gave the phosphate derivative 18 (41%). Careful deprotection of 18 with aqueous trifluoroacetic acid at low temperature afforded the target 1-deoxyribose-5-phosphate 10 (49%) that was subsequently converted to its tributylammonium salt.
Scheme 1. Synthesis of Modified Terminal Riboses 9 and 10.

Reagents and conditions: (a) 5-Ph-1-H-tetrazole, dibenzyl N,N-diisopropylphosphoramidite, DCM, 20 °C, 1 h; (b) triethylamine, H2O2, 0–20 °C, 1 h; (c) hydrogen (balloon), Pd/C, 5 h, TEAB (1 M), 20 °C; (d) Dowex D50 (H+), tributylamine; (e) tritylchloride, pyridine, 20 °C, 16 h; (f) sodium borohydride, ethanol, 0–20 °C, 2 h; (g) tosyl chloride, pyridine, 60 °C, 16 h; (h) HCOOH/diethylether, 20 °C, 16 h; (i) 5-Ph-1-H-tetrazole, di-tert-butyl N,N-diisopropylphosphoramidite, DCM, 20 °C, 1 h; (j) triethylamine, H2O2, 0–20 °C, 1 h; (k) aqueous TFA, 0–20 °C, 4 h; (l) Dowex D50 (H+), tributylamine.
Synthesis of the 2-deoxy version of the terminal ribose (Scheme 2) started from commercially available 2-deoxyribose-5-phosphate sodium salt 19 (Sigma-Aldrich) that was converted to its corresponding mono-tributylammonium salt 11 by using Dowex resin (H+ form) followed by titration with tributylamine to pH ≈ 7 (Scheme 2A). Partial decomposition (20–30%) of 19 was observed during the transformation to 11 via 31P NMR, suggesting that 2-deoxyribose-5-monophosphate is less stable toward changes in pH (see Supporting Information, S14–16) compared to its 1-deoxy- and 3-deoxy-counterparts. Synthesis of 3-deoxyribose-5-monophosphate 12 started from 1,2-O-isopropylidene xylose 20,34 which was protected at the 5-hydroxyl group as the TBDPS ether to give 21 (83%) (Scheme 2B). Reaction of 21 with 1,1′-thiocarbonyldiimidazole in DCM afforded compound 22 (63%). Reductive deoxygenation of 22 with tributyltin hydride led to the compound 23 (76%) which was subsequently deprotected in buffered TBAF to give 1,2-O-isopropylidene-3-deoxyribose 24 (82%). Phosphitylation and subsequent oxidation of 24 gave the desired di-tert-butyl phosphate derivative 25 (50%). However, several attempts to deprotect compound 25 with aqueous TFA led to a complex mixture, suggesting that di-tert-butyl protected phosphate 25 was not the optimal precursor for 12. Therefore, 24 was phosphitylated and subsequently oxidized to its dibenzylphosphate derivative 26 (72%) that was successfully deprotected to give 27 (68%). Hydrogenation of 27 followed by neutralization of the free phosphate using tributylamine afforded the target analogue 3-deoxyribose-5-phosphate as its mono-tributylammonium salt 12 (67%).
Scheme 2. Synthesis of Modified Terminal Riboses 11 and 12.

Reagents and conditions: (a) Dowex D50 (H+), tributylamine; (b) TBDPS chloride, DMAP, pyridine, 20 °C, 16 h; (c) 1,1′-thiocarbonyldiimidazole, DCM, reflux, 2 h; (d) tributyltinhydride, AIBN, toluene, 116 °C, 3 h; (e) TBAF·3H2O, acetic acid, 20 °C, 3 h; (f) 5-Ph-1-H-tetrazole, di-tert-butyl N,N-diisopropylphosphoramidite, DCM, 20 °C, 1 h; (g) triethylamine, H2O2, 0–20 °C, 1 h; (h) various conditions with aqueous TFA; (i) 5-Ph-1-H-tetrazole, dibenzyl N,N-diisopropylphosphoramidite, DCM, 20 °C, 1 h; (j) triethylamine, H2O2, 0–20 °C, 1 h; (k) aqueous TFA, 0 °C, 1.5 h; (l) hydrogen (balloon), Pd/C, 4 h, 20 °C; (m) tributylamine.
In previous work, we used morpholidate chemistry or 1,1′-carbonyldiimidazole-based methodology in coupling reactions, but these had been less than satisfactory during ADPR analogue preparation. To find the most suitable conditions here for pyrophosphate bond formation between the corresponding ribose-5-monophosphate and either AMP or 2′-dAMP, two different methods were tried. Activation of AMP tributylammonium salt with 1,1′-carbonyldiimidazole35 followed by addition of the triethylammonium salt of ribose 9 did show formation of the desired pyrophosphate product 1 by high-performance liquid chromatography (HPLC). However, several byproducts were also observed (see Supporting Information Figure S1a, S2). In contrast, following the Dabrowski-Tumanski procedure36 activation of the AMP tributylammonium salt using imidazole, 2,2′-dithiodipyridine (Aldrithiol) and triphenyl phosphine cleanly afforded the AMP-imidazolide that was isolated by precipitation with a cold solution of NaI in acetone. When stirred with the triethylammonium salt of 9 and magnesium chloride in dimethylformamide (DMF), the AMP-imidazolide gave almost exclusively the desired compound 1 (see Supporting Information Figure 1b, S2–S3). Finally, purified target ribose derivatives 9, 10, 11, and 12 were individually coupled to imidazolide-activated AMP or 2′-deoxy-AMP (Scheme 3). Adenosine-5′-monophosphate 28 and 2′-deoxy-adenosine-5′-monophosphate 29 were transformed to their imidazolides 30 and 31, by reaction of the corresponding mono-tributylammonium salt of 28 and 29 and imidazole using the Aldrithiol and triphenylphosphine condensation protocol.36 Imidazolides 30 and 31 were precipitated from the reaction mixture by addition of a 0.1 M solution of sodium iodide in cold, anhydrous acetone and were directly used for coupling. The target modified ADPR analogues 1–6 were then prepared by coupling36 the corresponding imidazolide 30 or 31 and mono-tributylammonium salt of the modified ribose-5-phosphate 9, 10, 11, or 12 to give variable yields of the desired ADPR analogues 1 (24%), 2 (66%), 3 (8%), 4 (17%), 5 (42%), and 6 (27%). Final compounds 1–6 were purified by semi-preparative HPLC using triethylammonium bicarbonate (TEAB) buffer and isolated as the corresponding triethylammonium salts.
Scheme 3. Synthesis of Target Terminal-Modified ADPR Analogues.

Reagents and conditions: (a) Aldrithiol, imidazole, triethylamine, triphenylphosphine, 20 °C, 16 h; (b) NaI, acetone (precipitation), 0 °C; (c) MgCl2, dimethylformamide, 20 °C, 3–16 h.
Stability of Terminal-Modified ADPR Analogues 1–6
The stability of ADPR analogues 1–6 was evaluated using analytical HPLC. Aqueous solutions of 1–6 stored at 5 °C were compared with freshly prepared standard solutions of 1–6 (from solid samples stored at −20 °C). All compounds except 3 were stable in aqueous solution for several days when stored in the fridge (5 °C). Compound 3, 2″-deoxy-ADPR, was found to be unstable under these conditions. We also observed decomposition of 3 during transport and sample preparation, so we could not do electrophysiological experiments with 3 (Supporting Information Figure 2, S4). The instability of 3 may correspond with the lower stability of 2-deoxyribose-5-monophosphate 11 that was sensitive toward changes in pH during conversion of the commercially available sodium salt to the tributylammonium salt (conversion of 19 to 11). This decomposition led to hydrolysis of the 5′-phosphate (approx. 20–30% de-phosphorylation of 11 was observed via 31P NMR). The corresponding decomposition by de-phosphorylation in 2″-deoxy-ADPR 3 might cleave the pyrophosphate bond and would be predicted to lead to inactive fragments.
Evaluation of Novel Analogues Against TRPM2
Recently, we investigated the importance of the ADPR terminal ribose for the activation of TRPM2.29 This can be investigated by whole cell patch-clamp experiments. During these experiments, a glass pipette with a diameter much smaller than a cell and filled with a solution mimicking the ionic composition of the cytosol is attached to the plasma membrane. By applying suction to the pipette, the membrane patch underneath the pipette is ruptured. In voltage-clamp experiments, a voltage is applied between an electrode in the pipette solution and a bath electrode in the bath solution surrounding the cells. This voltage results in ions moving through channels in the membrane resulting in a current that can be recorded.37,38 In HEK293 cells expressing TRPM2, addition of ADPR to the pipette solution results in a current that is absent in wild type HEK293 cells. ADPR analogues can be tested for their ability to activate the channel, and they can also be tested for antagonist activity by adding them in addition to ADPR. We found that adenosine 5′-monophosphate (AMP) and adenosine 5′-diphosphate (ADP) neither activated or inhibited the channel even when applied in large excess over ADPR.29 Interestingly, replacement of the terminal ribose of ADPR with small substituents led to analogues that did not activate the channel, but inhibited activation by ADPR, indicating that they compete with ADPR for the ligand binding site of TRPM2. Thus, activation of TRPM2 may be attributed to specific interactions between the hydroxyl groups of the terminal ribose of ADPR and the ligand binding NUDT9H domain of TRPM2. We evaluated the deoxy analogues 2 and 4 and an ADPR analogue 1 where all hydroxyl groups of the terminal ribose are masked and could no longer act as hydrogen bond donors (Figure 3). It was not possible to evaluate 3 biologically because of instability issues (see above and SI). During the experiments, we included 8-Br-ADPR as a control. 8-Br-ADPR has previously been shown to inhibit activation of TRPM2 by ADPR most likely by competing with the agonist.10
Figure 3.

Effect of ADPR analogues on whole cell currents in TRPM2 expressing HEK293 cells. Conditions: (a) Effect of ADPR analogues on whole cell currents in TRPM2 expressing HEK293 cells. Experiments were done as outlined in the Experimental Section. ADPR or the indicated ADPR analogue was added to the pipette solution at a concentration of 100 μmol/L. (b) Effect of ADPR analogues on TRPM2 whole cell currents elicited by ADPR. In this case, the pipette solution contained either no nucleotide (buffer), 100 μM ADPR or a combination of 100 μM ADPR with 900 μM of the indicated ADPR analogue. 8-Br-ADPR was included as the inhibitor control. Points indicate the maximum current from individual cells. The number of cells for each condition is indicated at the bottom. Bars represent the mean of the log-transformed currents (ns = non-significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). Control conditions on both panels include overlapping sets of data because on some days, both agonist and antagonist experiments were performed.
As expected, the masked analogue 1″-β-O-Me-2″,3″-O-iPr-ADPR 1 did not activate TRPM2 on its own (Figure 3a). Because it is more space-filling than ADPR, we were unsure whether it would compete with ADPR for binding to the NUDT9H domain. When applied in excess over ADPR, however, it indeed inhibited activation of TRPM2 (Figure 3b), showing that further steric constraints than those simply at the 1″-position of the terminal ribose are tolerated and may potentially be exploitable for TRPM2 antagonist optimization.
Interestingly, an analogue 32 (Figure 4) of the low-affinity partial TRPM2 agonist α-β-methylene ADPR (AMPCPR)39 masked in the same way as 1 was recently described to be a high affinity TRPM2 antagonist that inhibits ADPR-elicited TRPM2 currents with an IC50 of 5.7 μM.40 Luo et al. here did not explicitly mention whether they tested this compound for TRPM2 activation but, because it fully inhibits the channel at 100 μM, it is unlikely to exhibit agonist activity. Methylene analogues may show increased stability toward cellular pyrophosphatases like NUDT9 and NUDT5 which may be retained in the cell during whole cell patch-clamp experiments. Earlier reports that indicated an ADPR pyrophosphatase activity of the NUDT9H domain of TRPM2,1 that might contribute to degradation of agonists with pyrophosphate bridge have recently been disputed.3 The observed difference could also result from different experimental conditions as we buffered the intracellular Ca2+ concentration to 200 nmol/L with 10 mmol/L of EGTA, whereas Luo et al. left the Ca2+ concentration largely unbuffered (50 μmol/L EGTA) which would result in a much steeper dependence of the current on the agonist concentration and a more pronounced inhibition by the antagonist.
Figure 4.

Structures of masked ADPR 1, α-β-methylene ADPR (AMPCR) 32, 2″-O-acetyl-ADPR 33, and 3″-O-acetyl-ADPR 34.
Contrary to our expectations, both ADPR analogues 2 and 4 lacking hydroxyl groups at the terminal ribose did not induce significant whole cell currents in TRPM2-expressing HEK cells at 100 μmol/L (Figure 3a). This was unexpected as O-acetyl-ADPR, a product of NAD-dependent histone deacetylases of the Sir2 family, is an effective activator of TRPM2 that binds to the NUDTH domain with similar affinity as ADPR, as shown by UV cross-linking experiments and activates TRPM2 with a comparable concentration dependence as ADPR.41 2″-O-acetyl-ADPR 33, the product of the deacetylases, undergoes in neutral solution rapid intramolecular transesterification resulting in roughly equal amounts of 2″-O-acetyl-ADPR 33 and 3″-O-acetyl-ADPR 34.42 Either the nucleotide binding site of TRPM2 is indifferent to the position of the acetyl group, which would indicate that neither the 2″- nor the 3″-hydroxyl group is essential as hydrogen bond donor, or there is a specificity for one of the isomers which would mean that only half of the O-acetyl-ADPR molecules in solution are effective agonists. Because the latter seems unlikely, this may suggest that the 2″-OH and 3″-OH groups are hydrogen-bond acceptors, a feature that could be retained even when acetylated. We have previously shown that α-1″-O-methyl-ADPR and β-1″-O-methyl-ADPR (Figure 1) do not activate TRPM2, but both inhibit channel activation by ADPR.29 This indicates that the 1″-hydroxyl group in ADPR could not be masked or that there is no space in this region to accommodate even a relatively small methyl group, without losing agonist activity in contrast to the 2″- and 3″-hydroxyl groups. So why does 3″-deoxy-ADPR 4 not activate the channel? Possible explanations may be that the conformation of the furanose ring or the ratio between the two anomeric forms of the ribose is affected by the absence of the 3″-hydroxyl group in a way that prevents the proper hydrogen bonding necessary for channel activation or that this hydroxyl is required as a hydrogen bond acceptor.
