

Abstract
The genetic, biological, and environmental backgrounds of an organism fundamentally influence the balance between risk and resilience to stress. Sex, age, and environment transact with responses to trauma in ways that can mitigate or exacerbate the likelihood that post-traumatic stress disorder will develop. Translational approaches to modeling affective disorders in animals will ultimately provide novel treatments and a better understanding of the neurobiological underpinnings behind these debilitating disorders. The extant literature on trauma/stress has focused predominately on limbic and cortical structures that innervate the hypothalamic–pituitary–adrenal axis and influence glucocorticoid-mediated negative feedback. It is through these neuroendocrine pathways that a self-perpetuating fear memory can propagate the long-term effects of early life trauma. Recent work incorporating translational approaches has provided novel pathways that can be influenced by early life stress, such as the glucocorticoid receptor chaperones, including FKBP51. Animal models of stress have differing effects on behavior and endocrine pathways; however, complete models replicating clinical characteristics of risk and resilience have not been rigorously studied. This review discusses a four-factor model that considers the importance of studying both risk and resilience in understanding the developmental response to trauma/stress. Consideration of the multifactorial nature of clinical populations in the design of preclinical models and the application of preclinical findings to clinical treatment approaches comprise the core of translational reciprocity, which is discussed in the context of the four-factor model.
Keywords: Stress, Trauma, PTSD, adolescent, preclinical, animal model
INTRODUCTION
Given the overall negative impact that traumatic experiences have on youth, identification of risk factors for symptom maintenance and the development of post-traumatic stress disorder (PTSD) is integral in efforts to decrease rates of new cases of PTSD and to mitigate the powerful biological and psychological effects of trauma on youth. While preventive efforts offer one avenue by which to improve psychological health in youth samples, the translation of animal models to the treatment of affected youth offers another exciting possibility. Many pre-trauma risk factors are biological in nature; in addition, the experience of trauma seems to negatively impact numerous aspects of an individual’s biology. Therefore, studies investigating both the influence of biological predisposition on the evolution of PTSD as well as mechanisms through which physiological and psychological responses to trauma become maladaptive, and the influence of biological predisposition on this evolution, are warranted. Although adequately powered studies of pre-trauma risk and resilience factors in humans are largely untenable for logistical reasons, animal models offer a potent springboard for deriving personalized, individually-tailored early interventions based on biological and psychological pre-trauma risk. Here we review the existing clinical data regarding risk and treatment of PTSD, discuss animal models available for mechanistic assessment of risk and resilience, identify the current gaps in the field, and provide a framework by which to advance a mechanism-based research and treatment approach for stress disorders in adolescents with a four-factor model.
Traumatic events are experiences in which an individual perceives a serious threat to the life or well-being of oneself or someone else and during which the observer feels a sense of intense fear or helplessness (APA, 2000). Although epidemiological reports vary, studies generally have found that around 60% of men and 52% of women will experience a trauma at some point in their lifetimes; only 5–10% of these individuals, however, will go on to experience significant distress in the wake of a trauma, suggesting that a host of other factors (e.g., elements of the trauma, individual differences) likely play a crucial role in the development of PTSD (Kessler et al., 1995). One of the critical factors in the determination of whether or not PTSD will manifest is age, with adolescents much more likely to develop PTSD compared to adults exposed to similar traumatic stressors (van der Kolk, 1985).
A particularly strong illustration of this phenomenon comes from a longitudinal study of the passengers on the Ehime Maru, a Japanese fishing vessel that was used for teaching in the Pacific Ocean. In 2001, the boat was involved in a horrific sea accident in which nine of the 35 passengers died. Of the survivors, 78% of the adolescent passengers went on to develop PTSD, while only 12% of the adult passengers developed PTSD (Maeda et al., 2009). This increased susceptibility to PTSD in adolescents may be explained by both psychological and physiological characteristics of the adolescent compared to the adult. These variables will be discussed in detail below with particular attention to the need to design research and treatment strategies with this uniquely vulnerable subset of our population in mind.
A range of psychological and physiological reactions may occur in response to a traumatic event. Typical acute behavioral and psychological reactions include anxiety, heightened arousal, disturbed sleep patterns, emotional numbness, withdrawal from people and activities, and recurrent thoughts and worries about the traumatic event. While these initial experiences often attenuate over time, a subset of individuals experience ongoing symptoms that disrupt their lives (Rothbaum and Foa, 1993). The Diagnostic and Statistical Manual of Mental Disorders – Text Revision (DSM-IV-TR (APA, 2000)) distinguishes between two varying levels of response to trauma; individuals whose symptoms are disruptive but that remit within 30 days receive a diagnosis of Acute Stress Disorder, whereas individuals who experience prolonged distress in the face of a trauma generally meet criteria for PTSD. Moreover, the experience of a traumatic event has been linked to the development of a wide range of psychological difficulties either independent of or comorbid with PTSD, such as depression, anxiety, and substance abuse disorders (Kessler et al., 1995; Kilpatrick et al., 2003; Norrholm and Ressler, 2009). The manifestation of PTSD is multidimensional and interwoven with other stress-induced disorders, and the manifestation is somewhat dependent on clinically recognized risk factors.
CLINICALLY RECOGNIZED RISK FACTORS FOR PTSD
Given the short- and long-term adverse effects that trauma may have on an individual, identification of risk factors has important implications for both prevention and treatment efforts. Broadly, clinically recognized risk factors can be divided into two classes: pre-trauma and peri- or post-trauma, and both of those categories can be further divided into psychological, environmental, and physiological risk factors (see Fig. 1).
Fig. 1.
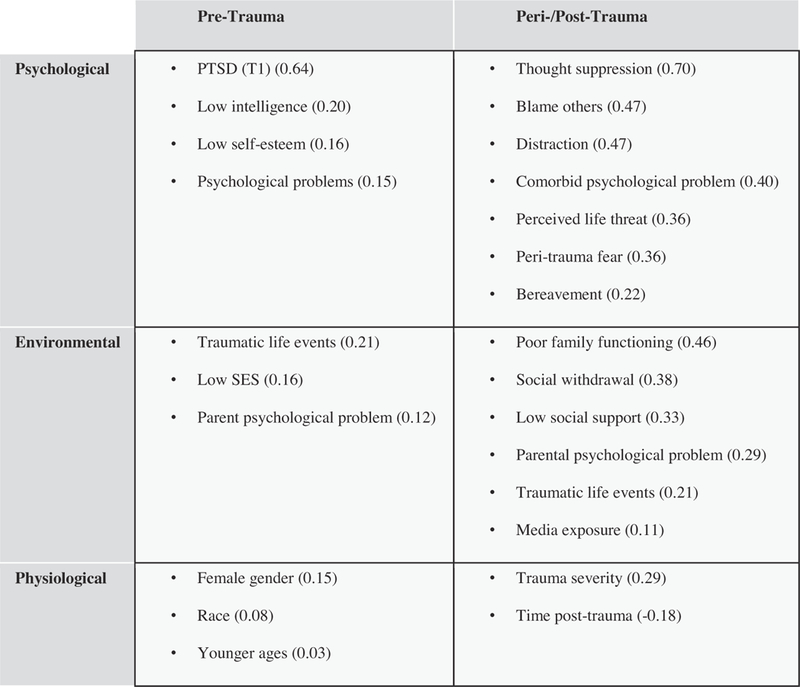
Risk factors for developing PTSD in children and adolescents. Clinically recognized risk factors for developing PTSD in children and adolescents are divided into two classes: pre-trauma and peri- or post-trauma. These categories can be further divided into psychological, environmental, and physiological risk factors. Numbers in parentheses refer to estimated population effect sizes in Trickey et al.’s (2012) meta-analysis.
Demographic and pre-trauma risk factors
Certain demographic and psychological correlates have been shown to be related to increased potential for the development of PTSD. As mentioned, various studies have reported that risk for PTSD increases based on earlier age at index trauma episode (Koenen et al., 2002; Schnurr et al., 2004); however, Trickey and colleagues (2012) reported a significant interaction between female gender and older children and adolescents, particularly when the index trauma was unintentional (e.g., a car accident). In a recent meta-analysis of 25 potential risk factors evaluated across 64 studies of PTSD symptoms in children and adolescents (total n = 32,238), Trickey and colleagues (2012) found that numerous other pre-trauma risk factors (e.g., low intelligence, low socioeconomic status, psychological functioning, parent psychological problems) also were associated with small-to-medium effect sizes in predicting PTSD symptoms for children and adolescents. Other studies have found that poor social support (both before and after the trauma) and poor previous psychological adjustment are associated with increased risk of developing PTSD (Brewin et al., 2000; Ozer et al., 2003). In addition, females develop PTSD following trauma approximately twice as often as males (Kessler et al., 1995). Other pre-trauma psychological variables also may impact the development of PTSD; for example, Gunderson and Sabo (Kessler et al., 1995) proposed that individuals with the emotion regulation difficulties often seen in borderline personality disorder (BPD) might be at increased risk for PTSD following trauma.
Another factor influencing the onset of PTSD is the occurrence of multiple traumatic events across the lifespan. Repeated trauma is relatively common; epidemiological research has found rates of multiple traumatic events in 25–38% of adults in the United States (Kessler et al., 1995; Green et al., 2000). The rate is higher in adolescents; approximately 55% of youth with a trauma history report having experienced multiple traumatic events (Macdonald et al., 2010), which is particularly important given the relationship between childhood trauma and the development of psychopathology over the course of the lifespan (Kilpatrick et al., 2003). Moreover, in a recent study of adolescents with at least one reported trauma, Macdonald and colleagues (2010) found that the number of victimizations experienced increased the odds of developing PTSD and at least one comorbid disorder, including substance use disorders (SUD) and major depressive disorder (MDD). Additionally, comorbid PTSD was more common than PTSD alone in the multiply traumatized group of adolescents. Importantly, pre-trauma risk factors interact with the trauma experience itself (i.e., type of trauma, severity, directness of threat to the individual) to magnify reactivity to the trauma and/or increase distress independent of the direct, reactive effects of the trauma (Foy et al., 1996; Trickey et al., 2012). This, in turn, can lead to elevated vulnerability for longer-lasting post-traumatic effects.
