

Abstract
The imino‐Diels‐Alder reaction is one of the most common strategies in organic chemistry and is an important tool for providing a broad spectrum of biologically active heterocyclic systems. A combined theoretical and experimental study of the imino‐Diels‐Alder reaction is described. The new phenanthroline‐tetrahydroquinolines were evaluated as cholinesterase inhibitors. Their cytotoxicity in human neuroblastoma SH‐SY5Y cells was also evaluated. The theoretical results suggest that compounds formation in stages can be explained by endo cycloadducts under the established reaction conditions, thereby confirming experimental results obtained for percentage yield. These results allowed us to establish that pyridine substituent remarkably influences activation energy and reaction yield, as well as in acetylcholinesterase (AChE) activity. Among these derivatives, compounds with 4‐pyridyl and 4‐nitrophenyl showed favorable AChE activity and proved to be non‐cytotoxic.
Keywords: Alzheimer's disease imino-Diels-Alder, acetylcholinesterase, tetrahydroquinolines, density functional theory
1. Introduction
Tetrahydroquinolines (THQs) derivatives are important pharmacological substructures that exhibit remarkable biological activities and industrial uses, including antibacterial,1 antitrypanosomal,2 antioxidant,3 anticancer,4 anti‐acetylcholinesterase (AChE).5 AChE is a cholinesterase responsible for catalyzing the hydrolysis of the neurotransmitter acetylcholine (ACh) in the cholinergic synapse.6 This enzyme is an attractive target for the treatment of many neurodegenerative disorders such as Alzheimer's disease (AD).7 Like the AChE, the Butyrylcholinesterase (BChE) enzyme called nonspecific cholinesterase, also belongs to cholinesterases family of enzymes, playing only a supportive role, being it more efficient in the hydrolysis at high ACh concentrations.8 Recent studies propose that AChE also plays a role in the speed up the Aβ peptide deposition, and in the aggregation of Aβ into insoluble fibrils.9,10 The inhibition of these enzymes could, therefore, improve symptoms of this disease. The inhibition of this enzyme has emerged as a promising strategy for the treatment of AD,7 being the tacrine the first AChE inhibitor approved for the treatment of AD.11 However, due to its toxicity, other molecules have been developed.12, 13 Currently, few drugs as rivastigmine, donepezil and galantamine are approved as AChE inhibitors (Figure 1).14 Due to the urgent need for potent and safer drugs, the design and synthesis of quinoline‐containing compounds is an active and growing field.13a,15 In this field, recent studies designed and synthesized dihydropyrimidine and quinazoline bicycle as selective inhibitors against AChE enzyme.16 Likewise, quinolinyl chalcones derivatives exhibited selective inhibition for BChE enzyme displayed significant inhibition for AChE,17 Similarly, cumarine‐quinolines compounds showed ACHE selective inhibition.15d The change of functional groups in basic structure demonstrated variance in inhibition potential against BChE and AChE.17
Figure 1.
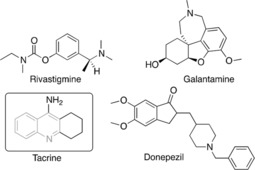
Chemical structures of some AChE inhibitors. Quinoline scaffold is highlighted in grey.
Considering the importance of THQs and quinolines in both chemical and biological fields, synthetic methods to prepare these molecules have received considerable attention.18 Reactions worth highlighting include conventional methods for the synthesis of the quinoline or THQ ring, which involve several classic routes such as Skraup, Doebne‐von Miller, Combes, Conrad‐Limpach, Pfitzinger, and Friedlander.19 However, the cycloaddition reaction [4+2], termed the imino‐Diels‐Alder (iDA), has been one of the most powerful strategies used in synthetic organic chemistry for the construction of these heterocycles.20 This reaction takes place between Schiff bases represented by imines that can react either as dienes or azadienes and almost always require a Lewis acid (LA) catalyst (Scheme 1).20a,21 Elucidating the molecular reaction mechanism by which the iDA reaction happens has been a topic of growing interest. Early evidence presented in the mid‐90s exhibited the presence of type‐THQs zwitterionic intermediaries between N‐arylimines and dihydrofuran (DHF) under LA‐catalyzed iDA.22 Nevertheless, very few theoretical studies exist for understanding the reaction mechanisms involving iDA cycloaddition.21,23 Cossio et al.24 presented a study of the reaction mechanism between N‐(3‐pyridyl) aldimines and styrene cyclopentadiene or indene using the BF3 ⋅ Et2O catalyst. Results suggested the formation of products arranged in an asynchronous process via the endo transition state, which is favored by the LA. Recently, Domingo et al.25 studied the molecular mechanism of the Povarov reaction occurring between N‐aryl‐imines and ethylene in acetonitrile (CH3CN), catalyzed by the BF3 LA. This iDA reaction presents a non‐concerted asynchronous mechanism through the presence of an intermediary zwitterionic initially associated with the nucleophilic attack of ethylene on imine carbon, constituting a decisive step for iDA. Therefore, Povarov reactions are domino processes involving two consecutive reactions: a) a LA‐catalyzed iDA reaction between an N‐arylimines (Shiff's bases, diene) and nucleophilic olefin (electron‐rich dienophiles) gives rise to the formal [4+2] cycloadduct; and b) a 1,3‐hydrogen shift in the formal [4+2] cycloadduct yield the final THQ. Nonetheless, the iDA mechanism can be more complex and can depend on variations in substituent nature on the diene, the dienophile system, or the LA catalyst.26,27
Scheme 1.
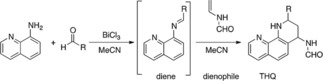
iDA reaction. This reaction takes place between imines as Schiff bases that can react as azadienes with an olefin that acts as dienophile to give of THQ.
In this study, we design new phenanthroline‐tetrahydroquinolines derivatives based on the potent inhibitory activity of quinolines against AChE.16b,28 We also explored two different approaches to accessing more derivatives of THQs and quinolines in the same molecule. First, we synthesized new THQs derivatives through the cycloaddition reaction iDA using aminoquinolines as reagent and BiCl3 as the catalyst. And as an alternative approach, we introduced of pyridine rings moieties attached to a quinoline core to evaluate the biological activity related to nitrogen position in pyridine ring.15c We established combining experimental results with theoretical assessments that helped us to elucidate the reaction mechanism and the main factors controlling the activation energy associated with the LA and pyridine substituent. The synthesized compounds were evaluated for their AChE inhibitory activity. Cytotoxicity study in human neuroblastoma SH‐SY5Y cells was also performed, aiming to discover a promising lead. Likewise, the binding mode studies of the compounds in the binding site of AChE were carried out using molecular docking calculations.
2. Results and Discussion
2.1. Experimental Studies
The iDA reaction is strongly influenced by the use of catalysts. The most widely used is BiCl3.14 Considering the above mentioned, we decided to carry out the iDA reaction at room temperature using CH3CN as the solvent and BiCl3 as the catalyst to obtain phenanthroline‐THQ 4 a–4 g (between aminoquinolines, benzaldehydes, and N‐vinylformamide (NVF, dienophile) (Scheme 1). The reaction conditions led to the successful synthesis of the designed products (Table 1).
Table 1.
Reaction conditions and physicochemical parameters obtained for new phenatroline‐tetrahydroquinolines.
| Compound | R | Conditions | Yield(%)a | M.p., °Cb |
|---|---|---|---|---|
| 4 a |
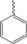
|
Without Catalyst | N.R. | |
| BF3−OEt | N.R. | |||
| 30 % BiCl3 | 31 | 227–229 | ||
| 4 b |
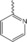
|
30 % BiCl3 | 72 | 231–233 |
| 4 c |
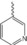
|
30 % BiCl3 | 27 | 110–112 |
| 4 d |
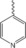
|
30 % BiCl3 | 60 | 148–150 |
| 4 e |
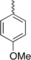
|
30 % BiCl3 | 63 | 237–238 |
| 4 f |
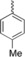
|
30 % BiCl3 | 61 | 207–209 |
| 4 g |
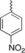
|
30 % BiCl3 | 65 | 214–216 |
aYields after column chromatography. bUncorrected. N.R. did not present reaction.