The five-membered ribose sugar ring is puckered because of nonbonded interactions, and the energetically most stable conformation has all substituents as far apart as possible.43 Thus, the different substitution patterns in the terminal ribose of ADPR, 1″-deoxy-ADPR 2, 2″-deoxy-ADPR 3, and 3″-deoxy-ADPR 4, might be expected to produce differing types of puckering. Extensive analysis of the ribose and 2′-deoxy-ribose rings in RNA and DNA nucleotides has shown that while ribose adopts a primarily C3′-endo configuration, 2′-deoxy-ribose adopts a primarily C2′-endo form.44 Such changes alter the orientation of the ribose ring substituents, and this may affect the orientation of the substituents and the way they are presented to the binding site, as well as the stability of a given ADPR analogue by bringing nucleophilic hydroxyl groups into proximity for intramolecular attack on the pyrophosphate. Unfortunately, the broad nature of the peaks in the 1H NMR spectra of the ADPR analogues means that it is not possible fully to determine the coupling constants and hence to analyze fully each terminal ribose ring conformation and suggests that rapid interconversion between configurations may be taking place in solution.
ADPR is a mixture of terminal ribose α- and β-anomers and it is not known whether one or both anomers activate TRPM2. Analysis of 1H NMR spectra demonstrates that both 2″-deoxy-ADPR 3 and 3″-deoxy-ADPR 4 are also mixtures of the α- and β-forms in the following ratios (α/β, 2″-deoxy-ADPR 3, 1:1.17; 3″-deoxy-ADPR 4; 1:4). Both analogues still adopt cyclic (as opposed to open chain form) structures for the terminal ribose. Given that both anomers (α- and β-forms) are present in both cases, it would seem unlikely that this could be the sole cause of their observed inactivity.
Surprisingly, di-deoxy ADPR analogues 5 and 6 did not have significant antagonist activity (data not shown). This was unexpected because the potency of our previous antagonist 8-Ph-ADPR (IC50 = 11 μM) was improved by the 2′-deoxy modification (8-Ph-2′-deoxy-ADPR IC50 = 3 μM).
The recent cryo-EM structures of human TRPM2 show that in the absence of ADPR interactions between the NUDT9H domain and MHR1/2 domains of the same and adjacent subunits lock the channel in a closed conformation.21 Binding of ADPR impacts the conformation of the NUDT9H domain of TRPM2 in a way that leads to disengagement of the inter-subunit interactions, priming the channel for full activation by binding of Ca2+ ions to a cytosolic site near the pore region.21 The NUDT9H domain of TRPM2 has a bi-lobed structure with the two lobes forming a cleft that is supposed to bind ADPR. While comparison of the apo and the ADPR bound state shows a narrowing of this cleft upon binding of ADPR,21 the low local resolution of the structure in the NUDT9H domain does not currently allow for localization of ADPR in this binding pocket or for identification of the interactions, which would trigger the conformational changes leading to the primed state. In the absence of a high-resolution structure, SAR data therefore remain essential for ligand based drug design. Our current data further support the role of interactions involving the terminal ribose of ADPR in the gating of TRPM2.
Recently, key differences between hTRPM2 and the invertebrate N. vectensis variant nvTRPM2 were identified. Thus, two reported synthetic ADPR analogue hTRPM2 antagonists were nvTRPM2 agonists.45 Moreover, a regulatory function of NUDT9H in nvTRPM2 opposed to that in hTRPM2 was uncovered through the action of another synthetic analogue, inosine 5′-diphosphate ribose IDPR. Thus, ADPR analogues, such as those reported here, are at the cutting edge of progress to unravel the mechanism of TRPM2 function.
Conclusions
Building on our previous study that showed the essential role of the terminal ribose of ADPR in gating of the Ca2+-permeable, nonselective cation channel TRPM2, we synthesized ribose and 2′-deoxyribose analogues of ADPR 1–6, each lacking one of the hydroxyl groups of the terminal ribose. The convergent synthesis of the targets 1–6 consisted of two separate steps: (a) synthesis of particular deoxyribose monophosphates 9–12 and (b) coupling analogues 9–12 to the AMP and 2′-deoxy-AMP imidazolides 30–31, following the Dabrowski-Tumanski procedure.36 Such an approach seems to be highly efficient in preparation of such complex molecules as modified ADPR analogues, offering improved yields in our hands compared to pyrophosphate formation using AMP-morpholidates or activation using CDI. While 2″-deoxy-ADPR 3 proved to be unstable, analogues 1, 2, 4, 5, and 6 showed acceptable stability and were tested electrophysiologically in patch-clamp cell experiments. Neither 1″-deoxy-ADPR 2 nor 3″-deoxy-ADPR 4 was able to activate TRPM2 significantly; instead, both 2 and 4 were weak antagonists of ADPR-mediated TRPM2 activation in whole cell experiments, further highlighting the sensitivity of channel activation to structural changes of the terminal ribose. Thus, 1″-β-O-Me-2″,3″-O-iPr-ADPR, 1″-deoxy-, and 3″-deoxy-ADPR (1, 2 and 4) were all antagonists of ADPR-mediated Ca2+-release in whole cell patch-clamp experiments. Unlike in our previous observations, where including an additional 2′-deoxy-modification generated a more potent TRPM2 antagonist, the corresponding 1″- and 3″, 2′-dideoxy-analogues 5 and 6 were less potent antagonists of TRPM2. Results further highlight the significance of the ADPR terminal ribose for activation of TRPM2 and the use of synthetic ADPR analogues in general. The synthesis of such ADPR analogues as chemical biology tools using the methods outlined here is proving invaluable in the wider TRPM2 field.45 Further analogues to interrogate individual hydroxyl group stereochemistry or individually mask each of the three terminal hydroxyl groups to probe hydrogen bonding interactions at the TRPM2 binding site will undoubtedly shed more light on the role of the terminal ribose.
Experimental Section
General
All reagents and solvents were of commercial quality and were used without further purification, unless described otherwise. Triethylamine was dried over potassium hydroxide, distilled, and then stored over potassium hydroxide pellets. H2O was of MilliQ quality. Unless otherwise stated, all reactions were carried out under an inert atmosphere of nitrogen. All 1H, 13C, and 31P NMR spectra of the final compounds were collected, either on a Varian Mercury 400 MHz or Bruker AVANCE III 500 MHz Spectrometer. Chemical shifts (δ) are reported in parts per million (ppm) and all 1H and 13C NMR assignments are based on COSY, HSQC, HMBC, and DEPT experiments. Abbreviations for splitting patterns are as follows: br, broad; s, singlet; d, doublet; t, triplet; m, multiplet etc. High-resolution time-of-flight mass spectra were obtained on a Bruker Daltonics micrOTOF mass spectrometer using electrospray ionisation. Analytical HPLC analyses were carried out on a Waters 2695 Alliance module equipped with a Waters 2996 Photodiode Array Detector (210–350 nm). The chromatographic system consisted of a Hichrom Guard Column for HPLC and a Phenomenex Synergi 4u MAX-RP 80A column (150 × 4.60 mm), eluted at 1 mL/min with a gradient of MeCN in 0.05 M TEAB. Semi-preparative HPLC was carried out on a Waters 2525 pump with manual FlexInject. The chromatographic system consisted of a Phenomenex Gemini, 5u, C18, 110A column (250 × 10.00 mm), eluted at 5 mL/min.
1-O-Methyl-2,3-O-isopropylidene-β-d-ribofuranose (7)30
Compound 7 was prepared according to the literature from d-ribose. Yield (4.28 g, 63%). NMR and HRMS confirmed. 1H NMR (400 MHz, CDCl3): δ 4.93 (s, 1H, H-1), 4.78 (d, J = 6.0 Hz, 1H, H-2), 4.54 (d, J = 6.0 Hz, 1H, H-3), 4.38 (br s, 1H, H-4), 3.66–3.55 (m, 2H, H-5a,b), 3.38 (s, 3H, OCH3), 3.19 (br s, 1H, OH), 1.44 (s, 3H, CH3), 1.27 (s, 3H, CH3).
Dibenzyl 1-O-methyl-2,3-O-isopropylidene-β-d-ribofuranose-5-phosphate (8)
1-O-methyl-2,3-O-isopropylidene-β-d-ribofuranose 7 (100 mg, 0.49 mmol) and 5-phenyl-1H-tetrazole (143 mg, 0.98 mmol) were co-evaporated with dry toluene (2×). Then, the solid mixture was suspended in dichloromethane (2 mL), and dibenzyl N,N-diisopropylphosphoramidite (250 μL, 0.669 mmol) was added dropwise. The mixture was stirred at 20 °C for 2 h. TLC analysis (petroleum ether/EtOAc, 2:1 v/v) showed complete phosphitylation. The reaction was cooled to 0 °C, and triethylamine (407 μL, 2.92 mmol) was added followed by aqueous hydrogen peroxide (35%, 109 μL, 1.24 mmol). The resulting mixture was stirred at 20 °C for 1 h. The mixture was diluted with EtOAc (20 mL) and extracted with aqueous Na2SO3 (10%, w/v). The organic phase was dried (Na2SO4), solids were removed by filtration, and the solvent was evaporated in vacuo. The crude product was purified by Isco-Flash chromatography using petroleum ether/EtOAc (1:0 → 0:1, v/v). This procedure afforded the title compound 8 as a colorless liquid (180 mg, 79%). 1H NMR (400 MHz, CDCl3): δ 7.40–7.31 (m, 10H, Bn-2, 3, 4), 5.10–5.0 (m, 4H, Bn-CH2), 4.93 (s, 1H, H-1), 4.60 (dd, 1H, J = 6.0 Hz, J = 0.4 Hz, H-2), 4.50 (d, 1H, J = 6.0 Hz, H-3), 4.28 (dt, 1H, J = 6.0 Hz, J = 0.4 Hz, H-4), 4.0–3.88 (m, 2H, H-5), 3.26 (s, 3H, OMe), 1.46 and 1.28 (2 × s, 2 × 3H, CH3iPr). 31P NMR (160 MHz, CDCl3): δ −1.23 (s, P). 13C{1H}NMR (100 MHz, CDCl3): δ 135.7 and 135.6 (2 × C-ipso-Bn), 128.5 and 127.9 (10 × C-Bn-o, m, p), 112.5 (C(CH3)), 109.3 (C-1), 84.9 (C-3), 84.6 (d, J = 9.0 Hz, C-4), 81.4 (C-2), 69.4 and 69.4 (CH2-Bn), 67.1 (d, J = 6.0 Hz, C-5), 55.0 (OMe), 26.3 and 24.9 (CH3iPr). HRMS (ES+) calcd for C23H29NaO8P, 487.1492 [M + Na]+; found, 487.1501.
1-O-Methyl-2,3-O-isopropylidene-β-d-ribofuranose-5-phosphate (9)
Dibenzyl 1-O-methyl-2,3-O-isopropylidene-β-d-ribofuranose-5-phosphate 8 (180 mg, 0.39 mmol) was dissolved in MeOH–water (17:3 v/v, 8 mL). After vacuum/argon deoxygenation of the reaction mixture, a catalytic amount of palladium on charcoal (10%, 20 mg) was added. The reaction was stirred under positive pressure of hydrogen atmosphere (balloon) at 20 °C for 5 h. Then, the reaction was flushed with argon and neutralized by addition of TEAB buffer (1 mL, 1 M). The solvent was evaporated and the crude product was co-evaporated (2×) with dry toluene and dried under high-vacuum to give a colourless solid film, compound 9 (1.17 × Et3N salt, 122 mg, 82%). 1H NMR (400 MHz, CD3OD): δ 4.83 (s, 1H, H-1), 4.77 (m, 1H, H-2), 4.53 (d, 1H, J = 8.0 Hz, H-3), 4.24–4.18 (m, 1H, H-4), 3.83–3.72 (m, 2H, H-5), 3.24 (s, 3H, OMe), 3.08 (q, 7H, J = 8.0 Hz, NCH2CH3), 1.37 (t, 3H, CH3iPr), 1.27–1.21 (m, 13.5 H, NCH2CH3, CH3iPr). 31P NMR (160 MHz, d4-MeOD): δ 0.87 (s, P). 13C{1H}NMR (100 MHz, CD3OD): δ 113.3 (C(CH3)), 110.7 (C-1), 86.9 (d, J = 9.0 Hz, C-4), 86.4 (C-3), 83.4 (C-2), 66.3 (d, J = 5.0 Hz, C-5), 55.1 (OMe), 47.5 (NCH2CH3), 26.7 and 25.0 (CH3iPr), 9.2 (NCH2CH3). HRMS (ES–) calcd for C9H16O8P 283.0588 [M – H]−; found 283.0575. (The product was transformed into its tributylammonium salt before the pyrophosphate coupling). Transformation of 9 to mono-tributylammonium salt: The triethylammonium salt of compound 9 (89 mg, 0.23 mmol) was dissolved in water (2 mL) and treated with Dowex D50 (H+). Then, the resin was removed by filtration and the solution of 9 (free acid) was titrated with tributylamine (54 μL, 0.23 mmol, 1 equiv, to pH 7). The solution of 9 was freeze-dried overnight to obtain a colorless film, compound 9 (mono-tributylammonium salt, quant.). 1H NMR (400 MHz, H2O, water suppression): δ 5.09 (s, 1H, H-1), 4.90 (d, 1H, J = 5.6 Hz, H-2), 4.65 (d, 1H, suppressed by H2O, H-3), 4.43–4.35 (m, 1H, H-4), 3.89–3.82 (m, 2H, H-5), 3.37 (s, 1H, OMe), 3.15–3.07 (m, 6H, NCH2), 1.69–1.59 (m, 6H, NCH2CH2), 1.49 (s, 3H, CH3iPr), 1.40–1.30 (m, 9H, N(CH2)2CH3, CH3iPr), 0.95–0.87 (m, 9H, N(CH2)3CH3). 31P NMR (160 MHz, D2O): δ 0.14 (s, P). 13C{1H}NMR (100 MHz, D2O): δ 113.2 (C(CH3)), 108.5 (C-1), 86.4 (C-3), 85.0 (d, J = 9.0 Hz, C-4, HSQC), 81.0 (C-2), 65.2 (d, J = 7.0 Hz, C-5, HSQC), 54.8 (OMe), 52.8 (NCH2), 25.3 (CH3iPr), 25.3 (NCH2CH2), 23.7 (CH3iPr), 19.3 (N(CH2)2CH2), 12.8 (N(CH2)3CH3).