Peri- and post-trauma risk factors
Peri-traumatic factors, such as type of trauma experienced, the severity of the trauma, and the degree to which the individual was exposed to the trauma also influence the likelihood of PTSD development (Kessler et al., 1995; Foy et al., 1996; Pine and Cohen, 2002). In meta-analyses of adult studies of PTSD (Ozer et al., 2003; Trickey et al., 2012), peri-traumatic factors showed the strongest predictive ability of risk for developing PTSD, above and beyond pre-trauma characteristics (Ozer et al., 2003). Similarly, Trickey and colleagues (2012) found that peri-traumatic fear and perception of threat to life had large effect sizes as predictors of PTSD onset. For clinical purposes, it is important to note that different individuals may experience differing levels of peri-traumatic fear and threat to life when experiencing very similar traumatic experiences (Trickey et al., 2012). Post-trauma individual factors are also predictive of the development of PTSD. Specifically, the ability to cognitively disengage from the trauma (Aupperle et al., 2012), degree of available post-trauma social support (Pine and Cohen, 2002), degree of acute post-trauma symptomatology, and comorbid psychological difficulties (Trickey et al., 2012) are strongly related to ongoing traumatic stress. These characteristics may account for part of the increased risk of PTSD in adolescents as compared to adults in that adolescents and adults differ in their ability to cope with traumatic stress (van der Kolk, 1985).
The biology of risk: Cause and consequence
Endogenous biological markers also have been shown to be associated with vulnerability to the development of PTSD in the wake of a traumatic experience. Multiple biological systems are both responsive to and altered by trauma/stress exposure. The most central system manipulated by exposure to trauma or stress is the hypothalamic–pituitary–adrenal (HPA) axis. Activation of this axis begins with release of corticotropin-releasing factor (CRF) from neurons in the paraventricular nucleus of the hypothalamus. The release of CRF precipitates the production of adrenocorticotropic hormone (ACTH) through proteolytic cleavage of proopiomelanocortin (POMC) in the pituitary. ACTH travels through the peripheral blood stream until it binds melanocortin type-2 receptors located on the adrenal gland. Binding of ACTH increases adrenal production of the glucocorticoid cortisol. The immediate implication of cortisol release is the mobilization of energy to facilitate the altered energetic demands of the stress response. Cortisol, however, is a pluripotent steroid hormone; in addition to energy production, cortisol impacts inflammation, growth, development, and protein and lipid catabolism (Owens and Nemeroff, 1991; Bourke and Owens, 2010).
The stress response of the HPA axis is modulated by a negative feedback loop encompassing the hippocampus, hypothalamus and anterior pituitary. Following cortisol secretion into the peripheral blood circulation, cortisol passes through the plasma membrane of cells in the pituitary, hypothalamus, and hippocampus where it binds to the glucocorticoid receptor (GR), a nuclear receptor transcription factor. Upon binding, the GR associates with another cortisol-bound GR to form a homodimer. Several chaperones are recruited to allow/facilitate the transport of the GR from the cytoplasm to the nucleus to influence GR sensitivity and mediate DNA binding (Gehring and Tomkins, 1974; Grad and Picard, 2007; Binder, 2009). The GR binds to specific glucocorticoid response elements to cause transcriptional regulation of CRF and POMC related genes, as well as many others, decreasing CRF and POMC production and release and thereby extinguishing the stress response.
Biological markers in PTSD point to a strong influence of the glucocorticoid receptor as a mediator of the long-term effects of stress. For example, individuals with decreased 24-h urinary cortisol excretion, potentially heightened lymphocyte glucocorticoid receptor number, and enhanced glucocorticoid responsiveness vis á vis the lysozyme IC50 appear to be more vulnerable to the development of PTSD (Yehuda et al., 1995, 2004). Such biological measures have been linked to other pre-traumatic risk factors, including age at the time of trauma and existence of parental PTSD (Yehuda et al., 2007; Trickett et al., 2010).
Genetic risk factors for PTSD also exist at the level of the molecular regulation of the GR. As mentioned above, GR requires co-regulators to translocate to the nucleus and bind to DNA. FK506-binding protein 51, also known as FKBP51, is one of these co-regulators. FKBP51 prevents GR translocation and GR-mediated gene expression, resulting in GR insensitivity to circulating glucocorticoids and prolongation of the stress response (Binder, 2009). In addition, some evidence points to the existence of a feedback loop whereby HPA axis activation alters FKBP51; specifically, glucocorticoid exposure increases FKBP51 expression in the hippocampus and hypothalamus, with a corresponding decrease in GR and CRF (Lee et al., 2010; Scharf et al., 2011). FKBP51 has been implicated in the pathogenesis of depression, anxiety disorders, and treatment response (Binder et al., 2004; Lekman et al., 2008; Horstmann et al., 2010). Specifically, genetic polymorphisms in FKBP51 have been shown to interact with early life abuse in humans to predict later onset of PTSD and suicidality (Binder et al., 2008; Brent et al., 2010; Roy et al., 2010; Xie et al., 2010). Taken together, these studies indicate that GR action may be influenced by early life stress. Moreover, the GR and mediators of GR action have been shown to be associated with or regulated by gonadal hormone pathways (Evaul et al., 2010; Weiser et al., 2010), suggesting that the interaction of early life stress and sex differences may alter mediators of GR action. Collectively, this work implicates the HPA axis in general, and FKBP51 specifically, as the potential biological mediators of the long-term changes catalyzed by developmental stress.
Underlying biological characteristics that confer risk may be further exacerbated by exposure to stress and trauma, thereby increasing future risk for PTSD. For example, co-regulators of GR recently have been demonstrated to differ in terms of expression in the brain between male and female adolescent rats (Bourke et al., 2012b). These differences in basal expression of GR co-regulators likely are due to the shared use of these co-regulators by the sex steroid progesterone combined with inherent differences in the male and female brain (Bourke et al., 2012a). These differences in co-regulatory expression appear to alter the responsiveness of the HPA axis to stress, and differences are further magnified by repeated stressful experiences with females acquiring a growing biological risk in the form of a dysregulated response to stress exposure (Bourke et al., 2012b). Such feed forward cycles demonstrated in basic research may underlie the clinical reports of increasing risk for PTSD with repeated trauma exposures and the susceptibility of females to these adverse effects of trauma.
Biological characteristics such as the aforementioned sex difference in GR co-regulators and susceptibilities produced by the rapidly developing brain are benign under normal conditions but can become detrimental in a state of repeated challenges in both human and animal models. Not surprisingly, children exposed to traumatic events and severe stress typically demonstrate a multitude of alterations in normative functioning, including delays in behavioral, cognitive, and emotional regulation (De Bellis et al., 1999). Results from imaging studies have shed some light on possible mechanisms by which these effects occur; for example, studies of maltreated children and adolescents compared to healthy controls have shown significant volumetric differences in numerous brain structures (De Bellis et al., 1999, 2002a,b; Dannlowski et al., 2012; Teicher et al., 2012). Notably, De Bellis and colleagues (1999) found that brain volume was positively correlated with PTSD age of onset and negatively correlated with duration of maltreatment. Furthermore, PTSD symptoms were positively correlated with ventricular volume (De Bellis et al., 1999). Studies of identical twins that are discordant for trauma exposure suggest that differences in brain volume precede post-traumatic behavioral disturbances and are likely a risk factor for the development of trauma-induced psychopathology (Pitman et al., 2006).
Research on model animals takes these imaging findings to more mechanistic levels. In addition to the HPA axis, physiological responses to trauma include activation of and increases in levels of catecholamines (which are associated with the acute stress response) in brain regions associated with memory and learning (McCarty and Gold, 1981; Roozendaal, 2002). Furthermore, the locus coeruleus (LC), which contains the catecholamine norepinephrine, serves a central function in the vigilance system of the brain, which has been implicated in post-trauma functioning. Activation of the LC promotes NE release in multiple brain regions, including the amygdala, hippocampus, striatum, and prefrontal cortex (Aston-Jones et al., 1998), which are influential in memory consolidation. Additionally, activation of the amygdala in intense, emotional situations plays a critical role in modulating the effects of epinephrine and glucocorticoids on memory storage (McGaugh et al., 1996; Quirarte et al., 1997). Under adverse conditions, the transactions between these multiple neural systems may set up a reverberating feedback loop by which the adverse effects of trauma/stress become self-perpetuating, multiplicative, and ultimately psychologically damaging.
CLINICALLY RECOGNIZED RESILIENCE FACTORS FOR PTSD
In addition to risk factors for the development of PTSD following a traumatic event, protective factors play an important role both in acute and prolonged response to trauma. Putative pre-trauma social and psychological factors that likely enhance resilience include positive affectivity, optimism, cognitive flexibility, active coping strategies, social support and social integration, the ability to regulate emotions (especially negative affect), and sense of mastery (Yehuda et al., 2006). Such factors have been associated with resilience across various domains, including lower rates of PTSD in individuals exposed to a variety of traumatic events and recovery from medical conditions, such as transplants (Jacobsen et al., 2002; Phipps et al., 2012). The degree of interaction among and the autonomous impact of these psychological and social characteristics in creating resilience in an individual is unclear, and further investigation is warranted.
THE BIOLOGY OF RESILIENCE: CAUSE AND CONSEQUENCE
Although the biology of resilience is far from established, there are multiple biological substrates that have been identified to play a role in preservation of function in the face of trauma/stress. In approximately 90% of cases, individuals are resilient to trauma/stress (Kessler et al., 1995), even when experiencing the same traumatic stressor (e.g., the Ehime Maru disaster; (Maeda et al., 2009)). This suggests that certain endogenous biological factors offer protection from exposure to trauma and that trauma/stress exposure is a necessary but not sufficient criterion for the development of a pathological response.
Of the limited available evidence regarding the biology of resilience, the forebrain has been the major focus. Specifically, biological factors associated with resilience involve the amygdala and medial prefrontal cortex (mPFC). In adult animal studies of fear responses, research has shown that the lateral nucleus of the amygdala influences the relationship between conditioned and unconditioned fear stimuli (Sotres-Bayon et al., 2004). In addition, although inescapable shock is not the only stressor which induces behavioral dysfunction (Forsyth, 1969; Natelson et al., 1976), adolescent rats exposed to repeated shock stress only develop behavioral dysfunction in conditions in which the shock is uncontrollable, and this effect is mediated by the mPFC (Kubala et al., 2012). Furthermore, human studies of adults with anxiety disorders have demonstrated a link between amygdala over-responsiveness and anxiety disorders (Yehuda et al., 2006). In human brain imaging studies of responses to provocation (e.g., exposure to upsetting pictures), reduced amygdala and elevated mPFC activity have been associated with emotion modulation through intentional cognitive processes (New et al., 2009). Research utilizing adult animal models has investigated the role of the mPFC in extinction (i.e., the process by which a conditioned response is reduced or extinguished as a function of withdrawing a previously administered reinforcer). Research on conditioned fear response suggests that mPFC activation is involved in extinction, and that abnormalities in the mPFC inhibit extinction (Phelps et al., 2004).