To evaluate the reaction under different conditions, the iDA reaction was carried out in the CH3CN, but without the catalyst, resulting in ineffective synthesis after 120 h. Similarly, BF3−OEt, a boron complex widely known due to its high catalytic power, was used in the differentially conditioned iDA reactions. This attempt resulted inefficient for this cycloaddition, leading to several byproducts.15 Due to this, all the reactions were performed using 30 % BiCl3, supporting the status of this catalyst as an economic, moisture‐tolerant, and often‐used alternative in this type of cycloaddition.
The efficiency of iDA reactions is related to the electronic properties of substituents on aldehyde rings. The electro‐withdrawing substituents, such as the nitro group of compound 4 g, as well as for nitrogen in position two of 4 b, increased the electro deficiency of the 2‐azadiene intermediary, allowing an increased yield. While electron‐donor groups, such as methoxy of compound 4 e, stabilized 2‐azadiene and decreased the yield (Table 1).
Structural characterization of the obtained compounds 4 a–4 g was performed using the following analytical techniques: 1D and 2D NMR spectroscopy; 2D homonuclear relationship (COSY and NOESY); heteronuclear (HMQC); and mass spectrometric data analysis. Through these methods, the cis configuration was confirmed for the substituents at positions 2 and 4 of the THQ ring. In the 1H‐NMR spectrum of the N‐(2‐(pyridin‐2‐yl)‐1,2,3,4‐tetrahydro‐1,10‐phenanthroline‐4‐yl) formamide 4 b, the CHO proton was observed at δ 8.17 ppm, as an acute and intense singlet, while the NH of the THQ was also presented as a broad singlet at δ 6.68 ppm. Furthermore, in the high field between δ 2.22 and 5.70 ppm, signals of aliphatic protons on the THQ ring were localized; whereas the δ 2.25 and 2.68 ppm field corresponded to the diasterotopics protons C‐3, 3‐Hb, and 3‐Ha, respectively, and the δ 5.66 and 4.90 ppm field showed signals for the 4‐H and 2‐H protons. According to the semi‐chair‐type disposal of THQ, we were able to conclude that this molecule could structurally behave as a cyclohexane in which the substituents at adjacent carbons (2, 3 and 4) of the aromatic ring are appropriately designated pseudo‐axial (ax) and pseudo‐equatorial (eq) (Figure 2). In the proton on C‐2, the signal at 4.91 ppm appeared as a double duplet, with a J=8.3 and 4.0 Hz, characteristics of ax/ax and ax/eq coupling with the methylene protons on the C‐3 of THQ. Likewise, the 4‐H signal appearing at δ 5.65 ppm and J=8.2 Hz (i. e. signal oriented to a medium‐low field due to an anisotropic effect of amide adjacent) was in an ax position with one of the protons on C‐3.
Figure 2.
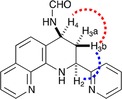
Key 2D NMR correlations for 4 b molecule. Semi‐chair‐type disposal of THQ, where adjacent carbons (2, 3 and 4) of the aromatic ring are appropriately designated pseudo‐axial (ax) and pseudo‐equatorial (eq).
All of the prior sustains that the preferably structure corresponded to a cis‐diastereomer (2 e, 4 e), thus leaving substituents on C‐2 and C‐4 in an eq position (pseudo‐equatorial). Stereochemistry analysis of the molecules was applied according to the endo principle of the iDA reaction. This was conserved for all of the synthesized molecules.
A NOESY spectrum of compound 4 b showed a spatial NOE effect between the aliphatic protons 2‐H and 4‐H. Furthermore, the relationship between these two protons (i. e. 2‐H and 4‐H) with the 3‐Ha proton (Heq) was confirmed by spatial proximity. At the same time, the 2‐H signal gives an additional NOE effect to the δ 5.70 ppm signal of 4‐H, which substantiates the information supplied by the coupling constants of the elucidated THQ cis conformation. Therefore, the arrangement of 2‐H and 4‐H protons was determined to be axial. Finally, the homonuclear COSY correlation spectrum (1H‐1H) complemented the complete characterization of the aromatic protons, thereby generating the correct assignment of the molecule under investigation. Consequently, the signals for 5‐H and 8‐H protons (multiple at 7.31 ppm) showed overlapping and were followed by the 4′‐H and 5′‐H proton signals at 7.19 and 7.66 ppm, which correlated together on the COSY spectrum, (see supplementary material).
2.2. Computational Studies
According to the experimental results, and taking into account previous iDA reaction studies (Domingo et al.13), which indicate that this reaction can proceed in a step‐wise pathway when LA‐catalyzed (Scheme 2), we decided to further study the mechanism for our reactions. Computational calculations were carried out using the density functional theory (DFT) with M06‐2X functional, 6‐311G** basis set,29 and a 2DZLAN pseudo potential for the bismuth (Bi). The computational calculations were implemented in Gaussian 09 software packages.30
Scheme 2.
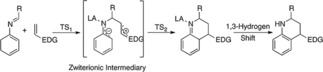
The simplified proposed theoretical mechanism for the iDA reaction.
iDA reaction between N‐arylimine (with a phenyl substituent) and N‐vinylformamide in the absence of catalyst and MeCN as the solvent, lead to an unsuccessful reaction without obtaining the expected product. Computer calculations confirmed this result due to excessive activation energy required to carry out this reaction, with energy values of 19.3 kcal/mol (TS1n) to the approach mode endo and 21.6 kcal/mol for exo (TS1x). These approaches endo and exo diene‐dienophile, are presented by the asymmetry of the starting reagents. From the activation energy values calculated, we could conclude that, although the reaction does not proceed efficiently in the absence of a catalyst, it could be a conformational preference for reactive channel endo (2.3 kcal/mol lower in energy) on the exo, demonstrating the high selectivity for this approach mode. The geometries associated to TS1n and TS1x are in Figure 3. The distance between C1 and C6 carbons atoms are of 1.970 and 1.922 Å for TS1n and TS1x, respectively. While, the length of the atoms involved in the process of closing the ring C4 and C5 has 2.375 and 2.412 Å values for TS1n and TS1x respectively, with asynchrony values of 0.41 in TS1n and 0.49 in TS1x Showing thus higher asymmetry in the exo approximation.
Figure 3.
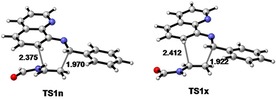
M06‐2X/6‐311G ** geometries, of the transition states of the iDA reaction between N‐quinoline imine and vinylformamide. Lengths are in Å.
Once the theoretical study of the iDA reaction without catalyst was verified, we proceeded to study through computer simulations the same process but in the presence of BiCl3, to establish the role of the LA catalyst in the IDA reaction. We determined the energetic and geometrical values for three specific reactions: a) with Ph substituent on the diene (1 a), b) with 2‐pyridine which was the highest percentage yield at experimental level (1 b); and finally, c) with 3‐pyridine which had lower percentage yield (1 c).
The computational results for the chemical reaction of these dienes in the presence of NVF as dienophile and BiCl3, indicated the [4+2] cycloadduct formation through two steps mechanism (Scheme 3). The first step went to a nucleophilic attack of the C6 dienophile (NVF) on the C1 complex 1a, through a molecular complex (MC 1 a, 1 b, 1 c) involving an exothermic energy activation of −9.3 kcal / mol for MC1a and −9.7 kcal/mol for MC1b (Scheme 3).
Scheme 3.