2,3-O-Isopropylidene-5-O-trityl-d-ribofuranose (14)
2,3-O-Isopropylidene-D-ribofuranose 13 (4 g, 21.1 mmol) was dissolved in pyridine (10 mL). Trityl chloride (7.05 g, 25.3 mmol) was added to the stirred solution, and the mixture was stirred at 20 °C for 16 h. Water (30 mL) was added, and the mixture was stirred additional 10 min. The aqueous phase was extracted with dichloromethane (3×). The combined organic phase was dried with MgSO4, solids were removed by filtration, and the solvent was evaporated in vacuo. The crude product was purified by silica gel chromatography petroleum ether/EtOAc (1:0 → 0:1, v/v). This procedure afforded the title compound 14 as a white amorphous solid (5.9 g, 65%). 1H NMR25 (400 MHz, DMSO-d6): δ 7.42–7.32 and 7.30–7.24 (2 × m, 12 + 3H, o, m, p-Tr), 6.41 (d, 1H, J = 4.0 Hz, OH), 5.15 (d, 1H, J = 4.0 Hz, H-1), 4.56 (dd, 1H, J = 6.0 Hz, J = 1.0 Hz, H-2), 4.36 (d, 1H, J = 6.0 Hz, H-3), 4.11–4.05 (m, 1H, H-4), 3.16–3.05 (m, 2H, H-5), 1.37 and 1.23 (2 × s, 2 × 3H, CH3iPr). HRMS (ES+) calcd for C27H28NaO5, 455.1829 [M + Na]+; found, 455.1842.
2,3-O-Isopropylidene-5-O-trityl-d-ribitol (15)
2,3-O-Isopropylidene-5-O-trityl-d-ribofuranose 14 (5.2 g, 11.98 mmol) was dissolved in EtOH and cooled to 0 °C. Sodium borohydride (460 mg, 12.1 mmol) was added to the solution in three portions over the period of 30 min. The mixture was stirred at 20 °C for 2 h. Water (80 mL) was added, and the solution was carefully acidified with acetic acid to pH 6. Then, the aqueous phase was extracted with dichloromethane (3×). The combined organic phase was dried (Na2SO4), solids were removed by filtration, and the solvent was evaporated in vacuo. The crude product was purified by Isco-Flash chromatography using CH2Cl2/acetone (1:0 → 1:1, v/v). This procedure afforded the title compound 15 as an amorphous white solid (4.35 g, 83%). 1H NMR (400 MHz, DMSO-d6): δ 7.48–7.41 (m, 6H, o-Tr), 7.35–7.28 (m, 6H, m-Tr), 7.26–7.20 (m, 3H, p-Tr), 5.15 (d, J = 5.6 Hz, OH-4), 4.80 (t, J = 5.6 Hz, OH-1), 4.17–4.10 (m, 1H, H-2), 4.08–4.01 (m, 1H, H-3), 3.78–3.66 (m, 2H, H-4 and H-1a), 3.51–3.43 (m, 1H, H-1b), 3.15–3.11 (m, 1H, H-5a), 3.06–3.01 (m, 1H, H-5b), 1.20 (s, 6H, CH3iPr). 13C{1H}NMR (100 MHz, DMSO-d6): δ 144.0 (3 × C-ipso-Tr), 128.4 (6 × C-o-Tr), 127.7 (6 × C-m-Tr), 126.8 (3 × C-p-Tr), 107.4 (C(CH3)), 85.7 (C(Tr)), 78.2 (C-2), 76.4 (C-3), 68.2 (C-4), 66.1 (C-1), 60.0 (C-5), 27.8 and 25.4 (CH3iPr). HRMS (ES+) calcd for C27H30NaO5, 457.1985 [M + Na]+; found, 457.2004.
1-Deoxy-2,3-O-isopropylidene-5-O-trityl-d-ribofuranose (16)
2,3-O-Isopropylidene-5-O-trityl-d-ribitol 15 (1.89 g, 4.35 mmol) was dissolved in pyridine (15 mL) and tosyl chloride (2.49 g, 13.06 mmol) was added. The mixture was stirred at 60 °C for 16 h. TLC analysis showed that the reaction was complete. The tosyl chloride surplus was decomposed by addition of water (50 mL) and the aqueous phase was extracted with dichloromethane (3×). The combined organic phase was dried (Na2SO4), solids were removed by filtration, and the solvent was evaporated in vacuo. The crude product was purified by Isco-Flash chromatography using petroleum ether/EtOAc (1:0 → 1:1, v/v), affording the title compound 16 as an amorphous white solid (1.44 g, 80%). 1H NMR (400 MHz, CDCl3): δ 7.44–7.39 (m, 6H, o-Tr), 7.33–7.27 (m, 6H, m-Tr), 7.26–7.21 (m, 3H, p-Tr), 4.91–4.86 (m, 1H, H-2), 4.65 (dd, 1H, J = 6.4 Hz, J = 1.2 Hz, H-3), 4.24–4.19 (m, 1H, H-4), 4.15 (dd, 1H, J = 10.4 Hz, J = 4.4 Hz, H-1a), 4.05 (dd, 1H, J = 10.0 Hz, J = 1.0 Hz, H-1b), 3.27 (dd, 1H, J = 10.0 Hz, J = 4.0 Hz, H-5a), 3.11 (dd, 1H, J = 10.0 Hz, J = 4.4 Hz, H-5b), 1.51 and 1.34 (2 × s, 6H, CH3iPr). 13C{1H}NMR (100 MHz, CDCl3): δ 143.6 (3 × C-ipso-Tr), 128.6 (6 × C-o-Tr), 127.8 (6 × C-m-Tr), 127.2 (3 × C-p-Tr), 112.3 (C(CH3)), 87.1 (C(Tr)), 84.0 (C-4), 83.0 (C-3), 81.6 (C-2), 74.3 (C-1), 64.5 (C-5), 26.6 and 25.1 (CH3iPr). HRMS (ES+) calcd for C27H28NaO4, 439.1880 [M + Na]+; found, 439.1868.
1-Deoxy-2,3-O-isopropylidene-d-ribofuranose (17)
1-Deoxy-2,3-O-isopropylidene-5-O-trityl-d-ribofuranose 16 (100 mg, 0.24 mmol) was dissolved in diethylether–HCOOH (2 mL, 1:1, v/v) and stirred at 20 °C for 16 h. TLC analysis (petroleum ether/EtOAc, 1:1, v/v) indicated the complete conversion of reaction and the mixture was evaporated to dryness. The dry residue was suspended in water and the suspension was filtered through a cotton pad. Evaporation of water afforded the title compound 17 as a colorless viscous liquid (14.5 mg, 32%). 1H NMR (400 MHz, CDCl3): δ 4.83–4.78 (m, 1H, H-2), 4.59 (dd, 1H, J = 6.4 Hz, J = 2.0 Hz, H-3), 4.13–4.09 (m, 1H, H-4), 3.98–3.95 (m, 2H, H-1a,b), 3.65 (dd, 1H, J = 11.6 Hz, J = 4.0 Hz, H-5a), 3.58 (dd, 1H, J = 11.6 Hz, J = 6.4 Hz, H-5b), 1.51 and 1.33 (2 × s, 6H, CH3iPr). 13C{1H}NMR (100 MHz, CDCl3): δ 113.0 (C(CH3)), 84.8 (C-4), 81.8 (C-3), 81.0 (C-2), 72.8 (C-1), 61.8 (C-5), 26.7 and 25.0 (CH3iPr). HRMS (ES+) calcd for C8H14NaO4 197.0784 [M + Na]+; found 197.0791.
1-Deoxy-2,3-O-isopropylidene-d-ribofuranose-5-O-di-tert-butylphosphate (18)
1-Deoxy-2,3-O-isopropylidene-d-ribofuranose 17 (94 mg, 0.49 mmol) and 5-phenyl-1H-tetrazole (145 mg, 0.99 mmol) were co-evaporated with dry toluene (2×). Then, the dry mixture was dissolved in dichloromethane (2 mL), and di-tert-butyl N,N-diisopropylphosphoramidite (234 μL, 0.74 mmol) was added dropwise. The mixture was stirred at 20 °C for 2 h after which TLC analysis (petroleum ether/EtOAc, 2:1, v/v) showed the reaction was complete. It was cooled to 0 °C and triethylamine (411 μL, 2.97 mmol) was added, followed by hydrogen peroxide (35% aq, 109 μL, 1.24 mmol). The resulting mixture was stirred at 20 °C for 1 h. The mixture was diluted with EtOAc (20 mL) and extracted with aqueous Na2SO3 (10%, w/v). The organic phase was dried (Na2SO4), solids were filtered off, and the solvent was evaporated in vacuo. The crude product was purified by Isco-Flash chromatography using petroleum ether/EtOAc (1:0 → 0:1, v/v), to afford the title compound 18 as a colorless liquid (74 mg, 41%). 1H NMR (400 MHz, CDCl3): δ 4.85–4.81 (m, 1H, H-2), 4.76 (dd, 1H, J = 6.4 Hz, J = 1.2 Hz, H-3), 4.23–4.18 (m, 1H, H-4), 4.2–3.94 (m, 4H, H-1, H-5), 1.51 (s, 3H, CH3iPr), 1.48 (s, 18H, CH3tBu), 1.33 (s, 3H, CH3iPr). 31P NMR (160 MHz, CDCl3): δ −10.1 (s, P). 13C{1H}NMR (100 MHz, CDCl3): δ 112.6 (C(CH3)), 83.1 (d, J = 8.0 Hz, C(tBu)), 82.6 (d, J = 7.0 Hz, C-4), 82.3 (C-3), 81.2 (C-2), 73.9 (C-1), 66.6 (d, J = 7.0 Hz, C-5), 29.8–29.7 (m, CH3tBu), 26.5 and 24.9 (CH3iPr)). HRMS (ES+) calcd for C16H31NaO7P, 389.1700 [M + Na]+; found, 389.1699.
Tributylammonium 1-deoxy-2,3-O-isopropylidene-d-ribofuranose-5-phosphate (10)
1-Deoxy-2,3-O-isopropylidene-D-ribosfuranose-5-O-di-tert-butyl phosphate 18 (36 mg, 0.098 mmol) was dissolved in aqueous methanol (1:1, v/v, 2 mL), and the solution was cooled to 0 °C. Trifluoroacetic acid (2 mL) was added dropwise and the solution was allowed to warm up to 20 °C and stirred for an additional 4 h. The solution was evaporated to dryness and the residue co-evaporated with water (3×) and with methanol (3×). The procedure afforded the pure phosphate derivative as a colorless gum (21 mg, quantitative) which was directly dissolved in water (2 mL), neutralized with tributylamine (33 μL, 0.14 mmol), the solvent evaporated, and the residue co-evaporated with EtOH (2×). The title compound 10 was obtained as the mono-tributylammonium salt (18.7 mg, 49%). 1H NMR (400 MHz, MeOH): δ 4.21–4.16 (m, 1H, H-2), 4.12–4.07 (m, 1H, H-3), 4.03–3.87 (m, 4H, H-1a, 4, 5a,b), 3.69 (dd, 1H, J = 9.6 Hz, J = 4.0 Hz, H-1b), 3.10–3.03 (m, 6H, NCH2), 1.73–1.64 (m, 6H, NCH2CH2), 1.41 (m, 6H, N(CH2)2CH2), 1.00 (t, 9H, J = 7.6 Hz, CH3Bu). 31P NMR (160 MHz, MeOH): δ 1.09 (s, P). 13C{1H}NMR (100 MHz, MeOH): δ 83.3 (d, J = 8.0 Hz, C-4), 73.7 (C-1), 73.6 (C-3), 72.5(C-2), 66.3 (d, J = 5.0 Hz, C-5), 54.0 (NCH2), 27.0 (NCH2CH2), 21.0 (N(CH2)2CH2), 14.0 (CH3Bu). HRMS (ES–) calcd for C5H11O7P, 213.0170 [M – H]−; found, 213.0162.