Studies of neurochemical indicators of resilience have focused on neuropeptide Y (NPY) and dehydroepiandrosterone (DHEA). NPY impacts the hippocampus and appears to have an anxiolytic effect. Mechanistically, release of NPY counteracts the anxiogenic properties of corticotropin-releasing hormone (Heilig, 2004), and research has shown that adult individuals with higher levels of NPY perform better under conditions of high stress (Morgan et al., 2000). Higher levels of plasma NPY were observed in veterans exposed to trauma both with and without current PTSD, and NPY levels were predicted by symptom improvement and less trauma exposure (Yehuda et al., 2006). Additionally, animal research suggests that NPY has a counter-regulatory impact on norepinephrine in brain areas implicated in fear, anxiety, and even depression in adult subjects (Thorsell et al., 1999; Hastings et al., 2001). Central administration of NPY in rats has been shown to prevent stress-induced alterations in anxiety-like behavior (Cohen et al., 2012b). Hence, evidence from adults indicates that elevated NPY may represent a neurochemical resilience factor.
Dehydroepiandrosterone (DHEA), another hormone secreted by HPA axis activation, has been shown to mediate stress and coping responses (Hawley et al., 2010; Bardi et al., 2011) and demonstrated to protect against the negative effects of glucocorticoid exposure (Morgan et al., 2004). An emerging theory in stress research posits that elevated DHEA-to-cortisol ratios confer an advantage in demanding environments (Wemm et al., 2010; Bardi et al., 2011). Specifically, increased DHEA and the DHEA-to-cortisol ratio have been associated with enhanced performance in adults under stress (Morgan et al., 2004). Due to the risk relationship between increased cortisol and poorer response to trauma, the DHEA-to-cortisol ratio may confer an interactive effect on degree of resilience, potentially serving as a biological influence on PTSD recovery (Yehuda et al., 2006). Animal studies investigating these neuroendocrine markers show that stress decreases plasma DHEA and increases plasma cortisol concentrations (Cohen et al., 2007). An animal study with early-life stress produced a similar finding: hypothalamic and entorhinal cortical Dihydropiandrosterone-Sulfate concentrations were increased in animals exposed to juvenile stress (Avital et al., 2006). Although more work is needed to fully understand the role of DHEA, evidence to date suggests that DHEA may play a role in resilience.
Some understanding of the line between resilience and stress-induced disorders may come from the field of stress inoculation. The theories of stress inoculation, resilience, and hormesis postulate that exposure to mild-to-moderate levels of stress (or, in this case, glucocorticoids) during development prepares the offspring for a stressful environment, thereby engendering a buffer against subsequent stress exposure (Edge et al., 2009). Relatedly, animal models of nonhuman primates have demonstrated that repeated maternal separation during early development serves as a protective factor in tolerating stress in later life (Lyons et al., 1998; Parker et al., 2006). Similar evidence has been generated from studies that exposed rats to glucocorticoids in utero and then assessed their behavioral and cerebrovascular responses to stress in adulthood (Neigh et al., 2010). Interestingly, the effects of in utero exposure to glucocorticoids were protective for some brain regions and detrimental for others; suggesting that the developmental exposure to stress hormone produced a series of adaptations as opposed to universal stress inoculation. Therefore, the response to trauma/stress may be a gestalt phenomenon of multiple psychological, environmental, and biological characteristics that further interact with stress type and intensity. Let us turn our attention at this point to the question of whether or not an understanding of these variables can generate a positive impact on the clinical treatment of trauma-exposed adolescents.
ANIMAL RESEARCH PARADIGMS RELEVANT TO TRAUMA-EXPOSED ADOLESCENTS
The preclinical data and available translational data demonstrate that identifiable biological substrates underlie the variable response to trauma/stress exposure. Although much work remains to fully elucidate the precise mechanisms by which risk and resilience transact for a given individual, acknowledgement of the substantial preclinical findings currently available may shape the way clinical treatment of trauma/stress in adolescents is approached. Conversely, available animal models do not take into account the complex patient with multiple trauma/stress exposures over multiple life phases. We will next review the available laboratory stress models for rodent research followed by a review of the trauma treatments for patients that are currently available. The take away message from these sections is that there is a mismatch between the laboratory and the clinic. We follow these sections with our proposal for facilitating translational reciprocity.
Laboratory stressors as models of PTSD
A stressor is typically defined as any stimulus that elicits a response of the HPA axis. Because of the limitations associated with conducting experiments in human subjects, animal models are invaluable in understanding the pathologic consequences and mediating mechanisms of stress and trauma exposure. A vast number of animal models have been developed to recapitulate the hallmarks of trauma exposure. Stress models may be used alone or in concert with subsequent shock paradigms to model the exaggerated response to stressors seen post-trauma. Fear conditioning, a type of classical conditioning, is a behavioral paradigm in which a neutral stimulus (i.e., light) is paired with an aversive stimulus (i.e., footshock). The subject learns to associate the light with the shock and eventually will demonstrate a fearful response to the light when presented alone. The manifestation of this fearful response to a previously neutral stimulus (i.e., the light) is termed ‘‘fear conditioning’’. In addition, exposure to stress can exaggerate the conditioning process, giving stress models face validity as models for PTSD (Phillips and LeDoux, 1992). Although a detailed review of extant stress paradigms is beyond the scope of this review, we will provide a brief overview of various tasks utilized in laboratory stress models.
Acute stress.
Acute stress can enhance classical conditioning in males (Shors et al., 1992) and working memory (Yuen et al., 2009) and as such is used as part of fear-conditioning paradigms, which are relevant for the evaluation of the effects of repeated trauma/stress exposures. Acute stress models typically use brief periods of exposure to physical or psychological stress to induce both physiological and behavioral changes, such as defensive immobility (‘‘freezing’’) or action (fight or flight; (Lang et al., 2000; Neumann et al., 2011)). The forced swim test is a classic acute stress paradigm originally used to model ‘‘behavioral despair’’ (Porsolt et al., 1977a,b). In this test, rodents are forced to swim in water from which they cannot escape, and they eventually cease to struggle more than necessary in order to remain afloat. While initially characterized as a screening mechanism for antidepressant therapies (Porsolt et al., 1977a,b), the forced swim test and other tasks using water immersion can potentiate fear behavior (reviewed in Korte and De Boer (2003)). Conversely, adult exposure to shock as the stressor can modify behavioral despair in the forced swim (Scott et al., 1996). A single session of acute restraint in adulthood enhances conditioned freezing in shock paradigms (Cordero et al., 2003) and increases anxiety-like behavior in the elevated plus maze (Gameiro et al., 2006).
Chronic stress.
Although the temporal distinction between acute and chronic stress is not always clear, chronic stress generally refers either to repeated exposure to the same stressor (homotypic stress) or to multiple days of exposure to a variety of stressors (heterotypic stress). Repeated restraint stress is an example of a chronic homotypic stress paradigm and has been shown to alter fear conditioning (Chan et al., 1993; Cullinan et al., 1995). Chronic mild or chronic unpredictable stress are examples of heterotypic stressors, and these paradigms consist of exposing rodents to repeated physical stressors (e.g., restraint, foot shock, cold temperature, etc.) for a set duration of time each day over a period from days to weeks (Nestler and Hyman, 2010). Whereas habituation can occur with chronic homotypic stress paradigms (Magarinos and McEwen, 1995), habituation does not occur in chronic heterotypic stress paradigms and these models are particularly effective at inducing long-lasting changes in aspects of HPA axis function (Cox et al., 2011) and also elevate secretion of ACTH, potentiating anxiety-like behavior (Heinrichs et al., 1994). Chronic stress facilitates fear conditioning even independent of hippocampal remodeling (Conrad et al., 1999) and can further impair recall of fear extinction (Miracle et al., 2006) with notable sex differences (Baran et al., 2009), making chronic stress a particularly powerful model for PTSD.
Social stress.
Social stress paradigms elicit long-lasting changes in behavior in the rat that can serve as excellent models for aspects of PTSD neurobiology and behavior. Predator exposure can be used to model both an immediate fear response as well as a conditioned response (Blanchard et al., 2001; Barnum et al., 2012). Common predator models expose the test subject to cat or fox odor, or to the presence of an actual predator (e.g., rat as subject exposed to cat as predator; mouse as subject exposed to rat as predator (Neumann et al., 2011; Cohen et al., 2012a). These models produce significant increases in anxiety-like behavior as well as changes in sexually-motivated and cognitive behavior (Cohen and Zohar, 2004; Cohen et al., 2012a). Social defeat stress similarly alters behavior and also produces changes in metabolism consistent with fundamental perturbations to physiologic homeostasis (Nestler and Hyman, 2010). In social defeat stress, an animal is subjected to bouts of social subordination by a larger, aggressive conspecific for a given time period (usually 10–60 min, occurring in one or multiple rounds). After these encounters, the defeated rat shows a variety of increased stress-reactivity and anxiety- and depressive-like behaviors for several weeks (for a review, see Stam (2007a)). Although most social defeat paradigms have demonstrated effects in males, female models of social defeat also exist (DeBold and Miczek, 1984; Bourke et al., 2012b) and the behavioral effects of social defeat are more evident in female than male adolescent rats (Bourke and Neigh, 2011).
Another social stress paradigm involves isolation. Social isolation during adolescence causes increased anxiety in adolescent females and adults of both sexes (McCormick et al., 2008; Weintraub et al., 2010). In addition, social buffering in the form of pair housing can buffer rodents against the development of stress-related dysfunction (Beck and Luine, 2002) and improves outcomes from a variety of health challenges (DeVries et al., 2007). When social pairing is not possible, environmental enrichment also has been shown to be effective in limiting stress effects (Lehmann and Herkenham, 2011). Manipulations of housing in these positive manners can produce a level of resilience in rodent models of stress exposure.
Early-life stress.
In addition to stressor type, the timing of stress in developmental animal models plays a particularly important role in adult behavioral and physiological responses to aversive stimuli (Teicher et al., 2006; Andersen and Teicher, 2008). Specifically, early-life stressors such as maternal separation (Kehoe et al., 1998; Popp et al., 2004) and abusive maternal care (Maestripieri et al., 2005) can alter stress-reactivity as well as hippocampal development (Kehoe et al., 1998; Popp et al., 2004). Although the effects of stress during puberty are not as widely studied as those earlier in life, puberty and adolescence appears to be an additional critical period for the potential development of PTSD. Rodent models indicate enhanced stress susceptibility during puberty (Bingham et al., 2011), suggesting a physiological rather than psychological element to this risk factor. Social stress exposure during puberty also alters the formation of agonistic behaviors in female golden hamsters (Taravosh-Lahn and Delville, 2004). In addition, social stress during puberty can alter subsequent stress responses (McCormick and Mathews, 2007), as well as substance abuse behaviors in animals (Ferris and Brewer, 1996; McCormick et al., 2004). Exposure to chronic variable stress during puberty enhances the acoustic startle response both at the end of puberty and in adulthood (Maslova et al., 2002). Furthermore, short-term exposure (3 days) to a psychogenic stressor (predator odor and elevated platform) during puberty produces sustained changes in fear-related behavior (Toledo-Rodriguez and Sandi, 2007). The effects of stress during puberty may be particularly detrimental for females (Bourke et al., 2012a,b). Given the role of estrogen in modulation of the neurobiological response to stress (McEwen, 2002) and the increased risk of violence and abuse exposure for women during this period, understanding the effects of stress during puberty in the female is of particular importance.