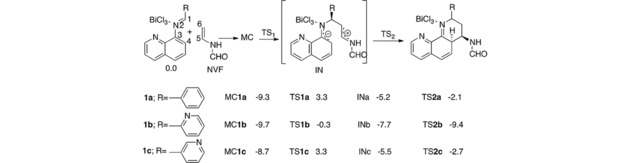
iDA reaction between N‐aryl imine and NVF using M06‐2X/6‐311G** level of theory. The relative energies are in kcal mol−1.29
The Activation energy associated with TS1a and TS1c is substantially equal to a value of 3.3 kcal/mol. By contrast, energy expense associated with TS1b corresponds to −0.3 kcal/mol, differing from the first two in 3.0 kcal/mol. It difference is probably given by Bi coordination with 3 nitrogen atoms of the molecule 4 b (represented as 1 b in the reaction mechanism)) which is more stable than compound 4 a (represented as 1 a in the reaction mechanism) and 4 c (represented as 1 c in the reaction mechanism) (Figure 4). The triple coordination of BiCl3 could provide great conformational stability in TS1b causing a decrease in the activation barrier, and accelerating the reaction velocity given by the increase the experimental percentage yield, of 72 % for 1 b over 1 a and 1 c (31 and 27 % respectively). In iDA reaction for the three compounds, the formation of a zwitterionic intermediate (IN) is presented with endothermic energy approximate of −5.2 kcal/mol for INa, −5.5 kcal/mol for INc, and less for INb (−7.7 kcal/mol) (Scheme 3). Finally, the theoretical studies showed that the molecular coordination between LA catalyst and nitrogen atoms allows the formation of a bond between C4 and C5 given by an attack of dienophile carbon (NVF) to the imine (Scheme 3). Thus the process of cycloaddition and ring closure is promoted, leading the formation of the products via transition structures TS2 with relative activation energies of −2.4 and −2.7 kcal/mol for TS2a and TS2c, significantly higher than required to TS2b (−9.4 kcal/mol); which confirms the data obtained experimentally in yield and reaction time that were lower for the latter molecule (4 b), compared with the other two in study.
Figure 4.
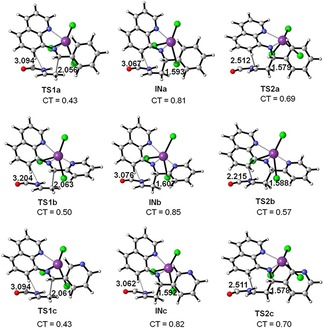
M06‐2X/6‐311G ** geometries of the optimized transition‐state structures involved in the BiCl3 catalyzed iDA reaction between N‐aryl imines 1 a, 1 b, 1 c and NVF. Lengths are in Angstroms Å. CT represents net charge transfer.
The results of activation energy for the formation of 1 a in the presence of BiCl3 catalyst is 16.9 kcal/mol more favorable than in the absence of this, which indicates that BiCl3 as LA could be in charge of coordinating the pair of nonbonding electron of imine group, facilitating interaction with the dienophile and thereby lowering the energy process. Finally, the iDA reactions were analyzed using the reactivity indices DFT‐based parameters.31 The values of reactivity index, particularly, chemical potential μ, hardnesses η, global electrophilicity ω, and global nucleophilicity N indices for the reagents involved in iDA reactions are in Table 2.
Table 2.
Reactivity index analysis: M06‐2X/6‐311G** level of theory. Electronic chemical potential μ, hardnesses η, global electrophilicity ω, and global nucleophilicity N indices, for the reagents and compounds involved in iDA reactions for phenanthroline‐THQ.
| Compounds | μ [a.u.] | η [a.u.] | ω [eV] | N [eV] | Δω |
|---|---|---|---|---|---|
| NVF | ‐0.1545 | 0.3273 | 0.99 | 2.91 | |
| 1 a | ‐0.1802 | 0.1988 | 2.22 | 2.71 | 1.23 |
| 1 b | ‐0.1848 | 0.1822 | 2.55 | 2.65 | 1.59 |
| 1 c | ‐0.1891 | 0.2251 | 2.16 | 2.62 | 1.17 |
The electronic chemical potential μ for NVF has a value of −0.1545 a.u., highest value that each of the complexes involved in the cycloaddition reaction, this value that indicates that along the polar reaction net charge transfer (CT) comes from the NVF towards each complex 1 a, 1 b and 1 c. Unlike of the molecular complexes with global electrophilicity (ω) values greater than 2.00 eV, the NVF or dienophile has values of ω of 0.99 eV which classify it as a moderate electrophile,25 while the molecular complexes are classified as strong electrophiles; especially 1 b complex that has the highest value, ω=2.55 eV. A change in the position of the pyridine nitrogen of two to three positions decreases the electrophilicity in 0.39 eV (1 c), which influence notably the activation energies and in experimental yields. The electrophilicity values of reagents indicate that the iDA reaction proceeds through of cycloaddition polar. This result suggests that the polarity of the process depends mostly on a high electrophilicity value at any of both fragments, thereby indicating that during an electrophile−nucleophile interaction the effect of electrophilicity outweigh the effect of nucleophilicity. This requisite of high electrophilicity value before the process can be labeled as polar may be traced to the role different that electrophiles and nucleophiles play in their mutual interactions. While the nucleophile is expected to be a good electron releasing molecule or fragment, a property mainly determined by a high value of its μ, the electrophilic group or molecule must be soft enough to accept the transferred charge and then to rearrange it within the electrophilic structure, a molecule in the case of intermolecular process, or a group in the case of an intramolecular process.
2.3. Biological Activity
Once studied the theoretical mechanism and chemically characterized the new phenanthroline‐THQs, we proceeded with the pharmacological research conducting the enzymatic assays against AChE and BuChE, due to the evident activity of some quinolinic analogs such as tacrine on AChE.32 The biological tests results described in Table 3 showed the moderate inhibitory activity of the phenanthroline‐THQ against one or both cholinesterases. Nitrogen‐containing compounds at position two, three and four of the phenyl rings (molecules 4 b, 4 c and 4 d), that acts as substituent in position two of the THQ, revealed a moderate inhibition in the μΜ range for AChE enzyme (143.66, 140.2, 63.63 μΜ for AChE, respectively) (table 3).
Table 3.
Determined AChE and BuChE inhibitory activities IC50 values compounds 4 a–4 g.
| Compound | AChE (IC50) μM | BuChE (IC50) μM |
|---|---|---|
| 8‐amino‐quinoline | 171.56±0.01 | 195.08±0.02 |
| 4 a | >500 | NA |
| 4 b | 143.66±0.02 | 152.99±0.01 |
| 4 c | 140.2±0.02 | NA |
| 4 d | 63.63±0.01 | 138.63±0.01 |
| 4 e | 362.11±0.03 | NA |
| 4 f | 208.07±0.02 | NA |
| 4 g | 96.52±0.01 | NA |
| Galantamine | 0.54±0.5 | 8.8±0.7 |
| Tacrine | 0.43±0.5 | 0.045±0.003 |
Each IC50 value is the mean±SEM of three independent measurements. 500 μM was the highest tested concentration. At higher concentrations the compounds precipitated.
The IC50 values obtained from the biological assays for these molecules revealed that the electron‐donating groups as nitrogen could induce effects of the activity, through the several interactions with the active site of the enzyme. The nitrogen present in pyridine molecule has high Lewis basicity,33 therefore, it can act as a base donating its electronic pair. The 4‐pyridine activity (4 d) could be related to the nitrogen electron pair are fully exposed and available for making some important interaction, while the nitrogen located in position two and three on pyridine could have a more steric hindrance to doing protein interaction. The explain previously described could be the cause by which molecule 4‐pyridine is the most active of the series synthesized against AChE and BuChE (63.63 μΜ and 138.63 μΜ, respectively), demonstrating its ability to act as a dual inhibitor against the two enzymes, but with selectivity towards AChE.