2-Deoxy-d-ribofuranose-5-phosphate sodium Salt (19)
Compound 19 (sodium salt) was purchased from Sigma-Aldrich (UK) and found to be a mixture of α and β anomers in the ratio 1/1.17 α/β). 1H NMR (500 MHz, D2O): δ 5.52–5.47 (m, 1.6H, H-1α, β, deuterium exchange), 4.51–4.46 (m, 1H, H-3β), 4.38–4.34 (m, 1H, H-3α), 4.25–4.20 (m, 1H, 4α), 4.04–3.99 (m, 1H, H-4β), 3.85–3.81 (m, 2H, H-5β), 3.81–3.75 (m, 2H, H-5α), 2.51–2.42 (m, 1H, H-2α), 2.22–2.14 (m, 2H, H-2β), 1.91–1.83 (m, 1H, H-2α). 31P NMR (202 MHz, D2O): δ 3.65 (P-β) and 3.68 (P-α). 13C{1H}NMR (126 MHz, D2O): δ 98.4 (C-1β), 98.0 (C-1α), 84.9 (d, J = 10.08 Hz, C-4β), 84.6 (d, J = 8.82 Hz, C-4α), 71.6 (C-3β), 71.1 (C-3α), 64.6 (d, J = 5.0 Hz, C-5β), 63.7 (d, J = 3.8 Hz, C-5α), 40.8 (C-2β), 40.6 (C-2α). Compound 19 was transformed to its tributylammonium salt.
2-Deoxy-D-ribofuranose-5-phosphate Mono-tributylammonium Salt (11)
Commercially available 2-deoxy-D-ribofuranose-5-phosphate sodium salt 19 (Sigma-Aldrich) (8 mg, 0.029 mmol) was dissolved in water (2 mL) and acidified to pH 1–2 by using the ion-exchange resin Dowex (D50 H+). The resin was filtered off and the free phosphate was neutralized by addition of one equivalent of tributylamine (6.9 μL, 0.029 mmol). The aqueous solution was freeze-dried (quant.). (NMR analysis showed partial decomposition (approx. 20–30%), of 2′-deoxyribose. The compound itself is sensitive to basic conditions (pH 9)). 1H NMR (500 MHz, D2O): δ 5.50–5.47 (m, 0.54H, H-1β), 5.43–5.41 (dd, 0.46H, J = 5.5 Hz, J = 2.5 Hz, H-1α), 4.43–4.39 (m, 0.53H, H-3β), 4.29–4.25 (m, 0.47H, H-3α), 4.15–4.1 (m, 0.46H, H-4α), 4.02–3.97 (m, 0.7H, H-5β), 3.94–3.85 (m, 1.32H, H-4β, 5α, 5β), 3.84–3.77 (m, 0.52H, H-5α), 3.09–3.01 (m, 10H, NCH2(CH2)2CH3), 2.38–2.30 (m, 0.5H, H-2α), 2.12–1.99 (m, 1H, H-2β), 1.86–1.79 (m, 0.5H, H-2α), 1.72–1.60 (m, 10H, NCH2CH2CH2CH3), 1.47–1.35 (m, 10H, NCH2CH2CH2CH3), 1.04–0.94 (m, 15H, NCH2(CH2)2CH3). 31P NMR (202 MHz, D2O): δ 1.10 (P-β) and 1.03 (P-α). 13C{1H}NMR (126 MHz, D2O): δ 100.0 (C-1β), 99.5 (C-1α), 86.3 (d, J = 8.4 Hz, C-4β), 85.6 (d, J = 8.7 Hz, C-4α), 73.5 (C-3β), 73.0 (C-3α), 67.3 (d, J = 5.4 Hz, C-5β), 66.1 (d, J = 5.3 Hz, C-5α), 53.8 (NCH2(CH2)2CH3), 43.0 (C-2β), 42.6 (C-2α), 26.9 (NCH2CH2CH2CH3), 21.1 (NCH2CH2CH2CH3), 14.0 (NCH2(CH2)2CH3).
1,2-O-Isopropylidene-d-xylofuranose (20)34
Finely powdered d-xylose (10 g, 67 mmol) was suspended in acetone (260 mL) containing sulphuric acid (10 mL) and stirred for 30 min until it was dissolved. The solution was cooled to 0 °C, and a solution of Na2CO3 (13 g in 112 mL H2O) was added. The mixture was stirred at 20 °C for 1 h, and then solid Na2CO3 (7 g) was added. The slurry was stirred at 20 °C for an further 30 min. Solids were removed by filtration and washed with acetone. Acetone was evaporated, and the water phase was extracted with EtOAc (3×) and dried with Na2SO4. Isco-Flash chromatography using petroleum ether/EtOAc (1:0 → 1:1, v/v) afforded the title compound 20 as a colorless viscous oil (4.35 g, 34%) and doubly protected 1, 2-O-isopropylidene-3,5-O-isopropylidene-d-xylofuranose (3.29 g, 21%).341H NMR (400 MHz, CDCl3): δ 5.98 (d, 1H, J = 3.6 Hz, H-1), 4.52 (d, 1H, J = 3.6 Hz, H-2), 4.32 (d, 1H, J = 2.4 Hz, H-4), 4.19–4.11 (m, 2H, H-3, 5a), 4.05 (dd, 1H, J = 12.4 Hz, J = 2.4 Hz, H-5b), 1.48 and 1.32 (CH3iPr). 13C{1H}NMR (100 MHz, CDCl3): δ 112.0 (C(CH3)2), 105.1 (C-1), 85.9 (C-2), 78.7 (C-3), 77.3 (C-4), 61.5 (C-5), 26.9 and 26.3 (CH3iPr). HRMS (ES+) calcd for C8H14NaO5, 213.0733 [M + Na]+; found, 213.0787.
5-O-tert-Butyl(diphenyl)silyl-1,2-O-isopropylidene-d-xylofuranose (21)
1,2-O-Isopropylidene-d-xylofuranose 20 (3.63 g, 19.08 mmol) and DMAP (5 mg) were dissolved in pyridine (15 mL). tert-Butyl(diphenyl)silyl chloride (4.89 mL, 19.08 mmol) was added dropwise, and the solution was stirred at 20 °C for 16 h. Solvent was evaporated and the crude product was purified by Isco-Flash chromatography using petroleum ether/EtOAc (1:0 → 1:1, v/v). This procedure afforded the title compound 21 as a white solid (6.58 g, 83%). 1H NMR (400 MHz, CDCl3): δ 7.74–7.65 (m, 4H, o-Ph), 7.48–7.38 (m, 6H, m, p-Ph), 6.00 (d, 1H, J = 3.6 Hz, H-1), 4.54 (d, 1H, J = 3.6 Hz, H-2), 4.39–4.35 (m, 1H, H-4), 4.16–4.12 (m, 1H, H-3), 4.12–4.09 (m, 2H, H-5), 3.99 (d, 1H, J = 2.4 Hz, 3-OH), 1.47 and 1.33 (CH3iPr), 1.06 (CH3tBu). 13C{1H}NMR (100 MHz, CDCl3): δ 135.8 and 135.7 (o-Ph), 132.6 and 132.1 (C-ipso-Ph), 130.2 and 128.1 (2 × d, J = 2.2 Hz, J = 1.7 Hz, m, p-Ph), 111.8 (C(CH3)2), 105.2 (C-1), 85.6 (C-2), 78.6 (C-3), 77.0 (C-4, obstructed by CDCl3 signal), 62.9 (C-5), 27.0 and 26.3 (CH3iPr), 26.9 (CH3tBu), 19.2 (C(CH3)3). HRMS (ES+) calcd for C24H32NaO5Si, 451.1911 [M + Na]+; found, 451.1929.
5-O-tert-Butyl(diphenyl)silyl-1,2-O-isopropylidene-3-thiocarbonylimidazolo-d-xylofuranose (22)
A mixture of 5-O-tert-butyl(diphenyl)silyl-1,2-O-isopropylidene-d-xylofuranose 21 (3.0 g, 7.01 mmol) and 1,1′-thiocarbonyldiimidazole (2.37 g, 13.32 mmol) was gently refluxed in dichloroethane at 85 °C for 2 h. The solvent was evaporated, and the crude product was purified by Isco-Flash chromatography using petroleum ether/EtOAc (1:0 → 0:1, v/v) to afford the title compound 22 as a white solid (2.4 g, 63%). 1H NMR (400 MHz, CDCl3): δ 8.24–8.21 (m, 1H, imidazole), 7.64–7.60 (m, 2H, Ph), 7.55–7.51 (m, 2H, Ph), 7.47 (t, 1H, J = 1.6 Hz, imidazole), 7.46–7.33 (m, 4H, Ph), 7.29–7.23 (m, 2H, Ph), 7.04 (q, 1H, J = 0.8 Hz, imidazole), 5.98 (d, 1H, J = 2.8 Hz, H-3), 5.94 (d, 1H, J = 4.0 Hz, H-1), 4.72 (d, 1H, J = 4.0 Hz, H-2), 4.61 (dq, 1H, J = 8.8 Hz, J = 2.8 Hz, H-4), 4.02 (dd, 1H, J = 10.0 Hz, J = 4.4 Hz, H-5a), 3.82 (dd, 1H, J = 10.0 Hz, J = 8.4 Hz, H-5b), 1.60 and 1.36 (2 × s, 2 × 3H, CH3iPr), 1.02 (s, 9H, CH3tBu). 13C{1H}NMR (100 MHz, CDCl3): δ 182.6 (C=S), 136.9 (imidazole), 135.6 and 135.4 (Ph), 132.8 and 132.7 (C-ipso-Ph), 131.2 (imidazole), 130.1 (Ph), 127.9 and 127.8 (Ph), 117.8 (Imz), 112.8 (C(CH3)2), 105.0 (C-1), 84.2 (C-3), 82.7 (C-2), 78.9 (C-4), 60.3 (C-5), 26.9 (CH3tBu), 26.7 and 26.4 (CH3iPr), 19.2 (C(CH3)3). HRMS (ES+) calcd for C28H34N2NaO5SSi, 561.1850 [M + Na]+; found, 561.1837.
5-O-tert-Butyl(diphenyl)silyl-1,2-O-isopropylidene-3-deoxy-d-ribofuranose (23)
Tributyltin hydride (1.07 mL, 3.99 mmol) was added into boiling toluene (90 mL) under argon followed by AIBN (13 mg, 0.08 mmol). Then, 5-O-tert-butyl(diphenyl)silyl-1,2-O-isopropylidene-3-thiocarbonylimidazolo-d-xylofuranose 22 (1.26 g, 2.35 mmol) was added dropwise in toluene (20 mL). The mixture was refluxed for 3 h. Solvent was evaporated and the crude product was purified by Isco-Flash chromatography using petroleum ether/EtOAc (1:0 → 0:1, v/v) to afford the title compound 23 as a white solid (731 mg, 76%). 1H NMR (400 MHz, CDCl3): δ 7.71 (m, 4H, o-Ph), 7.45–7.35 (m, 6H, m, p-Ph), 5.83 (d, 1H, J = 3.6 Hz, H-1), 4.75 (t, 1H, J = 4.0 Hz, H-2), 4.33 (dq, 1H, J = 10.4 Hz, J = 4.0 Hz, H-4), 3.82 (dd, 1H, J = 11.2 Hz, J = 4.4 Hz, H-5a), 3.76 (dd, 1H, J = 11.2 Hz, J = 4.0 Hz, H-5b), 3.08 (dd, 1H, J = 13.6 Hz, J = 4.8 Hz, H-3a), 1.88 (ddd, 1H, J = 13.6 Hz, J = 10.4 Hz, J = 4.8 Hz, H-3b), 1.52 and 1.34 (2 × s, 2 × 3H, CH3iPr), 1.06 (s, 9H, CH3tBu). 13C{1H}NMR (100 MHz, CDCl3): δ 135.8 and 135.7 (o-Ph), 133.7 and 133.5 (C-ipso-Ph), 129.8 and 129.8 (p-Ph), 127.8 and 127.8 (m-Ph), 111.3 (C(CH3)2), 105.9 (C-1), 80.6 (C-2), 78.7 (C-4), 64.9 (C-5), 35.1 (C-3), 27.1 (CH3iPr), 27.0 (CH3tBu), 26.5 (CH3iPr), 19.4 (C(CH3)3). HRMS (ES+) calcd for C24H32NaO4Si, 435.1962 [M + Na]+; found, 435.1995.
1,2-O-Isopropylidene-3-deoxy-d-ribofuranose (24)
A mixture of TBAF·3H2O (1.55 g, 5.0 mmol) and acetic acid (300 μL, 5.25 mmol) in DMF (10 mL) was stirred at room temperature for 30 min. The mixture was cooled to 0 °C and 5-O-tert-butyl(diphenyl)silyl-1,2-O-isopropylidene-3-deoxy-d-ribofuranose 23 (687 mg, 1.67 mmol) was added dropwise in DMF (5 mL). The resulting mixture was stirred at 20 °C for 3 h. Solvent was evaporated and the crude product was purified by Isco-Flash chromatography using petroleum ether/EtOAc (1:0 → 0:1, v/v). This procedure afforded the title compound 24 as white solid (260 mg, 82%). 1H NMR (400 MHz, CDCl3): δ 5.79 (d, 1H, J = 3.6 Hz, H-1α), 4.73 (t, 1H, J = 4.4 Hz, H-2), 4.31 (dtd, 1H, J = 10.8 Hz, J = 4.4 Hz, J = 3.2 Hz, H-4), 3.84 (dd, 1H, J = 12.0 Hz, J = 2.8 Hz, H-5a), 3.54 (dd, 1H, J = 12.0 Hz, J = 4.4 Hz, H-5b), 1.97 (dd, 1H, J = 13.6 Hz, J = 4.8 Hz, H-3a), 1.85–1.76 (m, 1H, H-3b), 1.48 and 1.29 (2 × s, 2 × 3H, CH3iPr). 13C{1H}NMR (100 MHz, CDCl3): δ 111.4 (C(CH3)2), 105.7 (C-1α), 80.9 (C-2), 78.7 (C-4), 63.1 (C-5), 34.0 (C-3), 26.9 and 26.3 (CH3iPr). HRMS (ES+) calcd for C8H14NaO4, 197.0784 [M + Na]+; found, 197.0793.