The wealth of laboratory stress models provides a sophisticated tool set that can be used to model the various types of trauma/stress experienced by the clinical population. We will revisit these various models and will examine the utility and applicability of these models for use in translational research. First, we review the current empirically supported treatments for trauma.
AVAILABLE TRAUMA TREATMENTS: FOCUS ON ADOLESCENTS
Existing trauma treatments for youth consist primarily of modifications made to trauma treatments designed for and originally implemented with adults (Cohen et al., 2000), and the most efficacious adult trauma treatments to date are psychotherapy (Cukor et al., 2010). Exposure-based therapies have generated the greatest empirical support in the literature and have been recommended by the Institute of Medicine (2008) as the first line of treatment for PTSD. Prolonged Exposure (PE; Foa and Rothbaum, 1998) is considered the gold standard treatment for trauma and has gathered an extensive evidence base across a variety of populations with diverse trauma histories. Cognitive processing therapy, which contains an exposure element, has demonstrated efficacy in treating PTSD, and both prolonged exposure and cognitive processing therapy have been shown to effectively treat PTSD and comorbid depressive symptoms in a sample of female rape victims (Resick et al., 2002). In recent meta- analyses (Bradley et al., 2005; Powers et al., 2010), cognitively-based interventions for trauma were associated with significant reductions in trauma-related symptomatology and outperformed waitlist controls and treatment as usual. While a variety of other treatments for PTSD exist (e.g., eye movement desensitization and reprocessing, stress management/relaxation groups, supportive/nondirective psychotherapy), they have shown comparatively less empirical support in the literature (Bradley et al., 2005; Bisson et al., 2007).
Despite the effectiveness of psychotherapy, a substantial number of participants in clinical trials remain symptomatic at the end of treatment (Cukor et al., 2010). A recent meta-analysis found that 56% of adults who enrolled in treatment and 67% of treatment completers no longer met criteria for PTSD (Bradley et al., 2005); hence, approximately 30–40% of patients either do not benefit significantly from treatment and/or do not complete treatment. Moreover, many randomized clinical trials exclude participants with more severe comorbid psychopathology or severe suicidality (Harned and Linehan, 2008). A recent meta-analysis of PTSD treatment outcome studies (Bradley et al., 2005) found that 46% of trials excluded individuals with risk for suicide, 62% excluded individuals with substance or alcohol abuse or dependence, and 62% excluded individuals identified as having a serious comorbid disorder. This limited generalizability is particularly problematic for adolescents, for whom comorbid PTSD is more common than PTSD alone (Kilpatrick et al., 2003; Macdonald et al., 2010).
Several of the aforementioned therapies have been modified or extended for use with youth samples. The basic cognitive principles used with traumatized adults were modified into a treatment called trauma-focused cognitive behavior therapy for youth (Cohen et al., 2000). Trauma-focused cognitive behavior therapy includes four components: exposure, cognitive processing/reframing, stress management, and parental interventions (Cohen et al., 2000) and has been shown to be effective in reducing trauma symptoms and depression in traumatized youth samples (for a review, see Dynegrov and Yule (2006)). Other adaptations of cognitive behavioral therapy for PTSD in adolescents have emphasized cognitive restructuring rather than exposure therapy based on the rationale that many traumatized adolescents continue to live in environments where they may witness or experience new traumas; thus, emphasizing cognitive restructuring is thought to provide a type of stress inoculation in the face of ongoing exposure to traumatic stressors. In a pilot study of this intervention among a community sample of adolescents, results indicated a significant reduction in PTSD and depressive symptoms at post-intervention and at three-month follow-up (Rosenberg et al., 2011).
There is limited research of prolonged exposure for adolescents in randomized controlled trials; however, preliminary evidence suggests that prolonged exposure reduces PTSD symptoms, depression, and anxiety (Feeny et al., 2004). Gilboa-Schechtman and colleagues (2010) recently compared prolonged exposure to psychodynamic therapy in adolescents who experienced a single traumatic event. While both interventions elicited reduction in PTSD and depressive symptoms, prolonged exposure for adolescents resulted in greater reductions in PTSD and depressive symptom severity and a higher increase in global functioning than did psychodynamic therapy, and gains achieved in prolonged exposure lasted through both six-month and 17-month follow-up assessments (Gilboa-Schechtman et al., 2010).
Finally, group and family therapies have been studied in youth (see Dynegrov and Yule (2006)). Group treatments typically involve exposure to the traumatic event through oral or written exposure-based techniques (Dynegrov and Yule, 2006). Preliminary support for group interventions utilizing cognitive behavioral therapy principles in traumatized youth exists; however, larger trials with more diverse trauma exposure are necessary for further generalizability (Feeny et al., 2004). Additionally, results from studies investigating parent– child therapy for sexual abuse survivors are equivocal (Lester et al., 2008).
An important theme arises from these clinical statistics. Namely, while a majority of PTSD sufferers benefit from existing treatments, a substantial minority of patients remain symptomatic or drop out of treatment. Moreover, patients commonly seen in outpatient practice (i.e., comorbid, multidiagnostic, suicidal patients) are often excluded from randomized clinical trials. The patient who is likely to manifest the most pervasive and treatment-resistant PTSD is the patient who will have faced multiple chronic developmental traumas in addition to the acute trauma that precipitated the current diagnosis of PTSD. In addition, this patient is likely to demonstrate hallmark symptoms of depression and anxiety along with PTSD. Importantly, these multidimensional aspects of the treatment-resistant clinical patient are not typically recapitulated in preclinical animal models of stress, leaving a major gap in the types of preclinical research most likely to benefit individuals who are particularly vulnerable to PTSD and who do not benefit from extant treatments.
FILLING THE GAPS IN THE TREATMENT LITERATURE WITH TRANSLATIONAL RESEARCH
Even with the aforementioned gold standard treatments for PTSD, 30–40% of individuals who seek treatment will be classified as non-responders or will be lost to attrition (Foa, 2000). A host of participant characteristics – including preexisting depression and anxiety as well as tendencies toward avoidance, guilt, shame, anger, numbing, and dissociation – have been shown to be associated with poorer treatment outcomes in standard treatments for PTSD. Moreover, in treatment research with adults diagnosed with other psychiatric disorders (e.g., major depression, borderline personality disorder), trauma history and comorbid PTSD is frequently a strong predictor of treatment non-response (Green et al., 2006; Campbell et al., 2007). Thus, intervening on the effects of early life stress and trauma are important for both proximal and distal psychological functioning, and understanding the impact of early life stress in general remains an area in need of ongoing investigation and study.
FOUR-FACTOR MODEL
Perhaps our focus, then, should be on the unique neurobiological and psychological characteristics that are associated with treatment response or failure. The goal of such studies would be to extend what has been learned from animals to the diagnosis and treatment of humans exposed to traumatic stressors. Specifically, preclinical research with animals could shed substantial light on the biological markers associated with risk and resilience that would better help clinical scientists classify and properly intervene on traumatic symptoms and functioning at an appropriate time. However, it is critical that animal models recapitulate the multidimensional nature of the treatment resistant patient.
We strongly suspect that animal and human studies would enjoy considerably greater translational reciprocity if supported by a common underlying structural theory that is mutually informative and beneficial. As a preliminary step toward improving personalized medicine for individuals with PTSD (see Mayberg (2003) and Gillespie et al. (2010)), we propose a four-factor model as a method of classifying varying levels of risk and resilience for individuals exposed to traumatic stressors that is accessible to animal researchers (see Fig. 2). Although the model is overly simplistic in that it does not capture the complex and widely variable cognitive, emotional, experiential, and clinical presentations that emerge in clinical practice with trauma survivors, we nevertheless propose its utility for the purpose of making preclinical animal models more broadly applicable to and reciprocal with treatment studies of humans exposed to traumatic stressors. We consider this simple 2 × 2 model a necessary first step in the process of developing a broader and more elegant framework in which clinical and preclinical researchers are able to effectively communicate and build toward a multi-dimensional model that will recapitulate the dynamic range of risk and resilience. The need for additional translational research is great and, therefore, we constructed a parsimonious model that was reflective of the translational literature to date.
Fig. 2.
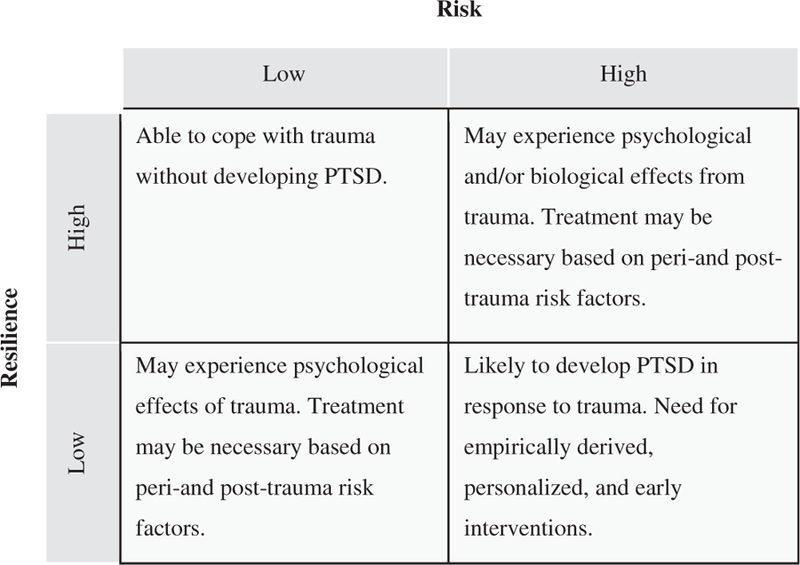
Four-factor risk/resilience model for the clinical population. The four-factor model is a method of classifying different levels of risk and resilience for individuals exposed to traumatic stressors. This classification system aims to yield clinically translatable information pertaining to direct and early intervention or, combined (i.e., medication and psychotherapy) versus singular (i.e., psychotherapy alone) interventions.
Here we propose a simple 2 × 2 model as the essential framework to initiate the translational reciprocity that is critical to designing better studies and, therefore better treatments. By way of illustration, we know that approximately 90% of individuals exposed to a traumatic stressor will not develop PTSD (Kessler et al., 1995). We hypothesize that those individuals can be broadly characterized as low risk/high resilience in Fig. 2. An example may be a child who is in a stable home environment with excellent social support and no biological risk factors. Of the 10% of individuals who do go on to develop PTSD in the wake of a trauma, we also know that approximately 60% of those individuals will respond to existing treatments (Foa, 2000). We hypothesize that those individuals likely can be characterized as high risk/high resilience or as low risk/ low resilience. For these individuals, we suspect that the host of risk factors outlined in Fig. 1 are particularly relevant vis á vis the development of PTSD. An example of a high risk/high resilience individual may be a female with a history of trauma (high risk) but who is living in a stable home environment with extensive social support (high resilience). Low risk/low resilience is exemplified by an individual with no prior trauma and no genetic risk (low risk) who lacks the necessary support to recover from a trauma (low resilience). Finally, we hypothesize that a majority of the individuals who develop PTSD and who do not respond to existing treatments likely can be characterized as high risk/low resilience. An individual classified as high risk/low resilience may have already experienced multiple traumas and carry a genetic risk (high risk) and lack the necessary support post-trauma (low resilience).