The compound 4 g, with an acceptor group of electrons (nitro group) at position four of the phenyl substituent, proved to have a good AChE inhibitory activity (IC50=96.52 μΜ), being entirely selective for this enzyme. These results indicate the importance of the kind of chemical group and their position; it the relationship is maintained in the compound 4 b, 4 c and 4 d, in which the activity was increased gradually according to the nitrogen position on the ring, being less active for the molecule with nitrogen at position two of the aromatic ring.
Three molecules were selected for further assays in cell‐based models, specifically in human neuroblastoma SH‐SY5Y cells to evaluate their cytotoxic and neuroprotective effects. This cell line is frequently used by their human origin, catecholaminergic neuronal properties, and their relationship with several neurodegenerative diseases.34 As shown in table 4, the evaluated compounds don't display negligible cytotoxicity to 1000 μM after 24 h incubations. Three of the compounds being assessed presented high cell viability at 10 μM, being non‐cytotoxic at effective concentrations, showing a tendency for neuroprotection at low concentrations (viability superior to 100 % at 10 μM). The compounds 4 d and 4 g showed good AChE activity and non‐cytoxicity, which is clear evidence of a new lead to optimizing to develop a drug for Alzheimer.
Table 4.
Cytotoxicity studies for a series of selected compounds.
| Compound | Cytotoxicity Cell viability (%) | IC50 [mM] | |
|---|---|---|---|
| 10 μM | 1 mM | ||
| 4 d | 132.5±25.6 | 102.3±18.9 | 5.5 |
| 4 e | 118.6±6.4 | 78.5±12.5 | 3.7 |
| 4 g | 116.4±5.0 | 21.9±3.9 | 0.3 |
The results correspond to the IC50 obtained when applying the function: log (inhibitor) vs normalized response. The analysis was performed with the program GraphPad Prism version 8.0.0 for Windows (GraphPad Software, La Jolla California, USA). A – Cell viability was, at all tested concentrations, higher than the control.
2.4. Molecular Docking
Molecular docking study was performed for compounds 4 d and 4 g to explain to molecular level the primary interactions with AChE. Docking studies were conducted using Maestro of Schrödinger and AChE enzyme (PDB ID: 4EY7).[35] As shown in figure 5 the compound 4 d the most active in the series, exhibited a π‐π stacking interaction between phenanthroline moiety and residue W286 to a distance of 3.36 Å, as well as one Hydrogen bond (H‐Bond) between the amine group of formamide moiety with Y341 to 1.91 Å. The residues W286 and Y341 are amino acids belonging peripheral anionic site (PAS) that are located at the entry to the active gorge and are responsible of several activities as the interaction with β‐amyloid.[36] [37] The amine group of THQ moiety forms an H‐Bond with the hydroxyl group of Y72, amino acid belonging to the PAS. The binding mode of the compound 4 d reveals a conformational disposition in the entry to the active site of the enzyme, with the pyridine moiety solvent‐exposed. In contrast, the compound 4 g showed another kind of interaction and different conformational disposition. Compound 4 g established firm cation‐π contact between NO2 group with F295 and Y124 to a distance of 2.55 and 2.37 Å respectively. Y124 belongs to PAS, while F295 is a residue of acyl pocket and together whit F297, prevents access of larger molecules to the catalytic center.38 In the molecule 4 g, the phenyl portion presented closeness to W286 interacting by π‐π staking to 3.74 Å, while the quinolone moiety is solvent‐exposed. All these contacts allow us to have an idea of the potential lead, as well as their pharmacological features and of how to improve them.
Figure 5.
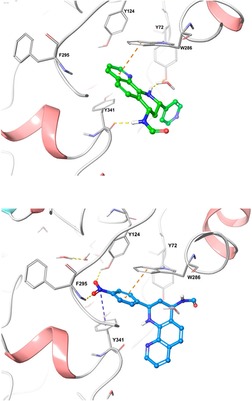
Binding molecular interactions for a). phenanthroline‐THQ 4 d (green), and b). phenanthroline‐quinoline 4 g (blue) into AChE binding site (PDB code: 4E7Y). New molecules are represented in sticks green and blue, while relevant AChE residues are in grey sticks. Orange dotted lines represent π‐π staking interactions between Quinolines and AChE enzyme. Yellow dotted lines represent Hydrogen bonds and blue dotted lines represent cation‐ π interactions.
2.5. In Silico Drug‐likeness Evaluation
We used QikProp as a tool for predictions of physicochemical properties based on cheminformatics data of structural fragments, moreover of the analysis of relationships between structure and property descriptors to determine some ADME properties. As shown in Table 5 all the physicochemical descriptors calculated for synthesized compounds are within the range set by Lipinski's Rule of Five, included the octanol/water partition coefficient (LogP)≤5. Besides, the bioavailability values showed that all molecules have values into the range for the predictive coefficient Blood/brain partition coefficient (QplogBB), which indicate an appropriate theoretical polarity to crossing the blood‐brain barrier. QplogBB is in according to results for the prediction of the apparent permeability of MDCK cells, which are considered a good imitator for the blood‐brain barrier (BBB), denoting values for a rapid BBB permeation, suggesting good brain permeation and CNS distribution. The presence of the basic pyridine moiety increases water solubility, according to low LogP values, lower values than for molecules without pyridine substituent. The LogP values also influence the percentage of human absorption, descriptor with values into the range of prediction. Taken these findings highlighted the potential of this class of compounds and particularly of compound 4 d as a potential lead for treatment of neurodegenerative diseases.
Table 5.
Drug and Bioactivity scores and the drug‐likeness properties calculated for molecules 4 a–4 g using the QikProp of Schrödinger Suite.
| Compound | LogPa | Molweight | Donor | Acceptor | QPlogBBb | QQPPMDCKc | CNSd | Percentage Human Oral Absorptione |
|---|---|---|---|---|---|---|---|---|
| 4 a | 2.632 | 303.363 | 2 | 4.5 | −0.459 | 539.84 | 0 | 92.14 |
| 4 b | 1.998 | 304.351 | 2 | 5.5 | −0.528 | 432.89 | 0 | 88.019 |
| 4 c | 1.757 | 304.351 | 2 | 6 | −0.759 | 262.08 | 0 | 81.62 |
| 4 d | 1.762 | 304.351 | 2 | 6 | −0.676 | 297.16 | 0 | 82.67 |
| 4 d | 2.714 | 333.389 | 2 | 5.25 | −0.474 | 605.89 | 0 | 93.60 |
| 4 d | 1.875 | 348.360 | 2 | 5.5 | −1.392 | 66.38 | −2 | 73.16 |
| 4 f | 2.865 | 317.390 | 2 | 4.5 | −0.433 | 590.56 | −2 | 94.30 |
| 4 g | 1.944 | 348.360 | 2 | 5.5 | −1.486 | 56.85 | −2 | 72.06 |
aLog P: Predicted octanol/water partition coefficient <5. bQPlogBB Predicted brain/blood partition coefficient: −3.0–1.2. cQPPMDCK Predicted apparent MDCK cell permeability in nm/sec. MDCK cells are considered to be a good mimic for the blood‐brain barrier: <500. dCNS Predicted central nervous system activity on a −2 (inactive) to +2 (active) scale. ePercent Human Oral Absorption: Predicted human oral absorption on 0 to 100 % scale.
3. Conclusions
In this study, a series of phenanthroline‐tetrahydroquinoline compounds were designed, synthesized, and their inhibitory activity AChE and BChE was evaluated. Reaction yields obtained experimentally were confirmed by Theoretical calculations, which revealed the differences in activation energy associated with the Bi coordination with nitrogen atoms of the molecules 4 a, 4 b, and 4 c. Combining enzymatic analysis and bioinformatic approaches, we identified two moderate AChE inhibitors as molecule 4 d (IC50=63.63 μM) and compound 4 g (IC50=96.52 μM), with the presence of 4‐pyridine ring and 4‐nitrophenyl ring respectively. These compounds no presented neurotoxicity according to studies on neuroblastoma SH‐SY5Y cells. The molecular docking studies revelated that interactions between protein‐ligands were given mainly by hydrophobic interactions and ionic contacts. Finally, studying the information obtained in this investigation, the compounds 4 d and 4 g could be considered as promising leads for the new design and development of therapeutics against AD.