1,2-O-Isopropylidene-3-deoxy-d-ribofuranose-5-O-di-tert-butylphosphate (25)
1,2-O-Isopropylidene-3-deoxy-d-ribofuranose 24 (100 mg, 0.52 mmol) and 5-phenyl-1H-tetrazole (154 mg, 1.05 mmol) were co-evaporated with dry toluene (2×). The solid mixture was then dissolved in dichloromethane (2 mL) and di-tert-butyl N,N-diisopropylphosphoramidite (249 μL, 0.79 mmol) was added dropwise. The mixture was stirred at 20 °C for 2 h. TLC analysis (petroleum ether/EtOAc, 2:1, v/v) showed complete phosphitylation. The reaction was cooled to 0 °C and triethylamine (437 μL, 3.15 mmol) added followed by hydrogen peroxide (35% aq, 116 μL, 1.32 mmol). The resulting mixture was stirred at 20 °C for 1 h. The mixture was diluted with EtOAc (20 mL) and extracted with aqueous Na2SO3 (10%, w/v). The organic phase was dried (Na2SO4), solids were filtered off, and the solvent was evaporated in vacuo. The crude product was purified by Isco-Flash chromatography using petroleum ether/EtOAc (1:0 → 0:1, v/v), followed by a second chromatography in dichloromethane/acetone (1:0 → 1:1, v/v). This procedure afforded the title compound 25 as a colorless viscous liquid (97 mg, 50%). 1H NMR (400 MHz, CDCl3): δ 5.81 (d, 1H, J = 4.0 Hz, H-1), 4.73 (t, 1H, J = 4.4 Hz, H-2), 4.04 (ddd, 1H, J = 8.8 Hz, J = 4.4 Hz, J = 1.2 Hz, H-4), 4.12–4.06 (m, 1H, H-5a), 4.02–3.96 (m, 1H, H-5b), 2.1 (dd, 1H, J = 13.6 Hz, J = 4.8 Hz, H-3a), 1.84–1.76 (m, 1H, H-3b), 1.50 (s, 3H, CH3iPr), 1.48 (s, 9H, CH3tBu) 1.31 (s, 3H, CH3iPr). 31P NMR (160 MHz, CDCl3): δ −9.91 (s, phosphate). 13C{1H}NMR (100 MHz, CDCl3): δ 111.4 (C(CH3)2), 105.8 (C-1), 82.7 and 82.6 (2 × d, J = 2.1 Hz, C(CH3)3), 80.7 (C-2), 76.6 (d, J = 8.7 Hz, C-4), 67.0 (d, J = 6.0 Hz, C-5), 35.2 (C-3), 30.01 and 29.97 (2 × CH3tBu), 27.0 and 26.4 (2 × CH3iPr). HRMS (ES+) calcd for C16H32O7P, 367.1880 [M + H]+; found, 367.1877; calcd for C16H31NaO7P, 389.1700 [M + Na]+; found, 389.1737.
1,2-O-Isopropylidene-3-deoxy-d-ribofuranose-5-O-dibenzylphosphate (26)
1,2-O-Isopropylidene-3-deoxy-d-ribofuranose 24 (120 mg, 0.63 mmol) and 5-phenyl-1H-tetrazole (185 mg, 1.26 mmol) were co-evaporated with dry toluene (2×). Then, the solid mixture was dissolved in dichloromethane (3 mL) and dibenzyl N,N-diisopropylphosphoramidite (353 μL, 0.95 mmol) was added dropwise. The mixture was stirred at 20 °C for 2 h. TLC analysis (petroleum ether/EtOAc, 2/1, v/v) showed complete phosphitylation. The reaction was cooled to 0 °C and triethylamine (524 μL, 3.79 mmol) added followed by hydrogen peroxide (35% aq, 139 μL, 1.58 mmol). The resulting mixture was stirred at 20 °C for 1 h. The mixture was diluted with EtOAc (20 mL) and extracted with Na2SO3 (10% aq, w/v). The organic phase was dried (Na2SO4), solids were removed by filtration, and the solvent was evaporated in vacuo. The crude product was purified by Isco-Flash chromatography using dichloromethane/acetone (1:0 → 1:1, v/v) followed by a second chromatography in petroleum ether/EtOAc (1:0 → 0:1, v/v) to afford the title compound 26 as a colorless viscous liquid (218 mg, 73%). 1H NMR (400 MHz, CDCl3): δ 7.37–7.32 (m, 10H, Bn), 5.76 (d, J = 3.6 Hz, H-1), 5.10–5.00 (m, 4H, CH2Bn), 4.68 (t, 1H, J = 4.4 Hz, H-2), 4.35 (ddd, 1H, J = 8.8 Hz, J = 4.4 Hz, J = 0.8 Hz, H-4), 4.16 (dq, 1H, J = 3.6 Hz, J = 2.8 Hz, H-5a), 4.00 (dq, 1H, J = 4.4 Hz, J = 2.8 Hz, H-5b), 2.00 (dd, 1H, J = 13.2 Hz, J = 4.4 Hz, H-3a), 1.72–1.64 (m, 1H, H-3b), 1.48 and 1.31 (2 × s, 2 × 3H, CH3iPr). 31P NMR (160 MHz, CDCl3): δ −1.00 (s, phosphate). 13C{1H}NMR (100 MHz, CDCl3): δ 136.0 (d, J = 2.0 Hz, C-ipso-Bn), 135.9 (d, J = 3.0 Hz, C-ipso-Bn), 128.8–128.7 and 128.2–128.1 (2 × m, C-Bn-o, m, p), 111.5 (C(CH3)2), 105.8 (C-1), 80.5 (C-2), 76.4 (d, J = 8.0 Hz, C-4), 69.6 and 69.5 (2 × d, J = 1.3 Hz and J = 1.4 Hz, CH2Bn), 68.8 (d, J = 5.6 Hz, C-5), 34.7 (C-3), 27.0 and 26.3 (CH3iPr). HRMS (ES+) calcd for C22H28O7P, 435.1567 [M + H]+; found, 435.1571; calcd for C22H27NaO7P, 457.1387 [M + Na]+; found, 457.1392.
3-Deoxy-d-ribofuranose-5-O-dibenzylphosphate (27)
An aqueous solution (4 mL) of 1, 2-O-isopropylidene-3-deoxy-d-ribofuranose-5-O-dibenzylphosphate 26 (67 mg, 0.154 mmol) was cooled to 0 °C and trifluoroacetic acid (4 mL) was added. The solution was stirred at 0 °C for 1.5 h and then allowed to warm to 20 °C. The solvent was evaporated in vacuo at 30 °C and co-evaporated with water. Crude material was purified by silica gel chromatography using petroleum ether/EtOAc (1:0 → 0:1, v/v) to give a colorless solid (mixture of α/β anomers 1:4, 41 mg, 68%). 1H NMR (400 MHz, CD3OD): δ 7.39–7.35 (m, 10H, Bn), 5.18 (s, 0.2H, C-1α), 5.17 (s, 0.8H, C-1β), 5.09–5.04 (m, 4H, CH2Bn), 4.45–4.39 (m, 0.8H, H-4β), 4.39–4.35 (m, 0.2H, H-4α), 4.13–4.07 (m, 1H, H-2α, β), 4.06–3.99 (m, 1.8H, H-5αa, βa,b), 3.90 (ddd, 0.2H, J = 12.8 Hz, J = 5.6 Hz, J = 4.0 Hz, H-5αb), 1.96–1.85 (m, 2H, H-3α, β). 31P NMR (160 MHz, CD3OD): δ −1.30 (s, α-phosphate), −1.54 (s, β-phosphate). 13C{1H}NMR (100 MHz, CD3OD): δ 137.2 and 137.2 (2 × C-ipso-Bn), 129.8–129.7 and 129.3–129.2 (2 × m, 10 × C-Bn-o, m, p), 104.3 (C-1β), 98.4 (C-1α), 78.6 (d, J = 6.2 Hz, C-4β), 77.1 (C-2β), 75.8 (d, J = 6.2 Hz, C-4α), 72.4 (d, J = 4.8 Hz, C-5β), 72.2 (C-2α), 71.0 (d, J = 4.8 Hz, C-5α), 70.9–70.8 (m, CH2Bn), 34.7 (C-3β), 33.9 (C-3α). HRMS (ES+) calcd for C19H24O7P, 395.1254 [M + H]+; found, 395.1256; calcd for C19H23NaO7P, 417.1074 [M + Na]+; found, 417.1079.
3-Deoxy-d-ribofuranose-5-phosphate Tributylammonium Salt (12)
3-Deoxy-d-ribofuranose-5-O-dibenzylphosphate 27 (39 mg, 0.098 mmol) was dissolved in MeOH–water (8 mL, 17:3 v/v). After vacuum/argon deoxygenation of the reaction mixture, palladium on charcoal (Pd/C 10%, 10 mg) was added. The reaction was stirred under positive pressure of hydrogen atmosphere (balloon) at 20 °C for 4 h. Then, the reaction was flushed with argon and solids were filtered off and washed with water. The aqueous solution (pH 1–2) was carefully titrated by portionwise addition of tributylamine (5 × 4 μL, 0.084 mmol, pH 6–7). The solvent was evaporated, and the crude product was co-evaporated with dry toluene (2×) and dried under high vacuum for 16 h to give a colorless solid film, compound 12 (mixture of α/β anomers 1:4, 1 × Bu3N salt, 29 mg, 67%). 1H NMR (500 MHz, CD3OD): δ 5.20 (d, 0.2H, J = 4.5 Hz, H-1α), 5.12 (s, 0.8H, H-1β), 4.48–4.41 (m, 0.8H, H-4β), 4.41–4.36 (m, 0.2H, H-4α), 4.25–4.20 (m, 0.2H, H-2α), 4.08 (d, 0.8H, J = 4.5 Hz, H-2β), 3.96–3.84 (m, 1.6H, H-5β), 3.83–3.79 (m, 0.4H, H-5α), 3.14–3.08 (m, 6H, NCH2), 2.12 (ddd, 0.2H, J = 12.5 Hz, J = 5.5 Hz, J = 2.5 Hz, H-3αa), 2.04 (ddd, 0.8H, J = 13.5 Hz, J = 8.5 Hz, J = 5.0 Hz, H-3βa), 2.01–1.97 (m, 0.2H, H-3αb), 0.92 (dd, 0.8H, J = 14.0 Hz, J = 7.0 Hz, H-3βa), 1.73–1.64 (m, 6H, NCH2CH2), 1.47–1.38 (m, 6H, N(CH2)2CH2), 10.14 (t, 9H, J = 7.5 Hz, N(CH2)3CH3). 31P NMR (202 MHz, CD3OD): δ 1.04 (s, α-phosphate), 0.97 (s, β-phosphate). 13C{1H}NMR (126 MHz, CD3OD): δ 104.3 (C-1β), 98.2 (C-1α), 79.7 (d, J = 8.8 Hz, C-4β), 77.4 (C-2β), 76.8 (d, J = 8.8 Hz, C-4α), 72.3 (C-2α), 69.7 (d, J = 5.03 Hz, C-5β), 68.4 (d, J = 5.0 Hz, C-5α), 53.8 (NCH2), 35.3 (C-3β), 34.3 (C-3α), 26.8 (NCH2CH2), 21.0 (N(CH2)2CH2), 14.0 (N(CH2)3CH3). HRMS (ES–) calcd for C5H10O7P, 213.0170 [M – H]−; found, 213.0159.
AMP Imidazolide: Adenosine-5′-phosphoryl Imidazolide (30)
AMP sodium salt (50 mg, 0.128 mmol, Sigma-Aldrich) was dissolved in water (2 mL) and treated with Dowex D50 (H+) until all the phosphate was transformed to free acid (pH 1–2). Then, the resin was filtered off and washed with water, and the solution of AMP (free acid) was titrated with tributylamine (30 μL, 0.13 mmol, 1 equiv, pH 7). The solution of adenosine-5′-phosphate mono-tributylammonium salt (28) was freeze-dried to obtain a light white powder (quantitative).
AMP tributylammonium salt (15 mg, 0.028 mmol) and imidazole (19 mg, 0.28 mmol) were co-evaporated with EtOH (2×) and with toluene (2×). Aldrithiol (18.6 mg, 0.085 mmol) was added followed by DMF (220 μL), and the suspension stirred until all solid matter was dissolved. Triethylamine (16 μL, 0.113 mmol, distilled-dry-stored over KOH in the dark) and triphenylphosphine (22.2 mg, 0.085 mmol) were added, and the solution was stirred at 20 °C for 16 h. Product precipitation: The reaction mixture was cooled to 5 °C and a cold solution of NaI (33.8 mg) in dry acetone (2.2 mL) was added at 5 °C. The white precipitate was filtered off and washed with cold acetone. The product was dried under high vacuum for 16 h and stored under argon (crude compound, hygroscopic, yield was not quantified). 1H NMR (400 MHz, CD3OD): δ 8.38 (s, H-8), 8.19 (s, H-2), 7.88–7.87 (m, 1H, imidazole), 7.28–7.26 (m, 1H, imidazole), 6.99–6.97 (m, 1H, imidiazole), 6.04 (d, 1H, J = 6.0 Hz, H-1′), 4.65 (t, 1H, J = 5.6 Hz, H-2′), 4.25 (dd, 1H, J = 5.2 Hz, J = 3.6 Hz, H-3′), 4.18–4.13 (m, 1H, H-4′), 4.06–3.96 (m, 2H, H-5a,b′). 31P NMR (160 MHz, CD3OD): δ −9.03 (s, P). 13C{1H}NMR (100 MHz, CD3OD): δ 157.4 (C-6), 153.9 (C-2), 150.9 (C-4), 140.9 (C-8), 140.8 (d, J = 5.5 Hz, imidazole), 129.7 (d, J = 10.4 Hz, imidazole), 121.3 (d, J = 5.7 Hz, imidazole), 120.3 (C-5), 89.2 (C-1′), 85.3 (d, J = 8.8 Hz, C-4′), 75.8 (C-2′), 72.19 (C-3′), 66.8 (d, J = 5.9 Hz, C-5′). HRMS (ES–) calcd for C13H15N7O6P, 396.0827 [M – H]−; found, 396.0853.