In some respects, resilience is the opposite side of the stress risk coin; however, we do not conceptualize risk and resilience in humans as purely orthogonal constructs (for a discussion, see Rutter (2006), Yehuda and Flory (2007), and Kolar (2011)). Moreover, we assert that animal laboratory studies that focus uniquely on stress-induced pathology likely miss the mark on allowing for the identification of the mechanisms of resilience. That is, if resilience is mediated by an active biological mechanism that is absent in those who succumb to the adverse effects of trauma/stress, then the current dogma of assessing pathology alone necessarily misses the mark. Nevertheless, such a risk/ resilience model generally maps onto the basic extant paradigms in animal research and provides a springboard for the development of a more elegant, empirically-derived classification system that more appropriately captures the human experience of trauma responding. As the interrelatedness of risk and resilience to trauma is better understood, such studies could ultimately yield clinically translatable information as to the necessity of direct and early intervention or, alternatively, combined (i.e., medication and psychotherapy) versus monotherapeutic (i.e., psychotherapy alone) interventions.
The identification of associated neural correlates in PTSD may similarly lend insight into the most direct and effective ways of intervening on PTSD symptoms. Knowledge gained from preclinical studies of memory and trauma responding in animals could inform the development of treatments designed to remediate the neurological effects of trauma exposure in humans. Alternatively, animal studies can dovetail with the existing literature on human risk/resilience along with what is known about the developmental trajectory of adolescents to create animal models to test various interventions and combinations thereof (i.e., psychotherapy and pharmacotherapy) that are tailored to the developmental needs, strengths, and weaknesses of youth rather than relying on the downward extension of adult treatments to youth samples. This, in turn, greatly enhances our ability to derive personally-tailored approaches to the treatment of PTSD by matching endogenous patient characteristics on the risk and resilience dimensions to treatments designed to capitalize on patient strengths and bolster deficits (see Gillespie et al., 2010).
As discussed, the relationship between resilience and stress as it relates to the development of psychopathology can be divided into resistance and recovery (Yehuda et al., 2006). Resistance refers to ‘‘the psychological and/or biological characteristics that may be associated with being relatively impervious to the deleterious effects of stress’’ (Yehuda et al. 2006, p. 382), or conversely, the characteristics that put one at risk for the adverse effects of stress. Recovery is defined as ‘‘an individual’s ability to mend or restore psychological and/or physical damage that may have resulted from trauma exposure’’ (Yehuda et al. 2006, p. 383); that is, the extent to which the organism is resilient in the face of a stressor. In terms of human recovery, this may relate to one’s ability to respond to psychotherapy, because if learning/memory processes are impaired, the efficacy of psychotherapy may be limited (Dunlop et al., 2012).
animal studies through the combination of established stress paradigms and social buffering (Fig. 3). As we have discussed, the most difficult clinical group with regard to treatment seems to fall into the category of high risk/low resilience. Notably, in addition to a paucity of work regarding high resilience, current animal models fail to investigate many aspects of the multidimensional group of high risk/low resilience. However, combinations of multiple established paradigms could provide a more representative model. Developmental stress has been demonstrated repeatedly to produce a biological and behavioral risk for both humans and preclinical animal models (Neigh et al., 2009, 2010), and the use of developmental stressors can be used to produce animal models to fulfill the high risk division of the four-factor model. Low resilience in animals can be precipitated through the use of individual housing during stress exposure that removes the positive aspects of social buffering (Stanton et al., 1985; Beck and Luine, 2002; Hennessy et al., 2009) and the introduction of chronic stress minimizes the animal’s resilience to stress (Cohen and Zohar, 2004; Stam, 2007b). Other aspects of the four-factor model can be reproduced as detailed in Fig. 3. In addition to these social and environmental manipulations to model risk and resilience, genetic manipulations may also be advantageous. However, in order for a genetic manipulation to be clinically relevant, the manipulation should represent an established clinical finding, and it should be noted that generalizability of the data will not extend beyond those with the genetic deficit.
Fig. 3.
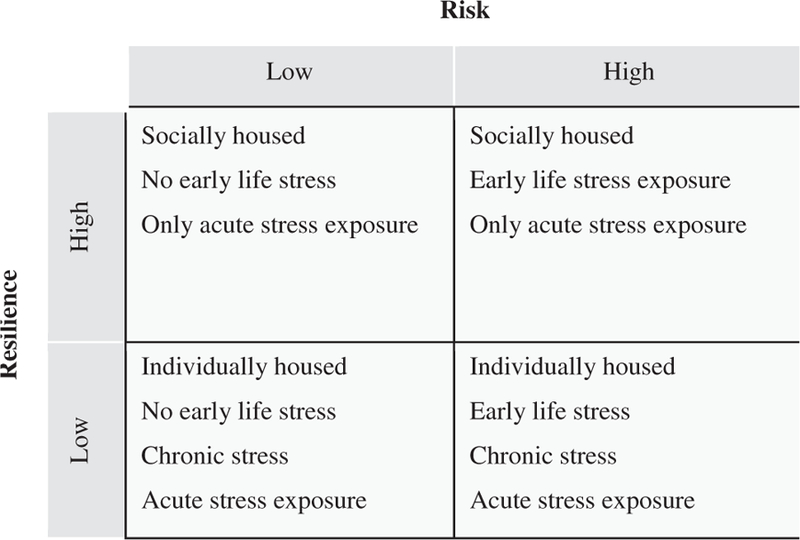
Four-factor risk/resilience model for guiding preclinical models. The four-factor risk/resilience model for the clinical population can be adapted to animal models through the combination of stress paradigms and social buffering. Current animal models fail to recapitulate many aspects of the high risk/low resilience group, but a combination of multiple paradigms may create a more representative model.
In order for translational reciprocity to be achieved, basic and clinical scientists must both be willing to attend to the findings of their counterpart’s field and adjust their paradigms and practices accordingly. The implication for the neuroscientist is to recognize that the patient most in need of novel therapeutics is likely one of high risk and low resilience; thus, such characteristics are critical in developing and testing translational animal models. By designing animal models that are most representative of clinical presentation, we will be able to bridge the gap between preclinical studies and clinical treatment and advance the treatment of stress effects on the adolescent brain.
Abbreviations:
- ACTH
adrenocorticotropic hormone
- BPD
borderline personality disorder
- CRF
corticotropin-releasing factor
- DHEA
dehydroepiandrosterone
- GR
glucocorticoid receptor
- HPA
hypothalamic–pituitary–adrenal
- LC
locus coeruleus
- MDD
major depressive disorder
- mPFC
medial prefrontal cortex
- NPY
neuropeptide Y
- POMC
proopiomelanocortin
- PTSD
post-traumatic stress disorder
- SUD
substance use disorders
REFERENCES
- Andersen SL, Teicher MH (2008) Stress, sensitive periods and maturational events in adolescent depression. Trends Neurosci 31:183–191. [DOI] [PubMed] [Google Scholar]
- APA (2000) Diagnostic and statistical manual of mental disorders – test revision Washington, DC: American Psychiatric Association. [Google Scholar]
- Aston-Jones G, Rajkowski J, Ivanova S, Usher M, Cohen J (1998) Neuromodulation and cognitive performance: recent studies of noradrenergic locus ceruleus neurons in behaving monkeys. Adv Pharmacol 42:755–759. [DOI] [PubMed] [Google Scholar]
- Aupperle RL, Melrose AJ, Stein MB, Paulus MP (2012) Executive function and PTSD: disengaging from trauma. Neuropharmacology 62:686–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avital A, Ram E, Maayan R, Weizman A, Richter-Levin G (2006) Effects of early-life stress on behavior and neurosteroid levels inz the rat hypothalamus and entorhinal cortex. Brain Res Bull 68:419–424. [DOI] [PubMed] [Google Scholar]
- Baran SE, Armstrong CE, Niren DC, Hanna JJ, Conrad CD (2009) Chronic stress and sex differences on the recall of fear conditioning and extinction. Neurobiol Learn Mem 91:323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardi M, Franssen CL, Hampton JE, Shea EA, Fanean AP, Lambert KG (2011) Paternal experience and stress responses in California mice (Peromyscus californicus). Comp Med 61:20–30. [PMC free article] [PubMed] [Google Scholar]
- Barnum CJ, Pace TW, Hu F, Neigh GN, Tansey MG (2012) Psychological stress in adolescent and adult mice increases neuroinflammation and attenuates the response to LPS challenge. J Neuroinflammation 9:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck KD, Luine VN (2002) Sex differences in behavioral and neurochemical profiles after chronic stress: role of housing conditions. Physiol Behav 75:661–673. [DOI] [PubMed] [Google Scholar]
- Binder EB (2009) The role of FKBP5, a co-chaperone of the glucocorticoid receptor in the pathogenesis and therapy of affective and anxiety disorders. Psychoneuroendocrinology 34(Suppl. 1):S186–S195. [DOI] [PubMed] [Google Scholar]
- Binder EB, Bradley RG, Liu W, Epstein MP, Deveau TC, Mercer KB, Tang Y, Gillespie CF, Heim CM, Nemeroff CB, Schwartz AC, Cubells JF, Ressler KJ (2008) Association of FKBP5 polymorphisms and childhood abuse with risk of posttraumatic stress disorder symptoms in adults. JAMA 299:1291–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder EB, Salyakina D, Lichtner P, Wochnik GM, Ising M, Putz B, Papiol S, Seaman S, Lucae S, Kohli MA, Nickel T, Kunzel HE, Fuchs B, Majer M, Pfennig A, Kern N, Brunner J, Modell S, Baghai T, Deiml T, Zill P, Bondy B, Rupprecht R, Messer T, Kohnlein O, Dabitz H, Bruckl T, Muller N, Pfister H, Lieb R, Mueller JC, Lohmussaar E, Strom TM, Bettecken T, Meitinger T, Uhr M, Rein T, Holsboer F, Muller-Myhsok B (2004) Polymorphisms in FKBP5 are associated with increased recurrence of depressive episodes and rapid response to antidepressant treatment. Nat Genet 36:1319–1325. [DOI] [PubMed] [Google Scholar]