Experimental Section
Chemistry
Melting points were measured using a Büchi apparatus and are uncorrected. The purity of compounds was checked by means of analytical TLC (Thin Layer Chromatography) on silica gel plates (Merck 60 F254) and they were purified by column chromatography, when were necessary. Chemicals were bought from Aldrich and used without further purification. FT‐IR spectra were recorded in potassium bromide pellets using a Thermo Nicolet NEXUS 670 FT‐IR spectrophotometer, with 0.125 cm−1 spectral resolution. 1H NMR (200 MHz) and 13C NMR (101 MHz) spectra were recorded in CDCl3 or DMSO‐d6, with a Bruker AMX or a Bruker AM‐400 spectrometers. High‐resolution mass spectrometry ESI‐MS and ESI‐MS/MS analyses were conducted in a high‐resolution hybrid quadrupole (Q) and orthogonal time‐of‐flight (TOF) mass spectrometer (Waters/Micromass Q‐TOF micro, Manchester, UK) with a constant nebulizer temperature of 100 °C. The samples were directly infused into the ESI source, via a syringe pump, at flow rates of 5 μL/min, via the instrument's injection valve.
General Procedure for the Synthesis Phenanthroline‐tetrahydroquinolines
A mixture of aromatic amine (3 mmol) and aromatic aldehydes with different substitution patterns (3 mmol) in anhydrous CH3CN (5 mL) under N2 were stirred at room temperature for 1 hour. After this time was added a solution of BiCl3 (30 % mol) in CH3CN. After 20 min under stirring, was added dropwise a solution of the N‐vinylformamide (NVF) (3.1 mmol) in CH3CN (5 mL). The resulting mixture was stirred for 8–15 hours, monitoring by TLC the reactants consumption. After the reaction was complete, the mixture was diluted with water and extracted with ethyl acetate (3×10 mL). The organic layer was separated and dried with anhydrous Na2SO4. The organic solvent was removed under vacuum and the resulting product was purified by column chromatography (silica gel, petroleum ether/ethyl acetate) to finally obtain the pure substances, phenanthroline‐THQs 4 a–4 g.
N‐(2‐phenyl‐1,2,3,4‐tetrahydro‐1,10‐phenanthroline‐4‐yl) formamide (4 a). Product was obtained from 8‐aminoquinoline (3 mmol), benzaldehyde (3 mmol), BiCl3 30 % and NVF (3.1 mmol). White solid; M.p. 227–229 °C; Yield, 31 %.1H NMR (200 MHz, CDCl3) δ 8.73 (dd, J=8.4, 3.4 Hz, 1H, 9‐H), 8.25 (s, 1H, CHO), 8.07 (dd, J=8.3, 1.6 Hz, 1H, 7‐H), 7.51–7.47 (m, 2H, 2′‐H y 6′‐H), 7.43–7.34 (m, 5H, 3′‐H, 4′‐H, 5′‐H, 5‐H y 8‐H), 7.11 (d, J=8.5 Hz, 1H, 6‐H), 6.44 (s, 1H, NH), 5.81‐5.69 (m, 1H, 4‐H), 5.53 (m, 1H, NH), 4.86 (dd, J=10.1, 2.6 Hz, 1H, 2‐H), 2.69–2.57 (m, 1H, 3‐H), 2.30–2.19 (m, 1H, 3‐H). 13C NMR (101 MHz, CDCl3) δ 156.41, 152.59, 150.50, 148.71, 142.79, 141.89, 135.99, 133.41, 129.60, 128.87, 126.98, 126.49, 121.65, 118.83, 114.56, 99.98, 65.99, 54.48, 29.38. MS (ESI, m/z): 304.27 ([M+H])+, 526.10 ([M+Na])+.
N‐(2‐(pyridin‐2‐yl)‐1,2,3,4‐tetrahydro‐1,10‐phenanthroline‐4‐yl) formamide (4 b). Product obtained from 8‐aminoquinoline (3 mmol), 2‐pyridine‐aldehyde (3 mmol), BiCl3 30 % and NVF (3.1 mmol). Yellow solid; M.p. 231–233 °C; Yield, 72 %.IR (cm−1): 3006, 1680, 1523, 1244, 757 cm−1.1H NMR (400 MHz, CDCl3) δ 8.68 (d, J =4.2 Hz, 1H, 9‐H), 8.58 (d, J=4.7 Hz, 1H, 3′‐H), 8.17 (s, 1H, CHO), 8.00 (d, J=8.1 Hz, 1H, 7‐H), 7.66 (t, J=7.7 Hz, 1H, 5′‐H), 7.41 (d, J=8.1 Hz, 1H, 6′‐H) 7.31 (m, 2H, 8‐H, 5‐H), 7.19 (m, 1‐H, 4′‐H), 7.01 (d, J=8.4 Hz, 1H, 6‐H) 6.67 (s, 1H, NH), 6.14 (d, 1H, NH), 5.65 (dd, J=14.7, 8.2 Hz, 1H, 4‐H), 4.91 (dd, J=8.3, 4.0 Hz 1H, 2‐H), 2.68–2.62 (m, 1H, 3‐H), 2.30‐2.22 (m, 1H, 3‐H). 13C NMR (101 MHz) δ: 161.49, 160.77, 149.51, 147.58, 140.75, 137.61, 137.07, 135.93, 128.13, 126.57, 122.61, 121.58, 120.80, 115.74, 114.59, 77.35, 77.03, 76.71, 55.19, 44.21, 34.97. MS (ESI, m/z): 305.19 ([M+H])+, 327.17 ([M+Na])+.
N‐(2‐(pyridin‐3‐yl)‐1,2,3,4‐tetrahydro‐1,10‐phenanthroline‐4‐yl) formamide (4 c). Product obtained from 8‐aminoquinoline (3 mmol), 3‐pyridinaldehyde (3 mmol), BiCl3 30 % and NVF (3.1 mmol). Cream solid; M.p. 110–112 °C, Yield, 27 %.1H NMR (200 MHz, DMSO‐d6) δ 8.70–8.59 (m, 3H, 9‐H, 2′‐H, 4′‐H), 8.30 (m, 1H, CHO), 8.09–8.00 (m, 1H, NH), 7.85–7.77 (m, 1H, 7‐H), 7.36–7.04 (m, 5H, 5′‐H, 6′‐H, 5‐H, 6‐H y 8‐H), 6.31 (m, 1H, NH), 5.71 (m, 1H, 4‐H), 4.87‐4.76 (m, 1H, 2‐H), 2.65‐2.07 (m, 2H, 3‐H). 13C NMR (101 MHz) δ: 164.27, 162.28, 155.70, 150.84, 148.29, 147.65, 139.77, 137.79, 136.64, 129.51, 129.14, 126.28, 126.22, 121.61, 114.50, 110.38, 57.85, 42.81, and 39.36. MS (ESI, m/z): 305.36 ([M+H])+, 327.18 ([M+Na])+.