1″-β-O-Methyl-2″,3″-O-isopropylidene adenosine diphosphoriboside (1)
Compound 9 (4.3 mg, 0.009 mmol) and MgCl2 (1.8 mg, 0.018 mmol) were dissolved in DMF (1 mL), co-evaporated with dry toluene (2×) and evaporated to dryness. Dry DMF (500 μL) was added to the solid residue and the mixture was stirred at 20 °C until all solids were dissolved (5–10 min). Then, imidazolide 30 (3.5 mg, 0.008 mmol) was added dropwise to the mixture in DMF (400 μL). The mixture was stirred at 20 °C for 3 h, after which conversion to product was 80–90% by HPLC. Solvent was evaporated in vacuo and crude product was dissolved in TEAB buffer (3 mL, 0.1 M) and purified by semi-preparative, reverse-phase HPLC using a gradient of TEAB (0.1 M)–acetonitrile (95:5 → 35:65, v/v). The title compound 1 was obtained as a colorless glass, 1.7 × Et3N salt, (2 μmol, 1.57 mg, 24%). 1H NMR (500 MHz, D2O): δ 8.48 (s, 1H, H-8), 8.20 (s, 1H, H-2), 6.06 (d, 1H, J = 5.0 Hz, H-1′), 4.89 (s, 1H, H-1″), 4.70–4.63 (m, 2H, H-2′, 2″), 4.50 (d, 1H, J = 5.0 Hz, H-3″), 4.45 (t, 1H, J = 5.0 Hz, H-3′), 4.33–4.28 (m, 1H, H-4′), 4.21–4.11 (m, 3H, H-4″, 5a,b′), 3.82–3.75 (m, 2H, H-5a,b), 3.21 (s, 3H, OMe), 3.12 (m, 9H, NCH2CH3), 1.31 (s, 3H, CH3-iPr), 1.19 (m, 16.5H, NCH2CH3, CH3-iPr). 31P NMR (202 MHz, D2O): δ −11.47 (m, pyrophosphate). 13C{1H}NMR (126 MHz, D2O): δ 155.1 (C-6), 152.0 (C-2), 149.1 (C-4), 140.0 (C-8), 118.3 (C-5), 112.9 (C(CH3)2), 108.4 (C-1″), 87.0 (C-1′), 84.7 (d, J = 10.1 Hz, C-4″), 84.1 (C-3″), 83.9 (d, J = 6.3 Hz, C-4′), 80.9 (C-2″), 74.3 (C-2′), 70.3 (C-3′), 65.8 (d, J = 3.8 Hz, C-5″), 65.1 (d, J = 6.3 Hz, C-5′), 54.7 (OMe), 46.6 (NCH2CH3), 25.1 and 23.5 (CH3-iPr), 8.2 (NCH2CH3). HRMS (ES–) calcd for C19H28N5O14P2, 612.1114 [M – H]−; found, 612.1143. UV (H2O, pH 7.4) λmax 259 nm (ε 16 900 L/mol·cm). HPLC tR = 5.52 min.
1″-Deoxyadenosine diphosphoriboside (1″-deoxy-ADPR) (2)
1-Deoxy-d-ribofuranose-5-phosphate tributylammonium salt 10 (5.8 mg, 0.0145 mmol) and MgCl2 (2.8 mg, 0.029 mmol) were dissolved in DMF (1 mL), co-evaporated with dry toluene (2×) and evaporated to dryness. Dry DMF (500 μL) was added to the solid residue and the mixture was stirred at 20 °C until all solids were dissolved (5–10 min). Then, AMP-imidazolide 30 (5.5 mg, 0.013 mmol) was added dropwise to the mixture in DMF (400 μL). The mixture was stirred at 20 °C for 3 h. Solvent was evaporated in vacuo and the crude product was dissolved in TEAB buffer (3 mL, 0.1 M) and purified by semi-preparative, reverse-phase HPLC using a gradient of TEAB (0.1 M)–acetonitrile (95:5 → 35:65, v/v). The title compound 2 was obtained as a colorless glass, 1.7 × Et3N salt, (9.6 μmol, 6.86 mg, 66%). 1H NMR (500 MHz, D2O): δ 8.54 (s, 1H, H-8), 8.26 (s, 1H, H-2), 6.14 (d, 1H, J = 5.0 Hz, H-1′), 4.79–4.76 (m, 1H, H-2′, obstructed by H2O peak), 4.56–4.52 (m, 1H, H-3′), 4.42–4.38 (m, 1H, H-4′), 4.26–4.21 (m, 3H, H-5a,b, H-2″), 4.17–4.14 (m, 1H, H-3″), 4.13–4.07 (m, 1H, H-5″a), 4.03–3.92 (m, 3H, H-1″a, 4″, 5″b), 3.74 (dd, 1H, J = 8.5 Hz, J = 3.5 Hz, H-1b″), 3.10 (m, 10H, NCH2CH3), 1.18 (m, 15H, NCH2CH3). 31P NMR (202 MHz, D2O): δ −11.35 (m, pyrophosphate). 13C{1H}NMR (126 MHz, D2O): δ 155.2 (C-6), 152.3 (C-2), 149.1 (C-4) HMBC, 140.0 (C-8), (C-5) not observed (HMBC), 86.8 (C-1′), 83.9 (d, J = 7.6 Hz, C-4′), 80.3 (d, J = 7.6 Hz, C-4″), 74.2 (C-2′), 72.2 (C-5a,b), 71.6 (C-3″), 70.9 (C-2″), 70.4 (C-3′), 65.5 (d, J = 3.8 Hz, C-5a,b″), 65.2 (d, J = 3.8 Hz, C-5′), 46.6 (NCH2CH3), 8.2 (NCH2CH3). HRMS (ES–) calcd for C15H22N5O13P2, 542.0695 [M – H]−; found, 542.0716. UV (H2O, pH 7.4) λmax 259 nm (ε 15 958 L/mol·cm). HPLC tR = 2.93 min.
1″-α/β-2″-Deoxyadenosine diphosphoriboside (2″-deoxy-ADPR) (3)
2-Deoxyribose-5-phosphate tributylammonium salt 11 (8 mg, 0.029 mmol) and MgCl2 (5.6 mg, 0.058 mmol) were dissolved in DMF (1.5 mL), co-evaporated with dry toluene (2×), and evaporated to dryness. Dry DMF (500 μL) was added to the solid residue and the mixture was stirred at 20 °C until all solids dissolved (5–10 min). Then, AMP-imidazolide 30 (9 mg, 0.0215 mmol) was added dropwise to the mixture in DMF (700 μL). The mixture was stirred at 20 °C for 16 h (HPLC showed a complex reaction mixture comprising 3–4 products, including AMP and AMP dinucleotide). The solvent was evaporated in vacuo and the crude product was dissolved in TEAB buffer (3 mL, 0.1 M) and purified by semi-preparative, reverse-phase HPLC using a gradient of TEAB (0.1 M)–acetonitrile (95:5 → 35:65, v/v). The title compound was obtained as a colorless glass as a mixture of anomers β/α 0.45:0.55, 1.7 × Et3N salt, (1.72 μmol, 1.23 mg, 8%). 1H NMR (500 MHz, D2O): δ 8.43 (s, 1H, H-8), 8.19 (s, 1H, H-2), 6.06 (d, 1H, J = 10.0 Hz, H-1′), 5.49–5.46 (m, 0.45H, H-1″β), 5.42–5.39 (m, 0.55H, H-1″α), 4.70–4.68 (m, 1H, H-2′, obstructed by H2O peak), 4.47–4.43 (m, 1H, H-3′), 4.37–4.33 (m, 0.45H, H-3″β), 4.33–4.29 (m, 1H, H-4′), 4.23–4.19 (m, 0.55H, H-3″α), 4.16–4.10 (m, 2.45H, H-4″α and H-5′), 3.93–3.89 (m, 1.45H, H-4″β and H-5″β), 3.87–3.83 (m, 1H, H-5″α), 3.12 (q, 10H, J = 7.5 Hz, NCH2CH3), 2.30–2.23 (m, 0.55H, H-2a″-α), 2.06–2.01 (m, 0.9H, H-2a,b-β), 1.77–1.70 (m, 0.55H, H-2b″-α), 1.19 (t, 15H, J = 7.5 Hz, NCH2CH3). 31P NMR (202 MHz, D2O): δ −11.34 (m, pyrophosphate). 13C{1H}NMR (126 MHz, D2O): δ 155.7 (C-6), 152.9 (C-2), 149.1 (C-4), 139.8 (C-8), 118.6 (C-5), 98.4 (C-1″β), 98.1 (C-1″α), 86.7 (C-1′, 84.0 and 83.9 (m, C-4′, 4″α, 4″β), 74.2 (C-2′), 71.4 (C-3″β), 71.0 (C-3″α), 70.4 (C-3′), 66.4 (d, J = 3.8 Hz, C-5″β), 65.6 (d, J = 2.5 Hz, C-5″α), 65.2 (d, J = 3.8 Hz, C-5′), 46.6 (NCH2CH3), 40.6 (C-2″α, β), 8.3 (NCH2CH3). HRMS (ES–) calcd for C15H22N5O13P2, 542.0695 [M – H]−; found 542.0690; (ES+) calcd for C15H22N5NaO13P2, 565.0587 [M + Na]+; found, 565.0579. HPLC tR = 3.10 min.
1″-α/β-3″-Deoxyadenosine diphosphoriboside (3″-deoxy-ADPR) (4)
3-Deoxy-d-ribofuranose-5-phosphate tributylammonium salt 12 (10 mg, 0.025 mmol) and MgCl2 (4.9 mg, 0.05 mmol) were dissolved in DMF (1 mL), co-evaporated with dry toluene (2×), and evaporated to dryness. Dry DMF (600 μL) was added to the solid residue, and the mixture was stirred at 20 °C until all solids were dissolved (5–10 min). Then, AMP-imidazolide 30 (9.4 mg, 0.023 mmol) was added dropwise to the mixture in DMF (400 μL). The mixture was stirred at 20 °C for 16 h. Solvent was evaporated in vacuo, and the crude product was dissolved in TEAB buffer (3 mL, 0.1 M) and purified by semi-preparative, reverse-phase HPLC using a gradient of TEAB (0.1 M)–acetonitrile (95:5 → 35:65, v/v). The title compound 4 was obtained as a colorless glass, 1.7 × Et3N salt, (3.88 μmol, 2.8 mg, 17%). 1H NMR (500 MHz, D2O, water suppression exp.): δ 8.37 (s, 1H, H-8), 8.11 (s, 1H, H-2), 5.98 (d, 1H, J = 6.0 Hz, H-1′), 5.10 (d, 0.3H, J = 4.0 Hz, H-1″α), 5.06 (s, 0.7H, H-1″β), 4.63 (t, 1H, J = 5.5 Hz, H-2′), 4.40–4.37 (m, 1H, H-3′), 4.34–4.27 (m, 1H, H-4″α, β), 4.26–4.22 (m, 1H, H-4′), 4.12–4.04 (m, 2.3H, H-2″α, 5a,b′), 4.04–4.01 (m, 0.7H, H-2″β), 3.91–3.85 (m, 0.7H, H-5″βa), 3.81–3.71 (m, 1H, H-5″αa, βb), 3.69–3.63 (m, 0.3H, H-5″αb), 3.04 (q, 10H, J = 7.5 Hz, NCH2), 1.90 (ddd, 0.3H, J = 13.0 Hz, J = 7.5 Hz, J = 2.0 Hz, H-3″αa), 1.84–1.78 (m, 1.7H, H-3″αb, βa,b), 1.12 (t, 15H, J = 7.5 Hz, NCH2CH3). 31P NMR (202 MHz, D2O): δ −11.17 (m, α-pyrophosphate), −11.46 (m, β-pyrophosphate). 13C{1H}NMR (126 MHz, D2O): δ 155.1 (C-6), 152.1 (C-2), 149.0 (C-4), 139.9 (C-8), 118.5 (C-5), 102.0 (C-1″β), 96.5 (C-1″α), 86.7 (C-1′), 83.9 (d, J = 8.7 Hz, C-4′), 78.2 (d, J = 8.4 Hz, C-4″β), 75.5 (d, J = 8.4 Hz, C-4″α), 75.3 (C-2″β), 74.2 (C-2′), 70.3 (C-3′), 70.3 (C-2″α), 68.8 (d, J = 5.2 Hz, C-5″β), 67.8 (d, J = 4.8 Hz, C-5″α), 65.1 (d, J = 5.0 Hz, C-5′), 46.5 (NCH2), 32.3 (C-3″β), 31.5 (C-3″α), 8.1 (NCH2CH3). HRMS (ES–) calcd for C15H22N5O13P2, 542.0695 [M – H]−; 542.0716. HPLC tR = 2.87 min.
2′-Deoxy AMP Imidazolide: 2′-Deoxy-adenosine-5′-phosphoryl Imidazolide (31)
2′-Deoxy-AMP was treated in the same way as AMP to generate the mono Bu3N salt (29).