- Bingham B, McFadden K, Zhang X, Bhatnagar S, Beck S, Valentino R (2011) Early adolescence as a critical window during which social stress distinctly alters behavior and brain norepinephrine activity. Neuropsychopharmacology 36:896–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisson JI, Ehlers A, Matthews R, Pilling S, Richards D, Turner S (2007) Psychological treatments for chronic post-traumatic stress disorder. Systematic review and meta-analysis. Br J Psychiatry 190:97–104. [DOI] [PubMed] [Google Scholar]
- Blanchard RJ, Yang M, Li CI, Gervacio A, Blanchard DC (2001) Cue and context conditioning of defensive behaviors to cat odor stimuli. Neurosci Biobehav Rev 25:587–595. [DOI] [PubMed] [Google Scholar]
- Bourke CH, Harrell CS, Neigh GN (2012a) Stress-induced sex differences: adaptations mediated by the glucocorticoid receptor. Horm Behav 62(3):210–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourke CH, Neigh GN (2011) Behavioral effects of chronic adolescent stress are sustained and sexually dimorphic. Horm Behav 60:112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourke CH, Owens MJ (2010) Corticotropin-releasing factor. In: Encyclopedia of psychopharmacology Springer; p. 355–360. [Google Scholar]
- Bourke CH, Raees MQ, Malviya S, Bradburn CA, Binder EB, Neigh GN (2012b) Glucocorticoid sensitizers Bag1 and Ppid are regulated by adolescent stress in a sex-dependent manner. Psychoneuroendocrinology [Epub ahead of print]. [DOI] [PMC free article] [PubMed]
- Bradley R, Greene J, Russ E, Dutra L, Westen D (2005) A multidimensional meta-analysis of psychotherapy for PTSD. Am J Psychiatry 162:214–227. [DOI] [PubMed] [Google Scholar]
- Brent D, Melhem N, Ferrell R, Emslie G, Wagner KD, Ryan N, Vitiello B, Birmaher B, Mayes T, Zelazny J, Onorato M, Devlin B, Clarke G, DeBar L, Keller M (2010) Association of FKBP5 polymorphisms with suicidal events in the Treatment of Resistant Depression in Adolescents (TORDIA) study. Am J Psychiatry 167:190–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewin CR, Andrews B, Valentine JD (2000) Meta-analysis of risk factors for posttraumatic stress disorder in trauma-exposed adults. J Consult Clin Psychol 68:748–766. [DOI] [PubMed] [Google Scholar]
- Campbell DG, Felker BL, Liu CF, Yano EM, Kirchner JE, Chan D, Rubenstein LV, Chaney EF (2007) Prevalence of depression- PTSD comorbidity: implications for clinical practice guidelines and primary care-based interventions. J Gen Intern Med 22:711–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan RK, Brown ER, Ericsson A, Kovacs KJ, Sawchenko PE (1993) A comparison of two immediate-early genes, c-fos and NGFI-B, as markers for functional activation in stress-related neuroendocrine circuitry. J Neurosci 13:5126–5138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen H, Kozlovsky N, Alona C, Matar MA, Joseph Z (2012a) Animal model for PTSD: from clinical concept to translational research. Neuropharmacology 62:715–724. [DOI] [PubMed] [Google Scholar]
- Cohen H, Liu T, Kozlovsky N, Kaplan Z, Zohar J, Mathe AA (2012b) The neuropeptide Y (NPY)-ergic system is associated with behavioral resilience to stress exposure in an animal model of post-traumatic stress disorder. Neuropsychopharmacology 37:350–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen H, Maayan R, Touati-Werner D, Kaplan Z, A Mater M, Loewenthal U, Kozlovsky N, Weizman R (2007) Decreased circulatory levels of neuroactive steroids in behaviourally more extremely affected rats subsequent to exposure to a potentially traumatic experience. Int J Neuropsychopharmacol 10:203–209. [DOI] [PubMed] [Google Scholar]
- Cohen H, Zohar J (2004) An animal model of posttraumatic stress disorder: the use of cut-off behavioral criteria. Ann N Y Acad Sci 1032:167–178. [DOI] [PubMed] [Google Scholar]
- Cohen JA, Mannarino AP, Berliner L, Deblinger E (2000) Trauma-focused cognitive behavioral therapy for children and adolescents: an empirical update. J Interpers Violence 15:1202–1223. [Google Scholar]
- Conrad CD, LeDoux JE, Magarinos AM, McEwen BS (1999) Repeated restraint stress facilitates fear conditioning independently of causing hippocampal CA3 dendritic atrophy. Behav Neurosci 113:902–913. [DOI] [PubMed] [Google Scholar]
- Cordero MI, Venero C, Kruyt ND, Sandi C (2003) Prior exposure to a single stress session facilitates subsequent contextual fear conditioning in rats. Evidence for a role of corticosterone. Horm Behav 44:338–345. [DOI] [PubMed] [Google Scholar]
- Cox BM, Alsawah F, McNeill PC, Galloway MP, Perrine SA (2011) Neurochemical, hormonal, and behavioral effects of chronic unpredictable stress in the rat. Behav Brain Res 220:106–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cukor J, Olden M, Lee F, Difede J (2010) Evidence-based treatments for PTSD, new directions, and special challenges. Ann N Y Acad Sci 1208:82–89. [DOI] [PubMed] [Google Scholar]
- Cullinan WE, Herman JP, Battaglia DF, Akil H, Watson SJ (1995) Pattern and time course of immediate early gene expression in rat brain following acute stress. Neuroscience 64:477–505. [DOI] [PubMed] [Google Scholar]
- Dannlowski U, Stuhrmann A, Beutelmann V, Zwanzger P, Lenzen T, Grotegerd D, Domschke K, Hohoff C, Ohrmann P, Bauer J, Lindner C, Postert C, Konrad C, Arolt V, Heindel W, Suslow T, Kugel H (2012) Limbic scars: long-term consequences of childhood maltreatment revealed by functional and structural magnetic resonance imaging. Biol Psychiatry 71:286–293. [DOI] [PubMed] [Google Scholar]
- De Bellis MD, Keshavan MS, Clark DB, Casey BJ, Giedd JN, Boring AM, Frustaci K, Ryan ND (1999) A.E. Bennett Research Award. Developmental traumatology. Part II: Brain development. Biol Psychiatry 45:1271–1284. [DOI] [PubMed] [Google Scholar]
- De Bellis MD, Keshavan MS, Frustaci K, Shifflett H, Iyengar S, Beers SR, Hall J (2002a) Superior temporal gyrus volumes in maltreated children and adolescents with PTSD. Biol Psychiatry 51:544–552. [DOI] [PubMed] [Google Scholar]
- De Bellis MD, Keshavan MS, Shifflett H, Iyengar S, Beers SR, Hall J, Moritz G (2002b) Brain structures in pediatric maltreatment-related posttraumatic stress disorder: a sociodemographically matched study. Biol Psychiatry 52:1066–1078. [DOI] [PubMed] [Google Scholar]
- DeBold JF, Miczek KA (1984) Aggression persists after ovariectomy in female rats. Horm Behav 18:177–190. [DOI] [PubMed] [Google Scholar]
- DeVries AC, Craft TK, Glasper ER, Neigh GN, Alexander JK (2007) 2006 Curt P. Richter award winner: social influences on stress responses and health. Psychoneuroendocrinology 32:587–603. [DOI] [PubMed] [Google Scholar]
- Dunlop BW, Mansson E, Gerardi M (2012) Pharmacological innovations for posttraumatic stress disorder and medication-enhanced psychotherapy. Curr Pharm Des [DOI] [PubMed]
- Dynegrov A, Yule W (2006) A review of PTSD in children. Child Adolesc Ment Health 11:176–184. [DOI] [PubMed] [Google Scholar]
- Edge MD, Ramel W, Drabant EM, Kuo JR, Parker KJ, Gross JJ (2009) For better or worse? Stress inoculation effects for implicit but not explicit anxiety. Depress Anxiety 26:831–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evaul K, Li R, Papari-Zareei M, Auchus RJ, Sharifi N (2010) 3Beta-hydroxysteroid dehydrogenase is a possible pharmacological target in the treatment of castration-resistant prostate cancer. Mol Cell Endocrinol 151:3514–3520. [DOI] [PubMed] [Google Scholar]
- Feeny NC, Foa EB, Treadwell KH, March J (2004) Posttraumatic Stress Disorder in Youth: a critical review of the cognitive and behavioral treatment outcome literature. Prof Psychol Res Pract 35:466–476. [Google Scholar]
- Ferris CF, Brewer J (1996) Adolescent stress alters ethanol ingestion and agonistic behavior in the male golden hamster. Ann N Y Acad Sci 794:348–351. [DOI] [PubMed] [Google Scholar]
- Foa EB (2000) Psychosocial treatment of posttraumatic stress disorder. J Clin Psychiatry 61(Suppl. 5):43–48. discussion 49–51. [PubMed] [Google Scholar]
- Foa EB, Rothbaum B (1998) Treating the trauma of rape: cognitive-behavioral therapy for PTSD New York, NY: Guilford Press. [Google Scholar]
- Forsyth RP (1969) Blood pressure responses to long-term avoidance schedules in the restrained rhesus monkey. Psychosom Med 31:300–309. [DOI] [PubMed] [Google Scholar]
- Foy DW, Madvig BT, Pynoos RS, Camilleri AJ (1996) Etiologic factors in development of posttraumatic stress disorder in children and adolescents. J School Pyschol 34:133–145. [Google Scholar]
- Gameiro GH, Gameiro PH, Andrade Ada S, Pereira LF, Arthuri MT, Marcondes FK, Veiga MC (2006) Nociception- and anxiety-like behavior in rats submitted to different periods of restraint stress. Physiol Behav 87:643–649. [DOI] [PubMed] [Google Scholar]
- Gehring U, Tomkins GM (1974) A new mechanism for steroid unresponsiveness: loss of nuclear binding activity of a steroid hormone receptor. Mol Cell Endocrinol 3:301–306. [DOI] [PubMed] [Google Scholar]
- Gilboa-Schechtman E, Foa EB, Shafran N, Aderka IM, Powers MB, Rachamim L, Rosenbach L, Yadin E, Apter A (2010) Prolonged exposure versus dynamic therapy for adolescent PTSD: a pilot randomized controlled trial. J Am Acad Child Adolesc Psychiatry 49:1034–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie C, Binder EB, Holtzheimer P, Nemeroff CB (2010) Stress and the impact of personalized medicine. In: Gordon E, Koslow S, editors. Integrative neuroscience and personalized medicine . USA: Oxford University Press; p. 73–92. [Google Scholar]
- Grad I, Picard D (2007) The glucocorticoid responses are shaped by molecular chaperones. Mol Cell Endocrinol 275:2–12. [DOI] [PubMed] [Google Scholar]
- Green BL, Goodman LA, Krupnick JL, Corcoran CB, Petty RM, Stockton P, Stern NM (2000) Outcomes of single versus multiple trauma exposure in a screening sample. J Trauma Stress 13:271–286. [DOI] [PubMed] [Google Scholar]
- Green BL, Krupnick JL, Chung J, Siddique J, Krause ED, Revicki D, Frank L, Miranda J (2006) Impact of PTSD comorbidity on one-year outcomes in a depression trial. J Clin Psychol 62:815–835. [DOI] [PubMed] [Google Scholar]