N‐(2‐(pyridin‐4‐yl)1,2,3,4‐tetrahydro‐1,10‐phenanthroline‐4‐yl) formamide (4 d). Product obtained from 8‐aminoquinoline (3 mmol), 4‐pyridinaldehyde (3 mmol), BiCl3 30 % and NVF (3.1 mmol). White solid; M.p. 148–150 °C, Yield, 60 %.IR (cm−1): 3015, 1659, 1397, 1109, 743 cm−1. 1H NMR (200 MHz, CDCl3) δ 8.72 (d, J=4.5 Hz, 1H, 9‐H), 8.59 (d, J=4.7 Hz, 1H, 7‐H), 8.32 (s, 1H, CHO), 8.07 (d, J=8.2 Hz, 1H, 3′‐H), 7.83 (d, J=7.6 Hz, 1H, 5′‐H), 7.42–7.26 (m, 4H, 2′‐H, 5′‐H, 5‐H, 8‐H), 7.14 (d, J=8.5 Hz, 1H, 6‐H), 6.34 (s, 1H, NH), 5.72 (s, 1H, NH), 4.85 (d, J=10.0 Hz, 1H, 4‐H), 2.62 (d, J=13.2 Hz, 1H, 3‐H), 2.26–2.10 (m, 1H, 3‐H). 13C NMR (101 MHz, CDCl3) δ 161.03, 149.48, 148.55, 147.85, 141.14, 137.93, 136.01, 134.27, 127.85, 125.76, 123.71, 121.76, 115.58, 115.36, 77.33, 77.01, 76.70, 53.10, 44.90, 38.01. MS (ESI, m/z): 305.37 ([M+H])+.
N‐(2‐(4‐methoxyphenyl)‐1,2,3,4‐tetrahydro‐1,10‐phenanthroline‐4‐yl) formamide (4 e). Product obtained from 8‐aminoquinoline (3 mmol), 4‐methoxy‐benzaldehyde (3 mmol), BiCl3 30 % and NVF (3.1 mmol). White solid; M.p. 237–238 °C, Yield, 63 %. IR (cm−1): 3004, 1664, 1508, 1245, 747 cm−1.1H NMR (200 MHz, CDCl3) δ 8.72 (dd, J=4.2, 1.7 Hz, 1H, 9‐H), 8.28 (s, 1H, CHO), 8.09–8.04 (d, 1H, J=8.3 Hz 7‐H), 7.44‐7.27 (m, 4H, 2′‐H, 6′‐H, 5‐H y 8‐H), 7.10 (d, J=8.5 Hz, 1H, 6‐H), 6.94 (d, J=8.7 Hz, 2H, 3′‐H, 5′‐H), 6.37 (s, 1H, NH), 5.78–5.69 (m, 1H, 4‐H), 5.59–5.54 (m, 1H, NH), 4.80 (d, J=9.5 Hz, 1H, 2‐H), 3.84 (s, −OCH3), 2.64–2.54 (m, 1H, 3‐H), 2.21‐2.14 (m, 1H, 3‐H). 13C NMR (101 MHz, CDCl3) δ 161.18, 159.09, 154.77, 148.00, 145.37, 136.35, 127.71, 127.57, 127.38, 121.54, 120.87, 120.17, 114.20, 97.40, 55.34, 52.41, 46.42, 38.04.MS (ESI, m/z): 334.39 ([M+H])+, 356.17 ([M+Na])+.
N‐(2‐(4‐methyl)‐1,2,3,4‐tetrahidro‐1,10‐phenanthroline‐4‐yl)formamide (4 f). Product obtained from 8‐aminoquinoline (3 mmol), 4‐methyl benzaldehyde (3 mmol), BiCl3 30 % and NVF (3.1 mmol). Black solid; M.p. 207–209 °C yield, 61 %. 1H NMR (200 MHz, CDCl3) δ 8.72 (dd, J=4.2, 1.7 Hz, 1H, 9‐H), 8.26 (s, 1H, CHO), 8.07 (dd, J=8.3, 1.6 Hz, 1H, 7‐H), 7.42‐7.33 (m, 4H), 7.24–7.08 (m, 3H), 6.40 (s, 1H, NH), 5.81–5.68 (m, 1H, 4‐H), 5.55 (d, J=10.4 Hz, 1H, NH), 4.82 (dd, J=9.6, 2.5 Hz, 1H, 2‐H), 2.79–2.44 (m, 1H, 2‐H), 2.38 (s, 3H, −CH3), 2.27‐2.11 (m, 1H, 2‐H). 13C NMR (101 MHz, CDCl3) δ 164.27, 162.28, 155.70, 150.84, 148.29, 147.65, 139.77, 137.79, 136.64, 129.51, 129.14, 126.28, 126.22, 121.61, 114.50, 110.38, 57.85, 42.81, 39.36, 21.09.
N‐(2‐(4‐nitrophenyl)‐1,2,3,4‐tetrahydro‐1,10‐phenanthroline‐4‐yl) formamide (4 g). Product obtained from 8‐aminoquinoline (3 mmol), 4‐nitrophenyl (3 mmol), BiCl3 30 % and NVF (3.1 mmol). Yellow solid; M.p. 214–216 °C, Yield, 65 %. IR (cm−1): 2927, 1666, 1514, 1347, 749 cm−1. 1H NMR (200 MHz, CDCl3) δ 8.75 (dd, J=4.1, 1.4 Hz, 1H, 9‐H), 8.29–8.08 (m, 4H, 7‐H, 3′‐H, 5′‐H, CHO), 7.74–7.70 (m, 2H, 6′‐H, 2′‐H), 7.54 (dd, J=5.7, 3.3 Hz, 1H, ), 7.56‐7.34 (m, 2H, 5H, 8‐H), 7.17 (d, J=8.5 Hz, 1H, 6‐H), 6.47 (d, J=12.5 Hz, 1H, NH), 5.82–5.62 (m, 1H, 4‐H), 4.95 (d, J=10.1 Hz, 1H, 2‐H), 2.67–2.06 (m, 2H, 3‐H). 13C NMR (101 MHz, CDCl3) δ 160.96, 154.86, 147.94, 147.60, 140.83, 136.14, 130.88, 128.80, 127.68, 127.39, 125.76, 124.15, 124.06, 122.04, 121.86, 115.55, 54.64, 44.81, 38.69. MS (ESI, m/z): 349 ([M+H])+.
Computational Details
DFT computations were carried out using the M06‐2X functional, together with the 6–311G(d,p) basis set for C, H, O, N, Cl and LANL2DZ for Bi.29,39 The optimizations were carried out using the Berny analytical gradient optimization method.40 The stationary points were characterized by frequency computations in order to verify that TSs have one and only one imaginary frequency. The IRC paths41 were traced in order to check the energy profiles connecting each TS to the two associated minima of the proposed mechanism using the second order González−Schlegel integration method.42 Solvent effects of toluene and dichloromethane on the thermodynamic calculations were considered by using a self‐consistent reaction field (SCRF)43 based on the polarizable continuum model (PCM) of the Tomasi's group.44 All computations were carried out with the Gaussian 09 suite of programs.30 The global electrophilicity index, ω, is given by the following expression: ω=(μ2/2η), in terms of the electronic chemical potential μ and the chemical hardness η.25 Both quantities may be approached in terms of the one‐electron energies of the frontier molecular orbitals HOMO and LUMO, ϵH and ϵL, as μ≈(ϵH+ϵL)/2 and η≈(ϵL−ϵH), respectively. Recently, we introduced an empirical (relative) nucleophilicity index, N, based on the HOMO energies obtained within the Kohn−Sham scheme,45 and defined as N=EHOMO(Nu)−EHOMO(TCE). The nucleophilicity is referred to tetracyanoethylene (TCE), because it presents the lowest HOMO energy in a large series of molecules already investigated in the context of polar cycloadditions.