2′-Deoxy-AMP mono-tributylammonium salt 29 (145 mg, 0.28 mmol) and imidazole (191 mg, 2.8 mmol) were put into the flask and co-evaporated with EtOH (2×) and with toluene (2×). Aldrithiol (186 mg, 0.84 mmol) was added to the flask followed by dry DMF (1.5 mL) and stirred until all solid matter was dissolved. Triethylamine (156 μL, 1.13 mmol, distilled-dry-stored over KOH in the dark) and triphenylphosphine (221 mg, 0.85 mmol) were added to the mixture, and the mixture was stirred at 20 °C for 16 h. The reaction mixture was cooled to 5 °C, and a cold solution of NaI (337 mg, 2.25 mmol) in dry acetone (15 mL) was added at 5 °C. The white precipitate was filtered off and washed with cold acetone. The product was dried under high vacuum and stored under argon (crude compound, hygroscopic, yield was not quantified). 1H NMR (400 MHz, CD3OD): δ 8.36 (s, H-8), 8.18 (s, H-2), 7.87–7.86 (m, 1H, imidazole), 7.26–7.24 (m, 1H, imidazole), 6.98–6.96 (m, 1H, imidazole), 6.44 (dd, 1H, J = 7.6 Hz, J = 1.2 Hz, H-1′), 4.49 (q, 1H, J = 2.8 Hz, H-3′), 4.08–4.04 (m, 1H, H-4′), 3.99–3.95 (m, 2H, H-5′), 2.75 (ddd, 1H, J = 13.6 Hz, J = 7.6 Hz, J = 1.6 Hz, H-2a′), 2.42 (dq, 1H, J = 13.6 Hz, J = 2.8 Hz, H-2b). 31P NMR (160 MHz, CD3OD): δ −9.00 (s, P). 13C{1H}NMR (100 MHz, CD3OD): δ 157.3 (C-6), 153.8 (C-2), 150.5 (C-4), 140.9 (C-8), 140.7 (d, J = 5.5 Hz, imidazole), 129.6 (d, J = 10.5 Hz, imidazole), 121.3 (d, J = 5.7 Hz, imidazole), 120.3 (C-5), 87.5 (d, J = 8.7 Hz, C-4′), 85.5 (C-1′), 72.9 (C-3′), 67.0 (d, J = 5.9 Hz, C-5′), 41.2 (C-2′). HRMS (ES–) calcd for C13H15N7O5P, 380.0878 [M – H]−; found, 380.0883.
1″,2′-Dideoxyadenosine diphosphoriboside (1″,2′-deoxy ADPR) (5)
1-Deoxy-d-ribofuranose-5-phosphate tributylammonium salt 10 (12 mg, 0.03 mmol) and MgCl2 (5.9 mg, 0.06 mmol) were dissolved in DMF (1 mL), co-evaporated with dry toluene (2×), and evaporated to dryness. Dry DMF (600 μL) was added to the solid residue and the suspension was stirred at 20 °C until all solids were dissolved (5–10 min). Then, 2′-dAMP-imidazolide 31 (10.8 mg, 0.027 mmol) was added dropwise in DMF (400 μL). The mixture was stirred at 20 °C for 3 h. HPLC showed consumption of all starting material. The solvent was evaporated in vacuo, and the crude product was dissolved in TEAB buffer (10 mL, 0.1 M) and purified by semi-preparative, reverse-phase HPLC using a gradient of TEAB (0.1 M)–acetonitrile (95/5 → 35/65, v/v). The title compound 5 was obtained as a colorless amorphous solid (11.3 μmol, 8.02 mg, 42%, 1.8 × Et3N salt). 1H NMR (500 MHz, D2O): δ 8.47 (s, 1H, H-8), 8.23 (s, 1H, H-2), 6.50 (t, 1H, J = 5.0 Hz, H-1′), 4.79–4.74 (m, 1H, H-3′, obstructed by water peak), 4.31–4.28 (m, 1H, H-4′), 4.25–4.21 (m, 1H, H-2″), 4.19–4.11 (m, 3H, H-3″, 5a,b′), 4.10–4.05 (m, 1H, H-5a), 4.02–3.92 (m, 3H, H-1a″, 4″, 5b), 3.74 (dd, 1H, J = 10.0 Hz, J = 2.5 Hz, H-1b″), 3.19 (q, 10H, J = 7.5 Hz, NCH2CH3), 2.83 (quint, 1H, J = 7.0 Hz, H-2a), 2.59 (dq, 1H, J = 14.0 Hz, J = 3.0 Hz, H-2b′), 1.27 (t, 15H, J = 7.0 Hz, NCH2CH3). 31P NMR (202 MHz, D2O): δ −11.33 (m, pyrophosphate). 13C{1H}NMR (126 MHz, D2O): δ 155.5 (C-6), 152.7 (C-2), 148.7 (C-4), 139.9 (C-8), 118.6 (C-5), 85.7 (d, J = 8.6 Hz, C-4′), 83.6 (C-1′), 80.3 (d, J = 8.4 Hz, C-4″), 72.2 (C-1″), 71.5 (C-3″), 71.2 (C-3′), 70.9 (C-2″), 65.5 and 65.4 (2 × d, J = 5.3 Hz, J = 4.7 Hz, C-5′, 5″), 64.6 (NCH2CH3), 39.0 (C-2′), 8.2 (NCH2CH3). HRMS (ES–) calcd for C15H22N5O12P2, 526.0746 [M – H]−; found, 526.0768. HPLC tR = 3.7 min.
1″-α/β-3″,2′-Dideoxyadenosine diphosphoriboside (3″,2′-dideoxy-ADPR) (6)
3-Deoxy-d-ribofuranose-5-phosphate tributylammonium salt 12 (12 mg, 0.03 mmol) and MgCl2 (5.9 mg, 0.06 mmol) were dissolved in DMF (1 mL), co-evaporated with dry toluene (2×), and evaporated to dryness. Dry DMF (600 μL) was added to the solid residue, and the mixture was stirred at 20 °C until all solids were dissolved (5–10 min). Then, 2′-deoxy-AMP-imidazolide 31 (10.8 mg, 0.027 mmol) was added dropwise in DMF (400 μL). The mixture was stirred at 20 °C for 16 h. HPLC showed consumption of all starting material. The solvent was evaporated in vacuo and the crude product was dissolved in TEAB buffer (10 mL, 0.1 M) and purified by semi-preparative, reverse-phase HPLC using a gradient of TEAB (0.1 M)–acetonitrile (95/5 → 35/65, v/v). The title compound 6 was obtained as a colorless amorphous solid (7.5 μmol, 5.32 mg, 27%, 1.8 × Et3N salt). 1H NMR (500 MHz, D2O): δ 8.48 (s, 1H, H-8), 8.24 (s, 1H, H-2), 6.51 (t, 1H, J = 7.0 Hz, H-1′), 5.26 (d, 0.3H, J = 4.0 Hz, H-1″α), 5.21 (s, 0.7H, H-1″β), 4.78–4.75 (m, 1H, H-3′, obstructed by water signal), 4.49–4.39 (m, 1H, H-4″α, β), 4.31–4.23 (m, 1.3H, H-2″α, 4′), 4.20–4.11 (m, 2.7H, H-2″β, 5a,b′), 4.05–3.98 (m, 0.7H, H-5″βa), 3.94–3.84 (m, 1H, H-5″αa, βb), 3.82–3.76 (m, 0.3H, H-5″αb), 3.19 (q, 11H, J = 7.5 Hz, NCH2), 2.84 (ddd, 1H, J = 7.5 Hz, J = 6.5 Hz, J = 1.0 Hz, H-2a), 2.63–2.57 (m, 1H, H-2b′), 2.10–2.01 (m, 0.3H, H-3″αa), 2.00–1.92 (m, 1.7H, H-3″αb, βa,b), 1.27 (t, 16.5H, J = 7.5 Hz, NCH2CH3). 31P NMR (202 MHz, D2O): δ −11.12 (m, α-pyrophosphate), −11.38 (m, β-pyrophosphate). 13C{1H}NMR (126 MHz, D2O): δ 155.6 (C-6), 152.7 (C-2), 148.7 (C-4), 139.9 (C-8), 118.6 (C-5), 102.1 (C-1″β), 97.6 (C-1″α), 85.8 (d, J = 8.8 Hz, C-4′), 83.6 (C-1′), 78.3 (d, J = 8.2 Hz, C-4″β), 75.5 (d, J = 8.4, C-4″α), 75.4 (C-2″β), 71.3 (C-3′), 70.4 (C-2″α), 68.8 (d, J = 5.4 Hz, C-5″β), 67.9 (d, J = 5.5 Hz, C-5″α), 65.4 (d, J = 4.9 Hz, C-5′), 46.6 (NCH2), 39.0 (C-2′), 32.4 (C-3′β), 31.7 (C-3′α), 8.2 (NCH2CH3). HRMS (ES–) calcd for C15H22N5O12P2, 526.0746 [M – H]−; 526.0761. HPLC tR = 3.68 min.
Pharmacology
Materials
ADPR was obtained from Sigma-Aldrich.
Cell Culture
HEK293 cells were kept at 37 °C and 5% CO2 in complete medium (DMEM with 4.5 g/L glucose, Glutamax-I, 10% FBS, 100 units/mL penicillin, and 100 μg/mL streptomycin) at 37 °C. For the maintenance of HEK293 clones with stable expression of TRPM2 400 μg/mL G418 sulfate was added to the medium.
Transfection and Generation of Cell Lines
Generation of the clonal HEK293 cell line with stable expression of human TRPM2 has been described previously.16 In brief, HEK293 were transfected with an expression vector encoding the full-length of human TRPM2 and EGFP (pIRES2-EGFP-TRPM2). Cells that successfully integrated the expression vector were then enriched by selection with 400 μg/mL G418 sulfate (Biochrom). Clonal cell lines were established from these cells by limiting dilution. Expression of TRPM2 was confirmed by Ca2+ measurement and whole cell patch clamp.
Patch-Clamp Measurements
The day before the experiments, cells from a clonal HEK293 cell line with stable expression of human TRPM2 were seeded to 35 mm tissue culture dishes at a low density. For the whole cell patch-clamp experiments, the culture medium was replaced by a bath solution containing 1 mM CaCl2, 140 mM NMDG, 5 mM KCl, 3.3 mM MgCl2, 1 mM CaCl2, 5 mM d-glucose, and 10 mM HEPES, pH 7.4. Patch pipettes were pulled from borosilicate glass capillaries with an outer diameter of 1.5 mm and an inner diameter of 1.05 mm using a Sutter P-97 and filled with a pipette solution containing 120 mM KCl, 8 mM NaCl, 1 mM MgCl2, 10 mM HEPES, 10 mM EGTA, and 5.6 mM CaCl2 (resulting in 200 nM free [Ca2+] as calculated by CaBuf software (G. Droogmans, formerly available from ftp://ftp.cc.kuleuven.ac.be/pub/droogmans/cabuf.zip). These pipettes had a resistance between 1.5 and 3.5 MΩ. Data were recorded using an EPC-10 amplifier and PatchMaster software (HEKA Elektronik, Germany). Fast and slow capacity transients were compensated, series resistance was compensated by 70%. After establishing whole cell configuration, cells were held at −50 mV and channel activation was followed by applying voltage ramps from −85 to +20 mV over 140 ms every 5 s for a total of 450 s. For further analysis, the maximum outward current at +15 mV during the course of the recording was taken as a measure of channel activity. To test for agonist activity, ADPR analogues were included in the pipette solution at 100 μM. To test for antagonist activity, the pipette solution contained 100 μM ADPR and 900 μM of the ADPR analogue under test. All experiments were performed at room temperature.
Statistical analysis
The data from patch-clamp experiments were analyzed using GraphPad Prism (version 7.04, GraphPad Software Inc.) Because the distribution of currents is skewed toward higher values, data were log-transformed. Log-transformed data were tested for significant differences using one-way-Anova followed by post hoc testing against the respective control (buffer for agonist experiments, ADPR for antagonist experiments) using Bonferroni correction for multiple testing (α = 0.05). In the charts, the horizontal bar indicates the mean of the log-transformed data.
Acknowledgments
B.V.L.P. is a Wellcome Trust Senior Investigator (grant 101010). This study was supported by the Deutsche Forschungsgemeinschaft (GU 360/16-1 and Projektnummer 335447717-SFB1328 project A01 to A.H.G., Projektnummer 335447717-SFB1328 project A05 to R.F.) and Landesforschungsförderung Hamburg (Research Group ReAd Me, project 01, to A.H.G.). The authors thank Andreas Bauche for technical support.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.9b00338.
1H NMR, 13C NMR, and 31P NMR spectra for compounds (PDF)
Author Present Address
∥ Department of Organic Chemistry, Faculty of Science, Charles University, Hlavova 2030/8, 128 43 Prague 2, Czech Republic.
Author Contributions
O.B. and J.M.W. equally contributed. R.F. and B.V.L.P. equally contributed. B.V.L.P. and A.H.G. devised the overall work area. B.V.L.P., J.M.W. and R.F. devised the focused strategy. O.B. synthesized the ADPR analogues supervised by J.M.W. M.D.R. carried out patch-clamp experiments with R.F., O.B., J.M.W., R.F. and B.V.L.P. wrote the manuscript with input from all authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Perraud A.-L.; Fleig A.; Dunn C. A.; Bagley L. A.; Launay P.; Schmitz C.; Stokes A. J.; Zhu Q.; Bessman M. J.; Penner R.; et al. ADP-Ribose Gating of the Calcium-Permeable LTRPC2 Channel Revealed by Nudix Motif Homology. Nature 2001, 411, 595–599. 10.1038/35079100. [DOI] [PubMed] [Google Scholar]
- Perraud A.-L.; Shen B.; Dunn C. A.; Rippe K.; Smith M. K.; Bessman M. J.; Stoddard B. L.; Scharenberg A. M. NUDT9, a Member of the Nudix Hydrolase Family, Is an Evolutionarily Conserved Mitochondrial ADP-Ribose Pyrophosphatase. J. Biol. Chem. 2003, 278, 1794–1801. 10.1074/jbc.m205601200. [DOI] [PubMed] [Google Scholar]
- Iordanov I.; Mihályi C.; Tóth B.; Csanády L. The Proposed Channel-Enzyme Transient Receptor Potential Melastatin 2 Does Not Possess ADP Ribose Hydrolase Activity. eLife 2016, 5, e17600 10.7554/elife.17600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonfria E.; Marshall I. C. B.; Benham C. D.; Boyfield I.; Brown J. D.; Hill K.; Hughes J. P.; Skaper S. D.; McNulty S. TRPM2 Channel Opening in Response to Oxidative Stress Is Dependent on Activation of Poly(ADP-Ribose) Polymerase. Br. J. Pharmacol. 2004, 143, 186–192. 10.1038/sj.bjp.0705914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buelow B.; Song Y.; Scharenberg A. M. The Poly(ADP-Ribose) Polymerase PARP-1 Is Required for Oxidative Stress-Induced TRPM2 Activation in Lymphocytes. J. Biol. Chem. 2008, 283, 24571–24583. 10.1074/jbc.m802673200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W.; Hirschler-laszkiewicz I.; Tong Q.; Conrad K.; Sun S.; Penn L.; Barber D. L.; Stahl R.; Carey D. J.; Cheung J. Y.; et al. TRPM2 Is an Ion Channel That Modulates Hematopoietic Cell Death through Activation of Caspases and PARP Cleavage. Am. J. Physiol. Cell Physiol 2006, 290, C1146–C1159. 10.1152/ajpcell.00205.2005. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Jiang G.; Zhang P.; Fan J. Programmed Cell Death and Its Role in Inflammation. Mil. Med. Res. 2015, 2, 1–12. 10.1186/s40779-015-0039-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonfria E.; Mattei C.; Hill K.; Brown J. T.; Randall A.; Benham C. D.; Skaper S. D.; Campbell C. A.; Crook B.; Murdock P. R.; et al. TRPM2 Is Elevated in the TMCAO Stroke Model, Transcriptionally Regulated, and Functionally Expressed in C13 Microglia. J. Recept. Signal Transduct. Res. 2006, 26, 179–198. 10.1080/10799890600637522. [DOI] [PubMed] [Google Scholar]
- Knowles H.; Li Y.; Perraud A.-L. The TRPM2 Ion Channel, an Oxidative Stress and Metabolic Sensor Regulating Innate Immunity and Inflammation. Immunol. Res. 2013, 55, 241–248. 10.1007/s12026-012-8373-8. [DOI] [PubMed] [Google Scholar]
- Partida-Sanchez S.; Gasser A.; Fliegert R.; Siebrands C. C.; Dammermann W.; Shi G.; Mousseau B. J.; Sumoza-Toledo A.; Bhagat H.; Walseth T. F.; et al. Chemotaxis of Mouse Bone Marrow Neutrophils and Dendritic Cells Is Controlled by ADP-Ribose, the Major Product Generated by the CD38 Enzyme Reaction. J. Immunol. 2007, 179, 7827–7839. 10.4049/jimmunol.179.11.7827. [DOI] [PubMed] [Google Scholar]
- Sumoza-Toledo A.; Lange I.; Cortado H.; Bhagat H.; Mori Y.; Fleig A.; Penner R.; Partida-Sánchez S. Dendritic Cell Maturation and Chemotaxis Is Regulated by TRPM2-Mediated Lysosomal Ca2+ Release. FASEB Journal 2011, 25, 3529–3542. 10.1096/fj.10-178483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G.; Cao L.; Liu X.; Bai C.; Malik A. B.; Xu Correspondence J.; Sieracki N. A.; Di A.; Wen X.; Chen Y.; et al. Oxidant Sensing by TRPM2 Inhibits Neutrophil Migration and Mitigates Inflammation In Brief Oxidant Sensing by TRPM2 Inhibits Neutrophil Migration and Mitigates Inflammation. Dev. Cell 2016, 38, 453–462. 10.1016/j.devcel.2016.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto S.; Shimizu S.; Kiyonaka S.; Takahashi N.; Wajima T.; Hara Y.; Negoro T.; Hiroi T.; Kiuchi Y.; Okada T.; et al. TRPM2-Mediated Ca2+ Influx Induces Chemokine Production in Monocytes That Aggravates Inflammatory Neutrophil Infiltration. Nat. Med. 2008, 14, 738–747. 10.1038/nm1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehrhahn J.; Kraft R.; Harteneck C.; Hauschildt S. Transient Receptor Potential Melastatin 2 Is Required for Lipopolysaccharide-Induced Cytokine Production in Human Monocytes. J. Immunol. 2010, 184, 2386–2393. 10.4049/jimmunol.0902474. [DOI] [PubMed] [Google Scholar]
- Melzer N.; Hicking G.; Göbel K.; Wiendl H. TRPM2 Cation Channels Modulate T Cell Effector Functions and Contribute to Autoimmune CNS Inflammation. PLoS One 2012, 7, e47617 10.1371/journal.pone.0047617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelderblom M.; Melzer N.; Schattling B.; Göb E.; Hicking G.; Arunachalam P.; Bittner S.; Ufer F.; Herrmann A. M.; Bernreuther C.; et al. Transient Receptor Potential Melastatin Subfamily Member 2 Cation Channel Regulates Detrimental Immune Cell Invasion in Ischemic Stroke. Stroke 2014, 45, 3395–3402. 10.1161/strokeaha.114.005836. [DOI] [PubMed] [Google Scholar]
- Belrose J. C.; Jackson M. F. TRPM2: A Candidate Therapeutic Target for Treating Neurological Diseases. Acta Pharmacol. Sin. 2018, 39, 722–732. 10.1038/aps.2018.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Gao Y.; Bao X.; Li F.; Yao W.; Feng Z.; Yin Y. TRPM2: A Potential Drug Target to Retard Oxidative Stress. Front. Biosci. (Landmark Ed.) 2017, 22, 1427–1438. 10.2741/4551. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Tóth B.; Szollosi A.; Chen J.; Csanády L. Structure of a TRPM2 Channel in Complex with Ca2+ Explains Unique Gating Regulation. eLife 2018, 7, e36409 10.7554/elife.36409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y.; Winkler P. A.; Sun W.; Lü W.; Du J. Architecture of the TRPM2 Channel and Its Activation Mechanism by ADP-Ribose and Calcium. Nature 2018, 562, 145–149. 10.1038/s41586-018-0558-4. [DOI] [PubMed] [Google Scholar]
- Wang L.; Fu T. M.; Zhou Y.; Xia S.; Greka A.; Wu H. Structures and Gating Mechanism of Human TRPM2. Science 2018, 362 (6421), eaav4809. 10.1126/science.aav4809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau C.; Kirchberger T.; Swarbrick J. M.; Bartlett S. J.; Fliegert R.; Yorgan T.; Bauche A.; Harneit A.; Guse A. H.; Potter B. V. L. Structure-Activity Relationship of Adenosine 5’-Diphosphoribose at the Transient Receptor Potential Melastatin 2 (TRPM2) Channel: Rational Design of Antagonists. J. Med. Chem. 2013, 56, 10079–10102. 10.1021/jm401497a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fliegert R.; Bauche A.; Wolf Pérez A.-M.; Watt J. M.; Rozewitz M. D.; Winzer R.; Janus M.; Gu F.; Rosche A.; Harneit A.; et al. 2′-Deoxyadenosine 5′-Diphosphoribose Is an Endogenous TRPM2 Superagonist. Nat. Chem. Biol. 2017, 13, 1036–1044. 10.1038/nchembio.2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan A. J.; Wang Y. K.; Roberts M. F.; Miller S. J. Chemistry and Biology of Deoxy-myo-Inositol Phosphates: Stereospecificity of Substrate Interactions within an Archaeal and a Bacterial IMPase. J. Am. Chem. Soc. 2004, 126, 15370–15371. 10.1021/ja047360x. [DOI] [PubMed] [Google Scholar]
- Fowler B. S.; Laemmerhold K. M.; Miller S. J. Catalytic Site-Selective Thiocarbonylations and Deoxygenations of Vancomycin Reveal Hydroxyl-Dependent Conformational Effects. J. Am. Chem. Soc. 2012, 134, 9755–9761. 10.1021/ja302692j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan P. A.; Miller S. J. An Approach to the Site-Selective Deoxygenation of Hydroxyl Groups Based on Catalytic Phosphoramidite Transfer. Angew. Chem. Int. Ed. 2012, 51, 2907–2911. 10.1002/anie.201109033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacios D. S.; Dailey I.; Siebert D. M.; Wilcock B. C.; Burke M. D. Synthesis-Enabled Functional Group Deletions Reveal Key Underpinnings of Amphotericin B Ion Channel and Anfifungal Activities. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 6733–6738. 10.1073/pnas.1015023108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szpilman A. M.; Manthorpe J. M.; Carreira E. M. Synthesis and Biological Studies of 35-Deoxy Amphotericin B Methyl Ester. Angew. Chem. Int. Ed. 2008, 47, 4339–4342. 10.1002/anie.200800590. [DOI] [PubMed] [Google Scholar]
- Fliegert R.; Watt J. M.; Schöbel A.; Rozewitz M. D.; Moreau C.; Kirchberger T.; Thomas M. P.; Sick W.; Araujo A. C.; Harneit A.; et al. Ligand-Induced Activation of Human TRPM2 Requires the Terminal Ribose of ADPR and Involves Arg1433 and Tyr1349. Biochem. J. 2017, 474, 2159–2175. 10.1042/bcj20170091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W.; Niu Y.; Xiong D.-C.; Cao X.; Ye X.-S. Highly Substituted Cyclopentane-CMP Conjugates as Potent Sialyltransferase Inhibitors. J. Med. Chem. 2015, 58, 7972–7990. 10.1021/acs.jmedchem.5b01181. [DOI] [PubMed] [Google Scholar]
- Graham S. M.; Pope S. C. Selective Phosphitylation of the Primary Hydroxyl Group in Unprotected Carbohydrates and Nucleosides. Org. Lett. 1999, 1, 733–736. 10.1021/ol990769k. [DOI] [PubMed] [Google Scholar]
- Bessodes M.; Komiotis D.; Antonakis K. Rapid and Selective Detritylation of Primary Alcohols Using Formic Acid. Tetrahedron Lett 1986, 27, 579–580. 10.1016/s0040-4039(00)84045-9. [DOI] [Google Scholar]
- Skorupa E.; Dmochowska B.; Pellowska-Januszek L.; Wojnowski W.; Chojnacki J.; Wiśniewski A. Synthesis and Structure of Selected Quaternary N-(1,4-Anhydro-5-Deoxy-2,3- O-Isopropylidene-D,L-Ribitol-5-yl)Ammonium Salts. Carbohydr. Res. 2004, 339, 2355–2362. 10.1016/j.carres.2004.06.025. [DOI] [PubMed] [Google Scholar]
- Moravcová J.; Čapková J.; Staněk J. One-Pot Synthesis of 1,2-O-Isopropylidene-α-d-Xylofuranose. Carbohydr. Res. 1994, 263, 61–66. 10.1016/0008-6215(94)00165-0. [DOI] [Google Scholar]
- Ko H.; Das A.; Carter R. L.; Fricks I. P.; Zhou Y.; Ivanov A. A.; Melman A.; Joshi B. V.; Kováč P.; Hajduch J.; et al. Molecular Recognition in the P2Y14 receptor: Probing the Structurally Permissive Terminal Sugar Moiety of Uridine-5′-Diphosphoglucose. Bioorg. Med. Chem. 2009, 17, 5298–5311. 10.1016/j.bmc.2009.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabrowski-Tumanski P.; Kowalska J.; Jemielity J. Efficient and Rapid Synthesis of Nucleoside Diphosphate Sugars from Nucleoside Phosphorimidazolides. Eur. J. Org. Chem. 2013, 2013, 2147–2154. 10.1002/ejoc.201201466. [DOI] [Google Scholar]
- Hamill O. P.; Marty A.; Neher E.; Sakmann B.; Sigworth F. J. Improved Patch-Clamp Techniques for High-Resolution Current Recording from Cells and Cell-Free Membrane Patches. Pflügers Arch. Eur. J. Physiol. 1981, 391, 85–100. 10.1007/bf00656997. [DOI] [PubMed] [Google Scholar]
- Molleman A.Patch Clamping: An Introductory Guide to Patch Clamp Electrophysiology; John Wiley & Sons, 2003. [Google Scholar]
- Tóth B.; Iordanov I.; Csanády L. Putative Chanzyme Activity of TRPM2 Cation Channel Is Unrelated to Pore Gating. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 16949–16954. 10.1073/pnas.1412449111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X.; Li M.; Zhan K.; Yang W.; Zhang L.; Wang K.; Yu P.; Zhang L. Selective Inhibition of TRPM2 Channel by Two Novel Synthesized ADPR Analogues. Chem. Biol. Drug Des. 2018, 91, 552–566. 10.1111/cbdd.13119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubisha O.; Rafty L. A.; Takanishi C. L.; Xu X.; Tong L.; Perraud A.-L.; Scharenberg A. M.; Denu J. M. Metabolite of SIR2 Reaction Modulates TRPM2 Ion Channel. J. Biol. Chem. 2006, 281, 14057–14065. 10.1074/jbc.m513741200. [DOI] [PubMed] [Google Scholar]
- Jackson M. D.; Denu J. M. Structural Identification of 2’- and 3’-O-Acetyl-ADP-Ribose as Novel Metabolites Derived from the Sir2 Family of Beta-NAD+-Dependent Histone/Protein Deacetylases. J. Biol. Chem. 2002, 277, 18535–18544. 10.1074/jbc.m200671200. [DOI] [PubMed] [Google Scholar]
- Neidle S.Principles of Nucleic Acid Structure; Elsevier, 2008, pp 20−33. [Google Scholar]
- Saenger W.Principles of Nucleic Acid Structure; Springer-Verlag, 1984, pp 51−104. [Google Scholar]
- Kühn F. J. P.; Watt J. M.; Potter B. V. L.; Lückhoff A. Different substrate specificities of the two ADPR interaction sites in TRPM2 channels of Nematostella vectensis and the role of IDPR. Sci Rep 2019, 9, 4985. 10.1038/s41598-019-41531-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.