- Harned MS, Linehan MM (2008) Integrating dialectical behavior therapy and prolonged exposure to treat co-occurring borderline personality disorder and PTSD: two case studies. Cogn Behav Pract 15:263–276. [Google Scholar]
- Hastings JA, McClure-Sharp JM, Morris MJ (2001) NPY Y1 receptors exert opposite effects on corticotropin releasing factor and noradrenaline overflow from the rat hypothalamus in vitro. Brain Res 890:32–37. [DOI] [PubMed] [Google Scholar]
- Hawley DF, Bardi M, Everette AM, Higgins TJ, Tu KM, Kinsley CH, Lambert KG (2010) Neurobiological constituents of active, passive, and variable coping strategies in rats: integration of regional brain neuropeptide Y levels and cardiovascular responses. J Trauma Stress 13:172–183. [DOI] [PubMed] [Google Scholar]
- Heilig M (2004) The NPY system in stress, anxiety and depression. Neuropeptides 38:213–224. [DOI] [PubMed] [Google Scholar]
- Heinrichs SC, Menzaghi F, Pich EM, Baldwin HA, Rassnick S, Britton KT, Koob GF (1994) Anti-stress action of a corticotropin-releasing factor antagonist on behavioral reactivity to stressors of varying type and intensity. Neuropsychopharmacology 11:179–186. [DOI] [PubMed] [Google Scholar]
- Hennessy MB, Kaiser S, Sachser N (2009) Social buffering of the stress response: diversity, mechanisms, and functions. Front Neuroendocrinol 30:470–482. [DOI] [PubMed] [Google Scholar]
- Horstmann S, Lucae S, Menke A, Hennings JM, Ising M, Roeske D, Muller-Myhsok B, Holsboer F, Binder EB (2010) Polymorphisms in GRIK4, HTR2A, and FKBP5 show interactive effects in predicting remission to antidepressant treatment. Neuropsychopharmacology 35:727–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Institute of Medicine (IOM) (2008) Treatment of posttraumatic stress disorder: an assessment of the evidence Washington, DC: The National Academies Press. [Google Scholar]
- Jacobsen PB, Sadler IJ, Booth-Jones M, Soety E, Weitzner MA, Fields KK (2002) Predictors of posttraumatic stress disorder symptomatology following bone marrow transplantation for cancer. J Consult Clin Psychol 70:235–240. [DOI] [PubMed] [Google Scholar]
- Kehoe P, Shoemaker WJ, Triano L, Callahan M, Rappolt G (1998) Adult rats stressed as neonates show exaggerated behavioral responses to both pharmacological and environmental challenges. Behav Neurosci 112:116–125. [DOI] [PubMed] [Google Scholar]
- Kessler RC, Sonnega A, Bromet E, Hughes M, Nelson CB (1995) Posttraumatic stress disorder in the National Comorbidity Survey. Arch Gen Psychiatry 52:1048–1060. [DOI] [PubMed] [Google Scholar]
- Kilpatrick DG, Ruggiero KJ, Acierno R, Saunders BE, Resnick HS, Best CL (2003) Violence and risk of PTSD, major depression, substance abuse/dependence, and comorbidity: results from the National Survey of Adolescents. J Consult Clin Psychol 71:692–700. [DOI] [PubMed] [Google Scholar]
- Koenen KC, Harley R, Lyons MJ, Wolfe J, Simpson JC, Goldberg J, Eisen SA, Tsuang M (2002) A twin registry study of familial and individual risk factors for trauma exposure and posttraumatic stress disorder. J Nerv Ment Dis 190:209–218. [DOI] [PubMed] [Google Scholar]
- Kolar K (2011) Resilience: revisiting the concept and its utility for social research. Int J Ment Health Addict 9:421–433. [Google Scholar]
- Korte SM, De Boer SF (2003) A robust animal model of state anxiety: fear-potentiated behaviour in the elevated plus-maze. Eur J Pharmacol 463:163–175. [DOI] [PubMed] [Google Scholar]
- Kubala KH, Christianson JP, Kaufman RD, Watkins LR, Maier SF (2012) Short- and long-term consequences of stressor controllability in adolescent rats. Behav Brain Res 234:278–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang PJ, Davis M, Ohman A (2000) Fear and anxiety: animal models and human cognitive psychophysiology. J Affect Disord 61:137–159. [DOI] [PubMed] [Google Scholar]
- Lee RS, Tamashiro KL, Yang X, Purcell RH, Harvey A, Willour VL, Huo Y, Rongione M, Wand GS, Potash JB (2010) Chronic corticosterone exposure increases expression and decreases deoxyribonucleic acid methylation of Fkbp5 in mice. Mol Cell Endocrinol 151:4332–4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann ML, Herkenham M (2011) Environmental enrichment confers stress resiliency to social defeat through an infralimbic cortex-dependent neuroanatomical pathway. J Neurosci 31:6159–6173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lekman M, Laje G, Charney D, Rush AJ, Wilson AF, Sorant AJ, Lipsky R, Wisniewski SR, Manji H, McMahon FJ, Paddock S (2008) The FKBP5-gene in depression and treatment response – an association study in the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) Cohort. Biol Psychiatry 63:1103–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lester P, Saltzman W, Vine V, Comulada WS, Goldstein R, Stuber M, Pynoos R (2008) Current practice of family based interventions for child traumatic stress: results from a national survey. J Child Adolesc Trauma 1:47–61. [Google Scholar]
- Lyons DM, Kim S, Schatzberg AF, Levine S (1998) Postnatal foraging demands alter adrenocortical activity and psychosocial development. Dev Psychobiol 32:285–291. [PubMed] [Google Scholar]
- Macdonald A, Danielson CK, Resnick HS, Saunders BE, Kilpatrick DG (2010) PTSD and comorbid disorders in a representative sample of adolescents: the risk associated with multiple exposures to potentially traumatic events. Child Abuse Negl 34:773–783. [DOI] [PubMed] [Google Scholar]
- Maeda M, Kato H, Maruoka T (2009) Adolescent vulnerability to PTSD and effects of community-based intervention: longitudinal study among adolescent survivors of the Ehime Maru sea accident. Psychiatry Clin Neurosci 63:747–753. [DOI] [PubMed] [Google Scholar]
- Maestripieri D, Lindell SG, Ayala A, Gold PW, Higley JD (2005) Neurobiological characteristics of rhesus macaque abusive mothers and their relation to social and maternal behavior. Neurosci Biobehav Rev 29:51–57. [DOI] [PubMed] [Google Scholar]
- Magarinos AM, McEwen BS (1995) Stress-induced atrophy of apical dendrites of hippocampal CA3c neurons: involvement of glucocorticoid secretion and excitatory amino acid receptors. Neuroscience 69:89–98. [DOI] [PubMed] [Google Scholar]
- Maslova LN, Bulygina VV, Popova NK (2002) Immediate and long-lasting effects of chronic stress in the prepubertal age on the startle reflex. Physiol Behav 75:217–225. [DOI] [PubMed] [Google Scholar]
- Mayberg HS (2003) Modulating dysfunctional limbic–cortical circuits in depression: towards development of brain-based algorithms for diagnosis and optimised treatment. Br Med Bull 65: 193–207. [DOI] [PubMed] [Google Scholar]
- McCarty R, Gold PE (1981) Plasma catecholamines: effects of footshock level and hormonal modulators of memory storage. Horm Behav 15:168–182. [DOI] [PubMed] [Google Scholar]
- McCormick CM, Mathews IZ (2007) HPA function in adolescence: role of sex hormones in its regulation and the enduring consequences of exposure to stressors. Pharmacol Biochem Behav 86:220–233. [DOI] [PubMed] [Google Scholar]
- McCormick CM, Robarts D, Gleason E, Kelsey JE (2004) Stress during adolescence enhances locomotor sensitization to nicotine in adulthood in female, but not male, rats. Horm Behav 46:458–466. [DOI] [PubMed] [Google Scholar]
- McCormick CM, Smith C, Mathews IZ (2008) Effects of chronic social stress in adolescence on anxiety and neuroendocrine response to mild stress in male and female rats. Behav Brain Res 187:228–238. [DOI] [PubMed] [Google Scholar]
- McEwen BS (2002) Sex, stress and the hippocampus: allostasis, allostatic load and the aging process. Neurobiol Aging 23:921–939. [DOI] [PubMed] [Google Scholar]
- McGaugh JL, Cahill L, Roozendaal B (1996) Involvement of the amygdala in memory storage: interaction with other brain systems. Proc Natl Acad Sci U S A 93:13508–13514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miracle AD, Brace MF, Huyck KD, Singler SA, Wellman CL (2006) Chronic stress impairs recall of extinction of conditioned fear. Neurobiol Learn Mem 85:213–218. [DOI] [PubMed] [Google Scholar]
- Morgan CA 3rd, Southwick S, Hazlett G, Rasmusson A, Hoyt G, Zimolo Z, Charney D (2004) Relationships among plasma dehydroepiandrosterone sulfate and cortisol levels, symptoms of dissociation, and objective performance in humans exposed to acute stress. Arch Gen Psychiatry 61:819–825. [DOI] [PubMed] [Google Scholar]
- Morgan CA 3rd, Wang S, Southwick SM, Rasmusson A, Hazlett G, Hauger RL, r DS (2000) Plasma neuropeptide-Y concentrations in humans exposed to military survival training. Biol Psychiatry 47:902–909. [DOI] [PubMed] [Google Scholar]
- Natelson BH, Krasnegor N, Holaday JW (1976) Relations between behavioral arousal and plasma cortisol levels in monkeys performing repeated free-operant avoidance sessions. J Comp Physiol Psychol 90:958–969. [DOI] [PubMed] [Google Scholar]
- Neigh GN, Gillespie CF, Nemeroff CB (2009) The neurobiological toll of child abuse and neglect. Trauma Violence Abuse 10:389–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neigh GN, Owens MJ, Taylor WR, Nemeroff CB (2010) Changes in the vascular area fraction of the hippocampus and amygdala are induced by prenatal dexamethasone and/or adult stress. J Cereb Blood Flow Metab 30:1100–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ, Hyman SE (2010) Animal models of neuropsychiatric disorders. Nat Neurosci 13:1161–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann ID, Wegener G, Homberg JR, Cohen H, Slattery DA, Zohar J, Olivier JD, Mathe AA (2011) Animal models of depression and anxiety: what do they tell us about human condition? Prog Neuropsychopharmacol Biol Psychiatry 35:1357–1375. [DOI] [PubMed] [Google Scholar]