Molecular Docking
Computational calculations were performed using the Schrödinger's Small‐Molecule Drug Discovery Suite. The initial setup of AChE for calculations was prepared using Protein Preparation Wizard of Schrödinger46 to add hydrogens, assign bond orders, and generate rotamers and protonation states. All compounds were prepared using the software LigPrep while ionization/tautomeric states were predicted using Epik.47 The AChE crystal structure complexed with the Donepezil (PDB code 4EY7)35 was used for the docking experiments. The docking calculations using rigid‐receptor and flexible‐ligand were performed with Glide through the Extra Precision (XP) mode.48 Docking grid box was centered on the donepezil co‐crystallized. Optimization of the resulting enzyme‐ligand complexes was conducted using Prime.49
Biological Assays
AChE/BuChE Inhibitory Assay
This assay was performed in 96‐well plates, where 50 μL of sample were dissolved in phosphate buffer (it contain: 8 mM K2HPO4, 2.3 mM NaH2PO4, 150 mM NaCl, and 0.05 % Tween 20 at pH 7.6) and a solution of 50 μL of AChE/BuChE (0.25 unit/mL) from Electroporus electricus and bovine serum, respectively, in the same phosphate buffer, was added. The assay solutions, without substrate, were incubated with the enzyme for 30 min at room temperature. After incubation, the substrate was added. The substrate solution consisted of Na2HPO4 (40 mM), acetylthiocholine/butyrylthiocholine (0.24 mM) and 5,5′‐dithio‐bis‐(2‐nitrobenzoic acid) (0.2 mM, DTNB, Ellman's reagent). Absorbance of the yellow anion product, due to the spontaneous hydrolysis of substrate, was measured at 405 nm for 5 min on a Microtiter plate reader (Multiskan EX, Thermo, Vanta, Finland). The AChE/BuChE inhibition was determined for each compound. The enzyme activity was calculated as a percentage compared to a control sample using only the buffer and enzyme solution. The compounds were assayed in the dilution interval of 500 to 15 μg/mL, and the alkaloid galantamine and tacrine were used as the reference compounds. Each assay was run in triplicate and each reaction was repeated at least three independent times. The IC50 values were calculated by means of regression analysis. The main reason for using galantamine as a reference compound is given, because, just like THQ, this compound belongs to the family of alkaloids.
Neurotoxicity Assays
To determine cell viability, the Neutral Red method was used.50 The experiments were performed on the human neuroblastoma cell line SH‐SY5Y in passages 18 to 23. The cells were suspended in DMEM culture medium supplemented with 5 % fetal bovine serum (FBS). They were incubated (density of 2×104 cells/well in 96‐well plates) at 37 °C under conditions of 5 % CO2 and 100 % humidity for 24 h (confluence between 40–60 %). The blank was prepared with 100 μL of culture medium without FBS plus acid ethanol solution (wells without cells). Viability control: cells in 200 μL medium supplemented with 5 % FBS. Death control: cells in 200 μL medium with K+ (120 mM). The studied molecules were dissolved in DMSO at a concentration of 1×10−2 M (stock). All the solutions were prepared in DMEM medium with 5 % FBS, immediately before using (1×10−7 to 1×10−4). The molecules were added in medium (200 μL) for the cytotoxicity assay. The plates were incubated for 24 h more. Cell viability measurement using the neutral red technique was performed after a total of 48 h. The cells were washed with PBS, the neutral red solution was added and the plates were incubated for 3 h at 37 °C. Subsequently, the extraction of the dye was carried out by applying an acidified ethanol solution for 20 min, under stirring. Absorbance at 540 nm was measured in a plate reader (VarioSkan Flash, Thermo Fisher Scientific, Waltham, MA, United States).
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This project was supported by the project PIEI QUIMBIO UTALCA of Universidad de Talca. YD thank to Universidad Andrés Bello.
Y. Duarte, M. Gutierrez, R. Álvarez, J. H. Alzate-Morales, J. Soto-Delgado, ChemistryOpen 2019, 8, 627.
References
- 1.
- 1a. Hoemann M. Z., Xie R. L., Rossi R. F., Meyer S., Sidhu A., Cuny G. D., Hauske J. R., Bioorg. Med. Chem. Lett. 2002, 12, 129–132; [DOI] [PubMed] [Google Scholar]
- 1b. Diaz G., Miranda I., Sartori S., Dias G., Kohlhoff M., Purgato G., Nogueira M., J. Braz. Chem. Soc. 2018, 29, 2646–2656. [Google Scholar]
- 2.
- 2a. Coa J. C., Castrillon W., Cardona W., Carda M., Ospina V., Munoz J. A., Velez I. D., Robledo S. M., Eur. J. Med. Chem. 2015, 101, 746–753; [DOI] [PubMed] [Google Scholar]
- 2b. Madkour H. M. F., El-Hashash M. A. E.-A. M., Salem M. S., Mahmoud A.-S. O. A., Res. Chem. Intermed. 2018, 44, 3349–3364. [Google Scholar]
- 3. Dorey G., Lockhart B., Lestage P., Casara P., Bioorg. Med. Chem. Lett. 2000, 10, 935–939. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Gedawy E. M., Kassab A. E., El-Malah A. A., Med. Chem. Res. 2015, 24, 3387–3397; [Google Scholar]
- 4b. Köprülü T. K., Ökten S., Tekin Ş., Çakmak O., J. Biochem. Mol. Toxicol. 2019, 33, e22260. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Duarte Y., Gutierrez M., Astudillo L., Alzate-Morales J., Valdes N., Molecules 2013, 18, 12951–12965; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Tomassoli I., Ismaili L., Pudlo M., de los Rios C., Soriano E., Colmena I., Gandia L., Rivas L., Samadi A., Marco-Contelles J., Refouvelet B., Eur. J. Med. Chem. 2011, 46, 1–10. [DOI] [PubMed] [Google Scholar]
- 6. Andreani A., Burnelli S., Granaiola M., Guardigli M., Leoni A., Locatelli A., Morigi R., Rambaldi M., Rizzoli M., Varoli L., Roda A., Eur. J. Med. Chem. 2008, 43, 657–661. [DOI] [PubMed] [Google Scholar]
- 7. Mishra P., Kumar A., Panda G., Bioorg. Med. Chem. 2019. [DOI] [PubMed] [Google Scholar]
- 8. Mushtaq G., Greig N. H., Khan J. A., Kamal M. A., CNS Neurol. Disord. Drug Targets 2014, 13, 1432–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pan L., Tan J. H., Hou J. Q., Huang S. L., Gu L. Q., Huang Z. S., Bioorg. Med. Chem. Lett. 2008, 18, 3790–3793. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Bartolini M., Bertucci C., Cavrini V., Andrisano V., Biochem. Pharmacol. 2003, 65, 407–416; [DOI] [PubMed] [Google Scholar]
- 10b. Hossain T., Saha A., Mukherjee A., J. Biomol. Struct. Dyn. 2018, 36, 1274–1285. [DOI] [PubMed] [Google Scholar]
- 11. Davis K. L., Powchick P., Lancet 1995, 345, 625–630. [DOI] [PubMed] [Google Scholar]
- 12. Deraeve C., Pitie M., Mazarguil H., Meunier B., New J. Chem. 2007, 31, 193–195. [Google Scholar]
- 13.
- 13a. Najafi Z., Saeedi M., Mahdavi M., Sabourian R., Khanavi M., Tehrani M. B., Moghadam F. H., Edraki N., Karimpor-Razkenari E., Sharifzadeh M., Foroumadi A., Shafiee A., Akbarzadeh T., Bioorg. Chem. 2016, 67, 84–94; [DOI] [PubMed] [Google Scholar]
- 13b. Chen Y., Bian Y. Y., Sun Y., Kang C., Yu S., Fu T. M., Li W., Pei Y. Q., Sun H. P., PeerJ 2016, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Anand a.P., Singh B., Arch. Pharmacal Res. 2013, 36, 375–399; [DOI] [PubMed] [Google Scholar]; Cheewakriengkrai b.L., Gauthier S., Expert Opin. Pharmacother. 2013, 14, 331–338. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Bohn P., Le Fur N., Hagues G., Costentin J., Torquet N., Papamicaël C., Marsais F., Levacher V., Org. Biomol. Chem. 2009, 7, 2612–2618; [DOI] [PubMed] [Google Scholar]
- 15b. Ujan R., Saeed A., Channar P., Larik F. Ali, Abbas Q., Alajmi M., El-Seedi H., Rind M. Ali, Hassan M., Raza H., Seo S.-Y., Molecules 2019, 24, 860; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15c. Son M., Park C., Rampogu S., Zeb A., Lee K., Int. J. Mol. Sci. 2019, 20, 2–15; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15d. Duarte Y., Fonseca A., Gutiérrez M., Adasme-Carreño F., Muñoz-Gutierrez C., Alzate-Morales J., Santana L., Uriarte E., Álvarez R., Matos M. J., ChemistrySelect 2019, 4, 551–558. [Google Scholar]
- 16.