- New AS, Fan J, Murrough JW, Liu X, Liebman RE, Guise KG, Tang CY, Charney DS (2009) A functional magnetic resonance imaging study of deliberate emotion regulation in resilience and posttraumatic stress disorder. Biol Psychiatry 66:656–664. [DOI] [PubMed] [Google Scholar]
- Norrholm SD, Ressler KJ (2009) Genetics of anxiety and trauma-related disorders. Neuroscience 164:272–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens MJ, Nemeroff CB (1991) Physiology and pharmacology of corticotropin-releasing factor. Pharmacol Rev 43:425–473. [PubMed] [Google Scholar]
- Ozer EJ, Best SR, Lipsey TL, Weiss DS (2003) Predictors of posttraumatic stress disorder and symptoms in adults: a meta-analysis. Psychol Bull 129:52–73. [DOI] [PubMed] [Google Scholar]
- Parker KJ, Buckmaster CL, Sundlass K, Schatzberg AF, Lyons DM (2006) Maternal mediation, stress inoculation, and the development of neuroendocrine stress resistance in primates. Proc Natl Acad Sci U S A 103:3000–3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelps EA, Delgado MR, Nearing KI, LeDoux JE (2004) Extinction learning in humans: role of the amygdala and vmPFC. Neuron 43:897–905. [DOI] [PubMed] [Google Scholar]
- Phillips RG, LeDoux JE (1992) Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav Neurosci 106:274–285. [DOI] [PubMed] [Google Scholar]
- Phipps S, Peasant C, Barrera M, Alderfer MA, Huang Q, Vannatta K (2012) Resilience in children undergoing stem cell transplantation: results of a complementary intervention trial. Pediatrics 129:e762–e770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pine DS, Cohen JA (2002) Trauma in children and adolescents: risk and treatment of psychiatric sequelae. Biol Psychiatry 51:519–531. [DOI] [PubMed] [Google Scholar]
- Pitman RK, Gilbertson MW, Gurvits TV, May FS, Lasko NB, Metzger LJ, Shenton ME, Yehuda R, Orr SP (2006) Clarifying the origin of biological abnormalities in PTSD through the study of identical twins discordant for combat exposure. Ann N Y Acad Sci 1071:242–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp S, Maurel P, Andersen JS, Margolis RU (2004) Developmental changes of aggrecan, versican and neurocan in the retina and optic nerve. Exp Eye Res 79:351–356. [DOI] [PubMed] [Google Scholar]
- Porsolt RD, Bertin A, Jalfre M (1977a) Behavioral despair in mice: a primary screening test for antidepressants. Arch Int Pharmacodyn Ther 229:327–336. [PubMed] [Google Scholar]
- Porsolt RD, Le Pichon M, Jalfre M (1977b) Depression: a new animal model sensitive to antidepressant treatments. Nature 266:730–732. [DOI] [PubMed] [Google Scholar]
- Powers MB, Halpern JM, Ferenschak MP, Gillihan SJ, Foa EB (2010) A meta-analytic review of prolonged exposure for posttraumatic stress disorder. Clin Psychol Rev 30:635–641. [DOI] [PubMed] [Google Scholar]
- Quirarte GL, Roozendaal B, McGaugh JL (1997) Glucocorticoid enhancement of memory storage involves noradrenergic activation in the basolateral amygdala. Proc Natl Acad Sci U S A 94:14048–14053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resick PA, Nishith P, Weaver TL, Astin MC, Feuer CA (2002) A comparison of cognitive-processing therapy with prolonged exposure and a waiting condition for the treatment of chronic posttraumatic stress disorder in female rape victims. J Consult Clin Psychol 70:867–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roozendaal B (2002) Stress and memory: opposing effects of glucocorticoids on memory consolidation and memory retrieval. Neurobiol Learn Mem 78:578–595. [DOI] [PubMed] [Google Scholar]
- Rosenberg HJ, Jankowski M, Fortuna LR, Rosenberg SD, Mueser KT (2011) A pilot study of a cognitive restructuring program for treating posttraumatic disorders in adolescents. Psychol Trauma Theory Res Pract Policy 3:94–99. [Google Scholar]
- Rothbaum BO, Foa EB (1993) Subtypes of posttraumatic stress disorder and duration of symptoms. In: Davidson JRT, EB Foa, editors. Posttraumatic stress disorder: DSM-IV and beyond Washington, DC: American Psychiatric Press. [Google Scholar]
- Roy A, Gorodetsky E, Yuan Q, Goldman D, Enoch MA (2010) Interaction of FKBP5, a stress-related gene, with childhood trauma increases the risk for attempting suicide. Neuropsychopharmacology 35:1674–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutter M (2006) Implications of resilience concepts for scientific understanding. Ann N Y Acad Sci 1094:1–12. [DOI] [PubMed] [Google Scholar]
- Scharf SH, Liebl C, Binder EB, Schmidt MV, Muller MB (2011) Expression and regulation of the Fkbp5 gene in the adult mouse brain. PLoS ONE 6:e16883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnurr PP, Lunney CA, Sengupta A (2004) Risk factors for the development versus maintenance of posttraumatic stress disorder. J Trauma Stress 17:85–95. [DOI] [PubMed] [Google Scholar]
- Scott PA, Cierpial MA, Kilts CD, Weiss JM (1996) Susceptibility and resistance of rats to stress-induced decreases in swim-test activity: a selective breeding study. Brain Res 725: 217–230. [DOI] [PubMed] [Google Scholar]
- Shors TJ, Weiss C, Thompson RF (1992) Stress-induced facilitation of classical conditioning. Science 257:537–539. [DOI] [PubMed] [Google Scholar]
- Sotres-Bayon F, Bush DE, LeDoux JE (2004) Emotional perseveration: an update on prefrontal-amygdala interactions in fear extinction. Learn Mem 11:525–535. [DOI] [PubMed] [Google Scholar]
- Stam R (2007a) PTSD and stress sensitisation: a tale of brain and body. Part 1: Human studies. Neurosci Biobehav Rev 31:530–557. [DOI] [PubMed] [Google Scholar]
- Stam R (2007b) PTSD and stress sensitisation: a tale of brain and body. Part 2: Animal models. Neurosci Biobehav Rev 31:558–584. [DOI] [PubMed] [Google Scholar]
- Stanton ME, Patterson JM, Levine S (1985) Social influences on conditioned cortisol secretion in the squirrel monkey. Psychoneuroendocrinology 10:125–134. [DOI] [PubMed] [Google Scholar]
- Taravosh-Lahn K, Delville Y (2004) Aggressive behavior in female golden hamsters: development and the effect of repeated social stress. Horm Behav 46:428–435. [DOI] [PubMed] [Google Scholar]
- Teicher MH, Anderson CM, Polcari A (2012) Childhood maltreatment is associated with reduced volume in the hippocampal subfields CA3, dentate gyrus, and subiculum. Proc Natl Acad Sci U S A 109:E563–E572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teicher MH, Tomoda A, Andersen SL (2006) Neurobiological consequences of early stress and childhood maltreatment: are results from human and animal studies comparable? Ann N Y Acad Sci 1071:313–323. [DOI] [PubMed] [Google Scholar]
- Thorsell A, Carlsson K, Ekman R, Heilig M (1999) Behavioral and endocrine adaptation, and up-regulation of NPY expression in rat amygdala following repeated restraint stress. Neuroreport 10:3003–3007. [DOI] [PubMed] [Google Scholar]
- Toledo-Rodriguez M, Sandi C (2007) Stress before puberty exerts a sex- and age-related impact on auditory and contextual fear conditioning in the rat. Neural Plast 2007:71203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trickett PK, Noll JG, Susman EJ, Shenk CE, Putnam FW (2010) Attenuation of cortisol across development for victims of sexual abuse. Dev Psychopathol 22:165–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trickey D, Siddaway AP, Meiser-Stedman R, Serpell L, Field AP (2012) A meta-analysis of risk factors for post-traumatic stress disorder in children and adolescents. Clin Psychol Rev 32:122–138. [DOI] [PubMed] [Google Scholar]
- van der Kolk BA (1985) Adolescent vulnerability to posttraumatic stress disorder. J Am Acad Child Adolesc Psychiatry 48: 365–370. [DOI] [PubMed] [Google Scholar]
- Weintraub A, Singaravelu J, Bhatnagar S (2010) Enduring and sex-specific effects of adolescent social isolation in rats on adult stress reactivity. Brain Res 1343:83–92. [DOI] [PubMed] [Google Scholar]
- Weiser MJ, Foradori CD, Handa RJ (2010) Estrogen receptor beta activation prevents glucocorticoid receptor-dependent effects of the central nucleus of the amygdala on behavior and neuroendocrine function. Brain Res 1336:78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wemm S, Koone T, Blough ER, Mewaldt S, Bardi M (2010) The role of DHEA in relation to problem solving and academic performance. Biol Psychol 85:53–61. [DOI] [PubMed] [Google Scholar]
- Xie P, Kranzler HR, Poling J, Stein MB, Anton RF, Farrer LA, Gelernter J (2010) Interaction of FKBP5 with childhood adversity on risk for post-traumatic stress disorder. Neuropsychopharmacology 35:1684–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yehuda R, Blair W, Labinsky E, Bierer LM (2007) Effects of parental PTSD on the cortisol response to dexamethasone administration in their adult offspring. Am J Psychiatry 164:163–166. [DOI] [PubMed] [Google Scholar]
- Yehuda R, Boisoneau D, Lowy MT, Giller EL Jr (1995) Dose– response changes in plasma cortisol and lymphocyte glucocorticoid receptors following dexamethasone administration in combat veterans with and without posttraumatic stress disorder. Arch Gen Psychiatry 52:583–593. [DOI] [PubMed] [Google Scholar]
- Yehuda R, Flory JD (2007) Differentiating biological correlates of risk, PTSD, and resilience following trauma exposure. J Trauma Stress 20:435–447. [DOI] [PubMed] [Google Scholar]
- Yehuda R, Flory JD, Southwick S, Charney DS (2006) Developing an agenda for translational studies of resilience and vulnerability following trauma exposure. Ann N Y Acad Sci 1071: 379–396. [DOI] [PubMed] [Google Scholar]
- Yehuda R, Golier JA, Yang RK, Tischler L (2004) Enhanced sensitivity to glucocorticoids in peripheral mononuclear leukocytes in posttraumatic stress disorder. Biol Psychiatry 55:1110–1116. [DOI] [PubMed] [Google Scholar]
- Yuen EY, Liu W, Karatsoreos IN, Feng J, McEwen BS, Yan Z (2009) Acute stress enhances glutamatergic transmission in prefrontal cortex and facilitates working memory. Proc Natl Acad Sci U S A 106:14075–14079. [DOI] [PMC free article] [PubMed] [Google Scholar]