- 16a. Sultana N., Sarfraz M., Tanoli S. T., Akram M. S., Sadiq A., Rashid U., Tariq M. I., Bioorg. Chem. 2017, 72, 256–267; [DOI] [PubMed] [Google Scholar]
- 16b. Sarfraz M., Sultana N., Rashid U., Akram M. S., Sadiq A., Tariq M. I., Bioorg. Chem. 2017, 70, 237–244. [DOI] [PubMed] [Google Scholar]
- 17. Shah M. S., Najam-ul-Haq M., Shah H. S., Rizvi S. U. Farooq, Iqbal J., Comput. Biol. Chem. 2018, 76, 310–317. [DOI] [PubMed] [Google Scholar]
- 18. Sridharan V., Suryavanshi P. A., Menendez J. C., Chem. Rev. 2011, 111, 7157–7259. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Kouznetsov V. V., Mendez L. Y. V., Gomez C. M. M., Curr. Org. Chem. 2005, 9, 141–161; [Google Scholar]
- 19b. Manske R., Chem. Rev. 1942, 30, 113–144. [Google Scholar]
- 20.
- 20a. Kouznetsov V. V., Tetrahedron 2009, 65, 2721–2750; [Google Scholar]
- 20b. Gómez C. M. Meléndez, Marsiglia M., Escarsena R., del Olmo E., Kouznetsov V. V., Tetrahedron Lett. 2018, 59, 22–25. [Google Scholar]
- 21. Espinoza-Hicks C., Montoya P., Bautista R., Jiménez-Vázquez H. A., Rodríguez-Valdez L. M., Camacho-Dávila A. A., Cossío F. P., Delgado F., Tamariz J., J. Org. Chem. 2018, 83, 5347–5364. [DOI] [PubMed] [Google Scholar]
- 22. Lucchini V., Prato M., Scorrano G., Stivanello M., Valle G., J. Chem. Soc. Perkin Trans. 2 1992, 259–266. [Google Scholar]
- 23.
- 23a. Rohling R. Y., Tranca I. C., Hensen E. J. M., Pidko E. A., J. Phys. Chem. C 2018, 122, 14733–14743; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23b. Quijano-Quiñones R. F., Castro-Segura C. S., Mena-Rejón G. J., Quesadas-Rojas M., Cáceres-Castillo D., Molecules (Basel, Switzerland) 2018, 23, 2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Palacios F., Alonso C., Arrieta A., Cossío F. P., Ezpeleta J. M., Fuertes M., Rubiales G., Eur. J. Med. Chem. 2010, 2091–2099. [Google Scholar]
- 25. Domingo L. R., Aurell M. J., Perez P., Contreras R., Tetrahedron 2002, 58, 4417–4423. [Google Scholar]
- 26. Domingo L. R., Theor. Chem. Acc. 2000, 104, 240–246. [Google Scholar]
- 27. Domingo L. R., Aurell M. J., Saez J. A., Mekelleche S. M., RSC Adv. 2014, 4, 25268–25278. [Google Scholar]
- 28. Duarte L. F. B., Barbosa E. S., Oliveira R. L., Pinz M. P., Godoi B., Schumacher R. F., Luchese C., Wilhelm E. A., Alves D., Tetrahedron Lett. 2017, 58, 3319–3322. [Google Scholar]
- 29. Kenneth B. Wiberg, J. Comput. Chem. 1986, 7, 379–379. [Google Scholar]
- 30.M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, D. J. Fox, Gaussian 09, Revision B.01 2009.
- 31. Daniel H. Ess, Gavin O. Jones, Houk K. N., Adv. Synth. Catal. 2006, 348, 2337–2361. [Google Scholar]
- 32. Wong K. Y., Mercader A. G., Saavedra L. M., Honarparvar B., Romanelli G. P., Duchowicz P. R., J. Biomed. Sci. 2014, 21, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Guo P., Joo J. M., Rakshit S., Sames D., J. Am. Chem. Soc. 2011, 133, 16338–16341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xicoy H., Wieringa B., Martens G. J. M., Mol. Neurodegener. 2017, 12, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cheung J., Rudolph M. J., Burshteyn F., Cassidy M. S., Gary E. N., Love J., Franklin M. C., Height J. J., J. Med. Chem. 2012, 55, 10282–10286. [DOI] [PubMed] [Google Scholar]
- 36. Johnson G., Moore S. W., Curr. Pharm. Des. 2006, 12, 217–225. [DOI] [PubMed] [Google Scholar]
- 37. Inestrosa N. C., Alvarez A., Pérez C. A., Moreno R. D., Vicente M., Linker C., Casanueva O. I., Soto C., Garrido J., Neuron 1996, 16, 881–891. [DOI] [PubMed] [Google Scholar]
- 38. Bajda M., Wieckowska A., Hebda M., Guzior N., Sotriffer C. A., Malawska B., Int. J. Mol. Sci. 2013, 14, 5608–5632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hay P. J., Wadt W. R., J. Phys. Chem. 1985, 82, 270–283. [Google Scholar]
- 40. Schlegel H. B., J. Comput. Chem. 1982, 3, 214–218. [Google Scholar]
- 41. Fukui K., Acc. Chem. Res. 1981, 14, 363–368. [Google Scholar]
- 42. Gonzalez C., Schlegel H. B., J. Phys. Chem. 1990, 94, 5523–5527. [Google Scholar]
- 43. Rauhut G., Clark T., Steinke T., J. Am. Chem. Soc. 1993, 115, 9174–9181. [Google Scholar]
- 44. Mennucci B., Tomasi J., Cammi R., Cheeseman J. R., Frisch M. J., Devlin F. J., Gabriel S., Stephens P. J., J. Phys. Chem. A 2002, 106, 6102–6113. [Google Scholar]
- 45. Stowasser R., Hoffmann R., J. Am. Chem. Soc. 1999, 121, 3414–3420. [Google Scholar]
- 46.
- 46a.S. Schrödinger Release 2018-4: Schrödinger Suite 2018-3 Protein Preparation Wizard; Epik, LLC, New York, NY, 2018; Impact, Schrödinger, LLC, New York, NY, 2018; Prime, Schrödinger, LLC, New York, NY, 2018;
- 46b. Sastry G. M., Adzhigirey M., Day T., Annabhimoju R., Sherman W., J. Comput.-Aided Mol. Des. 2013, 27, 221–234. [DOI] [PubMed] [Google Scholar]
- 47.S. LigPrep, LLC, New York, NY, 2018.
- 48.
- 48a.S. Glide, LLC, New York, NY, 2018;
- 48b. Friesner R. A., Murphy R. B., Repasky M. P., Frye L. L., Greenwood J. R., Halgren T. A., Sanschagrin P. C., Mainz D. T., J. Med. Chem. 2006, 49, 6177–6196. [DOI] [PubMed] [Google Scholar]
- 49.
- 49a.S. Prime, LLC, New York, NY, 2018;
- 49b. Jacobson M. P., Pincus D. L., Rapp C. S., Day T. J., Honig B., Shaw D. E., Friesner R. A., Proteins 2004, 55, 351–367. [DOI] [PubMed] [Google Scholar]
- 50.
- 50a. Repetto G., del Peso A., Zurita J. L., Nat. Protoc. 2008, 3, 1125; [DOI] [PubMed] [Google Scholar]
- 50b. Borenfreund E., Puerner J. A., J. Tissue Cult. Methods 1985, 9, 7–9; [Google Scholar]
- 50c. Borenfreund E., Babich H., Martin-Alguacil N., Toxicol. in Vitro 1988, 2, 1–6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary